
**f $1 33 CK »' so30 - Defense Technical Information Center · ONCF 2CF 2CF 2NO . 42 e. Attempted...
122
»' < 30 so > i 5 « 5? Q X t **f ( „<;* ' .KL* ncimcti BiPoiT $1 33 CK SYNTHESIS OF NEW FLUORINE-CONTAINING NITROSO COMPOUNDS, COPOLYMERS m TERPOLYMERS by Eugene C Stump and Calvin D. Padgett I Peninsular Chem Research, Inc I Gainesville, Florida I Contract No. DAI9-129 AMC-152 CN) n S i i November 1967 'ÜW1TEÖ ^T^TES ftRMV* Haticlk; MassachMseUs Q1760 ji «§ AKMj. Clsfhing anä Organe Materials Laboratory AJ \ \
Transcript of **f $1 33 CK »' so30 - Defense Technical Information Center · ONCF 2CF 2CF 2NO . 42 e. Attempted...

»'
< 30 so
> i 5 « 5?
Q X
t
**f (
„<;*■'■ .KL*
ncimcti BiPoiT $1 33 CK
SYNTHESIS OF NEW FLUORINE-CONTAINING
NITROSO COMPOUNDS, COPOLYMERS
m TERPOLYMERS
by
Eugene C Stump and Calvin D. Padgett
I Peninsular Chem Research, Inc
I Gainesville, Florida
I Contract No. DAI9-129 AMC-152 CN)
n S
■i i November 1967
'ÜW1TEÖ ^T^TES ftRMV*
Haticlk; MassachMseUs Q1760
ji Ǥ AKMj.
Clsfhing anä Organe Materials Laboratory
AJ \
\

DISTRT3OTI0S OF THIS DCCTMSHT IS URLBCHm
The findings in tola report are not to be „rostrueä as an official Department of the Artsy position unleff< so designated by- other authorised documents.
Citation of trade namee in this report dots not constitute an official indorsement or approval of the use of such item».
-cm
Destroy this report when no longer needed. Do not return it to the originator.
r ,' us i«t
, ^ MML »*'*

This document has been approved Au_ for public release and sale; its distribution is unlimited.
TECHNICAL REPORT
68-33-CM
SYNTHESIS OF NEW FLUORINE-CONTAINING NITROSO COMPOUNDS, COPOLYMERS AND TERPOLYMERS
By
Eugene C. Stump and Calvin D. Padgett
Peninsular ChemResearch, Inc. Gainesville, Florida
Contract No. DA19-129-AMC-152(N) (01 9116)
Project Reference: 1C024401A329 Series: C&OM-38
November 1967
Clothing and Organic Materials Laboratory L. S. ARMY NATICK LABORATORIES
Natick, Massachusetts 01760

FOREWORD
This is a final report covering research conducted by Peninsular ChemResearch, Inc., Gainesville, Florida from July 1, 1963 to November 26, 1966. The purpose of this project was to investigate the modification of nitroso rubber by incorporation of various other atoms and groups to give either enhanced physical properties or to provide reactive sites for cross-linking. As a necessary adjunct to achieve this purpose, a number of new fluorine-containing monomers were prepared and characterized.
This reporc was prepared by Eugene C. Stump and Calvin D. Padgett, both of Peninsular ChemResearch, Inc., under U. S. Army Contract DA19-129-AMC-152(N), with Mr. Charles B. Griffis and Mr. Frank H. Babers as project supervisors for the Army. E. C. Stump served as project director for Peninsular ChemResearc \. 0 .her workers a«. Peninsular ChemResearch who contributed to this research are Ward H. Oliver, Larry Sapp, Charles B. Wetzel, and Roy Higgenbotham.
S. J. KENNEDY Director Clothing and Organic Materials Division
APPROVED:
PALE H. SIEL1NG, Ph.D. Scientific Director
CLIFFORD T. RIORDAN Colonel, QMC Commanding
ii

CONTENTS
Pafle
LIST OF TABLES ix
LIST OF FIGURES ix
ABSTRACT x
I INTRODUCTION 1
II DISCUSSION 3
A. Monomer Synthesis 3
1. Synthesis of Sulfur-Containing Compounds ...... 3
2. Synthesis of Fluoroaromatic Compounds 10
3. Synthesis of Nitroso Esters and Derivatives 11
4. Synthesis of Trifluoronitrosomethanc and Other Nitroso Alkanes 14
5. Synthesis of Intermediates and Miscellaneous Compounds 16
B. Polymer Synthesis 17
C. NMR Analysis of CF3NO/CF2«CF2 Copolymers - 24
III EXPERIMENTAL 25
A. Monomer Synthesis 25
1. Synthesis of Sulfur-Containing Compounds 25
a. Thiocarbonyl Fluoride 25 b. Perfluorothioacetone 25
'■■■
:
iii

CONTENTS '.Cont'd)
g. Attempted Synthesis ot SF,
£a£e
c. TrifJuoromethyl Trifluorovmyi Sulfide . , 26
CF-SCi 26 CF-SCFC1CF Cl .... 26 CF^SCF-CF^ • • . . 27
d. Trifluorovinyisulfur Pentafluonda .... 27
SF.C1 . ............. 27 CF,C1CFHSFS 2 7 CF^-CFSF, ' 27
e. 2-Chloro-l-mtroso-l,2,2- trlfluoroethyl Trifluoromethyl Sulfide . . . . . . 28
f. Attempted Synthesis of Carboxysulfur Pentafluoride . 28
SF,CH CH Cl 28 SF^CH-CIH ............ 28 SF^COOH . 28
By Oxidation of SF CH=CH 28 By Reaction of Carbonyl Fluoride with i'.ulfur Tetrafluoride .... 29 By Reaction of SF.Cl with Butyl Lithium and Carbon Dioxide .... 29
NO ....... 29
Reaction or Disulfur Decatluorlde and Nitric Oxide at 150fc 29
Reaction of Disulfur Decafluoride and Nitric Oxide in Electric Discharge Tube . , 30 Reaction of Sulfur Tetrafluoride and Nitrcsyl Fluoride 30
Reaction of Nitrosyl Chloride and SF Cs . . . . 31
Reaction of Sulfur Tetrafluoride, Cesium Fluoride, and Dinitrogen Trloxide ......... 31
IV

CONTENTS (Cont'd)
Pafie
Reaction of SF Cl and Nitric Oxide : 31 Reaction of Disulfur Decafluoride and Nitrosyl Chloride 31
h. Attempted Synthesis of Trifluoromethyl Thlonitrite . . 31
Reaction of CF-SAg and Nitrosyl Chloride 31
Reaction of Bid(trifluoromethyl) Disulfide and Nitric Oxide 32 Reaction with Trifluoromethane- sulfenyl Chloride and Nitric Oxide , 32
Reaction of Trifluoromethane- sulfenyl Chloride and Nitrosyl Chloride 33 Reaction of Bis(trifluoromethyl) Disulfide and Nitrosyl Chloride ... 33
Reaction of Thiocarbonyl Fluoride and Nitrosyl Fluoride 34
i. Attempted Synthesis of Pentafluoro- phenyl Thionitrite .... 34
j. Attempted Synthesis of 2-Chloro-l- Nitroso-1,2,2-Trifluoroethylsulfur Pentafluoride ... ..... 34
k. Attempted Synthesis of 2-Chloro-2- Nitroso-1,1,2-Trifluoroethyl Trifluoromethyl Sulfide 35
2. Synthesis of Fluoroaromatic Compounds .... 35
a. Pentafluoronitrosobenzene 35 b. Attempted Preparation of
p-Nitrosotetrafluorobenzoic Acid 35 c. p-Aminotetrafluoronitrosobenzene 36 d. Attempted Synthesis of
4,4'-Dinitrosooctafluorobiphenyl 36
I
'-iWHwS'lgH

CONTENTS (Cont'd)
Page
3. Synthesis of Nltroso Esters and Derivatives 36
a. Synthesis of CH O.CICF-KCO-NO 36 b. Synthesis of CH^CCCFjKCCO'O 37 c. Synthesis of CH^ofcCCF^NO 37 d. Synthesis of CH^02C(CFp3NO 38 e. Isolation of
[CH302C(CF-)312N(KCF2)3C02CH3 39 f. Synthesis of
[H2NOC(CF.)3)2NOCCF2)3CONH2 39 g. Synthesis of
[NC(CF )J NO(CF ) CN . 39 h. Synthesis of HO.cfcF-KNO by
Hydrolysis of CH.O,C(CF2)3NO 39 I. Attempted Synthesis"of
CH2«CHCH202C(CF2)3C02NO , . . 40 j. Attempted Synthesis or
C10C(CF2)3C02NO 40
4. Synthesis of Trifluoronitrosomethane and Other Nitrosoalkanes 41
a. Trifluoronitrosomethane 41 b. CF2BrCF,N0 41 c. CH3OCFCtCF2NO 41 d. Attempted Synthesis of
ONCF2CF2CF2NO . 42 e. Attempted Synthesis of
CH3OCF2CF2NO . 42 f. Reaction of Methyl Nitrite
with Perfluorobutadiene 42
5. Synthesis of Intermediates and Miscellaneous Compounds 43
a. Sulfur Containing Compounds 43
2,2,4,4-Tetrachlorodithietane 43 2,2,4,4-Tetrafluorodlthietane 43 Trifluoromethylthiosilver . . 43 Trifluoromethyliminosulfur Difluoride . 43 Addition of Sulfur Chloride Pentafluoride to 1,1,-Dichloro- 2,2-difluoroethylene 44
1,1,2,2-Tetrafluoroethyl Methvl Sulfide ............ 44
vi

CONTENTS (Cont'd)
Page
b. Fluoroaromatie Compounds 45
Pentafluorophenyl Hydrazine 45 1,2,4,5-Tetrafluorobenzene 45 Tetrafluoroterephthalic Acid 45 1,4-Dihydrazinotetrafluorobenzene . . 46 l,4-Bis(acetophenone) Tetrafluoro- phenyldihydrazone 46
1,4-Diaminotetrafluorobenr.ene .... 46 1,2,4,5-Tetrafluorobenzene ...... 47
c. Nitroso Compound Precursors 47
1,2-Dilodotetrafluoroethane 47 1,3-Diiodohexafluoropropane 47 Attempted Synthesis of
(CH3)2NC(CF2)3COONO 47
Preparation of CF^SCF^FCII 48
d. Fluoroolefins 48
Tetrafluoroallene 48 Methyl Trifluorovinyl Ether 49 4-Methyl-a,P,6-Trifluorostyrene ... 49 Attempted Synthesis of Perfluoroketene 50
Attempted Synthesis of 1-Trifluoro- vinyl Perfluorocyclobutene ..... 50 Preparation of C3F70[CF(CF3)CF20]2CF-CF2 51
Preparation of C3F70[CF(CF3)CF20]3CF-CF2 ...... 51
e. Miscellaneous Compounds .... 52
Perfluoropropylene Epoxide. 52 Perfluoroglutaric Anhydride ..... 52 Perfluorosuccinic Anhydride 52 Perfluoro(2-Methyl-3,6-Dihydro- l,2,2H-Oxazine 52
Nitrosyl Bromide 52 Methyl Nitrite 53 Monosodium Hexafluoropentanadiol ... 53 Attempted Preparation of Disodium Hexafluoropentanediol 53
Attempted Epoxidation of Perfluorobutadiene 53
vii

CONTENTS (Cont'd)
B. Polymer Synthesis 5 5
IV ACKNOWLEDGMENTS 74
V LITERATURE CITED ... 75
VI APPENDICES . 79
A. Infrared Spectra 80
B. NMR Analysis of Monomers 95
C. NMR Analysis of Polymers LOO
D. Monomers from Outside Sources 106
E. Folymer Samples Submitted 107

LIST OF TABLES
Table Page
I Synthesis of CF..NO 14
II Properties of New Compounds Prepared 58
III Copolymers of CF.NO and CF2=CF2 59
IV Copolymers of CF NO with Other Olefins
and Dienes 60
V Copolymers of CF =CF„ with Other Nitroso
Compounds 62
VI Terpolymers 63
VII Miscellaneous Polymers 68
VIII Unsuccessful Polymerizations 70
IX NMR Analysis of CF3N0/CF2«CF2 Copolymers ....... 73
LIST OF FIGURES
Figure Page
1 Electric Discharge Apparatus .... 7
2 Low Temperature Polymerization Apparatus 56
ix

ABSTRACT
This report describes work carried out in the area of nitroso polymers. Two general classes were investigated. These were (1) sulfur- containing nitroso polymers and (2) nitroso polymers containing functional groups.
Synthesis of a wide variety of fluorine-containing nitroso compounds and olefins is described, as well as the synthesis of various riuorlne-containing intermediates.
Nitroso copolymers and terpolymers were prepared containing functional groups such as haloge". ester, double bonds, and methoxy. Properties of selected s./stems are described.
A terpolymer which could be cross-linked by peroxide was prepared using methyl 4-nitrosoperfluorobutyrate as the termonomer.
Infrared spectra and NMR analysis of the new monomers and polymers are included.

\ \ \
I. INTRODUCTION
As part of its program on the development of materials for use in extreme environmental conditions, the U. S. Army Natick Laboratories (NLABS) has sponsored several research projects in the area of so-called nitroso rubber, a copolymer of trifluoronitrosomethane and tetrafluoro- ethylene.
This copolymer was first described by Barr and Haszeldine, who reported that CF3NO and CF2=CF? reacted readily to give perfluoro-2- methyl~l,2-oxazetidine and a colorless, viscous oil which was identified as a 1-to-l alternating copolymer.
CFQNO + CF?=CF? > CF,N 0 - " "I I
CF;-CF,
Formation of the polymer was favored by a low temperature, liquid phase reaction. Russian workers^ had earlier reported polymer formation in the reaction of nitrosyl chloride with 1,1-difluoro-2- chloroethylene. Although this polymer probably resulted from reaction of the olefin with the initial product, CF^CldlClNO, or its dehydro- fluorinated derivative, CFC1=CC1N0, its structure was not elucidated and was not shown to contain the -N-O-(C) - repeating unit found in nitroso polymers.
C1N0 + CF2-CHC1 > CF2C1CHC1N0
CF2C1CHC1N0 > HF + CFC1=CC1N0
CFjClCHCINO and/cr CFC1-CC1N0 + CF2=CHC1 > polymer
The copolymer reported by Haszeldine was found to be unaffected by hot concentrated sulfuric acid and potassium hydroxide.■ In addition, the polymer chain contained heteroatoms which might confer good low temperature flexibility. On the basis of this information a research program was initiated in 1957 by the U. S. Army at Minnesota Mining and Manufacturing Company (3M) to study the preparation and properties of polymers derived from fluorine-containing nitroso compounds and fluoro- olef ins.3'1* Although a wide variety of polymers was studied under this program, the CF3N0/CF2"*CF2 system was given the greatest amount of attention. It was shown that, when high purity monomers were used, elastomerlc gums could he obtained with molecular weights in excess of 1,000,000, The solvent resistance and low temperature properties (Tg » -51°) of the copolymer were also demonstrated.

To evaluate the conoiymer mort extensively and Investigate end-use applications, a program was initiated at Reaction Motors Division ot ihiokoi Chemical Corporation in 1963. Under this contract a method of producing high purity trlfluoromtrosomethane at a rate of about 25 pounds per day was developed and 200 pounds ot the copolymer were produced.-
The research program at Peninsular ChemResearch was also started in 1963. The Initial objective ot this work was the incorporation of compounds ontaining sulfur into '.he nitroso polymer system. This objective was later modified F.O that the major portion of this research was concerned with tne synthesis ot new nitroso copolymers and terpoiymers. The only known cure tor the .opolymer, cross-linking by triethyxenetttramme and hexamethylene diamine carbamate,^ caused scotching and sponging with consequent low tensile strength of the vul- cani-ate For this reason the majority of the termonomers used in this work .ontamed labile groups which might be capable of providing a new cross-linking site without the use of the standard amine cure. The search for potential termonomers necessitated a major eftort in the area or monomer synthesis Both nitroso compounds and substituted f luor.'- ölet ins were investigated as termonomers. Certain monomers were als., supplied by the University of Florida and the University of Colorado under .ont.racts directed by Professor Paul Tarrant and Professor J D Park, respectively.
A complete description ot the work sponsored by the U. S. Army Nati.k Laboratories in this field has been presented by Henry and Gntiis

II. DISCUSSION
A. Monomer Synthesis
To prepar« „ew nitroso polymers, it was frequently necessary to synthesize both new and previously reported fluorine- containing monomers ana intermediates. Since these compounds varied greatly in structure and in functional groups, a discussion of their preparation will be divided into sections according to the characteristic feature of the Individual compounds. For convenience, the following classifications have been selected:
1. sulfur-containing compounds 2. fluoroaromatic compounds 3. nitroso-esters and derivatives 4. trifluoronitrosomethane and other nitroso compounds 5. intermediates and miscellaneous compounds
Physical properties and analyses of the new compounds prepared are listed in Table II, A reference to both the infrared and NMR spectra of these compounds is included in this table. These spectra are described in Appendix A and B, respectively. Parentheses, e.g. (Al), after a compound name or formula indicate that its infrared spectrum is shown in Appendix A.
1. Synthesis of Sulfur-Containing Compounds
The initial research carried out under this program was directed toward the preparation of nitroso polymers containing sulfur. This objective, it was felt, might be accomplished by polymerization using either (a) a monomer which would incorporate sulfur in the backbone of the polymer or (b) a monomer which would result in a pendent group containing sulfur. In the latter case either a sulfur-containing olefin or nitroso compound might be used.
In order to investigate method (a), small quantities of thio- carbonyl fluoride and perfluorothioacetone were prepared. These compounds were synthesized using reported procedures, as shown. Due to the tendency
Thiocarbonvl Fluoride8
S 1. C12C»S — -^—> C12C CC12
\ / s

/\ SbF3 /\ 2, C12C CC12 > F2C CF2 (Al)
V V 3. F2C CF2 —-—> F2C=S (A2)
\ / ^ S
Perfluorothioacetone9
/ \ * (CF02C C(CF3)2 —-—> (CF3)2C=S (A3)
NS
of these compounds to polymerize or cycllze, it was necessary to store them in acid-washed glass ampoules at -78s. Although the dithi- etane precursor to thiocarbonyl fluoride was washed until colorless with sodium hydroxide and hydrogen peroxide prior to distillation, this treatment is not strictly necessary and results in handling losses. It was later found that distillation of the crude dithietane results in samples of greater than 98% purity.
The investigation of polymerization using method (b) required the synthesis of both olefins and nitroso compounds. The previously unreported trifluoromethyl trifluorovinyl sulfide was prepared by the addition of trifluoromethanesulfenyl chloride to chlorotrifluoroethylene followed by dechlorination of the product mixture as shown. The addition
Tiifiuoromethyl Trifluorovinyl Sulfide10 (A4)
1. CCI3SCI + NaF —^> CF3SCI + CF3SSCF3 n
2. CF3SCI + CF2-CFCI U,V" > CF3SCFC1CF2C1 +
CF3SCF2CFC12 12
3. CF3SCFC1CF2C1/CF3SCF2CFC12 Zn
Dioxane
CF3SCF=CF2 + CF3SCF2CFCI2
of CF3SC1 to the olefin (step 2) was accomplished using a modification of the procedure described by Harris.12 Whereas Harris carried out the addition by irradiation in th? liquid phase, our reaction was run ir. the gas phase. This variation apparently results in a different product
* A sample of 2,2,4,4-tetrakis(trifluoromethyl)-l,3-dithietane was provided by C. G. Krespan of DuPont, Wilmington, Delaware.

distribution. Harris reports a ratio of 78:22 (by GLC) while a ratio of 57:43 CF3SCI'C1CF,C1:CF3SCF2CFC12 was determined by NMR for the mixture produced by gas-phase irradiation. Dechlorination using zinc in dioxane gave CF3SCF=»CF2 conversions as high as 83%. The structure was verified by infrared, NMR analysis, and molecular weight determination. Pure (95%) CF3SCF2CFCl2(A5) could also be distilled from the product mixture. Since separation of the isomers could not be achieved by distillation or GLC, this method provides a relatively simple means of obtaining this compound.
Trifluorovinylsulfur pentafluoride, CF2=CFSF5, was prepared by a reported procedure using the following sequence of reactions:
Trifluorovinylsulfur Pentafluoride (A6)
1. SFu + Cl2 + CsF > SFc,Cl ] "*
Bz202 2. SF5C1 + CF2=CFH ^ > CF2ClCFHSFt
3. CF2C1CFHSF5 —^^—> CF2=CFSF5
An attempt to add SF5C1 to CF2«CFH (seep 2) using ultraviolet irradiation in the gas phase produced no adduct. Reaction in an autoclave using benzoyl peroxide in carbon tetrachloride did, however, give reasonable conversions (58%). The dehydrochlorination reaction (step 3) was carried out using powdered potassium hydroxide in hexane. solvent.
Several methods were investigated for the synthesis of highly fluorinated, sulfur-containing nitroso compounds, previous examples of which had not been found in the literature. The first compound in this class was prepared by the addition of nitrosyl chloride to trifluoro- methyl trifluorovinyl sulfide as shown:
2-Chloro-l-nitroso-l,2,2-trifluoroethyl (A 7) Trifluoromethyl Sulfide
CF3SCF-CF2 + C1N0 -U,V' ? CF3SCF(N0)CF2C1
A similar attempt to add nitrosyl chloride to CF2=CFSFc by ultraviolet irradiation in the gas phase was not successful. When the reaction was carried out in dimethylformamide and AlC^a nitroso compound, identified as CF2C1CFC1N0, was produced. Formation of this compound indicates a cleavage of the sulfur-carbon bond with intermediate formation of CF2»CFC1, which then adds nitrosyl chloride to give the observed product.
A number of methods to synthesize nitrosylsulfur pentafluoride, SF5NO, were examined without success. These methods included:

1 reaction ot disulfur decatluonde with nitric oxide using both thermal activation and electric discharg
S2F L, •»• NO —/—> SF NO
2. reaction cl disulfur decatluoride with nitrosyl chloride in solution
S,F u + C1N0 —f SF:NO + SF;C1
3. reaction ot sulfur tetrafluoride with nitrosyl fluoride
SF + FNO —f ■> SF:NO
4 eacticn of ceslumsulfur pentatluorlde with nitrosyl chloride
CsSF- + C1N0 —/ SF;NO + CsCl
5. reaction ot sulfur chloride pentafluoride with nitric oxide using beneoyl peroxide
SFsCl + NO —t > SFcNO
6: rea-tion of dlnltrogen tiiioxide with sulfur tetraf luoride and cesium fluoride
SF. + CsF > lCsSF5] + N;03 —+—> SF5NO
Several previous attempts, all uns'ccessful, to prepare SF5NO have been reported. For example, a mixture ot disulfur decafluoride5 and nitric oxide was Irradiated by ultraviolet light In the gas phase without formation of the desired product.'"' Since S2F-o is known to cleave homolytically at 150' to give «SFs radicals, the thermally activated reactions with S,F j were carried out at this temperature. The products of the reaction between S^Fj0 and NO . glass were identified as NO2, SOF2, SO^F^, and SF^OSF>, while reaction in steel produced N20, SOF2, SOrF?, and SF,
The reaction of SF. and nitrosyl fluoride at 300-400* in a nickel cylinder produced SOF., NO, SFt, N2O, SO^F;, S0F2, NO2, and SO2.
The reaction of nitrosyl chloride with CsSF; at room tempera- ture in glass gave, in addition to SF5CI, N02, SiFH, SOF;, SO2F2, and SF A similar reaction, using dlnltrogen trloxlde rather than nitrosyl chloride, produced SOF^ and S0,F2-
Since alkyl nltroso compounds may be prepare4 by pyrolysis or photolysis of the. acyl nitrite,^3 as shown, It was proposed that
RrC00N0 U,V, or > R£N0 t CO2 f L 1
SF-N0 might be synthesized by the series of reactions below, This method

1. SF5C1 + CH2=CH2 —
2, SF5CH2CH2CI + KOH
SFbCH2CH2Cl
-> SFtjCH=CH2
3. SF5CH=CH2 -2^z->
-H20 SF5COOH
SF5COOH
(SF5CO)20
5. (SF5CO)20 + N203 > SF5COONO
6. SF5COONO U' V-'-> SF5NO + C02
could not be evaluated, however, due to difficulties in preparing SF5COOH (step 3). In addition to oxidation of SF5CH=CH2, other methods of pre- paring the acid were examined without success.
1. SF4 + COF2
2. SF5CI + BuLi
CsF y—> SF5CF
C02
H?0 SF5COOH
SF5COOH H
Since the failure to detect SF5NO as a product of these reactions may have been due to an inherent instability of the compound under the condi-ions employed, an attempt was made to produce "hot" SF5 and NO radicals by passing a mixture of S2Fio and nitric oxide through an electric discharge (Figure 1). The reaction was carried out both at ambient temperature ?nd at -78°. At ambient temperature, identified
transformer
vacuum system
nTtn
>»
c water-cooled electrodes
>«_
manometer >
Figure 1, Electric Discharge Apparatus
e
T reactants

products were SiF., N/), SO., SOF,, SF,:, SF (SO ,F) 2, CF4, CF3CI, and SF5CI. The last three compounds probably resulted by reaction with the Kel-F grease used on the joints in the discharge apparatus. At -78°, in addition to these same compounds, SO ^2 and SOF., were formed. In both cases an unidentified compound was isolated. From its infrared spectrum it appeared to contain an -SO;- group as well as SF bonds. No further efforts were made to identify this compound.
The various reactions used in the attempted synthesis of SF 5NO are described more fully in the experimental section.
The synthesis of SFcCF2COOH as a possible precursor to SF5CF2NO was also desired. Since the reaction of SFtCl with ketene has been reported to give SF-CH2C0C1,
2 - the reaction sequence below might give the the desired intermediate. Although difluoroketene has reportedly26 been
1. SF-C1 + CF^-OO > SFc,CF?COCl
2. SF-CF COC1 t H;0 > SF5CF2COOH
prepared by reaction of CF2ClC0Br with zinc in ether, attempts to repeat this synthesis gave none of the desired ketene. Similar unsuccessful attempts by both English^7 and Russian'8 workers were subsequently reported.
In the initial phase of this contract, one of the classes of sulfur-containing monomers, which was desired for our polymerization studies, was thionitrites, RfSNO, where R could be a perfluoroalkyl or perfluoroaryl group. Although the hydrocarbon analogues, RSNO, had been reported, " there was no mention in the literature of a successful synthesis of a perfluoroalkylthiomtrite, R SNO. An unsuccessful attempt to prepare trifluoromethylthionitrite, CF^SNO, had been described by Emeleus and Pugh, who reacted bis(trifluoromethylthio)mercury with nitrosyl chloride. Instead of the thionitrite, the major product was the disulfide.
(CF3S)2Hg + 2C1N0 CFjSSCF3 + HgCl2 + 2N0
In our laboratories a similar synthesis using trifluoromethylthiosilver and nitrosyl chloride gave similar results.
2CF;SAfe + 2C1NO > CF3SSCF? + 2AgCl -1- 2N0
Since the disulfide seemed to be the favored product in reactions of this type, the reaction of CF^SSCF-, with nitric oxide was attempted. The disulfide can be cleaved homolyf.ically by ultraviolet irradiation to give trifluoromethylthio radicals1' so that a coupling reaction might give the desired product as shown.
CF SSCF —-•--> CF.S- ——- CFiSNO r. t.

