
GUIDANCE DOCUMENT...GUIDANCE DOCUMENT Preparation of Regulatory Activities in the “Non-eCTD...
51
GUIDANCE DOCUMENT Preparation of Regulatory Activities in the “Non-eCTD Electronic-Only” Format Published by authority of the Minister of Health Date Adopted 2016/10/25 Effective Date 2016/10/25 Revised Date 2016/10/31 Health Products and Food Branch
Transcript of GUIDANCE DOCUMENT...GUIDANCE DOCUMENT Preparation of Regulatory Activities in the “Non-eCTD...

GUIDANCE DOCUMENT Preparation of Regulatory Activities in the “Non-eCTD Electronic-Only” Format
Published by authority of the Minister of Health
Date Adopted 2016/10/25 Effective Date 2016/10/25 Revised Date 2016/10/31
Health Products and Food Branch

Our mission is to help the people of Canada maintain and improve their health.
Health Canada
The Health Products and Food Branch's mandate is to take an integrated approach to the management of the risks and benefits to health related products and food by: • minimizing health risk factors to Canadians while
maximizing the safety provided by the regulatory system for health products and food; and
• promoting conditions that enable Canadians to make healthy choices and providing information so that they can make informed decisions about their health.
Health Products and Food Branch © Minister of Public Works and Government Services Canada 2016 Également disponible en français sous le titre : Ligne directrice : Préparation des activités réglementaires des drogues en format « électronique autre que le format eCTD »

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 i
FOREWORD Guidance documents are meant to provide assistance to industry and health care professionals on how to comply with governing statutes and regulations. Guidance documents also provide assistance to staff on how Health Canada mandates and objectives should be implemented in a manner that is fair, consistent and effective. Guidance documents are administrative instruments not having force of law and, as such, allow for flexibility in approach. Alternate approaches to the principles and practices described in this document may be acceptable provided they are supported by adequate justification. Alternate approaches should be discussed in advance with the relevant program area to avoid the possible finding that applicable statutory or regulatory requirements have not been met. As a corollary to the above, it is equally important to note that Health Canada reserves the right to request information or material, or define conditions not specifically described in this document, in order to allow the Department to adequately assess the safety, efficacy or quality of a product. Health Canada is committed to ensuring that such requests are justifiable and that decisions are clearly documented. This document should be read in conjunction with the accompanying notice and the relevant sections of other applicable guidance documents.

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 ii
REVISION HISTORY
Date Description
June 15, 2015 Notice: Health Canada’s requirements for filing a regulatory activity in “non-eCTD electronic-only” format. (DIN)
September 25, 2015 Guidance Document: Preparation of Drug Regulatory Activities in “Non-eCTD Electronic-Only” Format. (DIN, MF)
February 29, 2016 Guidance Document: Preparation of Drug Regulatory Activities in “Non-eCTD Electronic-Only” Format. (Div.1, Div.5, Div.8, DSUR, Post-market Vigilance, Level III, DNF and MF)
October 31, 2016 Guidance Document: Preparation of Regulatory Activities in “Non-eCTD Electronic-Only” Format. (Medical Devices, and Veterinary Drugs)
format, as soon as possible.non-eCTD guidance document to reflect the amended requirements for all products filed in non-eCTD document: Preparation of regulatory activities in the non-eCTD format. We intend to update the in Health Canada’s IMDRF ToC document supersedes the requirements indicated in this Guidance Medical Devices prepared using the Summary Technical Document (STED) structure. Theinformation structure. Since April 2019, Health Canada has no longer been accepting regulatory activities for website to provide guidance in preparing regulatory activities for Medical Devices using the ToC IMDRF table of contents for medical device applications guidance is available on the Health Canada structure to be used in preparing Medical Device regulatory activities. The Draft Health Canada developed by the International Medical Device Regulators Forum (IMDRF), as the recommended January 2, 2020 - Please note that Health Canada has adopted the Table of Contents (ToC) format,

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 iii
How to read this document: To facilitate the identification of areas of interest for the reader, colours with a dotted lined box around the information (see legend below) have been used to visually display specific requirements pertaining to individual or groups of regulatory activities and transactions. General requirements pertaining to all regulatory activities and transactions in scope of this guidance document have no colour nor a dotted lined box associated with them. Legend
Div
.1
Part C, Division 1 of the Food and Drug Regulations (e.g., DINA, DINB).
Div
.5
Part C, Division 5 of the Food and Drug Regulations (e.g., CTA, CTA-A, CTA-N, CTSI).
Div
.8
Part C, Division 8 of the Food and Drug Regulations (e.g., NDS, SNDS, ANDS, NC).
DS
UR
Developmental Safety Update Report (DSUR)
PV
-D
ata
Post-market Vigilance Data (e.g., PSUR, PBER, RMP).
Lev
.3 Post NOC Level III Changes Forms pursuant to Guidance Document: Post-Notice of Compliance
(NOC) Changes: Framework Document.
DN
F
Drug Notification Forms (DNF) pursuant to Part C, Division 1 and Division 8 of the Food and Drug Regulations.
MF
Master Files (MF)
MD
Medical Device Regulatory Activities pursuant to section 32 of the Medical Devices Regulations (e.g., Class II, III & IV licences & amendments, and Faxback amendments)
VD
Veterinary Drugs pursuant to Part C, Division 1 and Division 8 of the Food and Drug Regulations.

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 iv
TABLE OF CONTENTS
1 INTRODUCTION ...................................................................................................................... 1
1.1 Policy Objectives ............................................................................................................... 1
1.2 Policy Statements ............................................................................................................... 2
1.3 Scope and Application ....................................................................................................... 3
1.4 Background ........................................................................................................................ 8
2 STRUCTURE AND CONTENT OF A REGULATORY ACTIVITY ..................................... 9 2.1 Cover Letter ....................................................................................................................... 9 2.2 Folder Structure ............................................................................................................... 12
2.2.1 Top Level Folder and Dossier Identifier ............................................................ 15 2.2.2 Common Technical Document (CTD) Folder Structure .................................... 15 2.2.3 Medical Device Folder Structures...................................................................... 17 2.2.4 Veterinary Drugs Folder Structure ..................................................................... 18
2.3 File Naming Convention .................................................................................................. 18
3 TECHNICAL REQUIREMENTS FOR REGULATORY ACTIVITIES ............................... 19 3.1 File Format ....................................................................................................................... 19 3.2 Signature .......................................................................................................................... 21 3.3 Transmission of Electronic Data...................................................................................... 21
APPENDICES .............................................................................................................................. 26
Appendix A: Contacts .............................................................................................................. 26
Appendix B: Glossary of Terms ............................................................................................... 28
Appendix C: Division 1 Sample Folder Structure(s) ............................................................... 30
Appendix D: Division 5 Sample Folder Structure(s) ............................................................... 31
Appendix E: Division 8 Sample Folder Structure(s) ............................................................... 32
Appendix F: Master Files (MF) Sample Folder Structure(s) ................................................... 33
Appendix G: Distribution of MF Information between the Applicant and Restricted Parts .... 37
Appendix H: Medical Device Sample Folder Structures for IMDRF TOC format ................. 43

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 1
1 INTRODUCTION Health Canada is pleased to announce a revision to the Guidance Document: Preparation of Drug Regulatory Activities in the “Non-eCTD Electronic-Only” Format, published in February 2016, to include Medical Device and Veterinary Drug regulatory activities in “non-eCTD electronic-only” format. This document defines the filing requirements and provides guidance on the structure, content and format of regulatory activities filed in “non-eCTD electronic-only” format. Future refinements and subsequent iterations of this guidance document will continue to be necessary as a result of the transition from paper format to electronic format. Health Canada would like to provide an advanced notice that we are considering January 1st, 2018 for mandatory filing of all NDS, SNDS, SANDS and ANDS regulatory activities (for human drugs) in eCTD format. Once a sponsor files a regulatory activity in eCTD format, all additional information and subsequent regulatory activities (such as NC, PSUR, RMP, etc.) must also be filed in eCTD format. The mandatory eCTD requirement is established to keep aligned with other regulatory authorities such as the Food and Drug Administration (FDA) and the European Medicines Agency (EMA). Questions or comments regarding this advanced notice can be sent to [email protected]. 1.1 Policy Objectives To integrate the “non-eCTD electronic-only” format within the Canadian drug registration framework by describing the requirements for the preparation of: An application for Drug Identification Number (DIN) pursuant to Part C, Division 1 of the Food
and Drug Regulations A Clinical Trial involving human subjects pursuant to Part C, Division 5 of the Food and Drug
Regulations A human drug regulatory activity pursuant to Part C, Division 8 of the Food and Drug Regulations Developmental Safety Update Reports (DSUR) Post-market Vigilance Data Post-Notice of Compliance (NOC) Changes: Level III forms pursuant to Guidance Document:
Post-Notice of Compliance (NOC) Changes: Framework Document Drug Notification Forms (DNF) pursuant to Part C, Division 1 and Division 8 of the Food and
Drug Regulations Administrative Regulatory Activities pursuant to Part C, Division 8 of the Food and Drug
Regulations Master Files (MF) Medical Device regulatory activities pursuant to sections 32 and 82 of the Medical Devices
Regulations Veterinary drug regulatory activities pursuant to Part C, Division 1 and Division 8 of the Food and
Drug Regulations

