
Entropy in Bi-Substrate Enzymes: Proposed Role of an .... Mol. Biol. (1996) 255 , 522Ð535 Entropy...
14
J. Mol. Biol. (1996) 255, 522–535 Entropy in Bi-substrate Enzymes: Proposed Role of an Alternate Site in Chaperoning Substrate into, and Products out of, Thymidylate Synthase David L. Birdsall*, Janet Finer-Moore and Robert M. Stroud Three steps along the pathway of binding, orientation of substrates and Department of Biochemistry and Biophysics, University of release of products are revealed by X-ray crystallographic structures of California, San Francisco ternary complexes of the wild-type Lactobacillus casei thymidylate synthase CA 94143-0448, USA enzyme. Each complex was formed by diffusion of either the cofactor 5,10-methylene-5,6,7,8-tetrahydrofolate or the folate analog 10-propargyl- 5,8-dideazafolate into binary co-crystals of thymidylate synthase with 2'-deoxyuridine-5'-monophosphate. A two-substrate/enzyme complex is formed where the substrates remain unaltered. The imidazolidine ring is unopened and the pterin of the 5,10-methylene-5,6,7,8-tetrahydrofolate cofactor binds at an unproductive ‘‘alternate’’ site. We propose that the presence of the pterin at this site may represent an initial interaction with the enzyme that precedes all catalytic events. The structure of the 2'-deoxyuridine-5'-monophosphate and 10-propargyl-5,8-dideazafolate folate analog complex identifies both ligands in orientations favorable for the initiation of catalysis and resembles the productive complex. A product complex where the ligands have been converted into products of the thymidylate synthase reaction within the crystal, 2'-deoxythymidine-5'- monophosphate and 7,8-dihydrofolate, shows how ligands are situated within the enzyme after catalysis and on the way to product release. 1996 Academic Press Limited *Corresponding author Keywords: thymidylate synthase; X-ray crystallography; enzyme; catalysis Introduction In bi-substrate enzymatic reactions mutual align- ment of substrates against one another prior to catalysis is a major factor in catalytic rate enhancement. Thymidylate synthase (TS) closes around its substrates to sequester and orient them in a major conformational change evoked primarily by binding CH 2 H 4 folate (Kamb et al ., 1992). This change is also dependent on the carboxy terminal COO − group on the enzyme (Perry et al ., 1993). Thus mechanisms by which both substrates are bound and oriented prior to catalysis are under selective pressure for optimization. Yet such mechanisms for binding can rarely be delineated by structural analysis. Three structures of thymidylate synthase elucidate mechanisms of bi-substrate binding and suggest mechanisms by which selective expulsion of products takes place. Intermolecular reactions are catalyzed most effectively by the remarkably high effective concen- tration of reactive groups on the substrates once bound correctly such that catalysis becomes effectively an intramolecular reaction. This contri- bution to enzymatic reaction rate is entropic, and the rate enhancement, relative to the rate at which the same reaction would take place in solution, can be estimated by calculating the entropy change due to fixation of the second substrate against the first (Page and Jencks, 1971; Fersht, 1985). In TS, mutual orientation of substrates is followed by methylene transfer of (CH 2 ) 11 of CH 2 H 4 folate to C5 of dUMP, and finally by hydride transfer from cofactor to the transferred methylene to form the products dTMP and the oxidized form of the cofactor, H 2 folate (Stroud & Finer-Moore, 1993). Conformational changes accompany each step of this reaction, as Abbreviations used: TS, thymidylate synthase; LCTS, Lactobacillus casei TS; ECTS, Escherichia coli TS; CH2THF, CH2H4folate, 5,10-methylene-5,6,7,8- tetrahydrofolate; DHF, H2folate, 7,8-dihydrofolate; CB3717, 10-propargyl-5,8-dideazafolate; PABA, para-amino benzoic acid; dUMP, 2'-deoxyuridine- 5'-monophosphate; dTMP, 2'-deoxythymidine-5'- monophosphate; FdUMP, 5-fluoro-2'deoxyuridine- 5'-monophosphate; DTT, dithiothreitol; rms, root mean square; wt, wild-type. 0022–2836/96/030522–14 $12.00/0 1996 Academic Press Limited
Transcript of Entropy in Bi-Substrate Enzymes: Proposed Role of an .... Mol. Biol. (1996) 255 , 522Ð535 Entropy...

J. Mol. Biol. (1996) 255, 522–535
Entropy in Bi-substrate Enzymes: Proposed Role ofan Alternate Site in Chaperoning Substrate into, andProducts out of, Thymidylate SynthaseDavid L. Birdsall*, Janet Finer-Moore and Robert M. Stroud
Three steps along the pathway of binding, orientation of substrates andDepartment of Biochemistryand Biophysics, University of release of products are revealed by X-ray crystallographic structures ofCalifornia, San Francisco ternary complexes of the wild-type Lactobacillus casei thymidylate synthaseCA 94143-0448, USA enzyme. Each complex was formed by diffusion of either the cofactor
5,10-methylene-5,6,7,8-tetrahydrofolate or the folate analog 10-propargyl-5,8-dideazafolate into binary co-crystals of thymidylate synthase with2'-deoxyuridine-5'-monophosphate. A two-substrate/enzyme complex isformed where the substrates remain unaltered. The imidazolidine ring isunopened and the pterin of the 5,10-methylene-5,6,7,8-tetrahydrofolatecofactor binds at an unproductive ‘‘alternate’’ site. We propose that thepresence of the pterin at this site may represent an initial interactionwith the enzyme that precedes all catalytic events. The structure of the2'-deoxyuridine-5'-monophosphate and 10-propargyl-5,8-dideazafolatefolate analog complex identifies both ligands in orientations favorable forthe initiation of catalysis and resembles the productive complex. A productcomplex where the ligands have been converted into products of thethymidylate synthase reaction within the crystal, 2'-deoxythymidine-5'-monophosphate and 7,8-dihydrofolate, shows how ligands are situatedwithin the enzyme after catalysis and on the way to product release.
! 1996 Academic Press Limited
*Corresponding author Keywords: thymidylate synthase; X-ray crystallography; enzyme; catalysis
IntroductionIn bi-substrate enzymatic reactions mutual align-
ment of substrates against one another prior tocatalysis is a major factor in catalytic rateenhancement. Thymidylate synthase (TS) closesaround its substrates to sequester and orient themin a major conformational change evoked primarilyby binding CH2H4folate (Kamb et al., 1992). Thischange is also dependent on the carboxy terminalCOO− group on the enzyme (Perry et al., 1993). Thusmechanisms by which both substrates are boundand oriented prior to catalysis are under selective
pressure for optimization. Yet such mechanisms forbinding can rarely be delineated by structuralanalysis. Three structures of thymidylate synthaseelucidate mechanisms of bi-substrate binding andsuggest mechanisms by which selective expulsionof products takes place.Intermolecular reactions are catalyzed most
effectively by the remarkably high effective concen-tration of reactive groups on the substrates oncebound correctly such that catalysis becomeseffectively an intramolecular reaction. This contri-bution to enzymatic reaction rate is entropic, andthe rate enhancement, relative to the rate at whichthe same reaction would take place in solution, canbe estimated by calculating the entropy change dueto fixation of the second substrate against the first(Page and Jencks, 1971; Fersht, 1985). In TS, mutualorientation of substrates is followed by methylenetransfer of (CH2)11 of CH2H4folate to C5 of dUMP,and finally by hydride transfer from cofactor to thetransferred methylene to form the products dTMPand the oxidized form of the cofactor, H2folate(Stroud & Finer-Moore, 1993). Conformationalchanges accompany each step of this reaction, as
Abbreviations used: TS, thymidylate synthase; LCTS,Lactobacillus casei TS; ECTS, Escherichia coli TS;CH2THF, CH2H4folate, 5,10-methylene-5,6,7,8-tetrahydrofolate; DHF, H2folate, 7,8-dihydrofolate;CB3717, 10-propargyl-5,8-dideazafolate; PABA,para-amino benzoic acid; dUMP, 2'-deoxyuridine-5'-monophosphate; dTMP, 2'-deoxythymidine-5'-monophosphate; FdUMP, 5-fluoro-2'deoxyuridine-5'-monophosphate; DTT, dithiothreitol; rms, root meansquare; wt, wild-type.
