
ENDOSTATIN IN THE REGULATION OF ENDOTHELIAL CELL ΠMATRIX...
71
Helsinki University Biomedical Dissertations No. 51 ENDOSTATIN IN THE REGULATION OF ENDOTHELIAL CELL MATRIX INTERACTIONS AND PERICELLULAR PROTEOLYSIS Sara A. Wickstrm Departments of Pathology and Virology, Haartman Institute, Helsinki University Hospital and Biomedicum Helsinki University of Helsinki Finland Academic Dissertation To be presented, with the permission of the Faculty of Medicine, University of Helsinki, for public criticism, in the Lecture Hall 3 of the Biomedicum Helsinki, Haartmaninkatu 8, Helsinki, on March 26th, 2004, at 12 oclock noon Helsinki 2004
Transcript of ENDOSTATIN IN THE REGULATION OF ENDOTHELIAL CELL ΠMATRIX...

Helsinki University Biomedical Dissertations No. 51
ENDOSTATIN IN THE REGULATION OF ENDOTHELIAL CELL � MATRIX
INTERACTIONS AND PERICELLULAR PROTEOLYSIS
Sara A. Wickström
Departments of Pathology and Virology, Haartman Institute,
Helsinki University Hospital and Biomedicum Helsinki University of Helsinki
Finland
Academic Dissertation
To be presented, with the permission of the Faculty of Medicine, University of Helsinki, for public criticism, in the Lecture Hall 3 of the Biomedicum Helsinki, Haartmaninkatu 8,
Helsinki, on March 26th, 2004, at 12 o�clock noon
Helsinki 2004

4
Supervised by:
Professor Jorma Keski-Oja Departments of Pathology and Virology Biomedicum and Haartman Institute University of Helsinki Helsinki, Finland
Reviewed by:
Professor Ismo Virtanen Department of Biomedicine and Anatomy Biomedicum Helsinki University of Helsinki Helsinki, Finland
and
Docent Raija Soininen Department of Biochemistry Biocenter Oulu University of Oulu Oulu, Finland
Opponent:
Professor Lena Claesson-Welsh Department of Genetics and Pathology Rudbeck Laboratory University of Uppsala Uppsala, Sweden
ISSN 147-8433 ISBN 952-10-1723-6 (nid.) ISBN 952-10-1724-4 (PDF) http://ethesis.helsinki.fi YliopistopainoHelsinki 2004

5
ContentsOriginal publications ............................................................................................ 7
Abbreviations......................................................................................................... 8
Abstract .................................................................................................................. 9
Introduction ......................................................................................................... 10 Cell migration and extracellular matrix ............................................................ 10
Extracellular matrix....................................................................................... 10 Basement membranes................................................................................ 11
Mechanisms of cell motility.......................................................................... 13 Integrins and cell-matrix interactions............................................................ 15 Signaling pathways involved in cell migration............................................. 15
Pericellular proteolysis...................................................................................... 17 Plasminogen activators.................................................................................. 17
Urokinase-type plasminogen activator...................................................... 18 Urokinase receptor .................................................................................... 18 Inhibitors of plasminogen activators ......................................................... 19
Interplay between proteolysis, signaling, and migration .............................. 20 Vasculogenesis and angiogenesis ..................................................................... 22
Molecular mechanisms of angiogenesis........................................................ 22 Tumor angiogenesis and the angiogenic switch............................................ 23 Role of integrins in angiogenesis .................................................................. 24 Proteolytic cascades in angiogenesis ............................................................ 25
Basement membrane-derived inhibitors of angiogenesis ................................. 26 Fragments of type IV collagen...................................................................... 26 Endostatin...................................................................................................... 28
Structural features ..................................................................................... 28 Biological roles ......................................................................................... 29 Effects of endostatin on tumor growth...................................................... 30 Cell surface receptors ................................................................................ 32 Mechanisms of action ............................................................................... 33
Angiogenesis inhibitors in cancer therapy .................................................... 34
Aims of the present study ................................................................................... 36
Materials and methods ....................................................................................... 37 Cell culture and treatments............................................................................ 37 Expression and characterization of human endostatin .................................. 37 Growth factors, chemicals, and enzymes...................................................... 37 Antibodies ..................................................................................................... 38 SDS-PAGE and immunoblotting .................................................................. 38 Casein zymography and reverse zymography............................................... 38 Immunoprecipitation ..................................................................................... 39 Immunofluorescence ..................................................................................... 39 Cell adhesion assays...................................................................................... 40 In vitro chemotaxis assay .............................................................................. 40 Isolation of the extracellular matrix .............................................................. 40

6
Isolation of detergent-insoluble membrane fractions.................................... 41 Affinity precipitation of GTP-Rho................................................................ 41 Src family kinase autophosphorylation assay. .............................................. 41 Metabolic labeling......................................................................................... 42 Wound-induced migration assay................................................................... 42 Endothelial cell tube formation assay ........................................................... 42
Results .................................................................................................................. 43 Endostatin regulates levels of secreted soluble uPA/PAI-1 complex and induces changes in cell surface localization of uPA and uPAR (I)................................ 43 Loss of endothelial cell focal adhesions and actin cytoskeleton in endostatin-treated cells (I, II, III) ........................................................................................ 44 Association of endostatin with α5β1 integrin and caveolin-1 (II, III)................ 44 Tyrosyl phosphatase-dependent activation of caveolin-1- associated Src regulates endostatin-induced disassembly of cytoskeleton and decreased deposition of fibronectin matrix (II) ................................................................. 44 Endostatin and α5β1 integrin partition in lipid rafts in a heparan sulfate proteoglycan-dependent manner (III) ............................................................... 45 Endostatin downregulates RhoA activity via Src and p190 (III) ...................... 46 Endostatin-derived peptides promote cell adhesion via integrins and induce disassembly of the cytoskeleton (IV) ................................................................ 46 Endostatin-derived peptides inhibit endothelial migration and tubular morphogenesis of endothelial cells (IV) ........................................................... 47
Discussion............................................................................................................. 48 Effects of endostatin on endothelial cell pericellular proteolysis ..................... 48 Regulation of actin cytoskeleton and cell migration by endostatin .................. 49 Requirement of integrin α5β1, heparan sulfate proteoglycans and lipid rafts in endostatin signaling........................................................................................... 51 Identification of an integrin-binding peptide within endostatin........................ 52 A model for endostatin-induced signaling ........................................................ 53
Perspective ........................................................................................................... 55
Acknowledgements.............................................................................................. 56
References ............................................................................................................ 58

7
Original publications
This thesis is based on the following original articles, which are referred to by their Roman numerals in the text.
I. Wickström, S.A., Veikkola, T., Rehn, M., Pihlajaniemi, T., Alitalo, K., and Keski-Oja, J. Endostatin-induced modulation of plasminogen activation with concomitant loss of focal adhesions and actin stress fibers in human endothelial cell cultures. Cancer Res. 61: 6511-6516, 2001.
II. Wickström, S.A., Alitalo, K., and Keski-Oja, J. Endostatin associates with α5β1 integrin and caveolin-1 and activates Src via a tyrosyl phosphatase-dependent pathway in human endothelial cells. Cancer Res. 62: 5580-5589,2002.
III. Wickström, S.A., Alitalo, K., and Keski-Oja, J. Endostatin associates with lipid rafts and induces reorganization of the actin cytoskeleton via downregulation of RhoA activity. J. Biol. Chem. 278: 37895-37901, 2003.
IV. Wickström, S.A., Alitalo, K., and Keski-Oja, J. An endostatin-derived peptide interacts with integrins and regulates actin cytoskeleton and migration of endothelial cells. J. Biol. Chem. 279: in press, 2004.

8
Abbreviations
aa amino acid bFGF basic fibroblast growth factor BM basement membrane BSA bovine serum albumin DOC sodium deoxycholate ECM extracellular matrix FAK focal adhesion kinase GAP GTPase-activating protein GPI glycosyl-phosphatidyl inositol GPCR G-protein-coupled receptor HDMEC human dermal microvascular endothelial cell hsp27 heat shock protein 27 HSPG heparan sulfate proteoglycan kDa kilodalton MMP matrix metalloproteinase MT-MMP membrane-type matrix metalloproteinase NC non-collagenous PAGE polyacrylamide gel electrophoresis PAI plasminogen activator inhibitor PBS phosphate buffered saline pTyr phosphotyrosine RGD Arginine-Glycine-Aspartate SDS sodium dodecyl sulphate SPARC secreted protein, acidic and rich in cysteine tPA tissue type plasminogen activator uPA urokinase type plasminogen activator uPAR uPA receptor VEGF vascular endothelial growth factor

9
Abstract
The growth and survival of a malignant tumor is dependent on its ability to induce the formation and to maintain its own microvasculature. Inhibition of this process termed tumor angiogenesis is an emerging strategy in cancer therapy. The extracellular matrix surrounding the vascular endothelial cells contains cryptic domains, which are exposed and released by changes in the proteolytic homeostasis of the tumor microenvironment. These fragments transmit local signals regulating vascular endothelial cell proliferation and migration. Endostatin, the proteolytic fragment of collagen type XVIII, is a potent inhibitor of tumor angiogenesis in various mouse models and is currently in clinical trials for therapeutic use in human cancer.
To understand the nature of the inhibitory effects of human endostatin on angiogenesis, we studied the cell biological mechanisms contributing to the anti-migratory and anti-proliferative effects of endostatin on cultured human microvascular endothelial cells. We observed that endostatin effectively regulates the pericellular proteolysis of endothelial cells by downregulating the levels of soluble secreted urokinase-type plasminogen activator (uPA) and its inhibitor, plasminogen activator inhibitor-1 (PAI-1). In addition, the cell surface localization of uPA/PAI-complexes was altered, and they accumulated on the cell surface. These changes were accompanied by the disassembly of focal adhesions and actin stress fibers. The disassembly of the cytoskeletal structures was associated with the tyrosyl phosphatase-dependent activation of Src tyrosine kinase. Src activation resulted also in altered deposition of the endothelial cell fibronectin matrix and decreased migratory capacity of these cells. Our results further indicated that the intracellular events leading to the cytoskeletal changes were a consequence of endostatin interaction with integrin α5β1, caveolin-1 and a heparan sulfate proteoglycan. Facilitated by these interactions, a fraction of endostatin partitioned to lipid rafts, membrane microdomains specialized in signal transduction. The binding of endostatin to integrin α5β1 and cell surface heparan sulfates resulted in the Src-dependent activation of p190, subsequent downregulation of RhoA activity, and finally the disassembly of the actin cytoskeleton.
We generated synthetic peptides derived from the amino terminus of endostatin and analyzed their effects on endothelial cell adhesion and migration. The peptides promoted cell adhesion via integrin β1 and induced cytoskeletal changes comparable to the effects of full-length endostatin. The peptides also inhibited directional migration and tubular morphogenesis of endothelial cells.
The current results identify a novel mechanism, by which a fragment of an extracellular matrix component transmits signals to the endothelial cell cytoplasm and regulates multiple functions of endothelial cells.

10
Introduction
Angiogenesis, the formation of new blood vessels, is a hallmark of cancer. Without developing a functional vasculature, tumors are unable to grow beyond a microscopic size or metastasize to distant organs. The process of angiogenesis involves complex cellular and molecular mechanisms initiated by a shift in the balance of pro- and anti-angiogenic molecules. Subsequently, cellular programs regulating endothelial cell proliferation, migration, extracellular matrix (ECM) degradation, and differentiation are initiated, leading to vessel sprouting from existing vessels. Inhibition of these processes is an emerging strategy of cancer therapy. Endogenous anti-angiogenic molecules have been isolated and are currently in clinical trials to be used alone or in combination with conventional therapies in the treatment of cancer as well as other diseases involving pathological angiogenesis.
Cell migration and extracellular matrix Most cell types are able to move within their tissue compartment, while some
highly motile cells are capable of penetrating through tissue boundaries such as basement membranes (BM). In multicellular organisms cell migration plays an important role in fundamental processes such as embryonic development, immunological defense mechanisms, trophoblast invasion, and wound healing. In addition, it is an important cascade of events in pathological conditions such as tumor invasion and metastasis (Lauffenburger and Horwitz, 1996).
Extracellular matrix
Most cells in multicellular organisms are surrounded by an organized meshwork of macromolecules that constitute the extracellular matrix (ECM). The ECM functions as a structural framework and provides cells with positional and environmental information, but also forms specialized structures such as cartilage, tendons, BMs, bone and teeth. In addition, it regulates multiple aspects of cell behavior, such as their development, migration, proliferation, shape, and metabolic functions.
The ECM is not a static structure, but is continually produced and remodeled. The macromolecules of the ECM are secreted by local cells such as fibroblasts. Two of the main classes of extracellular proteins that make up the matrix are the collagens and the proteoglycans. The long collagen fibers strengthen and organize the matrix, while the polysaccharides of the proteoglycans form an aqueous phase, which permits the diffusion of nutrients, metabolites, and hormones between tissue compartments. Elastin is a component of fibers, which confer matrix resilience. In addition, two high molecular glycoproteins, fibronectin and laminin are among major components of the ECM. Fibronectin is widely distributed in

11
connective tissues, whereas laminin is found exclusively in the BM. Collagens are among the most abundant proteins in mammals and the most
abundant protein in the ECM. All the 23 vertebrate collagens are composed of three α-chains and contain at least one triple-helical domain of repeating glycines (Gly-X-Y motif). The triple helix is a rigid structure, but it is interrupted in many collagens by non-triple helical domains that provide flexibility (Prockop and Kivirikko, 1995).
Fibronectin is a large, dimeric glycoprotein that is produced by most cell types. It exists in two major forms, as a soluble form in the plasma and other body fluids and as an insoluble form in the ECM. Insoluble fibronectin plays a critical role in embryonic development, as adhesion of embryonic cells to fibronectin is essential for their migration (George et al., 1993). The deposition of fibronectin is a cell-dependent event initiated with binding of soluble fibronectin to cell surface integrins (Mosher et al., 1991; Schwarzbauer and Sechler, 1999; Wu, 1997). Fibronectin interacts with integrins mainly through the RGD-sequence, but also other sites are involved (Miyamoto et al., 1998). Fibronectin is also deposited to the ECM by endothelial cells to provide support in cell migration and adhesion during angiogenesis, and is thus important both in developmental and pathological angiogenesis (George et al., 1997; Kim et al., 2000a). Vitronectin, thrombospondin, tenascin, and SPARC (secreted protein, acidic and rich in cysteine) are other ECM glycoproteins involved in mediating both adhesive and anti-adhesive interactions and also in the regulation of angiogenesis (Adams, 2001; Bradshaw and Sage, 2001; Murphy-Ullrich, 2001; Schvartz et al., 1999).
Basement membranes
Basement membranes (BM) are dense sheets of extracellular matrix that function as structural barriers separating epithelial and endothelial cells as well as peripheral nerve axons, fat cells and muscle cells from the underlying tissue stroma. BMs provide structural support, separate tissues into compartments, and regulate cell behavior (Timpl, 1996). All cell types are known to produce components of BMs, which include type IV collagen, laminin, heparan sulfate proteoglycans and nidogen/entactin. Minor components include agrin, SPARC, fibulins, type XV collagen and type XVIII collagen. Fibronectin is present in fetal BMs (Erickson and Couchman, 2000; Ghohestani et al., 2001). The molecular composition of the BM varies among different tissues. The differences are believed to confer tissue specificity, which is important for defining the specialized functions of epithelial and endothelial cells in different organs.
Type IV collagen is the major collagen found in basement membranes. It is composed of three parallel α chains, which form a triple helical structure with interruptions. Six different α chains have been identified, and they can form 56 different combinations of collagen trimers (Hudson et al., 1993). Type IV forms a network-like structure that is associated with perlecan and, via nidogen, with the laminin network. Structural studies have indicated that type IV collagen network formation is crucial for BM stability and assembly (Kuhn, 1995; Timpl, 1996).

12
Laminins are major contributors to BM assembly and the resulting supramolecular structure. They are a family of at least 15 heterotrimeric glycoproteins composed of five α, three β, and three γ subunits. The various laminin isoforms have a cell and tissue-specific expression pattern and are differentially recognized by their integrin receptors. Via interactions with various integrin as well as non-integrin receptors, laminins display a large repertoire of biological functions, such as regulation of tissue morphogenesis, cell migration and differentiation and wound healing (Colognato and Yurchenco, 2000).
Type XVIII collagen is a component of several different types of epithelial and vascular BMs. The collagen molecule consists of 10 triple-helical domains that are separated by non-triple-helical (NC) regions (Fig. 1)(Oh et al., 1994). The collagen 18 gene encodes for two 1516 or 1336 amino acid residue variantα1(XVIII) chains, which are expressed in a tissue specific manner. The two chains have different signal peptides and variant N-terminal noncollagenous NC1 domains, but share multiple triple-helical domains that are separated by non-triple-helical regions. The longer form has a cystein-rich motif homologous to the extracellular part of the frizzled receptors involved in the Wingless signaling pathway in Drosophila. The longer splice variant is mainly expressed in the liver, whereas the shorter variant is virtually absent from the liver, but is a ubiquitous component of vascular and epithelial BMs throughout the body (Saarela et al., 1998a). α1(XVIII) mRNAs are produced by several cell types, including epithelial and endothelial cells, cardiac muscle cells, keratinocytes, and hepatocytes (Saarela et al., 1998b). Collagen XVIII is also a heparan sulfate proteoglycan, which serves as a ligand for the receptor tyrosine phosphatase σ(Aricescu et al., 2002).
Figure 1. Schematic structure of type XVIII collagen. Ten collagenous domains are interrupted by 11 NC domains. The long isoform contains the alternatively spliced Frizzled domain. The NC1 domain contains the trimerization domain, the protease sensitive hinge domain and the 20 kDa endostatin fragment (Oh et al., 1994).
Type XVIII collagen is believed to contribute to the normal development of vasculature in the retina. A mutation in human collagen XVIII has been associated

13
with Knobloch syndrome, a disease characterized by a failure in the development of retinal vasculature, retinal degeneration and blindness (Sertie et al., 2000). Mice deficient in type XVIII collagen are viable and fertile, and display no major vascular abnormalities. However, a defect in the regression of hyaloid vessels is observed (Fukai et al., 2002; Ylikärppä et al., 2003a). In addition, these mice develop age-dependent loss of vision, which is due to the deposition of electron dense material in the sub-retinal pigment epithelium. This results in disturbances in vitamin A metabolism and photoreceptor function, suggesting that type XVIII collagen is essential for retinal pigment epithelium function (Marneros et al., 2004).
The C-terminal NC domain of collagen XVIII contains the anti-angiogenic fragment known as endostatin. The endostatin domain is separated from an upstream trimerization region by a hinge domain. Proteolytic cleavage within the hinge region results in the release of monomeric endostatin (O'Reilly et al., 1997).
Type XV collagen is highly homologous to type XVIII collagen, consisting of a central triple-helical domain interrupted by NC domains (Muragaki et al., 1994; Myers et al., 1992). It is also expressed in the BM of blood vessels (Muragaki et al., 1995). The vasculature and the vascular BM develop normally in mice deficient in type XV collagen, but collapsed capillaries and endothelial-cell degeneration in the heart and skeletal muscle are observed, indicating that collagen XV plays a role in stabilizing the microvasculature and skeletal muscle cells (Eklund et al., 2001). Despite significant structural homology and overlapping expression patterns, collagens XV and XVIII seem to have separate biological roles. The double knockout mice for the two collagens do not have any additional major defects apart from the ones found in the single knockouts (Ylikärppä et al., 2003b).
Mechanisms of cell motility
Cell motility can be regulated at multiple levels. In general, it is regulated through interactions of molecules expressed on the cell surface with the surrounding tissue microenvironment. The initial step of cell motility is polarization, i.e. establishment of the front and rear of the cell. This involves redistribution of filamentous actin, cell adhesion molecules such as integrins, as well as chemokine receptors. In addition, membrane protrusions, lamellipodia and filopodia, extend at the cell front as a result of actin polymerization. Concomitantly, new cell-matrix adhesions are formed at the leading edge. The cell migrates over the adhesion complexes until they reach the cell rear, where they are disrupted and the adhesion subsequently detaches (Fig. 2). Two distinct types of force move the cell body forward: the protrusive force needed to extend lamellipodia and filopodia, and the contractile force needed to move the cell body forward. These forces are generated by actin filament contraction and traction from the cell-substratum adhesions (Lauffenburger and Horwitz, 1996). The strength of cell adhesion to the ECM regulates the choice between adhesion and migration. If the adhesion is too strong, the cell remains stationary, and if the

14
adhesion is too weak, enough traction cannot be generated for movement. Migration occurs thus at intermediate levels of adhesion, which can be achieved by regulating the expression levels of cell adhesion molecules or their extracellular ligand, as well as by modulating the activation status of the receptors (Schwarzbauer, 1997).
The strength of cell adhesion in migrating cells can also be regulated by proteins in the extracellular environment. While several ECM components promote cell adhesion, there are a number of proteins with anti-adhesive functions. Proteins with anti-adhesive properties include tenascin-C, thrombospondin-1, SPARC, and latent transforming growth factor-β binding protein-2. These proteins induce a rapid transition to an intermediate state of adhesiveness characterized by altered cell morphology and cytoskeletal structures (Hyytiäinen and Keski-Oja, 2003; Murphy-Ullrich, 2001).
The direction of cell movement is controlled by growth factors such as bFGF and VEGF. These growth factor signals are transmitted into the cytoplasm via receptor tyrosine kinases, which engage in extensive crosstalk with specific adhesion molecules such as integrins to induce changes in intracellular signaling, gene expression levels, and proteolytic cascades to enhance cell polarization and directed cell motility (Eliceiri, 2001; Schwartz and Baron, 1999).
Figure 2. Integrin-mediated adhesion and focal adhesion assembly in cell motility. Cycles of cell-ECM attachment and detachment together with the contraction of the actin cytoskeleton move the cell body forward. Ligand binding and clustering of integrins at the leading edge activate signaling proteins such as FAK, Src, and RhoA, which regulate the assembly and disassembly of focal adhesion complexes as well as actin stress fiber dynamics. New adhesion complexes are formed at the leading edge. The cell subsequently migrates over the complexes, which translocate to the cell rear where they are disassembled. Modified from (Gumbiner, 1996).

