HEMATOLOGIA-Javier Perez Bracchigglione
72
Integrado de Sistemas I Javier Pérez Bracchiglione HEMATOLOGÍA Coordinador: Dr. Carlos Merino Javier Pérez Bracchiglione III Medicina Universidad de Valparaíso ÍNDICE Métodos de estudio y exploración hematológica………………………………………… 1 3 6 10 14 19 24 28 28 29 30 34 36 37 39 41 43 45 50 57 59 64 67 70
-
Upload
milton-william -
Category
Documents
-
view
3.037 -
download
2
description
Hematologia
Transcript of HEMATOLOGIA-Javier Perez Bracchigglione

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 1/72
Integrado de Sistemas IJavier Pérez Bracchiglione
HEMATOLOGÍACoordinador: Dr. Carlos Merino
Javier Pérez BracchiglioneIII Medicina
Universidad de Valparaíso
ÍNDICE
Métodos de estudio y exploración hematológica…………………………………………
1
3
6
10
14
19
24
28
28
29
30
34
36
37
39
41
43
45
50
57
59
64
67
70

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 2/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Inmunohematología y grupos sanguíneos………………………………………………..
Transfusión sanguínea……………………………………………………………………
Anemias en pediatría………………………………………………………………….......
Evaluación del paciente con Anemia……………………………………………………
Anemias Hemolíticas…………………………………………………………………......
Mieloptisis (anemia asociada a infiltración de la médula ósea)……………………….
Anemia de la Insuficiencia Renal Crónica…………………………………………….
Anemia Aplástica………………………………………………………………………..
Alteraciones del Hemograma…………………………………………………………...
Neutropenia……………………………………………………………………………
Enfermedades del Bazo…………………………………………………………………
Enfermedad de Hodgkin………………………………………………………………..
Linfoma No Hodgkin…………………………………………………………………..
Síndrome Purpúrico………………………………………………………………….....
Leucemia Aguda…………………………………………………………………………
Síndromes Mieloproliferativos Crónicos………………………………………………
Gammapatías Monoclonales……………………………………………………………
Leucemia Linfática Crónica……………………………………………………………
Evaluación y Tratamiento del paciente con Diátesis Hemorrágica…………………….
Trombofilia…………………………………………………………………………….
Anticoagulantes…………………………………………………………………………
Coagulación Intravascular Diseminada (CID)…………………………………………
MÉTODOS DE ESTUDIO
Y EXPLORACIÓN HEMATOLÓGICA
2
5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 3/72
Leucemia mieloide crónica = 4:1 / 3:1
Integrado de Sistemas IJavier Pérez Bracchiglione
MIELOGRAMA
BIOPSIA DE MÉDULA ÓSEA
CITOQUÍMICA
3
Examen citológico de Médula Ósea
Evalúa
Celularidad Médula ÓseaFierro en Médula Ósea (hemosiderina)
Infiltración en Médula Ósea
Causas de Anemia o Pancitopenia
Indicaciones
Bicitopenia / Pancitopenia
Anemia secundaria
Anemia refractaria o Mielodisplasia (para hacer dg dif con anemia megaloblástica)
Sd. mieloproliferativos crónicosLeucemia aguda (también se podría hacer inmunofenotipo y citogenética)
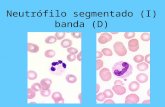
Mielogramanormal
Población celular heterogéneaPredominio seriegranulocítica (60% deltotal), predominandolos neutrófilos
(60% del total).Predominio de
neutrófilos
Muestra (-)Densidad celular elevada
Mielofibrosis
Metástasis
Leucemia
Examen histológico de Médula Ósea
Utilidad
Evalúa relación Mieloide/Eritroide
V.N. = 2:1
Evalúa tejido de sostén En Sd. mieloproliferativos crónicos está muy aumentado
Sd. mieloproliferativos crónicosIndicaciones
Anemia aplástica
Mielodisplasias
Sd. linfoproliferativos
Cáncer metastásico
TBC diseminada
Examen químico de Médula Ósea(No muy usado actualmente)
5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 4/72
Aplicaciones
Sd. linfoproliferaticos crónicos Diferencia Hodgkin de No Hodgkin
Leucemia aguda Diferencia Mieloide de Linfoide (99% sens. y especificidad)
Caracterización inmunológica de células plasmáticasEstudio de función de granulocitos
Integrado de Sistemas IJavier Pérez Bracchiglione
INMUNOCITOLOGÍA
Citometría de flujo
La inmunotipificación con anticuerpos monoclonales mediante citometría de flujo esespecífica de línea celular y de estado madurativo. Ejemplo:
Linfocito T: CD2, CD3, CD4, CD5, CD7, CD8Linfocito B: CD19, CD20, CD21, CD22
Antígenos de activación linfoide:
CD23, CD25, CD30
4
Evalúa
Enzimas oxidantes
Enzimas hidrolíticas
Compuestos inorgánicos
Tinción dePeroxidasa
Peroxidasa es propiade serie granulocítica
Leucemia aguda: Ayuda
a diferenciar origenmieloide de linfoide
Actividad deFosfatasa alcalina
Si hay recuentoaumentado de
blancos (neutrófilos)
Infección: Actividad aumentada
Tumor: Actividad disminuida
Reaccióncitoquímica de
PearlsTinción de fierroen médula ósea
Ausencia: Anemia Ferropriva
Exceso: Anemia Secundaria
Permitereconocer
Ag. de superficie
Ag. intracelulares
Ag. nucleares
Mediante anticuerposmonoclonales
Puederealizarse en
Sangre
Médulaósea
Presente en blastos y células T y B activadas
Utilizado en clínica para Linfoma células grandes B
Marcar células de Reed-Stenberg

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 5/72
Muestra de
Médula ósea (leucemias)
Linfonodos (linfomas)
Sangre periférica (leucemia linfática crónica)
Integrado de Sistemas IJavier Pérez Bracchiglione
Antígenos de diferenciación mieloide:CD13 (granulomonocítico)CD14 (monolítica)CD34 (Stem cell)
Antígenos marcadores de serie eritroide:CD71 (receptor de transferrina)
Antígenos marcadores de serie megacariocítica:CD41, CD61
CITOGENÉTICA
Alteraciones características:
1) Sd. mielodisplásico: 5q- ; 7q- ; +82) Leucemia mieloide crónica: t(9;22) Cromosoma Philadelphia3) Leucemia mieloide aguda: t(15;17)4) Leucemia linfoblástica aguda: t(12;21) / t(8;14) / 9p-5) Linfoma No Hodgkin:
a. Folicular: t(14;18)
b. Linfoplasmolítico: t(9;14)c. Del Manto: t(11;14)
BIOLOGÍA MOLECULAR
1) Hibridación in situ2) Reacción en cadena de la polimerasa (PCR): Detecta 1 célula maligna sobre
100.000 (el mielograma o la citogenética detectan 1:200)
INMUNOHEMATOLOGÍA Y
GRUPOS SANGUÍNEOS
5

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 6/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Los grupos sanguíneos están dados por los antígenos de membrana sobre las diferentescélulas y sobre los glóbulos rojos.
En total hay 400 antígenos sobre la superficie del glóbulo rojo. Cabe destacar que estosmismos antígenos se repiten en otras células, como son las plaquetas y los leucocitos.
Así, encontramos determinantes antigénicos de tipo eritrocitarios, plaquetarios,leucocitarios y séricos.
El ser humano en total tiene 23 grupos sanguíneos, de los cuales los más importantesson el ABO y el Rh; los otros antígenos no tienen mayor importancia desde el punto devista de la transfusión.
GRUPOS ERITROCITARIOS
Los antígenos y anticuerpos eritrocitarios toman importancia en: Reacción transfusional hemolítica Enfermedad hemolítica del recién nacido Transplante de órganos: Los antígenos de superficie de los grupos sanguíneos
están presentes en todas las células del organismo; tiene que ver con el sistemade histocompatibilidad. Si se transplanta, por ejemplo, un corazón, debe ser compatible desde el punto de vista ABO.
Anticuerpos eritrocitarios
Los anticuerpos eritrocitarios pueden ser IgM o, menos frecuentemente, IgG
IgM ABO IgG Rh
RegularesEspontáneos
6
Sistema Antígenos Reacción hemolítica
transfusionalABO A, B, AB, O SíRh D, C, c, E, e SíMNSs M, N, S, s SíLewis Le(a), Le(b) Muy raroLutheran Lu(a), Lu(b) RaroKell K, k, Kp SíDuffy Fy(a), Fy(b) SíKidd Jk(a), Jk(b) Sí
Grupos sanguíneos mayores
Grupos sanguíneos menores
Anti-ABO

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 7/72
Integrado de Sistemas IJavier Pérez Bracchiglione
o NaturalesIrregulares
Aloanticuerpos
(reconocen hematíesajenos)
Adquiridoso Inmunes
Anticuerpos
Autoanticuerpos(reconocen hematíes propios)
IgM: - Aglutinan los hematíes- Provocan lisis por activación del complemento- Se produce, por tanto, hemólisis intravascular. Clínicamente esto se expresacomo hemoglobinuria.
IgG: - Se fija a la membrana del glóbulo rojo- No provocan aglutinación ni activan el complemento en forma completa.- Por tanto, producen una hemólisis extravascular, o sea, es el sistemamononuclear fagocítico el que destruye al glóbulo rojo. Clínicamente esto seexpresa como hiperbilirrubinemia de predominio indirecto.
Antígenos eritrocitarios
1) Sistema ABO
7
Anti-Rh
Anti-LewisIgM
IgG
Por tanto:
ANTICUERPOS ANTI-ABO SON ESPONTÁNEOS O NATURALES,REGULARES, DEL TIPO IgM
(sin sensibilización previa; no atraviesan barrera placentaria)
ANTICUERPOS ANTI-Rh SON ADQUIRIDOS O INMUNES, DE TIPO IgG(con sensibilización previa; atraviesan barrera placentaria)

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 8/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Es el más importante en la práctica transfusional: Si se hace una transfusiónincompatible, se provoca la reacción hemolítica más severa.
Los genes A y B son codominantes. Los antígenos del sistema ABO aparecen entre el
3° y 6° mes de vida. Estos antígenos están ampliamente distribuidos en todos lostejidos, por lo que son parte importante del sistema de histocompatibilidad.
Grupo Sanguíneo Ag Hematíe Ac en SueroA A Anti-BB B Anti-AAB A y B -O - Anti-A y Anti-B
En el sistema ABO el fenotipo A presenta dos subgrupos: A1 y A2.
2) Sistema Rh
Los antígenos del sistema Rh son cinco: C, c, D, E, e (no existe un antígeno “d”),siendo el D el más inmunogénico. Por esta razón, recordemos, es que al hablar de Rh(+)nos estamos refiriendo a alguien D(+), y al hablar de Rh(-) nos estamos refiriendo aalguien D(-). En lo que respecta a este sistema, a los donantes se les estudia sólo la
presencia del antígeno D.
Como regla, nunca se debe transfundir a un receptor Rh(-) con glóbulos rojos Rh(+),sobre todo en mujeres jóvenes (pensando en una futura transfusión o embarazo). Un
problema se nos presenta en el siguiente ejemplo: Hay pacientes politraumatizadosgraves, en shock hipovolémico por hemorragia masiva, que son Rh(-). El banco de
8
Donante universal de Plasma: AB
Receptor universal de
Plasma: O
Receptor universal de
Glóbulos rojos: AB
Donante universal de
Glóbulos rojos: O
A1 80% de los individuos
A2 El 2% presenta anticuerpos Anti-A1
A2B El 25% presenta anticuerpos Anti-A1
Estos anticuerpos seríannaturales irregulares

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 9/72
Integrado de Sistemas IJavier Pérez Bracchiglione
sangre, en ese momento, tiene disponible sólo Rh(+). En este caso se puede transfundir al paciente Rh(-) con sangre Rh(+), pero será la única vez que se pueda transfundir, yaque luego el paciente sintetizará los anticuerpos correspondientes.
SISTEMAS ANTÍGENOS LEUCOCITARIOS Y PLAQUETARIOS
Plaquetas y leucocitos presentan también en su membrana antígenos del sistema ABO.Son parte del sistema HLA (Human Leukocyte Antigens) o sistema dehistocompatibilidad mayor.
Existen dos tipos de antígenos HLA:
HLA-I: A, B, C
En membrana de casi todas las células (excepto hematíes)
HLA-II: DR, DP, DQ
Linfocitos B Macrófagos Células dendríticas
Células de Kupffer Algunas células endoteliales
La función del sistema HLA es presentar péptidos en los que las células fraccionan losantígenos extraños. Así, el HLA-I presenta antígenos intracelulares (ej: virales), blancode células citotóxicas, mientras que el HLA-II presenta antígenos extracelulares (ej:
bacterianos), procesados por células presentadoras de antígenos.
El sistema HLA tiene importancia en la transfusión de plaquetas. Al transfundir plaquetas a una persona reiteradamente, se pueden producir anticuerpos contra el
sistema HLA y se pueden rechazar las plaquetas. Se debe considerar también al sistemaHLA cuando aparece la reacción transfusional no hemolítica febril. Esto se debe en el60% de los casos a anticuerpos anti-HLA pero que están presentes en los neutrófilos. El
paciente presenta fiebre y calofríos que ceden al detener la transfusión.
Por su parte, las plaquetas presentan dos tipos de antígenos: Antígenos no específicos: Sistema ABO, HLA-A y HLA-B Antígenos específicos, tales como las lipoproteínas 2A, 3A, 3B, etc.
TRANSFUSIÓN SANGUÍNEA
9

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 10/72
Se puede transfundir:
- Sangre total- Glóbulosrojos- Concentrados de plaquetas- Plasmafresco congelado- Plasma de
banco- Crioprecipitado
Integrado de Sistemas IJavier Pérez Bracchiglione
Es el traslado de elementos formes de la sangre o plasma, desde un donante a unreceptor.
Al transfundir glóbulos rojos de un donante a un receptor, sólo tomando en cuenta quesean del mismo grupo ABO, se tiene un 97% de probabilidades de que no existan
problemas inmunitarios graves. Si agregamos un estudio del grupo Rh D, lacompatibilidad será del 98%. El 2% presenta incompatibilidad dado que tienenanticuerpos séricos dirigidos hacia subgrupos menos importantes antigénicamente.
Estudios al Donante Estudios al Receptor - Encuesta detallada- Compatibilidad grupos ABO y Rh- Estudio serológico (buscando VIH 1 y2, Hepatitis B y C, HTLV-1, protozoos)
- Correcta identificación del paciente- Formulario de solicitud de transfusión- No deben existir discrepancias entre lainformación de los tubos y la solicitud
1.- Sangre total:
Recolección completa de una donación individual o “unidad” de 450 mlaproximadamente en una solución anticoagulante. La desventaja es que tiene granulocitosy plaquetas ineficientes, pero que aún conservan su potencial antigénico, pudiendo dar reacciones adversas en el receptor. Además, tiene baja cantidad de factores V, VIII ycomplemento, y tiene un hematocrito bajo (35% - 45%). Así, la única indicación para dar sangre total es la pérdida aguda masiva de sangre.
2.- Glóbulos rojos:
De la unidad de sangre se elimina la mayor parte del plasma y quedan los glóbulos rojossolamente, dando un hematocrito de 55% a 75% app. Dentro de las indicaciones paratransfundir glóbulos rojos se incluyen:• Mejorar la capacidad de transporte de O2 a los tejidos en un corto plazo• Concentración baja de hemoglobina• Pérdida de sangre por traumatismo o cirugía (transfundir glóbulos rojos juntocon expansores plasmáticos antes que sangre total)• Necesidad urgente de corregir una anemia severa• Insuficiencia medular: La indicación clave es transfusión de glóbulos rojos• Anemias congénitas, anemias aplásticas, anemias sideroblásticas
10
Elementos figurados
Plasma
Glóbulos rojos
Glóbulos blancos
Plaquetas
Proteínas plasmáticas
Agua
Sangre

