
FGFR Signaling Promotes the Growth of Triple-Negative and...
13
Human Cancer Biology FGFR Signaling Promotes the Growth of Triple-Negative and Basal-Like Breast Cancer Cell Lines Both In Vitro and In Vivo Rachel Sharpe 1 , Alex Pearson 1 , Maria T. Herrera-Abreu 1 , Damian Johnson 1 , Alan Mackay 1 , Jonathan C. Welti 1 , Rachael Natrajan 1 , Andrew R. Reynolds 1 , Jorge S. Reis-Filho 1 , Alan Ashworth 1 , and Nicholas C. Turner 1,2 Abstract Purpose: The oncogenic drivers of triple-negative (TN) and basal-like breast cancers are largely unknown. Substantial evidence now links aberrant signaling by the fibroblast growth factor receptors (FGFR) to the development of multiple cancer types. Here, we examined the role of FGFR signaling in TN breast cancer. Experimental Design: We examined the sensitivity of a panel of 31 breast cancer cell lines to the selective FGFR inhibitor PD173074 and investigated the potential mechanisms underlying sensitivity. Results: TN breast cancer cell lines were more sensitive to PD173074 than comparator cell lines (P ¼ 0.011), with 47% (7/15) of TN cell lines showing significantly reduced growth. The majority of TN cell lines showed only modest sensitivity to FGFR inhibition in two-dimensional growth but were highly sensitive in anchorage-independent conditions. PD173074 inhibited downstream mitogen-activated pro- tein kinase and PI3K–AKT signaling and induced cell-cycle arrest and apoptosis. Basal-like breast cancer cell lines were found to express FGF2 ligand (11/21 positive) and, similarly, 62% of basal-like breast cancers expressed FGF2, as assessed by immunohistochemistry compared with 5% of nonbasal breast cancers (P < 0.0001). RNA interference targeting of FGF2 in basal-like cell lines significantly reduced growth in vitro and reduced down stream signaling, suggesting an autocrine FGF2 signaling loop. Treatment with PD173074 significantly reduced the growth of CAL51 basal-like breast cancer cell line xenografts in vivo. Conclusions: Basal-like breast cancer cell lines, and breast cancers, express autocrine FGF2 and show sensitivity to FGFR inhibitors, identifying a potential novel therapeutic approach for these cancers. Clin Cancer Res; 17(16); 5275–86. Ó2011 AACR. Introduction Therapies that target the drivers of individual breast cancers have substantially improved the outcome of women with breast cancer, in particular endocrine thera- pies for luminal type cancers that express the estrogen receptor and trastuzumab for cancers with HER2 amplifi- cation (1). However, for the approximately 10% to 15% of breast cancers that are triple negative (TN), cancers that express neither the estrogen or progesterone receptors nor have amplification of HER2, the oncogenic drivers are poorly understood (2–5). This subgroup of cancers has a poor prognosis in the adjuvant setting (6, 7) and is highly proliferative with a short time from relapse to death (8). There is substantial overlap between TN breast cancers and the basal-like subtype of breast cancer, approximately 80% of TN breast cancers are basal-like (9), and therefore the two terms describe a broadly similar group of cancers. Identifying the oncogenic drivers of TN breast cancer and basal-like breast cancer is a priority, if the outcome of women with this group of cancers is to be improved. The oncogenic drivers and the factors that promote TN tumor growth are largely unclear with current evidence pointing to substantial heterogeneity (5, 10). Mutations of PIK3CA are found in less than 10% TN breast cancers (11), although the tumor suppressor PTEN may also be lost in a high proportion of these cancers (12), and no other high frequency kinase gene mutations have been identified (13, 14). Focal amplifications are found in the majority of TN cancers, although TN cancers often exhibit high levels of genomic instability (15, 16) and amplification of each individual genomic locus is only present in a small proportion of cancers (5). Significant progress has been made in identifying commonly activated signal transduc- tion pathways in TN and basal-like breast cancers. Deletion Authors' Affiliations: 1 The Breakthrough Breast Cancer Research Centre, Institute of Cancer Research; and 2 Breast Unit, Royal Marsden Hospital, London, United Kingdom Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/). R. Sharpe and A. Pearson have contributed equally to this work. Corresponding Author: Nicholas C. Turner, The Breakthrough Breast Cancer Research Centre, Institute of Cancer Research, 237 Fulham Road, London SW3 6JB, United Kingdom. Phone: 44-207-1535574; Fax: 44-0- 207-51535340; E-mail: [email protected] doi: 10.1158/1078-0432.CCR-10-2727 Ó2011 American Association for Cancer Research. Clinical Cancer Research www.aacrjournals.org 5275 on May 30, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from Published OnlineFirst June 28, 2011; DOI: 10.1158/1078-0432.CCR-10-2727
Transcript of FGFR Signaling Promotes the Growth of Triple-Negative and...

Human Cancer Biology
FGFR Signaling Promotes the Growth of Triple-Negative andBasal-Like Breast Cancer Cell Lines Both In Vitro and In Vivo
Rachel Sharpe1, Alex Pearson1, Maria T. Herrera-Abreu1, Damian Johnson1, Alan Mackay1,Jonathan C. Welti1, Rachael Natrajan1, Andrew R. Reynolds1, Jorge S. Reis-Filho1,Alan Ashworth1, and Nicholas C. Turner1,2
AbstractPurpose: The oncogenic drivers of triple-negative (TN) andbasal-like breast cancers are largely unknown.
Substantial evidence now links aberrant signaling by the fibroblast growth factor receptors (FGFR) to the
development of multiple cancer types. Here, we examined the role of FGFR signaling in TN breast cancer.
Experimental Design: We examined the sensitivity of a panel of 31 breast cancer cell lines to the
selective FGFR inhibitor PD173074 and investigated the potential mechanisms underlying sensitivity.
Results: TN breast cancer cell lines were more sensitive to PD173074 than comparator cell lines
(P ¼ 0.011), with 47% (7/15) of TN cell lines showing significantly reduced growth. The majority of
TN cell lines showed onlymodest sensitivity to FGFR inhibition in two-dimensional growth butwere highly
sensitive in anchorage-independent conditions. PD173074 inhibited downstream mitogen-activated pro-
tein kinase and PI3K–AKT signaling and induced cell-cycle arrest and apoptosis. Basal-like breast cancer cell
lines were found to express FGF2 ligand (11/21 positive) and, similarly, 62% of basal-like breast cancers
expressed FGF2, as assessed by immunohistochemistry compared with 5% of nonbasal breast cancers
(P < 0.0001). RNA interference targeting of FGF2 in basal-like cell lines significantly reduced growth in vitro
and reduced down stream signaling, suggesting an autocrine FGF2 signaling loop. Treatment with
PD173074 significantly reduced the growth of CAL51 basal-like breast cancer cell line xenografts in vivo.
Conclusions: Basal-like breast cancer cell lines, and breast cancers, express autocrine FGF2 and
show sensitivity to FGFR inhibitors, identifying a potential novel therapeutic approach for these cancers.
Clin Cancer Res; 17(16); 5275–86. �2011 AACR.
Introduction
Therapies that target the drivers of individual breastcancers have substantially improved the outcome ofwomen with breast cancer, in particular endocrine thera-pies for luminal type cancers that express the estrogenreceptor and trastuzumab for cancers with HER2 amplifi-cation (1). However, for the approximately 10% to 15% ofbreast cancers that are triple negative (TN), cancers thatexpress neither the estrogen or progesterone receptors norhave amplification of HER2, the oncogenic drivers are
poorly understood (2–5). This subgroup of cancers has apoor prognosis in the adjuvant setting (6, 7) and is highlyproliferative with a short time from relapse to death (8).There is substantial overlap between TN breast cancers andthe basal-like subtype of breast cancer, approximately 80%of TN breast cancers are basal-like (9), and therefore thetwo terms describe a broadly similar group of cancers.Identifying the oncogenic drivers of TN breast cancerand basal-like breast cancer is a priority, if the outcomeof women with this group of cancers is to be improved.
