
Fission Yeast Sap1 Protein Is Essential for Chromosome ...ec.asm.org/content/2/5/910.full.pdf ·...
12
EUKARYOTIC CELL, Oct. 2003, p. 910–921 Vol. 2, No. 5 1535-9778/03/$08.000 DOI: 10.1128/EC.2.5.910–921.2003 Copyright © 2003, American Society for Microbiology. All Rights Reserved. Fission Yeast Sap1 Protein Is Essential for Chromosome Stability Raynald de Lahonde `s, 1 Veronique Ribes, 2 and Benoit Arcangioli 1 * Dynamique du Genome, URA 1644 du CNRS, Institut Pasteur, 75724 Paris, Cedex 15, France, 1 and Institute of Interdisciplinary Research (IRIBHN), School of Medicine, University of Brussels, B1070 Brussels, Belgium 2 Received 31 March 2003/Accepted 8 July 2003 Sap1 is a dimeric sequence-specific DNA binding-protein, initially identified for its role in mating-type switching of the fission yeast Schizosaccharomyces pombe. The protein is relatively abundant, around 10,000 dimers/cell, and is localized in the nucleus. sap1 is essential for viability, and transient overexpression is accompanied by rapid cell death, without an apparent checkpoint response and independently of mating-type switching. Time lapse video microscopy of living cells revealed that the loss of viability is accompanied by abnormal mitosis and chromosome fragmentation. Overexpression of the C terminus of Sap1 induces minichromosome loss associated with the “cut” phenotype (uncoupling mitosis and cytokinesis). These phe- notypes are favored when the C terminus of Sap1 is overexpressed during DNA replication. Fluorescence in situ hybridization experiments demonstrated that the cut phenotype is related to precocious centromere separa- tion, a typical marker for loss of cohesion. We propose that Sap1 is an architectural chromatin-associated protein, required for chromosome organization. Eukaryotic chromosomes are organized into discrete do- mains or loops defining independent and functional units that are attached to a proteinaceous nuclear scaffold (27). This higher-order structural organization is not static but rather is dramatically remodeled during the cell cycle such as the DNA replication and mitotic phases. In addition, transcriptional reg- ulation and site-specific recombination are also linked to mod- ulation of the chromatin architecture during interphase (for reviews, see references 16, 24, and 38). It is generally accepted that during DNA replication, sister chromatids are aligned and held together by a tight chromo- somal connection that is conserved throughout the G 2 phase, until the metaphase/anaphase transition. The beginning of the mitotic phase is accompanied by chromosome condensation, which is essential for topoisomerase II-dependent DNA decat- enation of intertwined sister chromatids. It has been proposed that the disruption of both linkages between sister chromatids allows them to segregate to opposite poles of the cell (34). The main candidate for organizing sister chromatids to- gether is the cohesin-condensing complexes, which have also been implicated in recombination and DNA repair (36, 37) and more recently in postreplicative double-strand-break re- pair (48). Interestingly, Scc1p, known as Rad21 in Schizosac- charomyces pombe, was first identified as a DNA repair protein (13). Other players, not part of these complexes, interact with the replication machinery and have been proposed to partici- pate in the establishment of cohesion between sister chroma- tids (for a review, see reference 14). These recent findings indicate a direct connection between cohesion and the replication-repair machinery, supporting the notion that cohesion participates in maintaining genomic in- tegrity and ensuring segregation of two identical copies of the genetic material into daughter cells. The result is the produc- tion of two identical cells, the basis of clonal cell growth. To achieve this fidelity, cells have evolved surveillance mecha- nisms to coordinate repair of natural, unavoidable errors with cell cycle progression (60). Interestingly, the fission yeast S. pombe escapes from this clonal rule and exhibits recurrent asymmetric divisions. This organism profits from the intrinsic asymmetry of the DNA replication process to mark one of the two sister chromatids at the mating-type locus (mat1) by a single-strand DNA modifi- cation. Molecular and biochemical studies indicate that the DNA modification is a site- and strand-specific nick or an alkali-labile modification (2, 17). Furthermore, this stable DNA modification restricts mating-type switching to only one of the two sister chromatids during the subsequent round of DNA replication (3, 5). Mating-type switching consists of re- placing genetic information at the transcriptionally active mat1 locus with sequences copied from one of the silent donor loci, mat1P and mat3M. The accessibility of the donor is, in part, controlled at the chromatin level, and the gene conversion process requires specific and programmed folding of chromo- some II (30, 52). cis- and trans-acting elements are essential for establishing or maintaining the single-stranded DNA lesion at mat1.A mutation in the SAS1 or SAS2 (switching activating sequence 1 or 2) cis element, located 140 or 70 bp away from mat1, respectively, significantly reduces the DNA modification level (4). Four genes, swi1 , swi3 , swi7 , and sap1 , are also in- volved (4, 11, 22, 31). swi1 and swi3 encode proteins re- quired for pausing the DNA replication fork next to the mat1 locus, and they interact genetically with topoisomerase I (18, 19). swi7 encodes the DNA polymerase catalytic subunit (20, 47), required for DNA replication initiation and lagging- strand progression. Sap1 (switching activating protein 1) is a DNA-binding protein that interacts specifically with SAS1 and participates in the formation or maintenance of the single- strand DNA lesion at mat1 (4); it might be also involved in the chromosomal folding required for mating-type switching. sap1 * Corresponding author. Mailing address: Dynamique du Genome, URA 1644 du CNRS, Institut Pasteur, 25-28 rue du Dr Roux, 75724 Paris, Cedex 15, France. Phone: 33 14568 8454. Fax: 33 14568 8906. E-mail: [email protected]. 910 on June 10, 2018 by guest http://ec.asm.org/ Downloaded from
-
Upload
nguyencong -
Category
Documents
-
view
219 -
download
1
Transcript of Fission Yeast Sap1 Protein Is Essential for Chromosome ...ec.asm.org/content/2/5/910.full.pdf ·...

EUKARYOTIC CELL, Oct. 2003, p. 910–921 Vol. 2, No. 51535-9778/03/$08.00�0 DOI: 10.1128/EC.2.5.910–921.2003Copyright © 2003, American Society for Microbiology. All Rights Reserved.
Fission Yeast Sap1 Protein Is Essential for Chromosome StabilityRaynald de Lahondes,1 Veronique Ribes,2 and Benoit Arcangioli1*
Dynamique du Genome, URA 1644 du CNRS, Institut Pasteur, 75724 Paris, Cedex 15, France,1 and Institute ofInterdisciplinary Research (IRIBHN), School of Medicine, University of Brussels, B1070 Brussels, Belgium2
Received 31 March 2003/Accepted 8 July 2003
Sap1 is a dimeric sequence-specific DNA binding-protein, initially identified for its role in mating-typeswitching of the fission yeast Schizosaccharomyces pombe. The protein is relatively abundant, around 10,000dimers/cell, and is localized in the nucleus. sap1� is essential for viability, and transient overexpression isaccompanied by rapid cell death, without an apparent checkpoint response and independently of mating-typeswitching. Time lapse video microscopy of living cells revealed that the loss of viability is accompanied byabnormal mitosis and chromosome fragmentation. Overexpression of the C terminus of Sap1 inducesminichromosome loss associated with the “cut” phenotype (uncoupling mitosis and cytokinesis). These phe-notypes are favored when the C terminus of Sap1 is overexpressed during DNA replication. Fluorescence in situhybridization experiments demonstrated that the cut phenotype is related to precocious centromere separa-tion, a typical marker for loss of cohesion. We propose that Sap1 is an architectural chromatin-associatedprotein, required for chromosome organization.
Eukaryotic chromosomes are organized into discrete do-mains or loops defining independent and functional units thatare attached to a proteinaceous nuclear scaffold (27). Thishigher-order structural organization is not static but rather isdramatically remodeled during the cell cycle such as the DNAreplication and mitotic phases. In addition, transcriptional reg-ulation and site-specific recombination are also linked to mod-ulation of the chromatin architecture during interphase (forreviews, see references 16, 24, and 38).
It is generally accepted that during DNA replication, sisterchromatids are aligned and held together by a tight chromo-somal connection that is conserved throughout the G2 phase,until the metaphase/anaphase transition. The beginning of themitotic phase is accompanied by chromosome condensation,which is essential for topoisomerase II-dependent DNA decat-enation of intertwined sister chromatids. It has been proposedthat the disruption of both linkages between sister chromatidsallows them to segregate to opposite poles of the cell (34).
The main candidate for organizing sister chromatids to-gether is the cohesin-condensing complexes, which have alsobeen implicated in recombination and DNA repair (36, 37)and more recently in postreplicative double-strand-break re-pair (48). Interestingly, Scc1p, known as Rad21 in Schizosac-charomyces pombe, was first identified as a DNA repair protein(13). Other players, not part of these complexes, interact withthe replication machinery and have been proposed to partici-pate in the establishment of cohesion between sister chroma-tids (for a review, see reference 14).
