
Germinal mosaicism of GATA3 in a family with HDR syndrome
3
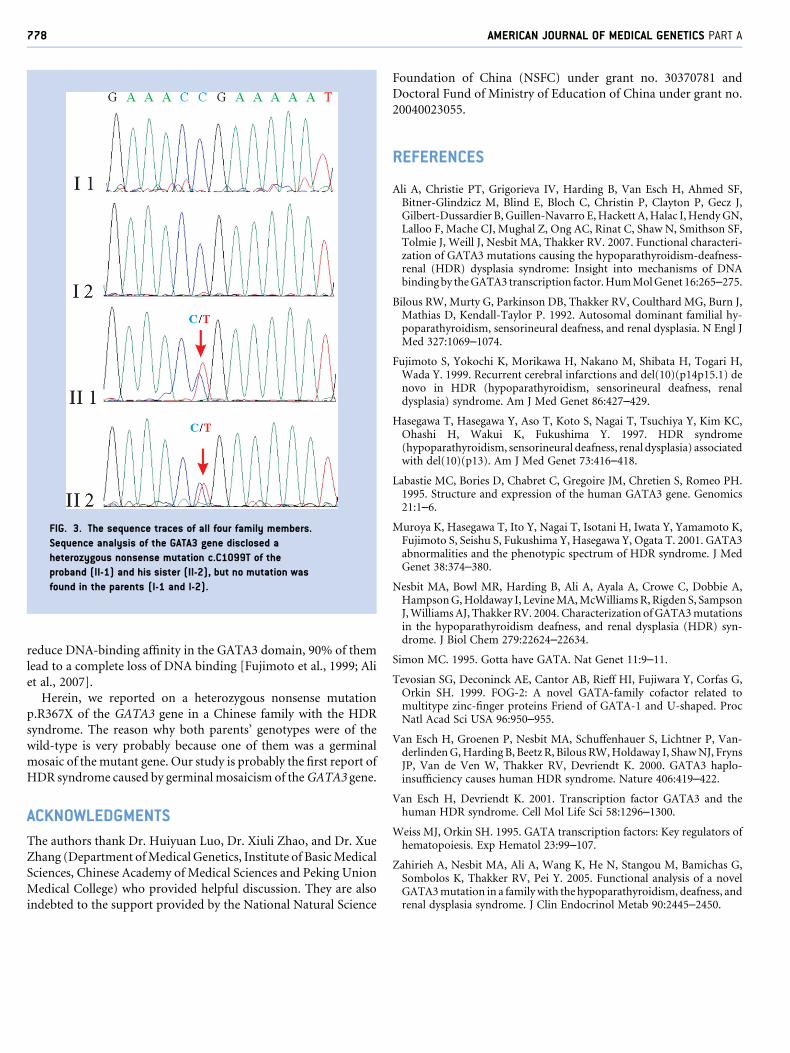
RESEARCH LETTER Germinal Mosaicism of GATA3 in a Family With HDR Syndrome Yue Sun, 1 Weibo Xia, 1 * Xiaoping Xing, 1 Mei Li, 1 Ou Wang, 1 Yan Jiang, 1 Yu Pei, 2 Ping Ye, 3 Huaicheng Liu, 1 Yingying Hu, 1 Xunwu Meng, 1 and Xueying Zhou 1 1 Department of Endocrinology, Key Laboratory of Endocrinology, Ministry of Health, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, Beijing, China 2 Department of Endocrinology, Second Artillery Forces General Hospital, People’s Liberation Army, Beijing, China 3 Dalian Red Cross Blood Center, Dalian, Liaoning Province, China Received 24 July 2008; Accepted 12 December 2008 TO THE EDITOR: The combination of hypoparathyroidism, sensorineural deafness and renal anomalies (HDR syndrome, OMIM 146255) was de- scribed in by Bilous et al. [1992]. The pattern of inheritance of this disorder was autosomal dominant. The gene mutated in patients with HDR syndrome was identified during a detailed study of individuals with a DiGeorge-like phenotype associated with dele- tions of chromosome 10p. Van Esch et al. [2000] performed deletion-mapping studies in two patients with HDR syndrome and defined a critical 200-kb region that contains the GATA3 gene (OMIM 131320). Haploinsufficiency of this gene causes HDR syndrome. A Chinese family of the Han ethnic group was diagnosed with HDR syndrome based on the clinical presentation of hypoparathy- roidism and hearing impairment. The propositus, an 18-year-old boy (Fig. 1, II-1), came to our clinic when he was 12, complaining of recurrent tetany over the preceding 11 years. From the age of 3, he suffered from bilateral sensorineural hearing loss, necessitating the use of a hearing aid. In addition, he had multiple bouts of seizures since the age of 1. At the age of 10, he was found to have a low serum calcium concentration, and a cranial CT showed multiple calcifi- cations. At the time, he was presumed to have hypoparathyroidism but received no medical treatment. At our hospital, electric re- sponse audiometry revealed sensorineural deafness, and biochemi- cal assays showed apparent hypocalcemia (serum total calcium 6.5 mg/dl; reference 8.5–10.8 mg/dl) and hyperphosphatemia (serum phosphorus 9.1 mg/dl; reference 2.5–4.88 mg/dl). His serum creatinine level was normal, but he had mild proteinuria. Ultrasound of his kidneys was normal. His sister (Fig. 1, II-2) was 5 years old and had hearing impairment, hypocalcemia (serum total calcium 8.0 mg/dl) and intracalvarial calcification similar to her brother’s. She was found to have paroxysmal tetany since the age of 5. Her serum creatinine level was normal, urine studies showed no proteinuria, and her renal ultrasound was normal. Both patients’ serum concentrations of intact PTH were normal (II-1 was 1.6 pg/ ml; II-2 was 3.0 pg/ml; reference value was 7–53 pg/ml). Based on these clinical findings, a diagnosis of HDR syndrome was made. Sequence analysis of the GATA3 gene disclosed a heterozygous nonsense