The oncogenic drivers and the factors that promote TNtumor growth are largely unclear with current evidencepointing to substantial heterogeneity (5, 10). Mutations ofPIK3CA are found in less than 10% TN breast cancers (11),although the tumor suppressor PTEN may also be lost in ahigh proportion of these cancers (12), and no other highfrequency kinase gene mutations have been identified(13, 14). Focal amplifications are found in the majorityof TN cancers, although TN cancers often exhibit highlevels of genomic instability (15, 16) and amplificationof each individual genomic locus is only present in a smallproportion of cancers (5). Significant progress has beenmade in identifying commonly activated signal transduc-tion pathways in TN and basal-like breast cancers. Deletion
Authors' Affiliations: 1The Breakthrough Breast Cancer Research Centre,Institute of Cancer Research; and 2Breast Unit, Royal Marsden Hospital,London, United Kingdom
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
R. Sharpe and A. Pearson have contributed equally to this work.
Corresponding Author: Nicholas C. Turner, The Breakthrough BreastCancer Research Centre, Institute of Cancer Research, 237 Fulham Road,London SW3 6JB, United Kingdom. Phone: 44-207-1535574; Fax: 44-0-207-51535340; E-mail: [email protected]
doi: 10.1158/1078-0432.CCR-10-2727
�2011 American Association for Cancer Research.
ClinicalCancer
Research
www.aacrjournals.org 5275
on May 30, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 28, 2011; DOI: 10.1158/1078-0432.CCR-10-2727

of the phosphatase PTPN12 may set up a permissiveenvironment for oncogenic tyrosine kinase signaling inTN cancer (17). TN cancer cell lines show high sensitivityto SRC inhibitors in vitro (18), and mitogen-activatedprotein kinase (MAPK) pathway activation is more pro-minent in these cancers than luminal type cancers in vitro(4, 19). In a subset of cancers, epidermal growth factorreceptor (EGFR) has potentially been shown to be onco-genic in vitro (19), and there is recent clinical trial datasupporting EGFR as a therapeutic target in a small pro-portion of TN cancers (20). The oncogenic drivers thatactivate the MAPK pathway in the remaining cancers areunknown.
We have previously suggested that amplification of thefibroblast growth factor receptor (FGFR) genes may repre-sent a therapeutic target in breast cancer, with amplificationof FGFR1 occurring in approximately 10% of breast cancers(21), predominantly of luminal subtype (22). Amplifica-tion of FGFR2 also occurs more rarely being found in onlyapproximately 1% to 2%of breast cancers overall, althoughapproximately 4% of TN breast cancer have FGFR2 ampli-fication (5). These data suggest that aberrant activation ofFGF signaling can play a role in breast tumorigenesis (23).In this study, we examine the prevalence of FGFR signalingas a driver in breast cancer, analyzing the sensitivity of apanel of breast cancer cell lines to PD173074, a potent andselective FGFR inhibitor (24). We find that TN and basal-like breast cancer cell lines frequently show sensitivity toFGFR inhibition, and analyze the potential mechanismsthat may explain this sensitivity.
Materials and Methods
Cell lines, materials, and antibodiesCell lines were obtained from American Type Culture
Collection or Asterand and maintained in phenol red–freeDulbecco’s modied Eagle’s medium or RPMI with 10% FBS(PAA gold) and 2mmol/L L-glutamine (Sigma-Aldrich). Allcell lines were banked in multiple aliquots on receipt to
reduce risk of phenotypic drift and identity confirmed byshort tandem repeat profiling with the PowerPlex 1.2System (Promega) and arrayCGH profiling. PD173074and recombinant FGF2 were from Sigma. siRNA were fromDharmacon: FGF2 siGenome SMARTpool (siFGF2, M-006695-00), FGF2 siGenome individual siRNA (siFGF2-A-C, D-006695-02/03/04, respectively), FGFR1 siGenomeSMARTpool (siFGFR1, M003131-03), FGFR1 siGenomeindividual siRNA (siFGFR1 A-C D003131-09/22/23,respectively), siGenome Non-Targeting siRNA Pool#1(siCON, D-001206-13), and PLK1 siGenome SMARTpool(siPLK1, M-003290-01). Antibodies used were phosphory-lated AKT-Ser473 (4058), AKT (4691), phosphorylatedERK1/2-Thr202/Tyr204 (4370), phosphorylated RSK-Thr359/Ser363 (9344), ERK1/2 (9102; all Cell SignallingTechnology), PARP-1 (sc-8007), FGF2 (sc-79) b-actin(sc-1616; all Santa-Cruz Biotechnology).
Allocation of molecular subtype and receptor statusBreast cancer cell lines were ascribed to be TN as
described by Neve and colleagues (25), with the exceptionof MDA-MB-453 which is HER2 amplified (26). CAL51,CAL120, MFM223, and SUM52PE are ER, PR, and HER2negative by Western blotting as previously described (5).S68 is ER positive by Western blotting and VP229 andJIMT1 are HER2 amplified as assessed by HER2 FISHand array Comparative Genomic Hybridization (data notshown). Cell line gene expression subtype was as describedby Neve and colleagues, with the exception of CAL51 andCAL120 that are of basal B subtype by using the cancer classprediction of Neve and colleagues (5).
Breast cancers in a tissue microarray (27) were classifiedinto the molecular subtypes by using the immunohisto-chemical surrogate described by Nielsen and colleagues(28) and Cheang and colleagues (29). TN cancers weredivided into core basal-like (positive for EGFR or CK5/6)and nonbasal TN (negative for EGFR and CK5/6).
Cell line drug sensitivity, siRNA transfection, andFGF2-neutralizing antibody
Cell lines were transfected with siRNA (50 nmol/Lfinal concentration) in 96-well plates with RNAiMax(Invitrogen) or Dharmafect4 (Dharmacon) accordingto manufacturers instructions, and survival was assess-ed with Cell Titre-Glo cell viability assay (Promega)after 5 days growth. For sensitivity to PD173074,cells were plated in 96-well plates and starting 24 hourspostplating were exposed for 72 hours to 1 mmol/LPD173074 or vehicle, and survival assessed by usingCell Titre-Glo. For assessment of activated caspase 3/7,cells were treated as for sensitivity to PD173074, asses-sed by using the Apo-ONE Homogeneous Caspase-3/7Assay (Promega) according to manufacturer’s instruc-tions, and the level of activated caspase 3/7 adjustedfor cell number as assessed by Cell Titre-Glo in cor-responding wells. For sensitivity to FGF2-neutralizingantibody, cells were plated in 96-well plates coatedwith polyHEMA [poly(2-hydroxyethylmethacrylate],
Translational Relevance
The oncogenic drivers of triple negative (TN) andbasal-like breast cancer are largely unknown. In thisstudy, we show that multiple TN and basal-like breastcancer cell lines are sensitive to a fibroblast growthfactor receptor (FGFR) inhibitor and provide evidencethat the growth of basal-like breast cancer cell lines ispromoted by autocrine FGF2 signaling. Basal-likebreast cancers are characterized by a poor prognosis,and this work identifies a potential novel therapeuticstrategy for these cancers. Potent and selective FGFRinhibitors are in early clinical development, and thisstudy provides a rational for assessing these inhibitors,or therapies targeting FGF2 ligand, in TN and basal-like breast cancer.
Sharpe et al.
Clin Cancer Res; 17(16) August 15, 2011 Clinical Cancer Research5276
on May 30, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 28, 2011; DOI: 10.1158/1078-0432.CCR-10-2727

and starting 24 hours postplating were exposed for 72hours to various concentrations of control normal GoatIgG (AB-108-C; R&D Systems) or FGF2-neutralizing anti-body (AB-233-NA) and survival assessed by using CellTitre-Glo.
Anchorage-independent growthUnless stated otherwise, assays of anchorage indepen-
dence were on plates coated with 1.2% polyHEMA (Sigma)in 95% ethanol, as previously described (30). Soft agarassays were done as previously described (22).
Western blotting and fluorescence-activated cellsortingIndicated cell lines were grown on 10-cm plates, treated
as indicated, and lysed in NP40 lysis buffer. Western blotswere carried out with precast TA or Bis-Tris gels (Invitro-gen) as previously described (22). Fluorescence-activatedcell sorting (FACS) analysis was done as previouslydescribed (22).