These recent findings indicate a direct connection betweencohesion and the replication-repair machinery, supporting thenotion that cohesion participates in maintaining genomic in-tegrity and ensuring segregation of two identical copies of thegenetic material into daughter cells. The result is the produc-
tion of two identical cells, the basis of clonal cell growth. Toachieve this fidelity, cells have evolved surveillance mecha-nisms to coordinate repair of natural, unavoidable errors withcell cycle progression (60).
Interestingly, the fission yeast S. pombe escapes from thisclonal rule and exhibits recurrent asymmetric divisions. Thisorganism profits from the intrinsic asymmetry of the DNAreplication process to mark one of the two sister chromatids atthe mating-type locus (mat1) by a single-strand DNA modifi-cation. Molecular and biochemical studies indicate that theDNA modification is a site- and strand-specific nick or analkali-labile modification (2, 17). Furthermore, this stableDNA modification restricts mating-type switching to only oneof the two sister chromatids during the subsequent round ofDNA replication (3, 5). Mating-type switching consists of re-placing genetic information at the transcriptionally active mat1locus with sequences copied from one of the silent donor loci,mat1P and mat3M. The accessibility of the donor is, in part,controlled at the chromatin level, and the gene conversionprocess requires specific and programmed folding of chromo-some II (30, 52).
cis- and trans-acting elements are essential for establishingor maintaining the single-stranded DNA lesion at mat1. Amutation in the SAS1 or SAS2 (switching activating sequence1 or 2) cis element, located 140 or 70 bp away from mat1,respectively, significantly reduces the DNA modification level(4). Four genes, swi1�, swi3�, swi7�, and sap1�, are also in-volved (4, 11, 22, 31). swi1� and swi3� encode proteins re-quired for pausing the DNA replication fork next to the mat1locus, and they interact genetically with topoisomerase I (18,19). swi7� encodes the DNA polymerase � catalytic subunit(20, 47), required for DNA replication initiation and lagging-strand progression. Sap1 (switching activating protein 1) is aDNA-binding protein that interacts specifically with SAS1 andparticipates in the formation or maintenance of the single-strand DNA lesion at mat1 (4); it might be also involved in thechromosomal folding required for mating-type switching. sap1
* Corresponding author. Mailing address: Dynamique du Genome,URA 1644 du CNRS, Institut Pasteur, 25-28 rue du Dr Roux, 75724Paris, Cedex 15, France. Phone: 33 14568 8454. Fax: 33 14568 8906.E-mail: [email protected].
910
on June 10, 2018 by guesthttp://ec.asm
.org/D
ownloaded from

null mutations have been shown to be essential for growth,independently of mating-type switching (7).
Despite our knowledge of numerous proteins interactingwith specific DNA sequences, Sap1 constitutes a novel type ofDNA-binding protein. Two dimerization interfaces flank theDNA-binding domain. The C terminus has a predicted coiled-coil structure allowing dimerization of the protein in solution,and the N terminus allows cooperative binding of two dimerson target DNA (6, 28). The last 40 residues of the proteincontain the tetrapeptide GANM, repeated four times, whosefunction is unknown. This portion is not required for DNAbinding or for self-oligomerization. We speculate that the long(12-nm) dimerization domain of Sap1 is used to separate thetarget DNA from its C terminus (8).
To investigate the essential role of sap1, we studied thephenotype of cells lacking or overexpressing the protein. Inboth cases the yeast cells die, due to abnormal chromosomesegregation and loss of DNA during anaphase. Because ofthese complex pleiotropic phenotypes, we overexpressed theC-terminal part of Sap1 (Sap1-Cter), which is incapable ofinteracting with the DNA and with the endogenous Sap1 pro-tein. Biochemical and cellular analyses indicate that Sap1-Cterinduces a “cut” phenotype (septation in the absence of normalnuclear division) (35) and minichromosome loss. Cell cycleanalysis indicates that the cut phenotype is induced when theC-terminal domain is expressed during S phase but not when itis expressed during the beginning of M phase. Fluorescence insitu hybridization (FISH) experiments show that abnormal an-aphase is related to precocious sister chromatid separation.
MATERIALS AND METHODS
Yeast strains. The following strains were used in this study: PB1 (h90 leu1-32ura4D-18), PB10 (Msmt0 leu1-32 ade6-210) (23), the minichromosome-contain-ing strain PB456 (h�) (39), the thermosensitive cdc25 (h�) mutant strain (45),the thermosensitive cut9 mutant strain (56), and the sap1�/sap1�::LEU2 hetero-zygote strain PB69 (h90/Msmt0) (7). Cells were grown in minimal medium com-plemented with adenine or uracil, depending on the auxotrophies. Rich medium(YE medium) and YES medium were supplemented with histidine, adenine,leucine, and uracil as described previously (1). Thiamine was added to a finalconcentration of 20 �M to repress expression of the nmt1 promoter.
Cell synchronization. Cells were presynchronized by using lactose gradients(9). Drugs were added as follows: 12 mM hydroxyurea (HU) diluted in water and100 �g of thiabendazole (TBZ)/ml diluted in dimethyl sulfoxide. Cells werewashed after 2 or 3 h for TBZ or HU, respectively, and then allowed to restartgrowth in fresh medium.
Plasmids and constructions. pREP3-Sap1 was constructed by insertion of thesap1 open reading frame by using primers BA1 (AAAAATGGCCATGGAAGCTCCCAAGATGGAACT) and BA3 (AAAAAGGATCCTTAATGGTCACCAAGATTAGG) (sequences given from 5� to 3�). This PCR DNA fragment wasdigested with BalI and BamHI and inserted into pREP3 (40) at BalI/BamHI. Forconstruction of pREP3-GFP/Sap1-Cter, green fluorescent protein (GFP) wasamplified from pUC-GFPmut2 (15) by using primers GFP-XhoIF (AAAAACTCGAGCAAAGGAGAACTTTTCACTGG) and GFP-SalIR (AAAAAAGTCGACTTGTATAGTTCATCCATGCC), digested with XhoI/SalI, and inserted intopREP3 at SalI. The Sap1-Cter coding sequence was amplified by using CTER-BamHIF (TTTTTGGATCCCTCTGCTTACGCTCCCCATGGCGTTAACATGGGC) and CTER-SmaIR (AAAAACCCGGGTTAATGGTCACCAAGATTAGGAGAGATGCTAGAATGG), digested with BamHI/SmaI, and insertedinto pREP3 at BamHI/SmaI. Finally, a nuclear localization signal (NLS) segmentwas amplified by using the complementing primers NLS2-BglIIF (GATCTTCCCAAAAAGAAACGCAAGGTCGGG) and NLS2-BamHIR (GATCCCCGACCTTGCGTTTCTTTTTGGGAA), corresponding to peptide PKKKRKV, andinserted at BamHI. pREP3-HA/Sap1-Cter was constructed in the same manner,except that for the first step we inserted a fragment, corresponding to tagscomprising 6 histidines and 3 (HA) molecules, amplified from plasmid pCM262(a gift from Enrique Herrero, University of Lleida, Lleida, Spain) by using
primers TAG-XhoIF (TTTTTCTCGAGACACCATCACCATCATCACG) andTAG-SalIR (TTTTTGTCGACCCAGCGTAATCTGGAACGTCAT). ThepIRT-Sap1 plasmid was constructed by insertion into pIRT1 at SmaI of a 3-kbPvuII genomic fragment containing sap1.
Proteins and DNA analysis. Gel shift assays were performed as reportedpreviously (4). The bacterially truncated Sap1(1-203) protein was obtained asdescribed previously (8, 28). Standard procedures were used for Western andSouthern blot analyses. Plasmid topology gel analysis was performed in theabsence of ethidium bromide, by using 0.6% agarose gels.
Time lapse photography. One milliliter of cell cultures was taken, and 1 �g ofHoechst 33342/ml was added. Cells were incubated for 2 min in the dark, spunfor 1 min, resuspended in 100 �l of fresh medium, and observed microscopicallyat 20°C with a Zeiss Axiophot microscope. Under these conditions, mitosis takesabout 30 min to be completed. Photographs were taken with a Hamamatsucharge-coupled device (CCD) camera piloted by a program developed by SergeGarbay under the Khoros developing environment, with a SunOS Sun Sparcworkstation. Photographs were taken every 30 s by using 0.1-s exposure times.
PFGE. DNA was prepared in agarose plugs (2). Pulsed-field gel electrophore-sis (PFGE) was carried out as recommended by the manufacturer (Rotaphortype IV; Biometra). DNA was separated on a 0.7% agarose gel (20 mM Tris-acetate–1 mM EDTA [pH 8]) for 96 h at 13°C by using 50 V with an alternatingcurrent of 90-min intervals.