0022–2836/96/030522–14 $12.00/0 ! 1996 Academic Press Limited

Thymidylate Synthase Ternary Complex Structures 523
revealed by comparison of crystal structures ofunliganded enzyme, substrate-bound binary com-plex, cofactor-bound binary complex, and ternarycomplexes with both substrate and cofactor boundto the enzyme (Montfort et al., 1990; Matthews et al.,1990b; Kamb et al., 1992; Stroud & Finer-Moore,1993). The largest conformational change occurswhen cofactor binds. Thus CH2H4folate-bound TSgenerally crystallizes in different crystal forms thanunliganded or nucleotide-bound TS. When cofactor(or anti-folate analogs) is soaked into crystals ofTS·dUMP, the crystals usually crack or dissolve. Thepresence of cofactor shifts the equilibrium from theinitial loose ‘‘adsorption’’ ternary complex in orderto stabilize a tight, covalent complex where theactive site cysteine becomes bonded to C6 of dUMPthrough Michael addition (Ivanetich & Santi, 1992)and the 11-methylene group of CH2H4-folate isbonded to C5 of dUMP. With antifolates such asCB3717, TS can be trapped after the Michaeladdition at C6 of dUMP (Montfort et al., 1990) orprior to addition (this work). The covalent complexof TS with CH2H4folate and an analog of thesubstrate, FdUMP, becomes a covalent transitionstate analog (Matthews et al., 1990b).The three-dimensional structures of three steps in
alignment of dUMP and either folate or antifolateshow ligands bound to TS prior to complete closureof the active site cavity and after enzymaticallycatalyzed product formation in the crystals. Thesestructures define a mechanism for sandwichingsubstrate and CH2H4folate, and a mechanism forselective expulsion of products.
Results
A structure approximating initial ternarycomplex formationTo determine the structure of a product complex,
data were collected on a wild-type TS·dUMPco-crystal showing the least topical damage aftersoaking with folate cofactor. The resulting complexrevealed from the X-ray analysis, however, approxi-mates an initial interaction of folate with TS·dUMPwhere the pterin moiety of the folate moleculeretains a closed imidazolidine ring and binds at analternate site (Figures 2(a) and 4(a)). The torsionangles relating the pterin and PABA rings,N5–C6–C9–N10, C6–C9–N10–C11, and C9–C10–C11–C12, are 39°, 108°, and 152°, respectively,compared to torsion angles of !30°, !50°, and!180° for bound folates with no imidazolidine ring(Matthews et al., 1990a). In other words, the twoconformations differ primarily by a !50° rotationabout the C9–N10 bond. In addition, the entirecofactor is shifted towards dUMP by 2.5 A. Thisconformation for CH2THF places the pterin andimidazolidine rings close to, and overlapping, thebinding of the quinazoline moiety of folate analogCB3717 for an Escherichia coli wild-type TS ternarycomplex formed under non-reducing conditions(Montfort et al., 1990). In this case the complex wasformed under reducing conditions via the additionof DTT to the crystallization and cofactor solutions,which leads us to conclude that this alternate sitemay be mechanistically important rather than anartifact due to the oxidation of cysteine residues.A form (Figure 1) of the pterin with a closed
five-membered imidazolidine ring, including aC11–N10 bond, could best be refined at this positionto match the X-ray data. The reduced pterin issurrounded by the side-chains of very highlyconserved hydrophobic residues, Ile81, Trp82, Trp85and Leu195. The exocyclic oxygen and amine of thepterin can make hydrogen bonds to His199. Theexocyclic amine may also hydrogen bond to anordered water molecule (Figure 4(a)). With thepterin at this position the pyrimidine base is pushedfarther away from the protein, prohibiting covalentattachment. The nucleotide, however, is in an anticonformation and the ribose positioning is stabil-ized by a hydrogen bond between the O3' atom andthe hydroxyl group of Tyr261, as in other binary andternary complexes of TS. Water molecules replacethe pterin at its normal productive binding site. ThePABA ring may form hydrophobic contacts withresidues Phe228 and Ile81, and is perpendicularto the Phe228 side-chain. The methylene carbon
Figure 1. Chemical structures of anti-folate CB3717,dihydrofolate (DHF), and 5-methylene tetrahydrofolate(CH2THF) with a closed imidazolidine ring. The exocylichydroxyl oxygen atoms for the quinazoline and CH2THFpterin moieties were considered to be ionized duringX-PLOR refinements.

(a)
(b)
(c)
Figure 2. ‘‘Wall-eyed’’ stereo drawings of LCTS active sites with ligands: 2.5 A Fo − Fc omit map densities for the ligandsare shown. Ligands and water molecules were removed and protein was subjected to positional refinement to eliminatephase biases. (a) LCTS·dUMP·CH2THF substrate complex (map contoured to 2.3!). (b) LCTS·dUMP·CB3717 folate analogcomplex (map contoured to 2.3!). (c) LCTS·dTMP·DHF product complex (map contoured to 2.0!). Water molecules aredepicted as filled spheres. Each figure was rendered using the MOLSCRIPT graphics program (Kraulis, 1991).

Thymidylate Synthase Ternary Complex Structures 525
(a) (b)
(c) (d)
Figure 3. Comparisons of orientations of TS ternary complex ligands. LCTS ligands are gray; ECTS ligands are black.(a) LCTS substrate and ECTS folate analog complexes. (b) LCTS and ECTS folate analog complexes. (c) LCTS folateanalog and ECTS product complexes. (d) LCTS and ECTS product complexes. Figures 3 and 4 were created using theMIDAS graphics display system (Ferrin et al., 1988) with space-filling rendition (Huang et al., 1991).
atom C11 faces, at a distance of 4.4 A, the C5 atomof dUMP to which it is transferred at the time of theopening of the imidazolidine ring to form an openpterin. Aside from the adjustment of the dUMPbase orientation, the pterin binding at the alternatesite does not appear to alter the over-all structure ofthe active site cavity very much and the C terminusis pulled towards a ‘‘closed’’ position (Table 1). A
hydrogen bond between the side-chains of residuesCys198 and Arg218 is observed, polarizing thereactive thiol group of Cys198. The guanidiniumgroup of Arg23 helps to coordinate the dUMPphosphate, and also interacts with the C terminus,mediated by a water molecule.The binding of the pterin at the alternate site is
probably responsible for the lack of product

Thymidylate Synthase Ternary Complex Structures526
formation within the co-crystal, since no structuralevidence of dTMP formation could be found. Thiswould be characteristic of a treated crystal showingonly minimal lattice disruption. Difference Fourierelectron density maps also suggest binding of thepterin at the normal site at some occupancy, but ithas not yet been determined whether the imidazo-lidine ring has opened in the folate that is boundthere.