XenograftsCAL51 cells were injected into the flank of 20 nude mice
(3 � 106 cells per injection). After 21 days to establishtumors, half of the mice were treated with PD170374 25mg/kg by daily intraperitoneal injection in 50 mmol/Llactate buffer pH 5.0 and half with lactate buffer. Tumorswere bidimensionally measured twice weekly, and tumorvolume of each cohort expressed relative to baseline ofvehicle only group [tumor volume ¼ 0.5 � (l � w2), inwhich l ¼ tumor length and w ¼ tumor width)]. Thegrowth of tumors treated with PD173074 and vehicle werecompared with ANOVA.
Quantitative reverse transcriptase PCRcDNAwas synthesized from RNA by using Superscript III
and random hexamers (Invitrogen). Quantitative PCR(qPCR) was done as previously described (22). Expressionof FGF2 (Hs00241111_m1) was expressed relative to themean of endogenous controls S18 (4310893E), MRPL19(Hs00608519_m1), and b-actin (4310881E). Tumor sam-ples for FGF2 mRNA expression analysis were onlyincluded if more than 50% cells in the section were tumorcells.
FGF2 ELISAFGF2 ELISA was done with DUoSet DY233 (R&D Sys-
tems) according to manufacturer’s instructions with serialdilutions of recombinant FGF2 for a standard curve.
FGF2 immunohistochemistryFGF2 imunohistochemistry (IHC) was done on a tissue
microarray, extensive characterization of which has beenreported previously (27). Antigen retrieval was with pH 6.0citrate buffer for 30 minutes at 90�C, before FGF2 antibody(rabbit polyclonal 500-P18; PeproTech) 1:100 dilution for60 minutes and development with dual Envision kit(Dako). FGF2 was scored by 2 observers (JSRF and
NCT), blinded to the clinicopathologic data. Data wererecorded separately for nuclear and cytoplasmic FGF2expression. FGF2 expression was considered positive forcytoplasmic staining if unequivocally malignant cellsexpressed FGF2 and for nuclear staining with a Quick scoremore than 2 (31).
Statistical analysis and analysis of gene expressiondata
All statistical tests were done with GraphPad Prismversion 5.0. Unless stated otherwise, P values were 2 tailedand considered significant if P < 0.05. Error bars representSEM of 3 experiments.
Results
Sensitivity to PD173074 in breast cancer cell linesWe examined the sensitivity of a panel of 31 breast cancer
cell lines to the FGFR selective inhibitor PD173074 (24,32), treating cell lines plated in 96-well plates for 72 hourswith 1 mmol/L PD173074. Eight breast cancer cell linesshowed a significant reduction in growth after 72 hourstreatment (P < 0.01 Student’s t test). We noted that 7 ofthese cell lines were of TN phenotype (Fig. 1A) and that TNcell lines were more sensitive to PD173074 than other celllines (P ¼ 0.011, Mann–Whitney U Test; Fig. 1A). Thesensitivity of the majority of cell lines to PD173074 wasonly modest (Fig. 1A). We have previously shown that oneof the sensitive cell lines, CAL120, was substantially moresensitive to PD173074 growing in anchorage-independentconditions (22). Therefore, we assessed the sensitivity ofthe moderately sensitive cell lines in anchorage-indepen-dent conditions. Substantially increased sensitivity toPD173074 under these conditions was shown for TN celllines (Fig. 1B), with no sensitivity seen in control cell linesthat were ER and/or HER2 positive (Fig. 1C). Confirmingthese results, the growth of CAL51 cells in soft agar wasblocked by PD173074, with no effect of PD170374 on thegrowth of control T47D ER-positive cell line (Fig. 1D andSupplementary Fig. S1).
We have previously identified FGFR gene amplificationin 3 of the sensitive TN cell lines: FGFR2 amplifications inSUM52 and MFM223 (5) and FGFR1 amplification inCAL120 (22). MDA-MB-453, a HER2 amplified non-TNcell line that was sensitive to PD173074, has been shown tohave an activating FGFR4 mutation (26). The data shownabove suggested a more pervasive sensitivity to PD173074in TN breast cancer, andwe, therefore, examined the factorsunderlying the sensitivity of the remaining TN breast cancercell lines.
Cellular consequences of FGFR inhibitionWe examined downstream signaling in the TN breast
cancer cell lines CAL51, Hs578T, and BT549 after exposureto PD173074 (Fig. 2A). We have previously shown thatMAPK signaling is inhibited by PD173074 in the CAL120cell line (22), and we confirmed that phosphorylationof RSK and ERK1/2 was substantially decreased by
FGFR Dependency of Triple-Negative Breast Cancer
www.aacrjournals.org Clin Cancer Res; 17(16) August 15, 2011 5277
on May 30, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 28, 2011; DOI: 10.1158/1078-0432.CCR-10-2727

PD173074 in all cell lines, with a decrease in AKT phos-phorylation in Hs578T under anchorage-independentconditions (Fig. 2A). We note that CAL51 has previouslybeen shown to have an activating mutation in PIK3CA(c.1624G>A, p.E542K), and BT549 has a homozygousmutation of PTEN (c.821delG, p.V275fs*1) that resultsin a truncated nonfunctional protein (www.sanger.ac.uk/cosmic), which might explain the lack of modulation ofAKT in these cell lines. Mirroring the increase in sensitivityseen with PD173074 in anchorage-independent condi-tions (Fig. 1B), there was an increase in the FGFR depen-dence of downstream signaling in anchorage-independentconditions. To confirm inhibition of FGFR signaling byPD173074, we showed that FGFR-Tyr653/654 autopho-sphorylation and phosphorylation of FRS2-Tyr196, a keyFGFR adapter protein required for MAPK pathway activa-tion (33), was decreased by PD173074 in CAL120 andCAL51 cells (Supplementary Fig. S2).
We examined the consequences of FGFR inhibition.CAL51 and BT549 cells exhibited G1 cycle arrest inresponse to PD173074 (Fig. 2B) and Hs578T arrested at4N (Fig. 2B). There was a substantial increase in subG1 cellsin CAL120 and CAL51 in response to PD173074, suggest-ing the induction of apoptosis, but only aminor increase inHs578T. To confirm that the increase in subG1 representedapoptosis, we measured PARP cleavage in response toPD173074, with evidence of increased PARP cleavage inCAL120 and Hs578T cell lines (Fig. 2C). In addition,CAL120 and Hs578T showed increased levels of activatedcaspase 3/7 on exposure to PD173074 (Fig. 2D). We couldtherefore confirm apoptosis in CAL120 and Hs578T butnot in CAL51.
Therefore, multiple basal-like TN breast cancer cell linesare sensitive to FGFR inhibitor PD173074 with a decreasein downstream signaling, and this is associated with induc-tion of both cell-cycle arrest and apoptosis.
Figure 1. The growth of multiple TN cell lines is dependent on FGFR signaling. A, indicated cell lines were grown in 1 mmol/L PD173074 for 72 hours, or vehicle,and growth of PD173074 exposed cells expressed relative to controls. *, P < 0.01 Student's t test. Black bars indicate TN cell lines and gray bars cell lines withexpression of estrogen receptor (ER) or HER2 amplification. Below is the cell line subtype as defined by Neve and colleagues (25). TN breast cancer cell linesare more sensitive to PD173074 than other cell lines (P ¼ 0.011 Mann–Whitney U Test). B, relative growth of indicated TN cells lines exposed to 1 mmol/LPD173074 for 72 hours, or vehicle, growing in conventional 2D conditions on cell culture plastic or in anchorage-independent conditions on polyHEMA-coatedplates. C, relative growth of indicated cell lines exposed to a range of PD173074 concentrations for 72 hours in anchorage-independent conditions onpolyHEMA-coated plates. Black–TN cell lines, Gray–ER and/or HER2 positive. D, growth of CAL51 (TN–sensitive) and T47D (ER positive–insensitive)cells in soft agar exposed to 1 mmol/L PD173074 or vehicle continuously for 2 weeks.
Sharpe et al.