15
Integrins and cell-matrix interactions
Integrins are heterodimeric transmembrane receptors, which bind to ligands in the extracellular matrix or counter-receptors on adjacent cells. The mammalian family of integrins consists of at least 16 α and 8 β subunits, which are able to noncovalently heterodimerize into approximately 24 receptors with distinct, but often overlapping substrate specificities (Hynes, 2002a). ECM or cell surface proteins containing the Arg-Gly-Asp (RGD) sequence constitute the major class of integrin ligands. Integrins that recognize this sequence include the major fibronectin receptor α5β1 integrin and vitronectin receptors αvβ3 and αvβ5
(Ruoslahti, 1996). Integrins function as transmembrane links between the ECM and the actin cytoskeleton. In addition to regulating cell adhesion to the ECM, integrins are involved in transmitting signals, which regulate cell shape, survival, proliferation, gene transcription, and migration (Aplin et al., 1999).
Integrins not occupied by a ligand are distributed diffusely over the cell surface and are not linked to the cytoskeleton. Upon ligand binding, integrins cluster into specialized adhesive structures called focal contacts and become associated with the actin cytoskeleton. Monovalent ligand occupancy can in some cases trigger an intracellular response, but for efficient accumulation of cytoskeletal proteins clustering is additionally required (Miyamoto et al., 1995). The assembled focal contacts undergo tyrosine phosphorylation events and subsequently mature into stable multiprotein complexes called focal adhesions (Luna and Hitt, 1992; Sastry and Horwitz, 1993; Schoenwaelder and Burridge, 1999). Focal adhesions comprise of a large number of proteins with both structural and signaling function. A large number of the proteins bind directly to actin and function at different stages of the adhesion complex maturation cycle. The most abundant structural focal adhesion proteins include vinculin, talin, and paxillin. The most common integrins found in these adhesions are α5β1 and αvβ3,although others are present on substrates like collagen (Jockusch et al., 1995). The multiprotein adhesion complexes regulate cell attachment, migration, and signaling on extracellular substrates as well as the composition and assembly of the ECM (Geiger et al., 2001).
Signaling pathways involved in cell migration
Cell migration involves dynamic changes in cell - matrix interactions. Focal contacts are constantly assembled and disassembled as the cell moves across the ECM. This regulation requires complex signaling downstream of the adhesion receptors. Intracellular signaling is often mediated by covalent modifications, which activate or inactivate proteins, and thus act as molecular switches. Such modifications include protein phosphorylation and conformational changes that expose cryptic sites for subsequent protein-protein interactions (Hunter, 2000). Focal adhesion kinase (FAK) and Src are molecular switches involved in the transmission of intracellular signals from extracellular cues via direct interaction

16
with the cytoplasmic tails of integrins (Fig. 2)(Parsons et al., 2000; Thomas and Brugge, 1997). They form a molecular complex involving regulation of activity both via autophosphorylation and phosphorylation of each other. Both molecules subsequently phosphorylate various focal adhesion target proteins. Src-FAK- mediated phosphorylation has a dual role in regulating focal adhesion dynamics and turnover, as it can both stimulate the recruitment of proteins in to the complex and induce the disassembly of these structures (Felsenfeld et al., 1999; Volberg et al., 2001).
The key molecules regulating actin polymerization and focal adhesion assembly downstream of FAK and Src are the Rho family of GTPases. At least 10 members are known to exist in mammals: RhoA-E, RhoG, Rac1-2, Cdc42 and TC10 (Kaibuchi et al., 1999). The Rho proteins cycle between two conformational states: the active GTP-bound and inactive GDP-bound form. In the active form they are able to interact with downstream target proteins and generate signaling responses, after which they return to the inactive state through intrinsic GTPase activity. The GTPase activity is enhanced by their interaction with GTPase activating proteins (GAPs). These molecules are in turn regulated by tyrosine phosphorylation by kinases such as Src (Bishop and Hall, 2000). The most extensively characterized members of the Rho family are RhoA, Rac and Cdc42. Each of them acts in a spatially and temporally coordinated manner to regulate separate aspects of contractile actin and myosin filament assembly.
Rac induces actin polymerization at the cell periphery, leading to the formation of membrane protrusions called lamellipodia. Rac also promotes the formation of integrin-containing adhesion complexes at the leading edge of migrating cells (Ridley et al., 1992). These processes are essential for the migration of all cell types, and loss of Rac results in the inhibition of cell motility (Small et al., 2002). RhoA in turn stimulates the formation of actin stress fibers and focal adhesions (Ridley and Hall, 1992). The balance of RhoA activity in migrating cells is tightly regulated in order to maintain the state of intermediate cell adhesion. RhoA activity thus plays a dual role in both promoting and inhibiting migration. Cdc42 promotes the formation of thin, finger-like membrane protrusions called filopodia (Nobes and Hall, 1995). In addition, Cdc42 has an important role in directed cell movement, possibly through stabilization of Rac at the cell front. Loss of Cdc42 activity does not block cell motility, but results in random migration (Allen et al., 1998).
In addition to bioactive lipids, peptides and growth factors, RhoGTPases are activated via integrin-mediated cell adhesion. Binding of integrins to their ECM substrates promotes actin polymerization and formation of filopodia and membrane ruffles via Rac and Cdc42. RhoA is also activated in response to integrin ligation (Clark et al., 1998; Price et al., 1998).

17
Pericellular proteolysis Various physiological processes require coordinated tissue remodeling.
Degradation of the ECM is involved in tissue morphogenesis and growth as well as angiogenesis, bone remodeling wound healing, trophoblast implantation, and involution of the postpartum uterus or postlactation mammary gland. In addition, proteolysis is involved in pathological conditions such as tumor growth, invasion, and metastasis, chronic wounds, arthritis and other autoimmune diseases (Johnsen et al., 1998; Werb, 1997). The ECM is not the only target for pericellular proteolysis, as cell surface proteins, receptors, and transmembrane ECM proteins are also regulated by proteolysis (Werb et al., 1999).
ECM degradation in vivo occurs in a spatially confined manner in the immediate pericellular environment of cells. This can be achieved by targeting the proteolytic enzymes to the cell membranes and into specific microdomains within the membranes. The targeting occurs via distinct mechanisms. Soluble enzymes are bound to specific receptors on the cell surface. Proteinases with transmembrane domains contain sequences that localize them to adhesion sites or invasive protrusions. In addition, interactions with other cell surface molecules such as integrins modulate the localization of cell surface proteinases (Basbaum and Werb, 1996).
Degradation of ECMs and BMs involves concerted activity of numerous proteinases. They are classified into exo- or endopeptidases according to the terminal or internal cleavage site on their substrate proteins. The endopeptidases are further divided into subgroups of serine, cysteine, aspartic and metalloproteinases according to sequence homology and cofactors determining their catalytic activity.
Plasminogen activators
The plasminogen activator / plasmin system is composed of plasminogen and two plasminogen activators, urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA), plasminogen activator inhibitors (PAI), and receptors. The activation of the zymogen plasminogen into the active proteinase plasmin occurs through proteolytic cleavage of a single peptide bond by the plasminogen activators. Plasmin is primarily involved in the proteolysis of the fibrin clot, but is also capable of degrading various components of the ECM.
All the components of the plasminogen activator system are anchored to the cell surface via specific receptors, enabling their function in targeted pericellular proteolysis and processes such as tissue morphogenesis, angiogenesis, arteriosclerosis and tumorigenesis (Ellis, 2003).
The activity of tPA is stimulated by and localized to the fibrin matrix due to a high-affinity interaction between the finger-kringle domains of tPA and the polymerized fibrin. This property makes tPA important in intravascular fibrinolysis (Stubbs et al., 1998). In contrast, uPA is mainly involved in cell

18
migration, invasion, and tissue remodeling due to its specific targeting to the cell surface (Ellis et al., 1989).
Urokinase-type plasminogen activator
The serine proteinase uPA is a 55 kDa polypeptide consisting of two disulfide-linked chains, a C-terminal B chain containing the serine proteinase domain, and an N-terminal A chain containing the growth factor-like domain, a kringle domain and an interdomain linker region. The single-chain form of uPA is an inactive zymogen, which is proteolytically cleaved to yield the active two-chain form. The cleavage can be carried out by plasmin and several other proteinases (Andreasen et al., 2000).
Through the activation of plasminogen and its intrinsic proteolytic activity uPA is able to degrade fibrin, laminin, fibronectin, proteoglycans, gelatin and thrombospondin. Importantly, native collagens are resistant to proteolysis by uPA or plasmin (Danø et al., 1985; Kwaan, 1992). In addition, uPA/plasmin catalyze the activation of several growth factors such as hepatocyte growth factor and transforming growth factor-β. They also activate other proteolytic enzymes, such as matrix metalloproteinases (MMP). The zymogens for MMP-3, -9, and -8 have been shown to be physiological substrates for uPA/plasmin using plasminogen activator deficient mice (Andreasen et al., 2000).
Most tissues express low levels of uPA. The expression occurs mostly in tissues undergoing remodeling. Epithelial cells in the kidney produce measurable amounts of uPA, while alveolar epithelial cells produce uPAR that can recruit uPA. In these compartments the uPA system may be used to catalyze the lysis of microthrombi (Solberg et al., 2001; Wagner et al., 1996). uPA is constitutively expressed in endothelial cells of various tissues and it is up-regulated by various angiogenic growth factors, transforming growth factor-β and hypoxia (Graham et al., 1998; Laiho et al., 1986; Laiho and Keski-Oja, 1989; Pinsky et al., 1998).
Urokinase receptor
uPA is targeted to the cell membrane via a specific cell surface receptor, uPAR. Pro-uPA and active two-chain uPA bind uPAR with similar affinity (Kd< 1 nM). The binding of pro-uPA to uPAR facilitates its activation to uPA, localizing plasminogen activation to the cell surface (Fig. 3)(Andreasen et al., 2000). uPAR binds also the plasma and ECM protein vitronectin via an interaction dependent on the presence of uPA (Waltz and Chapman, 1994). The uPAR protein is composed of three internally repeated sequence motifs (D1, D2, and D3), which are attached to the plasma membrane by a glycosylphosphatidylinositol (GPI) moiety. The GPI anchor can be cleaved to release soluble uPAR. In addition, cleavage can occur between D1 and D2 domains to yield D1 and D2D3 fragments. D2D3 has been shown to have chemotactic properties (Blasi, 1999).
The GPI anchor as a mode of membrane attachment has a critical impact on the functional properties of uPAR. Interactions with various transmembrane proteins to form functional complexes compensate for the lack of direct

19
interaction between uPAR and the cytoplasm. The internalization of the receptor-ligand complexes is mediated via interaction with the low-density lipoprotein-receptor-related protein (Czekay et al., 2001) In addition, the GPI-anchor regulates the partitioning of uPAR into lipid rafts, which are specialized membrane microdomains that serve as foci for the recruitment of transmembrane proteins and intracellular signaling molecules (Simons and Toomre, 2000). Intracellular signaling by uPA and uPAR, on the other hand, are mediated mainly by direct interactions of uPAR with integrins, caveolin, and G-protein-coupled receptors (GPCR) (Resnati et al., 2002; Wei et al., 1999; Wei et al., 1996).
uPAR not only functions as a receptor for uPA, but is also involved in the regulation of cell migration, adhesion, differentiation, and proliferation. This occurs mainly through the specialized properties of the uPA/uPAR complex as a transducer of intracellular signaling. The signaling functions are mostly independent of the proteolytic activity of uPA, since catalytically inactive derivatives of uPA are as effective as active two-chain form in inducing intracellular signals. However, the binding of uPA to uPAR is required for these functions. Downstream targets of uPAR include protein kinases such as Src, FAK, and mitogen-activated protein kinase (Blasi and Carmeliet, 2002).
Inhibitors of plasminogen activators
There are three plasminogen activator inhibitors, PAI-1, PAI-2, and PAI-3, of which PAI-1 is thought to be physiologically the most important. PAI-1 is a 50 kDA protein capable of inhibiting both uPA and tPA. PAI-1 is secreted by endothelial cells and is abundant in the blood (Ginsburg et al., 1986; Juhan-Vague et al., 1984). It is also secreted by other cell types, such as fibroblasts, smooth muscle cells, hepatocytes, adipocytes, as well as tumor cells (Feinberg et al., 1989; Quax et al., 1990; Samad et al., 1996).
The three dimensional structure of the uPA/PAI-1 complex has not been solved yet, but PAI-1 is known to bind uPA in a 1:1 stoichiometric ratio via a 20-aa sequence motif termed the reactive center (Wind et al., 2002). Free PAI-1 is unstable, and is rapidly converted to a latent form with low inhibitory capacity. The binding of PAI-1 to vitronectin stabilizes the active conformation. PAI-1 in complex with uPA is not able to bind vitronectin (Seiffert et al., 1994).
PAI-2 is expressed mainly in monocytes and placental cells (Feinberg et al., 1989; Wohlwend et al., 1987). It also inhibits both uPA and tPA, but is less potent in its inhibition than PAI-1. High PAI-2 levels have been found in the sera of pregnant women, as well as in malignancy-associated ascites, which may contribute to the increased occurrence of thrombosis frequently associated with these conditions (Nilsson et al., 1986; Quax et al., 1990).
PAI-3 (protein-C inhibitor) is expressed by colon and breast carcinoma cells, but its role in the regulation of plasminogen activation and thrombosis is still unclear (Costantini et al., 1991).

20
Interplay between proteolysis, signaling, and migration
Cell migration is an important process in normal tissue morphogenesis and repair, but when deregulated, it is also a major contributor to numerous pathological conditions. Proteases are involved in cell migration via several different pathways. In response to chemoattractants, pericellular matrix is degraded by cell surface associated proteases to clear a path for the migrating cell. Recent studies have implicated, however, that this targeted proteolysis plays a subtler role in cell migration. Changes in cell shape and adhesion are required for cell motility. Proteases act in coordination with cell surface adhesion molecules and components of the ECM to reorganize the cytoskeleton and to regulate cell-cell and cell-matrix interactions. In addition, cell surface associated proteases not only respond to, but also induce and regulate multiple intracellular signaling pathways. This regulation occurs through interactions with cell surface receptors and their intracellular partners (Blasi and Carmeliet, 2002; Stefansson and Lawrence, 2003). Proteases can also activate and release growth factors sequestered in the ECM, which subsequently promote cell migration (Lyons et al., 1988; Saksela and Rifkin, 1990; Taipale and Keski-Oja, 1997). Limited proteolysis also exposes cryptic fragments in the ECM that locally regulate cell behavior (Giannelli et al., 1997; Xu et al., 2001). The uPA/PAI-1/uPAR system plays an important role in cell migration. Expression of uPAR has been linked to cell migration important to inflammation and tumor metastasis (Andreasen et al., 2000; Bianchi et al., 1996; Huang et al., 2000).
The effects of uPAR on cell migration are mediated via its interactions with several transmembrane proteins, which include integrins, GPCRs, and caveolin-1. Ligand-activated uPAR regulates integrin-dependent cell migration and adhesion via direct association with the α subunit of integrins (Fig. 3). The fibronectin receptor α5β1 integrin and the laminin receptor α3β1 integrin seem to bind uPAR with the highest affinity (Bohuslav et al., 1995; Simon et al., 2000; Wei et al., 2001; Wei et al., 1996; Yebra et al., 1996). Inhibition of the interaction of uPAR with integrins leads to the inhibition of integrin-mediated cell spreading and migration via inhibition of the downstream tyrosine kinases FAK and Src (Aguirre Ghiso et al., 1999; Wei et al., 2001).
Gradients of uPA and pro-uPA are also chemotactic for cells expressing uPAR. Binding of uPA to uPAR induces unmasking of a chemotactic epitope between domains D1 and D2 of the receptor. This epitope can also be exposed via direct proteolytic shedding of the D2D3 fragment (Fazioli et al., 1997; Resnati et al., 1996). The chemotactic effect is transduced via a GPCR, which binds the cryptic epitope and acts as a transmembrane adaptor (Fig. 3)(Resnati et al., 2002).
The subcellular localization of uPAR is critical in determining its transmembrane binding partners and subsequent cellular functions. uPAR interacts directly with caveolin-1, a scaffold protein that through its cholesterol binding properties and propensity to oligomerize regulates the formation of membrane microdomains termed caveolae (Simons and Ikonen, 1997; Wei et al., 1999). The localization of uPAR into lipid rafts is essential for its ability to bind vitronectin (Cunningham et al., 2003). On the other hand, uPAR can be localized

21
to the leading edge of migrating cells, where it associates with focal adhesions and regulates targeted proteolysis, cell-matrix interactions, and cytoskeletal rearrangement (Degryse et al., 1999; Gomez-Mouton et al., 2001; Kroon et al., 1999; Myöhänen et al., 1993).
Figure 3. uPAR regulates pericellular proteolysis, cell adhesion and chemotaxis. A. uPAR localizes the proteolytic activity of uPA to the pericellular area. PAI-1 binds uPAR-bound uPA on the cell surface and inhibits its proteolytic activity. B. uPAR binds vitronectin via an interaction dependent on uPA and modulates cell adhesion. In addition, uPAR directly interacts with integrins, mediates their affinity to ECM ligands and induces intracellular signaling. C. Binding of uPA to uPAR unmasks a chemotactic epitope between domains D1 and D2 of the receptor. The epitope can also be exposed by proteolytic shedding of the D2D3 fragment. The chemotactic effects are mediated via G-protein-coupled receptors (GPCR), which bind the cryptic epitope.
PAI-1 affects cell behavior via multiple mechanisms. Direct inhibition of plasminogen activation by PAI-1 results in decreased pericellular proteolysis with subsequent effects on cell adhesion and migration. Binding of PAI-1 to uPAR-bound uPA induces internalization of the complex, which affects the turnover and recycling of uPAR. Via its effect on uPA/uPAR internalization PAI-1 can also affect uPAR-mediated signaling. PAI-1-mediated endocytosis has been observed to enhance the phosphorylation of ERK induced by uPAR (Webb et al., 2001). In addition, PAI-1 is capable of inhibiting the interactions of both uPAR and integrins with vitronectin, and can induce the detachment of cells adhering to vitronectin (Stefansson and Lawrence, 1996). On the other hand it promotes α5β1
integrin-mediated migration from vitronectin toward fibronectin (Isogai et al., 2001). The effects of PAI-1 are highly concentration dependent. It promotes cell migration by reducing the amount of cell adhesion sites, but the saturation of these sites effectively prevents migration by complete inhibition of cell attachment (Loskutoff et al., 1999). Addition of uPA to this system decreases the affinity of PAI-1 to vitronectin (Seiffert et al., 1994). Thus, uPA could indirectly promote cell migration by removing of PAI-1 from the matrix, which leads to the exposure of integrin binding sites in the ECM.

22
Vasculogenesis and angiogenesis The first functional organ system in mammalian embryonic development is
the circulatory system. Mammalian cells require oxygen and nutrients for their survival, and the circulatory system functions in the delivery of these substances as well as in the removal of waste products. Blood vessel formation in the developing organism involves endothelial cell precursors, which assemble into a primitive vascular plexus of veins and arteries in response to growth factor stimulus. This process is termed vasculogenesis. This primitive vascular network is further remodeled and expanded by sprouting, vessel splitting, and fusion to form the mature vascular network in a process collectively termed angiogenesis. Finally, depending on the specialized function and host tissue of the vessels, the endothelial cells variably recruit pericytes and smooth muscle cells to form the periendothelial cell layer. Extracellular matrix is also produced to form the vascular BM, which stabilizes the vessel structures. In the adult, new vessels are produced mainly through angiogenesis, which occurs in conditions like tissue regeneration, inflammation, and cancer (Carmeliet, 2000; Klagsbrun and D'Amore, 1991; Risau, 1997).
Molecular mechanisms of angiogenesis
Mammalian cells must be located within the diffusion limit for oxygen (100 to 200 µm) of blood vessels to survive. Growth of a tissue beyond this limit leads to hypoxia. Under hypoxic conditions a transcriptional complex composed of hypoxia inducible factors is stabilized, leading to transcription of hypoxia inducible genes. These genes commonly encode for angiogenic growth factors such as vascular endothelial growth factor (VEGF) and angiopoietin-2 (Semenza, 2001). VEGF promotes vascular permeability and angiopoietin-2 loosens endothelial cell-matrix interactions, which are initial steps in angiogenic sprouting. This is followed by the release of proteases to degrade the surrounding ECM and extravasation of plasma proteins. These events facilitate endothelial cell migration by removing tissue boundaries, by liberating additional matrix-bound growth factors, and by providing a provisional matrix for cell attachment. Other important growth factors involved in the stimulation of endothelial cell migration include basic fibroblast growth factor (bFGF) and platelet derived growth factor (PDGF) (Conway et al., 2001; Ferrara, 2002; Yancopoulos et al., 2000).
The migrating endothelial cells subsequently adhere to the surrounding ECM, form tubular structures, and acquire a lumen. These processes are regulated by synergistic activity of various growth factors as well as integrin-mediated signaling and modulation of cell-matrix interactions (Bayless et al., 2000; Koolwijk et al., 1996). Finally, the vessel structures are stabilized and the endothelial cells acquire a quiescent phenotype (Fig. 4). Vascular integrity is maintained through survival signals generated by growth factors and shear stress (Carmeliet, 2000).

23
Figure 4. Cellular events in angiogenesis. Tumor cells, immune cells and stromal fibroblasts secrete growth factors and proteases, which induce degradation of the vascular BM and formation of the provisional matrix. These changes promote the proliferation and migration of endothelial cells. The migrating endothelial cells adhere to the surrounding ECM, form tubular structures, and acquire a lumen. The vessel structures are stabilized by the formation of the mature BM and the recruitment of pericytes. Fragments of the degraded BM are important regulators of these processes. Modified from (Kalluri, 2003).
Tumor angiogenesis and the angiogenic switch
An expanding tumor becomes rapidly hypoxic and undergoes necrotic cell death unless it becomes capable of developing its own vasculature. Tumor vessels develop by sprouting or splitting from pre-existing vessels (Carmeliet and Jain, 2000). In addition, recruited circulating endothelial cell precursors or stem cells mobilized directly from the bone marrow may contribute to the process (Rafii et al., 2002). Tumor vessels are abnormal in both structure and function. They are tortuous and dilated, with excessive branching and high permeability. In addition, the tumor endothelial cells often lack the perivascular cell lining, resulting in the lack of stability of the vessels. The tumor vessel wall is often a mosaic of endothelial cells and cancer cells, or it can lack endothelial cells entirely (Chang et al., 2000). The abnormal conditions result in variable blood flow and transient hypoxic events, which subsequently affect the production of angiogenic growth factors, promote the selection of cancer cell clones more resistant to hypoxia, and lower effectiveness of anti-cancer therapy (Eberhard et al., 2000).
The ability of tumors to induce angiogenesis is acquired in a stepwise process described as an angiogenic switch. Analysis of various transgenic mouse models has revealed that oncogene expression and hyperproliferation are not sufficient to transform a carcinoma in situ-lesion into an expanding tumor. Instead, accumulating evidence suggests that the rate-limiting step of tumorigenesis is the ability of the lesions to acquire an angiogenetic phenotype. This occurs through a shift in the balance of pro- and anti-angiogenic agents, which can be derived from

24
cancer cells, endothelial cells, or stromal cells (Hanahan and Folkman, 1996). Several tumors express increased levels of VEGF and/or bFGF, and decreased levels of endogenous inhibitors of angiogenesis, such as thrombospondin-1. These changes in expression levels may be a consequence of mutations in oncogenes or tumor suppressor genes (Rak et al., 2002). Another source of pro-angiogenic factors are inflammatory cells infiltrating the tumor, which secrete VEGF, angiopoietin-1, bFGF, PDGF, and many others (Yu and Rak, 2003). In addition, enhanced secretion of proteases leads to increased bioavailability of angiogenic growth factors through their release from the ECM (Bergers and Benjamin, 2003; Taipale and Keski-Oja, 1997). On the other hand, excess proteolysis may also contribute to the generation of anti-angiogenic molecules by exposing cryptic fragments of matrix molecules (Kalluri, 2003).
Role of integrins in angiogenesis
Endothelial cells express multiple integrins, but endothelial cell integrins αvβ3, αvβ5, and α5β1 are considered central to the regulation of angiogenesis. Integrins αvβ3 and αvβ5 become dramatically induced in activated endothelial cells in the tumor vasculature. In addition, inhibitors of integrins αvβ3, and αvβ5 disrupt pathological angiogenesis in various model systems (Brooks et al., 1994a; Brooks et al., 1994b; Hammes et al., 1996). However, mice lacking β3 integrins have no defects in developmental angiogenesis, but instead display enhanced tumor angiogenesis (Reynolds et al., 2002), suggesting that β3 integrins can also function as negative regulators of angiogenesis (Hynes, 2002b). A putative mechanism of negative regulation via αvβ3 integrin is its interaction with endogenous angiogenesis inhibitors such as tumstatin (Hamano et al., 2003; Maeshima et al., 2002). In addition, negative regulation could be mediated via integrin�mediated cell death, where expression of αvβ3 integrin in the absence of proper ligands results in endothelial cell apoptosis (Stupack et al., 2001). Furthermore, extensive crosstalk occurs between different integrin heterodimers, suggesting that they could functionally substitute for each other during angiogenesis (Hynes, 2002b; Kim et al., 2000b; Simon et al., 1997).
The role of integrin α5β1 seems to be different from that of the αv integrins. Like αv integrins, α5β1 integrin is upregulated on endothelial cells in response to angiogenic growth factors (Kim et al., 2000a). In contrast to αv integrins, the loss of α5 leads to severely impaired developmental angiogenesis (Yang et al., 1993). In addition, neutralization of the function of this integrin by specific antibodies or synthetic peptides inhibits bFGF-induced angiogenesis in vitro and in vivo (Kim et al., 2000a).