A folate analog ternary complex structuremimics a stage in the TS reaction justpreceding catalysisThe LCTS·dUMP·CB3717 active site structure
shows the ligands in orientations favorable for theinitiation of catalysis. The L. casei† enzyme presentsitself in a conformation where the C terminus ismoved into, and partially closes, down the activesite and sequesters substrates from solution (Fig-ure 4(b)). The dUMP nucleotide is in the lessconstrained anti conformation normally found in TSternary complexes. Its phosphate is coordinated byArg218, Arg23 and Ser219 of one monomer (Table 2)and Arg178' and Arg179' of the other monomer. Theplane of the pyrimidine base runs almost parallel tothat of the quiniazoline ring of the CB3717, withsome overlapping of the planar ring systems (Fig-ure 3(b)). Unlike ternary complexes formed byco-crystallizations, the nucleotide is not covalentlyattached to the protein. The C6 atom of thepyrimidine base lies more than 3 A from thereactive thiol of Cys198 and difference electrondensity maps do not suggest covalency. This allowsthe pyrimidine ring to lie closer over thequinazoline ring than for the corresponding E. coilcomplex (Figure 3(b)).The CB3717 molecule makes many of the same
electrostatic and hydrophobic contacts with theprotein observed in E. coli TS ternary complexco-crystals. The glutamate residue of CB3717 makeselectrostatic contacts to basic residue Lys50. ThePABA ring moiety lies under and angled to theside-chain of Phe228 and is surrounded byhydrophobic residues Ile81, Gly225, and Leu224.Isoleucines 57 and 81 along with Phe228 cluster overthe PABA and propargyl moieties of the folateanalog. The propargyl moiety points towards thePhe228 side-chain and lies above the pyrimidinebase. The quinazoline ring is surrounded on oneside by the aromatic side-chain of Trp82 and Trp85;however, the indole groups are not close enough tomake direct contacts with it. The quinazoline ringspoints in a direction towards the C terminus and theArg23 guanidinium group, and away from theglutamate moiety to give the folate analog moleculea roughly trans configuration. As in the E. coli TSco-crystals the exocyclic amine of the quinazolinering can hydrogen bond to the backbone carboxyl
oxygen of Ala315 (3.4 A). The Tyr261 side-chainhydroxyl and the O3' atom of the nucleotide ribose(Figure 4(b)) are also close enough to the exocyclicamine to hydrogen bond to it. This helps to stabilizeits positioning in the active site. N1 of the pterin ishydrogen bonded to N"2 of Arg23, while in E. coliTS co-crystals an ordered water mediates the contactbetween N1 and the guanidinium group of Arg23.In summary, the folate analog is stacked against thedUMP base and makes many of the same proteincontracts as seen in the covalent E. coli TS·-dUMP·CB3717 complex, but there is no covalentbond formed between Cys198 and C6 of thesubstrate dUMP.
The enzyme/product complex, on the way toproduct releaseVariable results occur upon the soaking of
cofactor with wild-type TS co-crystallized with itssubstrate, dUMP, as judged by the deterioration ofthe crystals shown by data collection statistics. Thesource of this variation may be due to differencesin the ease of diffusion of cofactor into binaryco-crystals due to crystal size and minor fluctu-ations of room temperature (which may affect theviscosity of the crystallization solution), variablesthat were not controlled during the experiments. Amore perturbed crystal structure represents greaterconformational changes (from the binary complex)within the protein, which may be related to changesin ligand binding and orientation.In one case, when cofactor is added to crystals of
the TS/substrate complex, the products, dTMP andH2folate, are clearly present at the active site in thecrystal structure. Analysis of the X-ray structuraldata reveals the conversion of dUMP andCH2H4folate to dTMP and DHF at the active site(Figure 2(c)). Thus, the enzyme reaction mayproceed at a slow rate even in the absence ofcomplete closure of the active site cavity, aftersoaking of binary complex crystals of L. casei TS inlow concentrations of cofactor.The active site cavity is ‘‘closed’’ off by movement
of the C-terminal residues (313 to 316) towards theactive site region (Table 1). The terminal carboxylicacid forms a possible intermolecular contact withthe dihydrofolate (DHF) pterin (Table 2). The Cterminus forms a salt bridge with the side-chain ofTyr261. Like the other two ternary complexesstudied, a water molecule hydrogen bonds to one ofthe terminal carboxylic acid oxygen atoms (Fig-ure 4(c)) to help anchor the C terminus.Modification of the nucleotide does not disturb its
over-all binding at the active site. The dTMPproduct is situated like the dUMP substrate with noapparent movement of the phosphate moiety (Fig-ure 4(c)). The dTMP nucleotide is in an unstrainedconformation with the pyrimidine base anti to aC-3'-endo ribose sugar. The plane of the dTMP baselies parallel to that of the pterin rings but with onlyvery slight overlapping of the ring systems. Thenucleotide phosphate is firmly coordinated by a
† L. casei protein residue numbering is used.Residues contributed from the second monomer of thedimer are shown by a prime.
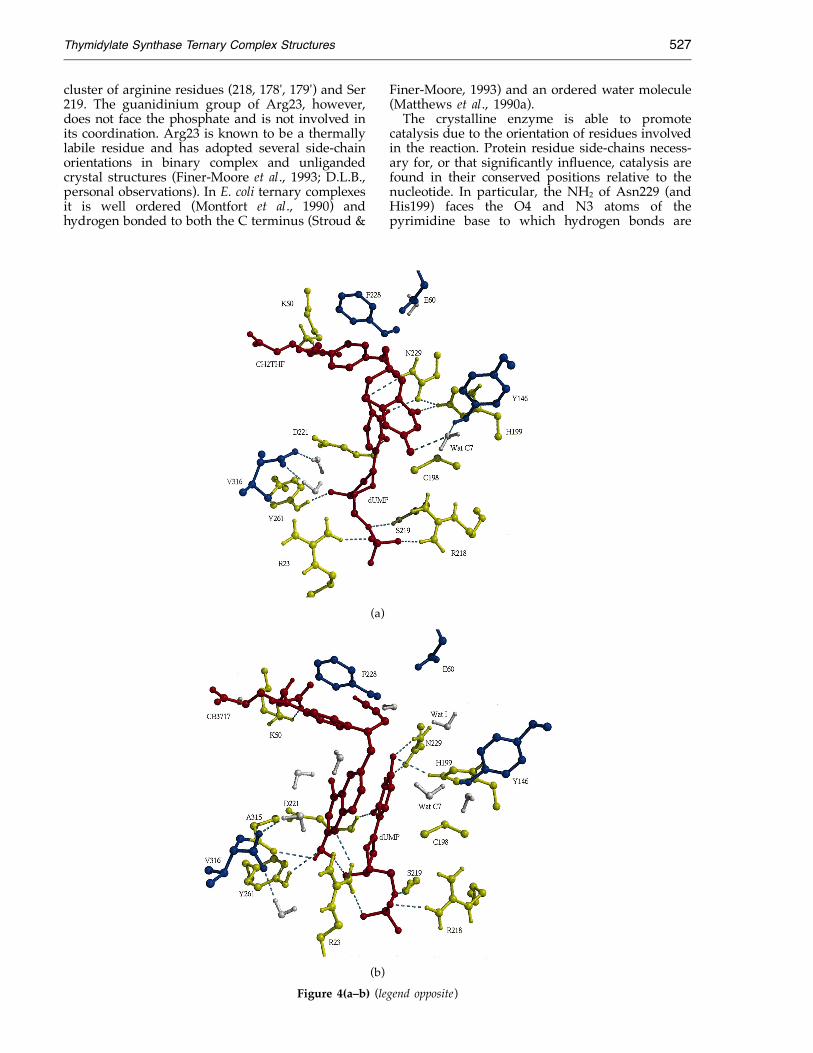
Thymidylate Synthase Ternary Complex Structures 527
cluster of arginine residues (218, 178', 179') and Ser219. The guanidinium group of Arg23, however,does not face the phosphate and is not involved inits coordination. Arg23 is known to be a thermallylabile residue and has adopted several side-chainorientations in binary complex and unligandedcrystal structures (Finer-Moore et al., 1993; D.L.B.,personal observations). In E. coli ternary complexesit is well ordered (Montfort et al., 1990) andhydrogen bonded to both the C terminus (Stroud &
Finer-Moore, 1993) and an ordered water molecule(Matthews et al., 1990a).The crystalline enzyme is able to promote
catalysis due to the orientation of residues involvedin the reaction. Protein residue side-chains necess-ary for, or that significantly influence, catalysis arefound in their conserved positions relative to thenucleotide. In particular, the NH2 of Asn229 (andHis199) faces the O4 and N3 atoms of thepyrimidine base to which hydrogen bonds are
(a)
(b)
Figure 4(a–b) (legend opposite)

Thymidylate Synthase Ternary Complex Structures528
Figure 4. Some intermolecularinteractions between ligands andprotein for the three wild-typeLCTS ternary complexes. (a) LCTS·-dUMP·CH2THF (closed imidazo-lidine). (b) LCTS·dUMP·CB3717. (c)
(c) LCTS·dTMP·DHF.