Clin Cancer Res; 17(16) August 15, 2011 Clinical Cancer Research5278
on May 30, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 28, 2011; DOI: 10.1158/1078-0432.CCR-10-2727

Basal-like breast cancer cell lines express FGF2 ligandWe investigated the potential mechanisms underlying
sensitivity to PD173074, in the cell lines in which themechanism of sensitivity was unknown (MDA-MB-157,CAL51, BT549, and Hs578T). As well as being TN, wenoted that all these cell lines were basal-like in phenotypeand, in particular, the basal B cell line subtype as describedby Neve and colleagues (25). Basal B like cell lines arereported to be the cell line subtype most enriched in "stemcell-like" gene expression patterns, as well as having highlevels of mesenchymal markers (25). Assessing PD173074sensitivity data according to cell line subtype, basal B celllines were more sensitive to PD173074 compared with celllines of other subtypes (P ¼ 0.0328 Mann–Whitney U test,Fig. 1A).We first examined the presence of amplifications by
microarray-based comparative genomic hybridization
(aCGH) and showed that none of these cell lines exhibitedamplification of either the FGF receptors or FGF ligands(Supplementary Table S1). We next examined the correla-tion between sensitivity to PD173074 and whole genomegene expression in the panel of 31 cell lines, as assessed byIlumina human WG6 gene expression microarrays, exam-ining for components of FGFR signaling pathways (Sup-plementary Table S2). We noted that FGF2 mRNAexpression correlated significantly, but modestly, with sen-sitivity to PD173074 (r ¼ �0.43 P ¼ 0.024 Spearman’scorrelation coefficient; Supplementary Fig. S3).
To extend this observation, we assessed FGF2 mRNAexpression by quantitative PCR in the panel of breast cancercell lines. The majority of cell lines had undetectable FGF2mRNA. Basal-like breast cancer cell lines expressed sub-stantially greater FGF2mRNA levels than luminal cell lines(P ¼ 0.0001 Mann–Whitney U Test, Fig. 3A). Within the
Figure 2. FGFR signaling engages down stream signal transduction pathways promoting cell-cycle progression and inhibition of apoptosis. A, indicated celllines either growing on plastic or on polyHEMA-coated plates were treated for 1 hour prior to lysis with 1 mmol/L PD173074 (þ) or vehicle (�). Lysates weresubject to SDS-PAGE and Western blotting with antibodies against phosphorylated-RSK-Thr359/Ser363, phosphorylated-AKT-Ser473, phosphorylated-ERK1/2-Thr202/Tyr204, total AKT, total ERK1/2, and b-actin. B, quantification of propidium iodide FACS profiles in BT549, Hs578T, CAL51, and CAL120cells growing in polyHEMA-coated plates and treated for 48 hours with 1 mmol/L PD173074 (þ) or no treatment (�). C, lysates of CAL120, CAL51, and Hs578Tcells growing in polyHEMA-coated plates and treated for 48 hours with 1 mmol/L PD173074 or no treatment (�), blotted for PARP1 and b-actin. Arrowindicates cleaved PARP1. D, indicated cell lines growing in polyHEMA-coated plates were treated for 48 hours with a range of PD173074 concentrations orno treatment (�), and apoptosis assessed with an assay of activated caspase3/7. Activated caspase 3/7 was expressed relative to the level in cellswith no treatment (�).
FGFR Dependency of Triple-Negative Breast Cancer
www.aacrjournals.org Clin Cancer Res; 17(16) August 15, 2011 5279
on May 30, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 28, 2011; DOI: 10.1158/1078-0432.CCR-10-2727
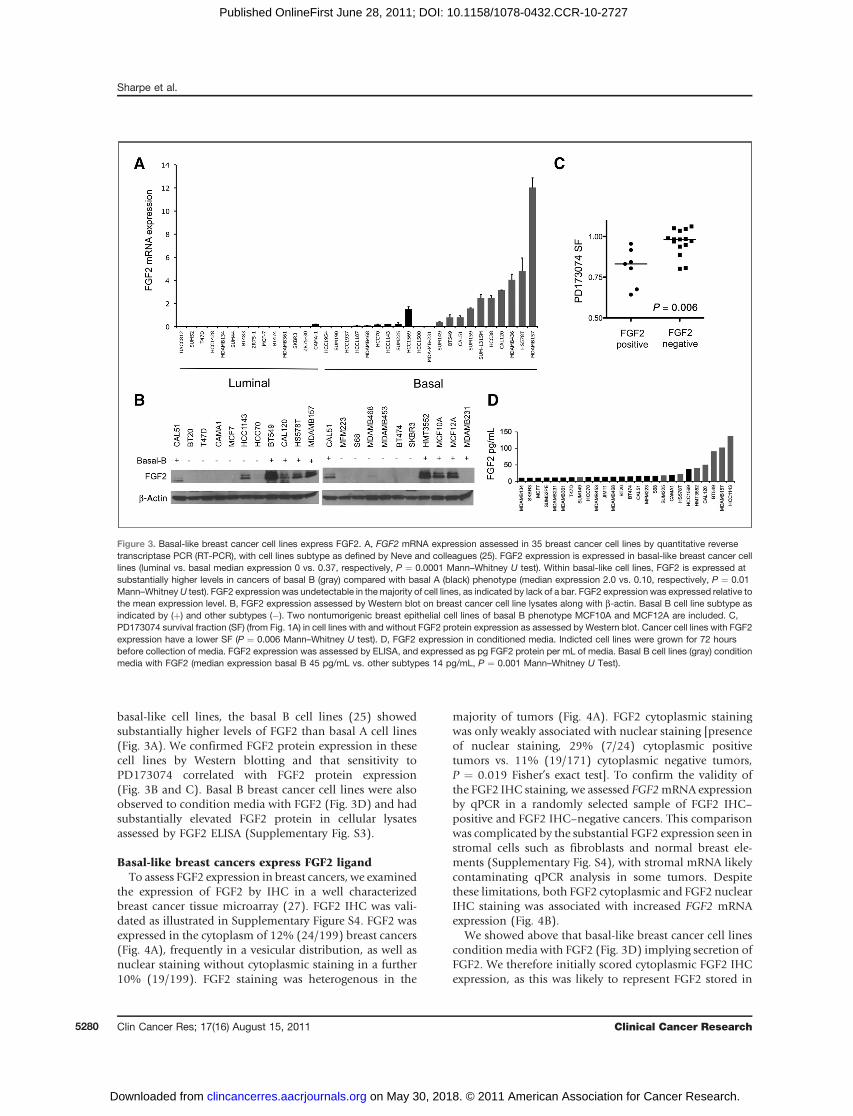
basal-like cell lines, the basal B cell lines (25) showedsubstantially higher levels of FGF2 than basal A cell lines(Fig. 3A). We confirmed FGF2 protein expression in thesecell lines by Western blotting and that sensitivity toPD173074 correlated with FGF2 protein expression(Fig. 3B and C). Basal B breast cancer cell lines were alsoobserved to condition media with FGF2 (Fig. 3D) and hadsubstantially elevated FGF2 protein in cellular lysatesassessed by FGF2 ELISA (Supplementary Fig. S3).
Basal-like breast cancers express FGF2 ligandTo assess FGF2 expression in breast cancers, we examined
the expression of FGF2 by IHC in a well characterizedbreast cancer tissue microarray (27). FGF2 IHC was vali-dated as illustrated in Supplementary Figure S4. FGF2 wasexpressed in the cytoplasm of 12% (24/199) breast cancers(Fig. 4A), frequently in a vesicular distribution, as well asnuclear staining without cytoplasmic staining in a further10% (19/199). FGF2 staining was heterogenous in the
majority of tumors (Fig. 4A). FGF2 cytoplasmic stainingwas only weakly associated with nuclear staining [presenceof nuclear staining, 29% (7/24) cytoplasmic positivetumors vs. 11% (19/171) cytoplasmic negative tumors,P ¼ 0.019 Fisher’s exact test]. To confirm the validity ofthe FGF2 IHC staining, we assessed FGF2mRNA expressionby qPCR in a randomly selected sample of FGF2 IHC–positive and FGF2 IHC–negative cancers. This comparisonwas complicated by the substantial FGF2 expression seen instromal cells such as fibroblasts and normal breast ele-ments (Supplementary Fig. S4), with stromal mRNA likelycontaminating qPCR analysis in some tumors. Despitethese limitations, both FGF2 cytoplasmic and FGF2 nuclearIHC staining was associated with increased FGF2 mRNAexpression (Fig. 4B).
