
Future of Non Thermal Ablation: Is the Future of Endovenous Ablation

of February 14, 2018.This information is current as
Diet-Induced ObesityImproves Insulin Sensitivity in Mice with GPR105 Ablation Prevents Inflammation and
Olefsky and Jane J. KimM.Ai Chen, Saswata Talukdar, Eduardo Lazarowski, Jerrold
Jianfeng Xu, Hidetaka Morinaga, Dayoung Oh, Pingping Li,
ol.1103207http://www.jimmunol.org/content/early/2012/07/09/jimmun
published online 9 July 2012J Immunol
MaterialSupplementary
7.DC1http://www.jimmunol.org/content/suppl/2012/07/09/jimmunol.110320
average*
4 weeks from acceptance to publicationSpeedy Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Errata
/content/190/3/1380.full.pdfor:
next pageAn erratum has been published regarding this article. Please see
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2012 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on February 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on February 14, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on February 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

The Journal of Immunology
GPR105 Ablation Prevents Inflammation and ImprovesInsulin Sensitivity in Mice with Diet-Induced Obesity
Jianfeng Xu,* Hidetaka Morinaga,* Dayoung Oh,* Pingping Li,* Ai Chen,*
Saswata Talukdar,* Eduardo Lazarowski,† Jerrold M. Olefsky,* and Jane J. Kim‡,x
GPR105, a G protein-coupled receptor for UDP-glucose, is highly expressed in several human tissues and participates in the innate
immune response. Because inflammation has been implicated as a key initial trigger for type 2 diabetes, we hypothesized that
GPR105 (official gene name: P2RY14) might play a role in the initiation of inflammation and insulin resistance in obesity. To
this end, we investigated glucose metabolism in GPR105 knockout (KO) and wild-type (WT) mice fed a high-fat diet (HFD). We
also examined whether GPR105 regulates macrophage recruitment to liver or adipose tissues by in vivo monocyte tracking and
in vitro chemotaxis experiments, followed by transplantation of bone marrow from either KO or WT donors to WT recipients.
Our data show that genetic deletion of GPR105 confers protection against HFD-induced insulin resistance, with reduced mac-
rophage infiltration and inflammation in liver, and increased insulin-stimulated Akt phosphorylation in liver, muscle, and adipose
tissue. By tracking monocytes from either KO or WT donors, we found that fewer KO monocytes were recruited to the liver of
WT recipients. Furthermore, we observed that uridine 5-diphosphoglucose enhanced the in vitro migration of bone marrow-
derived macrophages from WT but not KO mice, and that plasma uridine 5-diphosphoglucose levels were significantly higher in
obese versus lean mice. Finally, we confirmed that insulin sensitivity improved in HFD mice with a myeloid cell-specific deletion of
GPR105. These studies indicate that GPR105 ablation mitigates HFD-induced insulin resistance by inhibiting macrophage
recruitment and tissue inflammation. Hence GPR105 provides a novel link between innate immunity and metabolism. The
Journal of Immunology, 2012, 189: 000–000.
Insulin resistance is a major metabolic feature of obesity anda key factor in the pathogenesis of type 2 diabetes. Manylines of evidence show that the activation of proinflammatory
pathways within insulin target tissues can impair insulin signaling,and chronic, low-level tissue inflammation is now recognized asan important cause of systemic insulin resistance (1). Severalstudies have shown that macrophages, a key component of theinnate immune defense against pathogens, are critical effectorcells in this proinflammatory process.Macrophages present in noninflamed tissues (resident macro-
phages) help maintain homeostasis and participate in tissue remod-
eling (2). However, in obese states, macrophages accumulate infat, liver, and muscle tissues, secreting proinflammatory cytokinessuch as TNF-a and IL-6 that induce cellular insulin resistance
by the activation of JNK or IkB kinase b signaling with serine
phosphorylation of insulin receptor substrate proteins. Macro-
phages that are newly recruited after high-fat diet (HFD) exposure
have been shown to differ from resident macrophages in adipose
tissue, showing increased proinflammatory properties (3). Al-
though both innate and adaptive immune systems have been im-
plicated in macrophage activation and recruitment in obesity (4),
the precise mechanisms that regulate the recruitment of new
macrophages to insulin target tissues are unclear.G protein-coupled receptors (GPCRs) mediate a variety of
physiological functions, which poise these proteins as therapeutic
targets in metabolic disease. They are also the target of approxi-
mately half of all modern medicinal drugs (5). GPR105 (also
known as the P2Y14 receptor) is a member of a GPCR subgroup
potently activated by pyrimidine UDP-sugars, especially uridine
5-diphosphoglucose (UDP-Glc) (6). GPR105 is broadly expressed
in several human tissues, including the placenta, adipose tissue,stomach, intestine, and discrete brain regions (6). Notably,
GPR105 is also prominently expressed in immune cells, including
macrophages, lymphocytes, and neutrophils (7–9). Previous studies
have revealed that GPR105 regulates leukocyte chemotaxis in
response to UDP-Glc (10, 11).Because GPR105 participates in the innate immune response,
we hypothesized that GPR105 may contribute to the initiation ofobesity-induced insulin resistance. In this study, we have assessed
the effects of whole-body and bone marrow-specific deletion of
GPR105 (official gene name: P2ry14) on insulin action and glu-
cose homeostasis. We show that GPR105 in immune cells medi-
ates macrophage recruitment to the liver in chronic HFD feeding
with subsequent local inflammation and insulin resistance. Thus,
*Department of Medicine, University of California San Diego, La Jolla, CA 92093;†Department of Medicine, University of North Carolina, Chapel Hill, NC 27599;‡Department of Pediatrics, University of California San Diego, La Jolla, CA 92093; andxRady Children’s Hospital-San Diego, San Diego, CA 92123
Received for publication November 9, 2011. Accepted for publication June 14, 2012.
This work was supported by National Institutes of Health Grants DK075479 (toJ.J.K.), DK033651, DK074868, and DK063491 (to J.M.O.), and by the EuniceKennedy Shriver National Institute of Child Health and Human Development/National Institutes of Health through cooperative agreement U54 HD 012303-25as part of the specialized Cooperative Centers Program in Reproduction andInfertility Research. Additional research support was provided by Merck & Co.(to J.M.O.).
Address correspondence and reprint requests to Dr. Jane J. Kim, University ofCalifornia San Diego, 9500 Gilman Drive, MC 0673, La Jolla, CA 92093-0673.E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: ATM, adipose tissue macrophage; BMDM, bonemarrow-derived macrophage; BMT, bone marrow transplantation; eWAT, epididymalwhite adipose tissue; GDR, glucose disposal rate; GIR, glucose infusion rate; GPCR,G protein-coupled receptor; GTT, glucose tolerance test; HFD, high-fat diet; HGP,hepatic glucose production; IS-GDR, insulin-stimulated GDR; ITT, insulin tolerancetest; KO, knockout; NC, normal chow; 32PPi, [32P]-pyrophosphate; RHM, recruitedhepatic macrophage; TG, triglyceride; UDP-Glc, uridine 5-diphosphoglucose; WT,wild-type.
Copyright� 2012 by The American Association of Immunologists, Inc. 0022-1767/12/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1103207
Published July 9, 2012, doi:10.4049/jimmunol.1103207 by guest on February 14, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

GPR105 may provide a novel link between innate immunity and
insulin resistance.