Fluorescence microscopy. Rabbit polyclonal antibodies directed against thepurified full-length Sap1 protein (residues 1 to 254), the N-terminal part (resi-dues 1 to 136), and the C-terminal part (residues 134 to 254) were prepared.Cells were fixed by using 3.6% paraformaldehyde for immunofluorescence and1.8% for FISH. Cells were then processed as previously described (32) forimmunofluorescence, with the exception that no glutaraldehyde was added.FISH was performed as previously described (12). FISH probes were made fromcenII unique cosmid sequences (26). Photographs were taken either with thesame system as that for time lapse photography or with a Hammamatsu CCDdevice piloted with the Metaview system (Universal Imaging) on a Pentium IIpersonal computer. Image treatment was carried out using the Gimp on aDebian GNU/Linux workstation.
Cell counting and FACS analysis. Cell counting and viability were determinedwith a Coulter Counter Z1. Fluorescence-activated cell sorting (FACS) analysiswas performed on a Beckman-Coulter FACS machine. For live FACS, cells weredirectly diluted in 50 mM sodium citrate and analyzed by FACScan. GFP signalswere measured logarithmically, revealing two separate cell populations: thosewith and those without GFP signals. FACS analysis was performed as previouslydescribed (1).
RESULTS
Sap1 is a stable chromatin-associated protein. We raised apolyclonal rabbit antiserum against the Sap1 protein in orderto study its cellular concentration by Western blot analysis andits localization by indirect immunofluorescence. The specificantiserum reacted strongly with a polypeptide of about 30 kDa,the size expected for the Sap1 protein (Fig. 1). Under expo-nential cell growth conditions, 2 � 107 cells contain approxi-mately 2 ng of Sap1. With a molecular size of 30 kDa, weestimate that each cell contains about 10,000 Sap1 dimers.
FIG. 1. Endogenous Sap1 levels. Quantitative Western blot analy-sis was performed with an anti-Sap1 antibody raised against the N-terminal part of the protein. In lanes 1 to 5, crude protein whole-cellextracts corresponding to 2 � 107 cells were mixed with increasingquantities of purified, truncated Sap1(1-203) protein expressed inEscherichia coli. The level of Sap1 was estimated to be 2 ng for 2 � 107
cells (lanes 3 and 4). No signals are observed with the preimmuneserum (data not shown).
VOL. 2, 2003 CHROMOSOME STABILITY 911
on June 10, 2018 by guesthttp://ec.asm
.org/D
ownloaded from

Since S. pombe spends 70% of its cell cycle in G2, this corre-sponds to 1 dimer/20 nucleosomes.
We fixed the cells with paraformaldehyde and used rabbitanti-Sap1 antibodies and 4�,6�-diamidino-2-phenylindole (DAPI)staining for immunolocalization of Sap1. Most of the fluores-cence appeared in the nucleus, with slightly more intensity atthe nuclear periphery (data not shown). No major changes inlocalization or intensity were observed during the differentstages of the cell cycle.
Mitotic sap1 loss induces abnormal mitosis. We trans-formed heterozygous sap1�/� diploid cells with a full-lengthsap1-containing plasmid, controlled by its own promoter, andinduced sporulation. sap1� germinating cells, containing a mi-totically unstable plasmid, were selected, formaldehyde fixed,and analyzed by immunofluorescence with anti-Sap1 antibod-ies. About 20% of the cells exhibited faint or absent Sap1nuclear staining, consistent with the plasmid loss rate. Twoexamples are shown in Fig. 2A, in which a reduced Sap1 signalis associated with abnormal genomic DNA staining. Quantita-tive analysis (Fig. 2B) indicated that the loss of Sap1 is accom-panied by abnormal chromosome segregation. We also noticedthat cells which were negative for Sap1 fluorescence were notelongated, indicating cell death without a significant cell cycledelay prior to mitosis. Unfortunately, the sap1� null mutationis rescued only by a plasmid expressing Sap1 from its ownpromoter. Therefore, Sap1 depletion using the thiamine-re-pressible nmt1 promoter was not possible.
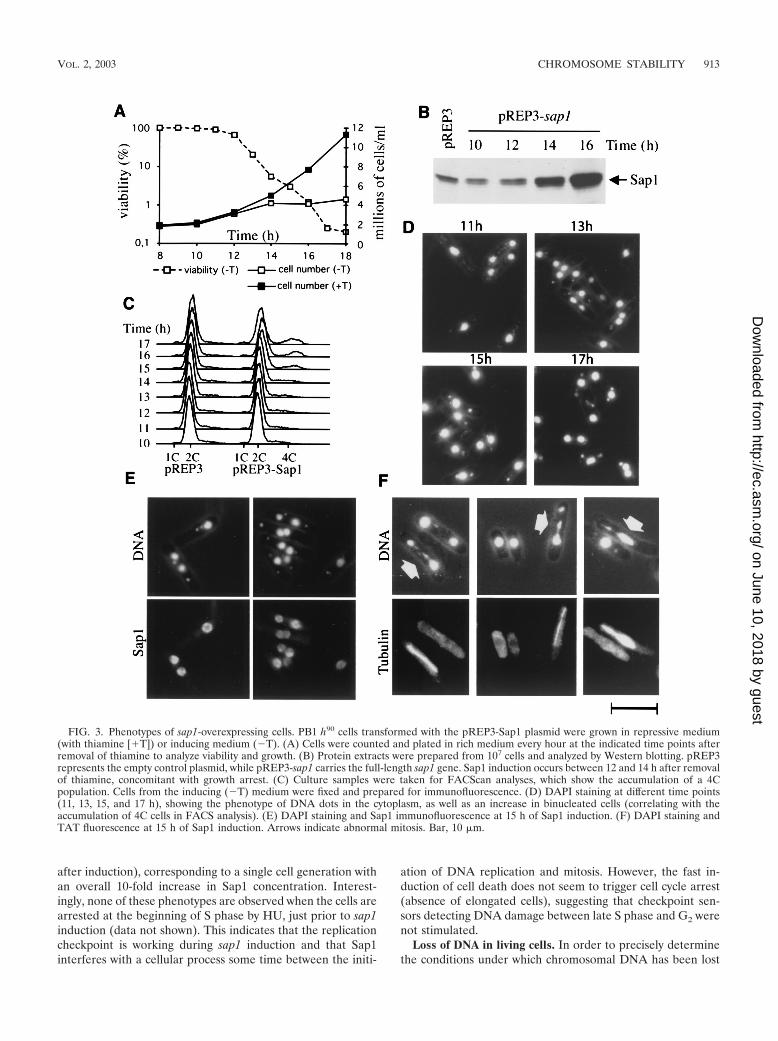
Sap1 overexpression is toxic and induces chromosome frag-mentation. Since lack of Sap1 causes abnormal mitosis, wedecided to examine the effect of overproduction of Sap1. Weused the thiamine-repressible nmt1 promoter carried by plas-mid pREP3 (or the attenuated plasmid pREP81 [data notshown]) to overexpress Sap1 at a high or moderate level (10,25, 40) (see Materials and Methods for constructions). Trans-formed cells (wild type) were grown in thiamine-containingmedium to repress ectopic sap1 overexpression and allow fur-ther investigation. Figure 3A shows that cells ceased dividing
about 14 h after thiamine withdrawal and rapidly lost viability.To determine the timing and the level of expression, whole-cellextracts were prepared and analyzed by immunoblotting withthe antibody against Sap1 (Fig. 3B) and by gel retardationassays using radiolabeled SAS1 as a DNA-binding target (datanot shown). Sap1 products started to accumulate between 12and 14 h after thiamine withdrawal. From these results, weconclude that an increase in the intracellular Sap1 concentra-tion causes division arrest and cell death within one genera-tion.
We next analyzed the phenotype of the dying cells by mi-croscopy. Cell aliquots were taken at different times duringsap1 induction, fixed with paraformaldehyde, and stained withthe DNA-binding dye DAPI. We observed an increased pro-portion (�50%) of cells containing DNA spots in the cyto-plasm (Fig. 3D). The number of spots per cell was consistentlybetween 1 and 2, and their intensity appeared relatively con-stant. We also observed binucleated cells, which accumulate toabout 20% of the dead cell population at 15 to 16 h postin-duction. These phenotypes are observed in wild-type and un-switchable mutant cells and thus are not related to mating-typeswitching. Flow cytometry (FACScan) analysis (Fig. 3C)showed a slight increase in DNA content from 2 to 4 cellequivalents (2C to 4C) for about 20% of the cell population,consistent with DNA replication of the binucleated cells andarrest at the end of S phase or in G2. Immunofluorescenceanalysis showed a redistribution of Sap1 along the chromo-somes, resulting in nonuniform staining of nuclei by DAPI andan absence of DNA dots (Fig. 3E). By using the TAT1 anti-body directed against microtubules (55), we observed abnor-mal mitosis, as shown by asymmetric segregation of the geneticmaterial (Fig. 3F). Time lapse video microscopy of living cellsconfirmed that more than half of the mitotic cells clearly ex-hibit a nonequal distribution of their chromosomes (data notshown). The kinetics of appearance of the different phenotypesare complex and indicate pleiotropic effects. All of the pheno-types described above appear within 3 h (between 12 and 15 h
FIG. 2. Plasmid loss experiment. Haploid sap1� spores complemented by plasmid pIRT-Sap1 were selected and allowed to grow withoutplasmid selection. Cells that lost the plasmid died. (A) (Left) Two cells (arrows) display fragmented (upper arrow) and decondensed (lower arrow)DNA (DAPI staining). (Right) The same cells (arrows) exhibit no Sap1 staining. Bar, 10 �m. (B) The proportion of cells with no detectable levelof Sap1 is 15%. Among the sap1� cells (n � 56), we observed the following phenotypes: no apparent phenotype (10%), faint DAPI staining (20%),and chromosome fragmentation (70%).
