
Genetic Disorders of Adipose Tissue Development, Differentiation, and Death
27
Genetic Disorders of Adipose Tissue Development, Differentiation, and Death Anil K. Agarwal and Abhimanyu Garg Division of Nutrition and Metabolic Diseases, Department of Internal Medicine and the Center for Human Nutrition, University of Texas Southwestern Medical Center at Dallas, Dallas, Texas 75390-9052; email: [email protected] Annu. Rev. Genomics Hum. Genet. 2006. 7:175–99 First published online as a Review in Advance on May 24, 2006 The Annual Review of Genomics and Human Genetics is online at genom.annualreviews.org This article’s doi: 10.1146/annurev.genom.7.080505.115715 Copyright c 2006 by Annual Reviews. All rights reserved 1527-8204/06/0922-0175$20.00 Key Words mandibuloacral dysplasia, acyltransferases, lamins, zinc metalloproteinase, PPAR gamma, AKT2 Abstract Lack of adipose tissue, either complete or partial, is the hallmark of disorders known as lipodystrophies. Patients with lipodystro- phies suffer from metabolic complications similar to those associ- ated with obesity, including insulin resistance, type 2 diabetes, hy- pertriglyceridemia, and hepatic steatosis. The loss of body fat in inherited lipodystrophies can be caused by defects in the develop- ment and/or differentiation of adipose tissue as a consequence of mutations in a number of genes, including PPARG (encoding a nu- clear hormone receptor), AGPAT2 (encoding an enzyme involved in the biosynthesis of triglyceride and phospholipids), AKT2 (encod- ing a protein involved in insulin signal transduction), and BSCL2 (encoding seipin, whose role in the adipocyte biology remains un- clear). The loss of body fat can also be caused by the premature death of adipocytes due to mutations in lamin A/C, nuclear lamina proteins, and ZMPSTE24, which modifies the prelamin A post- translationally. In this review, we focus on the molecular basis of in- herited lipodystrophies as they relate to adipocyte biology and their associated phenotypic manifestations. 175 Annu. Rev. Genom. Human Genet. 2006.7:175-199. Downloaded from www.annualreviews.org by Otterbein University on 08/24/13. For personal use only.
Transcript of Genetic Disorders of Adipose Tissue Development, Differentiation, and Death

ANRV285-GG07-08 ARI 12 August 2006 17:8
Genetic Disordersof Adipose TissueDevelopment,Differentiation, and DeathAnil K. Agarwal and Abhimanyu GargDivision of Nutrition and Metabolic Diseases, Department of Internal Medicine andthe Center for Human Nutrition, University of Texas Southwestern Medical Center atDallas, Dallas, Texas 75390-9052; email: [email protected]
Annu. Rev. Genomics Hum. Genet. 2006.7:175–99
First published online as a Review inAdvance on May 24, 2006
The Annual Review of Genomics and HumanGenetics is online atgenom.annualreviews.org
This article’s doi:10.1146/annurev.genom.7.080505.115715
Copyright c© 2006 by Annual Reviews.All rights reserved
1527-8204/06/0922-0175$20.00
Key Words
mandibuloacral dysplasia, acyltransferases, lamins, zincmetalloproteinase, PPAR gamma, AKT2
AbstractLack of adipose tissue, either complete or partial, is the hallmarkof disorders known as lipodystrophies. Patients with lipodystro-phies suffer from metabolic complications similar to those associ-ated with obesity, including insulin resistance, type 2 diabetes, hy-pertriglyceridemia, and hepatic steatosis. The loss of body fat ininherited lipodystrophies can be caused by defects in the develop-ment and/or differentiation of adipose tissue as a consequence ofmutations in a number of genes, including PPARG (encoding a nu-clear hormone receptor), AGPAT2 (encoding an enzyme involved inthe biosynthesis of triglyceride and phospholipids), AKT2 (encod-ing a protein involved in insulin signal transduction), and BSCL2(encoding seipin, whose role in the adipocyte biology remains un-clear). The loss of body fat can also be caused by the prematuredeath of adipocytes due to mutations in lamin A/C, nuclear laminaproteins, and ZMPSTE24, which modifies the prelamin A post-translationally. In this review, we focus on the molecular basis of in-herited lipodystrophies as they relate to adipocyte biology and theirassociated phenotypic manifestations.
175
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
CGL: congenitalgeneralizedlipodystrophy
INTRODUCTION
Several rare diseases manifest clinical fea-tures observed in many common conditions.Studying rare genetic diseases, thus, has im-plications for understanding pathogenesis ofcommon disease conditions and provides theopportunity to identify new biochemical path-ways that when defective result in disease.For example, identifying the molecular basisof familial lipodystrophies (disorders of adi-pose tissue development, differentiation anddeath) has provided new insights into sev-eral biochemical pathways in adipocyte biol-ogy (41). A major factor contributing to thedifficulties in identifying the genetic defectsresponsible for these disorders is the rela-tive rarity and lack of standardized definitionsand nomenclature for these disorders. Recentrapid advances in the capability to sequencegenes and careful phenotyping have resultedin the discoveries of the molecular basis ofmany of these disorders. For years, the studyof these rare disorders was negelected; de-spite, the fact that affected subjects developmany more common features associated withinsulin resistance, including hyperglycemia,hyperinsulinemia, hypertriglyceridemia, lowhigh-density lipoprotein (HDL) levels, hep-atic steatosis, hyperuricemia, acanthosis nigri-cans, hirsutism, polycystic ovarian syndrome,and coronary heart disease (44). Some formsof lipodystrophies are also characterized byan increased prevalence of hypertension (44).The severity of insulin resistance and otherassociated complications correlates with theextent of loss of adipose tissue.
Identification of the molecular basis oflipodystrophies has provided the promise ofadvancing our understanding of the biochem-ical defects in type 2 diabetes mellitus, hyper-lipidemia, and nonalcoholic hepatic steatosis.These advances have heightened the interestand increased the recognition of these syn-dromes. Pediatricians, endocrinologists, ge-neticists, reproductive endocrinologists, andgynecologists are increasingly diagnosing pa-tients with lipodystrophy.
Genetic lipodystrophies can be broadlyclassified into two main types of inheri-tance patterns: (a) autosomal recessive and (b)autosomal dominant. In some patients, thedisorder is sporadic and due to de novo muta-tions. For some syndromic disorders, the pre-cise pattern of inheritance remains to be char-acterized. A brief classification is provided inTable 1 and a detailed description of key clin-ical features is presented in Table 2.
AUTOSOMAL RECESSIVELIPODYSTROPHIES
Congenital GeneralizedLipodystrophy
Congenital generalized lipodystrophy(CGL), also known as Berardinelli-Seip syn-drome, is named after physicians Berardinellifrom Brazil and Seip from Norway, who pro-vided the original descriptions (16, 83) of thisdisorder. It becomes manifest in the perinatalperiod so pediatricians or neonatologiststypically diagnose the disorder. However,some cases are discovered later during life.Approximately 300 patients with this disorderhave been reported to date, with an estimatedworldwide prevalence of ∼1 in 10 million.Clusters of patients have been reported fromsome regions of Brazil and Lebanon, whereconsanguinity is more common.
Clinical features. Affected babies are bornwith a near complete absence of adipose tis-sue, which manifests as prominent muscles(Figure 1a). Affected children have increasedenergy intake supporting an increased rate ofgrowth. Bone age is typically advanced be-yond their chronological age and many pa-tients have enlarged hands, feet, and mandible(“acromegaloid” features). Umbilical herniasare frequently present in this disorder. Laterin childhood, patients develop severe acan-thosis nigricans affecting multiple areas of thebody.
Almost all subjects with CGL have hep-atomegaly (+/− splenomegaly) due to a fatty
176 Agarwal · Garg
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
Table 1 Classification of genetic lipodystrophies
1. Autosomal recessivea. Congenital generalized lipodystrophy (CGL)b. Mandibuloacral dysplasia (MAD)c. SHORT syndromed. Neonatal progeroid syndrome
2. Autosomal dominanta. Familial partial lipodystrophyb. Pubertal-onset generalized lipodystrophyc. Hutchinson-Gilford Progeria syndrome–associated lipodystrophyd. SHORT syndrome
liver, which can result in cirrhosis. Irregularmenstruation, oligo-amenorrhea, hirsutism,clitoral enlargement, and polycystic ovariesare frequent in affected women. Only a fewaffected women have had successful pregnan-cies. Some patients develop multiple focallytic lesions in the long bones of the body afterpuberty (37), most likely due to an inability toreplace hematopoeitic marrow with adiposetissue. Some patients also have cardiomyopa-thy and mild mental retardation (10, 84, 98).
Extreme insulin resistance is a majorcharacteristic of this disease and is asso-ciated with severe hypoleptinemia and hy-poadiponectinemia (55). Most patients haveelevated fasting and postprandial plasma lev-els of insulin. Although most patients donot develop diabetes mellitus until puberty,some neonates develop hyperglycemia, whichrequires high doses of insulin to control.Severe hypertriglyceridemia can predisposepatients to recurrent episodes of acute pan-creatitis. Plasma levels of low-density lipopro-tein (LDL) cholesterol are typically normalor reduced, and plasma levels of high-densitylipoprotein (HDL) cholesterol are often low.It is not clear if these patients are prone todevelop atherosclerotic vascular disease
Molecular basis. Positional cloning strate-gies using the conventional genome-widelinkage analysis identified two loci for CGL.We identified one locus on human chromo-some 9q34 (48) and Magre et al. (66) dis-
AGPAT2:1-acylglycerol-3-phosphate-O-acyltransferase 2gene
BSCL2:Berardinelli-SeipCongenitalLipodystrophy 2gene
covered the second locus on chromosome11q13. The defective gene on chromosome 9was 1-acylglycerol-3-phosphate-O-acyltran-sferase 2 (AGPAT2). Several mutations in-cluding nonsense, frameshift, deletion, andmissense mutations in AGPAT2 have nowbeen reported in affected patients (2,3).AGPATs are members of the acyltransferasefamily of enzymes with eight known iso-forms, each of which is encoded by a differentgene (4, 62, 63, 103). AGPATs play a criti-cal role in the biosynthesis of glycerophos-pholipids and triacylglycerols. The enzymecatalyzes the conversion of lysophosphatidicacid (1-acylglycerol-3-phosphate) to phos-phatidic acid (1,2 diacylglycerol-3-phosphate)by adding a fatty acyl group at the sn-2 po-sition (Figure 2) (62, 100). AGPAT2 is pre-dominantly expressed in the omental adiposetissue (2, 32, 33, 100); it is likely that re-duced levels or absence of AGPAT2 activ-ity causes lipodystrophy either by impairingtriglyceride synthesis in adipocytes or by af-fecting adipocyte differentiation and functionby interfering with the biosynthesis of phos-pholipids.
