
Evaluation of quality and interchangeability of medicinal products Training workshop for evaluators...
64
Evaluation of quality and interchangeability of medicinal products Training workshop for evaluators from National Medicines Regulatory Authorities in East Africa Community 10-14 September 2007, Dar Es Salaam, Tanzania Presented by Rutendo Kuwana Active pharmaceutical ingredients
-
Upload
dwayne-stone -
Category
Documents
-
view
215 -
download
0
Transcript of Evaluation of quality and interchangeability of medicinal products Training workshop for evaluators...

Evaluation of quality and interchangeability of medicinal productsTraining workshop for evaluators from National Medicines
Regulatory Authorities in East Africa Community
10-14 September 2007, Dar Es Salaam, Tanzania
Presented by
Rutendo Kuwana
Active pharmaceutical ingredients

Active Pharmaceutical Ingredients 2 |
What is an API?What is an API?
Active Pharmaceutical Ingredient (API)
A substance or compound that is intended to be used in the manufacture of a pharmaceutical product as a therapeutically active compound (ingredient)

Active Pharmaceutical Ingredients 3 |
Presentation approachPresentation approach
Collect and interpret available information on the APIs (pre dossier studies), such as:
Literature, all aspects (chemical/physical) Monographs in pharmacopoeia (example: ARVs)

Active Pharmaceutical Ingredients 4 |
Some definitionsSome definitions
Enantiomer
cpds with same molecular formula as substance but differ in spatial arrangement of atoms and are non-superimpossable mirror images
Polymorphism – occurrence of different crystalline forms of the same substance
Degradation product – molecule resulting from chemical change in substance due to e.g. light, temperature, pH, water, reaction with excipient, immediate container/closure

Active Pharmaceutical Ingredients 5 |
Some definitions (2)Some definitions (2)
Impurity – any component of the medicinal product which is not the chemical entity defined as the active substance or an excipient of the product
Identified Impurity – an impurity for which structural characterisation has been achieved
Unidentified degradation product – an impurity defined only by qualitative properties e.g. Rt

Active Pharmaceutical Ingredients 6 |
Available information on APIAvailable information on API
Applicants should collect and analyse available information of the API in a systematic approach
Some outcomes: Sound scientific understanding of the API, with respect to
properties, stability, specifications, etc. Assists in API manufacture and DMF compilation Sound choice of API manufacturer (source) Assists in dossier compilation Important for FPP pharmaceutical development Reduction of time / cost

Active Pharmaceutical Ingredients 7 |
Literature information on APILiterature information on API
Standard works / series / books – such as: (Analytical) Profiles of Drug Substances and Excipients [eds:
Florey / Brittain – 31 volumes] The Merck Index (for structures, properties) Pharmaceutical Codex (12th edition) (“old” APIs)
Journals through search facilities such as International Pharmaceutical Abstracts, Chemical Abstracts,
Analytical Abstracts & internet
Pharmacopoeial monographs (current)
Analysis of structure & stereochemistry

Active Pharmaceutical Ingredients 8 |
Information from literature and structures
Information from literature and structures
APIs which are organic compounds, have unique chemical structures & stereochemistry
These structures, together with the solid/liquid state conditions, are basically responsible for chemical and physical properties of the APIs

Active Pharmaceutical Ingredients 9 |
Information from literature & structure: Rifampicin
Information from literature & structure: Rifampicin
hydrolysis (to 25-desacetyl)
oxidation hydrolysis (to quinone) (to 3-
formyl)
Oxidation to N oxide
light sensitive

Active Pharmaceutical Ingredients 10 |
Information from literature & structure Rifampicin (discussion - 1)
Information from literature & structure Rifampicin (discussion - 1)
Oxidation
Hydroquinone group Main degradation of API (to rifampicin quinone) Enhances solubility in alkaline medium
Tertiary amine Moderately prone towards oxidation (to N-oxide) Enhances solubility in acid medium
Oxidation enhanced by Metal ions Low pH