25
Proteolytic cascades in angiogenesis
Proteases are involved in multiple aspects of angiogenesis. Proteolyic degradation of sub-endothelial BM is required for endothelial cell migration into the underlying ECM. In addition, proteases stimulate endothelial cell migration through activation of pro-angiogenic growth factors and through their release from the ECM. In addition, proteolytic cleavage of the ECM unmasks cryptic adhesion sites and liberates bioactive degradation products (Pepper et al., 1996; Werb et al., 1999). On the other hand, proteases negatively regulate angiogenesis by generation of endogenous inhibitors of angiogenesis from the components of the ECM (Kalluri, 2003). Physiological angiogenesis requires a balance in proteolysis. Excessive proteolysis results in inhibition of angiogenesis due to destruction of the matrix scaffold that is needed for endothelial cell migration and tubular morphogenesis and generation of endogenous inhibitors of angiogenesis. Insufficient proteolysis, on the other hand, inhibits migration and tubular morphogenesis due to limited degradation of ECM barriers and release of proangiogenic growth factors (Bergers and Benjamin, 2003; Pepper, 2001). The most relevant protease systems involved in angiogenesis are the plasminogen activators and the MMPs.
uPA, uPAR, and PAI-1 are not detectable in the quiescent endothelium, but are expressed during active angiogenesis (Bacharach et al., 1992; Larsson et al., 1984). In vitro, the expression of these proteins is regulated by angiogenic growth factors such as bFGF and VEGF (Mignatti and Rifkin, 1996). In addition, at least uPAR and PAI-1 are upregulated by hypoxia, a major inducer of angiogenesis (Kroon et al., 2000; Uchiyama et al., 2000). uPA induces neovascularization in the rabbit cornea and promotes growth, chemotaxis, and matrix invasion of cultured endothelial cells. These effects require interaction of uPA with uPAR, which can be prevented by monoclonal antibodies against either of the proteins (Fibbi et al., 1998). Despite the evidence implicating a role for the plasminogen activator system in angiogenesis, no abnormalities in vascular development are seen in mice lacking uPA, tPA, or uPAR (Carmeliet et al., 1994; Dewerchin et al., 1996). Several studies have, however, indicated a role for plasminogen activators in other processes involving angiogenesis. The uPA/uPAR interaction has been observed to be important for tumor-associated neovascularization, primary tumor growth, and metastasis (Crowley et al., 1993; Min et al., 1996; Ossowski, 1996). Studies on PAI-1 deficient mice have revealed it to be crucial for tumor vascularization, and that the proteolytic activity of plasmin is involved in this process. The role of PAI-1 is to maintain controlled proteolytic breakdown of the ECM (Bajou et al., 2001; Bajou et al., 1998). tPA is important for the maintenance of vascular patency in uninjured arteries, but does not seem to play an important role in angiogenesis (Loskutoff et al., 1995).
Endothelial cells express MMP-1, -2, -3, -9, and �14. The basal expression levels of these proteases are relatively low, but they are upregulated in a variety of physiological and pathological conditions involving neovessel formation. These

26
include embryogenesis, wound healing, rheumatoid arthritis, and cancer (John and Tuszynski, 2001; Pepper, 2001). MMP-9 is upregulated in angiogenic lesions, and in a transgenic mouse model of pancreatic islet carcinomas, it has been shown to induce the transition of benign carcinoma in situ lesions into angiogenic carcinomas, via an increase in the bioavailabilty of vascular endothelial growth factor VEGF (Bergers et al., 2000). On the other hand, MMP-9 is essential in generating the anti-angiogenic fragment tumstatin from type IV collagen. When tumor growth exceeds a critical size, accelerated tumor growth is observed in mice lacking MMP-9 (Hamano et al., 2003). MMP-2 has been observed to play a role in angiogenesis in a rat Swarm chondrosarcoma model, in which increased MMP-2 activity was associated with the switch to the angiogenic phenotype (Fang et al., 2000). In addition, the C-terminal hemopexin-like domain of MMP-2 inhibits bFGF induced angiogenesis and tumor growth. It also inhibits the targeting of MMP-2 to the cell surface via αvβ3 integrin (Brooks et al., 1998; Silletti et al., 2001). MMP-2 deficient mice display reduced tumor angiogenesis in a dorsal sac assay with B16-BL6 melanoma cells (Itoh et al., 1998). MT1-MMP is expressed at high levels in the endothelium of the developing embryo (Apte et al., 1997). MT1-MMP deficient mice display defects in angiogenesis in the cartilage. In addition, an impaired angiogenic response to bFGF stimulus is seen in the corneal angiogenesis assay of these mice (Holmbeck et al., 1999; Zhou et al., 2000). Analysis of the fibrinolytic activity in an ex vivo model has revealed MT1-MMP to be an extremely potent fibrinolytic enzyme, suggesting an important role for this protease in endothelial cell invasion of the provisional ECM (Hiraoka et al., 1998). In addition, the ability of MT1-MMP to activate MMP-2 may provide additional roles for this enzyme in angiogenesis.
Basement membrane-derived inhibitors of angiogenesis Vascular integrity and endothelial cell quiescence is maintained in part via
interaction of endothelial cells with the underlying intact basement membrane. However, as a result of angiogenesis-associated assembly or disassembly of the BM, endothelial cells interact with the different domains of these proteins. Thus, the same molecules, depending on their structural configuration, differently regulate endothelial cell behavior at various stages of the angiogenic process and cancer progression. As the angiogenic switch is turned on, targeted proteolysis induces degradation of the BM, and cryptic domains of partially degraded collagens become exposed. Fragments of perlecan, laminin, SPARC, type XV collagen and type XVIII collagen are molecules, which contain both anti- and pro-angiogenic cues (Mongiat et al., 2003; Ortega and Werb, 2002; Sage et al., 2003).
Fragments of type IV collagen
Type IV collagen isolated from the vascular BM contains no anti-angiogenic activity. However, when the collagen is further degraded with a mixture of tumor microenvironment-associated proteolytic enzymes, the liberated cryptic fragments have anti-angiogenic activity (Petitclerc et al., 2000). Three of the fragments
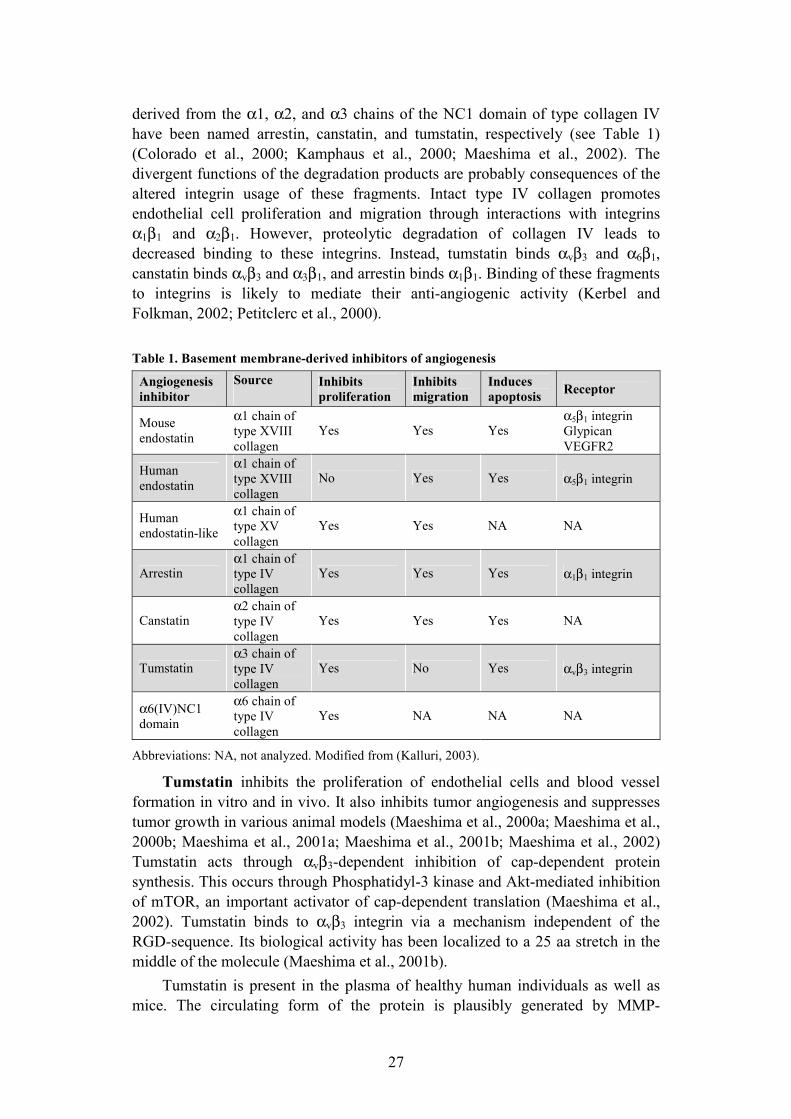
27
derived from the α1, α2, and α3 chains of the NC1 domain of type collagen IV have been named arrestin, canstatin, and tumstatin, respectively (see Table 1) (Colorado et al., 2000; Kamphaus et al., 2000; Maeshima et al., 2002). The divergent functions of the degradation products are probably consequences of the altered integrin usage of these fragments. Intact type IV collagen promotes endothelial cell proliferation and migration through interactions with integrins α1β1 and α2β1. However, proteolytic degradation of collagen IV leads to decreased binding to these integrins. Instead, tumstatin binds αvβ3 and α6β1,canstatin binds αvβ3 and α3β1, and arrestin binds α1β1. Binding of these fragments to integrins is likely to mediate their anti-angiogenic activity (Kerbel and Folkman, 2002; Petitclerc et al., 2000).
Table 1. Basement membrane-derived inhibitors of angiogenesis
Abbreviations: NA, not analyzed. Modified from (Kalluri, 2003).
Tumstatin inhibits the proliferation of endothelial cells and blood vessel formation in vitro and in vivo. It also inhibits tumor angiogenesis and suppresses tumor growth in various animal models (Maeshima et al., 2000a; Maeshima et al., 2000b; Maeshima et al., 2001a; Maeshima et al., 2001b; Maeshima et al., 2002) Tumstatin acts through αvβ3-dependent inhibition of cap-dependent protein synthesis. This occurs through Phosphatidyl-3 kinase and Akt-mediated inhibition of mTOR, an important activator of cap-dependent translation (Maeshima et al., 2002). Tumstatin binds to αvβ3 integrin via a mechanism independent of the RGD-sequence. Its biological activity has been localized to a 25 aa stretch in the middle of the molecule (Maeshima et al., 2001b).
Tumstatin is present in the plasma of healthy human individuals as well as mice. The circulating form of the protein is plausibly generated by MMP-
Angiogenesis inhibitor
Source Inhibits proliferation
Inhibits migration
Induces apoptosis Receptor
Mouse endostatin
α1 chain of type XVIII collagen
Yes Yes Yesα5β1 integrin Glypican VEGFR2
Human endostatin
α1 chain of type XVIII collagen
No Yes Yes α5β1 integrin
Human endostatin-like
α1 chain of type XV collagen
Yes Yes NA NA
Arrestin α1 chain of type IV collagen
Yes Yes Yes α1β1 integrin
Canstatin α2 chain of type IV collagen
Yes Yes Yes NA
Tumstatin α3 chain of type IV collagen
Yes No Yes αvβ3 integrin
α6(IV)NC1 domain
α6 chain of type IV collagen
Yes NA NA NA

28
mediated proteolysis as part of the regular turnover process of BMs (Kalluri, 2003). Studies on mice lacking the α3 chain of collagen IV/tumstatin suggest, that the physiological levels of circulating tumstatin suppress tumor angiogenesis through interaction with the αvβ3 integrin expressed in the tumor endothelium. Physiological processes involving angiogenesis, such as pregnancy, development, and wound healing occur normally in these mice. MMP-9 deficient mice have reduced levels of circulating tumstatin, and display accelerated tumor growth after the tumors exceed 500 mm3 in size. Tumor growth can be sustained with the delivery of exogenous tumstatin (Hamano et al., 2003).
Endostatin
Endostatin was isolated in 1996 from the conditioned media of a murine hemangioendothelioma (EOMA) cell line. Sequence analysis revealed it to be the C-terminal, 20 kDa fragment of the NC1 domain of type XVIII collagen. In the pioneer study, soluble baculovirally produced endostatin was observed to be an endothelial cell-specific inhibitor of endothelial cell proliferation and migration (O'Reilly et al., 1997). The growth of primary tumors, such as the Lewis lung carcinoma, T241 fibrosarcoma, EOMA hemangioendothelioma and B16F10 melanoma planted in syngeneic mice, were inhibited by systemic administration of endostatin, produced as an insoluble precipitate in E. coli. No evidence of any toxicity, drug resistance, or regrowth of tumors during treatment was observed (O'Reilly et al., 1997).
Structural features
Endostatin is cleaved C-terminally of the trimerization domain of type XVIII collagen to yield monomeric endostatin. The cleavage can be executed by various proteinases, including cysteine proteinases such as cathepsin L, MMPs, and the serine proteinase elastase. Proteolytic processing of type XVIII collagen generates both NC1 trimers and endostatin monomers in vivo, and both forms can be detected in tissues and serum. MMPs generate a 30-kDa fragment, but cathepsin L directly releases the 20-kDa endostatin fragment. Endostatin itself is resistant to proteolysis by MMPs, but is degraded by the other proteinases. Protease activity thus regulates both the generation and stability of endostatin (Felbor et al., 2000; Ferreras et al., 2000; Sasaki et al., 1998; Wen et al., 1999). Proteolytically cleaved endostatin remains associated with the vascular BM, where it colocalizes with the heparan sulfate proteoglycan perlecan (Miosge et al., 1999). Endostatin has also been detected from platelets and the plasma of healthy individuals (Ma et al., 2001; Zorick et al., 2001).

29
Figure 5. Model of the three dimensional structure of the mouse collagen XVIII C-terminal domain, endostatin. The endostatin domain has a compact globular fold that is distantly related to the C-type lectins. The four cysteines of endostatin are linked 1�4, 2�3. A zinc ion is bound near the N-terminus, but the zinc binding site appears not to be functionally important. Endostatin contains an arginine rich extensive basic patch responsible for its binding to heparan sulfate. The basic arginine (R) residues and the solvent-exposed side chains of phenylalanine (F) residues are labeled (Hohenester and Engel, 2002; Hohenester et al., 1998).
The structure of the endostatin domain is related to the C-type lectin carbohydrate-recognition domain (Fig. 5). It is characterized by a compact globular fold and a basic patch of 11 arginine residues, which act as binding sites for heparin (Hohenester et al., 1998; Kreuger et al., 2002). Endostatin binds also Zn++ at a 1:1 molar ratio, but this property does not seem to be essential for the activity of endostatin (Ding et al., 1998; Hohenester et al., 2000; Yamaguchi et al., 1999). The molecule is stabilized by two intramolecular disulfide bridges (Standker et al., 1997).
The NC1 domain of type XV collagen is highly homologous to that of type XVIII with 60 % sequence identity. Its proteolytic fragment has also been observed to have anti-angiogenic activity, although it is less potent than endostatin. Interestingly, the fragment of type XV collagen, termed restin or endostatin-like, lacks the zinc- and heparin-binding domains (Sasaki et al., 2002).
Biological roles
The basement membrane location of the endostatin fragment suggests a local regulatory role of endostatin in vessel growth. However, endostatin appears not to be a critical regulator of angiogenesis, since type XVIII collagen/endostatin-deficient mice display no major vascular abnormalities. No significant changes in the growth of primary tumors or tumor angiogenesis compared to the wild-type mice have been observed (Fukai et al., 2002). In addition, patients with Knobloch syndrome do not display vascular abnormalities or increased incidence of cancer (Sertie et al., 2000).
The physiological levels of circulating endostatin in the plasma of healthy individuals ranges from 10-50 ng/ml (0.5-2.5 nM), with a wide range of variation within the population (Hefler et al., 1999; Zorick et al., 2001). The biological

30
functions of circulating endostatin are currently unclear. It might participate in the regulation of angiogenesis, but the circulating concentrations are lower than those that efficiently inhibit endothelial cell migration in vitro. A number of studies have reported elevated levels of circulating endostatin in various types of human cancer. The elevated levels of endostatin correlate to tumor aggressiveness and poor prognosis (Feldman et al., 2000b; Feldman et al., 2001b; Feldman et al., 2001c; Feldman et al., 2002; Shaarawy and El-Sharkawy, 2001; Suzuki et al., 2002). Interestingly, a point mutation in endostatin has been reported to predispose to the development of prostate adenocarcinoma (Iughetti et al., 2001).
Down syndrome is a complex developmental disorder most commonly caused by a duplication of one of the copies of chromosome 21, which is the chromosome where the COL18A1 gene is located. Down syndrome patients have decreased incidence for solid tumors and concomitantly increased serum levels of endostatin. No recognized tumor suppressor genes localize to this chromosome, implying that circulating endostatin might act as a protective agent for the development of solid tumors (Zorick et al., 2001).
Studies on cle-1, the Caenorhabditis elegans homologue of type XVIII collagen, have revealed a putative role for the NC1 domain/endostatin in cell motility. The protein is expressed at high levels in the nervous system, and deletion of the domain leads to multiple defects in axon guidance and migration of neural and non-neural cells. Interestingly, this phenotype can be rescued by ectopic expression of the trimeric NC1, but not monomeric endostatin (Ackley et al., 2001). The motogenic activity of the trimeric NC1 domain of human type XVIII collagen and its inhibition by monomeric endostatin is also observed cell culture experiments. The activity of the NC1 domain is not specific for endothelial cells, but has also been observed with various non-endothelial cells, whereas the activity of endostatin is restricted to endothelial cells. The motogenic activity of NC1 is dependent on the presence of the ECM and of rac, cdc42 and the MAP kinase pathway (Kuo et al., 2001).
Endostatin may also regulate tissue morphogenesis. The ureteric bud produces the endostatin fragment, which inhibits hepatocyte growth factor-induced migration and branching morphogenesis of renal epithelial cells and the ureteric bud. These effects are dependent on the presence of the heparan sulfate proteoglycan syndecan-3 (Karihaloo et al., 2001).
Effects of endostatin on tumor growth
While the biological role of the endostatin fragment is still unclear, numerous studies have indicated recombinant endostatin to be a very potent inhibitor of tumor angiogenesis. The lack of dramatic vascular effects of endogenous endostatin could be a result of its sequestration in the basement membranes, where it is not accessible for interaction with cell surface receptors (Fukai et al., 2002). In addition, the concentrations of endostatin used to achieve anti-tumor effects are 10-fold higher than the levels of endostatin in the circulation, suggesting that the pharmacological effects of high concentrations of endostatin might be distinct from its physiological effects.
31
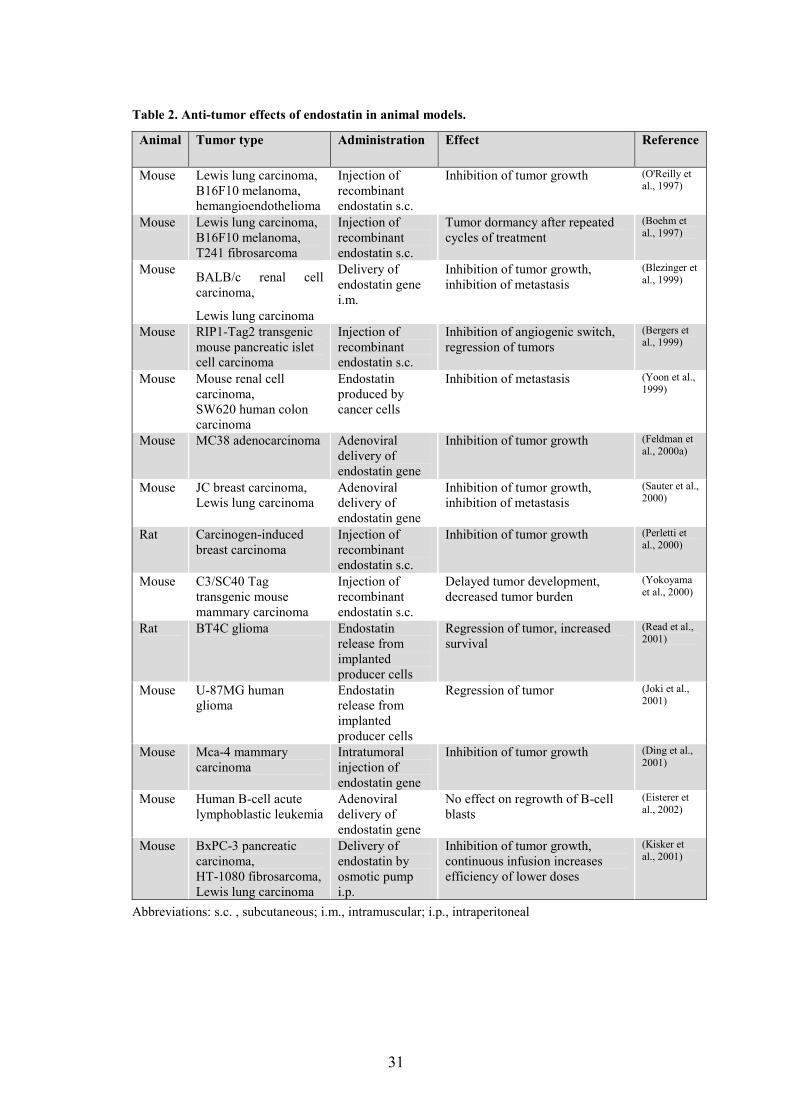
Table 2. Anti-tumor effects of endostatin in animal models.
Abbreviations: s.c. , subcutaneous; i.m., intramuscular; i.p., intraperitoneal
Animal Tumor type Administration Effect Reference
Mouse Lewis lung carcinoma, B16F10 melanoma, hemangioendothelioma
Injection of recombinant endostatin s.c.
Inhibition of tumor growth (O'Reilly et al., 1997)
Mouse Lewis lung carcinoma, B16F10 melanoma, T241 fibrosarcoma
Injection of recombinant endostatin s.c.
Tumor dormancy after repeated cycles of treatment
(Boehm et al., 1997)
Mouse BALB/c renal cell carcinoma,
Lewis lung carcinoma
Delivery of endostatin gene i.m.
Inhibition of tumor growth, inhibition of metastasis
(Blezinger et al., 1999)
Mouse RIP1-Tag2 transgenic mouse pancreatic islet cell carcinoma
Injection of recombinant endostatin s.c.
Inhibition of angiogenic switch, regression of tumors
(Bergers et al., 1999)
Mouse Mouse renal cell carcinoma, SW620 human colon carcinoma
Endostatin produced by cancer cells
Inhibition of metastasis (Yoon et al., 1999)
Mouse MC38 adenocarcinoma Adenoviral delivery of endostatin gene
Inhibition of tumor growth (Feldman et al., 2000a)
Mouse JC breast carcinoma, Lewis lung carcinoma
Adenoviral delivery of endostatin gene
Inhibition of tumor growth, inhibition of metastasis
(Sauter et al., 2000)
Rat Carcinogen-induced breast carcinoma
Injection of recombinant endostatin s.c.
Inhibition of tumor growth (Perletti et al., 2000)
Mouse C3/SC40 Tag transgenic mouse mammary carcinoma
Injection of recombinant endostatin s.c.
Delayed tumor development, decreased tumor burden
(Yokoyama et al., 2000)
Rat BT4C glioma Endostatin release from implanted producer cells
Regression of tumor, increased survival
(Read et al., 2001)
Mouse U-87MG human glioma
Endostatin release from implanted producer cells
Regression of tumor (Joki et al., 2001)
Mouse Mca-4 mammary carcinoma
Intratumoral injection of endostatin gene
Inhibition of tumor growth (Ding et al., 2001)
Mouse Human B-cell acute lymphoblastic leukemia
Adenoviral delivery of endostatin gene
No effect on regrowth of B-cell blasts
(Eisterer et al., 2002)
Mouse BxPC-3 pancreatic carcinoma, HT-1080 fibrosarcoma, Lewis lung carcinoma
Delivery of endostatin by osmotic pump i.p.
Inhibition of tumor growth, continuous infusion increases efficiency of lower doses
(Kisker et al., 2001)