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 11/72
Integrado de Sistemas IJavier Pérez Bracchiglione
• Hay situaciones que pueden ser manejadas con tratamiento médico, pero queeventualmente podrían llegar a requerir transfusión de glóbulos rojos, como la anemiaferropriva y la insuficiencia renal.
Con cada unidad de glóbulos rojos transfundida, se espera un aumento de 1 gr/dl de
hemoglobina. Por tanto, es mala práctica dar en el adulto la indicación de menos de 2unidades. En los niños se ocupa normalmente 10 a 20 ml por kg de peso.
Problemas con la transfusión de glóbulos rojos:
• Inmediatos:- Sobrecarga circulatoria- Hiperkalemia (el potasio sale de los hematíes que se dañan)- Reacción hemolítica, con fiebre, taquicardia, dolor lumbar, intranquilidad,rigores, vómitos, diarrea, cefalea, disnea, hipotensión, shock e insuficienciarenal aguda. Esto sobre todo en caso que no se identifique bien al donante o alreceptor - Reacciones no hemolíticas de tipo alérgicas, generalmente a proteínas del
plasma o a antígenos leucocitarios y plaquetarios, apareciendo urticaria yfiebre principalmente.
• Mediano plazo:- Flebitis local- Infección transmitida por la transfusión
• Largo plazo:
- Pacientes dependientes de transfusiones presentan sobrecarga de hierro yterminan con una hemosiderosis, lo cual daña páncreas, hígado y corazónentre otros.
Otros preparados de glóbulos rojos:
• Glóbulos rojos pobre en leucocitos: Para pacientes sensibilizados a antígenosleucocitarios o plaquetarios, para los que son candidatos a transplante renal o de médulaósea, y en especial para inmunodeficientes, para disminuir el riesgo de transmisión deCMV• Glóbulos rojos lavados. Para pacientes sensibilizados a proteínas del plasma o que
presentan hemoglobinuria paroxística nocturna.• Glóbulos rojos congelados y descongelados: En Chile no se usan
3.- Plaquetas: Se pueden obtener de 2 formas:
1. Obteniendo una bolsa de 30 a 40 ml de plasma, con la mayor parte de las plaquetas que tenía la bolsa de sangre del donante. Así, se requerirían variosdonantes para un número suficiente de plaquetas para un receptor
2. Conectar al donante a un equipo de aféresis, el cual saca sangre y va
separando los elementos formes del plasma, obteniéndose así los elementosdeseados de esta centrifugación. Por tanto, de un sólo donante se pueden extraer el equivalente a 6 u 8 unidades de plaquetas
11

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 12/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Características del concentrado plaquetario:- 50 a 70 ml por unidad- Número: 0,5 a 1,1 * 1011 / litro- Vida media es de 5 días app.
- No pueden ser congeladas. Deben guardarse en un agitador y a temperaturaambiente para que no se aglutinen.- Al estar a temperatura ambiente, tienen el riesgo de transmitir una posible
bacteremia del donante al receptor.
La idea de transfundir plaquetas es, en general, mantener un recuento de plaquetas sobre20.000/μl, para así prevenir hemorragias graves. Se requieren con más frecuencia encasos de pacientes con trombocitopenia asociada a infección, hemorragia, CID,esplenomegalia, aloanticuerpos, anticuerpos antiplaquetarios y anti-HLA (idealmente las
plaquetas deben ser del mismo grupo ABO y Rh del paciente), ya que son situaciones quedisminuyen la vida media de las plaquetas.
Se requiere una unidad (obtenida de una unidad de sangre) por cada 10 kg. paraaumentar las plaquetas en 20.000/ μl, siempre y cuando el paciente no tenga ninguna otracondición asociada.
Indicaciones:• Falla medular • Anomalías de la función de las células (en especial en algunas trombopatíascongénitas)• Pacientes que reciben una transfusión masiva por hemorragia si ésta es más de 2
veces su volumen sanguíneo, ya que ocurre dilución de las plaquetas.• Operación con bypass cardiopulmonar.• En pacientes con PTI no hay indicación de transfusión de plaquetas, a menos quehaya riesgo de muerte (por ejemplo, si hay hemorragia intracraneana).
Complicaciones:• Refractariedad, es decir, a pesar de la transfusión no hay un aumento de las
plaquetas, probablemente porque ya se montó una respuesta de anticuerpos.• En pacientes extremadamente inmunodeprimidos, puede darse una enfermedad deinjerto contra el huésped
4.- Plasma fresco congelado:
Se obtiene por separación de plasma de sangre total o por plasmaféresis, congelandoluego lo más rápido posible, para evitar la pérdida de los factores lábiles V y VIII.
Se usa en general para corregir trastornos de coagulación como deficiencia congénita defactores de coagulación o exceso de dosis de TACO. El plasma fresco congelado estácontraindicado como expansor plasmático.5.- Crioprecipitado:
12

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 13/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Se obtiene descongelando el plasma fresco congelado a 4 – 6 °C. Al hacer esto, vaquedando un botón flotando dentro del plasma; a esto le llamamos crioprecipitado, siendoel resto plasma de banco.
El plasma de banco es pobre en factor VIII, fibrinógeno y factor Von Willebrandt, pero
tiene el resto de los componentes del plasma, por lo cual se puede utilizar por ejemplo endeficiencias de factor V o IX. El crioprecipitado, por su parte, tiene en altasconcentraciones todo lo que le falta al plasma de banco: Factor VIII, Von Willebrandt,factor XIII, fibrinógeno y fibronectina.
Complicaciones inmunológicas de la transfusiónGlóbulos Rojos Reacciones hemolíticas inmediatas o
retardadasLeucocitos Reacciones febriles, daño pulmonar
agudo
Plaquetas Púrpura post transfusiónProteínas plasmáticasnativas
Anafilaxia
Ingeridas Fiebre, urticaria
Complicaciones infecciosas de la transfusión
Bacterianas: Raras, asociadas a la transfusión de plaquetasVirales: CMVParasitarias: Chagas
ANEMIAS EN PEDIATRÍA
13
5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 14/72
ClasificaciónFisiopatológica
Anemia aneritroblástica congénita
IRC
Enfermedades endocrinas (hipotiroidismo)
Integrado de Sistemas IJavier Pérez Bracchiglione
Se define como la disminución bajo 2 desviaciones estándar de la hemoglobina o delhematocrito, según sexo, edad y condición fisiológica (es más precisa la hemoglobina).
HB ( G/ DL ) HTO
MEDIA - 2 DS MEDIA -2 DSNACIMIENTO(SG CORDON)
16.5 13.5 51 42
1-3 DÍAS 18.5 14.5 56 451 SEMANA 17.5 13.5 54 422 SEMANAS 16.5 12.5 51 391 MES 14.0 10.0 43 312 MESES 11.5 9.0 35 283-6 MESES 11.5 9.5 35 290.5 – 2 AÑOS 12.0 11,0 36 33
2 – 6 AÑOS 12.5 11.0 37 347 – 12 AÑOS 13.5 11.5 40 3513 – 18 AÑOS♀
14.0 12.0 41 36
13 – 18 AÑOS♂
14.5 13.0 43 37
A) DEFECTOS DE LA PRODUCCIÓN
1.- Alteración proliferación / maduración célula madre
• Alteración célula troncal Pancitopenia Anemia aplástica
• Alteración progenitor eritroide
2.- Alteración de la maduración eritroide
14
Defectos en la producción
Alteración en la capacidad de proliferación y maduración de célulasmadres hematopoyéticas
Alteración en la maduración eritroide
Mecanismos múltiples o desconocidos
Anemias por destruccióno pérdida de glóbulosrojos
Anemias hemolíticas
Anemias por hemorragia

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 15/72
Falta de fierro
Síndromes Talasémicos
Intoxicación por Plomo
Esferocitosis familiar hereditaria
Deficiencia Glucosa-6-fosfatodeshidrogenasa; Defecto Piruvatokinasa
Defectos en Hemoglobina (Ejemplo:Drepanocitosis en raza negra)
Globinuria paroxística nocturna
Genéticas
ÚnicaAdquirida
Alteraciones inmunitariasPasivas (Ej: Lactancia)
Activas
Toxinas (Ej: Loxocelismo)
Infecciones
Fenómenos microangiopáticos (Ej: CID)
Integrado de Sistemas IJavier Pérez Bracchiglione
• Alteración nuclear Falta de vitamina B12 o de ácido fólico
• Alteración maduración citoplásmica
3.- Mecanismos múltiples o desconocidos
• Anemia de enfermedades crónicas
• Anemia asociada a infiltración medular por tumores
• Anemia asociada a carencia nutricional proteica
•
Anemias sideroblásticas
B) ANEMIAS POR DESTRUCCIÓN O PÉRDIDA DE GLÓBULOS ROJOS
1.- Anemias Hemolíticas
• Anomalías intrínsecas
• Anomalías extrínsecas
2.- Anemias por Hemorragia
15
Clasificación segúnMorfología y Tamaño
Microcíticas Hipocromas
Macrocíticas
Normocíticas normocromas
Con anormalidades morfológicas específicas

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 16/72
Integrado de Sistemas IJavier Pérez Bracchiglione
A) ANEMIAS MICROCÍTICAS HIPOCROMAS
• Falta de Fierro: - Nutricional en niños pequeños- Problema de absorción- Hemorragia
• Síndromes Talasémicos: Enfermedad genética de la producción de cadenas deglobina
• Anemias Sideroblásticas: Fierro se acumula en eritroblastos, habiendo undefecto en su utilización
• Anemia secundaria a inflamación crónica o a intoxicación por Plomo
B) ANEMIAS MACROCÍTICAS
• Con Médula Megaloblástica: Defecto madurativo del núcleo de eritroblastos y también de otras
series, muy evidente morfológicamente Ejemplo: Carencia de vitamina B12 o de ácido fólico.
Más raro: Secundario a alteraciones congénitas o adquiridas de lasíntesis de DNA
• Sin Médula Megaloblástica: Enfermedad Hepática Hipotiroidismo Mielodisplasia Comienzo de Anemia Aplástica Drogas
C) ANEMIAS NORMOCÍTICAS NORMOCRÓMICAS
• Infecciones agudas• Anemias aplásticas• Por infiltración medular • Anemias hemolíticas (ojo: reticulocitos pueden dar un VCM elevado)• Hemorragia reciente• Enfermedades crónicas: Renales, endocrinas, inflamatorias, neoplásicas
D) ANEMIAS CON ANORMALIDADES ESPECÍFICAS
16
Niño: VCM < 74 HCM < 25Adulto: VCM < 80 HCM < 27
VCM > 100 fl

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 17/72
Esferocitosis familiar hereditaria
Anemia hemolítica mediada por anticuerpos
Glóbulos rojos fragmentadoscuando se rompen
mecánicamente
Anemias microangiopáticas
SHU
PTT
CID
HTA maligna
Talasemia
Enfermedad hepática
Pre-Parto: Hemorragia feto-materna ofeto-fetal
Examen Físico:
- Palidez de piel y mucosas- Taquicardia, soplos cardiacos
- Hepatoesplenomegalia- Signos de enfermedad subyacente- Desarrollo pondo-estatural alterado
Laboratorio:
- Hematocrito, hemoglobina, recuentoreticulocitario
- VCM (Hto / rcto. GR * 100)- CHCM (Hb / Hto * 100)
- HCM (Hb / rcto GR * 100)- Estado de las otras series
hematológicas- Frotis
Anamnesis:
- Astenia, adinamia, angor,orina oscura, ictericia
- Fatigabilidad- Irritabilidad- Cefaleas, mareos- Anorexia, Pica
- Antecedentes familiares de ictericia- Antecedentes alimentarios- Infecciones- Alteraciones digestivas- Exposición a drogas o tóxicos
DIAGNÓSTICO DE ANEMIA
Índice reticulocitario:
Integrado de Sistemas IJavier Pérez Bracchiglione
Esferocitos
Eliptocitos Si hay más de un 20% corresponde habitualmente a una Esferocitosis
familiar hereditaria
Esquistocitos
Glóbulos rojos falciformes (forma de plátano) Pacientes con Hb S (raza negra)
Dianocitos (target cells)
17
Rcto. reticulocitos x Hto. actualHto. deseado
1% en una persona sana promedio
En anemia:> 2% Médula ósea funcionando
5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 18/72
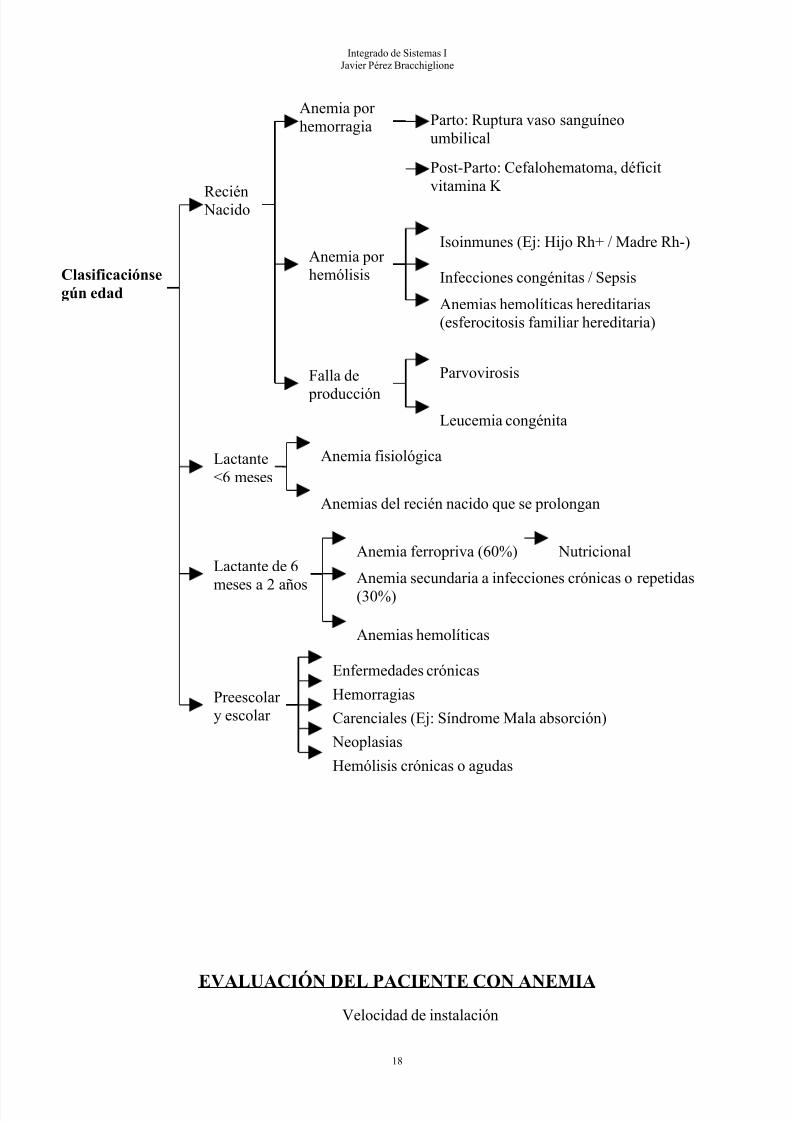
Clasificaciónsegún edad
Recién Nacido
Anemia por hemorragia Parto: Ruptura vaso sanguíneo
umbilical
Post-Parto: Cefalohematoma, déficitvitamina K
Velocidad de instalación
Integrado de Sistemas IJavier Pérez Bracchiglione
EVALUACIÓN DEL PACIENTE CON ANEMIA
18
Anemia por hemólisis
Isoinmunes (Ej: Hijo Rh+ / Madre Rh-)
Infecciones congénitas / Sepsis
Anemias hemolíticas hereditarias(esferocitosis familiar hereditaria)
Falla de
producción
Parvovirosis
Leucemia congénita
Lactante<6 meses
Anemia fisiológica
Anemias del recién nacido que se prolongan
Lactante de 6
meses a 2 años
Anemia ferropriva (60%) Nutricional
Anemia secundaria a infecciones crónicas o repetidas(30%)
Anemias hemolíticas
Preescolar y escolar
Enfermedades crónicas
Hemorragias
Carenciales (Ej: Síndrome Mala absorción)
Neoplasias
Hemólisis crónicas o agudas

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 19/72
Síntomas dependen de Edad y sexo
Presencia de enfermedades intercurrentes
Integrado de Sistemas IJavier Pérez Bracchiglione
ANEMIA FERROPRIVA
Descenso de la Hemoglobina secundario a baja en la concentración de fierro en elorganismo.
Clínica:
• Síndrome anémico (Palidez, fatigabilidad, disnea, angor, debilidad, taquicardia ysoplos cardiacos, signos de IC, curva ponderal inadecuada)
• Pica• Glositis, lengua repapilada, queilitis angular • Coiloniquia (casos severos)
Labortatorio:
19
Distribución de Fierro en el cuerpo:
Fierro en Hb 67% (2000 mg)
Fierro en depósitos
27% (1000 mg)Fierro mioglobina 3,5%Pool lábil 2,2%Fierro otros tejidos 0,2%Fierro de transporte 0,08%
Fuentes de Fierro:
Ricas: Hígado, legumbres (Fierroinorgánico)
Medianos: Carnes, pescado, ave
Ausentes: Fruta
Metabolismo del Fierro:
Equilibrio ingesta / excreción (ante un déficitde fierro, sube la absorción y baja la
excreción)
Fierro es absorbido en duodeno Requiere pH ácido
Fierro para glóbulos rojos proviene denuestro propio pool. La ingesta (1-2 mg/día)es para compensar las pérdidas
Causas de Anemia Ferropriva:
1) Pérdida Sanguínea:- Gastrointestinal
- Ginecológica
2) Ingesta inadecuada(vegetarianos)
3) Absorción defectuosa:- Gastrectomía- Síndrome Mala absorción