Active Pharmaceutical Ingredients 11 |
Information from literature & structure Rifampicin (discussion - 2)
Information from literature & structure Rifampicin (discussion - 2)
Hydrolysis
Hydrazone (imine) group Hydrolysis to 3-formyl rifamycin
25-acetyl (ester) group Hydrolysis to 25-desacetyl rifampicin (minor)
Light sensitive Due to conjugation in molecule (unsaturated)
Storage of bulk raw material (BP/Ph.Eur.): Store under nitrogen in an airtight container, protected from light at
temperature of ≤ 25ºC

Active Pharmaceutical Ingredients 12 |
Information from literature & structure Isoniazid
Information from literature & structure Isoniazid
Small molecule (quite stable) Basic amino functions Primary amine - reacts with aldehydes/lactose see presentation: FPPs – formulation problems? Can hydrolyze under stress conditions to e.g. isonicotinic acid & hydrazine Oxidize in presence of strong oxidants (e.g. permanganate), with metals as
catalyst
Active Pharmaceutical Ingredients 13 |
Information from literature & structure
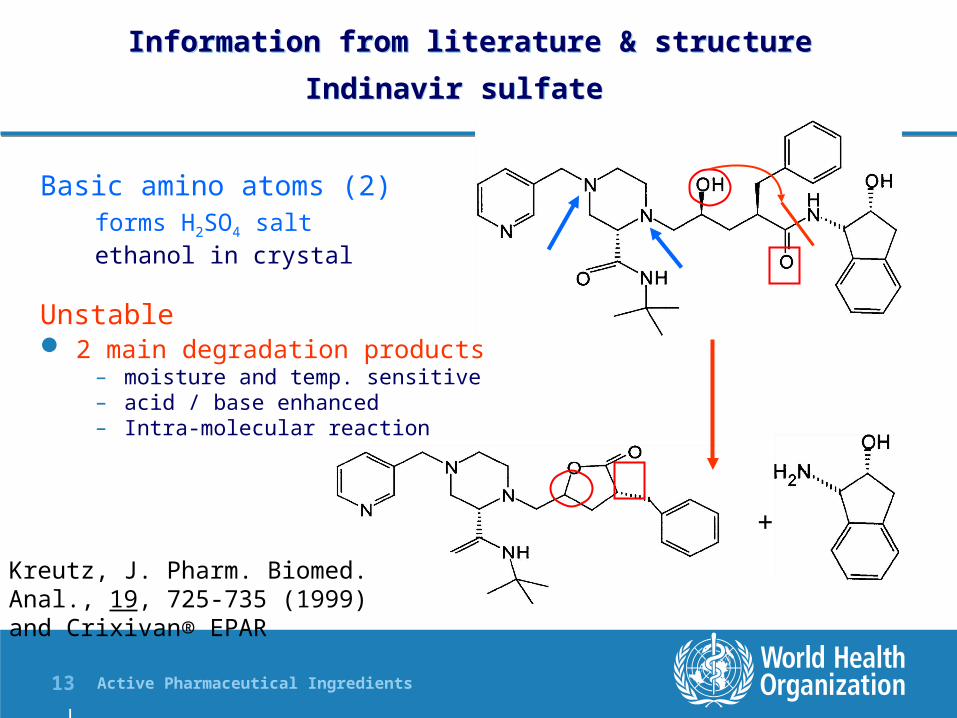
Indinavir sulfate Information from literature & structure
Indinavir sulfate
Basic amino atoms (2)forms H2SO4 saltethanol in crystal
Unstable 2 main degradation products
– moisture and temp. sensitive– acid / base enhanced– Intra-molecular reaction
+
Kreutz, J. Pharm. Biomed. Anal., 19, 725-735 (1999) and Crixivan® EPAR

Active Pharmaceutical Ingredients 14 |
Literature supportLiterature support
Literature information used in the dossier should always be accompanied by
Full traceable reference citations, for instance: Devani, M.B., Shishoo, C.J., Doshi, K.J. & Patel, H.B. Kinetic
studies of the interaction between isoniazid and reducing sugars. Journal of Pharmaceutical Sciences, 74, 427-432 (1985)
Photocopies of publication or relevant pages