formed. The side-chain of Asn229 forms a saltbridge with the imidizole of His199, implicatinga role for His 199 in aiding catalysis byinfluencing the orientation of Asn229. The back-bone hydrogen of Asp221 hydrogen bonds to thedTMP base’s O2 atom, and the hydroxyl group ofthe Tyr261 side-chain may form a long hydrogenbond to O3' of the ribose. Similar to other TSternary complex structures, the imidizole ofHis259 helps to influence the orientation of theribose via a hydrogen bond to the O3' atom(Finer-Moore et al., 1993). The dTMP nucleotide isnot judged to be covalently linked to the proteinbecause its C6 atom lies more than 3 A from thereactive thiol group of Cys198, although electron
density maps show continuous density betweenthe two.Water molecules within the active site cavity are
probably involved in the enzymatic reaction. TheX-ray structure of the product complex reveals acoordination of water molecules to protein residuesthat is important for the catalytic conversion ofdUMP to dTMP. Mutation of Glu60 greatlycompromises enzymatic activity for both E. coli andLactobacillus casei TS (Zapf et al., 1993; Huang &Santi, 1994). While distant from the nucleotide inthe active site, the side-chain of Glu60 exerts itscatalytic effects through the mediation of awell-ordered water molecule (labeled Wat I inFigure 4(c) located between it and the Asn229,
Table 1. Segmental shifts in protein structure for L. casei TS ternarycomplexes
Averaged atomic shifts in complexes (A)a
Segment Residues CB3717 Product SubstrateHelix A 3–14 0.87 1.15 0.99Phosphate-binding loop 20–22 1.21 1.24 1.02Beta-strand i 23–30 2.12 2.52 2.15Folate-binding loop 44–54 0.68 0.88 0.70Helix J 219–231 0.64 0.83 0.72Beta-strand ii 256–261 0.71 1.04 0.69Helix K 262–271 0.77 1.10 0.94C-terminal loop 272–281 0.80 1.01 0.83C-terminal loop 302–307 0.64 0.94 0.91C-terminal loop 308–313 1.25 1.71 1.23C-terminal loop 314–316 2.52 3.67 3.89
a Comparison of protein from wild-type L. casei TS, dUMP binary complex(Finer-Moore et al., 1993) to protein of ternary complex: distances between atomicpositions of equivalent atoms within each segment were added together and this sumwas divided by the number of atoms within the segment.

Thymidylate Synthase Ternary Complex Structures 529
Table 2. Intermolecular interactions under 3.5 A between proteinmonomer and ligandsSubstrate complex
Distance DistanceCH2THF Protein/dUMP (A) dUMP Protein (A)N3 OH Tyr146 3.4 P H21 Arg23 2.7N3 H Cys198 3.0 P HG Ser219 3.0N3 SG Cys198 3.4 OP1 NH1 Arg218 3.1N8 O4 dUMP 3.3 OP1 NH2 Arg218 2.7NA2 O Pro196 3.2 OP1 OG Ser219 3.4NA2 SG Cys198 3.4 OP2 NH2 Arg23 3.0OA4 NE2 His199 2.7 OP3 H21 Arg23 3.0OA4 N3 dUMP 3.4 O5' H21 Arg23 2.9N10 O4 dUMP 3.2 O5' OG Ser219 3.2O1 N Gly225 2.5 O1' SG Cys198 3.4O2 NZ Lys50 3.0 O2 NE2 Gln217 3.2O2 O Phe223 3.2 O2 N Asp221 2.8.
N3H OD1 Asn229 2.6O3' HE2 His259 2.8O3' OH Tyr261 2.9O3' O water 2.7
Folate analog complexaDistance Distance
CB3717 Protein (A) dUMP Protein (A)N1 H22 Arg23 2.7 P NH1 Arg218 3.4NA2 OH Tyr261 3.4 P HG Ser219 3.3NA2 O Ala315 3.4 (3.1) OP1 NH2 Arg23 3.1CP3 O water 2.3 OP1 NH1 Arg218 3.4N O water 3.5 OP2 NH2 Ar23 3.4O H2 water 2.8 OP3 NH1 Arg218 2.9 (2.5)
O5' NH1 Arg218 3.3O5' OG Ser219 2.8O2 N Asp221 2.8 (2.9)N3 OD1 Asn229 3.0 (2.9)O4 ND2 Asn229 3.5 (3.1)O4 O water 3.4 (3.1)O3' NE2 His259 3.0 (2.7)
Product complexbDistance Distance
DHF Protein/dTMP (A) dTMP Protein (A)N1 O1' dTMP 3.4 P H12 Arg218 2.7N1 O3' dTMP 3.4 P Ser219 3.0N3 OD1 Asp221 2.8 (2.7) OP1 NH1 Arg218 3.1 (3.3)N5 N1 dTMP 3.2 OP1 OG Ser219 3.1N8 OT2 Val316 3.3 OP2 NH1 Arg218 3.2 (2.8)N8 O1' dTMP 3.5 OP3 O water 2.7NA2 OD1 Asp221 3.0 O5' OG Ser219 3.4NA2 O3' dTMP 2.9 O2 H Asp221 2.8N10 H2 water 2.9 N3 OD1 Asn229 3.2 (2.9)O1 O Leu224 3.2 C6 SG Cys198 3.2O2 O Leu224 3.1 O3' NE2 His259 3.2 (2.7)OE1 N Ala312 3.3
a Distance for corresponding ECTS ternary complex (Montfort et al., 1990).b Distance for corresponding ECTS ternary complex (Fauman et al., 1994).