mutation in exon 6 (c.C1099T, which predicts p.R367X) of the proband and his sister (Fig. 2A). This mutation was con- firmed by AlwNI digestion of the PCR product of exon 6 (Fig. 2B). This c.C1099T mutation of GATA3 was first reported by Muroya et al. [2001] in a Japanese family with HDR syndrome. Interestingly, in this Chinese family, the phenotypes of both parents were normal. We repeated the relevant laboratory inves- tigations of the parents in different clinical centers, ending up with the same results. Furthermore, the sequencing of the GATA3 gene and subsequent restriction enzymatic digestion showed that the genotypes of both parents were of the wild-type (Fig. 3). In order to prove that the parents in this family were actually the biological parents of the two afflicted children, we performed haplotype analysis. The results showed that the affected sibling pair shared the same alleles at locus D10S1751, D10S1779, D10S1728, and Grant sponsor: National Natural Science Foundation of China (NSFC); Grant number: 30370781; Grant sponsor: Ministry of Education of China; Grant number: 20040023055. *Correspondence to: Weibo Xia, M.D., Department of Endocrinology, Key Laboratory of Endocrinology, Ministry of Health, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, Shuaifuyuan No. 1, Wangfujing, Dongcheng District, Beijing 100730, China. E-mail: [email protected] Published online 27 February 2009 in Wiley InterScience (www.interscience.wiley.com) DOI 10.1002/ajmg.a.32706 How to Cite this Article: Sun Y, Xia W, Xing X, Li M, Wang O, Jiang Y, Pei Y, Ye P, Liu H, Hu Y, Meng X, Zhou X. 2009. Germinal mosaicism of GATA3 in a family with HDR syndrome. Am J Med Genet Part A 149A:776–778. Ó 2009 Wiley-Liss, Inc. 776
Transcript of Germinal mosaicism of GATA3 in a family with HDR syndrome

RESEARCH LETTER
Germinal Mosaicism of GATA3 in a Family WithHDR SyndromeYue Sun,1 Weibo Xia,1* Xiaoping Xing,1 Mei Li,1 Ou Wang,1 Yan Jiang,1 Yu Pei,2 Ping Ye,3 Huaicheng Liu,1
Yingying Hu,1 Xunwu Meng,1 and Xueying Zhou1
1Department of Endocrinology, Key Laboratory of Endocrinology, Ministry of Health, Peking Union Medical College Hospital, Chinese Academy ofMedical Sciences, Beijing, China2Department of Endocrinology, Second Artillery Forces General Hospital, People’s Liberation Army, Beijing, China3Dalian Red Cross Blood Center, Dalian, Liaoning Province, China
Received 24 July 2008; Accepted 12 December 2008
TO THE EDITOR:
The combination of hypoparathyroidism, sensorineural deafness
and renal anomalies (HDR syndrome, OMIM 146255) was de-
scribed in by Bilous et al. [1992]. The pattern of inheritance of this
disorder was autosomal dominant. The gene mutated in patients
with HDR syndrome was identified during a detailed study of
individuals with a DiGeorge-like phenotype associated with dele-
tions of chromosome 10p. Van Esch et al. [2000] performed
deletion-mapping studies in two patients with HDR syndrome
and defined a critical 200-kb region that contains the GATA3 gene
(OMIM 131320). Haploinsufficiency of this gene causes HDR
syndrome.