Instead of the thionitrite, the major product of this reaction was found to be CF3NO, formed in 38% conversion. Sulfur was also formed. Appar- ently, the trifluoromethylthio radicals decompose under these, conditions to give trifluoromethyl radicals which then combine with nitric oxide.
CF3SSCF3 U-V' -> CF5S S + CM- —^2—> CF 3N0
Some bis(trifl oromethyDsulfide was also formed, indicating a combination of CF3S* and Lf3* radicals. A^ lower temperatures (-78°), CF3SCF3 was the only product.
The reaction of CF3SCI, which also cleaves homolytically to give a CF3S' radical,12 with excess nitric oxide produced a red solid when the reactants were condensed and frozen. This color existed only in the solid phase and could be regenerated by refreezing. The red color was probably not due to nitrosyl chloride since this compound was not detected in the reaction mixture on warming to room temperature, suggest- ing that CF3SNO was formed in a solid matrix and is unstable under normal conditions. The few known thionitrites which have been reported1 ~;9
(RSNO, where R=CH3, C2H5, C6H5) are described as red, relatively unstable liquids.
Reaction of both CF3SCI and CF3SSCF3 with nitrosyl chloride resulted in products (CF3CI, CF3NO2) which showed that carbon-sulfur bond cleavage occurs rather easily in the CF3S" radical to give sulfur and a CF3* radical.
An attempt to react thiocarbonyl fluoride with nitrosyl fluoride, as shown, gave inconclusive results. An infrared spectrum
F2C=S + FNO ■> CF3SNO
of the volatile product mixture, which was found to contain SiFt., COS and N2O, exhibited absorption peaks between 8 and 92 microns and at 13.17 microns, indicating a CF3S group. Other anassigned peaks occurred at 6.15, 6.25 and 6.52 microns. Although the peak at 6.52 can probably be attributed to carbon disulfide, the other two peaks are in the region in which typical highly fluorinated nitroso compounds absorb. This spectrum changed over a period of time, indicating decomposition of the sample at room temperature. Due to the difficulty in preparing thio- carbonyl fluorides, this method was not investigated further.
The synthesis of pentafluorophenyl thionitrite, C5F5SNO, was attempted using a procedure similar to that reported for the preparation of alkyl thionitrites, as shown.
C6F5SH + C1N0 —i > C6F5SNO + HC1

At -40" a solid identified as bis(pentafluorophenyl)disulfide (A8) was formed in quantitative yield. Known thionitrites have been found to form the corresponding disulfides very readily at room temperature so that C^FsSNO, if formed, was converted under these conditions to the disulfide as shown.
2C6F5SNO -> 2NO + C6F5SSC6F5
2. Synthesis of Fluoroaromatic Compounds
Fluorine atoms in pentafluorophenyl derivatives, C5F5R, may be displaced by nucleophilic reagents to give disubstituted products.29»30
Substitution is generally directed to the position para to the R group.31
Consequently, the incorporation of pentafluoroaromatic compounds into nitroso polymer would offer a potential cross-linking site. Functional groups capable of cross-linking would also be of interest as substituents on a perfluoroaromatic monomer.
The synthesis of pentafluoronitrosobenzene was carried out initially by a reported procedure,3^ as shown. Yields were generally around 40%.
Pentafluoronitrosobenzene (A9)
CH2C12 C6F5NH2 + HCOOH/H202 > C6F5NO
The product could be purified by distillation at ret iced pressure and by sublimation to give blue crystals, m.p. 44-45*. The major by-product was found to be C6F5NO2. The synthesis of CgFsNO by several other routes, shown below, was also examined, but without success. Only the third method gave any indication of the formation of the nitroso compound.
1. C6FSNHN-C(CH3)C6H5 + HCOOH/H202 —i > C6F5NO
riNo 2. C6F5H + BuLi > C6F5Li / > C6F5NO
3. C6F5COOAg + C1N0 > C6F5COONO —--—> C6F5Nr
Pentafluorobenzoyl nitrite (A 10) was prepared and isolated as an amber liquia. Although pyrolysis of the nitrite gave inconclusive results, a sample sealed in £n NMR tube at room temperature under partial vacuum formed a green solid believed to be C15F5NO.
Decomposition of the aryl nitrite was also used in attempts to prepare p-nitrosotetrafluorobenzoic acid. The monosilver salt of tetra- fluoroterephthalic acid was prepared by reaction of the dicarboxylic acid with silver oxide. Reaction of the silver salt with nitrosyl chloride gave
10

1. HOOC ÜJj) COOH +1/2 Ag20 > HOOC (("F^ C02Ag
2. HOOC (TM) C02Ag + C1N0
COONO -/■ •> HOOC OJJ
a product mixture from which a yellow, crystalline product, believed to be the mononitrite, was sublimed. Evidence for this structure was obtained by infrared analysis and by its hydrolysis with water to form nitric oxide and tetrafluoroterephthalic acid. Heating the nitrite at 200° under vacuum produced no noticeable color change. Subsequc .t to this work, the synthesis of p-nitrosotetrafluorobenzoic acid by another method was reported.3
The synthesis of r -aminotetrafluoronitrosobenzene was accom- plished, as shown, by the performic acid oxidation of 1,4-diaminotetra-
p-Aminotetrafluoronitrosobenzene
jps\ CH2C12 r^, H2N ((F)) NH2 + HC02H/H202 > H2N ((?)) NO (A 12)
fluorobenzene. The product, after separation from the reaction mixture by sublimation, was a green solid which gave a green solution with methylene chloride. Further evidence for its structure was obtained by infrared analysis, which shows a peak near 1555 cm-1 attributed to N-0 absorption, as well as N-H peaks below 3 microns. In addition, the compound reacted with tetrafluoroethylene to give a solid polymer whose infrared spectrum showed peaks attributable to N-H, C-F, and fluoro- aromatic absorption.
The oxidation of 4,4idiaminooctafluorobiphenyl was also attempted using performic acid. Although a yellow solid was obtained in good yield, it was not identified. Its infrared spectrum (A 13) indicates that it still contains N-H bonds.
3. Synthesis of Nitroso-Esters and Deriva.ives
The previously unreported reaction of both perfluorosuccinic and perfluoroglutaric anhydrides with methyl nitrite was found to produce a nitrite-ester, as shown. The reaction occurs easily and exothermically
J>0 + U«C(CF2)2 3<">0 + CH30N0 > CH302C(CF2)2)3COONO (A 14, A 15)
to give near-quantitative yields. Structure of the two nitrite-esters was established by NMR, elemental and infrared analysis. The infrared spectra show typical methyl, nitrite, and ester absorption, as well as strong C-F absorption.
11

The nitrite-esters were decarbcxylated by both pyrolysis and ultraviolet irradiation, as shown, to give the corresponding nit.roso-
CH^O ,C(CF,)£) .COONO — V'°Z > CO. -f- CH302C(CF2) 2) 3NO (A 16, \ 17)
esters. Conversions ranged from about 20% to 47% with the average conver- sion from over ten reactions close to 21%. The compounds were identified by NMR, elemental, and infrared analysis. The blue liquid nitroso-esters wsre purified by distillation at low pressures to give high-purity (>99%) mor.nmers. Due to the high cost of perfluorosuccmic anhydride, most of the reactions were carried oat with the pert"luoroglutaric anhydiide derivatives
Ä Decaibcxylation by pyrolysis was generally carried out at about 250' by either dropping the nitrite-ester into a packed, heated column under vacuum or by dropping the nitrite-ester into a heated pot at the bottom of a heated wCiaran ander vacuum and removing the product from the top In the latter procedure a distillation head was placed on the column to withdraw non-vclatlie products In the pyrolysis of both nitrite- esters appreciable amounts of colorless, high-boiling liquid were obtained. The high boiling liquid produced in the pyrolysis of CH302C(CF2)3COONO was identified by NMR, elemental, and infrared analysis as the triester, [CH 0;C(CF,) ,),N0(CF )?C0?CH? IA 18). This compound could be formed by the following reactions:
CH;0 C(CF ) .NO .£'Y-,.91:...o NO + CH<02C(.CF,) 3«
CH<0,C(CF;).• + CHj02C(CF2)3NO —> [CH302C(CF2)3]2NO«
[CH.0,C(CF;)31?N0- + CH.02C(CF2)3* —> [CH302C(CF2)3]2N0(CF2)3CO2CH3
A similar compound, (C_<F-,) ;N0C <F , ha3 been obtained by the pyrolysis or photolysis of C3F-NO ■ ** High-boiling liquid formed in the pyrolysis of CH.O,C CF;) COONO was subsequently identified by NMR and infrared analysis as (CH 0 CiCF ,V 1,N0(CF2),X0(CH< (A 19).
Decarboxyiation of the nitrite-esters to nitroso-p»ters could aiso be accomplished by ultraviolet irradiation. Photolysis also produced the hlgh-boilmg liquids mentioned above. Both methods give approximately the same conversions, but the photclytic decomposition is preferred since it requires less attention and regulation.
Attempts to increase the conversion to nitroso-ester by passage of pure mtti_ oxide through the pyrolysis tube at 5 mm. pressure were not successful-
Pyrolysis at higher temperatures (350'-400") results in the formation of a high-boiling purple liquid, possibly a nitroxide.
12

The triester by-product was converted to the trlamide and tri- nitrile, as shown,
NH3 || II tCH302C(CF2) 3]2NO(CF2) 3C02CH3 > [H2NC(CF2) 3] 2NO(CF2) 3CNH2 (A20)
S if »^ [H2NC(CF2)3]2NO(CF2)3CNH2 > [NC(CF2)3]2NO(CF2)3CN (A21)
which were identified by NMR, elemental and infrared analysis.
An Initial attempt to synthesize a nitroso-ester by pyrolysis of a nitrite-ester, prepared by the reaction of a monosilver salt with
I—0 — I
0=CCF2CF2C=0 + CH3OH > CH302CCF2CF2COOH
CH302CCF2CF2COOH + Ag20 > CH302CCF2CF2C02Ag
CH302CCF2CF2C02Ag + C1N0 > AgCl + CH ,02CCF2CF2COONO
CH302CCF2CF2COONO ——■> CH302CCF2CF2NO
nitrosyl chloride, produced a volatile blue liquid. This approach was not pursued, however, due to the concurrent discovery of the more facile method previously discussed.
The attempted synthesis of CH2=CHCH202C(CF2)3COONO by reaction of allyl nitrite with perfluoroglutaric anhydride resulted in a vigorous, exothermic reaction and, in one case, a detonation. When the reaction was carried out at 0° in an open system,a viscous, dark brown material was obtained. Infrared analysis showed that this material was not the desired nitrite-ester.
The synthesis of C10C(CF2)2 3C00N0 was attempted to provide a precursor to nitroso acids. No evidence of reaction was obtained,
I 0—I OC(CF2)2j3C=0 + C1NC —+ > C10C(CF2)2, .C00N0
C10C(CF2)2) 3C00N0 ^•°r > -> HOOC(CF2)2<3NO
however, when the reactants were mixed at room temperature or irradiated by ultraviolet light. Samples of 4-nitrosoperfluorobutyric acid (A22) were subsequently prepared both by a reported procedure' and by the hydrolysis of methyl 4-nitrosoperfluorobutyrate.
H20 CH302C(CF2)3NO > HOOC(CF2)3NO
Hydrolysis can be accomplished using water, acid, or dilute (0,1 N) base. When a stronger (5% NaOH) base was used, the blue color of the nitroso compound faded after 10 minutes at room temperature. Consequently, neutral or acidic hydrolysis is preferred. The blue product was then isolated
13
either by extraction or by salting out with sodium chloride and then dried. Although hydrolysis occurs more rapidly in acidic media (1 day) than in neutral media (4-5 days), conversions as high as 75% were obtained with the latter method
4. Synthesis of Trlfluoronltrosomethane and Other Nitrosoalkanes
In the initial stage oü this contract it was necessary to prepare working quantities of trifluoronltrosomethane in the laboratory. Subsequently, this compound was made available from stock. The procedure used for this synthesis was as follows:
(CF3CO)20 + N;03 -> 2CF3COONO 3?
CFXOONO —-> CF3NO + C02 38
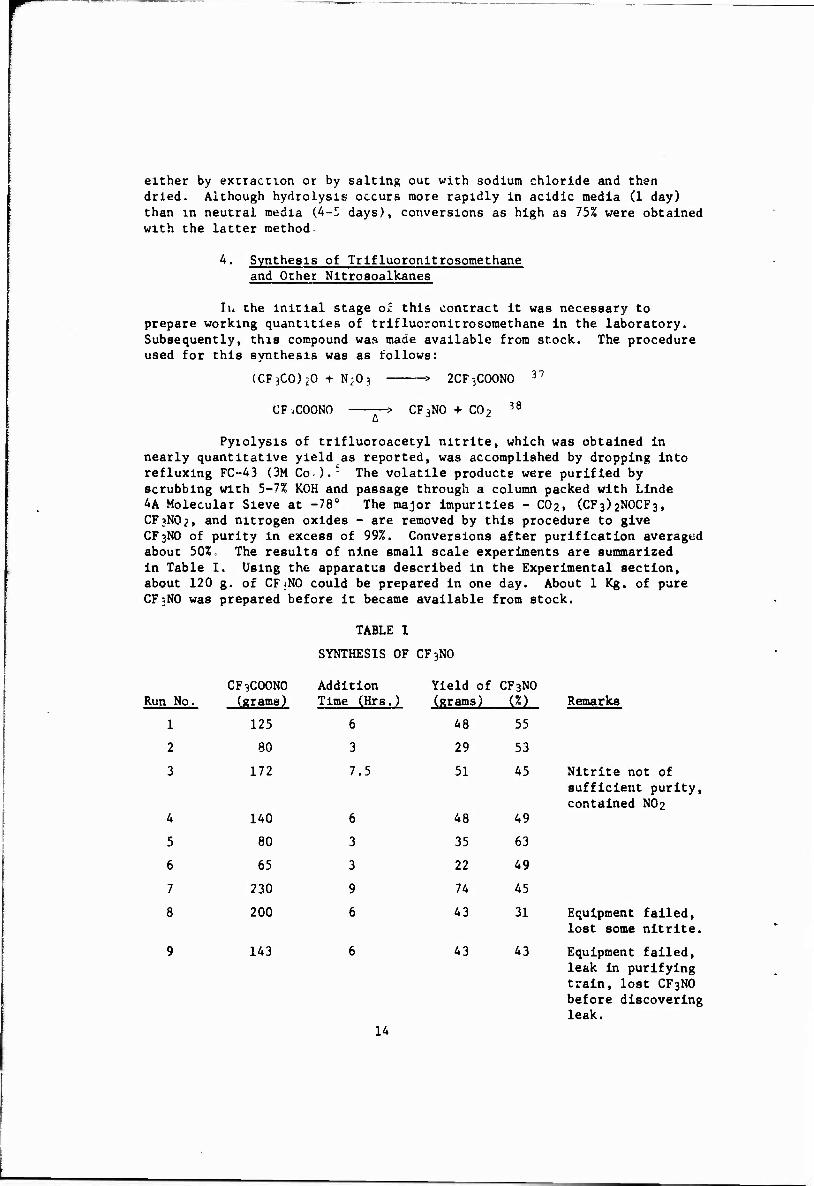
Pyiolysis of trifluoroacetyl nitrite, which was obtained in nearly quantitative yield as reported, was accomplished by dropping Into refluxlng FC-43 (3M Co ).- The volatile products were purified by scrubbing with 5-7% KOH and passage through a column packed with Linde 4A Molecular Sieve at -78° The major impurities - CO2, (CF3)2NOCF3, CF3NO2, and nitrogen oxides - are removed by this procedure to give CF3NO of purity in excess of 99%. Conversions after purification averaged about 50%. The results of nine small scale experiments are summarized in Table I. Using the apparatus described in the Experimental section, about 120 g. of CF-NO could be prepared in one day. About 1 Kg, of pure CF3NO was prepared before it became available from stock.
TABLE I
SYNTHESIS OF CF3NO
Run No. CF3COONO («rams)
Addition Time (Hrs )
Yl eld of rams)
CF3NO (%) Remarks
1 125 6 48 55
2 80 3 29 53
3 172 7.5 51 45 Nitrite not of
4 140 6 48 49
sufficient purity, contained NO2
5 80 3 35 63
6 65 3 22 49
7 230 9 74 45
8 200 6 43 31 Equipment failed, lost some nitrite.
9 143 6 43 43 Equipment failed, leak in purifying train, lost CF3NO before discovering leak.
14

A detailed description of the synthesis of CF3NO on a larger scale ('^1 lb./hr.) may be '.ound elsewhere.-
1-Bromo-l,1,2,2-tetrafluoro-2-nitrosoethane was prepared by the ultraviolet-catalyzed, vapor phase addition of nitrosyl bromide to tetrafluoroethylene. In addition to the desired product, a small amount
BrNO + CF2=CF2 -^J-> CF2BrCF>NO (A23)
of CF2brNO was also formed. This compound could result from addition of nitrosyl bromide to difluoromethylene or by carbon-carLon bond cleavage followed by combination with NO or Br. NMR analysis of CF2BrCF2NO is given in Appendix B.
Methyl l-chloro-l,2,2-trifluoro-2-nitrosoethyl ether, CH3OCFCICF2NO, was prepared by the addition of nitrosyl chloride to methyl trifluorovinyl ether.3'3 The addition is carried out in CF2C12 at -78° and
C1N0 + CH3OCF=CF2 > CH30CFC1CF2N0 (A24)
is unusually rapid. Another noteworthy aspect of this reaction is tnat the nitrosonium ion adds to the terminal difluoromethylene group to give a primary nitroso group. In most cases of addition of nitrosyl chloride tr> a fluoröolefin containing one group or atom other than fluorine attached to the olefinic carbon atoms, the nitroso group will attack the carbon atom with fewer fluorine atoms. For example, nitrosyl chloride adds to chlorotrifluoroethylene and perfluoropropylene, as shown. Assuming the
C1N0 + CF2=CFC1 > CF2C1CFC1N0 u°
C1N0 + CF3CF>«CF2 > CF3CF(N0)CF2C1 M
addition of nitrosyl chloride to alkyl trifluorovinyl ethers to be ionic, the reversed nature of the addition might be explained by contribution of the structure resulting from donation of electrons to the system through the oxygen atom:
+ - CH3OCF=CF2 ^ CH30=CFCF;
An attempt has been made to prepare a functional nitroso compound by the reaction of 1,3-dliodohtixafluoropropane with nitric oxide.
ICF2CF2CF2I + NO > ICF2CF2CF2NO
and/or ONCF2CF2CF2NO
15

This reaction .ould give either .in lodonltroso propane, which could be used as n reactive termonomer, or a dinitroso propane, which might be valuable as a cress-linking agent. This reaction was attempted both with and without mer^uiy in the presence of sunlight, but no evidence for the formation ot a nitroso compound was obtained in either case.
An attempt was made to prepare an unsaturated nitroso compound by reaction ol methyl nitrite with perfluorobutadiene. Although no reaction was apparent in the absence of light, exposure to sunlight produced a light blue-green liquid, which rapidly lost its color during attempted purification An infrared spectrum of this material showed C-H absorption as well as peaks which might be attributed to C=C, -NO;, and -0N0 gioap» The desired reaction is shown below
CH-ONO f CF =CFCF*CF CrT'OCF . CF=CFCF<NO
and/or CH-OCFvCFCF-CF?
NO
A similar reaction was also attemptel using methyl nitrite and tetrafiuoroethylene. The only nitroso compound isolated from the product mixture was identified as ONCF^CF.NO,.
5. Synthesis cf Intermediates and Miscellaneous Compounds
During the course ot this contract it was necessary to prepare a wide variety cf intermediates which were not commercially available. These compounds, for convenience, have been classified as sulfur-containing compounds, fluoroaromatic compounds, nitroso compound precursors, fluoro- "refins, and miscellaneous. In some cases a particular synthesis was carried out several nmts and, in describing these in the Experimental section, a typical reaction or the reaction providing the best yield is given References for ea^h synthesis, when available, are also given.
16

B. Polymer Synthesis
Initial efforts under this contract were directed toward the incorporation of sulfur atoms into the nitroso system. This might be accomplished in either of two ways. First, by polymerization with a sulfur-containing nitroso compound or fluoroolefin as exemplified by Eq. 1 and 2, and second, by polymerisation with a compound such as thio- carbonyl fluoride or hexafluoroethioacetone, as exemplified by Eq. 3. The latter method would give a polymer containing sulfur in the backbone.
1. CF2=CF2 + R ,N0 > «N0CF2CF2> sf I *• c n R c sf
2. CF3NO + CF9-CFR r > «NOCFoCF* and/ot sf j I n
CF3 R , 0 sf
«NO—CFCF2> 1 I n CF3 R 3 "sf
Rf 3. CF3NO + (R )2OS > «NO—CS>
IF, I n f
where R = a sulfur-containing perfluoroalkyl group sx
R, = fluorine anJ/or a perfluoroalkyl group
Although it was found that copolymers of thiocarbonyl fluoride and hexafluorothioacetone could not be prepared under normal conditions (only the homopolymer of the sulfur compound was formed), a copolymer of trifljoronitrosomethane and trifluoromethyl trifluorovinyl sulfide was prepared (Table IV, No. 15). The olefin appeared to be much less reactive than tetrafluoroethylena and a relatively low conversion and molecular weight were obtained. The glass transition temperature of the copolymer was found to be -48°, about that of the copolymer of CF3NO and CF2=CF2. An infrared spectrum of this copolymer is shown in Appendix A (A26).
Small amounts of another sulfur-containing monomer, CF3SCF(N0)CF2C1, were also incorporated as a termonomer but, due to the difficulty in obtaining adequate quantities of tie monomer, this system was not examined further.
17

The second and major area of our polymerization work was concerned with the incorporation of monomers containing funccional groups into the mtroso system One of the major problems in the development of nitroso rubber was in finding an adequate means of curing It. Although a cure may be obtained using triethylenetetramlne and hexamethylene- diamine dlcarbamate, the vulcanizates are generally weak and Inconsistent in properties
At the time this research was undertaken, very few nitroso tetpolymers containing functional groups had been reported. Under an early NI.ABS-sponscied nitroso program at 3M, a terpolymer containing p-trifluorovinyl benzole acid was prepared and cross-linked with barium hydroxide. Of more significance, however, was the preparation of a terpolymei containing either 3-nitrosoperfluoropropionic acid or 4-mtroso- perf luorobutyrlc add ' -'ö The 3M researchers found that these terpolymers ccuid be . ured by metal salts such as chromium trifluoroacetate, diols, and diepoxides The chromium trifluoroacetate cured material was found to txhibit good tensile strength and elongation and had excellent resistance tc Nt0 , nitric £cid, sulfuric acid, and sodium hydroxide.
As part of the work at PCR, we investigated the incorporation of both olefmlc and nitroso monomers.
It was found that bromotrifluoroethylene would copolymerize readily with trifluoronitrosomethane to give a tough, almost insoluble gum in conversions in the order of 90': NMR and elemental analysis
CF,N0 + CF -CFBr «NOCF^CFBr» (A27) 90% conversion I n CF. Tg 3°
CF,N0 + CF;-CF + CF -CFBr —-> «N0CF,CF,N0CF?CFBr>
CF. CF.
Charge (mmoles)
35 29 35 23 35 17.5
6 12 17.5
In FC-43
CF^=CFBr in polymer (mole %)
9.4 21.7 31.0
Jg lull -45 0.15 -35 0.13 -21
indicated a 1-to-l copolymer with the alternating structure shown in the first equation predominating. As in the case of the copolymer of CF,N0 with tetrafluoroethylene, about 15% "abnormal" addition was observed.
18
J
Intrinsic viscosities of the terpolymers were relatively low, possibly due to chain transfer process involving abstraction of a bromine atom from the olefin. As expected, the glass temperature ses with increasing amounts of BTFE, as shown. At 9 mole "'■ the Tg is practically unchanged, but rises to 3° in the copolymer. These samples were found to decompose around 200°. By way of comparison, the decomposition temperature of CF H0/rF2"^'; copolymer is about 270°. It was felt that this system might be cured by abstraction of bromine using a peroxide, but no cross-linking was observed using DiCup 40.5?
A series of polymers containing pentafluoronitrosobenzene was also prepared as shewn below. Incorporation of this compound drastically
CF2»CF2 + ON (Cjj) -> «N0CF^CF2>
e CF3NO + CF2-CF2 + C6F5NO — > 'ww\JNOCF2CF2NO'wv\-'\.
CF3 C6F5
Charge (mole %) T _£.
-43
Td
47.5 50 2.5 166 45.0 50 5,0 -39 158 40.0 50 10.0 -35 152
CA28)
reduced tie thermal stability of the system and raised the glass temperature, The terpolymer containing about 2.5 mole % pentafluoronitrosobenzene decomposed about 100° lower than the copolymer. Attempts to cross-link the terpolymer through the normally labile para fluorine atom were unsuccessful. 57
As shown below, a copolymer and terpolymers containing perfluoro- butadiene were prepared. In all these polymerizations, appreciable quantities of the Diels-Alder addact (A31) were formed. NMR and infrared analysis indicates that polymerization occurs by a 1,2 and 1,4 mechanism with the 1,4 predominating. An infrared spectrum of the terpolymer shows the trifluorovinyl group at 5.61 microns and the CF-CF group at 5,80 microns, About 5-10% of the diene undergoes 1,2 polymerization to give polymers containing a pendent trifluorovinyl group.
19

CF.NÜ f CF =CFCF«CF : -fNOCF-CF=CFCF > -(NOCF;CF> + CF JNCF?CF=CFCF I ■ x i i y i i * CF j CF) CF 0
II CF2
Tg = -43J
CF;NO + CF,*CF, + (p =CFCF=CF,. > terpolymer containing -CF-CF-
and -CF=CF2 (A29)
Copoiymet + CF OF * +NOCF;CFCF >CF p» «NOCF2CF> (A30) I | - - x | | y CF, OCF . CF< CF2
CF; I OCF<
Copolymer + C1NO -(NOCF ,CFC1CF(N0)CF2* 4NOCF2CF* (A32) i x i i y CFs CF? CF(NO)
CF2C1
Blue polymer
Tg = -85"
ln order to determine the reactivity of the double bonds, both trifluoiomethyl hypotluorite and nltrosyl chloride were added to the dissolved copolymer. NMR analysis of the CF^OF adduct shown in the third equation again confirmed that the predominant structure was formed by 1,4 polymerization- The addition of nltrosyl chloride produced a light blue polymer whose infrared spectrum showed a peak, at 6,2 microns. This may be attributed to the formation of nitroso groups as shown in the fourth equation. The glass tempe.-ature of this polymer was found to be -85'. Since the glass temperature of the copolymer is -43°, this unexpected v/alue is difficult to explain. The addition of bis(fluoroxy)difluoro- metharu , CF,> (.OF).-, to the terpolymer was also examined in an attempt to cross-link the poiymer, but the results were Inconclusive.
As shown below, polymers containing trifiaorobutadiene were also prepared. As in tht previous case, the 1,4 mechanism predominates and part of the monomer charge is consumed to give the Dlels-Alder adduct. As before, the reactivity of the double bonds was demonstrated by the addition of CF?0F to give a saturated terpolymer.
20

CF3NO + CF2=CFCH=CH2 •(NOCFoCF-CHCH,* +NOCF2CF» + CF .NCF >CF-=CHCH; i x ! i y i i CF CF, CH
' II
CF3NO + CF2-CF2 + CF2=CFCH=«CH2 -> terpolymer (-CF-CH- & -CH«CH2)
(A3 3)
Terpolymer + CF3OF -> terpolymer (saturated) (A34)
Attempts to cure the unsaturated terpolymers prepared from perfluorobutadiene and 1,1,2-trifluorobutadiene were unsuccessful.57
In addition to the polymers prepared from perfluorobutadiene and trifluorobutadiene mentioned previously, unsaturated nitroso polymers have been prepared, as shown below, by terpoJymerization of CF3NO and tetrafluoroethylene with tetrafluoroallene, difluoroallene, and 3-nitroso- perfluoropropene.
CF3NO + CF2-CF2 + CF2-C-CF2
2 1 1
[n] - 0.13
Tg = -35°
-> 4NOCF2CF2> 4NOCF2C> (A35) 1 "i 11 y CF3 CF3 CF2
CF3NO + CF2-CF2 + CF2=C-CH2 -> 4NOCF2CF2> 4NOCF2C> 1 * 1 11 y CF3 CF3 CH2
dec. 70°
CF3NO + CF2=CF2 + CF2-CFCF2NO <NOCF?CF2» 4NOCF2CF2* -(NOCF2CF> I x 1 y , '| 2 CF3 CF2 CF3 CF2
CF II CF2
NO
(A36)
A low molecular weight gum with an intrinsic viscosity of 0.13 was obtained using tetrafluoroallene. An infrared spectrum of the polymer showed a strong peak at 5.68 microns probably due to C«CF2absorption.
21

The terpolymer obtained from difluoroaliene was found to decompose at /0 . A copolyrner of dlfluoroaliene with trIfluoronitrosomethane decomposed iapldly at room temperature. The terpolymer with 3-nitroso- perfluoropropene gave, in addition to material soluble in Freon 113, a small amount of insoluble gel, indicating that some cross-linking had o^curr<.d. The infrared spectrum uf the reprecipltated polymer exhibited strong absorption at 5,38 microns due to the double bond in the pendent perfluoroallyl group In addition, a peak at 6,2 microns indicates that some polymerization occurred through the trifluorovinyl group, giving pendent -CF NO groups.
Probably ehe most sigmlicant development during this work was tht synthesis and polymerization of the nifoso esters, CHjOpCCCF^) 2^0 and CH-OCtCF ' NO. Since the latter compound was more easily prepared and the starting anhydride less expensive, the majority of the polymeri- sations were carried out with this compounu
As shown below, terpolymers containing the nitroso ester were
CF NO t CF -CF v CH 0:C(CF^jN0 •
4N0CF CF 4 +N0CF CF NO* (A37) i x , | y CF. CF (CF,,),
COtCH
Conditions: bulk, -30 , 2'4 hrs Conversions. 70-80% I ,i = 0.25 to 0.50
prepared containing from 2,5 to 10 mole % ester. Incorporation of the nitroso ester was established by infrared and NMR analysis The polymers were eiastome:iv. gums With the same appearance as the copolymer of CF NO/CF *CF, and .louid bf cured using a standard peroxide cure developed at the Natick Laboratories The gum handles easily on the mill and shows nc signs of scorching.
Finally, as shown below, a polymer of potential utility was prepared u?tng trifiuorovinyl methyl ether as a comonomer or termonomer.
CF NO -r CH 0CF*CF -(N0CF CF-» and/or 4N0-CFCF;>
CF OCH !;F OCH3
6? mole % ether
insoluble in Freon 113 and FC-43
CF-N0 + CF..---CF t rH.OCr-CF Terpolymer (A39)
5 «. 1 1 J - 0.64 (in FC-43)
22

The trifluorovinyl ether is extremely reactive with trifluoronltroso- methane, copolymerizing at -78' in a few hours. By comparison, tetra- fluoroethylene and CFjNO require periods of about one month at this temperature. In addition, elemental analysis indicates that the copolymer contains greater than 67 mole % ether. As far as can be determined, this is the first time a nitroso copolymer containing more or less than the equivalent amount of one of the monomers has been prepared.
The copolymer is a tough, colorless gum which is insoluble in Freon 113 and FC-43. A terpolymer containing about 10 mole X ether was soluble in FC-43 and had an intrinsic viscosity of 0.64. Attempts to cure the terpolymers using peroxide have to date been unsuccessful.
Other copolymerizations and terpolymerizations, including unsuccessful attempts, are described in Tables III to VIII.
23

C NMR Analysis of CF NO/CF -CF Copoiymers
On the basis ot NMR data, researchers at 3M" reported that a small percet.Lage ot abnormal" linkages appear to be present in the nitroso polymer backbone These abnormalities could result from a deviation in the normal :iead-to-tall structu e to give linkages such as:
+NOCF.CF ON? and +ONCF CF N0> I CF. CF< CF. CF
NMR spectra ot nitroso polymers prepared in our laboratories have consistently shown additional fluorine NMR peaks, occasionally in appreciable amounts In addition to the normal peaks found at -12.0 (CF3), +11.4 .CF.) and +23.7 (CF,) ppm with reference to trifluoroacetic acid, peaks are aimc.-,t invariably found at +14 0 and +21.5 ppm. These peaks amount tc anywnere trcm 14 to 16% of the area attributed to CF2. Also, se-.eia- polymers have shown additional CF, and CF > peaks suggesting not only abnormal linkages, but also the possibility that a certain amount ot non-alternating polymerization may occur to give units such as:
«NOCF.CF.CF C?J and +CF,CFdN 0 N 0 >
CFj CF, CFj
Although these additional peaks may be due to higher molecular weight cyclic compounds, it has been definitely established that they are not due tc dissolved oxazetidme, CF-N0CF;CF, NMR results are summarized in
I II Table IX Another unusual observation can be made in that the total CF3 content ranges from 41. 7 to 58.0%, The peak area for CF? in an alternating 1-to-l copolymer should be about 43%. Also, polymers prepared under practicall> identical -ondltions sometimes exhibited gross differences in spectra as exemplified by samples 78.4 2 and 78.5.2. It should be roted that the polymers listed in the table are all intentionally of relatively low molecular weight to allow the possible detection of end groups by NMR.
Due to the complexity of the problem of analyzing these polymers by NMR, and because of the increased interest in the functional lerpolymer systems, this study was not pursued.
24

III. EXPERIMENTAL
A. Monomer Synthesis
1, Synthesis of Sulfur-Containing Compounds
a. Thiocarbonyl Fluoride8
2,2,4,4-Tetrafluorodithietane (4.0 g.) was ^dded dropwise over a period of 15 minutes through a Pyrex pyrolysis tube consisting of a 12-inch Vigreaux column with a 6-inch heated zone. The tube was maintained at 52CT-540' and a slow stream of nitrogen (25-30cc/min) was passed through the system during pyrolysis« Products were caught in traps at -183c. Trap to trap distillation indicated that an appreciable amount of material had passed through unreacted. The more volatile portion was primarily F2C=S. An early attempt tc store this material in stainless steel at tuom temperature resulted in decomposition No difficulty was encountered, however, when the material was stored at -78° in a glass Fischer-Potter tube. The infrared spectrum of this compound is shown in Figure A2.
b, Perfluorothloacetone
Reaction of bis(perfluoroisopropyl) mercury with sulfur
Two attempts were made to prepare (CFO^CS from [ (CF <) £CFj 2Hg and sulfur. The first time a water-cooled condenser was used, and it soon plugged. In the second attempt, described below, an air-^joled tube one inch in diameter was used as a condenser
A 1-llter, 4-neck flask was half tilled with melted sulfur and fitted with a condenser, thermometer, stirrer, and addition funnel containing [(CFj^CF]^Hg, The sulfur was heated to its boiling point and the mercury compound slowly added. The condenser was vented through a trap cooled to -78°. Some blue material was caught, but after standing for several days it turned to a white solid and, on warming to room temperature, to a colorless liquid Perfluorothioacetone is reported to be a blue liquid which easily dimenzes, and it appears that (CFO^CS was made in this reaction but dinerized «raring storage.
25

9a Pytolysis ^t 2.2 .-«,^-tecrakis j it llluotcmethyl )-l, 3-dtthiet.ane
Fetlluorothioa~etcne was piepazed by the pyrolysis of 2,2 ,i,<*-te:rakis (tt it luorumethyl >-l,3-dithietane. An addition funnel containing iCF-) CSClCF IS was connected to a 12-inch Vigreaux column
heated to 600 C The column was vented to a ttap cooled to -78°. The dlthietane was slowly dtopped into the heated column as it was being swept with a stream ot N; The product was trapped as a blue liquid. An infrared spectrum of the compound is shown in Figure A3.
Ttitlootomethyl it ifluotovlnyl Sulfide
CF -SCi
Sodium tiuc-tide \400 g. , 9.52 moies) and tetramethylenesulfone U000 ml > were placed in a 2-liter, 4-ne^.k flask fitted with a thermometer, stlrrer, spiral .^ndenset, and addition tunnel The funnel contained CC1 SCi '404 g. , 1 19 moles) which was added to the reaction flask over a ten minuit period. The contents of the flask were stirred and heated at 160-170 , and the volatile products were trapoed at -78°. These products were distilled, and CF ,SCl» v89 g., b.p 0-9') and CF3SSCF3* (69 g., b p l~> -34 ; were obtained,
Irifluoromethanesultenyl chloride was also prepared by the gas phase reaction ot bisltrifluoromethyl) disulfide and chlorine. The dlsulfide »180 g , 0.891 mole) was bled into a 72-liter evacuated flask. Chlorine '68.06 g , 0,891 mole) was added and the mixture irradiated two hours by ultraviolet light using a Vycor immersion well. The product was removed and distilled to give CF.SC1 (.204 g., 84% conversion).
CF.SCFC1CFC1
A 72-iiter liask was evacuated and charged with CFjSCl (243 g., 1.78 moles 1 and CF =CFC1 (86 g., 0.74 mole) and irradiated for three hours by a Hanovla model 8A36 high-pressure mercury vapor lamp suspended in a Vycor immersion well extending into the flask The products from two experiments were combined and distilled to give recovered CFjSCl (220 g., 1.62 moles), material boiling neat 44 (100 ml ) and assumed to be a mixture ot CF.SSCF and CF C1CFC1 , product (95.3 g.; boiling at 78<"-83°, ind poc residue 80 g ) assumed to be telomers with the composition CF.S'C F Cl< Cl. NMR analysis of the tra:tion boiling at 78'-83° showed it to be D^.XCF SCFC1CF Ci'and 43% CF.SCF CFC1 ,.
These compounds have been reported to be highly toxic.
26