912 DE LAHONDES ET AL. EUKARYOT. CELL
on June 10, 2018 by guesthttp://ec.asm
.org/D
ownloaded from
after induction), corresponding to a single cell generation withan overall 10-fold increase in Sap1 concentration. Interest-ingly, none of these phenotypes are observed when the cells arearrested at the beginning of S phase by HU, just prior to sap1induction (data not shown). This indicates that the replicationcheckpoint is working during sap1 induction and that Sap1interferes with a cellular process some time between the initi-
ation of DNA replication and mitosis. However, the fast in-duction of cell death does not seem to trigger cell cycle arrest(absence of elongated cells), suggesting that checkpoint sen-sors detecting DNA damage between late S phase and G2 werenot stimulated.
Loss of DNA in living cells. In order to precisely determinethe conditions under which chromosomal DNA has been lost
FIG. 3. Phenotypes of sap1-overexpressing cells. PB1 h90 cells transformed with the pREP3-Sap1 plasmid were grown in repressive medium(with thiamine [�T]) or inducing medium (�T). (A) Cells were counted and plated in rich medium every hour at the indicated time points afterremoval of thiamine to analyze viability and growth. (B) Protein extracts were prepared from 107 cells and analyzed by Western blotting. pREP3represents the empty control plasmid, while pREP3-sap1 carries the full-length sap1 gene. Sap1 induction occurs between 12 and 14 h after removalof thiamine, concomitant with growth arrest. (C) Culture samples were taken for FACScan analyses, which show the accumulation of a 4Cpopulation. Cells from the inducing (�T) medium were fixed and prepared for immunofluorescence. (D) DAPI staining at different time points(11, 13, 15, and 17 h), showing the phenotype of DNA dots in the cytoplasm, as well as an increase in binucleated cells (correlating with theaccumulation of 4C cells in FACS analysis). (E) DAPI staining and Sap1 immunofluorescence at 15 h of Sap1 induction. (F) DAPI staining andTAT fluorescence at 15 h of Sap1 induction. Arrows indicate abnormal mitosis. Bar, 10 �m.
VOL. 2, 2003 CHROMOSOME STABILITY 913
on June 10, 2018 by guesthttp://ec.asm
.org/D
ownloaded from

upon overexpression of Sap1, we used time lapse video micros-copy analysis, in which the cellular DNA is stained withHoechst 33342 (see Materials and Methods). To increase theproportion of sap1-overexpressing cells going through mitosis,the thermosensitive cdc25 mutant strain was transformed withthe plasmid overexpressing sap1. Figure 4A depicts a cell un-dergoing unequal partitioning of the two chromosome massesat the beginning of observation (t � 0 min); 3 min later, thelarger nucleus releases a broken piece of a chromosome arm,and from this point the separation of the nuclei appears to bearrested. However, the chromosome arms lagging behind arestill pulled back to the main chromosome mass. A similarscenario is observed in the cell shown in Fig. 4B. For this cell,the aneuploidy is not obvious and the nuclear separation seemscorrect for the first 12 min, with the exception of the two
lagging chromosome arms. At 14 min, a chromosome fragmentis released from the right nucleus, followed by an arrest ofanaphase progression. The observations from video micros-copy of living cells allowed us to visualize chromosomal DNAloss in real time and indicated that at least some of the DNAdots observed during sap1 overexpression appeared during an-aphase and were due to loss of chromosome pieces. Further-more, the correlation between loss of DNA during anaphase Band arrest of mitotic segregation indicates that chromosomeloss probably triggers a mitotic checkpoint apparatus. It hasrecently been proposed that another checkpoint control mightoperate during chromosome loss in anaphase (43, 57). How-ever, the situation described here is slightly different, since onlya portion of the chromosome is lost, probably due to chromatidbreakage. Furthermore, the lost piece of chromatid, probablydevoid of a centromere, cannot be reattached to the kineto-chore, leading to cell death.
Importantly, the presence of DNA dots is not related tomating-type switching, since very similar phenotypes are ob-served in switching and non-switching-proficient strains (datanot shown). This conclusion was confirmed by FISH using themat region as a probe (data not shown).
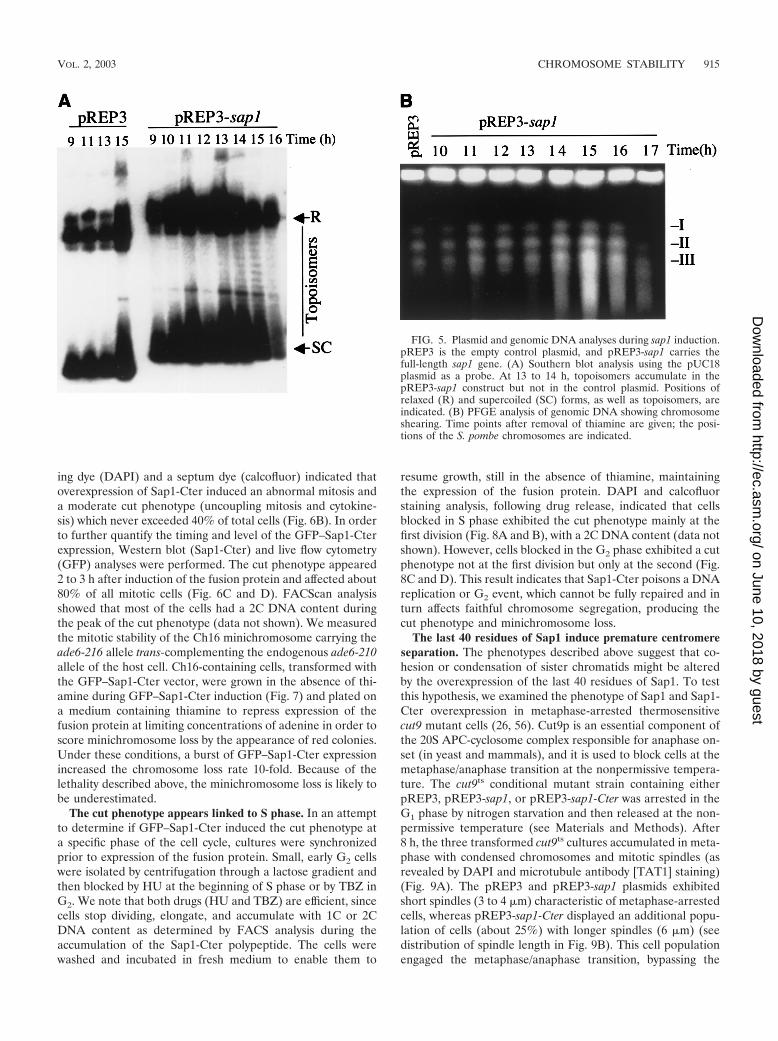
Sap1 overexpression affects plasmid supercoiling and in-duces chromosome breakage. We took advantage of the sap1overexpression plasmid to analyze its topology by Southernblot analysis during sap1 induction. We compared the plasmidtopology of pREP3 with or without sap1 after thiamine re-moval (Fig. 5). For both plasmids, two major migrating DNAbands were observed, corresponding to supercoiled and re-laxed, or dimer, plasmid forms. As expected, transcriptionalinduction did not change the migration pattern of pREP3 (Fig.5A, first four lanes). In contrast, when pREP3 drove sap1expression, new bands were observed between the supercoiledand relaxed forms during Sap1 induction, indicative of topo-logical plasmid change. The effects were observed after 13 h ofthiamine withdrawal and were concomitant with the increasein intracellular Sap1 levels. Since sap1 codes for a DNA-bind-ing protein, it is tempting to suggest that the Sap1 protein isdirectly involved in this plasmid DNA relaxation. This molec-ular effect may explain, at least partially, the general pheno-types observed in the course of this study. We also noticed thatbefore or after sap1 induction, the circular plasmid did notshow catenated forms, suggesting no major defects of the en-dogenous topoisomerase II activity.
To confirm the chromosome breakage in sap1-overexpress-ing cells at the molecular level, we used PFGE. Figure 5Bshows results for the three S. pombe chromosomes during Sap1overexpression. Following sap1 induction (t � 15 h), chromo-somal shearing was observed, consistent with the cellular phe-notypes previously described.