We showed above that basal-like breast cancer cell linescondition media with FGF2 (Fig. 3D) implying secretion ofFGF2. We therefore initially scored cytoplasmic FGF2 IHCexpression, as this was likely to represent FGF2 stored in
Figure 3. Basal-like breast cancer cell lines express FGF2. A, FGF2 mRNA expression assessed in 35 breast cancer cell lines by quantitative reversetranscriptase PCR (RT-PCR), with cell lines subtype as defined by Neve and colleagues (25). FGF2 expression is expressed in basal-like breast cancer celllines (luminal vs. basal median expression 0 vs. 0.37, respectively, P ¼ 0.0001 Mann–Whitney U test). Within basal-like cell lines, FGF2 is expressed atsubstantially higher levels in cancers of basal B (gray) compared with basal A (black) phenotype (median expression 2.0 vs. 0.10, respectively, P ¼ 0.01Mann–WhitneyU test). FGF2 expression was undetectable in themajority of cell lines, as indicated by lack of a bar. FGF2 expression was expressed relative tothe mean expression level. B, FGF2 expression assessed by Western blot on breast cancer cell line lysates along with b-actin. Basal B cell line subtype asindicated by (þ) and other subtypes (�). Two nontumorigenic breast epithelial cell lines of basal B phenotype MCF10A and MCF12A are included. C,PD173074 survival fraction (SF) (from Fig. 1A) in cell lines with and without FGF2 protein expression as assessed by Western blot. Cancer cell lines with FGF2expression have a lower SF (P ¼ 0.006 Mann–Whitney U test). D, FGF2 expression in conditioned media. Indicted cell lines were grown for 72 hoursbefore collection of media. FGF2 expression was assessed by ELISA, and expressed as pg FGF2 protein per mL of media. Basal B cell lines (gray) conditionmedia with FGF2 (median expression basal B 45 pg/mL vs. other subtypes 14 pg/mL, P ¼ 0.001 Mann–Whitney U Test).
Sharpe et al.
Clin Cancer Res; 17(16) August 15, 2011 Clinical Cancer Research5280
on May 30, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 28, 2011; DOI: 10.1158/1078-0432.CCR-10-2727

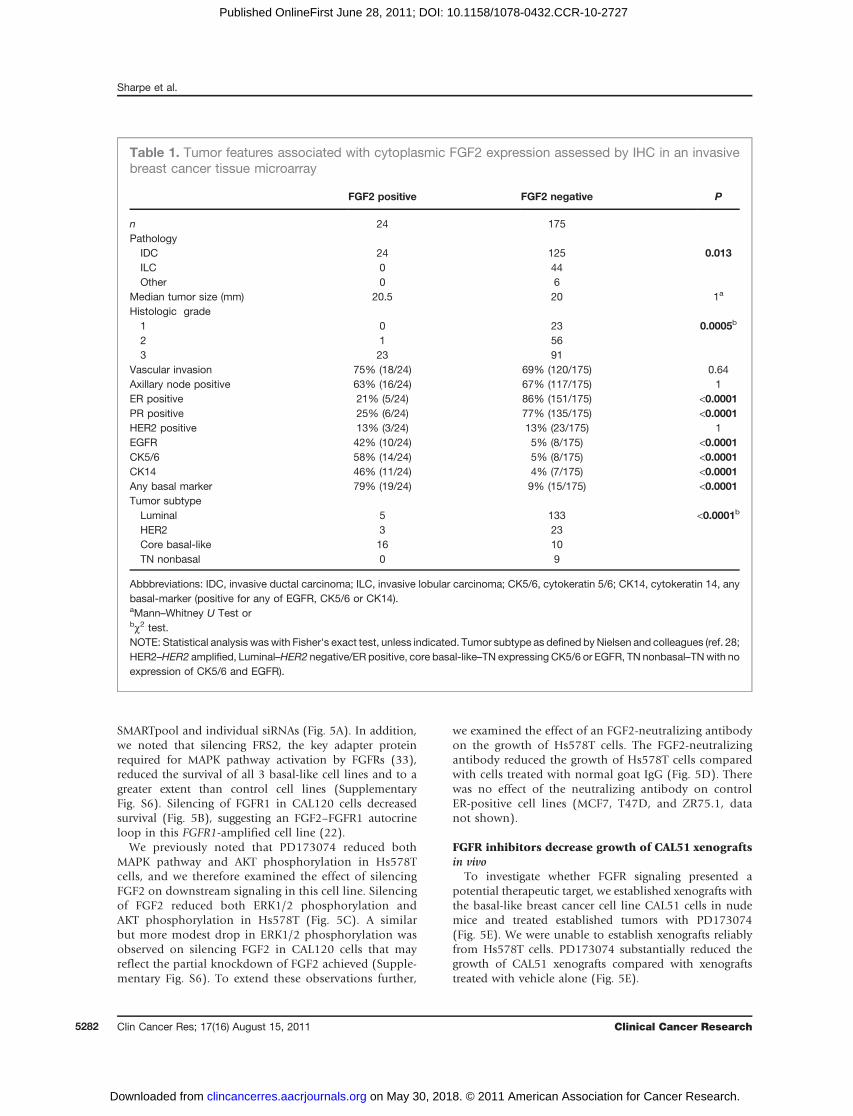
secretary granules. Cytoplasmic FGF2 expression wasstrongly associated with core basal-like breast cancers,TN cancers positive for EGFR or CK5/6 (28, 29), with62% (binomial 95% CI: 41–80) of core basal-like breastcancers expressing FGF2 compared with 5% (95% CI: 2.0–8.9) of nonbasal–like breast cancers (P < 0.0001 Fisher’sexact test, Table 1 and Fig. 4C). Within TN breast cancers,FGF2 expression was associated specifically with cancers ofa core basal-like phenotype (TN: core basal-like 62% vs.nonbasal 0% P ¼ 0.0015 Fisher’s exact test). FGF2 expres-sion was associated with an adverse prognosis because ofthe relatively early relapse of FGF2-expressing cancers(Fig. 4D). However, it is unclear whether this simplyreflects an association with the basal-like subtype, a sub-type that itself has a poor prognosis, or whether FGF2 has acausal role in the poor prognosis.To extend these observations, we interrogated the pub-
licly available TransBIG dataset (34). In this dataset, basal-like breast cancers, as defined by Parker and colleagues(35), had higher FGF2 expression compared with other
breast cancer subtypes (Supplementary Fig. S5). Within thebasal-like subset of cancers, high FGF2 expression wasassociated with poor outcome (HR ¼ 2.924, 95% CI:1.019–8.393, P ¼ 0.046 log-rank test, SupplementaryFig. S5). However, only 41 basal-like cancers were includedin this analysis and therefore the results should be inter-preted with appropriate caution.
Nuclear FGF2 IHC staining, in the absence of cytoplas-mic staining, was not associated with any tumor phenotype(Supplementary Table S3), suggesting a distinct biologicalrole for nuclear FGF2. Interestingly, the myoepithelial cellsof normal breast ducts were observed to strongly expressFGF2 in both nuclear and cytoplasmic compartments(Supplementary Fig. S4).