CF;SCF=CF;
1 1-liter, 3-neck flask was fitted with a stirrer, addition funnel and reflux condenser leading to a cold trap. The flask was charged with dioxane (50C ml.), powdered 2inc (.75 g,), and a trace of zinc chloride. The mixtu-e was heated to reflux and a mixture of 57% CF3SCFC1CF,C1 - 4J% CF3SCF2CFCI2 (95 3 g. containing 0.215 mole CF^SCFCICF.CI) was added over 90 minutes. Volatile material was caught in tha cold trap and distilled using an 18-mch spinning band column. Material boiling from 13t-20° was collected with the major fraction boiling at 18J-20 A total of 32.4 g. t.83% conversion) of CFjSCF-CF, was obtained. Further distillation of the reaction mixture gave 22 g. of 95% pure CF3SCF2CFCI2.
Anal, Calcd. for C3F6S: M W 182, Found: M.W. 180.
An infrared spectrum of pure CF,SCF=CF2 is shown in Figure A4 and the NMR analysis in Appendix B. A spectrum of 95% pure CF -SCF.CFCl, is shown in Figure A5.
d. Trifluorovinylsulfur Pentafluoride
SF5CI
Cesium fluoride (344 g., 2.26 moles), SF C216 g., 2,0 moles) and chlorine (142 g,, 2.0 moles) were reacted in a 1,5-liter autoclave according to a reported procedure.1" The product mixture was washed by passing through two columns of water about 4 feet in length and 225 g. (69% conversion) of SF;C1 obtained. GLC showed it to be 98% pure,
CF2CICFHSF5'3
A 12-liter flask was charged with SF:C1 (31 i g,, 0.192 nole) and CF;«CFH (15,7 g., 0.192 mole) and irradiated with u.V. light for 10.5 hours. No liquid product was formed and infrared spectra of the overgas showed no evidence of reaction. Product was obtained by carrying out the reaction as described below,
A 300-ml. autoclave was charged with CC1» (15 ml.), benzoyl peroxide (0.6 g.), SF5CI (60.2 g., 0.37 mole), and CF;-CFH (37 I g., 0.45 mole) and heated at 150' for six hours. The liquid product was distilled and 64.2 g, of material was obtained over the range 58"-61". Analysis by GLC showed it to contain 82% SFcCHFCF.-Cl (.52.4 g., 0,215 mole) Conversion was 58%
CF2-CFSF$U
Heptane (215 ml.) and SFSCFHCF2C1 (.52,4 g., 0,215 mole) were placed in a 500-ml., 3-neck flask fitted with a stirrer, tube for addition of solid KOH, and a reflux condenser vented to a trap cooled to -186". The solution was heated to reflux, and powdered 10H (38.5 g., 0.69 mole) was added in small amounts. A white solid was trapped in the -186' trap
27

Ihis jicuiu. was diötixitd aüd an lntraied apt.cram cf the product boiling at Is -18 shewed it tc De SF-CF-CF v20.j g , 0 098 mole) of 80% purity by GLC
t. 2-Ch.LGio-l-riit iüSü-1,2,/-i i itiuoroethyl Inf lüiromethyi Sulfide
Iwo preparations were arried out as follows. A 1-liter Vycor flask wa; charged with CF SCF-CF (2 6« g , 0.02 molej and C1N0 (1.31 g., Ü 02 mole» and placed in sunlight IOI several hours. At the end of this rime it .ontained a blue liquid <ind a blue overgas. This product was separated by GLC using an 8-mch column packed with Kel-F Acid 8114 ethyl toter and pure CF bCF.NO/CF Cl (0 4 g., 0 0016 mole) was obtained. An infrared spe.tr-m >-t this material la shown in Figure A7 and the NMR analysis if Apptncux B
t Attempted synthesis of Carboxysuifur Pentafluoride
SF C.H CH Cx
A 12-litei tidSk titted with a V> .or immersion well was evacuated and naiged with SF Cl ;60 3 g., 0.371 m<le) and CH.=CH> (7,70 g., 0.2"5 mcie» Iht mixture was irradiated lor six hours and then removed and distilled, giving 32 g \bj.Z .onversion) of SF-CH CH^Cl.
SF CH«CH
A 300-mi., J-ne^K riask wa& titted with an addition funnel, stlrrer, and reflux .jnatnser ented to a trap cooled to -78°. Potassium hydroxide < i8 0 g , 0.32a. mole I wao placed in the flask and h^O (20 ml.) was added As =oun as the KOH dissolved, C,H OH (60 ml.) was added. The solution was heated to retux and SF CH,CH,Ci (32 0 g. , 0.168 mole) was added from the ^dditicn tunnel Ihe mixtjte was retluxed for one hour «?:»d distilled giving 3.5 g <2lZ ;onver»ion ot sF CH=CH , b.p. 38"-43t;.
SF COOH
By oxidation ot SF CH-CH : \lnylsulfur pentafluoride (1,35 g., 8. " mracies wns p*.a.ed in a 3-ne.k, 100-ml tlask fitted with a stirrer, retlux -cndenstt, and addition tunnel i_ontaining a solution of KMn04 2 5 g , lb mmoies) in acetone i>? ml ) and H\0 (12 ml ). The KMnOu
sciuticn was tiowly dropped into the reaction pot and MnO<> formed almost lmroedlcuely Alter stirring tor several hours the MnO, was filtered off,
and 'he filtrate was a.idified with H SO A white solid formed immediately. This solid was filtered eft and an infrared spectrum was made, but it did not shew the presence et ar. S-F group The filtrate was extracted with ethyl ether and au inflated spe^ti^m made ot the extracts, but it did net show S-F absorption
28

By reaction of carbonyl fluoride wich sulfur tetraf luonde: Two attempts were made to react SF4 with COFt using CH3CN as a solvent and one with tetramethylene sulfone as a solvent. The results of all three reactions were similar. One of them is described below.
A 300-ml. autoclave was charged with CsF (3.5 g., 0,023 mole), CH3CN (50 ml.), SF. (28.0 g., 0.26 mole), and COF^ (17.0 g., 0.26 mole) ana heated at 200° for eight hours. The volatile products were removed and examined by infrared spectra. There was no evidence for the presence of any compound containing an S-F group. When the autoclave was opened it was found to be partially filled with carbonaceous material. Tt appears that reaction occurred with solvent.
By reaction of SFsCl with outyl lithium and carbon dioxide: A solution of butyl lithium (3.5 g., 55 mmoles) in hexane (^1,7 molar) was placed in a small 4-neck flask fitted with a stirrer, gas inlet, gas outlet, and low temperature thermometer. The reaction flask was cooled to -78" and SF5CI (8.72 g., 54 mmoles) was bubbled in at a rate so that the temperature did not rise above -55c. After the SFsCl was added, CO- (0.2 mole) was oubbled In at the rate of 80 cc/min. The solution was then allowed to warm to room temperature with continued carbonation. The solution was poured into 100 ml. of 6N HC1 and a 2-layer mixture resulted. The hexane was evaporated and a layer of H^O over a small amount of orange liquid was left. This liquid was separated and dried and an infrared and nuclear magnetic resonance spectrum were made. Nei:her spectrum showed evidence for SF5, The water layer was extracted wir.h ethyl ether; the extract was dried and an infrared spectrum made, but it showed no SF^. absorption. The remainder of the water layer was then neutralized with NaOH. A solid was filtered off, but again it showed no evidence for the presence of a sulfur-fluorine compound.
g, Attempted Synthesis of SF- NO
Reaction of disulfur decafluoride and nitric oxide at 150°
An 80-ml. Fischer-Porter tube was charged with StFi0* (2.06 g., 8.1 mmoles) and NO (0.486 g., 16.2 mmoles) and heated at 150c for three hours. During the reaction the tube became etched. The volatlles were separated by VPC and identified as 1TO2, SO_Ft, SOF2, S02, GFcOSFe, NO, and S2F,0.
A 300-ml., stainless steel autoclave was charged with S 2 F J 0 (2.80 g., 11 mmoles) and NO (0.66 g., 22 mmoles) and heated at 150° for two hours. An infrared spectrum of the overgas showed N20, SOF2, SO2F2, and SF-. It was then heated for an additional 24 hours. Another infrared spectrum showed that SFL, had disappeared and that the major product was SOF2. There was no absorption to indicate the presence of SF5NO.
Sample donated by Dr. James W. Dale, Monsanto Research Corporation.
29

Reaction of disulfur decafluorlde and nitric oxide in electric discharge tube
A 1-llter flask was charged with S>F g (2.80 g,, 11 mmoles) and NO (0.66 g,, 22 mmoles) and connected to an electric discharge apparatus (Fig. 1) powered by a 15 kv, 30 mamp. transformer. A pressure of 5-7 mm was maintained during reaction. The products were caught at. -196" and later separated by VPC. They were identified by infrared spectra as CF ; TCI; SiF.; NO; SFb; 50* ; SF5C1; S0t>: SF..(S0 ,F) 2; and an unknown with infrared absorption at 6.8, 8.12, 12 10, and 15.65 microns. The major product was SO.;. The CF4, CFiCl, and SF5C1 probably resulted from reaction with Kel-F grease used on the -joints of the discharge apparatus A small amount or sulfur was deposited in the discharge tube.
A 1-llter flask was charged with SOF,Q (2.80 g., 11 mmoles) and NO (0.66 g., 22 mmoles) and connected to the electric discharge apparatus powered by a 7.5 kv, 13 mamp. transformer, A pressure of 2-3 mm was maintained during reaction \ stream of air from an air gun was directed across the discharge tube during reaction, but it still warmed above room temperature. One-half of the material was reacted under these conditions, and the products were caught at -196'', These products were separated by VPC, and infrared analysis showed them to be CF^, CF3C] : C1C(0)F; SiF_; SFf; N 0; S0^; S2FJQ; unknown with infrared absorption at 6.8, 8 12, 12.10, and 15.65 microns; and unknown with strong absorption at 8.61 and 9.06 and very strong absorption aroui' 11.0 and 11.7 microns.
One-half of the material was reacted with the discharge tube cooled to -78". These products were caught at -196° and separated by VPC. Infrared analysis showed them to be SiF.; SF6; N^O; S02F^; SOF2; S02; SF j; and an unknown showing absorption at 6.8, 8.12, 12.10, and 15.65 microns.
Reaction of sulfur tetrafluorlde and nitrosyl fluoride
A 2075-ml. nickel reactor was evacuated and charged with sulfur tetrafluorlde (250 mm., 3.0 g., 0.0277 mole) and nitrosyl fluoride (500 mm, 2 7 g. , 0.0554 mole) The reactor was heated at 300° for two hours with iltcle apparent reaction as determined by infrared analysis. Additional heating for two hours at 400", however, resulted in a reaction. Infrared analysis showed the presence of SOF.. Gas Chromatographie separation of the 1 roduct mixture on a Chromosorb-Kel-F 8114 ester column gave six peaks identified (in order of elution) as NO, SFb, N20, S02F^, S0F2 and N02-S02.
Sulfur tetrafluorlde (5.8 g., 0.054 mole) was condensed into a trap ac -78' Nitrosyl fluoride (5.5 g., 0.112 mole) was condensed into this yellow liquid, resulting in a green solution which was separated by warming to -40' to remove unreacted FNO. The higher boilers were washed free of SF by bubbling through water. Infrared analysis of the products showed the presence of NO, SOF,, NO,, and SFcCl. Chlorine may have been introduced into the system either by Kel-F grease or by reaction with the NaCl windows of the infrared cell.
30

Reaction cf nltrosyl chloride and SF Cs_
A 300-ml flask containing a mixture of CsF ('0,8 g., 0.025 mole) and CsSFs ( >-6.6 g. , .025 mole) was charged witn NOC1 (.1,67 g, , 0.025 mole), and the contents stirred. The volatile produ^.s were removed to a trap in liquid air and later separated by GLC They were identified by infrared spectra as CINO, NO.., SiF., SOF,, S0,F , SF., and SFtCl. There was no evidence for the formation of SF^NO.
Reaction of sulfur tetrafluoride, cesium fluoride, and dlnitrogen trioxide
Two reactions of CsF, SF , and N^0< were carried out as described below
A 1.4-liter autoclave was charged with SF, (23.5 g. , 0,218 mole), CsF (34.4 g. , 0.226 mole), and N.O, (20.7 g , 0,2?/ mole) and heated at 100^ for one hour, 150 for one hour, and i75* tor two hoars, The only product isolated other than starting material was SOF,.
A 75-ml. monel cylinder was charged wi SF. (4.32 g,, 0.04 mole), and CsF (6.08 g. , 0.04 mole) and heated at 150" for five hours. The cylinder was then charged with N.-Oj (3 04 g. , 0.04 mole)and placed in an ice bath. The cylinder was allowed to warm to room temperature over the weekend and the volatile products were analyzed The only products other than starting material were SOF, and a trace of S0£F .
Reaction of SF-Cl and nitric oxide
A 300-ml. autoclave was charged with SF<-,C1 (1.86 g., 0.0115 mole), NO (0.345 g. , 0.0115 mole), and benzoyl peroxide (0.3 g ) and heated at 100" for three hours. An infrared spectrum of the overgas showed that the only product formed was a small amount of SOF;
Reaction of dlsulfut decafluoride and nitrosyl chloride
A 1-liter flask was charged with DMF «20 ml.), A1C1. (1.0 g.), S2F o (2.92 g., 0.0115 moie), and CINO (0.75 g., 0 0115 mole), and the mixture was stirred for 0.5 hour at room temperature. An orange solution ever a yellow solid developed. An infrared spectrum of the overgas showed only unreacted starting material. The reaction vessel was placed in a 70-85° bath for two hours with no further change.
h. Attempted Synthesis of Tritiuoromethyl Thionltrite
Reaction of CFJSAR and nitrosyl chloride
An 80-ml. Fischer-Porter tube was charged with CF.SAg (1.00 g., 4.78 mmoles) dissolved in 1.0 ml. of dimethyl!ormamlde and C1N0 (0.244 g., 3.76 mmoles). Nitrosyl chloride was charged into the tube by freezing it in liquid air, evacuating, and condensing from a tared flask.
31
After the tube was sealed it was allowed to warm to room temperature, but before it had warmed completely a reaction took place, The mixture was allowed to stand overr; ?ht it room temperature before an infrared spectrum was made of the overgas. It showed only CF^SSCF^. A white solid, probably AgCl, was also produced.
Reacticn of bis(trlfluoromethyl) disulfide and nitric oxide
BisUrlfluoromethyl) disulfide and nitric oxide were reacted using both u.v. light and electric discharge. Three attempts were made to prepare CF ;SNO by reaction of CF>SSCF3 wlth N0 ln the presence of u.v. light. The following is a typical reaction,
A 300-ml. ouartz flask was charged with CF3SSCF3 (0.354 g., 1." mmoAes
> and NO (0.105 g., 3.5 mmoles) and irradiated with u.v. light trom a Kanovia utility lamp, Type 30620, for 32 hours. The following compounds were round to be present in the indicated ratios (by gas -hromacogram):
Relative Relative Relative Compound Ratio Compound Ratio Compound Ratio
NO 190 Unknown 5 S0? 470
CF jNO 630 CF3SCF3 40 CS2 330
NO liO CF 3N0 2 30 CF2SSCF2 470
It is very likely that the CS2 was originally present in the CF3SSCF3 as an Impurity.
The reaction produced CF3NO (1.3 mmoles) in 38% conversion. This amount represents 90% of the material formed from CF3SSCF3.
A small amount of CFjSSCF. was placed in the bottom of the exectrlc discharge tube which was cooled to -78*. A 1-liter flask charged with 500 mm. cf NO was connected to the apparatus, and NO was passed over the CF SSCF- while a discharge was produced by a 15 kv, 30 mamp. transformer. The volatile products were trapped in liquid air and later separated by VPC. A small amount of very viscous material, insufficient for analysis, was left ln the discharge tube. Infrared spectra of the volatiles showed them to be CF3CI; C1FC-0; CF3SCI; N0C1; NO; N02; CTV, N20; S02F2; S0F2: CF.NO^; SO,; CF3SSCF3; unknown with absorption at 6.8, 8.12, 12.10, and 15.65 microns; and an unknown eluted from the VPC column about 45 minutes after CF3SSCF3 with absorption at 8.1, 8.3(s), 9,0(s) and 13.1 microns.
Reaction with trifluoromethanesulfenyl chloride and nitric oxide
Two reactions of CF3SCI with NO were run. In the first reaction a 1-llter Vycor 7910 flask was charged with CF3SCI (1,46 g,, 10.7 mmoles) and purified NO (0.32 g. , 10.7 mmoles). The materials 1 ere frozen into the flask as a white solid, allowed to warm to room temperature, and

irradiated with ultraviolet light for 16 hours, An infrared spectrum of the product mixture showed N0?, SO;, CF3SCI, CF.NO, CF-NO,, and SlF .. An attempt was made to separate the mixture by GLC, but unsatisfactory resolution was obtained.
In the second reaction a 1-liter flask was charged with purified NO (0.642 g., 0,021 mole) by freezing it in as a white solid. As soon as the CF3SCI (1.46 g., 0,011 mole) was condensed in, the solid material became bright red. On warming above the freezing point the color disappeared, but when the sample was solidified again the bright red color returned. Infrared spectra of the gas phase showed only starting materials, The reactants were transferred to a flask containing absolute ethanol and cooled in an attempt to develop the red color in a liquid phase, However, no color developed even when the alcohol was cooled to its freezing point, An infrared spectrum still showed CF .SCI and NO.
Reaction of trlfluoromechanesulfenyl chloride and nitrosyl chloride
Two reactions of CFiSCl with N0C1 were run. The following is a description of one reaction.
A magnetic stirring bar and several ml, of Hg were placed in a 1-llter flask which was then evacuated. This flask was charged with CF3SCI (1.18 g., 0.009 mole) and NOC1 (1.71 g., 0,026 mole), immersed in ice-water, and the contents stirred for three hours. The volatile products were removed to a trap in liquid air and later separated by a co--distillation. Some of the products were identified by infrared spectra as NO2, C1N0, SiFu, CF3SCI, CFjCl, COr'2, and CF^NO,, In addition to these products there was a continuous elution of material which gave absorption in the CF region and in the NO? region, evidence that sample decomposition was occurring.
Reaction of bls(trifluoromethyl) disulfide and nitrosyl chloride
Three reactions of CFsSSCFj with N0C1 were run. The following is a description of < ne of them.
A 1-liter Vycor 7910 flask was charged with C. SSCF, (2.16 g,, 0.011 mole) and C1N0 (0.53 g , 0.011 mole) and placed in sunlight for four hours. As soon as the flask was placed in sunlight the brown color 01 the C1N0 began to fade. At the end of four hours the flask was filled with a light yellow gas and covered with a thin, white, solid coating. The volatile products were separated by GLC and identified by their Infrared spectra as C1N0, CF3SSCF3, NO, N02, S02, CF3SCI, CF3CI, CFjNO, CF3NO;, and an unknown eluted after CF3SSCF;.
33

Rea~:lcn it tfu.^drbcnyi tlurnde and mtiosyl fluoride
Niuosyl fluoride U g,) was condensed inco a Pyrex Fischer- Potter :ubt . i.ntaining ab^ut x g. oi impure thiocarbonyl fluoride. ihe tube was mi^wtd to waim and an lnirared spectrum was made of the overgas, whi_h was t.und to contain $1$ , COS and N 0. Absorption between 8 and ■*.2 microns and dt iJ.i" microns indicated a CF ,S group, Other unassigned peaks occurred at. 6,15, 6,25, and 6,52 microns, The latter peak is prcbabiy due to CS whiie the first two peaks may be due to NO, The »pe.trum -har.ged over a period of time, indicating decomposlton of the sample at room temperature.
l. Attempted Synthesis of Pentafluorophenyl Th.-pnit r ite
At. 80-nu, Fis^her-Pcrter tube was charged with pentafluorothie- phetu- i8.0 g , 0.0^0 mcie> and sealed. Ihe contents were frozen in liqu.1 nitrogen, trie tube was evacuated, and nitrosyl chloride (2.8 g. , 0.0^.3 ra^it) condensed in. The tub* was placed in a -A0C bath where it liq-it^ed and then lap'd.v solidified. The tube was warmed to room temperature An lnUared spectrum of tne overgas showed NO, HN0, SiFu, and HC1, The cvergas \ ^s pumped off and the solid (8.0 g.; wat, washed with ben^enc and dried. it was identified as CgF^SSC-Ft, m.p, 49.5°-51° 'rptd. m p 30 -5i ), m.wt. it.p. in benzene) 388 ualcd, 398.2). Corivtisicn uas 100% An infrared spectrum is shown in Figure A8.
j. Attempt d Synthesis ot 2-Chloro-L-Nltroso- 1,2,2 ltluoroethylsultur Pentafluocide
Trltlucrovinylsuitur pentafluoride was reacted with nitrosyl htcilde in b^th the gas phase and in solution .• iescribed below.
A 1-iittr V>coi riask was charged with SF:CF=CF; (2.39 g., 0.01x5 moie» and C1N0 i0 ?5 g., 0,0115 mcle) and placed in sunlight. After abo^t :r.e hour the gas began to turn green, An infrared spectrum of the mixture at this point showed absorption at 6.2 microns in addition to new peaks in the C-F region. Ihe flask remained in sunlight over the weekend.
this time there was a light green liquid preü^.nt, but no colored overgas, AU lr.tiaied spe.u-tt showed only very weak absorption in the 6-6.5 micron region.
An 80-m-. tube was charged wich dimethyl tormamide (8.0 g,), A1CI (1.0 g i, Sf CF-CF (2 08 g., 0.01 mole), and C1N0 (0.65 g., 0.01 mole' As soon as the tube warmed to room temperature, a green solution with a blue overgas developed. After standing tor about one hour the mixture developed into a blue gas over a two phase liquid (green over blue), The blue overgas was trapped in another container and the boctom layer of blue liquid bodied oct with l'. The product was purified by GLC and .Jentnltd by its infrared sKc.trum as CF ClCTCiNO. No evidence was found id tne presence ot SF CF(NO)CF Cl.

k• Attempted Synthesis of 2-Chloro-2-Nittoso-l,1.2t-Tnf luoroethyl Trifluoromethyl Sulfide
A 1-liter Vycor flask was charged with CF3SCF2CFC1I (4.50 g,, 0.013 mole) and NO (0.51 g., 0.017 mole) and placed in sunlight for four days. At the beginning of the reaction the liquid reactant was almost colorless. It continuouslv turned a darker red and iodine began depositing on the walls of the flask. An Infrared spectrum of the overgas showed absorption in the -NO regioa, 6,2 microns, as well as the C=F region 8-9 microns. It appeared that the major reaction was the decomposition of the starting material.
2. Synthesis of Fluoroaromatic Compounds
a. Fentdfluoronitrosobenzene '
A solution of 90% H202 (100 ml.) and 90% HCOOH (400 ml.) in CH2C12 (2 1.) was placed in a 5-liter, 3-neck flask fitted with addition funnel, stirrer, and ice-water-cooled condenser. A solution of CgFsNH^ (102 g., 0.55 mole) in CH2C12 (500 ml.) was rapidly added dropwise to the stirred solution, Shortly after addition was begun, the solrtion turned blue, then deep blue-green. Heating to reflux was begun end after about five minutes the solution was brown. In 15 minutes the color was green- brown and after 30 minutes the solution was again green. Refluxing was continued for five hours and the solution was then washed with six 500-ml. portions of water. The organic layer was dried over anhydrous magnesium sulfate and the solvent stripped until a pot temperature of 77s was reached. The remaining liquid was then distilled on an 18-inch spinning band column giving 48.8 g. of product, b.p. 51,J/20mm. The pot residue, presumably C6F5NO2, weighed 40.5 g. Recrystallizatlon from pentane (2 ml./g.) gave 39,9 g. of product, m.p. 42t'-44c. The compound may also be purified by sublimation.
b. Attempted Preparation of p-Nitrosotetrafluorobenzoic Acid
Tetrafluoroterephthalic acid (10 g., 0.042 mole) and water (500 ml.) were placed in a flask and heated 'o 60' while Ag C (4.98 g,, 0.021 mole) was added in small amounts as it appeared to react. After about two hours the solution was evaporated to half its volume and then cooled. Some solid formed and was filtered ofi. The i;ltrate was washed with acetone and more precipitate formed and was filtered off. These filter ca^es were recrystaliized twice by dissolving in hot water and precipitating in excess acetone. The final product was a white solid which was oven dried nnd found to weigh 7 g. Analysis gave 32% Ag. Theory for H02CC6F/.C02Ag is 31.3% Ag.
35

Several grams of the silver salt were placed In an ampoule and »haken at icom temperature with excess C1NC. When the C1N0 was removed, a yellow soijd remained. This solid was heated at 200" under complete ^a^uum with almost no noticeable change. An attempt was then made to isol <te the product from the AgCl which was formed during its preparation by dissolving it in ethyl tther. However, the ether appeared to be wet as the product immediately turned white and gave off a gas which infrared showed to be NO. An infrared spectrum of the solid residue showed it to be tetratluoroterephthalic a«_id.
p-Amino r,e tr at 1 uo tonitroso benzene
Mcthyiene chloride (175 mi.), 90% Ht,0v (20 ml.) and 90% HC02H (0 ml.) were (.laced in a flask fitted with a stirrer, reflux condenser, and addition lunnel containing p-NH. C..F NH '9 g,, 0.05 mole) in Cl^Cl? <200 ml ' This solution was added dropwise while the reaction mixture was heated tu reiiux. The solution turned orange-brown and remained that coj.Gr during the ij.ve-hour reduxing. The mixture was washed with water .and dried over Na.SO. It was then transferred to a sublimation apparatus and the solvent removed under vacuum. After the solvent was removed, the soiid was heated at 100 under total vacuum, and a green solid deposited on the cold finger (0.4 g.) Left in the sublimation apparatus was 4 g, of a red-brewn solid which had an odor similar to Ct-F-NO^. An infrared spectrum of the product J.S shown in Figure AJ 2.
d, Attempted Synthesis of 4,^'-Dinittosooctafluorobiphenyl
A solution oi 4,4'-dlamlnooctafluorobiphenyl (23.5 g., 0.07 mole) in CH;C1 16OO ml.> was added rapidly to a stirred solution of 90% formic acid (100 ml.), 90% hydrogen peroxide (25 ml.), and CH^Cl^ (5C0 ml,). After initial formation of a deep blue-green color, the color faded to reddish-brown After two hours at reflux a green color reappeared. Reflux was .ontmued tor five hours. The yeliow-green solution was washed with six 500-mi portions of water and dried over MgS0_. The solution was stripped of solvent to a volume of about 100 ml. On cooling, 15.1 g, of yellow solid precipitated. The meeting point of the sample depends on age, purity, ana temperature at which the sample is placed in the melting point apparatus. The true melting point appears to be between 175c-185°. A second run using only 1 g. of the amine gave identical results.
3. Synthesis of Nitroso Esters and Derivatives
a Synthesis .t CH.0,C(CF I^CO.NO
The nitrite was prepared by two methods - reaction of CH.O C(CF ) CO.Ag with nitrosyl chic -ide and reaction of perfluorosuccinic anhydride with methyi nitrite. The iacter method is described here.
36

Methyl nitrite (12,2 g., 0,2 mole) and perfluorosuccinic anhydride (34,2 g., 0.2 mole) were condensed into a Fischer-Porter aerosol compati- bility tube and allowed to warm to room temperature, The tube was shaken to insure thorough mixing and soon became warm, After about 1-2 hours the tube had cooled and contained an amber liquid. Unreacted starting material was removed at reduced pressure. Yield was nearly quantitative,
Anal.: Calcd. for C5H3FuN05: %C, 25.76; %H, 1.29; %F, 32.60. Found: %C, 25.84; %H, 1.33; %F, 45.25, 36.91.
An infrared spectrum of this compound is shown in Figure A14 and the NMR analysis in Appendix B.
b. Synthesis of CH30?C(CF2)3C02NO
Methyl nitrite (12.2 g., 0.2 mole) and perfluoroglutaric anhydride (44.4 g., 0.2 mole) were reacted as described above with similar results.
Anal.: Calcd for CfH3F6N05: %C, 25.44; %H, 1,06; %F. 40.28. Found: %C, 25.70; %H, 1.28; %F, 40.53.
An infrared spectrum of this compound is shown in Figure A15 and the NMR analysis in Appendix B.
c. Synthesis of CH302C(CF2)2NO
A 250-ml,, 2-neck flask was fitted with an addition funnel containing CH302C (CF2)2COONO (52 g. , 0.223 mole) and a 15-inch Vigreaux column which was fitted with an air-cooled condenser constructed on the order of a Dean-Stark apparatus. The condenser was vented to a vacuum system through a -183° trap, and a total vacuum was maintained rhoughout the system as the nitrite was dropped into the flask which was heated to 200°. The Vigreaux column was heated to 250°. After the pyrolysis had been going for several minutes, a blue product collected in the -183° trap and a colorless liquid began to condense in the air-cooled cc^-ien- t, This liquid was periodically removed. The -183° trap was allowed . ;m to room temperature and the remaining liquid product was washed water. The blue product was separated and distilled twice by <1 ■>■ _. ling from one trap to another under vacuum, discarding the last several ml. of liquid each time. A GLC of the final product (8.3 g,, 20% conversion) showed it to be 100% pure.
37

