
Evidence Dossier to support COPD formulary decision making ... · Prescribing and adverse event...
36
Prescribing and adverse event reporting information can be found on the final page of this document. Relvar and Ellipta Trademarks are owned by or licensed to the GSK Group of Companies. ©2017 GSK Group of Companies or its licensor. UK/FFT/0094/17(2) Date of preparation: September 2018 Evidence Dossier to support COPD formulary decision making and guideline development
Transcript of Evidence Dossier to support COPD formulary decision making ... · Prescribing and adverse event...

Prescribing and adverse event reporting information can be found on the final page of this document. Relvar and Ellipta Trademarks are owned by or licensed to the GSK Group of Companies. ©2017 GSK Group of Companies or its licensor.
UK/FFT/0094/17(2) Date of preparation: September 2018
Evidence Dossier to support COPD formulary decision making
and guideline development

UK/FFT/0094/17(2) Date of preparation: September 2018 2
Table of Contents
Clinical study information ..................................................................................................................................................... 4
Dosing information .................................................................................................................................................... 4
Licensing information ................................................................................................................................................ 4
Interpreting clinical trial results – statistical hierarchy ............................................................................................ 4
Vestbo et al., 2016 (Salford Lung Study - COPD)............................................................................................................ 5
Study design ............................................................................................................................................................... 5
Patient population ..................................................................................................................................................... 6
Endpoints ................................................................................................................................................................... 6
Results ........................................................................................................................................................................ 6
Safety .......................................................................................................................................................................... 8
Siler et al., 2015 (placebo-controlled study) .................................................................................................................. 9
Study design ............................................................................................................................................................... 9
Patient population ..................................................................................................................................................... 9
Endpoints ................................................................................................................................................................... 9
Results ...................................................................................................................................................................... 10
Safety ........................................................................................................................................................................ 12
Agusti et al., 2014 (active comparator study) .............................................................................................................. 14
Study design ............................................................................................................................................................. 14
Patient population ................................................................................................................................................... 14
Endpoints ................................................................................................................................................................. 15
Results ...................................................................................................................................................................... 16
Safety ........................................................................................................................................................................ 17
Boscia et al., 2012 (dose-ranging study)....................................................................................................................... 18
Study design ............................................................................................................................................................. 18
Patient population ................................................................................................................................................... 18
Endpoints ................................................................................................................................................................. 18
Results ...................................................................................................................................................................... 19
Safety ........................................................................................................................................................................ 20
Dransfield et al., 2013 (dose-ranging study) ................................................................................................................ 22
Study design ............................................................................................................................................................. 22
Patient population ................................................................................................................................................... 22
Endpoints ................................................................................................................................................................. 23
Results ...................................................................................................................................................................... 23
Safety ........................................................................................................................................................................ 24
Crim et al., 2017 (SUMMIT pre-specified analyses) ..................................................................................................... 26
Study design ............................................................................................................................................................. 26
Patient population ................................................................................................................................................... 26
Analyses .................................................................................................................................................................... 27
Results ...................................................................................................................................................................... 27
Van der Palen et al., 2016 (critical errors) .................................................................................................................... 30
Study design ............................................................................................................................................................. 30
Patient population ................................................................................................................................................... 30
Endpoints ................................................................................................................................................................. 30
Results ...................................................................................................................................................................... 31

UK/FFT/0094/17(2) Date of preparation: September 2018 3
References ........................................................................................................................................................................... 34
How to request additional information from GSK ....................................................................................................... 34
Incruse▼Ellipta (umeclidinium) Prescribing Information ................................................................................................. 35
Relvar Ellipta (fluticasone furoate/vilanterol) Prescribing Information ............................................................................36

4
Clinical study information
Dosing information
Each single inhalation of Relvar Ellipta (fluticasone furoate/vilanterol) provides a delivered dose (the dose leaving the
mouthpiece) of 92 mcg of fluticasone furoate and 22 mcg of vilanterol (as trifenatate). This corresponds to a pre-
dispensed dose of 100 mcg of fluticasone furoate and 25 mcg of vilanterol (as trifenatate).1 Please note that the licensed
dose of fluticasone furoate/vilanterol (FF/VI) is 92/22 mcg in COPD.1
Licensing information
Relvar Ellipta (fluticasone furoate/vilanterol 92/22 mcg) is indicated for the symptomatic treatment of adults with COPD
with a FEV1 <70% predicted normal (post-bronchodilator) with an exacerbation history despite regular bronchodilator
therapy.1 Relvar Ellipta 184/22 mcg is not indicated for patients with COPD. There is no additional benefit of the 184/22
mcg dose compared with the 92/22 mcg dose and there is a potential increased risk of pneumonia and systemic
corticosteroid-related adverse reactions.1
Incruse Ellipta (umeclidinium 55 mcg) efficacy and safety is also included in this Evidence Dossier. Incruse Ellipta is
indicated as a maintenance bronchodilator treatment to relieve symptoms in adult patients with COPD.2
Umeclidinium (UMEC) 113 mcg, UMEC/VI 113/22 mcg, VI 22 mcg, FF 92 mcg, FF/VI 44/22 mcg and FF/VI 184/22 mcg
were included in studies described in this Evidence Dossier, and are not licensed in the UK. The results have not been
included in the efficacy section but have been included in the safety section for transparency purposes. Safety studies
with un-licensed doses have also been included for transparency.
Interpreting clinical trial results – statistical hierarchy
When reviewing the outcomes of clinical trials it is important to understand the relevance of statistical hierarchy and how
that impacts the conclusions that can be derived from a study. This concept is explained below.
Statistical hierarchy sequentially tests the significance of a number of endpoints in a study programme in a predetermined
order. For each endpoint, a determination of significance can only be made if all prior endpoints were also significant.
Treatment comparisons for the primary endpoints are required to be statistically significant in order to infer significance
for the secondary endpoints. Therefore, if the trial does not meet its primary endpoint, the secondary endpoints cannot
be statistically analysed; those results are described as ‘descriptive only’.
UK/FFT/0094/17(2) Date of preparation: September 2018

5
Vestbo et al., 2016 (Salford Lung Study - COPD)
Effectiveness of fluticasone furoate-vilanterol for COPD in clinical practice3,4
Relvar Ellipta 92/22 mcg significantly reduced moderate/severe COPD exacerbations vs. twice daily ICS/LABA, of which a high proportion were receiving fluticasone propionate/salmeterol
Relvar Ellipta (FF/VI 92/22 mcg) is indicated for the symptomatic treatment of adults with COPD with a FEV1 <70%
predicted normal (post-bronchodilator) with an exacerbation history despite regular bronchodilator therapy.1
Study design
The Salford Lung Study (SLS-COPD) was an open-label, randomised, controlled study comparing the effectiveness and
safety of FF/VI 92/22 mcg OD with usual care (GP or investigator’s free choice of COPD maintenance treatment) in a large
population intended to reflect patients with COPD seen in everyday practice.
* ICS monotherapy not indicated in COPD** Patient allowed to remain on LAMA in addition to their randomised treatment if already receiving LAMA therapy at randomisation COPD = chronic obstructive pulmonary disease; FF/VI = fluticasone furoate/vilanterol; ICS = inhaled corticosteroid; LABA = long-acting β2-agonist; LAMA = long-acting muscarinic antagonist; OD = once daily; R = randomisation
• Usual care was determined by the GP:
o Non-ICS containing treatment: n=391 (14%)
o ICS, ICS/LABA or ICS + LAMA: n=958 (34%)
o Triple therapy in multiple inhalers: n=1,450 (52%)
• Randomisation stratified by recent exacerbation status and existing COPD maintenance therapy at baseline
• Patients randomly assigned to FF/VI 92/22 mcg OD who were previously treated with two long-acting
bronchodilators and an ICS were allowed to continue taking a LAMA in addition to FF/VI 92/22 mcg
• If at months 3, 6 and 9, patients had no contact with their general practice within the previous 8 weeks, they were
contacted for assessments of adverse events or adverse drug reactions
• Treatment adjustment was at the GP’s discretion; patients could switch from FF/VI 92/22 mcg to usual care, but
patients in the usual care group could not switch to the FF/VI 92/22 mcg group
• Patients in each treatment group were trained in the correct inhaler techniques and dosing
• No follow-up assessments for treatment adherence were conducted in either treatment group
UK/FFT/0094/17(2) Date of preparation: September 2018

6
Patient population
• ≥40 years with a GP diagnosis of COPD
• ≥1 moderate/severe COPD exacerbation in the previous 3 years
• Regular maintenance inhaler therapy (ICS and/or LAMA and/or LABA; note ICS monotherapy is not indicated in
COPD)
• There were no restrictions regarding smoking history or spirometric values; the very few exclusion criteria were
intended to represent the real-world patient population, irrespective of co-morbidities
Endpoints
Primary endpoint
• Mean annual rate of moderate or severe exacerbations, defined as any worsening of respiratory symptoms that led
to treatment with antibiotic agents or systemic glucocorticoids (or both), to hospital admission, or to scheduled or
unscheduled hospital visits (assessed in the modified ITT population [patients who had undergone randomisation,
received a prescription of trial medication and had ≥1 exacerbation in the preceding year])
Secondary endpoints (assessed in the ITT population [all patients who underwent randomisation and received a
prescription of trial medication])
• Rate of first exacerbations, as assessed by time-to-event analysis
• Annual rates of primary and secondary healthcare contacts
• COPD Assessment Test (CAT) score
• European Quality of Life-5 Dimensions (EQ-5D) questionnaire results
Safety and tolerability
• Safety and tolerability were assessed by monitoring serious adverse events of pneumonia, the frequency and type
of other serious adverse events, and adverse drug reactions
Demographics and baseline characteristics
Characteristic
Entire trial population Entire trial population (n=2,799)
Modified ITT population (n=2,269)
FF/VI 92/22 mcg (n=1,396)
Usual care (n=1,403)
Mean age, years ± SD 67 ± 10 67 ± 10 67 ± 10 67 ± 10
Female gender, n (%) 698 (50) 671 (48) 1,369 (49) 1,122 (49)
Current smoking, n (%) 623 (45) 666 (47) 1289 (46) 1046 (46)
Coexisting condition, n (%) 1069 (77) 1076 (77) 2145 (77) 1758 (77)
Number of exacerbations during 12 mo before randomisation, mean ± SD
1.98 ± 1.90 2.04 ± 2.08 2.01 ± 1.99 2.48 ± 1.93
Postbronchodilator FEV1 (L), mean ± SD
1.62 ± 0.64 1.62 ± 0.65 1.62 ± 0.64 1.59 ± 0.64
Values are mean ± SD unless specified otherwise FEV1 = forced expiratory volume in 1 second; FF/VI = fluticasone furoate/vilanterol; mo = months
Results
Primary endpoint
• Mean annual rate of moderate or severe exacerbations
o The rate of moderate or severe exacerbations in the modified ITT population was 1.74 exacerbations per
year in the FF/VI 92/22 mcg group compared with 1.90 per year in the usual care group, indicating an
8.4% (95% CI 1.1, 15.2) lower rate in the FF/VI 92/22 mcg group (p=0.02)
UK/FFT/0094/17(2) Date of preparation: September 2018

