
Exploring the Ciprofloxacin Resistome - DiVA portal
62
ACTA UNIVERSITATIS UPSALIENSIS UPPSALA 2018 Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 1495 Exploring the Ciprofloxacin Resistome LINNÉA GAROFF ISSN 1651-6206 ISBN 978-91-513-0448-9 urn:nbn:se:uu:diva-361204
Transcript of Exploring the Ciprofloxacin Resistome - DiVA portal

ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2018
Digital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 1495
Exploring the CiprofloxacinResistome
LINNÉA GAROFF
ISSN 1651-6206ISBN 978-91-513-0448-9urn:nbn:se:uu:diva-361204

Dissertation presented at Uppsala University to be publicly examined in B42, BMC,Husargatan 3, Uppsala, Friday, 9 November 2018 at 13:15 for the degree of Doctor ofPhilosophy (Faculty of Medicine). The examination will be conducted in English. Facultyexaminer: Professor Jesús Blázquez (Spanish National Research Council CSIC, SpanishNational Center for Biotechnology Madrid).
AbstractGaroff, L. 2018. Exploring the Ciprofloxacin Resistome. Digital Comprehensive Summariesof Uppsala Dissertations from the Faculty of Medicine 1495. 61 pp. Uppsala: ActaUniversitatis Upsaliensis. ISBN 978-91-513-0448-9.
This thesis presents an exploration of the resistance evolution in Escherichia coli towardsthe antibiotic ciprofloxacin. High level ciprofloxacin resistance is typically acquired by anaccumulation of mutations and plasmid borne genes reducing drug target binding, increasingdrug efflux, and modifying the drug.
Paper I describes the finding that novel mutations in tRNA synthetase gene leuS conferredresistance to ciprofloxacin. We also provided evidence for a mechanism, where the leuSmutations induced global changes in transcription that generated a net effect of increased drugefflux.
In Paper II we observed that the evolutionary trajectory towards high level ciprofloxacinresistance in E. coli is repeatable and predictable in in vitro evolution experiments. However, thetypes and order of appearance of selected mutations was highly dependent on the bottleneck sizeused. In addition to the findings in Paper I, we found that mutations involved in transcription andtranslation were repeatedly selected upon subjection to high concentrations of ciprofloxacin.
Paper III explored the resistance capacity of the plasmid-borne gene qnr, which reducesciprofloxacin susceptibility by a target protection mechanism. We found that upon increasedexpression, the gene qnrS was able to bring E. coli to clinically resistant levels of ciprofloxacinwithout the addition of other resistance elements.
In Paper IV we aimed for a similar study as described above but with another plasmid-bornegene, the inner-membrane efflux pump qepA. However, we ran into the interesting finding ofa potentially undescribed regulatory mechanism of qepA expression, which we are currentlyinvestigating.
The work in this thesis presents a new addition of mutations causing ciprofloxacin resistance,and evidence that the dogma of accumulative mutations being a requirement to develop clinicalresistance to ciprofloxacin in E. coli can be circumvented. This shows that there is still much toexplore, even with a drug used for several decades with an already well documented resistome.We need to learn more about the evolutionary trajectories leading to antibiotic resistance, inorder to slow down its development towards existing and future antibiotics to the furthest extentpossible.
Keywords: Antibiotic resistance, Experimental evolution, Ciprofloxacin, Escherichia coli
Linnéa Garoff, Department of Medical Biochemistry and Microbiology, Box 582, UppsalaUniversity, SE-75123 Uppsala, Sweden.
© Linnéa Garoff 2018
ISSN 1651-6206ISBN 978-91-513-0448-9urn:nbn:se:uu:diva-361204 (http://urn.kb.se/resolve?urn=urn:nbn:se:uu:diva-361204)

To my Family, Friends and Workmates


List of Papers
This thesis is based on the following papers, which are referred to in the text by their Roman numerals.
I Garoff, L., Huseby, DL., Praski Alzrigat, L., Hughes, D. (2018) Effect of aminoacyl-tRNA synthetase mutations on susceptibility to ciprofloxacin in Escherichia coli. J Antimicrob Chemother, doi:10.1093/jac/dky356
II Pietsch, F*., Garoff, L*., Huseby, DL*., Lilja, T., Brandis, G., Hughes, D. (2018) Evolutionary trajectories dependent on bottle-neck size and a new class of genes selected during the develop-ment of ciprofloxacin resistance in Escherichia coli. Manuscript
III Garoff, L., Yadav, K., Hughes, D. (2018) Increased expression of Qnr is sufficient to conver clinical resistance to ciprofloxacin in Escherichia coli. J Antimicrob Chemother, 2018 Feb 1;73(2):348-352
IV Garoff, L., Crone, L., Broom, KB., Hughes, D. (2018) The qepA gene is dependent on upstream sequences to reduce susceptibility to ciprofloxacin. Manuscript
Not part of the thesis:
Huseby, DL., Pietsch, F., Brandis, G., Garoff, L., Tegehall, A., Hughes, D. (2017) Mutation supply and relative fitness shape the genotypes of ciprofloxacin-resistant Escherichia coli. Mol Bio Evol, 2017 May 1;34(5):1029-1039
* These authors contributed equally
Reprints were made with permission from the respective publishers.


Contents
Introduction ............................................................................................... 11Antibiotic resistance ............................................................................. 11
Medical impact of resistance in the past, present, and future .......... 11Ciprofloxacin ........................................................................................ 13
The targets in E. coli: DNA gyrase and Topoisomerase IV ............ 14Mechanism of action ....................................................................... 16Medical importance ......................................................................... 17
Escherichia coli .................................................................................... 18The commensal and the pathogen ................................................... 18The experimental model organism .................................................. 20
Experimental evolution ........................................................................ 20Chromosomal mutation rates in E. coli ........................................... 21Transfer bottlenecks ........................................................................ 22Bacterial fitness ............................................................................... 23
The ciprofloxacin resistome ...................................................................... 25Part I - Chromosomal mutations .......................................................... 26
Reducing drug target interaction ..................................................... 27Reducing drug target access ............................................................ 28Stress responses ............................................................................... 29Present investigation Paper I ........................................................... 31Present investigation Paper II: ......................................................... 33Future perspectives Part I: ............................................................... 36
Part II – Plasmid mediated resistance .................................................. 37Drug target protection ...................................................................... 38Drug efflux ...................................................................................... 39Drug modification ............................................................................ 40Could PMQR genes provide short-cuts to ciprofloxacin resistance? ........................................................................................ 41Present investigation: Paper III ........................................................ 42Present investigation: Paper IV ....................................................... 44Future perspectives Part II: .............................................................. 46
Concluding remarks .................................................................................. 48
Acknowledgements ................................................................................... 50
References ................................................................................................. 54


Abbreviations
ATP cfu CIP DNA ds ECDC E. coliISLPSMICmRNAPMQRRNARNDROSsstRNAUTIWHO
Adenosine triphosphate Colony forming units Ciprofloxacin Deoxyribonucleic acid Double-stranded European Centre for Disease Control and Prevention Escherichia coli Insertion sequence Lipopolysaccharides Minimal inhibitory concentration Messenger ribonucleic acid Plasmid-mediated quinolone resistance Ribonucleic acid Resistance-nodulation-division Reactive oxygen species Single-stranded Transfer ribonucleic acid Urinary tract infection World Health Organisation


11
Introduction
This thesis concerns the bacterium Escherichia coli and its development to resistance against the antibiotic ciprofloxacin. As such it relates to one of to-day’s greatest health concerns, which is briefly described next.
Antibiotic resistance An antibiotic is a chemical substance inhibiting the progression of bacterial growth. Resistance is the acquired or inherent ability of a bacterium to resist the effects of an antibiotic. When put together, the term antibiotic resistance commonly refers to the acquired ability of a bacterium to withstand the effects of an antibiotic, to which it was once sensitive.
Antibiotic resistance development is part of an ancient natural selection process, wherein organisms producing and organisms targeted by antibiotics have been in an arm’s race against each other for millions of years (D'Costa, McGrann et al. 2006, Wright and Poinar 2012, Andersson and Hughes 2017). Not only do resistance mechanisms evolve in the target organisms to facilitate their survival, but producers might also need to co-evolve self-protection mechanisms towards the compound they are producing. Furthermore, genetic material is readily transferred between microorganisms, implying that any ge-netic information that encodes the means to withstand antibiotics, originating from either producers or targets, could have spread and evolved long before antibiotic use in medicine. Thus, bacteria are naturally provided with a large arsenal of countermeasures to toxic compounds (D'Costa, McGrann et al. 2006, Wright and Poinar 2012). As it turns out, the capacity of bacteria to adapt is far from exclusive to antibiotics produced naturally by co-habitant organisms. Rather, it extends to an inherent ability to develop resistance to-wards man-made antibiotics, including semi- and fully synthetic antibiotics, upon recurrent exposure.
Medical impact of resistance in the past, present, and future The finding of antibiotic resistance development as a potential medical issue was encountered shortly after the introduction of sulphonamides in 1937 (Davies and Davies 2010). In the case of penicillin, resistance was detected in 1940: three years before its introduction to medical use in 1943 (Davies and

12
Davies 2010, Ventola 2015). For each class of drug discovered and introduced to medical practice from the 1940’s and onwards, resistance was detected within 10-20 years of the antibiotics’ introduction to the market. Nevertheless, several new antibiotic drug classes were discovered around the 1950’s (known as the golden age of antibiotic discovery), providing confidence that antibiotic resistance would be overcome by the continued discovery and use of novel antibiotic drug classes (Ventola 2015).
Consequently, a tradition of heavy and at times misconducted usage of an-tibiotics has been established in the medical, veterinary and agricultural fields, with widespread resistance development, and some treatment failures, as a re-sult. The previous confidence in continuously finding novel drug classes de-creased as we entered a void of antibiotic discovery, with an absence of novel drug classes being translated to medical use for the past 30 years (Silver 2011, Fair and Tor 2014).
In 2014, the World Health Organization (WHO) made an estimation of the extent and impact of antibiotic resistance, by gathering available surveillance data from nations covering all six WHO regions. Most of the WHO regions’ national reports presented frequencies of resistance of 50 % or more to several antibiotics used against bacteria commonly causing infections in hospitals and in the community (WHO 2014). Surveillance for antimicrobial resistance in the EU/EAA countries is continuously conducted by the European Centre for Disease Control and Prevention (ECDC). In their latest report for 2016 they state numbers similar to those in the WHO report. The sampled clinical iso-lates of bacteria being resistant to at least one antimicrobial group exceeded 30-50 % on average, although with large regional differences; in certain cases, percentages varied by 0-60 % depending on the country (ECDC 2017). How-ever, both WHO and ECDC mention several biases in these reports; i) the results are usually limited to clinical isolates obtained from hospital settings, so the overall population is not well represented; ii) the lack of a consistent methodology for antibiotic resistance sampling and surveillance; and iii) the lack of world-wide coordinated surveillance. This makes it difficult to get an unbiased picture of the extent of antibiotic resistance in clinically relevant pathogens (WHO 2014, ECDC 2017). But irrespective of exact numbers, an-tibiotic resistance is a current (not future) and serious threat to public health, with potentially great financial burdens as a consequence. Antibiotics are not only saving lives by curing established bacterial infections, but they also ena-ble the prevention of infection in immunocompromised individuals, as well as in patients undergoing surgeries and transplants.
If the situation is not improved, a report conducted by request of the British government estimated that by the year 2050, antibiotic resistance could be re-sponsible for up to 10 million deaths globally per year. From now until 2050, this would be accompanied by a substantial economic loss of 100.2 trillion US dollars in gross domestic product (O’Neill 2014). These numbers were based on two modelled scenarios with the assumptions of either 100 % antimicrobial

13
resistance levels, or an increase in resistance by 40 % from the levels of 2014. Although being an important wake-up call to implement strategies aimed to avoid a so called “post-antibiotic era”, the O’Neill report has been met with some criticism for having presented a worst-case scenario that does not con-sider interventions shown to reduce the spread of antibiotic resistance. Im-proved diagnostics and drug administration regimens to reduce overall antibi-otic exposure to bacteria, rigid hygiene procedures to prevent the spread of resistant pathogens, and initiatives to speed up development and clinical eval-uations of novel antibiotics are some of the strategies that if implemented might reduce the probability of all bacteria becoming resistant in future dec-ades (Barbier, Lipman et al. 2016).
A cautious optimism is also found in the recent advances of culturing tech-niques of microorganisms, which has allowed for an expansion in the search of antibacterial agents (Piddock 2015). In 2015, a compound named teixobac-tin was found to inhibit cell wall synthesis of Gram-positive bacteria by a pre-viously undescribed mode of action, making it a finding of a new drug class (Ling, Schneider et al. 2015). It remains to be seen if teixobactin, or derivates of this molecule, succeed as a treatment for multidrug-resistant Gram-positive bacterial infections. The future will also tell if advanced laboratory techniques will help bridge the antibiotic discovery void. However, with the continued heavy use of antibiotics resistance development appears to be inevitable. Therefore, there is a great need for continuous discovery of antibiotics, as well as to preserve the functionality of current antibiotics to the longest extent pos-sible. In order to do the latter, a continued research to understand the mecha-nisms, evolution and spread of antibiotic resistance is essential.
Today we know that antibiotics typically need to enter a bacterial cell and bind to a target within it. Sufficient binding to the target will consequently disturb a molecular process in the bacterial cell, with the final outcome of cell-divi-sion inhibition and/or killing. For the bacterial cell, there are generally three ways to prevent such a final outcome from occuring; i) to reduce access to the target, ii) to reduce the drug-target binding affinity, or iii) to incapacitate the drug (Blair, Webber et al. 2015). The next sections will consider these features for the subjects of this thesis, the antibiotic ciprofloxacin and the bacterium Escherichia coli.
Ciprofloxacin Ciprofloxacin belongs to the class of fluoroquinolone antibiotics. The devel-opment of quinolone antibiotics started in the early 1960’s with a fortunate finding, that an impurity generated from the chemical process of producing the anti-malarial drug chloroquine, was found to have antibacterial properties

14
(Mitscher 2005). From this impurity, the compound nalidixic acid was pa-tented (Lesher, Froelich et al. 1962) and eventually used for oral treatment against urinary tract infections (UTI’s) caused by E. coli (Mitscher 2005). Other derivatives of nalidixic acid soon followed. The development of nor-floxacin (patented in 1978) with the additions of a 6-fluor, and 7-piperazine ring, led to a breakthrough of broadened spectrum of activity against both Gram-negative and Gram-positive bacteria (Ito, Hirai et al. 1980, Wolfson and Hooper 1985, Appelbaum and Hunter 2000) (Figure 1). It was suggested that the fluorine increased drug uptake as well as drug target potency (Mitscher 2005). The next improvement came with the second-generation fluoroquin-olones, where ciprofloxacin (patented in 1981) with an additional cyclopropyl side chain was found to have an increased activity, spectrum, and bioavaila-bility (Figure 1). This established ciprofloxacin as a broad-spectrum antibiotic used in treatments of several systemic infections, in addition to UTI’s (Wise, Andrews et al. 1983, Chin and Neu 1984, Eliopoulos, Gardella et al. 1984, Wolfson and Hooper 1985, Appelbaum and Hunter 2000, Guan, Xue et al. 2013).
It was believed from start that the targets among different quinolone anti-biotics was shared, as susceptibility to several different quinolones commonly decreased (although to a varying degree) if a strain was known to be resistant to one of them (Wise, Andrews et al. 1983, Wolfson and Hooper 1985). The quinolone research actually assisted in the discovery and continued molecular research of their targets, the type II topoisomerases, and their functions, which are described next (Mitscher 2005).
Figure 1. Structural development of ciprofloxacin. Dashed circles indicate structural changes made from nalidixic acid and norfloxacin.
The targets in E. coli: DNA gyrase and Topoisomerase IV Early on it was discovered that quinolone antibiotics inhibited bacterial DNA replication. The first proposed target came with the discovery that the drugs novobiocin and coumermycin inhibited an enzyme essential for replication, DNA gyrase. The proposition was enforced by the finding that DNA gyrase isolated from coumermycin-resistant E. coli strains were less affected by the drugs novobiocin and coumermycin (Gellert, O'Dea et al. 1976). Shortly after, a proposed mechanism of action for nalidixic acid was published indicating