Anal . Ca...a Ui C H F Nu.: :.C, 25.aO: %H, 1.60; %F, 40.21,
Found: %C, .^.bb; ZH, ..2; %F, .0 kl.
An intrar.cd spectrum oi eras compound is shown In Figure A16 and
the NMR analysis in Appendix B
d. Synthesis ct CH .0 C CF ) .NO
By py r yl y' l 3t ■Jiiibcm^i_ai ion
A 250-nu 2-r.t.K tiask was fjtted with an addition funnel v.iuai.nmft CH 0 C'CF / CuONO ,n.O g. , O.1I6 mole) and a 15-inch Vigreaux clumn vented to a -a....um syotem through .=. trao cooled Co -183°. A
'-jaipAc'.t vaLuutn was mairicained if.roughout the system as the nitrite was
dropped into .ne tiasK whi_n was heater* to ^00'. The column was heated r. 2"0 Attet r ht pyrolysii was -lompleted, the material in the -1831" trap was allowed tw wairr. to toom temperatute, the nitrogen, oxides were
removed ander vd-uum, and the blue, liquid residue was washed with water.
Ihe blue ptoau.it was separated and distilled twice by belling from one
ttap to anoch-r onder vacuum, discarding the last several ml. of liquid each t^mc. A oLC or the final product showed it to be 100% pure. A total
ci 13 g ci pure CH0 C'CF ) NO was obtained. Conversion was 47%.
Anal.: Caivd tor C HF.N0.: %C, 25 11; %H, 1,25; %F, 47.70. Found: K, i; 30; %H, 1.x/; %F, <<7.40.
B> phot^yti. de^ot boxy idt ion
^-Car bemet hcxyper nuorobutyryl nitrite (40 g,, 0,14 mole) was pia^cd in a i-iitei t.ask equipped with a Vycor immersion well and an outlet -onne^ted t.. a va.-um system through a trap cooled in liquid air, Iht nitrite w=,<- irradiated by a No. 8AJ6 Hanovia lamp suspended in the
immersion weii., Irradiation was continued tor ^8 hours at 0.1 mm pressure. The blue liquid pruduvit was removed turn the trap and combined with the
product ticm a similar experiment ubing 33 g. ot nitrite. The combined produ.t.- were Ira tionated to give 12 g. (20% conversion) of pure methyl '»-mi rosope t tluor obut yr ate , b p. 24 -26 /ib mm.
An infrared spectrum ci this ..ormound is shown in Figure A17 and the NMR and.iy.-is in Appendix B
38

e. Isolation of [CH ;.0;C(CF;) 3] 2NO(CF2) CO _.CH<
iligh-boiling liquid by-products from the preparation of CH3O2C (CF, .'• (NO were combined and fractionated using a 24-mch glass helix packed column. The major portion of this material was removed at 137°-140°/0.1 mm. Infrared (Figure A18) and NHR analysis (Appendix B) were consistent with the structure [CH 30?C(CF?) 3] ;NO(CF2,> .CO ,CH3.
Anal.: Calcd. for C, SH9F, 8N'0 -: %C, 27.45; %H, 1.37: %F, 52.00- %N, 2.13. Found: %C, 27.80; %H, 1.60; %F, 52.98: %N, 2.40.
f. Synthesis of [H2N0C(CF2) ,12N0(CF2)JCONH,
Ethyl ether (150ml.) and [CH302C(CF,).],N0(CF2)3C0,CH< (113.5 g., 0,173 mole) were placed in a 250-ml. flask and cooled in ice-water. Ammonia was bubbled into the solution with rapid stirring until uptake of ammonia ceased. Ether was removed under vacuum and the residue was ground to a powder and dried in a vacuum oven at 50" to give 76 g. (72% conversion) of a white solid, m.p. 157"-160':. Infrared (Figure A20) and NMR analysis (Appendix B) were consistent with the structure [H?N0C(CF?)3]2N0(CF2)3CONH2.
Anal.: Calcd. for Cj2H6Fi8N^0U: %C, 23.50; %H, 0.98; %F, 55.90; %N, 9.15. Found: %C, 23.59; %I1, 1.03; %F, 55.54; %N, 9.19.
g. Synthesis of [NC(CF2)3]2NO(CF2):CN
A 250-ml. flask containing thoroughly mixed P20t, (150 g,) and the triamide (72.6 g., 0.119 mole) from the preceeding reaction was heated to 200° under vacuum. A liquid product distilled from the flask and was caught in a cold trap. Distillation gave 21.7 p. (33% conversion) of [NC(CF2)3l2N0(CF2)3CN boiling at 110°-115
J/60 mm. Infrared (Figure A21) and NMR analysis (Appendix B) confirmed the assigned structure,
Anal.: Calcd for C;2F:0N„0: %C, 25,82: %F, 61.28; ZN, 10.04. Found: %C, 25.68; %F, 61.12; %N, 10.16.
h. Synthesis of H02C(CF2)3N0 by
Hydrolysis of CH-J02C(CF2) 3NO
Methyl 4-nitrosoperfluorobutyrate (10 g., 0.042 mole > was placed in an Erlenmeyer flask containing distilled water (60 ml.) and a magnetic stirring bar. Tht aqueous layer gradually acquired a blue color as the eater hydrolyzed. After five days the lower, organic layer was no longer present. The product, 4-nitrosoperfluorobutyrlc acid (7.0 g., 75% conversion), was isolated by salting out with sodium chloride.

l, A:tempted Synthe^i.-. 01 CH -CHCH 0 CvCF ) CONO
Anyi ni'.iUt
Iwc e>nthcito ci axivi nicnte were arried out, In the first synthesis no wate, «as used, and although some allyl nitrite was prepared,
tht tto.utn nii»tuie became badly charred. Ihe toilowing is a description
el the tfctond synthesis,
A i-iiies, >-rc.K tiask was fitted with a stirrer, a gas outlet
veriitd thiCLgh a tiap ..o.itd t^. -i83 to a v/avaum pump, and an addition t jiint x .ntd.diiii; i :0 m± of 9M H SO . Int.. ttie ilask wcte placed CH «CHCH OH ul6 g. , 2 moles;, NaNO; (175 g , 2.3 moles), and H,0 (100 ml.). The pressure Ud; tedu.ed iu approximately 600 mm and the H SO, was added
dropwiot. Atiti aD„jt O'' oi the H SO. had been added, the mixture began
giving .ii ctcup l.met and the reaction was stopped The material caught in tr»« -i8i trap w=^ o.stiiicd to give 1 ?5 ml. ol a paj.e yeliow liquid
belling a u- An .nt.jrcO spectrum showed typical nitrite absorption.
CH »CHCH U C CF CO NO
Iwc ua.'Kns wert .arried out In the first reaction perfluoro- glwtariv cfhydtide \t'' g , 0.12 mole) and allyl nitrite (IJ. g., 0.13 mole) wtrt enden^ed into an bC-nu tube at -183 After it was sealed the
tubt Wei a.^wtd :o warm to room temperature It detonated abcut 45 minutes alter wa;mmg began
in the st.^nd leactlon anhydride ( 39 °., 0,170 mole) was placed in a <:50-mi , J-ne^K iiaak containing a stirririg bai and fitted with a nitrogen inie. arid -utiet and an addition tunnel containing allyl nitrite
■ 22 g , 0.25 mole) Ihe tlask and its contents i^ie maintained at 0°
while the iiitutc wab äddtd aropvise Afttr tne addition was completed, the mixture \»a- otirred tci about one minute betöre a vigorous reaction took pia.e, leading a viscous, dark brown unidentified product
j. A', tempted Synthesis of ClOCiCF ) .CO,NO
Three attempts were made tc ieaa pertluotoglutaric anhydride with C1N0 to prepare: C10C(CF )-CO NO. in the first rea;tion a 20-ml. ampcL.it was harged with anhydride ( IO g., .05 mole; and C1N0 (6.5 g,,
0,10 mere1 and allowed to stand it room temperature tor two days.
Removal -f th more volatile material under vacuum left only the unreacted anhjdtide with no evidence tor the formation of the nitrite.
In the e.cnd tea^tion a 1-lite:, Vy^or flask was charged with anhydr.de b 6 g., 0,03 mciei and CiNC (0.2' g., .04 mole) and placed in sunligh On warming iv. ambient temperature the liquid phase became red and the octigas wa = paie yellow. After several days the overgas acquired
a s.igt.tly gcetn J;„I . Shortly atter this the stopper popped from the
tiask and tiic .or.ttnu- were lost.

In the third reaction a 12-liter tlask was charged with anhydride (30.0 g., 0.13") mole) and C1N0 (8 0 g. , 0 120 mole) and irradiated overnight with u.v. light through a Vycor immersion well Before irradiation the pressure was 280 mm The more volatile material was removed under vacuum and the highest boiling product was again found to be the anhydride.
4. Synthesis of Trifluoronitrosomethane and Other Nlttosoalkanes
a. Tritluoronitrosomethane
Trifluoroacetyl nitrite is added at a constant rate to a 5-liter flask containing refluxing FC-43 ;3000 g.) The flask is fitted with a 2 x 24-inch column topped by a water-cooled -endenser Ihe products are passed through the condenser to a wash column containing Raschig ring packing and circulating 7% aqueous NaOH, then through a CaCi drying tube, a 2 x 40-cm. column containing Linde ^A Molecular Sieve, and finally into a trap immersed in liquid air
In a typical run, 352 g. of CF,N0 was prepared from 945 g, of CF;C00N0, a conversion of 54%. About 120 g. of CF ,N0 is prepared per day using this apparatus.
b. CF.BrCF.NO
A 72-liter flask was charged with CF,=CF, (127 g., 1.27 moles) and BrNO (140 g., 1.27 moles) and irradiated with u.v. light for 24 hours. The mixture which was originally brown had now turned blue-green and was transferred to a tlask for distillation. The mixture was distilled on a glass-packed column to give approximately 10 g. of CF.BrNO boiling at 0" and 69 g, of CF,BrCF^N0 boiling at 13'-20v. NMR analysis is given in Appendix B and an infrared spectrum is shown in Figure A23.
c. CH.OCFCICF.NO
Approximately 100 ml, of CF2CI,, was condensed into a 300-ml. Fischer-Porter aerosol compatibility tube at -78" and stirred using a magnetic stirring bar Nitrosyl chloride (7.5 g., 0.12 mole) was condensed into the tube and thoroughly mixed with the solvent. Methyl tiltlucro- vinyl ether (10.5 g., 0.095 mole) was slowly condensed into the solution, with rapid color change occurring. Stirring was continued tor two hours. The solvent was then evaporated from the tube on warming to room temperature. The tube was then connected through a trap in liquid oxygen to a vacuum system and evacuated. A total of 1<*. 6 g. of blue liquid was collected. Trap to trap distillation gave 10.6 g. of CH;0CFC1CF;N0, which was about 97% pure by GLC. The product was stored at 0' NMR analysis is given in Appendix B and an infrared spectrum is shown in Figure A2^
41

d Attempted Synthesis of ONCF;CF2CF2NO
A magnetic stirring bar, mercury (5 ml.), NO (0.10 g, , 0.035 mole), and HCF?)-! (14.1 g,, 0.035 mole) were placed in a 1-liter Vycor flask and the mixture was stirred in sunlight for eight hours. During this time a large amount of red solid deposited on the walls of the flask. The volatile material was removed to a trap in liquid air, forming a white solid with a trace of blue coloring. An infrared spectrum gave no evidence tor norma) nitroso absorption.
The reaction was repeated without the presence of Hg, The only difference in the product was that crystals of li were deposited on the walls of the flask. There was still no evidence for the formation of a nitroso compound.
e. Attempted Synthesis of CH3OCF2CF2NO
Tetrafluoroethylene (25 g., 0.25 mole) and methyl nitrite (13.7 g., 0.225 mole) were charged to a 12-liter flask equipped with a quartz immersion well. After u.v. irradiation for 48 hours with a No. 8A36 Hanovia lamp, trap to trap distillation gave mostly unreacted tetrafluoro- ethylene. A higher boiling blue product was also partially purified. Further ourlfication of this product on VPC gave a pure sample. The compound was identified as ONCF/CF2NO2 by NMR and infrared analysis.
f. Reaction of Methyl Nitrite with Perfluorobutadiene
Methyl nitrite (1.2 g., 20 mmoles) and perfluorobutadlene (1.6 g , 10 mmoles) were charged to a 20-ml. ampoule. No reaction was apparent at room temperature in the absence of light. The ampoule was placed in sunlight for a total of 16 hours. A light green liquid resulted. After the unreacted material was removed, an infrared spectrum of tne product indicated the reaction product was an unsaturated nitro compound.
Methyl nitrite (1.2 g., 20 mmoles) and perfluorobutadiene (1.6 g., 10 mmoles) were charged to a 1-liter flask and irradiated with u.v. light from a Hanovia lamp type 8A36 for four hours. A light blue-green liquid was formed. Concentration of the blue liquid by trap to trap distillation gave only a trace of product, but the color quickly faded. The inlra d spectrum was similar to the product obtained above.
42

5. Synthesis of Intermediates and Miscellaneous Compounds
a. Sulfur Containing Compounds
2,2,4.4-Tetrachlorodithietane -? Cl,c(^ XCC1 VS
Thiophosgene (275 g., 2.4 moles) was placed in a dry, nitrogen- flushed 1-liter Vycor 7910 flask which was then fitted with an adapter and stopcock and placed in sunlight. After one day exposure the contents had solidified to the solid dlmer. The flask was opened in the hood and 72 g, unreacted thiophosgene was decanted. The solid crystals were washed with hexana leaving 148 g. of the desired product. An additional 42 g, was recovered from the hexane wash.
2,2,4,4-TetrafluorodithietaneJ F<?C\ /CF;
Tetrachlorodithietane (108 g., 0.47 mole), antimony trifluoride (179 g., 1.00 mole), and tetramethylene sulfone (250 ml.) were placed in a 1-liter, 3-neck flask fitted with a stirrer and outlet to traps cooled to 0s and -183°. The mixture was heated with stirring to 90-100° and maintained at this temperature for one hour. The 0° trap was removed and the condensate (50.6 g,) distilled through a 70-cm. vacuum-jacketed column packed with glass helices. A cut (35.3 g.) boiling at 43-48" was obtained and washed until colorless with a solution of 10% NaOH (25 ml.) and 50^ H;0^ (.3 ml.) The organic layer was separated, dried over silica gel and refractlonated to give 17.4 g. of product boiling at 48-48.3".
Trifluoromethylthiosilveru'< CF^SAg
A 300-ml. stainless steel autoclave was charged with AgF (14,88 g,, 0.117 mole) and CS2 (18,9 g., 0.248 mole) and heated at 140" for 12 hours. The volatiles were removed, and the remaining solid was extracted with dry acetone and filtered. A fine, brown suspension remained with the filtrate and could not be removed. The acetone was evaporated, and 5,5 g. of solid remained. An infrared spectrum of a Nujol mull of this solid showed absorption in the region 8.8-9.3 and 13.2-13,3 microns as reported for T3SAg,
Triflüoromethyliminosulfur dlfluorlde CF)N=SF,,
Sulfur tetrafluoride (70 g. , CG" mole) and NaSCN (14 g. , 0.17 mole) were reacted according to a reported procedure " "a Tne products were separated by trap to trap distillation in a vacuum line, A portion of the product-rich fraction was subjected to preparative-scale chromatography using a 40-foot column packed with the ethyl ester of Kel F Acid 8114 on HMDS-treated Chromosorb to giv.. 25 g. of pure CF^N-SFj.
43

a
Addition jt aaitui chloride pentaflaorlde Co 1,1-di.hioifw ,2-diflaotoechylene
l. Pfcioxidfc initiation
A 300-mi stainless steel autoclave was charged with SF5CI (18.92 g., 0 life mole), CF.-CC1, ('2 33 g., 0,0927 mole), CC1, (15 ml.), and benzoyi peroxide (0 5 g.) and jated at IOC" for 10 hours. Almost all ct the starting material was recovered unreacted.
11 Ultraviolet initiation
The recovered rea^tants from the above reaction were placed in 12-Mter tlask and irradiated with u.v. light over the weekend. Some liquid product was formed, and it was distilled on a spinning band column. One tta.tlwn was obtained at x06-114 (0,9 g.) and another at 114-115" (3.9 g.). Infrared spt.tra of these two fractions were identical;
*n a st.ond add.:ion a 12 liter flask was charged with SF5CI (31 2 g , 0.192 mole and CF .-CC1; <25.5 g., 0,i92 mole) and irradiated overnignt with u v. .tight Distillation of the liquid product gave 5.58 g. of material boiling at 11«*--115
Ihe reaction of equlmolar amounts of SF^Cl and CF2"CCl2 was repeated several times until about 60 gc of liquid product was obtained. This product was distilled on a spinning band column and a small amount of material was oDtained at 100-104" The material in the pot then appeared to undergo a reaction and vapors began passing through the condenser. An mtrared »pe.ttum ct these vapors showed them to be SOF;, SiFi», and SF6. The distillation pot was ccoied to room temperature and the pressure reduced to 2 mm. At this pressure the major fraction (21 g.) boiled at 48-56°. An infrared spe.trum of this material was similar to the spectrum of the. proposed SF CF CCi ., but not identical.
1.1.2.2-Tetraiiaoroethyi methyl sultide
A 12-iicer Pyrex tlask was fitted with a Vycor immersion well and inlet valve and evaluated. Methyl mercaptan (9.2 g., 0.19L mole), and tetratiuoroethyiene 16.4 g , 0.064 mole) w- re bled into the Tlask and irradiated by an Hanovia type SOL lamp for focr hours. The contents were then remcveH und partially purified by passing through traps at -8° and -133 The .untents ot the -8' trap were distilled using an 18-inch spinning band vOinmii A ^leat, colorless fraction (4.1 g.) was obtained boiling at 65-68 and identified as CH SCF CFH. Conversion was 42%.
uU

b. Fluoroaromatlc Compounds
Pentafluorophenyl hydrazine
Two separate reactions gave a total yield of 542 g. of CgFsNHNH^ The following is a description of one reaction. Hexafluoro- benzene (250 g. , 1.34 moles) anc* uydrazine hydrate (141 g. , _.82 moles) were refluxed overnight with stirring in 600 ml. of tettahydrofuran, About 500 ml. of solvent was stripped off, and the remaining hot solution was poured into 3 liters of H^O. A pale yellow solid formed. This material was washed several times by stirring with watei and decanting; finally, it was filtered, washed with water, and dried in a vacuum desiccator to yield 217 g. of pentafluorophenylhydrazine as a white powder (82°1 conversion*
1.2 ,4.5-Tetraf luorob .izane"
Three reactions were run. The following is a description of one of them.
A 5-liter flask was fitted with a stirrer, reflux condenser, and tubing for addition of solids. The flask was filled with 3200 ml. of 3N NaOK and the solution heated to reflux. Pentafluorophenylhydrazine (217 g., 1.1 moles) was added in small amounts over a one hour period. After the last addition the solution was refluxed an additional 2 1/2 hours. The mixture was distilled and a two phase product was obtained. The organic layer was separated and dried over MgSOt,. The water layer was extracted several times with xylene and these extracts were combined with the organic distillate over MgSO.. After drying, the MgSO« was filtered off and the filtrate was distilled on a spinning band column. Fractions were taken as follows:
20 B.P. -C "D Wt. K.
90-92 1.4088 55.4 92-93 1.4098 29.1 93-98 1.4149 16.7
Infrared spectra were tne same for all fractions,
Tetrafluoroterephthalic acid1-6
A 3-liter, 5-neck flask was fitted with a stirrer, gas inlet, gas outlet, thermometer, and addition funnel and cooled to -78°. Butyl lithium (48 g. in 320 g. of hexane solution, 0.74 mole) was placed in the flask and cooled to jelow -70°. Tetrahydrofuran (I liter) was cooled to -70° and poured into the BuLi. The addition funnel was filled with p-C6F^H2 (55.4 g., 0.37 mole) in THF (75 ml.) and this solution was slowly dropped into the reaction mixture so that, the temperature did not rise above -65°. The solution was stirred for two hours after the addition was
45

completed. Tht mixture was then .arbonated by bubbling COi through at a iace su.h that ehe temperature did not rise above -65v At the end of three hours the now rate was i'JO A/hr The mixture was allowed to warm to room temperature with contl'ced .arbcnatlon. After It reached room temperature the mixture was poured mtc i200 ml of 6N HC1, The two phase mixture which developed was strippeo ot IHF and about 600 ml. of water before a solid began dropping out As the residue became thicker the distillation was stopped Ihe mixture was cooied In ice water and filtered, The filter cake was dissolved in hot water ana decolorized with activated charcoal. This soi-uti^n was tittered and ^^oled to 0 The white solid which formed was tiitered otr and dried ever P,0 to give 72 g. v81% conversion) of tccraf ldorcierepr thali- a.id, m p * -;60-28j-.
1 .t»-Dihvdrazir.c.tetrallaorobeniene "
Hcxaiiuorobeniene U&6 g., 1,0 m^ie), 95£ hydrazine (141 g,, i* .k moles.1, and tet rahydr«ji.ran '300 ml,) were stirred and refluxed for 45 hcurs The mixture was ucicd in ice and ehe precipitate removed by liitr^iion After washing with 500 ml. ot water, 39 g. of crude 1,4-dihydra- 2inobenzene <m.p i53-it,4 ; remained The remainder of the material (96 g.) was re.oveitd tr^m .he THF solution, but consisted essentially of pentafluoro- phen>lh\droiAne and i,3-dihydra2inobenzene as determined by infrared analysis.
An initial experiment en a 0.5 mole scale gave 17 g, of product.
1.4-BiS acetuphenonc > *etrai.iuorotihenyldilivdia?one~ "
A solution cr A,<*-dihydra2inotetralluorobenzene (39 g., 0,22 mole), a.etophenone <1?5 mi ;, absolute ethanoi (1500 ml.), and glacial acetic acid '15 ml.) was stirred at retiux tor 16 hours. The solution was cooled in ice and the product «54.5 g , 69 8% ..enversion) separated by filtration. Recry&tallization trcm lsopropanoi gave 50 5 g ot yellow plates, m.p. 160- 162 vRpt. i67 5-168 )
An mit iai experiment en a O.i mole scale gave 22 g, of product.
l ,^-Diamir.otetratiu>.robenzene
i.^-Bis;a.etophen.rifc.tetralluorcphen>ldlhydrazone (45.0 g., i0.8 mmcles' v»as reacted with 2in. \1HI g , 2,i6 moles) in acetic a.id i860 mi ■ at retiux temperature tor 3 i/2 nours. The mixture was then filtered, wasied with water <l250 mi.) and extracted with ether in a Scxhlet. The ether extra-1 i*as iabelied Sol A The water washings were combined with the original filtrate and extracted with benzene (five 375-ml. portions) This benzene extract was then washed with H;0 (five 250-ml. portions». Alter separation from H,,0 the oenzene extract was labelled Sei B The aqueous washings trom Soi. B were neutralized with NaOH and extracted with ethyl ether This ether extract was labelled Sol, C. Solutions A, B, and C were combined, dried over MgSO,, filtered, and stripped ut solvent. Tr.t dark red-brown residue was sublimed under vacuum at 120-130° to give 2.3 g of a light tan Solid. A melting poinr determination (95-120c) showed f.hat the pr-du.t wa» stni impure.

1.2.4,5-Tetrafluorobenzene
Two reactions were run. The following is a description of one of them. A 5C0-ml., 3-necV flask was fitted with a stirrer, reflux condenser, and addition funnel and charged with 65% fuming f^SO^ (65.5 ml.), Br2 (60.2 ml.), and AlBr3 (2.2 g.). The addition funnel was filled with p-C6FuH2 (43.0 g.f 0.29 iole) which was slowly added to the solution in the flask. After the addition of CgF4H2 was completed the solution was heated at 50-60" for four hours. The mixture was then poured over 4 liters of cracked ice. After the ice melted, the pale yellow precipitate which had formed was filtered off and washed several times with water followed by washings with Na2C0 3 solution. The filter cake was then dissolved in methanol and precipitated by pouring into H2O. The product (50 g.) was filtered and dried, m.p. 73-75°.
c. Nitroso Compound Precursors
1.2-Diiodotetrafluoroethane
The reaction was carried out twice in a similar manner with similar results. The following is a description of one reaction,
A 300-ml. autoclave was charged with C^F4 (45 g., 0.45 mole) and 12 (127 g., 0.50 mole) and heated at 250° for 10 hours. A red liquid product was removed and washed with Na2S20? until colorless. It was then dried over Na2C03 and distilled to give a light pink liquid (96 g., 60% yield of CF2ICF2I) boiling at 67° at 166 mm. Hg.
1,3-Diiodohexafluoropropane
A 300-ml. autoclave was charged with perfluoroglutaryl chloride (100 g., 0.36 mole) and potassium iodide (130 g., 0.78 mole) and heated at 200° for 20 hours. A red liquid product was removed and washed several times with waLer. This was followed by washings with Na2S20 3, then dilute NaOH, and finally several more washings with water. The product was then dried over Na2S0.« and distilled. This gave 52 g. (36% yield) of ICF2CF2CF2I boiling at 126-132°.
0 II
Attempted Synthesis of (CH3)2NC(CF2)3COONO
Perfluoroglutaric anhydride (22.2 g,, 0,1 mole) and N-nitroso- dimethylamine (7.4 g., 0.1 mole) were charged to a Fischer-Porter tube cooled to -183°. After melting had occurred the reactants were mixed thoroughly by shaking. The tube exploded soon after this latter step.
47

Preparation of CFJSCF2CFC1I
A 12-liter flask was charged with CF3SCI (34.3 g., G.252 mole) and CF2-CFI (24.2 g., 0.126 mole), and irradiated with u.v. light for three hours. During this* time 22 g. of a red liquid was formed. This liquid was distilled on a spinning band column under reduced pressure (40 mm.)» A large portion of the sample decomposed to give iodine. Two small fractions, both containing iodine, were obtained at 24-28° and 70-75". Both fractions were washed with aqueous Na2S2Ü3 to give colorless products; however, after the Na2S^03 solution was removed they both reverted to a deep red color. Nuclear magnetic resonance analysis of each sample gave inconclusive results. An infrared spectrum of the higher boiling material showed strong absorption at 8.48-8.68, 9.02, 9.32, 9.91, 12.61, and 14.30 microns, and weaker absorption at 11.52, 13.18, and 15.8-15.9 microns, indicating the presence of both CFjS- and Cl-C groups- No further analyses were carried out because of its instability. The sample was reacted as soon as possible after isolation
d, Fluoroolefins
Tetrafluoroallene
1. CF2BrCH2CF2Br'9
Five similar preparations were carried out in a 1.4-liter auto- clave. Another preparation on a larger scale was carried out in c 3-liter autoclave. The following is a description of a series of three preparations.
Dibromodifluoromethane (4.5 moles), 1,1-difluoroethylene (l.Om^le), and benzoyl peroxide (10.0 g.) were heated in a 1,4-liter stainless-steel rocking autoclave at 110° for five hours. CF2Br2 and CF2"CH2 were recovered by low-temperature distillation, leaving a residue containing the product, CF;BrCH2CF2Br, and telomers of CF2Br(CH2CF2) Br. After two additional runs utilizing recovered material, combination ana distillation of the residue gave 270 g. of CF2BrCH2CF2Br (b.p. 72-75°/300 mm.) from a total of 132 g. of CH2-CF2 consumed (49.5% yield).
ii. CF^BrCH-CF;
Four preparations were carried out, the most successful being similar to a recent literature method.50
l,3-Dibromo-l,l,3,3-tetrafluoropropane (90 g., 0.33 mole) was added drop-wise to potassium hydroxide pellets (200 g.) in a 500-ml. flask fitted with a stain! ^s-steel chain stirrer driven by a high torque motor. A nitrogen pressure of 300 to 350 mm. was maintained in the system with a slow sweep into traps at -78 and -196°. The flask was heated to 60". As dehydrobromination occurred, the reaction mixture became dark and sticky.
48

Stirring became less efficient and the temperature rose tj M.00*. Distillation of the contents of the cold traps gave 6% tetrafluoroallene (2.3 g., 0.02 mole) and 45% 3-bromo-l,l,3,3-tetraflucr'->oropene (30 g., 0.15 mola). The latter was further distilled through a small glass helix- packed column (b.p. 34°).
iii. CF2-OCF2
Six reactions were carried out using a variety of different conditions. The .results were poor. In some cases only a trace of product was obtained and ntified by I.R. analysis. When powdered 90% potassium hydroxide was used at 100° the mixture became molten and no volatile products could be trapped. The following is a description of a more successful reaction.
In a 300-ml. flask fitted with an addition funnel and stainless- steel chain stirrer driven by a high torque motor, and connected to two traps at -183°, was placed 150 g. of pelleted potassium hydroxide. The flask was heated to 80° by a water bath while a slow stream of dry nitiogen was passed through the system. 3-Bromo-l,1,3,3-tetrafluoropropene (30 g. , 0.15 moles) was added drop-wise. The low boiling product in the first trap was separated by a vaporization distillation and then purified by distillation on a low temperature still. Tetrafluoroallene (1.8 g., 0.15 moles) was obtained in 10% conversion (b.p, -38°).
Dehydrobromination of 275 g. of CF2BrCH2CF2Br in two steps by the method of Jacobs and Bauer5 gave 24 g. of CF2~OCF2. Conversion was 24% based on the starting alkane.
Methyl trmugrpYinyl 9ther_ 52
A total of 105 gt of CH30CF-CF2 was prepared in two reactions. The following describes the larger reaction.
Dioxane (450 ml, dried over sodium, refluxed over and distilled from CaH2), sodium methoxide (125 g., 98% commercial material), was charged to a 2180-ml. Monel cylinder fitted with a pressure gauge, a-Pinene (1 g.) was included to act as an inhibitor. Tetrafluoroethylene (180 g., 1.3 moles) was pressurized into the cylinder at ^270 psi. The reactor was placed in an autoclave rocker. A strong exotherm and pressure drop were noted 1 1/2 hours later. After 40 hours the low b ^ling products were stripped. Distillation on a low-temperature column gave S5 g. of material, b.p, 10-12.5°, VPC showed the product to be 95% pure.
4-Methvl-a.6,8-trIfluorostyrene
4-Bromotoluene (10 g., 0.062 mole) was reacted with lithium in ether to form the lithium reagent. The solution was placed in a 300-ml. Fischer-Porter bottle and cooled in liquid nitrogen. The bottle was exhausted and excess tetrafluoroethylene was condensed into the bottle.
49

The bottle was sealed and placed in a cold bath at -30J for four hours. The mixture was hydrolyzed and the ether layer dried. Evaporation of solvent and vacuum distillation gave 4-methyl-a,ß,ß-trifluorostyrene (3 g.( 30%), b p. 90-92
c/70 mm. nj° - 1.478. Reported b.p. 91t5°/70 mm, n*0 - 1.481. a
a
Attempted synthesis of perfluoroketene
0 II
i. ClCF2CBr
53
Into a 3-neck flask fitted with a stirrer, thermometer, and reflux condenser were placed CF2CICOOH (65.7 g., 50.1 mmoles), and PBr^ (72.0 g., 16.7 mmoles). The mixture was stirred for one hour at room temperature and 3.5 hours at 70°. The mixture was then distilled on a glass-packed column and 68.9 g. of material was obtained, b.p. 46.5C, This represents a 71% conversion to CF2ClC0Br.
ii. Reaction of CF^ClCOBr with Zinc
Two attempts were made to synthesize CF2*C«0. No low-boiling product was obtained from either reaction. The following is a description of the second reaction.
Zinc (65.4 g., 1.0 mole) and 16 ml. of 6N HC1 were stirred together in a 300-ml., 3-neck flask. The flask was then heated under vacuum overnight to remove excess HC1 and H2O. The flask was fitted with an addition funnel and a reflux condenser vented to a trap in liquid oxygen, and a solution of CF2ClC0Br (68.9 g., 35.9 mmoles) of ethyl ether was added. At first vigorous bubbling occurred, but this soon subsided. The flask was then heated with no apparent reaction taking place. Suddenly the contents of the flask began to undergo an apparent polymerization to produce a charred material which swelled out of the flask. No volatiles werp. caught in the -183° trap. Similar unsuccessful attempts to prepare this compound by this procedure were subsequently reported in the litera- ture. 5^5
Attempted synthesis of 1-trifluorovinyl perfluorocyclobutene
i. with CF2=CFLi
Ethyl ether (75 ml.) and lithium (1.4 g., 0.2 mole) were placed in a small 3-neck flask fitted with a stirrer, gas outlet vented to a trap in liquid oxygen, and a gas inlet opening beneath the surface of the ether. Methyl bromide (12 g., 0.126 mole) was bubbled in at room temperature.
50