7
o The rate of moderate or severe exacerbations in the ITT population was 1.50 exacerbations per year in the
FF/VI 92/22 mcg group compared with 1.64 per year in the usual care group, indicating an 8.4% (95% CI
1.4, 14.9) lower rate in the FF/VI 92/22 mcg group (p=0.02)
o In a pre-specified analysis of patients receiving ICS/LABA in the modified ITT population, the rate of
moderate or severe exacerbations was 1.87 exacerbations per year in the FF/VI 92/22 mcg group
compared with 2.03 exacerbations per year in the usual care group, indicating an 8.0% (95% CI 0.11, 15.4)
lower rate in the FF/VI group (p=0.047)
▪ FF/VI was shown to help prevent one additional moderate or severe COPD exacerbation for every
seven patients treated over 12 months compared with twice daily ICS/LABA, a high proportion of
which were on FP/SAL (NNT=6.25)
Figure 1: Treatment effect on moderate or severe exacerbations
Adapted from Vestbo et al, 2016 CI = confidence interval; FF/VI = fluticasone furoate; ITT = intention-to-treat
Secondary endpoints
• Rate of first exacerbations
o There was no significant difference in the rate of first moderate or severe exacerbation in the time-to-
event analysis in the ITT population (probability of event 68.9% [n=947] vs 70.4% [n=997]; HR [FF/VI 92/22
mcg vs usual care] 0.93; 95% CI 0.85, 1.02)
o There was no significant difference in the rate of first severe exacerbation in the time-to-event analysis
(probability of event 8.9% [n=122] vs 7.0% [n=97]; HR [FF/VI 92/22 mcg vs usual care] 1.27; 95% CI 0.98,
1.66; p=0.08)
o There was no significant difference in the rate of severe exacerbations between the FF/VI 92/22 mcg
group (0.09 exacerbations per year) and the usual care group (0.08 exacerbations per year); the rate was
9.7% higher in the FF/VI 92/22 mcg group (95% CI –16.9, 44.7; p=0.52)
• Annual rates of primary and secondary healthcare contacts
o The annual rate of COPD-related contact was 1.7% lower (95% CI –5.1, 8.0) in the FF/VI 92/22 mcg group
(2.42) than in the usual care group (2.46); there was no significant difference between groups
o The annual rate of all primary care contacts was slightly higher (12.3%; 95% CI 5.4, 19.6) in the FF/VI 92/22
mcg group (21.2) than in the usual care group (18.9); there was no significant difference between groups
o The LS mean annual rate of on-treatment COPD-related secondary care contacts was 1.57 in the FF/VI
92/22 mcg group and 1.48 in the usual care group; there was no significant difference in the rates of
secondary healthcare contacts
• CAT score
o A total of 45% of patients in the FF/VI 92/22 mcg group (n=1,317) had a decrease of ≥2 points (indicating
an improvement in COPD-related health status) in their CAT score, compared with 36% in the usual care
group (n=1,325; OR 1.51; 95% CI 1.28, 1.77; p<0.001)
• EQ-5D questionnaire results
o There was no significant between-group difference in the change from baseline in the EQ-5D score
UK/FFT/0094/17(2) Date of preparation: September 2018

8
Safety
• The incidence of SAEs was similar in the FF/VI 92/22 mcg group and the usual care group, with events occurring in
404 patients (29%) and 383 patients (27%), respectively
• There was no notable difference in occurrence of AEs of special interest (AESI) between treatment groups
• There was no notable difference in the occurrence of pneumonia: a total of 94 patients (7%) in the FF/VI 92/22 mcg
group had ≥1 pneumonia SAE compared with 83 (6%) in the usual care group (incidence ratio [IR] 1.1; 95% CI
0.9, 1.5)
o For the incidence of SAEs of pneumonia, the non-inferiority margin for the ratio of the proportions of
patients with ≥1 SAE of pneumonia on FF/VI versus usual care was set at 2
o A total of 13 patients (1%) in each treatment group had an event of pneumonia (AESI) with a fatal
outcome
• One patient in each group died from a SAE that was recorded as being related to the study medication (pneumonia
in 1 patient in the usual care group, and pulmonary embolism and deep-vein thrombosis in 1 patient in the FF/VI
92/22 mcg group)
Table 1. Summary of adverse events
FF/VI 92/22 mcg (n=1,396)
Usual care (n=1,403)
On-treatment AEs, n (%) 211 (15) 97 (7)
Most common AE type (≥3% in any group), n (%) Infections and infestations Respiratory, thoracic and mediastinal disorders
74 (5) 78 (6)
51 (4) 20 (1)
AEs leading to permanent discontinuation/withdrawal, n (%) 59 (4) 33 (2)
On-treatment SAEs, n (%) 404 (29) 383 (27)
AEs of special interest, n (%) Cardiovascular event
Cardiac arrhythmia Cardiac failure Cardiac ischemia Hypertension Stroke
Pneumonia‡ LRTI excluding pneumonia Decreased bone mineral density and associated fracture Effects on glucose level Hypersensitivity Effects on potassium level Glucocorticoid-associated eye disease Local effects from glucocorticoids
‡Incidence ratio 1.1 (95% CI 0.9, 1.5); non-inferiority margin was set at 2
108 (8) 52 (4) 28 (2) 34 (2)
0 21 (2) 94 (7) 64 (5) 45 (3)
23 (20) 10 (1) 2 (<1) 2 (<1)
0
107 (8) 54 (4) 28 (2) 33 (2) 1 (<1) 25 (2) 83 (6) 58 (4) 45 (3) 16 (1) 10 (1) 2 (<1) 2 (<1) 1 (<1)
All-cause mortality, n (%)* 45 (<1) 30 (<1)
Drug-related death from an SAE,** n (%) 1 (<1) 1 (<1)
* The most frequent (≥2 subjects within either treatment arm) on-treatment fatal SAEs were infections and infestations (FF/VI: 15[1%]; usual care: 18 [1%]), cardiac disorders (FF/VI: 12 [<1%]; usual care: 7 [<1%]), respiratory, thoracic and mediastinal disorders (FF/VI: 9 [<1%]; usual care: 8 [<1%]), neoplasms benign, malignant and unspecified (FF/VI: 13 [<1%]; usual care: 3 [<1%]), nervous system disorders (FF/VI: 4 [<1%]; usual care: 3 [<1%]) and renal and urinary disorders (FF/VI: 3 [<1%]; usual care: 2 [<1%]) ** Two subjects experienced fatal SAEs that were considered related to the study treatment in the opinion of the investigator (pneumonia in one patient in the usual care group, and pulmonary embolism and deep-vein thrombosis in one patient in the FF/VI group) AE = adverse event; FF/VI = fluticasone furoate/vilanterol; LRTI = lower respiratory tract infection; SAE = serious adverse event
UK/FFT/0094/17(2) Date of preparation: September 2018

9
Siler et al., 2015 (placebo-controlled study)
Efficacy and safety of umeclidinium added to fluticasone furoate/vilanterol in chronic obstructive pulmonary disease:
Results of two randomized studies5
Incruse in combination with FF/VI provides significant improvements in lung function compared with placebo + FF/VI in patients with COPD, with a similar safety profile
Study design
Two replicate Phase III, randomised, multicentre, double-blind, placebo-controlled, parallel-group, 12-week lung
function studies comparing UMEC 55 mcg OD, UMEC 113 mcg OD or placebo added to open-label FF/VI 92/22 mcg OD
in patients with COPD.
UMEC 113 mcg is not licensed for the treatment of COPD in the UK and therefore the results have not been included in the efficacy section but have been included in the safety section FEV1 = forced expiratory volume in 1 second; FF/VI = fluticasone furoate/vilanterol; FVC = forced vital capacity; MRC = Medical Research Council; OD = once daily; R = randomisation; UMEC = umeclidinium
• Inhaled corticosteroids were permitted to Visit 1 (screening visit)
• LAMA use required a 7-day exclusion period and the use of ICS/LABA combination therapies required a 48-hour
exclusion period
Patient population
• ≥40 years with clinical history of COPD
• Smoking history ≥10 pack-years
• Pre- and post-salbutamol FEV1/FVC ratio <0.70
• Predicted FEV1 ≤70%
• MRC dyspnoea score ≥2
Endpoints
Primary endpoint
• Trough FEV1 at Day 85, defined as the mean of FEV1 values obtained 23h and 24h after dosing on Day 84
Secondary endpoints and other endpoints
• Weighted mean (wm) FEV1 over 0–6h post-dose on Day 84
• Proportion of patients achieving an increase of ≥0.1 L above baseline in trough FEV1
• Proportion of patients achieving an increase in FEV1 of ≥12% and ≥0.2 L above baseline at any time during 0–6h
post-dose on Day 1
UK/FFT/0094/17(2) Date of preparation: September 2018

10
• Serial FEV1 assessments
• Peak FEV1 at Days 1, 28 and 84
• Time to onset of treatment response
• Serial and trough FVC at each timepoint
• Rescue medication use
• COPD Assessment Test (CAT)
• St George’s Respiratory Questionnaire (SGRQ)
Safety and tolerability
• AEs, vital signs, and COPD exacerbations
Demographics and baseline characteristics
Study 1 Placebo + FF/VI 92/22 mcg
(n=206) UMEC 55 mcg + FF/VI 92/22 mcg
(n=206)
Mean age, years ± SD 64.7 ± 7.9 64.9 ± 8.7
Female gender, n (%) 65 (32) 67 (33)
Smoking pack-year history, years ± SD 50.6 ± 24.8 50.1 ± 24.9
Post-salbutamol % predicted FEV1, mean ± SD 45.9 ± 13.0 44.2 ± 13.4
Post-salbutamol FEV1/FVC, mean ± SD 48.0 ± 10.8 47.8 ± 10.2
Study 2 Placebo + FF/VI 92/22 mcg
(n=206) UMEC 55 mcg + FF/VI 92/22 mcg
(n=206)
Mean age, years ± SD 62.6 ± 9.0 62.6 ± 8.1
Female gender, n (%) 81 (39) 71 (34)
Smoking pack-year history, years ± SD 46.2 ± 25.7 46.8 ± 27.0
Post-salbutamol % predicted FEV1, mean ± SD 47.4 ± 12.5 46.3 ± 12.2
Post-salbutamol FEV1/FVC, mean ± SD 49.0 ± 10.2 48.1 ± 10.3
FEV1 = forced expiratory volume in 1 second; FF/VI = fluticasone furoate/vilanterol; FVC = forced vital capacity; SD = standard deviation; UMEC = umeclidinium
Results
Primary endpoint
• Pre-dose (trough) FEV1 on Day 85, defined as the mean of FEV1 values obtained 23h and 24h after dosing on Day 84
o Trough FEV1 at Day 169 (LSM) was significantly improved from baseline with UMEC 55 mcg + FF/VI 92/22
mcg (122–124 mL) compared with placebo + FF/VI 92/22 mcg (p≤0.001)
o Statistically significant improvements with UMEC 55 mcg + FF/VI 92/22 mcg versus placebo + FF/VI 92/22
mcg were also observed for trough FEV1 at Days 2, 28, 56 and 84 (p<0.001 at each timepoint in both
studies)
UK/FFT/0094/17(2) Date of preparation: September 2018