Active Pharmaceutical Ingredients 15 |
Properties of APIsProperties of APIs
Scenarios:
API not described in BP, Ph., JP, Ph.Eur., or USP (non - compendial)
API described in BP, Int.Ph., JP, Ph.Eur.,or USP (compendial)
Information from literature (important)

Active Pharmaceutical Ingredients 16 |
Properties: non-compendial APIsProperties: non-compendial APIs
Proof of structure/stereochemistry correctness– Single crystal X-ray structure (sufficient) or– Spectrometric data (IR, 1H & 13C NMR, MS, etc.): QA certified
copies of the spectra and tabulated data with• assignments against structure
or• correlation against API spectral data from peer reviewed literature,
preferable innovator publication (in tabulated form!!). Strongly recommended
Physico-chemical properties

Active Pharmaceutical Ingredients 17 |
Properties: Compendial APIsProperties: Compendial APIs
Physico-chemical and other relevant properties, e.g.– Solubility in water (effect of pH), other solvents such as ether,
ethanol, acetone and dichloromethane– pKa, partition coefficient– Existence/absence of polymorphs and pseudo-polymorphs e.g.
solvates (with XRPD, DSC, IR)• e.g. Rifampicin polymorphs I and II• See Nevirapine (later in this presentation)
– Hygroscopicity e.g. Ethambutol hydrochloride in FDC tablet– Particle size

Active Pharmaceutical Ingredients 18 |
Properties for Compendial APIsExample: solubility of TB APIs
Properties for Compendial APIsExample: solubility of TB APIs
API Water CHCl3* Ethanol
Rifampicin Water: Slightly 1,2
pH 7.5: 0.3% 2
pH 5.3: 0.4% 2
pH 2.0: 10% 2
Freely 1,2 Slightly 2
Ethambutol 2HCl 50% 2 0.1% 2 20% 2
Ethambutol base Sparingly 2 Very 2
Isoniazid 14% 1 0.1% 1 2% 1
Pyrazinamide 1.5% 1 0.7% 1 0.6% 2
1 Merck Index 13th ed 2 Pharmaceutical Codex 12th ed
* Dichloromethane has similar properties to chloroform as solvent, but preferred for safety reasons

Active Pharmaceutical Ingredients 19 |
Properties APIsExample: solubility protease inhibitors (mg/ml)
Properties APIsExample: solubility protease inhibitors (mg/ml)
GC Williams & PJ Sinko, Advanced Drug Delivery Reviews, 39, 211-238 (1999)
Medium Saquinavir
mesilate
Ritonavir Indinavir
sulfate
Nelfinavir
mesilate
Amprenavir
mesilate
Water 2.2 0.001 >100 4.5 0.19
pH 7.4 0.036 0.005 0.07 very low 0.06
pH 6.8 0.19
pH 6.5 0.073
pH 4.8 0.3
pH 4.0 0.007
pH 3.5 60 0.5
pH 2.6 4.5

Active Pharmaceutical Ingredients 20 |
Properties Compendial APIsPseudo-polymorphism nevirapine
Properties Compendial APIsPseudo-polymorphism nevirapine
Int. Ph. monograph Nevirapine (anhydrous & hemihydrate)
Identification test C
Carry out the examination as described under “Spectrophotometry in the infrared region”.– For the anhydrous substance, the infrared (IR) absorption spectrum
is concordant with the spectrum obtained from anhydrous nevirapine RS or with the reference spectrum of anhydrous nevirapine
– For the hemihydrate, the IR absorption spectrum shows a characteristic sharp absorbance at about 3503 cm−1; after heating the test substance for one hour at 140°C and cooling, the IR absorption spectrum is concordant with the spectrum obtained from anhydrous nevirapine RS or with the reference spectrum of anhydrous nevirapine