15
that the drug induced double stranded DNA breaks by binding to DNA gyrase (Gellert, Mizuuchi et al. 1977, Sugino, Peebles et al. 1977). More than 15 years later, a second target of quinolones also involved in replication, Topoi-somerase IV, was discovered in Staphylococcus aureus and proposed to be the primary target of the drugs in this bacterium (Ferrero, Cameron et al. 1994). Not long after, it was found that Topoisomerase IV in E. coli constituted a secondary target to DNA gyrase upon treatment with ciprofloxacin (Khodursky, Zechiedrich et al. 1995, Chen, Malik et al. 1996).
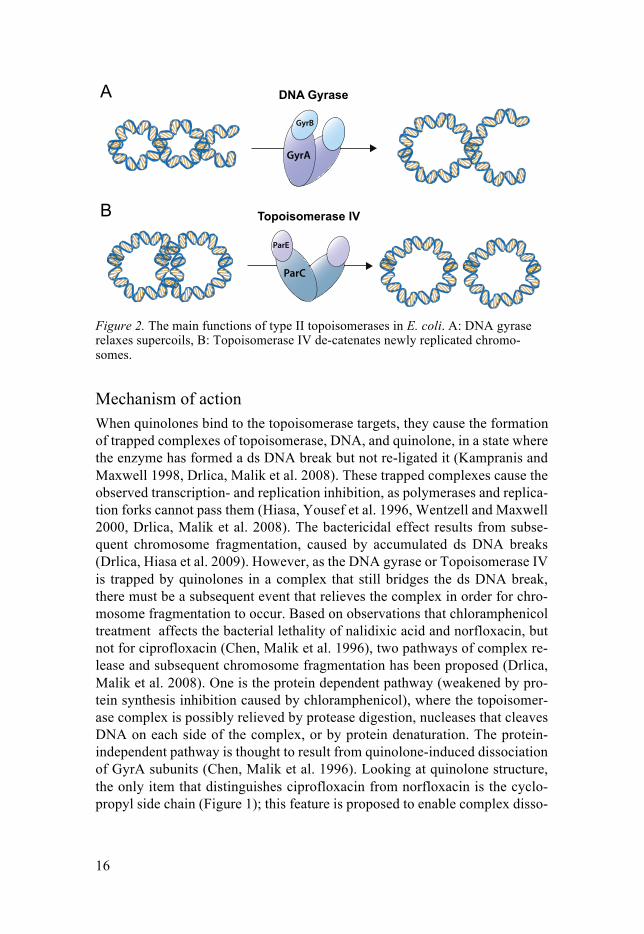
DNA-gyrase and Topoisomerase IV each consist of four subunits; in E. coli two each of GyrA and GyrB for DNA gyrase, and of ParC and ParE for Topoi-somerase IV (Figure 2). They are each essential enzymes involved in bacterial replication and the maintenance of DNA topology, commonly referred to as type II topoisomerases. The double-stranded (ds) helix of DNA in bacteria is highly dynamic and subjected to a phenomenon called supercoiling, where the ds DNA forms loops around itself due to tensions formed in the helical struc-ture. Imagine this as if you are separating two intertwined pieces of rope with attached ends – with enough constraint, the intertwining’s will start to twist around each other (Figure 2A). This allows for the cell to pack the DNA in a condensed way, saving a lot of space. However, the cell needs to relax super-coils in order to make the DNA accessible for replication, transcription and recombination processes; processes that in turn leads to supercoiling in the adjacent DNA (Schoeffler and Berger 2008, Blair, Webber et al. 2015).
Both type II topoisomerases are able to relax supercoils, although this func-tion is mainly exerted by DNA gyrase, in order to maintain a functional DNA structure (Figure 2A)(Schoeffler and Berger 2008, Pommier, Leo et al. 2010). Another feature of the circular bacterial chromosome is that the two daughter chromosomes become catenated at the end of replication, linked together as in a chain (Pommier, Leo et al. 2010). Topoisomerase IV primarily acts as a de-catenation enzyme, separating the two daughter chromosomes prior to cell division (Figure 2B) (Schoeffler and Berger 2008, Pommier, Leo et al. 2010, Blair, Webber et al. 2015). The type II topoisomerases exert these functions in a similar manner, by ATP-utilization: they preferably bind to DNA crosso-vers and cleave one of the ds DNA segments, but remains attached to the 5’ ends of the DNA so that the ds DNA break is bridged, and not left open. Then, the enzyme associates with the intact ds DNA segment and transfers it through the ds DNA break, untangling the supercoil or catenation. Once the strand passage is completed, the ds DNA break is re-ligated and left without scars (Drlica, Malik et al. 2008, Schoeffler and Berger 2008).
16
GyrA
GyrB
ParC
ParE
Figure 2. The main functions of type II topoisomerases in E. coli. A: DNA gyrase relaxes supercoils, B: Topoisomerase IV de-catenates newly replicated chromo-somes.
Mechanism of action When quinolones bind to the topoisomerase targets, they cause the formation of trapped complexes of topoisomerase, DNA, and quinolone, in a state where the enzyme has formed a ds DNA break but not re-ligated it (Kampranis and Maxwell 1998, Drlica, Malik et al. 2008). These trapped complexes cause the observed transcription- and replication inhibition, as polymerases and replica-tion forks cannot pass them (Hiasa, Yousef et al. 1996, Wentzell and Maxwell 2000, Drlica, Malik et al. 2008). The bactericidal effect results from subse-quent chromosome fragmentation, caused by accumulated ds DNA breaks (Drlica, Hiasa et al. 2009). However, as the DNA gyrase or Topoisomerase IV is trapped by quinolones in a complex that still bridges the ds DNA break, there must be a subsequent event that relieves the complex in order for chro-mosome fragmentation to occur. Based on observations that chloramphenicol treatment affects the bacterial lethality of nalidixic acid and norfloxacin, but not for ciprofloxacin (Chen, Malik et al. 1996), two pathways of complex re-lease and subsequent chromosome fragmentation has been proposed (Drlica, Malik et al. 2008). One is the protein dependent pathway (weakened by pro-tein synthesis inhibition caused by chloramphenicol), where the topoisomer-ase complex is possibly relieved by protease digestion, nucleases that cleaves DNA on each side of the complex, or by protein denaturation. The protein-independent pathway is thought to result from quinolone-induced dissociation of GyrA subunits (Chen, Malik et al. 1996). Looking at quinolone structure, the only item that distinguishes ciprofloxacin from norfloxacin is the cyclo-propyl side chain (Figure 1); this feature is proposed to enable complex disso-

17
ciation and release of free, lethal, ds DNA breaks without depending on pro-tein synthesis. However, ciprofloxacin is also believed to induce the protein-dependent pathway at lower drug concentrations (Drlica, Hiasa et al. 2009, Wang, Zhao et al. 2010).
Apart from the accumulation by ds DNA breaks, the DNA damage caused by quinolones induces the cellular SOS-stress response. This causes inhibition of cell-division and, paradoxically, also induces DNA break-repair mecha-nisms. However, an overload of SOS induction by accumulated quinolone-caused DNA damage could add to the lethality of quinolones by preventing cell-division and causing the formation of filamentous cells. (Piddock and Walters 1992, Lopez, Elez et al. 2007, Drlica, Malik et al. 2008, Blair, Webber et al. 2015). In addition, a consequence of DNA damage is increased amounts of reactive oxygen species (ROS) that have been shown to contribute to fluo-roquinolone lethality (Wang, Zhao et al. 2010, Erental, Kalderon et al. 2014, Machuca, Recacha et al. 2017). But the suggestion that ROS are responsible for antibiotic-associated bactericidal activity in E. coli is debated, in certain studies even disproven, implying that the effects of ROS production by anti-biotic treatments and its effects are more complex, and possibly depend on the level of DNA damage caused (Baharoglu and Mazel 2014, Erental, Kalderon et al. 2014, Zhao, Hong et al. 2015). DNA damage by quinolone treatment has also been suggested to cause cell death via the activity of the toxin-antitoxin module MazEF, where MazE causes a preferential translation of gene prod-ucts involved in mediating cell death (Erental, Kalderon et al. 2014, Machuca, Recacha et al. 2017).
Medical importance Even though there are still details left uncovered about how ciprofloxacin kills bacteria, the fact that it targets not only one but two essential enzymes in a wide range of bacterial species has made it of great use in the treatment of several types of infections. Today’s medical importance of ciprofloxacin is crucial, as shown by WHO’s listing of it as a “highly prioritized critically im-portant antimicrobial”. With this listing, WHO encourages the implementa-tion of management strategies to maintain ciprofloxacin functionality (i.e. pre-venting resistance spread and development). Ciprofloxacin meets a number of criteria that summarizes its importance in human medicine. It is one of a lim-ited number of available therapies for serious bacterial infections (such as Sal-monella and Escherichia coli), in certain cases because antibiotic resistance has rendered other antimicrobials useless. It is also used very widely, not only for serious infections. Additionally, it is used to treat infections with evident transmission of resistance from non-human sources (WHO 2017).

18
Escherichia coli The commensal and the pathogen E. coli is a Gram-negative, facultative anaerobic and non-sporulating bacte-rium. It is foremost a widespread, commensal bacteria residing in the intes-tines of mammals, birds and reptiles. In humans, E. coli resides in the mucus layer of the large intestine, where it is shielded from outside stresses and ob-tains a steady nutritional supply. Colonization by harmless commensals such as E. coli is in turn advantageous to the host by preventing gut colonization of harmful pathogens (Tenaillon, Skurnik et al. 2010).
There is a huge variation within the E. coli species, due to its genomic plas-ticity; the core genome constitutes about 3100 gene families whereas the pan-genome constitutes at least 89000 gene families (Land, Hauser et al. 2015). Not only is the bacterial chromosome subject to modification by point muta-tions, and recombination events including inversions, duplications, insertions and deletions; it is also prone to receive genetic material, a process known as horizontal gene transfer, by the action of plasmids, bacteriophages and mobile genetic elements such as integrases and transposases (Leimbach, Hacker et al. 2013). Although mutation and horizontal gene transfer is commonly associ-ated with antibiotic resistance development, pathogenic strains of E. coli have also developed via the same processes by the acquisition of virulence factors. As with antibiotic resistance, pathogenicity is the result of E. coli adapting to withstand and replicate in ever changing environments, both within and out-side of the host gut, with the result that some variants can cause illness to the host (Croxen and Finlay 2010, Tenaillon, Skurnik et al. 2010, Leimbach, Hacker et al. 2013). Accordingly, pathogenic E. coli have been demonstrated to have higher recombination rates than commensal E. coli, implying a selec-tion for increased genomic plasticity (Rodriguez-Beltran, Tourret et al. 2015).
Pathogenic E. coli are classified as intestinal or extra-intestinal. The intes-tinal pathogens cause diarrhoea of varying severity, whereas the extra-intesti-nal pathogens can cause neonatal meningitis and urinary tract infections (UTIs) (Croxen and Finlay 2010). In Europe, E. coli is the most common cause of complicated and uncomplicated UTIs and is the leading cause of blood-stream infections by Gram-negative bacteria (ECDC 2017), and fluoroquino-lone treatments apply to several of the conditions caused by E. coli.
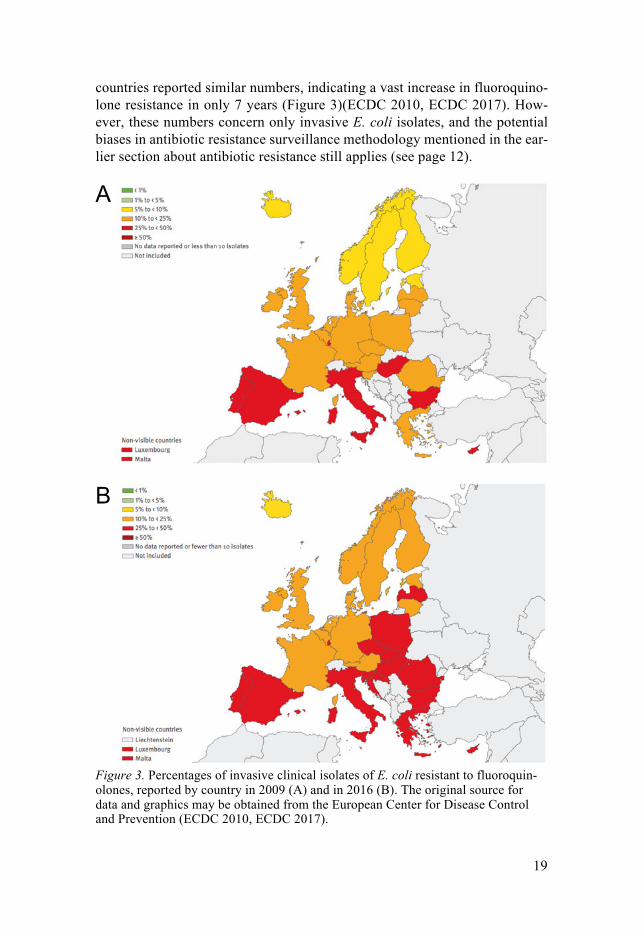
Fluoroquinolone resistance According to the ECDC annual surveillance report from 2016, more than half of the E. coli isolates reported in Europe were resistant to at least one of the antibiotics under regular surveillance. Of these, the population-weighted mean resistance percentage for fluoroquinolones was at 21 %; however, 16 countries reported percentages of fluoroquinolone resistant E. coli isolates in excess of 25 %. In a similar surveillance report published in 2009, only half as many
19
countries reported similar numbers, indicating a vast increase in fluoroquino-lone resistance in only 7 years (Figure 3)(ECDC 2010, ECDC 2017). How-ever, these numbers concern only invasive E. coli isolates, and the potential biases in antibiotic resistance surveillance methodology mentioned in the ear-lier section about antibiotic resistance still applies (see page 12).
Figure 3. Percentages of invasive clinical isolates of E. coli resistant to fluoroquin-olones, reported by country in 2009 (A) and in 2016 (B). The original source for data and graphics may be obtained from the European Center for Disease Control and Prevention (ECDC 2010, ECDC 2017).

20
The experimental model organism The model organism that has been used throughout the works in this thesis is E. coli K-12 MG1655. It is a commensal strain that was isolated in 1922 from the stool of a diphtheria patient, and deposited with the designation K-12. With properties of being prototrophic (i.e. able to produce nutrients from inorganic material), easy to grow in the laboratory, and having a fast generation time, the strain was deemed suitable for genetic studies. E. coli K-12 generated find-ings such as the F+ plasmid used for sexual recombination, as well as the dis-covery of bacteriophage lambda and sensitivity to bacteriophage P1, leading to the development of these three elements as tools for genetic manipulation and strain construction. Especially the latter two have been used extensively in the present investigations of this thesis. The descendant strain MG1655 is closely related to K-12, but has been cured from the F+ factor as well as of phage lambda by Guyer et al in 1981 (Guyer, Reed et al. 1981, Blattner, Plunkett et al. 1997, Hayashi, Morooka et al. 2006). It also has some distinc-tive mutations such as a frameshift in rph, causing a reduced expression of the downstream gene pyrE which consequently causes a mild pyrimidine starva-tion phenotype, and a mutation in ilvG disrupting an isoleucine-valine biosyn-thesis pathway. The chromosome of MG1655 still contains several transpos-able IS and phage elements implicated in casuing spontaneous mutations, which also has resulted in MG1655 having an IS5 insertion causing the loss of the O-antigen in the bacteria’s lipopolysaccharide (LPS), referred to as the rfb-50 mutation (Blattner, Plunkett et al. 1997). Together, these traits summa-rize the full wild-type description of E. coli K-12 MG1655: F- lambda- ilvG- rfb-50 rph-1.
The plasticity of the MG1655 genome, in combination with the continuous use and re-stocking of MG1655 since the 1980’s, has led to several variations in stock cultures used throughout the world (Freddolino, Amini et al. 2012). Some of these variations have been mistakenly thought of as mutations result-ing from experimental evolution. Therefore, it is always important to consider the genotype of the founding strain of an experiment before comparing ge-nomic changes directly to a reference sequence of MG1655 (Freddolino, Amini et al. 2012).
Experimental evolution Experimental evolution is a controlled way of studying adaptation by natural selection. Thanks to the fast generation time and large population size of mi-crobes, they provide a solid and reproducible approach to study questions and hypotheses raised by Darwin’s theories (Lenski 2017).
Natural selection can be observed due to the spontaneous occurrence of mutations, which ensures that a population of organisms contains individuals

21
with genetic variations from one another. When subjected to a selection pres-sure, certain individuals in the population might by chance already carry a genetic variation that render them better equipped to survive the selection. As such, these individuals are able to pass on their genetic traits and accumulate in the succeedings generations. The realization that mutations can occur ran-domly before organisms are subjected to selection was shown by the fluctua-tion tests of Luria and Delbrück, and by the replica-plating conducted by the Lederbergs (Luria and Delbruck 1943, Lederberg and Lederberg 1952).
In this thesis experimental evolution of E. coli towards increasing resistance to ciprofloxacin sets the foundations of papers I and II. E. coli developing high level resistance to ciprofloxacin has, from an evolutionary perspective, the neat prerequisite of a stepwise selection and accumulation of several re-sistance determinants. Therefore, to select for high-level resistant E. coli in the lab the bacteria needs to be subjected to step-wise increases of the antibi-otic, with sufficient growth at each selection-step to enable mutant cells to emerge and accumulate. However, the details of the approach used to conduct an evolution experiment can affect the resulting outcome. For example, hori-zontal gene transfer is not included as a parameter in the evolutionary experi-ments presented in papers I and II, so the selections towards increased re-sistance is purely asexual and only dependent on the accumulation of chromo-somal mutations. As such, it is important to consider how parameters such as mutation rates, population transfer bottlenecks, and relative bacterial fitness (generations of growth in each selection) affect the selection process.
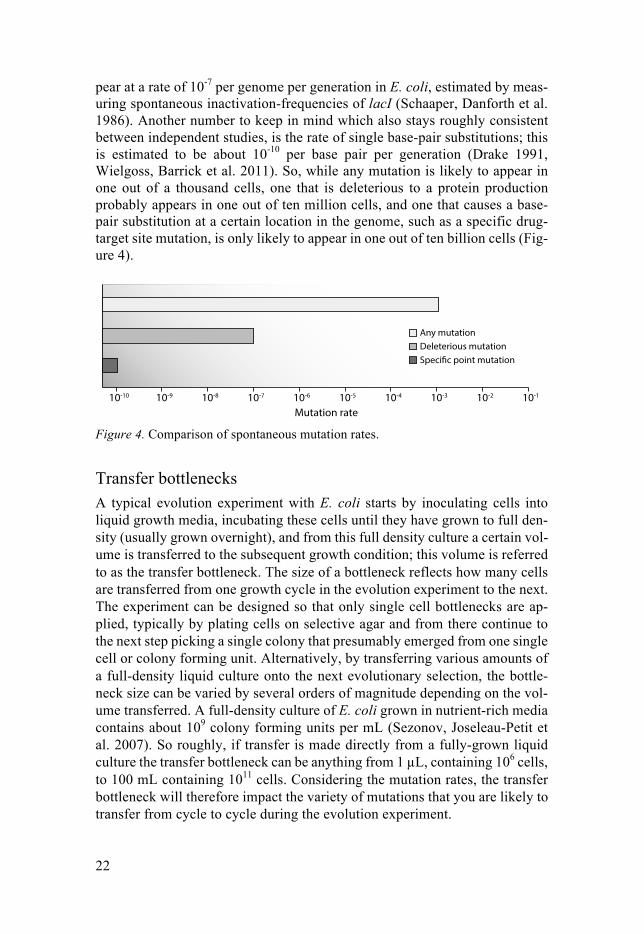
Chromosomal mutation rates in E. coli A mutation rate is the likelihood of a mutation occurring within one generation of growth of a cell. Chromosomal mutations can occur by mistakes during replication, and include single nucleotide base substitutions (also called point mutations), small insertions, deletions and duplications of part of the DNA sequence. Several methods of estimating mutation rates have been conducted, each with their own biases and with slightly varying results (Williams 2014). But, the overall chance of any mutation occurring in a genome is roughly con-sistent throughout studies, being about 10-3 per genome per generation (Drake 1991, Andersson and Hughes 1996, Williams 2014). This means that approx-imately one in a thousand clonal bacterial cells is likely to have acquired a mutation. This mutation might be either neutral, detrimental, or advantageous, depending on the type of mutation, its position, and on the selective condition. It is more likely that insertion- or deletion-mutations occurring within a tran-scriptional region of the DNA will be deleterious to its protein production, as any deletion or insertion could disrupt the regulation or reading frame of the protein coding sequence. One estimate is that such deleterious mutations ap-
22
pear at a rate of 10-7 per genome per generation in E. coli, estimated by meas-uring spontaneous inactivation-frequencies of lacI (Schaaper, Danforth et al. 1986). Another number to keep in mind which also stays roughly consistent between independent studies, is the rate of single base-pair substitutions; this is estimated to be about 10-10 per base pair per generation (Drake 1991, Wielgoss, Barrick et al. 2011). So, while any mutation is likely to appear in one out of a thousand cells, one that is deleterious to a protein production probably appears in one out of ten million cells, and one that causes a base-pair substitution at a certain location in the genome, such as a specific drug-target site mutation, is only likely to appear in one out of ten billion cells (Fig-ure 4).
10-10 10-9 10-8 10-7 10-6 10-5 10-4 10-3 10-2 10-1
Any mutationDeleterious mutationSpecific point mutation
Mutation rate Figure 4. Comparison of spontaneous mutation rates.
Transfer bottlenecks A typical evolution experiment with E. coli starts by inoculating cells into liquid growth media, incubating these cells until they have grown to full den-sity (usually grown overnight), and from this full density culture a certain vol-ume is transferred to the subsequent growth condition; this volume is referred to as the transfer bottleneck. The size of a bottleneck reflects how many cells are transferred from one growth cycle in the evolution experiment to the next. The experiment can be designed so that only single cell bottlenecks are ap-plied, typically by plating cells on selective agar and from there continue to the next step picking a single colony that presumably emerged from one single cell or colony forming unit. Alternatively, by transferring various amounts of a full-density liquid culture onto the next evolutionary selection, the bottle-neck size can be varied by several orders of magnitude depending on the vol-ume transferred. A full-density culture of E. coli grown in nutrient-rich media contains about 109 colony forming units per mL (Sezonov, Joseleau-Petit et al. 2007). So roughly, if transfer is made directly from a fully-grown liquid culture the transfer bottleneck can be anything from 1 µL, containing 106 cells, to 100 mL containing 1011 cells. Considering the mutation rates, the transfer bottleneck will therefore impact the variety of mutations that you are likely to transfer from cycle to cycle during the evolution experiment.