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 20/72
Alta
Baja
Benigna
Maligna
Integrado de Sistemas IJavier Pérez Bracchiglione
Hemograma: Anemia arregenerativa
• Microcitosis (bajo VCM)• Hipocromía (baja HCM)• Eliptocitos• Anisocitosis (RDW aumentado)• Leucocitos normales; Plaquetas normales o aumentadas
Estudio del Fierro:• Ferremia (transferrina con fierro): Baja (V.N: 70 - 140 μg/dl)• UIBC (transferrina sin fierro): Alta• TIBC (Ferremia + UIBC): Alta (V.N: 250 - 350)• % saturación transferrina: Bajo (V.N:20% - 45%)• Ferritina sérica (fierro depositado): Baja (V.N: 15 – 200 μg/dl)
Mielograma:• Hiperplasia serie roja (por aumento de la eritropoyetina)• Hemosiderina medular ausente
Estudio de etiología:
• Causa: Probablemente hemorragia digestiva
• Naturaleza
Exámenes para determinar el sitio de la hemorragia digestiva:- Test de hemorragia oculta en deposiciones (90% sensibilidad)- Endoscopía alta- Colonoscopía- Tránsito intestinal- Cintigrama con glóbulos rojos marcados
Causas de fracaso de tratamiento:- No ingiere medicamentos
20
Tratamiento:
1) Sulfato ferroso: Ferro F, Iberol
105 mg Fierro elemental, 1-2 tabletas/día
2) Fumarato ferroso: Confer, Ferramin
109 mg Fierro elemental, 1-2 tabletas/día
3) Protein-succilinato ferroso: Fisiofer, Legofer
40 mg Fierro elemental, 15 ml/día
Duración: 3-6 meses No olvidar tratar enfermedad de base
Criterios de Respuesta:
Laboratorio:
- Normalización ferritina sérica:2 a 3 semanas- Normalización Hto: 6 semanasa 2 meses.
Clínicos:- Desaparición síntomas: 3 a 7días- Desaparición depapilaciónlingual: 3 meses- Desa arición coiloni uia: 3 a 6
5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 21/72
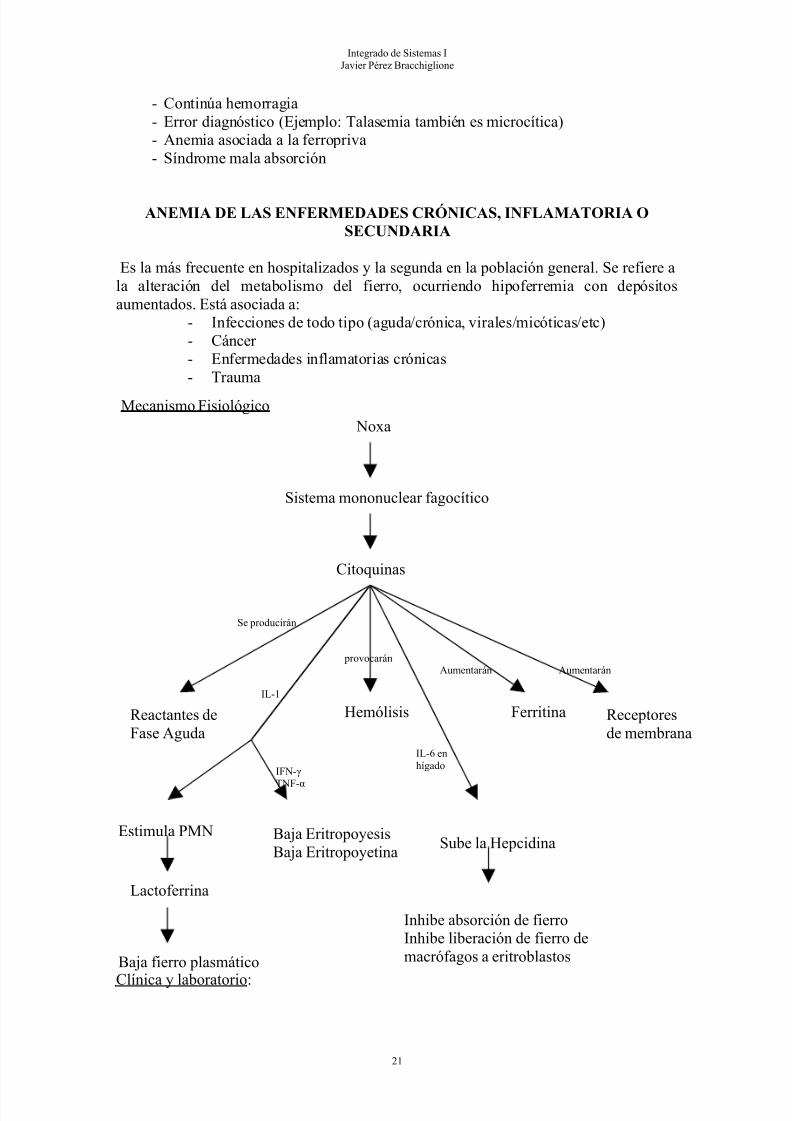
Mecanismo Fisiológico
Integrado de Sistemas IJavier Pérez Bracchiglione
- Continúa hemorragia- Error diagnóstico (Ejemplo: Talasemia también es microcítica)- Anemia asociada a la ferropriva- Síndrome mala absorción
ANEMIA DE LAS ENFERMEDADES CRÓNICAS, INFLAMATORIA OSECUNDARIA
Es la más frecuente en hospitalizados y la segunda en la población general. Se refiere ala alteración del metabolismo del fierro, ocurriendo hipoferremia con depósitosaumentados. Está asociada a:
- Infecciones de todo tipo (aguda/crónica, virales/micóticas/etc)- Cáncer - Enfermedades inflamatorias crónicas- Trauma
Clínica y laboratorio:
21
Noxa
Sistema mononuclear fagocítico
Citoquinas
Reactantes deFase Aguda
Se producirán
Estimula PMN Baja EritropoyesisBaja Eritropoyetina
Lactoferrina
Baja fierro plasmático
IL-1
IFN-γ
TNF-α
Hemólisis
provocarán
Sube la Hepcidina
IL-6 enhígado
Inhibe absorción de fierroInhibe liberación de fierro demacrófagos a eritroblastos
Ferritina Receptoresde membrana
Aumentarán Aumentarán

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 22/72
Lo ocupan las bacterias
Hay depósitos aumentados No se absorbe
Integrado de Sistemas IJavier Pérez Bracchiglione
• Clínica según cuadro de base• Normocítica / Normocrómica. Puede llegar a ser microcítica e hipocrómica leve• Arregenerativa• Laboratorio:
- Hipoferremia
- TIBC bajo- % saturación fierro: normal o bajo- Depósitos de fierro: aumentado (por la ferritina sérica)- Proteína C reactiva: aumentada
Tratamiento:
• De la enfermedad de base
• No dar fierro
• Transfusión de glóbulos rojos o uso de eritropoyetina si Hto < 21%
ANEMIA MEGALOBLÁSTICA
Se debe al déficit de vitamina B12 o de ácido fólico. Afecta a las líneas mieloide yeritroide, y también a epitelios respiratorio, urogenital y gastrointestinal.
22
Vitamina B12:
- Origen animal, ausente en vegetales,granos, frutas.- Requiere del factor intrínseco para suabsorción en el íleon- Reservas duran 2 a 4 años
Anemia Perniciosa (75%) (Ac anti – factor intrínseco)DéficitVit. B12 Gastrectomía
Resección ileal
Ácido Fólico:
- Fuente: vegetales verdes, hígado- Reservas duran 3 a 4 meses- La etiología del déficit de ácido fólico
puede ser:
Cocción de vegetales Nutricional Alcoholismo
Hemodiálisis
Enfermedad celíacaMala Enteropatía diabéticaAbsorción Anticonvulsivantes
Requerimientos Anemia hemolíticaaumentadosClínica:
- Síndrome anémico- Ictericia (predominio indirecto)- Glositis / queilitis angular - Canicie precoz- Neuropatías: Parestesias,demencia, pérdida de controlesfinteríano. Esto se da sólo endéficit de vitamina B12.
Laboratorio:
Hemograma:- Anemia o pancitopenia- Macrocitosis VCM > 110 fl- Macroovalocitos- Neutrófilos hipersegmentados- LDH aumentada- Hiperbilirrubinemia de
predominio indirecto
Endoscopía alta: Atrofia gástrica(anemia perniciosa)
Biopsia gástrica: Gastritis atrófica(anemia perniciosa)
Tratamiento:
- Vitamina B12 intramuscular:100 μg/día por 7 días100 μg bisemanal por 1 mes
100 μg mensual de por vida(anemia perniciosa)
- Ácido fólico:1-5 mg por 5 mesesTransfusión de glóbulos rojosen casos graves
Diagnóstico diferencial:- Alcoholismo (sin déficit de folatos)VCM < 110 fl- Hepatopatías- Reticulocitosis- Mielodisplasia- Hipotiroidismo

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 23/72
Integrado de Sistemas IJavier Pérez Bracchiglione
ANEMIAS HEMOLÍTICAS
23

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 24/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Características glóbulo rojo:Vida media: 120 días Producción diaria: 2x108
Diámetro: 8 micrones Función depende de: Integridad de membrana,enzimas y estructura de la hemoglobina
Hemólisis: Es la destrucción prematura del glóbulo rojo circulante
Laboratorio:
24
AnemiasHemolíticas
Congénitas
Adquiridas
Alteracionesde membrana
Microesferocitosis hereditaria
Eliptocitosis hereditaria
Hemoglobinopatías
Enzimopatías
Mecánica
Inmunológica
Mediada por agentes físicos, químicos ymicroorganismos
Anamnesis:
Anemia hemolítica congénita:
- Anemia refractaria a tratamientoconvencional médico.
- Historia familiar de anemia- Ictericia (predominio indirecto)- Esplenectomía
- Colecistectomía
Anemia hemolítica adquirida:
- Crisis hemolítica: Rápidaaparición de síndrome anémico
Examen Físico:
Depende de:
- Velocidad de comienzo- Sitio de destrucción celular:
Intravascular: En anemiahemolítica adquirida. Dahemoglobinuria
Extravascular: A cargo delsistema mononuclear fagocítico. Daictericia de predominio indirecto.
- Enfermedad asociada
Signos: Síndrome anémico,Esplenomegalia leve a moderada (sobretodo en la congénita), úlceras a nivel detobillos (propio de membranopatías)

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 25/72
Ejemplo: Microesferocitosishereditaria encontramosesferocitos, eliptocitosis
encontramos eliptocitos, etc.
Haptoglobina se une a hemoglobinalibre. Así, cuando hay hemólisis seforma el complejo haptoglobina-Hb que
va a la médula hematopoyética parareutilizar el fierro
Integrado de Sistemas IJavier Pérez Bracchiglione
- Anemia - Reticulocitosis - Hiperbilirrubinemia no conjugada- Frotis característico - Aumento LDH - Disminución haptoglobina sérica
ANEMIA HEMOLÍTICA CONGÉNITA
1) Membranopatías (los glóbulos rojos tienen forma característica que puede ser
vista al frotis)
Esferocitosis hereditaria:
- Autosómica dominante- Destrucción prematura en el bazo- 25% de la hemólisis compensada por la médula ósea- Historia familiar de anemia, ictericia, esplenectomía, colecistectomía- Puede haber crisis hemolítica por infección intercurrente, con anemia e ictericia másseveras y con dolor abdominal- Hemograma con microesferocitos y policromacia- Test de resistencia globular osmótica (+), es decir, hematíes poco resistentes a loscambios de osmolaridad- Tratamiento: Ácido fólico para evitar megaloblastosis. Si continúa con anemia severa,hacer esplenectomía
Eliptocitosis hereditaria
Estomatocitosis hereditaria
Acatocitosis hereditaria
2) Hemoglobinopatías
Hemoglobinopatías estructurales
Talasemias:
- Problema en la síntesis de globina.- En Chile sólo hay portadores, es decir, personas con sólo un pequeño déficit de lacadena α o β, que harán anemias leves microcíticas e hipocromas (diagnósticodiferencial de anemia ferropriva). Lo que las distingue es que en Talasemia el estudiodel fierro es normal.- Para hacer el diagnóstico de talasemia se pide una electroforesis de hemoglobina
3) Enzimopatías
25

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 26/72
Lo más frecuente es quecoexistan ambosmecanismos
Integrado de Sistemas IJavier Pérez Bracchiglione
Déficit glucosa-6-fosfato deshidrogenasa: Las más frecuente de las enzimopatías.
Déficit de piruvato-kinasa
ANEMIA HEMOLÍTICA ADQUIRIDA
1) Hemólisis por trauma físico
Valvulopatías: Por ejemplo, en el caso de una estenosis aórtica puede haber hemólisis
Prótesis valvulares cardiacas
Anemias hemolíticas microangiopáticas: Estas enfermedades se caracterizan por laformación de coágulos en los capilares. Ejemplos son la CID, PTT y SHU.
2) Hemólisis por infecciones
Por ejemplo: Clostridium perfinges, Citomegalovirus, Micoplasma pneumoniae (quedesencadena una anemia hemolítica autoinmune) y VIH
3) Anemia Hemolítica Autoinmune (AHAI)
- Es la destrucción prematura del glóbulo rojo debido a la presencia de autoanticuerpossobre la membrana eritrocitaria. Es bastante frecuente.- Los mecanismos por los que ocurre AHAI son:
Hemólisis intravascular: Inmunoglobulinas activan el complemento IgM, IgG1, IgG2, IgG3 Hemoglobinemia Hemoglobinuria
Hemólisis extravascular Destrucción por sistema mononuclear fagocítico IgG4, IgA Hiperbilirrubinemia de predominio indirecto
- El diagnóstico de AHAI se realiza teniendo un Test de Coombs directo (+). En un paciente con sospecha de AHAI se ponen los glóbulos rojos con el suero de Coombs(suero con anticuerpos anti-IgG o anti-complemento). Si el paciente tuviera AHAI,tendría inmunoglobulinas o complemento unido a la membrana de sus glóbulos rojos,
por lo cual habría aglutinación al juntarlo con el suero de Coombs, dando un resultado positivo.
26
Presencia oausencia deenfermedadsubyacente
AHAI Primaria
AHAI Secundaria
Enf. Autoinmune(LES, AR)
Sd. linfoproliferativo(LNH, EH, LLC)
Drogas α -metildopa
Por infecciones(micoplasma)
ClasificaciónAHAI
Segúncaracterísticasserológicas
Autoanticuerpos calientes: activaciónmáxima a 37°C (70% de los pacientes)
Autoanticuerpos fríos: activación amenos de 37°C (15% de los pacientes)
Mixta

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 27/72
AHAI anticuerpo caliente:- Signos y síntomas: Síndrome anémico (comienzo insidioso o
súbito) Fiebre Icterícia Dolor abdominal Orina oscura
Esplenomegalia (leve): 50-60% Hepatomegalia: 30% Linfoadenopatía (muy raro)
- Laboratório: Anemia leve a moderada Leucocitosis Aumento de reticulocitos (hipercromos al
frotis, ya que el macrófago puede fagocitar parte de la membrana, manteniendo el glóbulorojo su contenido de hemoglobina)
Hiperplasia eritroide al mielograma Test de coombs directo (+)
- Pronóstico: Curso crónico, con mortalidad de 15% a
AHAI anticuerpo frío o Sd. deaglutininas en Frío:- Peak a los 50 – 60 años
-Signos y síntomas: Síndrome anémico crónico Hemoglobinuria (raro; la
destrucción es principalmenteextravascular)
Acrocianosis Ictericia Esplenomegalia leve Hepatomegalia leve
- Laboratorio Anemia leve a moderada Autoaglutinación Test de coombs directo (+) Falso VCM elevado (130-
140, poco creíbles): Esto porque la máquina toma laaglutinación de glóbulos
Integrado de Sistemas IJavier Pérez Bracchiglione
MIELOPTISIS
27
Diagnósticodiferencial: Colangitis
Tratamiento AHAI
Corticoides: Prednisona 1 mg/kg/día en principio, después va
bajando la dosis a 5mg por semana
Esplenectomía: Hay 50%-75% de remisiónInmunosupresores: Imuran (azatioprina) 50-200 mg/día
Ciclofosfamida 50-150 mg/día