Active Pharmaceutical Ingredients 21 |
Properties Compendial APIsPseudo-polymorphism nevirapine (2)
Properties Compendial APIsPseudo-polymorphism nevirapine (2)
Interpretation of this IR identification test:
Nevirapine anhydrous (one test)– IR spectrum against nevirapine anhydrous RS
Nevirapine hemihydrate (two tests, conform to both)1. IR spectrum shows signal at 3503 cm-1 (water) and2. Heat converts the hemihydrate to the anhydrous form
• IR spectrum against nevirapine anhydrous RS
- ½H20
Nevirapine, ½H20 ————> Nevirapine
heat
The reaction is not reversible at room temperature

Active Pharmaceutical Ingredients 22 |
IR-spectraIR-spectra
Nevirapine ½H20
3503 cm-1
O-H signal (water)
Spectra not concordant
(do not match)
Nevirapine anhydrous
no crystal water
no O-H signal
434.5
0
464.4
3
500.0
3
548.2
3
588.5
8619.5
1
652.2
3
698.4
071
1.8
6
767.1
1780.8
279
4.2
6
819.4
583
0.5
684
4.9
2894.6
8
939.8
995
9.8
497
9.3
699
8.5
110
27.0
010
50.9
610
71.5
71092
.50
1107
.16
1117
.27
1144
.79
1168
.40
1221
.94
1244
.441272
.70
1295
.94
1351
.84
1381
.78
1411
.84
1459
.55
1491
.86
1582
.33
1600
.74
1654
.00
1913
.05
1945
.48
1961
.46
1983
.68
2070
.73
2218
.44
2515
.27
2599
.59
2861
.41
2916
.77
3007
.45
3062
.78
3193
.26
3502
.85
3893
.86
40
45
50
55
60
65
70
75
80
85
90
95
%T
rans
mitta
nc
e
500 1000 1000 2000 3000 4000
Wavenumbers (cm-1)
436.3
2
462.3
9
499.6
5
523.6
754
0.2
655
1.8
8
576.0
6
621.4
9
655.8
8
697.5
671
0.8
7
761.6
6
789.1
280
3.6
2
829.4
4
885.0
3
941.7
595
6.6
396
5.3
397
5.1
3
999.2
410
25.4
01048
.30
1074
.82
1091
.16
1107
.27
1153
.13
1167
.32
1210
.77
1243
.06
1258
.94
1289
.19
1354
.95
1384
.34
1415
.3214
65.3
514
87.8
6
1568
.92
1586
.57
1647
.72
1927
.40
1950
.21
2065
.49
2088
.38
2320
.66
2456
.46
2505
.28
2583
.59
2698
.58
2861
.26
2914
.76
3011
.59
3062
.45
3124
.51
3189
.89
3294
.95
10
20
30
40
50
60
70
80
90
100
110
%T
rans
mitta
nc
e
500 1000 1000 2000 3000 4000
Wavenumbers (cm-1)

Active Pharmaceutical Ingredients 23 |
Route(s) of synthesisRoute(s) of synthesis
Scenarios:
API not described in BP, Int.Ph., JP,Ph.Eur., or USP (non-compendial APIs)
Specifications of raw materials and intermediates used in the synthesis of non-compendial APIs
API described in BP, Int.Ph., JP, Ph.Eur., or USP (compendial APIs)

Active Pharmaceutical Ingredients 24 |
Route(s) of synthesis (cont.)Route(s) of synthesis (cont.)
Requirements: The synthesis should– lead to the correct structure, stereochemistry and crystal
form & size (if relevant)– be well controlled and validated (GMP)– produce an API which meets acceptable standards of
quality, including limits of impurities (organic, inorganic, residual solvents)

Active Pharmaceutical Ingredients 25 |
The information required for the synthesis of the API may depend onThe information required for the synthesis of the API may depend on
Is a valid CEP is available? - no synthesis information required. CEP must however have all appendices and applicant to submit other info not covered by CEP
Is the quality of the API controlled by a monograph in an acknowledged pharmacopoeia?
No official monograph is available for quality control
- Detailed information required e.g. Open Part of DMF (from API manufacturer)
- Also signed declaration from API manuf that synthesis and purification are as described in the dossier