The defective gene on chromosome 11q13that causes CGL is the Berardinelli-Seip Con-genital Lipodystrophy 2 (BSCL2) gene, whichwas predicted to encode a 398–amino acidprotein called seipin (66). Based on sequencesimilarities among different species, seipin has64 additional residues at the amino termi-nus that were not included in the original
www.annualreviews.org • Genetic Disorders of Adipose 177
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
Tab
le2
Clin
ical
feat
ures
and
mol
ecul
arba
sis
ofge
neti
clip
odys
trop
hies
Inhe
rita
nce
Dis
ease
OM
IML
ocus
/gen
eG
ene
prod
uct
Func
tion
sP
heno
type
Oth
erfe
atur
esA
utos
omal
Rec
essi
veC
onge
nita
lge
nera
lized
lipod
ystr
ophy
,typ
e1
6085
949q
34.3
/AG
PAT
2A
GPA
T2
Tri
glyc
erid
ebi
osyn
thet
icen
zym
eG
ener
aliz
edlip
odys
trop
hyat
birt
h
Pre
serv
atio
nof
mec
hani
cal
adip
ose
tissu
e,hy
pole
ptin
emia
,an
dse
vere
insu
linre
sist
ance
Con
geni
tal
gene
raliz
edlip
odys
trop
hy,t
ype
2
2697
0011
q13/
BSC
L2Se
ipin
Tra
nsm
embr
ane
prot
ein
ofun
know
nfu
nctio
n
Gen
eral
ized
lipod
ystr
ophy
atbi
rth
Car
diom
yopa
thy,
mild
men
tal
reta
rdat
ion,
lack
ofm
echa
nica
lad
ipos
etis
sue
hypo
lept
inem
ia,
and
seve
rein
sulin
resi
stan
ceM
andi
bulo
acra
ldy
spla
sia–
asso
ciat
edlip
odys
trop
hy,t
ype
1
2483
701q
21.2
/LM
NA
Lam
insA
and
CN
ucle
arla
min
apr
otei
nsP
artia
llip
odys
trop
hy,
smal
lman
dibl
ean
dre
sorp
tion
ofcl
avic
les
and
phal
ange
s
Pro
gero
idfe
atur
essu
chas
alop
ecia
,be
aked
nose
,mild
insu
linre
sist
ance
Man
dibu
loac
ral
dysp
lasi
a–as
soci
ated
lipod
ystr
ophy
,typ
e2
6086
121p
34/Z
MPS
TE
24Z
inc
met
allo
-pr
otei
nase
Pos
t-tr
ansl
atio
nal
proc
essi
ngof
prel
amin
Ato
mat
ure
lam
inA
Gen
eral
ized
lipod
ystr
ophy
,sm
all
man
dibl
ean
dre
sorp
tion
ofcl
avic
les
Pro
gero
idfe
atur
essu
chas
alop
ecia
,be
aked
nose
,ch
roni
cre
nal
failu
re
178 Agarwal · Garg
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
Neo
nata
lpro
gero
idsy
ndro
me
2640
90N
otkn
own
Gen
eral
ized
lack
ofbo
dyfa
tand
prog
eroi
dap
pear
ance
atbi
rth
Spar
ing
ofgl
utea
lfa
t,m
ultip
lesk
elet
al,a
ndey
eab
norm
aliti
esA
utos
omal
rece
ssiv
e/D
omin
ant
SHO
RT
synd
rom
e26
9880
Not
know
n-
Par
tiall
ipod
ystr
ophy
Shor
tsta
ture
,hy
per-
exte
nsib
ility
,he
rnia
s,oc
ular
depr
essi
on,R
iege
ran
omal
y,te
ethi
ngde
lay
Aut
osom
aldo
min
ant
Fam
ilial
part
ial
lipod
ystr
ophy
,D
unni
gan
vari
ety
1516
601q
21.2
/LM
NA
Lam
insA
and
CN
ucle
arla
min
apr
otei
nsP
artia
llip
odys
trop
hyon
setd
urin
gpu
bert
yIn
crea
sed
fat
accu
mul
atio
nin
the
face
and
neck
.So
me
have
card
iom
yopa
thy.
Fam
ilial
part
ial
lipod
ystr
ophy
asso
ciat
edw
ithPP
AR
Gm
utat
ions
6043
673p
25/P
PAR
GP
PAR
γN
ucle
artr
ansc
ript
ion
fact
orin
volv
edin
adip
ogen
esis
Par
tiall
ipod
ystr
ophy
Hyp
erte
nsio
n,hy
perl
ipid
emia
Fam
ilial
part
ial
lipod
ystr
ophy
asso
ciat
edw
ithA
KT
2m
utat
ions
1647
3119
q13.
1-q1
3.2/
AK
T2
AK
T2
Pho
spho
inos
itide
seri
ne/t
hreo
nine
kina
sein
volv
edin
post
rece
ptor
insu
linsi
gnal
ing
Par
tiall
ipod
ystr
ophy
Mild
insu
linre
sist
ance
Pub
erta
l-on
set
gene
raliz
edlip
odys
trop
hy
6080
561q
21.2
/LM
NA
Lam
insA
and
CN
ucle
arla
min
apr
otei
nsG
ener
aliz
edlip
odys
trop
hyon
set
duri
ngpu
bert
y
Pro
gero
idfe
atur
essu
chas
lack
ofha
ir,be
aked
nose
,and
lack
ofbr
east
deve
lopm
ent
www.annualreviews.org • Genetic Disorders of Adipose 179
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.
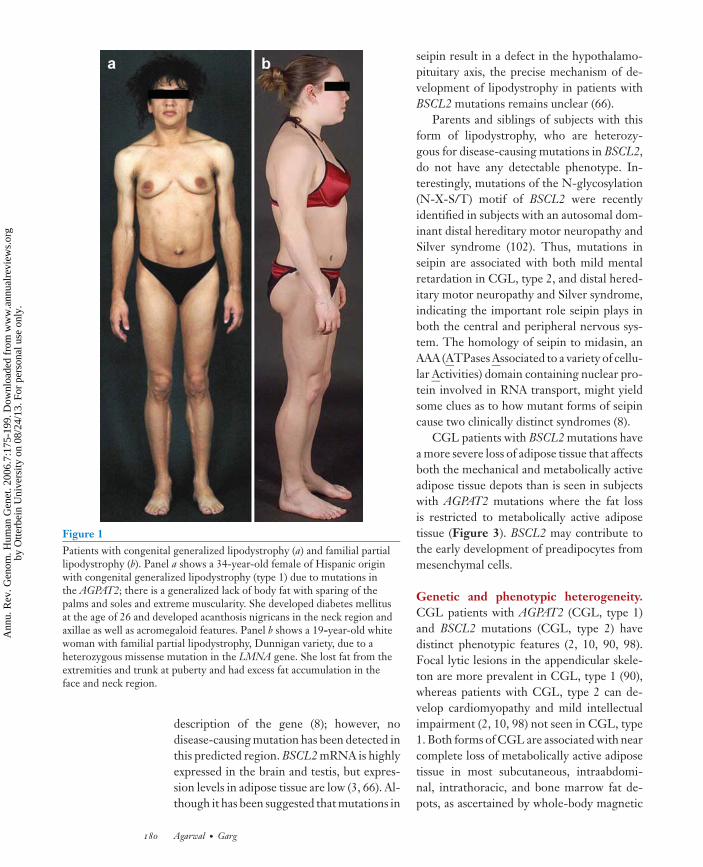
ANRV285-GG07-08 ARI 12 August 2006 17:8
Figure 1Patients with congenital generalized lipodystrophy (a) and familial partiallipodystrophy (b). Panel a shows a 34-year-old female of Hispanic originwith congenital generalized lipodystrophy (type 1) due to mutations inthe AGPAT2; there is a generalized lack of body fat with sparing of thepalms and soles and extreme muscularity. She developed diabetes mellitusat the age of 26 and developed acanthosis nigricans in the neck region andaxillae as well as acromegaloid features. Panel b shows a 19-year-old whitewoman with familial partial lipodystrophy, Dunnigan variety, due to aheterozygous missense mutation in the LMNA gene. She lost fat from theextremities and trunk at puberty and had excess fat accumulation in theface and neck region.
description of the gene (8); however, nodisease-causing mutation has been detected inthis predicted region. BSCL2 mRNA is highlyexpressed in the brain and testis, but expres-sion levels in adipose tissue are low (3, 66). Al-though it has been suggested that mutations in
seipin result in a defect in the hypothalamo-pituitary axis, the precise mechanism of de-velopment of lipodystrophy in patients withBSCL2 mutations remains unclear (66).
Parents and siblings of subjects with thisform of lipodystrophy, who are heterozy-gous for disease-causing mutations in BSCL2,do not have any detectable phenotype. In-terestingly, mutations of the N-glycosylation(N-X-S/T) motif of BSCL2 were recentlyidentified in subjects with an autosomal dom-inant distal hereditary motor neuropathy andSilver syndrome (102). Thus, mutations inseipin are associated with both mild mentalretardation in CGL, type 2, and distal hered-itary motor neuropathy and Silver syndrome,indicating the important role seipin plays inboth the central and peripheral nervous sys-tem. The homology of seipin to midasin, anAAA (ATPases Associated to a variety of cellu-lar Activities) domain containing nuclear pro-tein involved in RNA transport, might yieldsome clues as to how mutant forms of seipincause two clinically distinct syndromes (8).
CGL patients with BSCL2 mutations havea more severe loss of adipose tissue that affectsboth the mechanical and metabolically activeadipose tissue depots than is seen in subjectswith AGPAT2 mutations where the fat lossis restricted to metabolically active adiposetissue (Figure 3). BSCL2 may contribute tothe early development of preadipocytes frommesenchymal cells.