32
Studies on various animal models have shown that endostatin inhibits the growth of primary tumors. In addition, endostatin can inhibit or reduce the formation of metastases (Table 2). Strikingly, when delivered in cycles where the tumors are allowed to regrow between repeated treatments, endostatin is still effective and induces prolonged tumor dormancy without drug resistance (Boehm et al., 1997).
In the adult, endostatin seems to specifically act on tumor vessels. Physiological processes involving angiogenesis, such as wound healing are essentially unaffected by endostatin treatment, even though some disturbances in vessel maturation can be seen (Berger et al., 2000; Bloch et al., 2000). This might be a result of altered expression of cell surface proteins in tumor blood vessels compared to the normal vasculature (Ruoslahti, 2002). Endostatin may also target endothelial cell progenitors, and inhibit their mobilization and clonogenic potential. Endothelial cell progenitors play a role in tumor angiogenesis at least in certain tumor types (Capillo et al., 2003; Schuch et al., 2003).
Cell surface receptors
A number of different putative cell surface receptors for endostatin have been identified. Immobilized endostatin promotes cell adhesion via α5β1 and αvβ3
integrins (Rehn et al., 2001). Soluble endostatin has been observed to inhibit α5β1-mediated signaling. In addition, blocking the interaction between endostatin and integrin α5β1 leads to the loss of endostatin-induced effects on cell migration, indicating that α5β1 integrin is a functional receptor for endostatin (Sudhakar et al., 2003). However, both mouse and human endostatin lack RGD-sequences, and specific integrin-binding sites have not been identified yet.
Endostatin is also a heparin-binding protein. Studies using chick chorioallantoic membrane assay as a model for growth factor induced angiogenesis indicate that mutations of arginine residues critical for heparin binding results in the loss of endostatin function (Sasaki et al., 1999). In addition, endostatin binds with low affinity to glypicans 1 and 4, cell surface heparan sulfate proteoglycans expressed on endothelial cells (Karumanchi et al., 2001). The recognition motif of endostatin on glycosaminoglycans is highly specific and distinct from known growth factor binding domains, providing a putative explanation for the preferential interaction of endostatin with specific cell surface proteoglycan species (Kreuger et al., 2002).
Endostatin has also been reported to bind to VEGF receptor 2, resulting in competitive inhibition of receptor activity (Kim et al., 2002). Other studies have, however, concluded that endostatin does not interfere with major growth factor signaling pathways (Dixelius et al., 2000; Eriksson et al., 2003; Karihaloo et al., 2001).
An intracellular actin-interacting protein tropomyosin has been identified as an endostatin binding protein by using a yeast two-hybrid approach. A peptide that inhibited the interaction between endostatin and tropomyosin-3 decreased the inhibitory effect of endostatin on the formation of metastases in a mouse melanoma model (MacDonald et al., 2001).

33
Mechanisms of action
The possible cell biological mechanisms underlying the anti-angiogenic effects of endostatin include inhibition of endothelial cell migration, induction of cell cycle arrest, and promotion of apoptosis (Dhanabal et al., 1999; Dixelius et al., 2000; Hanai et al., 2002a; O'Reilly et al., 1997). The most consistent effects seem to be the inhibition of migration and proliferation. However, in contrast to mouse endostatin, human endostatin does not seem to affect endothelial cell proliferation (Sudhakar et al., 2003; Yamaguchi et al., 1999). The reason for this major difference in activity is unclear. Endostatin seems to act differently depending on the cell culture conditions. Endostatin induces apoptosis and inhibits cell migration in the absence of serum, but in the presence of serum only inhibition of migration is observed (Shichiri and Hirata, 2001).
Endostatin inhibits bFGF in VEGF induced migration in several different endothelial cell types in vitro (Dhanabal et al., 1999; Dixelius et al., 2000; O'Reilly et al., 1997; Taddei et al., 1999; Yamaguchi et al., 1999), but the molecular targets of endostatin have remained unclear. Endostatin has various effects on intracellular signaling pathways in different types of endothelial cells. However, although endostatin is a potent inhibitor of bFGF- and VEGF-induced endothelial cell migration, it does not seem to affect the key signaling pathways involved in growth factor stimulated cell migration (Eriksson et al., 2003). Migration of endothelial cells involves concerted assembly and disassembly of focal adhesions in cooperation with integrin signaling. Immobilized endostatin promotes and soluble endostatin inhibits α5β1 integrin-mediated endothelial cell migration (Rehn et al., 2001; Sudhakar et al., 2003). In addition, endostatin targets the cytoskeleton by binding tropomyosin or affecting the assembly of the cytoskeleton (Dixelius et al., 2002; MacDonald et al., 2001). It also affects the stability and sub-cellular localization of β-catenin, a protein involved in the regulation of cell-cell interactions and cell motility (Dixelius et al., 2002). ß-catenin has also been identified as a target for endostatin in studies using Xenopusembryogenesis as a model for endostatin signaling. Endostatin RNA blocks the axis duplication induced by ß-catenin, partially inhibits Wnt-dependent transcription, and stimulates degradation of ß-catenin (Hanai et al., 2002b). Inhibition of VEGF-induced migration induces dephosphorylation of eNOS via an Akt-mediated pathway (Urbich et al., 2002).
In addition to its anti-migratory effect, endostatin can act as a proteinase inhibitor. Endostatin inhibits MMP-2 activity by binding to its catalytic domain. The affinity of endostatin binding to MMP-2 is significantly weaker than that of the endogenous inhibitor tissue inhibitor of metalloproteinases (TIMP)-2 (Kim et al., 2000c; Lee et al., 2002). Inhibition of the activity of MMPs -9 and -13 has also been observed (Nyberg et al., 2003).
The other putative cell biological effects of endostatin include induction of apoptosis via phosphorylation of the adaptor protein Shb (Dixelius et al., 2000), induction of endothelial cell cycle arrest (Hanai et al., 2002a), induction of intracellular calcium signaling (Jiang et al., 2001; Shichiri and Hirata, 2001), downregulation of genes involved in endothelial cell migration and proliferation

34
(Shichiri and Hirata, 2001; Yin et al., 2002), and induction of protein kinase A activity via cAMP production (Shichiri and Hirata, 2001).
Angiogenesis inhibitors in cancer therapy
Various inhibitors of angiogenesis have been developed to target vascular endothelial cells in order to inhibit tumor angiogenesis and thus prevent tumor growth. Targeting endothelial cells rather than the tumor cells themselves is promising, since endothelial cells are genetically stable and therefore less prone to accumulate mutations leading to drug resistance (Boehm et al., 1997). The mechanisms of action of angiogenesis inhibitors can be direct or indirect. Direct angiogenesis inhibitors such as endostatin and tumstatin target the tumor endothelial cells and inhibit their migration or proliferation (Table 3). Indirect angiogenesis inhibitors such as the receptor tyrosine kinase inhibitors target the tumor cells and inhibit the expression of pro-angiogenic proteins. They can also target the endothelial cells and inhibit the expression of the receptors for the pro-angiogenic proteins (Kerbel and Folkman, 2002).
Table 3. Direct inhibitors of angiogenesis.
Drug Mechanisms of action Clinical trials Reference
Angiostatin Inhibits endothelial cell proliferation and migration Phase I (O'Reilly et al.,
1994)
Bevacizumab A humanized monoclonal antibody against VEGF Phase I and III (Ferrara, 2002)
Arresten Binds to integrin α1β1 and inhibits endothelial cell proliferation and migration
No (Colorado et al., 2000)
Canstatin Binds to integrin αvβ3 and inhibits endothelial cell proliferation and migration
Phase I (Kamphaus et al., 2000)
Combretastatin Binds tubulin and causes microtubule depolymerization
Completed Phase I
(Kanthou and Tozer, 2002)
Endostatin Binds integrin α5β1 and inhibits endothelial cell migration Phase I and II (O'Reilly et al.,
1997)
NM-3 Small molecule inhibitor of VEGF Phase I (Reimer et al., 2002)
Thrombospondin Binds CD36 and inhibits endothelial cell proliferation and migration
No (Lawler, 2000)
Tumstatin Binds to integrin αvβ3 and inhibits endothelial cell proliferation No (Maeshima et al.,
2002)
2-methoxy-estradiol
Induces endothelial cell apoptosis by inhibiting microtubule function Phase I and II (D'Amato et al.,
1994)
Vitaxin A humanized monoclonal antibody against integrin αvβ3
Phase I and II (Gutheil et al., 2000)
Modified from (Kerbel and Folkman, 2002).

35
Angiogenesis inhibitors exert their anti-tumor effects via multiple mechanisms. Most anti-angiogenic agents inhibit the sprouting of new microvessels from the existing vasculature, but also induce regression of recently developed microvessels. This subsequently leads to tumor cell death (Kieran et al., 2003). However, angiogenesis inhibitors also transiently decrease vessel leakiness, which results in increased tumor blood flow and decreased intratumor pressure. Angiogenesis inhibitors may thus enhance the efficacy of radiation therapy as well as increase the delivery of cytotoxic drugs into the tumors (Jain, 2001). Despite an increase in blood flow in the early course of anti-angiogenic therapy, the net effect seems to be a gradual decrease in tumor blood flow. This effect has also been observed in endostatin-treated cancer patients (Herbst et al., 2002b).
Studies on the effects of recombinant endostatin in various animal tumor models have indicated that continuous administration of endostatin is more efficient than interval or bolus treatment (Kisker et al., 2001). In addition, the anti-angiogenic effects of endostatin are achieved most effectively by maintaining constant serum levels of 300-500 ng/ml, and concentrations below or above this window may result in decreased efficacy (Kerbel and Folkman, 2002). Several strategies for the constant and local delivery of endostatin have been successfully applied, including adenoviral gene transfer and implantation encapsulated endostatin producer cells (Feldman et al., 2001a; Joki et al., 2001; Read et al., 2001; Sauter et al., 2000).
The Phase I clinical trials of endostatin concluded that the recombinant protein can be administered to patients at high doses concentrations no signs of toxicity (Herbst et al., 2002a). Endostatin is currently in Phase II clinical trials, but the results are still unavailable.

36
Aims of the present study
When this study was initiated, endostatin had been identified as an inhibitor of angiogenesis and tumor growth. The aims of the current study were to elucidate the cell biological mechanisms underlying the anti-angiogenic potential of endostatin and to understand the mechanisms by which a fragment of the ECM regulates cell-matrix interactions. As pericellular proteolysis is an important process in angiogenesis and endothelial cell migration, the first aim of this study was to analyze changes in endothelial cell plasminogen activation in response to endostatin treatment. During the course of these studies we found that endostatin induces the disruption of actin stress fibers and focal adhesions. This finding led us to further characterize the mechanisms by which endostatin associates with cell surface adhesion molecules and induces changes in cell adhesion and migration.
Specific questions addressed in these studies include the following:
1) How does endostatin regulate uPA and PAI-1 at the levels of protein expression, and how is their cell surface localization altered?
2) How does endostatin affect the cytoskeletal structures involved in cell migration and what are the molecular mechanisms behind these effects?
3) What kind of molecular interactions regulate the endothelial cell surface association of endostatin, and what are the consequences resulting from these interactions?
4) Which specific domains or amino acid sequences of the endostatin protein account for its cell surface and integrin binding properties?

37
Materials and methods
Cell culture and treatments
Human dermal microvascular endothelial cells (HDMEC) were purchased from Promocell (Heidelberg, Germany) and were cultured in Endothelial Cell Growth Medium (Promocell), at 37 °C in a humidified 5 % CO2 atmosphere. The cells used for the experiments were from passages 3-6. All experiments were carried out under serum-free conditions, in which the cells were washed twice and incubated in serum-free medium-199 for at least 8 h prior to treatment with the various proteins or chemicals. The cells were treated with endostatin (40-50 nM), bFGF (50 ng/ml), PP1 (10-100 µM), orthovanadate (50-100 µM), heparinase III (20 mU/ml), Filipin III (2.5 µg/ml), methyl-β-cyclodextrin (5 mM) or endostatin-derived peptides (500 nM) for various periods of time.
Expression and characterization of human endostatin
Recombinant human endostatin used in studies I and II was expressed and purified as described (Rehn et al., 2001). A fragment of human collagen XVIII that corresponds to mouse endostatin sequences (O'Reilly et al., 1997) was cloned to the vector pQE-31 (Qiagen, Inc., Santa Clarita, CA) and expressed as an N-terminal His-tagged protein in E. coli strain M15 according to the manufacturer�s protocol (Qiagen). Bacterial pellets were lysed and the supernatant was lightly sonicated and applied to a ProBond column (Invitrogen, San Diego, CA). Bound protein was eluted by an imidazole gradient, and the endostatin fractions were pooled and refolded in vitro. Refolded soluble endostatin was separated by centrifugation and applied to a HiTrap SP cation-exchange column (Amersham Pharmacia, Piscataway, NJ). Endostatin fractions were eluted by a NaCl gradient and applied to a heparin-Sepharose CL-6B column (Amersham Pharmacia). Bound endostatin was eluted by a NaCl gradient. Endostatin fractions were pooled and dialyzed against PBS, pH 7.4, and passed through a Polymyxin agarose column using PBS buffer. No endotoxin was found in the purified fractions. In studies III and IV, endostatin was from Calbiochem.
Growth factors, chemicals, and enzymes
bFGF was from R&D Systems Inc. (Minneapolis, MN). Sodium orthovanadate was from Sigma Chemical Co. (St. Louis, MO). PP1 was purchased from Biomol Research Laboratories Inc. (Plymouth Meeting, PA). Human plasminogen was from Chromogenix (Mölndal, Sweden) and HMW-uPA from Calbiochem (San Diego, CA).

38
Antibodies
The following antibodies have been used in the experiments as specified in the original publications.
Name: Source/ Clone: Application: Reference: α5 integrin Mouse / P1D6 BLK Chemicon αv integrin Mouse / M9 BLK Chemicon α5β1 integrin Mouse / JBS5 IF Chemicon α5β1 integrin Goat IB, IP Chemicon β1 integrin Mouse / P5D2 BLK Chemicon β3 integrin Rabbit BLK Chemicon Caveolin-1 Mouse IB Transduction
Laboratories Caveolin-1 Rabbit IF, IP Transduction
Laboratories Endostatin Rabbit IB Chemicon Fibronectin Rabbit IB, IF, IP Sigma Glypican-1 Mouse / N-16 IB Santa Cruz p190 Mouse / 30 IB, IP Transduction
Laboratories PAI-1 Mouse IP American Diagnostica Paxillin Mouse IF Transduction
Laboratories pSrc Rabbit IB, IF New England Biolabs RhoA Mouse IB Santa Cruz Src Rabbit IB, IP Santa Cruz Transferrin Receptor Mouse / 2 IB Transduction
Laboratories uPA Rabbit IF, IP American Diagnostica uPAR Mouse IF (Rønne et al., 1991) Vinculin Mouse IF Sigma
SDS-PAGE and immunoblotting
SDS-PAGE (polyacrylamide gel electrophoresis in the presence of sodium dodecyl sulphate) was carried out using 4-15 % gradient polyacrylamide gels (Bio-Rad Co., Richmond, VA). The proteins were then transferred onto nitrocellulose filters, which were saturated with 5 % bovine serum albumin (BSA) in PBS/Tween-20 (0.05 %) to prevent non-specific binding. The filters were then incubated with primary antibodies in 50 mM Tris-HCl buffer, pH 8.5, containing 500 mM NaCl, 0.05 % Tween-20, and 0.1 % bovine serum albumin. After extensive washing the bound antibodies were detected using biotinylated anti-mouse and anti-rabbit antibodies, peroxidase-conjugated streptavidine and enhanced chemiluminescence detection system (Amersham).
Casein zymography and reverse zymography
Zymography and reverse zymography were used to identify the molecular forms of uPA and PAI (Granelli-Piperno and Reich, 1978). The polypeptides of the conditioned media or acid eluates were separated under non-reducing

39
conditions by SDS-PAGE (4-15 %). SDS was removed by washing the gels with PBS, pH 7.4 /Triton X-100 (2.5%) 4x15 min. For reverse zymography, reducing conditions were used and uPA (2 IU/ml) was added to the final wash. The gels were then placed on caseinolysis gels containing plasminogen and casein in 1.2% agarose and incubated at 37 °C until the lytic bands correlating to the PA-activity became visible. A lysis resistant band indicated the migration of PAI-1.
Analysis of membrane-bound PA activity was performed essentially as described (Stoppelli et al., 1986). Endothelial cells were washed with 0.2% BSA in PBS, pH 7.4, and treated with 50 mM Glycine-HCl buffer, pH 3.0, containing 0.1 M NaCl at room temperature for 10 min. The buffer was collected, immediately neutralized, and subjected to casein zymography.
Immunoprecipitation
Cells were lysed with RIPA-DOC lysis buffer (50 mM Tris-HCl buffer, pH 8.0, containing 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate, 10 mM EDTA, 10 µg/ml aprotinin, 1 µg/ml pepstatin A, 1 µg/ml aminoethylbenzene sulphonyl fluoride, 1 mM sodium orthovanadate, and 10 mM sodium fluoride) and subjected to immunoprecipitation. Cell lysates were preabsorbed by incubation with preimmune serum and Gamma-bind Sepharose (Amersham Pharmacia, Uppsala, Sweden) at 4 ºC in an end-over mixer for 2 h. The beads were removed by centrifugation and the supernatants were incubated with the indicated antibodies on ice for 1 h. Gamma-bind Sepharose was then added and the samples were incubated in an end-over mixer at 4 ºC for 1 h. The beads were subsequently collected by centrifugation and washed three times with RIPA-DOC buffer. The bound proteins were eluted with Laemmli´s sample buffer at 95 ºC and separated by SDS-PAGE (4-15% acrylamide monomer gradient), transferred onto nitrocellulose and subjected to immunoblot analysis. Intensity of the detected bands was quantified from scanned immunoblots using Adobe Photoshop software.
Immunofluorescence
Cells cultured on glass coverslips were washed with PBS and fixed with 3 % paraformaldehyde at 4 ºC for 10 min. When required, the cells were permeabilized with 0.2 % Triton X-100 in PBS for 3 min. Nonspecific protein binding sites were then saturated with 5 % BSA in PBS for 30 min. The cells were then washed with PBS, and incubated with antibodies for 1 h. Rhodamine-conjugated phalloidin (Sigma) was used for the staining of the actin cytoskeleton. Unbound proteins were removed by washing, followed by incubation with fluorescein isothiocyanate or rhodamine -labeled secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h. The coverslips were finally washed and mounted on glass slides using Vectashield (Vector Laboratories, Burlingame, CA). The fluorescent images were obtained using an epifluorescent microscope and logarithmic image aquisition.

