
Gene Prediction: Past, Present, and Future Sam Gross.
53
Gene Prediction: Gene Prediction: Past, Present, and Past, Present, and Future Future Sam Gross
-
date post
20-Dec-2015 -
Category
Documents
-
view
228 -
download
2
Transcript of Gene Prediction: Past, Present, and Future Sam Gross.

Gene Prediction:Gene Prediction:Past, Present, and FuturePast, Present, and Future
Sam Gross

GenesGenes
ATG
• Gene RNA Protein• Proteins are about 500 AA long
• Genes are about 1500bp long
TAGTAATGA

ORF ScanningORF Scanning
In “lower” organisms, genes are contiguous
We expect about 1 stop codon per 64bpIf we see a long ORF, it’s probably a
gene!– And conversely, all genes are long ORFs

IntronsIntrons
GT GTAG AG
ATG TGATAATAG
• Drosophila:• 3.4 introns per gene on average• mean intron length 475, mean exon length 397
• Human:• 8.8 introns per gene on average• mean intron length 4400, mean exon length 165
• ORF scanning is defeated

SplicingSplicing
GT GTAG AG
ATG TGATAATAG
GT GTAG AG
ATG TGATAATAG
AG

Needles in a HaystackNeedles in a Haystack
Human genome is about 3.2Gbp20,000 – 25,000 genes78% intergenic, 20% introns, 2% coding

Gotta Find ‘Em AllGotta Find ‘Em All
60-85% of all human genes have been found, mostly by random EST sequencing– This probably won’t work for the rest
For most genes, only one splice variant is known
If we can computationally predict a gene, we have a cheap experiment (RT-PCR) to verify

Looking For CluesLooking For Clues
Signals used by the cell– 99% of introns begin with GT, end with AG– 0.8% of introns begin with GC, end with AG– Gene begins with ATG– Gene ends with TAG, TAA, or TGA
Other properties of genes– Exons have characteristic lengths– Base composition of exons is characteristic due to genetic
code– Exons tend to be conserved between species
• Pattern of conservation is three-periodic

Three-PeriodicityThree-Periodicity
Most amino acids can be coded for by more than one DNA triplet (codon)
Usually, the degeneracy is in the last position
Human CCTGTT (Proline, Valine)Mouse CCAGTC (Proline, Valine)Rat CCAGTC (Proline, Valine)Dog CCGGTA (Proline, Valine)Chicken CCCGTG (Proline, Valine)

Hidden Markov ModelsHidden Markov Models
The de facto standard for gene prediction Probabilistic finite state machine Transition to a state, emit a character, transition to a
new state– Many independence assumptions
CDS NC
ACG )|()|()|()|()|()( CDSGPNCCDSPNCCPNCNCPNCAPNCP

HMMs For Gene PredictionHMMs For Gene Prediction
Generative model– Define P(X, Y) as a product of many independent
termsP(ACG) = P(start in noncoding) *P(noncoding emits A) *P(noncoding transitions to noncoding) *P(noncoding emits C) *P(noncoding transitions to coding) *P(coding emits A)• Terms are of the forms P(yi | yi-1) and P(xi | yi)
– Trained by collecting counts

HMMs For Gene PredictionHMMs For Gene Prediction
To predict genes given a sequence X, calculateargmaxY P(Y | X) = argmaxY P(X, Y) / P(X) =
argmaxY P(X, Y)

Generalized Hidden Markov Generalized Hidden Markov ModelsModels Like a HMM, but state durations are explicit Transition to a state, pick a duration d, emit d
characters, transition to a new state Dynamic programming algorithm complexity
goes from O(N2L) to O(N2LK)– K is the maximum state duration– Not so bad in practice

Predicting Genes With HMMsPredicting Genes With HMMs
Given a sequence, we can calculate the most likely annotation
InternalExon
Intron
Inter-genic
FinalExon
InitialExon
SingleExon
GGTGAGGTGACCAAGAACGTGTTGACAGTAGGTGAGGTGACCAAGAACGTGTTGACAGTAGGTGAGGTGACCAAGAACGTGTTGACAGTAGGTGAGGTGACCAAGAACGTGTTGACAGTA