Genetic and phenotypic heterogeneity.CGL patients with AGPAT2 (CGL, type 1)and BSCL2 mutations (CGL, type 2) havedistinct phenotypic features (2, 10, 90, 98).Focal lytic lesions in the appendicular skele-ton are more prevalent in CGL, type 1 (90),whereas patients with CGL, type 2 can de-velop cardiomyopathy and mild intellectualimpairment (2, 10, 98) not seen in CGL, type1. Both forms of CGL are associated with nearcomplete loss of metabolically active adiposetissue in most subcutaneous, intraabdomi-nal, intrathoracic, and bone marrow fat de-pots, as ascertained by whole-body magnetic
180 Agarwal · Garg
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
Glycerol-3-phosphate
Fatty acid-CoA
Lysophosphatidic acid
Phosphatidic acid
Pi
Diacylglycerol
Triacylglycerol
2-monoacylglycerol
AGPATs
GPATs
PAPs
DGATs
MGATs
AGPAT2
CoA
Phosphatidyl inositolCardiolipin
Phosphatidyl cholinePhosphatidyl ethanolaminePhosphatidyl serine
DGK
Fatty acid-CoA
CoA
Fatty acid-CoA
CoA
Fatty acid-CoA
CoA
Figure 2Pathways for the biosynthesis of triacylglycerol. Glycerol-3-phosphate undergoes acylation at the sn-1position by glycerol-3-phosphate acyltransferase (GPAT), to form 1-acylglycerol-3-phosphate orlysophosphatidic acid (LPA). LPA is then acylated at the sn-2 position by 1-acylglycerol-3-phosphateacyltransferase (AGPAT) to form phosphatidic acid (PA). In the next step, the phosphate group isremoved by phosphatidate phosphohydrolase (PAP) to produce diacylglycerol (DAG). DAG is thenacylated at the sn-3 position by diacylglycerol acyltransferase (DGAT) to produce triacylglycerol (TG).DAG kinase (DGK) can phosphorylate DAG to synthesize PA. TG can also be synthesized via theacylation of 2-monoacylglycerol by the enzyme monoacylglycerol-acyltransferase (MGAT), which isexpressed at high levels in the small intestine. PA and DAG are also substrates for the synthesis ofglycerophospholipids. PA is the substrate for synthesis of phosphatidylinositol and cardiolipin.Phosphatidylcholine, phosphatidylethanolamine, and phosphatidylserine are synthesized from DAG.LPA and related lipid molecules are also involved in signal transduction and are ligands for Gprotein-coupled receptors. Figure 2 is reproduced from Reference 7. Copyright 2003, with permissionfrom Elsevier.
resonance imaging (90). Patients with CGL,type 1 have preserved mechanical adipose tis-sue depots in the palms, soles, scalp, retro-orbital, and peri-articular regions, whereas inCGL, type 2 there is no fat sparing in theseregions (Figure 4) (24, 43, 90). As expecteddue to the more complete lipodystrophy inCGL, type 2, these patients have lower levelsof serum leptin (10).
Most patients with CGL are either ho-mozygous or compound heterozygotes formutations in AGPAT2 or BSCL2. However,occasionally only a single mutation in oneof these genes has been identifed. The dis-ease is not linked to either locus in a few
pedigrees of patients with CGL, suggestingas yet unidentified loci for CGL (10). Otherlipodystrophic disorders, such as acquiredgeneralized lipodystrophy, leprechaunism,childhood-onset generalized lipodystrophydue to rare LMNA mutations, and neona-tal progeroid syndrome, can present as phe-nocopies of CGL, so must be excluded. Itremains possible that CGL, type 3 patientsmay harbor heterozygous mutations in twoacyltransferases catalyzing different steps oftriglyceride biosynthetic pathway. For ex-ample, a patient may develop generalizedlipodystrophy due to being heterozygous formutations in both AGPAT2 and another gene
www.annualreviews.org • Genetic Disorders of Adipose 181
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
AGPAT,
Myocytes Osteoblasts
stem cells
Fasting Feeding
C/EBPβC/EBPδ
C/EBP
PPARγ /RXRα
BSCL2?
Transcriptionfactors?
Adipogenic factors(insulin, cortisol, etc.)
SREBP1cLipogenesis(FAS, ACC,
GPAT, DGAT)
Apoptosis
aitnereffiDtnempoleveD sisotpopa/htaeDnoit
Mesenchymal Pre-adipocyte Adipocyte Cell deathMature adipocyte
Figure 3Pathways involved in the development, differentiation, and death of adipocytes. The pluripotentmesenchymal stem cells respond to yet-to-be fully characterized cues to commit to formingpreadipocytes. Preadipocytes respond to hormonal signals (insulin and steroids) to initiate changes inlevels of expression of selected transcription factors leading to the differentiation to mature adipocytes.In vitro and in vivo models have established that C/EBPβ/δ are the initial genes upregulated in responseto hormones, etc. These transcription factors stimulate other transcription factors such as PPARγ,C/EBPα, and SREBP1c. Other genes that are downregulated in this process include cellular factors likePREF1, an adipogenesis inhibitor. Lipogenic genes, such as FAS, ACC, GPAT, AGPAT, and DGAT, areupregulated, resulting in increased biosynthesis of triglycerides and phospholipids. Lipid droplet size isreduced upon fasting and increases with increased substrate availability. BSCL2 (seipin) is likely involvedin adipocyte development and early differentiation, whereas the AGPAT2 gene more likely affectstriglyceride synthesis but may also contribute to adipocyte differentiation. Clinical evidence fromlipodystrophy patients harboring LMNA or ZMPSTE24 mutations suggests that nuclear dysfunction mayaccelerate apoptosis/death of mature adipocytes.
involved in triglyceride biosynthesis, such asother AGPATs, DGATs, or GPATs. Examplesof such digenic inheritance resulting in com-plex phenotypes have been described for othermetabolic disorders such as cortisone reduc-tase deficiency (30) and severe insulin resis-tance (81).
Genotype and phenotype variation. Al-though the functional defects in the enzy-matic activities of many of the AGPAT2 mu-tations have been characterized in vitro (53)(Figure 5), there appears to be no correla-tion between the genotype and phenotype.All missense mutations characterized to datehave some residual functional activity (>20%of the wild type). Only one homozygous mis-sense mutation, E172K (affecting one of theconserved motifs, EGTR, which most likelyparticipates in fatty acid binding), has been
identified in AGPAT2 in CGL, type 1 patients(4, 66a). All other individuals with a missensemutation in one allele have a null mutation inthe other allele. Subjects who are homozygous(or compound heterozygous) for other mis-sense mutations of AGPAT2 may have a moresubtle phenotype that could range from beinglean and resistant to diet-induced obesity tohaving mild generalized lipodystrophy and in-sulin resistance. Individuals heterozygous fornull mutations, predicted to have ∼50% of thewild-type AGPAT2 activity, have no obviousphenotype. These individuals may have moresubtle metabolic perturbations, such as hy-pertriglyceridemia, low plasma levels of HDLcholesterol, or impaired glucose tolerance.
Most of the BSCL2 mutations reportedin patients with CGL have been null muta-tions due to deletions, insertions, frameshifts,or premature terminations. A single missense
182 Agarwal · Garg
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
mutation (A212P) has been identified (66, 98).Individuals homozygous for the A212P muta-tion (two women and three men) are clinicallyindistinguishable from subjects homozygousfor null mutations in BSCL2. The five subjectshomozygous for the A212P mutation had on-set of lipodystrophy at birth, mild to moder-ate intellectual impairment, and hypertrophiccardiomyopathy, three died prematurely be-tween 24–35 years of age (98).
Mandibuloacral Dysplasia–AssociatedLipodystrophy
Mandibuloacral dysplasia (MAD) is extremelyrare and has been reported in only ∼40 pa-tients. Based on genotyping and phenotyping,there appears to be more than two types ofMAD. Most of the initial patients describedwere from consanguineous pedigrees fromItaly; however, patients of other ethnicitieshave also been reported.
Clinical features. This disorder presentswith mandibuloacral dysplasia (71, 89), whichis characterized by mandibular hypoplasiaand resorption of the clavicles and termi-nal phalanges (acro-osteolysis). Some patientspresent with progeroid features and have abird-like facies with a beaked nose. These pa-tients characteristically have a high-pitchedvoice, and ectodermal defects, such as skinatrophy, alopecia, and nail dysplasia. Otherclinical features include delayed closure ofcranial sutures, joint contractures, mottledcutaneous pigmentation, and short stature.Patients occasionally present with hypogo-nadism and sensorineural deafness. These pa-tients also have an associated lipodystrophy,which presents in two patterns: partial loss ofsubcutaneous fat from the extremities but nor-mal or excess fat in the face, neck, and truncalregions similar to that seen in patients withautosomal dominant, familial partial lipodys-trophy of the Dunnigan variety (FPLD)(type A), and more generalized loss of sub-cutaneous fat in the face, trunk, and extrem-ities (type B) (89). Along with lipodystrophy,
Figure 4Axial magnetic resonance T1-weighted images through the orbits andsagittal magnetic resonance images through the orbits and foot in a patientwith CGL, type 1, (a, d, g) CGL, type 2 (b, e, h), and a normal subject(c, f, i ). Body fat appears as regions of increased signal intensity. The CGL,type 1 patient has normal amounts of fat in the retro-orbital region,temporal region, and subcutaneous area of the scalp, and plantar region, butlack subcutaneous fat in the submental, anterior, and posterior neck regions.The CGL, type 2 patient had a near total lack of retro-orbital and plantarfat as well as subcutaneous fat from the temporal region, scalp, and neck.Normal fat distribution in the retro-orbital, temporal, scalp, submental,cervical, and plantar regions is shown in the images of the normal subject.Reprinted from Reference 90. Copyright 2005, The Endocrine Society.
MAD:mandibuloacraldysplasia
FPL: familial partiallipodystrophy
some patients also develop hyperinsulinemia,insulin resistance, impaired glucose tolerance,diabetes mellitus, and hyperlipidemia (84).
Molecular basis. The pattern of lipodystro-phy among patients with MAD and FPLDwas reported to be similar (89), suggest-ing that these two disorders may be dueto defects in the same gene. Novelli et al.(71) identified 5 Italian offsprings of consan-guineous matings with MAD (type A) whowere homozygous for a missense mutation(R527H) in the LMNA gene. Additional ho-mozygous (A529V and K542N) or compound
www.annualreviews.org • Genetic Disorders of Adipose 183
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
Median enzymatic activity
(% of wild type)
0
10
20
30
40
50
60
70
80
90
100
A239V
G136R
L228P
140delF
221delGT 252delMRT
R68X V167fsX183
D180fsX251
Figure 5In vitro enzymatic activity of AGPAT2 mutants. The enzymatic activity of various mutants of AGPAT2was determined by expressing the recombinant protein in Chinese hamster ovary (CHO) cells andmonitoring the conversion of 3H-lysophosphatidic acid (LPA) to 3H-phosphatidic acid (PA). The medianvalues are plotted as percentages of the wild-type activity (53). The missense mutants are shown inyellow, deletion mutants in blue, and null mutants in purple.
heterozygous (R472C/R527C) LMNA muta-tions in other patients with MAD have beendescribed by us and others (22, 42, 78). Allof these mutations cluster in the C-terminalregion of LMNA. Subjects heterozygous forthese mutations reveal no evidence of skeletalabnormalities or lipodystrophy.
The LMNA gene is located on chro-mosome 1q21-22 and contains 12 exons.Through alternative splicing in exon 10, theLMNA gene encodes for two major isoforms,lamin A and C (64). Lamin A is produced bypost-translational modification of its precur-sor prelamin A (91). Prelamin A is encoded byall 12 exons of LMNA and contains a CAAXmotif at the C terminal. Post-translationalmodification of prelamin A occurs in astep-wise process including (a) farnesyla-tion, (b) proteolytic cleavage of the terminalthree amino acids (AAX), (c) carboxymethy-lation of cysteine residue, and (d) anotherproteolytic cleavage of 15 carboxy-terminal
amino acids, resulting in formation of maturelamin A. The enzyme, zinc metalloproteinase(ZMPSTE24), is responsible for the first andsecond step of the proteolysis. Both maturelamin A and lamin C share the N-terminal566 amino acids, but lamin A has 98 uniquecarboxy-terminal amino acids and lamin C has6 unique C-terminal amino acids.