Active Pharmaceutical Ingredients 26 |
Synthesis non-compendial APIsSynthesis non-compendial APIs
A flow diagram of the synthesis process including structures & stereochemistry of starting materials & intermediates;
reagents; catalysts; solvents
A full description of each step / process, including: Reaction conditions (temp., time, moisture control, etc.) Quantities of reagents/solvents Size of production scale Purification of intermediates Final API purification method / crystallisation / solvent(s) Reprocessing (has to be justified, validated) Process controls Validation of critical steps, e.g. aseptic processes Discussion of (possible) process impurities
Organic, residual solvents and catalysts/inorganic

Active Pharmaceutical Ingredients 27 |
Specifications of raw materials and intermediates used in synthesisSpecifications of raw materials and intermediates used in synthesis
Provide specifications for starting materials and intermediates (if isolated) reagents, solvents & catalysts
Class 1 solvents should not be used (ICH Q3C) Benzene, Carbon tetrachloride, 1,2-Dichloroethane,
1,1-Dichloroethene & 1,1,1-Trichloroethane
Provide a declaration on the use/non-use of material of animal or human origin (TSE) Risk of Transmitting Animal Spongiform Encephalopathy Agents
(WHO TRS 908, Annex 1 or EMEA/410/01 Rev.2)
To limit impurities in the API (Safety reasons)

Active Pharmaceutical Ingredients 28 |
WHAT IS A STARTING MATERIAL?WHAT IS A STARTING MATERIAL?
Contributes an important structural part of the API
Available in free trade
Compound well defined in chemical literature (name, chemical structure, chemical and physical properties, and impurity profile)
Synthesized by commonly known process

Active Pharmaceutical Ingredients 29 |
RE-DEFINITION OF STARTING MATERIALRE-DEFINITION OF STARTING MATERIAL
MARKS THE START OF THE MANUFACTURING PROCESS DESCRIBED IN AN APPLICATION Manufacturing steps before are not described Manufacturing steps before need not be performed in accordance
with GMP Changes in manufacturing steps before need not be reported to
Agency
EACH BRANCH OF A SYNTHESIS WILL BEGIN WITH ONE OR MORE STARTING MATERIALS

Active Pharmaceutical Ingredients 30 |
INDINAVIRINDINAVIR

Active Pharmaceutical Ingredients 31 |
CHEMICAL SYNTHESISCHEMICAL SYNTHESIS
• Indinavir is a chiral molecule with 5 stereogenic centers
• Only stereoisomer observed in the API is the 4-(R)-epimer.
• It is stereoselectively prepared in six steps.
• The enantiomeric purity of the API and other ingredients is ensured by the route of manufacture and quality control on intermediate products (starting materials and intermediate indinavir free base) rather than a test for specific rotation.

Active Pharmaceutical Ingredients 32 |
CHEMICAL EQUIVALENCE (1)CHEMICAL EQUIVALENCE (1)
The stereochemistry is well under control during the synthesis
Racemization after the synthesis is extremely unlikely
Formation of epimers cannot be excluded but should be detectable by the purity tests applied.
Potential impurities from synthesis, stereoisomeric impurities and degradants have been identified. Minimised or removed by control on the reaction parameters and in-process controls
High humidity, which leads to formation of degradants, is avoided.

Active Pharmaceutical Ingredients 33 |
CHEMICAL EQUIVALENCE (2)CHEMICAL EQUIVALENCE (2)
The API is very pure
The limit for any single impurity is not more than 0.1 %
limit for the sum of all impurities is not more than 0.5 %
Due to the high doses to be given in clinical use (> 2 g/day), the qualification threshold as defined in the ICH guideline on impurities, is 0.05 %.