His199 side-chains. This water molecule is hydro-gen bonded to the O#1 oxygen of the Glu60side-chain and also forms a long hydrogen bond tothe side-chain amine of Asn229 (3.7 A) as well as tothe O4 of dTMP (3.7 A). A long polar interaction(4.8 A) between Wat I and the His199 side-chaincompletes clustering of Glu60, Asn229, and His199side-chains about the pyrimidine base. Anotherwater molecule is relatively close (<3.5 A) to the C7methyl group of dTMP and is displaced by about1 A relative to the analogous water molecule in thesubstrate/dUMP complex. This water molecule(labeled Wat C7 in Figure 4(c)) is hydrogen bondedto the Tyr146 hydroxyl group. Steric conflict
between Wat C7 and the transferred methyleneduring catalysis may be used to distinguish dUMPfrom dTMP (Fauman et al., 1994). This watermolecule may also be involved in the dehalogina-tion reaction of dUMP brominated at the C5position (Liu & Santi, 1993), since it is close enoughto allow for the formation of the correspondinghalogen acid (Garret et al., 1979). The C7 methylgroup of dTMP is relatively close to one of theLeu195 side-chain carbon atoms (3.4 A). This maysuggest that Leu195 is involved in stabilizing theformation of dTMP via hydrophobic interactionswith the nascently transferred methylene group toC5 of the pyrimidine base. Leu195 may also be a

Thymidylate Synthase Ternary Complex Structures530
Table 3. Data collection and refinement statisticsData collection wt-dUMP-CB3717 wt-dUMP-CH2THP wt-dTMP-DHPUnit cell lengths (P6122) (A)a, b 78.7 78.9 78.6c 230.1 229.7 229.4
No. reflectionsa 8992 9061 8043R-factorb 0.185 0.204 0.220Rmergec 0.108 0.098 0.196CompletenessdTo 2.5 A 58.9 58.5 52.62.5 A shell (I/!) 26.5 (1.70) 27.9 (1.78) 28.7 (1.60)To 3.0 A 76.5 74.5 66.73.0 A shell (I/!) 64.1 (2.73) 62.3 (3.31) 52.5 (2.11)
Average B-factors after refinement (A2)Backbone 14.1 12.1 6.7Side-chain 14.9 13.2 4.3Nucleotide 21.7 18.2 2.5Folate 26.2 29.7 2.6Water (no.) 14.0 (125) 13.4 (30) 10.4 (39)
rms deviations from idealityBonds >0.06 A 0.022 0.024 0.029Number found 49 58 118
Angles >10° 4.262 4.559 4.963Number found 113 156 211
Dihedrals >90° 26.66 26.90 27.42Violations None None None
Impropers >20° 1.737 1.724 1.950Violations None None None
rms differences from initial protein structureBackbone atoms 0.655 0.761 0.919Side-chain atoms 1.304 1.510 1.711
a Resolution range 15 A to 2.5 A with 1.0 sigma-cutoff.b R-factor = $%(%Fo% − %Fc%)%hkl/$%Fo%hkl.c R-merge = ($$%F2(i ) − &F2(h)'%)/$$F2(i ), where F2(i ) = ith reflection intensity,
&F2(h)' = mean intensity of h, k and l.d Percentage of total reflections collected in resolution range 15 A to 3.0 A and 2.5 A and
for the resolution shells 3.0 A and 2.5 A.
factor in orienting the dTMP pyrimidine base forsubsequent release from the active site.Diffusion of CH2H4folate into the co-crystal
allows it to reach the enzyme’s active site and bindin a manner favorable for catalysis. The pterinmoiety of the cofactor binds to the enzyme in a waythat favors the methylene and hydride transfers. TheN5 atom of DHF faces the plane of the dTMP basein an orientation that suggests prior methylenetransfer to C5 of dUMP. This is also true for thepterin’s C6 atom, which once held the hydridetransferred to reduce the methylene. The N10 atomof the pterin lies directly over and in a plane almostperpendicular to C7 of the dTMP base. The indolerings of Trp82 and Trp85 stack around the pterin,and the exocyclic amine of the pterin is hydrogenbonded to Asp221 O(. Trp82 stabilizes the position-ing of the Glu60 side-chain by making a hydrogenbond at N#1–H (Finer-Moore et al., 1993; Montfortet al., 1990), although this interaction is notnecessary for catalysis, since E60A and E60Lmutants still show some activity (Huang & Santi,1994).The glutamate and PABA moieties of the cofactor
are not directly involved in catalysis. These twomoieties are distant from the nucleotide and are
most probably involved in the general positioningof the molecule within the active site cavity. Theglutamate of DHF appears firmly bound to theprotein at its binding site and is surrounded byseveral basic residues that counter its negativecharge. The PABA moiety is surrounded byhydrophobic residues Leu224, Ile8 and Phe228 andits orientation in the active site, while making itparallel to the Phe228 side-chain plane, gives it andthe entire folate molecule a sense of fluidity as ifthe cofactor had been trapped (within the crystalstructure) in a moment of intermolecular motion.The region of the PABA ring yields the leastconvincing electron density for the complex ingeneral. When the positions of DHF and dTMP aretaken together, one gets the impression that bothmolecules are approximating a stage in the TSreaction where they are about to leave the activesite (DHF more so), in contrast to the E. coliwild-type TS·dUMP·CB3717 ternary structure,where there is much more overlap of thepyrimidine base and pterin ring systems (Montfortet al., 1990). Stacking of the nucleotide base to thepterin ring aids proper orientation of both tofacilitate enzymatic catalysis (Montfort et al., 1990;Perry et al., 1993).

Thymidylate Synthase Ternary Complex Structures 531
Discussion
The initial non-productive binding sitefor folateWe propose that TS binds the cofactor at a
chaperone site, and that interactions betweencofactor and protein aid in transferring the pterinring into the productive catalytic site whilefacilitating ring opening in a critical step in thecatalytic reaction. This alternate site is similar to thatfound for CB3717 in E. coli TS complexes. However,in that structure a cysteine residue that is not in theactive sites of the dimer, present in E. coli TS but notin the L. casei TS analyzed here, had been eitheroxidized or modified by reaction with )-mercap-toethanol. In our case there is no equivalent cysteineresidue present in the L. casei enzyme andcontinuous maintenance of reducing potential, byDTT, precludes this particular modification. There-fore, binding at the alternate site by the cofactor, ina highly conserved cavity deeply buried in theenzyme, argues that this site is functional, and notartifactual as it could have been in the E. colianalysis. The hypothesis suggesting that this is abiologically meaningful initial binding site may befurther tested by studying mutations within theregion that, with this hypothesis, would be expectedto decrease significantly the rate of cofactor binding.It has long been suggested that CH2H4folate bindsto TS with an intact imidazolidine ring, and thatstrain incurred by less-than-ideal fit into thecofactor binding site contributes to ring opening(Santi et al., 1987; Matthews et al., 1990b). Asevidence for such a mechanism, it was noted thatwhen CH2H4folate is docked with its solutionconformation (Poe et al., 1979) into the E. coli TSactive site, with its pterin ring in a productiveorientation relative to the dUMP pyrimidine base asseen in crystal structures of TS ternary complexes,there are unacceptable contacts between the proteinand the PABA-glutamate moiety (Matthews et al.,1990b). These contacts, it was postulated, could berelieved by breaking the N10 to C11 bond to formthe iminium ion intermediate (Matthews et al.,1990b), close to the rate-limiting step of the reaction(Bruice & Santi, 1982).The structure of the TS complex shows that the
CH2H4folate molecule with the initially closedimidazolidine ring can bind the enzyme with itspterin ring at a highly conserved alternate site. Thisconformation differs from the proposed NMR/sol-ution conformation in that the PABA-glutamatemoieties are oriented towards the pterin rather thanaway from it. The six-membered tetrahydropy-razine ring, however, is still found in a half-chairconformation. Since the closed conformation ofCH2H4folate in the crystal to some degreeresembles its solution conformation, the alternatepterin site is likely to be part of the initial bindingsite for the cofactor. The carboxyl oxygen atom ofthe Glu60 side-chain is 3.5 A from N10 of theimidazolidine ring and is well-oriented to assist in
protonation of this atom, which necessarily accom-panies opening of the five-membered ring. ThusGlu60, which assists in breakdown of the transitionstate complex (Huang & Santi, 1994), may also havea role in the opening of the imidazolidine ring, asproposed earlier (Matthews et al., 1990b). Openingof the imidazolidine ring and movement of thepterin ring into a productive mode for catalysis arepresumably accompanied by the conformationalchange that completely closes the active site cavityand blocks the space occupied by the pterin ring inthe initial ternary complex.The initial, alternate site for the pterin may
therefore serve as a chaperone site for the pterinbefore the initiation of catalysis. Binding at thisposition may presage catalysis, since this forces thesubstrate nucleotide too far from the active siteCys198 for covalent bond formation. The observednon-competitive inhibition of TS by folate analogssuch as CB3717 (Pogolotti et al., 1986) andmethotrexate (Chello et al., 1976) may be due tobinding at the alternate site of one monomer of thedimer. The binding of these analogs at one activesite might transmit structural changes to the activesite of the other monomer that may hinder catalysis.While a monomer is the asymmetric unit for this
crystal’s space group, the enzyme itself exists asan obligate dimer and there is chemical evidencethat each active site can operate independently(Bradshaw & Dunlap, 1992), although both carboxytermini of the dimer must be intact (Cisneros et al.,1993). It is therefore possible that pterin binding tothe alternate site may be preferred for one monomerwhile the other is actively engaged in catalysis. Thisis indirectly supported by experimental resultsshowing that the nucleotide inhibitor FdUMP(which mimics dTMP) dissociates from the dimerasymmetrically with respect to active sites(Pogolotti et al., 1986) and that full activity ismeasured for asymmetric E. coli TS mutant dimerswith only one active site able to catalyze the reaction(Maley et al., 1995). It would be easier for a pterinmoiety to bind at the alternate site and affect thepyrimidine base positioning of dUMP beforeformation of the Michael adduct. Alternate sitebinding of the pterin would then precede productformation.