The last 40 residues of Sap1 induce abnormal anaphase andchromosome instability. Because of the complex phenotypesdescribed above and the oligomerization properties of Sap1,we decided to examine the phenotype induced by overexpres-sion of Sap1-Cter. The last 40 residues were fused to GFP andto an NLS motif to restrict the localization of the protein to thenucleus. During expression of GFP–Sap1-Cter, the cellsstopped dividing and partially lost viability (Fig. 6A). The samephenotype has been observed with an HA-tagged Sap1-Cterfusion protein (data not shown). Staining with the DNA-bind-
FIG. 4. Chromosomal shearing. The thermosensitive cdc25 mutantstrain, transformed with the pREP3-Sap1 plasmid and induced (with athiamine-deficient medium) at the permissive temperature, was ar-rested for 4 h at the nonpermissive temperature (36°C) and thentransferred at the permissive temperature (26°C) for observation ofchromosome segregation. Two cells (A and B) appeared in the samecamera field during time-lapsed photography. Time is indicated inminutes, and 0 min corresponds to the beginning of the observation.Bar, 10 �m.
914 DE LAHONDES ET AL. EUKARYOT. CELL
on June 10, 2018 by guesthttp://ec.asm
.org/D
ownloaded from
ing dye (DAPI) and a septum dye (calcofluor) indicated thatoverexpression of Sap1-Cter induced an abnormal mitosis anda moderate cut phenotype (uncoupling mitosis and cytokine-sis) which never exceeded 40% of total cells (Fig. 6B). In orderto further quantify the timing and level of the GFP–Sap1-Cterexpression, Western blot (Sap1-Cter) and live flow cytometry(GFP) analyses were performed. The cut phenotype appeared2 to 3 h after induction of the fusion protein and affected about80% of all mitotic cells (Fig. 6C and D). FACScan analysisshowed that most of the cells had a 2C DNA content duringthe peak of the cut phenotype (data not shown). We measuredthe mitotic stability of the Ch16 minichromosome carrying theade6-216 allele trans-complementing the endogenous ade6-210allele of the host cell. Ch16-containing cells, transformed withthe GFP–Sap1-Cter vector, were grown in the absence of thi-amine during GFP–Sap1-Cter induction (Fig. 7) and plated ona medium containing thiamine to repress expression of thefusion protein at limiting concentrations of adenine in order toscore minichromosome loss by the appearance of red colonies.Under these conditions, a burst of GFP–Sap1-Cter expressionincreased the chromosome loss rate 10-fold. Because of thelethality described above, the minichromosome loss is likely tobe underestimated.
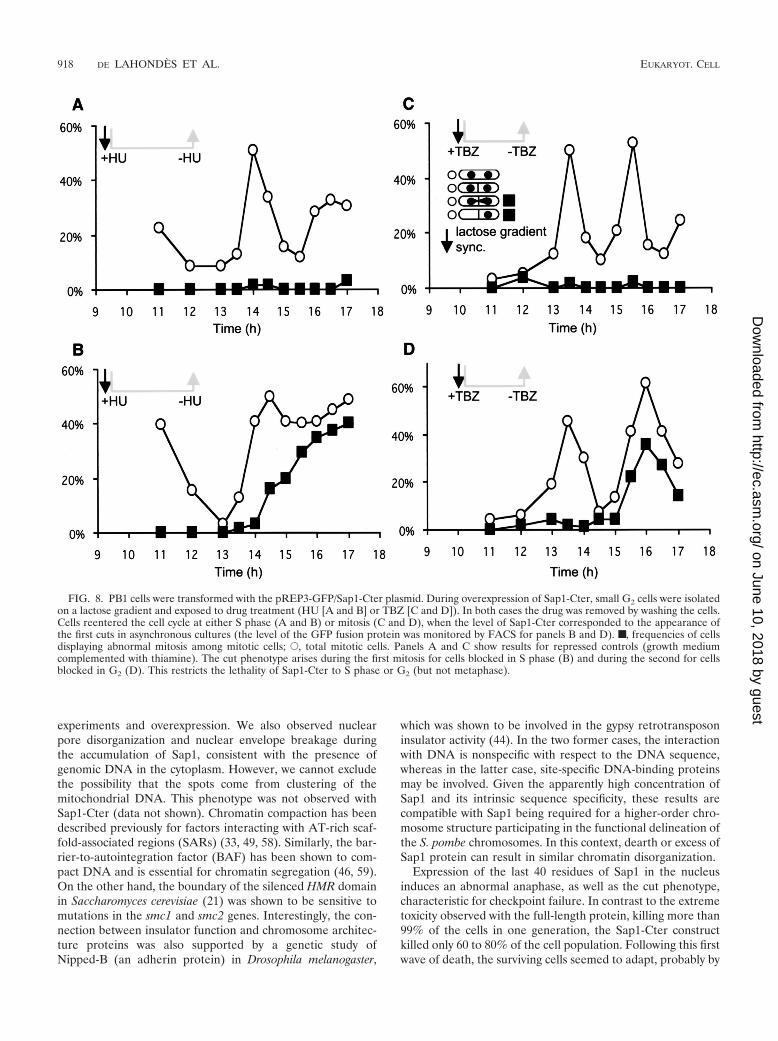
The cut phenotype appears linked to S phase. In an attemptto determine if GFP–Sap1-Cter induced the cut phenotype ata specific phase of the cell cycle, cultures were synchronizedprior to expression of the fusion protein. Small, early G2 cellswere isolated by centrifugation through a lactose gradient andthen blocked by HU at the beginning of S phase or by TBZ inG2. We note that both drugs (HU and TBZ) are efficient, sincecells stop dividing, elongate, and accumulate with 1C or 2CDNA content as determined by FACS analysis during theaccumulation of the Sap1-Cter polypeptide. The cells werewashed and incubated in fresh medium to enable them to
resume growth, still in the absence of thiamine, maintainingthe expression of the fusion protein. DAPI and calcofluorstaining analysis, following drug release, indicated that cellsblocked in S phase exhibited the cut phenotype mainly at thefirst division (Fig. 8A and B), with a 2C DNA content (data notshown). However, cells blocked in the G2 phase exhibited a cutphenotype not at the first division but only at the second (Fig.8C and D). This result indicates that Sap1-Cter poisons a DNAreplication or G2 event, which cannot be fully repaired and inturn affects faithful chromosome segregation, producing thecut phenotype and minichromosome loss.
The last 40 residues of Sap1 induce premature centromereseparation. The phenotypes described above suggest that co-hesion or condensation of sister chromatids might be alteredby the overexpression of the last 40 residues of Sap1. To testthis hypothesis, we examined the phenotype of Sap1 and Sap1-Cter overexpression in metaphase-arrested thermosensitivecut9 mutant cells (26, 56). Cut9p is an essential component ofthe 20S APC-cyclosome complex responsible for anaphase on-set (in yeast and mammals), and it is used to block cells at themetaphase/anaphase transition at the nonpermissive tempera-ture. The cut9ts conditional mutant strain containing eitherpREP3, pREP3-sap1, or pREP3-sap1-Cter was arrested in theG1 phase by nitrogen starvation and then released at the non-permissive temperature (see Materials and Methods). After8 h, the three transformed cut9ts cultures accumulated in meta-phase with condensed chromosomes and mitotic spindles (asrevealed by DAPI and microtubule antibody [TAT1] staining)(Fig. 9A). The pREP3 and pREP3-sap1 plasmids exhibitedshort spindles (3 to 4 �m) characteristic of metaphase-arrestedcells, whereas pREP3-sap1-Cter displayed an additional popu-lation of cells (about 25%) with longer spindles (6 �m) (seedistribution of spindle length in Fig. 9B). This cell populationengaged the metaphase/anaphase transition, bypassing the
FIG. 5. Plasmid and genomic DNA analyses during sap1 induction.pREP3 is the empty control plasmid, and pREP3-sap1 carries thefull-length sap1 gene. (A) Southern blot analysis using the pUC18plasmid as a probe. At 13 to 14 h, topoisomers accumulate in thepREP3-sap1 construct but not in the control plasmid. Positions ofrelaxed (R) and supercoiled (SC) forms, as well as topoisomers, areindicated. (B) PFGE analysis of genomic DNA showing chromosomeshearing. Time points after removal of thiamine are given; the posi-tions of the S. pombe chromosomes are indicated.