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 28/72
Causas
No hematológicas(metastásicas)
Hematológicas
Pulmón
RiñónMelanomaPróstata / MamaGastrointestinal
Leucemia Mieloide Aguda
Leucemia Linfática Aguda
Metaplasia Mieloide Agnogénica
Linfoma No Hodgkin
Enfermedad de Hodgkin
Mieloma múltiple
Células en lágimaEritroblastosCélulas mieloides inmaduras
Integrado de Sistemas IJavier Pérez Bracchiglione
(Anemia asociada a infiltración de la Médula Ósea)
Es la infiltración medular por células tumorales que alteran la estructura medular. Se produce una pancitopenia.
Hemograma:- Pancitopenia- Frotis: Reacción leucoeritroblástica
ANEMIA DE LA INSUFICIENCIA RENAL CRÓNICA
Se manifiesta cuando el clearence de creatinina está bajo el 50%
Mecanismos:
• Baja la producción de eritropyetina• Baja la vida media del glóbulo rojo•
Déficit de fierro por sangramiento• Déficit de ácido fólico por hemodiálisis
Tratamiento:
• Fierro• Ácido fólico• Eritropoyetina (2000 U subcutánea, 3 veces a la semana)
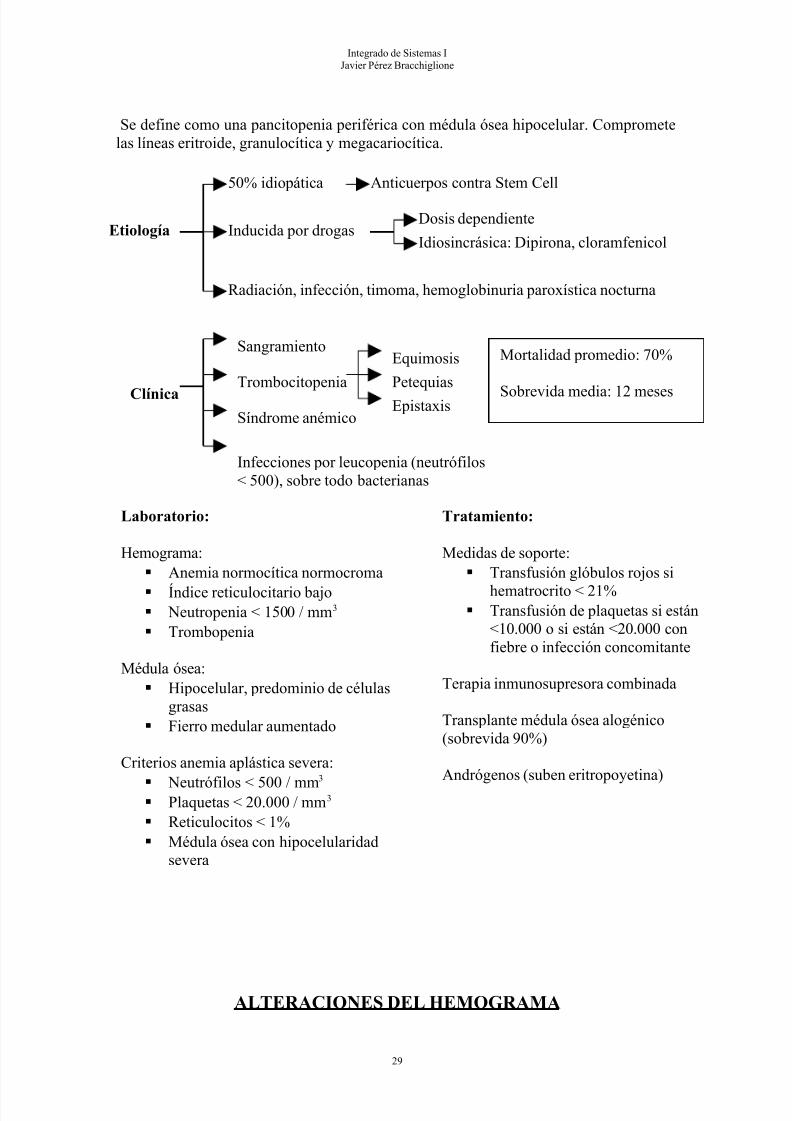
ANEMIA APLÁSTICA
28
Método diagnóstico: Biopsia
Tratamiento: De la enfermedadde base
5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 29/72
Etiología
50% idiopática Anticuerpos contra Stem Cell
Inducida por drogasDosis dependiente
Idiosincrásica: Dipirona, cloramfenicol
Radiación, infección, timoma, hemoglobinuria paroxística nocturna
Laboratorio:
Hemograma: Anemia normocítica normocroma Índice reticulocitario bajo Neutropenia < 1500 / mm3
Trombopenia
Médula ósea: Hipocelular, predominio de células
grasas Fierro medular aumentado
Criterios anemia aplástica severa:
Neutrófilos < 500 / mm3
Plaquetas < 20.000 / mm3
Reticulocitos < 1% Médula ósea con hipocelularidad
severa
Tratamiento:
Medidas de soporte: Transfusión glóbulos rojos si
hematrocrito < 21% Transfusión de plaquetas si están
<10.000 o si están <20.000 confiebre o infección concomitante
Terapia inmunosupresora combinada
Transplante médula ósea alogénico(sobrevida 90%)
Andrógenos (suben eritropoyetina)
Integrado de Sistemas IJavier Pérez Bracchiglione
Se define como una pancitopenia periférica con médula ósea hipocelular. Comprometelas líneas eritroide, granulocítica y megacariocítica.
ALTERACIONES DEL HEMOGRAMA
29
Clínica
Sangramiento
Síndrome anémico
Infecciones por leucopenia (neutrófilos< 500), sobre todo bacterianas
Trombocitopenia
Equimosis
Petequias
Epistaxis
Mortalidad promedio: 70%
Sobrevida media: 12 meses

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 30/72
Policromacia: Traduce presencia dereticulocitos, por tanto, anemia regenerativa.
Causas:- Sangramiento- Hemólisis
Neutrófilo hipersegmentado:Causado por anemia megaloblástica
Microcitosis: VCM < 80Hipocromía: glóbulo rojo más pálido. Seda en anemia ferropriva.
Causas:- Déficit fierro- Talasemia
Eliptocitos: glóbulos rojos en forma de puroCausas: Déficit de fierro, eliptocitosishereditaria
Esferocito: Glóbulo rojo pequeño sincentro pálido
Causas:-Esferocitosishereditaria- AHAI
Esquistocito: Glóbulo rojo fragmentado
Causas: Anemias hemolíticasmicroangiopáticas y mecánicas
Target cell o célula diana: glóbulo rojocon centro denso rodeado de halo claro
Causas:- Cirrosis- Talasemias- Hemoglobinopatías- Postesplenectomía- Enf. Hepática crónica
Corpúsculo de Howell-Jolly:Remanente nuclear en el glóbulo rojo.
Causas: Postesplenectomía(se usa para saber si se extirpó por completo el bazo)
Aglutinación de glóbulos rojos
Causas:- AHAI (Ig bloquean las cargas negativasde la membrana y hacen que los glóbulosrojos se puedan juntar)
Roleaux: Aglutinaciónde los glóbulos rojos enforma de pila demonedasCausas:- Mieloma múltiple
Integrado de Sistemas IJavier Pérez Bracchiglione
GLÓBULOS ROJOS
30
Anemia:Hombre: Hb < 13 g/dl
Mujer: Hb < 12 g/dl
Poliglobulia o eritrocitosis:Hombre: Hb > 18 g/dl
Mujer: Hb > 16 g/dl
Alteraciones:Anisocitosis: Glóbulos rojos de diferente tamañoPoiquilocitosis: Glóbulos rojos de diferente forma
Anisocromía: Glóbulos rojos de diferente intensidad de coloración
Macrocitosis: VCM > 100
Causas:
- Déficit vit. B12- Déficit ác. Fólico- Reticulocitosis- Hipotiroidismo
Macroovalocito: glóbulo rojo demayor tamaño que lo normal. Típico deanemia megaloblástica
Células en lágrima: Glóbulo rojo enforma de gota.Causas: Mielofibrosis, mieloptisis
Crenocitos: Glóbulo rojo con proyecciones de membrana regularesCausa: Falla renal
Acantocitos: Glóbulo rojo con proyecciones de membrana irregularesCausas: Enfermedad hepática severa
(cirrosis)Punteado basófilo: Pesencia de gránulosazulados (ribosomas) en el citoplasma delos glóbulos rojosCausas: Alcoholismo, reticulocitosis,talasemias, anemias hemolíticascongénitas o adquiridas

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 31/72
Integrado de Sistemas IJavier Pérez Bracchiglione
LEUCOCITOS
31
Leucocitosis
Aumento del recuento total de leucocitos sobre lo normal >10000Incremento puede incluir uno o más subgrupos: Neutrofilia: aumento del recuento absoluto de neutrófilos > 7500 /mm3 (más común)Linfocitosis: aumento del recuento absoluto de linfocitos > 4000 /mm3
Monocitosis: aumento del recuento absoluto de monocitos > 800 /mm3
Eosinofilia: aumento del recuento absoluto de eosinófilos > 450 /mm3
Reacción leucemoide: Leucocitosis > 50.000, en infecciones graves o abscesos (no tienen quever con leucemias)Neutrofilia
Causas:
- Infecciones: comúnmente bacterianas o por hongos
- Inflamación y necrosis: trauma,infarto, artritis reumatoidea,quemaduras
- Tumores
LinfocitosisCausas:
- Infecciones: comúnmente virales- Enfermedades autoinmunes
(menos frecuente)- Neoplasias hematológicas: por
ejemplo, leucemia linfáticacrónica (raro)
MonocitosisCausas:
- Infecciones:
Virales: CMV, VVZ TBC Endocarditis bacterianasubaguda
- Enfermedades inflamatorias yautoinmunes (LES, colitisulcerosa
EosinofiliaCausas:
- Infecciones: parásitos(triquinosis, isosporosis,diastomatosis)
- Reacciones alérgicas (rinitisalérgica, dermatitis atópica,urticaria, asma)
Neutropenia: recuento absoluto de neutrófilos < 1500 mm3Linfopenia: recuento absoluto de linfocitos < 1500 mm3Eosinopenia: recuento absoluto de eosinófilos < 10-50 mm3
5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 32/72
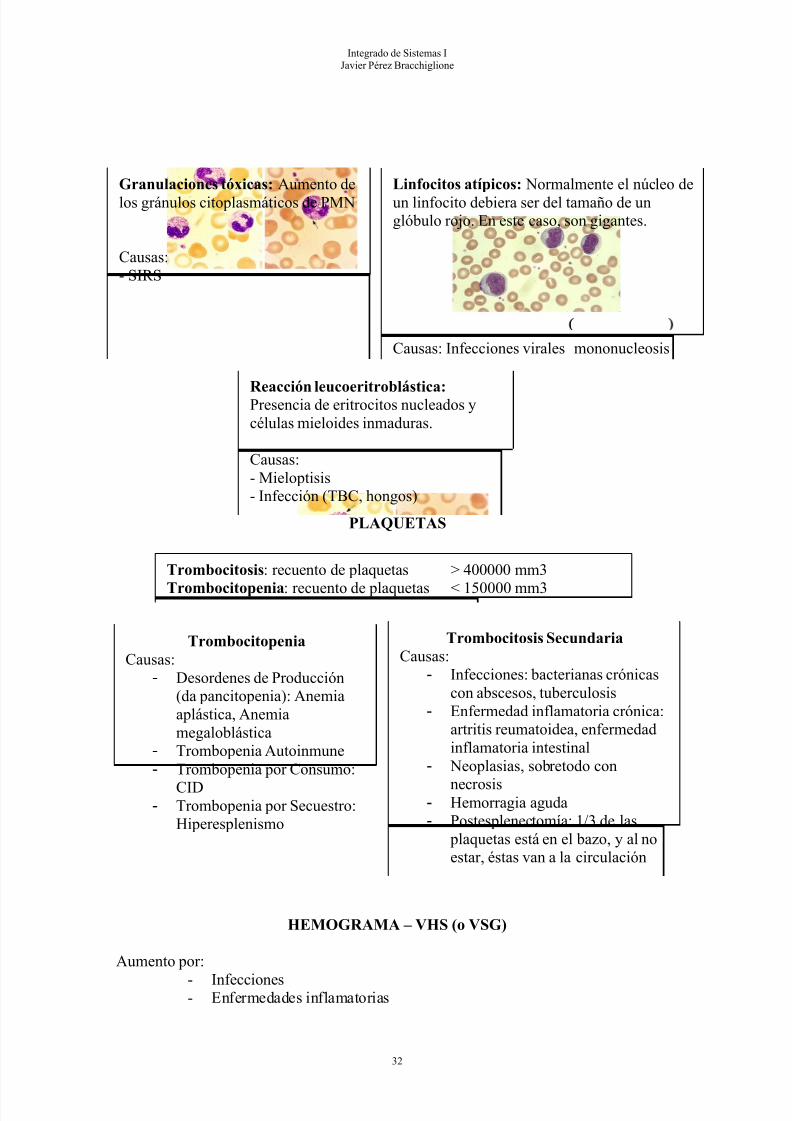
Granulaciones tóxicas: Aumento delos gránulos citoplasmáticos de PMN
Causas:- SIRS
Linfocitos atípicos: Normalmente el núcleo deun linfocito debiera ser del tamaño de unglóbulo rojo. En este caso, son gigantes.
Causas: Infecciones virales mononucleosis
Reacción leucoeritroblástica:Presencia de eritrocitos nucleados ycélulas mieloides inmaduras.
Causas:- Mieloptisis- Infección (TBC, hongos)
Integrado de Sistemas IJavier Pérez Bracchiglione
PLAQUETAS
HEMOGRAMA – VHS (o VSG)
Aumento por:
- Infecciones- Enfermedades inflamatorias
32
Trombocitosis: recuento de plaquetas > 400000 mm3
Trombocitopenia: recuento de plaquetas < 150000 mm3
Trombocitosis SecundariaCausas:
- Infecciones: bacterianas crónicascon abscesos, tuberculosis
- Enfermedad inflamatoria crónica:artritis reumatoidea, enfermedadinflamatoria intestinal
- Neoplasias, sobretodo con
necrosis- Hemorragia aguda- Postesplenectomía: 1/3 de las
plaquetas está en el bazo, y al noestar, éstas van a la circulación
TrombocitopeniaCausas:
- Desordenes de Producción(da pancitopenia): Anemiaaplástica, Anemiamegaloblástica
- Trombopenia Autoinmune- Trombopenia por Consumo:
CID- Trombopenia por Secuestro:
Hiperesplenismo
5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 33/72
Causas
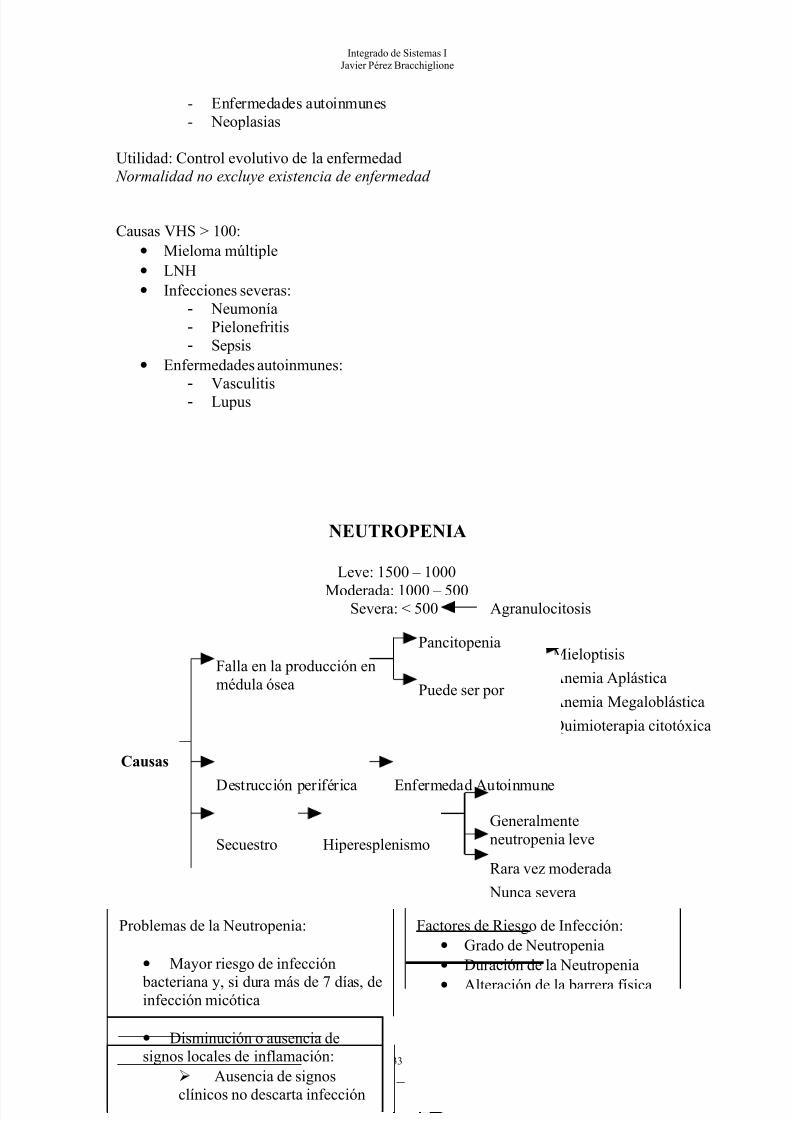
Falla en la producción enmédula ósea
Pancitopenia
Puede ser por
Mieloptisis
Anemia Aplástica
Anemia Megaloblástica
Quimioterapia citotóxica
Destrucción periférica Enfermedad Autoinmune
Secuestro Hiperesplenismo
Generalmenteneutropenia leve
Rara vez moderada
Nunca severa
Consumo Sepsis
Integrado de Sistemas IJavier Pérez Bracchiglione
- Enfermedades autoinmunes- Neoplasias
Utilidad: Control evolutivo de la enfermedad Normalidad no excluye existencia de enfermedad
Causas VHS > 100:• Mieloma múltiple• LNH• Infecciones severas:
- Neumonía- Pielonefritis- Sepsis
• Enfermedades autoinmunes:-
Vasculitis- Lupus
NEUTROPENIA
Leve: 1500 – 1000
Moderada: 1000 – 500Severa: < 500 Agranulocitosis
33
Problemas de la Neutropenia:
• Mayor riesgo de infección
bacteriana y, si dura más de 7 días, deinfección micótica
• Disminución o ausencia designos locales de inflamación:
Ausencia de signosclínicos no descarta infección
Factores de Riesgo de Infección:• Grado de Neutropenia• Duración de la Neutropenia• Alteración de la barrera física• Origen del microorganismo
Foco de Infección:• Difícil de localizar • Generalmente está en la cavidadorofaríngea