Materials and MethodsAnimals
GPR105 (P2ry14) null mice were purchased from Taconic and have beenpreviously described (12). In brief, embryonic stem cells from 129P2 micewere manipulated by replacement of the P2ry14 gene open reading frameby a LacZ-neo cassette and subsequently injected into C57BL/6J blasto-cytes to generate GPR105 knockout (KO) mice. In our animal facility,these mice were backcrossed to C57BL/6N mice obtained from HarlanLaboratories for six generations. Heterozygous mice from the seventhgeneration were then used to breed wild-type (WT) and GPR105 KO lit-termates used for all experiments described in this article. Mice were fedeither a normal chow (NC) or HFD (60% kcal from fat; D12492; ResearchDiets), and maintained on a 12-h light/dark cycle with free access to foodand water. For bone marrow transplantation (BMT) studies, 10-wk-oldC57BL/6N mice (Harlan, CA) were exposed to 10 Gy Co-60 irradiationand injected with bone marrow cells from WT or GPR105 KO mice within24 h. Eight weeks after transplantation, mice were fed either NC or HFDfor 12 wk to generate lean or obese/insulin-resistant phenotypes. Allmouse procedures conformed to the Guide for the Care and Use of Lab-oratory Animals of the National Institutes of Health and were approved bythe Animal Subjects Committee of the University of California, San Diego.
Metabolic studies
Glucose and insulin tolerance tests (GTT and ITT, respectively) wereperformed on 7-h fasted mice. Animals were given i.p. or oral dextrose (1 g/kg; Hospira) for the GTT, or i.p. insulin (0.6 U/kg, Novolin R, Novo-Nordisk) for the ITT. Glucose levels after injection were monitored overtime. Blood samples were drawn by tail nick at basal and indicated times,and glucose was measured using a One-Touch glucose meter (Lifescan).Hyperinsulinemic-euglycemic clamp studies were performed as previouslydescribed (13, 14). In brief, two jugular catheters were inserted underanesthesia, tunneled to the midscapular region, and externalized. Five dayslater, the clamp experiments began with the infusion of an equilibrationsolution of D-[3-3H] glucose (NEN, Boston, MA) for 90 min at a constantrate of 5 mCi/h. Then insulin was infused at 8 mU/kg/min while the glu-cose infusion rate (GIR) was adjusted as necessary until euglycemia wasachieved (∼120 mg/dl for .20 min). Blood samples were collected beforeinsulin infusion and on euglycemia to confirm steady-state. At steady-state,the rate of glucose disappearance (total glucose disposal rate [GDR])equals the sum of hepatic glucose production (HGP) and the GIR. Theinsulin-stimulated GDR (IS-GDR) is equal to the total GDR minus thebasal glucose turnover rate. Serum insulin levels were analyzed by ELISA(Alpco Diagnostics).
Triglyceride and glycogen measurements
Triglyceride (TG) content was measured in liver homogenates in PBScontaining 5% Triton X-100 and in plasma samples (EnzyChrom, BioAssaySystems, Hayward, CA). Glycogen levels were measured by the method ofSeifter et al. (15).
Histology
H&E staining in liver and adipose tissue sections was conducted by theUniversity of California, San Diego histology core at the Moore’s CancerCenter. Anti-Mac2 (Cedarlane Labs) and anti-F4/80 (Abcam, Cambridge,MA) Abs were used for immunohistochemistry.
Immunoblot analysis
For insulin-stimulated phospho-Akt analysis, mice were fasted for 7 h andthen injected with 0.2 U/kg insulin via the inferior vena cava under anes-thesia. Tissues were taken before or after injection (3min for liver and 10minfor epididymal white adipose tissue [eWAT] and skeletal muscle), snapfrozen, and subjected to Western blot analyses performed according tostandard techniques. Abs against p-Akt (Ser473), Akt, and a-tubulin wereobtained from Cell Signaling Technologies (Beverly, MA). To measure Aktphosphorylation, we immunoblotted solubilized extracts containing equalamounts of tissue protein with rabbit polyclonal Abs against either Akt orphospho-Ser473 Akt (Cell Signaling Technology, Beverly, MA), followed bysecond Ab detection. For quantification, the relative intensities of proteinbands were measured with ImageJ and expressed as arbitrary units.
RNA isolation and quantitative RT-PCR
Total RNA was isolated from cells and tissues using TRIzol Reagent(Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions.First-strand cDNA was synthesized using a High-Capacity cDNA ReverseTranscription Kit (Applied Biosystems, Foster City, CA). The sampleswere run in 20-ml reactions using an MJ Research PTC-200 96-wellthermocycler coupled with the Chromo 4 Four-Color Real-Time System(GMI, Ramsey, MN). Gene expression levels were calculated after nor-malization to the standard housekeeping gene RNA polymerase II (RPII)using the DDCT method as described previously (16). Primer sequences areavailable in Supplemental Table I.
Monocyte tracking
Blood collected from the retro-orbital sinus ofWTor GPR105 KOmice wassubjected to RBC lysis, and monocyte subsets were enriched with theEasySep mouse monocytes enrichment kit (Stem Cell Technologies,Vancouver, BC, Canada). Isolated monocytes from 10–15 mice of eachgenotype were pooled together. These monocytes (5 3 105 to 10 3 105)were then washed in serum-free RPMI and suspended in 2 ml Diluentsolution C. PKH26 (Sigma-Aldrich, St. Louis, MO) dissolved at 2 3 1023
M in Diluent C was added, and the cells were incubated for 10 min at roomtemperature in the dark. The staining reaction was halted by the addition ofan equal volume of medium supplemented with 10% FBS. The mixturewas centrifuged, and the cells were washed once and resuspended inserum-containing medium. Subsequent to labeling with PKH26, themonocytes were counted and ∼1 3 105 viable cells were suspended in 0.2ml PBS and injected into the femoral vein of WT mice who were receivingHFD. Five days after injection, adipose tissue macrophages (ATMs) andliver nonparenchymal cells were isolated and analyzed by FACS analysis.
FACS analysis of macrophages from the adipose stromalvascular fraction and liver
Epididymal fat pads wereweighed, rinsed in PBS, and then minced in FACSbuffer (PBS/1%BSA). Adipocytes and stromal vascular cells were preparedfrom collagenase-digested adipose tissue (17). Liver macrophage cellswere prepared by a two-step liver collagenase digestion and fractionationon a density gradient (18). FACS analyses of stromal vascular cells formacrophage content and subtypes were performed as previously described(17). The Abs used for surface staining were F4/80 (BM8), CD11b (M1/70), and CD11c (N418; eBioscience, San Diego, CA).
Bone marrow-derived macrophage culture and in vitrochemotaxis assay
Bone marrow-derived macrophage (BMDM) culture was performed as pre-viously described (17). In brief, bone marrow cells were flushed from thefemurs and tibias of 10-to 12-wk-old WT and GPR105 KO mice. The cellswere then differentiated into BMDMs in RPMI medium containing rM-CSF,low-endotoxin FBS, and streptomycin/penicillin. Five days after differentia-tion, cells were harvested and suspended in DMEM with 1 g/l glucose. Thein vitro chemotaxis assay was performed as previously described (19). Ap-proximately 105 cells were seeded into the upper chamber of an 8-mM pol-ycarbonate filter (24-transwell format; Corning, Lowell, MA), and DMEMwith indicated concentrations of UDP-Glc was placed in the lower chamber.After 3 h, the cells that had migrated to the lower chamber were counted.