23
Bacterial fitness For new mutations to occur the cells must be allowed to replicate for a number of generations following each transfer, so that new mutations can appear be-fore the next selection step. This growth phase enables certain genotypes to enrich in the population, if they confer a growth advantage in the selective media relative to cells without this genotype. Thus, the proportions of different clones in the population might be skewed, and clones present at a higher pro-portion are more likely to become transferred to the next selection step, irre-spective of the mutation rates. This describes the concept of bacterial fitness.
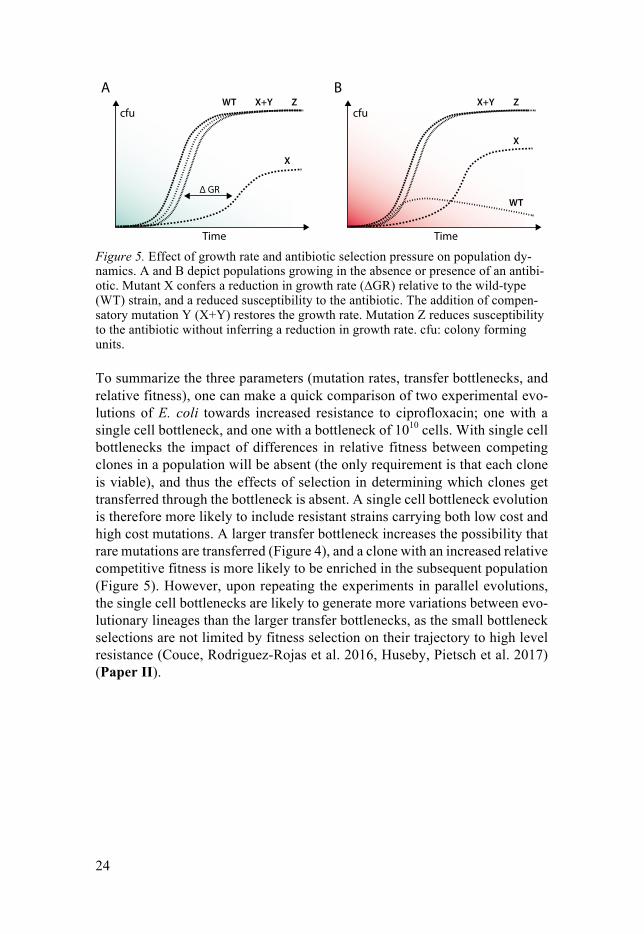
There are different definitions of fitness, but it general it can be described as the ability of a bacterium to survive and proliferate. Fitness is always rela-tive to the environment, as a mutation might be beneficial in one condition while conferring a fitness cost in another (Andersson and Hughes 2010). One way of measuring fitness is to compare growth rates during the exponential growth phase (Andersson and Levin 1999, Andersson and Hughes 2010). Pic-ture a population containing bacteria with and without a mutation X. The mu-tation causes the bacteria to grow slower in the exponential phase. Conse-quently, the wild-type bacteria divide faster and will reach a higher proportion in the population (Figure 5A). Under these conditions, mutation X confers a relative fitness cost. However, if an antibiotic is added to the environment, fitness is no longer dependent on the speed of proliferation in the absence of the antibiotic but on the ability to grow in the presence of a toxic compound. Imagine that the fitness cost in growth conferred by mutation X was due to a reduced protein functionality, but this consequently reduced susceptibility to the antibiotic. If the same two bacteria are now exposed to an environment containing the antibiotic, cells with mutation X will reach a higher proportion in the population despite their fitness cost in growth in drug-free medium (Fig-ure 5B). Because these cells are able to survive and proliferate in the presence of the antibiotic, mutation X confers a selective advantage over the wild-type; this is known as competitive fitness (Andersson and Levin 1999, Andersson and Hughes 2010). The relative competitive fitness can become improved in a continued evolutionary trajectory, if cells with mutation X where to acquire a second, compensatory mutation Y. A compensatory mutation might restore the cost in relative growth fitness inferred by mutation X in drug-free medium without affecting the mutant strains ability to withstand the antibiotic. An al-ternative way to reach the competitive ability of mutant X+Y could also be by acquisition of mutation Z; a mutation that confers a decreased antibiotic sus-ceptibility but without inferring a cost in relative growth rate in drug-free me-dium (Figure 5). (Marcusson, Frimodt-Moller et al. 2009, Andersson and Hughes 2010, Brandis and Hughes 2013).
24
TimeTime
WT
X
X+Y ZWT X+Y
Δ GR
Zcfu cfu
A B
X
Figure 5. Effect of growth rate and antibiotic selection pressure on population dy-namics. A and B depict populations growing in the absence or presence of an antibi-otic. Mutant X confers a reduction in growth rate (∆GR) relative to the wild-type (WT) strain, and a reduced susceptibility to the antibiotic. The addition of compen-satory mutation Y (X+Y) restores the growth rate. Mutation Z reduces susceptibility to the antibiotic without inferring a reduction in growth rate. cfu: colony forming units.
To summarize the three parameters (mutation rates, transfer bottlenecks, and relative fitness), one can make a quick comparison of two experimental evo-lutions of E. coli towards increased resistance to ciprofloxacin; one with a single cell bottleneck, and one with a bottleneck of 1010 cells. With single cell bottlenecks the impact of differences in relative fitness between competing clones in a population will be absent (the only requirement is that each clone is viable), and thus the effects of selection in determining which clones get transferred through the bottleneck is absent. A single cell bottleneck evolution is therefore more likely to include resistant strains carrying both low cost and high cost mutations. A larger transfer bottleneck increases the possibility that rare mutations are transferred (Figure 4), and a clone with an increased relative competitive fitness is more likely to be enriched in the subsequent population (Figure 5). However, upon repeating the experiments in parallel evolutions, the single cell bottlenecks are likely to generate more variations between evo-lutionary lineages than the larger transfer bottlenecks, as the small bottleneck selections are not limited by fitness selection on their trajectory to high level resistance (Couce, Rodriguez-Rojas et al. 2016, Huseby, Pietsch et al. 2017) (Paper II).

25
The ciprofloxacin resistome
The term ‘antibiotic resistome’ refers to the collection of all antibiotic re-sistance genes in microorganisms, including genes providing only modest in-creases in resistance (Wright 2007). The concept was formulated in 2006 with the realization that antibiotic resistance genes are far from exclusive to clini-cally relevant strains, but are extensively prevalent in environmental microor-ganisms prior to any obvious antibiotic exposure (D'Costa, McGrann et al. 2006). Here, I expend the definition of resistome to include also all variants of genes and their regulatory sequences that reduce susceptibility to the drug. Accordingly, the ciprofloxacin resistome applies to any gene, or variant thereof, contributing to increased resistance to this fluoroquinolone, and the scope of this thesis concerns genes prevalent in E. coli.
It is important to consider the meaning of ‘resistance’, and what is meant by it being modest, increased, high, or clinically significant. In the case of ciprofloxacin, there is a gradient from susceptible to clinically resistant E. coli. This gradient is generated since several mutations or mobile resistance ele-ments are needed to reach clinical resistance, as ciprofloxacin targets two es-sential replication enzymes. Clinical resistance is defined as the breach of a breakpoint value, a defined minimal inhibitory concentration (MIC) of an an-tibiotic. The clinical breakpoint value for ciprofloxacin in E. coli is at MIC >0.5 mg/L; an MIC above this value defines E coli as clinically resistant (EUCAST 2018). The value is picked based on the likelihood of treatment success, and on MIC distributions of E. coli isolates that are continuously col-lected by The European Committee on Antimicrobial Susceptibility Testing, EUCAST (EUCAST 2018). According to these MIC distributions, the epide-miological cut-off value for wild-type susceptibility of ciprofloxacin is de-fined as MIC <0.064 mg/L. So, E. coli with an MIC in between 0.064 and 0.5 mg/L could be described as having a reduced susceptibility from the wild-type population or as having an increased resistance (modest or high), but it is not clinically resistant. In papers I-IV, the wild-type MIC level of E. coli K-12 MG1655 to ciprofloxacin was usually measured to be ≤ 0.016 mg/L; thus, any increase from this value was interpreted as an increase in ciprofloxacin re-sistance.
There are generally three ways in which a bacterium can reduce its antibiotic susceptibility: to reduce the drug-target binding, to reduce access to the target,

26
or to incapacitate the drug (Blair, Webber et al. 2015): the ciprofloxacin resis-tome in E. coli covers all three of these aspects (Figure 6, Figure 9). The fol-lowing sections have been divided into two main parts. Part I covers resistance mechanisms conferred by chromosomal mutations and stress responses. This part includes results from present investigations Paper I and Paper II where novel mutational targets are discussed. Part II will focus on plasmid-mediated resistance genes, in which present investigations Paper III and Paper IV ex-amines the possibility of potential short-cuts towards high level resistance to ciprofloxacin.
Part I - Chromosomal mutations Upon finding that ciprofloxacin was active against strains resistant to the older quinolone nalidixic acid, the risk of resistance development was presumed to be low (Chin and Neu 1984). However, after introducing ciprofloxacin in 1987 clinical resistance was soon observed; for example, it was detected in tuberculosis patients already in 1991 (Sullivan, Kreiswirth et al. 1995). Chro-mosomal mutations typically confer resistance to ciprofloxacin by either re-ducing the drug-target binding, or by reducing the intracellular concentration of the drug by increased efflux and decreased influx through the membranes (Figure 6)(Hooper and Jacoby 2015).
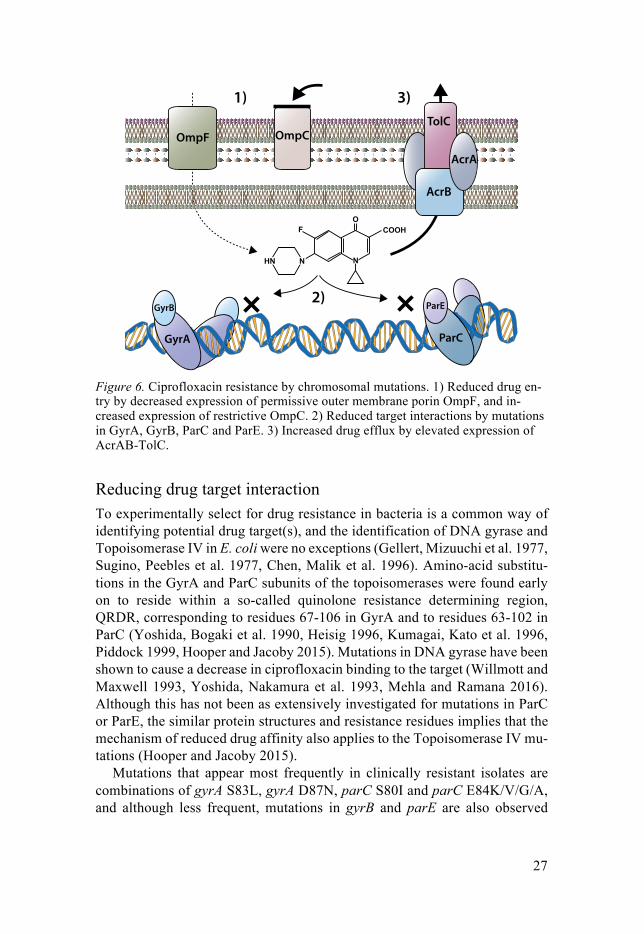
27
OmpF
AcrB
AcrA
TolC
GyrA ParC
GyrB ParE
OmpC
1)
2)
3)
Figure 6. Ciprofloxacin resistance by chromosomal mutations. 1) Reduced drug en-try by decreased expression of permissive outer membrane porin OmpF, and in-creased expression of restrictive OmpC. 2) Reduced target interactions by mutations in GyrA, GyrB, ParC and ParE. 3) Increased drug efflux by elevated expression of AcrAB-TolC.
Reducing drug target interaction To experimentally select for drug resistance in bacteria is a common way of identifying potential drug target(s), and the identification of DNA gyrase and Topoisomerase IV in E. coli were no exceptions (Gellert, Mizuuchi et al. 1977, Sugino, Peebles et al. 1977, Chen, Malik et al. 1996). Amino-acid substitu-tions in the GyrA and ParC subunits of the topoisomerases were found early on to reside within a so-called quinolone resistance determining region, QRDR, corresponding to residues 67-106 in GyrA and to residues 63-102 in ParC (Yoshida, Bogaki et al. 1990, Heisig 1996, Kumagai, Kato et al. 1996, Piddock 1999, Hooper and Jacoby 2015). Mutations in DNA gyrase have been shown to cause a decrease in ciprofloxacin binding to the target (Willmott and Maxwell 1993, Yoshida, Nakamura et al. 1993, Mehla and Ramana 2016). Although this has not been as extensively investigated for mutations in ParC or ParE, the similar protein structures and resistance residues implies that the mechanism of reduced drug affinity also applies to the Topoisomerase IV mu-tations (Hooper and Jacoby 2015).
Mutations that appear most frequently in clinically resistant isolates are combinations of gyrA S83L, gyrA D87N, parC S80I and parC E84K/V/G/A, and although less frequent, mutations in gyrB and parE are also observed

28
(Piddock 1999, Hooper and Jacoby 2015, Huseby, Pietsch et al. 2017). Com-binations of mutations in gyrA and parC were found to increase the MIC to fluoroquinolones substantially more than mutations in a single target (Khodursky, Zechiedrich et al. 1995, Chen, Malik et al. 1996). The most com-mon combination of target mutations is gyrA S83L, gyrA D87N and parC S80I (Huseby, Pietsch et al. 2017), which together brings the MIC of ciprofloxacin of a wild-type E. coli up by 2000-fold, way beyond the clinical resistance breakpoint (Marcusson, Frimodt-Moller et al. 2009). The high MIC increase and the lack of fitness costs associated with this combination of mutations is probably the explanation for why it is highly prevalent (Marcusson, Frimodt-Moller et al. 2009, Huseby, Pietsch et al. 2017). However, this genotype is not exclusive to ciprofloxacin resistant isolates of E. coli; mutations that enhance drug efflux are also observed.
Reducing drug target access E. coli being a Gram-negative bacterium has an intrinsic resistance against several toxic compounds thanks to its double-membrane structure. Ciproflox-acin needs to pass through the outer membrane to reach the periplasmic space, and it also needs to pass through the inner membrane to reach the cytoplasm and the topoisomerase targets (Figure 6). The outer membrane consists of an asymmetric bilayer with an inner leaflet of Lipid A, preventing free diffusion of hydrophilic compounds such as ciprofloxacin, and an outer leaflet of core sugars attached to LPS that provides a strong barrier to hydrophobic com-pounds (Silver 2016). However, some nutrients necessary to be taken up by the bacteria are, like ciprofloxacin, small and hydrophilic molecules. These are able to pass the outer membrane via barrel-shaped proteins called porins. The two major porins in E. coli are OmpC and OmpF, where OmpF allows for slightly larger molecules to pass, and as such is more permissible to fluo-roquinolones (Li, Plesiat et al. 2015). Once in the periplasmic space, ciprof-loxacin is able to diffuse through the symmetric inner phospholipid membrane to reach the cytoplasm, possibly due to changes in ionic composition that ren-ders the molecule more capable of diffusion (Silver 2016). Once inside the cytoplasm, the type II topoisomerase targets are accessible.
E. coli can actively reduce the intracellular concentration of ciprofloxacin by chromosomal mutations that up-regulate the expression of efflux pump proteins. What is commonly observed in clinical isolates are mutations that increase the expression of the AcrAB-TolC multidrug-efflux pump. (Komp Lindgren, Karlsson et al. 2003, Li, Plesiat et al. 2015, Huseby, Pietsch et al. 2017). AcrAB-TolC is a multi-component efflux transporter. AcrB constitutes the pump that binds drugs from the periplasmic leaflet of the inner membrane. AcrA acts as a periplasmic adaptor protein that couples the pump to the third component; the outer membrane channel TolC (Yu, Aires et al. 2003, Li, Plesiat et al. 2015). The expression of AcrA, AcrB and TolC is negatively