VOL. 2, 2003 CHROMOSOME STABILITY 915
on June 10, 2018 by guesthttp://ec.asm
.org/D
ownloaded from

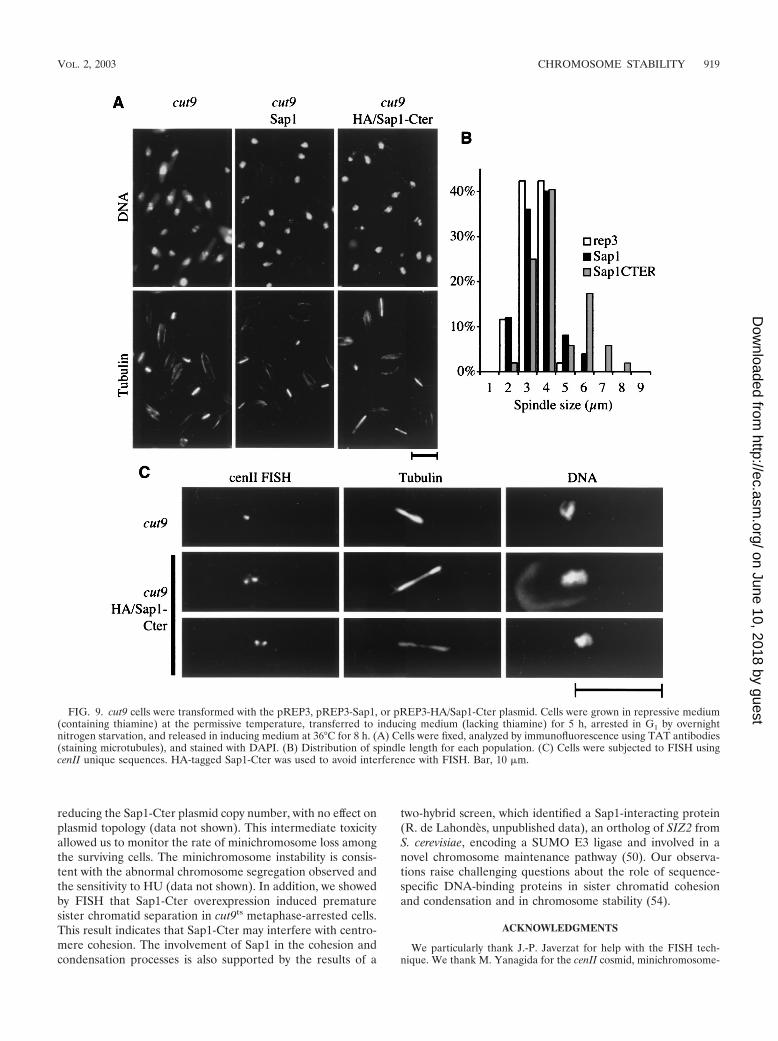
cut9ts cell cycle arrest phenotype, but retained a second blockduring a later stage in anaphase (phase 3 [41]), possibly due toactivation of the spindle checkpoint. A similar phenotype hasbeen described for the Mis4/Scc2p chromatid cohesion mole-cule, part of the adherin family (26, 29).
These observations raise the question of whether sister chro-matids are prematurely separated in the presence of Sap1-Cter. To address this question, we used the FISH methodology(53) with a probe derived from the centromere II (cenII)unique sequence. Figure 9C shows an example of cut9ts ar-rested cells transformed with pREP3, with a typical single cenIIsignal, whereas two examples of cut9ts arrested cells trans-formed with pREP3-sap1-Cter exhibit two cenII signals. Theseparation of the two sister centromeres was observed in mostof the elongated-spindle-containing cells and rarely in cellswith typical metaphase spindle lengths. The loss of cohesion atthe centromeres triggers the metaphase/anaphase transition,allowing sister chromatids to be pulled apart by the opposingforces exerted by the mitotic microtubules. This demonstratespremature loss of centromere cohesion, even though the spin-dle force might be responsible for the separation.
DISCUSSION
Our study introduces the sequence-specific DNA-bindingprotein Sap1 as a novel player in the chromosome dynamics offission yeast. Sap1 is a relatively abundant chromatin-associ-ated protein that is essential for viability and participates inmating-type switching. Its localization and concentration ap-pear stable throughout the cell cycle. The connection betweenmating-type switching and chromosome organization is stilltenuous. However, we hypothesize that mating-type switchingevolved by recruiting existing cellular components used inchromosome morphogenesis. Moreover, insights into themechanism of one process may shed light on the mechanism ofthe other.
Implication of Sap1 in chromosome organization. Severallines of evidence indicate that a particular Sap1 protein con-centration is essential for the maintenance of chromosomalintegrity and obeys a threshold-sensing process. First, germi-nating sap1� spores generated from a sap1�/� heterozygotediploid strain cannot divide, indicating that Sap1 must be newlysynthesized from the zygote genome early during germination.Second, plasmid loss experiments indicate that a decrease inthe Sap1 concentration is associated with abnormal chromo-some segregation. Third, Sap1 overexpression affects plasmidsuperhelicity, anaphase, and chromosomal integrity. The ab-sence of phenotypes in HU-arrested cells during overexpres-sion of full-length Sap1 indicates that the S-phase checkpoint is
FIG. 6. Phenotypes of GFP–Sap1-Cter-overexpressing cells. PB1h90 cells transformed with the pREP3-GFP/Sap1-Cter plasmid (seeMaterials and Methods) were grown in repressive (�T) or inducing(�T) medium. (A) Cells were counted and plated in rich mediumevery hour at the indicated time points after the removal of thiamineto analyze viability and growth. (B) The associated phenotype corre-sponds to a cut phenotype at 15 h of Sap1-Cter induction. GenomicDNA and the septum were stained with DAPI and calcofluor, respec-tively. Bar, 10 �m. (C) Protein extracts were prepared and analyzed byWestern blotting. The antibody recognizes the C-terminal domain ofendogenous Sap1 (30 kDa) and the ectopically expressed GFP–Sap1-Cter (35 kDa), as indicated. (D) GFP quantification and proportion ofcells with abnormal anaphase. At 12 h of induction, 80% of cellsshowed a GFP signal, 3 h before the abnormal anaphase peak. Thelevel of GFP was estimated by FACScan on living cells (see Materialsand Methods).
916 DE LAHONDES ET AL. EUKARYOT. CELL
on June 10, 2018 by guesthttp://ec.asm
.org/D
ownloaded from

operational and suggests that cell cycle progression is requiredto reveal the cellular Sap1-induced cell death phenotype.Fourth, the sap1�/� heterozygote diploid has a slow growthphenotype due to an apparently extended S phase, is hyper-sensitive to DNA-damaging agents, and exhibits a higher rateof chromosome loss than the parental diploid (V. Ribes, B.Arcangioli, and B. Xhemalce, unpublished data). Finally, onlythe construction containing the endogenous sap1 promoterwas able to complement the sap1� strain, while other versions,using the repressible nmt1 promoters to drive sap1 expression,failed to complement the null mutant strain. A gene dosagephenomenon has also been reported for the cohesin factorPds5p, in which loss of function and overexpression producedthe same effect with regard to the establishment of cohesion(51). The apparent sensitivity to the Sap1 dosage is consistentwith the cooperative DNA-binding property of the protein(28). Biochemical and structural studies have shown that Sap1oligomerizes and forms an unusual elongated shape in solu-tion. This rigid structure maintains the C-terminal part of the
protein 12 nm from the DNA (8) and may play an importantrole in Sap1 activity. Although this is speculative, we proposethat excess Sap1 protein will cooperatively bind to ectopicweak DNA-binding sites along the chromosomes, interferingwith normal chromosomal organization. We propose that Sap1has a structural rather than an enzymatic function for organiz-ing the nuclear chromatin.
The effects of Sap1 on plasmid superhelicity and genomicDNA are the early events observed following induction of thefull-length protein (Fig. 3E and 5A); interestingly, these phe-notypes were not observed during overexpression of Sap1-Cter, which instead induced the cut phenotype. This observa-tion indicates that the effect on plasmid topology induced bysap1 is not due to its transcription, as was expected (42). It isconceivable that an excess of Sap1 induces a signal to remodelthe chromatin, which in turn excludes Sap1 from its cognateDNA-binding sequences. The exclusion of Sap1 might be theinitial event triggering abnormal anaphase. This interpretationis consistent with similarities observed during the plasmid loss
FIG. 7. The minichromosome strain (PB456) was transformed by the pREP3 plasmid or the pREP3-GFP/Sap1-Cter plasmid. Cells were grownin minimal medium with thiamine up to time zero; then they were washed and grown in the absence of thiamine for the following hours. Cells wereplated on YE medium, and the appearance of red colonies was measured. (A) Photograph of plates at �22 h. (Left) Plasmid pREP3. (Right)pREP3-GFP/Sap1-Cter. (B) Incidence of minichromosome loss and viability during induction.