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 2
To consolidate several notices regarding different product lines that are published in different locations into one single document for ease of access by stakeholders. Completing the consolidation of the published notices and increasing the scope of regulatory activities accepted in “non-eCTD electronic-only” format will result in several iterations of this guidance document. 1.2 Policy Statements
As part of an ongoing efficiency measure and efforts to reduce regulatory burden on industry as well as to transition to an electronic environment, Health Canada has established the following options that are available immediately for filing regulatory activities in scope of this document and their related transactions: Human Drugs
A. Health Canada has been accepting regulatory activities in electronic Common Technical Document (eCTD) format since 2004. Therefore, the recommended option is to file regulatory activities/transactions in the eCTD format (where applicable). Refer to the Guidance Document: Preparation of Regulatory Activities in the electronic Common Technical Document (eCTD) Format, for further information.
B. In the situation that a regulatory activity/transaction is not yet accepted in eCTD format by Health Canada, an interim option is to file the regulatory activity/transaction in the “non-eCTD electronic-only” format until it is accepted in either the eCTD format or in the Regulated Product Submission (RPS1) format.
C. If a stakeholder is not ready to file in the eCTD format, they may file regulatory activities/ transactions in the “non-eCTD electronic-only” format as an interim option until their transition to the eCTD format has been completed.
When filing using options B and C, refer to this guidance document for further information on preparing your regulatory activity/transaction.
Medical Devices
Health Canada has been accepting Class III and IV medical device applications in electronic and paper format since November 2014. Effective immediately, Health Canada is accepting all medical device applications in scope of this guidance document to be provided in “non-eCTD electronic-only” format. Until the Regulated Product Submission (RPS1) format has been implemented, “Non-eCTD electronic-only” will be the only format accepted by Health Canada for medical devices applications as of April 1st, 2017. Therefore paper will no longer be accepted as of this date.
Veterinary Drugs
1 RPS format is a Health Level Seven standard. Refer to URL (http://www.hl7.org) for more information.

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 3
Regulatory Activities provided in electronic-only format have been accepted by Health Canada since 2014. Effective April 1st, 2017, Health Canada will no longer accept the paper copies. Therefore, “non-eCTD electronic-only” will be the only format accepted.
Electronic documents filed using any of the above options will be uploaded onto the Health Canada viewing tool, where they will be immediately accessible to Health Canada staff involved in the review of the regulatory activities. This will contribute to effective record management and ensure authenticity, integrity, availability, traceability, and non-repudiation of the data.
Table 1: Dates for phasing out paper filing
Regulatory Activity (and subsequent transactions)
Paperd will no longer be accepted as of:
Division 1a (e.g.: DINA, DINB…)
Already in effect
Division 5 (e.g : CTA, CTA-N…)
Division 8a (e.g.: NDS, ANDS…)
DSUR
Post-market Vigilance Data
Level III forms
DNF
Master Files (New MFs, MF Updatesb, Transactions related to existing MFsc)
Medical Devices (e.g.: Class II, III, & IV licences & amendments) April 1, 2017
Veterinary Drugs April 1, 2017
a. Including administrative regulatory activities and transactions. b. The first update, for the existing MF in paper format, must include a complete MF conversion in an electronic-only
format. Failure to provide the complete electronic copy of the MF will result in the MF being suspended (no further access for review will be granted and no update will be accepted for the MF).
c. For example: letter of access, administrative information, CEP and Attestation, Responses to clarification requests. d. Any paper received after the date indicated will be shredded or returned at the owner’s expense.
1.3 Scope and Application The following regulatory activity types are eligible for filing in “non-eCTD electronic-only” format:
Div
.1
Application for Drug Identification Number (DINA) Application for Drug Identification Number - Biologic (DINB) Application for Drug Identification Number - Disinfectant Product (DIND) Application for Drug Identification Number - Category IV Product (DINF) Post-Authorization Division 1 Change (PDC) Post-Authorization Division 1 Change - Biologics (PDC-B)

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 4
Div
.5
Clinical Trial Application (CTA) Clinical Trial Application - Amendment (CTA-A) Clinical Trial Application - Notification (CTA-N) Clinical Trial Site Information (CTSI) Forms Pre-CTA Meeting Information
Div
.8
New Drug Submission (NDS)2 Extraordinary Use New Drug Submission (EU NDS) Abbreviated New Drug Submission (ANDS)2 Supplement to a New Drug Submission (SNDS)2 Extraordinary Use Supplement to a New Drug Submission (EU SNDS) Supplement to a New Drug Submission-Confirmatory (SNDS-C) Supplement to an Abbreviated New Drug Submission (SANDS)2 Notifiable Change (NC)2 Request for Priority Review Status for NDS or SNDS Yearly Biologic Product Report (YBPR)3 Periodic Safety Update Report - Confirmatory (PSUR-C) or Periodic Benefit Risk
Evaluation Report - Confirmatory (PBRER-C) when provided to TPD, BGTD or NNHPD Pre-Submission Meeting Information4 (MPNDS, MPSNDS, MPDIN, or MPNC) Undefined Regulatory Activity (UDRA)
o Response to Advisement Letter to update the Product Monograph, when an NC (or other Division 8 regulatory activity) is not filed
o Notification of Discontinued Sale (DIN Cancellation)
DS
UR
Development Safety Update Report (DSUR) when provided as a standalone regulatory activity to Therapeutic Products Directorate (TPD), Biologics and Genetic Therapies Directorate (BGTD) or Natural and Non-prescription Health Products Directorate (NNHPD).
PV
-Data
Periodic Safety Update Report (PSUR) or Periodic Benefit Risk Evaluation Report (PBRER) when provided to the Marketed Health Products Directorate (MHPD)5
Risk Management Plan (RMP), when provided to MHPD5 Other Post-market Vigilance data6 (Undefined Data Post-market Vigilance (UDPV)) requested
by MHPD5 o Risk communication document (e.g., Dear Health Care Professional Letter, dissemination
lists, Proposed Dissemination Strategy) should be sent to the Office of Submissions and
2 Including regulatory activities for administrative changes 3 YBPRs should not be filed in the same regulatory transaction with Level III change forms 4 Pre-Submission Meeting Information includes the Pre-Submission Meeting Request 5 Must be filed as a separate regulatory activity even if a single request for multiple Post-market Vigilance
data has been issued by MHPD 6 Also referred to as “Pharmacovigilance Data”