29
regulated by the global and local regulatory gene products of marR, acrR, and soxR; any mutation that inactivates or reduces the function of any one of these three genes will cause an increased expression of AcrAB-TolC, with decreas-ing intracellular concentrations of ciprofloxacin as a result (Everett, Jin et al. 1996, Oethinger, Podglajen et al. 1998, Webber and Piddock 2001, Li, Plesiat et al. 2015, Praski Alzrigat, Huseby et al. 2017). It has also been shown that inactivating mutations in marR have downstream effects of reduced expres-sion of the outer membrane porin OmpF, potentially causing a decrease in ciprofloxacin influx (Li, Plesiat et al. 2015). In contrast to drug target muta-tions, increased expression of AcrAB-TolC is usually associated with de-creased fitness in the absence of antibiotic (Marcusson, Frimodt-Moller et al. 2009, Li, Plesiat et al. 2015, Praski Alzrigat, Huseby et al. 2017). However, as any mutation that reduces the functionality of marR, acrR or soxR will gen-erate an increase of drug efflux and a decrease in influx, the likelihood of se-lecting bacteria with an efflux enhancing mutation in a population is substan-tially higher than that of a drug target mutation, upon ciprofloxacin selection (see section about mutation rates on page 21). Another significant difference to drug target mutations is that efflux-enhancing mutations by themselves do not confer as big a reduction in ciprofloxacin susceptibility of E. coli MG1655 as target mutations, but in combination with drug target mutations they can increase the MIC substantially (Marcusson, Frimodt-Moller et al. 2009).
Stress responses Apart from the direct selection of mutations, there is evidence that certain stress responses in bacteria provide them with temporary tools to survive and increase the chances to develop resistance.
The SOS response It is generally accepted that fluoroquinolones, when they cause DNA lesions and replication fork arrest, will stimulate the initiation of the SOS-response. The SOS response is induced by RecA, a protein that is recruited to portions of single-stranded (ss) DNA either prevalent or created from lesions or gaps, by RecBCD or RecFOR, respectively. When bound to ss DNA, RecA pro-motes the auto-proteolysis of the SOS-repressor LexA. LexA represses the transcription of about 40 SOS-response genes in E. coli by binding to specific sequences on their promoters. Consequently, LexA proteolysis via RecA in-teraction induces the SOS response, which promotes DNA repair by homolo-gous recombination and continued replication by the action of several alterna-tive DNA polymerases (Baharoglu and Mazel 2014). Although suggested to have downstream effect contributing to lethality (see page 17), SOS induction at sub-lethal levels of antibiotic has been implied to provide bacteria with sev-eral advantages. Ciprofloxacin treatment was shown to increase intra- and in-tergenic recombination rates in E. coli via RecA (although partly independent

30
from SOS induction by LexA). Increased recombination rates could potentiate the success of horizontal gene transfer of antibiotic resistance genes, and thereby provide the cells with a selective advantage upon antibiotic exposure (Lopez, Elez et al. 2007, Lopez and Blazquez 2009). A LexA binding site was found upstream of the plasmid-born quinolone resistance genes qnrB and qnrD, and SOS-response induction by ciprofloxacin was shown to induce their expression and increase the E. coli MIC (Da Re, Garnier et al. 2009, Briales, Rodriguez-Martinez et al. 2012). Another feature of the SOS response is the induction of DNA-repair by error-prone polymerases, which increase muta-genesis and thereby accelerates the selection of resistant mutants (Blazquez 2003, Cirz and Romesberg 2006, Hughes and Andersson 2012, Baharoglu and Mazel 2014). Apart from the error prone polymerases, cell filamentation by SOS induction has been suggested to promote mutations by increased recom-bination events between sister-chromosomes contained in the same filamen-tous cells (Bos, Zhang et al. 2015).
It has been speculated that the differential outcomes associated with SOS induction are due to fine-tuning of the response to the level of DNA damage (Baharoglu and Mazel 2014). Findings that quinolone resistance mutations cause an altered transcriptional profile in E. coli that downregulates SOS-genes, such as recA (Machuca, Recacha et al. 2017), provides further evidence that manipulation of this system is an advantageous approach, favouring pro-cesses that enable bacterial survival and thus resistance development to fluo-roquinolones. Lastly, a weak induction of the SOS response by ciprofloxacin may also induce a dormant stage in a subpopulation of bacteria that allows them to survive the antibiotic attack by simply avoiding replication until the antibiotic pressure is removed – a non-inheritable trait known as tolerance or persistence (Dorr, Lewis et al. 2009, Theodore, Lewis et al. 2013). Antibiotic persistence is also associated with the onset of another stress response, de-scribed below.
The stringent response The stringent response is initiated in response to amino acid starvation (and several other environmental cues), where the binding of uncharged tRNAs to ribosomes evokes a ribosome-associated protein, RelA, to produce ppGpp. This molecule is under normal physiological conditions both synthesized and degraded by SpoT in E. coli, but with the additional activation of RelA the cellular levels of ppGpp can increase hugely. This leads to global changes in transcriptional patterns of the cell, as ppGpp binds directly to RNA polymer-ase and acts as a repressor or activator of transcription (depending on the GC-content between the -10 region and the transcriptional start site of a gene) (Durfee, Hansen et al. 2008, Osterberg, del Peso-Santos et al. 2011). Another function of ppGpp is to stabilize the interaction of RNA polymerase and sigma-factors associated with cellular stress responses (Jishage, Kvint et al.

31
2002, Gaca, Colomer-Winter et al. 2015). The resulting change in cell physi-ology shifts the focus of transcription from growth to that of biosynthesis, in-hibiting processes like replication and synthesis of ribosomal RNA, while in-ducing synthesis of amino acids and stress responses. Among those, the SOS-response is induced; probably by stalled transcription complexes that causes replication fork arrests and exposes ss DNA to induction by RecA (Durfee, Hansen et al. 2008, Gaca, Colomer-Winter et al. 2015). Whereas the stringent response induction has been connected to tolerance or persistence towards several antibiotics, such as penicillins and fluoroquinolones, (Rodionov and Ishiguro 1995, Amato, Orman et al. 2013), we found that stringent response induction by novel mutations in tRNA synthetase genes can cause a decreased susceptibility to ciprofloxacin by the induction of drug efflux components. This is presented in Paper I.
Present investigation Paper I The question initiating this project was if there were previously undescribed mutations that could decrease ciprofloxacin susceptibility, in addition to ca-nonical drug target or efflux mutations. Clinical isolates display a variety of mutation rates, which facilitates the selection of highly fluoroquinolone re-sistant strains (Komp Lindgren, Karlsson et al. 2003, Huseby, Pietsch et al. 2017). Therefore, strains of E. coli with an impaired mutS gene (rendering them hyper-mutators) were used to provide a large mutation supply, and were put through a single-cell bottleneck evolution with stepwise increasing ciprof-loxacin concentrations. Whole genome sequencing revealed the recurrent se-lection of mutations in tRNA synthetase genes. Two such mutations where independently selected in one of these synthetase genes, leuS. By constructing isogenic strains with these leuS mutations in different genetic contexts (with and without combinations of canonical resistance mutations), we concluded that the leuS mutations conferred a decrease in ciprofloxacin susceptibility. The magnitude thereof depended on the presence or absence of pre-existing canonical resistance mutations, being at most 3- to 4-fold. The leuS mutations also caused a growth rate reduction of about 20 %. However, competition as-says in the presence of ciprofloxacin showed that with increasing drug con-centrations, the leuS mutations conferred a selective advantage relative to iso-genic leuS+ strains.
We reasoned that if the leuS mutations induced the stringent response via RelA induction, the cause of decreased ciprofloxacin susceptibility might lie within the resulting transcriptional changes. This hypothesis was confirmed by deleting relA, which completely reversed the resistance phenotype of the leuS mutants. We observed that the leuS mutations conferred a transcriptional pattern corresponding to stringent response induction, by RNA sequencing and transcript quantification. Among these transcriptional changes, three genes involved in ciprofloxacin efflux were upregulated, as well as the outer

32
membrane porin ompF. The results of subsequent strain constructions and sus-ceptibility assays generated the following explanation (Figure 7): 1. The mutations in leuS decreased the aminoacylation of leucine tRNA,
causing uncharged tRNA to bind to the ribosomes. 2. This led to activation of RelA and stringent response induction, with
global changes in the mRNA expression profile of the cell as a result. 3. These changes included up-regulated expression of inner membrane ef-
flux components MdtK (a multi-drug efflux pump), AcrZ (an AcrAB-TolC associated protein) and YdhIJK (a putative efflux pump), as well as the outer membrane porin OmpF.
4. Even if increased expression of OmpF imposes increased drug influx, the net effect was that of drug efflux enabled by MdtK, AcrZ and YdhIJK.
Furthermore, the resistance effect of the leuS mutations is likely potentiated by the AcrAB-TolC efflux pump. Since MdtK, AcrZ and YdhIJK would only increase efflux of ciprofloxacin to the periplasm (from where it can readily diffuse back to the cytoplasm via the inner membrane), AcrAB-TolC is needed for the complete efflux of the drug out of the periplasm and past the outer membrane (Figure 7) (Yu, Aires et al. 2003).
OmpF
AcrB
AcrA
TolC
AcrZ
MdtK YdhIJK
Figure 7. Mutations in leuS generate a net effect of ciprofloxacin efflux. Transcrip-tional changes inferred by the leuS mutations increase the expression of inner mem-brane effluc pump MdtK, the putative inner membrane efflux pump YdhIJK, and the AcrAB-TolC associated protein AcrZ. Despite an associated increase in transcrip-tion of the ciprofloxacin-entry porin OmpF, the combined activities of MdtK, YdhIJK and AcrZ are able to reduce ciprofloxacin susceptibility.

33
Mutations in tRNA synthetase genes are already known to decrease suscepti-bility to the penicillin mecillinam, and to the gyrase-inhibitor novobiocin. With both antibiotics, the phenotypes conferred by the tRNA synthetase mu-tations are RelA dependent (Vinella, D'Ari et al. 1992, Jovanovic, Lilic et al. 1999, Thulin, Sundqvist et al. 2015). We found, as expected, that the leuS mutations were able to decrease susceptibility to mecillinam and novobiocin. In addition, the leuS mutations also reduced susceptibility to rifampicin, chlo-ramphenicol, trimethoprim and ampicillin. Based on previous work, increased MdtK expression and stringent response induction could explain the decreased susceptibilities to chloramphenicol, trimethoprim and ampicillin (Rodionov and Ishiguro 1995, Nishino and Yamaguchi 2001, Kwon, Higgins et al. 2010). In addition to leuS, mutations in thrS and aspS, as well as chemical induction of the stringent response by the IleS inhibitor mupirocin, also reduced E. coli susceptibility to ciprofloxacin.
With these findings we have described a resistance mechanism connected to a novel and potentially large mutational target, that can affect susceptibility not only to ciprofloxacin but to multiple antibiotics. One can even hypothesize that any cue that induces the stringent response, mutational or environmental, would potentiate increased efflux of drugs and enable E. coli survival and re-sistance development.
Present investigation Paper II: In addition to the finding that tRNA synthetase genes constitute non-canonical mutational targets for ciprofloxacin resistance, earlier results by Pietsch et al showed that mutations in rpoB also reduced ciprofloxacin susceptibility. In both cases, resistance was mediated by increased transcription of efflux pump gene(s), and fitness constraints in terms of reduced growth rate were observed (Pietsch, Bergman et al. 2017, Garoff, Huseby et al. 2018). The reduced growth rate poses a selective disadvantage, as these mutations could be out-competed by faster-growing genotypes. Hence, we wondered: would these mutations be improbable to fixate under more competitive selections? Fur-thermore, a previous study revealed that the typical clinical genotype observed with mutations in gyrA S83, D87 and parC S80, is probably selected on the basis of generating high-level resistance to ciprofloxacin with the least possi-ble fitness cost. According to modelling, this genotype would require a large bottleneck size (or a large mutational supply) to be selected for in evolutionary experiments (Huseby, Pietsch et al. 2017).
We decided to investigate how frequently mutations in the transcrip-tional/translational machinery would be selected, and simultaneously put the model of large mutational supply rendering high-ciprofloxacin resistance by low-cost mutations to the test.

34
Experimental evolutions with E. coli subjected to stepwise increases to ciprof-loxacin were made using three different transfer bottlenecks: i) single cell on agar, ii) ≈ 3x108 colony forming units (cfu) and iii) ≈ 3x1010 cfu, the latter two in liquid cultures. For each bottleneck size, a number of 20, 10 and 5 individ-ual lineages were set up to assess reproducibility, respectively. Liquid bacte-rial populations were whole genome sequenced at intermittent steps of the evolution so that the trajectory of selection could be followed, and the propor-tions of mutations in the populations were indicated by the percentage of reads obtained from the whole genome sequencing.
Irrespective of the bottleneck size, all evolutions selected mutations in gyrA. The liquid evolutions revealed that mutations in gyrA were the first to appear, out of which one mutation reached fixation (i.e. occurring in 100 % of the reads). The mutations that followed as ciprofloxacin concentrations in-creased with the single cell- and medium sized bottlenecks, concerned genes regulating drug efflux (marR, acrR, soxR), and subsequently genes involved in transcription and translation (e.g. rpoB, thrV, mnmA, and tRNA synthetase genes). Thereby we answered our first question: we showed that mutations in genes affecting transcription and translation were repeatedly selected with bottleneck sizes ≤108 cfu, strongly suggesting that these mutations constitute a novel class of genes that can confer a competitive advantage with increasing ciprofloxacin concentrations, despite their potential costs in growth rate. The last mutational event observed in the single cell- and medium sized bottleneck evolutions occurred in additional drug target genes (Figure 8).
With the large transfer bottleneck, the initial mutations in gyrA were typi-cally followed by mutations in parC and additional mutations in gyrA (closely resembling the mutations in gyrA S83, D87, and parC S80 frequently found in resistant clinical isolates), with rare occurrences of mutations affecting genes regulating efflux or transcription/translation (Figure 8). With this, we also support that the model of a high mutational supply generating high-level and low-cost ciprofloxacin resistance mutations holds true.

35
Increasing CIP [mg/L]
Transfer bottleneck [cfu]
1010
108
gyrA
gyrA gyrA parC
parCparEgyrB
Efflux Transcr.Transl.
parCparE
Figure 8. Sequential appearance of mutational targets selected at increasing ciprof-loxacin concentration. The evolutionary trajectory is dependent on the bottleneck size used. CIP: ciprofloxacin, cfu: colony forming units.
The patterns of mutations selected throughout the evolutions were consistent among the independent respective lineages, indicating that the trajectories to ciprofloxacin resistance in E. coli are highly predictable and strongly influ-enced by the bottleneck size used. Clones isolated from the single cell bottle-neck evolutions displayed the highest variability of mutations between line-ages, and the lack of competing clones at each transfer allowed for genotypes with a high fitness cost in terms of reduced growth rate to develop. Populations from the medium-sized bottleneck of ≈ 3x108 cfu displayed a more consequent set of mutations established in each lineage at end of the evolutions. However, this bottleneck size is also theoretically limiting for the transfer of specific, low-cost drug target mutations, with mutation rates of ≈10-9. It is therefore more likely that genetic events with higher mutation rates, such as inactivating mutations upregulating drug efflux or mutations with a large target size such as those involved in transcription and translation, become selected although they confer a fitness cost in reduced growth rate. With the largest bottleneck of ≈ 3x1010 cfu, the chances of transferring rare and low-cost drug target mu-tations increase. Mutations with a lower cost in bacterial growth rate should be able to increase as a proportion of the population by the time of next trans-fer (dependent on the number of generations of growth). In agreement with this, the bacterial populations from the ciprofloxacin evolutions with the larg-est transfer bottleneck carry almost exclusively drug target mutations.
The result of the bottleneck parameter on population dynamics is neatly illustrated already in the early selection steps, by observing the differences in gyrA mutations selected. Among the single-cell bottlenecks evolutions, there were 8 different types of mutation in gyrA represented among the isolates. With the medium- and large-sized bottlenecks, only two different mutations in gyrA (changing amino acids S83 and D87) reached fixation in the popula-tions, indicative of a selection pressure for gyrA mutations conferring a higher