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 34/72
Pueden asociarse a Vancomicina (sospecha deS. aureus multirresistente)
Integrado de Sistemas IJavier Pérez Bracchiglione
Paciente Neutropénico Febril:• Es una emergencia médica• Se define como el paciente con agranulocitosis y peak de temperatura >38,3°C,o como el paciente con agranulocitosis y 38°C por una hora• Ante este paciente:
- Tomar cultivo de todo- Iniciar tratamiento antibiótico empírico- Puede manejarse en sala común mientras no hayan pacientes infectados- Dar alimentos cocidos- Evitar procedimientos productores de bacteremia
• Tratamiento antibiótico:- Ceftazidima + Amikacina- Imipenem- Meropenem- Cefepime
• El paciente neutropénico febril se clasifica en: Grupo 1: Hospitalizado con neoplasia hematológica. Altamortalidad Grupo 4: Neutropenia < 7 días, ambulatorios. Gérmenesde la comunidad
(multisensibles)
• El diagnóstico de infección en neutropenia se hace por PCR (que es inespecífica pero muy sensible). En un paciente sano, es infección un valor > 20 mg%. En el paciente neutropénico se considera infección > 10 mg%.
34
Sigue con fiebre a los 4 díasAgregar vancomicina
Cambiar régimen antibiótico
Sigue con fiebre a los 7 días Agregar Anfotericina B (para cándida y aspergillus)

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 35/72
Integrado de Sistemas IJavier Pérez Bracchiglione
ENFERMEDADES DEL BAZO
HIPOESPLENISMO• Causa: Esplenectomía• Complicaciones: Gérmenes capsulados (S. pneumoniae, H. influenzae, N.meningitidis)• Diagnóstico: Se hace a través del Hemograma
Corpúsculos de Howell-Jolly en glóbulos rojos Eritroblastos Target cells Trombocitosis
• Tratamiento: Profilaxis
HIPERESPLENISMO• Generalmente asociado a Esplenomegalia• Clínica:
Puede haber anemia, trombopenia o leucopenia, ocualquiera de sus combinaciones Médula ósea responde con hiperplasia compensatoria
• Causas de Esplenomegalia:
35

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 36/72
90% del total de casos
Integrado de Sistemas IJavier Pérez Bracchiglione
Infecciosas Enfermedades inmunológicas
Neoplásicas: Linfoma no Hodgkin Enfermedad de Hodgkin Sd. mieloproliferativos crónicos Leucemia de células velludas
Congestivas: Cirrosis (90%)
INDICACIONES ESPLENECTOMÍA
- Anemia Hemolítica Congénita- AHAI- PTI- Infarto esplénico- Como procedimiento diagnóstico- Nunca en esplenomegalia congestiva
ENFERMEDAD DE HODGKIN
Definición: Neoplasia originada del sistema linfático, cuyo marcador anatomopatológico es la presencia de células de Reed-Sternberg. Ésta es la célulatumoral del linfoma de Hodgkin, y se origina del linfocito B.
Etiología: Se asocia en un 40% a VEB (mononucleosis)
Epidemiología: Mayor incidencia a los 20 años, luego nuevo peak a los 50 años.
Clasificación histológica:
1.- De predominio linfocitario2.- Esclerosis nodular (la más frecuente en el mundo)3.- Celularidad mixta (la más frecuente en Chile)4.- Depleción linfocitaria
La de predominio linfocitario y de depleción linfocitariatienen marcadores No Hodgkin.
Importancia de saber el tipo histológico: Índice pronóstico:
Esclerosis nodular y celularidad mixta son curables Depleción linfocitaria es prácticamente incurable
36
Esplenomegalia leve, sin hiperesplenismo
Esplenomegalia gigante:Hiperesplenismo
Esplenomegalia leve, con hiperesplenismo

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 37/72
Síntomas sistémicos (40%):• Fiebre intermitente sobre 38°C• Sudocración nocturna (profusa)• Pérdida de Peso
Basta 1 para decir que el paciente
tiene síntomas sistémicos
Adenopatía superficial (en orden):• Cervical• Axilar • Inguinal• Sólo bajo el diafragma
Otros:• Sd. Vena Cava superior por masa tumoral mediastínica• Prurito• AHAI• Trombocitopenia autoinmune• Dolor ganglionar asociado aingesta de alcohol• Esplenomegalia (puede ser gigante)• Hepatomegalia
Recordar que célulastumorales provienen dellinfocito B
Integrado de Sistemas IJavier Pérez Bracchiglione
Diagnóstico Histopatológico de Linfoma de Hodgkin: Células de Reed-Sternbergrodeadas de células reactivas benignas
Clínica:
Hemograma:• Anemia: De enfermedades crônicas y/oautoinmune• Neutrofilia• Monocitosis• Posible eosinofilia• VHS aumentada (70%)
Biopsia ganglionar o del tejido comprometido: Confirma el diagnósticoInmunofenotipo característico: CD15+ / CD30+ / CD45-
Además, se le agregan letras segúncorresponda:A: Sin síntomas sistémicosB: Con síntomas sistémicosX: Masa tumoral > 10 cms
Anamnesis: Buscar síntomas sistémicos
Examen Físico: Buscar adenopatías, anillode Waldayer, esplenomegalia yhepatomegalia.
Laboratorio: Incluye hemograma, pruebashepáticas, creatinina, LDH, electroforesis de
proteínas, calcemia, test de ácido úrico,coombs directo, uricemia, VIH, y, por sobretodo, biopsia ganglionar y de médula ósea
37
Clasificación según diseminación, 4estadios:
I: Compromiso una única regiónganglionar, o también cuando haycompromiso de un solo organoextralinfático (si no hay compromisode órganos linfáticos, se le agrega laletra “E”. Ejemplo: Estadio I E)
II: Compromiso de 2 o más regionesde un sólo lado del diafragma
III: Compromiso ganglionar a amboslados del diafragma. Se le llama III Ssi además hay compromiso esplénico
IV: Compromiso extraganglionar asociado a adenopatías. Si haycompromiso de Hígado o MédulaÓsea, pasa a ser estadio IV deinmediato.

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 38/72
I o II A(tumor no
voluminoso)
2 ciclos quimioterapia ABVD+
RadioterapiaCuración: 95%
III o IV
6-8 ciclos quimioterapia ABVD+
RadioterapiaCuración: 70%
LNHindolentes
Lento crecimiento
Incurables
Sobrevida 7 años
LNHagresivos
Rápido crecimiento
Curables en un 50% (si no
responden a quimioterapia, haycorta sobrevida)
Integrado de Sistemas IJavier Pérez Bracchiglione
Imágenes: TAC de tórax, abdomen y pelvis para ver compromiso ganglionar profundo.También se pueden hacer cintigrama con galio o, en lo posible, tomografía por emisiónde positrones.
Tratamiento:
Reacaída:
Antes de un año: Transplante médula óseaDespués de un año: Quimioterapia de 2° línea
LINFOMA NO HODGKIN
Definición: Grupo de neoplasias linfoides de gran heterogeneidad histológica, clínica yevolutiva. Es una detención y proliferación indefinida de linfocitos en distintas fases desu proceso de maduración.
Etiología: Casi nunca se puede establecer. Está asociado a VEB, VIH, HTLV-1 einmunodeficiencias.
Epidemiología: Edad de aparición a los 54 años en promedio. Más frecuente enmujeres que en hombres (1,5:1)
Clasificación:
“-cítico” o “-citoide”: Bajo grado de malignidad “-blástico”: Alto grado de malignidad
38
Factores pronósticos desfavorables
Antiguos: Nuevos:- > 60 años - Tumor mediastínico > 1/3 del diámetro torácico- Depleción linfocitaria - Tumor original > 10 cms- Síntomas B - Más de 2 localizaciones- Estadio III B o IV - Síntomas B + VHS > 30
- Sin síntomas B, pero con VHS > 70
OMS: 5 parámetros.
1) Histología
2) Inmunofenotipo: LB: 81% (curable) LT (incurable)
3) Citogenética: El 90% estáasociado a un oncogen.
4) Biología molecular (buscar oncogen)
5) Características clínicas
Esta clasificación se basa en el
momento de maduración en que
se detiene y empieza a proliferar
indefinidamente

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 39/72
Síntomas sistémicos (30%): Fiebre, sudoraciónnocturna, baja de peso.
Adenopatías Superficiales: Cervicales,inguinales, axilares, sitios múltiples.
Presentación extranodal: Anillo de Waldayer,Estómago/colon, Hueso, Piel, glándulassalivales, tiroides, testículo
Tumor de mediastino Linfoma difusode células grandes B
Compromiso SNC Linfomalinfoblástico
Anamnesis
Ritmo de crecimiento
Síntomas B
Indolente (6 meses – 1 año)
Agresivo (1 – 2 meses)
Linfoma diseminado
Laboratorio
HemogramaVHS elevadaLDH: A mayor masa tumoral, más sube (igual con la β-2 microglobulina)Función renalFunción hepáticaBiopsia ganglionar y de médula ósea
Integrado de Sistemas IJavier Pérez Bracchiglione
Según estadísticas de la OMS, el Linfoma de células B comprende el 88% del total de
linfomas, siendo los más frecuentes el Linfoma difuso de células B grandes (31%) y elLinfoma folicular (22%). El Linfoma de células T comprende el 12% restante.
Clínica:
Clínica específica:
Linfomas Foliculares Linfoma Células del Manto Linfoma de CélulasGrandes
• Indolentes,incurables• 20-30% de todoslos linfomas•
Aparición: 55 años• 2/3 de los pacientesconsultan en estadioavanzado• Nacen de ganglioslinfáticos• Quimiosensibles,
pero con continuasrecaídas• Sobrevida: 7-10años
• 6-8% de todoslos linfomas• Aparición: 50-60años•
Linfoadenopatías• Esplenomegalia(puede ser gigante)• Infiltraciónmedular • Quimiosensible,
pero con continuasrecaídas• Sobrevida: 3años
• 25-40% de todos loslinfomas (el másfrecuente)• Aparición: Cualquier
edad• Presentaciónganglionar oextraganglionar • Crecimiento rápido• Quimiosensible,supervivencia del 40-50%a largo plazo• Recaídas <10%después de 3 años deremisión
39
ExamenFísico
Linfonodos
Anillo de Waldayer
Hepatomegalia
Esplenomegalia
Piel
Imágenes
TAC tórax, abdomeny pelvis
Cintigrama óseo
Cintigrama con galio

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 40/72
Traumático
Vasculares
InmunológicasSd. Schönlein-HenochVasculitis secundaria a enfermedad del tejidoconectivo
A.R.L.E.S.
Trombocitopenias periféricas
Trombopatías
Congénitas
Adquiridas
Bernard Soulier Wiscott AldrichSd. plaquetas grises
Por drogas (aspirina)Por insuficiencia renalPor enfermedades hepaticasPor mielodisplasias
Trombocitopenia con ausencia deradioPúrpura AmegacariocíticoLeucemia congénita
No InmunesInmunes
SHU, PTT, CIDHiperesplenismoHemangioma giganteHipotermia
PTIPor infeccionesPor enfermedad autoinmune (LES)Por drogas
Integrado de Sistemas IJavier Pérez Bracchiglione
Siempre para confirmar diagnóstico: Histología, inmunofenotipo y citogenética.
SÍNDROME PURPÚRICO
Definición: Conjunto de síntomas y signos caracterizados principalmente por hemorragias de piel y mucosas, que clínicamente se manifiesta como petequias,
púrpuras y equimosis.
Clasificación:
40
Infecciosas
MeningococcemiaEndocarditis bacteriana subagudaVRS, CMV, EBV, etc.
Por defectos del tejido conectivo
Púrpura senilEhler-DanlosOsler Rendü Weber
Por defectos plaquetarios

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 41/72
Trombocitopeniascentrales
Congénitas
Adquiridas
Leucemia
Anemia aplásticaHemoglobinuria paroxística nocturnaMieloptisisMielodisplasiaCarencia de vit. B12 o ácido fólico
Clínica:• Púrpura palpable (“respeta el tronco”)• Compromiso articular (artalgia/artritis)
• Compromiso abdominal (dolor)
• Compromiso renal• Compromiso SNC• Compromiso testicular
Tríada
clásica
Etiología:Desconocida, pero
habitualmente hayantecedente de infección previa (bacteriana, viral,vacunas, alimentos, por insectos, medicamentos)
Diagnóstico: Clínico
Laboratorio: InespecíficoHemograma: Normal o con aspecto de proceso inflamatorio
Plaquetas normales
Pedir examen de orina, uremia y creatinina para evaluar compromiso renal
Tratamiento: Sintomático, con reposo, antiespasmódicos, AINEs, corticoides.
Integrado de Sistemas IJavier Pérez Bracchiglione
Síndrome de Schönlein Henoch (púrpura vascular)
Púrpura Trombocitopénico Inmune (niños): Es igual clínicamente al PTA del adulto.
Clínica:• Comienzo brusco y antecedente de infección
41

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 42/72
Cuadro clínico:
Sd. Anémico
Sd. Hemorrágico
Sd. Febril
Sd. Tumoral
Diagnóstico:
Demostración de presencia de más del20% de célulasleucémicas o blastos enla médula ósea
Diferenciación LLA de LMA:
•
Cuadro clínico• Morfología de blastos (bastones
de Auer: propio de leucemiasmieloblásticas)
• Tinciones citoquímicas(peroxidasa)
• Inmunofenoti o
Integrado de Sistemas IJavier Pérez Bracchiglione
• Hemorragias cutáneo-mucosas (no siempre; plaquetas muy eficaces)
Fisiopatología: Antígeno viral muy similar a antígeno plaquetario
Diagnóstico:• Por descarte• Paciente con púrpura, sin otra molestia• Hemograma normal, salvo por trombopenia• Mielograma normal o megacariocitosaumentados
Tratamiento:• Reposo, sin aspirina ni AINEs• Corticoides orales y endovenosos• Factor VII recombinante
Evolución:• 70% - 80% mejoran en menos de 6 meses (en el resto, pensar un probable PTA)• Dar tratamiento si plaquetas <20.000 o si son muy sintomáticos
LEUCEMIA AGUDA
Definición: Enfermedad clonal, maligna o neoplásica que se origina en una célula precursora hematopoyética en la médula ósea. Ésta inicia la proliferación y reemplaza a
los elementos medulares normales, llevando esto a diferentes intensidades deneutropenia, anemia y trombocitopenia.
Epidemiología: 5 de cada 100.000 adultos por añoLeucemia linfática aguda es más frecuente en niñosLeucemia mieloide aguda es más frecuente en adultosEn el caso de las crónicas, LLC (40%) es el doble que la LMC (20%)en adultos
Etiología: Factores genéticos, inmunológicos y ambientales• Varias enfermedades genéticas tienen mayor frecuencia de
leucemia. Ejemplo: Sd. de Down, Sd. de Klinefelter • También en enfermedades donde hay un defecta de utilización delDNA. Ejemplo: Ataxia telangectasia• También en enfermedades congénitas con inmunodeficiencias
primarias• Importancia de agentes físicos, químicos, biológicos yambientales
42
Diagnóstico diferencial:
Leucemia y aplasia Drogas Trombopatía LES Sd. antifosfolípidos Déficit IgA /

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 43/72
Morfológica
L1: Células pequeñas, núcleo denso y redondo, sin nucléolo visible.Poco citoplasma
L2: Células más grandes, más citoplasma. Núcleo de contornoirregular, clivado, con uno o más nucléolos
L3: Típico de células B anormales. Células medianas, de núcleoredondo, com uno o más nucléolos. Citoplasma moderadamenteabundante, con vacuolas
Citogenética Menor recuento de cromosomas indica peor pronóstico
T (9;22) o cromosoma Philadelphia: Presente en el 95% de lasLMC, pero también en el 25% de las LLA del adulto
Común (CD10 o CALLA +)
Pre Pre B, Nulas o Pro B (CALLA -)
PreB
Pre T / T
B
Laboratorio:
• Hemograma:- Anemia- Con o sin trombocitopenia- Generalmente neutropenia- Con o sin blastos circulantes
• Mielograma• Inmunofenotipo• Citogenética de médula ósea• Citoquímico de LCR • Radiografía de Tórax• LDH aumentado
Tratamiento: Poliquimioterapia
Complicaciones:• Resistencia de la enfermedad• Recaída• Aplasia• Toxicidad de las drogas
Pronóstico (sobrevida):- 70%-75% en niños- 30%-40% LLA adultos- 20%-30% LMA adultos
Integrado de Sistemas IJavier Pérez Bracchiglione
LEUCEMIA LINFÁTICA AGUDA (LLA)
Clasificación:
43
Inmunofenotipo
Riesgo
Standard o Bajo: >1 año a <6 años; <20.000 leucocitos/mm3;leucemia no T; responde a prednisona al día 7; remisión completa día33; sin compromiso SNC; sin t(9;22) ni t(4;11)
Medio: <1 año o >6 años; <20.000 leucocitos/mm3; responde a prednisona; remisión completa día 33; sin t(9;22) ni t(4;11);
compromiso SNC o testicular
Alto: Mala respuesta a prednisona; no hay remisión al día 33; cont(9;22) o t(4;11)