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 5
Intellectual Property (OSIP) with an electronic convenience copy being provided directly to MHPD via email
o Post-market Surveillance (e.g., Issue-related Summary Reports, Council For International Organizations Of Medical Sciences (CIOMS), Line Listings, Registry Reports, Clinical Study Reports, or Patient Exposure Data)
o Benefit Risk Assessment o Response to MHPD Requests for Additional Information7 o Notification of Change in benefit-risk profile (under sections C.01.018(3) and (4) of the
Food and Drug Regulations) o Meetings regarding post marketing issues with MHPD
Lev
.3 Post NOC - Level III Changes Form
MF
MF Type I - Drug Substance MF Type II - Container Closure Systems and Components MF Type III - Excipients MF Type IV- Drug Product
MD
Licence Applications for Class II, III & IV Licence Amendments for Class II, III & IV Licence Amendment Fax-back Forms for Class II, III & IV Private Label Licence Applications for Class II, III & IV Private Label Licence Amendments for Class II, III & IV Post-market Vigilance Data (Responses MHPD Requests for Additional Information for Class I,
II, III & IV)
VD
Application for Drug Identification Number (DIN) New Drug Submission (NDS)8 Abbreviated New Drug Submission (ANDS)8 Supplement to a New Drug Submission (SNDS) 8 Supplement to an Abbreviated New Drug Submission (SANDS) 8 Notifiable Change (NC) 8 Request for Priority Review Status for NDS or SNDS Periodic Safety Update Report (PSUR) Pre-Submission Meeting Information9 (eg: MPNDS, MPSNDS, MPDIN, or MPANDS) Change to DIN Submission Experimental Studies Certificate (ESCs) and their amendments
7 Throughout this guidance document, when additional information is referred to, solicited and unsolicited
information should be understood (See Appendix B: Glossary of Terms) 8 Including regulatory activities for administrative changes 9 Pre-Submission Meeting Information includes the Pre-Submission Meeting Request
Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 6
Investigational New Drug (IND) Investigational New Drug (IND) amendments Protocol Review Veterinary Drugs Master Files (MF)
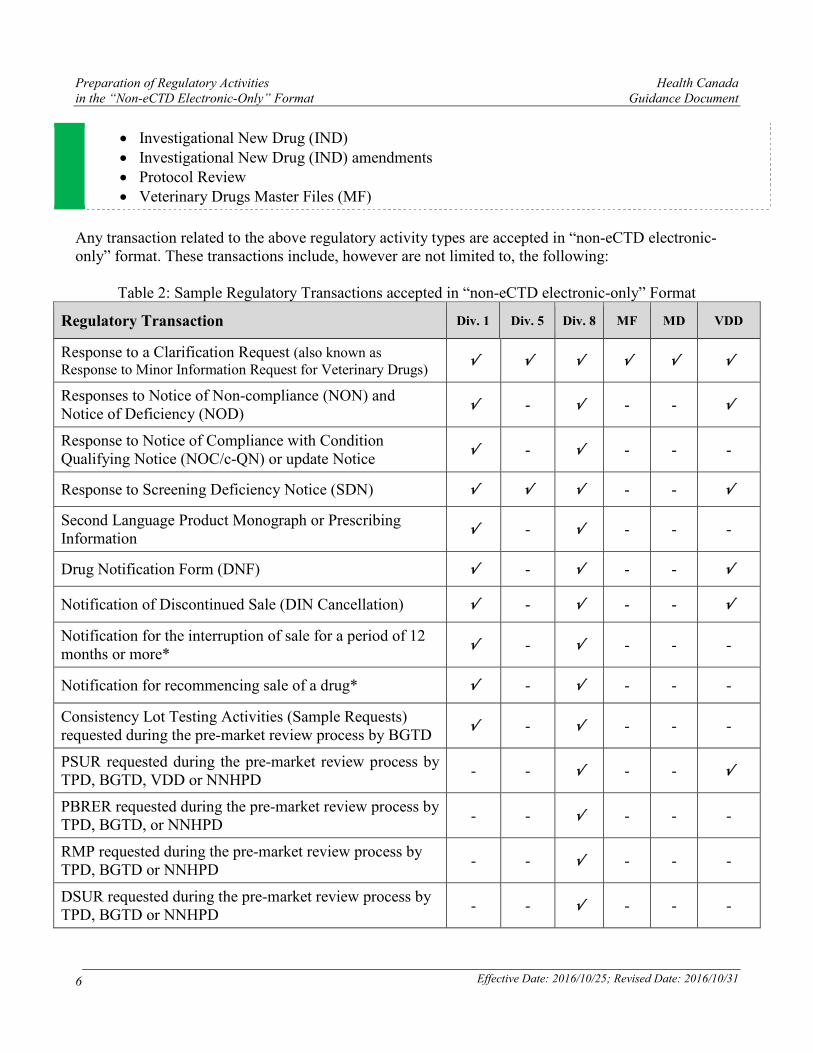
Any transaction related to the above regulatory activity types are accepted in “non-eCTD electronic-only” format. These transactions include, however are not limited to, the following:
Table 2: Sample Regulatory Transactions accepted in “non-eCTD electronic-only” Format
Regulatory Transaction Div. 1 Div. 5 Div. 8 MF MD VDD
Response to a Clarification Request (also known as Response to Minor Information Request for Veterinary Drugs)
√ √ √ √ √ √
Responses to Notice of Non-compliance (NON) and Notice of Deficiency (NOD)
√ - √ - - √
Response to Notice of Compliance with Condition Qualifying Notice (NOC/c-QN) or update Notice
√ - √ - - -
Response to Screening Deficiency Notice (SDN) √ √ √ - - √
Second Language Product Monograph or Prescribing Information
√ - √ - - -
Drug Notification Form (DNF) √ - √ - - √
Notification of Discontinued Sale (DIN Cancellation) √ - √ - - √
Notification for the interruption of sale for a period of 12 months or more*
√ - √ - - -
Notification for recommencing sale of a drug* √ - √ - - -
Consistency Lot Testing Activities (Sample Requests) requested during the pre-market review process by BGTD
√ - √ - - -
PSUR requested during the pre-market review process by TPD, BGTD, VDD or NNHPD
- - √ - - √
PBRER requested during the pre-market review process by TPD, BGTD, or NNHPD
- - √ - - -
RMP requested during the pre-market review process by TPD, BGTD or NNHPD
- - √ - - -
DSUR requested during the pre-market review process by TPD, BGTD or NNHPD
- - √ - - -

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 7
Regulatory Transaction Div. 1 Div. 5 Div. 8 MF MD VDD
Comments to the Summary Basis of Decision/ Notice of Decision
- - √ - - -
Written correspondence related to the Patented Medicines (Notice of Compliance) Regulations
√ - √ - - -
Notice of allegation and related materials (i.e. notice of allegation, proof of service, certification of filing date) under the Patented Medicines (Notice of Compliance) Regulations
√ - √ - - -
Form IV, including updates, filed in accordance with the Patented Medicines (Notice of Compliance) Regulations
√ - √ - - -
Form V, including updates, filed in accordance with the Patented Medicines (Notice of Compliance) Regulations
√ - √ - -
-
Consent Letter (Patent Information) √ - √ - - -
Written correspondence related to data protection under section C.08.004.1 of the Food and Drug Regulations
- - √ - - -
Consent Letter (Data Protection Information) - - √ - - -
Documents related to data protection under section C.08.004.1 of the Food and Drug Regulations
- - √ - - -
Authorization for Sharing Information (Consent Letter) - √ √ - - -
Reconsideration of Decisions and Other Related Information
√ - √ - - -
MF Updates (updates or amendments) - - - √ - √
MF Response to Deficiency Letter or Incomplete Letter - - - √ - √
CEP and Attestations - - - √ - √
CEP Revisions and Attestations - - - √ - √
MF Administrative Information - - - √ - √
MF Authorization to Access - - - √ - √
MF Statement of Commitment - - - √ - √
MF BSE/TSE Certificate - - - √ - √

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 8
Regulatory Transaction Div. 1 Div. 5 Div. 8 MF MD VDD
Response to Additional Information Letter - - - - √ -
Response to Screening Deficiency Letter (SDL) - - - - √ -
(‘√’ = Accepted / ‘-’ = Not Applicable)
*C.01.014.12 is a new provision that is part of the Regulations Amending the Food and Drug Regulations (Shortages of Drugs and Discontinuation of Sale of Drugs) that will come into force on March 29, 2017.
1.4 Background A first iteration of this document was developed to include DIN applications and published in June 2015. A second iteration of the document introduced MFs in “non-eCTD electronic-only” format and was published in September 2015. In February 2016, the third iteration widening the scope to introduce Part C, Division 5 and Division 8 (human drugs) of the Food and Drugs Regulations was published. This fourth iteration again expands the scope of regulatory activities accepted to include Medical Device applications as well as veterinary drugs by incorporating information from the following notices: Notice - Guidance for Industry: Formatting of Class III and Class IV License Applications
(Electronic and Paper Formats) Guidance for industry - Preparation of electronic submissions for the Veterinary Drugs
Directorate This guidance should be read in conjunction with the following documents:
Div
.1
Guideline on Preparation of Drug Identification Number Submissions Guidance Document on Post-Drug Identification Number (DIN) Changes
Div
.5
Guidance Document for Clinical Trial Sponsors: Clinical Trial Applications
Div
.8
Guidance Document: Preparation of Drug Regulatory Activities in the Common Technical Document (CTD) Format
Guidance for Industry: Management of Drug Submissions Draft Guidance Document: Quality (Chemistry and Manufacturing) Guidance: New Drug
submissions (NDSs) and Abbreviated New Drug Submissions (ANDSs) Guidance Document: Post-Notice of Compliance (NOC) Changes: Quality Document Guidance Document: Post-Notice of Compliance (NOC) Changes: Safety and Efficacy Document Guidance Document: Post-Notice of Compliance (NOC) Changes: Framework Document Changes in Manufacturer’s Name and/or Product Name

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 9
MF
Draft Guidance Document: Drug Master Files (DMFs) Draft Guidance Document: Master Files (MFs) – Procedures and Administrative Requirements Drug Master File (DMF) Application Form Drug Master File (DMF) Application Fee Form
MD
Guidance on supporting evidence to be provided for new and amended licence applications for Class III and Class IV medical devices, not including in vitro diagnostic;
Guidance on supporting evidence to be provided for Class III and IV in vitro diagnostic device licence applications and amendments;
Guidance Document: Preparation of the Summary Technical Documentation (STED)-based Class III and Class IV Premarket Medical Device Licence Applications;
Preparation of Summary Technical Document (STED)-based Class III and IV Premarket in vitro diagnostic device licence applications and amendments.
Management of Applications for Medical Device Licences and Investigational Testing Authorizations
VD
Guidance for Industry - Preparation of Veterinary New Drug Submissions, Guidance for Industry - Management of Regulatory Submissions Guidance for Industry - Preparation of Veterinary Abbreviated New Drug Submissions - Generic
Drugs Also refer to the Post-NOC Changes guidance documents listed under Division 8
Stakeholders that choose to file a regulatory activity or transaction in eCTD format must comply with the scope and the specifications included in the Guidance Document: Preparation of Drug Regulatory Activities in Electronic Common Technical Document (eCTD) Format. 2 STRUCTURE AND CONTENT OF A REGULATORY ACTIVITY Health Canada would like to emphasize that the requirements highlighted below are not limited to initial regulatory activities as defined in section “1.3 Scope and Application”. The requirements also apply to all regulatory transactions (see Table 2 in section 1.3 “Scope and Application”). Any transactions not formatted as per this guideline, will be rejected. 2.1 Cover Letter Regulatory activities in “non-eCTD electronic-only” format provided on media must include a cover letter, in both electronic format and paper format. A paper cover letter is required as this is the only means by which to quickly identify the content of the media. In addition, the following are exceptions to this requirement:

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 10
Div
.5
A cover letter should not be provided with a CTSI form when submitted via email.
Lev
.3
A cover letter in electronic format should not be provided with a Level III Changes form.
MD
A cover letter, for medical device class II regulatory activities, is only required when including relevant information that does not have a placeholder in the application form or structure.
Cover letters may be submitted in electronic-only format
Health Canada requires the following information for all cover letters: Clearly state what is being provided and the reason for filing, for example:
o Type of transaction as per section “1.3 Scope and Application”, o Notification for the interruption of sale for a period of 12 months or more o Notification for recommencing sale of a drug o Special requests (DNF) o Request for Review reports o Request for Pre-Submission Meeting
Include reference to correspondence with Health Canada prior to filing Include reference to a request letter (including an Advisement Letter), if applicable. Stakeholder10 Name and Role (e.g. Sponsor, Manufacturer, DIN / MF Owner, or Agent). Brand name, MF name or Device name Control number, DSTS number, MF number or Application number (if known) Dossier identifier (if known) Regulatory activity type Any cross-referenced regulatory activity should be clearly stated (date the regulatory activity was
approved).
In addition to the above general requirements, the cover letters for:
Div
.1
DINAs should indicate if there is a labelling reference product.
Div
.5
Clinical Trial regulatory activities should include relevant protocol number(s)
DS
UR
DSUR (when provided to TPD or BGTD) should also indicate which of the following applies: o VOLUNTARY - unsolicited information o REQUESTED - provided as a response to a request made by Health Canada
10 See definition in Appendix B

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 11
PV
-Data
PSUR or PBRER (when provided to MHPD) should also indicate any of the following that applies: o Significant change in what is known about the risks and benefits of the health product o VOLUNTARY PSUR/PBRER - unsolicited information o REQUESTED PERIODIC PSUR/PBRER - requested by Health Canada (for example RMP
follow-up or post-authorization commitment) o REQUESTED AD HOC PSUR/PBRER - provided as a one-time request made by either the
pre-market review directorate reviewing the associated regulatory activity or by MHPD (the requester should be specified)
RMP (when provided to MHPD) should also indicate which of the following applies: o VOLUNTARY RMP - unsolicited information o REQUESTED AD HOC RMP - provided as a one-time request made by MHPD (the
requester should be specified).
MF
MFs in “non-eCTD electronic-only” format filed as a replacement for an existing MF in paper format should include a confirmation that the entire MF is being included.
MF conversions including a new Letter of Access, that has not previously been authorized, should be identified so it can be processed and acknowledged. Failure to identify a new Letter of Access in the cover letter, could delay access to the MF due to the absence of an authorized Letter of Access.
Response to a request for clarification:
o Should clearly indicate the name of the requester. o Should not contain any scientific information. Supporting data should be placed in the
appropriate module/section/part o Should not include the summary response in a Question and Answer format and the Note to
Reviewer. For human drugs, these should be placed in folders 1.0.4 and 1.0.7 respectively. Regulatory transactions for human drugs, that contain an HC-SC3011 form should include a
table, structured as below, placed at the end of the cover letter. The information required to fill the table should be taken from the indicated box numbers of the completed HC-SC3011 form.
Table 3: Information from the HC-SC3011 Form
Information HC-SC3011 Form Box Number 1. Regulatory Activity Type < Box 5 > 2. Brand Name < Box 8 > 3. Common Name < Box 9 > 4. Stakeholder Name < Box 11 > 5. Stakeholder Legal Address < Boxes 12-16 > 6. Dosage Form < Box 60 > 7. Route(s) of Administration < Box 63 > 8. Drug Product < Box 64 >

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 12
Information HC-SC3011 Form Box Number 9. Proposed Indication/Use < Box 67 >
10. Phase of Clinical Trial* < Box 86 > Phase I - bioequivalence study (7-day administrative target) Phase I - study in healthy humans (30-day default) Phase I - other (30-day default) Phase II (30-day default) Phase III (30-day default)
Other (please specify): _
11. Medicinal (Active)
Ingredient(s) Ingredient Name Strength Unit Per
Calculated as Base?
(Yes/No)
< Box 56 >
*Required only for Clinical Trial regulatory activities.
2.2 Folder Structure The content of the electronic media should be organized in folders. Table Sample folder structures for specific regulatory activity types are illustrated in the locations indicated below: Division 1 Appendix C Division 5 Appendix D Division 8 Appendix E Level III Changes Section 2.2 Folder Structure Master Files (MF) Appendix F Medical Devices Appendix H Veterinary Drugs The attached zipped folder structure (available in the HTML version
of this document) can be used by adding documents in their respected folders. Empty folders must be deleted before filing to Health Canada.
L
Level III changes forms should be provided at the time of the changes are implemented.

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 13
Figure 1: Sample folder structure for Level III Changes Form
In accordance with section “3.3 Supporting Data - Level III Changes” in the Guidance Document: Post-Notice of Compliance (NOC) Changes: Quality Document, supporting data should not be provided with a level III changes form. If additional information is required, it will be requested. Any supporting data provided without request, will be destroyed or returned to the sponsor at their own expense.
Sample folder structures for specific regulatory transaction are illustrated below:
Figure 2: Sample folder structure for a Response to a Clarification Request for human drugs

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 14
DN
F
Figure 3: Sample folder structure for a DNF transaction for human drugs
The following are common folders structure errors to avoid: The top level folder should not contain any files; it should only contain the required folders. Multiple documents provided as a single PDF file is not acceptable. Information provided in previous transactions should not be provided again, unless it is affected
by a change (such as a MF update, MF letter of access, administrative change, response to NON, response to NOD or response to SDN).
Empty folders should not be included in the structure (i.e.: if a Fee Form is not applicable for a regulatory transaction, then folder 1.2.2 should not be created in the structure).
Leading sheets at the beginning of sub-folders (indicating a folder is not applicable or describing the content) must not be provided.

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 15
Figure 4: Leading Sheet
2.2.1 Top Level Folder and Dossier Identifier
The top level folder of a dossier in “non-eCTD electronic-only” format contains all other folders and their content. The name of the top level folder should be a segment of the Dossier Identifier11, if known; otherwise the product name or protocol number can be used. Regulatory activities and additional information in “non-eCTD electronic-only” format for the same dossier should retain the same Dossier Identifier. The top level folder should be included every time a regulatory transaction is provided to Health Canada.
2.2.2 Common Technical Document (CTD) Folder Structure
2.2.2.1 Module 1 Folders
The structure and name of the Module 1 folders are defined in Appendix A of Health Canada’s Guidance Document: Preparation of Drug Regulatory Activities in the Common Technical Document (CTD) Format. Examples of content for specific folders:
Companies must ensure that the Advanced Payment Details for Drug Submissions and Master Files form containing payment information is not be included within an electronic transaction as the information cannot be deleted and will remain as part of the electronic record for all reviewers. This form should be sent via mail or fax separately.
The copy of an original request issued by Health Canada, such as a request for clarification, SDN, NOD, and NON, should be filed in folder “1.0.3 copy of Health Canada issued correspondence”.
Div
. 5 When filing clinical trial applications, stakeholders may also refer to “Table 1: Contents of
Submission Package in accordance with CTD Format” of the Guidance Document for Clinical Trial Sponsors: Clinical Trial Applications for further clarity.
11 See definition in Appendix B

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 16
DS
UR
A DSUR should be a single file in folder “1.3.8.4 Other Pharmacovigilance Information”. A DSUR Checklist should be in folder “1.2.3 Certification and Attestation Forms”.
MF
When providing MF Types I, II, III & IV, the folders in Module 1 will be considered as the Restricted Part (RP). See Appendix F for illustrations.
A side by side comparison table listing all changes as per Draft Guidance Document: Master Files (MFs) - Procedures and Administrative Requirements should be provided in folder “1.0.7 General Note to Reviewer” for each: o MF update o MF conversion provided in “non-eCTD electronic-only” format as a complete replacement
for an existing MF in paper format.
When providing an update, a new MF application form should be provided. Any changes to the contact information should be indicated on the application form to ensure that all correspondence is sent to the appropriate contact.
When providing a Letter of Access for a MF, section numbers should be listed as the reference for the electronic MFs.
When providing a MF conversion, identical copies of all previously filed Letters of Access are recommended to be provided (one letter per pdf file with the name of the company, to whom access is granted, as the pdf file name). The letters should be provided in folder “1.2.6 Authorization for Sharing Information”.
When providing company name change, ownership change, or product name change, they should be included in a document in folder “1.2.1 Application Forms”.
VD
When providing a master file for a veterinary drug, the MF version number should be provided. When providing a revision to the MF the version number should be updated.
2.2.2.2 Modules 2 to 5 Folders
The structure and names of the Modules 2 to 5 folders are defined in Appendix D of Health Canada’s Guidance Document: Preparation of Drug Regulatory Activities in the Common Technical Document (CTD) Format. Note: The folder names as defined in the above indicated appendix should exclude information in parentheses. Examples of content for specific folders:
Div
. 5 When filing clinical trial applications, stakeholders may also refer to “Table 1: Contents of
Submission Package in accordance with CTD Format” of the Guidance Document for Clinical Trial Sponsors: Clinical Trial Applications for further clarity.
MF
When providing MF Types I & IV, two separate documents should be included in the folder “2.3 Quality Overall Summary”, a “QOS (RP)” and a “QOS (AP)” files.