The Past: GENSCANThe Past: GENSCAN
Chris Burge, Stanford, 1997Before the Human Genome Project
– No alignments available– People still thought there were 100,000
human genes

The GENSCAN ModelThe GENSCAN Model

The GENSCAN ModelThe GENSCAN Model
Output probabilities for NC and CDS depend on previous 5 bases (5th-order)– P(Xi | Xi-1, Xi-2, Xi-3, Xi-4, Xi-5)
Each CDS frame has its own model Special 2nd-order positional models for start
codon, stop codon, and acceptor site Even fancier model for donor sites
– Maximal dependence decomposition (MDD)– Long-range dependencies
Separate model for different isochores

GENSCAN PerformanceGENSCAN Performance
First program to do well on realistic sequences– Multiple genes in both orientations
Pretty good sensitivity, poor specificity– 70% exon Sn, 40% exon Sp
Not enough exons per geneWas the best gene predictor for about 4
years

Comparative Gene PredictionComparative Gene Prediction
ExonIntron
ExonIntron
-3 -2 -1 +1 +2 +3
Human A A G G T G
-3 -2 -1 +1 +2 +3
Human A A G G T G
Mouse A A G G T G Mouse A A T G T G
Chicken A A G G T G Chicken A A _ A C G
A B

The Recent Past: TWINSCANThe Recent Past: TWINSCAN
Korf, Flicek, Duan, Brent, Washington University in St. Louis, 2001
Uses an informant sequence to help predict genes– For human, informant is normally mouse
Informant sequence consists of three characters– Match: |– Mismatch: :– Unaligned: .
Informant sequence assumed independent of target sequence

The TWINSCAN ModelThe TWINSCAN Model
Just like GENSCAN, except adds models for conservation sequence
5th-order models for CDS and NC, 2nd-order models for start and stop codons and splice sites– One CDS model for all frames
Many informants tried, but mouse seems to be at the “sweet spot”

TWINSCAN PerformanceTWINSCAN Performance
Slightly more sensitive than GENSCAN, much more specific– Exon sensitivity/specificity about 75%
Much better at the gene level– Most genes are mostly right, about 25%
exactly rightWas the best gene predictor for about 4
years

The Present: N-SCANThe Present: N-SCAN
Gross and Brent, Washington University in St. Louis, 2005
If one informant sequence is good, let’s try more!
Also several other improvements on TWINSCAN

N-SCAN ImprovementsN-SCAN Improvements
Multiple informants
Richer models of sequence evolution
Frame-specific CDS conservation model
Conserved noncoding sequence model
5’ UTR structure model

GENSCAN
TWINSCAN
N-SCAN
HMM OutputsHMM Outputs
Target GGTGAGGTGACCAAGAACGTGTTGACAGTA
Target GGTGAGGTGACCAAGAACGTGTTGACAGTAConservation |||:||:||:|||||:||||||||......sequence
Target GGTGAGGTGACCAAGAACGTGTTGACAGTAInformant1 GGTCAGC___CCAAGAACGTGTAG......Informant2 GATCAGC___CCAAGAACGTGTAG......Informant3 GGTGAGCTGACCAAGATCGTGTTGACACAA.
..
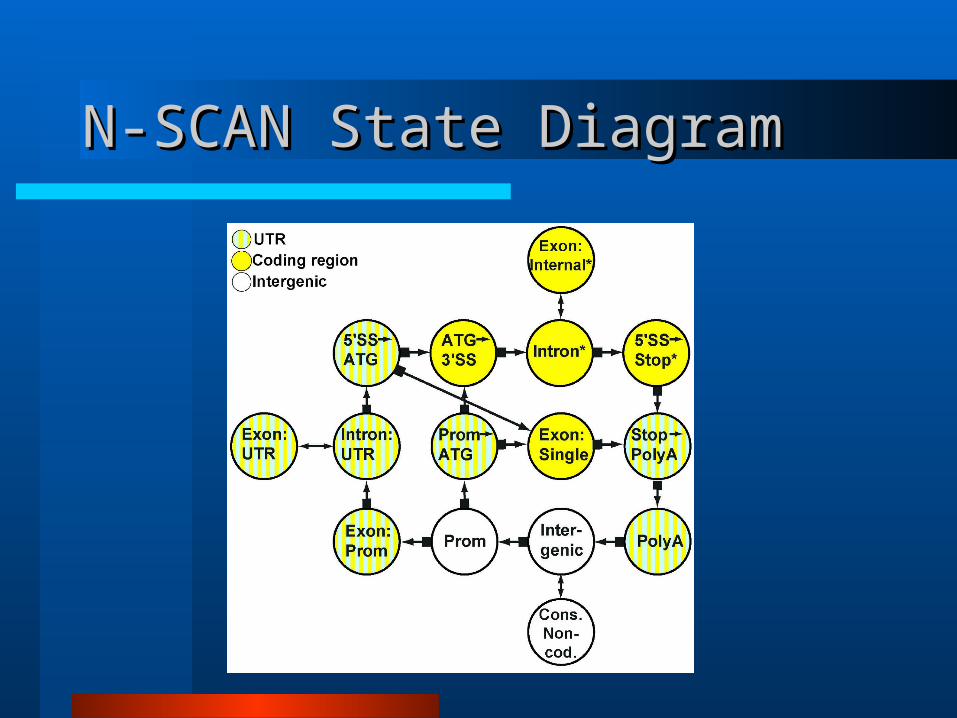
N-SCAN State DiagramN-SCAN State Diagram