After this the flask was cooled to -78" and CP;=CFBr \18 g., Ü.11 mole.) iid.s babbled In. This was followed bv the addition of pert luorocyciobutene (17 g., Ü.10 mole). The solution was allowed to warm to room temperature over the weekend. The only r>rodurt obtained was a dark brown suspension of solids in the ether. Nothing in the product mixture contained the -CF=CF group as determined by infrared analysis.
j i. with CF2=CFMgBr
Magnesium (A.8 g. , 0,2 mole) and tetrahydroturan (.200 ml.) fres'ilv distilled from LiAlH. were placed in a 500-ml. flask fitted with a stirrer, gas inlet, and low-temperature head vented to a trap in liquid oxygen. Throughout the reaction ;. stream of dry N was passed through the mixture. The mixture was he,-ted and CF -CFBr (32 g., 0,2 mole) was bubbled in until reaction began to take place1 it was then cooled in ue while the remainder of the CF,=CFBr was added. After this the perfluoro- cyclobutene (16 g., 0.1 mole) was bubbled in, and the mixture was allowed to warm to room temperature overnight. No volatiles were obtained. As before, the product was a suspended brown solid. All of the liquid was removed to another flask by vacuum and then distilled, but nothing other than starting material was obtained.
Preparation of C5F ;0[CF(CF <)CF-,0] ?CF*CF;>
C,F 0[CF(CF.|)CF,0],CFHCF} (56.1 g., 0.1 mole) and dry ether (500 ml.) were placed in a 1-liter, 3-neck flask fitted with a stirrer and dropping funnel. Methyllithium (5% in ether, 55 ml., 0.11 mole) was added dropwise to the stirred solution. After addition was complete C«-l hour) the mixture was stirred for one hour. Methanol was added dropwise to destroy unreacted mtthyllithium and water was then added to dissolve the precipitated lithium fluoride. The ether layer was separated and the water lcyer extracted with ether. The ether layers were combined, dried over Na^SO,, and stripped under vacuum. The remaining crude product was distilled using a spinning band column. The major fraction (18.1 g,), b.p. 153.5-155", was identified as the desired trifluorovmyl ether. The yield based on the single fraction was 33.5%.
Preparation of C3F OiCF(CF .)CF?0 hCF=CF;. (A25)
The preceding reaction was repeated using C F 0|CF(CFJCF ,01 CFHCF (72.7 g., 0.1 mole) and methyl!ithium (0.1 mole). Crude material (65 g.) was distilled to give 45 g. of product boiling at 188'/45 mm. The infrared spectra of the two ethers were nearly identical.
51

fe Miscellaneous Compounds
Pert 1joropropyiene epoxide'fc
Water (150 ml.), KOH (84 g.), CH.OH (.1000 ml.), and 50% H20, (473 ml.) were stirred together in a flask and cooled to -60'. Liquid CF<CF«CF^ (105 g , 0.70 mole) was added and the mixture was stirred at -60" for two hours. It was then allowed to warm slowly to room temperature and the product (54.4 g,) was collected in a -78" trap. Analysis by VPC showed the product to be 80% perfluoropropylene epoxide with unreacted perfluoropropylene as the impurity. A small amount of this product mixture was shaken in an ampoule with excess Br,;, An infrared spectrum of the overgas showed pure epo:.ide.
Perfluoroglutarlc anhydride
Perrluorogiutaryl chloride (910 g., 3,3 moles) was slowly added with stirring to 150 ml, water in a 3-liter flask. After the acid chloride nad been added, the water was removed by azeotroping with benzene. The benzene was distilled off at atmospheric pressure, then by aspirator Excess P-0 was then mixed thoroughly with the perfluoroglutaric acid and heated. The anhydride was collected (600 g., 83%) directly by distillation.
Perfluorosucclnic anhydride
Pertluorosuccmic acid (200 g. , 1.05 moles) was mixed with excess Pj.05 in a 1-liter flask. The mixture was heated and perf luorosucclnic anhydride (141 g., 0.82 mole) was removed by distillation as formed.
Perfluoro .2-methyl-3,6-dlhydro-l.2,2H-oxa?ine)
Trifluoronitrosomethane (4 3 g., 0.043 mole) and perfluorobutadiene (6.8 g., 0.043 mole) were condensed into a Fischer-Porter tube and kept at -25" for 72 hours. After this time the blue color of the nitroso compound had disappeared The volatile material (^5 g.) was removed from the tube by vacuum distillation and found to consist essentially of CF<NCF^CF=CFCF;0,
Yields on several runs averaged 50% The following properties were observed: b.p 51-52 , rip- 1.2789, d<° 1 6075.
Anal: Calcd tor C^NO: %C, 23.01; %H, 0.00; %¥, 65.52 Found: %C, 23 32; %H, 0.00; %F, 65.38.
Nitrosyl bromide
A 500-ml., 3-neck flask was fitted with a stirrer, a gas inlet rube, and an outlet tube vented to traps at -78'' and -183". Water (80 ml.) and NaNO (383 g., 5.5 moles) were stirred in the flask as HBr was bubbled in. It appears that no reaction takes place until the HBr concentration reaches a certain point. The product was caught in the -78' trap and distilled on a glass-packed column to give 42 g. of material boiling at 8°.
52

It was found that the following l? the better method of producing BrNO A 1-liter flask containing 567 ml. of concentrated HBr solution (7 moles) was fitted with a stirrer, gas outlet vented to -78" and -183" traps, and an addition funnel containing 345 ml. of a solution of NaNO^ ;3 moles). The flask was maintained at ambient temperature with a water bath and the NaNO; solution was slowly added The product was trapped at -78'.
Methyl nitrite
Methanol (32 g., 1 mole) was dissolved in an equal volume of watei and added to a flask containing 70 g. NaN02. The mixture was stirred and concentrated H;S0i, slowly added. The evolved gas was passed through a CaCl2 tube and collected in a trap cooled in liquid air. The product (57 g., 93%) was condensed into a cylinder and stored under refrigeration.
Monosodiam hexafluoropentanedlol
Hexafluoropentanediol (21.2 g., 0,10 mole) and sodium (1.15 g, , 0.05 mole) in 200 ml. of dry ether were stirred by means of an air motor for 48 hours. Formation of a white solid occurred slowly, After filtering and drying under vacuum for six hours, 12 g. of NaOCH2CF;CF2CF<:>CH..OH was collected. Evaporation of ether solution yielded 9 g, of the unreacted diol.
Attempted preparation of disodium hexafluoropentanediol
Three attempts were made to obtain the disodium salt. In the first, hexafluoropentanediol (5 g.) was dissolved in 50 ml, dry ether and refluxed over sodium for one week. The white solid which precipitated was dried and analyzed by titration. Found: eq. wt 225; 236 Theory for monosodium salt, 234; for disodium salt, 128,
Hexafluoropentanediol (5 g.) was dissolved in 50 ml diglyme and heated with sodium overnight just below the boiling point. A black tar was obtained.
Attempted epoxldation of perfluorobutadlene
1. by H20?
Water (150 ml.), KOH (84 g., 1.5 moles), and CH30H (1.1 liter) were placed in a 3-neck flask fitted with a gas inlet, stirrer, and addition funnel containing 45% H202 (480 ml,). When the KOH had dissolved the mixture was cooled to -40° and the H2O2 slowly added. The addition funnel was then replaced with a low-temperature (-78") head and CF^CF-CF-CF: (50 g,, 0.31 mole) was added through the gas inlet. This mixture was stirred for 2 1/2 hour3 at ^O" and then allowed to warm to room temperature,
53

The volatiles were caught in a trap coded to -183°. Only a small amount of material was caught and this proved to bs mostly CO; with a trace of C F.. Distillation of the reaction mixture which was found to have a pH ^5 gave only 1 8 g. of material boiling below the boiling point of methanoi. This material was separated by GLC. Methanol, tetrafluoroethylene, and perfluorobutadiene were isolated as well as several unidentified compounds. Infrared spectra of these volatile compounds showed no peaks in the 6-7 micron region where a perfluorinated oxirane usually absorbs. NMR analysis indicated that one of the products was CF?CH=CFCF20.
11. by Oxygen
A 1-llter Vycor flask was charged with CF^-CFCF-CF; (2.4 g., 0.015 mole) and 0; (0 48 g , 0.015 mole) and placed in sunlight for five days At the end of this time the flask contained a water-white liquid. An infrared spectrum of the overgas showed some u'Teacted diene and a peak
at 6.6 microns (thought to be -CF - C?^) as well as other peaks. A spectrum of the liquid also showed absorption at 6.5-6,6 microns.
The reaction was repeated on a larger scale. A 12-lit.r flask was charged with C.Fe (20.7 g., 0.128 mole) and 0;- (8.19 g. , 0.^6 mole) and irradiated with a u.v. lamp through a Vycor immersion well. After 24 hours an Infrared spectrum of the vapor phase showed a peak at 6.6 microns and the reaction was stopped. Although the products were not isolated and identified, it appears that epoxidatlon can be effected by this method.
54

D Polymer Synthesis
Polymers were prepared uaing bulK, -oiuiiun, and suspension techniques and are described in Tables 3-8
Bulk polymerizations were came' out in either glass ampculfee (20-80 ml.) or mud steel cylinders ö00 and 900 ml./, generally wich agitation at the temperatures indicated. Solution polymerizations were carried out in sealed glass ampouies, while suspension polymerizations were carried out in glass ampouies, mild steei . ylmders, and a starred autoclave.
Liquid monomers were weighed and added iu the tubes; gasecas monomers were expanded into an evacuated 1-liter riasit at measured pressure and condensed into the polymerization tube;: . Ire tubes wtre then pia^ed in a ;old bath lor the indicated time. It a suspension polymerization was run, the tube was shaken vigorously The 3Jc.pcr.a1rg medium was water -cntaining .3 g L1B1 and 3 5 g MgCü per i00 mi In working ..p ehe products, the jr.rfca~ted gases were vented and tht vo^ctile materiai.» recovered by vacuum distillation, The polymer was then removed and weighed. In the _ase cf suspension polymerizations, the mixture was first treated vith ..undent rated HCi to remove tne MgCO -, and washed several times betöre vacuum drying
Polymers were frequently purified by dissolving in a suitable solvent such as Freon 113 and reprecipitatmg with methanoi. They were then dried under vacuum.
In order to ^arry out these low temperature polymerizations, a system including a refrigerated bath and stirred reactor was constructed, The apparatus (shown in Fig. 2) consists of five components. The compressor (A) cools a reservoir <B) of CaCl, solution whi„h is circulated ty a pump (C) through a polymerization bath vD) and an auto-lave (Ej.
(A) The compressor is a commercial unit, a 1.5 horse-power Copeiameti«., which uses R-5Ü2 as a refrigerant. The compressor is connected to two large copper coils sealed inside the reservoir.
(B) The reservoir is a tank 12 inches x 18 inches x 36 inches containing two cooling coils from the compressor bathed in the ^oolant which if circulated throughout the system. Near the bottom the reservoir has an outlet tube which leads to the pump and at the top an inlet tube for the return of the coolant. The reservoir is filled with 20 gallons of a 29 6% CaCl, solution which is gravity-ted to the pump and can be maintained at any temperature between ambient and -40~ within 1.5'F- It is insulated on all sides with six inches of polystyrene and encased in a vapor sealed plywood box
55
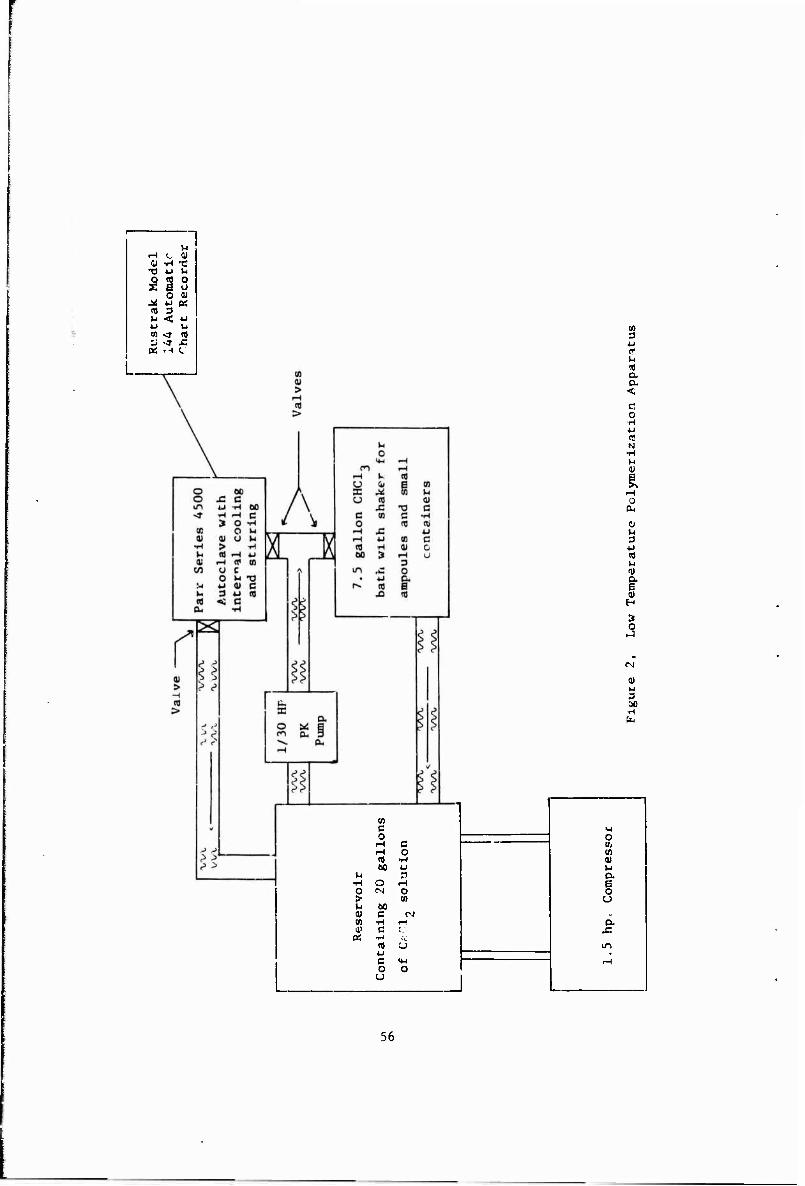
t ■
SJ
-H I, 0) 01 •H -c
T3 U u
£S 0 o
O 01 .* «J « (fl D VJ < *-> 4J u 0) <t <fl £> ••5 £.
OS *-( t
3 <r >-i
<d o. a < c 0
■H 4-1
(9 N
t a a, o D
a)
3 o
30
fa
c o
rH c r-t o n) •H 60 *-l
M n ■H o iH o M o > 10 »-( 00 <u C c U) •H r—f
a» •H
C
m O 4-> c M-l 0 0
<_)
o UJ CO 0) M
f a
56

(C) The first pump used in the system was an ODerdorftr centr lt-jgal pump connected directly to a 3450 RPM motor, bat it was found that this unit was transferring too much heat to the ^ooiing system. The situation was remedied by adapting an ordinary pump used tor circulating water in lab-scale distillations, a 1/30 horse-power PK pump. This pump was connected to the system through 5/8" copper tubing and then insulated with polyurethane foam. The pump is gravity-fed from the reservoir and forces the coolant through the polymerization bath and autoclave and then through the return inlet at the top of the .reservoir
*.D) The polymerization bath is a steel tank 12 inches x 18 inches x 18 inches surrounded by a water jacket 9 inches high and 11/2 inches wide. The CaCl solution from the reservoir is circulated through the jacket which is baffled so that the coolant must circulate completely around the tank which is filled with CHC1 to a depth of six inches. Three inches from the top of the tank a 3 '< inch brass rod mounted in air-tight bearings at ea:h wall extends the length of the tank and six inches beyond one end This extended end of the shaft is connected by a rocker arm to a pin and bushing mounted off-center in a pully powered by a I/18 horse-power Bodine motor The motor is controlled by a Varlac to give the degree of agitation desired on the brass rod. Several wire baskets which hold the polymerization vessels are suspended from the brass rod into the CHC1 bath. The entire tank is insulated with three inches of polyurethane and encased in a wooden box with a removable, insulated lid.
(E) The autoclave used is a 2000-ml., Parr Series 4500, stirred, pressure reaction vessel with an internal cooling coll. As received from the factory, the cooling coil was made of 1/4 inch O.D. stainless steel, but after the first run Jt was apparent that this coil was too small to provide sufficient coding. It was, therefore, replaced first with a 1/4 inch O.D. copper coll (which also proved unsatisfactory) and finally a 3/8 inch O.D. stainless steel coil was used. The autoclave, stirring motor, and holder are encased in a plywood box insulated with five inches of polystyrene and a temperature of -30"C was easily obtained
The temperature on the autoclave is monitored with a Rustrak automatic chart temperature recorder, Model 144.
5T
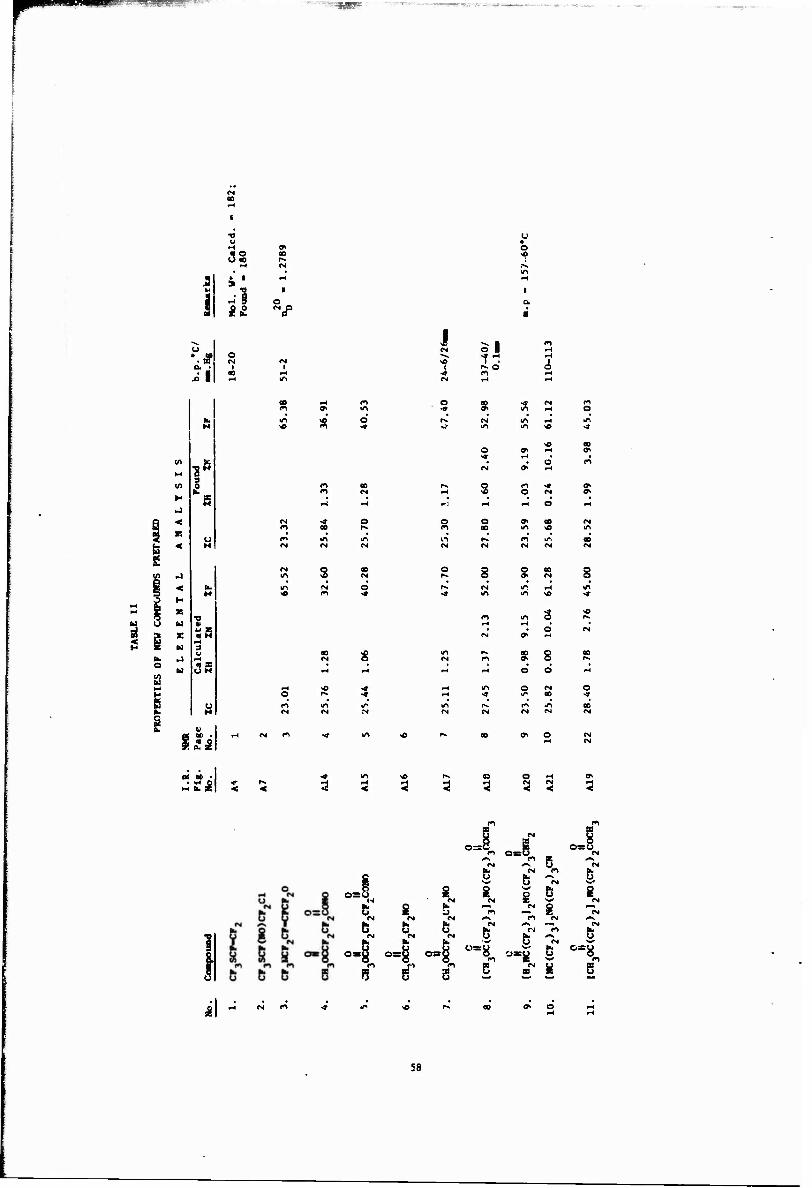
''"s^'7T1HK?^SSr- "w
Ü »
•1 i&
00 o
V
I 58 1-1
w
< R •<
< H z u r w J w
a.
g
UN
3 o
o S s CN -< o
m ^ « in
o •» <N CH*
PH
O *-*
00
s o CN 0"i
r-l r* .H o H
O o 09
0» in
OS CN in
IN in CN
00 <N
O 8 O 0>
00 CN 8
r-* <N in H m
(*" O
93 en 8 O o
O in
CN 00
M oo 3 3
O H CN CN ■< <
0=t
6 G
£ G DM 4 6
en SB
,J en 0=0
/-. n at IN <-> U
u
CN
G
csj en
I S -r s en (N
iN ^s *-«
G ?«N
■qfi 5
£\ M
58
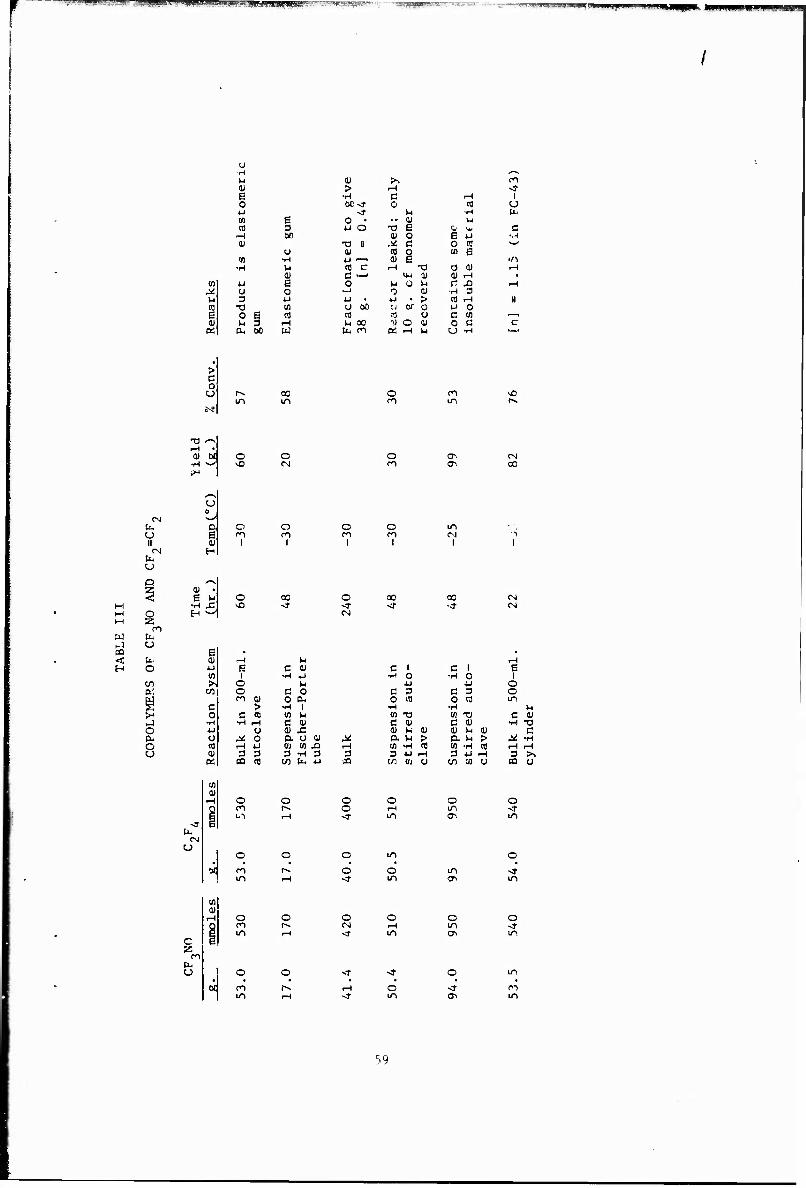
r . ■ fWBf^SSM ^T^r?^"^"3"" r== -'igsr-gH* 41» JUW
u OJ >■> n OJ > r~i vT B ■H C rH 1 0 6C<J 0 cr] u u -cf u H t>4 Ul E O < 01 M
(S 3 (-1 O T) B Cv w c rH 60 0) o E 4-1 •,H ai "0 II .X c o « ^-^
u CU cö o CO E en •H 4-1 i—i 1) E i/1
■H M CO c H -o 0 0) rH OJ C w—■* 14-1 V 4) .-i •
CO 4-1 B o l-i 0 u « £> i-H
^ o o —1 O 01 H 3 u 3 ■u 4-1 ■ 4-1 • > CO ^H H «3 T3 in u 00 Ci or o u O e 0 e «9 a) cd CJ c in i—-.
01 M 3 rH )J CO <y c 01 0 a C a Bu DC W tn ro pi ^H u u •r-l *_.
00 m
o n in
01 (X o o CN
o r-i
CM oo
O. B
H
o O O O m
1 c-i
1 1 o-,
1 CN
1
01 • e u •H JC
o 00 o 00 00 CN vO -»
CN <f ■* CN
w CO < E •
01 H V4 4J B c oi W 1 •H _> > O u
en O C 0 ro 4) o a.
n > •H i 0 c CO tn v-i
•H •IH rH C 01 4J o 01 .C U M o o. u 01 Jd «1 rH 4-1 tu en x> rH 01 3 3 3 i-t 3 3
öS CO CO en fc u »
C 1 c 1 E •H o
4J •rl 0
4-1 i
O c 3 c 3 o 0 « 0 « m
■H ■H t-i tn ■v U! TJ c 01 C OJ c 01 *H 13 Oi rJ 0) 0> M 0» C a M > a )-l > ^; •H en •H cO en •rH cfl r-H rH 3 4-1 •-* 3 4J rH 3 >s
en Uj o C/l en CJ 03 O
i* CN
o
o o O o o O d r-~ o ^ m ~tf O rH <r m CTN in
in o o
m iri
in
C z
CJ
0£
O m
C-I
o O O o CN iH m <f sf m o> m
o rH o
m in
59
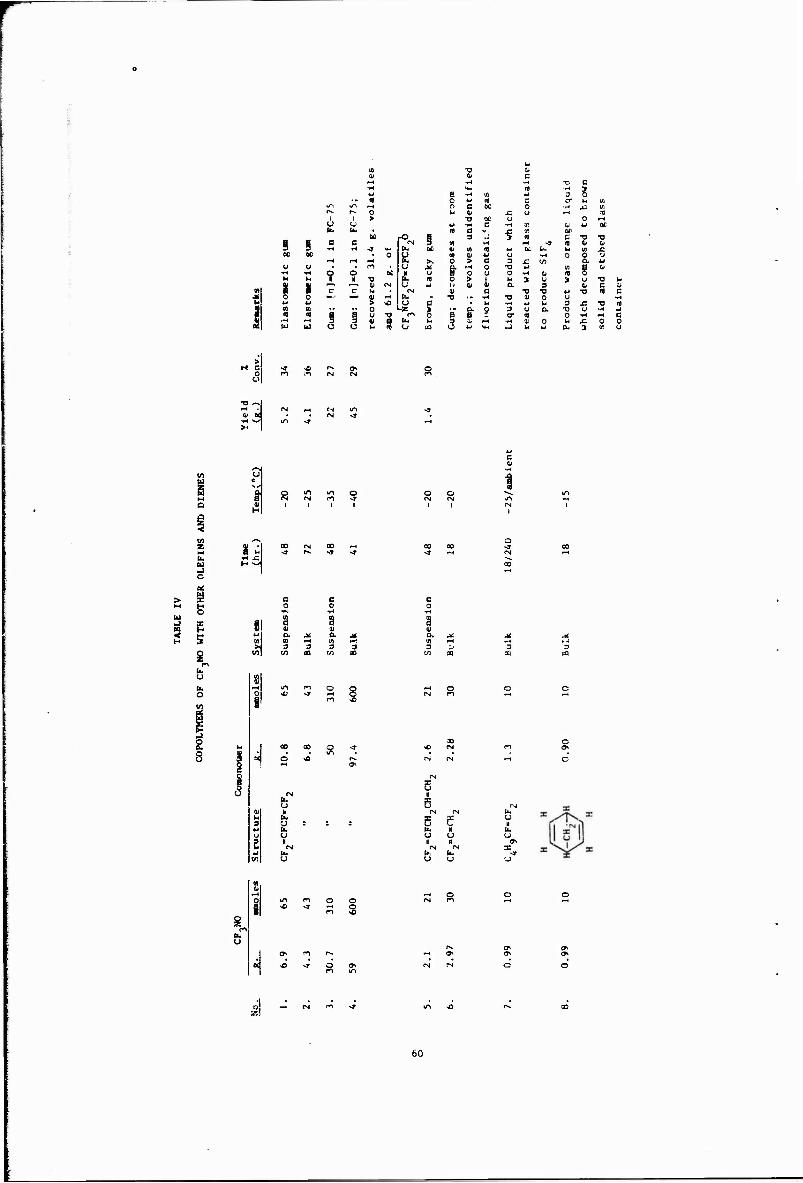
§ ! a ■H
c bo oc ^ _ u u u
■H M =f o
II
OS % 1 CT c .al o 0 u hi u
I tfi ■ # a ! 1 « W w u O
R '** ■Jt 4J 0 u m B o c ec 0 u oi
71 M u u
■H c ■H « r« I
n C 3
", ^ U5
m .-H «. u. no it <t w ft*. u.
id w «■ w u •H X 0 > C 3 £ tn
u J< fr *-< 0 TJ ä I u 0 u O •** 01 a 0 > I U "» u
u u 11 «1 a 3 tM 01 Q TJ T>
lb TJ T> V 0 u B U -H u U
-z i • * c O 3 u a m 0 a B z CT (0
b. u 3 w 1-1 •-< - o U oa o u **-. _] 1- w
c 73 « 01 w tfl
0 0 a
Ul B fl o
> 0 O
ä 00 f*J
T. § -I
c c 0 o
-H
§ s 01
a 01
w a M a .* a> 3 X 3 3 3
to tn a w « 3
1 2
to. U 1 I» U to. O
o o -I o
0->
60

T) 0 0 8 M 0 U O I** m ä -» u. 0 n 0 W t' u Ml in «• 1-1 0 3
•r-< H K u ■o c C* 1-1 X RJ « ■H r
R ►, m it in to. 3 -H u -T 91 -H 3 0 ■ » V] U nt. ffl e 1 D ÖC U) 0 01
C u X. "3 00 r-< c 0) n u u. ¥ "O •H u c -T u ~M
w U 3 •H s 0 •H .—i (Ni c O ffl cr M 0 «H 01 r: a th ■H M £ •H V k< •u 0 ^H
T *J u ~H J3 01 V J
■8 c *«s, 0 u H x: a o F-H V & TJ u •. 3 J= -H
» * 0 u « .* T3 ¥ 0 ii ffl X ■IH 3 15 M 0 I s 0 —4 u. c 5 £ —< i—i n M 0
H 1 u ■■H u- UJ Q Q. o H ■f-t
01 01
cn oj . z S ^
•H J= I». H ^
r*^ u ■o
o u c « 0 C) S w
H O > I § s-
ÜJ s to t/3 < o H z
3 CO
3 EC
3 CO
3 a
o o
O -l
PD to. to. to, u u u m to,
CJ
to. U i 8
o NO
o
61