Measurement of UDP-glucose levels in plasma
Plasma samples were deproteinized with 5% trichloroacetic acid followedby ethyl ether extraction and neutralization. The UDP-Glc content in theextracts was quantified using the assay that is based on enzymatic con-version of [32P]-pyrophosphate (32PPi) + UDP-glucose into [32P]UTP +glucose-1P, as previously described (20). In brief, samples were incubatedfor 1 h in the presence of 1 U/ml UDP-Glc pyrophosphorylase frombaker’s yeast (Sigma) and 100 nM 0.1 mCi 32PPi (Perkin Elmer). Incu-bations were terminated by the addition of 0.5 mM PPi and subsequentheating (2 min at 95˚C). The resulting [32P] species were separated byHPLC, via a Nova-Pack C18 column (Waters), and quantified online witha Flo-One 500TR Radiomatic analyzer (Packard). A calibration curveusing known amounts of UDP-glucose (Fluka) was performed in parallel.
Statistical analysis
All values are expressed as means6 SEM unless otherwise noted. We usedStudent two-tailed t test or ANOVA to determine differences betweengroups, and repeated-measures ANOVA testing for comparisons over time.The p values ,0.05 were considered significant.
2 GPR105-MEDIATED INFLAMMATION AND INSULIN RESISTANCE
by guest on February 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

ResultsGPR105 deletion protects mice from HFD-induced insulinresistance
To investigate the functional role of GPR105 in HFD-inducedobesity, we fed 8-wk-old GPR105 KO and their WT littermatesa NC or 60% HFD for 16 wk. Whole-body weights did not differbetween GPR105 KO mice and their WT controls on either diet(Fig. 1A). HFD food intake was also similar between genotypes(Fig. 1B).On the NC diet, there were no differences in glucose tolerance
(Fig. 1C) or fasting insulin levels (Fig. 1D) between WT andGPR105 KO mice. After HFD feeding, WT mice developed se-vere glucose intolerance, as expected (Fig. 1C). However, HFD-fed GPR105 KO mice were protected from HFD-induced defectsin glucose tolerance, with significantly lower glucose excursionsduring the IPGTT (Fig. 1C). Despite similar basal glucose values,fasting insulin levels were also lower in HFD GPR105 KO micecompared with WT controls (Fig. 1D, 1E), suggesting that theimproved glucose tolerance was secondary to reduced insulin re-sistance. This finding was further supported by decreased insulinvalues measured 15 min after an oral dextrose challenge (Fig. 1E)and a greater hypoglycemic response during ITT (Fig. 1F) inHFD-fed GPR105 KO mice. These data indicate improved insulinsensitivity in HFD-fed obese GPR105 KO mice.
Improved insulin sensitivity in HFD-fed GPR105 KO mice
To further investigate in vivo insulin sensitivity in HFD-fedWTandGPR105 KO mice, we performed hyperinsulinemic-euglycemicglucose clamp studies. These studies further supported our GTT
and ITT findings with a 3-fold increase in the GIR in GPR105 KOmice compared with controls (Fig. 2A). Total glucose turnover(Fig. 2B) and IS-GDRs (Fig. 2C) were also significantly enhancedin GPR105 KO mice. Because 70–80% of the IS-GDR is attrib-utable to skeletal muscle glucose uptake, these results suggest thatthe deletion of GPR105 leads to improved skeletal muscle insulinsensitivity.GPR105 KO mice also demonstrated significantly lower rates
of HGP under both basal and steady-state clamp conditions(Fig. 2D), indicating that the ability of insulin to suppress HGPwas enhanced in HFD-fed GPR105 KO mice (Fig. 2E). Con-sistent with the improved hepatic and skeletal muscle insulinsensitivity found in the clamp studies, AKT phosphorylation wassignificantly increased in liver, adipose, and skeletal muscletissues of GPR105 KO mice after insulin stimulation (Fig. 2F).Taken together, these in vivo results support the conclusion thatGPR105 KO mice exhibit improved systemic insulin sensitivityafter HFD with enhanced insulin action in liver, skeletal muscle,and adipose tissues.
Reduced liver steatosis and inflammation in GPR105 KO mice
Liver weights were decreased in the GPR105 KO mice (Fig. 3A)despite the similar body weights of GPR105 KO and WT mice.GPR105 KO mice also demonstrated lower liver TG and glycogencontent (Fig. 3B, 3C), as well as a lower grade of hepatic steatosisby histology (Fig. 3D).
FIGURE 1. Improved glucose homeostasis of GPR105 KO HFD-fed
mice. (A) Body weight gain of WT and GPR105 KO mice on NC and HFD
(n = 12 per group). (B) Food intake of WT and GPR105 KO mice on HFD
(n = 12). (C) Glucose tolerance testing in NC and HFD mice. Glucose (1 g/
kg) was injected i.p. after a 7-h fast, and tail blood was collected for
glucose measurement (n = 8). (D) Fasting insulin levels of WT and
GPR105 KO mice on NC and HFD (n = 8). (E) Acute insulin secretion
measured in HFD-fed WT and GPR105 KO mice at basal (7 h fast) and 15
min after oral glucose challenge (n = 8). (F) Insulin tolerance testing in
HFD mice. Intraperitoneal insulin (0.6 U/kg) was injected to 7-h–fasted
HFD WT and GPR105 KO mice. Blood glucose was measured at the in-
dicated time points. All values are expressed as means 6 SEM. *p , 0.05
versus diet-matched WT.
FIGURE 2. Improved insulin sensitivity of GPR105 KO HFD-fed mice.
In vivo insulin sensitivity as determined by hyperinsulinemic-euglycemic
clamp in WT (n = 6) and GPR105 KO (n = 6) mice fed HFD. GIR (A),
GDR (B), IS-GDR (C), basal and clamp HGP (D), and suppression of HGP
(E) are presented as means6 SEM. *p, 0.05 versus WT. (F) Immunoblot
analyses of Ser473 phosphorylation of Akt, as well as total Akt and HSP90
in liver, adipose tissue, and skeletal muscle. Tissues were collected before
or at indicated time after insulin injection (0.2 U/kg) into the inferior vena
cava.