VOL. 2, 2003 CHROMOSOME STABILITY 917
on June 10, 2018 by guesthttp://ec.asm
.org/D
ownloaded from
experiments and overexpression. We also observed nuclearpore disorganization and nuclear envelope breakage duringthe accumulation of Sap1, consistent with the presence ofgenomic DNA in the cytoplasm. However, we cannot excludethe possibility that the spots come from clustering of themitochondrial DNA. This phenotype was not observed withSap1-Cter (data not shown). Chromatin compaction has beendescribed previously for factors interacting with AT-rich scaf-fold-associated regions (SARs) (33, 49, 58). Similarly, the bar-rier-to-autointegration factor (BAF) has been shown to com-pact DNA and is essential for chromatin segregation (46, 59).On the other hand, the boundary of the silenced HMR domainin Saccharomyces cerevisiae (21) was shown to be sensitive tomutations in the smc1 and smc2 genes. Interestingly, the con-nection between insulator function and chromosome architec-ture proteins was also supported by a genetic study ofNipped-B (an adherin protein) in Drosophila melanogaster,
which was shown to be involved in the gypsy retrotransposoninsulator activity (44). In the two former cases, the interactionwith DNA is nonspecific with respect to the DNA sequence,whereas in the latter case, site-specific DNA-binding proteinsmay be involved. Given the apparently high concentration ofSap1 and its intrinsic sequence specificity, these results arecompatible with Sap1 being required for a higher-order chro-mosome structure participating in the functional delineation ofthe S. pombe chromosomes. In this context, dearth or excess ofSap1 protein can result in similar chromatin disorganization.
Expression of the last 40 residues of Sap1 in the nucleusinduces an abnormal anaphase, as well as the cut phenotype,characteristic for checkpoint failure. In contrast to the extremetoxicity observed with the full-length protein, killing more than99% of the cells in one generation, the Sap1-Cter constructkilled only 60 to 80% of the cell population. Following this firstwave of death, the surviving cells seemed to adapt, probably by
FIG. 8. PB1 cells were transformed with the pREP3-GFP/Sap1-Cter plasmid. During overexpression of Sap1-Cter, small G2 cells were isolatedon a lactose gradient and exposed to drug treatment (HU [A and B] or TBZ [C and D]). In both cases the drug was removed by washing the cells.Cells reentered the cell cycle at either S phase (A and B) or mitosis (C and D), when the level of Sap1-Cter corresponded to the appearance ofthe first cuts in asynchronous cultures (the level of the GFP fusion protein was monitored by FACS for panels B and D). ■, frequencies of cellsdisplaying abnormal mitosis among mitotic cells; E, total mitotic cells. Panels A and C show results for repressed controls (growth mediumcomplemented with thiamine). The cut phenotype arises during the first mitosis for cells blocked in S phase (B) and during the second for cellsblocked in G2 (D). This restricts the lethality of Sap1-Cter to S phase or G2 (but not metaphase).
918 DE LAHONDES ET AL. EUKARYOT. CELL
on June 10, 2018 by guesthttp://ec.asm
.org/D
ownloaded from
reducing the Sap1-Cter plasmid copy number, with no effect onplasmid topology (data not shown). This intermediate toxicityallowed us to monitor the rate of minichromosome loss amongthe surviving cells. The minichromosome instability is consis-tent with the abnormal chromosome segregation observed andthe sensitivity to HU (data not shown). In addition, we showedby FISH that Sap1-Cter overexpression induced prematuresister chromatid separation in cut9ts metaphase-arrested cells.This result indicates that Sap1-Cter may interfere with centro-mere cohesion. The involvement of Sap1 in the cohesion andcondensation processes is also supported by the results of a
two-hybrid screen, which identified a Sap1-interacting protein(R. de Lahondes, unpublished data), an ortholog of SIZ2 fromS. cerevisiae, encoding a SUMO E3 ligase and involved in anovel chromosome maintenance pathway (50). Our observa-tions raise challenging questions about the role of sequence-specific DNA-binding proteins in sister chromatid cohesionand condensation and in chromosome stability (54).
ACKNOWLEDGMENTS
We particularly thank J.-P. Javerzat for help with the FISH tech-nique. We thank M. Yanagida for the cenII cosmid, minichromosome-
FIG. 9. cut9 cells were transformed with the pREP3, pREP3-Sap1, or pREP3-HA/Sap1-Cter plasmid. Cells were grown in repressive medium(containing thiamine) at the permissive temperature, transferred to inducing medium (lacking thiamine) for 5 h, arrested in G1 by overnightnitrogen starvation, and released in inducing medium at 36°C for 8 h. (A) Cells were fixed, analyzed by immunofluorescence using TAT antibodies(staining microtubules), and stained with DAPI. (B) Distribution of spindle length for each population. (C) Cells were subjected to FISH usingcenII unique sequences. HA-tagged Sap1-Cter was used to avoid interference with FISH. Bar, 10 �m.
VOL. 2, 2003 CHROMOSOME STABILITY 919
on June 10, 2018 by guesthttp://ec.asm
.org/D
ownloaded from

containing strains, and the cut9 thermosensitive strain; S. Garbay forsoftware development, help with time lapse photography, and generalcontributions to informatics; V. Doye and E. Herrero for providingplasmids; and A. Holmes and J. Weitzman for critical reading of themanuscript.
This work was supported in part by the Association pour la Recher-che sur le Cancer and the Human Frontiers Science Program (to B.A.).R.D.L. was supported by fellowships from La Ligue Contre le Cancer.
REFERENCES
1. Alfa, C., P. Fantes, J. Hyams, J. McLeod, and E. Warbrick. 1993. Experi-ments with fission yeast. Cold Spring Harbor Laboratory Press, Cold SpringHarbor, N.Y.
2. Arcangioli, B. 1998. A site- and strand-specific DNA break confers asym-metric switching potential in fission yeast. EMBO J. 17:4503–4510.
3. Arcangioli, B. 2000. Fate of mat1 DNA strands during mating-type switchingin fission yeast. EMBO Rep. 1:145–150.
4. Arcangioli, B., and A. J. Klar. 1991. A novel switch-activating site (SAS1)and its cognate binding factor (SAP1) required for efficient mat1 switching inSchizosaccharomyces pombe. EMBO J. 10:3025–3032.
5. Arcangioli, B., and R. de Lahondes. 2000. Fission yeast switches mating typeby a replication-recombination coupled process. EMBO J. 19:1389–1396.
6. Arcangioli, B., M. Ghazvini, and V. Ribes. 1994. Identification of the DNA-binding domains of the switch-activating-protein Sap1 from S. pombe byrandom point mutations screening in E. coli. Nucleic Acids Res. 22:2930–2937.
7. Arcangioli, B., T. D. Copeland, and A. J. Klar. 1994. Sap1, a protein thatbinds to sequences required for mating-type switching, is essential for via-bility in Schizosaccharomyces pombe. Mol. Cell. Biol. 14:2058–2065.
8. Bada, M., D. Walther, B. Arcangioli, S. Doniach, and M. Delarue. 2000.Solution structural studies and low-resolution model of the Schizosaccharo-myces pombe sap1 protein. J. Mol. Biol. 300:563–574.
9. Barbet, N. C., and A. M. Carr. 1993. Fission yeast wee1 protein kinase is notrequired for DNA damage-dependent mitotic arrest. Nature 364:824–827.
10. Basi, G., E. Schmid, and K. Maundrell. 1993. TATA box mutations in theSchizosaccharomyces pombe nmt1 promoter affect transcription efficiency butnot the transcription start point or thiamine repressibility. Gene 123:131–136.
11. Beach, D. H. 1983. Cell type switching by DNA transposition in fission yeast.Nature 305:682–688.
12. Bernard, P., K. Hardwick, and J. P. Javerzat. 1998. Fission yeast bub1 is amitotic centromere protein essential for the spindle checkpoint and thepreservation of correct ploidy through mitosis. J. Cell Biol. 143:1775–1787.
13. Birkenbihl, R. P., and S. Subramani. 1992. Cloning and characterization ofrad21, an essential gene of Schizosaccharomyces pombe involved in DNAdouble-strand-break repair. Nucleic Acids Res. 20:6605–6611.
14. Carson, D. R., and M. F. Christman. 2001. Evidence that replication forkcomponents catalyze establishment of cohesion between sister chromatids.Proc. Natl. Acad. Sci. USA 98:8270–8275.
15. Cormack, B. P., R. H. Valdivia, and S. Falkow. 1996. FACS-optimizedmutants of the green fluorescent protein (GFP). Gene 173:33–38.
16. Cremer, T., and C. Cremer. 2001. Chromosome territories, nuclear archi-tecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2:292–301.
17. Dalgaard, J. Z., and A. J. Klar. 1999. Orientation of DNA replicationestablishes mating-type switching pattern in S. pombe. Nature 400:181–184.
18. Dalgaard, J. Z., and A. J. Klar. 2000. swi1 and swi3 perform imprinting,pausing, and termination of DNA replication in S. pombe. Cell 102:745–751.
19. Dalgaard, J. Z., and A. J. Klar. 2001. Does S. pombe exploit the intrinsicasymmetry of DNA synthesis to imprint daughter cells for mating-typeswitching? Trends Genet. 17:153–157.
20. Damagnez, V., J. Tillit, A. M. de Recondo, and G. Baldacci. 1991. The POL1gene from the fission yeast, Schizosaccharomyces pombe, shows conservedamino acid blocks specific for eukaryotic DNA polymerases alpha. Mol. Gen.Genet. 226:182–189.