■c U. ■ „ s 1_> to* K 41 ■ M M C
cs M 0 u V tfc • * • «• V *J
I «J u r-* r- -a <u •*4
■» v» 8 a
u a
C 0 u-i Z z c o a 41 u. M M u t-t •*H
rH %o *J «> •§ u •- ■ n 1« ■o ^ f— r-t rg -H 1'
•s rH 1» -a > o 0 0 c-j rsj •-* H
f* 2 m m tn a 0 • ■ n to M ■ •H • •■ U u 3 to U
T) •X M ■H 10
£ « 00 § u ■o
u 0 T> T3 3 •s 3 00 O a fJ § ■c OJ (N r- ! 0 >H >
Z O .-H ,3 0 b.
u a. J 00
41 3
CO
P
ll
8 i
00
•< 5 6
4 o
o
s -* IK i (J t C*
h. U t
5
62
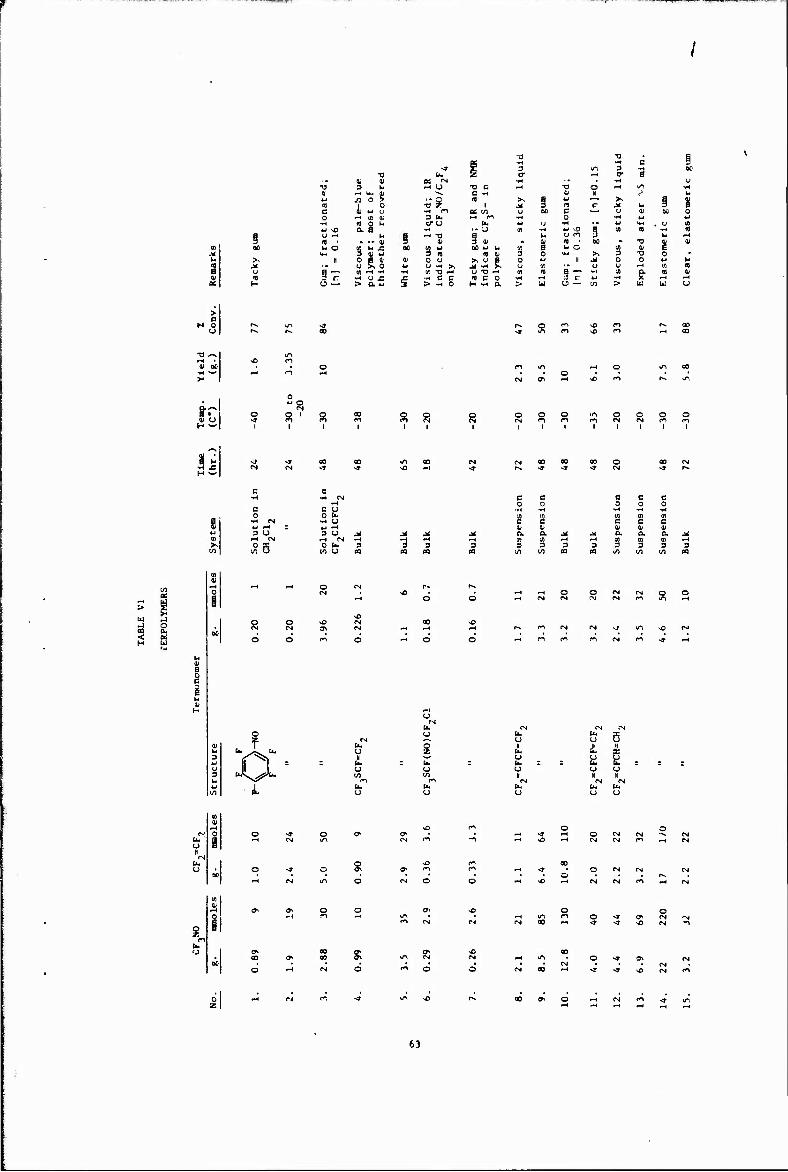
wmmfmm^mm^
<T t 3 •D b. 2 o- • » 01 01 as IN ■>H
TJ 3 U M U ■o c i—i -a 1' ~« w. OJ — C -rt 0
a ^2 O >
0> *-» CJ «g 1 M 1 4J
rj a T4 M Da </) U DO c 0 —i « a: 3 b. *-l fl •-< 0 f4 n o ti er u b. t-i cj ■H
W *0 a B •* • • u to •H •J >c U fH u § — T3 1« u u rn « • . .. 4| 01 * o> i« • M O
9 01 4J ac 3 -S M *J l- tfl B w o
144 fl> 0> 3 0 i*4
II 0 B 4< o 5, 0
V 0 u .* -H S*
0 w N <-> u -. >, u a
17 VI »-» -H ■H «■OH U "O ^H CA to 17 -1 Ü £ 5 •-C C C (fl C 0 •^ a — > a u > -H O H -H a > U o —
s
-o 01
■o 0
! %
—i 5< —< .—t
> M O
-I oo
■a —.
U 00 ■H S^
IT o 4J O
s
■H £
c B U 0 O tv
1-1 CN •H U U iH I W -H 3 U 3 O
rM CM Fl r<
W U O b. to u
1 rH 3 3
CO 3
to
o a. OS u
o
o
o T
00 r- ft CM
H m m
b.
b. u > b. o Ik o
b. u
u
&
b. U
b. U
R 2
* O ■H sO .-I
O M <N r-j
<N CM m FH CM
oo g d
CM
d
o i-H in fO CM ao —i
«-I in CM
CM 00 •-•
O >* <7» CM 'N ■i» -» ■£> CM ""I
O •*» >7» CN
•* -» so es* m
II CO CT> o tH CM »-n NJ IA
63
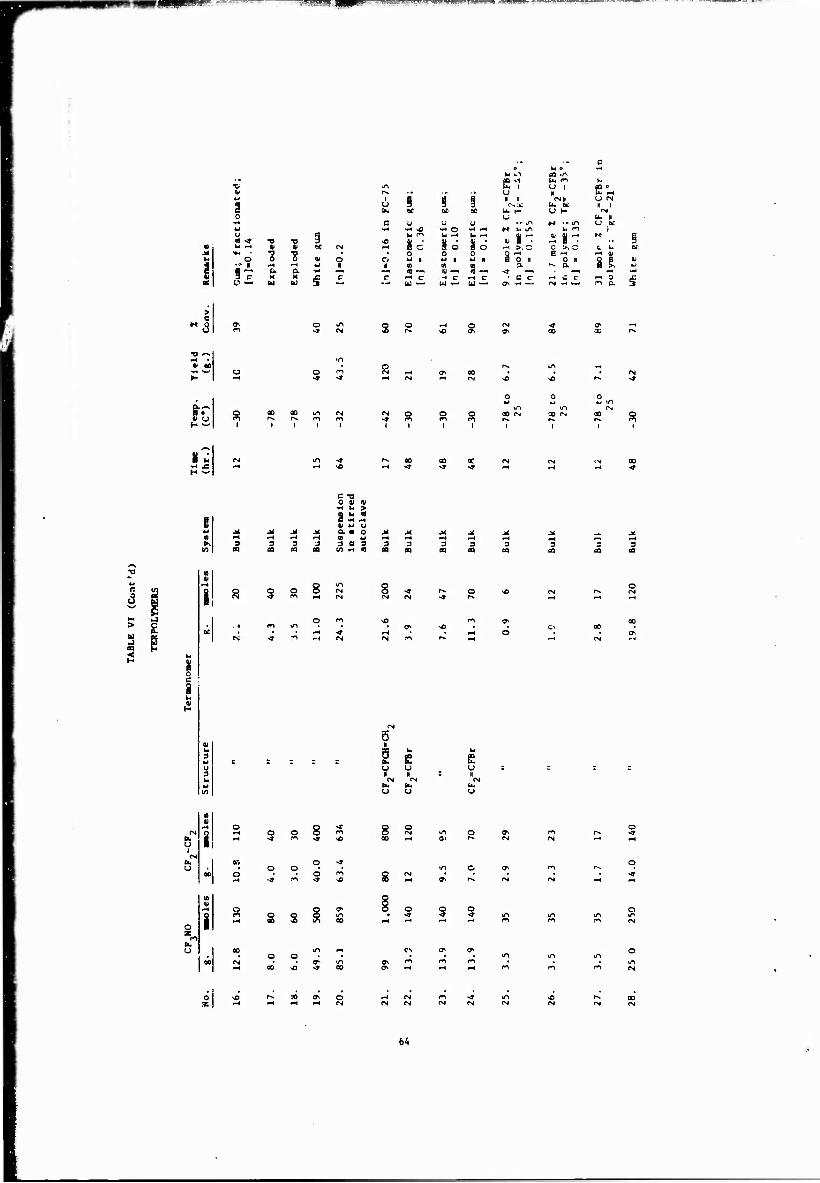
b ~i IM
o
17 o —
T3 V •3 0
■o V
0
£ £
D 1* o ■*4
U Li CO iA u. m Et fcni
ul U 1 Etn r- ■» • r ■ k o 1 i ^ % II II MI
ii i U. 0£ ac &c U. f U H IN
a Ü u u ■ •■ in M •• lA U oc ■fH ■* ^> ■* o ■<H -H H U LA |M r*l i
u tn <u l-t k- « % i 8 M *0 lo in it <H a o <-t >. o c >, Q & u
0 0 0 1-3 B ~« -H 0 o « • u a *J H N 0 1 it B. 1 en W a a f* a
«I — «1 —. -» cr M C -H C ,—t r • c C —i tZ c: -• O •-* U — w — UJ —■ CT1 -H '—' (N -H k— <n Q.
> c M3 2 rv
•H • 4» 00
•ri w 8 n
«l u
0 o O fcJ aW *W lA
W> lA CN 00 <N oo fN 00 O r-. rv r- n
KM LT» -» r-» 00 00 or CN .-4
ens
Ion
6
tlrr
ed
clav
e
^H ■» -J -»
J< a a o -* M M j< .* » w -H -4 -H .H *-.
3 3 C 3 3 3 3 3 3 a c/) i-l ■) ID CO oo 00 00
CO
3
s 8 g * <N (N
2
• 0>
CN tn
to.
Ö (N CO -i
to. O o
o
o
to. u
s o 00 3 ft 00 •-4 •~4
o
oö
O a/S Ft
CO H
.-1 i-4 O (M
6A

f vgfOKwmmsm ^'BüWüiffr'
s a 0 0
*-» (S u .C *C u ro u rn CJ ffl • i • M O
1*4 - o
IM O
a 1 »
s o
H
17 97
H no a
01 •» tl > H N «J m T3 O
V h H >
£ g~ >K 0 c fl O '—'
■a t. •- « u
■O ■- 01 ü « i «■
>-* M >, r a 3 —
•o -o «I HI w I- u V HI > «I c
W X o o y ~ a. ».
C in ac •H M
01 HI r-i tJ
Sir •§"s 1 Of C »H
0 o • 0£ u x m
■■H t* r^ tfl M s >>
it < —' t-i a s u -1 IS •—* 0 <g t-. " m •M m 0 t/i 1 «J *T «i
1« o I- 1 > n <_> a
u UJ (A ab. 13
41 k* 0< 3 'fl
K • at o w
01 *j a. tn O -I c -*
13 o O ~H N I*H 3 C O 01 o ü J3 O
o J
8 £. 00 (ft fM
os o
u —
> B
«*< 0 I U I
T3 ". iH • 01 60
•»■4 ^
in o
8T 01 u
00 I»! rt
01
3
J!
3 rH 3 3
03
J4
3 3 3 i-H 3 =5
<
i b. u
«^
o z (N
b. CJ
(N b. U
u
b. u
s o
in o
b. U
u. U
o 3:
b. U
b. u
o I u
g
b. u
r» n
o o
o
65
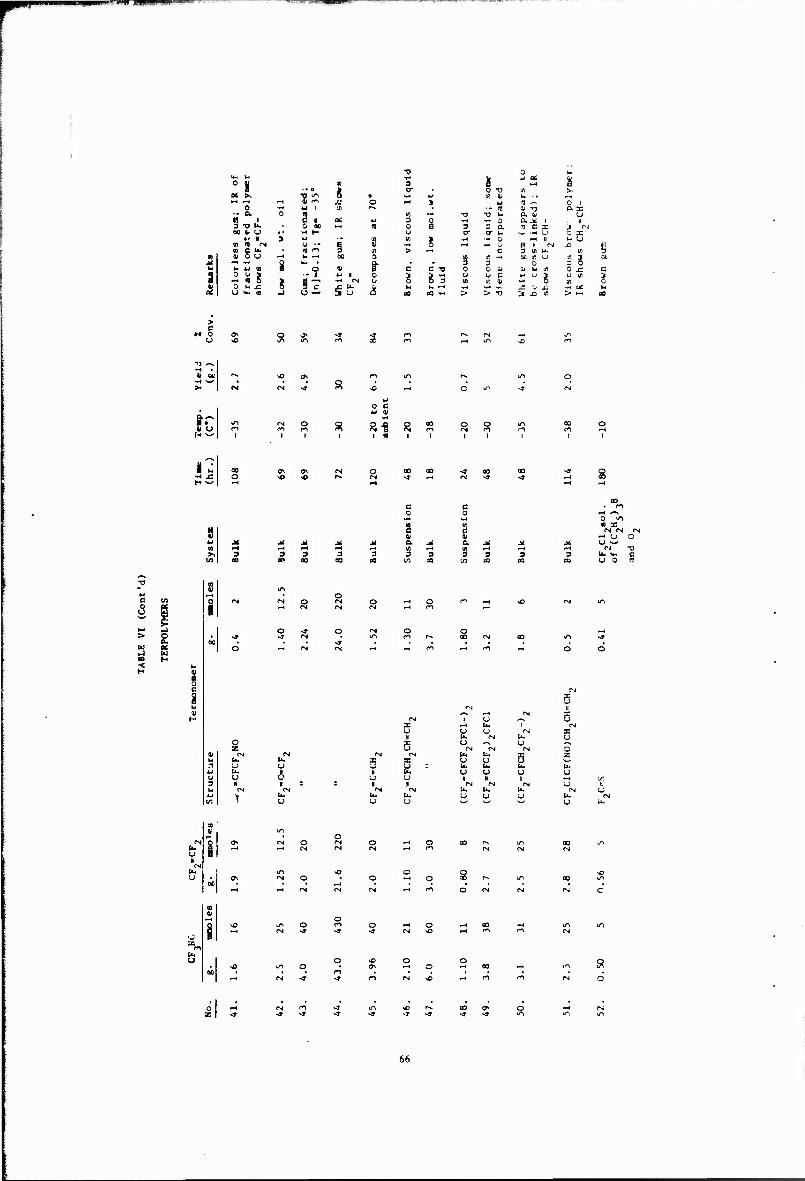
wm
•O 0 u 0 | BO
1-4 3 1 u Ä %
■ •■ a * cr o -o Ift > x >, 4» m
0 tH *J if) 4J t-. — •-« —« ~H JC O 1-4 3 U <u -- 0 1 0 «H U 1 85 r^ .. $ <u -a a s ■ - a 0 S . 3 —4 T) ■a u. Q. 01
H t at u 0 -H 0 a jc II 3 tJ u. 0 ftC ►-« 1» 0 B 3 3 a (fl c X J* rj ac l> u t' ■* H CJ <T cr ki -.- ><-4 U 0 5 *J B > 4J « (A 8 ■H o II u « <q CM U *- I s •H wH n i r-j -D (~; « s «1 0
U. cfl r*> J) > -H c 3 (ft (i, (ft 3
i u r-^ k) r-t 00 D 3 i/i -i~* at' </) (J 'J) 3 (JC *■* *H 2 W, 1 » * 3 o 3 0
u ^ M iO o 41 c c TJ 0 0 i U u en 0 jr c
i 0 SJ 8 ■- 1 w H 0 8 8 u u c t-t u 8 (J Efl 8 -. « 8 §7 CN u 3 (/I (A ii ■Jl
a 0 v. JZ § U. & hi *M n •^1 T-t -fj j^ o j^. ■rA uC t-. u <c (ft -i o — U CO a > > n :« J3 IT: > 33
> c 0 0< s JN <r 3 m r- IN iTl u o lA r*> ro —. U~1 x- fl
8-r HI u Si 8
oo o
M r-* 3
C c 01 S
J< Jl ,* a M a 3 3 3 3 3 3
SO a OQ V5 aa VI
a X CN CN r
H U f) O w
r4 ■n [x. «^ c (J 0 B
■o
c
> 2 M u oc J u ID H < 1-
8 c
8
o z CN la. t.
X U L) U ; u. U- E u CJ u II II i CN r
CN U. u. U. u u
-I o
CX) c^
*n irv
U. O ■H O S
o
66
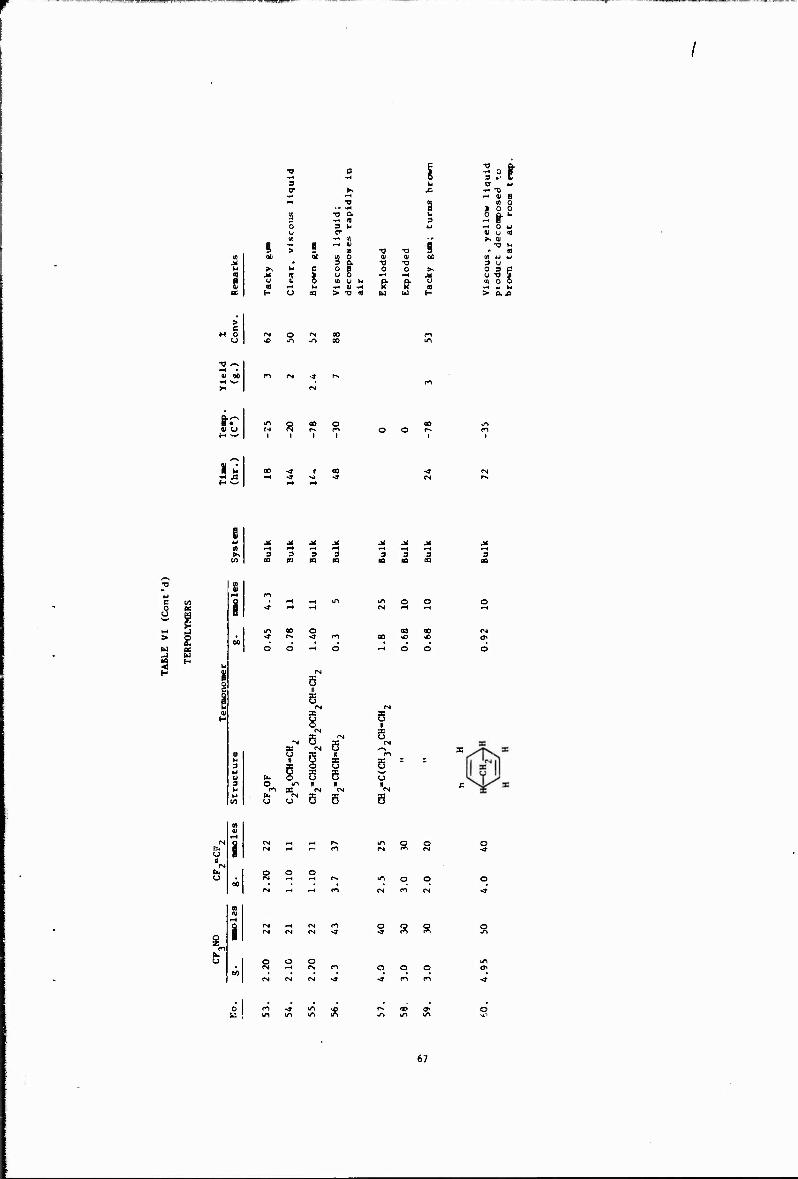
c ■o a ■o 0 5 X oi •~t ■H 8 3 .- B 3 u tr w a- ►> X ■H X -* — — ma H ■o
3
CA 0 • «> 1-1 10 0
§ -a in 5 fr"
0 3 N W -I 0 <J u CT cj u eg 1« fi 3 >, i
5 9 -H 3 ■o v. > 00 T! •a «J HI M DC 3 o li X oc m w *J M - a ■o "O 3 u M >* u a 0 1 o 0 >. 0 3 p
«00 1 it 3 s u 0 u w
t—
& -s a IM 0 ■»■ « ■rt M fci
at H CJ CO > ■a « w u H > Q..O
> c
M 0 <N o <N CO m u vC in irt ao ifl
•o *-. »H • (1 oc fl (N -* K
•H w • m S» CM
frr p 00 o r* r- n l I l
CO O i»
3 3 3 00
3
Jt •-4 3
C O u
u £
<r <H in
in ao O f>
o O <-H O
n Q
rM X
X CM
5 r
5 • S ■
X X o Q
u. X X u 9 o *n 1 H m X (N r
u. o
CM U 5 5
00
d
.H »-4 ro
tt. U o
ri
67
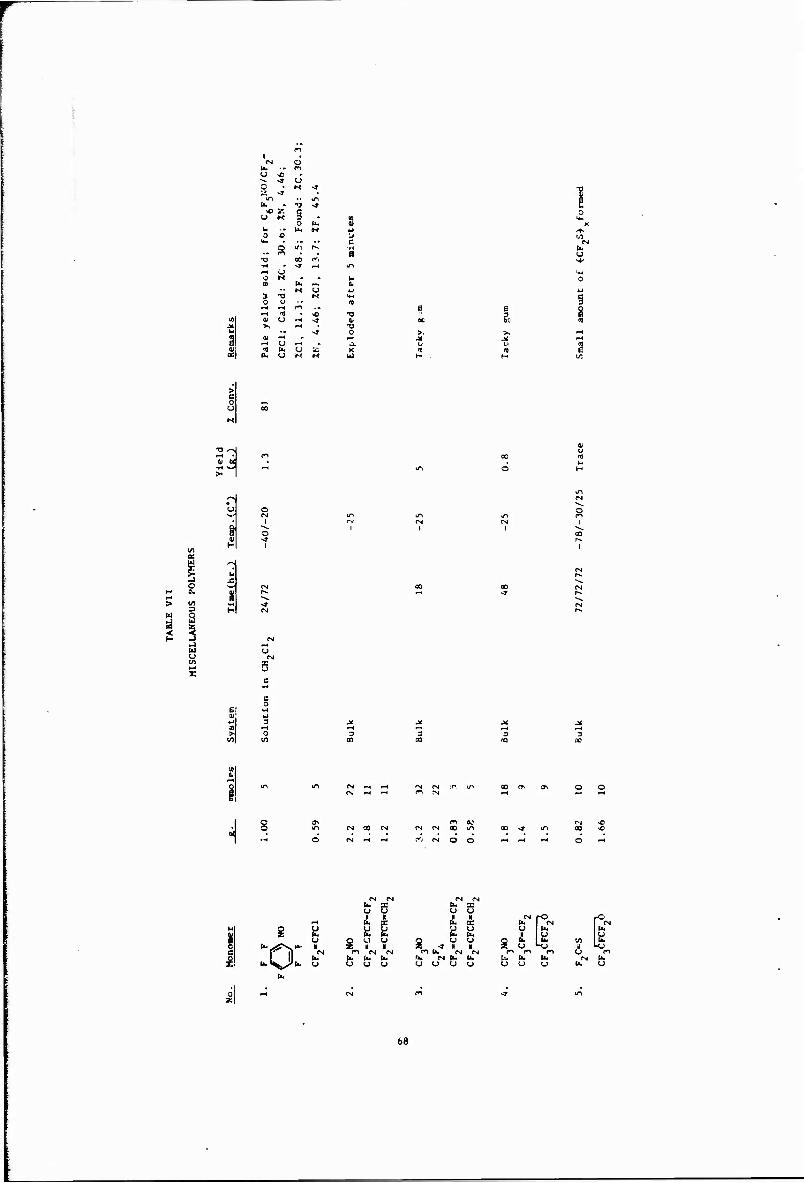
r
u
o u •• ■-* ^H m
4) i~4 -I u
a. u ■3
o w e.
o PI
I ^~ oo r>
I
5
■X
3 3 3
so ^ o
00
d OO <t ü-1
I c i :o:
ft, U s ft.
U s H £ > £ X
PN ft. -o fN r
u • 0 i -T
id u a
o ft. u • i £ ft.
a
ft. U ft.
lb PN f» rl u f» 0. ft. ft. ft. ft. fN ft. ft. ft. ft. ft. fN ft. u U U u u u u u u u (J ft. u
68
i
3 a
T 11 U c 3 •H •fl n * u H a a. •3 0
•H a *H W 0 0 3 ■a •** 01 *H •H u 0 3 U ►s a) cr 0 .-t ■H V !» g -H u fl
cH •O 3 V) ■*
■a u 0
PH 0 -H
ti 3 a l-t u 0 «J 3 0 ... 4J £
T> u -J 00 n w u, o •H fc •H ■H (N ^ u > VI X w
J
s o-
> c 3
w CO
c o u ■-. oo
<
3 3 as
b. o
s fM
s
m ft
5 B
CM (N
U
I* IN O ■"<-> B *
b. Q tt, u 5 Q
o 7.
<N U. U r*
(N b. •M U u >
r* b. 1« O a X 8
1*1 (N p
5 m b. u u
o
uT* 5 b. U
=-8
69

r ?-
V I >
r. o
c o
■H ■H
«J 4J
■ ffl N N
■H ■rt
ki h a o g a a a C 0 l a t a a a a 41 a a 0
•H 0 o .2 0 0 0 o 0 0 0 o I
u w u W w *i 4J W »-I U u *J «J W 4J H u u u U u u u U
8 U u O U u U 0 « « 3 8 a
5 ? 3 • i V
a « 91 cr u 3 3 O.
0 M h h u M M M M h U u M ki u
£ £ 2 £ 2 £ £ i £ 0 z 2 s 0
z 0 2
0 z £
i « 0 u
«I M
z o M
5 HHHtillHHH
C V
•H 4J u 4J Jl 4J *-- u c B P * C o c c «1 11 V 0J u o 111 •H ■H •H ■H ^■^ ■H
in A J} ^1 o J 1 -O ao in o Xi i O CM
1 i 5 5 •-4
1 1 5 1 Csl
1 B
1
o -» CN
*o OJ 00 in o vO *T> O o J5 rj CO 0^ -I
f CY a M^
o •a c
cr C^ !X
C
~r r
O in -o
.O 0 a) •H o c IT, 3 8 IX n
w ~t c m C c C O <J n m 0 4) o <•
TM -*H •* C o*^i *•*
.J or.' u. in u 0 -* u •. .* Ji J< ^~ u 3 X -Ü J< j; 3 *J .v jr U 3 I ^H r-4 rH u •~4 x .-< PH «1 ^ PH (^*-i p
<g i 3 •--* 4J 5 s U 3 3 3 0 u 3 3 b- 0 U CO c « H •s^ CO S3 03 en to cfi CO u ä ■*-*
•< r-
tn u tJ u «1
4 2 00 -»
d PH
tu X V ft. u
u u
o ■
p-t u CJ
X
o II u
z
X u
CJ I o u i z z
o W X CJ
s o
O *-H r*» u-^ «H ^ iH
O fH r*- *n
w <* iH O
I*.
4 p4 O r-:
r-l f* O
£1 ^
70

9 "l
ki «I 1
c 0
w 0
& M 11 « 5 w u I« 0 V
b >• w 8 2 N b o 1/1
c I 2 M 0
II • » h . - 0 a V
& *1
0 a ■o & ■H
0 ' 0
U & 3 i i a 0 ■o c T3
a a y •H 0 0" <r u *H 01 0 0 0 b -4 0 •* *J 3 u -4 •H c 0 «J
■H
4J ■H I kl U § 2 o- a : -» * § ■"'
■H n § OS
u U 1 M 0 rH IA «i w 3 T) 0 M 0 n tg Ul «H * & V 3 e u ~* e a V V lb 0 w k
9 J 0 tl 0 0 u QJ u
k« u t/1 a a ä Ji 0 u k> 0 (. 1« .. -o w 0 u u w £ g en a »
3 *■* •H tj
£ i £ c ~* V. Q a *-i *H *4 •H o 5 c *H U 01 H -H U (0 > 01 iH > c Ü «9 T4
a H o
i_>
T) •H tl w
•H >■
5 s IM
u U. J Wl Kl i/i < w H U
u 3 w
3 no
3 S3
3 3
a* Q m ^ in
-HOC
H 3
c a: 0
c o 0 >r\ s •" I *J cs V
-X Ji 3 <J a w -H *H 3 3 o c 3
M 0) W « w
b. U
PL, £ <N U u (J
U £ fS £ £ o n u
csj I r IN u tu o
lb -N X r u [X TH X £ u r>i t/1 U
*g 5 o in -*
LO c-i (*i X hi 33 h fN en Ö U U u
3
r* ^ OJ
-too
00
o z 8
71
IS r°~ *i
0 1 I5 (3 0
■Ö
c ■o •H t- <N rN W |0 u £ 0 a L5 O 4. pN u *J ex 0 to -X U. 1»
■o <g c M o «I ■H h a 0 o 0 87 IS V- a 0 1-4 ■ » u BTS c -o 5 a" & i c V « en
i-< 14, ■H • •• 4J .» w .*. u u. "H 0 B w
s 5
§ 2 12 9 2 u •o 41
§ H H 3 M 0 n. "SL oü 0
0. 0 41 0 0 >, k. ►. k« >, t* > u vC u «J K Jf 0 J« 0 ■8 S
M o » a. ■J) (N! u u y u O u •w -» 0 ■H g « a « a <0 B o «I > u z > H -H H ■* H •H <-t M
*-« B 0 U
-o !X>
>■
a. B 01 H
9 u
o in t m
i o
v. z o 1-4
IM
M O
l-J a.
W > nj w
B w U* 0 ~l w a
3 0
1 H u o u 3
13" E CJ l(
E
r° CN
0 Lg
u.
u o
z r- u. 'J
'I c.
72

TA LBLE IX
NMR ANALYSIS
(Peak areas in pe rcent of total area)
Sample Solvent
78.4.2 Neat
78.5. Neat
2 83.1. Neat
2 78.6.2 Freon 113
78.7.1 Freon 11
78.8 Neat
Shift ppm (CF3COOH)
-20.0 1.6
-18.2 4.3 2.7
-15.0 1.7 1.6
-12.0 21.1 46.1 44.0 41,7 39.^ 44.4
-9.9 25.2 6.3 5.7 6.2
-8.9 -6.4 5.7 1.2 1.4
2.4 1.6
Total CF3 58.0 53.6 51.1 41.7 52.3 47.6
+9.6 1.4
+11.4 10.9 20.6 22.8 25.8 19.5 22.6
fl4.0 3.7 1.2 1.4 2.9 1.4 4.4
+21.5 7.7 5.5 5.7 4.4 4.9 4.0
+23.7 6.0 16.5 17.4 23.4 17.6 21.4
+26,3 2.3 0.8
+27.1 i.4 1.6
+28.2 6.0 2.3 1.4 2,2
Total CF2 42.0 46.1 48.7 58.1 57.8 52 4
Description of sample polymerization:
78.4.J Bulk, 25°, 1:1 CF3N0: CF2»CF2
78.5.2 Same as above
83.1.2 Terpolymer with CF„Br2, bulk, 25°, 1:1:1 monomer ratio
78.6.2 In Freon 113, -15° to 25', 5:1 CF3NO:CF2«CF2
78.7.1 In Freon 113, -15° to 25°, 1:5 CF3NO:CF2-CF2
78.8 In cyclohexane, 25', 1:1 CF3N0:CF2=CF2, Ziegler catalyst
73

IV ACKNOWLEDGMENTS
NMR analysis and interpretation were carried out by Dr. Wallace Brey of the Department of Chemistry of the University 01 Fic-rida. Many helpful ideas and suggestions during this program were contributed by Dr. Paul Tarrant and Dr. George Butler, consultants tor Peninsular ChemResearch, inc Polymerlzations using .cbalt-60 as an irradiation source were „arried Oat through the cooperation of Dr Robert Hanrahan ct tfe Department of Chemistry of the University of Florida.
■it

V. LITERATURE CITED
1. Barr, D. A. and R N. Haszeldine, J, Chem Soc, 1881 (1955).
2. Yakubovich. A. Ya , V. A. Shpansk.il, and A, L. Lemke, Dokl, Akad. Nauk SSSR, 96, 774 (195'.).
3. Crawford, G, H., D. E. Rice, and W. J. Frazer, "Arctic Rubber," Final Report under Contract DA-19-129-QM-1043 for the period 15 October 1957 to 15 August 1960 (AD 2467CA),
4. Brown, H A,, N. Knoll, and D E. Rice, "Arctic Rubber," Final Report under Contract DA-19-129-QM-1684 for the period 24 August 1960 to 23 December 1962.
5. Paustian, J., et al., "Nitroso Rubber Research, Development and Production," Final Report under Contract DA-19-129-AMC-69(X) for the period 27 February 1963 to 28 February 1965.
6. Criffis, C. B., "Research Compounding of Nitroso Rubber," Technical Report C & 0M-13, September 1965.
7. Henry, M. C. and C. B. Griffis, "Nitroso Rubber Handbook," Technical Report 66-2-CM, January 1966.
8. Middleton, W. I., E. G. Howard, and W. H. Sharkey, J. Am. Chem. Soc, 83, 2589 (1961).
9. a. Martin, K. V., J. Chem. Soc,, 2944 (1964) and C. G. Krespan, DuPont, Wilmington, Delaware, personal communication.
b. Middleton, W- J., E. G. Howard, and W, H. Sharkey, J. Am. Chem. Soc. 83, 2589 (1961).
10. Durrell, W. S,, E. C. Stump, and C. D. Padgett, J. Poly Sei. A3, 4065 (1965).
11. Tullock, C. W., U. S. Patent 2,884,453 (28 April 1959).
12. Harris, J. F., Jr., J. Am. Chem. Soc. . 84, 3148 (1962),
13. Case, J. R., N. H. Ray, and H. L. Roberts, J. Chem. Soc., 2070 (1961).
75