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 44/72
Integrado de Sistemas IJavier Pérez Bracchiglione
LEUCEMIA MIELOIDE AGUDA (LMA)
Clasificación: Morfológica / Citoquímica: M0 (blasto), M1, M2, M3,M4, M5, M6, M7 Riesgo:
- Bajo: Menos del 5% de blastos em médula ósea al día 15, y fenotipo M1-M2 y/o t(8;21); o M3 y/o t(15;17); o M4 con eosinofilia e i16
- Alto: Todos los demás
Cuadro clínico: Además del anteriormente descrito, se agrega la presencia de cloromas(tumoraciones por granulocitos), leucemides (infiltración de la piel), infiltración deencías y trastornos hemorrágicos especiales (CID)
Tratamiento: Quimioterapia muy intensa, sin necesidad de tratamiento prolongado ode mantención.
SÍNDROMES MIELOPROLIFERATIVOS CRÓNICOS
Definición: Enfermedades hematológicas clonales que nacen de una transformación dela stem-cell (neoplasia). Se caracterizan por sobreproducir células sanguíneas maduras yfuncionales, con predominio de una línea celular específica, asociados frecuentemente a
basofilia y eosinofilia, esplenomegalia, aumento del ácido úrico y con tendencia adesarrollar fibrosis medular. Se diagnostican en general con una biopsia de médulaósea.
Clasificación:Enfermedad Línea celular
predominanteOncogen Historia Natural de la
Enfermedad
Policitemia Vera Poliglobulia JAK-2 Fase crónica
Fase acelerada
Transformación leucémica o
crisis blástica
TrombocitemiaEsencial
Trombocitosis JAK-2
Leucemia MieloideCrónica
Leucocitosis BCR-ABL(Filadelfia)
Metaplasia MieloideAgnogénica
(Mielofibrosis)
Disminución engeneral
Desconocido
44

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 45/72
Integrado de Sistemas IJavier Pérez Bracchiglione
A medida que va avanzando la enfermedad, más difícil se hace de tratar. Una vez quese instala la crisis blástica o transformación leucémica (también llamada leucemiasecundaria), se hace por definición incurable.
La enfermedad en que se distinguen más fácilmente estas fases es la LeucemiaMieloide Crónica, ya que desde que se instala demora sólo 6 años en llegar a crisis
blástica, mientras que en las demás pueden pasar incluso 25 años
POLICITEMIA VERA
Definición: Proliferación anormal de una célula madre pluripotente con predominio dehiperplasia eritroide. Puede afectarse también en un comienzo la serie granulocítica (nomás de 20.000 leucocitos) y las plaquetas (no más de 800.000).
Epidemiología: Incidencia de 2/100.000 habitantes por año. Igual relaciónhombre/mujer. Aparece a los 60 años aproximadamente.
Clínica:Síntomas Signos
Cefalea: Es lo más frecuenteVértigoAlteraciones visualesDisnea y Angor Otros: Prurito acuógeno, baja de peso,sudoración, tendencia a presentar gota
Aspecto pletóricoInyección conjuntival (conjuntivas rojas)Esplenomegalia leveHepatomegaliaHTA
Los síntomas están dados principalmente por la proliferación excesiva de líneas
hematopoyéticas. Así, el aumento del hematocrito producirá hiperviscosidad. Esto hace por un lado que la sangre fluya más lento, provocando vasodilatación, y, por otro lado,que aumente el riesgo de trombosis. En un 20% de los casos, la primera manifestaciónde la enfermedad son complicaciones trombóticas.
Laboratorio:
Hemograma : Hematocrito muy elevado (60-65%)Leucocitosis leveBasofiliaTrombocitosis leve
Mielograma: Hipercelularidad
Niveles: LDH aumentado, Hiperuricemia, Eritropoyetina disminuida o normal,fierro disminuido en médula ósea (dando un frotis microcítico ehipocromo)
Biopsia de Médula Ósea : Confirma el diagnóstico.
Diagnóstico Diferencial: El diagnóstico de poliglobulia (no necesariamente dePolicitemia Vera) se puede hacer sólo con el hemograma. Se diagnostica si hay unHto>54% o Hb>18 en hombres, o si hay Hto>48% o Hb>16 en mujeres.
45

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 46/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Al evaluar a un paciente que presente características de Policitemia Vera hay queconsiderar inmediatamente la eritrocitosis secundaria como diagnóstico diferencial, lacual generalmente se da por problemas de oxigenación o tumores. Las principalescausas de Eritrocitosis secundaria son:
1. Hipoxemia arterial
2. Disminución de la liberación de O2 tisular (tabaquismo): Es lo más frecuente3. Lesiones Renales (principalmente hipernefroma y riñón poliquístico)4. Lesiones Hepáticas (como el hematoma)5. Tumores (ovario, bronquio, cerebelo)
La clínica nos puede orientar bastante para hacer el diagnóstico diferencial:- Prurito y trombosis son más sensibles y específicos de Policitemia Vera- Paciente con poliglobulia más esplenomegalia tiene Policitemia Vera- Eritrocitosis secundaria eleva sólo la serie roja. Policitemia Vera eleva
todas, aunque levemente de plaquetas y leucocitos- Policitemia Vera disminuye la eritopoyetina, mientras que en
eritrocitosis secundaria debiera estar elevada.
Tratamiento: Flebotomía (sangría), para inducir ferropenia Agente Mielosupresor: Hidroxiurea Aspirina: Para reducir riesgo de trombosis
Pronóstico: Sin tratamiento hay sobrevida de 18 meses, con tratamiento es de hasta 25años. Se describen 3 causas de muerte:
Trombosis Mielofibrosis (que terminará produciendo pancitopenia) Leucemia aguda (raro)
TROMBOCITEMIA ESENCIAL
Clínica:- Asintomático en el 30% de los casos- 35% presenta Hemorragia digestiva, ya que a pesar de que está aumentado elrecuento de plaquetas, está alterada su funcionalidad.
- 25-40% debuta como trombosis de vasos mayores.- Puede haber esplenomegalia leve en un 80%, y a veces hepatomegalia leve.
Laboratorio:
Hemograma : Hto normal, leucocitosis leve en 25-40%.Plaquetas > 600.000
Biopsia Medula Ósea : Hipercelular, con marcado aumento de megacariocitos.Confirma el diagnóstico.
Diagnóstico: Debe existir un aumento en el recuento de plaquetas por sobre los 600.000en al menos dos controles. Podemos descartar otros Síndromes mieloproliferativos
46

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 47/72
Integrado de Sistemas IJavier Pérez Bracchiglione
crónicos, analizando el fierro en médula ósea que debiera estar normal (descartandoPolicitemia Vera), confirmando la ausencia del cromosoma Filadelfia (descartando laLeucemia mieloide crónica).
Diagnóstico Diferencial: Se debe hacer con las trombocitosis secundarias (que causan
el 90% de las trombocitosis). Dentro de éstas destacan:1. Déficit de Fierro2. Esplenectomía3. Enfermedad neoplásica con metástasis (si hay necrosis, hay trombocitosis)4. Infección o inflamación crónica (artritis reumatoide, lupus eritematoso, etc.)
Tratamiento: Hidroxiurea para bajar el recuento de plaquetas Aspirina para evitar trombosis
Pronóstico: Sobrevida en general es de 10 años. Las 3 causas de muerte son: Trombosis Hemorragias (por déficit funcional, no de cantidad) Leucemia aguda
LEUCEMIA MIELOIDE CRÓNICA
Definición: Síndrome mieloproliferativo crónico causado por la traslocación de loscromosomas 9 y 22 – t(9;22) -, también llamada cromosoma Filadelfia.Se diferencia de la leucemia aguda por el grado de maduración celular. En este caso, las
células tumorales terminan siendo polimorfonucleares. Se diferencia de los otros
desórdenes mieloproliferativos crónicos en que esta enfermedad llegará siempre atransformarse en leucemia aguda.
Epidemiología: Corresponde al 15-20% de las leucemias del adulto. Incidencia de1-2/100.000 habitantes por año. Se presenta en promedio a los 50 años.
Clínica:- 40% asintomático- Fatigabilidad y anorexia (ya que es un cáncer)- Esplenomegalia gigante, pudiendo llegar a pesar 5 kg (bazo normal: 150-200 gr)- Dolor en hipocondrio izquierdo, por la esplenomegalia
- Hepatomegalia
Curso Clínico:1. Fase Crónica: En esta fase se pesquisan el 85% de los casos. Hayleucocitosis periférica (con todas las etapas de maduración) y trombocitosis. Lasobrevida es de 1 a 5 años, y con tratamiento el paciente remite fácilmente laenfermedad.2. Fase de Aceleración: Dura entre 3 y 8 meses, y comienza a presentarsecon anemia, trombocitopenia y leucocitosis pero ahora con predominio deformas inmaduras.3. Fase Final: Es la crisis blástica, vale decir, una transformación aleucemia aguda con presencia igual o mayor a 30% de blastos en la sangre
47

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 48/72
Integrado de Sistemas IJavier Pérez Bracchiglione
periférica. El 70% se transformará a leucemia mieloide aguda, y el 30% aleucemia linfática aguda. Esta etapa es refractaria al tratamiento.
Laboratorio:
Hemograma : Leucocitos > 100.000, a veces asociado a trombocitosis y basofilia.Hay presencia de todas las etapas de maduración que debiesenestar en la médula ósea en el frotis periférico. Esto nos sirve paradiferenciarlo con la leucemia aguda, donde sólo se encuentran
blastos en el frotis periférico.
Niveles: Puede verse disminuida la actividad de la fosfatasa alcalina de losneutrófilos (en infecciones estaría alta por activación de los
neutrófilos).Se presenta además hiperuricemia
Biopsia de Médula Ósea : Hipercelularidad. Aumento relación mieloide/eritroide.Cantidad de blastos y promielocitos <10%, lo quesirve para determinar si se está transformando en unaleucemia aguda (que tiene blastos igual o mayor a 20%en médula ósea).Aumento considerable de la serie granulocítica
Diagnóstico: Ante la sospecha, determinar la presencia del cromosoma Filadelfia o delgen BCR-ABL, mediante citogenética y biología molecular respectivamente. Si seencuentran, se confirma el diagnóstico
Tratamiento: Imatinib (nombre comercial: Glivec), 400 mg/día. Con este tratamiento, el
hemograma se ve completamente normal, ya que el medicamento actúa a nivelcitogenético y molecular. El problema es que cuesta dos millones de pesos almes.
Hidroxiurea, que mejora los parámetros hematológicos (baja recuentoleucocitario). Tiene un 90% de remisión, pero transitoria y, por tanto, paliativa.
Transplante de Médula Ósea alogeneico durante la fase crónica. Tiene una tasade curación del 50%.
METAPLASIA MIELOIDE AGNOGÉNICA (MIELOFIBROSIS)
Definición: Fibrosis de la médula ósea como resultado de la hiperactividad y proliferación fibroblástica inducida por factores de crecimiento anormalmente derivadosde megacariocitos expandidos clonalmente. Produce anemia, que evoluciona a
pancitopenia. Es el más raro de todos los síndromes mieloproliferativos crónicosexplicados en este capítulo.
Clínica: Poco específica- Fatigabilidad- Esplenomegalia gigante, que produce saciedad precoz- Disminución de peso
48

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 49/72
Gammapatía monoclonal: Neoplasia. Se presenta un peak estrecho en la posicióngamma.
Gammapatía policlonal: Este peak ancho yno muy grande significa que hay muchos
tipos de inmunoglobulinas sintetizándose.Traduce un síndrome de respuestainflamatoria sistémica, no un tumor.
Integrado de Sistemas IJavier Pérez Bracchiglione
- Diaforesis nocturna- Fiebre de predominio vespertino- Dolor óseo
Laboratorio:
Hemograma: Anemia con células en lágrimaLeucocitos normales (o elevados muy al principio de laenfermedad)Reacción leucoeritroblásticaEn etapas avanzadas produce pancitopenia.
Tratamiento: Hidroxiurea, para la trombocitosis Transfusión de glóbulos rojos Transplante de médula ósea alogeneico. Es el único tratamiento potencialmente
curativo, pero se puede hacer sólo en gente joven, por lo cual muy poca gentecalifica para realizárselo.
Pronóstico: Sobrevida entre 3 y 10 años. Las causas de muerte pueden ser:1. Trombosis (en la etapa de recuento de plaquetas aumentadas)2. Hemorragia (por trombopenia, en etapas avanzadas de la enfermedad)3. Infección (por avance de la fibrosis y baja de leucocitos4. Transformación leucémica (5-22% de los casos)
GAMMAPATÍAS MONOCLONALES
Definición: Neoplasias hematológicas que se caracterizan por tener un peak estrecho enla posición gama en la electroforesis de proteínas, dado por el marcador tumoralllamado paraproteína, proteína M o proteína monoclonal, que es un subtipo deinmunoglobulina producido por un linfocito B tumoral. La proteína M tiene 2 cadenas
polipeptídicas pesadas de la misma clase o subclase y dos cadenas livianas del mismotipo.
Cadenas pesadas: gamma, alfa, mu, delta, epsilon
Cadenas livianas: kappa, lambda
49

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 50/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Se puede hacer una inmunoelectroforesis para identificar si la inmunoglobulina tieneexclusivamente cadena liviana kappa o lambda, buscando en sangre o en orina(proteinuria Bence-Jones)
MIELOMA MÚLTIPLE
Definición: Es la proliferación neoplásica de un clon de células plasmáticas (derivadode un linfocito B) que se caracteriza por producir una proteína monoclonal. Esta últimaes útil para hacer el diagnóstico y el tratamiento.
Hay varios tipos de Mieloma múltiple (en orden de frecuencia): IgG (kappa o lambda) / IgA (kappa o lambda) / Productor de cadenas
livianas (kappa o lambda) / IgD (kappa o IgD lambda)
Siempre produce sólo un subtipo de cadena liviana
Epidemiología: 1% de las neoplasias, 10% de las neoplasias hematológicas, incidenciade 4/100.000 habitantes por año, aparición a los 70 años aproximadamente.
Clínica:
50
Gammapatíasmonoclonales
De significaciónincierta (IgG, IgA,IgD, IgM)
Malignas
Mieloma Múltiple(IgG, IgA, IgD,cadenas livianas)
Plasmocitoma
Enfermedadeslinfoproliferativas
Amiloidosis
Macroglobulinemia deWaldenstrom
Mieloma Múltiple
Mieloma MúltiplequiescenteMieloma osteoesclerótico(Síndrome de POEMS)
Plasmocitoma óseosolitario
Plasmocitomaextramedular
Linfoma Maligno
Primaria
Con Mieloma
Tríada clásica
Dolor óseo (66%)
Fatigabilidad (anemia)
Insuficiencia Renal (50%)

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 51/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Otros: Dolor por compresión de raíces nerviosas, aumento de la susceptibilidad ainfecciones (neumonía neumocócica, Gram [-], Herpes Zoster), sangramiento por trombopenia.
Laboratorio e Imágenes: Los exámenes se piden en este orden Hemograma: Anemia normocítica normocrómica
VHS > 100Formación de RoleauxOcasionalmente leucopenia y trombopenia
Electroforesis de proteínas: Peak monoclonal (80%)Hipogammaglobulinemia (10%)Sin alteraciones (10%)
Radiografía: Lesiones osteolíticas o fracturas patológicas(en columna, cráneo, pelvis, fémur, húmero)
Diagnóstico: Se ocupan los siguientes criterios
10% de células plasmáticas atípicas o inmaduras en el mielograma
+
Clínica sugerente más uno de los siguientes criterios:1. Proteína monoclonal > 3 g/dl2. Proteína monoclonal en orina3. Lesiones osteolíticas
o
Recuento > 30% de células plasmáticas
51Clasificación
SegúnEstadio
I(baja masa celular)Cumple todos estos
parámetros
II
III(alta masa celular)
Cumple al menos unode estos parámetros
Hb > 10 g/dlCalcio sérico normalIgG < 5 g/dl o IgA < 3 g/dlProteinuria Bence-Jones <4g/24hAusencia de lesionesosteolíticas
Hb < 8.5 g/dlCalcio sérico > 12 g/dlIgG > 7 g/dl o IgA > 5 g/dl
proteinuria de Bence-Jones >12g/24hlesiones osteolíticas (+)
Según Creatininemia A < 2 mg/dlB > 2 mg/dl