Two-Component Output Two-Component Output DistributionsDistributions
Target sequence model
Phylogenetic model for informants
Product gives the probability of a multiple alignment column
),...,,...,|( 1 oiioiii TTP III
),...,|( 1 oiii TTTP
),...,,,...,|,( 11 oiioiiii TTTP III

Phylogenetic Bayesian Network Phylogenetic Bayesian Network ModelsModels
)|()|()|(
)|()|()|()(),,,,,,(
3323
21211321
ARPAMPAAP
AHPAAPACPAPAAARMCHP
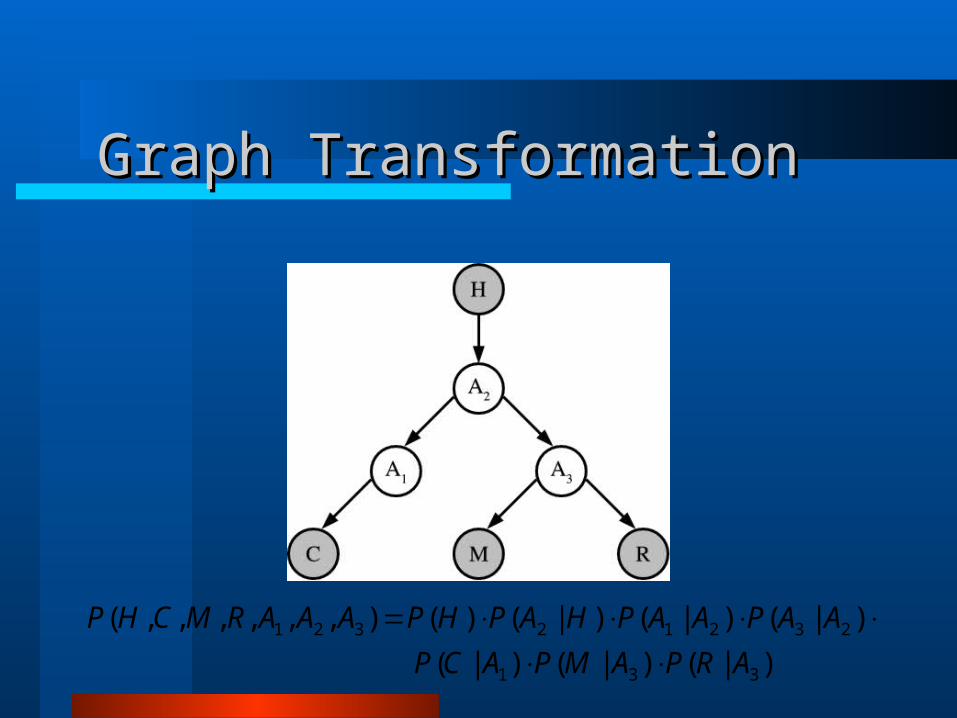
)|()|()|(
)|()|()|()(),,,,,,(
331
23212321
ARPAMPACP
AAPAAPHAPHPAAARMCHP
Graph TransformationGraph Transformation