The Journal of Immunology 3
by guest on February 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
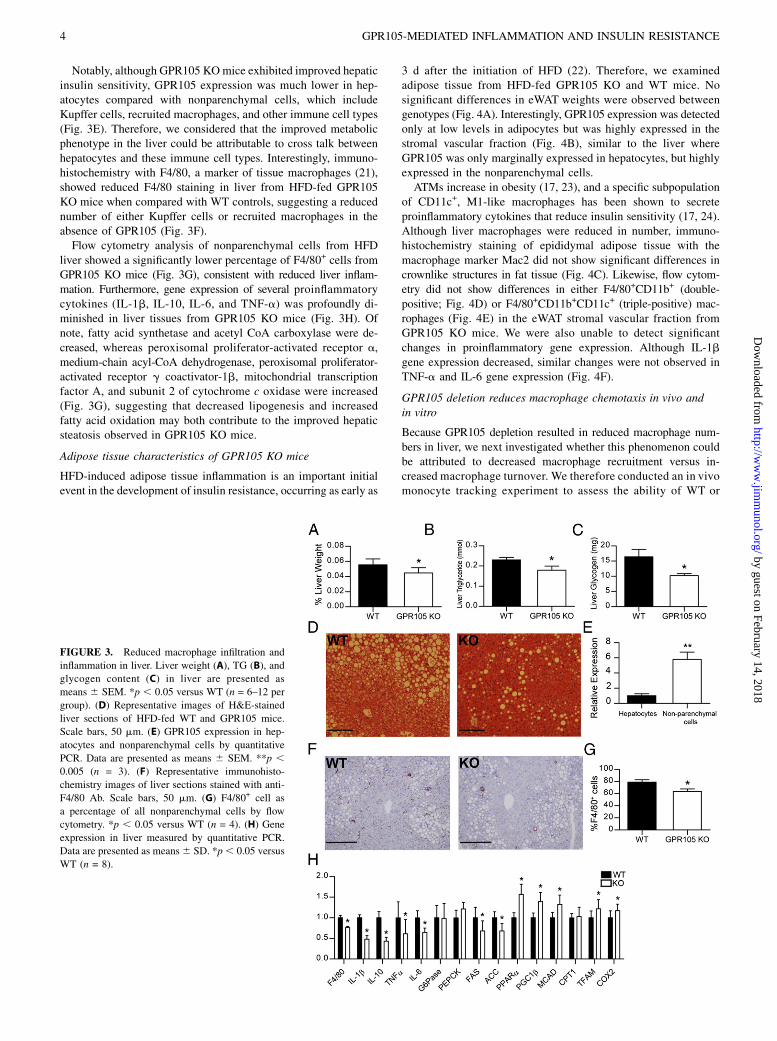
Notably, although GPR105 KOmice exhibited improved hepaticinsulin sensitivity, GPR105 expression was much lower in hep-atocytes compared with nonparenchymal cells, which includeKupffer cells, recruited macrophages, and other immune cell types(Fig. 3E). Therefore, we considered that the improved metabolicphenotype in the liver could be attributable to cross talk betweenhepatocytes and these immune cell types. Interestingly, immuno-histochemistry with F4/80, a marker of tissue macrophages (21),showed reduced F4/80 staining in liver from HFD-fed GPR105KO mice when compared with WT controls, suggesting a reducednumber of either Kupffer cells or recruited macrophages in theabsence of GPR105 (Fig. 3F).Flow cytometry analysis of nonparenchymal cells from HFD
liver showed a significantly lower percentage of F4/80+ cells fromGPR105 KO mice (Fig. 3G), consistent with reduced liver inflam-mation. Furthermore, gene expression of several proinflammatorycytokines (IL-1b, IL-10, IL-6, and TNF-a) was profoundly di-minished in liver tissues from GPR105 KO mice (Fig. 3H). Ofnote, fatty acid synthetase and acetyl CoA carboxylase were de-creased, whereas peroxisomal proliferator-activated receptor a,medium-chain acyl-CoA dehydrogenase, peroxisomal proliferator-activated receptor g coactivator-1b, mitochondrial transcriptionfactor A, and subunit 2 of cytochrome c oxidase were increased(Fig. 3G), suggesting that decreased lipogenesis and increasedfatty acid oxidation may both contribute to the improved hepaticsteatosis observed in GPR105 KO mice.
Adipose tissue characteristics of GPR105 KO mice
HFD-induced adipose tissue inflammation is an important initialevent in the development of insulin resistance, occurring as early as
3 d after the initiation of HFD (22). Therefore, we examinedadipose tissue from HFD-fed GPR105 KO and WT mice. Nosignificant differences in eWAT weights were observed betweengenotypes (Fig. 4A). Interestingly, GPR105 expression was detectedonly at low levels in adipocytes but was highly expressed in thestromal vascular fraction (Fig. 4B), similar to the liver whereGPR105 was only marginally expressed in hepatocytes, but highlyexpressed in the nonparenchymal cells.ATMs increase in obesity (17, 23), and a specific subpopulation
of CD11c+, M1-like macrophages has been shown to secreteproinflammatory cytokines that reduce insulin sensitivity (17, 24).Although liver macrophages were reduced in number, immuno-histochemistry staining of epididymal adipose tissue with themacrophage marker Mac2 did not show significant differences incrownlike structures in fat tissue (Fig. 4C). Likewise, flow cytom-etry did not show differences in either F4/80+CD11b+ (double-positive; Fig. 4D) or F4/80+CD11b+CD11c+ (triple-positive) mac-rophages (Fig. 4E) in the eWAT stromal vascular fraction fromGPR105 KO mice. We were also unable to detect significantchanges in proinflammatory gene expression. Although IL-1bgene expression decreased, similar changes were not observed inTNF-a and IL-6 gene expression (Fig. 4F).
GPR105 deletion reduces macrophage chemotaxis in vivo andin vitro
Because GPR105 depletion resulted in reduced macrophage num-bers in liver, we next investigated whether this phenomenon couldbe attributed to decreased macrophage recruitment versus in-creased macrophage turnover. We therefore conducted an in vivomonocyte tracking experiment to assess the ability of WT or
FIGURE 3. Reduced macrophage infiltration and
inflammation in liver. Liver weight (A), TG (B), and
glycogen content (C) in liver are presented as
means 6 SEM. *p , 0.05 versus WT (n = 6–12 per
group). (D) Representative images of H&E-stained
liver sections of HFD-fed WT and GPR105 mice.
Scale bars, 50 mm. (E) GPR105 expression in hep-
atocytes and nonparenchymal cells by quantitative
PCR. Data are presented as means 6 SEM. **p ,0.005 (n = 3). (F) Representative immunohisto-
chemistry images of liver sections stained with anti-
F4/80 Ab. Scale bars, 50 mm. (G) F4/80+ cell as
a percentage of all nonparenchymal cells by flow
cytometry. *p , 0.05 versus WT (n = 4). (H) Gene
expression in liver measured by quantitative PCR.
Data are presented as means6 SD. *p, 0.05 versus
WT (n = 8).