1^ IuiAv-k, C W D D Celt man, and E L Huctttuies, J, Am Chen. So. , 86, 35 ■ (196^ •
13 Cohen, B and A C> Ma.Diarmid, Cntm, and Ind , i.866 .i9tv)
16, Dale, J W , N A Edison, and G J O'Neill, Monsanto Research Corporation, "Dcv eicpment or Fluoiinated bjaui and Aromatic Nituso Monomeis," Final Report under Contract DA-19-i29-QM-1989, ' December 1962
1 . Ia=kfei, H and H J„ncs>, J Chem So. , 95, i«i- \x90Q».
18 Rcinbolt, H , Bc_r_ . _59, i3,x i926
19. Le.hti H and H ^ttktn, Sei , _59_, i;l- and ^594 «19*6
20. Emeieus, » J and H Fugh, J or.em. a... , liC8 1960
21. Haszeldine, R N and J M Kiad, J , Ctuir, bo , >2x9 u953.
22. Stamp, E C and C D Padge-Ct, inot^ Chem , _J_, 6j.0 i964,
23. Taylor , L to , T. J Bri>.c, and R L Wear, J 0i£> Cr.cm , 21, i06i (1962,
24 Case, J R , N H Ray, H L Roberts, J, Chtm Si -. . ^066 (.1961),
25 Colrman, D and C TUIIOCK, U S Fatent 3,4.02,903 3 September 1963)
26 ^arovenko, N N , S P. Motomyl, and L N Kirtn^kaya, Zhur. Ocschei Khim , 21» <?9b '195?,; C A , 52_, 8042a u938*
z1. Banks, R E , R N Has2e.rH ■ ne, and D R Iaylor, J. Cnem Soc , 5602 u965>
28. Cheburkov, iu A , Fluorine Chem Revs-, 1 10? >196 )
29. Tatlow, J C , Endeavour , 21, (86;, 89 a%3/-
30 Coe, F L , Chem Prod , 2b_, 1 i 196<>.
31. Mwloughlin, VCR and J Thrower, "Fiuor^aromatl. f^lvmers," Royai Air^rait Establishment Report No CPM 11, Mar.h j.96i
32. Brooke, G M , J Burdon, and J C Tauow, Chem lnd , 8:z 1961)
33. Castellano, J A , J. Green, and J M Kaufman, J Org Chem , 31, 821 1X966)
3^ Bank». R E , et al , J Cnem S^_ , "'203 u965)

35. Case, J. R., N. H. Ray, and H. L. Roberts, J. Chem. Soc. , 2066 (1961).
36. Robson, P., M. Stacey, R. Stephens, and J. C. Tatlow, J. Chem. Soc, 4754 (1960).
37. Rice, D. E. and G. E. Crawford, J. Chem. Soc, 28, 872 (1963).
38. Banks, R. E., M. G. Barlow, R. N. Haszeldine, and M. K. McCreath, J. Chem. Soc, 1350 (1966).
39. Dyatkin, B. L., et al., Dokl. Akad. Nauk SSSR, 165 (6), 1305 (1965); CA.. 6±, 9581b (1966).
40. Ginsburg, V. A., et al., Zh. Obshch. Khim., 30, 2409 (1960).
41. Park, J. D., A. P. Stefani, and J. R. Lacher, J. Org, Chem,, 26, 4017 (1961).
42. Schonberg. A. and A. Stephenson, Be_£., 66B, 567 (1933).
43. Middleton, W. J., E. G. Howard, and W. H. Sharkey, J. Am Chem. Soc., 83, 2589 (JnSl) and W. J. Middleton, private communication.
44. Emeleus, H. J. and D. E. MacDuffie, J. Chem. Soc, 2597 (1961).
44a. Smith, W. C, et al.. J. Am. Chem. Soc, 82, 551 (1960).
45. Holland, D. G., G. J. Moore, and C. Tamborski, J. Org. Chem.. 29, 3042 (1964).
46. Harper, R. J., Jr., F. J. Sokolski, and C. Tamborski, J. Org. Chem., 29, 2385 (1964).
47. Holland, D. G., G. J. Moore, and C. Tamborski, J. Org. Chem., 29, 1562 (1964).
48. Tamborski, C, private communication.
49. Tarrant, P., A. Lovelace, and M. Lilyquist, J. am. Chem. Soc. . 77, 2783 (1955).
50. Banks, R. E., R. N. Haszeldine, and D. R. Taylor, J. Chem. Soc., 978 (1965).
51. Jacobs, T. L. and R. S. Bauer, J. Am. Chem. Soc. , 81_, 608 (1959).
52. Dixon, S., U. S. Patent 2,917,548 (15 December 1959).
77

-3. Yaro.pnko, N N , S P Motornyl, and L N Klrenskaya, Zhur. Obah>h. Khlm , 2]_, 2796 (193?); CA., 52_, 8042a (1938).
54. Cherburkov, Yu A., A. M, Platoshkm, and 1 L. Knunvants, Dokl. Akad. Nauk SSSR. 173, (5), (1967).
55 Banks, R E , R. N Haszeldine, and D. R Taylor, J. Chem, Soc., 5602 (1965)
56. British Patent 904,877 (5 September l9o2>
57. Henry, M C , C. B. Griliis, and E. C. Stump, Fluorine Chem, Revs. , 1. (1), 1 (1967).
58. Crawford, G H and D. E Rice, U. S Patent 3,321,454 (23 May 196?),
78

APPENDICES
79

APPENDIX A
Infrared Spectra
Figure
Al . 2,2,<=*,4-Ietrafluoro-l,3-dlthietane
A2 Thlccarbonyl Fluoride
A3. Pertluorotruoacetone
A4, Tritluoromechyl TrIflucrovinyl Sulfide
A5 Triflucrome'hyl 2,2-Dichloro-l,1,2-trlfluoroethyl Sulfide
A6. Tritluorovmylsulfur Peritafluorlde
A7. 2-Chloro-l-nicroso-l,2,2-trifluoroethyl Trifluoromethyl Sulfide
A8 BIStpentafluorophenyl) Disulfide
A9 Pfentafl'iorünitrosobenzene
A10. Pentatluorobenzoyl Nitrite
All. Tetrafluoroterephthalyl Mononitrite
A12. p-Aminotetrafluoronitrosobenzene
A13. A,','-Diaminooctafluorobiphenyl Oxidation Product
A.IA 3-Caibomethoxyperfluoropropionyl Nitrite
A15 4-Carbomethoxyperiluorobutyryl Nitrite
A16. Methyl 3-Nitrosoperfluoroproplonate
Al?. Methyj 4-Nitrosoperfluorobutyrate
A18. ICH302C (CF.,) 31 2N0 (CF2) 3C02CH3
A19 |CH302C(CF,)2)2N0(CF2)2CC
2CH3
A20. IH,NC(CF,)J ,N0vCF,)-CNH, Z Z 5 Z Z j Z
A21 iNC<CF,) J0NO(CF,),CN L J L i. J
A22. ^-N\trosopertlu< robutyric Acid
A2 3. i-Bromo-1,1,2,2-tfctrafiuoro-2-nitrosoethane
A24 Methyl i-Chloro-l,2,2-tr it Luoro-2-mtrosoethyl Ether
A25. C3F70(CF(CF3)CF20]^Cr-CF2
A26. CF NO/CF3SCF=CF' Copolymer
A27. CF^NO/Cf2-CFBr Copolymer
A28. :F.NO/CF~=CF./C,FtNO Terpolymer 3 2 <: 6 5 Y J
A29. CF NO/CF2-CF2/CF2-CFCF-CF2 TeL?olymer
80

APPENDIX A (Cont'd)
Figure
A30. CF-NO/CF2=CFCF«CF2 Copolymer after Reaction with CF.
A31. CF0NCF„CF=CFCF. 3| 2 I 2 0 1
OF
A32. CF3NO/CF2=CFCF-CF2 Copolymer after Reaction with C1N0
A33. CF3NO/CF2=CF2/CF2=CFCH=CH2 Terpolymer
A34. CF3NO/CF2=CF2/CF2»CFCH=CH2 Terpolymer after Reaction
with CF OF
A35 Cf3NO/CF2=CF2/CF «C-CF, Terpolymer
A36. CF3NO/CF2=CF2/CF2=CFCF2NO Terpolymer
A37. CF3NO/CF2=CF2/CH302C(.CF2)3NO Terpolymer
A38. CF3NO/CF2=CF2/CH OCF=CF2 Terpolymer
81

f-'-rf
ra» r :~T ■■•—{-
Fig. Al. 2,2,^,^-Tetrafluoro-1,3-dithietane (gas, k and 8 mm)
;'■•• ;'•'• y »"'" ^- ! I i "I ' I
Fig. A2. Thiocarbonyl Fluoride (gas, 6 and 90 mm)
iqjJJ^'il^IITi^lL: 1J1LL1J JL1,4JJLILLL1| I I T I T I
Fig. A3. Perfluorothioacetone (gas, 12 mm)
82

Fig. A^4. Trif luoromethyl Tri f luorovinyl Sulfide (gas, k and 60 mm)
Fig. A5. Trifluoromethyl 2,2-Dichloro-l,] .2- Trifluoroethy! Sulfide (Iiquid)
Fig. A6. Trifluorovinylsulfur Pentafluoride (gas, 15 mm)
83
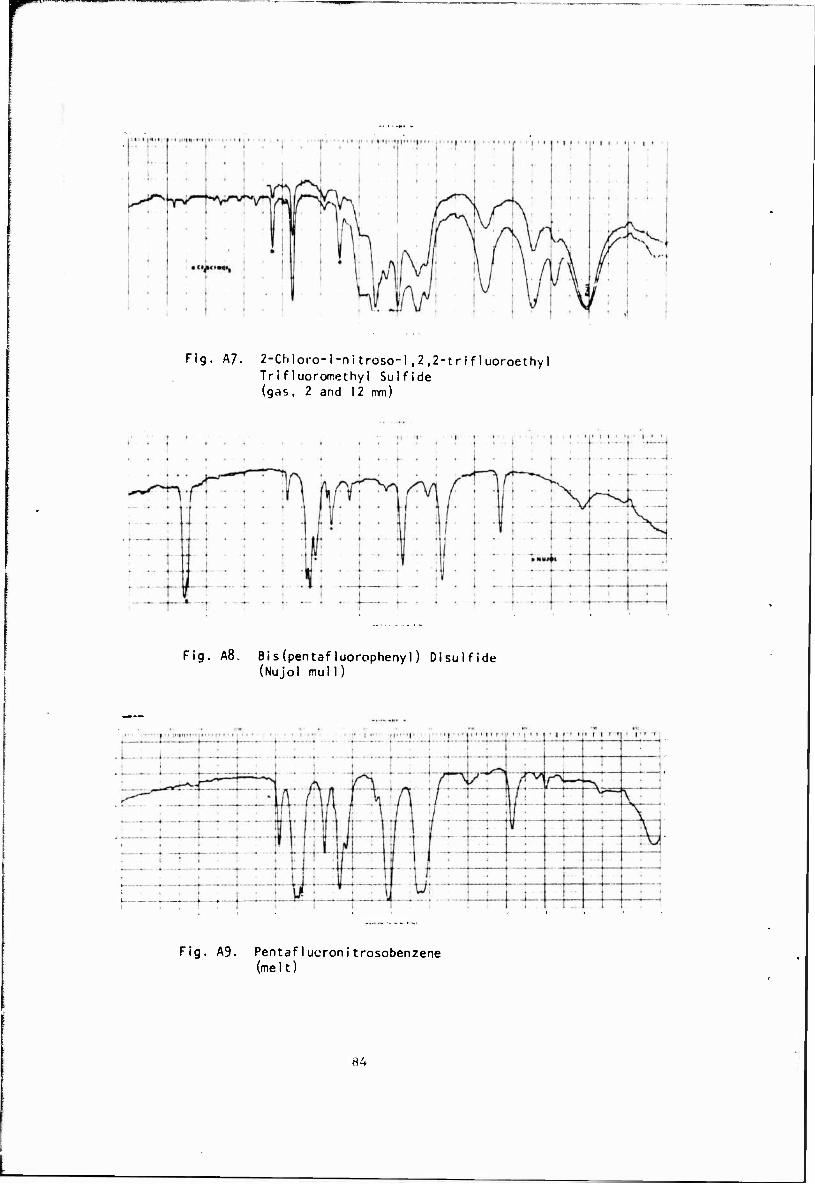
Fig. A7. 2-Chloro-l-nitroso-l,2,2-trIfIuoroethy1 Trifluoromethy! Sulfide (gas, 2 and 12 mm)
Fig. A8. Bis(pentafluorophenyl) Disulfide (Nujol muH)
-+-T-+
Fig. A9. Pentaflucronitrosobenzene (me 11)
84
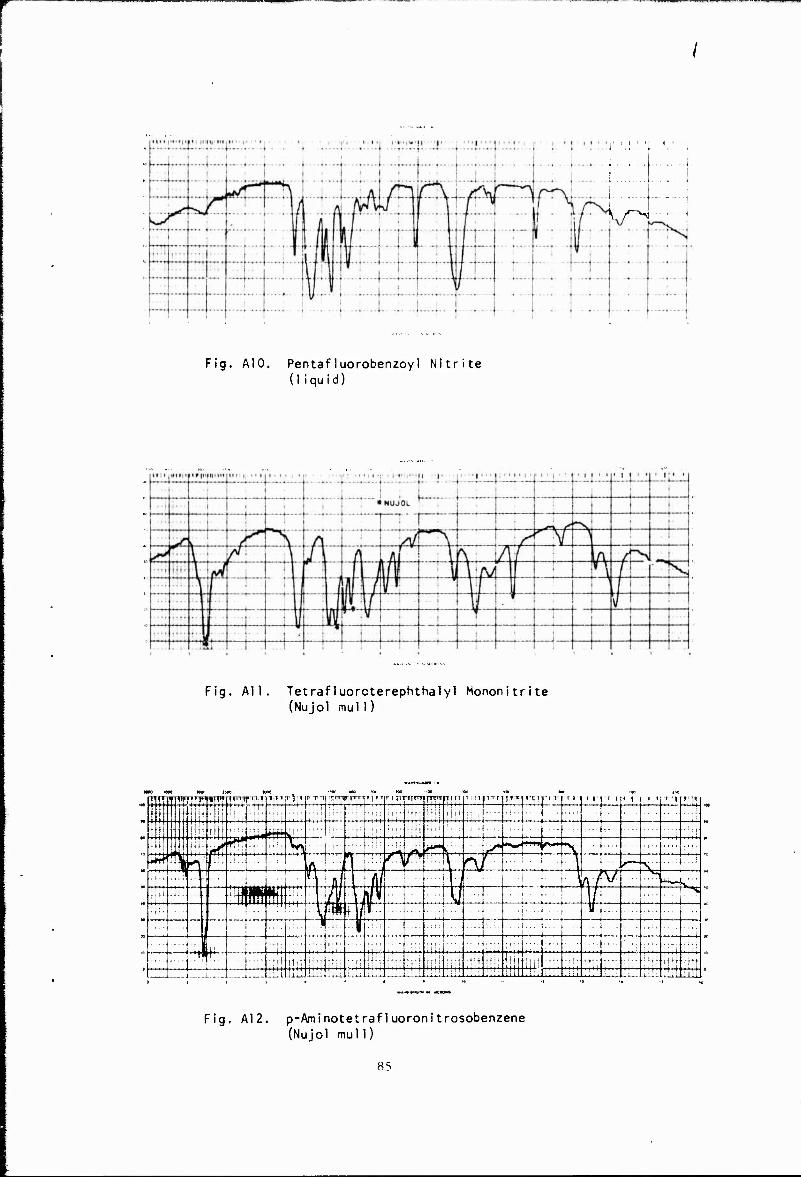
'II1' i -.
......
i ■
\ r^ . . 4
Fig. A10. Pentafluorobenzoyl Nitrite (1 iqu id)
Fig. All. Tetrafluoroterephthalyl Mononitrite (Nujol mul1)
«O «0«)
f»PW1" p-n:f DC
f -Vi ill I 11 in irijTtr rji M 1 t ! ! TIT l,7r|*"F ! 1-tT "'" 1 ' ' ' 1 ' ' 1 1 irji
1 1 i ii !i IT: jtil r ■ • Hi.:-;- .,:. . 1 ■
it I ■ frf] tVif
IH ......
*£■ ! !: ■■>■ lit* «-<+~n\ . j.. .
M\ •*-F' A ;' " ^; r .. . . 1 k - ;]...! MM ;iij
-■»I . !:;:. ,.::!. : 1 ' Al 1 / w/r ..j.:^ "Hv«
'ftjfm Hfl] üitt i ■ ■ y i • )
/ ■ i ■ .... |
•'•■ ■ 11/ i ...... L
• - i ■ i ..! = ... . .j..
iiBl; ....
i i ii iiM j plil'i 'I*)!' Jptd hi ILLL |*i
■:—■
llij Ü4 Tui m {)j| |H1 4M'
-*■
+H+ t-j-14 £+ iu. ■ ■ill i ..
44 ilü
Fig. A12. p-Aminotetraf1uoronitrosobenzene (Nujol mull)
83
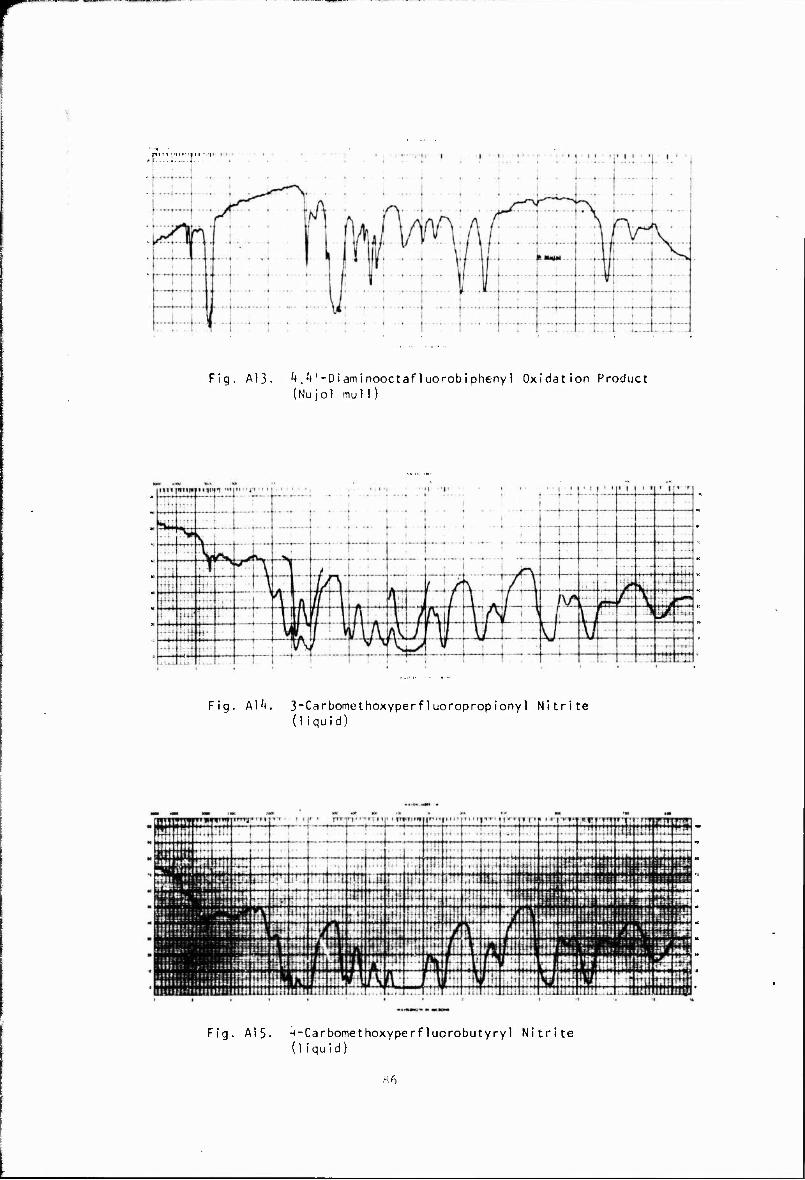
.pra:."!
Fig. A13 - ^ .^'-Diaminooctaf1uorobipheny1 Oxidation Product (Nu jol mu11)
Fig. Al'». 3~Carbomethoxyperf! uoropropiony 1 Nitrite (li qu id)
Fig. AI 5 • •» - Ca r borne thoxyperf luorobutyry 1 Nitrite ()iqui d)
86
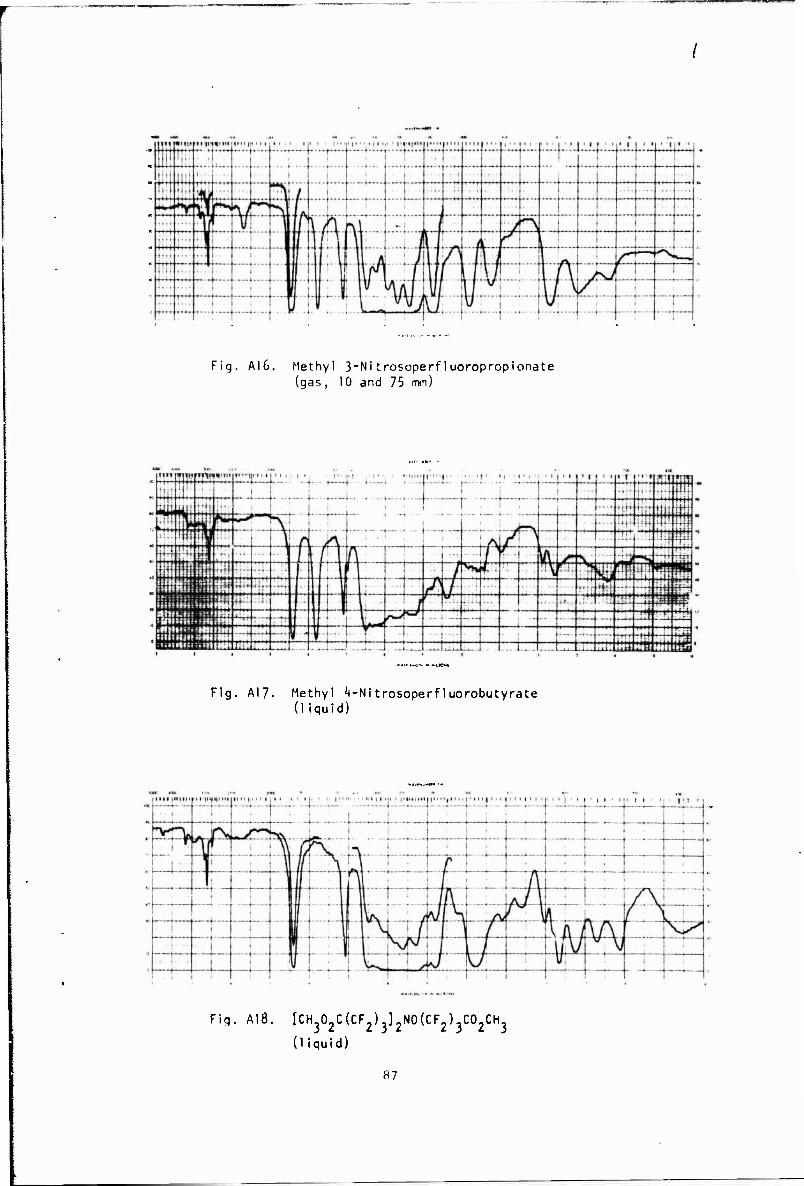
Fig. A16. Methyl }-U\ trosoperf1uoropropionate (gas, 10 and 75 mm)
Fig. Al 7. Methyl ^—NItrosoperfluorobutyrate (liguid)
Fig. A18. [CH 02C(CF2)J2N0(CF2)3C02Ch
(1iquid)
87
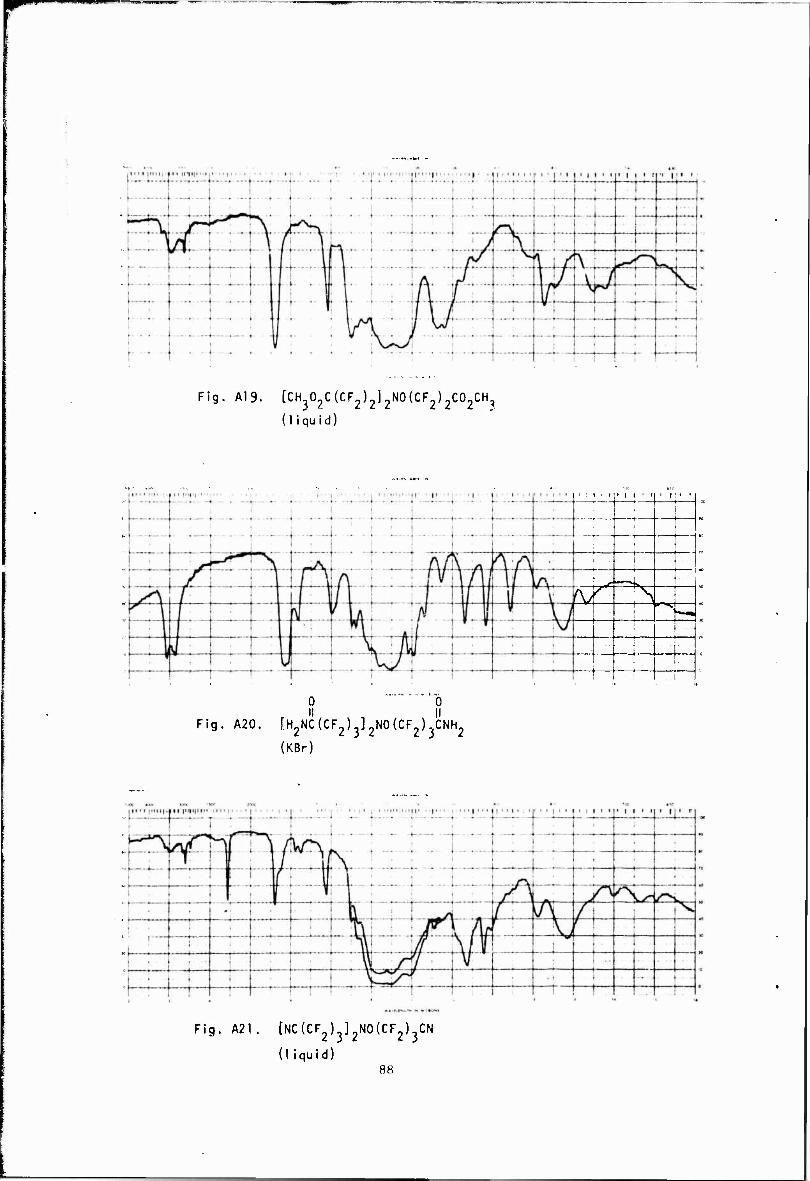
^IWW1IIII,H
Fig. A19. [CH302C(CF2)2J2N0(CF2)2C02CH?
(liquid)
1 ,' I I I
^TTX
0 0 II II
Fig. A20. fH2NC(CF2)3]2NO(CF2)3CNh2
(KBr)
Fig. A2I. [NC(CF2)3]2NO(CF2)3CN
(1iquid) 88

IHM inii|iiii |lii
Fig. A22. ^ — Nftrosoperf1uorobutyric Acid (1iquid)
Fig. A23. I-Bromo-1,1,2,2-tetrafluoro-2-nitrosoethane (gas, 15 mm)
; '' I ■ ) i ■ 'I' ' i • 'I ' i;1 * i
Fig. A2^. Methyl 1-CMoro-l ,2,2-tr i f luoro-2-ni trosoethyl Ether (gas, 3 and 2k mm)
89