Lamins form hetero- or homo-dimericcoiled-coil structures intercalated betweenchromatin and the inner nuclear membrane(20, 36). Besides lamins A and C, laminsB1 and B2, which are products of two dif-ferent genes, also participate in the forma-tion of nuclear lamina. Mutations in lamins Aand C cause many other syndromes includingcardiomyopathy, muscular dystrophies, neu-ropathy, and progeria (18, 25, 27, 34, 70).The precise mechanisms by which defectivelamins A and C cause these various syndromesremain unclear. It is hypothesized thatdefective interactions of lamins A and C with
184 Agarwal · Garg
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
chromatin or other nuclear lamina proteinsduring cell division leads to apoptosis and pre-mature cell death. Consistent with this hy-pothesis, skin fibroblasts from patients withvarious types of laminopathies show abnormalnuclear morphology including nuclear blebsand multilobulated nuclei (Figure 6). How-ever, why specific LMNA mutations cause pre-dominantly skeletal and cutaneous dystrophybut less severe lipodystrophy in patients withMAD remain unclear.
Various mouse models have been devel-oped to better understand the pathogenesisof syndromes due to LMNA mutations. TheLmna null mice, though not embryonic lethal,show severe growth retardation and die pre-maturely at 5–6 weeks of age due to muscu-lar dystrophy and cardiomyopathy (95). Theyhave little adipose tissue but are not insulinresistant, presumably due to insufficient timeto develop the metabolic disorder (26).
In addition, three knock-in mouse mod-els for Lmna have been created, althoughnone of them are specific for either MADor FPLD (13, 68, 69). The first one withL530P mutation was intended to recreate fea-tures of autosomal dominant Emery-Dreifussmuscular dystrophy (68). The heterozygousLmnaL530P/+ mouse did not show any phe-notype, but the homozygous LmnaL530P/L530P
mouse replicated features of human progeria(68). These mice died by 4 weeks of age andhad bone, muscle, and skin pathology. Thedefect did not appear to be due to defectsin adipocyte differentiation since myoblastsand fibroblasts from the LmnaL530P/L530P micewere capable of differentiation into adipocytesin vitro (68). This suggests that structural cel-lular defects may cause cell death by a combi-nation of altered transcription, and mislocal-ization of nuclear proteins.
Another murine model was developed torecreate Emery-Dreifuss muscular dystro-phy by inserting a H222P substitution intoLMNA(13). The heterozygous LmnaLH222P/+
mice were indistinguishable from wild-typemice but the homozygous LmnaH222P/H222P
mice developed muscle degeneration, dislo-
Figure 6Nuclear morphology of skin fibroblasts in patients with LMNA mutations.(a) Indirect immunofluorescence microscopy using lamin A/C antibody inskin fibroblasts obtained from a control subject shows normal nuclearmorphology. (b) Nuclear bleb in one of the nuclei in a patient withmandibuloacral dysplasia with homozygous A529V LMNA mutation.(c) Multilobulated nuclei were seen in a patient with R133L heterozygousLMNA mutation who had generalized lipodystrophy, diabetes, and atypicalprogeroid syndrome. (d ) Occasional nuclear blebs were observed in apatient with R133L heterozygous LMNA mutation who presented withmild peripheral lipodystrophy and atypical progeroid syndrome. Reprintedwith permission from References 42, 59. Copyright 2005, The EndocrineSociety.
cation of heterochromatin, and activation of atranscription factor, Smad, in both the heartand skeletal muscle. The LmnaN195K/N195K
mouse generated to replicate the cardiomy-opathy (69) showed all the features of the dis-ease. The transcription factor Hf1b/sp4 andthe gap junction protein connexin 40 and 43were both misexpressed or mislocalized in thecardiomyocytes of these mice.
We identified a second locus that wasdefective in a patient with MAD (5). Thisseverely affected patient from Belgium,who had progeroid features and general-ized lipodystrophy, had compound heterozy-gous mutations (F361fsX379 and W340R)in ZMPSTE24 (5). This young woman died
www.annualreviews.org • Genetic Disorders of Adipose 185
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
ZMPSTE24: zincmetalloproteinasegene
at age 27 due to complications of chronicrenal failure resulting from focal segmentalglomerulosclerosis (5). Subsequently, another2-year and 9-month-old girl who died prema-turely with MAD, sclerodermatous skin, andprogeroid features was reported to have com-pound heterozygous ZMPSTE24 mutations(F361fsX379 and N265S) (87). Recently, a 5-year-old Turkish boy with overlapping fea-tures of MAD and progeria was reported tohave a heterozygous R654X LMNA mutationand a homozygous V402SfsX403 ZMPSTE24mutation (28). Another patient of Europeanorigin with MAD, who required renal trans-plantation due to focal segmental glomeru-losclerosis, was reported to have compoundheterozygous F361fsX379 and N265S muta-tions (11). Because ZMPSTE24 is critical forthe post-translational proteolytic processingof prelamin A to mature lamin A, it is likelythat accumulation of farnesylated prelamin Aor lack of mature lamin A is responsible forthe phenotypic effects of these mutations (91).The embryonic fibroblasts obtained fromZMPSTE24 null mice show accumulation ofprelamin A (17, 75). In addition, these miceshow retarded growth and premature death,lipodystrophy, progeria, and skeletal defectsincluding multiple spontaneous bone frac-tures (17, 75). Recent studies show improve-ment in phenotype of Zmpste24−/− mice withfarnesyl transferase inhibitor therapy suggest-ing primary role of accumulation of farne-sylated prelamin A in causing the pathology(38, 104).
Genetic and phenotypic heterogeneity.Patients with ZMPSTE24 mutations appear tohave a more severe phenotype than MAD pa-tients with LMNA mutations (71, 88) includ-ing early onset of skeletal defects such as acro-osteolysis, more progeroid appearance, anddevelopment of subcutaneous calcified nod-ules on the phalanges. In addition, progres-sive glomerulopathy has not been observed inMAD patients with LMNA mutations (5).
Some patients with MAD have mandibu-lar hypoplasia along with other clinical fea-
tures but do not develop clavicular resorp-tion or acro-osteolysis. These patients alsodo not harbor LMNA or ZMPSTE24 vari-ants, suggesting the possibility that there areother genes that are defective in MAD (5).Patients with MAD need to be differentiatedfrom those with Hutchinson-Gilford Proge-ria, atypical progeroid, Hajdu-Cheney, con-genital insensitivity to pain with anhidro-sis, Haim-Munk, and Papillon-Lefevre syn-dromes because of some overlapping features.
Genotype and phenotype variation. MADpatients who are homozygous for the K542NLMNA mutation have severe progeroid fea-tures, including alopecia, lack of eyebrowsand eyelashes, early-onset (age 1–2) acro-osteolysis, and absent or impaired sexual mat-uration. Two of these patients died at age10 and 16 (78). A Turkish girl who was ho-mozygous for an A529V missense mutationin LMNA lacked breast development despitehaving normal menarche and menses (42).These findings are in contrast to patientswith the homozygous R527H LMNA mu-tation who usually live beyond the age of30 and have normal breast development andsexual maturation (71, 89). Other subtle phe-notypic differences in MAD patients withdifferent LMNA mutations remain to be char-acterized. These phenotypic differences in pa-tients with mutations of different residues arelikely attributable to the differences in the ter-tiary structures of lamins A and C.
SHORT Syndrome–AssociatedLipodystrophy
Approximately 30 patients have been re-ported to have SHORT syndrome, anacronym for features such as short stature,hyperextensibility of joints and/or inguinalhernia, ocular depression, Rieger anomaly andteething delay (52, 85, 93). Rieger anomaly in-cludes eye abnormalities such as hypoplasia ofiris stroma, prominent Schwalbe ring, irido-corneal synechiae, micro- or megalo-cornea,
186 Agarwal · Garg
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
and teeth abnormalities such as hypodontia,microdontia, enamel hypoplasia, and atypicalteeth. Lipodystrophy in these patients com-monly affects the face, upper extremities, andsometimes the trunk. Both autosomal reces-sive (52, 85) and autosomal dominant pat-terns of inheritance have been reported (1, 14,93). Some investigators have reported a differ-ent pattern of lipodystrophy in patients withan autosomal dominant variety affecting onlythe face, gluteal region, and elbows (1, 14),but others found similar loss of fat as seen inthe autosomal recessive variety (93). Some pa-tients develop diabetes in the second and thirddecade. The precise molecular basis of thesedisorders remains unclear.
Neonatal Progeroid Syndrome(Wiedemann-RautenstrauchSyndrome)-AssociatedLipodystrophy
This syndrome has been reported in about 30patients (77, 79, 101) and is characterized bya progeroid appearance consisting of a trian-gular, aged-looking face with a relatively largeskull, prominent subcutaneous veins, particu-larly in the scalp, sparse scalp hair, large ante-rior fontanelle, and generalized lipodystrophyat birth. Accumulation of subcutaneous fat oc-curs in the sacral and gluteal areas (60, 77).About 50% of the patients die before the ageof 6 (77). The genetic basis of this syndromeremains unknown.
AUTOSOMAL DOMINANTLIPODYSTROPHIES
Familial Partial Lipodystrophy,Dunnigan Type
FPLD is named after Dunnigan, who alongwith his colleagues provided a detailed de-scription of the syndrome (31). So far, approx-imately 300 men and women mostly of Euro-pean origin have been reported to have FPLD(40, 54).
Clinical features. Affected children do notshow any abnormalities in body fat distri-bution.The lipodystrophy manifests at thetime of puberty with a gradual loss of adi-pose tissue from the extremities and trunk(Figure 1b). With increasing age, the loss offat continues from other regions of the body,particularly the anterior truncal region (45).Some regions such as the face, chin, neck, andintra-abdominal region are spared. Excess fataccumulates in the spared areas, resulting in a“double chin” and a Cushingoid appearance.There is also preservation of intermuscularand bone marrow fat (45). It is easier to rec-ognize FPLD in women than men, due to theincrease in muscle definition in the extremi-ties. About 20–33% of patients have acantho-sis nigricans, hirsutism, menstrual abnormal-ities, and polycystic ovaries (40).
Metabolic complications such as diabetes,hyperlipidemia and atherosclerotic vasculardisease also occur more often in affectedwomen than men (40, 56). In our experience(40), women have twice the prevalence of di-abetes and more than a three-times-higherprevalence of atherosclerotic vascular diseasethan men with this disorder. Women also hadsignificantly higher plasma levels of triglyc-erides and significantly lower high-densitylipoprotein cholesterol concentrations thanmen (40). Several explanation may exist forthe predisposition of women with FPLD todevelop complications of insulin resistance.First, women have increased peripheral sub-cutaneous fat deposition compared with men,consequently women with FPLD are subjectto more fat loss than men. Second, pregnancyis a time of efficient fat storage and as womenwith FPLD may not be able to accumulate fatin most subcutaneous regions, they may accu-mulate fat in the intra-abdominal region, pre-disposing them to metabolic complications.Consistent with these, among women withFPLD, the risk factors for diabetes includedmultiparity and excess fat deposition in thenonlipodystrophic regions such as the chin(54). Some patients develop cardiac problems
www.annualreviews.org • Genetic Disorders of Adipose 187
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
PPARG: peroxisomeproliferator-activatedreceptor-γ gene
and occasionally patients develop myopathy(46).