36
competitive advantage. The mutation in gyrA shown to generate the highest MIC increase to ciprofloxacin in combination with the best competitive ad-vantage, is gyrA S83L (Huseby, Pietsch et al. 2017), and this particular muta-tion went to fixation in populations with the largest transfer bottleneck-size.
Future perspectives Part I: In Paper I we identified and provided a mechanism for how mutations in a tRNA synthetase gene, leuS, reduce susceptibility to ciprofloxacin. In Paper II we showed that mutations in transcriptional and translational genes are fre-quently selected in in vitro evolution of E. coli to high-level ciprofloxacin re-sistance.
It remains to be determined if the novel mutations occurring in some of the transcriptional/translational genes observed in paper II (e.g. mutations in thrV and mnmA) reduce susceptibility to ciprofloxacin in E. coli. It is also of inter-est to investigate if reduced susceptibility occurs via a mechanism similar to those observed for mutations in leuS and rpoB (Pietsch, Bergman et al. 2017, Garoff, Huseby et al. 2018). Analysis of the transcriptomes of evolved clones would reveal if the transcriptional/translational mutations are coupled with in-creases in RNA levels, such as in genes involved with the stringent response and efflux as found in Paper I. Additional genetic engineering and subsequent phenotypic tests of the mutation’s effect on ciprofloxacin resistance, and growth fitness, should be assayed in order to test mechanistic hypotheses.
Another subject of interest is the clinical relevance of the described novel mutational targets. No identical mutations to the ones found in Paper I or Paper II have yet been found in genome sequences of ciprofloxacin-resistant clinical isolates. However, the search for these mutations in genomes of clinical iso-lates is non-trivial, for several reasons. Firstly, we have identified several dif-ferent genes, and even different mutations within the same genes, that increase resistance to ciprofloxacin (Pietsch, Bergman et al. 2017, Garoff, Huseby et al. 2018) (Paper II). The fact that none of the particular mutations of this class were selected in more than one lineage each (apart from ∆thrV in Paper II), indicates that the mutational target size is probably very large. Secondly, there is a large degree of sequence variation within genes encoding tRNA synthe-tases in clinical isolates; their sequences differ from the tRNA synthetase se-quences in the reference strain E. coli MG1655 as well as between clinical isolates, including codon differences predicted to alter several amino acids per protein sequence. Without knowing the ancestry of each clinical isolate, it is difficult to distinguish genetic polymorphisms (possibly selectively neutral, or possibly selected for other reasons) from mutations that have been selected in response to ciprofloxacin or other antibiotic exposure.
One counter-argument for the clinical relevance of mutations in e.g. leuS is that they are costly in terms of growth rate, and would be easily out-competed by faster growing strains. Nevertheless, the mutations that were characterized

37
in Paper I did confer a selective advantage in the presence of ciprofloxacin. Evidence that costly genotypes are able to manifest in clinical isolates is the frequent observation of mutations increasing efflux pump expression by inac-tivating or reducing the functions of marR and acrR (Komp Lindgren, Karlsson et al. 2003, Marcusson, Frimodt-Moller et al. 2009, Praski Alzrigat, Huseby et al. 2017), indicating that the selection of costly resistance mutations are not impossible.
With these facts in mind, it does not seem entirely improbable to find mu-tations in transcriptional or translational genes that reduce ciprofloxacin sus-ceptibility among clinical isolates – but with no specific genetic markers to look for, it currently leaves the question of clinical relevance unanswered. A possible (although elaborate) search could be done by cloning, for example by transferring leuS genes from resistant clinical isolates into E. coli MG1655 and screening for decreased ciprofloxacin susceptibility. This would enable identification of alleles possibly selected by antibiotic pressure.
Part II – Plasmid mediated resistance With ciprofloxacin being i) a fully synthetic antibiotic, with no natural pro-ducers, ii) where high-level resistance is achieved by accumulated chromoso-mal mutations, and iii) where some of the resistance mutations were reported to decrease plasmid transfer, it was thought that plasmid-born resistance would not emerge toward this antibiotic (Neu 1984, Wolfson and Hooper 1985). As often, nature proved this assumption wrong. Plasmid-mediated quinolone resistance (PMQR) genes are now frequently observed in clinical isolates (Piddock 2014, Rodriguez-Martinez 2016), and they work by provid-ing drug target protection, drug efflux, or drug modification (Figure 9).
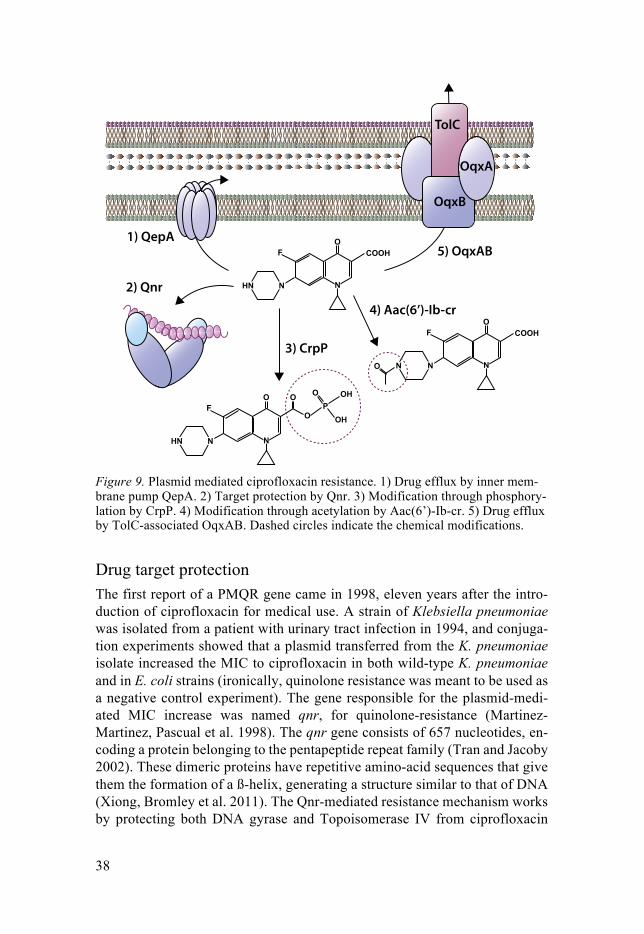
38
OqxB
3) CrpP
2) Qnr
1) QepA5) OqxAB
4) Aac(6’)-Ib-cr
OqxA
TolC
Figure 9. Plasmid mediated ciprofloxacin resistance. 1) Drug efflux by inner mem-brane pump QepA. 2) Target protection by Qnr. 3) Modification through phosphory-lation by CrpP. 4) Modification through acetylation by Aac(6’)-Ib-cr. 5) Drug efflux by TolC-associated OqxAB. Dashed circles indicate the chemical modifications.
Drug target protection The first report of a PMQR gene came in 1998, eleven years after the intro-duction of ciprofloxacin for medical use. A strain of Klebsiella pneumoniae was isolated from a patient with urinary tract infection in 1994, and conjuga-tion experiments showed that a plasmid transferred from the K. pneumoniae isolate increased the MIC to ciprofloxacin in both wild-type K. pneumoniae and in E. coli strains (ironically, quinolone resistance was meant to be used as a negative control experiment). The gene responsible for the plasmid-medi-ated MIC increase was named qnr, for quinolone-resistance (Martinez-Martinez, Pascual et al. 1998). The qnr gene consists of 657 nucleotides, en-coding a protein belonging to the pentapeptide repeat family (Tran and Jacoby 2002). These dimeric proteins have repetitive amino-acid sequences that give them the formation of a ß-helix, generating a structure similar to that of DNA (Xiong, Bromley et al. 2011). The Qnr-mediated resistance mechanism works by protecting both DNA gyrase and Topoisomerase IV from ciprofloxacin

39
binding. Qnr binds to the type II topoisomerases irrespective of the presence of ATP, DNA, or ciprofloxacin, and the binding reduces inhibition of the topoisomerases by ciprofloxacin. This suggests that the drug-target protection is mediated by topoisomerase-Qnr binding before formation of the DNA-en-zyme-quinolone cleavage complex (Tran, Jacoby et al. 2005, Tran, Jacoby et al. 2005). The presence of Qnr slightly reduces the DNA-binding affinity of DNA gyrase, suggesting that the presence of Qnr also reduces the number of enzyme-DNA complexes that constitute the target for quinolone binding (Tran, Jacoby et al. 2005). A second theory of how the protective mechanism mediated by Qnr works has emerged from structural analysis of Qnr and DNA gyrase proteins. This hypothesis states that formation of the gyrase-DNA-quinolone cleavage complex stabilizes a conformation of the gyrase protein favourable for Qnr binding, and that Qnr binding subsequently disrupts the cleavage complex. A rationale for this second hypothesis is that the protection conferred by Qnr against ciprofloxacin occurs at much lower concentrations that that required for Qnr to inhibit the enzymatic function of the gyrase (Xiong, Bromley et al. 2011). Since the initial finding of what is now referred to as qnrA, several genetic variants have been characterised (qnrB, qnrC, qnrD, qnrS, qnrVC) also encoding Qnr proteins that reduce susceptibility to fluoroquinolones (Hooper and Jacoby 2015, Rodriguez-Martinez, Machuca et al. 2016). Homologues of Qnr-proteins have been found in more than 90 di-verse bacterial species, indicating that their time of existence vastly exceeds the usage of fluoroquinolone antibiotics; but the native functions of the Qnr proteins are still unknown (Jacoby and Hooper 2013).
Drug efflux The first PMQR genes that increased ciprofloxacin efflux were initially iden-tified as a resistance element towards the swine growth enhancer olaquindox in 2004 (Hansen, Johannesen et al. 2004). Olaquindox was introduced to ag-riculture in the early 1980s, but was banned in Europe about 20 years later in 1999. E. coli isolated from swine faeces was found to contain a plasmid in which an operon of two genes conferred resistance to olaquindox by increas-ing efflux. The two genes were named oqxA and oqxB, and they belong to the resistance-nodulation-cell-division (RND) family of efflux pumps, as does AcrAB. The efflux activity of OqxAB is also dependent on the presence of TolC (Hansen, Johannesen et al. 2004). The substrate specificity of OqxAB efflux is not limited to olaquindox, but it also increases the MICs to chloram-phenicol, nalidixic acid, fluoroquinolones and tripmethoprim (Hansen, Jensen et al. 2007). In 2009, screening for oqxAB among human clinical isolates of E. coli obtained from blood samples identified the plasmid borne efflux genes in an isolate obtained in 1999. The oqxAB genes were also found on the chro-mosome of a Klebsiella pneumoniae isolate (Kim, Wang et al. 2009).

40
In 2007, clinical isolates of E. coli were found to harbour plasmids that increased the MICs for hydrophilic fluoroquinolones (such as ciprofloxacin); however, these plasmids did not contain any at the time known PMQR genes (Perichon, Courvalin et al. 2007, Yamane, Wachino et al. 2007). By restriction enzyme-cloning, the sequence responsible for the MIC increase was identified as a 1536 bp long gene, encoding for a protein with high structural similarity to inner membrane efflux pumps. With the finding that resistance conferred by this gene was diminished by subjection to efflux pump inhibitors, the gene was designated QepA: quinolone efflux pump A (Perichon, Courvalin et al. 2007, Yamane, Wachino et al. 2007). As with the qnr genes, several variants of QepA were subsequently found in clinical isolates of E. coli throughout the world: QepA2, QepA3, and QepA4, differing from QepA1 by two to five amino acid changes (Cattoir, Poirel et al. 2008, Hooper and Jacoby 2015, Rahman, Islam et al. 2017, Wang, Huang et al. 2017).
Drug modification In 2006, a finding was presented in which a plasmid from clinical isolates of E. coli carrying qnrA as the sole quinolone resistance determinant generated an unusually high MIC to ciprofloxacin. By subjecting the plasmid to random transposon mutagenesis, this unusually high MIC was found to be caused by a gene causing resistance to aminoglycosides; aac-(6’)-Ib (Robicsek, Strahilevitz et al. 2006). This gene encodes an aminoglycoside N-acetyltrans-ferase that reduces the activities of aminoglycoside antibiotics by the enzy-matic addition of an acetyl-group to the compound (Ramirez and Tolmasky 2010). This gene is widespread among clinically relevant Gram-negative bac-teria, and usually found in resistance gene cassettes together with qnr, qepA and beta-lactamase genes (Robicsek, Strahilevitz et al. 2006, Ramirez and Tolmasky 2010). The particular aac-(6’)-Ib gene found in 2006 had two amino acid changes, Trp102Arg and Asp179Tyr, that generated the ability to acety-late the amino nitrogen N4 on the piperazinyl substituent of fluoroquinolones such as ciprofloxacin; and so the gene was named aac-(6’)-Ib-cr (Robicsek, Strahilevitz et al. 2006, Maurice, Broutin et al. 2008, Vetting, Park et al. 2008). The acetylation reduces the antibacterial activity of ciprofloxacin, with N-acetylated ciprofloxacin generating 4-fold higher MIC values than regular ciprofloxacin (Robicsek, Strahilevitz et al. 2006).
Interestingly, the finding of another ciprofloxacin-modifying enzyme was published recently in early 2018. A plasmid from a Pseudomonas aeruginosa isolate was able to reduce resistance to ciprofloxacin without carrying any of the above presented PMQR genes. Analysis of the plasmid sequence revealed an open reading frame with homology to genes for aminoglycoside phos-photransferase enzymes, and cloning demonstrated that this gene generates a MIC increase in E. coli specific to ciprofloxacin (the susceptibility remained unchanged for levofloxacin, moxifloxacin, norfloxacin and nalidixic acid and

41
to aminoglycosides). The gene was named crpP, for ciprofloxacin resistance protein on a plasmid. Enzymatic assays combined with mass-spectrometry analysis led to the conclusion that CrpP increases ciprofloxacin resistance by phosphorylation of the drug’s carboxyl group, utilizing ATP. In addition, mass spectrometry analysis of ciprofloxacin from cell-extracts of E. coli overex-pressing crpP revealed that the phosphorylated form of ciprofloxacin is pos-sibly subjected to stepwise degradation (Chavez-Jacobo, Hernandez-Ramirez et al. 2018).
Could PMQR genes provide short-cuts to ciprofloxacin resistance? As with chromosomal ciprofloxacin resistance mutations, each of the PMQR genes described individually only generates a small increase in MIC in the absence of other resistance determinants. We wondered if it would be possible for these non-essential, extrinsic genes to evolve or be placed in a genetic con-text so that they could provide high-level resistance to ciprofloxacin in E. coli in one step. Even before PMQR genes were discovered, a somewhat similar thought was presented in a mini-review by P. Courvalin in 1990. The author reasoned that the absence of PMQR, 30 years after extensive usage of quin-olones, reflected a lack of need for bacteria to acquire extra-genomic se-quences conferring fluoroquinolone resistance. However, he hypothesized that selection of plasmid borne quinolone resistance would not be unlikely if it “would give rise to quinolone-resistant cells at a much higher frequency than would chromosomal mutations, in particular, when two independent events are required for the latter” (Courvalin 1990).
With this in mind, we sought to determine if PMQR genes could provide a short-cut in the evolutionary trajectory to ciprofloxacin resistant E. coli. We decided to clone genes providing drug-target protection and drug-efflux be-hind constitutive or inducible promoters of varying strengths, to determine if changes in expression levels could impact the PMQR gene’s effect on re-sistance. That expression levels of resistance determinants such as the RND-efflux pumps influences the level or resistance has been clearly shown, not least with AcrAB-TolC (Praski Alzrigat, Huseby et al. 2017). Additionally, in a selection for increased ciprofloxacin resistance using strains harbouring qnr, it was shown that amplification of this gene increased its transcription levels and thereby decreased ciprofloxacin susceptibility even further (Vinue, Corcoran et al. 2015). The drug target modification enzyme aac(6’)-Ib-cr was deliberately excluded from these experiments, since it was shown that treat-ment with N-acetylated ciprofloxacin only increased the MIC to ciprofloxacin in E. coli by 4-fold (Robicsek, Strahilevitz et al. 2006). This means that even if there was enough aac(6’)-Ib-cr transcribed to acetylate every single ciprof-loxacin molecule within a cell, the MIC would still not increase further than