11
Figure 2: Change from baseline in trough FEV1 (ITT population) in Study 1 (a) and Study 2 (b)
Adapted from Siler et al., 2015 CI = confidence interval; FEV1 = forced expiratory volume in 1 second; FF/VI = fluticasone furoate/vilanterol; ITT = intention-to-treat; LS = least squares; UMEC = umeclidinium
Secondary and other endpoints
• wmFEV1 over 0–6h post-dose on Day 84
o wmFEV1 at Day 84 (LSM) was significantly improved from baseline with UMEC 55 mcg + FF/VI 92/22 mcg
(147–153 mL) compared with placebo + FF/VI 92/22 mcg (p<0.001)
• Proportion of patients achieving an increase of ≥0.1 L above baseline in trough FEV1
o The proportion of patients achieving an increase of ≥0.1 L above baseline in trough FEV1 was greater with
UMEC 55 mcg + FF/VI 92/22 mcg (43–46%) compared with placebo + FF/VI 92/22 mcg (13–14%)
o The odds of achieving ≥0.1 L increase in FEV1 above baseline was higher for UMEC 55 mcg + FF/VI 92/22
mcg (OR 4.8–5.6) compared with placebo + FF/VI 92/22 mcg (p<0.001)
• Proportion of patients achieving an increase in FEV1 of ≥12% and ≥0.2 L above baseline at any time during 0–6h
post-dose on Day 1
o The proportion of patients achieving an increase in FEV1 of ≥12% and ≥0.2 L above baseline at any time
during 0–6h post-dose on Day 1 was greater with UMEC 55 mcg + FF/VI 92/22 mcg (45–46%) compared
with placebo + FF/VI 92/22 mcg (13–14%)
o The odds of achieving an increase in FEV1 of ≥12% and ≥0.2 L above baseline at any time during 0–6h post-
dose on Day 1 was higher for UMEC 55 mcg + FF/VI 92/22 mcg (OR 5.0–6.1) compared with placebo +
FF/VI 92/22 mcg, all p<0.001
UK/FFT/0094/17(2) Date of preparation: September 2018

12
• Serial FEV1 assessments
o Serial FEV1 on Day 1 showed a rapid onset of FEV1 improvements (statistically significantly greater after
~15 min) with UMEC 55 mcg + FF/VI 92/22 mcg, which was maintained at 24h and through to Day 84 in
both studies (all p<0.001)
• Peak FEV1
o Peak FEV1 increases from baseline (LSM) at Day 84 were significantly greater with UMEC 55 mcg + FF/VI
92/22 mcg (154–156 mL) compared with placebo + FF/VI 92/22 mcg (p<0.001)
• Trough FVC
o Improvements in trough FVC change from baseline (LSM) at Day 85 were significantly greater with UMEC
55 mcg + FF/VI 92/22 mcg (186–199 mL, p<0.001) compared with placebo + FF/VI 92/22 mcg
• Rescue medication use
o Over the 12-week study period, UMEC 55 mcg + FF/VI 92/22 mcg resulted in less rescue salbutamol use
compared with placebo (UMEC 55 mcg + FF/VI 92/22 mcg: –0.4 puffs/day, p≤0.001; –0.3 puffs/day,
p≤0.01 in Study 1 and Study 2, respectively, compared with placebo + FF/VI 92/22 mcg)
• CAT
o Numerical improvements from baseline in CAT scores (where a negative number denotes an
improvement) were observed with UMEC 55 mcg + FF/VI 92/22 mcg (–0.6 to –1.1) at Day 84. In both
studies, the placebo + FF/VI 92/22 mcg treatment group showed a numerical deterioration (0.1–0.3) at
Day 84.
• SGRQ
o No significant differences in SGRQ scores (where a negative number denotes an improvement) were
observed between the treatment arms at Days 28 and 84 in Study 1. In Study 2, patients treated with
UMEC 55 mcg + FF/VI 92/22 mcg showed statistically significant improvements in SGRQ score versus
placebo + FF/VI 92/22 mcg at Day 84 (–2.16; p<0.05) and also higher odds of being an SGRQ responder
(defined by a reduction from baseline of 4 units in SGRQ score) versus being an SGRQ non-responder
(OR: 2.01, p≤0.01)
Safety
• On-treatment AEs were similar across treatment groups (30–39%)
• Treatment-related AEs occurred in 3–7%, 3–7%, and 3–12% in the UMEC 55 mcg + FF/VI 92/22 mcg, UMEC 113
mcg + FF/VI 92/22 mcg, and placebo + FF/VI 92/22 mcg groups, respectively
• The most frequent on-treatment AE was nasopharyngitis (3–11%)
• Six deaths were reported during both studies; however, none was considered related to the study treatment. Five
deaths were in the placebo + FF/VI groups (three due to myocardial infarction; one due to cardio-respiratory arrest;
one due to pneumonia) and one death was in the UMEC 55 mcg FF/VI group (Study 2; due to gastric ulcer
hemorrhage, atrial fibrillation, cardiogenic shock and myocardial infarction)
• COPD exacerbations were infrequent (≤8% in any group) in both studies
• No clinically relevant treatment-related changes in vital signs were reported across any treatment group in either
study
UK/FFT/0094/17(2) Date of preparation: September 2018

14
Agusti et al., 2014 (active comparator study)
A comparison of the efficacy and safety of once-daily fluticasone furoate/vilanterol with twice-daily fluticasone
propionate/salmeterol in moderate to very severe COPD6
Relvar Ellipta (FF/VI 92/22 mcg OD) showed clinically meaningful improvement in lung function and demonstrated a comparable safety profile to FP/SAL 460/47 mcg BD*
Study design
A 12-week, randomised, multicentre, double-blind, double-dummy, parallel-group study comparing FF/VI 92/22 mcg
OD against FP/SAL 460/47 mcg BD in patients with moderate to very severe COPD.
BD = twice daily; FEV1 = forced expiratory volume in 1 second; FF/VI = fluticasone furoate/vilanterol; FP/SAL = fluticasone propionate/salmeterol; FVC = forced vital capacity; OD = once daily; R = randomisation
• Following screening, patients entered a 2-week placebo run-in period, after which they were randomised 1:1 to
FF/VI 92/22 mcg OD via the Ellipta inhaler or FP/SAL 460/47 mcg BD via the Accuhaler
o Patients receiving FF/VI also took placebo via the Accuhaler once in the morning and once in the evening,
and patient receiving FP/SAL also took placebo via the Ellipta inhaler once in the morning
• Salbutamol was supplied to patients for symptomatic relief during the study
• Randomisation was stratified by patient’s reversibility, defined as a change in FEV1 of ≥12% and ≥200 mL 10–15
minutes after four inhalations of salbutamol
Patient population
• ≥40 years
• Smoking history ≥10 pack-years
• Post-salbutamol FEV1/FVC ratio ≤0.70 and FEV1 ≤70% of predicted
• Patients must have experienced ≥1 moderate COPD exacerbations (requiring treatment with oral
corticosteroid/antibiotic) or severe exacerbations (leading to hospitalisation) within the past 3 years
• Patients with a current diagnosis of asthma, serious underlying disease or infection, hospitalisation due to COPD
within 12 weeks of screening or acute worsening of COPD within 6 weeks of screening were not included
* Equivalent to FP/SAL 500/50 mcg pre-dispensed dose
UK/FFT/0094/17(2) Date of preparation: September 2018

15
Endpoints
Primary endpoint (ITT population)
• Change from baseline in wmFEV1 on Day 84
o The predetermined treatment difference for superiority of one treatment compared with the other was
60 mL
Secondary and other endpoints (ITT population)
• Change from baseline in trough FEV1 on Day 85
• Time to 100 mL increase from baseline from 0–4h on Day 1
• SGRQ for COPD score on Day 84
• Rescue-free 24h periods on Day 1 and Day 84
• Post-hoc analysis of the difference in lung function 0–4h, 0–12h and 12–24h post-dose on Day 84
Safety and tolerability (ITT population)
• Incidence of AEs, severe AEs and adverse events of special interest (AESIs)
Demographics and baseline characteristics
Characteristic FF/VI 92/22 mcg
(n=266) FP/SAL 460/47 mcg
(n=262)
Mean age, years ± SD 63.0 ± 8.1 62.9 ± 9.1
Female gender, n (%) 54 (20.3) 41 (15.6)
Post-bronchodilator FEV1/FVC, mean ± SD 0.29 ± 0.10 0.48 ± 0.10
Post-bronchodilator FEV1 % predicted, mean ± SD 47.94 ± 11.50 47.64 ± 11.91
FEV1 reversibility %, mean ± SD 11.7 ± 11.9 11.9 ± 11.8
Comorbid conditions, n (%)
Vascular disorders
Metabolism and nutrition disorders
Cardiac disorders
112 (42)
54 (20)
37 (14)
125 (48)
63 (24)
25 (10)
COPD exacerbations, n (%)
Requiring antibiotics/corticosteroids in prior 3 yearsa
0
1
2
3
≥4
Requiring hospitalisation in prior 3 yearsa
0
1
2
≥3
29 (11)
125 (47)
52 (20)
32 (12)
28 (11)
200 (75)
53 (20)
12 (5)
1 (<1)
29 (11)
126 (48)
52 (20)
36 914)
19 (7)
190 (73)
60 (23)
11 (4)
1 (<1) a percentages do not sum to 100 due to rounding COPD = chronic obstructive pulmonary disease; FEV1 = forced expiratory volume in 1 second; FF/VI = fluticasone furoate/vilanterol; FP/SAL = fluticasone propionate/salmeterol; FVC = forced vital capacity; SD = standard deviation
UK/FFT/0094/17(2) Date of preparation: September 2018

13
Table 2: Summary of adverse events
Study 1 Placebo + FF/VI
92/22 mcg (n=206)
UMEC 55 mcg + FF/VI 92/22 mcg
(n=206)
UMEC 113 mcg + FF/VI 92/22 mcg
(n=207)
On-treatment AE, n (%) 72 (35) 75 (36) 80 (39)
On-treatment SAE, n (%) 6 (3) 2 (<1) 8 (4)
On-treatment fatal AEs, n (%) None was considered related to the study treatment*
1 (<1) 0 0
Treatment-related AEs, n (%) 15 (7) 15 (7) 24 (12)
AEs leading to permanent discontinuation of study drug or withdrawal from study, n (%)
5 (2) 3 (1) 6 (3)
Most common AE type, n (%) Headache Nasopharyngitis
6 (3) 7 (3)
7 (3) 9 (4)
9 (4) 10 (5)
AEs of special interest, n (%) Cardiovascular, any event 6 (3) 5 (2) 3 (1)
On-treatment COPD exacerbation,** n (%) 7 (3) 6 (3) 14 (7)
Study 2 Placebo + FF/VI
92/22 mcg (n=206)
UMEC 55 mcg + FF/VI 92/22 mcg
(n=206)
UMEC 113 mcg + FF/VI 92/22 mcg
(n=207)
On-treatment AE, n (%) 81 (39) 67 (33) 62 (30)
On-treatment SAE, n (%) 11 (5) 8 (4) 3 (1)
On-treatment fatal AEs, n (%) None was considered related to the study treatment*
4 (2) 1 (<1) 0
Treatment-related AEs, n (%) 7 (3) 6 (3) 7 (3)
AEs leading to permanent discontinuation of study drug or withdrawal from study, n (%)
9 (4) 7 (3) 2 (<1)
Most common AE type, n (%) Headache Nasopharyngitis
5 (2) 22 (11)
11 (5) 8 (4)
4 (2) 19 (9)
AEs of special interest, n (%) Cardiovascular, any event 6 (3) 2 (<1) 3 (1)
On-treatment COPD exacerbation,** n (%) 17 (8) 6 (3) 4 (2)
* Six deaths were reported during both studies, none of which were considered related to the study treatment. Five deaths were inthe placebo + FF/VI groups (three due to myocardial infarction; one due to cardio-respiratory arrest; one due to pneumonia) and one death was in the UMEC 55 mcg FF/VI group (Study 2; due to gastric ulcer haemorrhage, atrial fibrillation, cardiogenic shock and myocardial infarction) ** Incruse Ellipta is indicated as a ‘maintenance bronchodilator to relieve symptoms in adult patients with chronic obstructive pulmonary disease (COPD)’ AE = adverse event; BD = twice daily; FEV1 = forced expiratory volume in 1 second; FF = fluticasone furoate; UMEC = umeclidinium; VI = vilanterol
UK/FFT/0094/17(2) Date of preparation: September 2018