4 GPR105-MEDIATED INFLAMMATION AND INSULIN RESISTANCE
by guest on February 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
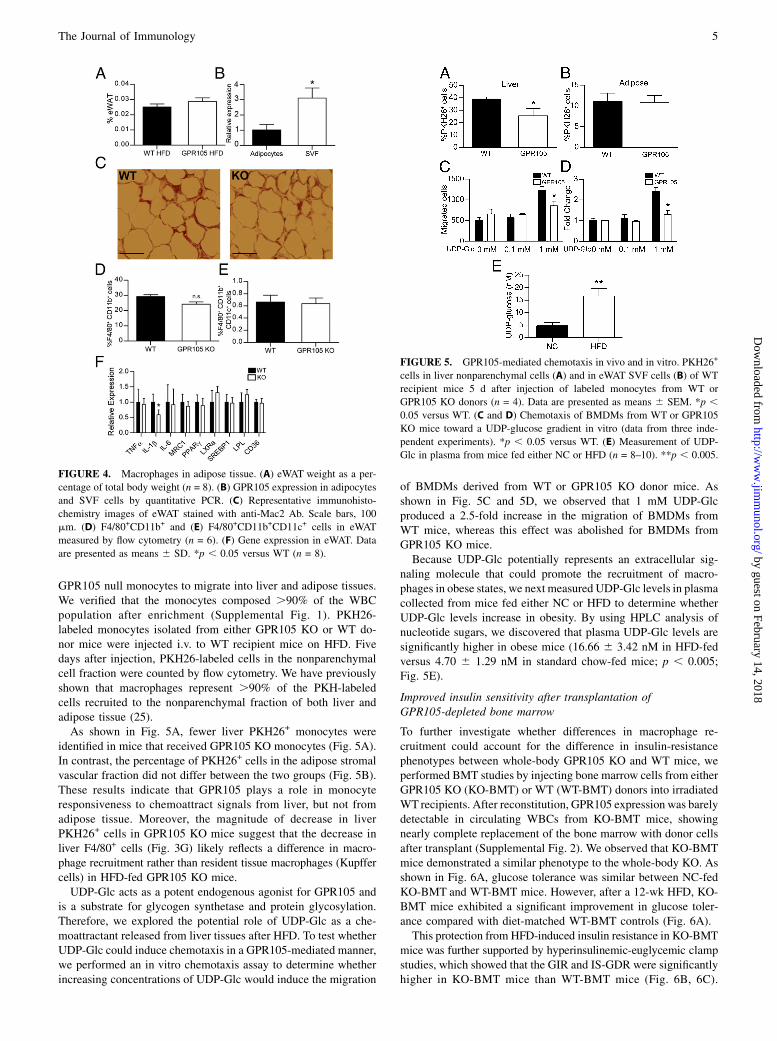
GPR105 null monocytes to migrate into liver and adipose tissues.We verified that the monocytes composed .90% of the WBCpopulation after enrichment (Supplemental Fig. 1). PKH26-labeled monocytes isolated from either GPR105 KO or WT do-nor mice were injected i.v. to WT recipient mice on HFD. Fivedays after injection, PKH26-labeled cells in the nonparenchymalcell fraction were counted by flow cytometry. We have previouslyshown that macrophages represent .90% of the PKH-labeledcells recruited to the nonparenchymal fraction of both liver andadipose tissue (25).As shown in Fig. 5A, fewer liver PKH26+ monocytes were
identified in mice that received GPR105 KO monocytes (Fig. 5A).In contrast, the percentage of PKH26+ cells in the adipose stromalvascular fraction did not differ between the two groups (Fig. 5B).These results indicate that GPR105 plays a role in monocyteresponsiveness to chemoattract signals from liver, but not fromadipose tissue. Moreover, the magnitude of decrease in liverPKH26+ cells in GPR105 KO mice suggest that the decrease inliver F4/80+ cells (Fig. 3G) likely reflects a difference in macro-phage recruitment rather than resident tissue macrophages (Kupffercells) in HFD-fed GPR105 KO mice.UDP-Glc acts as a potent endogenous agonist for GPR105 and
is a substrate for glycogen synthetase and protein glycosylation.Therefore, we explored the potential role of UDP-Glc as a che-moattractant released from liver tissues after HFD. To test whetherUDP-Glc could induce chemotaxis in a GPR105-mediated manner,we performed an in vitro chemotaxis assay to determine whetherincreasing concentrations of UDP-Glc would induce the migration
of BMDMs derived from WT or GPR105 KO donor mice. Asshown in Fig. 5C and 5D, we observed that 1 mM UDP-Glcproduced a 2.5-fold increase in the migration of BMDMs fromWT mice, whereas this effect was abolished for BMDMs fromGPR105 KO mice.Because UDP-Glc potentially represents an extracellular sig-
naling molecule that could promote the recruitment of macro-phages in obese states, we next measured UDP-Glc levels in plasmacollected from mice fed either NC or HFD to determine whetherUDP-Glc levels increase in obesity. By using HPLC analysis ofnucleotide sugars, we discovered that plasma UDP-Glc levels aresignificantly higher in obese mice (16.66 6 3.42 nM in HFD-fedversus 4.70 6 1.29 nM in standard chow-fed mice; p , 0.005;Fig. 5E).
Improved insulin sensitivity after transplantation ofGPR105-depleted bone marrow
To further investigate whether differences in macrophage re-cruitment could account for the difference in insulin-resistancephenotypes between whole-body GPR105 KO and WT mice, weperformed BMT studies by injecting bone marrow cells from eitherGPR105 KO (KO-BMT) or WT (WT-BMT) donors into irradiatedWT recipients. After reconstitution, GPR105 expression was barelydetectable in circulating WBCs from KO-BMT mice, showingnearly complete replacement of the bone marrow with donor cellsafter transplant (Supplemental Fig. 2). We observed that KO-BMTmice demonstrated a similar phenotype to the whole-body KO. Asshown in Fig. 6A, glucose tolerance was similar between NC-fedKO-BMT and WT-BMT mice. However, after a 12-wk HFD, KO-BMT mice exhibited a significant improvement in glucose toler-ance compared with diet-matched WT-BMT controls (Fig. 6A).This protection from HFD-induced insulin resistance in KO-BMT
mice was further supported by hyperinsulinemic-euglycemic clampstudies, which showed that the GIR and IS-GDR were significantlyhigher in KO-BMT mice than WT-BMT mice (Fig. 6B, 6C).
FIGURE 4. Macrophages in adipose tissue. (A) eWAT weight as a per-
centage of total body weight (n = 8). (B) GPR105 expression in adipocytes
and SVF cells by quantitative PCR. (C) Representative immunohisto-
chemistry images of eWAT stained with anti-Mac2 Ab. Scale bars, 100
mm. (D) F4/80+CD11b+ and (E) F4/80+CD11b+CD11c+ cells in eWAT
measured by flow cytometry (n = 6). (F) Gene expression in eWAT. Data
are presented as means 6 SD. *p , 0.05 versus WT (n = 8).
FIGURE 5. GPR105-mediated chemotaxis in vivo and in vitro. PKH26+
cells in liver nonparenchymal cells (A) and in eWAT SVF cells (B) of WT
recipient mice 5 d after injection of labeled monocytes from WT or
GPR105 KO donors (n = 4). Data are presented as means 6 SEM. *p ,0.05 versus WT. (C and D) Chemotaxis of BMDMs from WT or GPR105
KO mice toward a UDP-glucose gradient in vitro (data from three inde-
pendent experiments). *p , 0.05 versus WT. (E) Measurement of UDP-
Glc in plasma from mice fed either NC or HFD (n = 8–10). **p , 0.005.
The Journal of Immunology 5
by guest on February 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

Insulin-stimulated suppression of hepatic glucose output wassomewhat enhanced in KO-BMT mice, although this differencedid not reach statistical significance (Fig. 6D). Consistent withimproved insulin sensitivity, we found that insulin-stimulatedAKT phosphorylation significantly increased in liver, adipose,and skeletal muscle of KO-BMT mice (Fig. 6E).