21. Donze, D., C. R. Adams, J. Rine, and R. T. Kamakaka. 1999. The boundariesof the silenced HMR domain in Saccharomyces cerevisiae. Genes Dev. 13:698–708.
22. Egel, R., D. H. Beach, and A. J. Klar. 1984. Genes required for initiation andresolution steps of mating-type switching in fission yeast. Proc. Natl. Acad.Sci. USA 81:3481–3485.
23. Engelke, U., L. Grabowski, H. Gutz, L. Heim, and H. Schmidt. 1987. Mo-lecular characterization of h� mutants of Schizosaccharomyces pombe. Curr.Genet. 18:535–540.
24. Felsenfeld, G. 1996. Chromatin unfolds. Cell 86:13–19.25. Forsburg, S. L. 1993. Comparison of Schizosaccharomyces pombe expression
systems. Nucleic Acids Res. 21:2955–2956.26. Furuya, K., K. Takahashi, and M. Yanagida. 1998. Faithful anaphase is
ensured by Mis4, a sister chromatid cohesion molecule required in S phaseand not destroyed in G1 phase. Genes Dev. 12:3408–3418.
27. Gasser, S. M., and U. K. Laemmli. 1987. A glimpse at chromosomal order.Trends Genet. 3:16–22.
28. Ghazvini, M., V. Ribes, and B. Arcangioli. 1995. The essential DNA-bindingprotein Sap1 of Schizosaccharomyces pombe contains two independent oli-gomerization interfaces that dictate the relative orientation of the DNA-binding domain. Mol. Cell. Biol. 15:4939–4946.
29. Goshima, G., S. Saitoh, and M. Yanagida. 1999. Proper metaphase spindlelength is determined by centromere proteins Mis12 and Mis6 required forfaithful chromosome segregation. Genes Dev. 13:1664–1677.
30. Grewal, S. I., and A. J. Klar. 1997. A recombinationally repressed regionbetween mat2 and mat3 loci shares homology to centromeric repeats andregulates directionality of mating-type switching in fission yeast. Genetics146:1221–1238.
31. Gutz, H., and H. Schmidt. 1985. Switching genes in Schizosaccharomycespombe. Curr. Genet. 9:325–331.
32. Hagan, I. M., and J. S. Hyams. 1988. The use of cell division cycle mutantsto investigate the control of microtubule distribution in the fission yeastSchizosaccharomyces pombe. J. Cell Sci. 89:343–357.
33. Henikoff, S., and D. Vermaak. 2000. Bugs on drugs go GAGAA. Cell 103:695–698.
34. Hirano, T. 2000. Chromosome cohesion, condensation, and separation.Annu. Rev. Biochem. 69:115–144.
35. Hirano, T., S. C. Funahashi, T. Uemura, and M. Yanagida. 1986. Isolationand characterization of Schizosaccharomyces pombe cut mutants that blocknuclear division but not cytokinesis. EMBO J. 5:2973–2980.
36. Jessberger, R., B. Riwar, H. Baechtold, and A. T. Akhmedov. 1996. SMCproteins constitute two subunits of the mammalian recombination complexRC-1. EMBO J. 15:4061–4068.
37. Lehmann, A. R., M. Walicka, D. J. Griffiths, J. M. Murray, F. Z. Watts, S.McCready, and A. M. Carr. 1995. The rad18 gene of Schizosaccharomycespombe defines a new subgroup of the SMC superfamily involved in DNArepair. Mol. Cell. Biol. 15:7067–7080.
38. Marshall, W. F., A. Straight, J. F. Marko, J. Swedlow, A. Dernburg, A.Belmont, A. W. Murray, D. A. Agard, and J. W. Sedat. 1997. Interphasechromosomes undergo constrained diffusional motion in living cells. Curr.Biol. 7:930–939.
39. Matsumoto, T., K. Fukui, O. Niwa, N. Sugawara, J. W. Szostak, and M.Yanagida. 1987. Identification of healed terminal DNA fragments in linearminichromosomes of Schizosaccharomyces pombe. Mol. Cell. Biol. 7:4424–4430.
40. Maundrell, K. 1993. Thiamine-repressible expression vectors pREP andpRIP for fission yeast. Gene 123:127–130.
41. Nabeshima, K., T. Nakagawa, A. F. Straight, A. Murray, Y. Chikashige,Y. M. Yamashita, Y. Hiraoka, and M. Yanagida. 1998. Dynamics of centro-meres during metaphase-anaphase transition in fission yeast: Dis1 is impli-cated in force balance in metaphase bipolar spindle. Mol. Biol. Cell 9:3211–3225.
42. Pederson, D. S., and R. H. Morse. 1990. Effect of transcription of yeastchromatin on DNA topology in vivo. EMBO J. 9:1873–1881.
43. Pidoux, A. L., S. Uzawa, P. E. Perry, W. Z. Cande, and R. C. Allshire. 2000.Live analysis of lagging chromosomes during anaphase and their effect onspindle elongation rate in fission yeast. J. Cell Sci. 113:4177–4191.
44. Rollins, R. A., P. Morcillo, and D. Dorsett. 1999. Nipped-B, a Drosophilahomologue of chromosomal adherins, participates in activation by remoteenhancers in the cut and Ultrabithorax genes. Genetics 152:577–593.
45. Russell, P., and P. Nurse. 1986. cdc25� functions as an inducer in the mitoticcontrol of fission yeast. Cell 45:145–153.
46. Segura-Totten, M., A. K. Kowalski, R. Craigie, and K. L. Wilson. 2002.Barrier-to-autointegration factor: major roles in chromatin decondensationand nuclear assembly. J. Cell Biol. 158:475–485.
47. Singh, J., and A. J. Klar. 1993. DNA polymerase-alpha is essential formating-type switching in fission yeast. Nature 361:271–273.
48. Sjogren, C., and K. Nasmyth. 2001. Sister chromatid cohesion is required forpostreplicative double-strand break repair in Saccharomyces cerevisiae. Curr.Biol. 11:991–995.
49. Strick, R., and U. K. Laemmli. 1995. SARs are cis DNA elements of chro-mosome dynamics: synthesis of a SAR repressor protein. Cell 83:1137–1148.
50. Strunnikov, A. V., L. Aravind, and E. V. Koonin. 2001. Saccharomyces cer-evisiae SMT4 encodes an evolutionarily conserved protease with a role inchromosome condensation regulation. Genetics 158:95–107.
51. Tanaka, K., Z. Hao, M. Kai, and H. Okayama. 2001. Establishment andmaintenance of sister chromatid cohesion in fission yeast by a unique mech-anism. EMBO J. 20:5779–5790.
52. Thon, G., and A. J. Klar. 1993. Directionality of fission yeast mating-typeinterconversion is controlled by the location of the donor loci. Genetics134:1045–1054.
53. Uzawa, S., and M. Yanagida. 1992. Visualization of centromeric and nucle-olar DNA in fission yeast by fluorescence in-situ hybridization. J. Cell Sci.101:267–275.
54. van Heemst, D., E. Kafer, T. John, C. Heyting, M. van Aalderen, and D.Zickler. 2001. BimD/SPO76 is at the interface of cell cycle progression,chromosome morphogenesis, and recombination. Proc. Natl. Acad. Sci. USA98:6267–6272.
55. Woods, A., T. Sherwin, R. Sasse, T. H. MacRae, A. J. Baines, and K. Gull.
920 DE LAHONDES ET AL. EUKARYOT. CELL
on June 10, 2018 by guesthttp://ec.asm
.org/D
ownloaded from

1989. Definition of individual components within the cytoskeleton of Trypano-soma brucei by a library of monoclonal antibodies. J. Cell Sci. 93:491–500.
56. Yamada, H., K. Kumada, and M. Yanagida. 1997. Distinct subunit functionsand cell cycle regulated phosphorylation of 20S APC/cyclosome required foranaphase in fission yeast. J. Cell Sci. 110:1793–1804.
57. Yang, S. S., E. Yeh, E. D. Salmon, and K. Bloom. 1997. Identification of amid-anaphase checkpoint in budding yeast. J. Cell Biol. 136:345–354.
58. Zhao, K., E. Kas, E. Gonzales, and U. K. Laemmli. 1993. SAR-dependent
mobilization of histone H1 by HMG-I/Y in vitro: HMG-1/Y is enriched inH1-depleted chromatin. EMBO J. 12:3237–3247.
59. Zheng, R., R. Ghirlando, M. S. Lee, K. Mizuuchi, M. Krause, and R. Craigie.2000. Barrier-to-autointegration factor (BAF) bridges DNA in a discrete,higher-order nucleoprotein complex. Proc. Natl. Acad. Sci. USA 97:8997–9002.
60. Zhou, B. B., and S. J. Elledge. 2000. The DNA damage response: puttingcheckpoints in perspective. Nature 408:433–439.
VOL. 2, 2003 CHROMOSOME STABILITY 921
on June 10, 2018 by guesthttp://ec.asm
.org/D
ownloaded from


