FGF2 promotes the growth of basal-like cell linesTo examine whether FGF2 potentially acted in an auto-
crine fashion, we silenced FGF2 expression by using siRNAin CAL120, CAL51, and Hs578T cells which caused asignificant reduction in cell line growth with both siFGF2
Figure 4. FGF2 expression is associated with the core basal-like phenotype in breast cancers. A, assessment of FGF2 expression by IHC, with acancer negative for FGF2 expression (left), with FGF2 cytoplasmic expression (middle), and FGF2 nuclear expression (right). B, validation of FGF2 IHC staining,with assessment of FGF2 expression by quantitative RT-PCR on mRNA extracted from a random selection of tumors positive and negative for FGF2expression (n ¼ 28). FGF2 mRNA was expressed at higher levels in FGF2 IHC–positive cancers (P < 0.0001 Kruskal–Wallis one way ANOVA), and at higherlevels in cancers with FGF2 cytoplasmic expression (n ¼ 6, P ¼ 0.018) and FGF2 nuclear expression (n ¼ 3, P ¼ 0.013 Mann–Whitney U test) compared withFGF2 negative cancers (n ¼ 15). C, Cytoplasmic FGF2 expression according to tumor subtype, defined using the IHC criteria of Nielsen and colleagues (28),with the percentage of tumors in each subtype positive for cytoplasmic FGF2 expression (P < 0.0001 comparison of all subtypes with c2 test and P ¼ 0.0015comparison of core basal-like and nonbasal TN cancers with Fisher's exact test). Number in each group luminal n¼ 138, HER2 n¼ 26, core basal-like n¼ 26,TN nonbasal n ¼ 9. D, Kaplan–Meier curves of overall survival for breast cancers with cytoplasmic FGF2 expression (n ¼ 24) compared with cancers withoutcytoplasmic FGF2 expression (n ¼ 175; P ¼ 0.035 log-rank test, P ¼ 0.007 Gehan–Breslow–Wilcoxon test).
FGFR Dependency of Triple-Negative Breast Cancer
www.aacrjournals.org Clin Cancer Res; 17(16) August 15, 2011 5281
on May 30, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 28, 2011; DOI: 10.1158/1078-0432.CCR-10-2727
SMARTpool and individual siRNAs (Fig. 5A). In addition,we noted that silencing FRS2, the key adapter proteinrequired for MAPK pathway activation by FGFRs (33),reduced the survival of all 3 basal-like cell lines and to agreater extent than control cell lines (SupplementaryFig. S6). Silencing of FGFR1 in CAL120 cells decreasedsurvival (Fig. 5B), suggesting an FGF2–FGFR1 autocrineloop in this FGFR1-amplified cell line (22).
We previously noted that PD173074 reduced bothMAPK pathway and AKT phosphorylation in Hs578Tcells, and we therefore examined the effect of silencingFGF2 on downstream signaling in this cell line. Silencingof FGF2 reduced both ERK1/2 phosphorylation andAKT phosphorylation in Hs578T (Fig. 5C). A similarbut more modest drop in ERK1/2 phosphorylation wasobserved on silencing FGF2 in CAL120 cells that mayreflect the partial knockdown of FGF2 achieved (Supple-mentary Fig. S6). To extend these observations further,
we examined the effect of an FGF2-neutralizing antibodyon the growth of Hs578T cells. The FGF2-neutralizingantibody reduced the growth of Hs578T cells comparedwith cells treated with normal goat IgG (Fig. 5D). Therewas no effect of the neutralizing antibody on controlER-positive cell lines (MCF7, T47D, and ZR75.1, datanot shown).
FGFR inhibitors decrease growth of CAL51 xenograftsin vivo
To investigate whether FGFR signaling presented apotential therapeutic target, we established xenografts withthe basal-like breast cancer cell line CAL51 cells in nudemice and treated established tumors with PD173074(Fig. 5E). We were unable to establish xenografts reliablyfrom Hs578T cells. PD173074 substantially reduced thegrowth of CAL51 xenografts compared with xenograftstreated with vehicle alone (Fig. 5E).
Table 1. Tumor features associated with cytoplasmic FGF2 expression assessed by IHC in an invasivebreast cancer tissue microarray
FGF2 positive FGF2 negative P
n 24 175Pathology
IDC 24 125 0.013ILC 0 44Other 0 6
Median tumor size (mm) 20.5 20 1a
Histologic grade1 0 23 0.0005b
2 1 563 23 91
Vascular invasion 75% (18/24) 69% (120/175) 0.64Axillary node positive 63% (16/24) 67% (117/175) 1ER positive 21% (5/24) 86% (151/175) <0.0001PR positive 25% (6/24) 77% (135/175) <0.0001HER2 positive 13% (3/24) 13% (23/175) 1EGFR 42% (10/24) 5% (8/175) <0.0001CK5/6 58% (14/24) 5% (8/175) <0.0001CK14 46% (11/24) 4% (7/175) <0.0001Any basal marker 79% (19/24) 9% (15/175) <0.0001Tumor subtype
Luminal 5 133 <0.0001b
HER2 3 23Core basal-like 16 10TN nonbasal 0 9
Abbbreviations: IDC, invasive ductal carcinoma; ILC, invasive lobular carcinoma; CK5/6, cytokeratin 5/6; CK14, cytokeratin 14, anybasal-marker (positive for any of EGFR, CK5/6 or CK14).aMann–Whitney U Test orbc2 test.NOTE: Statistical analysis waswith Fisher's exact test, unless indicated. Tumor subtype as defined by Nielsen and colleagues (ref. 28;HER2–HER2 amplified, Luminal–HER2 negative/ER positive, core basal-like–TN expressing CK5/6 or EGFR, TN nonbasal–TNwith noexpression of CK5/6 and EGFR).
Sharpe et al.
Clin Cancer Res; 17(16) August 15, 2011 Clinical Cancer Research5282
on May 30, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 28, 2011; DOI: 10.1158/1078-0432.CCR-10-2727

Discussion
TN breast cancers have a poor prognosis and the factorsthat drive the proliferation of these breast cancers havebeen largely unclear. Here, we provide evidence thatFGFR signaling may promote the growth of a proportionof TN breast cancers. Multiple TN cell lines show sensi-tivity to FGFR inhibitor PD173074, with evidence thatMAPK pathway signaling in particular is under control ofFGFR signaling. We further showed that TN breast cancercell line CAL51 xenografts are sensitive to PD173074in vivo.The underlying mechanism of sensitivity to PD173074
seems to be heterogenous in TN breast cancer cell lines,although autocrine FGF2 ligand expression seems to be
the predominant mechanism (Supplementary Table S4).Cytoplasmic FGF2 expression is found specifically in corebasal-like TN cancers, and not in TN cancers that are notbasal-like (Fig. 4C), and we provide data that suggests FGF2acts in an autocrine fashion as both siRNA targeting FGF2and an FGF2-neutralizing antibody reduce the growth ofbasal-like cell lines. These data suggest that it isspecifically basal-like cancers, as opposed to TN per se,whose growth is promoted by autocrine FGF2 signaling,further adding to the evidence that TN cancers are aheterogenous group of cancers.
In cancer cell lines, FGF2 expression was found athighest levels in cancer cell lines of basal B cell linesubtype (Fig. 3A). Recent data has suggested thatthe basal B cell line subtype shows high similarity with
Figure 5. FGF2 autocrine signaling promotes growth and downstream signaling. A, CAL120, CAL51, and Hs578T cell lines were transfected withsiCON nontargeting control, 2 individual siRNA targeting FGF2 (siFGF2-A and siFGF2-B) and SMARTpool targeting FGF2 (siFGF2), with survival assessedafter 5 days. Comparison between siCON and FGF2 siRNA with Student's t test *, P < 0.01, þ, P < 0.05. B, CAL120 cells were transfected with siCON, 3individual siRNA targeting FGFR1 (siFGFR1 A–C), and FGFR1 SMARTpool (siFGFR1) and survival was assessed 5 days post-transfection. Comparisonbetween siCON and FGFR1 siRNA with Student's t test *, P < 0.01. C, Hs578T cells were transfected with siCON and siFGF2, lysates were made 72 hourspost-transfection and subject to Western blotting with indicated antibodies. D, Hs578T cells in polyHEMA-coated plates were exposed to variousconcentrations of control IgG antibody or FGF2-neutralizing antibody, with survival assessed after 72 hours exposure. E, FGFR signaling presents apotential therapeutic target. CAL51 cells were injected into the flanks of 20 nude mice (3 � 106 cells per flank), and xenografts established over 21 days. Halfmice were treated with 25 mg/kg PD173074 and half vehicle, for 5 days of 7, and tumor growth assessed over 2 weeks. PD173074 substantially reduced thegrowth of CAL51 xenografts (P < 0.0001 ANOVA). Arrows indicate the treatment days with PD173074 or vehicle.