DiscussionChronic tissue inflammation is now recognized as a key cause ofsystemic insulin resistance in the context of obesity and type 2diabetes. A substantive literature has emerged indicating thatthe accumulation of activated macrophages in tissues provides amechanism for inflammation-induced insulin resistance, becausethese cells secrete a variety of cytokines that can directly impairinsulin sensitivity in insulin target cells. Other immune cell typessuch as lymphocytes and eosinophils can also participate in theseevents, but their effects are likely mediated by modulating mac-rophage chemotaxis and activation. Thus, the macrophage wouldbe the effector cell, causing decreased insulin sensitivity. In thisarticle, we report the role of a macrophage-expressed GPCR termedGPR105, which heretofore has not been implicated in obesity-associated chronic tissue inflammation and insulin resistance.GPR105 (also known as the P2Y14 receptor) is a member of the
P2Y purinergic receptor subfamily. P2Y receptor subtypes havebeen identified in immune cells such as monocytes and macro-phages, and exert effects on inflammatory signaling (26). Forexample, P2Y2 receptors regulate macrophage chemotaxis towardcomplement C5a in response to ATP release via autocrine puri-
nergic signaling (26). Unlike other P2Y receptors that are activatedby adenine and/or uracil nucleotides, GPR105 is a membrane re-ceptor for UDP-glucose.GPR105 expression has been reported in various human tissues,
including the placenta, adipose tissue, spleen, gut, and discretebrain regions, by microarray or quantitative PCR (6, 7). However,because immune cells are widely distributed among several tissuetypes, it was previously unknown whether GPR105 is expressedby parenchymal or nonparenchymal cells. In this report, wedemonstrate that GPR105 is highly expressed in the stromalvascular fraction but not in the parenchymal fraction of liver andadipose tissue. Therefore, the tissue-specific role of GPR105 re-quires careful interpretation because cross talk between residentimmune and parenchymal cells within tissues and cross talk withcirculating leukocytes may account for the in vivo phenotypes.We show that GPR105 KO mice are more insulin sensitive with
reduced liver steatosis and inflammation after HFD feeding. Inaddition, GPR105 mediates macrophage chemotaxis to UDP-Glcin vitro and macrophage recruitment to liver tissues in responseto HFD in vivo. Notably, the most impressive phenotypic changesin HFD GPR105 KO mice were observed in the liver. Hepaticinsulin resistance in obesity is associated with increased expressionof inflammatory mediators and massive accumulation of intra-cellular lipid within hepatocytes. Recruited hepatic macrophages(RHMs) and resident Kupffer cells are important immune cell typesin the liver and reside in the sinusoids, where they are poised tocommunicate with hepatocytes. Improved hepatic insulin sensi-tivity in HFD GPR105 KO mice was associated with decreased
FIGURE 6. Improved glucose homeostasis and in-
sulin sensitivity of GPR105 KO bone marrow-trans-
planted mice. (A) Glucose tolerance testing in WT-
BMT and KO-BMT mice on NC or HFD (n = 12).
*p , 0.05, **p , 0.005, versus diet-matched WT.
(B–D) Hyperinsulinemic-euglycemic clamp studies in
WT-BMT (n = 4) and KO-BMT (n = 4) mice fed with
HFD. GIR (B), IS-GDR (C), and suppression of HGP
(D) are presented as means 6 SEM. *p , 0.05 versus
WT. (E) Immunoblot analyses of Ser473 phosphoryla-
tion of Akt and HSP90 in liver, adipose tissue, and
skeletal muscles of WT-BMT and KO-BMT. Tissues
were collected at the indicated times before and after
insulin injection (0.2 U/kg). *p , 0.05 versus WT.
6 GPR105-MEDIATED INFLAMMATION AND INSULIN RESISTANCE
by guest on February 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

recruited macrophage numbers, reduced tissue inflammation, andreduced hepatic steatosis. Our monocyte tracking studies corrob-orated these data, showing reduced migration of macrophages tothe liver in HFD mice.We also demonstrated that macrophage chemotaxis was medi-
ated by GPR105, as BMDMs obtained from GPR105 KO micedisplayed significantly impaired migration toward a UDP-Glcgradient in an in vitro transwell assay. GPR105 has been previ-ously implicated in the migration of immune cells. For example,GPR105 expressed in primitive hematopoietic cells regulates thechemotaxis of these cells to conditioned media from the bonemarrow stroma (10). GPR105 expressed in endometrial tissues ofthe female reproductive tract also facilitates the chemotaxis ofneutrophils in response to UDP-Glc (11). In our study, we ob-served that fewer monocytes from GPR105 KO mice wererecruited to the liver in response to HFD in vivo.Interestingly, increased hepatic UDP-Glc has been reported in
association with the development of obesity (27, 28). UDP-Glchas been shown to be a functional agonist for GPR105-mediatedcellular responses in various cell types (29–34), and the endo-plasmic reticulum and Golgi apparatus provide the major route forextracellular release of UDP-Glc (35). Furthermore, damagedcells may release nucleotides such as ATP and UDP-Glc duringinflammation and mechanical stress (11). Because HFD-inducedlipotoxicity and inflammation have been shown to increase he-patocellular apoptosis and necrosis (36–38), we hypothesize thatUDP-Glc may act as an extracellular signaling molecule andchemoattractant for macrophage recruitment via GPR105. To ourknowledge, we were able to demonstrate for the first time thatUDP-Glc levels in plasma are significantly higher in obese com-pared with lean mice. Although further investigation is required toaddress this hypothesis, our results suggest that UDP-Glc releasefrom hepatocellular injury may trigger innate immune responsesand promote the development of hepatic insulin resistance.In addition to improved hepatic insulin sensitivity, we observed
improved global insulin sensitivity in whole-body GPR105 KOmice, with enhanced insulin action not only in liver, but also inskeletal muscle and adipose tissue. GPR105 is expressed at onlyvery low levels in murine muscle (7). Furthermore, we were unableto detect significant changes in proinflammatory gene expressionin adipose tissue. Prior studies have shown that strategies thatexclusively target insulin action in the liver can alter insulinsensitivity in extrahepatic tissues (39, 40) by modifying circulat-ing glucose. Thus, the improved insulin sensitivity in muscle andfat of GPR105 KO mice could be secondary to indirect effectsfrom the liver. However, myeloid cells have been shown to beresponsible for cross talk involving proinflammatory cytokinesbetween insulin-responsive tissues (41). Because we observedsignificantly improved insulin sensitivity in mice with a myeloid-specific GPR105 deletion obtained by BMT, myeloid cells ap-pear to play a primary role in GPR105-mediated inflammationand insulin resistance in our study as well. BMT results in thereconstitution of several immune cell types in addition tomacrophages. Therefore, we cannot conclude that GPR105 inlymphocytes or other immune cells does not contribute to ourobserved phenotype.We have recently shown that RHMs comprise more than half of
the population of liver macrophages under HFD conditions (25).These cells are radiosensitive and turn over in a matter of weeks sothat the RHMs in the KO-BMT mice are essentially donor derived.Conversely, the majority of resident Kupffer cells are radio-resistant and turn over more slowly (42, 43), so they are probablynot fully replaced at the time of our studies (20 wk after BMT). Incontrast, nearly all of the ATMs are donor derived after 8-wk
reconstitution and 12-wk high-fat feeding (Supplemental Fig. 3).To the extent that chimerism of the Kupffer cell population existsat the time of our studies, this would tend to underestimate theeffect of GPR105 ablation on the hepatic phenotype, consistentwith the results of our clamp studies showing a smaller im-provement in hepatic glucose suppression in the KO-BMT micethan that observed in the whole-body GPR105 KOs. Thus, ourdata overall suggest that GPR105-mediated macrophage recruit-ment to the liver after HFD propagates strong inflammatory sig-nals in the liver that may enter the circulation to cause peripheralinsulin resistance in other tissues such as muscle and fat.In this study, we have expanded our understanding of GPR105
in vivo by demonstrating that GPR105 null mice are protected fromHFD-induced insulin resistance. This improvement in systemicinsulin sensitivity was accompanied by decreased macrophageinfiltration and a reduction in inflammation in the liver. We alsoshowed that GPR105 mediates monocyte migration to the liver inresponse to HFD, and that myeloid-specific deletion of GPR105results in improved insulin sensitivity. Taken together, these datasupport the conclusion that GPR105 participates in macrophagerecruitment to the liver in chronic HFD feeding with subsequentlocal inflammation and insulin resistance. Novel therapeutic strat-egies that inhibit GPR105 and prevent macrophage migration mayprove to be protective to the development of insulin resistance andtype 2 diabetes.