A Chinese family of the Han ethnic group was diagnosed with
HDR syndrome based on the clinical presentation of hypoparathy-
roidism and hearing impairment. The propositus, an 18-year-old
boy (Fig. 1, II-1), came to our clinicwhen hewas 12, complaining of
recurrent tetany over the preceding 11 years. From the age of 3, he
suffered from bilateral sensorineural hearing loss, necessitating the
use of a hearing aid. In addition, he had multiple bouts of seizures
since the age of 1. At the age of 10, he was found to have a low serum
calcium concentration, and a cranial CT showed multiple calcifi-
cations. At the time, he was presumed to have hypoparathyroidism
but received no medical treatment. At our hospital, electric re-
sponse audiometry revealed sensorineural deafness, and biochemi-
cal assays showed apparent hypocalcemia (serum total calcium
6.5 mg/dl; reference 8.5–10.8 mg/dl) and hyperphosphatemia
(serum phosphorus 9.1 mg/dl; reference 2.5–4.88 mg/dl). His
serum creatinine level was normal, but he had mild proteinuria.
Ultrasound of his kidneys was normal. His sister (Fig. 1, II-2) was 5
years old and had hearing impairment, hypocalcemia (serum total
calcium 8.0 mg/dl) and intracalvarial calcification similar to her
brother’s. She was found to have paroxysmal tetany since the age of
5. Her serum creatinine level was normal, urine studies showed no
proteinuria, and her renal ultrasound was normal. Both patients’
serum concentrations of intact PTH were normal (II-1 was 1.6 pg/
ml; II-2 was 3.0 pg/ml; reference value was 7–53 pg/ml). Based on
these clinical findings, a diagnosis of HDR syndrome was made.
Sequence analysis of the GATA3 gene disclosed a heterozygous
nonsense mutation in exon 6 (c.C1099T, which predicts p.R367X)
of the proband and his sister (Fig. 2A). This mutation was con-
firmed by AlwNI digestion of the PCR product of exon 6 (Fig. 2B).
This c.C1099Tmutation ofGATA3was first reported byMuroya et
al. [2001] in a Japanese family with HDR syndrome.
Interestingly, in this Chinese family, the phenotypes of both
parents were normal. We repeated the relevant laboratory inves-
tigations of the parents in different clinical centers, ending up with
the same results. Furthermore, the sequencing of the GATA3 gene
and subsequent restriction enzymatic digestion showed that the
genotypes of both parents were of the wild-type (Fig. 3). In order to
prove that the parents in this family were actually the biological
parents of the two afflicted children, we performed haplotype
analysis. The results showed that the affected sibling pair shared
the same alleles at locus D10S1751, D10S1779, D10S1728, and
Grant sponsor: National Natural Science Foundation of China (NSFC);
Grant number: 30370781; Grant sponsor: Ministry of Education of China;
Grant number: 20040023055.
*Correspondence to:
Weibo Xia, M.D., Department of Endocrinology, Key Laboratory of
Endocrinology, Ministry of Health, Peking Union Medical College
Hospital, Chinese Academy of Medical Sciences, Shuaifuyuan No. 1,
Wangfujing, Dongcheng District, Beijing 100730, China.