Molecular basis. We mapped the gene caus-ing FPLD to chromosome 1q21-22 (76) andthen Cao and Hegele (21) identified a mis-sense (R482Q) mutation in the LMNA gene ina Canadian pedigree. Since then, several mis-sense mutations in the LMNA gene have beenreported in patients with FPLD (46, 54, 86,94, 99). It is still unclear how these LMNA mu-tations promote lipodystrophy and why theadipose tissue in the face, neck, and intra-abdominal regions is spared.
Genotype and phenotype variation. Ap-proximately three fourths of FPLD patientsharbor missense mutations involving the argi-nine residue at position 482 (R482Q, R482W,or R482L) (54). Other mutations in the globu-lar C-terminal (tail) portion of the protein en-coded by exons 8–11 have also been described(21, 54, 86, 94, 99). We reported a milder formof FPLD in patients with the R582H muta-tion in exon 11, which affects lamin A andnot lamin C (47). Some patients with R28Wand R62G mutations in exon 1 have an as-sociated cardiomyopathy, which presents aspremature congestive heart failure or with ar-rhythmias, such as atrial fibrillation and con-duction system disturbances, requiring pace-maker implantation (46). A few patients alsohave a mild proximal myopathy (46, 99). Thus,LMNA mutations cause a multisystem dystro-phy syndrome affecting adipose tissue, car-diac, skeletal muscle, nerve, cutaneous, andskeletal tissue (46). However, why some mu-tations cause more pathology in skeletal mus-cles and others in nerve, cutaneous, or skeletaltissue remains unclear.
Familial Partial Lipodystrophy Dueto Peroxisome Proliferator-ActivatedReceptor-γ Mutation
Following our initial report of a heterozygousmissense mutation, R397C, in the PPARγ
gene in a 64-year-old woman of European
descent with partial lipodystrophy, diabetes,hypertriglyceridemia, hypertension, and hir-sutism (6), nine other subjects (five fe-males and four males) have been reportedto have FPL due to PPARG missense muta-tions (V290M, F360L, P467L, and PPARG4promoter-14A>G) (12, 57, 82). These pa-tients have variable manifestations of insulinresistance such as diabetes, hypertension, andhypertriglyceridemia. The onset of lipodys-trophy seems to vary from puberty to mid-adulthood. All patients have fat loss from theextremities; however, facial fat loss has beenvariable (6, 57, 82). It appears that lipodys-trophy mainly affects the distal regions of theextremities such as the forearms and calvesmore than the proximal regions (6).
Given the essential role that PPARγ playsin adipocyte differentiation (80), and the highlevel of expression of this gene in the adi-pose tissue, it is likely that PPARG missensemutations are dominant negative mutationscausing lipodystrophy by reducing adipo-genesis. Because the affected patients donot develop lipodystrophy during childhood,it appears that PPARγ-mediated adipocytedifferentiation continues during adulthood.
The homozygous deletion of Pparg in miceis embryonic lethal (15, 61). However, the het-erozygous mice have reduced adipose tissueand develop insulin resistance (61, 67). In ad-dition, tissue-specific knockout mice for Pparghave also been generated such that the geneis inactivated only in the liver, muscle, or adi-pose tissue. All of these tissue-specific knock-outs are insulin resistant (97), but it was notreported whether the mice have lipodystro-phy. Recently, the missense mutation P465Lwas introduced into the mouse Pparg gene(96). The heterozygous mice (PpargP465L/+)have selective loss of adipose tissue from theretroperitoneal and gonadal fat depots, withsparing of inguinal fat. The PpargP465L/+ miceare insulin resistant and develop hyperten-sion. Although there is a consensus for thecritical role of PPARγ in adipocyte differen-tiation, the depot-specific sparing of adiposetissue remains unclear. It is likely that there
188 Agarwal · Garg
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
are other nuclear receptors besides PPARγ
that may play a role in differentiation in otheradipose tissue depots.
Familial Partial Lipodystrophy Dueto the v-AKT Murine ThymomaOncogene Homolog 2Gene Mutation
Recently, George et al. (50) reported a het-erozygous missense mutation, R274H, in thev-AKT murine thymoma oncogene homolog2(AKT2) gene in a family in which affectedsubjects developed insulin resistance, diabetesmellitus, and hypertension. The proband, a34-year-old female, had partial lipodystro-phy affecting her extremities (Figure 7)(S. O’Rahilly, personal communica-tion). AKT2 belongs to the familyof phosphoinositide-dependent serine/threonine kinases, also known as proteinkinase B (PKB). AKT2 is predominantlyexpressed in insulin-sensitive tissues and isinvolved in postreceptor insulin signaling(Figure 8). In mammals, there are threeknown isoforms, AKT-1, 2, and 3, eachencoded by a different gene, which sharesignificant homology among themselves (92).These kinases also have very similar sub-strates but display tissue-specific expressionand function. Akt2−/− mice display featuresof insulin resistance and develop diabetes(49). These mice have reduced adiposetissue mass in the inguinal, subcutaneousand epididymal/gonadal depots with mildhepatomegaly. It is possible that reducedadipocyte differentiation, as seen in 3T3-L1cells expressing the mutant form of AKT2, ordysfunctional postreceptor insulin signaling,may be responsible for the developmentof lipodystrophy in patients with AKT2mutations.
Other Types of FamilialPartial Lipodystrophy
No molecular defect in the LMNA, PPARG,or AKT2 genes has been identified in a subset
Figure 7Lipodystrophy of the lower extremities in a patientheterozygous for a missense mutations in AKT2(R274H) AKT2. Panel a shows the posterior viewof the left hip, indicating marked loss ofsubcutaneous fat with prominent muscularity ofthe gluteal muscle and skin creases. Panel b showsthe posterior view of the lower extremities withincreased muscular definition due to loss ofsubcutaneous fat from the thighs and calves.Photographs courtesy of Stephen O’Rahilly.
AKT2: v-AKTmurine thymomaoncogene homolog 2gene
of FPL patients, suggesting that there are yet-to-be-determined additional genes that aredefective in FPL (A. Agarwal & A. Garg, per-sonal observation; 58). These patients seemto have a different pattern of fat loss thanthose with FPLD, but the distinctive pheno-typic differences between these patients andthose with FPL due to PPARG or AKT2 mu-tations remain to be identified (Figure 9).FPL patients should be carefully distin-guished from those with Cushing’s syndrome,truncal obesity, multiple symmetric lipomato-sis due to alcohol intake, and acquired gener-alized lipodystrophy.
Pubertal-OnsetGeneralized Lipodystrophy
Two unrelated patients of European originwho are heterozygous for a missense muta-tion in LMNA (R133L) have been describedwith this type of lipodystrophy (23, 59). Bothpatients had a normal fat distribution at birthbut developed generalized lipodystrophy,
www.annualreviews.org • Genetic Disorders of Adipose 189
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
PIP2P P
P P
P P
PIP3
P110
GSK3β
Active AKT2(PKB)
FKHR
(Cell survival mode)Transcriptional activation
(glucose metabolism)
P85
Insulin receptor
Insulin
PH
Figure 8Role of AKT2 in intracellular insulin receptor signaling pathway. Insulin binding to the insulin receptoractivates phosphoinositol-3-kinase (PI3K), which phosphorylates membrane-bound phosphatidylinositol 4,5 phosphate (PIP2) to phosphatidyl inositol 3,4,5 phosphate (PIP3). AKT2, by virtue of itspleckstrin homology (PH) domain, is then recruited to the membrane, where it is phosphorylated by thephosphatidyl inositol-dependent kinase 1 (PDK1). The active form of AKT2 upregulates genes involvedin glucose metabolism and downregulates proteins like glycogen synthase kinase 3 β(GSK3β) andforkhead/FoxO transcription factor (FKHR).
diabetes, and hypertriglyceridemia after pu-berty. The patients have atypical progeroidfeatures, such as early graying of the hairwith thinning of eyebrows, beaked nose, andatrophic skin over the hands and feet. Skinfibroblasts from one of these patients hada markedly abnormal nuclear morphology(Figure 6b). However, another patient witha similar R133L heterozygous mutation ofLMNA of African-American origin had onlymild lipodystrophy involving only the distalextremities; skin fibroblasts from this patientdid not display a major alteration in nuclearmorphology (Figure 6d ). The reason for thedifference in phenotype in this subject, whencompared with the others identified withthis same mutation remains unclear. Varia-
tions in phenotypic expression in this disor-der may be due to differences in genetic back-ground, in methylation and imprinting, orin intra-uterine and post-natal environmentalfactors.
Other Rare Varieties
An autosomal dominant generalized lipodys-trophy with acromegaloid features and onsetafter 18 years of age was reported in a pedigreefrom Brazil (74). In another pedigree, lipodys-trophy of the lower extremities was observedin two siblings with unbalanced transloca-tion involving chromosome 8p and 10p. Re-cent data from Hutchinson-Gilford proge-ria syndrome patients with heterozygous
190 Agarwal · Garg
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
Figure 9Axial T1-weighted magnetic resonance images through the thigh and calf in patients with familial partiallipodystrophy, Dunnigan (FPLD) variety due to a heterozygous LMNA mutation; a subject with FPL dueto a heterozygous PPARG mutation; a subject with another type of FPL; and a normal subject. Body fatappears as increased signal intensity. The FPLD patient has a near complete loss of subcutaneous fat, butfat in the intermuscular fasciae and bone marrow is preserved. The patient with FPL due to a PPARGmutation has decreased amounts of subcutaneous fat particularly in the antero-lateral and posterior thighand calf region, and the patient with another type of FPL (not due to LMNA, PPARG, and AKT2mutations) has asymmetrical loss of subcutaneous fat, particularly from the anterior, lateral, and posteriorregions of the thigh and calf, sparing the subcutaneous fat in the medial parts. Reprinted withmodification from Reference 6. Copyright 2005, The Endocrine Society.
G608G mutation in LMNA reveal generalizedlipodystrophy and mild insulin resistance.
Mechanisms of Lipodystrophies
We propose a working hypothesis for the lossof adipose tissue in these disorders, which isbased on our current understanding of the lifecycle of adipocyte. After preadipocytes differ-entiate into mature adipocytes they can un-dergo apoptosis and cell death (Figure 3).The process of adipocyte differentiation andcell death likely continues throughout life.Genetic defects in the BSCL2 gene likelyaffect an early step in the development ordifferentiation of adipose tissue, resulting ingeneralized lipodystrophy. AGPAT2 defectslikely affect differentiation processes down-stream of this step, resulting in sparing ofmechanical adipose tissue depots in patientswith CGL, type 1. Mutations in PPARγ
may also affect terminal differentiation steps.