16
Results
Primary endpoint
• Change from baseline in wmFEV1 on Day 84
o An improvement from baseline in 0–24h wmFEV1 (± SD) was observed with FF/VI 92/22 mcg (130 ± 222
mL) and FP/SAL 460/47 mcg (108 ± 221 mL)
o The treatment difference of 22 mL (95% CI –18, 63) did not reach statistical significance (p=0.282)
Secondary endpoints can only be considered descriptive, due to the statistical hierarchy
Secondary and other endpoints
• Change from baseline in trough FEV1 on Day 85
o FF/VI 92/22 mcg: 111 mL vs FP/SAL 460/47 mcg: 88 mL (mean treatment difference 23 mL;
95% CI –20, 66)
• Time to 100 mL increase from baseline from 0–4h on Day 1
o FF/VI 92/22 mcg: 16 minutes vs FP/SAL 460/47 mcg: 28 minutes
• SGRQ for COPD score on Day 84
o At Day 84, the minimum clinically important difference was achieved in the FF/VI 92/22 mcg group (mean
± SD: –4.3 ± 11.8) but not in the FP/SAL group (–3.0 ± 11.8)
o Treatment difference: –1.3 (95% CI –3.5, 0.8)
• Rescue-free 24h periods on Day 1 and Day 84
o Proportion of rescue-free periods was similar between treatment groups
▪ First week of study: FF/VI 92/22 mcg, 61.6% vs FP/SAL 460/47 mcg, 58.5%
▪ Entire study duration: FF/VI 92/22 mcg, 62.5% vs FP/SAL 460/47 mcg, 59.8%
• Post-hoc analysis of the difference in lung function 0–4h, 0–12h and 12–24h post-dose on Day 84
o There was a difference from 0–4h and 0–12h (daytime) but not from 12–24h post-dose
▪ wm (0–4h) FEV1: FF/VI 92/22 mcg, 205 ± 226 vs FP/SAL 460/47 mcg, 162 ± 227
▪ wm (0–12h) FEV1: FF/VI 92/22 mcg, 175 ± 225 vs FP/SAL 460/47 mcg, 128 ± 225
▪ wm (12–24h) FEV1: FF/VI 92/22 mcg, 88 ± 229 vs FP/SAL 460/47 mcg, 87 ± 229
Figure 3: Change from baseline in FEV1 on Day 84 (ITT population)
Adapted from Agusti et al., 2014 FEV1 = forced expiratory volume in 1 second; FF/VI = fluticasone furoate/vilanterol; FP/SAL = fluticasone propionate/salmeterol
UK/FFT/0094/17(2) Date of preparation: September 2018
17
Safety
• On-treatment AEs and drug-related AEs were similar between the treatment groups
• The most frequent on-treatment AEs were headache (7–8%) and nasopharyngitis (3–5%)
• The occurrence of AEs of special interest was generally low and similar across treatment groups, although more
patients in the FF/VI 92/22 mcg group experienced cardiovascular AEs (9 vs 1) and more patients in the FP/SAL
460/47 mcg group experienced local steroid effects (10 vs 3)
• COPD exacerbations occurred in 6 (2%) patients in the FF/VI 92/22 mcg group and 7 (3%) patients in the FP/SAL
460/47 mcg group
• Three patients experienced four pneumonia events during the study (one patient in the FF/VI 92/22 mcg group and
two patients in the FP/SAL 460/47 mcg group)
Table 3. Summary of adverse events
FF/VI 92/22 mcg (n=266) FP/SAL 460/47 mcg (n=262)
On-treatment AEs, n (%) 73 (27) 68 (26)
On-treatment SAEs, n (%) 6 (2) 3 (1)
AEs leading to permanent discontinuation or treatment withdrawal, n (%)
6 (2) 3 (1)
On-treatment AEs occurring in ≥2% of patients in any treatment group, n (%)
Headache
Nasopharyngitis
Back pain
Cough
Oral candidiasis
20 (8)
8 (3)
10 (4)
3 (1)
2 (<1)
18 (7)
12 (5)
3 (1)
7 (3)
4 (2)
On-treatment fatal AEs,a n (%) 0 0
Drug-related AEs,b n (%) 4 (2) 9 (3)
AEs of special interest, n (%)
Cardiovascular effects
Local steroid effects
Pneumonia
9 (3)c
3 (1)
1 (<1)
1 (<1)
10 (4)d
2 (<1) a One fatal event occurred during the post-treatment follow-up period of FF/VI treatment (congestive heart failure) that was not considered to be treatment related; b as determined by the investigator; c one patient had angina pectoris and coronary artery disease; d one patient had dysphonia and oral candidiasis AE = adverse event; FF/VI = fluticasone furoate/vilanterol; FP/SAL = fluticasone propionate/salmeterol; SAE = serious adverse event
UK/FFT/0094/17(2) Date of preparation: September 2018

18
Boscia et al., 2012 (dose-ranging study)
Effect of once-daily fluticasone furoate/vilanterol on 24-hour pulmonary function in patients with chronic obstructive
pulmonary disease: a randomized, three-way, incomplete block, crossover study7
Relvar resulted in significant improvements in lung function, with a 24 hr duration of action vs placebo, in patients with COPD
Study design
A Phase III, randomised, multicentre, double-blind, placebo-controlled, 3-way, incomplete block, crossover study of
three dose combinations of FF/VI (44/22 mcg OD, 92/22 mcg OD or 184/22 mcg OD) in patients with COPD.
FF/VI 44/22 mcg and FF/VI 184/22 mcg are not licensed in the UK and therefore no results have been included in the efficacy section but have been included in the safety section COPD = chronic obstructive pulmonary disease; FEV1 = forced expiratory volume in 1 second; FF/VI = fluticasone furoate/vilanterol; FVC = forced vital capacity; mMRC = modified Medical Research Council; OD = once daily; R = randomisation
• Patients who completed the 2-week placebo run-in period were randomised to 1 of 18 three-course sequences of
placebo and 2 of 3 dose combinations of FF/VI (44/22 mcg, 92/22 mcg or 184/22 mcg)
• Treatment periods were separated by 2-week, single-blind, placebo wash-out periods, and a safety follow-up visit
was conducted 7 days after the last treatment day
Patient population
• ≥40 years with documented clinical history of COPD
• Post-bronchodilator FEV1 of ≤70% predicted and a FEV1/FVC ratio of ≤0.70
• Current smoker or smoking history ≥10 pack-years
• Modified Medical Research Council (mMRC) dyspnoea score ≥2
Endpoints
Primary endpoint (ITT population)
• 0–24h wmFEV1 (AUC) at the end of each 28-day treatment period (period Days 28–29)
Secondary and other endpoints (ITT population)
• 0–25h serial FEV1 at period Days 28–29
• Change from treatment period baseline in trough FEV1 (period Day 29)
• Change from baseline in 0–4h peak FEV1 (period Day 28)
• Pharmacokinetic parameters, including Cmax, Tmax and AUC, over Days 28 and 29 (PK population)
UK/FFT/0094/17(2) Date of preparation: September 2018

19
Safety and tolerability (ITT population)
• Incidence of AEs, COPD exacerbations and pneumonias
• Other safety evaluations included assessment of serum glucose and potassium levels, oropharyngeal examinations,
change from baseline in vital signs, QTc, and serum cortisol levels at Days 28–29
Demographics and baseline characteristics
Characteristic Total (N=54)
Mean age, years ± SD 57.9 ± 9.2
Female gender, n (%) 29 (54)
Smoking status at screening, n (%)
Current smoker
Former smoker
45 (83)
9 (17)
Duration of COPD, n (%)
<1 year
≥1 year to <5 years
≥5 years to <10 years
≥10 years
6 (11)
19 (35)
17 (31)
12 (22)
Post-bronchodilator % predicted FEV1, mean ± SD 49.8 ± 10.6
Post-bronchodilator FEV1/FVC, %, mean ± SD 52.9 ± 9.5
Percent reversibility FEV1, mean ± SDa 8.8 ± 17.0 a patients could select both chronic bronchitis and emphysema COPD = chronic obstructive pulmonary disease; FEV1 = forced expiratory volume in 1 second; FVC = forced vital capacity; SD = standard deviation
Results
Primary endpoint
• 0–24h wmFEV1 (AUC) at the end of each 28-day treatment period (period Days 28–29)
o FF/VI 92/22 mcg demonstrated significantly higher 0–24h wmFEV1 than placebo at the end of the 28-day
treatment period. The difference vs placebo was 220 mL (95% CI 165, 275; p<0.001)
Secondary and other endpoints
• 0–25h serial FEV1 at period Days 28–29
o Significantly higher with FF/VI 92/22 mcg at all timepoints tested; adjusted mean differences 147–283 mL
compared with placebo (all p<0.001)
UK/FFT/0094/17(2) Date of preparation: September 2018

20
Figure 4: Change from baseline in 0–25h FEV1 over Days 28–29
Adapted from Boscia et al., 2012 A) LS mean and B) treatment differences from placeboFEV1 = forced expiratory volume in 1 second; FF/VI = fluticasone furoate/vilanterol; LS = least squares
• Change from treatment period baseline in trough FEV1 (period Day 29)
o Significantly greater change with FF/VI 92/22 mcg compared with placebo; adjusted mean difference
177mL (95% CI 97, 257; p<0.001)
• Change from baseline in 0–4h peak FEV1 (period Day 28)
o Significantly greater with FF/VI 92/22 mcg compared with placebo; adjusted mean difference 207mL (95%
CI 137, 276; p<0.001)
• Pharmacokinetic parameters, including Cmax, Tmax and AUC, over Days 28 and 29 (PK population)
o The extent of systemic exposure to FF and VI at steady state was low for the FF/VI 92/22 mcg group
Safety
• The overall incidence of AEs was low (10–12% with FF/VI and 4% with placebo)
• One patient in the FF/VI 92/22 mcg experienced a COPD exacerbation; the episode was resolved with
corticosteroids and antibiotics and led to study withdrawal
• No cases of pneumonia were reported
• There were no new safety signals with increasing dose of FF, and none of the reported AEs or SAEs, either on-
treatment or during washout, was considered to be treatment related by the study investigator
UK/FFT/0094/17(2) Date of preparation: September 2018

