
FRACP LECTURE 2010.ppt - Royal Children's Hospital · FRACP LECTURE 2010 IMMUNE DEFICIENCY 3 DR...
41
FRACP LECTURE 2010 IMMUNE DEFICIENCY 3 DR MARNIE ROBINSON PAEDIATRIC PAEDIATRIC IMMUNOLOGIST/ALLERGIST
Transcript of FRACP LECTURE 2010.ppt - Royal Children's Hospital · FRACP LECTURE 2010 IMMUNE DEFICIENCY 3 DR...

FRACP LECTURE 2010IMMUNE DEFICIENCY 3
DR MARNIE ROBINSON
PAEDIATRICPAEDIATRIC IMMUNOLOGIST/ALLERGIST

IMMUNOLOGY LECTURE 3IMMUNOLOGY LECTURE 3
• Neutrophil defectsNeutrophil defects
• INTERFERON –Y /IL‐12 pathway defect
l i d fi i i• Dysregulatory immune deficiencies

NEUTROPHIL DEFECTSNEUTROPHIL DEFECTS• Neutropaenia
All i / t i– Alloimmune/autoimmune
– Kostmann, WHIM
l l– cyclical
• Chronic granulomatous disease
• Leukocyte adhesion deficiency
• Neutrophil specific granule deficiencyNeutrophil specific granule deficiency
• Chediak – higashi syndrome

AUTOIMMUNE NEUTROPAENIAAUTOIMMUNE NEUTROPAENIA• Antibodies against different neutrophil antigen
l k• Aeitiology unknown
• Slightly more common in females
• Present with skin and upper respiratory tract infections (pneumonia/meningitis/sepsis less (p / g / pcommon)
• Neutrophil count usually <0 5 but mayNeutrophil count usually <0.5 but may increase during infection
• Treatment with G CSF (IVIG)• Treatment with G‐CSF (IVIG)
• Usually remits spontaneously by <24 months

ALLOIMMUNE NEUTROPAENIAALLOIMMUNE NEUTROPAENIA• Caused by transplacental transfer of maternal against the FcyRIIIb isotypes of NA 1 and NA2against the FcyRIIIb isotypes of NA 1 and NA2 causing immune destruction of neutrophils
d f /• Incidence of 1/500
• Usually presents in first weeks of life
• Present with omphalitis, cellulitis, pneumonia
• Diagnosed by detection of neutrophil specificDiagnosed by detection of neutrophil specific alloantibodies in maternal blood
• Treat with G CSF• Treat with G‐CSF
• resolves with waning of maternal antibodies

KOSTMANN’S SYNDROMEKOSTMANN S SYNDROME
• Bone marrow granulocyte arrest atBone marrow granulocyte arrest at promyeolocyte or myelocyte stage
• Present early in life (usually <6 months)• Present early in life (usually <6 months)
• Present with omphalitis , respiratory tract i f i ki d li binfections, skin and liver abscesses
• Increased susceptibility to AML
• Treatment is with G‐CSF

CYCLICAL NEUTROPAENIACYCLICAL NEUTROPAENIA
• Defect in elastase 2
• Sporadic 2/3, familial 1/3(AD)
• Neutropaenia occurs classically at intervals of• Neutropaenia occurs classically at intervals of 21 days
U ll b i i hildh d ( 30% 1 )• Usually begins in childhood (~30% <1year)
• Recurrent episodes of malaise, fever , aphthous stomatitis , cervical lymphadenopathy (episodes 5‐6 days)
• May treat with G‐CSF

WHIM SYNDROMEWHIM SYNDROME• Warts
• Hypogammaglobulinaemia
• InfectionsInfections
• Melokathexis :chronic neutropaenia but hyercellularity on BMA +/‐ lymphopaeniahyercellularity on BMA +/‐ lymphopaenia
• Autosomal recessive due to mutation in h ki t CXCR4chemokine receptor CXCR4
• Warts , recurrent sinopulmonary infections
• Treatment : steroids , G‐CSF