42
4-fold. The results of overexpressing qnr and qepA are presented in Paper III and Paper IV.
Present investigation: Paper III Earlier studies reported that the presence of a qnr gene in an otherwise sus-ceptible E. coli strain generates an MIC to ciprofloxacin of 0.125-0.25 mg/L, while the breakpoint of clinical resistance is 0.5 mg/L (Martinez-Martinez, Pascual et al. 1998, Nazir, Cao et al. 2011, Hooper and Jacoby 2015, Rodriguez-Martinez, Machuca et al. 2016, EUCAST 2018). In order to deter-mine if variations in expression level affected the MIC, we amplified and cloned the genes qnrS and qnrB from our collection of ciprofloxacin resistant E. coli isolates (Nazir, Cao et al. 2011) onto the chromosomes of a series of isogenic E. coli MG1655 strains. Each of the E. coli strains carried a constitu-tive promoter of varying strength in the galK loci, and each qnr gene was placed under the control of eight different promoters, generating a total of 16 strains. The constitutive promoters belong to the J23-series of promoters, where specific nucleotide changes in the promoter sequence generate varia-tions of expression levels differing by at least a 100-fold (iGEM). A linear relationship was observed between J23-promoter strength and qnr mRNA level when measured by qPCR. A range of expression levels from the weakest to the strongest promoter was achieved, with the extreme differences in mRNA levels varying by 102-fold for qnrB and 104-fold for qnrS. We were able to show a positive correlation between expression level and MIC for ciprofloxacin. MIC values associated with expression of qnrB ranged from 0.03-0.375 mg/L, whereas the MIC values for qnrS ranged from 0.015 to 1 mg/L: thus, we proved that an increased expression level of qnrS is sufficient to increase the E. coli MIC above the clinical breakpoint for resistance (Figure 10).
During the cloning process, a lexA binding sequence was introduced be-tween the promoters and the start codon of qnrB, which in principle might have affected qnrB transcription levels. However, upon measurement of MIC to ciprofloxacin it was evident that the MIC increase stagnated at 0.375 mg/L with the four strongest promoters regulating qnrB expression, despite a 5-fold difference in mRNA levels generated between them. This renders it unlikely that even higher mRNA levels would have increased the MIC further. The MIC increase observed with overexpression of qnrS reached a maximum value at a higher level, 1 mg/L, with the three strongest promoters used. We observed that even at the highest MIC values obtained, expression of qnrB or qnrS did not generate any significant fitness costs when comparing growth rates and cfu levels relative to an isogenic E. coli strain lacking qnr. In con-clusion, not only was increased expression of qnrS sufficient to elevate the ciprofloxacin MIC of E. coli 2-fold higher than the clinical breakpoint, it was also achieved without inferring a fitness cost.
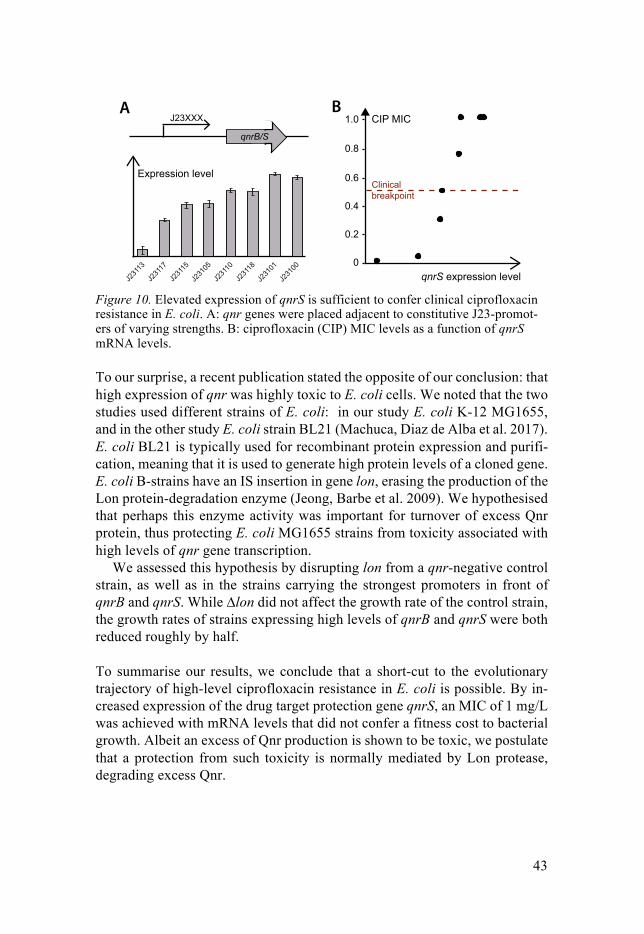
43
A B
Figure 10. Elevated expression of qnrS is sufficient to confer clinical ciprofloxacin resistance in E. coli. A: qnr genes were placed adjacent to constitutive J23-promot-ers of varying strengths. B: ciprofloxacin (CIP) MIC levels as a function of qnrS mRNA levels.
To our surprise, a recent publication stated the opposite of our conclusion: that high expression of qnr was highly toxic to E. coli cells. We noted that the two studies used different strains of E. coli: in our study E. coli K-12 MG1655, and in the other study E. coli strain BL21 (Machuca, Diaz de Alba et al. 2017). E. coli BL21 is typically used for recombinant protein expression and purifi-cation, meaning that it is used to generate high protein levels of a cloned gene. E. coli B-strains have an IS insertion in gene lon, erasing the production of the Lon protein-degradation enzyme (Jeong, Barbe et al. 2009). We hypothesised that perhaps this enzyme activity was important for turnover of excess Qnr protein, thus protecting E. coli MG1655 strains from toxicity associated with high levels of qnr gene transcription.
We assessed this hypothesis by disrupting lon from a qnr-negative control strain, as well as in the strains carrying the strongest promoters in front of qnrB and qnrS. While ∆lon did not affect the growth rate of the control strain, the growth rates of strains expressing high levels of qnrB and qnrS were both reduced roughly by half.
To summarise our results, we conclude that a short-cut to the evolutionary trajectory of high-level ciprofloxacin resistance in E. coli is possible. By in-creased expression of the drug target protection gene qnrS, an MIC of 1 mg/L was achieved with mRNA levels that did not confer a fitness cost to bacterial growth. Albeit an excess of Qnr production is shown to be toxic, we postulate that a protection from such toxicity is normally mediated by Lon protease, degrading excess Qnr.

44
Present investigation: Paper IV In this study we aimed to extend the question from Paper III, asking if high expression levels of a single PMQR gene other than qnrS could provide E. coli with clinically significant resistance to ciprofloxacin. We set out to make this investigation with the inner membrane efflux-pump gene qepA, cloned behind the arabinose inducible promoter pBAD, in the araBAD loci of E. coli MG1655. However, the findings presented below led us onto a different ex-perimental path.
Reports on the magnitude of increased MIC levels to ciprofloxacin associated with the presence of qepA in E. coli (either on native plasmids from clinical isolates or as cloned fragments containing the qepA region) vary from 1 to 64-fold increases, possibly due to differences in genetic contexts (Perichon, Courvalin et al. 2007, Yamane, Wachino et al. 2007, Liu, Deng et al. 2008, Rincon, Radice et al. 2014, Machuca, Briales et al. 2015). However, we were surprised to find that when we cloned the qepA coding sequence onto the chro-mosome of E. coli behind the pBAD promoter it did not generate any increase in MIC, irrespective of the level of arabinose present in the growth medium. To control for pBAD promoter function, the coding sequence of a reporter gene was cloned in the same position as qepA, and expression levels measured by fluorescence as a function of arabinose concentration indicated that the arabinose induction of pBAD worked as expected. To control for the induction of transcription in our qepA-strain, qPCR analysis of qepA mRNA levels were measured at different arabinose concentrations. Even though qepA mRNA was detected, it was only present in low levels relative to those of the control housekeeping genes, and only small and insignificant increases in mRNA lev-els were measured with higher arabinose concentrations.
Looking back at previous publications, we noticed that strains with MIC increases to ciprofloxacin resulting from the cloning of qepA always carried additional DNA sequences from the region upstream of the qepA coding se-quence. The coding sequence of qepA is 1,536 bp, whereas the cloned frag-ments that conferred an increase in MIC from the native qepA plasmid were either 2.1 kilobases (kb) or 3.2 kb (Perichon, Courvalin et al. 2007, Yamane, Wachino et al. 2007). We decided to clone the equivalent 3.2 kb fragment to the one published by Yamane et. al. in the araBAD loci (Figure 11A), and with this construct we observed an 8-fold MIC increase to ciprofloxacin independ-ent of arabinose concentration. This MIC increase depended on qepA tran-scription, as a disruption of the qepA gene in this context reduced the MIC back to an E. coli wild-type susceptible level. Also, mRNA levels of qepA from the 3.2 kb construct was about 100-fold higher than mRNA levels with the qepA coding sequence alone placed behind pBAD. The independence of arabinose concentration implied that a promoter other than pBAD was respon-sible for transcription of qepA in the context of the 3.2 kb fragment. A putative

45
transcriptional start-site has been previously identified 24 bp upstream of the qepA start codon, associated with a promoter sequence extending to 60 bp upstream of qepA, which we named putative promoter 1 (pP1) (Cattoir, Poirel et al. 2008), and by sequence analysis we now identified an additional putative promoter sequence, putative promoter 2 (pP2), with a -10 region 482 bp up-stream of the qepA start codon. We also identified two putative open reading frames (orf1 and orf2) located between pP2 and the qepA coding sequence (Figure 11B).
By constructing deletions and disruptions in the cloned 3.2 kb fragment, we were able to demonstrate the following requirements for qepA to generate a MIC increase to ciprofloxacin: The increase in MIC was dependent on the pP2 sequence 482 bp upstream from the qepA start codon. With this promoter sequence still intact, the downstream orf1 sequence could be deleted without affecting the elevated MIC. In contrast, deletion of orf2 reduced ciprofloxacin MIC to the wild-type susceptible level. The orf 2 sequence overlapped with pP1 upstream of qepA, identified by Cattoir et. al., raising the possibility that this sequence is an active promoter required for qepA expression. Therefore, a second deletion was made, removing the first half of orf2 but keeping the pP1 sequence intact. Still, the MIC was reduced to wild-type levels even if only the first half of orf2 was deleted, and pP1 upstream of qepA was intact (Figure 11B).
Conclusively, these findings tell us that a previously unidentified promoter is responsible for qepA expression, and that this expression is dependent on a sequence element in between this promoter and the qepA start codon, a region we named orf2. Work is ongoing to elucidate if the dependence of the orf2 region is of a sequence-structural nature, of if translation of orf2 is required to enable the MIC increase conferred by qepA. Once we have disentangled the expression regulation of this inner membrane efflux pump, we will modify the transcriptional levels of this gene so as to answer the original question that initiated this project.
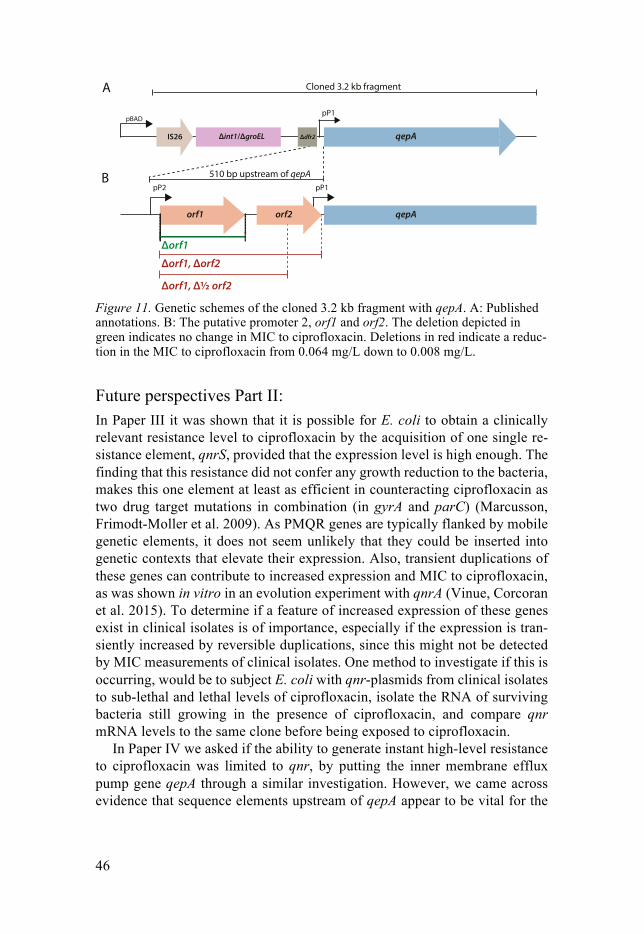
46
A
B
pBAD
Cloned 3.2 kb fragment
510 bp upstream of qepA
IS26 Δdfr2Δint1/ΔgroEL qepA
qepAorf2orf1
pP2 pP1
pP1
Δorf1
Δorf1, Δorf2
Δorf1, Δ½ orf2 Figure 11. Genetic schemes of the cloned 3.2 kb fragment with qepA. A: Published annotations. B: The putative promoter 2, orf1 and orf2. The deletion depicted in green indicates no change in MIC to ciprofloxacin. Deletions in red indicate a reduc-tion in the MIC to ciprofloxacin from 0.064 mg/L down to 0.008 mg/L.
Future perspectives Part II: In Paper III it was shown that it is possible for E. coli to obtain a clinically relevant resistance level to ciprofloxacin by the acquisition of one single re-sistance element, qnrS, provided that the expression level is high enough. The finding that this resistance did not confer any growth reduction to the bacteria, makes this one element at least as efficient in counteracting ciprofloxacin as two drug target mutations in combination (in gyrA and parC) (Marcusson, Frimodt-Moller et al. 2009). As PMQR genes are typically flanked by mobile genetic elements, it does not seem unlikely that they could be inserted into genetic contexts that elevate their expression. Also, transient duplications of these genes can contribute to increased expression and MIC to ciprofloxacin, as was shown in vitro in an evolution experiment with qnrA (Vinue, Corcoran et al. 2015). To determine if a feature of increased expression of these genes exist in clinical isolates is of importance, especially if the expression is tran-siently increased by reversible duplications, since this might not be detected by MIC measurements of clinical isolates. One method to investigate if this is occurring, would be to subject E. coli with qnr-plasmids from clinical isolates to sub-lethal and lethal levels of ciprofloxacin, isolate the RNA of surviving bacteria still growing in the presence of ciprofloxacin, and compare qnr mRNA levels to the same clone before being exposed to ciprofloxacin.
In Paper IV we asked if the ability to generate instant high-level resistance to ciprofloxacin was limited to qnr, by putting the inner membrane efflux pump gene qepA through a similar investigation. However, we came across evidence that sequence elements upstream of qepA appear to be vital for the

47
expression of its resistance phenotype. Therefore, we are in the process of dis-entangling the expression regulation of qepA by further analysis of its native sequence context. By qPCR we will measure the mRNA levels of qepA in relation to the various deletions and other mutations of upstream sequence elements, i) to verify that the putative promoter identified regulates transcrip-tion of qepA, and ii) to clarify if the sequence region named orf2 is also essen-tial for qepA transcription. To get a further understanding on the importance of orf2, we plan to mutate the sequence by a single nucleotide (to mutate the start codon or introduce in-frame stop codons) in order to elucidate if the qepA MIC increase is dependent of orf2 translation, or rather on RNA structural features provided by the orf2 sequence.

48
Concluding remarks
In this thesis, the subject of study has been to explore the evolutionary trajec-tories of E. coli to ciprofloxacin resistance. This exploration has further em-phasized the seemingly endless arsenal of tools available to develop antibiotic resistance, both those that lie within a single bacterial genome, as well as those provided by horizontal gene transfer.
An unexpected mutational target in genes of the translation machinery was found to increased resistance to an antibiotic inhibiting DNA replication, by causing an onset of a nutrient-starvation response, that in turn happened to have an advantageous net-effect by increasing drug efflux. We demonstrated that mutations in equivalent and additional genes involved in transcription and translation are repeatedly selected, in a highly predictable evolutionary trajec-tory of E. coli subjected to increasing concentrations of ciprofloxacin. How-ever, the patterns of mutations selected was strongly influenced by the bottle-neck-size used when transferring bacteria to fresh medium.
In the second part, we asked whether it was possible to develop a short-cut in the trajectory towards high-level resistance to ciprofloxacin, which nor-mally requires an accumulation of mutations and/or plasmid born resistance genes. We learned that this is indeed possible, by increasing the expression of a plasmid-born gene providing drug target protection. We set out to make a similar analysis of another plasmid borne quinolone resistance gene, an inner membrane efflux pump. However, with this gene we discovered that upstream sequences of the gene’s coding region are apparently required to generate the phenotypic increase in MIC. This expression regulation is currently being dis-entangled before the initial question can be answered.
These findings underline the importance of thoroughly designing, model-ling, and repeating selections and in vitro evolutions aimed at detecting and evaluating antibiotic resistance mutations. Resistance selections or evolutions should be adapted to cover several possibilities, so that mutations (and com-binations of them) that are rare, costly, or that only generate minor decreases in susceptibility are not ignored. To include the impact of horizontally trans-ferred genes in experimental evolution experiments is challenging, as they could interfere at any step of the evolutionary trajectory. Nonetheless, our findings that a horizontally transferred gene has the capability to speed up the development of high-level resistance to a drug enlisted as being of critical im-portance only emphasizes the importance of taking on this challenge.

49
Although the lack of internationally coordinated surveillance complicates ac-curate estimations of its exact impact, antibiotic resistance remains a serious threat to the current and future public health, as common infections turn deadly and untreatable. This is due to the extent and ways of usage that anti-biotics have been exploited in, forcing pathogenic microorganisms to adapt at an increasing rate, whilst development of new antibiotics has been stalling for decades. In order to reduce the rate of resistance emergence for novel and cur-rently useful antibiotics, the study of evolutionary trajectories toward antibi-otic resistance is of crucial importance in order to avoid repeating the same mistakes.

50
Acknowledgements
“The only thing I am absolutely sure of, is that I will never ever become a PhD student.”
Me, in 2012 This was the only definite decision I had made about my career at the time. Luckily, after 2 years in the Master program in Infection Biology at Uppsala University, I felt the complete opposite. I would like to thank all of you that changed my mind, and that supported and encouraged me throughout my stud-ies; you have all ensured that this work was completed without the slightest of regrets.
First and foremost, thank you Diarmaid. You were an inspiring teacher during my Master’s studies, and I felt so lucky and excited when I got to do my exam-project in your research group. Even more so when you employed me as a PhD student. As a supervisor I find you to be supportive and encouraging, but you can also be challenging in order to push your students towards becoming more independent. Something that I have valued greatly is the generosity you provide in terms of your time, for teaching, discussions, guidance and feed-back: whenever needed.
To my co-supervisor Dan: even though you might not remember being my co-supervisor, I really appreciate that you are always attentive at presenta-tions, generous with advice and feedback, and encouraging the PhD-students in the whole corridor to think bigger.
Also, thank you Linus: I’ve come to think of you as an unofficial co-su-pervisor after all the career- and science talks during commuting. And not least, for reminding of the importance of whiskey in between successes and failures.
To the former and present members of the DH-group: I consider myself very lucky to have spent these years in such a nice lab, and that is all thanks to you. Doug, you where such a patient and pedagogic lab-supervisor during my Mas-ter’s, and you have continued that role throughout my PhD (and provided lots of additional advice about cats). Gerrit, another unofficial supervisor: you were always very generous with good advice and clear explanations, when-ever I had questions about anything from lambda-red to statistics. Cao Sha, you always bring a smile to the lab, despite handling so many things at once