21
Table 4. Summary of adverse events
Placebo (n=51)
FF/VI 44/22 mcg (n=34)
FF/VI 92/22 mcg (n=33)
FF/VI 184/22 mcg (n=31)
On-treatment AEs, n (%) 2 (4) 4 (12) 4 (12) 3 (10)
AEs during washout, n (%) 3 (6) 4 (12) 0 1 (3)
AEs leading to permanent withdrawal from study, n (%)
0 1 (3)a 0 0
Serious AEs, n (%) 1 (2) 1 (3)a 0 0
Fatal AEs, n (%) 0 0 0 0 a Single patient who experienced a serious AE of transient ischemic attack (with a history of strokes), accompanied by nausea and hyponatraemia, which occurred during the second washout period and led to withdrawal from treatment AE = adverse event; FF/VI = fluticasone furoate/vilanterol
UK/FFT/0094/17(2) Date of preparation: September 2018

22
Dransfield et al., 2013 (dose-ranging study)
Once-daily inhaled fluticasone furoate and vilanterol versus vilanterol only for prevention of exacerbations of COPD: two
replicate double-blind, parallel-group, randomised controlled trials8
Relvar Ellipta is effective in the prevention of moderate and severe COPD exacerbations, in patients with a history of exacerbations
Relvar Ellipta (FF/VI 92/22 mcg) is indicated for the symptomatic treatment of adults with COPD with a FEV1 <70%
predicted normal (post-bronchodilator) with an exacerbation history despite regular bronchodilator therapy.1
Study design
Two identical 52-week randomised, multicentre, double-blind, parallel-group studies comparing FF/VI 44/22 mcg OD,
FF/VI 92/22 mcg OD and 184/22 mcg FF/VI OD against VI 22 mcg OD in patients with COPD.
FF/VI 44/22 mcg, FF/VI 184/22 mcg and VI 22 mcg are not licensed in the UK and therefore no results have been included in the efficacy section but have been included in the safety section COPD = chronic obstructive pulmonary disease; FEV1 = forced expiratory volume in 1 second; FF = fluticasone furoate; FF/VI = fluticasone furoate/vilanterol; FVC = forced vital capacity; OD = once daily; R = randomisation
• Patients received open-label FP/SAL 231/47 mcg† BD for 4 weeks to establish adherence to treatment and a stable
baseline
• Eligible patients were randomised 1:1:1:1 to receive FF/VI 44/22 mcg, FF/VI 92/22 mcg, FF/VI 184/22 mcg or
VI 22 mcg, delivered once daily via the Ellipta inhaler
• Randomisation was stratified by smoking status, i.e. current or former smoker
Patient population
• ≥40 years with a history of COPD (ATS or ERS definition)
• Smoking history ≥10 pack-years
• Post-bronchodilator FEV1/FVC ratio ≤0.70 and FEV1 ≤70% of predicted
• Documented history of ≥1 COPD exacerbation, for which they received systemic or oral corticosteroids or
antibiotics or were admitted to hospital, in the year prior to screening
† Equivalent to FP/SAL 250/50 mcg pre-dispensed dose
UK/FFT/0094/17(2) Date of preparation: September 2018

23
Endpoints
Primary endpoint
• Yearly rate of moderate and severe exacerbations (moderate exacerbations were defined as worsening symptoms
of COPD [≥2 consecutive days] necessitating treatment with oral corticosteroids or antibiotics, or both; severe
exacerbations were similar events that necessitated hospital admission)
Secondary and other endpoints
• Time to first moderate or severe exacerbation
• Yearly rate of exacerbations necessitating systemic or oral corticosteroids
• Change from randomisation in trough FEV1 at Week 52
• Yearly rate of severe exacerbations
• Number of night-time awakenings due to COPD symptoms
• Frequency of use of daily rescue medicines, i.e. salbutamol
• Dyspnoea score (dyspnoea was score on a scale of –2 to +2, with –2 indicating ‘much less than usual’ and +2
indicating ‘much more than usual’)
Safety and tolerability
• Incidence of AEs and AESIs
Demographics and baseline characteristics
Study 1 FF/VI 92/22 mcg (n=403)
Mean age, years ± SD 63.6 ± 9.1
Female gender, n (%) 172 (42.7)
Post-bronchodilator FEV1 (L), mean ± SD 1.3 ± 0.5
Predicted post-bronchodilator FEV1 (%), mean ± SD 45.7 ± 12.9
FEV1 reversibility (%), mean ± SD 14.8 ± 16.1
Study 2 FF/VI 92/22 mcg (n=403)
Mean age, years ± SD 64.0 ± 9.3
Female gender, n (%) 181 (44.9)
Post-bronchodilator FEV1 (L), mean ± SD 1.3 ± 0.5
Predicted post-bronchodilator FEV1 (%), mean ± SD 46.4 ± 13.9
FEV1 reversibility (%), mean ± SD 14.5 ± 14.9
FEV1 = forced expiratory volume in 1 second; FF/VI = fluticasone furoate/vilanterol; FVC = forced vital capacity; SD = standard deviation; VI = vilanterol
Results
Primary endpoint
• Yearly rate of moderate and severe exacerbations
o In Study 1, the LS mean yearly rate of exacerbations was 0.70 in the FF/VI 92/22 mcg group
o In Study 2, the LS mean yearly rate of exacerbations was 0.90 in the FF/VI 92/22 mcg group
Secondary and other endpoints
• Time to first moderate or severe exacerbation
o In Study 1, the HR for risk in time to first moderate or severe exacerbation was 0.7 (95% CI 0.6, 0.9) in the
FF/VI 92/22 mcg group
o In Study 2, the HR for risk in time to first moderate or severe exacerbation was 0.8 (95% CI 0.7, 1.0) in the
FF/VI 92/22 mcg group
UK/FFT/0094/17(2) Date of preparation: September 2018

24
• Yearly rate of exacerbations necessitating systemic or oral corticosteroids
o In Study 1, the yearly rate of exacerbations necessitating corticosteroids was 0.71 in the FF/VI 92/22 mcg
group
o In Study 2, the yearly rate of exacerbations necessitating corticosteroids was 0.66 in the FF/VI 92/22 mcg
group
• Change from randomisation in trough FEV1 at Week 52
o In Study 1, the change in trough FEV1 was 0.02 L in the FF/VI 92/22 mcg group
o In Study 2, the change in trough FEV1 was 0.01 L in the FF/VI 92/22 mcg group
• Yearly rate of severe exacerbations
o Very few severe exacerbations were reported in the individual studies and so analysis is presented only for
the pooled population. The rate of severe exacerbations did not differ significantly between the treatment
groups
• Number of night-time awakenings due to COPD symptoms
o In the pooled analysis, the LS mean night-time awakenings over each 4-week treatment interval during the
52-week treatment period was 0.42 in the FF/VI 92/22 mcg group
• Frequency of use of daily rescue medicines, i.e. salbutamol
o In the pooled analysis, the LS mean rescue medication use during a 24h period averaged over each 4-week
treatment interval during the 52-week treatment period was 1.95 in the FF/VI 92/22 mcg group
• Dyspnoea score
o In the pooled analysis, the LS mean dyspnoea score was –0.25 in the FF/VI 92/22 mcg group
Safety
• The overall incidence of AEs was similar across all four treatment groups (70.3–77.0%), as was the incidence of
SAEs (15.3–16.6%)
• The most frequently reported AE in both Studies 1 and 2 was nasopharyngitis
• In the pooled analysis, AEs leading to withdrawal were similar across all treatment groups, and were mainly caused
by disease exacerbations and episodes of pneumonia
• Pneumonia was noted in approximately twice as many patients in the FF/VI groups as in the VI 22 mcg group in
Studies 1 and 2 and in the pooled analysis
UK/FFT/0094/17(2) Date of preparation: September 2018

25
Table 5. Summary of adverse events (pooled population)
VI 22 mcg (n=818)
FF/VI 44/22 mcg (n=820)
FF/VI 92/22 mcg (n=806)
FF/VI 184/22 mcg (n=811)
Any AE, n (%) 575 (70.3) 620 (75.6) 621 (77.0) 622 (76.6)
Any AE leading to discontinuation or withdrawal, n (%)
45 (5.5) 53 (6.5) 62 (7.7) 61 (7.5)
Any SAE, n (%) 126 (15.4) 13 (16.6) 123 (15.3) 124 (15.3)
An on-treatment or post-treatment fatal AE,a n (%)
13 (1.6) 16 (2.0) 10 (1.2) 14 (1.7)
AEs of special interest, n (%)
Local corticosteroid effects
Cardiovascular effects
Lower-respiratory-tract infection (excl. pneumonia)
Pneumonia
Hypersensitivity
Bone disorders (incl. fractures)
Effects on glucose
Ocular events
Effects on potassium
Tremor
96 (11.7)
99 (12.1)
64 (7.8)
27 (3.3)
26 (3.2)
9 (1.1)
14 (1.7)
9 (1.1)
8 (1.0)
3 (0.4)
142 (17.3)
108 (13.2)
57 (7.0)
48 (5.9)
38 (4.6)
24 (2.9)
18 (2.2)
7 (0.9)
5 (0.6)
1 (0.1)
121 (15.0)
97 (12.0)
60 (7.4)
51 (6.3)
37 (4.6)
27 (3.3)
15 (1.9)
12 (1.5)
1 (0.1)
2 (0.2)
140 (17.3)
85 (10.5)
63 (7.8)
55 (6.8)
29 (3.6)
21 (2.6)
22 (2.7)
7 (0.9)
2 (0.2)
2 (0.2) a At the time of death, none of the fatal AEs were considered potentially related to the study drug AE = adverse event; FF/VI = fluticasone furoate/vilanterol; SAE = serious adverse event; VI = vilanterol
UK/FFT/0094/17(2) Date of preparation: September 2018

26
Crim et al., 2017 (SUMMIT pre-specified analyses)
Pneumonia risk with inhaled fluticasone furoate and vilanterol in COPD patients with moderate airflow limitation:
The SUMMIT trial9
SUMMIT was done with unlicensed doses and in an unlicensed patient population (history of exacerbations not
required) but has been included in the Evidence Dossier for safety transparency purposes.
Increased pneumonia risk with inhaled corticosteroid use was not evident in COPD patients with moderate airflow limitation and heightened cardiovascular risk
Study design
SUMMIT was a prospective, event-driven, randomised, multicentre, double-blind, parallel-group study comparing FF/VI
92/22 mcg OD against placebo, FF 92 mcg OD or VI 22 mcg OD in patients with moderate COPD and heightened CV risk.
The pre-specified analysis reported in this publication was to determine the incidence of pneumonia and risk factors for
COPD patients with moderate airflow limitation who had, or were at high risk for cardiovascular disease.
FF 92 mcg and VI 22 mcg are not licensed in the UK Patient numbers correspond to safety population COPD = chronic obstructive pulmonary disease; CVD = cardiovascular disease; FEV1 = forced expiratory volume in 1 second; FF = fluticasone furoate; FF/VI = fluticasone furoate/vilanterol; FVC = forced vital capacity; mMRC = modified Medical Research Council; OD = once daily; VI = vilanterol
• Patients were allowed to continue short-acting bronchodilators and/or theophylline; use of inhaled corticosteroids
and long-acting bronchodilators was discontinued at least 48h before study entry
• This was an event-driven study in which follow-up continued until at least 1,000 deaths had occurred, and
maximum follow-up was four years. Secondary endpoints were assessed after 12 months of treatment
Patient population
• 40–80 years with COPD
• Current or past smoker with smoking history ≥10 pack-years
• Post-salbutamol FEV1/FVC ratio ≤0.70 and FEV1 50–70% of predicted
• mMRC score ≥2
• History, or at increased risk, of cardiovascular disease (CVD). CVD was defined as coronary artery disease,
peripheral arterial disease, stroke, myocardial infarction or diabetes mellitus with target organ disease. Increased
CV risk was defined as aged 60 years or older and receiving medication for more than two of the following:
hypercholesterolaemia, hyper tension, diabetes mellitus or peripheral arterial disease
UK/FFT/0094/17(2) Date of preparation: September 2018