FGFR Dependency of Triple-Negative Breast Cancer
www.aacrjournals.org Clin Cancer Res; 17(16) August 15, 2011 5283
on May 30, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 28, 2011; DOI: 10.1158/1078-0432.CCR-10-2727

the claudin-low breast tumor subtype (36). Basal B breastcancer cell lines and claudin-low tumors are characterizedby high levels of mesenchymal markers, low or no expres-sion of E-cadherin and claudins at the mRNA level, and"stem cell-like" features, in addition to expression ofbasal markers and the TN phenotype (25, 36). It isnow generally held that claudin-low tumors are best seenas one end of the spectrum of basal-like cancers, ratherthan a distinct entity, as these features are also present,but to a lesser extent, in molecular subtype basal-liketumors. The nontumorigenic breast epithelial cell linesMCF10A and MCF12A expressed FGF2 (Fig. 3B), whichalso express a basal B transcriptional program (25), andalso myoepithelial cells from normal breast ductsexpress FGF2 by IHC (Supplementary Fig. S4). Thissuggests that FGF2 expression may be a manifestationof the basal/myoepithelial phenotype. With FGF2 expres-sion observed at the highest level in cell lines with ahigh expression of "stem cell-like" features, and theevidence that in vitro FGF2 supports the growth ofembryonic stem cells (37), this raises the possibility thatFGF2 autocrine signaling could be a lineage-specifictrophic factor associated with a stem/propagating celltype.
Our data on FGF2 expression does initially seem tocontrast previous observations measuring FGF2 by ELISAin tumor lysates, in which FGF2 expression in wholetumor lysates was associated with ER-positive breastcancer and low histologic grade (38). Potentially,FGF2 in whole tumor lysates represents increased expres-sion of FGF2 by tumor associated fibroblasts andincreased release of FGF2 via extracellular matrix pro-teolysis, as opposed to tumor-specific cytoplasmic FGF2expression which we have shown to be specific to corebasal-like breast cancer. It is also likely that high stromalelement FGF2 expression complicates the assessmentof FGF2 mRNA by gene expression arrays. Althoughnormal breast duct myoepithelial cells expressed bothcytoplasmic and nuclear FGF2, the majority of basal-likecancers expressed only cytoplasmic FGF2. FGF2 proteinis expressed as multiple different isoforms reflectingtranslation from differing noncanonical, translationstart sites (39), with the lower molecular weight iso-form being secreted through a nongolgi/endoplasmicreticulum–mediated process (40). Potentially, this obser-vation suggests that expression of the nuclear FGF2isoforms may be suppressed during basal-like tumordevelopment.
It will be interesting in future research to identifythe FGF receptor that mediates the FGF2 autocrine signal-ing. We show that FGFR1 likely mediates the autocrinesignaling in the CAL120 cell line (Fig. 5), althoughCAL120 is the only basal-like cell line that harborshigh level FGFR1 amplification and the FGF2–FGFR1autocrine loop could be specific to this cell line. We notethat FGFR2 mRNA expression is higher in TN and basal-like breast cancers, compared with other breast cancers,in external datasets (Supplementary Fig. S7). Recently, it
has also been suggested that an FGF9–FGFR3 paracrineloop may promote breast cancer stem cell expansion,suggesting a potential role for FGFR3 (41).
What might be the significance of our data for thetreatment of TN and basal-like breast cancer? Alter-native tyrosine kinase receptors are expressed as part ofthe basal-like phenotype. EGFR is expressed as part of thebasal-like phenotype, and clinical trials have examinedthe efficacy of the EGFR targeting antibody cetuximabwith evidence of modest levels of activity (20). In onephase II randomized trial, the response rates increasedfrom 10.3% to 20% with the addition of cetuximabto cisplatin (20). Overexpression of the HGF receptorc-MET drives the development of mammary cancer inmouse models, including cancers of a basal-like pheno-type, and c-MET is expressed in basal-like breast cancer(42). It will therefore be interesting to establish therelationship between FGF2 autocrine signaling andthese other pathways. For example, does FGFR signalingand EGFR/c-MET occur mutually exclusively, or is therecooperation between signaling pathways? In endothelialcells, there is evidence of cross-talk between FGFRand VEGFR signaling (43), as well as between FGFR4and HER2 in the FGFR4 mutant and HER2-amplifiedMDA-MB-453 breast cancer cell line (44). This raisesthe possibility that FGFR signaling could mediate resis-tance to drugs targeting alternative receptor tyrosinekinases, and it will be interesting to examine combinationapproaches in vitro.
Here, we show that PD173074, as a potent FGFRinhibitor, has activity in vitro on basal-like breast cancercell lines that express autocrine FGF2, as well as singleagent activity against CAL51 basal-like cell line xeno-grafts, suggesting that FGFR signaling may present atherapeutic target in basal-like breast cancer. A numberof potent inhibitors of the FGF receptors are in early phaseclinical trials (33) and the data presented here support theinvestigation of these agents in TN and basal-like breastcancer.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Kay Savage for assistance with FGF2 immunohistochemistry.
Grant Support
This work was supported by grants from Cancer Research UK andBreakthrough Breast Cancer. Dr. Nicholas C. Turner is a CRUK clinicianscientist. We acknowledge NHS funding to the NIHR Biomedical ResearchCentre.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received October 14, 2010; revised June 7, 2011; accepted June 13, 2011;published OnlineFirst June 28, 2011.
Sharpe et al.
Clin Cancer Res; 17(16) August 15, 2011 Clinical Cancer Research5284
on May 30, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 28, 2011; DOI: 10.1158/1078-0432.CCR-10-2727

References1. Turner NC, Jones AL. Management of breast cancer–part II. BMJ
2008;337:a540.2. Schneider BP, Winer EP, Foulkes WD, Garber J, Perou CM, Richard-
son A, et al. Triple-negative breast cancer: risk factors to potentialtargets. Clin Cancer Res 2008;14:8010–8.
3. Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ,Krishnamurthy S, Lee JS, et al. Characterization of a naturally occur-ring breast cancer subset enriched in epithelial-to-mesenchymaltransition and stem cell characteristics. Cancer Res 2009;69:4116–24.
4. MirzoevaOK, Das D, Heiser LM, Bhattacharya S, Siwak D, GendelmanR, et al. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinosi-tide 3-kinase feedback signaling determine susceptibility of breastcancer cells to MEK inhibition. Cancer Res 2009;69:565–72.
5. Turner N, Lambros MB, Horlings HM, Pearson A, Sharpe R, NatrajanR, et al. Integrativemolecular profiling of triple negative breast cancersidentifies amplicon drivers and potential therapeutic targets. Onco-gene 2010:29;2013–23.
6. Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, et al. Thetriple negative paradox: primary tumor chemosensitivity of breastcancer subtypes. Clin Cancer Res 2007;13:2329–34.
7. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA,et al. Molecular portraits of human breast tumours. Nature 2000;406:747–52.
8. Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, SpeersCH, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol2010: 28;3271–7.
9. Rakha EA, Elsheikh SE, Aleskandarany MA, Habashi HO, Green AR,Powe DG, et al. Triple-negative breast cancer: distinguishing betweenbasal and nonbasal subtypes. Clin Cancer Res 2009;15:2302–10.
10. Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, KuoWL,et al. Genomic and transcriptional aberrations linked to breast cancerpathophysiologies. Cancer Cell 2006;10:529–41.
11. Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL,Davies M, et al. An integrative genomic and proteomic analysis ofPIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res2008;68:6084–91.
12. Saal LH, Gruvberger-Saal SK, Persson C, Lovgren K, Jumppanen M,Staaf J, et al. Recurrent gross mutations of the PTEN tumor sup-pressor gene in breast cancers with deficient DSB repair. Nat Genet2008;40:102–7.
13. Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, HumphraySJ, Greenman CD, et al. A comprehensive catalogue of somaticmutations from a human cancer genome. Nature 2010;463:191–6.
14. Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, et al.Diverse somatic mutation patterns and pathway alterations in humancancers. Nature 2010;466:869–73.
15. Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, SimpsonJT, et al. Complex landscapes of somatic rearrangement in humanbreast cancer genomes. Nature 2009;462:1005–10.
16. Russnes HG, Vollan HK, Lingjaerde OC, Krasnitz A, Lundin P, NaumeB, et al. Genomic architecture characterizes tumor progression pathsand fate in breast cancer patients. Sci Transl Med 2010;2:38ra47.