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 52/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Pronóstico: Sobrevida promedio de 2,5 a 3 años Peor en estadio IIIB Peor con baja albúmina y alta β-2 microglobulina Es incurable con quimioterapia
Tratamiento: Quimioterapia: Melfalan – Prednisona oral (1° línea)
Ciclos cada 4-6 semanas. Respuesta 50-60%. Dura hasta alcanzar plateau
VAD endovenoso (Vincristina, Adriamicina, Dexometasona)
Talidomida (3° línea)
Transplante de médula ósea antólogo: Tratamiento actual en menor de 65 años
Complicaciones
Complicación Cuadro clínico TratamientoHipercalcemia Náuseas y vómitos
ConstipaciónDebilidad muscular PoliuriaConfusión, incluso coma
HiperhidrataciónFurosemidaCorticoidesBifosfonatos
Hiperuricemia Nefropatía por ácido úricoGota
Alopurinol
Insuficiencia Renal Depósito de IgHipercalcemia
Nefropatía por ácido úricoAmiloidosisPielonefritis crónica
Lesión Osteolítica Compresión médulaespinal
Radioterapia +quimioterapia
Hiperviscosidad Síntomas neurológicos Plasmaféresis
52

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 53/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Visión borrosaInsuficiencia cardiacacongestiva
MIELOMA MÚLTIPLE QUIESCENTE
Definición: Es un mieloma que no llena los criterios diagnósticos. No tiene anemia, nohay insuficiencia renal ni lesiones osteolíticas, sino que sólo presente proteína Melevada o mielograma con células plasmáticas atípicas > 10%. No se trata
MIELOMA MÚLTIPLE OSTEOESCLERÓTICO
Es muy raro. Se caracteriza por el Síndrome de POEMS:P: Polineuropatía O: Organomegalia E: Endocrinopatía M: Proteína M
S: Cambios cutáneos (skin)
Tratamiento: Radioterapia a lesiones osteolíticas y quimioterapiaPronóstico: Sobrevida de muchos años; disminución de la neuropatía
PLASMOCITOMA ÓSEO SOLITARIO
Es un tumor de células plasmáticas localizado, sin lesiones osteolíticas en otras zonas .El diagnóstico es histológico, y el mielograma se presenta sin evidencias de mieloma
múltiple.Tratamiento: RadioterapiaPronóstico: 50% de sobrevida a los 10 años. Progresa a mieloma múltiple
GAMMAPATÍA MONOCLONAL DE SIGNIFICACIÓN INCIERTA
Hay proteína monoclonal < 3g/dl, existiendo un peak monoclonal. El mielogramarevela <10% de células plasmáticas, obligatoriamente sin anemia ni lesionesosteolíticas.
Pronóstico: Evolución a mieloma múltiple o a linfoma; por esta razón se llama“incierta”. Un 50% de los pacientes no evoluciona a nada.Tratamiento: Sólo segumiento
MACROGLOBULINEMIA DE WALDENSTROM
Definición: Es un desorden linfoproliferativo maligno, un tipo de Linfoma No Hodgkin,que se caracteriza por la proliferación de linfocitos B y células plasmáticas que
producen una proteína monoclonal IgM, invadiendo la médula ósea, linfonodos y el
bazo. Peak monoclonal de IgM es casi sinónimo de Macroglobulinemia de Waldenstrom
53

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 54/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Clínica: - Comienzo insidioso. Paciente de 70 años aproximadamente- Fiebre, sudoración nocturna, baja de peso- Palidez, sangramiento oronasal
- Lo más importante son los síntomas de hiperviscosidad (ya que la gammaglobulinaIgM es muy grande): Visión borrosa, cefalea, vértigo, parestesia, insuficiencia cardiaca,compromiso de conciencia desde somnolencia hasta el coma.- Esplenomegalia, Hepatomegalia, adenopatías periféricas, polineuropatía periférica,rara vez da dolor óseo.
Laboratorio: Hemograma: Anemia normocítica normocrómica
Formación de Rouleaux, VHS > 100
Electroforesis de Proteínas: Peak monoclonal por IgM (75% son kappa)
Mielograma: Infiltración por células plasmáticas y linfoidesAumento de basófilos tisulares
Test de Coombs directo: (+) ocasionalmente (ya que una de las complicacioneses la anemia hemolítica autoinmune)
Tratamiento: Quimioterapia oral: Clorambucil Quimioterapia endovenosa: Fludarabina; 2-cloro-deoxiadenosina
Plasmaferésis (hiperviscosidad)
Pronóstico: Sobrevida de 5 años. Es incurableAMILOIDOSIS
Es una enfermedad en la que el amiloide (proteína rara que normalmente no está presente en el cuerpo) se acumula en varios tejidos.
1) Amiloidosis Primaria
Definición: Proceso infiltrativo en el cual hay depósito extracelular de una proteína fibrilar, formada por cadenas livianas kappa o lambda.
Clasificación: Puede ser amiloidosis primaria propiamente tal o amiloidosis primaria asociada a mieloma múltiple (esta última con mal pronóstico)
Sospecha de enfermedad: El 98% de los casos ocurre sobre los 40 años. Dadala masiva infiltración del amiloide, el paciente puede debutar con una ICC, unsíndrome nefrótico, una neuropatía periférica, un síndrome del túnel carpiano,un síndrome de mala absorción.
Clínica: Es inespecífica. Aparte de los casos recién nombrados, presentanfatigabilidad, baja de peso, disnea (por compromiso cardiaco), edema (por compromiso cardiaco y renal), parestesias (por compromiso nervioso
54

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 55/72
Integrado de Sistemas IJavier Pérez Bracchiglione
periférico), y posible síncope e hipotensión ortostática (por compromisocardiovascular).
El examen físico revela hepatomegalia (25%), esplenomegalia (5%),macroglosia (13%), compromiso cutáneo en cualquier forma (petequias,
equimosis, pápulas, alopecia, engrosamiento de la piel de manera muyinespecífica)
Laboratorio:
Hemograma: AnemiaVHS aumentadoTrombocitosis (10%)
Electroforesis de proteínas: Peak monoclonal (45%)Hipogammaglobulinemia
Inmunoelectroforesis en orina o sangre
Confirmación Diagnóstica:
Biopsia subcutánea de pared abdominal: Es la prueba de mayor rendimiento; en un 80% de los casos se encuentra el amiloide
Biopsia rectal: Positiva en un 70% de los casos
Biopsia de médula ósea: Positiva en un 55% de los casos
Sobrevida: En general 2 años, pero depende del síndrome asociado
Tratamiento: Melfalan-Prednisona, tanto para primaria propiamente tal como para la asociada a mieloma múltiple. Con esto se espera sobrevida de hasta 3años.
2) Amiloidosis Secundaria
Es la más frecuente desde el punto de vista clínico. Se desarrolla en pacientes quetienen un estímulo antigénico crónico. Así, se puede ver asociado a:
- Artritis reumatoidea- Espondilitis anquilosante- Artropatía psoriática- Síndrome de Reiter
- Bronquiectasias- Osteomielitis- Tuberculosis
55
Enfermedadesinflamatoriasautoinmunes
Infecciones

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 56/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Aparte de estar relacionada con una enfermedad de base, la amiloidosissecundaria se diferencia de la primaria en que en la secundaria un 90% de los
pacientes presenta síndrome nefrótico que puede estar asociado a insuficienciarenal.
LEUCEMIA LINFÁTICA CRÓNICA
Definición: Es la proliferación clonal y acumulación de linfocitos B neoplásicos en lasangre, médula ósea, linfonodos y bazo.
Epidemiología: Corresponde al 30% de todas las leucemias (es la más frecuente), y larelación hombre:mujer es de 2:1
Clínica:- Asintomático (30-40%)- Fatigabilidad- Baja de peso- Poliadenopatías- Esplenomegalia- Fenómenos autoinmunes (AHAI y PTI)
Laboratorio:
Hemograma: Linfocitosis absoluta > 15.000
56

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 57/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Linfocitos pequeños y maduros en un 95%Restos de Gumprecht (restos de linfocitostumorales reventados en un frotis, por ser más lábiles)
Estudio Médula ósea: Puede hacerse por mielograma o por biopsiaInfiltración por linfocitosSospecha diagnóstica: >30% de linfocitos (V.N: 15%)
Inmunofenotipo: Confirma el diagnósticoCD5 (+); CD19 (+); CD20 (+); CD23 (+)
El más importante es el CD23, que está siempre presente y es el
que confirma el diagnóstico
Citogenética: Anormalidades en el 90% de los pacientesAlteración más frecuente: 13q- (sobrevida sobre 140 meses)Otras: 11q- y +12q (ambas con sobrevida de 79 meses); 17p-(sobrevida 32 meses)
Clasificación de Rai:Etapa Características PronósticoEstadio 0 Linfocitosis sola > 10 añosEstadio I Linfocitosis, linfoadenopatía 7 añosEstadio II Linfocitosis, espleno y/o
hepatomegalia7 años
Estadio III Linfocitosis, anemia (Hb <
11g/dL)
3 años
Estadio IV Linfocitosis, trombopenia(<100000)
3 años
Estadio 0: Sobrevida habitualmente de 15 o 20 años, pero si hay alteración citogenética de mal pronóstico, puede haber sobrevida de 3añosEstadio III: La anemia debe estar producida por infiltración de la médula ósea, no por otra causa
57
Tipo de complicaciones ConsecuenciasAutoinmunes Anemia Hemolítica
Trombocitopenia Neutropenia
Hipogammaglobulinemia Infecciones
Transformaciones Leucemia prolinfocíticaLinfoma de células grandesLeucemia aguda

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 58/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Tratamiento:
- Tratar estadios III y IV.
- Quimioterapia oral con clorambucil. Si no responde, dar quimio-terapia endovenosa confludarabina y 2-clorodeoxiadenosina. No es curativo.
- Corticoides sólo en caso de enfermedad autoinmune.
- Transplante de médula ósea sólo en gente joven. Curación del 40%.
EVALUACIÓN Y TRATAMIENTO DEL PACIENTE CONDIÁTESIS HEMORRÁGICA
Definición: Entendemos por diátesis hemorrágica el sangramiento de un paciente enforma espontánea o ante traumatismos mínimos
58

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 59/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Clínica:- Sangramiento espontáneo- Sangramiento excesivo (por ejemplo, gingivorragia al cepillarse los dientes)-
Sangramiento tardío (debido a injuria, por ejemplo, sangrar horas o días despuésde una extracción dental)- Anemia ferropriva secundaria a hemorragias- Historia de transfusiones (post-cesárea, post-parto)
Una historia exclusivamente de hematuria, melena o metrorragia no es sinónimo de
diátesis hemorrágica, pues un paciente con la enfermedad antes de presentar estos
síntomas ha tenido todos los anteriormente dichos.
Si no hay historia de sangramiento anormal en respuesta a trauma u operaciones, se
descarta diátesis hemorrágica, ya que estas enfermedades son congénitas.
Causas:- Alteración de la integridad de la pared vascular - Alteración en el número o función plaquetaria- Alteración en factores de coagulación: Déficit en factores, hemofilia A (déficitfactor VIII)Los desórdenes hemorrágicos congénitos se caracterizan porque el sangrado comienza
en la infancia y porque es muy frecuente encontrar historia familiar asociada. Sinembargo, una historia negativa no descarta la enfermedad congénita (por ejemplo, lahemofilia A tiene un 30% de ausencia de historia familiar).
Si un paciente no tiene historia familiar ni antecedentes de hemorragia en la infancia, pensar en drogas asociadas, por ejemplo, aspirina.
59
Componentes de la Coagulación:
Cascada de la Coagulación Anticoagulantes Naturales:
Antitrombina III, ProteínaC y Proteína F Sistema de la Fibrinolisis Plaquetas
La principal vía de activación de lacascada de coagulación es la víaextrínseca. Cuando hay lesióntisular, se expone el factor tisular,que se une al factor VII. Elcomplejo desencadena la cascada
hasta formar el coágulo mismo.
La vía intrínseca a través del factor XII es sólo un potenciador de la víaextrínseca.

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 60/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Es importante diferenciar entre desorden primario y desorden secundario de lahemostasia:
Desorden primario : Alteración de la plaqueta o del vaso sanguíneo Desorden secundario : Alteración de los factores de coagulación
Característica Defecto Plaqueta Déficit FactoresCoagulación
Sitio sangrado Piel, Mucosas Profundo, Tejidos Blandos(musculares, articulaciones)
Petequias Presentes Ausentes
Equímosis Pequeñas, superficiales Grandes, Palpables
Hemartrosis, Hematomasmusculares
No existe Frecuente
Sangramientos Post-Quirúrgicos
Inmediato Tardío
Laboratorio: Las pruebas básicas para evaluar la coagulación son:
Recuento de plaquetas
Test de Sangría de Ivy: Se hace una incisión en el antebrazo de 1 cm de largo por
1 mm de profundida. El valor normal es antes de los 9 minutos con 30 segundos. Untiempo de sangría de Ivy prolongado lo encontraremos en:- Trombopenia con recuento < 50.000- Anormalidad cualitativa de las plaquetas (propio del Síndromeurémico)- Enfermedad de Von Willebrand
Tiempo de Protrombina: Evalúa la vía extrínseca de la coagulación (factores VII – X – V – II). El valor normal es de 70 a 120% y de 11 a 17 segundos.
Tiempo de tromboplastina parcial activado (TTPa o TTPk): Evalúa la víaintrínseca (factores XII – XI – IX – VIII). El valor normal es de 27 + 5
Fibrinógeno
HEMOFILIA A
Definición: Es el déficit del factor VIII, hereditario, ligado al sexo. Es la deficienciacongénita grave de la coagulación más común
Epidemiología: Prevalencia de 1/10.000 habitantes
60

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 61/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Clasificación CuantificaciónFactor VIII
Clínica
Leve 6-25% Tendencia a sangramiento excesivo con traumasevero o cirugía
Moderado 2-5% Hemorragia con traumatismo leve
Severo <1% Sangramiento espontáneo y severo (articulaciones,músculos). Habitualmente quedan con secuelas.
Diagnóstico: TTPa Prolongado (mientras más prolongado, más severo) Cuantificación factor VIII: Disminuido Otros exámenes normales (Tiempo de protrombina, Tiempo de sangría de Ivy,
Fibrinógeno)Tratamiento:- Crioprecipitados (ricos en factor VIII coagulante y factor Von Willebrand)- Liofilizados de Factor VIII (mejor, ya que es sólo factor VIII). Dar la dosis cada12 hrs por 5-14 días según la siguiente fórmula:
- Siempre dar un antifibrinolítico para disminuir la fibrinolisis primaria y asegurar la actividad del coágulo, previniendo un sangramiento tardío. Podemos dar ácido
tranexámico en dosis de 10 mg/kg cada 12 hrs
Complicaciones: Hemartrosis, transmisión viral de Hepatitis y VIH (especialmente elrecibir los crioprecipitados)
HEMOFILIA B
Definición: Es el déficit del factor IX, con una transmisión ligada al cromosoma X.
Epidemiología: Prevalencia de 1/60.000 habitantes
Clínica: Igual a la de la Hemofilia A
Diagnóstico: TTPa prolongado Cuantificación factor IX: Disminuido Otros exámenes normales (Tiempo de protrombina, Tiempo de sangría de Ivy,Fibrinógeno)
Tratamiento:-
Plasma fresco congelado. Dosis de 10-15 ml/kg- Liofilizado de factor IX: Se da cada 24 hrs. Las unidades se calculansegún:
61
Peso (kg) x % de aumento deseado (dosis)2
Peso (kg) x % de aumento deseado (dosis)

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 62/72
Integrado de Sistemas IJavier Pérez Bracchiglione
ENFERMEDAD DE VON WILLEBRAND
Definición: Defecto de la hemostasia primaria de transmisión autosómica dominante, condéficit del factor de Von Willebrand (encargado de la adhesión plaquetaria). Es undesorden de sangramiento congénito, el más común, habitualmente leve.
Clínica: La de un desorden de la hemostasia primaria. Habitualmente se presenta conepistaxis, gingivorragia y menorragia. La enfermedad se puede desenmascarar con el usode AINEs.
Laboratorio: Tiempo de sangría de Ivy: Prolongado Otros exámenes normales (Tiempo de protrombina, TTPa, Fibrinógeno)
Tratamiento: Crioprecipitados. 1U / 10kg por dosis, cada 8-12 hrs por 5-10 días.
DEFICIENCIA DE FACTORES ADQUIRIDA: ENFERMEDAD HEPÁTICA
Todos los factores de la coagulación (salvo el Von Willebrand) se sintetizan en elhígado. Así, el prototipo de esta deficiencia es la cirrosis. Toman especial importancia los
factores vitamina K dependientes: II, VII, IX y X.Laboratorio: Tiempo de Protrombina: Disminuido (<70%) TTPa: Prolongado (>32 seg) Factores V, VII, IX y X: Disminuidos (vit. K dependientes) Factor VIII y Fibrinógeno: Aumentados, normales o disminuidos, dado que son
proteínas de fase aguda
Tratamiento: Requiere tratamiento cuando llega con hemorragia o cuando requiere unacirugía de urgencia. Administrar:
- Plasma fresco congelado: 10-15 ml/kg por dosis (2 a 3 unidades)- Vitamina K: Tiempo de acción de 4-24 hrs, en dosis de 10-20 mg, en inyecciónintramuscular (es liposoluble)- Crioprecipitados sólo si el fibrinógeno < 75-100 mg/dl, en dosis de 1U / 10 kg por dosis
ANTICOAGULANTES ORALES
En el comercio chileno hay dos: Warfarina y Acenocumarol. Ambos actúan mediante elantagonismo de la vitamina K, produciendo déficit de los factores II – VII – IX – IX (por tanto, afectan vías intrínseca y extrínseca).
62