27
Analyses
• Adverse event reports of pneumonia
• Serious adverse event reports of pneumonia
• Exacerbations and pneumonia events
• Pneumonia deaths
• Risk factors for pneumonia
Baseline characteristics (safety population)
Characteristic Placebo
(n=4,131) FF 92 mcg (n=4,157)
VI 22 mcg (n=4,140)
FF/VI 92/22 mcg (n=4,140)
Mean age, years ± SD 65 ± 8 65 ± 8 65 ± 8 65 ± 8
Female gender, n (%) 1,050 (25) 1,098 (26) 1,071 (26) 1,019 (25)
BMI (kg/m2), mean ± SD 28 ± 6 28 ± 6 28 ± 6 28 ± 6
Current smokers, n (%) 1,949 (47) 1,952 (47) 1,941 (47) 1,875 (45)
Smoking pack years, mean ± SD 41 ± 25 41 ± 24 41 ± 24 41 ± 24
% predicted FEV1, mean ± SD 60 ± 6 60 ± 6 60 ± 6 60 ± 6
Exacerbation history, n (%)
0
1
≥2
2,454 (59)
1,049 (25)
628 (15)
2,552 (61)
997 (24)
608 (15)
2,510 (61)
994 (24)
636 (15)
2,532 (61)
1,007 (24)
601 (15)
mMRC
1
2
3
4
0
3,051 (74)
1,040 (25)
39 (<1)
1 (<1)
3,123 (75)
983 (24)
50 (1)
1 (<1)
3,027 (73)
1,071 (26)
40 (<1)
1 (<1)
3,057 (74)
1,020 (25)
60 (1)
Diabetes mellitus history 1,244 (30) 1,260 (30) 1,235 (30) 1,281 (31)
Congestive heart failure history 877 (21) 849 (20) 886 (21) 861 (21)
BMI = body mass index; FEV1 = forced expiratory volume in 1 second; FF = fluticasone furoate; FF/VI = fluticasone furoate/vilanterol; mMRC = modified Medical Research Council dyspnea score; SD = standard deviation; VI = vilanterol
Results
As the treatment effect on the primary endpoint for SUMMIT was not statistically significant, the statistical testing
reported here should be interpreted as descriptive only
• Adverse event reports of pneumonia
o The percentage of patients who experienced a pneumonia event was similar across treatment groups
▪ Placebo: 5.2% (3.84/100 treatment years)
▪ FF 92 mcg: 5.5% (3.84/100 treatment years)
▪ VI 22 mcg: 3.9% (2.77/100 treatment years)
▪ FF/VI 92/22 mcg: 5.7% (4.24/100 treatment years)
UK/FFT/0094/17(2) Date of preparation: September 2018
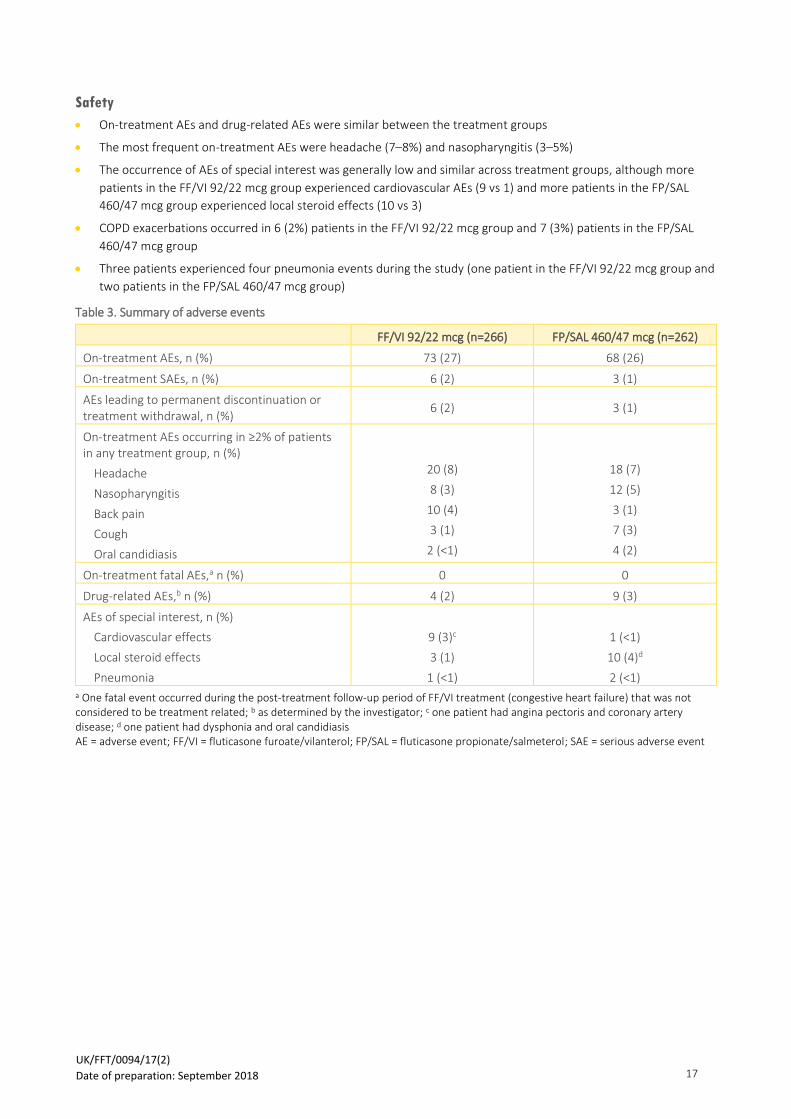
28
o The time to first pneumonia analysis showed that the risk was similar in the FF-containing and placebo
groups, and lower in the VI 22 mcg group; HRs of time to first AE pneumonia of special interest showed
that compared with placebo the risk was similar with the FF 92 mcg and FF/VI 92/22 mcg groups, but
lower with the VI 22 mcg group
▪ FF 92 mcg: HR 1.035; 95% CI 0.859, 1.247; p=0.716
▪ VI 22 mcg: HR 0.722; 95% CI 0.589, 0.886; p=0.002
▪ FF/VI 92/22 mcg: HR 1.038; 95% CI 0.863, 1.249; p=0.693
Figure 5: Time to first on-treatment adverse pneumonia event
Reproduced from Crim et al., 2017 FF = fluticasone furoate; FF/VI = fluticasone furoate/vilanterol; VI = vilanterol
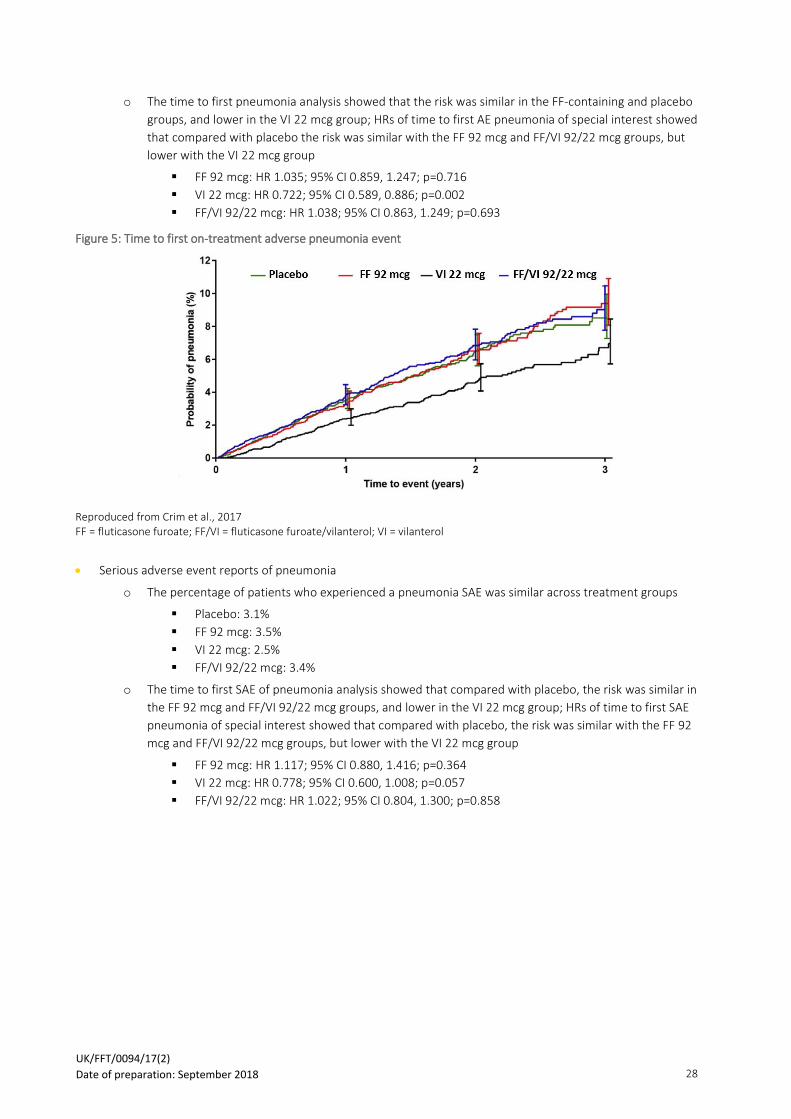
• Serious adverse event reports of pneumonia
o The percentage of patients who experienced a pneumonia SAE was similar across treatment groups
▪ Placebo: 3.1%
▪ FF 92 mcg: 3.5%
▪ VI 22 mcg: 2.5%
▪ FF/VI 92/22 mcg: 3.4%
o The time to first SAE of pneumonia analysis showed that compared with placebo, the risk was similar in
the FF 92 mcg and FF/VI 92/22 mcg groups, and lower in the VI 22 mcg group; HRs of time to first SAE
pneumonia of special interest showed that compared with placebo, the risk was similar with the FF 92
mcg and FF/VI 92/22 mcg groups, but lower with the VI 22 mcg group
▪ FF 92 mcg: HR 1.117; 95% CI 0.880, 1.416; p=0.364
▪ VI 22 mcg: HR 0.778; 95% CI 0.600, 1.008; p=0.057
▪ FF/VI 92/22 mcg: HR 1.022; 95% CI 0.804, 1.300; p=0.858
UK/FFT/0094/17(2) Date of preparation: September 2018
29
Figure 6: Time to first on-treatment serious adverse pneumonia event
Adapted from Crim et al., 2017 FF = fluticasone furoate; FF/VI = fluticasone furoate/vilanterol; VI = vilanterol
• Exacerbations and pneumonia events
o The risk of a moderate or severe COPD exacerbation or AE pneumonia of special interest was reduced in
the VI 22 mcg and FF/VI 92/22 mcg groups; HRs vs placebo were:
▪ FF 92 mcg: HR 0.977; 95% CI 0.904, 1.057; p=0.563
▪ VI 22 mcg: HR 0.920; 95% CI 0.850, 0.996; p=0.039
▪ FF/VI 92/22 mcg: HR 0.804; 95% CI 0.742, 0.872; p<0.001
o The risk of a severe COPD exacerbation or serious pneumonia was reduced in the FF/VI and VI treatment
groups compared with placebo; HRs vs placebo were:
▪ FF 92 mcg: HR 0.896; 95% CI 0.774, 1.038; p=0.142
▪ VI 22 mcg: HR 0.839; 95% CI 0.723, 0.974; p=0.021
▪ FF/VI 92/22 mcg: HR 0.798; 95% CI 0.687, 0.927; p=0.003
• Pneumonia deaths
o Investigator-reported pneumonia events present at the time of death occurred in <1% of patients,
whether on-treatment or after study drug discontinuation; the pneumonia-related fatal SAE event rate
per 100 patient years (number of events) was 0.15 (10) in the placebo group, 0.25 (17) in the FF 92 mcg
group, 0.07 (5) in the VI 22 mcg group and 0.23 (16) in the FF/VI 92/22 mcg group
o Deaths within 30 days of the last on-treatment pneumonia event occurred in 19 (8.9%) patients in the
placebo group, 22 (9.6%) patients in the FF 92 mcg group, 15 (9.2%) patients in the VI 22 mcg group and
20 (8.4%) patients in the FF/VI 92/22 mcg group
o A similar proportion of on-treatment adjudicated pneumonia deaths occurred in each treatment group,
although a lower on-treatment adjudicated pneumonia death rate per 100 patient years was evident in
the VI 22 mcg group
▪ Placebo: 9 patients (0.2%); rate = 0.14
▪ FF 92 mcg: 10 patients (0.2%); rate = 0.15
▪ VI 22 mcg: 6 patients (0.1%) rate = 0.09
▪ FF/VI 92/22 mcg: 13 patients (0.3%); rate = 0.18
• Risk factors for pneumonia
o Factors associated with a clear increased risk of pneumonia were:
▪ Prior exacerbation history (p<0.001)
▪ BMI <25 kg/m2 (p=0.006)
▪ Lung function impairment (FEV1 <60% predicted; p=0.017)
UK/FFT/0094/17(2) Date of preparation: September 2018