40
Cell adhesion assays
Plastic 96-well cell culture plates were coated with endostatin or endostatin-derived peptides at concentrations indicated (50 µl/well) at 4 °C for 16 h. Nonspecific adhesion sites were saturated by incubating the wells with 1 % heat inactivated BSA at 22 °C for 1 h. After washing the wells with PBS, 0.5x105 cells in 100 µl of prewarmed serum free medium were seeded into each well. After 90 min of incubation at 37 °C, nonattached cells were removed by washing with PBS. The cells were then fixed and stained in 30 % methanol, 10 % acetic acid containing 0.1 % Coomassie Blue. Subsequently, the cells were washed extensively with 30 % methanol, 10 % acetic acid and once with PBS. The cells were then lysed in 1 % SDS, after which the amount of lysed cells were quantified by measuring their absorbance at 620 nm using a Multiscan plate reader (Labsystems, Finland).
In vitro chemotaxis assay
Cell migration assays were performed in modified Boyden chambers containing polyethylene membranes (8 µm pore size; BD Falcon, Franklin Lakes, NJ) coated with 50 µg/ml fibronectin at 4 ºC for 16 h. After washing the membranes with PBS, nonspecific adhesion sites were saturated with 1 % heat inactivated BSA at 22 ºC for 1 h. Cells suspended in serum free medium at a concentration of 5x103/ml were placed in the upper compartment of the chambers. Medium containing 50 ng/ml bFGF was added to the lower compartment. The chambers were then incubated at 37 ºC for 4 h. Subsequently, the membranes were fixed and stained in 30 % methanol, 10 % acetic acid containing 0.1 % Coomassie Blue. The cells migrated to the lower surface were counted at 200x magnification. Three random fields from each pair of wells were counted. Values represent three independent experiments.
Isolation of the extracellular matrix
Extracellular matrices were prepared according to (Hedman et al., 1979). Cells were seeded on plastic culture dishes in serum-containing culture medium and allowed to attach for 1 h. After this the medium was withdrawn and replaced with serum free medium containing endostatin or various chemicals at concentrations indicated. After 4 h of incubation the cells were washed with PBS and treated three times with 0.5 % sodium deoxycholate in 10 mM Tris-HCl buffer, pH 8.0, for 10 min. Subsequently, the plates were washed with PBS and allowed to dry. The resulting extracellular matrices were extracted in reducing Laemmli´s sample buffer, solubilized by boiling and separated by SDS-PAGE. The proteins were then transferred onto nitrocellulose and detected by immunoblot analysis.

41
Isolation of detergent-insoluble membrane fractions
Cells were lysed in 1 % Triton X-100 in MES-buffered saline (25 mM MES, 150 mM NaCl, pH 6.5). Cell extracts were homogenized using a 22 G needle and adjusted to a final density of 40 % by the addition of OptiPrep Density Gradient Medium (Sigma). Samples were then placed in a centrifuge tube and overlaid with a discontinuous 30 % / 5 % OptiPrep density gradient. Samples were then centrifuged at 39,000 rpm for 18 h, after which 1-ml fractions were collected and analyzed by immunoblotting as described previously (Taipale et al., 1992).
Affinity precipitation of GTP-Rho
Affinity precipitation of GTP-Rho. GTP-Rho affinity precipitation assays were performed essentially as described earlier (Ren et al., 1999). HDMECs treated with the indicated chemicals were washed with ice-cold Tris-buffered saline and lysed in 50 mM Tris-HCl buffer, pH 7.2, containing 500 mM NaCl, 10 mM MgCl2, 1 % Triton X-100, 0.5 % sodium deoxycholate, 0.1 % SDS, 10 µg/ml leupeptin, 10 µg/ml aprotinin, and 100 µg/ml aminoethylbenzene sulphonyl fluoride. The lysates were then incubated with Rhotekin Rho Binding Domain-agarose (Upstate Biotechnology Inc., Lake Placid, NY) at 4 °C for 45 min and washed three times with 50 mM Tris-HCl buffer, pH 7.2, containing 150 mM NaCl, 10 mM MgCl2, 1 % Triton X-100, 10 µg/ml leupeptin, 10 µg/ml aprotinin, and 100 µg/ml aminoethylbenzenesulphonyl fluoride. The bound proteins were eluted with Laemmli´s sample buffer, separated by SDS-PAGE and detected by immunoblotting using a monoclonal antibody against RhoA (Santa Cruz Biotechnology Inc., Santa Cruz, CA). The intensity of the RhoA signal was quantified from scanned immunoblots using Adobe Photoshop software.
Src family kinase autophosphorylation assay.
HDMECs were lysed and subjected to immunoprecipitation with pan-Src antibodies (Src-2, Santa Cruz Biotechnology Inc., Santa Cruz, CA) as described above. The immunoprecipitates were washed three times with RIPA lysis buffer and twice with Src kinase buffer (20 mM Hepes, pH 7.35, 3 mM MnCl2). Autophosphorylation was initiated by adding 5 µCi of [γ-32P] ATP (5000 Ci/mmol; Amersham Pharmacia Biotech) and carried out in 40 µl of kinase buffer at 30 ûC for 15 min. The reactions were terminated by adding 10 mM EDTA (final concentration) on ice. The pellets were then washed twice with lysis buffer followed by SDS-PAGE and autoradiography. Incorporated radioactivity was detected and quantified with Fujifilm BAS-2500 Image Analyzer and MacBAS 2.5 program.

42
Metabolic labeling
Cells were seeded on plastic culture dishes in complete culture medium and allowed to attach for 90 min. After washing, the cells were incubated with endostatin for 3 h in serum-free medium containing [35S]-methionine (80 µCi/ml, 1175 Ci/mmol; Amersham Pharmacia Biotech, Uppsala, Sweden). Subsequently, aliquots of conditioned media were subjected to immunoprecipitation with antibodies against fibronectin as described above. The eluted polypeptides were separated by SDS-PAGE (4-15 % gradient) under reducing conditions and visualized by autoradiography.
Wound-induced migration assay
Confluent HDMECs on glass coverslips in complete culture medium were wounded with a sterile pipet tip and washed with serum free medium. The cells were treated with 50 ng/ml bFGF and with 50 nM endostatin or 50 nM endostatin in combination with sodium orthovanadate immediately after wounding and again after 12 h. After 24 h of incubation the cells were fixed with paraformaldehyde and subjected to immunofluorescence analysis as described above. The migration distance was measured using an inverted phase contrast microscope and UltraView 4.0 software (Perkin Elmer, Fremont, CA). The mean migration distances were calculated from three reference points on each coverslip by subtracting the distance between the migrating cell fronts measured after 24 h of incubation from that measured at the time of wounding.
Endothelial cell tube formation assay
24-well cell culture plates were coated with 200 µl growth factor-reduced Matrigel basement membrane matrix (BD Biosciences, Bedford, MA) and allowed to form a gel at 37 ºC for 45 min. The cells (4 x 105) were suspended in 600 µl pre-warmed serum free Medium 199 containing endostatin or endostatin-derived peptides. The cells were then seeded on Matrigel-coated plates, treated with 10 ng/ml bFGF and incubated at 37 ºC for 16 h. The cells were subsequently fixed, and tube formation was assessed using an inverted phase-contrast microscope. Tubular branches were counted from three random fields. The values represent the results of three independent experiments.

43
Results
Endostatin regulates the levels of secreted soluble uPA/PAI-1 complex and induces changes in the cell surface localization of uPA and uPAR (I)
At the beginning of these studies endostatin had been identified as a potent inhibitor of angiogenesis. Endostatin inhibited migration of endothelial cells, but had no effect on other cell types. As proteolytic systems have been implicated to play an important role in cell migration and invasion, it became of interest to characterize the effects of endostatin on uPA-mediated cell pericellular proteolysis.
To analyze the effects of recombinant endostatin on the levels of soluble, secreted uPA and PAI-1, HDMECs were treated with increasing concentrations of endostatin. Analysis of the conditioned media using casein zymography indicated that uPA appeared mostly as a complex with PAI-1. The levels of this complex were significantly decreased in response to endostatin treatment. The effect became visible after 3 h of treatment and at concentrations of 500 ng/ml (25 nM) of endostatin. PAI-1 levels were also independently assessed using reverse zymography. The analysis revealed down-regulated levels of soluble secreted PAI-1 in endostatin-treated cells. The decrease in uPA and PAI-1 levels did not involve changes in the corresponding mRNA levels of these proteins.
To further characterize the endostatin-induced changes in uPA/PAI-1 levels, we analyzed the levels of cell-surface associated uPA/PAI-1-complexes. Casein zymography analysis of acid-eluted cell surface-bound proteins revealed a marked increase in the levels of uPA/PAI-1 complexes of endostatin-treated cells. Blocking the cell surface receptor uPAR by monoclonal antibodies inhibited the accumulation of uPA/PAI-1 complexes on the cell surface. This was observable both in untreated control cells and endostatin-treated cells indicating that uPAR is involved in the endostatin-induced accumulation of uPA/PAI- complexes on the cell surface.
In addition to the controlled secretion of soluble proteases, pericellular proteolysis is tightly regulated through localization of the proteases to specific cellular compartments (Blasi and Carmeliet, 2002). To address the possible endostatin-induced changes in the localization of uPA and uPAR, immunofluorescence analysis was carried out. Under basal conditions, both of the proteins were found to localize in focal adhesions. In response to endostatin treatment, uPA and uPAR became redistributed, acquiring a diffuse cell surface localization.

44
Loss of focal adhesions and actin cytoskeleton in endostatin-treated endothelial cells (I, II, III)
Focal adhesions and actin stress fibers are functional protein complexes involved in cell adhesion and migration (Burridge and Chrzanowska-Wodnicka, 1996; Gumbiner, 1996). Since endostatin was observed to induce redistribution of uPA and uPAR from focal adhesions, we sought to investigate the effect of endostatin treatment on focal adhesions. Immunofluorescence analysis of focal adhesion proteins vinculin and paxillin revealed that focal adhesions were rapidly disassembled in response to endostatin treatment. This effect was reversible, as focal adhesion plaques reappeared after 6 h of treatment.
Association of endostatin with αααα5ββββ1 integrin and caveolin-1 (II, III) Previous results had indicated that immobilized endostatin mediates
endothelial cell adhesion via interaction with integrin α5β1 (Rehn et al., 2001). To investigate whether soluble endostatin binds to α5β1 integrin and whether this interaction affects the cell surface localization of this adhesion receptor, co-immunoprecipitation and immunofluorescence analyses was carried out. We observed that endostatin treatment induced redistribution of α5β1 integrin into clusters distinct from focal adhesions. These clusters colocalized with caveolin-1 staining. In addition, endostatin and caveolin-1 were found to co-immunoprecipitate with α5β1 integrin. The interaction between caveolin and integrin α5β1 was dependent on the concentration of endostatin administered to the cells.
Tyrosyl phosphatase-dependent activation of caveolin-1- associated Src regulates endostatin-induced disassembly of cytoskeleton and decreased deposition of fibronectin matrix (II)
Phosphorylation signals generated as a result of growth factor stimulation or through interaction with the surrounding matrix or adjacent cells regulate cellular events such as the assembly and disassembly of focal adhesions and reorganization of the actin cytoskeleton (Giancotti and Ruoslahti, 1999; Schwartz and Baron, 1999). As members of the Src family of membrane-associated tyrosine kinases are involved in integrin-mediated signaling and are thought to control actin stress fibers and focal adhesion turnover (Thomas and Brugge, 1997), we sought to determine the effect of endostatin on the tyrosine kinase activity of Src. Src activity is regulated via dephosphorylation of the inhibitory phosphorylation site Tyr-527 and phosphorylation of the activating Tyr-416 (Piwnica-Worms et al., 1987). In vitro kinase assays revealed an increase in the autophosphorylation activity of the fraction of Src associated with caveolin-1 in response to endostatin treatment. An increase in the amount of Src phosphorylated at the activating Tyr-416 was also observed using immunoblot analysis. The increase in activity was dependent on tyrosine phosphatase activity, since the tyrosyl phosphatase inhibitor orthovanadate inhibited these effects.

45
To investigate whether the effects of endostatin on actin stress fibers and focal adhesions would involve tyrosyl dephosphorylation, we administered the phosphatase inhibitor orthovanadate to HDMECs in combination with endostatin and analyzed cytoskeletal structures using immunofluorescence. Cells treated with endostatin in combination with orthovanadate displayed intact focal adhesions and actin stress fibers, whereas in cells treated with endostatin alone these structures were disrupted.
To determine whether the disruption of actin stress fibers and focal adhesions was due to increased activity of Src, we treated the cells with endostatin in combination with PP1, a specific inhibitor of Src family kinases (Hanke et al., 1996). Loss of actin stress fibers and focal adhesions were seen in cells treated with endostatin or PP1 alone, but in cells treated with both of the chemicals intact cytoskeletal structures were observed.
Both α5β1 integrins and Src kinase are involved in the regulation of fibronectin matrix assembly. Since endostatin binds α5β1 integrin and increases Src activity, we sought to investigate the effect of endostatin on the deposition of the fibronectin matrix. Both immunoblot and immunofluorescence analyses of the deoxycholate-insoluble ECM deposited by the endostatin-treated endothelial cells revealed a decrease in fibronectin deposition compared to untreated control cells. This was not due to decreased secretion of soluble fibronectin. In contrast, an increase in the levels of soluble fibronectin was observed in the conditioned media of endostatin-treated cells.
Endostatin and αααα5ββββ1 integrin partition in lipid rafts in a heparan sulfate proteoglycan-dependent manner (III)
Lipid rafts are specialized membrane microdomains enriched in glycosphingolipids and cholesterol, which serve as scaffolds for the recruitment of transmembrane proteins and intracellular signaling molecules at the plasma membrane (Simons and Ikonen, 1997). Caveolin is a major constituent of a subclass of lipid rafts. To investigate whether the previously observed interaction of endostatin with caveolin-1 involves the association of endostatin with the lipid rafts, we isolated Triton-X insoluble membrane fractions of cells treated with 50 nM endostatin (1 µg/ml) using a discontinuous OptiPrep density gradient. We found that a fraction of endostatin localized in the caveolin- and glypican-1-enriched top fractions of the flotation gradient, indicating raft localization. Transferrin receptor was used as a marker for the non-raft fractions.
Treatment of cells with heparinase III (5 mU/ml) for 30 min, prior to the addition of endostatin to remove the cell surface exposed heparan sulfates, significantly reduced the amount of endostatin associated with the low-density fractions without appreciably affecting the levels of endostatin associated in the non-raft fraction. This indicated that heparan sulfate proteoglycans mediated the raft localization of exogenous endostatin. Further support for this finding was provided by the observed loss of the heparan sulfate-containing form of glypican-1 from the raft fractions after heparinase III treatment. Chondroitin sulfate treatment was used as a negative control.

46
To investigate the role of α5β1 integrin in the cell surface association of endostatin, recombinant endostatin (1 µg/ml, 50 nM) was preincubated with purified soluble α5β1 integrin (5 µg/ml, 33 nM) for 12 h to saturate the putative integrin binding sites of endostatin. Solid-phase ligand binding analysis revealed that immobilized, purified α5β1 integrin bound endostatin in a saturable and dose-dependent manner, and that saturation was reached beyond the 50 nM concentration of endostatin. Endostatin preincubated with soluble integrin α5β1
displayed decreased association with both high- and low-density fractions of the endothelial cell membrane. Non-interacting soluble α3β1 integrin was used as a negative control.
Very low levels of α5β1 integrin were visible in the raft fraction of the untreated control cells, while the non-raft fraction displayed intensive staining. In endostatin treated cells, the levels of α5β1 integrin in the raft fraction were increased. Quantification of the immunoblots revealed an >2-fold increase in the ratio of raft versus non-raft associated integrins in response to endostatin treatment. When cells were pretreated with heparinase III (5 mU/ml) for 30 min prior to the addition of endostatin, the amount of α5β1 integrin in the raft fraction was decreased to the level of control cells.
Importantly, inhibition of lipid raft formation using the cholesterol-binding agent filipin III or cholesterol chelating agent methyl-β-cyclodextrin inhibited the trans-localization of endostatin into the raft fraction. In addition, immunofluorescence analysis revealed that interaction of endostatin with lipid rafts, α5β1 integrin, as well as an unidentified heparan sulfate proteoglycan were required for the endostatin-induced disassembly of the actin cytoskeleton.
Endostatin downregulates RhoA activity via Src and p190 (III) RhoA activity induces the formation of focal adhesions and actin stress fibers
(Ridley and Hall, 1992). To investigate whether changes in RhoA activity were associated with the endostatin-induced disassembly of focal adhesions and actin stress fibers, RhoA activity assays were carried out. These experiments revealed a dose-dependent decrease in RhoA activity in response to endostatin treatment. The Src inhibitor PP1 could partly reverse this inhibition.
Src regulates Rho activity by tyrosine phosphorylation of p190RhoGAP, a GTPase activating protein with high specificity for Rho (Arthur et al., 2000; Ridley et al., 1993). Endostatin treatment led to an increase in the tyrosine phosphorylation of p190RhoGAP. This effect could also be inhibited using PP1.
Endostatin-derived peptides promote cell adhesion via integrins and induce disassembly of the cytoskeleton (IV)
The crystal structure of mouse endostatin revealed a compact globular fold and a basic patch of 11 arginine residues, which have been suggested to act as binding sites for heparin (Hohenester et al., 1998; Sasaki et al., 1999). To define its critical cell interaction domains and to investigate the role of the basic amino acids for endostatin function we constructed endostatin-derived synthetic peptides

47
containing 12 out of the total 18 surface exposed basic amino acids. When immobilized, an arginine-rich peptide of 11 amino acids, termed ES-2, efficiently promoted endothelial cell adhesion. Antibodies against β1 integrin as well as heparin efficiently inhibited endothelial cell adhesion to this peptide. Peptides ES-3 and ES-5 also promoted cell adhesion. Heparin efficiently inhibited cell adhesion to these peptides, while antibodies against α5 or β1 integrins had no effect.
Actin staining of cells attached on ES-2 revealed the formation of membrane ruffles at the cell periphery. No formation of actin stress fibers was observed. The actin-rich membrane protrusions were also positive for vinculin staining. Integrin α5β1 was also observed to localize to the membrane ruffles in the cell periphery. Cells plated on ES-2 displayed limited cell spreading when compared to cells plated on fibronectin, the major substrate for α5β1 integrin.
When soluble, the ES-2 peptide induced the loss of focal adhesions and actin stress fibers in these cells. Substitution of the arginine residues with alanines resulted in the loss of these properties.
Endostatin-derived peptides inhibit endothelial migration and tubular morphogenesis of endothelial cells (IV)
Since the endostatin-derived peptides induced changes in cell-matrix interactions, we explored next their effects on directional cell motility using a modified Boyden chamber assay. Cell migration through a fibronectin matrix towards bFGF was inhibited approximately 50 %, when the endothelial cells were treated with 500 nM ES-2 peptide. This was comparable with the effect of 50 nM full length endostatin. The mutated peptide lacking all of the arginine residues did not affect cell migration.
To assess the effect of the endostatin-derived peptides on the formation of vascular structures, HDMECs were seeded on growth factor reduced Matrigel basement membrane matrix in the presence of bFGF. In cells treated with 50 nM endostatin or 500 nM ES-2 peptide, tube formation was significantly decreased and the cells remained as an adherent monolayer with only a few tubelike structures. The mutated peptide lacking all of the arginine residues had no inhibitory effect on the tubular morphogenesis of HDMECs.

48
Discussion
The process of angiogenesis is regulated by shifts in the balance between endogenous pro- and anti-angiogenic factors. When this work was initiated, endostatin, a novel negative regulator of angiogenesis, had been identified and characterized (O'Reilly et al., 1997). Further studies elucidated several possible cell biological mechanisms underlying the anti-angiogenic effects of endostatin. These included inhibition of endothelial cell migration, induction of cell cycle arrest, and promotion of apoptosis (Dhanabal et al., 1999; Dixelius et al., 2000; Hanai et al., 2002a; O'Reilly et al., 1997). The current work contributes to the effort of understanding the basic cell biological mechanisms underlying anti-angiogenic effects of endostatin in vivo. The central findings of this study are the following: (1) endostatin regulates pericellular proteolysis by downregulating the levels of secreted uPA and PAI-1, and by increasing the levels of uPA/PAI-1 complex bound to uPAR at the cell surface; (2) endostatin induces disruption of the actin cytoskeleton and loss of focal adhesions via Src-dependent activation of p190 and inactivation of RhoA; (3) endostatin signaling is dependent on lipid raft microdomains, and involves interactions with α5β1 integrins as well as cell surface heparan sulfate proteogycans; (4) identification of an endostatin-derived synthetic peptide, which interacts with α5β1 integrins and modulates the actin cytoskeleton and endothelial cell migration.
Effects of endostatin on endothelial cell pericellular proteolysis Proteolytic activity is an important process during endothelial cell sprouting
and tube formation (Koolwijk et al., 1996; Risau, 1997). In this work, pericellular proteolytic activity was observed to be redistributed and down-regulated in response to endostatin treatment (I). Several studies have indicated a role for plasminogen activators in tumor-associated neovascularization, primary tumor growth, and metastasis (Crowley et al., 1993; Min et al., 1996; Ossowski, 1996). For efficient proteolysis, proteases are targeted to distinct locations at the cell surface and, in particular, changes in uPA and PAI-1 secretion and localization can induce changes in cell migration and adhesion (Deng et al., 1996; Planus et al., 1997; Yebra et al., 1996). Endostatin induced the translocation of uPA/uPAR from focal adhesion structures into a diffuse cell surface localization. At the same time, the uPA/PAI-1 complexes isolated from the cell surface by acid treatment were increased in a dose-dependent manner. Interestingly, no intracellular or matrix accumulation of these proteins was observed (I). Interactions with various transmembrane proteins are crucial for the function of uPAR. The internalization of the uPA/PAI-1/uPAR complexes is mediated via interaction with the low-density lipoprotein-receptor-related protein (Czekay et al., 2001). The increased levels of this complex at the cell surface could be a consequence of its displacement from the focal adhesions, as the subcellular localization of uPAR is critical in determining its interaction partners. The relocalization could result in

49
altered internalization and recycling of the proteinase complex. Inhibition of uPAR internalization results in the increase in the levels of cell surface uPA/PAI-1 complexes. This is associated with decreased recycling of unoccupied uPAR to the cell surface with concomitant decrease in the migratory capacity of the cell (Degryse et al., 2001; Li et al., 2002). The relocalization of uPA and uPAR induced by endostatin may be a consequence of the focal adhesion disassembly, or an independent effect.
The levels of secreted uPA and PAI-1 were decreased in endothelial cells after endostatin treatment. However, no changes in the mRNA levels of uPA or PAI-1 were observed (I). The mechanism by which the post-transcriptional regulation of plasminogen activator levels by endostatin occurs is currently not understood. It is possible that the altered distribution and increased levels of uPA/PAI-1 on the cell surface could be linked to the decreased secretion of these proteins. Cell adhesion on a specific ECM ligand, such as vitronectin, directly regulates the secretion of uPA and influences its localization into focal adhesions (Wilcox et al., 1996). This suggests that an extensive network of crosstalk exists between secreted and uPAR-bound uPA/PAI-1 complexes, cell adhesion and migration.
Regulation of actin cytoskeleton and cell migration by endostatin The speed of cell migration is determined by the strength of cell adhesion to
the ECM. On the other hand, the direction of the motility is controlled by growth factors such as bFGF, which in coordination with cell adhesion molecules enhance cell polarity (Eliceiri, 2001). We observed that treatment of endothelial cell monolayers with soluble endostatin resulted in the loss of focal adhesions and actin stress fibers (I-IV). Disruption of actin stress fibers and focal adhesions by endostatin could result in decreased adhesion and increased random migration. On the other hand, the loss of these structures at the leading edge could inhibit directional migration towards a chemotactic stimulus. Various other endogenous inhibitors of angiogenesis target the endothelial cell cytoskeleton. Thrombospondin-1 interacts with the cell surface receptors CD36 and heparan sulfate proteoglycan, and induces the disassembly of actin stress fibers and focal adhesions (Dawson et al., 1997; Orr et al., 2002). Angiostatin, the antiangiogenic fragment of plasminogen, induces actin stress fiber reorganization via ceramide and RhoA (Gupta et al., 2001; O'Reilly et al., 1994). Like endostatin, these proteins negatively regulate endothelial cell migration (Dawson et al., 1997; O'Reilly et al., 1994; Troyanovsky et al., 2001).
Protein phosphorylation and dephosphorylation serve as molecular switches in the control of the migration-associated assembly and disassembly of focal adhesions and actin stress fibers (Burridge and Chrzanowska-Wodnicka, 1996). Important regulators of these phosphorylation events are the Src family kinases that are activated by integrin ligand binding. Their kinase activity directly regulates the turnover of the cytoskeletal structures (Thomas and Brugge, 1997). We observed that endostatin treatment lead to increased activation of a Src kinase directly associated with caveolin-1. Consequently, the endostatin-induced