AcknowledgmentsWe thank Merck Pharmaceuticals for providing the GPR105 KO mice.
DisclosuresThe authors have no financial conflicts of interest.
References1. Olefsky, J. M., and C. K. Glass. 2010. Macrophages, inflammation, and insulin
resistance. Annu. Rev. Physiol. 72: 219–246.2. Gordon, S., and P. R. Taylor. 2005. Monocyte and macrophage heterogeneity.
Nat. Rev. Immunol. 5: 953–964.3. Lumeng, C. N., S. M. Deyoung, J. L. Bodzin, and A. R. Saltiel. 2007. Increased
inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes 56: 16–23.
4. Thewissen, M. M., J. G. Damoiseaux, A. M. Duijvestijn, M. M. van Greevenbroek,C. J. van der Kallen, E. J. Feskens, E. E. Blaak, C. G. Schalkwijk, C. D. Stehouwer,J. W. Cohen Tervaert, and I. Ferreira. 2011. Abdominal fat mass is associatedwith adaptive immune activation: the CODAM Study. Obesity (Silver Spring)19: 1690–1698.
5. Filmore, D. 2004. It’s a GPCR world. Modern Drug Discov. 7: 24–28.6. Chambers, J. K., L. E. Macdonald, H. M. Sarau, R. S. Ames, K. Freeman,
J. J. Foley, Y. Zhu, M. M. McLaughlin, P. Murdock, L. McMillan, et al. 2000. AG protein-coupled receptor for UDP-glucose. J. Biol. Chem. 275: 10767–10771.
7. Freeman, K., P. Tsui, D. Moore, P. C. Emson, L. Vawter, S. Naheed, P. Lane,H. Bawagan, N. Herrity, K. Murphy, et al. 2001. Cloning, pharmacology, andtissue distribution of G-protein-coupled receptor GPR105 (KIAA0001) rodentorthologs. Genomics 78: 124–128.
8. Moore, D. J., P. R. Murdock, J. M. Watson, R. L. Faull, H. J. Waldvogel,P. G. Szekeres, S. Wilson, K. B. Freeman, and P. C. Emson. 2003. GPR105,a novel Gi/o-coupled UDP-glucose receptor expressed on brain glia and pe-ripheral immune cells, is regulated by immunologic challenge: possible role inneuroimmune function. Brain Res. Mol. Brain Res. 118: 10–23.
9. Lattin, J. E., K. Schroder, A. I. Su, J. R. Walker, J. Zhang, T. Wiltshire, K. Saijo,C. K. Glass, D. A. Hume, S. Kellie, and M. J. Sweet. 2008. Expression analysisof G protein-coupled receptors in mouse macrophages. Immunome Res. 4: 5.
10. Lee, B. C., T. Cheng, G. B. Adams, E. C. Attar, N. Miura, S. B. Lee, Y. Saito,I. Olszak, D. Dombkowski, D. P. Olson, et al. 2003. P2Y-like receptor, GPR105(P2Y14), identifies and mediates chemotaxis of bone-marrow hematopoieticstem cells. Genes Dev. 17: 1592–1604.
11. Arase, T., H. Uchida, T. Kajitani, M. Ono, K. Tamaki, H. Oda, S. Nishikawa,M. Kagami, T. Nagashima, H. Masuda, et al. 2009. The UDP-glucose receptorP2RY14 triggers innate mucosal immunity in the female reproductive tract byinducing IL-8. J. Immunol. 182: 7074–7084.
12. Bassil, A. K., S. Bourdu, K. A. Townson, A. Wheeldon, E. M. Jarvie, N. Zebda,A. Abuin, E. Grau, G. P. Livi, L. Punter, et al. 2009. UDP-glucose modulatesgastric function through P2Y14 receptor-dependent and -independent mecha-nisms. Am. J. Physiol. Gastrointest. Liver Physiol. 296: G923–G930.
The Journal of Immunology 7
by guest on February 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

13. Oh, D. Y., S. Talukdar, E. J. Bae, T. Imamura, H. Morinaga, W. Fan, P. Li,W. J. Lu, S. M. Watkins, and J. M. Olefsky. 2010. GPR120 is an omega-3 fattyacid receptor mediating potent anti-inflammatory and insulin-sensitizing effects.Cell 142: 687–698.
14. Lu, M., D. A. Sarruf, S. Talukdar, S. Sharma, P. Li, G. Bandyopadhyay,S. Nalbandian, W. Fan, J. R. Gayen, S. K. Mahata, et al. 2011. Brain PPAR-gpromotes obesity and is required for the insulin-sensitizing effect of thiazolidi-nediones. Nat. Med. 17: 618–622.
15. Seifter, S., and S. Dayton, et al. 1950. The estimation of glycogen with theanthrone reagent. Arch. Biochem. 25: 191–200.
16. Yoshizaki, T., J. C. Milne, T. Imamura, S. Schenk, N. Sonoda, J. L. Babendure,J.-C. Lu, J. J. Smith, M. R. Jirousek, and J. M. Olefsky. 2009. SIRT1 exerts anti-inflammatory effects and improves insulin sensitivity in adipocytes. Mol. Cell.Biol. 29: 1363–1374.
17. Nguyen, M. T., S. Favelyukis, A. K. Nguyen, D. Reichart, P. A. Scott, A. Jenn,R. Liu-Bryan, C. K. Glass, J. G. Neels, and J. M. Olefsky. 2007. A subpopulationof macrophages infiltrates hypertrophic adipose tissue and is activated by freefatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol.Chem. 282: 35279–35292.
18. Nnalue, N. A., A. Shnyra, K. Hultenby, and A. A. Lindberg. 1992. Salmonellacholeraesuis and Salmonella typhimurium associated with liver cells after in-travenous inoculation of rats are localized mainly in Kupffer cells and multiplyintracellularly. Infect. Immun. 60: 2758–2768.
19. Patsouris, D., J. G. Neels, W. Fan, P.-P. Li, M. T. A. Nguyen, and J. M. Olefsky.2009. Glucocorticoids and thiazolidinediones interfere with adipocyte-mediatedmacrophage chemotaxis and recruitment. J. Biol. Chem. 284: 31223–31235.
20. Lazarowski, E. R., D. A. Shea, R. C. Boucher, and T. K. Harden. 2003. Releaseof cellular UDP-glucose as a potential extracellular signaling molecule. Mol.Pharmacol. 63: 1190–1197.
21. Austyn, J. M., and S. Gordon. 1981. F4/80, a monoclonal antibody directedspecifically against the mouse macrophage. Eur. J. Immunol. 11: 805–815.
22. Lee, Y. S., P. Li, J. Y. Huh, I. J. Hwang, M. Lu, J. I. Kim, M. Ham, S. Talukdar,A. Chen, W. J. Lu, et al. 2011. Inflammation is necessary for long-term but notshort-term high-fat diet-induced insulin resistance. Diabetes 60: 2474–2483.
23. Xu, H., G. T. Barnes, Q. Yang, G. Tan, D. Yang, C. J. Chou, J. Sole, A. Nichols,J. S. Ross, L. A. Tartaglia, and H. Chen. 2003. Chronic inflammation in fat playsa crucial role in the development of obesity-related insulin resistance. J. Clin.Invest. 112: 1821–1830.