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 17
When providing a MF12:
o Type I - Drug Substance (see figure F-1 in Appendix F for an illustration): The folder “3.2.S Drug Substance” should be duplicated and each clearly
identified as either the Applicant’s Part or the Restricted Part using the abbreviations “AP” or “RP” respectively.
The folder “3.2.A Appendices” will be considered as the Restricted Part (RP). o Type II - Container Closure Systems, there are two possible options for structuring the
folders in Module 3. (See figure F-2 in Appendix F for an illustration). A separate subfolder under “3.2.P.7 Container Closer System” can be used for
each component provided, or The folder “3.2.P Drug Products” (with all its subfolders) can be used for
information that is common to all components and separate subfolders under “3.2.P.7 Container Closer System” can be used for information specific to each component.
o Type III - Excipients (See figure F-3 in Appendix F for an illustration): The folder “3.2.P.4 Control of Excipients” in Module 3 should be duplicated for
each excipient provided. o Type IV - Drug Products (see figure F-4 in Appendix F for an illustration):
The folder “3.2.P Drug Products” should be duplicated and each clearly identified as either the Applicant’s Part or the Restricted Part using the abbreviations “AP” or “RP” respectively.
The folders “3.2.A Appendices” and “3.2.R Regional Information” will be considered as the Restricted Part (RP).
2.2.3 Medical Device Folder Structures
MD
The structure and name of the folders must be provided using one of the following formats:
Table 4: Accepted Formats for Devices
Table of Contents (ToC) Structure
Health Canada Structure Summary Technical
Documentation (STED) Structure
Regulatory Activities
Class II New and Amendments
Class II, III, IV (Private Labels and Faxbacks)
Class I, II, III & IV Post-Market Requests
Class III & IV only (New and Amendments)
Class III & IV only (New and Amendments)
12 Including MF updates

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 18
Refer to: Appendix H of this guidance for sample structure and Table H-1 for the requirements
The zipped sample structures in Appendix H;
Guidance on supporting evidence to be provided for new and amended licence applications for Class III and Class IV medical devices, not including in vitro diagnostic;
Guidance on
supporting evidence to be provided for Class III and IV in vitro diagnostic device licence applications and amendments.
The zipped sample structures in Appendix H;
Guidance Document: Preparation of the Summary Technical Documentation (STED)-based Class III and Class IV Premarket Medical Device Licence Applications;
Preparation of
Summary Technical Document (STED)-based Class III and IV Premarket in vitro diagnostic device licence applications and amendments.
Manufacturers submitting any subsequent information must clearly identify the Application Number of the relevant application. Responses to Screening Deficiency Letters, Clarification Requests, and Additional Information Letters must be provided in a question and answer format and be accompanied by a copy of the original Health Canada letter. This information is to be filed under section 1.0 Cover Letter and the supporting information must be structured using the same format as the initial application.
2.2.4 Veterinary Drugs Folder Structure
VD
The structure and name of the folders for veterinary drug regulatory activities are defined in Appendix V: “Master Index” of Health Canada’s Guidance for Industry: Preparation of Veterinary New Drug Submissions.
2.3 File Naming Convention With the exception of the file extension, the file naming convention within each folder is left to the stakeholder preparing the regulatory transaction. However, Health Canada suggests that the file names be kept as brief and meaningful as possible: File names should describe the content in a meaningful way and must be limited to a maximum of
50 characters, including the file extension.

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 19
Commonly used and meaningful abbreviations, such as QOS for Quality Overall Summary, may be used to shorten file names.
Files provided electronically should not be password protected. 3 TECHNICAL REQUIREMENTS FOR REGULATORY ACTIVITIES 3.1 File Format Portable Document Format (PDF) (versions 1.7, PDF/A-2 and PDF/A-3)13 is the recommended format for electronic documents although other formats such as Microsoft® Office 2010 (.docx, .xlsx) may also be accepted. PDF versions of documents should be generated from electronic source documents and not from
scanned materials, except where access to an electronic source document is unavailable or where a signature is required.
To ensure that PDF files can be accessed efficiently, they should be no larger than 150 megabytes.
It is important that PDF files be properly bookmarked. Rules of thumb for good bookmarking include:
o Documents of ten pages or more should be bookmarked. o Bookmarks are equivalent to and should be organized like a document table of contents,
and should not include the regulatory activity level. o Sections, subsections, tables, figures, and appendices should all be bookmarked. o Too many levels of bookmarks are inefficient; in most instances, four levels of bookmarks
should be sufficient:
1 Heading 1.1 Subheading
1.1.1 Sub-subheading 1.1.1.1 Sub-Sub-Subheading
Health Canada recognizes that bookmarks are generated automatically from document headings, but nevertheless recommends they be kept concise.
It is important that PDF files be properly Hyperlinked:
o Hyperlinks within the same PDF document are acceptable but links between different documents are not to be used.
o It is the responsibility of the stakeholder preparing the regulatory transaction to ensure that hyperlinks are functioning.
o Links must also include references to the specific section or page in the event the link is broken.
13 PDF documents with attachments are not allowed

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 20
o Note that the required hyperlinks to related information should be included only in the PDF version of files.
Specific formats requirements: The documents in table 5 below, must be provided in PDF and/or Microsoft® Word 2010 format(s)
as specified:
Table 5: Specific File Format Requirements for human drugs
List of Documents File Format* PDF Word
Certified Product Information Document (CPID)
Annotated √ - Non-annotated - √
Comprehensive Summary: Bioequivalence √ √
Dear Healthcare Professional Letter √ √
Fee Form √ - HC-SC3011 Form √ - Label Safety Assessment Update - Sponsor Attestation √ √
Product Monograph (PM)
Annotated √ √
Non-annotated - √
Pristine - √
Second language √ - PSEAT-CTA - √
Public Communication √ √
Quality Overall Summary (QOS) Clinical Trial Applications - √
All other regulatory activities √ √
Responses to clarification requests for: SDN, NON, NOD or NOC/c-QN √ √
Sponsor Attestation Checklist for ANDS √ √
Summary Basis of Decision Clean √ - Annotated (redlined) - √
(‘√ ’ = Required / ‘-’ = Not Applicable)
* When PDF and Word are selected, the document should be provided in both formats
Presentations for meetings with Health Canada (e.g., pre-submission meetings), can be provided
in Microsoft® PowerPoint 2010 (.pptx) format.
Div
. 1
The “BE data sets” must be provided in ASCII format. For more information see Health Canada’s Guidance for Industry: Preparation of Comparative Bioavailability Information for Drug Submissions in the CTD Format, Appendix B: “Computer Format for the Submission of Data for Comparative Bioavailability Studies”.
Div
. 8

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 21
File formats that are not accepted include but are not limited to: Thumbnail Cache Files (Thumbs.db) Outlook item (.msg) PDF files with attachments Backup file (~*.docx) Image file (e.g. jpeg, .bmp, .tiff) Documents containing macros (e.g. .docm) Contact OSIP for other file formats that may be acceptable at the time of filing. See Appendix A for full contact information. 3.2 Signature The documents that legally require a signature should be printed, signed, scanned, saved as PDF files and included in the regulatory transaction, unless instructed otherwise in electronic PDF fillable forms available on the Health Canada website. If only one page of a multi-page document requires a signature, the sponsor should scan that page and then include the scanned page at the same location in the PDF file of the document. Each document should have only one PDF file.
Lev
.3
Unsigned level III changes forms will not be accepted. The stakeholder will be requested to refile the signed form(s).
MD
Medical Device license application forms may be signed with an electronic signature (e.g. an image of the official’s wet ink signature), or the signature page can be signed and scanned as indicated above.
VD
Documents may be signed with an electronic signature (e.g. an image of the official’s wet ink signature), or the signature page can be signed and scanned as indicated above.
3.3 Transmission of Electronic Data On media Regulatory transactions should be provided on media, unless specified otherwise. Media should be sent to the appropriate address as indicated in Appendix A. The media formats acceptable when providing electronic regulatory transactions are:
o Compact Disc-Recordable (CD-R) conforming to the Joliet specification o Digital Versatile Disc-Random Access Memory (DVD-RAM) Universal Disc Format (UDF)
standard o Single and dual layer Recordable Digital Versatile Discs