50
disassembly of focal adhesions and actin stress fibers was antagonized by the Src inhibitor PP1, which alone is capable of inducing disassembly of the cytoskeleton (II).
In addition, we observed Src dependent activation of p190RhoGAP by endostatin (III). This occurred in parallel with Src dependent downregulation of the small GTPase RhoA (III). Src-mediated transient downregulation of RhoA activity, accompanied by the disassembly of the cytoskeleton has also been observed to be an initial event following ligand binding of integrins This process promotes efficient cell spreading and is followed by an increase in Rho activity, resulting in reformation of actin stress fibers and focal adhesions (Arthur et al., 2000). However, the kinetics of both the activation of Src as well as the downregulation of RhoA activity were significantly slower than that resulting from the interaction of ECM-deposited fibronectin with integrin α5β1 (Arthur et al., 2000). This suggests that the downstream signaling induced by endostatin is not completely analogous to that of other ECM ligands binding integrins. Rho possesses a dual role in the regulation of cell migration. High levels of active Rho promote cell adhesion to the substrate, which decreases cell motility. On the other hand, decreased levels of Rho activity inhibit cell motility, possibly through reducing membrane ruffling and tail-retraction of the migrating cell, and delaying the turnover of actin stress fibers and focal adhesions (Nobes and Hall, 1999; Ridley et al., 1995; Santos et al., 1997). Since endostatin induces reversible loss of stress fibers persisting for several hours (I), the endostatin-induced downregulation of RhoA could significantly reduce the speed of endothelial cell migration by delaying the dynamics of actin stress fiber and focal adhesion turnover.
Since our primary observation of endostatin-induced disassembly of actin stress fibers and focal adhesions, several studies have provided further evidence of the actin cytoskeleton to be regulated by endostatin treatment. By comparing two-dimensional gel electrophoresis patterns of endostatin-treated and untreated cells, heat shock protein 27 (hsp27) and cofilin were identified as targets for endostatin (Keezer et al., 2003). Hsp27 regulates actin polymerization, and the phosphorylation of hsp27 is associated with changes in the actin cytoskeleton in VEGF-stimulated endothelial cell motility (Rousseau et al., 1997). Endostatin induced phosphorylation of hsp27 and cofilin, accompanied by an increase in actin stress fibers and paxillin-positive focal adhesions (Keezer et al., 2003). Interestingly, RhoA activity phosphorylates and inactivates cofilin, leading to the stabilization of actin stress fibers (Worthylake and Burridge, 2003).
In another study, endostatin was observed to induce the formation of actin stress fibers and focal adhesions in the absence of bFGF, but when the growth factor was present, disassembly of these structures was observed (Dixelius et al., 2002). This suggests that the apparent differences on the effects of endostatin on the cytoskeleton may arise from differences in the cell culture conditions before and during treatment, as well as from variation in the endogenous production of growth factors by different endothelial cell lines.

51
Requirement of integrin αααα5ββββ1, heparan sulfate proteoglycans and lipid rafts in endostatin signaling
When this work was initiated, two putative receptors for endostatin had been identified. Studies using bovine pulmonary arterial endothelial cells indicated, that endostatin binds endothelial cells with two distinct affinities, a high-affinity Kd of 18 pM (1.7 x 104 binding sites/cell) and a lower-affinity Kd of 200 pM (8.4 x 104
binding sites/cell). The low-affinity receptor was identified as the heparan sulfate proteoglycan glypican. Endostatin also binds to renal tubular epithelial cells via this receptor (Karumanchi et al., 2001). Glypican-4 is highly expressed in the developing brain and is the major glypican expressed in renal tubular cells, whereas the predominant glypican in endothelial cells is glypican-1 (Gengrinovitch et al., 1999; Watanabe et al., 1995). Glypican-1 expression is critical for endostatin inhibition of VEGF-induced endothelial cell migration, indicating that it is a functional receptor for endostatin. Interaction of endostatin with glypican seems to be necessary for its subsequent binding to the high-affinity site, suggesting that heparan sulfate proteoglycans present endostatin to another cell surface receptor. The high-affinity receptor was not identified (Karumanchi et al., 2001).
Cell adhesion on immobilized endostatin is mediated mainly by α5β1 and in part by αvβ3 integrins (Rehn et al., 2001). In this context, endostatin acts as an integrin ligand promoting cell adhesion, formation of focal adhesions and actin stress fibers and inducing tyrosine phosphorylation (Rehn et al., 2001). Another study confirmed that α5β1 integrin serves as a functional receptor also for soluble endostatin, and that the anti-migratory effects of endostatin are transduced in an integrin-dependent manner (Sudhakar et al., 2003).
We observed that endostatin associated directly with α5β1 integrin and the membrane scaffold protein caveolin-1 (II). In addition, α5β1 integrin directly mediated the cell surface association of endostatin. Heparan sulfate proteoglycans, on the other hand, mediated the translocation of endostatin and α5β1 integrin into lipid raft microdomains. Both of these interactions as well as the lipid raft microdomain per se were necessary for the cytoskeletal rearrangement induced by endostatin (III). Taken together, these findings imply that both heparan sulfate proteoglycans as well as α5β1 integrins act as functional receptors for endostatin. The signaling functions of endostatin are probably mediated via α5β1 integrin. Heparan sulfate proteoglycans act as coreceptors, presenting endostatin to integrins and regulating the localization of the endostatin-integrin complex into membrane rafts. Glypican belongs to the GPI-anchored proteins, which are a major class of proteins in the lipid rafts (Brown and Rose, 1992; Mayor et al., 1994). In addition, membrane lipids and raft microdomains have been observed to regulate integrin and integrin-associated protein activity (Green et al., 1999; Pande, 2000).
The presence of caveolin-1 in a complex with endostatin and integrin α5β1
suggest a role for caveolae, which are a specialized subclass of lipid rafts, in the effects of endostatin. Integrins are generally considered not to localize in caveolae (Lisanti et al., 1994). However, it has been suggested that caveolae serve as

52
temporary scaffolding domains for clusters of integrins, caveolin-1 and Src kinases, and that these structures are disassembled in response to integrin engagement by insoluble ECM ligands and subsequent recruitment to focal adhesions (Wei et al., 1999). An open question remains whether the lipid raft localization results in the internalization of endostatin via caveolae or a related raft pathway, and whether the internalization is required for the cell biological effects of endostatin. Some endothelial cell types apparently internalize recombinant endostatin, but the functional consequences of this process have not been characterized further (Dixelius et al., 2000).
Identification of an integrin-binding peptide within endostatin The structure of endostatin is characterized by a compact globular fold and a
basic patch of 11 clustered arginine residues, which have been suggested to act as binding sites for heparin (Hohenester et al., 1998; Sasaki et al., 1999). The roles of the arginine residues are unclear, since both decreased and unaltered anti-migratory potential has been reported in endostatin mutants unable to bind heparin (Sasaki et al., 1999; Yamaguchi et al., 1999). In addition, endostatin binds integrins, but no specific integrin-binding sites have been identified so far. The ligands of α5β1 integrins consist of ECM or cell surface proteins containing the RGD-sequence (Ruoslahti, 1996), but both mouse and human endostatins lack this sequence motif.
In the current study we observed that peptides derived from areas of human endostatin containing multiple surface exposed basic amino acid residues were able to promote endothelial cell adhesion and spreading via both heparin- and integrin β1-dependent interactions. In contrast to the integrin ligand fibronectin, which contains the RGD-sequence, ES-2 peptide promoted only a limited degree of cell spreading and did not induce the formation of actin stress fibers or abundant cytoplasmic focal adhesions (IV). These observations imply that the binding of endostatin or ES-2 peptide to the integrin could occur at a site distinct from the ligand binding pocket. On the other hand, endostatin exists as a monomer, and monovalent ligand occupancy of integrins is apparently insufficient for efficient accumulation of cytoskeletal proteins (Miyamoto et al., 1995). Adhesion of endothelial cells to full-length endostatin has been suggested, however, to occur in an RGD-dependent manner. Cyclic RGD peptides inhibit the attachment of human umbilical endothelial cells to fibronectin and also endostatin-coated plates. This implies that endostatin competes for the RGD binding sites within fibronectin (Sudhakar et al., 2003). We did not observe significant inhibition of endothelial cell adhesion on fibronectin by the ES-2 peptide or endostatin (IV). In addition to integrins, the adhesion of endothelial cells to ES-2 occurred in part via a mechanisms involving heparin (IV). The activity was critically dependent on the specific arginine residues within the peptides, which have been reported to be crucial for the heparin binding as well as the anti-migratory activity of endostatin. This finding further supports the conclusion that heparan sulfate proteoglycans may act as co-receptors for endostatin.

53
Synthetic peptides from other regions of full-length endostatin have been observed to contain anti-angiogenic activity. A peptide containing amino acids 6-49 is capable of inhibiting human umbilical vein-derived endothelial cell proliferation and migration, and is also effective in inhibiting angiogenesis in vivo (Chillemi et al., 2003). In that report, no activity was observed for the 42 aa peptide covering the amino acids 50-92, although it contains the sequence for the ES-2 peptide (11 aa) used in the current study. The variation between the results of the two studies may be a result of differences in the biochemical properties of peptides of different lengths. Studies using peptide fragments from another basement membrane-derived angiogenesis inhibitor, tumstatin, suggest that the length of the peptide is a critical determinant of biological activity. This is most likely due to differences in the secondary structure of the peptides and in their propensity to form aggregates (Floquet et al., 2003). The folding and aggregation of the peptides is likely to affect the exposure of specific aa residues, and thus mask putative cell surface interaction sites.
A model for endostatin-induced signaling Endostatin has been reported to induce a plethora of intracellular signals,
which regulate multiple aspects of endothelial cell behavior. The large variation in the observed effects may be a result of multiple factors. The endostatin used in both the in vivo and in vitro studies is derived from various sources. It has been produced as an insoluble precipitate in E. coli or as a soluble protein in Pichiapasteuris or a baculoviral expression system. The soluble and insoluble endostatins seem to differ in their biochemical properties and cell biological effects (Kranenburg et al., 2003). In addition, the cell culture models used in the studies range from macrovascular aortic endothelial cells to microvascular endothelial cells. Macro- and microvascular endothelial cells differ in various aspects, such as their responsiveness to growth factor stimulus (Lang et al., 2001; Lang et al., 2003). Unexpectedly wide range of concentrations have been used in various studies, ranging from 10 ng/ml to 10 µg/ml. Finally, human and mouse endostatins seem to have distinct cell biological effects, despite their close sequence homology (Sudhakar et al., 2003; Yamaguchi et al., 1999).
The most consistent effect of human endostatin seems to be its ability to inhibit growth factor-induced endothelial cell migration. As endostatin does not affect the major intracellular pathways stimulated in growth factor-induced cell motility (Eriksson et al., 2003), it is probable that endostatin has a unique mechanism of action, by which it modulates endothelial cell migration. The modulation could occur via direct effects on endothelial cell - matrix interactions. The inability of endostatin to perturb major intracellular signaling pathways may also be associated with its lack of effect on the normal quiescent vasculature. This may also explain the low toxicity of these agents in the clinical trials (Dixelius et al., 2003).
The results of the current study suggest that endostatin binds to α5β1 integrin on the cell surface. Its simultaneous or subsequent interaction with an unidentified heparan sulfate proteoglycan leads to the translocation of endostatin and α5β1

54
integrin into lipid rafts, where they associate with caveolin-1. These interactions induce the activation of caveolin-associated Src tyrosine kinase. Src then phosphorylates and activates p190RhoGAP, which in turn induces the autocatalytic inactivation of RhoA. Finally, the decrease in RhoA activity leads to the disassembly of actin stress fibers and focal adhesions resulting in reduced migratory capacity of the endothelial cell (Fig. 6). Other putative mechanisms of decreased cell migration include the phosphorylation of FAK and subsequent rearrangement of the cytoskeleton as well as inhibition of FAK and the ERK/p38-pathway (Dixelius et al., 2002; Sudhakar et al., 2003).
Figure 6. Intracellular signaling by endostatin. Endostatin binds α5β1 integrin on the cell surface. Its simultaneous or subsequent interaction with a heparan sulfate proteoglycan (HSPG) leads to the association of endostatin, α5β1 integrin, and caveolin-1 (cav-1) in the lipid rafts. These interactions induce the activation of the fraction of Src tyrosine kinase that is associated with caveolin-1. Src then phosphorylates and activates p190RhoGAP, which in turn catalyzes the conversion of the active GTP-bound Rho into the inactive GDP-bound form. Finally, the downregulation of Rho activity leads to the disassembly of actin stress fibers and focal adhesions. Activation of FAK followed by disassembly of the actin cytoskeleton, as well as inhibition of FAK and ERK/p38 activity by endostatin have also been described. These signals result in reduced adhesive and migratory capacity of endothelial cells.

55
Perspective
Proteolytic fragments of the basement membrane collagens are a recently discovered group of endogenous inhibitors of angiogenesis. They are also novel pharmacological agents developed to target vascular endothelial cells and to inhibit pathological angiogenesis. Since its isolation in 1997, endostatin has been extensively studied, both in the laboratory and in the clinic. Animal studies have concluded that recombinant endostatin is capable of effectively reducing tumor volume, and both animal studies and phase I human trials have reported negligible toxicity. Despite the promising results of the in vivo studies, the mechanisms of endostatin action are far from clear.
The results of the current study as well as previous data identify both heparan sulfate proteoglycans and α5β1 integrins as receptors for endostatin. These two proteins are expressed by multiple non-endothelial cell types in various tissues, but endostatin appears to function in a highly endothelial cell specific manner. A major question to be answered is which molecular mechanisms underlie this specificity. In addition, it is unclear, how endostatin associates with these cell surface receptors, and what are the precise molecular mechanisms by which the intracellular signals are generated. Determining the crystal structure of endostatin in complex with its putative integrin receptor would provide important information on the mechanisms of endostatin action. In addition, better understanding of the mechanisms of endostatin function could yield novel, more specific molecular targets for pharmacological intervention in disorders involving aberrant angiogenesis.
The vascular BM provides not only structural support to the vessel wall, but is also a regulator of angiogenesis and tumor growth. Its components regulate cell fate differently when in assembled or degraded form. Despite the potency of recombinant endostatin to reduce tumor volume in experimental animal models, the exact biological roles of type XVIII collagen and its endostatin fragment are still unclear. Analysis of mice deficient of type XVIII collagen in conditions involving aberrant angiogenesis, such as cancer or rheumatoid arthritis, could bring valuable new information on the role of the endostatin fragment in the regulation of angiogenesis.

56
Acknowledgements
These studies were carried out at the Departments of Virology and Pathology, Haartman Institute and Biomedicum Helsinki, University of Helsinki. The current and former heads of these departments, professors Antti Vaheri, Eero Saksela, and Veli-Pekka Lehto, as well as professor Olli Jänne, director of Biomedicum Helsinki are acknowledged for providing excellent research facilities.
I wish to express my gratitude to my supervisor professor Jorma Keski-Oja for his guidance and support during these years. I am grateful for his patience and encouragement especially in the beginning of these studies when the progress of the projects was slowed down by my medical studies as well as the independence he has given me in my work.
I warmly thank professor Kari Alitalo, the head of the molecular and cancer biology program, for creating a research program with such high scientific standards and excellent working facilities. I also wish to thank him for fruitful collaboration as well as for guidance and interest in my work.
I want to thank all the members of the Keski-Oja research group: Juha Hakulinen, Marko Hyytiänen, Sara Illman, Anna Kantola, Katri Koli, Mariliina Lifländer, Kaisa Lehti, Jouko Lohi, Carita Penttinen, Victor Solovyan, Olga Tatti, and Piia Vehviläinen, as well as all the past members of the lab, for creating a pleasant working atmosphere and for invaluable help and advice. It has been a pleasure to know you and to share the lab with you. I especially want to thank Kaisa Lehti and Heli Valtanen for patiently introducing me to laboratory work. I am grateful for Sami Starast and Anne Remes for excellent technical assistance. Terhi Kulmala is warmly thanked for secretarial assistance.
I wish to thank professor Taina Pihlajaniemi for collaboration in the early phases of this work. Tanja Veikkola and Marko Rehn are acknowledged for co-authorship.
Professor Ismo Virtanen and docent Raija Soininen are acknowledged for their expert review of this thesis.
The Helsinki Biomedical Graduate School and its head professor Tomi Mäkelä are acknowledged for providing financial support as well as scientific education. I also want to thank professor Antti Vaheri and docent Klaus Hedman for their advice and interest in my work as members of my thesis committee.
I wish to thank all my friends for all the joyful moments and support over the years. I especially want to thank my dear friends Hanna, Juha, Annina, Jaakko, Lotta, Timo, Vera, and Katja. Without you I wouldn�t be here. Members of Liitokiekkoseura Discus ry are thanked for friendship and for the numerous memorable occasions we have shared together both on and off the playing field.

57
Finally, I thank my parents Päivi and Hans, my sister Laura, and my brother Henrik for their love, encouragement and support. My special thanks go to Isoäiti for her support, and for being the person most interested in my work.
Financial support for this work was provided by the Finnish Cancer Organizations, Biomedicum Helsinki Foundation, Finnish Medical Foundation, Ida Montin Foundation, Paulo Foundation, Research and Science Foundation of Farmos, Emil Aaltonen Foundation, Helsinki Biomedical Graduate School, Helsinki University Central Hospital and University of Helsinki.
Helsinki, March 2004

58
References
Ackley, B.D., Crew, J.R., Elamaa, H., Pihlajaniemi, T., Kuo, C.J. and Kramer, J.M. (2001) The NC1/endostatin domain of Caenorhabditis elegans type XVIII collagen affects cell migration and axon guidance. J Cell Biol, 152, 1219-1232.
Adams, J.C. (2001) Thrombospondins: multifunctional regulators of cell interactions. Annu Rev Cell Dev Biol, 17, 25-51.
Aguirre Ghiso, J.A., Kovalski, K. and Ossowski, L. (1999) Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol, 147, 89-104.
Allen, W.E., Zicha, D., Ridley, A.J. and Jones, G.E. (1998) A role for Cdc42 in macrophage chemotaxis. J Cell Biol, 141, 1147-1157.
Andreasen, P.A., Egelund, R. and Petersen, H.H. (2000) The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sci, 57, 25-40.
Aplin, A.E., Howe, A.K. and Juliano, R.L. (1999) Cell adhesion molecules, signal transduction and cell growth. Curr Opin Cell Biol, 11, 737-744.
Apte, S.S., Fukai, N., Beier, D.R. and Olsen, B.R. (1997) The matrix metalloproteinase-14 (MMP-14) gene is structurally distinct from other MMP genes and is co-expressed with the TIMP-2 gene during mouse embryogenesis. J Biol Chem, 272, 25511-25517.
Aricescu, A.R., McKinnell, I.W., Halfter, W. and Stoker, A.W. (2002) Heparan sulfate proteoglycans are ligands for receptor protein tyrosine phosphatase σ. Mol Cell Biol, 22, 1881-1892.
Arthur, W.T., Petch, L.A. and Burridge, K. (2000) Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Curr Biol, 10, 719-722.
Bacharach, E., Itin, A. and Keshet, E. (1992) In vivo patterns of expression of urokinase and its inhibitor PAI-1 suggest a concerted role in regulating physiological angiogenesis. Proc Natl Acad Sci U S A, 89, 10686-10690.
Bajou, K., Masson, V., Gerard, R.D., Schmitt, P.M., Albert, V., Praus, M., Lund, L.R., Frandsen, T.L., Brunner, N., Danø, K., Fusenig, N.E., Weidle, U., Carmeliet, G., Loskutoff, D., Collen, D., Carmeliet, P., Foidart, J.M. and Noel, A. (2001) The plasminogen activator inhibitor PAI-1 controls in vivo tumor vascularization by interaction with proteases, not vitronectin. Implications for antiangiogenic strategies. J Cell Biol, 152, 777-784.
Bajou, K., Noel, A., Gerard, R.D., Masson, V., Brunner, N., Holst-Hansen, C., Skobe, M., Fusenig, N.E., Carmeliet, P., Collen, D. and Foidart, J.M. (1998) Absence of host plasminogen activator inhibitor 1 prevents cancer invasion and vascularization. Nat Med, 4, 923-928.
Basbaum, C.B. and Werb, Z. (1996) Focalized proteolysis: spatial and temporal regulation of extracellular matrix degradation at the cell surface. Curr Opin Cell Biol, 8, 731-738.
Bayless, K.J., Salazar, R. and Davis, G.E. (2000) RGD-dependent vacuolation and lumen formation observed during endothelial cell morphogenesis in three-dimensional fibrin matrices involves the αvβ3 and α5β1 integrins. Am J Pathol, 156, 1673-1683.
Berger, A.C., Feldman, A.L., Gnant, M.F., Kruger, E.A., Sim, B.K., Hewitt, S., Figg, W.D., Alexander, H.R. and Libutti, S.K. (2000) The angiogenesis inhibitor, endostatin, does not affect murine cutaneous wound healing. J Surg Res, 91, 26-31.
Bergers, G. and Benjamin, L.E. (2003) Tumorigenesis and the angiogenic switch. Nat Rev Cancer, 3, 401-410.
Bergers, G., Brekken, R., McMahon, G., Vu, T.H., Itoh, T., Tamaki, K., Tanzawa, K., Thorpe, P., Itohara, S., Werb, Z. and Hanahan, D. (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol, 2, 737-744.
Bergers, G., Javaherian, K., Lo, K.M., Folkman, J. and Hanahan, D. (1999) Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science, 284, 808-812.