Because insulin is a key hormone involvedin adipocyte differentiation, disruption ofAKT2, which is a downstream insulin signal-ing molecule, may also affect adipocyte dif-ferentiation. LMNA and ZMPSTE24 muta-tions cause the development of an abnormalnuclear lamina, resulting in premature deathof adipocytes. Alternatively, these genetic de-fects may hinder nuclear transport of specificRNA and proteins, explaining the cell type–specific functional abnormalities seen in thesedisorders.
Phenotypic Variationsin Lipodystrophies
Phenotypic variation occurs at a variety of lev-els. Different mutation(s) in the same genemay alter the activity of the protein or in-terfere differently in the protein-protein orprotein-chromatin interactions and thus mayresult in different phenotypes, as seen in
www.annualreviews.org • Genetic Disorders of Adipose 191
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
patients with LMNA mutations. Phenotypicdifferences are also observed among individ-uals with same mutations. In this regard, in-dividual genetic variation(s) may play a cru-cial role in the development of the phe-notype. Epigenetics, although not so welldefined, suggests that the same genetic in-formation is treated differently within thecell or an organ (65). This might includevariation in chromatin modulation, suchas histone acetyl transferases (HATs) anddeacetylases (HDACs) or histone methyl-transferases (HMTs) or chromatin remodel-ing by SWI/SNF factors (35). The DNAmethylation pattern between two or moresubjects carrying the same mutation could alsoalter the phenotype. Recently, it was shownthat as identical twins grow older, they havedifferent pattern of DNA methylation and hi-stone acetylation, resulting in dissimilar geneexpression (39). Alternatively, in some sub-jects the variation is affected by the maternalinheritance of the mitochondrial DNA alongwith the mutations (73).
Future Challenges
The search for additional loci for the few rareforms of lipodystrophies is complicated by thedifficulty of locating large enough families toperform linkage analysis or sufficent num-bers of unrelated individuals to use linkagedisequilibrium. The present strategy has beento sequence candidate genes known to playcritical roles in adipocyte development, dif-ferentiation or maintenance or in genes thatcause lipodystrophy when they are inactivatedor overexpressed in mice.
Other approaches to identify other geneticdefects causing recessive forms of lipodystro-phy have been to use global transcriptional
(cDNA chip) or protein (proteomics) analy-sis in cells and tissues from affected subjects,parents, and siblings (51). Detecting changesin the protein expression might reveal/reflectperturbation of the cellular pathway involved.Although the analysis of cDNA expressionusing chips is getting better and cheaper,the proteomic approach still needs significantimprovements in automation and analysis,requiring development of tools to analyze hy-drophobic and/or membrane proteins. Dis-covering additional loci may lead us to otherpreviously unrecognized pathways involved inthe development and maintenance of adiposetissues. Furthermore, as suggested recently, a“syndrome family” approach was successfullyapplied for determining loci for skeletal dys-plasias (19), and may also be useful for geneticlipodystrophies.
Our understanding of the mechanisms in-volved in various types of lipodystrophies mayassist in devising strategies for clinical man-agement of these patients (9). We still strug-gle with controlling the metabolic complica-tions associated with lipodystrophy. A recenttwo-center collaborative trial demontratedthat leptin replacement therapy markedlyimproved glycemic control and hypertriglyc-eridemia in hypoleptinemic patients with gen-eralized and partial lipodystrophies (72). Be-cause patients with lipodystrophies are rare,conducting clinical trials in these patients totest new treatment modalities is very difficultand will require the establishment of interna-tional clinical research centers. The explosionof knowledge regarding the molecular basis ofvarious types of genetic lipodystrophies willhopefully provide the opportunity to developnew effective therapies to treat this group ofoften debilitating and disfiguring disorders inwhich therapeutic options are very limited.
SUMMARY POINTS
1. Careful phenotyping of the patients with genetic lipodystrophies has defined distinctautosomal recessive and autosomal dominant disorders.
192 Agarwal · Garg
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
2. Autosomal recessive, congenital generalized lipodystrophy is caused by defects inAGPAT2 and BSCL2. AGPAT2 encodes 1-acylglycerol-3-phosphate O-acyltransferase2, which is involved in triacylglycerol and phospholipid biosynthesis. BSCL2 encodesseipin, a protein of unknown function.
3. The autosomal recessive, mandibuloacral dysplasia–associated lipodystrophies arecaused by mutations in the LMNA and ZMPSTE24 genes, which encode lamins A andC, integral components of nuclear lamina, and zinc metalloproteinase enzyme, whichis involved in post-translational proteolytic processing of prelamin A, respectively.
4. Autosomal dominant, familial partial lipodystrophies are genetically and phenotyp-ically heterogeneous disorders linked to LMNA, peroxisome proliferator-activatedreceptor γ (PPARG), a transcription factor playing a central role in adipocyte differen-tiation, and AKT2, which encodes a serine/threonine kinase involved in postreceptorinsulin signaling.
5. The molecular basis of lipodystrophies associated with SHORT syndrome and neona-tal progeroid syndrome remains to be elucidated.
6. Additional loci for congenital generalized lipodystrophy, mandibuloacral dysplasia–associated lipodystrophy, as well as for familial partial lipodystrophy have not yet beenidentified.
7. Understanding the underlying molecular mechanisms of genetic lipodystrophies willassist in the development of new therapeutic strategies to treat this class of geneticdisorders.
ACKNOWLEDGMENTS
The authors were supported in part by the National Institute of Health grants R01-DK54387and M01-RR00633 and by the Southwestern Medical Foundation. The authors thank StephenO’Rahilly for providing unpublished images of the proband with AKT2 mutation and MeredithMillay for the graphics.
LITERATURE CITED
1. Aarskog D, Ose L, Pande H, Eide N. 1983. Autosomal dominant partial lipodystrophyassociated with Rieger anomaly, short stature, and insulinopenic diabetes. Am. J. Med.Genet. 15:29–38
2. First paperreporting theAGPAT2 gene asthe locus forcongenitalgeneralizedlipodystrophy, type1.2. Agarwal AK, Arioglu E, de Almeida S, Akkoc N, Taylor SI, et al. 2002. AGPAT2
is mutated in congenital generalized lipodystrophy linked to chromosome 9q34.Nat. Genet. 31:21–23
3. Agarwal AK, Barnes RI, Garg A. 2004. Genetic basis of congenital generalized lipodys-trophy. Int. J. Obes. Relat. Metab. Disord. 28:336–39
4. Agarwal AK, Barnes RI, Garg A. 2006. Functional characterization of human 1-acylglycerol-3-phosphate acyltransferase isoform 8: cloning, tissue distribution, genestructure and enzymatic activity. Arch. Biochem. Biophys. 449:64–76
5. First paperreportingmutations in theZMPSTE24 gene ina patient withmandibuloacraldysplasia–associatedlipodystrophy.
5. Agarwal AK, Fryns JP, Auchus RJ, Garg A. 2003. Zinc metalloproteinase, ZMP-
STE24, is mutated in mandibuloacral dysplasia. Hum. Mol. Genet. 12:1995–2001
www.annualreviews.org • Genetic Disorders of Adipose 193
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
6. First paperreporting mutationin the PPARG genein a patient withfamilial partiallipodystrophy.
6. Agarwal AK, Garg A. 2002. A novel heterozygous mutation in peroxisomeproliferator-activated receptor-γ gene in a patient with familial partial lipodys-trophy. J. Clin. Endocrinol. Metab. 87:408–11
7. Agarwal AK, Garg A. 2003. Congenital generalized lipodystrophy: significance of triglyc-eride biosynthetic pathways. Trends Endocrinol. Metab. 14:214–21
8. Agarwal AK, Garg A. 2004. Seipin: a mysterious protein. Trends Mol. Med. 10:440–449. Agarwal AK, Garg A. 2006. Genetic basis of lipodystrophies and management of
metabolic complications. Annu. Rev. Med. 5:297–31110. Agarwal AK, Simha V, Oral EA, Moran SA, Gorden P, et al. 2003. Phenotypic and
genetic heterogeneity in congenital generalized lipodystrophy. J. Clin. Endocrinol. Metab.88:4840–47
11. Agarwal AK, Zhou XJ, Hall RK, Nicholls K, Bankier A, Van Esch H, Fryns JP,Garg A.2006. Focal segmental glomerulosclerosis in patients with mandibuloacral dysplasia dueto ZMPSTE24 deficiency. J. Investig. Med. In press
12. Al-Shali K, Cao H, Knoers N, Hermus AR, Tack CJ, Hegele RA. 2004. A single-basemutation in the peroxisome proliferator-activated receptor gamma4 promoter associ-ated with altered in vitro expression and partial lipodystrophy. J. Clin. Endocrinol. Metab.89:5655–60
13. Arimura T, Helbling-Leclerc A, Massart C, Varnous S, Niel F, et al. 2005. Mouse modelcarrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopa-thy similar to human striated muscle laminopathies. Hum. Mol. Genet. 14:155–69
14. Bankier A, Keith CG, Temple IK. 1995. Absent iris stroma, narrow body build and smallfacial bones: a new association or variant of SHORT syndrome? Clin. Dysmorphol. 4:304–12
15. Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, et al. 1999. PPAR gamma isrequired for placental, cardiac, and adipose tissue development. Mol. Cell 4:585–95
16. Berardinelli W. 1954. An undiagnosed endocrinometabolic syndrome: report of two cases.J. Clin. Endocrinol. Metab. 14:193–204
17. Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, et al. 2002. Zmpste24 deficiency inmice causes spontaneous bone fractures, muscle weakness, and a prelamin A processingdefect. Proc. Natl. Acad. Sci. USA 99:13049–54
18. Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, et al. 1999. Mutationsin the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss musculardystrophy. Nat. Genet. 21:285–88
19. Brunner HG, van Driel MA. 2004. From syndrome families to functional genomics. Nat.Rev. Genet. 5:545–51
20. Burke B, Stewart CL. 2002. Life at the edge: the nuclear envelope and human disease.Nat. Rev. Mol. Cell Biol. 3:575–85
21. First paperreporting acompoundheterozygousmutation in theLMNA gene inpatients withfamilial partiallipodystrophy ofthe Dunniganvariety.