E-mail: [email protected]
Published online 27 February 2009 in Wiley InterScience
(www.interscience.wiley.com)
DOI 10.1002/ajmg.a.32706
How to Cite this Article:Sun Y, XiaW, Xing X, LiM,Wang O, Jiang Y,
Pei Y, Ye P, Liu H, Hu Y, Meng X, Zhou X.
2009. Germinal mosaicism of GATA3 in a
family with HDR syndrome.
Am J Med Genet Part A 149A:776–778.
� 2009 Wiley-Liss, Inc. 776
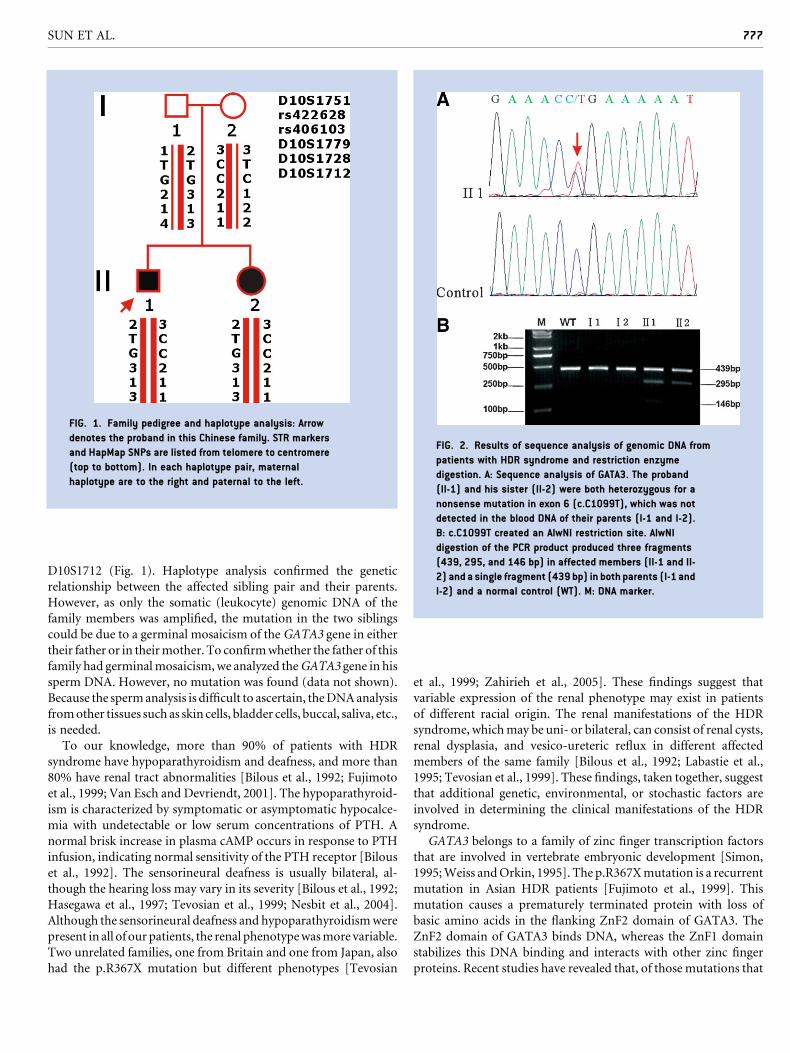
D10S1712 (Fig. 1). Haplotype analysis confirmed the genetic
relationship between the affected sibling pair and their parents.
However, as only the somatic (leukocyte) genomic DNA of the
family members was amplified, the mutation in the two siblings
could be due to a germinal mosaicism of the GATA3 gene in either
their father or in theirmother. To confirmwhether the father of this
family had germinalmosaicism, we analyzed theGATA3 gene in his
sperm DNA. However, no mutation was found (data not shown).
Because the spermanalysis is difficult to ascertain, theDNAanalysis
fromother tissues such as skin cells, bladder cells, buccal, saliva, etc.,
is needed.