59
Bianchi, E., Ferrero, E., Fazioli, F., Mangili, F., Wang, J., Bender, J.R., Blasi, F. and Pardi, R. (1996) Integrin-dependent induction of functional urokinase receptors in primary T lymphocytes. J Clin Invest, 98, 1133-1141.
Bishop, A.L. and Hall, A. (2000) Rho GTPases and their effector proteins. Biochem J, 348 Pt 2, 241-255.
Blasi, F. (1999) The urokinase receptor. A cell surface, regulated chemokine. Apmis, 107,96-101.
Blasi, F. and Carmeliet, P. (2002) uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol, 3, 932-943.
Blezinger, P., Wang, J., Gondo, M., Quezada, A., Mehrens, D., French, M., Singhal, A., Sullivan, S., Rolland, A., Ralston, R. and Min, W. (1999) Systemic inhibition of tumor growth and tumor metastases by intramuscular administration of the endostatin gene. Nat Biotechnol, 17, 343-348.
Bloch, W., Huggel, K., Sasaki, T., Grose, R., Bugnon, P., Addicks, K., Timpl, R. and Werner, S. (2000) The angiogenesis inhibitor endostatin impairs blood vessel maturation during wound healing. Faseb J, 14, 2373-2376.
Boehm, T., Folkman, J., Browder, T. and O'Reilly, M.S. (1997) Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature, 390, 404-407.
Bohuslav, J., Horejsi, V., Hansmann, C., Stockl, J., Weidle, U.H., Majdic, O., Bartke, I., Knapp, W. and Stockinger, H. (1995) Urokinase plasminogen activator receptor, β2-integrins, and Src-kinases within a single receptor complex of human monocytes. J Exp Med, 181, 1381-1390.
Bradshaw, A.D. and Sage, E.H. (2001) SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. J Clin Invest, 107, 1049-1054.
Brooks, P.C., Clark, R.A. and Cheresh, D.A. (1994a) Requirement of vascular integrin αvβ3for angiogenesis. Science, 264, 569-571.
Brooks, P.C., Montgomery, A.M., Rosenfeld, M., Reisfeld, R.A., Hu, T., Klier, G. and Cheresh, D.A. (1994b) Integrin αvβ3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell, 79, 1157-1164.
Brooks, P.C., Silletti, S., von Schalscha, T.L., Friedlander, M. and Cheresh, D.A. (1998) Disruption of angiogenesis by PEX, a noncatalytic metalloproteinase fragment with integrin binding activity. Cell, 92, 391-400.
Brown, D.A. and Rose, J.K. (1992) Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell, 68, 533-544.
Burridge, K. and Chrzanowska-Wodnicka, M. (1996) Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol, 12, 463-518.
Capillo, M., Mancuso, P., Gobbi, A., Monestiroli, S., Pruneri, G., Dell'Agnola, C., Martinelli, G., Shultz, L. and Bertolini, F. (2003) Continuous infusion of endostatin inhibits differentiation, mobilization, and clonogenic potential of endothelial cell progenitors. Clin Cancer Res, 9, 377-382.
Carmeliet, P. (2000) Mechanisms of angiogenesis and arteriogenesis. Nat Med, 6, 389-395. Carmeliet, P. and Jain, R.K. (2000) Angiogenesis in cancer and other diseases. Nature, 407,
249-257. Carmeliet, P., Schoonjans, L., Kieckens, L., Ream, B., Degen, J., Bronson, R., De Vos, R.,
van den Oord, J.J., Collen, D. and Mulligan, R.C. (1994) Physiological consequences of loss of plasminogen activator gene function in mice. Nature, 368, 419-424.
Chang, Y.S., di Tomaso, E., McDonald, D.M., Jones, R., Jain, R.K. and Munn, L.L. (2000) Mosaic blood vessels in tumors: frequency of cancer cells in contact with flowing blood. Proc Natl Acad Sci U S A, 97, 14608-14613.
Chillemi, F., Francescato, P., Ragg, E., Cattaneo, M.G., Pola, S. and Vicentini, L. (2003) Studies on the structure-activity relationship of endostatin: synthesis of human endostatin peptides exhibiting potent antiangiogenic activities. J Med Chem, 46, 4165-4172.
Clark, E.A., King, W.G., Brugge, J.S., Symons, M. and Hynes, R.O. (1998) Integrin-mediated signals regulated by members of the rho family of GTPases. J Cell Biol, 142, 573-586.
Colognato, H. and Yurchenco, P.D. (2000) Form and function: the laminin family of heterotrimers. Dev Dyn, 218, 213-234.

60
Colorado, P.C., Torre, A., Kamphaus, G., Maeshima, Y., Hopfer, H., Takahashi, K., Volk, R., Zamborsky, E.D., Herman, S., Sarkar, P.K., Ericksen, M.B., Dhanabal, M., Simons, M., Post, M., Kufe, D.W., Weichselbaum, R.R., Sukhatme, V.P. and Kalluri, R. (2000) Anti-angiogenic cues from vascular basement membrane collagen. Cancer Res, 60, 2520-2526.
Conway, E.M., Collen, D. and Carmeliet, P. (2001) Molecular mechanisms of blood vessel growth. Cardiovasc Res, 49, 507-521.
Costantini, V., Zacharski, L.R., Memoli, V.A., Kudryk, B.J., Rousseau, S.M. and Stump, D.C. (1991) Occurrence of components of fibrinolysis pathways in situ in neoplastic and nonneoplastic human breast tissue. Cancer Res, 51, 354-358.
Crowley, C.W., Cohen, R.L., Lucas, B.K., Liu, G., Shuman, M.A. and Levinson, A.D. (1993) Prevention of metastasis by inhibition of the urokinase receptor. Proc Natl Acad Sci U S A,90, 5021-5025.
Cunningham, O., Andolfo, A., Santovito, M.L., Iuzzolino, L., Blasi, F. and Sidenius, N. (2003) Dimerization controls the lipid raft partitioning of uPAR/CD87 and regulates its biological functions. Embo J, 22, 5994-6003.
Czekay, R.P., Kuemmel, T.A., Orlando, R.A. and Farquhar, M.G. (2001) Direct binding of occupied urokinase receptor (uPAR) to LDL receptor-related protein is required for endocytosis of uPAR and regulation of cell surface urokinase activity. Mol Biol Cell, 12, 1467-1479.
D'Amato, R.J., Lin, C.M., Flynn, E., Folkman, J. and Hamel, E. (1994) 2-Methoxyestradiol, an endogenous mammalian metabolite, inhibits tubulin polymerization by interacting at the colchicine site. Proc Natl Acad Sci U S A, 91, 3964-3968.
Danø, K., Andreasen, P.A., Grøndahl-Hansen, J., Kristensen, P., Nielsen, L.S. and Skriver, L. (1985) Plasminogen activators, tissue degradation, and cancer. Adv Cancer Res, 44, 139-266.
Dawson, D.W., Pearce, S.F., Zhong, R., Silverstein, R.L., Frazier, W.A. and Bouck, N.P. (1997) CD36 mediates the In vitro inhibitory effects of thrombospondin-1 on endothelial cells. JCell Biol, 138, 707-717.
Degryse, B., Resnati, M., Rabbani, S.A., Villa, A., Fazioli, F. and Blasi, F. (1999) Src-dependence and pertussis-toxin sensitivity of urokinase receptor-dependent chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. Blood, 94, 649-662.
Degryse, B., Sier, C.F., Resnati, M., Conese, M. and Blasi, F. (2001) PAI-1 inhibits urokinase-induced chemotaxis by internalizing the urokinase receptor. FEBS Lett, 505, 249-254.
Deng, G., Curriden, S.A., Wang, S., Rosenberg, S. and Loskutoff, D.J. (1996) Is plasminogen activator inhibitor-1 the molecular switch that governs urokinase receptor-mediated cell adhesion and release? J Cell Biol, 134, 1563-1571.
Dewerchin, M., Nuffelen, A.V., Wallays, G., Bouche, A., Moons, L., Carmeliet, P., Mulligan, R.C. and Collen, D. (1996) Generation and characterization of urokinase receptor-deficient mice. J Clin Invest, 97, 870-878.
Dhanabal, M., Volk, R., Ramchandran, R., Simons, M. and Sukhatme, V.P. (1999) Cloning, expression, and in vitro activity of human endostatin. Biochem Biophys Res Commun, 258, 345-352.
Ding, I., Sun, J.Z., Fenton, B., Liu, W.M., Kimsely, P., Okunieff, P. and Min, W. (2001) Intratumoral administration of endostatin plasmid inhibits vascular growth and perfusion in MCa-4 murine mammary carcinomas. Cancer Res, 61, 526-531.
Ding, Y.H., Javaherian, K., Lo, K.M., Chopra, R., Boehm, T., Lanciotti, J., Harris, B.A., Li, Y., Shapiro, R., Hohenester, E., Timpl, R., Folkman, J. and Wiley, D.C. (1998) Zinc-dependent dimers observed in crystals of human endostatin. Proc Natl Acad Sci U S A, 95, 10443-10448.
Dixelius, J., Cross, M., Matsumoto, T., Sasaki, T., Timpl, R. and Claesson-Welsh, L. (2002) Endostatin regulates endothelial cell adhesion and cytoskeletal organization. Cancer Res, 62,1944-1947.
Dixelius, J., Larsson, H., Sasaki, T., Holmqvist, K., Lu, L., Engstrom, A., Timpl, R., Welsh, M. and Claesson-Welsh, L. (2000) Endostatin-induced tyrosine kinase signaling through the shb adaptor protein regulates endothelial cell apoptosis. Blood, 95, 3403-3411.
Dixelius, J., Cross, M.J., Matsumoto, T. and Claesson-Welsh, L. (2003) Endostatin action and intracellular signaling: beta-catenin as a potential target? Cancer Lett, 196, 1-12.

61
Eberhard, A., Kahlert, S., Goede, V., Hemmerlein, B., Plate, K.H. and Augustin, H.G. (2000) Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res, 60, 1388-1393.
Eisterer, W., Jiang, X., Bachelot, T., Pawliuk, R., Abramovich, C., Leboulch, P., Hogge, D. and Eaves, C. (2002) Unfulfilled promise of endostatin in a gene therapy-xenotransplant model of human acute lymphocytic leukemia. Mol Ther, 5, 352-359.
Eklund, L., Piuhola, J., Komulainen, J., Sormunen, R., Ongvarrasopone, C., Fassler, R., Muona, A., Ilves, M., Ruskoaho, H., Takala, T.E. and Pihlajaniemi, T. (2001) Lack of type XV collagen causes a skeletal myopathy and cardiovascular defects in mice. Proc Natl Acad Sci U S A,98, 1194-1199.
Eliceiri, B.P. (2001) Integrin and growth factor receptor crosstalk. Circ Res, 89, 1104-1110. Ellis, V. (2003) Plasminogen activation at the cell surface. Curr Top Dev Biol, 54, 263-312. Ellis, V., Scully, M.F. and Kakkar, V.V. (1989) Plasminogen activation initiated by single-
chain urokinase-type plasminogen activator. Potentiation by U937 monocytes. J Biol Chem, 264,2185-2188.
Erickson, A.C. and Couchman, J.R. (2000) Still more complexity in mammalian basement membranes. J Histochem Cytochem, 48, 1291-1306.
Eriksson, K., Magnusson, P., Dixelius, J., Claesson-Welsh, L. and Cross, M.J. (2003) Angiostatin and endostatin inhibit endothelial cell migration in response to FGF and VEGF without interfering with specific intracellular signal transduction pathways. FEBS Lett, 536, 19-24.
Fang, J., Shing, Y., Wiederschain, D., Yan, L., Butterfield, C., Jackson, G., Harper, J., Tamvakopoulos, G. and Moses, M.A. (2000) Matrix metalloproteinase-2 is required for the switch to the angiogenic phenotype in a tumor model. Proc Natl Acad Sci U S A, 97, 3884-3889.
Fazioli, F., Resnati, M., Sidenius, N., Higashimoto, Y., Appella, E. and Blasi, F. (1997) A urokinase-sensitive region of the human urokinase receptor is responsible for its chemotactic activity. Embo J, 16, 7279-7286.
Feinberg, R.F., Kao, L.C., Haimowitz, J.E., Queenan, J.T., Jr., Wun, T.C., Strauss, J.F., 3rd and Kliman, H.J. (1989) Plasminogen activator inhibitor types 1 and 2 in human trophoblasts. PAI-1 is an immunocytochemical marker of invading trophoblasts. Lab Invest, 61, 20-26.
Felbor, U., Dreier, L., Bryant, R.A., Ploegh, H.L., Olsen, B.R. and Mothes, W. (2000) Secreted cathepsin L generates endostatin from collagen XVIII. Embo J, 19, 1187-1194.
Feldman, A.L., Alexander, H.R., Hewitt, S.M., Lorang, D., Thiruvathukal, C.E., Turner, E.M. and Libutti, S.K. (2001a) Effect of retroviral endostatin gene transfer on subcutaneous and intraperitoneal growth of murine tumors. J Natl Cancer Inst, 93, 1014-1020.
Feldman, A.L., Alexander, H.R., Jr., Bartlett, D.L., Kranda, K.C., Miller, M.S., Costouros, N.G., Choyke, P.L. and Libutti, S.K. (2001b) A prospective analysis of plasma endostatin levels in colorectal cancer patients with liver metastases. Ann Surg Oncol, 8, 741-745.
Feldman, A.L., Alexander, H.R., Jr., Yang, J.C., Linehan, W.M., Eyler, R.A., Miller, M.S., Steinberg, S.M. and Libutti, S.K. (2002) Prospective analysis of circulating endostatin levels in patients with renal cell carcinoma. Cancer, 95, 1637-1643.
Feldman, A.L., Pak, H., Yang, J.C., Alexander, H.R., Jr. and Libutti, S.K. (2001c) Serum endostatin levels are elevated in patients with soft tissue sarcoma. Cancer, 91, 1525-1529.
Feldman, A.L., Restifo, N.P., Alexander, H.R., Bartlett, D.L., Hwu, P., Seth, P. and Libutti, S.K. (2000a) Antiangiogenic gene therapy of cancer utilizing a recombinant adenovirus to elevate systemic endostatin levels in mice. Cancer Res, 60, 1503-1506.
Feldman, A.L., Tamarkin, L., Paciotti, G.F., Simpson, B.W., Linehan, W.M., Yang, J.C., Fogler, W.E., Turner, E.M., Alexander, H.R., Jr. and Libutti, S.K. (2000b) Serum endostatin levels are elevated and correlate with serum vascular endothelial growth factor levels in patients with stage IV clear cell renal cancer. Clin Cancer Res, 6, 4628-4634.
Felsenfeld, D.P., Schwartzberg, P.L., Venegas, A., Tse, R. and Sheetz, M.P. (1999) Selective regulation of integrin--cytoskeleton interactions by the tyrosine kinase Src. Nat Cell Biol, 1, 200-206.
Ferrara, N. (2002) Role of vascular endothelial growth factor in physiologic and pathologic angiogenesis: therapeutic implications. Semin Oncol, 29, 10-14.

62
Ferreras, M., Felbor, U., Lenhard, T., Olsen, B.R. and Delaisse, J. (2000) Generation and degradation of human endostatin proteins by various proteinases. FEBS Lett, 486, 247-251.
Fibbi, G., Caldini, R., Chevanne, M., Pucci, M., Schiavone, N., Morbidelli, L., Parenti, A., Granger, H.J., Del Rosso, M. and Ziche, M. (1998) Urokinase-dependent angiogenesis in vitro and diacylglycerol production are blocked by antisense oligonucleotides against the urokinase receptor. Lab Invest, 78, 1109-1119.
Floquet, N., Pasco, S., Ramont, L., Derreumaux, P., Laronze, J.Y., Nuzillard, J.M., Maquart, F.X., Alix, A.J. and Monboisse, J.C. (2003) The anti-tumor properties of the α3(IV) 185-203 peptide from the NC1 domain of type IV collagen (Tumstatin) are conformation-dependent. J Biol Chem.
Fukai, N., Eklund, L., Marneros, A.G., Oh, S.P., Keene, D.R., Tamarkin, L., Niemela, M., Ilves, M., Li, E., Pihlajaniemi, T. and Olsen, B.R. (2002) Lack of collagen XVIII/endostatin results in eye abnormalities. Embo J, 21, 1535-1544.
Geiger, B., Bershadsky, A., Pankov, R. and Yamada, K.M. (2001) Transmembrane crosstalk between the extracellular matrix--cytoskeleton crosstalk. Nat Rev Mol Cell Biol, 2, 793-805.
Gengrinovitch, S., Berman, B., David, G., Witte, L., Neufeld, G. and Ron, D. (1999) Glypican-1 is a VEGF165 binding proteoglycan that acts as an extracellular chaperone for VEGF165. J Biol Chem, 274, 10816-10822.
George, E.L., Baldwin, H.S. and Hynes, R.O. (1997) Fibronectins are essential for heart and blood vessel morphogenesis but are dispensable for initial specification of precursor cells. Blood,90, 3073-3081.
George, E.L., Georges-Labouesse, E.N., Patel-King, R.S., Rayburn, H. and Hynes, R.O. (1993) Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development, 119, 1079-1091.
Ghohestani, R.F., Li, K., Rousselle, P. and Uitto, J. (2001) Molecular organization of the cutaneous basement membrane zone. Clin Dermatol, 19, 551-562.
Giancotti, F.G. and Ruoslahti, E. (1999) Integrin signaling. Science, 285, 1028-1032. Giannelli, G., Falk-Marzillier, J., Schiraldi, O., Stetler-Stevenson, W.G. and Quaranta, V.
(1997) Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science,277, 225-228.
Ginsburg, D., Zeheb, R., Yang, A.Y., Rafferty, U.M., Andreasen, P.A., Nielsen, L., Danø, K., Lebo, R.V. and Gelehrter, T.D. (1986) cDNA cloning of human plasminogen activator-inhibitor from endothelial cells. J Clin Invest, 78, 1673-1680.
Gomez-Mouton, C., Abad, J.L., Mira, E., Lacalle, R.A., Gallardo, E., Jimenez-Baranda, S., Illa, I., Bernad, A., Manes, S. and Martinez, A.C. (2001) Segregation of leading-edge and uropod components into specific lipid rafts during T cell polarization. Proc Natl Acad Sci U S A, 98, 9642-9647.
Graham, C.H., Fitzpatrick, T.E. and McCrae, K.R. (1998) Hypoxia stimulates urokinase receptor expression through a heme protein- dependent pathway. Blood, 91, 3300-3307.
Granelli-Piperno, A. and Reich, E. (1978) A study of proteases and protease-inhibitor complexes in biological fluids. J Exp Med, 148, 223-234.
Green, J.M., Zhelesnyak, A., Chung, J., Lindberg, F.P., Sarfati, M., Frazier, W.A. and Brown, E.J. (1999) Role of cholesterol in formation and function of a signaling complex involving αvβ3, integrin-associated protein (CD47), and heterotrimeric G proteins. J Cell Biol, 146, 673-682.
Gumbiner, B.M. (1996) Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell, 84, 345-357.
Gupta, N., Nodzenski, E., Khodarev, N.N., Yu, J., Khorasani, L., Beckett, M.A., Kufe, D.W. and Weichselbaum, R.R. (2001) Angiostatin effects on endothelial cells mediated by ceramide and RhoA. EMBO Rep, 2, 536-540.
Gutheil, J.C., Campbell, T.N., Pierce, P.R., Watkins, J.D., Huse, W.D., Bodkin, D.J. and Cheresh, D.A. (2000) Targeted antiangiogenic therapy for cancer using Vitaxin: a humanized monoclonal antibody to the integrin αvβ3. Clin Cancer Res, 6, 3056-3061.

63
Hamano, Y., Zeisberg, M., Sugimoto, H., Lively, J.C., Maeshima, Y., Yang, C., Hynes, R.O., Werb, Z., Sudhakar, A. and Kalluri, R. (2003) Physiological levels of tumstatin, a fragment of collagen IV α3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via αvβ3integrin. Cancer Cell, 3, 589-601.
Hammes, H.P., Brownlee, M., Jonczyk, A., Sutter, A. and Preissner, K.T. (1996) Subcutaneous injection of a cyclic peptide antagonist of vitronectin receptor-type integrins inhibits retinal neovascularization. Nat Med, 2, 529-533.
Hanahan, D. and Folkman, J. (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell, 86, 353-364.
Hanai, J., Dhanabal, M., Karumanchi, S.A., Albanese, C., Waterman, M., Chan, B., Ramchandran, R., Pestell, R. and Sukhatme, V.P. (2002a) Endostatin causes G1 arrest of endothelial cells through inhibition of cyclin D1. J Biol Chem, 277, 16464-16469.
Hanai, J., Gloy, J., Karumanchi, S.A., Kale, S., Tang, J., Hu, G., Chan, B., Ramchandran, R., Jha, V., Sukhatme, V.P. and Sokol, S. (2002b) Endostatin is a potential inhibitor of Wnt signaling. J Cell Biol, 158, 529-539.
Hanke, J.H., Gardner, J.P., Dow, R.L., Changelian, P.S., Brissette, W.H., Weringer, E.J., Pollok, B.A. and Connelly, P.A. (1996) Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem, 271,695-701.
Hedman, K., Kurkinen, M., Alitalo, K., Vaheri, A., Johansson, S. and Hook, M. (1979) Isolation of the pericellular matrix of human fibroblast cultures. J Cell Biol, 81, 83-91.
Hefler, L., Tempfer, C., Kainz, C. and Obermair, A. (1999) Serum concentrations of endostatin in patients with vulvar cancer. Gynecol Oncol, 74, 151-152.
Herbst, R.S., Hess, K.R., Tran, H.T., Tseng, J.E., Mullani, N.A., Charnsangavej, C., Madden, T., Davis, D.W., McConkey, D.J., O'Reilly, M.S., Ellis, L.M., Pluda, J., Hong, W.K. and Abbruzzese, J.L. (2002a) Phase I study of recombinant human endostatin in patients with advanced solid tumors. J Clin Oncol, 20, 3792-3803.
Herbst, R.S., Mullani, N.A., Davis, D.W., Hess, K.R., McConkey, D.J., Charnsangavej, C., O'Reilly, M.S., Kim, H.W., Baker, C., Roach, J., Ellis, L.M., Rashid, A., Pluda, J., Bucana, C., Madden, T.L., Tran, H.T. and Abbruzzese, J.L. (2002b) Development of biologic markers of response and assessment of antiangiogenic activity in a clinical trial of human recombinant endostatin. J Clin Oncol, 20, 3804-3814.
Hiraoka, N., Allen, E., Apel, I.J., Gyetko, M.R. and Weiss, S.J. (1998) Matrix metalloproteinases regulate neovascularization by acting as pericellular fibrinolysins. Cell, 95,365-377.
Hohenester, E. and Engel, J. (2002) Domain structure and organisation in extracellular matrix proteins. Matrix Biol, 21, 115-128.
Hohenester, E., Sasaki, T., Mann, K. and Timpl, R. (2000) Variable zinc coordination in endostatin. J Mol Biol, 297, 1-6.
Hohenester, E., Sasaki, T., Olsen, B.R. and Timpl, R. (1998) Crystal structure of the angiogenesis inhibitor endostatin at 1.5 A resolution. Embo J, 17, 1656-1664.
Holmbeck, K., Bianco, P., Caterina, J., Yamada, S., Kromer, M., Kuznetsov, S.A., Mankani, M., Robey, P.G., Poole, A.R., Pidoux, I., Ward, J.M. and Birkedal-Hansen, H. (1999) MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell, 99, 81-92.
Huang, S., New, L., Pan, Z., Han, J. and Nemerow, G.R. (2000) Urokinase plasminogen activator/urokinase-specific surface receptor expression and matrix invasion by breast cancer cells requires constitutive p38alpha mitogen-activated protein kinase activity. J Biol Chem, 275, 12266-12272.
Hudson, B.G., Reeders, S.T. and Tryggvason, K. (1993) Type IV collagen: structure, gene organization, and role in human diseases. Molecular basis of Goodpasture and Alport syndromes and diffuse leiomyomatosis. J Biol Chem, 268, 26033-26036.
Hunter, T. (2000) Signaling--2000 and beyond. Cell, 100, 113-127. Hynes, R. (2002a) Integrins: bidirectional, allosteric signaling machines. Cell, 110, 673-687.