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 22
o Single and dual layer Blu-ray discs o Universal Serial Bus (USB) 2.0 or 3.0 drive o Portable External Hard Drive with USB 2.0 or 3.0 interfaces Contact OSIP for other media formats that may be acceptable at the time of filing. See Appendix A for full contact information.
Media and files should not be password protected. Files stored on the media should not be zipped. The complete regulatory transaction should be provided on a single disc/drive. Duplicate copies are
not required. Media should be scanned using current virus-scanning software and should be certified virus-free. All media should be labelled. The label on the disc/drive should contain the following information:
o Stakeholder14 Name o Brand Name or MF Name o Dossier Identifier (if known) o Control/DSTS Number or MF Number (if known) o “Protected B”15 o “This media has been virus-scanned and we certify that it is virus free” o Month and year of filing
Subsequent to burning the CD/DVD or transferring data to a drive, stakeholders should ensure that all files can be opened, no files are corrupt, and that “Thumb.db” files are removed.
Transactions for human drugs provided on approved media formats should be sent to OSIP regardless of the review Directorate except in the below cases:
Div
. 5
Clinical Trial regulatory transactions should be sent directly to the appropriate Directorate13.
Lev
. 3
The completed and signed Level III Changes forms for all drug products should be sent to: o Office of Submissions and Intellectual Property (OSIP)16 for human drugs. o Veterinary Drugs Directorate (VDD)17 for veterinary drugs.
MD
Medical device regulatory transactions should be sent directly to device licensing services division. Response to information request should be sent directly to the appropriate directorate.17
VD
Veterinary drug regulatory transactions should be sent directly to the veterinary drugs directorate (VDD)17
By email
14 See definition in Appendix B 15 “Protected” status identifies information the unauthorized disclosure of which could reasonably be
expected to cause injury to private interests. “Protected B” indicates a medium degree of potential injury. See Government Security Policy (July 2009), Section 10.6, “Identification of Assets”.
16 See Appendix A for contact information

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 23
The below specified regulatory transactions may/should be provided to Health Canada via email. Regulatory transactions provided by email should meet the following requirements: The sponsor assumes the risk of transmitting Protected B information through email. The maximum email size accepted by the corporate mail server is 20 megabytes, anything larger
should be sent on media. If the regulatory transaction is provided via email, a duplicate copy should not be provided by
mail. The regulatory transaction should be organised in folders (see section 2.2) and provided as a
zipped file. The body of the email should only contain the zipped regulatory transaction; no other documents
or related information should be included. Zipped files and documents contained in the email should not be password protected.
Additional specific requirements for transactions accepted via email include:
Div
.1 Second language product monographs or prescribing information should only be provided to
Health Canada via email to [email protected] o The subject line of the email should include one of the following statements:
"French PM - <Brand Name>, <Control Number>” o The zipped file should be named :
“French PM - <Product Name>” Div
.8
Div
.5
Responses to a Clarification Request (IR) for Clinical Trials can be sent via email: o Email should be addressed to the requestor(s) identified in the clarification request. o The subject line of the email should include the statement:
"Division 5 - IR (<Brand Name(s)>, < Protocol Number(s)>, <Control Number(s)>)”. o The zipped file should be named:
“IR (<Brand Name(s)>, <Protocol Number(s)>, <Control Number(s)>)”.
Responses to a No Objection Letter (NOL) for Clinical Trials can be sent via email to: o [email protected] for biologic and radiopharmaceutical drugs. o The subject line of the email should include the statement:
"Division 5 – Response to CTA/CTA-A NOL (<Brand Name(s)>, <Protocol Number(s)>, <Control Number(s)>)”.
o The zipped file should be named: “Response to CTA/CTA-A NOL (<Brand Name(s)>, <Protocol Number(s)>, <Control
Number(s)>)”.

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 24
Clinical Trial Application Notifications (CTA-N) can be sent via email to: o [email protected] for biologic and radiopharmaceutical drugs. o [email protected] for pharmaceutical drugs. o The subject line of the email should include the statement:
"Division 5 – CTA-N (<Brand Name(s)>, <Protocol Number(s)>, <Parent CTA Control Number(s)>)”.
o The zipped file should be named: “CTA-N (<Brand Name(s)>, <Protocol Number(s)>, <Parent CTA Control
Number(s)>)”.
Clinical Trial Site Information (CTSI) Forms can be sent via email to: o [email protected] for biologic and radiopharmaceutical drugs. o [email protected] for pharmaceutical drugs. o The subject line of the email should include the statement:
“Division 5 - CTSI (<Brand Name(s)>, <Protocol Number(s)>, <Parent CTA Control Number(s)>)”.
o The zipped file should be named: “CTSI (<Brand Name(s)>, <Protocol Number(s)>, <Parent CTA Control Number(s)>)”.
DN
F
DNF transactions should only be sent via email to [email protected]. o The subject line of the email should include one of the following statements:
"Division 1 - DNF <Brand Name>” “Division 8 - DNF <Brand Name>”
o The zipped file should be named : “DNF - <Product Name>”
MD
Medical device transactions provided via email should be directed to [email protected] o The subject line of the email should include one of the following statements:
Class II new application – <device name> Do not submit a new Class II application by e-mail if you intend to pay your fees
by credit card Class II amendment application – <licence number> Private Label new application – <device name> Private Label amendment – <licence number> Fax-back – addition Class III/IV – <licence number> Fax-back – Addition, Deletion, Change (other than addition of Class III/IV) – <licence
number> Fax-back – change in device/licence name – <licence number> Fax-back – change in manufacturers name/address – <Manufacturer’s name>
MHPD Medical Device Information Request should be directed to [email protected]

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 25
Note that only one regulatory activity is to be submitted per email. If multiple regulatory activities or transactions are being submitted, each will require a separate email
VD
Regulatory transactions (with the exception of DNFs) provided for veterinary drugs via email should be directed to [email protected].

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 26
APPENDICES Appendix A: Contacts Office of Submissions and Intellectual Property (OSIP) Finance Building 101 Tunney’s Pasture Driveway Address Locator: 0201A1 Ottawa, ON, Canada K1A 0K9 email to: [email protected]
Div
. 5
Clinical Trial Applications - Pharmaceuticals Office of Clinical Trials Therapeutic Products Directorate Health Canada 5th Floor, Holland Cross, Tower B 1600 Scott Street, Address Locator 3105A Ottawa, ON, Canada K1A 0K9 General enquiries email: [email protected] CTA-N email : [email protected] Clinical Trial Site Information Form email: [email protected]
Div
. 5
Clinical Trial Applications - Biologics and Radiopharmaceuticals Office of Regulatory Affairs Biologics and Genetic Therapies Directorate Ground Floor, Health Canada Building #6 100 Eglantine Driveway Address Locator 0601C Ottawa, ON, Canada K1A 0K9 General Enquiries email: [email protected] CTA-N email: [email protected] Clinical Trial Site Information Form email: [email protected]
MD
Device Licensing Services Division Medical Devices Bureau 11 Holland Avenue Tower A, Second Floor, Postal Locator: 3002A Ottawa, ON, Canada K1A 0K9

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 27
Telephone: 613-957-7285 Facsimile: 613-957-6345 email: [email protected] Marketed Health Product Directorate Marketed Pharmaceuticals and Medical Devices Bureau Regulatory Management Section Health Canada Address Locator 0702L 200 Tunneys Pasture Driveway Ottawa, Ontario K1A 0K9 Telephone: 613-948-8523 Facsimile: 613-952-6011 Email: [email protected]
Lev
.3 Veterinary Drugs Directorate (VDD)
Holland Cross Building Tower A, Ground Floor 14-11 Holland Ave., Address Locator 3000A Ottawa, ON, Canada K1A 0K9 email: [email protected]
VD

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 28
Appendix B: Glossary of Terms Stakeholder: Company, Sponsors/DIN owner/Manufacturer of pharmaceutical or biological drug for regulatory activities filed according the Food and Drug Act and Regulation, Owner/Agent/Manufacturer for Master File, and Manufacturer of Medical Devices for regulatory activities filed according to the Medical Devices Regulations and the Food and Drugs Act. Confidential Business Information: Information which provides a business advantage as a result of the fact that it is kept confidential. This is true whether the information is tangible or intangible. Confidential business information is broad enough to encompass trade-secrets. Dossier: A collection of all regulatory activities throughout the life cycle of a product/device for a stakeholder. Note: for clinical trials it is a collection of all regulatory activities throughout the life cycle of a single clinical trial protocol. Dossier Identifier: A code created by Health Canada to uniquely identify the dossier. The dossier identifier consists of two segments:
Functional Classification Standard Code: The “HC” stands for “Health Canada”, the number “6” refers to the “Health Risk Protection” function and the number “024” refers to the “Compliance Notification” activity.
Top Level Folder Name: Lowercase letter followed by six (6) or seven (7) unique numbers depending on the regulatory activity type.
Drug Identification Number (DIN): An eight (8) digit numerical code assigned to each drug product approved under the Food and Drugs Act and Regulations. Master File (MF): is a reference that provides information about specific processes or components used in the manufacturing, processing, and packaging of a drug. Drug Product (Dosage Forms): The finished product (e.g., tablets, capsules, injections, etc.) Drug Substance (Drug Substances or Intermediates in the Production of Drug Substances): A pharmacologically active ingredient.