24. Lumeng, C. N., J. L. Bodzin, and A. R. Saltiel. 2007. Obesity induces a phenotypicswitch in adipose tissue macrophage polarization. J. Clin. Invest. 117: 175–184.
25. Oh, D. Y., H. Morinaga, S. Talukdar, E. J. Bae, and J. M. Olefsky. 2012. Increasedmacrophage migration into adipose tissue in obese mice. Diabetes 61: 346–354.
26. Ferrero, M. E. 2012. Purinoceptors in inflammation: potential as anti-inflammatory therapeutic targets. Front. Biosci. 17: 2172–2186.
27. van de Werve, G. 1990. Fasting enhances glycogen synthase activation in hep-atocytes from insulin-resistant genetically obese (fa/fa) rats. Biochem. J. 269:789–794.
28. Veerababu, G., J. Tang, R. T. Hoffman, M. C. Daniels, L. F. Hebert, Jr.,E. D. Crook, R. C. Cooksey, and D. A. McClain. 2000. Overexpression ofglutamine: fructose-6-phosphate amidotransferase in the liver of transgenic miceresults in enhanced glycogen storage, hyperlipidemia, obesity, and impairedglucose tolerance. Diabetes 49: 2070–2078.
29. Skelton, L., M. Cooper, M. Murphy, and A. Platt. 2003. Human immaturemonocyte-derived dendritic cells express the G protein-coupled receptorGPR105 (KIAA0001, P2Y14) and increase intracellular calcium in response toits agonist, uridine diphosphoglucose. J. Immunol. 171: 1941–1949.
30. Scrivens, M., and J. M. Dickenson. 2005. Functional expression of the P2Y14receptor in murine T-lymphocytes. Br. J. Pharmacol. 146: 435–444.
31. Scrivens, M., and J. M. Dickenson. 2006. Functional expression of the P2Y14receptor in human neutrophils. Eur. J. Pharmacol. 543: 166–173.
32. Krzemi�nski, P., P. Pomorski, and J. Bara�nska. 2008. The P2Y14 receptor activityin glioma C6 cells. Eur. J. Pharmacol. 594: 49–54.
33. Dovlatova, N., Y. D. Wijeyeratne, S. C. Fox, P. Manolopoulos, A. J. Johnson,A. E. White, M. L. Latif, V. Ralevic, and S. Heptinstall. 2008. Detection of P2Y(14) protein in platelets and investigation of the role of P2Y(14) in plateletfunction in comparison with the EP(3) receptor. Thromb. Haemost. 100: 261–270.
34. Gao, Z. G., Y. Ding, and K. A. Jacobson. 2010. UDP-glucose acting at P2Y14receptors is a mediator of mast cell degranulation. Biochem. Pharmacol. 79:873–879.
35. Sesma, J. I., C. R. Esther, Jr., S. M. Kreda, L. Jones, W. O’Neal, S. Nishihara,R. A. Nicholas, and E. R. Lazarowski. 2009. Endoplasmic reticulum/golgi nu-cleotide sugar transporters contribute to the cellular release of UDP-sugar sig-naling molecules. J. Biol. Chem. 284: 12572–12583.
36. Wang, Y., L. M. Ausman, R. M. Russell, A. S. Greenberg, and X. D. Wang. 2008.Increased apoptosis in high-fat diet-induced nonalcoholic steatohepatitis in ratsis associated with c-Jun NH2-terminal kinase activation and elevated proapo-ptotic Bax. J. Nutr. 138: 1866–1871.
37. Wu, X., L. Zhang, E. Gurley, E. Studer, J. Shang, T. Wang, C. Wang, M. Yan,Z. Jiang, P. B. Hylemon, et al. 2008. Prevention of free fatty acid-induced hepaticlipotoxicity by 18b-glycyrrhetinic acid through lysosomal and mitochondrialpathways. Hepatology 47: 1905–1915.
38. Wanless, I. R., and K. Shiota. 2004. The pathogenesis of nonalcoholic steato-hepatitis and other fatty liver diseases: a four-step model including the role oflipid release and hepatic venular obstruction in the progression to cirrhosis.Semin. Liver Dis. 24: 99–106.
39. Saberi, M., D. Bjelica, S. Schenk, T. Imamura, G. Bandyopadhyay, P. Li,V. Jadhar, C. Vargeese, W. Wang, K. Bowman, et al. 2009. Novel liver-specificTORC2 siRNA corrects hyperglycemia in rodent models of type 2 diabetes. Am.J. Physiol. Endocrinol. Metab. 297: E1137–E1146.
40. Park, S. Y., Y. R. Cho, H. J. Kim, E. G. Hong, T. Higashimori, S. J. Lee,I. J. Goldberg, G. I. Shulman, S. M. Najjar, and J. K. Kim. 2006. Mechanism ofglucose intolerance in mice with dominant negative mutation of CEACAM1.Am. J. Physiol. Endocrinol. Metab. 291: E517–E524.
41. Arkan, M. C., A. L. Hevener, F. R. Greten, S. Maeda, Z. W. Li, J. M. Long,A. Wynshaw-Boris, G. Poli, J. Olefsky, and M. Karin. 2005. IKK-beta linksinflammation to obesity-induced insulin resistance. Nat. Med. 11: 191–198.
42. Klein, I., J. C. Cornejo, N. K. Polakos, B. John, S. A. Wuensch, D. J. Topham,R. H. Pierce, and I. N. Crispe. 2007. Kupffer cell heterogeneity: functionalproperties of bone marrow derived and sessile hepatic macrophages. Blood 110:4077–4085.
43. Kennedy, D. W., and J. L. Abkowitz. 1997. Kinetics of central nervous systemmicroglial and macrophage engraftment: analysis using a transgenic bone mar-row transplantation model. Blood 90: 986–993.
8 GPR105-MEDIATED INFLAMMATION AND INSULIN RESISTANCE
by guest on February 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

Corrections
Xu, J., H. Morinaga, D. Oh, P. Li, A. Chen, S. Talukdar, E. Lazarowski, J. M. Olefsky, and J. J. Kim. 2012. GPR105 ablation preventsinflammation and improves insulin sensitivity in mice with diet-induced obesity. J. Immunol. 189: 1992–1999.
Three authors were omitted from the article. The corrected author and affiliation lines are shown below.
Jianfeng Xu,* Hidetaka Morinaga,* Dayoung Oh,* Pingping Li,* Ai Chen,* Saswata Talukdar,* Yael Mamane,† Joseph A. Mancini,†
Andrea R. Nawrocki,‡ Eduardo Lazarowski,x Jerrold M. Olefsky,* and Jane J. Kim{,‖
*Department of Medicine, University of California San Diego, La Jolla, CA 92093; †Merck Frosst Canada, Kirkland, Quebec H3H3L1, Canada; ‡Merck Research Laboratories, Rahway, NJ 07065; xDepartment of Medicine, University of North Carolina, Chapel Hill,NC 27599; {Department of Pediatrics, University of California San Diego, La Jolla, CA 92093; and ‖Rady Children’s Hospital-San Diego,San Diego, CA 92123
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1290082
Copyright � 2013 by The American Association of Immunologists, Inc. 0022-1767/13/$16.00
The Journal of Immunology


