To our knowledge, more than 90% of patients with HDR
syndrome have hypoparathyroidism and deafness, and more than
80% have renal tract abnormalities [Bilous et al., 1992; Fujimoto
et al., 1999; Van Esch and Devriendt, 2001]. The hypoparathyroid-
ism is characterized by symptomatic or asymptomatic hypocalce-
mia with undetectable or low serum concentrations of PTH. A
normal brisk increase in plasma cAMP occurs in response to PTH
infusion, indicating normal sensitivity of the PTH receptor [Bilous
et al., 1992]. The sensorineural deafness is usually bilateral, al-
though the hearing loss may vary in its severity [Bilous et al., 1992;
Hasegawa et al., 1997; Tevosian et al., 1999; Nesbit et al., 2004].
Although the sensorineural deafness and hypoparathyroidismwere
present in all of our patients, the renal phenotypewasmore variable.
Two unrelated families, one from Britain and one from Japan, also
had the p.R367X mutation but different phenotypes [Tevosian
et al., 1999; Zahirieh et al., 2005]. These findings suggest that
variable expression of the renal phenotype may exist in patients
of different racial origin. The renal manifestations of the HDR
syndrome, whichmay be uni- or bilateral, can consist of renal cysts,
renal dysplasia, and vesico-ureteric reflux in different affected
members of the same family [Bilous et al., 1992; Labastie et al.,
1995; Tevosian et al., 1999]. These findings, taken together, suggest
that additional genetic, environmental, or stochastic factors are
involved in determining the clinical manifestations of the HDR
syndrome.
GATA3 belongs to a family of zinc finger transcription factors
that are involved in vertebrate embryonic development [Simon,
1995;Weiss andOrkin, 1995]. The p.R367Xmutation is a recurrent
mutation in Asian HDR patients [Fujimoto et al., 1999]. This
mutation causes a prematurely terminated protein with loss of
basic amino acids in the flanking ZnF2 domain of GATA3. The
ZnF2 domain of GATA3 binds DNA, whereas the ZnF1 domain
stabilizes this DNA binding and interacts with other zinc finger
proteins. Recent studies have revealed that, of those mutations that
FIG. 1. Family pedigree and haplotype analysis: Arrow
denotes the proband in this Chinese family. STR markers
and HapMap SNPs are listed from telomere to centromere
(top to bottom). In each haplotype pair, maternal
haplotype are to the right and paternal to the left.
FIG. 2. Results of sequence analysis of genomic DNA from
patients with HDR syndrome and restriction enzyme
digestion. A: Sequence analysis of GATA3. The proband
(II-1) and his sister (II-2) were both heterozygous for a
nonsense mutation in exon 6 (c.C1099T), which was not
detected in the blood DNA of their parents (I-1 and I-2).
B: c.C1099T created an AlwNI restriction site. AlwNI
digestion of the PCR product produced three fragments
(439, 295, and 146 bp) in affected members (II-1 and II-
2) and a single fragment (439 bp) in both parents (I-1 and
I-2) and a normal control (WT). M: DNA marker.
SUN ET AL. 777
reduce DNA-binding affinity in the GATA3 domain, 90% of them
lead to a complete loss of DNA binding [Fujimoto et al., 1999; Ali
et al., 2007].
Herein, we reported on a heterozygous nonsense mutation
p.R367X of the GATA3 gene in a Chinese family with the HDR
syndrome. The reason why both parents’ genotypes were of the
wild-type is very probably because one of them was a germinal
mosaic of the mutant gene. Our study is probably the first report of
HDR syndrome caused by germinal mosaicism of theGATA3 gene.
ACKNOWLEDGMENTS
The authors thank Dr. Huiyuan Luo, Dr. Xiuli Zhao, and Dr. Xue
Zhang (Department ofMedical Genetics, Institute of BasicMedical
Sciences, Chinese Academy of Medical Sciences and Peking Union
Medical College) who provided helpful discussion. They are also
indebted to the support provided by the National Natural Science
Foundation of China (NSFC) under grant no. 30370781 and
Doctoral Fund of Ministry of Education of China under grant no.
20040023055.