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 29
Leading Sheet: A document describing the information being provided (e.g. a document stating “this sub-folder contains the following documents…”). Regulatory Activity: A collection of all regulatory transactions throughout the process of a specific activity. Regulatory Transaction: Any information package sent by the stakeholder as part of a regulatory activity such as initial data, unsolicited and solicited data (e.g. response to a clarification request, NON, NOD, pristine PM, DNF, etc.). Additional Information: Both solicited and unsolicited information.
Solicited Information such as SDN, NOD, NON, or Response to a Clarification Request (response to telephone request, response to email request, response to screening Acceptance Letter).
Unsolicited information such as safety information and changes in the name of the company or product during review.
Note: For more details about solicited and unsolicited information, see Section 5.4, “Screening of Information and Material” and Section 5.5, "Evaluation of Submissions" in Health Canada's Guidance for Industry: Management of Drug Submissions

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 30
Div
.1
Appendix C: Division 1 Sample Folder Structure(s)
Figure C-1: DIN Applications

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 31
Div
.5
Appendix D: Division 5 Sample Folder Structure(s)
Figure D-1: Clinical Trial Applications
The attached zipped folder structure (available in the HTML version of this document) can be used by adding documents in their respected folders. Empty folders must be deleted before filing to Health Canada.

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 32
Div
.8
Appendix E: Division 8 Sample Folder Structure(s)
Figure E-1: Division 8 Regulatory Activities

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 33
MF
Appendix F: Master Files (MF) Sample Folder Structure(s)
Figure F-1: MF Type I - Drug Substance
1. All documents in this folder will be considered as Restricted Part (RP) of the MF. 2. Two separate documents should be included in the folder “2.3 Quality Overall Summary”, a “QOS (RP)” and a
“QOS (AP)” files.

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 34
MF
Figure F-2: MF Type II - Container Closure Systems and Components

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 35
MF
Figure F-3: MF Type III - Excipients

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 36
MF
Figure F-4: MF Type IV - Drug Product
1. All documents in this folder will be considered Restricted Part (RP) of the MF. 2. Two separate documents should be submitted in the folder “2.3 Quality Overall Summary”, a “QOS (RP)” and a
“QOS (AP)” files.

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 37
Appendix G: Distribution of MF Information between the Applicant and Restricted Parts
Table G-1: MF Type 1 - Drug Substance
Module/ Folder Names Proposed 2015
Applicant’s Part Proposed 2015 Restricted Part
Module 1: Administrative and Product Information
1.0 Correspondence
1.0.1 Cover Letter - √
1.0.2 Life Cycle Management Table (Only required for eCTD) - √
1.0.3
Copy of Health Canada Issued Correspondence Clarification Request (Clarifax) Notice of Deficiency Screening Deficiency Notice
- √
1.0.4
Health Canada Solicited Information Q&A response to Clarification Request (Clarifax) Q&A response to Notice of Deficiency
- √
1.0.7 General Note to Reviewer - √
1.1 Table of Contents (Not required for eCTD) - √
1.2 Administrative Information
1.2.1
Application Forms DMF Application/Amendment Form Can include the following:
o Agent Appointment Letter o Agent Withdrawal Letter o Agent Name Change
- √
1.2.2 Fee Forms
DMF Application/Amendment Fee Form - √
1.2.3
Certification and Attestation Forms BSE/TSE Attestation Form Certification of Suitability (CEP) CEP - Update CEP - Attestations
Statement of Commitment
- √
1.2.5 Compliance and Site Information
1.2.5.2 Establishment Licensing - √
1.2.5.5 Good Manufacturing Practices
Certificate of Compliance - √
1.2.6
Authorization for Sharing Information Letters of Access
Withdrawal of Authorization - √
1.2.7 International Information - √
1.3 Product Information

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 38
Module/ Folder Names Proposed 2015
Applicant’s Part Proposed 2015 Restricted Part
1.3.6 Certified Product Information Document - √
Module 2: Common Technical Document Summary
2.3 Quality Overall Summary (QOS)1 √ √
Module 3: Quality
3.1 Table of Contents of Module 3 (Not required for eCTD)
√ √
3.2 Body of Data
3.2.S Drug Substance
3.2.S.1 General Information
3.2.S.1.1 Nomenclature √ -
3.2.S.1.2 Structure √ -
3.2.S.1.3 General Properties √ -
3.2.S.2 Manufacture
3.2.S.2.1 Manufacturer(s) √ -
3.2.S.2.2 Description of Manufacturing Process and Process Controls
√ 2 √ 3
3.2.S.2.3 Control of Materials - √
3.2.S.2.4 Controls of Critical Steps and Intermediates √ 4 √ 5
3.2.S.2.5 Process Validation and /or Evaluation - √
3.2.S.2.6 Manufacturing Process Development - √
3.2.S.3 Characterisation
3.2.S.3.1 Elucidation of Structure and other Characteristics √ -
3.2.S.3.2 Impurities √ √ 6
3.2.S.4 Control of Drug Substance
3.2.S.4.1 Specification √ -
3.2.S.4.2 Analytical Procedures √ -
3.2.S.4.3 Validation of Analytical Procedures √ -
3.2.S.4.4 Batch Analyses √ -
3.2.S.4.5 Justification of Specification √ √ 7
3.2.S.5 Reference Standards or Materials √ -
3.2.S.6 Container Closure System √ -
3.2.S.7 Stability
3.2.S.7.1 Stability Summary and Conclusions √ -
3.2.S.7.2 Post-approval Stability Protocol and Stability Commitment
√ -
3.2.S.7.3 Stability Data √ -
3.2.A Appendices
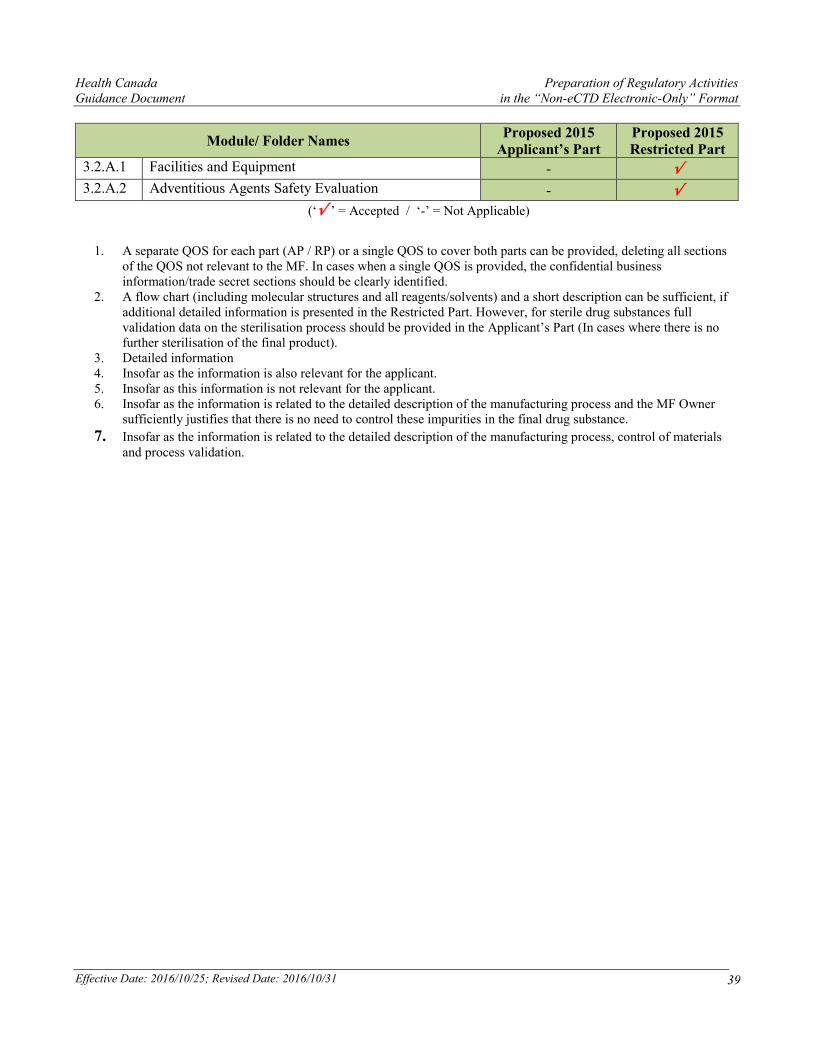
Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 39
Module/ Folder Names Proposed 2015
Applicant’s Part Proposed 2015 Restricted Part
3.2.A.1 Facilities and Equipment - √
3.2.A.2 Adventitious Agents Safety Evaluation - √
(‘√ ’ = Accepted / ‘-’ = Not Applicable)
1. A separate QOS for each part (AP / RP) or a single QOS to cover both parts can be provided, deleting all sections of the QOS not relevant to the MF. In cases when a single QOS is provided, the confidential business information/trade secret sections should be clearly identified.
2. A flow chart (including molecular structures and all reagents/solvents) and a short description can be sufficient, if additional detailed information is presented in the Restricted Part. However, for sterile drug substances full validation data on the sterilisation process should be provided in the Applicant’s Part (In cases where there is no further sterilisation of the final product).
3. Detailed information 4. Insofar as the information is also relevant for the applicant. 5. Insofar as this information is not relevant for the applicant. 6. Insofar as the information is related to the detailed description of the manufacturing process and the MF Owner
sufficiently justifies that there is no need to control these impurities in the final drug substance.
7. Insofar as the information is related to the detailed description of the manufacturing process, control of materials and process validation.