21. Cao H, Hegele RA. 2000. Nuclear lamin A/C R482Q mutation in Canadian kin-dreds with Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 9:109–12
22. Cao H, Hegele RA 2003. LMNA is mutated in Hutchinson-Gilford progeria (MIM176670) but not in Wiedemann-Rautenstrauch progeroid syndrome (MIM 264090). J.Hum. Genet. 48:271–74
23. Caux F, Dubosclard E, Lascols O, Buendia B, Chazouilleres O, et al. 2003. A new clinicalcondition linked to a novel mutation in lamins A and C with generalized lipoatrophy,insulin-resistant diabetes, disseminated leukomelanodermic papules, liver steatosis, andcardiomyopathy. J. Clin. Endocrinol. Metab. 88:1006–13
194 Agarwal · Garg
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
24. Chandalia M, Garg A, Vuitch F, Nizzi F. 1995. Postmortem findings in congenital gen-eralized lipodystrophy. J. Clin. Endocrinol. Metab. 80:3077–81
25. Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, et al. 2003. LMNA mutationsin atypical Werner’s syndrome. Lancet 362:440–45
26. Cutler DA, Sullivan T, Marcus-Samuels B, Stewart CL, Reitman ML. 2002. Charac-terization of adiposity and metabolism in Lmna-deficient mice. Biochem. Biophys. Res.Commun. 291:522–27
27. De Sandre-Giovannoli A, Chaouch M, Kozlov S, Vallat JM, Tazir M, et al. 2002.Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, causeautosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2)and mouse. Am. J. Hum. Genet. 70:726–36
28. Denecke J, Brune T, Feldhaus T, Robenek H, Kranz C, et al. 2006. A homozygousZMPSTE24 null mutation in combination with a heterozygous mutation in the LMNAgene causes Hutchinson-Gilford Progeria Syndrome (HGPS): Insights into the patho-physiology of HGPS. Hum. Mutat. 27:524–31
29. di Barletta M, Ricci E, Galluzzi G, Tonali P, Mora M, et al. 2000. Different mutationsin the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifussmuscular dystrophy. Am. J. Hum. Genet. 66:1407–12
30. Draper N, Walker EA, Bujalska IJ, Tomlinson JW, Chalder SM, et al. 2003. Mutations inthe genes encoding 11β-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphatedehydrogenase interact to cause cortisone reductase deficiency. Nat. Genet. 34:434–39
31. Dunnigan MG, Cochrane MA, Kelly A, Scott JW. 1974. Familial lipoatrophic diabeteswith dominant transmission. A new syndrome. Q. J. Med. 43:33–48
32. Eberhardt C, Gray PW, Tjoelker LW. 1997. Human lysophosphatidic acid acyltrans-ferase. cDNA cloning, expression, and localization to chromosome 9q34.3. J. Biol. Chem.272:20299–305
33. Eberhardt C, Gray PW, Tjoelker LW. 1999. cDNA cloning, expression and chromosomallocalization of two human lysophosphatidic acid acyltransferases. Adv. Exp. Med. Biol.469:351–56
34. Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, et al. 2003. Recurrent denovo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature423:293–98
35. Esteller M, Almouzni G. 2005. How epigenetics integrates nuclear functions. Workshopon epigenetics and chromatin: transcriptional regulation and beyond. EMBO Rep. 6:624–28
36. Fisher DZ, Chaudhary N, Blobel G. 1986. cDNA sequencing of nuclear lamins A andC reveals primary and secondary structural homology to intermediate filament proteins.Proc. Natl. Acad. Sci. USA 83:6450–54
37. Fleckenstein JL, Garg A, Bonte FJ, Vuitch MF, Peshock RM. 1992. The skeleton incongenital, generalized lipodystrophy: evaluation using whole-body radiographic sur-veys, magnetic resonance imaging and technetium-99m bone scintigraphy. Skeletal Radiol.21:381–86
38. Fong LG, Frost D, Meta M, Qiao X, Yang SH, et al. 2006. A protein farnesyltransferaseinhibitor ameliorates disease in a mouse model of progeria. Science 311:1621–23
39. Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, et al. 2005. Epigenetic differencesarise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 102:10604–9
40. Garg A. 2000. Gender differences in the prevalence of metabolic complications in familialpartial lipodystrophy (Dunnigan variety). J. Clin. Endocrinol. Metab. 85:1776–82
www.annualreviews.org • Genetic Disorders of Adipose 195
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
41. Garg A. 2004. Acquired and genetic lipodystrophies. N. Engl. J. Med. 350:1220–3442. Garg A, Cogulu O, Ozkinay F, Onay H, Agarwal AK. 2005. A novel homozygous
Ala529Val LMNA mutation in Turkish patients with mandibuloacral dysplasia. J. Clin.Endocrinol. Metab. 90:5259–64
43. Garg A, Fleckenstein JL, Peshock RM, Grundy SM. 1992. Peculiar distribution of adiposetissue in patients with congenital generalized lipodystrophy. J. Clin. Endocrinol. Metab.75:358–61
44. Garg A, Misra A. 2004. Lipodystrophies: rare disorders causing metabolic syndrome.Endocrinol. Metab. Clin. North Am. 33:305–31
45. Garg A, Peshock RM, Fleckenstein JL. 1999. Adipose tissue distribution pattern in pa-tients with familial partial lipodystrophy (Dunnigan variety). J. Clin. Endocrinol. Metab.84:170–74
46. Garg A, Speckman RA, Bowcock AM. 2002. Multisystem dystrophy syndrome due tonovel missense mutations in the amino terminal head and α-helical rod domain of thelamin A/C (LMNA) gene. Am. J. Med. 112:549–55
47. Garg A, Vinaitheerthan M, Weatherall P, Bowcock A. 2001. Phenotypic heterogeneityin patients with familial partial lipodystrophy (Dunnigan variety) related to the site ofmis-sense mutations in Lamin A/C (LMNA) gene. J. Clin. Endocrinol. Metab. 86:59–65
48. Usinggenome-widelinkage analysis,this paper reportedthe first geneticlocus for congenitalgeneralizedlipodystrophy onchromosome 9q34
48. Garg A, Wilson R, Barnes R, Arioglu E, Zaidi Z, et al. 1999. A gene for congenitalgeneralized lipodystrophy maps to human chromosome 9q34. J. Clin. Endocrinol.
Metab. 84:3390–9449. Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, et al. 2003. Severe diabetes, age-
dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKBbeta. J. Clin. Invest. 112:197–208
50. First paperreporting acompoundheterozygousmutation in theAKT2 gene in afamily with familialpartiallipodystrophy,insulin resistance,and diabetes.
50. George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, et al. 2004. A familywith severe insulin resistance and diabetes due to a mutation in AKT2. Science
304:1325–2851. Giallourakis C, Henson C, Reich M, Xie X, Mootha VK. 2005. Disease gene discovery
through integrative genomics. Annu. Rev. Genomics Hum. Genet. 6:381–40652. Gorlin RJ, Cervenka J, Moller K, Horrobin M, Witkop J. 1975. Rieger anomaly and
growth retardation (The S-H-O-R-T Syndrome). Birth Defects 11:46–4853. Haque W, Garg A, Agarwal AK. 2005. Enzymatic activity of naturally occurring 1-
acylglycerol-3-phosphate-O-acyltransferase 2 mutants associated with congenital gener-alized lipodystrophy. Biochem. Biophys. Res. Commun. 327:446–53
54. Haque WA, Oral EA, Dietz K, Bowcock AM, Agarwal AK, Garg A. 2003. Risk factorsfor diabetes mellitus in familial partial lipodystrophy, Dunnigan variety. Diabetes Care26:1350–55
55. Haque WA, Shimomura I, Matsuzawa Y, Garg A. 2002. Serum adiponectin and leptinlevels in patients with lipodystrophies. J. Clin. Endocrinol. Metab. 87:2395–98
56. Hegele RA. 2001. Premature atherosclerosis associated with monogenic insulin resis-tance. Circulation 103:2225–29
57. Hegele RA, Cao H, Frankowski C, Mathews ST, Leff T. 2002. PPARG F388L, atransactivation-deficient mutant, in familial partial lipodystrophy. Diabetes 51:3586–90
58. Herbst KL, Tannock LR, Deeb SS, Purnell JQ, Brunzell JD, Chait A. 2003. Kobber-ling type of familial partial lipodystrophy: an underrecognized syndrome. Diabetes Care26:1819–24
59. Jacob KN, Baptista F, Dos Santos HG, Oshima J, Agarwal AK, Garg A. 2005. Phenotypicheterogeneity in body fat distribution in patients with atypical Werner’s syndrome due toheterozygous Arg133Leu Lamin A/C mutation. J. Clin. Endocrinol. Metab. 90:6699–706.
196 Agarwal · Garg
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
60. Korniszewski L, Nowak R, Okninska-Hoffmann E, Skorka A, Gieruszczak-Bialek D,Sawadro-Rochowska M. 2001. Wiedemann-Rautenstrauch (neonatal progeroid) syn-drome: new case with normal telomere length in skin fibroblasts. Am. J. Med. Genet.103:144–48
61. Kubota N, Terauchi Y, Miki H, Tamemoto H, Yamauchi T, et al. 1999. PPAR gammamediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol. Cell4:597–609
62. Leung DW. 2001. The structure and functions of human lysophosphatidic acid acyl-transferases. Front. Biosci. 6:d944–53
63. Li D, Yu L, Wu H, Shan Y, Guo J, et al. 2003. Cloning and identification of the humanLPAAT-zeta gene, a novel member of the lysophosphatidic acid acyltransferase family. J.Hum. Genet. 48:438–42
64. Lin F, Worman HJ. 1993. Structural organization of the human gene encoding nuclearlamin A and nuclear lamin C. J. Biol. Chem. 268:16321–26
65. Mager J, Bartolomei MS. 2005. Strategies for dissecting epigenetic mechanisms in themouse. Nat. Genet. 37:1194–200
66. First paperreportingmutations inBSCL2 gene inpatients withcongenitalgeneralizedlipodystrophy, type2.
66. Magre J, Delepine M, Khallouf E, Gedde-Dahl T Jr, Van Maldergem L, et al. 2001.Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy onchromosome 11q13. Nat. Genet. 28:365–70
66a. Magre J, Delepine M, Van Maldergem L, Robert JJ, Maassen JA, et al. 2003. Prevalenceof mutations in AGPAT2 among human lipodystrophies. Diabetes 52:1573–638
67. Miles PD, Barak Y, He W, Evans RM, Olefsky JM. 2000. Improved insulin-sensitivityin mice heterozygous for PPAR-gamma deficiency. J. Clin. Invest. 105:287–92
68. Mounkes LC, Kozlov S, Hernandez L, Sullivan T, Stewart CL. 2003. A progeroid syn-drome in mice is caused by defects in A-type lamins. Nature 423:298–301
69. Mounkes LC, Kozlov SV, Rottman JN, Stewart CL. 2005. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice.Hum. Mol. Genet. 14:2167–80
70. Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, et al. 2000. Identification ofmutations in the gene encoding lamins A/C in autosomal dominant limb girdle musculardystrophy with atrioventricular conduction disturbances (LGMD1B). Hum. Mol. Genet.9:1453–59
71. First paperreportinghomozygousmutation in LMNA
gene in patientswithmandibuloacraldysplasia–associated partiallipodystrophy.