30
Van der Palen et al., 2016 (critical errors)
A randomised open-label cross-over study of inhaler errors, preference and time to achieve correct inhaler use in
patients with COPD or asthma: comparison of Ellipta with other commonly used inhaler devices10,11
The Ellipta inhaler is easy to use and significantly fewer patients made critical errors compared with other commonly used devices after reading the patient information leaflet
Study design
A randomised, multicentre, single visit, open-label, cross over study comparing the Ellipta inhaler device against other
devices in patients with asthma and patients with COPD. The results below focus only on COPD studies; asthma results
are not presented here.
COPD = chronic obstructive pulmonary disease; MDI = metered-dose inhaler; R = randomisation
• Patients entering the study were naïve to Ellipta and at least one other device to which they were assigned
• Each substudy was individually powered
• Critical errors were assessed by trained respiratory nurses after patients had read the patient information leaflet
(PIL). A critical error is defined as an error that is likely to result in the inhalation of significantly reduced, minimal or
no medication
• If the patient made errors, the investigator demonstrated the correct use of the inhaler, and the patient
demonstrated inhaler use again
Patient population
• ≥18 years with a physician diagnosis of COPD and currently receiving treatment for COPD
• Naïve to Ellipta inhaler use
Endpoints
Primary endpoint
• Critical errors using the devices after reading the PIL only
Secondary endpoints
• Overall errors using the devices after reading the PIL only
• Instruction from trained respiratory nurse on use of the devices
• Time to correct inhaler use
• Ease of use of the devices
• Patient preference for devices
UK/FFT/0094/17(2) Date of preparation: September 2018

31
Demographics and baseline characteristics
Characteristic Total
(N=567)
Ellipta vs Accuhaler (n=171)
Ellipta vs MDI
(n=80)
Ellipta vs Turbohaler
(n=100)
Ellipta vs HandiHaler
(n=118)
Ellipta vs Breezhaler
(n=98)
Mean age, years ± SD 67.3 ± 8.3 67.8 ± 7.4 65.6 ± 8.8 68.1 ± 8.7 67.3 ± 9.0 67.2 ± 8.2
Female gender, n (%) 225 (40) 68 (40) 27 (34) 33 (33) 46 (39) 51 (52)
COPD history, n (%)
6 months–10 years
10–25 years
≥25 years
422 (74)
123 (22)
22 (4)
135 (79)
29 (17)
7 (4)
59 (74)
18 (22)
3 (4)
62 (62)
32 (32)
6 (6)
92 (78)
23 (19)
3 (3)
74 (76)
21 (21)
3 (3)
COPD = chronic obstructive pulmonary disease; MDI = metered-dose inhaler
Results
Primary endpoint
• Critical errors using the devices after reading the PIL only
o After reading the PIL, the proportion of patients with COPD who made at least one critical error was
significantly lower with the Ellipta device compared with all others (all p<0.001)
o For the Ellipta inhaler, the most common critical error was exhaling directly into the mouthpiece (31/567
[5%] patients with COPD)
o For the Accuhaler device, the lever was not pushed back properly by 33% of patients with COPD (56/171)
o For the MDI device, poor press-and-breathe coordination was observed in 43% (34/80) of patients with
COPD
o For the Turbohaler device, 29% (29/100) patients with COPD did not twist the base properly and hear the
click
o For both the HandiHaler and the Breezhaler, the capsule did not rattle with 36% (42/118) and 43% (42/98)
of patients with COPD, respectively
Figure 7: Percentage of patients with COPD with at least one critical error after reading the PIL
Adapted from van der Palen et al., 2016 MDI = metered-dose inhaler; PIL = patient information leaflet
UK/FFT/0094/17(2) Date of preparation: September 2018

32
Secondary endpoints
• Overall errors using the devices after reading the PIL only
o In all five COPD groups, there were significantly fewer patients who experienced at least one overall error
with the Ellipta device compared with all the others
▪ Ellipta vs Accuhaler: 52 (30) vs 112 (65); p<0.001
▪ Ellipta vs MDI: 25 (31) vs 68 (85); p<0.001
▪ Ellipta vs Turbohaler: 31 (31) vs 71 (71); p<0.001
▪ Ellipta vs HandiHaler: 51 (43) vs 73 (62); p<0.001
▪ Ellipta vs Breezhaler: 30 (31) vs 55 (56); p<0.001
• Instruction from trained respiratory nurse on use of the devices
o After reading the PIL, the majority of patients with COPD made no errors using the Ellipta inhaler and did
not require instruction from a trained respiratory nurse. In contrast, for the other devices, the majority of
patients did require instruction from a trained respiratory nurse. Percentage of patients making no errors
and thus not requiring instruction:
▪ Ellipta vs Accuhaler: 70% (n=119) vs 35% (n=59)
▪ Ellipta vs MDI: 69% (n=55) vs 15% (n=12)
▪ Ellipta vs Turbohaler: 69% (n=69) vs 29% (n=29)
▪ Ellipta vs HandiHaler: 57% (n=67) vs 38% (n=45)
▪ Ellipta vs Breezhaler: 69% (n=68) vs 44% (n=43)
o The difference in the number of nurse instructions required until correct use was performed was
significant between the Ellipta inhaler and all the other devices (p<0.001 in all subgroups)
▪ When compared with the Ellipta inhaler, more patients required more than one instruction for
the use of the Accuhaler (7 versus 27 patients), MDI (3 versus 20 patients), Turbohaler (6 versus
19 patients), HandiHaler (13 versus 29 patients) and Breezhaler (2 versus 15 patients) devices
• Time to correct inhaler use
o In all substudies, median time to correct inhaler use without trained respiratory nurse support (reading
the PIL only) could only be determined for the Ellipta inhaler as for the other devices more than half of
patients with COPD could not perform correct use after reading the PIL only
o In all substudies, median time to correct inhaler use reading the PIL and receiving trained respiratory
nurse support was significantly shorter for patients using the Ellipta inhaler compared with those using:
▪ Accuhaler (2.75 versus 3.93 min; p<0.001)
▪ MDI (3.79 versus 6.30 min; p<0.001)
▪ Turbohaler (2.87 versus 7.80 min; p<0.001)
▪ HandiHaler (4.32 versus 8.50 min; p<0.001)
▪ Breezhaler (3.15 versus 8.44 min; p<0.001)
• Ease of use of the devices
o A larger proportion in each group rated the Ellipta inhaler very easy or easy compared with the other
devices
▪ Ellipta vs Accuhaler: 97% (n=165) vs 60% (n=104)
▪ Ellipta vs MDI: 92% (n=73) vs 44% (n=35)
▪ Ellipta vs Turbohaler: 96% (n=96) vs 55% (n=55)
▪ Ellipta vs HandiHaler: 98% (n=115) vs 38% (n=38)
▪ Ellipta vs Breezhaler: 94% (n=92) vs 55% (n=54)
UK/FFT/0094/17(2) Date of preparation: September 2018

33
o Between 96% and 100% of COPD patients reported they found it very easy to determine how much
medication was left in the Ellipta inhaler; 61%, 63%, 45%, 42% and 60% of patients reported they found it
very easy to determine how much medication was left in Accuhaler, MDI, Turbohaler, HandiHaler and
Breezhaler devices, respectively
• Patient preference for devices
o The majority of patients with COPD preferred Ellipta over other commonly used inhalers (all p<0.001)
Figure 8: Overall device preference reported in each substudy
Adapted from van der Palen et al., 2016 MDI = metered-dose inhaler
o The majority of patients preferred the Ellipta inhaler for most individual criteria:
▪ Number of steps for correct use, time taken to use, size of device, dose counter, comfort of
mouthpiece and ease of opening (p<0.001) with some exceptions where there was no difference
(listed below)
o Criteria for which there was no difference in patient preference between devices:
▪ Size of the inhaler, % patients preferring:
• Ellipta: 39% or MDI: 33%
• Ellipta: 44% or Turbohaler: 38%
• Ellipta: 41% or Breezhaler: 44%
▪ Comfort of mouthpiece, % patients preferring:
• Ellipta: 33% or HandiHaler: 31%
• Ellipta: 40% or Breezhaler: 37%
UK/FFT/0094/17(2) Date of preparation: September 2018