51
– I really admire and appreciate your positive attitude! Katrin, you always surpassed my mistakes as a supervisor with a laugh, and you bring such en-thusiasm over things I usually forget to appreciate (such as pretty gradient plates). Lisa, thank you for carrying on with a tricky project in such great spirits, even at times of confusing supervision and with your other tasks in the lab piling up. Rachel, you taught me what you can achieve with some grit towards tech-support, and gave me many good laughs over a drink! Vicky, I’ve heard that you make as good cakes as you generate results in the lab: I hope I won’t be so unlucky as to miss the next time you bring a cake! Eva, I miss your chatting at the lab bench across mine: thank you for all the shared thoughts about science, life, and TV-series. Anna, your contagious laughter and warmth is greatly missed in the lab! Franzi and Jessica, you were great role models with your positive and easy-going attitudes as senior PhD stu-dents, and you were always eager to help your colleagues out! Kavita, thank you for all the weeks you put into constructing the strains that are now used by so many of us. Lisa PA, if not for one of your projects, I might not have initiated my PhD studies. And to my former master students: Kenesha, you brought as much joy to the lab as you brought cakes and cookies (and that was a lot!), I hope to see you back in Scandinavia or at least Europe soon. And to Tua, my very first master student, thank you for all the hard work you spend on those evolutions.
To my fellow Masters of Infection: Fredrika, thank you for always providing hugs, advice, encouragement and that mischievous humour I enjoy so much. And Dennis, well… schlghrochshc (a nice compliment in Dutch). All this time from Bangladesh and onwards wouldn’t have been half as fun without the two of you!
To the other past and present members of the DA, LS, JN and LG groups that make up the awesome D7:3 corridor: Michael, congratulations on a great dis-sertation, no one expected any less! Looking forward to many more beers in the future. Ulrika and Karin, one could only imagine what condition the lab would be in without you! Thank you for keeping us in check, for always in-viting to a game of cards, and for your wonderful sense of humour. The SquarEriks, masters of movie-editing: Erik WY, I’m glad you have stayed in this corridor and put up with all of us ‘animals’ that might not wash our coffee-cups every day, and Erik L, thanks for all your efforts with the messy MACS, and congratulations: you’re up next! Marius, you are so brave to fol-low your ECDC goal to Helsinki, I’m sure you will do as great there as you did here! Nina, thanks for all the coffee breaks and your great capability of sharing laughs at your own expense! Tiffaine, with that hard-working mindset and French charm you will surely succeed with anything you put your mind to. Po, I suppose the secret to your constant smiling and laughing is all that sugar! 3 semlor, I still can’t believe it… Lisa Alb, I’ve really enjoyed our

52
sincere conversations and your honest opinions at the fika nook, about every-thing from microbiology to feminism. Anna, the lab is perhaps a bit less messy but also not quite the same without you! Apart from being such a warm person, you really wrote a kick-ass kappa that I learned a lot from. Omar, always so calm, friendly, and ready to help! Thanks for all the great input you provide at presentations, and for keeping M in check. Jocke, the mastermind of genetic manipulations: apart from being thankful for all your scientific in-put, I do hope that you will also pick up on that much appreciated beer-brew-ing again! Hind, you always claim that no one takes you seriously, but you’re getting there! Hearing your laugh spread through the corridor always brings a smile. Hervé, your ironic comments never cease to bring a laugh, and your constant questions and suggestions at seminars are as scary for a new student as they are appreciated by an older one. Arianne, thank you for providing me with my new motto: establish dominance! Lisa T, I hope you will keep check-ing in for AWs and fika every now and then. Jenny, ambitious, super-friendly and easy going, and without hesitation you always speak your honest opinions during discussions at the fika nook: there’s no doubt that you will do great. Greta, a natural in making posters, and I’m sure that you will bring about a whole new biofilm-platform to our corridor! Serhiy, you have such impres-sive work ethics to show up in the lab no matter how badly the bugs got to you. Marie, I’m sure that many are happy to see you back in the corridor, and I’m very glad to have got to know this immensely nice person that you are! Lisa All, with refraining so much of your holidays for research, I hope to see you in the PhD position that you deserve soon! Thanks for the good company at the hours of commuting. Lionel, probably every corridor could use a bioin-formatician. We were lucky to get someone so eager in sharing their knowledge, and someone that brought even more nice people to the lab. Isa, always cracking a spot-on joke at the coffee breaks, and daring to bring eu-karyotes to a microbiology lab! Next time we bump into each other in London we should have a beer. Susan, thank you for the rescue at Noor’s Slott: I’m looking forward to more chats with you! And Dennis, my Dutch translation: thank you for always reminding of and inviting to fun things to do outside of the lab, I hope we attend at least one more Gröna Lund concert!
Finally, without some of the tremendous support and encouragement I have been lucky to obtain from people outside of work, these years passed wouldn’t have passed as smoothly. Johanna, and Uppsala Ju-Jutsu Klubb: thank you for all the bruises, broken lips and muscle aches, for providing an outlet for my frustrations and boosting my confidence! Carlos, that we’re still such good friends though we spent most of our PhDs apart says it all! Malin, älskade vän, where would I be without your pep-talks?
To my Family: Pappa, Mamma, Niklas, Li, Viktor, Elina, Sanna, Alex, Malena, Hannah, Lovis, Maria, Rickard, Emma, Isak: I am so grateful,

53
and I know you would have encouraged and supported me just as much in anything I would have set my mind to!
And last, but certainly not least: Daniel. The one person that had to endure with most of my stress and frustrations. For putting up with that, for all your encouragement and reassuring, the hundreds (maybe thousands?) of lunch-boxes, for being my never receding rock: I love you.

54
References
Amato, S. M., M. A. Orman and M. P. Brynildsen (2013). "Metabolic control of persister formation in Escherichia coli." Mol Cell 50(4): 475-487.
Andersson, D. I. and D. Hughes (1996). "Muller's ratchet decreases fitness of a DNA-based microbe." Proc Natl Acad Sci U S A 93(2): 906-907.
Andersson, D. I. and D. Hughes (2010). "Antibiotic resistance and its cost: is it possible to reverse resistance?" Nat Rev Microbiol 8(4): 260-271.
Andersson, D. I. and D. Hughes (2017). "Selection and Transmission of Antibiotic-Resistant Bacteria." Microbiol Spectr 5(4).
Andersson, D. I. and B. R. Levin (1999). "The biological cost of antibiotic resistance." Curr Opin Microbiol 2(5): 489-493.
Appelbaum, P. C. and P. A. Hunter (2000). "The fluoroquinolone antibacterials: past, present and future perspectives." Int J Antimicrob Agents 16(1): 5-15.
Baharoglu, Z. and D. Mazel (2014). "SOS, the formidable strategy of bacteria against aggressions." FEMS Microbiol Rev 38(6): 1126-1145.
Barbier, F., J. Lipman and M. J. M. Bonten (2016). "In 2035, will all bacteria be multidrug-resistant? No." Intensive Care Med 42(12): 2017-2020.
Blair, J. M., M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. Piddock (2015). "Molecular mechanisms of antibiotic resistance." Nat Rev Microbiol 13(1): 42-51.
Blattner, F. R., G. Plunkett, 3rd, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau and Y. Shao (1997). "The complete genome sequence of Escherichia coli K-12." Science 277(5331): 1453-1462.
Blazquez, J. (2003). "Hypermutation as a factor contributing to the acquisition of antimicrobial resistance." Clin Infect Dis 37(9): 1201-1209.
Bos, J., Q. Zhang, S. Vyawahare, E. Rogers, S. M. Rosenberg and R. H. Austin (2015). "Emergence of antibiotic resistance from multinucleated bacterial filaments." Proc Natl Acad Sci U S A 112(1): 178-183.
Brandis, G. and D. Hughes (2013). "Genetic characterization of compensatory evolution in strains carrying rpoB Ser531Leu, the rifampicin resistance mutation most frequently found in clinical isolates." J Antimicrob Chemother 68(11): 2493-2497.
Briales, A., J. M. Rodriguez-Martinez, C. Velasco, J. Machuca, P. Diaz de Alba, J. Blazquez and A. Pascual (2012). "Exposure to diverse antimicrobials induces the expression of qnrB1, qnrD and smaqnr genes by SOS-dependent regulation." J Antimicrob Chemother 67(12): 2854-2859.
Cattoir, V., L. Poirel and P. Nordmann (2008). "Plasmid-mediated quinolone resistance pump QepA2 in an Escherichia coli isolate from France." Antimicrob Agents Chemother 52(10): 3801-3804.
Chavez-Jacobo, V. M., K. C. Hernandez-Ramirez, P. Romo-Rodriguez, R. V. Perez-Gallardo, J. Campos-Garcia, J. F. Gutierrez-Corona, J. P. Garcia-Merinos, V.

55
Meza-Carmen, J. Silva-Sanchez and M. I. Ramirez-Diaz (2018). "CrpP Is a Novel Ciprofloxacin-Modifying Enzyme Encoded by the Pseudomonas aeruginosa pUM505 Plasmid." Antimicrob Agents Chemother 62(6).
Chen, C. R., M. Malik, M. Snyder and K. Drlica (1996). "DNA gyrase and topoisomerase IV on the bacterial chromosome: quinolone-induced DNA cleavage." J Mol Biol 258(4): 627-637.
Chin, N. X. and H. C. Neu (1984). "Ciprofloxacin, a quinolone carboxylic acid compound active against aerobic and anaerobic bacteria." Antimicrob Agents Chemother 25(3): 319-326.
Cirz, R. T. and F. E. Romesberg (2006). "Induction and inhibition of ciprofloxacin resistance-conferring mutations in hypermutator bacteria." Antimicrob Agents Chemother 50(1): 220-225.
Couce, A., A. Rodriguez-Rojas and J. Blazquez (2016). "Determinants of Genetic Diversity of Spontaneous Drug Resistance in Bacteria." Genetics 203(3): 1369-1380.
Courvalin, P. (1990). "Plasmid-mediated 4-quinolone resistance: a real or apparent absence?" Antimicrob Agents Chemother 34(5): 681-684.
Croxen, M. A. and B. B. Finlay (2010). "Molecular mechanisms of Escherichia coli pathogenicity." Nat Rev Microbiol 8(1): 26-38.
D'Costa, V. M., K. M. McGrann, D. W. Hughes and G. D. Wright (2006). "Sampling the antibiotic resistome." Science 311(5759): 374-377.
Da Re, S., F. Garnier, E. Guerin, S. Campoy, F. Denis and M. C. Ploy (2009). "The SOS response promotes qnrB quinolone-resistance determinant expression." EMBO Rep 10(8): 929-933.
Davies, J. and D. Davies (2010). "Origins and evolution of antibiotic resistance." Microbiol Mol Biol Rev 74(3): 417-433.
Dorr, T., K. Lewis and M. Vulic (2009). "SOS response induces persistence to fluoroquinolones in Escherichia coli." PLoS Genet 5(12): e1000760.
Drake, J. W. (1991). "A constant rate of spontaneous mutation in DNA-based microbes." Proc Natl Acad Sci U S A 88(16): 7160-7164.
Drlica, K., H. Hiasa, R. Kerns, M. Malik, A. Mustaev and X. Zhao (2009). "Quinolones: action and resistance updated." Curr Top Med Chem 9(11): 981-998.
Drlica, K., M. Malik, R. J. Kerns and X. Zhao (2008). "Quinolone-mediated bacterial death." Antimicrob Agents Chemother 52(2): 385-392.
Durfee, T., A. M. Hansen, H. Zhi, F. R. Blattner and D. J. Jin (2008). "Transcription profiling of the stringent response in Escherichia coli." J Bacteriol 190(3): 1084-1096.
ECDC, E. C. f. D. P. a. C. (2010). Antimicrobial resistance surveillance in Europe 2009. Annual Report of the European Antimicrobial Resistance Surveillance Network (EARS-Net).
ECDC, E. C. f. D. P. a. C. (2017). Surveillance of antimicrobial resistance in Europe 2016. Annual Report of the European Antimicrobial Resistance Surveillance Network (EARS-Net).
Eliopoulos, G. M., A. Gardella and R. C. Moellering, Jr. (1984). "In vitro activity of ciprofloxacin, a new carboxyquinoline antimicrobial agent." Antimicrob Agents Chemother 25(3): 331-335.
Erental, A., Z. Kalderon, A. Saada, Y. Smith and H. Engelberg-Kulka (2014). "Apoptosis-like death, an extreme SOS response in Escherichia coli." MBio 5(4): e01426-01414.
EUCAST (2018). The European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters. Version 8.1,

56
valid from 2018-05-15, Electronic publication, available from http://www.eucast.org/clinical_breakpoints/.
EUCAST. (2018). "MIC distributions and ECOFFs." Retrieved May 23, 2018, from http://www.eucast.org/mic_distributions_and_ecoffs/.
Everett, M. J., Y. F. Jin, V. Ricci and L. J. Piddock (1996). "Contributions of individual mechanisms to fluoroquinolone resistance in 36 Escherichia coli strains isolated from humans and animals." Antimicrob Agents Chemother 40(10): 2380-2386.
Fair, R. J. and Y. Tor (2014). "Antibiotics and bacterial resistance in the 21st century." Perspect Medicin Chem 6: 25-64.
Ferrero, L., B. Cameron, B. Manse, D. Lagneaux, J. Crouzet, A. Famechon and F. Blanche (1994). "Cloning and primary structure of Staphylococcus aureus DNA topoisomerase IV: a primary target of fluoroquinolones." Mol Microbiol 13(4): 641-653.
Freddolino, P. L., S. Amini and S. Tavazoie (2012). "Newly identified genetic variations in common Escherichia coli MG1655 stock cultures." J Bacteriol 194(2): 303-306.
Gaca, A. O., C. Colomer-Winter and J. A. Lemos (2015). "Many means to a common end: the intricacies of (p)ppGpp metabolism and its control of bacterial homeostasis." J Bacteriol 197(7): 1146-1156.
Garoff, L., D. L. Huseby, L. Praski Alzrigat and D. Hughes (2018). "Effect of aminoacyl-tRNA synthetase mutations on susceptibility to ciprofloxacin in Escherichia coli." J Antimicrob Chemother.
Gellert, M., K. Mizuuchi, M. H. O'Dea, T. Itoh and J. I. Tomizawa (1977). "Nalidixic acid resistance: a second genetic character involved in DNA gyrase activity." Proc Natl Acad Sci U S A 74(11): 4772-4776.
Gellert, M., M. H. O'Dea, T. Itoh and J. Tomizawa (1976). "Novobiocin and coumermycin inhibit DNA supercoiling catalyzed by DNA gyrase." Proc Natl Acad Sci U S A 73(12): 4474-4478.
Guan, X., X. Xue, Y. Liu, J. Wang, Y. Wang, J. Wang, K. Wang, H. Jiang, L. Zhang, B. Yang, N. Wang and L. Pan (2013). "Plasmid-mediated quinolone resistance--current knowledge and future perspectives." J Int Med Res 41(1): 20-30.
Guyer, M. S., R. R. Reed, J. A. Steitz and K. B. Low (1981). "Identification of a sex-factor-affinity site in E. coli as gamma delta." Cold Spring Harb Symp Quant Biol 45 Pt 1: 135-140.
Hansen, L. H., L. B. Jensen, H. I. Sorensen and S. J. Sorensen (2007). "Substrate specificity of the OqxAB multidrug resistance pump in Escherichia coli and selected enteric bacteria." J Antimicrob Chemother 60(1): 145-147.
Hansen, L. H., E. Johannesen, M. Burmolle, A. H. Sorensen and S. J. Sorensen (2004). "Plasmid-encoded multidrug efflux pump conferring resistance to olaquindox in Escherichia coli." Antimicrob Agents Chemother 48(9): 3332-3337.
Hayashi, K., N. Morooka, Y. Yamamoto, K. Fujita, K. Isono, S. Choi, E. Ohtsubo, T. Baba, B. L. Wanner, H. Mori and T. Horiuchi (2006). "Highly accurate genome sequences of Escherichia coli K-12 strains MG1655 and W3110." Mol Syst Biol 2: 2006 0007.
Heisig, P. (1996). "Genetic evidence for a role of parC mutations in development of high-level fluoroquinolone resistance in Escherichia coli." Antimicrob Agents Chemother 40(4): 879-885.
Hiasa, H., D. O. Yousef and K. J. Marians (1996). "DNA strand cleavage is required for replication fork arrest by a frozen topoisomerase-quinolone-DNA ternary complex." J Biol Chem 271(42): 26424-26429.

57
Hooper, D. C. and G. A. Jacoby (2015). "Mechanisms of drug resistance: quinolone resistance." Ann N Y Acad Sci 1354: 12-31.
Hughes, D. and D. I. Andersson (2012). "Selection of resistance at lethal and non-lethal antibiotic concentrations." Curr Opin Microbiol 15(5): 555-560.
Huseby, D. L., F. Pietsch, G. Brandis, L. Garoff, A. Tegehall and D. Hughes (2017). "Mutation Supply and Relative Fitness Shape the Genotypes of Ciprofloxacin-Resistant Escherichia coli." Mol Biol Evol 34(5): 1029-1039.
iGEM. "Registry of Standard Biological Parts." Retrieved September 13, 2018, from http://parts.igem.org/Promoters/Catalog/Anderson.
Ito, A., K. Hirai, M. Inoue, H. Koga, S. Suzue, T. Irikura and S. Mitsuhashi (1980). "In vitro antibacterial activity of AM-715, a new nalidixic acid analog." Antimicrob Agents Chemother 17(2): 103-108.
Jacoby, G. A. and D. C. Hooper (2013). "Phylogenetic analysis of chromosomally determined qnr and related proteins." Antimicrob Agents Chemother 57(4): 1930-1934.
Jeong, H., V. Barbe, C. H. Lee, D. Vallenet, D. S. Yu, S. H. Choi, A. Couloux, S. W. Lee, S. H. Yoon, L. Cattolico, C. G. Hur, H. S. Park, B. Segurens, S. C. Kim, T. K. Oh, R. E. Lenski, F. W. Studier, P. Daegelen and J. F. Kim (2009). "Genome sequences of Escherichia coli B strains REL606 and BL21(DE3)." J Mol Biol 394(4): 644-652.
Jishage, M., K. Kvint, V. Shingler and T. Nystrom (2002). "Regulation of sigma factor competition by the alarmone ppGpp." Genes Dev 16(10): 1260-1270.
Jovanovic, M., M. Lilic, R. Janjusevic, G. Jovanovic and D. J. Savic (1999). "tRNA synthetase mutants of Escherichia coli K-12 are resistant to the gyrase inhibitor novobiocin." J Bacteriol 181(9): 2979-2983.
Kampranis, S. C. and A. Maxwell (1998). "The DNA gyrase-quinolone complex. ATP hydrolysis and the mechanism of DNA cleavage." J Biol Chem 273(35): 22615-22626.
Khodursky, A. B., E. L. Zechiedrich and N. R. Cozzarelli (1995). "Topoisomerase IV is a target of quinolones in Escherichia coli." Proc Natl Acad Sci U S A 92(25): 11801-11805.
Kim, H. B., M. Wang, C. H. Park, E. C. Kim, G. A. Jacoby and D. C. Hooper (2009). "oqxAB encoding a multidrug efflux pump in human clinical isolates of Enterobacteriaceae." Antimicrob Agents Chemother 53(8): 3582-3584.
Komp Lindgren, P., Å. Karlsson and D. Hughes (2003). "Mutation rate and evolution of fluoroquinolone resistance in Escherichia coli isolates from patients with urinary tract infections." Antimicrob Agents Chemother 47(10): 3222-3232.
Kumagai, Y., J. I. Kato, K. Hoshino, T. Akasaka, K. Sato and H. Ikeda (1996). "Quinolone-resistant mutants of escherichia coli DNA topoisomerase IV parC gene." Antimicrob Agents Chemother 40(3): 710-714.
Kwon, Y. K., M. B. Higgins and J. D. Rabinowitz (2010). "Antifolate-induced depletion of intracellular glycine and purines inhibits thymineless death in E. coli." ACS Chem Biol 5(8): 787-795.
Land, M., L. Hauser, S. R. Jun, I. Nookaew, M. R. Leuze, T. H. Ahn, T. Karpinets, O. Lund, G. Kora, T. Wassenaar, S. Poudel and D. W. Ussery (2015). "Insights from 20 years of bacterial genome sequencing." Funct Integr Genomics 15(2): 141-161.
Lederberg, J. and E. M. Lederberg (1952). "Replica plating and indirect selection of bacterial mutants." J Bacteriol 63(3): 399-406.
Leimbach, A., J. Hacker and U. Dobrindt (2013). "E. coli as an all-rounder: the thin line between commensalism and pathogenicity." Curr Top Microbiol Immunol 358: 3-32.