InferenceInference
Slightly-modified version of Felsenstein’s algorithm
At each of the O(N) nodes, we calculate 6o+1 summations over 6o+1 values
Total time complexity is O(N • 62(o+1))

TrainingTraining
Simple with labeled multiple alignment of all sequences
Can use known genes as a labeling
Don’t know ancestral genome sequences– Treat them as missing data and use EM

CPD ParameterizationsCPD Parameterizations Each Bayesian network of order o has
(2N-1)(6o+1)(6o+1-1) free parameters
We can reduce this number by restricting the form of the CPDs
Partially reversible models– Relative frequency of DNA k-mers remains constant as sequence
evolves– Gaps and unaligned regions introduced over time

N-SCAN Phylogenetic Models N-SCAN Phylogenetic Models vs. Traditional Phylogenetic vs. Traditional Phylogenetic ModelsModels
Root (target) node is observed
– Can use existing single-sequence models
– Can use higher-order models
– Can estimate target sequence model optimally

No assumption of homogeneous substitution process
– Gaps and unaligned regions can be treated naturally
– Robust against
• Function-changing mutation
• Alignment error
• Sequencing error
– The price is many more parameters
N-SCAN Phylogenetic Models N-SCAN Phylogenetic Models vs. Traditional Phylogenetic vs. Traditional Phylogenetic ModelsModels

Conservation Score CoefficientConservation Score Coefficient
N-SCAN uses log-likelihood scores internally. The score of a position i under state S is
Values of k between 0.3 and 0.6 result in the best performance– Performance is roughly constant in this range
)|(
)|(log
)|(
)|(log
NullP
SPk
NullTP
STP
i
i
i
i
I
I

Whole-Genome Human Gene Whole-Genome Human Gene PredictionPredictionAnnotations used were cleaned
RefSeqs– 16,259 genes
– 20,837 transcripts
N-SCAN used human, mouse, rat, chicken alignment

Exact Exon AccuracyExact Exon Accuracy
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
Exon Sn Exon Sp
GENSCAN EXONIPHY SGP2 TWINSCAN 2.0 N-SCAN

Exact Gene AccuracyExact Gene Accuracy
0
0.1
0.2
0.3
0.4
0.5
Gene Sn Gene Sp
GENSCAN SGP2 TWINSCAN 2.0 N-SCAN

Intron Sensitivity By LengthIntron Sensitivity By Length
0
0.2
0.4
0.6
0.8
1
0-10
10-2
0
20-3
0
30-4
0
40-5
0
50-6
0
60-7
0
70-8
0
80-9
0
90-1
00Length (Kb)
N-SCANSGP2GENSCANTWINSCAN

Human Informant EffectivenessHuman Informant Effectiveness
00.10.20.30.40.50.60.70.80.9
Gene Sn Gene Sp Exon Sn Exon Sp
Chicken Rat Mouse All

Drosophila Drosophila Informant EffectivenessInformant Effectiveness
0.3
0.4
0.5
0.6
0.7
0.8
0.9
Gene Sn Gene Sp Exon Sn Exon Sp
A. gambiae D. yakuba D. pseudoobscura All

The Future(?): CONTRASTThe Future(?): CONTRAST
New gene predictor currently in the works
Based not on a generalized HMM, but a semi-Markov conditional random field (SCRF)

HMMs For Gene PredictionHMMs For Gene Prediction
Generative model– Define P(X, Y) as a product of many independent
termsP(ACG) = P(start in noncoding) *P(noncoding emits A) *P(noncoding transitions to noncoding) *P(noncoding emits C) *P(noncoding transitions to coding) *P(coding emits A)• Terms are of the forms P(yi | yi-1) and P(xi | yi)
– Trained by collecting counts

HMMs For Gene PredictionHMMs For Gene Prediction
To predict genes given a sequence X, calculateargmaxY P(Y | X) = argmaxY P(X, Y) / P(X) =
argmaxY P(X, Y) Advantage: simplicity
– Extremely fast training, efficient inference Disadvantage: simplicity
– Makes many unwarranted independence assumptions
– Inaccurate model will get us into trouble