Movement of the C terminus can occurindependently of more global conformationalchanges that close the active site for catalysisDiffusion of CB3717 into crystalline TS-bound
dUMP induces a change in the C terminus of TS,but fails to evoke the major conformational changethat accompanies folate binding in solution. Apartfrom movement of the C terminus, only relativelyminor conformational changes in highly conservedresidues within the protein, such as His199 andTrp85, are necessary to allow for non-disruptivediffusion of CB3717 into binary complex crystalsand binding at the active site. In contrast, thestructures of co-crystallized ternary complexes,

Thymidylate Synthase Ternary Complex Structures532
where C6 of dUMP is covalently bound to Cys198,show multiple shifts of segments of the proteinrelative to unliganded TS, which serve to close theactive site and sequester the ligands from bulksolvent (Montfort et al., 1990). The C terminus is butone of many segments of chain that move, althoughits movement is the most dramatic.Initially, CB3717 acts as a competitive inhibitor
of CH2H4folate during a rapid pre-equilibriumformation of an initial ternary complex. Later,non-competitive inhibition kinetics are observedand a ternary complex is isolatable on nitrocellulosefilters (Pogolotti et al., 1986). The current LCTS·-dUMP·CB3717 crystal structure may representa stage of TS/ligand interactions indicative of theinitially observed competitive inhibition againstthe binding of folate cofactor in its catalyticallyfunctional site. The diffusion complex may alsorepresent a point on the reaction pathway thatwould just precede the formation of a covalentternary complex.Tykarska et al. (1986) diffused CH2H4folate
into P6122 crystals of L. casei TS-FdUMP with cellconstants a = 78 A and c = 242 A. In that case,crystals turned yellow and the c-axis decreasedin length to 235 A suggesting that the folate wasbound somehow in the crystals. The change inunit cell suggests that a larger conformationalchange occurred in this diffusion complex thanin the complexes whose structures are reportedhere. However, no structure was reported forthe Tykarska et al. (1986) diffusion complex.
Catalysis can take place within the L. caseiTS crystals
In one experiment substrates were presentedto the crystalline enzyme, but products were seenin the crystal structure. The diffusion methodemployed in these experiments assumes thatcofactor or folate analog molecules are able todiffuse into TS·dUMP co-crystals and bind atthe enzyme’s active site (productively for folate).It is most probable that dTMP was formed in thecrystal and not in the hanging drop solution:since (1) a low concentration of folate wasadded, so that any non-crystalline dTMP wouldhave been competed out by the great excessof dUMP; and (2) dUMP binds to TS threeto seven times more strongly than does dTMP(Santi & Danenberg, 1984) so it would be difficultfor dTMP to dislodge dUMP from TS ligandedat both monomers before and after crystallization.This implies that the nucleotide conversionoccurred within the crystal. Thus, whilethe global conformational changes that close theactive site are essential for normal catalysis,the reaction may occur at a slow rate in theabsence of these changes. This result suggeststhat the non-covalent diffusion complexes aremechanistically relevant species along the reactionpathway.
Product complex structure indicates howproducts may dissociate from the protein
The wild-type E. coli (Fauman et al., 1994) and ourL. casei TS product complexes share similarities inactive site structure, particularly for ordered watermolecules and orientation of dTMP. Wat C7 of theE. coli product complex influences the binding ofdTMP to the enzyme, since its displacement, via thedTMP C7 methyl, decreases the enthalpic energy ofthe complex by a loss of hydrogen bonds (to thewater) with only a small increase in the entropy ofthe water positioning. This may favor the release ofthe product nucleotide from the enzyme (Faumanet al., 1994). The L. casei product complex has asimilar water molecule positioned near the dTMPC7 group. This water is close enough to aneighboring water molecule to allow for hydrogenbonding or networking between the two, and it isalso within hydrogen-bonding distance of theTyr146 side-chain hydroxyl group. Therefore, in theL. casei diffusion complex this water molecule maynot have as great a propensity to disrupt dTMPbinding at the active site, or else the L. casei structurerepresents the product complex as the ligands are inthe process of being released. It is possible,however, that the departure of dTMP is inhibiteddue to the stability of the crystal lattice, which couldfavor movement of the Wat C7 relative to movementof dTMP. The structure trapped in the L. casei crystalrepresents a fairly stable environment, emphasizedby the relatively low temperature factors of theproduct complex for the nucleotide (Table 3), whichcould be a prerequisite to its leaving the active site.The dTMP orientation at the active site suggests
that a substrate nucleotide (dUMP) is held in asimilar position during catalysis. An apparent causeof inactivity of L. casei TS with its C terminustruncated by one residue, based on the ternarycomplex structure, is the inability of folate bindingto induce rotation of the nucleotide base to form theusual conformation. In that mutant ternary complexthe nucleotide is pivoted !20° about the 3'-hy-droxyl group compared to dUMP in wild-type TSbinary and ternary complexes (Perry et al., 1993).This contrasts sharply with the favorable anti baseorientation of dTMP in the L. casei product complex,even though the folate positioning is similar and theC terminus is in a partially ‘‘closed’’ orientation.The two complexes taken together show that theC-terminal change that accompanies folate bindingis necessary to allow pterin stacking against the baseplane to initiate catalysis, but does not inhibitfurther folate movement during and after productformation.The ECTS product complex (Fauman et al., 1994)
shows the DHF molecule to be in a more extendedorientation of the glutamate with the pterincompared to the folate conformation seen in theLCTS product complex. The DHF conformation inthe L. casei product complex resembles that foundfor CH2H4folate in the ternary complex with LCTSand FdUMP where the TS lacks the key carboxy-

Thymidylate Synthase Ternary Complex Structures 533
terminal COO− (Perry et al., 1993). There is muchless overlapping of the pterin and the pyrimidinerings compared to that found in other TS ternarycomplexes. The PABA ring of the folate does notmake van der Waals’ contact with the Phe228side-chain, a hydrophobic interaction usually seenin TS ternary complex structures formed byco-crystallization (Stroud & Finer-Moore, 1993),even while the monoglutamate moiety remainsanchored to its binding site. Therefore, the structuresuggests possible initial stages of folate release fromthe enzyme and supports the chemical finding that(in an ordered reaction) folate leaves the active sitebefore dTMP (Danenberg & Danenberg, 1978). Theconformation adopted by folate in the preformedLCTS·dUMP co-crystal (Figure 4(c)) might berelated to the formation of the necessary 5-iminiumion (pterin) intermediate at the active site, or itmight be a consequence of the diffusion process. Itmay also be specific for folate binding to L. casei TScomplexes in general.The Arg23 and Tyr261 side-chains, key conserved
interacting groups that participate in the setup ofthe active conformation, are also found in unusualpositions. The guanidinium group of Arg23interacts with neither the dTMP nucleotide nor thefolate pterin, remaining close to the C terminus anddistal to the nucleotide phosphate. Its normalposition in a ‘‘closed’’ active site is taken by awell-ordered water molecule that hydrogen bondsto one of the terminal carboxylic acid oxygen atoms.The position of Arg23 in the current complexresembles that taken by this residue in the LCTSternary complex where the TS has a C-terminaldeletion (Perry et al., 1993), which is characterizedas having a partially open enzyme conformationthat is not competent for catalysis. Tyr261, whichnormally hydrogen bonds to the O3' hydroxyl groupof the nucleotide ribose, has rotated farther awayfrom dTMP and is hydrogen bonded to the Thr24side-chain hydroxyl group. The pivotal roles ofArg23 and Tyr261 in TS are demonstrated by thefact that mutations of these residues severelycompromise enzymatic activity (Climie et al., 1990;Michaels et al., 1990; Zhang et al., 1990). Wetherefore propose that the movement of the Arg23and Tyr261 side-chains tracks a path of exit for thefolate and thus signals the initial stages of the‘‘reopening’’ of the enzyme for product release.