Active Pharmaceutical Ingredients 34 |
PHYSICOCHEMICAL EQUIVALENCEPHYSICOCHEMICAL EQUIVALENCE
Indinavir Sulfate Ethanolate
Freely Soluble In Aqueous Solutions
PARTICLE SIZE Not Critical
No POLYMORPHISM
POOR FLOWABILITY
Relatively Loose Bulk Density

Active Pharmaceutical Ingredients 35 |
API STABILITY TESTSAPI STABILITY TESTS
Indinavir is highly hygroscopic at relative humidity above 60 %
In the presence of moisture and/or elevated temperatures, the API undergoes conversion to an amorphous material or to a hydrate crystal form and to the formation of degradation products i.e. lactone and several unidentified impurities occur
HVAC SYSTEM SHOULD MAINTAIN A RELATIVE HUMIDITY OF ≤ 33% AT 25OC

NEVIRAPINE
Active Pharmaceutical Ingredients 37 |
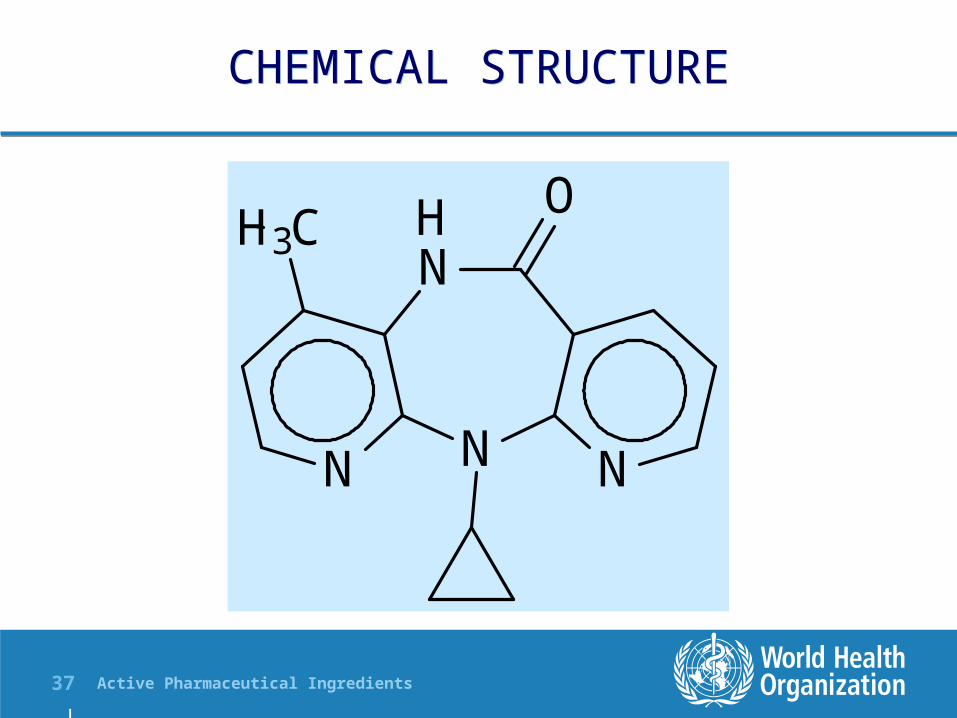
CHEMICAL STRUCTURECHEMICAL STRUCTURE
N
N
O
N
H3C
N
H

Active Pharmaceutical Ingredients 38 |
CHEMICAL STRUCTURECHEMICAL STRUCTURE
Nevirapine does not contain an assymetric carbon atom (a chiral centre)
The nitrogen in position 11 shows weekly basic properties
Other functional groups are not very reactive under everyday manufacturing environmental conditions

Active Pharmaceutical Ingredients 39 |
CHEMICAL INFORMATIONCHEMICAL INFORMATION
C15H14N4O (anhydrate for tablets) 266.30
C15H14N4O·1/2 H2O (hemihydrate
for oral suspension) 275.35
CAS number: 129618-40-2
NEVIRAPINE is lipophilic (partition coefficient 83) and is essentially nonionized at physiologic pH. As a weak base (pKa 2.8), NEVIRAPINE is known to be soluble at acidic pH
values.

Active Pharmaceutical Ingredients 40 |
PHYSICO-CHEMICAL INFORMATIONPHYSICO-CHEMICAL INFORMATION
Aqueous solubility (anhydrate) (90 μg/ml at 25°C).
NEVIRAPINE anhydrous is a white to off-white crystalline powder.
No potential toxicity was found in intermediates found in the synthesis of NEVIRAPINE
NEVIRAPINE is milled in order to obtain an acceptable particle size distribution.