34
References 1. GlaxoSmithKline UK. Relvar Ellipta (fluticasone furoate/vilanterol 92/22 mcg) Summary of Product Characteristics.
2. GlaxoSmithKline UK. Incruse Ellipta (umeclidinium 55 mcg) Summary of Product Characteristics.
3. Vestbo J, Leather D, Diar Bakerly N, et al. Effectiveness of Fluticasone Furoate-Vilanterol for COPD in Clinical Practice. The New England journal of medicine. 2016;375(13):1253-1260.
4. GlaxoSmithKline. Study 115151 Clinical Study Report. 2017; Online. Accessed November, 2017.
5. Siler TM, Kerwin E, Sousa AR, Donald A, Ali R, Church A. Efficacy and safety of umeclidinium added to fluticasone furoate/vilanterol in chronic obstructive pulmonary disease: Results of two randomized studies. Respiratory medicine. 2015;109(9):1155-1163.
6. Agusti A, de Teresa L, De Backer W, et al. A comparison of the efficacy and safety of once-daily fluticasone furoate/vilanterol with twice-daily fluticasone propionate/salmeterol in moderate to very severe COPD. The European respiratory journal. 2014;43(3):763-772.
7. Boscia JA, Pudi KK, Zvarich MT, Sanford L, Siederer SK, Crim C. Effect of once-daily fluticasone furoate/vilanterol on 24-hour pulmonary function in patients with chronic obstructive pulmonary disease: a randomized, three-way, incomplete block, crossover study. Clinical therapeutics. 2012;34(8):1655-1666.e1655.
8. Dransfield MT, Bourbeau J, Jones PW, et al. Once-daily inhaled fluticasone furoate and vilanterol versus vilanterol only for prevention of exacerbations of COPD: two replicate double-blind, parallel-group, randomised controlled trials. The Lancet Respiratory medicine. 2013;1(3):210-223.
9. Crim C, Calverley PMA, Anderson JA, et al. Pneumonia risk with inhaled fluticasone furoate and vilanterol in COPD patients with moderate airflow limitation: The SUMMIT trial. Respiratory medicine. 2017;131:27-34.
10. GlaxoSmithKline. Clinical Study Report for Study 200301. 2017; Online. Accessed November, 2017.
11. van der Palen J, Thomas M, Chrystyn H, et al. A randomised open-label cross-over study of inhaler errors, preference andtime to achieve correct inhaler use in patients with COPD or asthma: comparison of ELLIPTA with other inhaler devices. NPJ primary care respiratory medicine. 2016;26:16079.
How to request additional information from GSK
Should you require any further information, please contact the GSK customer contact centre:
• By phone on 0800 221 441. Lines are open from Monday to Friday 8.30am to 5.30pm. Outside these hours and on
bank holidays, an answer phone service is available.
• By email at [email protected]
UK/FFT/0094/17(2) Date of preparation: September 2018

35
Incruse▼Ellipta (umeclidinium bromide) Prescribing Information
(Please consult the full Summary of Product Characteristics (SmPC) before prescribing)
Incruse Ellipta 55mcg (umeclidinium) inhalation powder. Each single inhalation provides a delivered dose (the dose leaving the mouthpiece of the inhaler) of 55 micrograms umeclidinium (equivalent to 65 micrograms of umeclidinium bromide). Indications: Incruse is indicated as a maintenance bronchodilator treatment to relieve symptoms in adult patients with chronic obstructive pulmonary disease (COPD). Dosage and administration: Inhalation only. One inhalation once daily of Incruse Ellipta at the same time of the day each day. Contraindications: Hypersensitivity to the active substances or to any of the excipients (lactose monohydrate and magnesium stearate). Precautions: Incruse Ellipta should not be used in patients with asthma. Treatment with Incruse Ellipta should be discontinued in the event of paradoxical bronchospasm and alternative therapy initiated if necessary. Cardiovascular effects may be seen after the administration of muscarinic receptor antagonists, therefore Incruse Ellipta should be used with caution in patients with severe cardiovascular disorders, particularly cardiac arrhythmias. Incruse Ellipta should be used with caution in patients with urinary retention or narrow angle glaucoma. No dosage adjustment is required in renal or mild to moderate hepatic impairment. Acute symptoms: Incruse Ellipta is not indicated for acute episodes of bronchospasm. Warn patients to seek medical advice if short-acting inhaled
bronchodilator use increases, a re-evaluation of the patient and of the COPD treatment regimen should be undertaken. Interactions with other medicinal products: Co-administration with other long-acting muscarinic antagonists or medicinal products containing this active substance has not been studied and therefore, is not recommended. Fertility, pregnancy, and breast-feeding: No available human in vivo data. Balance risks against benefits. Side effects: Common (≥1/100 to <1/10): Nasopharyngitis, upper respiratory tract infection, urinary tract infection, sinusitis, headache, tachycardia, cough. Other Important side effects include. Uncommon (≥1/1,000 to <1/100): Atrial fibrillation, rhythm idioventricular, supraventricular tachycardia, supraventricular extrasystoles. Hypersensitivity reactions including rash, urticaria, pruritus. Not Known (cannot be estimated from available data): Glaucoma and vision blurred. Legal category: POM. Presentation and Basic NHS cost: Incruse Ellipta. 1 inhaler x 30 doses. Incruse Ellipta 55mcg - £27.50. Marketing authorisation (MA) nos. 55mcg 1x30 doses [EU/1/14/922/002]; MA holder: Glaxo Group Ltd, 980 Great West Road, Brentford, Middlesex TW8 9GS, UK. Last date of revision: July 2018. UK/INC/0001/17(1)a. Incruse and Ellipta are registered trademarks of the GlaxoSmithKline group of companies. All rights reserved.
Adverse events should be reported. Reporting forms and information can be found at www.mhra.gov.uk/yellowcard or search for MHRA Yellowcard in the Google Play or Apple App store. Adverse events should also be
reported to GlaxoSmithKline on 0800 221 441
UK/FFT/0094/17(2) Date of preparation: September 2018

36
Relvar Ellipta (fluticasone furoate/vilanterol [as trifenatate]) Prescribing Information
Relvar Ellipta Prescribing informationPlease consult the full Summary of Product Characteristics (SmPC) before prescribing. Relvar Ellipta (fluticasone furoate/vilanterol [as trifenatate]) inhalation powder. Each single inhalation of fluticasone furoate (FF) 100 micrograms (mcg) and vilanterol (VI) 25 mcg provides a delivered dose of 92 mcg FF and 22 mcg VI. Each single inhalation of FF 200 mcg and VI 25 mcg provides a delivered dose of 184 mcg of FF and 22 mcg of VI. Indications: Asthma: Regular treatment of asthma in patients ≥12 years where a long-acting β2-agonist (LABA) and inhaled corticosteroid (ICS) combination is appropriate; i.e. patients not adequately controlled on ICS and ''as needed” short-acting inhaled β2-agonists or patients already adequately controlled on both ICS and LABA. COPD: Symptomatic treatment of adults with COPD with a FEV1<70% predicted normal (post-bronchodilator) and an exacerbation history despite regular bronchodilator therapy. Dosage and administration: Inhalation only. Asthma: Adults and adolescents ≥12 years: one inhalation once daily of Relvar 92/22 mcg for patients who require a low to mid dose of ICS in combination with a LABA. If patients are inadequately controlled then the dose can be increased to one inhalation once daily Relvar 184/22 mcg. Relvar 184/22 mcg can also be considered for patients who require a higher dose of ICS in combination with a LABA. Regularly review patients and reduce dose to lowest that maintains effective symptom control. COPD: one inhalation once daily of Relvar 92/22 mcg. Relvar 184/22 mcg is not indicated for patients with COPD. Contraindications: Hypersensitivity to the active substances or to any of the excipients (lactose monohydrate & magnesium stearate). Precautions: Pulmonary tuberculosis, severe cardiovascular disorders or heart rhythm abnormalities, thyrotoxicosis, uncorrected hypokalaemia, patients predisposed to low levels of serum potassium, chronic or untreated infections, diabetes mellitus, paradoxical bronchospasm. In patients with moderate to severe hepatic impairment 92/22 mcg dose should be used. Acute symptoms: Not for acute symptoms, use short-acting inhaled bronchodilator. Warn patients to seek medical advice if short-acting inhaled bronchodilator use increases. Therapy should not be abruptly stopped without physician supervision due to risk of symptom recurrence. Asthma-related adverse events and exacerbations may occur during treatment. Patients should continue treatment but seek medical advice if asthma symptoms remain uncontrolled or worsen after initiation of Relvar. Systemic effects: Systemic effects of ICSs may occur, particularly at high doses for long periods, but much less likely than with oral corticosteroids. Possible Systemic effects include: Cushing’s syndrome,
Cushingoid features, adrenal suppression, decrease in bone mineral density, growth retardation in children and adolescents. Eye symptoms such as blurred vision may be due to underlying serious conditions such as cataract, glaucoma or central serous chorioretinopathy (CSCR); consider referral to ophthalmologist. More rarely, a range of psychological or behavioural effects including psychomotor hyperactivity, sleep disorders, anxiety, depression or aggression (particularly in children). Increased incidence of pneumonia has been observed in patients with COPD receiving inhaled corticosteroids. Risk factors for pneumonia include: current smokers, old age, patients with a history of prior pneumonia, patients with a body mass index <25 kg/m2 and patients with a FEV1<50% predicted. If pneumonia occurs with Relvar treatment should be re-evaluated. Patients with rare hereditary problems of galactose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption should not take Relvar. Interactions with other medicinal products: Interaction studies have only been performed in adults. Avoid-blockers. Caution is advised when co-administering with strong CYP3A4 inhibitors (e.g. ketoconazole, ritonavir, cobicistat-containing products). Concomitant administration of other sympathomimetic medicinal products may potentiate the adverse reactions of FF/VI. Relvar should not be used in conjunction with other long-acting β2-adrenergic agonists or medicinal products containing long-acting β2-adrenergic agonists. Pregnancy and breast-feeding: Experience limited. Balance risks against benefits. Side effects: Very Common (≥1/10): headache, nasopharyngitis. Common (≥1/100 to <1/10): candidiasis of the mouth and throat, dysphonia, pneumonia, bronchitis, upper respiratory tract infection, influenza, oropharyngeal pain, sinusitis, pharyngitis, rhinitis, cough, abdominal pain, arthralgia, back pain, fractures, pyrexia, muscle spasms. Other important side effects include: Uncommon (≥1/1,000 to <1/100); blurred vision, hyperglycaemia. Rare (≥1/10,000 to <1/1,000) paradoxical bronchospasm and hypersensitivity reactions including anaphylaxis, angioedema, rash, urticaria. See SmPC for other adverse reactions. Legal category: POM. Presentation and Basic NHS cost: Relvar Ellipta. 1 inhaler x 30 doses. Relvar Ellipta 92/22 - £22.00. Relvar Ellipta 184/22 - £29.50. Marketing authorisation (MA) nos. 92/22 mcg 1x30 doses [EU/1/13/886/002]; 184/22 mcg 1x30 doses [EU/1/13/886/005]. MA holder: Glaxo Group Ltd, 980 Great West Road, Brentford, Middlesex TW8 9GS, UK. Last date of revision: September 2018. UK/FFT/0227/15(6). Trademarks are owned by or licensed to the GSK group of companies. © 2018 GSK group of companies or its licensor. Relvar Ellipta was developed in collaboration with Innoviva Inc.
Adverse events should be reported. Reporting forms and information can be found at www.mhra.gov.uk/yellowcard or search for MHRA Yellowcard in the Google Play or Apple App
store. Adverse events should also be reported to GlaxoSmithKline on 0800 221 441
UK/FFT/0094/17(2) Date of preparation: September 2018


