MethodsCrystallization and data collection
Crystals of the wild-type L. casei TS·dUMP binarycomplex were grown in hanging drops at pH 7.4 andconcentrations of ammonium sulfate precipitate from 1%to 2% as we described (Hardy et al., 1987). The crystalsadopted a bipyramidal habit typically seen for ligandedL. casei TS, with space group P6122. Water dilutions ofstock solutions of either CB3717 (Ki = 1.1 nM; Jones et al.,1981) or (6-R)-CH2H4folate (Km = 10.0 *M; Liu & Santi,1993) at 10 mM DTT were made and 1 *l samples wereadded to 4 *l hanging drops. The highest folate or analog
concentrations were determined that did not eitherdissolve or degrade crystals, due to a conformationalchange in the protein to the point of disallowing X-raydata collection. The final concentration of CB3717 was!20 *M. The TS·dUMP crystals tended to tolerateconcentrations of CB3717 fivefold higher than those forfolate cofactor. Crystals that remained intact were allowedto stand for no more than two days after folate additionsor one week after CB3717 additions before mounting incapillaries and X-ray diffraction. Crystallographic intensi-ties were recorded at room temperature on an RAXIS IIimage plate detector system using CuK+ X-rays from aRigaku rotating anode generator. Crystal decay due tofolate or CB3717 diffusion resulted in the collection ofrelatively incomplete data for each crystal (Table 3) and,since the conditions for diffusing folates into the crystalswere not exactly reproducible, we did not merge data setsfrom different crystals. Data were reduced using softwareprovided by RAXIS (Higashi, 1990; Sato et al., 1992).Because structure factor data in each case are only 50 to60% complete, there is a poor sampling of high resolutiondata, which may contribute to an effective resolutionconsiderably lower than 2.5 A. The placement of somesolvent molecules may be suspect, although they arebased on the agreement of 2Fo − Fc and Fo − Fc omit mapsat 3.0 A.
Structure solution and refinement
The complex structures were solved by differenceFourier techniques (Chambers & Stroud, 1977) usingprotein phases (+calc) from the highly refined L. caseistructure of TS·dUMP (Finer-Moore et al., 1993). Evidencefor ligand positioning was found by densities in initial2Fo − Fc maps which also suggested a shift of the Cterminus, which is known to occur for TS upon folatebinding. Inclusion first of dUMP and later of folatemolecules was followed by successive rounds ofpositional and simulated annealing refinements usingX-PLOR (Brunger et al., 1987), combined with manualrebuilding using the Frodo graphics program (Jones,1985), and calculation of new 2Fo − Fc, Fo − Fc andsimulated annealing 2Fo − Fc omit maps to confirm ligandpositioning. Inclusion of water molecules and refinementof restrained individual thermal factors were done as laststeps. Table 3 gives a summary of the data collection andrefinement statistics.
Comparison of structures
For comparison of the ternary complexes describedhere with other complexes of E. coli or L. casei TS, acommon core between compared structures wasidentified and used for least-squares alignment. Differ-ence distance matrices were used to identify the commoncores, which are defined as the largest set of +-carbonatoms, each atom within 10 A of another atom in the core,whose distances from each other did not change by morethan !0.5 A between the compared structures (Montfortet al., 1990).
Deposition of coordinates and structure factors
Coordinates and structure factors have been depositedin the Brookhaven Protein Data Bank under the followingidentification codes: 1LCA and R1LCASF for the folateanalog complex, 1LCB and R1LCBSF for the productcomplex, 1LCE and R1LCESF for the substrate complex.

Thymidylate Synthase Ternary Complex Structures534
AcknowledgementsWe thank Ming Yu for preparation of the TS·dUMP
binary complex crystals and Weidung Huang for helpfuldiscussions on TS biochemistry. Molecular graphicsimages were produced using the Midas Plus programfrom the Computer Graphics Laboratory, University ofCalifornia, San Francisco (supported by NIH RR-01081).This work was supported by NIH grant CA-41323.
ReferencesBradshaw, T. P. & Dunlap, R. B. (1992). Catalysis
and ligand binding by thymidylate synthase immobi-lized on thiopropyl-sepharose 6B. Oncol. Res. 4,249–254.
Bruice, T. W. & Santi, D. V. (1982). Secondary +-hydrogenisotope effects on the interaction of 5-fluoro-2'-deoxyuridylate and 5,10-methylenetetrahydrofolatewith thymidylate synthase. Biochemistry, 21, 6703–6709.
Brunger, A. T., Kuriyan, J. & Karplus, M. (1987).Crystallographic R factor refinement by moleculardynamics. Science, 235, 458–560.
Chambers, J. & Stroud, R. M. (1977). Difference fourierrefinement of the structure of DIP-trypsin at 1.5 Awith a minicomputer technique. Acta Crystallogr. sect.B, 33, 1824–1837.
Chello, P. L., McQueen, C. A., DeAngelis, L. M. & Bertino,J. R. (1976). Elevation of dihydrofolate reductase,thymidylate synthetase, and thymidine kinase incultured mammalian cells after exposure to folateantagonists. Cancer Res. 36, 2442–2449.
Cisneros, R. J., Zapf, J. W. & Dunlap, R. B. (1993). Studiesof 5-fluorodeoxyuridine 5'-monophosphate bindingto carboxypeptidase A-inactivated thymidylate syn-thase from Lactobacillus casei. J. Biol. Chem. 268,10102–10108.
Climie, S., Ruiz-Perez, L., Gonzalez-Pacanowska, D.,Prapunwattana, P., Cho, S.-W., Stroud, R. & Santi,D. V. (1990). Saturation site-directed mutagenesisof thymidylate synthase. J. Biol. Chem. 265, 18776–18779.
Danenberg, P. V. & Danenberg, K. D. (1978). Effect of5,10-methylenetetra-hydrofolate on the dissociationof 5-fluoro-2'-deoxyuridylate from thymidylatesynthetase: evidence for an ordered mechanism.Biochemistry, 17, 4018–4024.
Fauman, E. B., Rutenber, E. E., Maley, G. F., Maley, F. &Stroud, R. M. (1994). Water-mediated substrate/product discrimination: the product complex ofthymidylate synthase at 1.83 A. Biochemistry, 33,1502–1511.