Active Pharmaceutical Ingredients 41 |
SPECIFICATION, STABILITYSPECIFICATION, STABILITY
Innovator results showed that Nevirapine is highly stable even under stressed conditions over a 24 month study period
No degradants were detected and all the results remained within the specifications.

Active Pharmaceutical Ingredients 42 |
DESK CONCLUSIONDESK CONCLUSION
Critical API parameters:
Particle size of the micronized drug substance

Active Pharmaceutical Ingredients 43 |
API specificationsAPI specifications
API not described in BP, Int.Ph., JP, Ph.Eur., or USP (non-compendial APIs)
API described in BP, Int.Ph., JP, Ph.Eur., or USP (compendial APIs)
General note
An API has only one set of specifications applicable at release and throughout the re-test period– an FPP may have two sets of specifications – release and shelf-life

Active Pharmaceutical Ingredients 44 |
Specifications: Non-Compendial APIsSpecifications: Non-Compendial APIs
ICH Q6A (new APIs and products) – for instance:
Requires justification for proposed specifications
Impurities to be characterised and limits set synthesis and degradation according to ICH Q3A(R) residual solvents according to ICH Q3C
Analytical methods with validation
Preparation and potency determination/specification of primary and secondary (working) standards, with CoAs
Valid CoAs for at least 2 batches

Active Pharmaceutical Ingredients 45 |
Non-compendial APIsTypical set of specifications
Non-compendial APIsTypical set of specifications
Appearance/description
Identification (at least one specific, e.g. IR spectrum)
Moisture content (or LOD: moisture + residual solvents)
Impurities - Related organic substances (synthesis or degradation)
specified unspecified and total organic impurities
- Inorganic impurities, including catalysts - Residual solvent(s)
Assay
Additional parameters important for specific API such as particle size, polymorphic form, microbial limits

Active Pharmaceutical Ingredients 46 |
Specs: Compendial APIsSpecs: Compendial APIs
The current monograph always applicable
Additional critical specifications that are not included in monograph e.g.– particle size & polymorphic form– synthesis related impurities resulting from specific process which
may be additional to monograph– residual solvents (specific to process)
Valid CoAs for at least 2 batches required
CEP normally states tests additional to the monograph – e.g. residual solvents & impurities

Active Pharmaceutical Ingredients 47 |
IMPURITIESIMPURITIES
Extraneous contaminant (foreign substances)
Toxic impurities
Concomitant components
Signal impurities

Active Pharmaceutical Ingredients 48 |
Classes of ImpuritiesClasses of Impurities
Organic
Inorganic
Residual solvents

Active Pharmaceutical Ingredients 49 |
Organic ImpuritiesOrganic Impurities
May arise during manufacturing process and storage
Starting materials
By products
Intermediates
Degradation products
Reagents, ligands and catalysts

Active Pharmaceutical Ingredients 50 |
Inorganic ImpuritiesInorganic Impurities
May be from manufacturing process and are normally known and identified:
Reagents, ligands and catalysts
Heavy metals
Inorganic salts
other materials (e.g. filter aids, charcoal etc.)

Active Pharmaceutical Ingredients 51 |
SolventsSolvents
Organic or inorganic liquids used during the manufacturing process
Toxicity generally known, therefore controls achievable
Limits to be based on pharmacopoeial standards or known safety data

Active Pharmaceutical Ingredients 52 |
IMPURITIESIMPURITIES
Identified impurity
Unidentified impurity
Specified impurity
Unspecified impurity

Active Pharmaceutical Ingredients 53 |
IMPURITY THRESHOLDSIMPURITY THRESHOLDS
Maximum daily dose
Reporting
threshold
Identification threshold
Qualification threshold
<= 2g/day 0.05%
0.10% or 1mg/day
intake
0.15% or 1mg/day
intake
>= 2g/day 0.03% 0.05% 0.05%