GLYCOGEN STORAGE TYPE 1BGLYCOGEN STORAGE TYPE 1B
• Autosomal recessive
• Hepatic incapacity to convert G‐6P to glucose and neutropaeniaand neutropaenia
• Present with hypoglycaemia , seizures , lactic acidosis hyperuriciaemia and hyperlipidaemiaacidosis, hyperuriciaemia and hyperlipidaemia
• neutropaenia : skin infections, l h d th l d l llymphadenopathy, oral and anal ulcers
• Treatment : prevention of hypoglycaemia and G‐CSF

CHRONIC GRANULOMATOUS DISEASECHRONIC GRANULOMATOUS DISEASE
GENETICS • X linked (70%)
– Tend to have earlier onset and more severe disease– x linked carriers : discoid lupus/mouth
ulcers/Raynauds
• Autosomal recessive– P47 phox mutation (ch7)p ( )– P67 phox mutation (ch1)– P 22 phox mutation (ch16)

CHRONIC GRANULOMATOUS DISEASECHRONIC GRANULOMATOUS DISEASE
• Caused by defects in the NADPH oxidaseCaused by defects in the NADPH oxidase which is responsible for the respiratory burst and generation of phagocyte superoxideand generation of phagocyte superoxide
• Inability to generate superoxide leads to failure to make the downstream reactivefailure to make the downstream reactive oxygen species hydrogen peroxide and hydroxyl radicalhydroxyl radical → defective microbial killing of catalase positive bacteria and fungibacteria and fungi

CHRONIC GRANULOMATOUS DISEASECHRONIC GRANULOMATOUS DISEASE
• PATHOPHYSIOLOGYPATHOPHYSIOLOGY

CHRONIC GRANULOMATOUS DISEASECHRONIC GRANULOMATOUS DISEASE
ORGANISMSORGANISMS• Aspergillus• Candida albicans• Candida albicans• Staph aureusN di• Nocardia
• E.coli• Serratia• Salmonella

CHRONIC GRANULOMATOUS DISEASE
PRESENTATION
(1) Early onset severe bacterial and fungal infections
• Skin abscesses/lymphadenitis
• Lung/splenic/liver abscesses• Lung/splenic/liver abscesses
• Recurrent pneumonia
• Osteomyelitis
• Peritonitis
• Gingivitis/mouth ulcers

CHRONIC GRANULOMATOUS DISEASE
PRESENTATION
(2) Granulomatous disease
• Skin granulomasSkin granulomas
• Granulomas of GIT Gastric outlet obstruction– Gastric outlet obstruction
– Granulomatous inflammatory bowel disease
l d• Genitourinary granulomatous disease– Urinary retention. dysuria
• Granulomatous disease of lungs

CHRONIC GRANULOMATOUS DISEASECHRONIC GRANULOMATOUS DISEASE
DIAGNOSISDIAGNOSIS
• Nitroblue tetrazloium test (NBT)N t hil i CGD bl t d d– Neutrophils in CGD are unable to reduce dye
– Should usually turn blue but in CGD does not changechange
• NEUTROPHIL FUNCTION
• GENETIC TESTING

CHRONIC GRANULOMATOUS DISEASECHRONIC GRANULOMATOUS DISEASETREATMENT
( )(1) AGGRESSIVE TREATMENT OF INFECTIONS
(2) PROPHYLAXIS AGAINST INFECTION
• Bacterial prophylaxis– Bactrim/ itrakonazoleBactrim/ itrakonazole
• IFN y
70% reduction in in infections‐ 70% reduction in in infections
(3) BONE MARROW TRANSPLANT
(4) GENETIC COUNSELLING

LEUKOCYTE ADHESION DEFICIENCYLEUKOCYTE ADHESION DEFICIENCY
TYPE 1TYPE 1
• AR
i i h d f C 8 2• Mutation in gene that codes for CD18 B2 leukocyte integrin subunit
• B2 subunit is responsible for adhesion of neutrophils to endothelial cell surface , migration from circulation and adhesionto C3b opsonised organisms