71. Novelli G, Muchir A, Sangiuolo F, Helbling-Leclerc A, D’Apice MR, et al. 2002.Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C.Am. J. Hum. Genet. 71:426–31
72. Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, et al. 2002. Leptin-replacementtherapy for lipodystrophy. N. Engl. J. Med. 346:570–578
73. Pakendorf B, Stoneking M. 2005. Mitochondrial DNA and human evolution. Annu. Rev.Genomics Hum. Genet. 6:165–83
74. Pardini VC, Victoria IM, Rocha SM, Andrade DG, Rocha AM, et al. 1998. Leptin levels,β-cell function, and insulin sensitivity in families with congenital and acquired generalizedlipoatrophic diabetes. J. Clin. Endocrinol. Metab. 83:503–8
75. Pendas AM, Zhou Z, Cadinanos J, Freije JM, Wang J, et al. 2002. Defective prelaminA processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet. 31:94–99
76. Usinggenome-widelinkage analysis,this paper reportedthe first geneticlocus for familialpartiallipodystrophy ofthe Dunniganvariety onchromosome1q21-22 in five wellcharacterizedpedigrees.
76. Peters JM, Barnes R, Bennett L, Gitomer WM, Bowcock AM, Garg A. 1998. Lo-calization of the gene for familial partial lipodystrophy (Dunnigan variety) to chro-mosome 1q21-22. Nat. Genet. 18:292–95
www.annualreviews.org • Genetic Disorders of Adipose 197
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
77. Pivnick EK, Angle B, Kaufman RA, Hall BD, Pitukcheewanont P, et al. 2000. Neonatalprogeroid (Wiedemann-Rautenstrauch) syndrome: report of five new cases and review.Am. J. Med. Genet 90:131–40
78. Plasilova M, Chattopadhyay C, Pal P, Schaub NA, Buechner SA, et al. 2004. Homozygousmissense mutation in the lamin A/C gene causes autosomal recessive Hutchinson-Gilfordprogeria syndrome. J. Med. Genet. 41:609–14
79. Rautenstrauch T, Snigula F, Krieg T, Gay S, Muller PK. 1971. Progeria: a cell culturestudy and clinical report of familial incidence. Eur. J. Pediatr. 124:101–11
80. Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, et al. 1999. PPAR gamma is requiredfor the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 4:611–17
81. Savage DB, Agostini M, Barroso I, Gurnell M, Luan J, et al. 2002. Digenic inheritanceof severe insulin resistance in a human pedigree. Nat. Genet. 31:379–84
82. Savage DB, Tan GD, Acerini CL, Jebb SA, Agostini M, et al. 2003. Human metabolic syn-drome resulting from dominant-negative mutations in the nuclear receptor peroxisomeproliferator-activated receptor-γ. Diabetes 52:910–17
83. Seip M. 1959. Lipodystrophy and gigantism with associated endocrine manifestations: anew diencephalic syndrome? Acta Paediatr. 48:555–74
84. Seip M, Trygstad O. 1996. Generalized lipodystrophy, congenital and acquired (lipoat-rophy). Acta Paediatrica. Suppl. 413:2–28
85. Sensenbrenner JA, Hussels IE, Levin LS. 1975. CC: a low birthweight syndrome, Riegersyndrome. Birth Defects 11:423–26
86. Shackleton S, Lloyd DJ, Jackson SN, Evans R, Niermeijer MF, et al. 2000. LMNA,encoding lamin A/C, is mutated in partial lipodystrophy. Nat. Genet. 24:153–56
87. Shackleton S, Smallwood DT, Clayton P, Wilson LC, Agarwal AK, et al. 2005. Com-pound heterozygous ZMPSTE24 mutations reduce prelamin A processing and result ina severe progeroid phenotype. J. Med. Genet. 42:e36
88. Simha V, Agarwal AK, Oral EA, Fryns JP, Garg A. 2003. Genetic and phenotypic het-erogeneity in patients with mandibuloacral dysplasia-associated lipodystrophy. J. Clin.Endocrinol. Metab. 88:2821–24
89. Simha V, Garg A. 2002. Body fat distribution and metabolic derangements in patients withfamilial partial lipodystrophy associated with mandibuloacral dysplasia. J. Clin. Endocrinol.Metab. 87:776–85
90. Simha V, Garg A. 2003. Phenotypic heterogeneity in body fat distribution in patientswith congenital generalized lipodystrophy due to mutations in the AGPAT2 or Seipingenes. J. Clin. Endocrinol. Metab. 88:5433–37
91. Sinensky M, Fantle K, Trujillo M, McLain T, Kupfer A, Dalton M. 1994. The processingpathway of prelamin A. J. Cell Sci. 107(Pt. 1):61–67
92. Song G, Ouyang G, Bao S. 2005. The activation of Akt/PKB signaling pathway and cellsurvival. J. Cell Mol. Med. 9:59–71
93. Sorge G, Ruggieri M, Polizzi A, Scuderi A, Di Pietro M. 1996. SHORT syndrome: anew case with probable autosomal dominant inheritance. Am. J. Med. Genet 61:178–81
94. Speckman RA, Garg A, Du F, Bennett L, Veile R, et al. 2000. Mutational and haplotypeanalyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrentmissense mutations in the globular C-terminal domain of lamin A/C. Am. J. Hum. Genet.66:1192–98
95. Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, et al. 1999. Loss of A-typelamin expression compromises nuclear envelope integrity leading to muscular dystrophy.J. Cell Biol. 147:913–20
198 Agarwal · Garg
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

ANRV285-GG07-08 ARI 12 August 2006 17:8
96. Tsai YS, Kim HJ, Takahashi N, Kim HS, Hagaman JR, et al. 2004. Hypertension andabnormal fat distribution but not insulin resistance in mice with P465L PPARgamma. J.Clin. Invest. 114:240–49
97. Tsai YS, Maeda N. 2005. PPARgamma: a critical determinant of body fat distribution inhumans and mice. Trends Cardiovasc. Med. 15:81–85
98. Van Maldergem L, Magre J, Khallouf TE, Gedde-Dahl T Jr, Delepine M, et al. 2002.Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J. Med.Genet. 39:722–33
99. Vigouroux C, Magre J, Vantyghem MC, Bourut C, Lascols O, et al. 2000. Lamin A/Cgene: sex-determined expression of mutations in Dunnigan-type familial partial lipodys-trophy and absence of coding mutations in congenital and acquired generalized lipoatro-phy. Diabetes 49:1958–62
100. West J, Tompkins CK, Balantac N, Nudelman E, Meengs B, et al. 1997. Cloning andexpression of two human lysophosphatidic acid acyltransferase cDNAs that enhancecytokine-induced signaling responses in cells. DNA Cell Biol. 16:691–701
101. Wiedemann HR. 1979. An unidentified neonatal progeroid syndrome: follow-up report.Eur. J. Pediatr. 130:65–70
102. Windpassinger C, Auer-Grumbach M, Irobi J, Patel H, Petek E, et al. 2004. Heterozygousmissense mutations in BSCL2 are associated with distal hereditary motor neuropathy andSilver syndrome. Nat. Genet. 36:271–76
103. Ye GM, Chen C, Huang S, Han DD, Guo JH, et al. 2005. Cloning and characteriza-tion a novel human 1-acyl-sn-glycerol-3-phosphate acyltransferase gene AGPAT7. DNASequation 16:386–90
104. Young SG, Fong LG, and Michaelis S. 2005. Prelamin A, Zmpste24, misshapen cell nu-clei, and progeria: new evidence suggesting that protein farnesylation could be importantfor disease pathogenesis. J. Lipid Res. 46:2531–58
www.annualreviews.org • Genetic Disorders of Adipose 199
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

Contents ARI 8 August 2006 1:10
Annual Review ofGenomics andHuman Genetics
Volume 7, 2006
Contents
A 60-Year Tale of Spots, Maps, and GenesVictor A. McKusick � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 1
Transcriptional Regulatory Elements in the Human GenomeGlenn A. Maston, Sara K. Evans, and Michael R. Green � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �29
Predicting the Effects of Amino Acid Substitutions on ProteinFunctionPauline C. Ng and Steven Henikoff � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �61
Genome-Wide Analysis of Protein-DNA InteractionsTae Hoon Kim and Bing Ren � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �81
Protein Misfolding and Human DiseaseNiels Gregersen, Peter Bross, Søren Vang, and Jane H. Christensen � � � � � � � � � � � � � � � � � � � � � 103
The Ciliopathies: An Emerging Class of Human Genetic DisordersJose L. Badano, Norimasa Mitsuma, Phil L. Beales, and Nicholas Katsanis � � � � � � � � � � � � � 125
The Evolutionary Dynamics of Human Endogenous RetroviralFamiliesNorbert Bannert and Reinhard Kurth � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 149
Genetic Disorders of Adipose Tissue Development, Differentiation,and DeathAnil K. Agarwal and Abhimanyu Garg � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 175
Preimplantation Genetic Diagnosis: An Overview of Socio-Ethicaland Legal ConsiderationsBartha M. Knoppers, Sylvie Bordet, and Rosario M. Isasi � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 201
Pharmacogenetics and Pharmacogenomics: Development, Science,and TranslationRichard M. Weinshilboum and Liewei Wang � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 223
Mouse Chromosome Engineering for Modeling Human DiseaseLouise van der Weyden and Allan Bradley � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 247
v
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.

Contents ARI 8 August 2006 1:10
The Killer Immunoglobulin-Like Receptor Gene Cluster: Tuning theGenome for DefenseArman A. Bashirova, Maureen P. Martin, Daniel W. McVicar,and Mary Carrington � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 277
Structural and Functional Dynamics of Human CentromericChromatinMary G. Schueler and Beth A. Sullivan � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 301
Prediction of Genomic Functional ElementsSteven J.M. Jones � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 315
Of Flies and Man: Drosophila as a Model for Human Complex TraitsTrudy F.C. Mackay and Robert R.H. Anholt � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 339
The Laminopathies: The Functional Architecture of the Nucleus andIts Contribution to DiseaseBrian Burke and Colin L. Stewart � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 369
Structural Variation of the Human GenomeAndrew J. Sharp, Ze Cheng, and Evan E. Eichler � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 407
Resources for Genetic Variation StudiesDavid Serre and Thomas J. Hudson � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 443
Indexes
Subject Index � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 459
Cumulative Index of Contributing Authors, Volumes 1–7 � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 477
Cumulative Index of Chapter Titles, Volumes 1–7 � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 480
Errata
An online log of corrections to Annual Review of Genomics and Human Genetics chaptersmay be found at http://genom.annualreviews.org/
vi Contents
Ann
u. R
ev. G
enom
. Hum
an G
enet
. 200
6.7:
175-
199.
Dow
nloa
ded
from
ww
w.a
nnua
lrev
iew
s.or
gby
Otte
rbei
n U
nive
rsity
on
08/2
4/13
. For
per
sona
l use
onl
y.


