K:
Fig. A25. C F 0[CF(CF )CF20] CF-CF2
('iqu id)
Fig. A26. CF NO/CF SCF»CF2 Copolymer
(solid)
'"Ji"1".' "' '<■<"" .,......, > ,
f ■ t
i.- .
, ■.,,,...
71 yrrl 1 ! r~f- "—K -r—'— -f f— • •
z:±::xx:r:7;:.: 1 [ 1 j U4
.... ±j i\"T\
_i_|__r_|_^J_-r.. T~\ ... -L ....... ; | ; j ;._[_„;._ ~-f -4- -*— - i
--f -
I'll
t
'I'll' '1 T~rT
inn -f-r-|-
"TT~M
Fig. A27. CF N0/CF2-CFBr Copolymer
(solid)
90
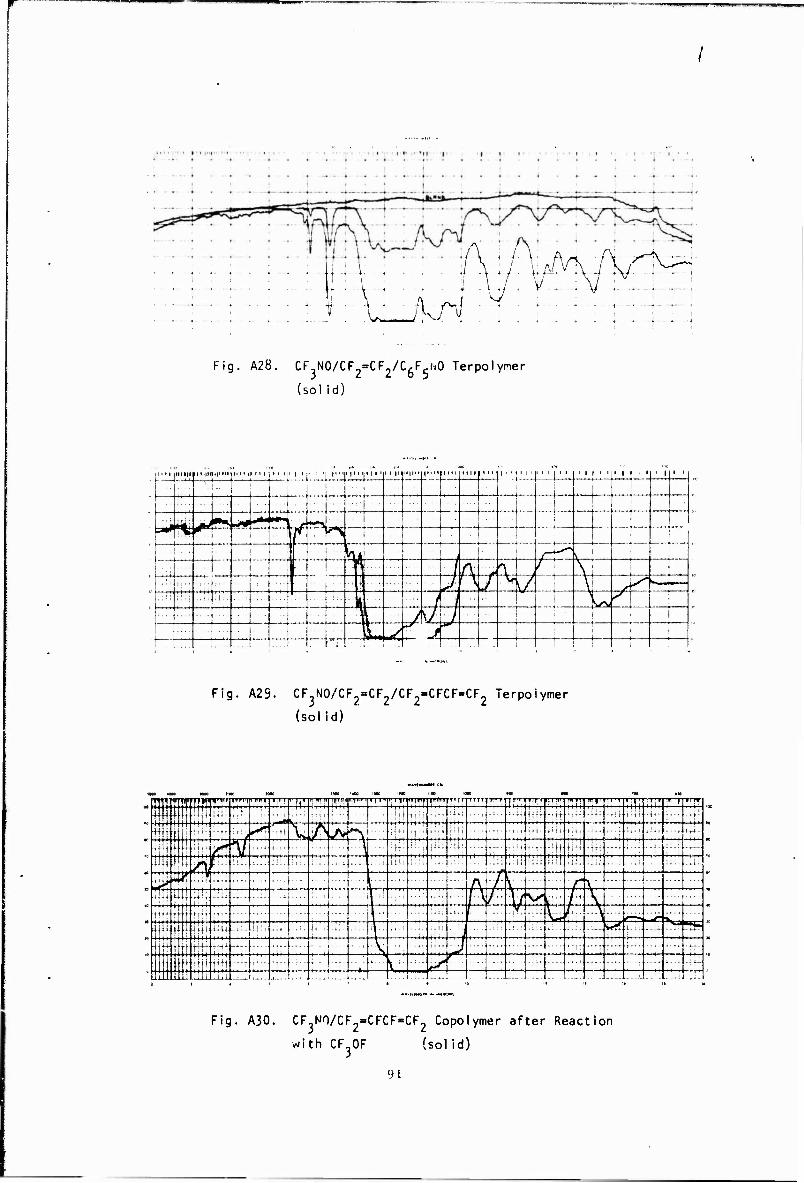
■ :
4. M- ■ A fkH m "<m
Fig. A28. CF NO/CF2=CF2/C6F hO Terpolymer
(sol id)
irrt "11(9 )■ -in 11 n ||l 11 1 *11 11 i .... ■; 'i«'" '!' i lit 111 11 II "i"* iv."!:,':: H11 . i. i ■' • 11 ■' i" 11111 i 1 j > i ' i i r
'■;;
.... 1.... i
i in i •■ r. T , ■ |
jüi i .. 1 , ....... 1 i , j. , ] I i ! J i
i 1 , I' rr 1 I
■ AN \1 } t t ..j. ' ii ■ ... j..,. \j. ^T r H/1 V . 1 .
' yf ;!i! !|i.
■ ■!■■
..,, l i r7! i
. •• \J' j ! i
_ i- --i-- i r. 1
Fig. A29. CF3NO/CF2-CF2/CF2-CFCF-CF2 Terpolymer
(solid)
itgc 'toe
!' 1 w w TIP1 »"" ini( rnrr Ill1 1 "|il tTTTTT TfTiTI Ffplfl T If FT n|H |!'l 'if i 1! 1 TP pnrrn R'f TT 'ii; ill? FT ■rmr
1 \ f -ii ! i tin ■H :,4 ; *! ■ |{ \ \ t; L!'/
y*t ■A **\ 1 ' ' " .;!, [Hi .-, .:,- .... .... :--+ W-
-I i * '!" iff y >, -• .... ..,. *tJ4 !''[ .:.. .... r-f —- ill! .{"! #t
im }!ll I") Oil !;..
IKt life V!f . .... A 1 / i. I.... A
Til liij i /" \l .... t....
H+" H ■H f —
r—*i P S-! mi LU-I ill: ... ji . **-! st ■■ ■ j
ttt~ rt+f ....... 4 — --- i j ■ ~s
! t i 1 ri+i ;U| •- ,i.. '•-- :.')... \
:......: .... .... 11 J liliiJ ..^^ 1 , . ■J.. >,,.■! „..
Fig. A30. CF3NO/CF2-CFCF-CF2 Copolymer after Reaction
with CF OF (solid)
91
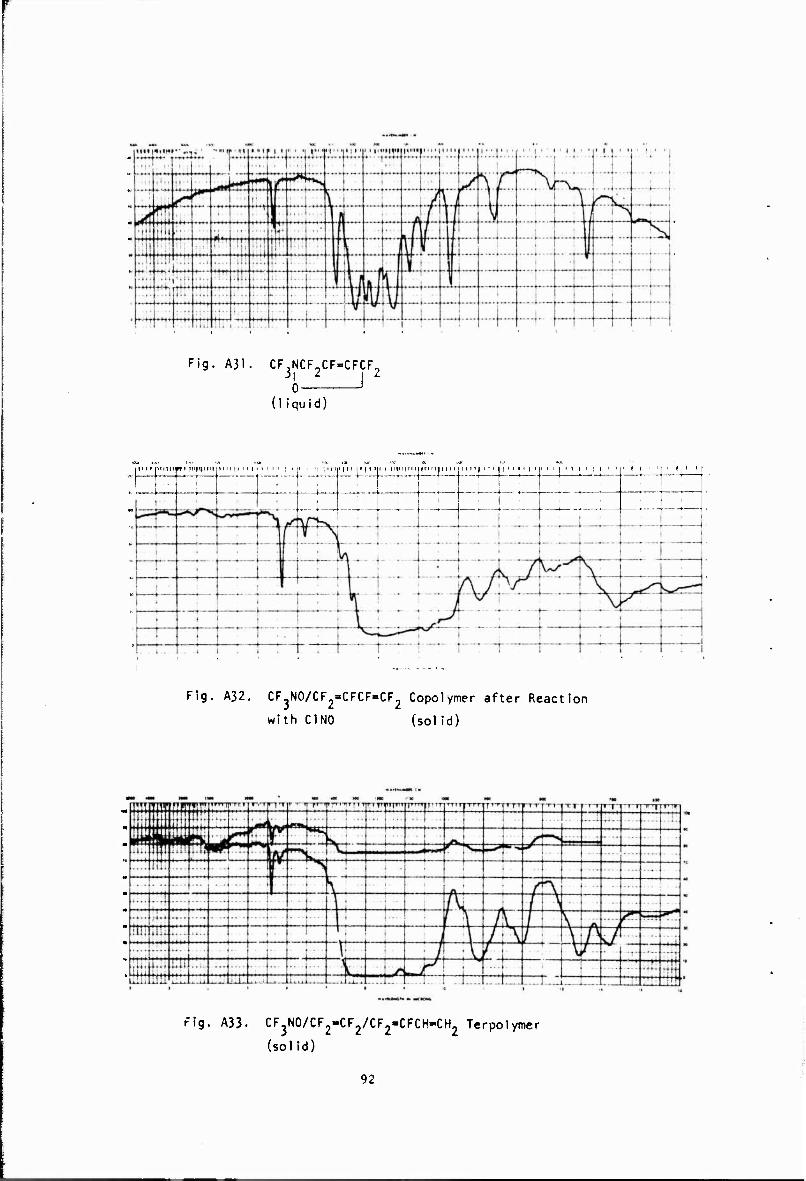
Fig. A31. CF,NCF,CF-CFCF, j| 2 | 2
0 1 (1 iquid)
I l< ' ? MMIII if' •! "r,|'rn i""j""rir"|""Ti"^.'i-
tTX:x: L^^UlU^-
Fig. A32. CF3NO/CF2=CFCF»CF2 Copolymer after Reaction
with C1N0 (solid)
rig. A33. CF3NO/CF2-CF2/CF2-CFCH-CH2 Terpolymer
(solid)
92
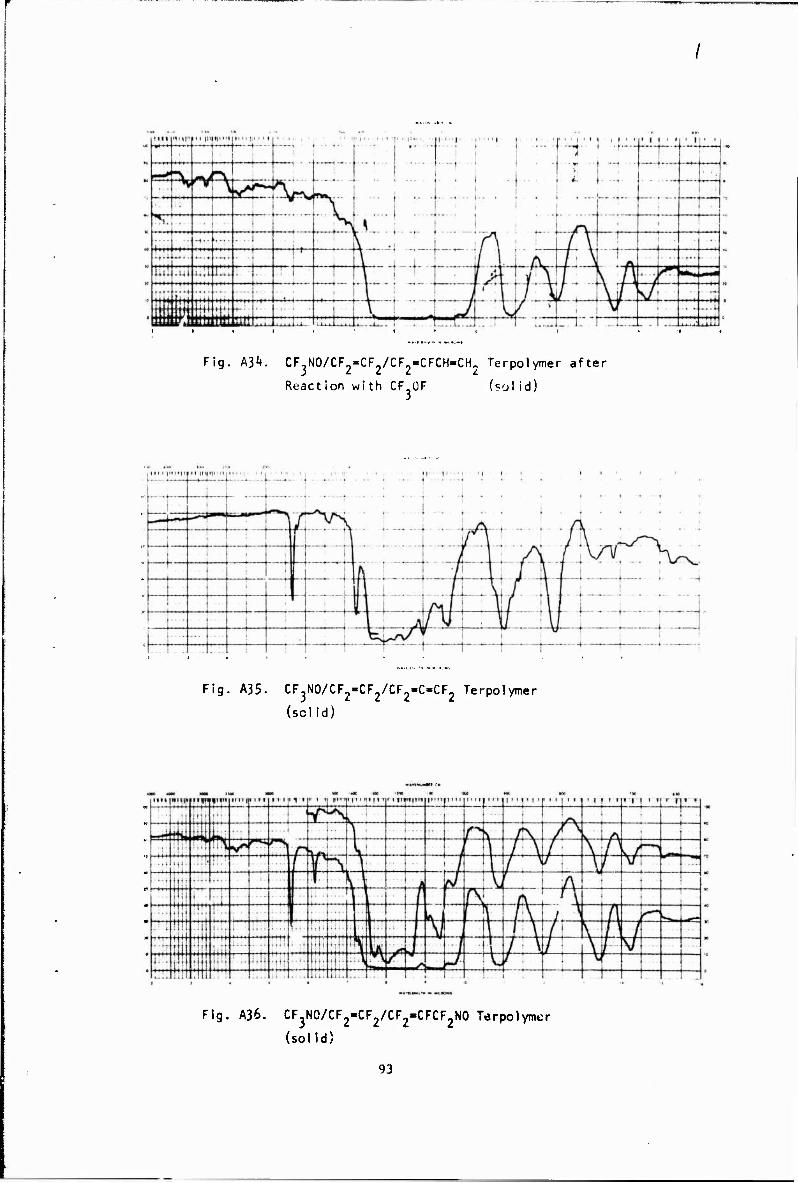
Fig. A3*». CF.NO/CF2-CF2/CF2-CFCH-CH2 Terpolymer after 2 v 2' Reaction wi th CF üF (solid]
Fig. A35. CF3NO/CF2-CF2/CF2-C-CF2 Terpolymer
(solid)
Fig. A36. CF3NC/CF2-CF2/CF2-CFCF2N0 Terpolymer
(solid)
93
Fig. Ä37. CF3NO/CF2-CF2/CH.02C(CF2)3NO Terpolymer
(5 mole % ester, sol id)
Flg. A38. CF3NO/CF2«CF2/CH 0CF=CF2 Terpolymer
(10 mole % ether, solid)
94
a I
•-* —* <*•
—I CO —< r-.
S
H 5
u u, u
u a u
's? a o. u.
+
O
CP
+ + -3 -J + +
M —t
o o O >H
o o
CP c iO CO
111 a. < a o Q
c
I u Q w
£ -
o
y U
ft. u
u
o o»u z p Q u.
0=0 u
K*l u. U 00 <->
CM\ • p u. (< b. tj J <J
o ^
95

r
<y o .a l- lfl 3
QQ H "O rH
CM*J
CM CO
|o=S 3
00
1 < u
IS
£ 3
o u
~s Q
+ CO (M 00
rj CM <N \fy 00 I-*-* C* m O — 00
O
4 + + r-i
+ + CO + + +
O CM
+ in
■*■■
Ifl II
(X < rsj o- <^ ul rsi
o
< m Q UJ U-
I g
CJ 00
U. (<
n
5 u
u O-t) o
X u
o=e u. Q u
U <N
U. < o z
N U
<NJ 00 U. u
__J u z
9t

U TJOD 3 a a
• O in vi -H
(Js —I 0">
m -^ <r
(N (N f*
8 M
CO
u a, 5! 3
c*S
01 •H 06 l/l ** 3 /-N <fl f> 4-t X
»w u X
s~'
X o m o r-* U 1 vT CM ^H m ■tf n sO
OKJ m o> -tf ON 00 n r-. ■—t .—< ■C
X + t—* eN -* -* lA -» 1 u + + + + + + i
+ _ -. + +
a ai
< M U
b. '_>
O
IN go b. U
b. u a u z
g- (N
b. U u 1/1
Ö *
b. u
b. U Z
b. U b. <_> Z
b. U
O O Z| H
97

r
a
*0
• -> <t <» ■" ry* _4 rn ft (-< -J .-« -* .-t ^H
fN <N <N <"v*
fN rg n* r*j (N fN
=*=5
o z (J
X
a.
n » o. «'!
o (N CO m CO u-\ o -* r-> ao o*> cr> o NO CO +■ + +
sO
+ + + + + 00 r*. CO >£> u"i CO fN
1 + + ~:
+ + + 3 +
8 m O o n rj ■—' ■^ -J rsi —« PH FH i-H —< .—I .^
tt.
tt. ->
•" J
tt, U it P
tt. -1 U CO
IN V « tt. (<
E * U U I
U. UJ
o ■ <N OQ tt.
o <
8 S 5 (I
g
9fl

a
8 3 r~n
i~) ^ *-< «H *0 >> >. >■ >, Oi C C C C >
0 o T) ID g o nj (fl fN CM 0 o • *
—i *S| t-4
5
oo
c 3 a
<
:cJ in fN r** <s
ao + +
O*. iTl H
r* <r ^ •—I <NJ
<f —t fN S 00 i^ H
K + + + +
O
+ +
1 O O o O o o "> ^ (N —• rl <N ~H fl
rg (N —( m f^ #H
<0QUOwU.UK»-i
P* P u u A u o
M
o <N < li. < 00 u
-u I
*J o a CM
» I«. < P0J u 1 O p ~M In a. lb
•H o 01 r"»
4 Id
0 o 2 .-•
(N < tb
U—'J X
0u
u. o o
99

r
01
ST o
VI S c
°) i> 0) *j
CO c > u *-• 0/
1 TJ t §
a. <0 *J
01 0 It c
m n u ■3 **i > u £• c
«1 01 01 01 > 3
r>0 <N f-j fj u. u u. U. U. U ft. u u u
O •-» O** <*1 "^ < O « O H r^ rn >H i-H
f*Vi psj rt <N (N rNffi tkHU.tmU.U.U.0
m rj r-i CM fN U. U. U, U. (*- u o u u u
n rn fN u. u- u.
■ i» *n »c o H ^ M (N <w a • ■
u^ O ""* "* °° ■J- n o o fM
o o 00 -J f>
gJ5 ■H OS •J 01 ■H a (fl
*J o -o 4-i 4J *J u w 41 a it 0J 0) 0; 0' oi a< 01
-O TJ r-t -O b 01) -o TJ TJ a TJ ft -* TJ TJ ■a TJ TJ «J nj ac (« 3; k- A3 OC OC ÖC « M m « ™ m m oil 0C oc nj m n> ifl n 0 0 C 0 a xi o c c c 0 C 0 0 0 0 0 c c c 0 0 o Ü u k. k, ■H ki 3 1-, •H 'p. *»H u u k. u k* A 05 tn « VI 03 V) W 0- 01 VI ee SO CO CO 03 vi vj VI CO CO CO CO CO
-a 0
U
X
a.
g
U-. V m
B R a o«j o r-> w h. u. u
vT) r». ü
sO 00 9.
CO N vO ■* >J
N M O f*1 O
I + + + +
00 CO -H i/l f-J o
Ml ifl 00 lA 0> «1
N *J * O ^ O n
^H ^ r-i <M (N I + + + + + +
vT DO ^ 0> H
CM o **» O <*1 H H H N N
I + + + +
CO <T 00
j*\ n -j
7 ) +
KasUQWu^oj:'-*^ < g| U Q UJ
u.
kl
1- o
CN ^H
u
E
o z
100

11 IA H C 1. Xi 0 u .a c o
■5 3" « co
-C 01 « 4, 0
>, u u —' X 4j 4i CCO 41 «H 0 k. P- u fl) 0 c a 3 ti u
*s
E u
z I
b. b. b. 1 u u u u o u u u u u o u u u f ■->
n n N M N rg b. U. b. tfc, U* b. 0 u u u o u
«*» CD CO r* O r^ rsi u- ao ^ N r j cr a- H PH J "1 PH pJP (\
B 0 U
Si
r 5
U-. Cv X
<■> X
ou b.
b. u
41 > O
(N iH r> O r* t*l e* H iH W (N (S
I + + + + +
C
H-jHffionpc 3 O rs *n <*> *0 »H r*i X *H H •-* *H (N (M </J I + + + + +
^\ ("i -^ I— p/1 vß
r-i ,-N m O tN O fN Q f"! •tf sj —* •3- ^H —i -H r-i e-i
t ! 1 ! I + + + •*■
<cuau fc < CO u c u b. u a b.
<BUU < a u a ui u.
e
b. u • b. U
b.
E u • b. u b,
«
b. U
b. u
to. O
b.
b.
b. u
101

■^g^WCT
> * T3 a
0 w W fc k< B O M Si
>.« >.
ü *: w -H ■-<
» (i r r c £ 'Q U U 4
n m IN (N N (N U. Uli. Ik Ik u. u u u u u u
c c a — - a ■^ -w a.
(VI (N (N r« .-^ r>4i. {k h. bk
U. kb to. CJ U O U o u u ■ ■ ■ ■
z z I I p>
U. U. CJ
--• 3 9
: o o o o o o -J >kj 80 -H
c
3 Li
X
a. Si
•5
kj «i
OS
H 5 a CKJ a n u. tv u
■0
0
■O T3 TJ T! 13 t3 (Q (Q >• (Q <4 <V (0 O O k- 0 o o o h M V L. U U U
co a) > (Q act act PQ
a« oo LO
«J (T: ,ü I I I
CON O
ro IA ^
lA .A CD ^ <—• <N
P* fN O <*1 -H <"** •cr —« -< "-" P*4 (Nj
I I + + + +
r-* in —. y. j-
<-« ^t •£ 00 <t —. ^H ^ f .
I + + + +
J3 00
+ +
HNj«TiHiJ«O0Nkfin w
__,■—■ -. H H tN N (N >J I I I +♦ + ++ + +
■31 ill a. I < ca u a u u. ouauiku <»ÜDU«.UI«1UJ
u. u 5 5
<- -a
3 %
$6 u kV
U m ,-«
51
102
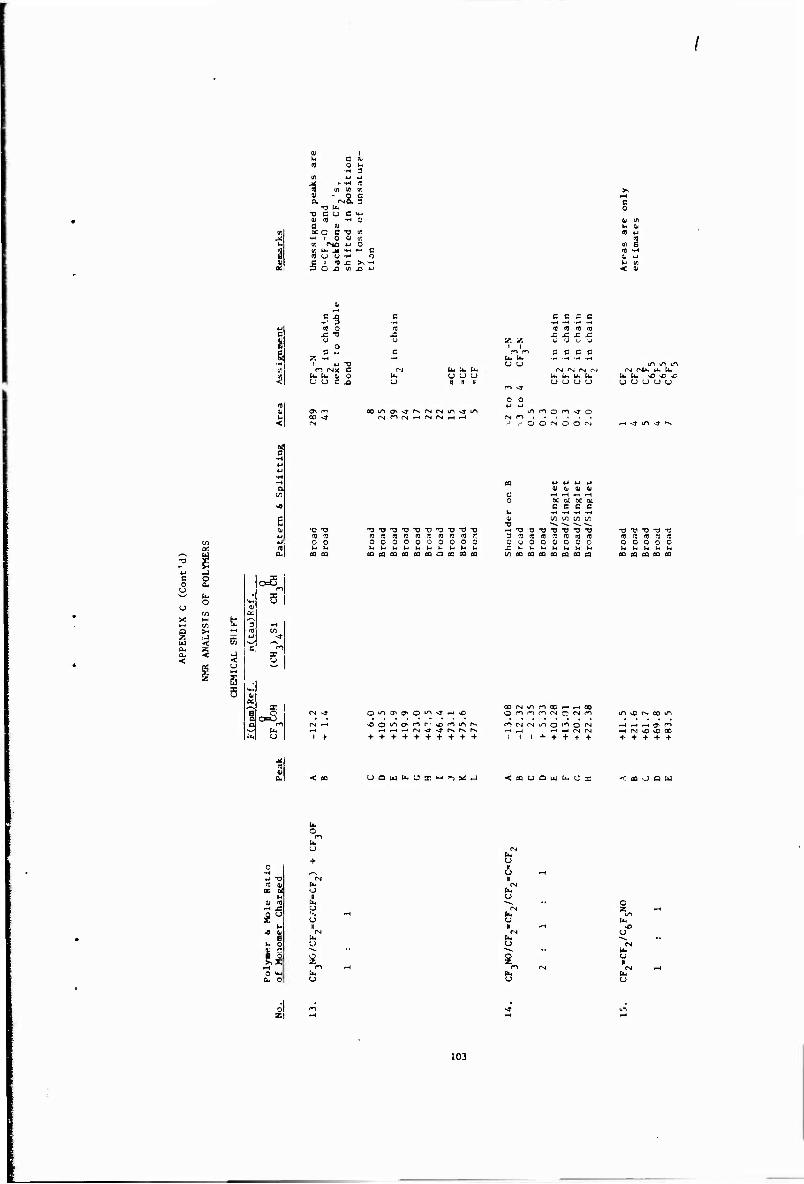
<t 1,1 [A »i
a •<N&§ ■0 U,
TJ c U C W-. 41 n -rH 0 c 0 be c a -o en
■^ 1 o 0 m wi INO *J o m >*- '♦I
P <J u >>
5 o X> en Xl
U» jg **-• ■""< C (T) -H Of w
< ai
8 5
5-8
r> fsix C U. U- Of O
u. u u. u u u
<t nj cfl nj X X X £ u y u u
(\ f\ (N fN U. w- I*. U. u u u u
m i/l o
CJ u o o u
in m o ^ ^r O O o (N o o rj -H ^T \r\ <s r*.
oo oooooooooo CQ cQ aiaacacQcucoeacaEtteQ
ai ai cu cu c ^ ,-t ,-., pH 0 M. ri M x
c c c c M -H fH <H -H ftj ^ w w w -o S **■* *■■* -v *H T3 n -o ■a -a -u T3 3 (fl "3 01 0 0 0 0 0 0 0 o X L, u u u u v. u c/i CO CO 0Q CQ S3 OQ CO
o o o o o u u u u u
CO CO CQ CQ CO
C O |c*g
\OOt/"»^<""lf''^f'",u"'r^
+ + + + + + + + + +
coiNinnoCHHflO
«-* t—i ^-t •-* CN CM
i I I ■*■ + + + +
>n <c is oo i/1
H M O >C CO + + + + +
UQWtfcCxw!-,^;..! <cQunujü-o:r
j- O u. u
+ 0
U 13 p <fl at (4-
a. a u u ii
V «1 u. H J= u
«U u M »
4 11 r
u
o z
01 SEI
;03

sa
b o. < • u 0
4) 4 C X O o u w A
*3 T3 35 (0 -H a J3 « C 4
i u. u. c c u U
s SB fN (N
•H 1 o o I C
■«* 1 IA in in SOOZZUU m u rl o o 35 N N -H OB n IN N N N rs r<h u. u. <N (N r»t) C1 f*l PT*i
a U. U. [h ('- u> u. u. u UOCJUUUUO
U. bu E*< U. VJ U u o u u u u U Q J!. u u
*n o ** <N & o *o 4 O^O^-COOO'-* 4> Cs|rO*H*HOOO-"tOOO
< >O0DOHN(MNN coooooooooo (N M CM »-4 <t -H m n rs*
31
■O TJT3"0T3T)*O*OTD
o oooooooo
T3 TJ TJ "0 ■o X) T3 ■o -a -T3 n <U fl) flj (9 m A tfl tU W n)
0 0 O 0 0 u 0 Ü o Li b. M 14 M '- V- )-. u t-
CQ CQ CO CQ A m CO </> 03 « 03 Cfl ca S3 CQ
C °*4 U. o «*H 6
o BC
X ►-* fe 3 w4 ►H 4 in
*■ R J X m
3» •<
1 5 S
8
5 4J IX
I (V
fc. u
J< 9 ii
O-J^HOOOH^ON^
I I I + + + + + + + +
<aauQi»)U.ux>--Jü
-» c*\ .o o ^o T-> m CM O rt *-■ rt ~-t m
I +
<B)<_>QUb.UX
. wO o **■»
I ■- + +
>» ?N CM • 00 -H *N N « H N H %} *
I I I + + + +
< n u Q u u. u
I*, o
u.
as
:i (V, o
a. u
X u
0- o
u
u M
u.
» u
o
01 >c asl -
10A

C J= o u
o o z
U.11.U.0.XU.U.I1- UUUUUUUSJ
CO O -H
r-* •» (N N <n N o
o.
o
TJ-8 D-O (9 ffl fl <fl nj (fl (fl 0 0 0 0 C 0 0 U »J ►- K u u u Ä 03 03 09. 03 GQ ec
T) -a -a -o >
O It
m c
c o
u
X t—I
w 0-
Si
o a.
5
•<
p-4 •-! >H <H ^4 CM <N *^ I ' I + + + + +
o
u +
d Si OC a u
it 9
g ■ 5
|6 I».
b r •4 4j
0 N 0 *>- 11 0 «W 0. O
10s

APPENDIX D
MONOMERS FROM OUTSIDE SOURCES
No. Compound
1. CF2-CFCF2OCH2CF3
Amount
0.5
Source
University of Florida
2. CF2-CFCF2COC2H5 Universltv of Florida
3.
F F 0
F F
University of Florida
CF „»C
0
25 University of Florida
5. CF2C1CF(N0)CH2CH»CH2
6. CF2-CFCF2NO
7. CF2-C-CH2
8. CF0-CFC.HC 2 4 9
9. CF2-CFCH2CH-CH2
10. CF2-CFSi(CH3)2OC2H5
11 CF2-CFCH2CF2CF2CH2CF-CF2
12. CF2-CHC.='2CH2CF2CH-CF2
13. CF2-CFCF2COCH3
14. CF2-CFCF2CFC1CF2CF-CF2
1 University of Florida
0.5 University of Florida
41 University of Florida
5 University of Florida
25 University of Florida
10 University of Florida
11 University of Colorado
6 University of Colorado
30 University of Colorado
29 University of Colorado
0 I!
15. CF2-CFCF?COK University of Colorado
106

APPENDIX E
POLYMER SAMPLES SUBMITTED
11.
12.
13.
14.
13.
i v.
20.
21,
22.
2 3.
24,
Designation
CO.-A
•;,().-B
V,'. O DJ
W.O.-D7
O.C.-60.4
Q.C. -60.5
Q.C.-4.3
Q.C.-C
.i.C. -8.1
O.C.-8.2
O.C.-8.3
Q.C.-8.9
O.C.-8.10
Q.C.-8.13
O.C.-8.12
O.C.-12..
W.O.-64.1
W.0.-56.1
W.O.-54.2
W.O.-46.1
W.G.-5 7.1
V.O.-68.1
W.O. -17.3
O.e.-35.2.1
o.e.-35.2.2
'-'.A. -51.1
■ 1 vmer
er,.N'n 'CF,.=CF., 1 / /
Charged lonomer Ratio Amount
CF N'O/CF
c;-\N'o/er
=CFBr
=CF !Q~ =CFBr
/ "
BrCFnCr.,
CF..NO/CF.
CF-NO/CF.
CF-NO/CF »CFCF
CF3NO/GF2-CF2/
/ /
/
/ /
/
=c- 'CF„=C^CF-G
; . i
j ■ i
i ■ -
i :1 9 V
1 L viw
1:1 5
l:i 7.<+
6:5: ' 5
3-2: . 5
2:1-1 5
6:5:1 20.4
3:2:1 18
2:1:1 27.3
2:1:1 41.6
1:1 2.6
1:1 16
v.F 2 6:5:: 5
:;o 1:1 + c: HO ■>
:H2 3:2.1
6:5:1
5:4:1
5:4. ',
19:14 '
5.5
5
7.5
230
23
19:14 : 20
4- Cr Op
3 ' 3:2.; + CF OF 6.5
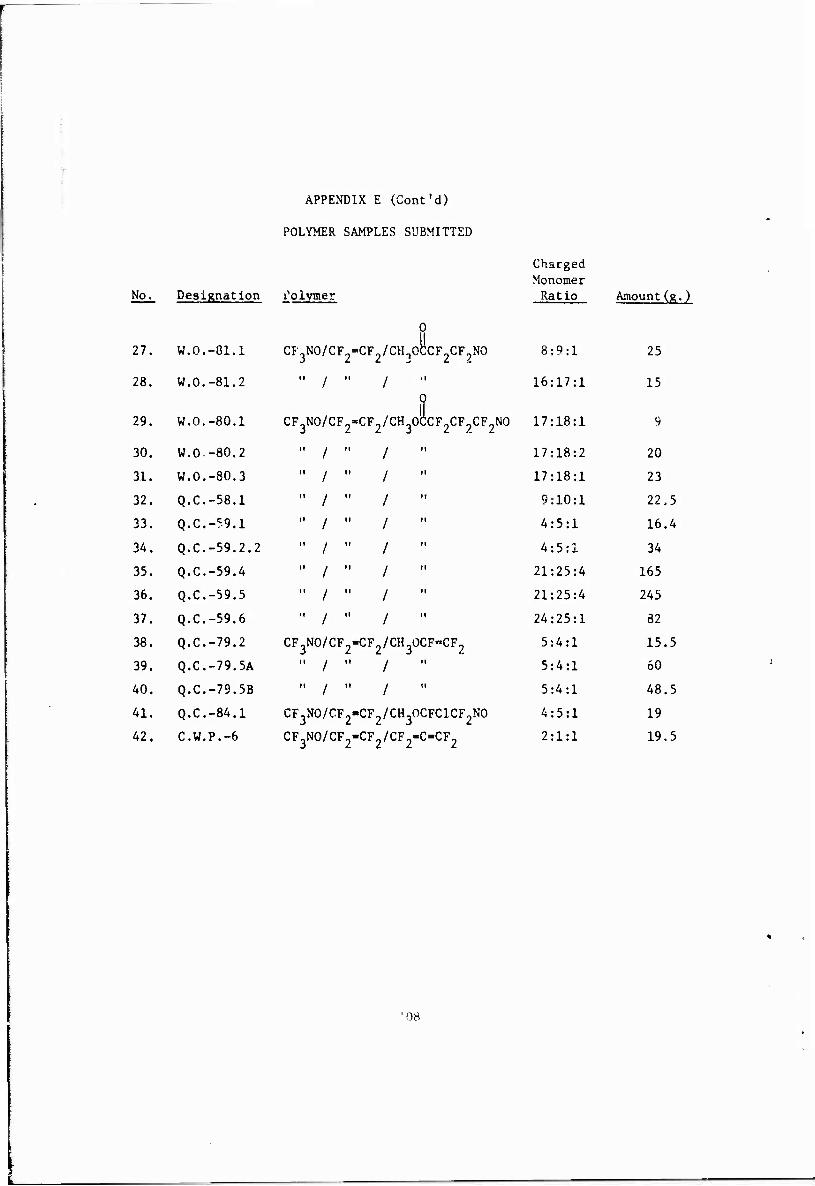
APPENDIX E (Cont'd)
POLYMER SAMPLES SUBMITTED
No. Designation
27. W.O.-81.1
28. W.O.-81.2
29. W.O.-80.1
30. W.O.-80.2
31. W.O.-80.3
32. Q.C-58.1
33. Q.C.-59.1
34. Q.C-59.2.2
35. Q.C-59.4
36. Q.C-59.5
37. Q.C.-59.6
38. Q.C-79.2
39. Q.C.-79.5A
40. Q.C.-79.5B
41. Q.C-84.1
42, C.W.P.-6
t'olymer
u II )CCF, CF3NO/CF2»CF2/CHQOCCF2CF2NO
CF3NO/CF2-CF2/CH3OCCF2CF?CF2NO
/
/
I
I
I
I
I
I
CF-NO/CF
I
I
CF3N0/CF2-CF2/CH30CFC1CF2N0
CF3NO/CF2-CF2/CF2-C-CF2
/ 11
/ tl
/ It
/ tl
/ tl
/ II
/ II
/ II
-CF2/CH OCF-CF2
/ II
/ II
Charged Monomer Ratio Amount (R.)
8:9:1 25
16:17:1 15
17:18:1 9
17:18:2 20
17:18:1 23
9:10:1 22.5
4:5:1 16.4
4:5:1 34
21:25:4 165
21:25:4 245
24:25:1 82
5:4:1 15.5
5:4:1 60
5:4:1 48.5
4:5:1 19
2:1:1 19.5
'OK

Unc lass i £ ied Security Classification
DOCUMENT CONTROL DATA -R&D (Security cttasWeatlon ol tltli. body ot mbitrmct mnd tndottng tmnolmtlon mm) b« tnltwrf whin IhQ ovrmU rmpott la clatMiUtd)
Tim. «SPOUT IICUPIITV CLAOSIFIC A TION
Unclassified
i ORIGINATING AC Ti vi T V (Corporal* Buthor)
Peninsular ChenResearch, In:. Gainesvi lie.,
Florida 32601
16. CROUP
5. REPORT TITLE
Synthesis of New Fluorine-Containing Nitroso Compounds, Copolymers and Terpolymers
4. DtlCBlPTlvt HOIK* (Typ* oi npoil and Inclutl** rlaf)
Final Report, July 1963 to November 1966 t- AUTHORUI (Flrtt rum«, mlddl. Inltltl. Im IMMJ
Stump, Eugene C. and Padgett, Calvin D.
• REPORT OATI
November 1967 7«. TOTAL NO. OP PAGE!
108 T6. NO. OP RIFS
58 U. CONTRACT OR GRANT NO
DA19-129-AMC-152(N) b. PROJECT NO.
ICO24401A329
U. ORIGINATOR'S REPORT NUKfBERI»!
•*. OTHER REPORT NOI«) (Any o«h«r niait-n tfiaf may thtm nport)
68-33-CM; C&OM-38
*• mmtlftmd
10. DISTRIBUTION STATEMENT
This document has been approved for public release and sale; its distribution is
unlimited.
II. SUPPLEMENTARY NOTES 11. SPONSORING MILITARY ACTIVITY
U. S. Army Natick Laboratories Natick Massachusetts 01760
I». ABSTRACT
This report describes work carried out in the area of nitroso polymers. Two general classes were investigated. These were (1) sulfur-containing nitroso polymers and (2) nitroso polymers containing functional groups.
Svnthesis of a wide variety of fluorine-containing nitroso compounds and olefins is described, as well as the synthesis of various fluorine-containing intermediates.
Nitroso copolymers and terpolymers were prepared containing functional groups such as halogen, ester, double bonds, and methoxy. Properties of selected systems
are described.
A terpolymer which could be cross-linked by peroxide was prepared using methyl
4-njtrosoperflucrcbutyrate as the termonomer.
Infrared spectra and NMR analysis of the new monomers and polymers are included.
DD,'r..1473 RSjPLAcas DO Pom« i« < JAH •«. «MI CM IS OBSOLETE PO« ASMV USE.
Unclassified ■acurity Classification '
Uii-d Security CUUTTTTc «tl
> l» ttOROt nOLC w MOLl MT
Synthesis (Chemistry) Copolymers Terpolymers Nicroso rubber Fluorine Sulfur Halogens Esters Physical properties Monomers
UncIHS .ified tocwity CUtilflcmion