LEUKOCYTE ADHESION DEFICIENCY
TYPE 1• Usually present within first months of life• Usually present within first months of life• Delayed separation of umbilical cord >21 days• Omphalitis• Omphalitis• Persistent leukocytosis• Severe gingivitis/periodontitis• Severe gingivitis/periodontitis• Recurrent infections
skin /airway /bowelPerirectal/labial– skin /airway./bowelPerirectal/labial– No pus /absence of neutrophils– Typical signs of inflammationTypical signs of inflammation absent(swelling/eythema etc
– Delayed healing

LEUKOCYTE ADHESION DEFICIENCY • TYPE 2• AR• AR • Mutation in GDP‐fucose transporter gene –ligand for E selectin – unable to make initialligand for E selectin unable to make initial attachment to endothelium
• Characteristic facial features :coarseCharacteristic facial features :coarse• Short stature• Mental retardation• Mental retardation• Increased infections:skin/gum/resp• Poor pus formation • Treatment :oral fucose supplemention

LEUKOCYTE ADHESION DEFCIENCYLEUKOCYTE ADHESION DEFCIENCY
DIAGNOSISDIAGNOSIS• Flow cytometry• Decreased chemotaxis• Decreased chemotaxis• FBE : marked neutrophiliaBi f t hil• Biopsy : few neutrophils
TREATMENT• Aggressive mx of infection/prophylaxis• BMT

NEUTROPHIL SPECIFIC GRANULE DEFICIENCY
• Autosomal recessiveAutosomal recessive
• Profound reduction or absence of neutrophil specific granules and their contentsspecific granules and their contents
• Recurrent infections : skin , ears , lungs and l h d GRAM ilymph nodes‐ GRAM + cocci
• Absent or very low specific granule contents on blood smear /EM

CHEDIAK –HIGASHI SYNDROMECHEDIAK HIGASHI SYNDROME
• ARAR
• LYST gene mutation:codes cytoplasmic protein involved in vascular formation function andinvolved in vascular formation, function and transport
D f i i b l hil ’• Defect in microtubules – neutrophils can’t orientate correctly during chemotaxis
• Oversized lysozymes , storage granules

CHEDIAK –HIGASHI SYNDROMECHEDIAK HIGASHI SYNDROME
• Partial oculocutaneous albinsmPartial oculocutaneous albinsm• Neuropathy :sensory or motor• Mild mental retardation• Mild mental retardation• NystagmusBl di• Bleeding
• Infection – mucous membranes, skin ‐ peridontal/respiratory
• Accelerated phase

CHEDIAK –HIGASHI SYNDROMECHEDIAK HIGASHI SYNDROME
DIAGNOSISDIAGNOSIS
• Blood film : large inclusions in all nucleated blood cellsblood cells
• TREATMENT
• BMT curative but does not alter neurological goutcome