Active Pharmaceutical Ingredients 54 |
IMPURITY EQUIVALENCEIMPURITY EQUIVALENCE
No new impurity is observed in the intermediate above 0.1%
No new impurity is observed in api above the qualification threshold
Each existing impurity is within its stated limit
Total impurities are within the stated limit

Active Pharmaceutical Ingredients 55 |
IMPURITY EQUIVALENCEIMPURITY EQUIVALENCE
Each existing residual solvent is within its stated limit
New residual solvents, in either an intermediate or the api, are at or below the levels recommended in the ich guide

Active Pharmaceutical Ingredients 56 |
IMPURITY EQUIVALENCEIMPURITY EQUIVALENCE
Ideally, impurities should be evaluated in isolated intermediates immediately following the process step in which they are produced
The impurity search can be extended to the next downstream intermediate and the evaluation process repeated until the final intermediate, even to the api

Active Pharmaceutical Ingredients 57 |
Stability testingStability testing
Stress testing of API (forced degradation) helps to identify the likely degradation products and pathways to establish stability of the molecule To verify specificity of stability assay method
Diode array detection for API peak purity !
Stability testing (regulatory) to provide evidence on how the quality of an API varies with time
under the influence of a variety of environmental factors such as temperature, humidity, and light; and
to establish a re-test period for the API and to recommended storage conditions

Active Pharmaceutical Ingredients 58 |
Stress testing (forced degradation) Typical conditions
Stress testing (forced degradation) Typical conditions
The conditions should
partially (e.g. 10-30%) decompose the API to primary degradation products
Conditions can be changed to get required degree of degradation
** Temperature should not come closer than 10°C from melting point
Stress factor Conditions (e.g.)
Humidity ≥ 75% RH (solid)
Heat ** ≥ 60°C (solid)
Heat water
Acid 0.1 M HCl
Base 0.1 M NaOH
Oxidative 3% H2O2
Photolytic ICH Q1B
Metal ions (optional)
0.05 M Fe2+ or Cu2+

Active Pharmaceutical Ingredients 59 |
Stress testing (forced degradation)Literature
Stress testing (forced degradation)Literature
Literature information and/or CEP– in support of and/or– to replace experimental data
Examples of literature information1. Rifampicin (earlier slides)
Oxidation, hydrolysis, light sensitivity2. Indinavir sulfate (earlier slide)
Intra-molecular reaction – heat, moisture, acid, base3. Efavirenz (see next slides)
Hydrolysis – pH dependent

Active Pharmaceutical Ingredients 60 |
Stress testing (forced degradation)Efavirenz (1)
Stress testing (forced degradation)Efavirenz (1)
Non-hygroscopic 4 Polymorphs
• Form 1 pharmaceutical(EPAR Sustiva®)
Hydrolysis main degradation– pH dependent
– Maximum stability at pH 4
– 2 Degradants isolated• structures elucidated
– Pathways postulated
Maurin, Pharm. Res. 19, 517 (2002)

Active Pharmaceutical Ingredients 61 |
Stress testing (forced degradation)Efavirenz (2)
Stress testing (forced degradation)Efavirenz (2)
The data (generated at 60°C) shows that
– Efavirenz is quite stable
– Maximum stability at pH 4 (Suspension possible?)
– Carbon dioxide formation
(30 mg/ml solution, 100 ml bottle:
1% decomposition ≈ 2 ml CO2

Active Pharmaceutical Ingredients 62 |
Efavirenz main route of degradation
Maurin (2002)
Efavirenz main route of degradation
Maurin (2002)
2nd route
1
2 + CO2 (g)

Active Pharmaceutical Ingredients 63 |
Important ElementsImportant Elements
The API must be of required structure & stereochemistry
The physical properties must be well understood, e.g.– hygroscopicity, crystal properties and solubility
The synthesis process must be according to GMP to– consistently produce an API of required chemical and physical
quality– limit impurities according to defined standards

Active Pharmaceutical Ingredients 64 |
Important Elements (2)Important Elements (2)
The set of specifications should be based on validated analytical methods with appropriate acceptance criteria to which an API should conform to be considered acceptable
for its intended use throughout the retest period in the proposed packaging


