REFERENCES
Ali A, Christie PT, Grigorieva IV, Harding B, Van Esch H, Ahmed SF,Bitner-Glindzicz M, Blind E, Bloch C, Christin P, Clayton P, Gecz J,Gilbert-Dussardier B,Guillen-Navarro E,Hackett A,Halac I,HendyGN,Lalloo F, Mache CJ, Mughal Z, Ong AC, Rinat C, Shaw N, Smithson SF,Tolmie J, Weill J, Nesbit MA, Thakker RV. 2007. Functional characteri-zation of GATA3 mutations causing the hypoparathyroidism-deafness-renal (HDR) dysplasia syndrome: Insight into mechanisms of DNAbindingby theGATA3 transcription factor.HumMolGenet 16:265–275.
Bilous RW, Murty G, Parkinson DB, Thakker RV, Coulthard MG, Burn J,Mathias D, Kendall-Taylor P. 1992. Autosomal dominant familial hy-poparathyroidism, sensorineural deafness, and renal dysplasia. N Engl JMed 327:1069–1074.
Fujimoto S, Yokochi K, Morikawa H, Nakano M, Shibata H, Togari H,Wada Y. 1999. Recurrent cerebral infarctions and del(10)(p14p15.1) denovo in HDR (hypoparathyroidism, sensorineural deafness, renaldysplasia) syndrome. Am J Med Genet 86:427–429.
Hasegawa T, Hasegawa Y, Aso T, Koto S, Nagai T, Tsuchiya Y, Kim KC,Ohashi H, Wakui K, Fukushima Y. 1997. HDR syndrome(hypoparathyroidism, sensorineural deafness, renal dysplasia) associatedwith del(10)(p13). Am J Med Genet 73:416–418.
Labastie MC, Bories D, Chabret C, Gregoire JM, Chretien S, Romeo PH.1995. Structure and expression of the human GATA3 gene. Genomics21:1–6.
Muroya K, Hasegawa T, Ito Y, Nagai T, Isotani H, Iwata Y, Yamamoto K,Fujimoto S, Seishu S, Fukushima Y, Hasegawa Y, Ogata T. 2001. GATA3abnormalities and the phenotypic spectrum of HDR syndrome. J MedGenet 38:374–380.
Nesbit MA, Bowl MR, Harding B, Ali A, Ayala A, Crowe C, Dobbie A,HampsonG,Holdaway I, LevineMA,McWilliamsR, Rigden S, SampsonJ,WilliamsAJ, Thakker RV. 2004. Characterization ofGATA3mutationsin the hypoparathyroidism deafness, and renal dysplasia (HDR) syn-drome. J Biol Chem 279:22624–22634.
Simon MC. 1995. Gotta have GATA. Nat Genet 11:9–11.
Tevosian SG, Deconinck AE, Cantor AB, Rieff HI, Fujiwara Y, Corfas G,Orkin SH. 1999. FOG-2: A novel GATA-family cofactor related tomultitype zinc-finger proteins Friend of GATA-1 and U-shaped. ProcNatl Acad Sci USA 96:950–955.
Van Esch H, Groenen P, Nesbit MA, Schuffenhauer S, Lichtner P, Van-derlindenG,Harding B, Beetz R, Bilous RW,Holdaway I, ShawNJ, FrynsJP, Van de Ven W, Thakker RV, Devriendt K. 2000. GATA3 haplo-insufficiency causes human HDR syndrome. Nature 406:419–422.
Van Esch H, Devriendt K. 2001. Transcription factor GATA3 and thehuman HDR syndrome. Cell Mol Life Sci 58:1296–1300.
Weiss MJ, Orkin SH. 1995. GATA transcription factors: Key regulators ofhematopoiesis. Exp Hematol 23:99–107.
Zahirieh A, Nesbit MA, Ali A, Wang K, He N, Stangou M, Bamichas G,Sombolos K, Thakker RV, Pei Y. 2005. Functional analysis of a novelGATA3mutation in a familywith the hypoparathyroidism, deafness, andrenal dysplasia syndrome. J Clin Endocrinol Metab 90:2445–2450.
FIG. 3. The sequence traces of all four family members.
Sequence analysis of the GATA3 gene disclosed a
heterozygous nonsense mutation c.C1099T of the
proband (II-1) and his sister (II-2), but no mutation was
found in the parents (I-1 and I-2).
778 AMERICAN JOURNAL OF MEDICAL GENETICS PART A


