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 40
Table G-2: MF Type IV - Drug Products
Module/Folder Names Proposed 2015
Applicant’s Part Proposed 2015 Restricted Part
Module 1: Administrative and Product Information
1.0 Correspondence
1.0.1 Cover Letter - √
1.0.2 Life Cycle Management Table (Only required for eCTD) - √
1.0.3
Copy of Health Canada Issued Correspondence Clarification Request (Clarifax)
Notice of Deficiency Screening Deficiency Notice
- √
1.0.4 Health Canada Solicited Information
Q&A response to Clarification Request (Clarifax)
Q&A response to Notice of Deficiency - √
1.0.7 General Note to Reviewer - √
1.1 Table of Contents (Not required for eCTD)
1.2 Administrative Information
1.2.1
Application Forms DMF Application/Amendment Form Can include the following:
o Agent Appointment Letter o Agent Withdrawal Letter o Agent Name Change
- √
1.2.2 Fee Forms
DMF Application/Amendment Fee Form - √
1.2.3
Certification and Attestation Forms BSE/TSE Attestation Form Certification of Suitability (CEP) CEP - Update CEP - Attestations
Statement of Commitment
- √
1.2.5 Compliance and Site Information
1.2.5.2 Establishment Licensing - √
1.2.5.5 Good Manufacturing Practices
Certificate of Compliance - √
1.2.6 Authorization for Sharing Information
Letter of Access
Withdrawal of Authorization - √
1.2.7 International Information - √
1.3 Product Information
1.3.6 Certified Product Information Document - √

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 41
Module/Folder Names Proposed 2015
Applicant’s Part Proposed 2015 Restricted Part
Module 2: Common Technical Document Summary
2.3 Quality Overall Summary (QOS)1 √ √
Module 3: Quality
3.1 Table of Contents of Module 3 √ √
3.2 Body of Data
3.2.P Drug Product
3.2.P.1 Description and Composition of the Drug Product √ √ 3
3.2.P.2 Pharmaceutical Development √ 4 √ 3
3.2.P.2.1 Components of the Drug Product* √ 5 √
3.2.P.2.2 Drug Product* - √
3.2.P.2.3 Manufacturing Process Development* - √
3.2.P.2.4 Container Closure System* - √
3.2.P.2.5 Microbiological Attributes* - √
3.2.P.2.6 Compatibility* - √
3.2.P.3 Manufacture
3.2.P.3.1 Manufacturer(s) √ √
3.2.P.3.2 Batch Formula √ √
3.2.P.3.3 Description of Manufacturing Process and Process Controls
√ 2 √ 3
3.2.P.3.4 Controls of Critical Steps and Intermediates √ 4 √ 6
3.2.P.3.5 Process Validation and /or Evaluation - √
3.2.P.4 Control of Excipients √ 4 √ 6
3.2.P.4.1 Specifications - √
3.2.P.4.2 Analytical Procedures - √
3.2.P.4.3 Validation of Analytical Procedures - √
3.2.P.4.4 Justification of Specifications - √
3.2.P.4.5 Excipients of Human or Animal Origin - √
3.2.P.4.6 Novel Excipients - √
3.2.P.5 Control of Drug Product
3.2.P.5.1 Specifications √ -
3.2.P.5.2 Analytical Procedures √ -
3.2.P.5.3 Validation of Analytical Procedures √ -
3.2.P.5.4 Batch Analyses √ -
3.2.P.5.5 Characterisation of Impurities √ √ 7
3.2.P.5.6 Justification of Specifications √ √ 8
3.2.P.6 Reference Standards or Materials √ -

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 42
Module/Folder Names Proposed 2015
Applicant’s Part Proposed 2015 Restricted Part
3.2.P.7 Container Closer System √ -
3.2.P.8 Stability
3.2.P.8.1 Stability Summary and Conclusions √ -
3.2.P.8.2 Post-approval Stability Protocol and Stability Commitment
√ -
3.2.P.8.3 Stability Data √ -
3.2.A Appendices
3.2.A.1 Facilities and Equipment - √
3.2.A.2 Adventitious Agents Safety Evaluation - √
3.2.A.3 Excipients - √
3.2.R Regional Information
3.2.R.1 Production Documentation
3.2.R.1.1 Executed Production Documents* - √
3.2.R.1.2 Master Production Documents* - √
(‘√ ’ = Accepted / ‘-’ = Not Applicable)
1. A separate QOS for each part (AP / RP) or a single QOS to cover both parts can be provided, deleting all sections
of the QOS not relevant to the MF. In cases when a single QOS is provided, the confidential business information/trade secret sections should be clearly identified.
2. A flow chart (including all manufacturing steps, excipients and processing agents) and a short description can be sufficient, if additional detailed information is presented in the Restricted Part.
3. Detailed information. 4. Insofar as the information is also relevant for the applicant. 5. Complete qualitative composition is provided to the applicant. 6. Insofar as this information is not relevant for the applicant. 7. Insofar as the information is related to the detailed description of the manufacturing process and the MF Owner
sufficiently justifies that there is no need to control these impurities in the final drug product. 8. Insofar as the information is related to the detailed description of the manufacturing process, control of materials
and process validation.
Note: Text with * refers to sections of MFs that are not defined in Appendix D of Health Canada’s Guidance Document: Preparation of Drug Regulatory Activities in the Common Technical Document (CTD) Format.

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 43
Appendix H: Medical Device Sample Folder Structures for IMDRF TOC format
Figure H-1: Sample folder structure for IMDRF TOC
Table H-1: Specific Content Requirements for Devices in TOC Format

Preparation of Regulatory Activities Health Canada in the “Non-eCTD Electronic-Only” Format Guidance Document
Effective Date: 2016/10/25; Revised Date: 2016/10/31 44
Folders
Licence Private Label
Fax-Back
Class II Class
II, III & IV Class III/IV
All Classes (except for additions to Class III/IV)
All Classes
New Licence
Licence Amendment
Applications & Amendments
Add a new Device
Identifier
Add, Delete or Change
Device Identifier
Change to Licence / Device Name
Change to Manufacturer’s Name/Address
1.01 Oa Oa Oa Oa Oa Oa Oa 1.04b X X X X X X X 1.06c X - - - - - X 1.09 Od Od Od Od Od Od Od 1.14 - - X e Xf Xf Xf Xf 2.4.4 - Og - Xg - - -
5 Z Z Z O O O O
‘X’ = always include ‘O’ = optional ‘Z’ = include all applicable folders ‘-’ = not applicable a. If information needs to be included that does not appear somewhere else in the application b. Includes fee form, where applicable c. ISO 13485:2003 certificate d. If applicable e. Letter of Authorization and Declaration of Compliance (for new private label applications) f. Copy of front page of licence license g. Comparison table showing similarities and differences to currently licenced devices
MD
The attached zipped folder structures (available in the HTML version of this document) can be used by adding documents in their respected folders. Empty folders must be deleted from structures before filing to Health Canada.
Summary Technical Documentation (STED)
Health Canada Structure

Health Canada Preparation of Regulatory Activities Guidance Document in the “Non-eCTD Electronic-Only” Format
Effective Date: 2016/10/25; Revised Date: 2016/10/31 45
MD
Figure H-2: Sample folder structure for Post-Market IMDRF TOC
Table H-2: Specific Content Requirements for Devices in TOC Format for Post-Market
Folders
Post-Market Transactions All Classes (I, II, III, IV)
Request for Additional Information
1.01 X 1.02 O 1.03 O
1.09 X
2.6 Z 3.2 Z
5 Z ‘X’ = always include ‘O’ = optional ‘Z’ = include all applicable folders


