THE INTERFERON‐Y/IL‐12 PATHWAY DEFECTS
Characterised by susceptibility to ;Characterised by susceptibility to ;
• BCG / other poor pathogenic mycobacteria
i i d• Disemminated TB
• Systemic and/or persistent non typhi salmonella
• Severe herpes virus (CMV/HSV/VZV)p ( / / )
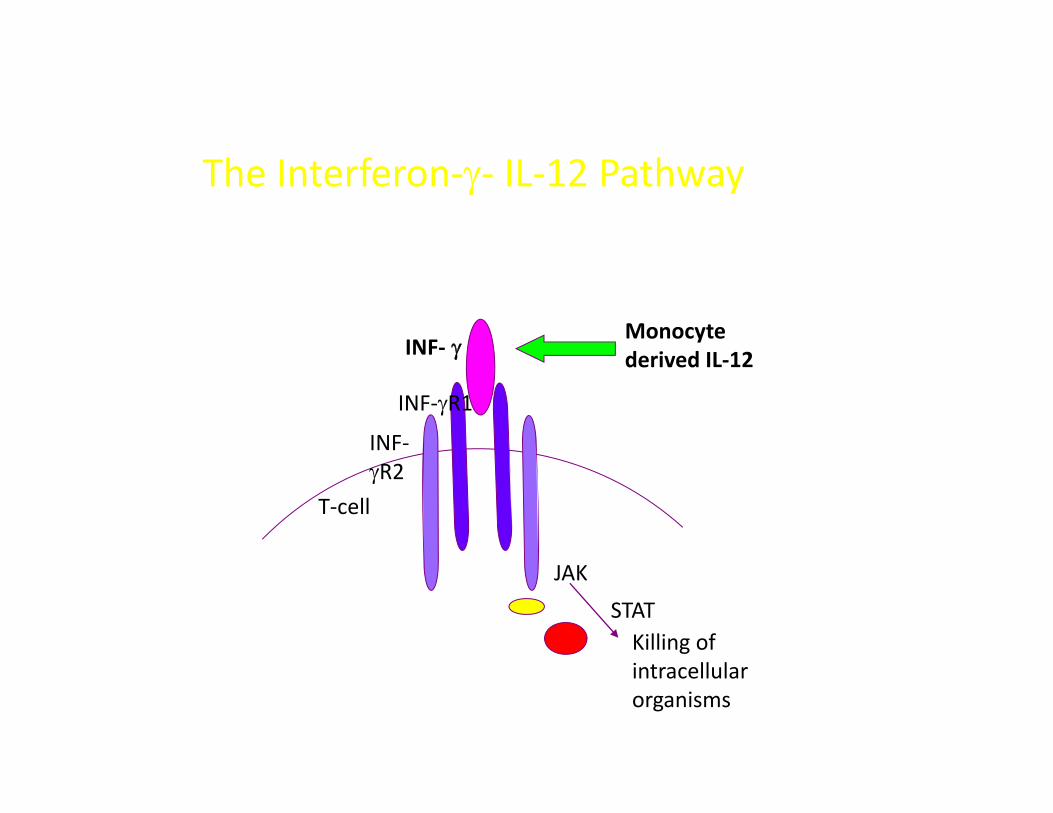
The Interferon‐γ‐ IL‐12 Pathway
INF‐ γMonocyte d i d IL 12γ
INF‐γR1
INF‐
derived IL‐12
γR2T‐cell
JAK
STATKilling of i ll lintracellular organisms

Defects of IFN‐g/IL‐12 AxisDefects of IFN g/IL 12 Axis
• Usually ARUsually AR
• Occas AD (partial IFNgRa; partial STAT1)
i l ( S )• Partial (IFNgRs; STAT1)
• Complete (IFNgR1s; IL‐12 p40; IL‐12RB1)
• Normal cellular and humoral IFNormal cellular and humoral IF

Defects of IFN‐g/IL‐12 AxisDefects of IFN g/IL 12 Axis
• Screening Ix – serum IFNg↑↑↑Screening Ix serum IFNg↑↑↑
GMANAGEMENT
• Some pt benefit from s/c IFN‐g (partial IFNgR; IL‐12p40; IL‐12BR1
• ?BMT (esp complete)( p p )

INTERFERON –Y RECEPTOR DEFICIENCIES
• Autosomal recessive and dominant formsAutosomal recessive and dominant forms (dominant tends to present later)
• Tend to develop severe mycobacterial disease• Tend to develop severe mycobacterial disease in early infancy or childhood
M b t i l t liti– Mycobacterial osteomyelitis
• May get disseminated infection from BCG vaccine

INTERFERON‐Y DEFECTSINTERFERON Y DEFECTS
• DiagnosisDiagnosis– Measurement of STAT 1 after stimulation with
IFN y stimulation requires Functional IFN yIFN‐y stimulation – requires Functional IFN‐y receptor
‐ Invitro TNF‐ alpha production by PBMC in‐ Invitro TNF‐ alpha production by PBMC in response to LPS – impaired
‐ geneticsgenetics
• Treatment : IFN Y– IFN –Y
– Mycobacterial prophylaxis