When HMMs Go WrongWhen HMMs Go Wrong
Normal HMM training optimizes wrong function– We use P(Y | X) for prediction, but we’re
optimizing P(X, Y) = P(Y | X) P(X)– This means we may prefer parameters that
lead to worse predictions if they assign a higher probability to the sequence

When HMMs Go WrongWhen HMMs Go Wrong
NCA 3%B 2%C 95%
CDSA 49%B 49%C 2%
NCA 3%B 2%C 95%
CDSA 3%B 95%C 2%
NNCA 2%B 2%C 96%
CNSA 96%B 2%C 2%
CDSA 49%B 49%C 2%
A = Conserved tripletB = Synonymous substitutionC = Nonsynonymous substitution
…CCCCCCCCCCCCCAAAAAAAAAACCCC…CCCCCCCBBABAAABBABBABCC…

Can We Fix It?Can We Fix It?
Directly optimize
No closed form solution– But function and gradient can be calculated
efficiently using DP If we’re going to numerically optimize anyway,
might as well switch to a more expressive model
),(
),()|(
YXP
YXPXYP
Y

CRFs For Gene PredictionCRFs For Gene Prediction
Discriminative model– Define P(Y | X) as a product of many terms
• Individual terms are not probabilities!• Terms are of the form fj(yi-1, yi, X, i) wj
The Good– Independence assumptions much weaker than in
HMMs– Inference complexity is the same as for HMM
The Bad– Training requires numerical optimization of (convex)
likelihood function

The MathThe Math
ji
iijj wiXyyfYXF ),,,(),( 1
jj YXF
XZXYP ),(exp
)(
1)|(
Y jj YXFXZ ),(exp)(
CRFs
i
iiiiba aybyPaybyYXT )|(log],[1),( 11,
i
iiiisa syaxPaxsyYXE )|(log],[1),(,
HMMs
sasa
baba YXEYXTYXP
,,
,, ),(),(exp),(

HMMs vs. CRFsHMMs vs. CRFs
y1
x1
y2
x2
y3
x3
y4
x4
y5
x5
y6
x6
…HMM
y1
x1
y2
x2
y3
x3
y4
x4
y5
x5
y6
x6
…CRF

HMMs vs. CRFsHMMs vs. CRFs
HMM-style “features”– Last state is exon, current state is intron– Current state is exon, current sequence character is “C”
CRF-style features– Current state is exon, CG percent in 100Kbp window is
between 40% and 50%, at least one CpG island predicted within 10Kbp
– Current state is exon, 3 unspliced ESTs with at least 95% identity aligned near current position
– Current state is exon, 1 spliced EST with at least 95% identity aligned near current position

Semi-Markov CRFsSemi-Markov CRFs
Semi-Markov CRFs are to CRFs as generalized HMMs (or semi-HMMs) are to HMMs
Instead of assigning labels to each position, assign labels to segments
Features are f(yi-1, yi, X, i, j)

Future DirectionsFuture Directions
SVM-based splice site models that use alignment information– Splice site models in current gene
predictors are pretty primitiveAlternative splicing!
– Not yet handled well– Very poor experimental coverage of
transcriptome


![AT07175: SAM-BA Bootloader for SAM D21ww1.microchip.com/downloads/en/DeviceDoc/Atmel-42366-SAM... · 2016-12-10 · AT07175: SAM-BA Bootloader for SAM D21 [APPLICATION NOTE] Atmel-42366A-SAM-BA-Bootloader-for-SAM-D21-ApplicationNote_082014](https://static.fdocuments.in/doc/165x107/5f38185f0481442629236b2e/at07175-sam-ba-bootloader-for-sam-2016-12-10-at07175-sam-ba-bootloader-for-sam.jpg)



![Welcome [] · Bladder outlet obstruction ... End of life care as per patient wishes Gross hematuria requiring continuous bladder irrigation # 2 PREDICTION: Nurses can apply criteria](https://static.fdocuments.in/doc/165x107/5e9e2940575a354c7d61fa46/welcome-bladder-outlet-obstruction-end-of-life-care-as-per-patient-wishes.jpg)