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 63/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Laboratorio: Tiempo de protrombina: Disminuido (<70%) TTPa prolongado (>32 seg)
Tratamiento: Como urgencia, en caso de hemorragia o cirugía
- Plasma fresco congelado: 10-15 ml/kg por dosis cada 12 hrs No dar vitamina K, sobre todo en paciente con prótesis valvular mecánica.
MONITOREO CLÍNICO
Sangremiento espontáneo inmediato:Pensar en problema de la hemostasia primaria
Trombopenia Enfermedad Von Willebrand
Alteración de la función plaquetaria (incluyendo AINEs)Sangramiento retardado (horas o días después de la injuria):
Pensar en problema de la hemostasia secundaria Déficit de factor de la coagulación
TROMBOFILIA
Definición: Predisposición individual a padecer episodios tromboembólicos. Es la
enfermedad en sí, y no un factor predisponente. Afecta con mayor frecuencia al territoriovenoso, pero puede afectar también al arterial.
63

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 64/72
Riesgo detrombosis venosa
Integrado de Sistemas IJavier Pérez Bracchiglione
Patogenia:1) Pérdida de endotelio y exposición de
subendotelio2) Activación plaquetaria por interacción con
agonistas circulantes3) Activación directa de la coagulación
4) Estasia
Situaciones de riesgo trombótico adquiridas:1. Neoplasia: Hay neoplasias que incluso antes de dar manifestaciones clínicas pueden dar eventos trombóticos, loque se conoce como el síndrome paraneoplásico. Los másfrecuentes son, en primer lugar, el cáncer de próstata, y ensegundo lugar el cáncer de páncreas.2. Embarazo y Puerperio3. Anticonceptivos orales4. Diabetes: Por la lesión a nivel endotelial. Riesgo de trombosis arterial5. Prótesis valvulares y vasculares artificiales6. Inmovilización prolongada7. Cirugías de todo tipo, especialmente las ortopédicas, por dos mecanismos:Primero por la inmovilización, y segundo por la lesión tisular que rápidamenteactiva la cascada de coagulación8. Obesidad9. Síndromes de hiperviscosidad, como la Policitemia10. Infecciones
TROMBOFILIA HEREDITARIA
Definición: Tendencia al tromboembolismo venoso genéticamente determinado. Tiene
la particularidad de que los eventos trombóticos recurren con frecuencia. Suele existir historia familiar.
64
Complicaciones de lasTrombosis
Efectos de la obstrucción del vaso
Embolización a distancia delmaterial trombótico ArterialVenosa
Necrosis
Embolia de pulmón
Mecanismos antitrombóticos:1. Endotelio intacto2. Inhibición por
anticoagulantes naturales(Antitrombina III, proteínaC, proteína S)
3. Sistema fibrinolítico
Congénita
Adquirida
Clasificación

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 65/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Causas:1. Mutación del factor V Leiden (también llamada resistencia a la proteína C
activada). Corresponde al 40%. La mutación del factor V lo hace resistente a suanticoagulante natural, la proteína C activada
2. Mutación del gen de la protrombina3. Déficit de proteína C4. Déficit proteína S5. Déficit antitrombina III
Sospecha diagnóstica:- Trombosis venosa profunda antes de los 45 años- Antecedente de trombosis venosa recurrente- Historia familiar de trombosis- Necrosis cutánea asociada a cumarínicos (acenocumarol, warfarina), queson anticoagulantes orales. Éstos inhiben los factores II, VII, IX y X, que tardan
72 horas en bloquearse, pero también interfieren con la síntesis deanticoagulantes (proteína S y C, que también dependen de vitamina K), quetienen vida media más corta y por ende se bloquean antes. Así, al principio, hayun estado inicial procoagulante que a los 3 días pasa a ser anticoagulante (por eso se receta heparina por 3 días antes que los anticoagulantes orales)- Trombosis arterial antes de los 30 años- Trombosis en lecho vascular inusual (ejemplo: portal, suprahepáticas,mesentérica, cerebrales)
El riesgo de trombosis varía según el defecto del paciente:
Defecto asociado Riesgo de padecer trombosisDéficit proteína S 8,5 veces
Antitrombina 8,1 vecesDéficit proteína C 7,3 vecesFactor V Leiden 2,2 veces
Tratamiento tromboembolismo venoso:- Inicialmente el tratamiento es igual para paciente con o sin trombofilia
Heparina no fraccionada o heparina estándar por 5 días o heparina de bajo peso molecular
Al 3° día superponer anticoagulante oral- A largo plazo: Prevenir la recurrencia. Anticoagulantes orales por 6-12 meses
Tratamiento de Mantención: Se evalúa si mantener un tratamiento indefinido según:- Número de recurrencias- Número de defectos trombóticos- Factores de riesgo asociados- Opinión del paciente
Indicaciones de tratamiento de por vida:- 2 o más eventos de trombosis espontáneas- Episodio de trombosis en déficit de antirombina III o de proteína S (son las más
trombogénicas)- Asociado a síndrome antifosfolípidos
65

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 66/72
Potenciación antitrombina III Heparina
Integrado de Sistemas IJavier Pérez Bracchiglione
- Episodio trombótico casi fatal (embolia pulmonar, trombosis cerebral,trombosis mesentérica, trombosis portal)
- Trombosis espontánea en sitio inusual- Episodio trombótico en presencia de más de un defecto genético (dos
trombofilias hereditarias
- Embarazo: En este caso, la anticoagulación se hace con heparina, ya que losanticoagulantes orales producen malformaciones congénitas
La warfarina usada de por vida como tratamiento anticoagulante es efectiva y no es peligrosa: no hay diferencia en la mortalidad entre anticoagulados de por vida yanticoagulados por 6 meses.
TROMBOFILIA ADQUIRIDASíndrome de Anticuerpos Antifosfolípidos
Definición: El síndrome de anticuerpos antifosfolípidos es el prototipo de trombofiliaadquirida, y se define como la presencia de autoanticuerpos dirigidos contra estructurasfosfolipídicas unidas a diferentes proteínas.
Se presentan dos tipos de anticuerpos:1. Anticardiolipinas: Son de tipo IgG e IgM, y pueden activar per se la cascada
de coagulación2. Anticoagulante lúpico: Es un anticuerpo que in vitro prolonga el TTPa
(siendo anticoagulante), pero que in vivo induce trombosis (siendo procoagulante).
Clínica: Se presenta con trombosis arterial o venosa, abortos a repetición (por trombosis a nivel placentario que producen una insuficiencia placentaria) ytrombocitopenia leve.
Tratamiento: Al hacer el evento trombótico, se debe dar heparina de bajo pesomolecular seguido de anticoagulantes orales, mínimo por 6 meses. A pesar de ser autoinmune, no es útil el uso de corticoides, citotóxicos ni inmunosupresores.
ESTUDIO DE LA TROMBOFILIA
Ante la sospecha de una trombofilia, solicitar: Resistencia a la proteína C activada / Factor V Leiden Mutación del gen de la protrombina (por técnica de PCR) Niveles de Antitrombina, Proteína C y Proteína S Anticardiolipinas IgG e IgM y anticoagulante lúpico
ANTICOAGULANTES
Definición: Conjunto heterogéneo de fármacos que producen disminución de lacoagulación.
66

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 67/72
Mecanismos deAcciónBloqueo directo de la trombina
Interferencia en la síntesis de factores decoagulación vitamina K dependientes (II,VII, IX, X)
Hirudina
Anticoagulantesorales
Integrado de Sistemas IJavier Pérez Bracchiglione
HEPARINAS Potenciador de la actividad de la antitrombina III
Heparina no fraccionada (HNF)
Compuesta porglucosaminoglucanos, exactamente50 sacáridos.
Potencia a la AT III al unirse einducir un cambio estructural.
La HNF inhibe a la trombina y alfactor Xa
Farmacocinética:- En cuanto a su eliminación, tieneuna captación muy rápida por partedel endotelio y los macrófagos.
Posteriormente sufre eliminaciónrenal- Por lo anterior, tiene una vidamedia muy variable: de 30 a 150minutos, con alta unión a proteínas- Por tanto, su biodisponibilidad esescasa y variable, lo cual determinauna variabilidad en el tratamientoanticoagulante. Debemos usar untratamiento que sea continuo ycontrolar con TTPa
Heparina de bajo peso molecular(HBPM)
Compuesta por glucosaminoglucanos,que según la HPBM puede tener 7-15sacáridos.
Potencia a la AT III al unirse e inducirun cambio estructural
Por tener la cadena más corta, inhibesólo al factor Xa
Farmacocinética:- Biodisponibilidad elevada por menortendencia a unirse a proteínas plasmáticas- Vida media de 2 a 4 veces mayor que la
HNF por escasa afinidad a célulasendoteliales y macrófagos. Por lo mismo,permite su utilización por vía subcutáneo 1 o2 veces al día- Cinética de eliminación regular- Su valor sanguíneo es predecible y conescasa variabilidad individual- No necesita un control analítico conexámenes de laboratorio
Indicaciones de las Heparinas:
1. Profilaxis Enfermedad tromboembólica venosa
67

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 68/72
Integrado de Sistemas IJavier Pérez Bracchiglione
HNF: Dosis de 5000 U c/12 horas vía subcutáneaEn cirugía, 2 horas previoPermite una disminución de 60% de TVP
HBPM: Dalteparina: 2500 U / Enoxaparina 2000 U / Nadroparina 2850 UTodas cada 24 horas, vía subcutáneaEficacia algo superior a HNF. Ventaja: Se da sólo 1 vez al día
2. Tratamiento TVP y TEP
HNF: Iniciar con bolo de 5000 U endovenosa, mantener con 400-500 U/kg cada24 horas durante 5-10 días. Controlar con TTPa
Disminuye mortalidad TEP de 25 a 10%. Disminuye TEP en pacientes con TVP a <5%
HBPM: Dalteparina 120 U/kg, Enoxaparina 100 U/kg, Nadroparina 85 U/kg Todas cada 12 horas, vía subcutánea
Ventajas: No requiere monitoreo con TTPa, es subcutánea, permite tratar al pacienteen el domicilio, sin ser hospitalizado
Complicaciones: Hemorragias, Trombocitopenia, Osteoporosis
INHIBIDORES DIRECTOS DE LA TROMBINA
Mecanismo de Acción: Bloquean directamente a la trombina, incluso estando unida ala fibrina.
Usos: Prevención y tratamiento de trombosis arterial y venosa
Medicamentos: Hirudina (tratamiento trombocitopenia), Argatroban (inducida por heparina), Bivalirudina (como alternativa a la heparina en intervención coronaria
percutánea), Diserudina (profilaxis de tromboembolismo venoso en reemplazo decadera)
No han demostrado aún una capacidad anticoagulante superior a la de la heparina. Se
usa sólo como alternativa
ANTICOAGULANTES ORALES
Mecanismo de acción: Inhibe los factores de coagulación vitamina K dependientes (II,VI, IX, X), pero también inhibe a los factores de anticoagulación vitamina K dependientes (proteína C, proteína S)
Medicamentos y Posología: Acenocumarol: 2 mg al díaWarfarina: 2,5 – 5 mg al día (es más estable)
Acción terapéutica: Demora hasta 72 horas. Se afectan primero los factores deanticoagulación que los de coagulación, por lo cual hay que administrar heparina por 3días antes de comenzar con anticoagulantes orales
68

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 69/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Usos: Tromboembolismo venoso (TVP Y TEP). Su función es prevenir la recidivaTVP: Usar por 3 mesesTEP: Usar por 3 meses a 1 añoSi el paciente posee prótesis valvular mecánica o fibrilación auricular, es
mejor usar de por vida
Control: A través de Tiempo de protrombina y a través del INR (razón normalizadainternacional).
Contraindicaciones: Diátesis hemorrágica, Hemorragia activa, HTA grave nocontrolada, Hemorragia intracraneal, Aneurisma intracerebral, Embarazo (producenmalformaciones congénitas). Evitar el uso concomitante con AINEs, ya que éstosinhiben la agregación plaquetaria, por lo cual aumento mucho el riesgo de hemorragia
COAGULACIÓN INTRAVASCULAR DISEMINADA (CID)
69

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 70/72
Es la más común. El 70% de los pacientes con sepsis presenta CID,siendo esta última índice pronóstico de la sepsis.
Inhibidores naturales de la coagulación: Disminuidos(proporcionalmente a la gravedad de la sepsis). Esto se da por Consumo por activación de la coagulación Disminución de la síntesis por disfunción hepática Inactivación proteolítica (elastas), secretada por los PMN
Actividad Fibrinolítica: Disminución relativa, dado que aumenta elnivel de inhibidor del activador del plasminógeno tisular (PAI-1)
Integrado de Sistemas IJavier Pérez Bracchiglione
Coagulación Normal: Su función es mantener la integridad del sistema circulatoriodespués de la injuria de un vaso sanguíneo. Recordemos que esto se logra gracias a lainteracción de los mecanismos regulatorios de la coagulación:
1. Sistema de coagulación sanguínea (factores de
coagulación)
2. Inhibidores naturales de la coagulación: La antitrombina,además de inhibir la trombina, inhibe al factor X. Las proteínas C y S inhibiránal factor V, y el inhibidor de la vía del factor tisular inhibirá la cascada decoagulación desde el inicio.
3. Sistema fibrinolítico: El plasminógeno es convertido, por acción del activador del plasminógeno tisular, en plasmina, la cual empieza adeshacer el coágulo de fibrina, formando productos de degradación. Dentro deéstos, es importante el dímero D en clínica, ya que puede ser medido en
laboratorio
Definición: La coagulación intravascular diseminada se define como un desordenasociado con trombosis y hemorragia (pero que favorece la trombosis), sistémico y queocurre cuando los mecanismos de coagulación fisiológica que guían a la formación detrombina y plasmina son gatillados por mecanismos patológicos (sepsis, cáncer ytrauma) de una manera diseminada y a través de toda la vasculatura.
Causas:
1) Sepsis:
2) Trauma
3) Cáncer
4) Complicaciones obstétricas
La cascada de coagulación se activa por un estímulo patológico que son las citoquinas.En los pacientes con infecciones, sepsis o cáncer, aumenta la secreción de citoquinas
proinflamatorias, dentro de las cuales es de suma importancia el TNF-α.
70

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 71/72
- Petequias y equimosis- Gangrena- Sangramiento por sitios de
punción o por heridas- Disfunción de órganos
- Fiebre- Hipotensión- Acidosis (dada la hipoperfusión)- Hipoxia
Integrado de Sistemas IJavier Pérez Bracchiglione
Fisiopatología: Hay trombosis micro y macrovascular, que al interferir el correcto flujosanguíneo producirán isquemia. Así, se puede presentar falla renal, hepática, respiratoriay por último, cuando se depletan los factores de coagulación y disminuye a nivelescríticos el número de plaquetas, aparece la hemorragia
Clínica: Dada principalmente por la enfermedad de base que desencadena la CID
Laboratorio: Antitrombina III: Disminuidos los niveles plasmáticos en el100% de los pacientes Productos de degradación de la fibrina: Aumentados. Podríanestar normales por supresión severa de la fibrinolisis Recuento de plaquetas: 2.000 – 100.000. Tiene directa relacióncon la severidad de la sepsis y de la CID Tiempo de trombina Fibrinógeno: Normalmente no se compromete mucho, ya que esuna proteína de fase aguda Tiempo de protrombina: Puede disminuir por consumo defactores de coagulación TTPa: Prolongada
Diagnóstico: Debemos tener evidencia de:1. Actividad procoagulante2. Evidencia de activación fibrinolítica (que en paciente CID aumenta, pero no en
la proporción correcta, por lo cual se dice que hay una disminución relativa) quese puede medir por recuento del dímero D
3. Evidencia de consumo de los inhibidores naturales de la coagulación, es decir,niveles plasmáticos disminuidos de AT III, proteína C, proteína S
Tratamiento: De la enfermedad de base. Luego, para la CID se han propuesto: Heparina: Sólo útil en CID crónica leve (neoplasia), donde existe unmínimo nivel de AT III para que funcione la heparina Transfusión de plasma fresco congelado y plaquetas: Sólo si hay unsangramiento activo, si hay alto riesgo de hemorragia o si va a ser sometido a un
procedimiento altamente invasivo Agentes antifibrinolíticos Anticuerpo monoclonal Anti TNF-α Antitrombina III recombinante Proteína C activada recombinante: Además de ser anticoagulante, tiene
efecto antiinflamatorio, lo que disminuye la síntesis y secreción de TNF-α-
71

5/14/2018 HEMATOLOGIA-Javier Perez Bracchigglione - slidepdf.com
http://slidepdf.com/reader/full/hematologia-javier-perez-bracchigglione 72/72
Integrado de Sistemas IJavier Pérez Bracchiglione
Además, tiene efecto antiapoptótico. Por tanto, mejora la CID y la sobrevida delos pacientes.
72