IL‐12 RECEPTOR DEFICIENCYIL 12 RECEPTOR DEFICIENCY
• Present with disseminated nontuberculousPresent with disseminated nontuberculous mycobacterial and salmonella infections or progressive BCG infection following BCGprogressive BCG infection following BCG vaccination
• Defect in IL 12 signalling leads to poor• Defect in IL‐12 signalling leads to poor production of IFN‐Y by T and NK cells
T IFN d i b i l• Treatment : IFN – y and antimycobacterials

STAT 1 DEFICIENCYSTAT 1 DEFICIENCY
• STAT1 is a critical molecule in the transduction or signal from both the IFN‐y.R and IFN a/BR
• Autosomal and recessive formsAutosomal and recessive forms – AD : IFN‐y mediated function impaired
– AR: : IFN‐y mediated function and IFN a/BAR: : IFN y mediated function and IFN a/B mediated function impaired
• Disseminated mycobacterim avium infectionDisseminated mycobacterim avium infection
• AD form associated with susceptibility to severe fatal mycobacterial infectionsevere fatal mycobacterial infection

IRAK 4 DEFICIENCYIRAK 4 DEFICIENCY
• Autosomal recessive ~ 25 described cases
• IRAK 4 (interleukin 1 receptor associated kinase 4 ) deficiency results in impairment in Toll receptor and Il‐1 receptor
di t d i llimediated signalling
• Recurrent invasive pyogenic infections with poor inflammatory responsesy p
• Infections classically involve S.pneumoniae and Staph aureus.
• Infections tend to decrease with advancing age (if survive)
• Normal immune function
• Treatment : IVIG and antibiotic prophylaxis


ALPS SYNDROMEALPS SYNDROME• Disorder of lymphocyte apoptosis (fas pathway)
PRESENTATION
• Lymphoproliferation• SplenomegalySplenomegaly
• Massive lymphadenopathy
• Autoimmune diseaseAutoimmune disease• Blood cells
• Autoimmune hepatitisp
• malignancy

ALPS SYNDROMEALPS SYNDROME
DIAGNOSISDIAGNOSIS• ↑ Double negative T cells (CD4‐/CD8‐)• APOPTOSIS studies• APOPTOSIS studies• autoantibodies
TREATMENT• Lymphoproliferation :steroids/chemotherapy• ?BMT

IPEX SYNDROMEIPEX SYNDROME
• IMMUNE DYSREGULATIONIMMUNE DYSREGULATION
• POLYENDOCRINOPATHY
O• ENTEROPATHY
• X LINKED
• Mutation in FOX P3 geneMutation in FOX P3 gene– Expressed in lymphoid tissue (thymus , spleen , lymph nodes) and CD4+CD25+ regulatory T cellslymph nodes) and CD4+CD25+ regulatory T cells

IPEX SYNDROME
• Present usually in first year of life with severe diarrhoea and FTT from enteropathydiarrhoea and FTT from enteropathy
• Dermatitis
• Endocrinopathy – Early onset type 1 diabetes
– Thyroid disease : hypo or hyperthyroidism
• Other autoimmune diseases– Cytopaenias
– Tubular nephropathyTubular nephropathy
– alopecia

IPEX SYNDROMEIPEX SYNDROME• ~50% have serious infections : sepsis , meningitis pneumonia osteomyelitismeningitis , pneumonia , osteomyelitis
• Most common pathogens Staphylococcus , d d dCMV and candida
• Diagnosis– Intermittent eosinophilia
– Markedly elevated IgE
– T AND B cell numbers normal , normal neutrophil function and complement
– Increased Th2 cytokines (IL‐4,5,10,130
– Decreased Th1 cytokines : IFN ‐Y

IPEX SYNDROMEIPEX SYNDROME
• ImmunsuppressionImmunsuppression
S ll l i• Stem cell transplant curative


