Ferrin, T. E., Huang, C. C., Jarvis, L. E., Langridge, R.(1988). The MIDAS display system. J. Mol. Graph. 6,13–27.
Fersht, A. (1985). Enzyme Structure and Mechanism,pp. 56–63, W. H. Freeman and Company, New York.
Finer-Moore, J., Fauman, E. B., Foster, P. G., Perry, K. M.,Santi, D. V. & Stroud, R. M. (1993). Refined structuresof substrate-bound and phosphate-bound thymidy-late synthase from Lactobacillus casei. J. Mol. Biol. 232,1101–1116.
Garret, C., Wataya, Y., Santi, D. V. (1979). Thymidylatesynthase. Catalysis of dehalogenation of 5-bromo-and 5-iodo-2'-deoxyuridylate. Biochemistry, 18, 2798–2804.
Hardy, L. W., Finer-Moore, J. S., Montfort, W. R., Jones,M. O., Santi, D. V. & Stroud, R. M. (1987). Atomic
structure of thymidylate synthase: target for rationaldrug design. Science, 235, 448–455.
Higashi, T. (1990). Auto-indexing of oscillation images.J. Appl. Crystallog. 23, 253–257.
Huang, W. & Santi, D. V. (1994). Isolation of a covalentsteady-state intermediate in glutamate 60 mutantsof thymidylate synthase. J. Biol. Chem. 269, 31327–31329.
Huang, C. C., Pettersen, E. F., Klein, T. E., Ferrin, T. E. &Langridge, R. (1991). Conic: a fast renderer forspace-filling molecules with shadows. J. Mol. Graph.9, 230–236.
Ivanetich, K. M. & Santi, D. V. (1992). 5,6-Dihydropyrim-idine adducts in the reactions and interactions ofpyrimidines with proteins. Prog. Nucl. Acid Res. Mol.Biol. 42, 127–156.
Jones, T. A. (1985). Interactive computer graphics:FRODO. Methods Enzymol. 115, 157–171.
Jones, T. R., Calvert, A. H., Jackman, A. L., Brown, S. J.,Jones, M. & Harrap, K. R. (1981). A potent antitumorquinazoline inhibitor of thymidylate synthetase:synthesis, biological properties, and therapeuticresults in mice. Eur. J. Cancer, 17, 11–19.
Kamb, A., Finer-Moore, J. S. & Stroud, R. M. (1992).Cofactor triggers the conformational change inthymidylate synthase: implications for an orderedbinding mechanism. Biochemistry, 31, 12876–12884.
Kraulis, P. (1991). MOLSCRIPT: a program to produceboth detailed and schematic plots of proteinstructures. J. Appl. Crystallog. 24, 946–950.
Liu, L. & Santi, D. V. (1993). Asparagine 229 inthymidylate synthase contributes to, but is notessential for, catalysis. Proc. Natl Acad. Sci. USA, 90,8604–8608.
Maley, F., Pedersen-Lane, J. & Changchien, L. (1995).Complete restoration of activity to inactive mutantsof Escherichia coli thymidylate synthase: evidence thatE. coli TS is a half-the-sites activity enzyme.Biochemistry, 35, 1469–1474.
Matthews, D. A., Appelt, K., Oatley, S. J. & Xuong,N. H. (1990a). Crystal structure of Escherichia coli thy-midylate synthase containing bound 5-fluoro-2'-deoxyuridylate and 10-propargyl-5,8-dideazafolate.J. Mol. Biol. 214, 923–936.
Matthews, D. A., Villafranca, J. E., Janson, C. A., Smith,W. W., Welsh, K. & Freer, S. (1990b). Stereochemicalmechanism of action for thymidylate synthase basedon the x-ray structure of the covalent inhibitoryternary complex with 5-fluoro-2'deoxyuridylate and5,10-tetrahydrofolate. J. Mol. Biol. 214, 937–948.
Michaels, M. L., Kim, C. W., Matthews, D. A. & Miller,J. H. (1990). Amino acid substitution analysis ofE. coli thymidylate synthase: the study of a highlyconserved region at the N terminus. Proteins: Struct.Funct. Genet. 13, 352–363.
Montfort, W. R., Perry, K. M., Fauman, E. B., Finer-Moore,J. S., Maley, G. F., Hardy, L., Maley, F. & Stroud, R. M.(1990). Structure, multiple site binding, and segmen-tal accommodation in thymidylate synthase onbinding dUMP and an anti-folate. Biochemistry, 29,6964–6977.
Page, M. I. & Jencks, W. P. (1971). Entropic contributionsto rate accelerations in enzymic and intramolecularreactions and the chelate effect. Proc. Natl Acad. Sci.USA, 68, 1678–1683.
Perry, K. M., Carreras, C. W., Chang, L. C., Santi, D. V. &Stroud, R. M. (1993). Structures of thymidylatesynthase with a c-terminal deletion: role of theC terminus in alignment of 2'-deoxyuridine 5'-

Thymidylate Synthase Ternary Complex Structures 535
monophosphate and 5,10-methylene-tetrahydrofo-late. Biochemistry, 32, 7116–7125.
Poe, M., Jackman, L. M., Benkovic, S. J. (1979).5,10-methylene-5,6,7,8-tetrahydrofolate. Conforma-tion of the tetrahydropyrazine and imidazolidinerings. Biochemistry, 18, 5527–5530.
Pogolotti, A. L., Danenberg, P. V. & Santi, D. V. (1986).Kinetics and mechanism of interaction of 10-propar-gyl-5,8-dideazafolate with thymidylate synthase.J. Med. Chem. 29, 478–482.
Santi, D. V. & Danenberg, P. V. (1984). In Folates and Pterin:vol. 1, Chemistry and Biochemistry of Folates (Blakely,R. L. & Benkovic, S. J., eds), pp. 345–399, John Wiley& Sons, New York.
Santi, D. V., McHenry, C. S., Raines, R. T. & Ivanetich,K. M. (1987). Kinetics and thermodynamics ofthe interaction of 5-fluoro-2'deoxyuridylate withthymidylate synthase. Biochemistry, 26, 8606–8613.
Sato, M., Yamamoto, M., Imada, K., Katsube, Y.,Tanaka, N. & Higashi, T. (1992). A high-speeddata-collection system for large-unit-cell crystals
using an imaging plate as a detector. J. Appl.Crystallog. 25, 348–357.
Stroud, R. M. & Finer-Moore, J. S. (1993). Stereochemistryof a multistep/bipartite methyl transfer reaction:thymidylate synthase. FASEB J. 7, 671–677.
Tykarska, E., Lebioda, L., Bradshaw, T. P. & Dunlap R. B.(1986). Crystallization and crystallographic data fornew forms of thymidylate synthase from Lactobacilluscasei. J. Mol. Biol. 191, 147–150.
Zapf, J. W., Weir, M. S., Emerick, V., Villafranca, J. E. &Dunlap, R. B. (1993). Substitution of glutamine forglutamic acid-58 in Escherichia coli thymidylatesynthase results in pronounced decreases in catalyticactivity and ligand binding. Biochemistry, 32, 9274–9281.
Zhang, H., Cisneros, R. J., Deng, W., Johnson, L. F. &Dunlap, R. B. (1990). Site-directed mutagenesis ofmouse thymidylate synthase: alteration of Arg 44 toVal 44 in a conserved loop guarding the active site hasstriking effects on catalysis and nucleotide binding.Biochem. Biophys. Res. Commun. 167, 869–875.
Edited by F. E. Cohen
(Received 26 June 1995; accepted 19 October 1995)


