17. Sun T, Aceto N, Meerbrey KL, Kessler JD, Zhou C, Migliaccio I, et al.Activation of multiple proto-oncogenic tyrosine kinases in breastcancer via loss of the PTPN12 phosphatase. Cell 2011;144:703–18.
18. Finn RS, Dering J, Ginther C, Wilson CA, Glaspy P, Tchekmedyian N,et al. Dasatinib, an orally active small molecule inhibitor of both the srcand abl kinases, selectively inhibits growth of basal-type/"triple-nega-tive" breast cancer cell lines growing in vitro. Breast Cancer Res Treat2007;105:319–26.
19. Hoeflich KP, O’Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, et al. Invivo antitumor activity of MEK and phosphatidylinositol 3-kinaseinhibitors in basal-like breast cancer models. Clin Cancer Res2009;15:4649–64.
20. Baselga J, Stemmer S, Pego A, Chan A, Goeminne J-C, Graas M-P,et al. Cetuximab þ cisplatin in estrogen receptor-negative, proges-terone receptor-negative, HER2-negative (triple-negative) metastaticbreast cancer: results of the Randomized Phase II BALI-1 Trial. SanAntonio Breast Cancer SymposiumSan Antonio, TX; 2010.
21. Courjal F, Cuny M, Simony-Lafontaine J, Louason G, Speiser P,Zeillinger R, et al. Mapping of DNA amplifications at 15 chromosomallocalizations in 1875 breast tumors: definition of phenotypic groups.Cancer Res 1997;57:4360–7.
22. Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-GarciaMA, et al. FGFR1 amplification drives endocrine therapy resistanceand is a therapeutic target in breast cancer. Cancer Res 2010;70:2085–94.
23. Dey JH, Bianchi F, Voshol J, Bonenfant D, Oakeley EJ, Hynes NE.Targeting fibroblast growth factor receptors blocks PI3K/AKT signal-ing, induces apoptosis, and impairs mammary tumor outgrowth andmetastasis. Cancer Res 2010;70:4151–62.
24. Pardo OE, Latigo J, Jeffery RE, Nye E, Poulsom R, Spencer-Dene B,et al. The fibroblast growth factor receptor inhibitor PD173074 blockssmall cell lung cancer growth in vitro and in vivo. Cancer Res 2009;69:8645–51.
25. Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. Acollection of breast cancer cell lines for the study of functionallydistinct cancer subtypes. Cancer Cell 2006;10:515–27.
26. Roidl A, Foo P, Wong W, Mann C, Bechtold S, Berger HJ, et al. TheFGFR4 Y367Cmutant is a dominant oncogene in MDA-MB453 breastcancer cells. Oncogene 2010;29;1543–52.
27. Lambros MB, Natrajan R, Geyer FC, Lopez-Garcia MA, Dedes KJ,Savage K, et al. PPM1D gene amplification and overexpression inbreast cancer: a qRT-PCR and chromogenic in situ hybridizationstudy. Mod Pathol 2010;23:1334–45.
28. Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, et al.Immunohistochemical and clinical characterization of the basal-likesubtype of invasive breast carcinoma. Clin Cancer Res 2004;10:5367–74.
29. Cheang MC, Voduc D, Bajdik C, Leung S, McKinney S, Chia SK, et al.Basal-like breast cancer defined by five biomarkers has superiorprognostic value than triple-negative phenotype. Clin Cancer Res2008;14:1368–76.
30. Pickl M, Ries CH. Comparison of 3D and 2D tumor models revealsenhanced HER2 activation in 3D associated with an increasedresponse to trastuzumab. Oncogene 2009;28:461–8.
31. Detre S, Saclani Jotti G, Dowsett M. A "quickscore" method forimmunohistochemical semiquantitation: validation for oestrogenreceptor in breast carcinomas. J Clin Pathol 1995;48:876–8.
32. Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, et al.Frequent and focal FGFR1 amplification associates with therapeuti-cally tractable FGFR1 dependency in squamous cell lung cancer. SciTransl Med 2010;2:62ra93.
33. Turner N, Grose R. Fibroblast growth factor signalling: from devel-opment to cancer. Nat Rev Cancer 2010;10;116–29.
34. Desmedt C, Piette F, Loi S, Wang Y, Lallemand F, Haibe-Kains B, et al.Strong time dependence of the 76-gene prognostic signature fornode-negative breast cancer patients in the TRANSBIG multicenterindependent validation series. Clin Cancer Res 2007;13:3207–14.
35. Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al.Supervised risk predictor of breast cancer based on intrinsic sub-types. J Clin Oncol 2009;27:1160–7.
36. Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al.Phenotypic and molecular characterization of the claudin-low intrinsicsubtype of breast cancer. Breast Cancer Res 2010;12:R68.
37. Xu C, Rosler E, Jiang J, Lebkowski JS, Gold JD, O’Sullivan C, et al.Basic fibroblast growth factor supports undifferentiated humanembryonic stem cell growth without conditioned medium. Stem Cells2005;23:315–23.
38. Smith K, Fox SB, Whitehouse R, Taylor M, Greenall M, Clarke J, et al.Upregulation of basic fibroblast growth factor in breast carcinoma andits relationship to vascular density, oestrogen receptor, epidermalgrowth factor receptor and survival. Ann Oncol 1999;10:707–13.
39. Yu PJ, Ferrari G, Galloway AC, Mignatti P, Pintucci G. Basic fibroblastgrowth factor (FGF-2): the high molecular weight forms come of age. JCell Biochem 2007;100:1100–8.
40. Zehe C, Engling A, Wegehingel S, Schafer T, Nickel W. Cell-surfaceheparan sulfate proteoglycans are essential components of the
FGFR Dependency of Triple-Negative Breast Cancer
www.aacrjournals.org Clin Cancer Res; 17(16) August 15, 2011 5285
on May 30, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 28, 2011; DOI: 10.1158/1078-0432.CCR-10-2727

unconventional export machinery of FGF-2. Proc Natl Acad Sci U S A2006;103:15479–84.
41. Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, LanderES, et al. Estrogen expands breast cancer stem-like cells throughparacrine FGF/Tbx3 signaling. Proc Natl Acad Sci U S A 2010;107:21737–42.
42. Ponzo MG, Lesurf R, Petkiewicz S, O’Malley FP, Pinnaduwage D,Andrulis IL, et al. Met induces mammary tumors with di-verse histologies and is associated with poor outcome and
human basal breast cancer. Proc Natl Acad Sci U S A 2009;106:12903–8.
43. Kanda S, Miyata Y, Kanetake H. Fibroblast growth factor-2-mediatedcapillary morphogenesis of endothelial cells requires signals via Flt-1/vascular endothelial growth factor receptor-1: possible involvement ofc-Akt. J Biol Chem 2004;279:4007–16.
44. Koziczak M, Hynes NE. Cooperation between fibroblast growth factorreceptor-4 and ErbB2 in regulation of cyclin D1 translation. J BiolChem 2004;279:50004–11.
Sharpe et al.
Clin Cancer Res; 17(16) August 15, 2011 Clinical Cancer Research5286
on May 30, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 28, 2011; DOI: 10.1158/1078-0432.CCR-10-2727

2011;17:5275-5286. Published OnlineFirst June 28, 2011.Clin Cancer Res Rachel Sharpe, Alex Pearson, Maria T. Herrera-Abreu, et al.
In Vivo and In VitroBasal-Like Breast Cancer Cell Lines Both FGFR Signaling Promotes the Growth of Triple-Negative and
Updated version
10.1158/1078-0432.CCR-10-2727doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2011/10/20/1078-0432.CCR-10-2727.DC1
Access the most recent supplemental material at:
Cited articles
http://clincancerres.aacrjournals.org/content/17/16/5275.full#ref-list-1
This article cites 41 articles, 23 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/17/16/5275.full#related-urls
This article has been cited by 13 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/17/16/5275To request permission to re-use all or part of this article, use this link
on May 30, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 28, 2011; DOI: 10.1158/1078-0432.CCR-10-2727


