58
Lenski, R. E. (2017). "What is adaptation by natural selection? Perspectives of an experimental microbiologist." PLoS Genet 13(4): e1006668.
Lesher, G. Y., E. J. Froelich, M. D. Gruett, J. H. Bailey and R. P. Brundage (1962). "1,8-Naphthyridine Derivatives. A New Class of Chemotherapeutic Agents." J Med Pharm Chem 91: 1063-1065.
Li, X. Z., P. Plesiat and H. Nikaido (2015). "The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria." Clin Microbiol Rev 28(2): 337-418.
Ling, L. L., T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schaberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis (2015). "A new antibiotic kills pathogens without detectable resistance." Nature 517(7535): 455-459.
Liu, J. H., Y. T. Deng, Z. L. Zeng, J. H. Gao, L. Chen, Y. Arakawa and Z. L. Chen (2008). "Coprevalence of plasmid-mediated quinolone resistance determinants QepA, Qnr, and AAC(6')-Ib-cr among 16S rRNA methylase RmtB-producing Escherichia coli isolates from pigs." Antimicrob Agents Chemother 52(8): 2992-2993.
Lopez, E. and J. Blazquez (2009). "Effect of subinhibitory concentrations of antibiotics on intrachromosomal homologous recombination in Escherichia coli." Antimicrob Agents Chemother 53(8): 3411-3415.
Lopez, E., M. Elez, I. Matic and J. Blazquez (2007). "Antibiotic-mediated recombination: ciprofloxacin stimulates SOS-independent recombination of divergent sequences in Escherichia coli." Mol Microbiol 64(1): 83-93.
Luria, S. E. and M. Delbruck (1943). "Mutations of Bacteria from Virus Sensitivity to Virus Resistance." Genetics 28(6): 491-511.
Machuca, J., A. Briales, P. Diaz-de-Alba, L. Martinez-Martinez, A. Pascual and J. M. Rodriguez-Martinez (2015). "Effect of the efflux pump QepA2 combined with chromosomally mediated mechanisms on quinolone resistance and bacterial fitness in Escherichia coli." J Antimicrob Chemother 70(9): 2524-2527.
Machuca, J., P. Diaz de Alba, E. Recacha, A. Pascual and J. M. Rodriguez-Martinez (2017). "Cytotoxic Effect Associated with Overexpression of QNR Proteins in Escherichia coli." Microb Drug Resist 23(7): 822-825.
Machuca, J., E. Recacha, A. Briales, P. Diaz-de-Alba, J. Blazquez, A. Pascual and J. M. Rodriguez-Martinez (2017). "Cellular Response to Ciprofloxacin in Low-Level Quinolone-Resistant Escherichia coli." Front Microbiol 8: 1370.
Marcusson, L. L., N. Frimodt-Moller and D. Hughes (2009). "Interplay in the selection of fluoroquinolone resistance and bacterial fitness." PLoS Pathog 5(8): e1000541.
Martinez-Martinez, L., A. Pascual and G. A. Jacoby (1998). "Quinolone resistance from a transferable plasmid." Lancet 351(9105): 797-799.
Maurice, F., I. Broutin, I. Podglajen, P. Benas, E. Collatz and F. Dardel (2008). "Enzyme structural plasticity and the emergence of broad-spectrum antibiotic resistance." EMBO Rep 9(4): 344-349.
Mehla, K. and J. Ramana (2016). "Structural signature of Ser83Leu and Asp87Asn mutations in DNA gyrase from enterotoxigenic Escherichia coli and impact on quinolone resistance." Gene 576(1 Pt 1): 28-35.
Mitscher, L. A. (2005). "Bacterial topoisomerase inhibitors: quinolone and pyridone antibacterial agents." Chem Rev 105(2): 559-592.
Nazir, H., S. Cao, F. Hasan and D. Hughes (2011). "Can phylogenetic type predict resistance development?" J Antimicrob Chemother 66(4): 778-787.

59
Nishino, K. and A. Yamaguchi (2001). "Analysis of a complete library of putative drug transporter genes in Escherichia coli." J Bacteriol 183(20): 5803-5812.
O’Neill, J. (2014). "Review on antimicrobial resistance. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations."
Oethinger, M., I. Podglajen, W. V. Kern and S. B. Levy (1998). "Overexpression of the marA or soxS regulatory gene in clinical topoisomerase mutants of Escherichia coli." Antimicrob Agents Chemother 42(8): 2089-2094.
Osterberg, S., T. del Peso-Santos and V. Shingler (2011). "Regulation of alternative sigma factor use." Annu Rev Microbiol 65: 37-55.
Perichon, B., P. Courvalin and M. Galimand (2007). "Transferable resistance to aminoglycosides by methylation of G1405 in 16S rRNA and to hydrophilic fluoroquinolones by QepA-mediated efflux in Escherichia coli." Antimicrob Agents Chemother 51(7): 2464-2469.
Piddock, L. J. (1999). "Mechanisms of fluoroquinolone resistance: an update 1994-1998." Drugs 58 Suppl 2: 11-18.
Piddock, L. J. (2015). "Teixobactin, the first of a new class of antibiotics discovered by iChip technology?" J Antimicrob Chemother 70(10): 2679-2680.
Piddock, L. J. and R. N. Walters (1992). "Bactericidal activities of five quinolones for Escherichia coli strains with mutations in genes encoding the SOS response or cell division." Antimicrob Agents Chemother 36(4): 819-825.
Pietsch, F., J. M. Bergman, G. Brandis, L. L. Marcusson, A. Zorzet, D. L. Huseby and D. Hughes (2017). "Ciprofloxacin selects for RNA polymerase mutations with pleiotropic antibiotic resistance effects." J Antimicrob Chemother 72(1): 75-84.
Pommier, Y., E. Leo, H. Zhang and C. Marchand (2010). "DNA topoisomerases and their poisoning by anticancer and antibacterial drugs." Chem Biol 17(5): 421-433.
Praski Alzrigat, L., D. L. Huseby, G. Brandis and D. Hughes (2017). "Fitness cost constrains the spectrum of marR mutations in ciprofloxacin-resistant Escherichia coli." J Antimicrob Chemother 72(11): 3016-3024.
Rahman, Z., A. Islam, M. U. Rashid, F. T. Johura, S. Monira, H. Watanabe, N. Ahmed, A. Camilli and M. Alam (2017). "Existence of a novel qepA variant in quinolone resistant Escherichia coli from aquatic habitats of Bangladesh." Gut Pathog 9: 58.
Ramirez, M. S. and M. E. Tolmasky (2010). "Aminoglycoside modifying enzymes." Drug Resist Updat 13(6): 151-171.
Rincon, G., M. Radice, M. Giovanakis, J. A. Di Conza and G. Gutkind (2014). "First report of plasmid-mediated fluoroquinolone efflux pump QepA in Escherichia coli clinical isolate ST68, in South America." Diagn Microbiol Infect Dis 79(1): 70-72.
Robicsek, A., J. Strahilevitz, G. A. Jacoby, M. Macielag, D. Abbanat, C. H. Park, K. Bush and D. C. Hooper (2006). "Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase." Nat Med 12(1): 83-88.
Rodionov, D. G. and E. E. Ishiguro (1995). "Direct correlation between overproduction of guanosine 3',5'-bispyrophosphate (ppGpp) and penicillin tolerance in Escherichia coli." J Bacteriol 177(15): 4224-4229.
Rodriguez-Beltran, J., J. Tourret, O. Tenaillon, E. Lopez, E. Bourdelier, C. Costas, I. Matic, E. Denamur and J. Blazquez (2015). "High Recombinant Frequency in Extraintestinal Pathogenic Escherichia coli Strains." Mol Biol Evol 32(7): 1708-1716.
Rodriguez-Martinez, J. M., J. Machuca, M. E. Cano, J. Calvo, L. Martinez-Martinez and A. Pascual (2016). "Plasmid-mediated quinolone resistance: Two decades on." Drug Resist Updat 29: 13-29.

60
Schaaper, R. M., B. N. Danforth and B. W. Glickman (1986). "Mechanisms of spontaneous mutagenesis: an analysis of the spectrum of spontaneous mutation in the Escherichia coli lacI gene." J Mol Biol 189(2): 273-284.
Schoeffler, A. J. and J. M. Berger (2008). "DNA topoisomerases: harnessing and constraining energy to govern chromosome topology." Q Rev Biophys 41(1): 41-101.
Sezonov, G., D. Joseleau-Petit and R. D'Ari (2007). "Escherichia coli physiology in Luria-Bertani broth." J Bacteriol 189(23): 8746-8749.
Silver, L. L. (2011). "Challenges of antibacterial discovery." Clin Microbiol Rev 24(1): 71-109.
Silver, L. L. (2016). "A Gestalt approach to Gram-negative entry." Bioorg Med Chem 24(24): 6379-6389.
Sugino, A., C. L. Peebles, K. N. Kreuzer and N. R. Cozzarelli (1977). "Mechanism of action of nalidixic acid: purification of Escherichia coli nalA gene product and its relationship to DNA gyrase and a novel nicking-closing enzyme." Proc Natl Acad Sci U S A 74(11): 4767-4771.
Sullivan, E. A., B. N. Kreiswirth, L. Palumbo, V. Kapur, J. M. Musser, A. Ebrahimzadeh and T. R. Frieden (1995). "Emergence of fluoroquinolone-resistant tuberculosis in New York City." Lancet 345(8958): 1148-1150.
Tenaillon, O., D. Skurnik, B. Picard and E. Denamur (2010). "The population genetics of commensal Escherichia coli." Nat Rev Microbiol 8(3): 207-217.
Theodore, A., K. Lewis and M. Vulic (2013). "Tolerance of Escherichia coli to fluoroquinolone antibiotics depends on specific components of the SOS response pathway." Genetics 195(4): 1265-1276.
Thulin, E., M. Sundqvist and D. I. Andersson (2015). "Amdinocillin (Mecillinam) resistance mutations in clinical isolates and laboratory-selected mutants of Escherichia coli." Antimicrob Agents Chemother 59(3): 1718-1727.
Tran, J. H. and G. A. Jacoby (2002). "Mechanism of plasmid-mediated quinolone resistance." Proc Natl Acad Sci U S A 99(8): 5638-5642.
Tran, J. H., G. A. Jacoby and D. C. Hooper (2005). "Interaction of the plasmid-encoded quinolone resistance protein Qnr with Escherichia coli DNA gyrase." Antimicrob Agents Chemother 49(1): 118-125.
Tran, J. H., G. A. Jacoby and D. C. Hooper (2005). "Interaction of the plasmid-encoded quinolone resistance protein QnrA with Escherichia coli topoisomerase IV." Antimicrob Agents Chemother 49(7): 3050-3052.
Ventola, C. L. (2015). "The antibiotic resistance crisis: part 1: causes and threats." P T 40(4): 277-283.
Vetting, M. W., C. H. Park, S. S. Hegde, G. A. Jacoby, D. C. Hooper and J. S. Blanchard (2008). "Mechanistic and structural analysis of aminoglycoside N-acetyltransferase AAC(6')-Ib and its bifunctional, fluoroquinolone-active AAC(6')-Ib-cr variant." Biochemistry 47(37): 9825-9835.
Vinella, D., R. D'Ari, A. Jaffe and P. Bouloc (1992). "Penicillin binding protein 2 is dispensable in Escherichia coli when ppGpp synthesis is induced." EMBO J 11(4): 1493-1501.
Vinue, L., M. A. Corcoran, D. C. Hooper and G. A. Jacoby (2015). "Mutations That Enhance the Ciprofloxacin Resistance of Escherichia coli with qnrA1." Antimicrob Agents Chemother 60(3): 1537-1545.
Wang, D., X. Huang, J. Chen, Y. Mou and Y. Qi (2017). "Characterization of a novel qepA3 variant in Enterobacter aerogenes." J Microbiol Immunol Infect 50(2): 254-257.

61
Wang, X., X. Zhao, M. Malik and K. Drlica (2010). "Contribution of reactive oxygen species to pathways of quinolone-mediated bacterial cell death." J Antimicrob Chemother 65(3): 520-524.
Webber, M. A. and L. J. Piddock (2001). "Absence of mutations in marRAB or soxRS in acrB-overexpressing fluoroquinolone-resistant clinical and veterinary isolates of Escherichia coli." Antimicrob Agents Chemother 45(5): 1550-1552.
Wentzell, L. M. and A. Maxwell (2000). "The complex of DNA gyrase and quinolone drugs on DNA forms a barrier to the T7 DNA polymerase replication complex." J Mol Biol 304(5): 779-791.
WHO, W. H. O. (2014). Antimicrobial resistance: global report on surveillance. WHO, W. H. O. (2017). Critially important antimicrobials for human medicine - 5th
Rev. Geneva: World Health Organization; 2017. Wielgoss, S., J. E. Barrick, O. Tenaillon, S. Cruveiller, B. Chane-Woon-Ming, C.
Medigue, R. E. Lenski and D. Schneider (2011). "Mutation Rate Inferred From Synonymous Substitutions in a Long-Term Evolution Experiment With Escherichia coli." G3 (Bethesda) 1(3): 183-186.
Williams, A. B. (2014). "Spontaneous mutation rates come into focus in Escherichia coli." DNA Repair (Amst) 24: 73-79.
Willmott, C. J. and A. Maxwell (1993). "A single point mutation in the DNA gyrase A protein greatly reduces binding of fluoroquinolones to the gyrase-DNA complex." Antimicrob Agents Chemother 37(1): 126-127.
Wise, R., J. M. Andrews and L. J. Edwards (1983). "In vitro activity of Bay 09867, a new quinoline derivative, compared with those of other antimicrobial agents." Antimicrob Agents Chemother 23(4): 559-564.
Wolfson, J. S. and D. C. Hooper (1985). "The fluoroquinolones: structures, mechanisms of action and resistance, and spectra of activity in vitro." Antimicrob Agents Chemother 28(4): 581-586.
Wright, G. D. (2007). "The antibiotic resistome: the nexus of chemical and genetic diversity." Nat Rev Microbiol 5(3): 175-186.
Wright, G. D. and H. Poinar (2012). "Antibiotic resistance is ancient: implications for drug discovery." Trends Microbiol 20(4): 157-159.
Xiong, X., E. H. Bromley, P. Oelschlaeger, D. N. Woolfson and J. Spencer (2011). "Structural insights into quinolone antibiotic resistance mediated by pentapeptide repeat proteins: conserved surface loops direct the activity of a Qnr protein from a gram-negative bacterium." Nucleic Acids Res 39(9): 3917-3927.
Yamane, K., J. Wachino, S. Suzuki, K. Kimura, N. Shibata, H. Kato, K. Shibayama, T. Konda and Y. Arakawa (2007). "New plasmid-mediated fluoroquinolone efflux pump, QepA, found in an Escherichia coli clinical isolate." Antimicrob Agents Chemother 51(9): 3354-3360.
Yoshida, H., M. Bogaki, M. Nakamura and S. Nakamura (1990). "Quinolone resistance-determining region in the DNA gyrase gyrA gene of Escherichia coli." Antimicrob Agents Chemother 34(6): 1271-1272.
Yoshida, H., M. Nakamura, M. Bogaki, H. Ito, T. Kojima, H. Hattori and S. Nakamura (1993). "Mechanism of action of quinolones against Escherichia coli DNA gyrase." Antimicrob Agents Chemother 37(4): 839-845.
Yu, E. W., J. R. Aires and H. Nikaido (2003). "AcrB multidrug efflux pump of Escherichia coli: composite substrate-binding cavity of exceptional flexibility generates its extremely wide substrate specificity." J Bacteriol 185(19): 5657-5664.
Zhao, X., Y. Hong and K. Drlica (2015). "Moving forward with reactive oxygen species involvement in antimicrobial lethality." J Antimicrob Chemother 70(3): 639-642.

Acta Universitatis UpsaliensisDigital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 1495
Editor: The Dean of the Faculty of Medicine
A doctoral dissertation from the Faculty of Medicine, UppsalaUniversity, is usually a summary of a number of papers. A fewcopies of the complete dissertation are kept at major Swedishresearch libraries, while the summary alone is distributedinternationally through the series Digital ComprehensiveSummaries of Uppsala Dissertations from the Faculty ofMedicine. (Prior to January, 2005, the series was publishedunder the title “Comprehensive Summaries of UppsalaDissertations from the Faculty of Medicine”.)
Distribution: publications.uu.seurn:nbn:se:uu:diva-361204
ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2018


