64
Hynes, R.O. (2002b) A reevaluation of integrins as regulators of angiogenesis. Nat Med, 8,918-921.
Hyytiäinen, M. and Keski-Oja, J. (2003) Latent TGF-β binding protein LTBP-2 decreases fibroblast adhesion to fibronectin. J Cell Biol, 163, 1363-1374.
Isogai, C., Laug, W.E., Shimada, H., Declerck, P.J., Stins, M.F., Durden, D.L., Erdreich-Epstein, A. and DeClerck, Y.A. (2001) Plasminogen activator inhibitor-1 promotes angiogenesis by stimulating endothelial cell migration toward fibronectin. Cancer Res, 61, 5587-5594.
Itoh, T., Tanioka, M., Yoshida, H., Yoshioka, T., Nishimoto, H. and Itohara, S. (1998) Reduced angiogenesis and tumor progression in gelatinase A-deficient mice. Cancer Res, 58,1048-1051.
Iughetti, P., Suzuki, O., Godoi, P.H., Alves, V.A., Sertie, A.L., Zorick, T., Soares, F., Camargo, A., Moreira, E.S., di Loreto, C., Moreira-Filho, C.A., Simpson, A., Oliva, G. and Passos-Bueno, M.R. (2001) A polymorphism in endostatin, an angiogenesis inhibitor, predisposes for the development of prostatic adenocarcinoma. Cancer Res, 61, 7375-7378.
Jain, R.K. (2001) Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med, 7, 987-989.
Jiang, L., Jha, V., Dhanabal, M., Sukhatme, V.P. and Alper, S.L. (2001) Intracellular Ca(2+) signaling in endothelial cells by the angiogenesis inhibitors endostatin and angiostatin. Am J Physiol Cell Physiol, 280, C1140-1150.
Jockusch, B.M., Bubeck, P., Giehl, K., Kroemker, M., Moschner, J., Rothkegel, M., Rudiger, M., Schluter, K., Stanke, G. and Winkler, J. (1995) The molecular architecture of focal adhesions. Ann Rev Cell Devel Biol, 11, 379-416.
John, A. and Tuszynski, G. (2001) The role of matrix metalloproteinases in tumor angiogenesis and tumor metastasis. Pathol Oncol Res, 7, 14-23.
Johnsen, M., Lund, L.R., Romer, J., Almholt, K. and Danø, K. (1998) Cancer invasion and tissue remodeling: common themes in proteolytic matrix degradation. Curr Opin Cell Biol, 10,667-671.
Joki, T., Machluf, M., Atala, A., Zhu, J., Seyfried, N.T., Dunn, I.F., Abe, T., Carroll, R.S. and Black, P.M. (2001) Continuous release of endostatin from microencapsulated engineered cells for tumor therapy. Nat Biotechnol, 19, 35-39.
Juhan-Vague, I., Moerman, B., De Cock, F., Aillaud, M.F. and Collen, D. (1984) Plasma levels of a specific inhibitor of tissue-type plasminogen activator (and urokinase) in normal and pathological conditions. Thromb Res, 33, 523-530.
Kaibuchi, K., Kuroda, S. and Amano, M. (1999) Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu Rev Biochem, 68, 459-486.
Kalluri, R. (2003) Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer, 3, 422-433.
Kamphaus, G.D., Colorado, P.C., Panka, D.J., Hopfer, H., Ramchandran, R., Torre, A., Maeshima, Y., Mier, J.W., Sukhatme, V.P. and Kalluri, R. (2000) Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J Biol Chem, 275, 1209-1215.
Kanthou, C. and Tozer, G.M. (2002) The tumor vascular targeting agent combretastatin A-4-phosphate induces reorganization of the actin cytoskeleton and early membrane blebbing in human endothelial cells. Blood, 99, 2060-2069.
Karihaloo, A., Karumanchi, S.A., Barasch, J., Jha, V., Nickel, C.H., Yang, J., Grisaru, S., Bush, K.T., Nigam, S., Rosenblum, N.D., Sukhatme, V.P. and Cantley, L.G. (2001) Endostatin regulates branching morphogenesis of renal epithelial cells and ureteric bud. Proc Natl Acad Sci U S A, 98, 12509-12514.
Karumanchi, S.A., Jha, V., Ramchandran, R., Karihaloo, A., Tsiokas, L., Chan, B., Dhanabal, M., Hanai, J.I., Venkataraman, G., Shriver, Z., Keiser, N., Kalluri, R., Zeng, H., Mukhopadhyay, D., Chen, R.L., Lander, A.D., Hagihara, K., Yamaguchi, Y., Sasisekharan, R., Cantley, L. and Sukhatme, V.P. (2001) Cell surface glypicans are low-affinity endostatin receptors. Mol Cell, 7, 811-822.
Keezer, S.M., Ivie, S.E., Krutzsch, H.C., Tandle, A., Libutti, S.K. and Roberts, D.D. (2003) Angiogenesis inhibitors target the endothelial cell cytoskeleton through altered regulation of heat shock protein 27 and cofilin. Cancer Res, 63, 6405-6412.

65
Kerbel, R. and Folkman, J. (2002) Clinical translation of angiogenesis inhibitors. Nat Rev Cancer, 2, 727-739.
Kieran, M.W., Folkman, J. and Heymach, J. (2003) Angiogenesis inhibitors and hypoxia. Nat Med, 9, 1104.
Kim, S., Bell, K., Mousa, S.A. and Varner, J.A. (2000a) Regulation of angiogenesis in vivo by ligation of integrin α5β1 with the central cell-binding domain of fibronectin. Am J Pathol, 156,1345-1362.
Kim, S., Harris, M. and Varner, J.A. (2000b) Regulation of integrin αvβ3 -mediated endothelial cell migration and angiogenesis by integrin α5β1 and protein kinase A. J Biol Chem,275, 33920-33928.
Kim, Y.M., Hwang, S., Pyun, B.J., Kim, T.Y., Lee, S.T., Gho, Y.S. and Kwon, Y.G. (2002) Endostatin blocks vascular endothelial growth factor-mediated signaling via direct interaction with KDR/Flk-1. J Biol Chem, 277, 27872-27879.
Kim, Y.M., Jang, J.W., Lee, O.H., Yeon, J., Choi, E.Y., Kim, K.W., Lee, S.T. and Kwon, Y.G. (2000c) Endostatin inhibits endothelial and tumor cellular invasion by blocking the activation and catalytic activity of matrix metalloproteinase. Cancer Res, 60, 5410-5413.
Kisker, O., Becker, C.M., Prox, D., Fannon, M., D'Amato, R., Flynn, E., Fogler, W.E., Sim, B.K., Allred, E.N., Pirie-Shepherd, S.R. and Folkman, J. (2001) Continuous administration of endostatin by intraperitoneally implanted osmotic pump improves the efficacy and potency of therapy in a mouse xenograft tumor model. Cancer Res, 61, 7669-7674.
Klagsbrun, M. and D'Amore, P.A. (1991) Regulators of angiogenesis. Annu Rev Physiol, 53,217-239.
Koolwijk, P., van Erck, M.G., de Vree, W.J., Vermeer, M.A., Weich, H.A., Hanemaaijer, R. and van Hinsbergh, V.W. (1996) Cooperative effect of TNFalpha, bFGF, and VEGF on the formation of tubular structures of human microvascular endothelial cells in a fibrin matrix. Role of urokinase activity. J Cell Biol, 132, 1177-1188.
Kranenburg, O., Kroon-Batenburg, L.M., Reijerkerk, A., Wu, Y.P., Voest, E.E. and Gebbink, M.F. (2003) Recombinant endostatin forms amyloid fibrils that bind and are cytotoxic to murine neuroblastoma cells in vitro. FEBS Lett, 539, 149-155.
Kreuger, J., Matsumoto, T., Vanwildemeersch, M., Sasaki, T., Timpl, R., Claesson-Welsh, L., Spillmann, D. and Lindahl, U. (2002) Role of heparan sulfate domain organization in endostatin inhibition of endothelial cell function. Embo J, 21, 6303-6311.
Kroon, M.E., Koolwijk, P., van der Vecht, B. and van Hinsbergh, V.W. (2000) Urokinase receptor expression on human microvascular endothelial cells is increased by hypoxia: implications for capillary-like tube formation in a fibrin matrix. Blood, 96, 2775-2783.
Kroon, M.E., Koolwijk, P., van Goor, H., Weidle, U.H., Collen, A., van der Pluijm, G. and van Hinsbergh, V.W. (1999) Role and localization of urokinase receptor in the formation of new microvascular structures in fibrin matrices. Am J Pathol, 154, 1731-1742.
Kuhn, K. (1995) Basement membrane (type IV) collagen. Matrix Biol, 14, 439-445. Kuo, C.J., LaMontagne, K.R., Jr., Garcia-Cardena, G., Ackley, B.D., Kalman, D., Park, S.,
Christofferson, R., Kamihara, J., Ding, Y.H., Lo, K.M., Gillies, S., Folkman, J., Mulligan, R.C. and Javaherian, K. (2001) Oligomerization-dependent regulation of motility and morphogenesis by the collagen XVIII NC1/endostatin domain. J Cell Biol, 152, 1233-1246.
Kwaan, H.C. (1992) The plasminogen-plasmin system in malignancy. Cancer Metastasis Rev, 11, 291-311.
Laiho, M. and Keski-Oja, J. (1989) Growth factors in the regulation of pericellular proteolysis: a review. Cancer Research, 49, 2533-2553.
Laiho, M., Saksela, O., Andreasen, P.A. and Keski-Oja, J. (1986) Enhanced production and extracellular deposition of the endothelial- type plasminogen activator inhibitor in cultured human lung fibroblasts by transforming growth factor-beta. J Cell Biol, 103, 2403-2410.
Lang, I., Hoffmann, C., Olip, H., Pabst, M.A., Hahn, T., Dohr, G. and Desoye, G. (2001) Differential mitogenic responses of human macrovascular and microvascular endothelial cells to cytokines underline their phenotypic heterogeneity. Cell Prolif, 34, 143-155.

66
Lang, I., Pabst, M.A., Hiden, U., Blaschitz, A., Dohr, G., Hahn, T. and Desoye, G. (2003) Heterogeneity of microvascular endothelial cells isolated from human term placenta and macrovascular umbilical vein endothelial cells. Eur J Cell Biol, 82, 163-173.
Larsson, L.I., Skriver, L., Nielsen, L.S., Grøndahl-Hansen, J., Kristensen, P. and Danø, K. (1984) Distribution of urokinase-type plasminogen activator immunoreactivity in the mouse. JCell Biol, 98, 894-903.
Lauffenburger, D.A. and Horwitz, A.F. (1996) Cell migration: a physically integrated molecular process. Cell, 84, 359-369.
Lawler, J. (2000) The functions of thrombospondin-1 and-2. Curr Opin Cell Biol, 12, 634-640.
Lee, S.J., Jang, J.W., Kim, Y.M., Lee, H.I., Jeon, J.Y., Kwon, Y.G. and Lee, S.T. (2002) Endostatin binds to the catalytic domain of matrix metalloproteinase-2. FEBS Lett, 519, 147-152.
Li, Y., Knisely, J.M., Lu, W., McCormick, L.M., Wang, J., Henkin, J., Schwartz, A.L. and Bu, G. (2002) Low density lipoprotein (LDL) receptor-related protein 1B impairs urokinase receptor regeneration on the cell surface and inhibits cell migration. J Biol Chem, 277, 42366-42371.
Lisanti, M.P., Scherer, P.E., Vidugiriene, J., Tang, Z., Hermanowski-Vosatka, A., Tu, Y.H., Cook, R.F. and Sargiacomo, M. (1994) Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. J Cell Biol, 126, 111-126.
Loskutoff, D.J., Curriden, S.A., Hu, G. and Deng, G. (1999) Regulation of cell adhesion by PAI-1. Apmis, 107, 54-61.
Loskutoff, D.J., van Aken, B.E. and Seiffert, D. (1995) Abnormalities in the fibrinolytic system of the vascular wall associated with atherosclerosis. Ann N Y Acad Sci, 748, 177-183; discussion 183-174.
Luna, E.J. and Hitt, A.L. (1992) Cytoskeleton--plasma membrane interactions. Science, 258,955-964.
Lyons, R.M., Keski-Oja, J. and Moses, H.L. (1988) Proteolytic activation of latent transforming growth factor-β from fibroblast-conditioned medium. J Cell Biol, 106, 1659-1665.
Ma, L., Elliott, S.N., Cirino, G., Buret, A., Ignarro, L.J. and Wallace, J.L. (2001) Platelets modulate gastric ulcer healing: role of endostatin and vascular endothelial growth factor release. Proc Natl Acad Sci U S A, 98, 6470-6475.
MacDonald, N.J., Shivers, W.Y., Narum, D.L., Plum, S.M., Wingard, J.N., Fuhrmann, S.R., Liang, H., Holland-Linn, J., Chen, D.H. and Sim, B.K. (2001) Endostatin binds tropomyosin. A potential modulator of the antitumor activity of endostatin. J Biol Chem, 276, 25190-25196.
Maeshima, Y., Colorado, P.C. and Kalluri, R. (2000a) Two RGD-independent αvβ3 integrin binding sites on tumstatin regulate distinct anti-tumor properties. J Biol Chem, 275, 23745-23750.
Maeshima, Y., Colorado, P.C., Torre, A., Holthaus, K.A., Grunkemeyer, J.A., Ericksen, M.B., Hopfer, H., Xiao, Y., Stillman, I.E. and Kalluri, R. (2000b) Distinct antitumor properties of a type IV collagen domain derived from basement membrane. J Biol Chem, 275, 21340-21348.
Maeshima, Y., Manfredi, M., Reimer, C., Holthaus, K.A., Hopfer, H., Chandamuri, B.R., Kharbanda, S. and Kalluri, R. (2001a) Identification of the anti-angiogenic site within vascular basement membrane-derived tumstatin. J Biol Chem, 276, 15240-15248.
Maeshima, Y., Sudhakar, A., Lively, J.C., Ueki, K., Kharbanda, S., Kahn, C.R., Sonenberg, N., Hynes, R.O. and Kalluri, R. (2002) Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science, 295, 140-143.
Maeshima, Y., Yerramalla, U.L., Dhanabal, M., Holthaus, K.A., Barbashov, S., Kharbanda, S., Reimer, C., Manfredi, M., Dickerson, W.M. and Kalluri, R. (2001b) Extracellular matrix-derived peptide binds to αvβ3 integrin and inhibits angiogenesis. J Biol Chem, 276, 31959-31968.
Marneros, A.G., Keene, D.R., Hansen, U., Fukai, N., Moulton, K., Goletz, P.L., Moiseyev, G., Pawlyk, B.S., Halfter, W., Dong, S., Shibata, M., Li, T., Crouch, R.K., Bruckner, P. and Olsen, B.R. (2004) Collagen XVIII/endostatin is essential for vision and retinal pigment epithelial function. Embo J, 23, 89-99.
Mayor, S., Rothberg, K.G. and Maxfield, F.R. (1994) Sequestration of GPI-anchored proteins in caveolae triggered by cross- linking. Science, 264, 1948-1951.

67
Mignatti, P. and Rifkin, D.B. (1996) Plasminogen activators and angiogenesis. Curr Top Microbiol Immunol, 213 ( Pt 1), 33-50.
Min, H.Y., Doyle, L.V., Vitt, C.R., Zandonella, C.L., Stratton-Thomas, J.R., Shuman, M.A. and Rosenberg, S. (1996) Urokinase receptor antagonists inhibit angiogenesis and primary tumor growth in syngeneic mice. Cancer Res, 56, 2428-2433.
Miosge, N., Sasaki, T. and Timpl, R. (1999) Angiogenesis inhibitor endostatin is a distinct component of elastic fibers in vessel walls. Faseb J, 13, 1743-1750.
Miyamoto, S., Akiyama, S.K. and Yamada, K.M. (1995) Synergistic roles for receptor occupancy and aggregation in integrin transmembrane function. Science, 267, 883-885.
Miyamoto, S., Katz, B.Z., Lafrenie, R.M. and Yamada, K.M. (1998) Fibronectin and integrins in cell adhesion, signaling, and morphogenesis. Ann N Y Acad Sci, 857, 119-129.
Mongiat, M., Sweeney, S.M., San Antonio, J.D., Fu, J. and Iozzo, R.V. (2003) Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J Biol Chem, 278, 4238-4249.
Mosher, D.F., Fogerty, F.J., Chernousov, M.A. and Barry, E.L. (1991) Assembly of fibronectin into extracellular matrix. Ann N Y Acad Sci, 614, 167-180.
Muragaki, Y., Abe, N., Ninomiya, Y., Olsen, B.R. and Ooshima, A. (1994) The human α1(XV) collagen chain contains a large amino-terminal non-triple helical domain with a tandem repeat structure and homology to α1(XVIII) collagen. J Biol Chem, 269, 4042-4046.
Muragaki, Y., Timmons, S., Griffith, C.M., Oh, S.P., Fadel, B., Quertermous, T. and Olsen, B.R. (1995) Mouse Col18α1 is expressed in a tissue-specific manner as three alternative variants and is localized in basement membrane zones. Proc Natl Acad Sci U S A, 92, 8763-8767.
Murphy-Ullrich, J.E. (2001) The de-adhesive activity of matricellular proteins: is intermediate cell adhesion an adaptive state? J Clin Invest, 107, 785-790
Myers, J.C., Kivirikko, S., Gordon, M.K., Dion, A.S. and Pihlajaniemi, T. (1992) Identification of a previously unknown human collagen chain, α1(XV), characterized by extensive interruptions in the triple-helical region. Proc Natl Acad Sci U S A, 89, 10144-10148.
Myöhänen, H.T., Stephens, R.W., Hedman, K., Tapiovaara, H., Ronne, E., Hoyer-Hansen, G., Dano, K. and Vaheri, A. (1993) Distribution and lateral mobility of the urokinase-receptor complex at the cell surface. J Histochem Cytochem, 41, 1291-1301.
Nilsson, I.M., Felding, P., Lecander, I., Lenner, C. and Astedt, B. (1986) Different types of plasminogen activator inhibitors in plasma and platelets in pregnant women. Br J Haematol, 62,215-220.
Nobes, C.D. and Hall, A. (1995) Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell, 81, 53-62.
Nobes, C.D. and Hall, A. (1999) Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol, 144, 1235-1244.
Nyberg, P., Heikkilä, P., Sorsa, T., Luostarinen, J., Heljäsvaara, R., Stenman, U.H., Pihlajaniemi, T. and Salo, T. (2003) Endostatin inhibits human tongue carcinoma cell invasion and intravasation and blocks the activation of matrix metalloprotease-2, -9, and -13. J Biol Chem, 278,22404-22411.
Oh, S.P., Kamagata, Y., Muragaki, Y., Timmons, S., Ooshima, A. and Olsen, B.R. (1994) Isolation and sequencing of cDNAs for proteins with multiple domains of Gly-Xaa-Yaa repeats identify a distinct family of collagenous proteins. Proc Natl Acad Sci U S A, 91, 4229-4233.
O'Reilly, M.S., Boehm, T., Shing, Y., Fukai, N., Vasios, G., Lane, W.S., Flynn, E., Birkhead, J.R., Olsen, B.R. and Folkman, J. (1997) Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell, 88, 277-285.
O'Reilly, M.S., Holmgren, L., Shing, Y., Chen, C., Rosenthal, R.A., Moses, M., Lane, W.S., Cao, Y., Sage, E.H. and Folkman, J. (1994) Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell, 79, 315-328.
Orr, A.W., Pallero, M.A. and Murphy-Ullrich, J.E. (2002) Thrombospondin stimulates focal adhesion disassembly through Gi- and phosphoinositide 3-kinase-dependent ERK activation. JBiol Chem, 277, 20453-20460.

68
Ortega, N. and Werb, Z. (2002) New functional roles for non-collagenous domains of basement membrane collagens. J Cell Sci, 115, 4201-4214.
Ossowski, L. (1996) Effect of antisense inhibition of Urokinase receptor on malignancy. Curr Top Microbiol Immunol, 213 ( Pt 3), 101-112.
Pande, G. (2000) The role of membrane lipids in regulation of integrin functions. Curr Opin Cell Biol, 12, 569-574.
Parsons, J.T., Martin, K.H., Slack, J.K., Taylor, J.M. and Weed, S.A. (2000) Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene, 19, 5606-5613.
Pepper, M.S. (2001) Extracellular proteolysis and angiogenesis. Thromb Haemost, 86, 346-355.
Pepper, M.S., Montesano, R., Mandriota, S.J., Orci, L. and Vassalli, J.D. (1996) Angiogenesis: a paradigm for balanced extracellular proteolysis during cell migration and morphogenesis. Enzyme Protein, 49, 138-162.
Perletti, G., Concari, P., Giardini, R., Marras, E., Piccinini, F., Folkman, J. and Chen, L. (2000) Antitumor activity of endostatin against carcinogen-induced rat primary mammary tumors. Cancer Res, 60, 1793-1796.
Petitclerc, E., Boutaud, A., Prestayko, A., Xu, J., Sado, Y., Ninomiya, Y., Sarras, M.P., Jr., Hudson, B.G. and Brooks, P.C. (2000) New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J Biol Chem, 275, 8051-8061.
Pinsky, D.J., Liao, H., Lawson, C.A., Yan, S.F., Chen, J., Carmeliet, P., Loskutoff, D.J. and Stern, D.M. (1998) Coordinated induction of plasminogen activator inhibitor-1 (PAI-1) and inhibition of plasminogen activator gene expression by hypoxia promotes pulmonary vascular fibrin deposition. J Clin Invest, 102, 919-928.
Piwnica-Worms, H., Saunders, K.B., Roberts, T.M., Smith, A.E. and Cheng, S.H. (1987) Tyrosine phosphorylation regulates the biochemical and biological properties of pp60c-src. Cell,49, 75-82.
Planus, E., Barlovatz-Meimon, G., Rogers, R.A., Bonavaud, S., Ingber, D.E. and Wang, N. (1997) Binding of urokinase to plasminogen activator inhibitor type-1 mediates cell adhesion and spreading. J Cell Sci, 110, 1091-1098.
Price, L.S., Leng, J., Schwartz, M.A. and Bokoch, G.M. (1998) Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol Biol Cell, 9, 1863-1871.
Prockop, D.J. and Kivirikko, K.I. (1995) Collagens: molecular biology, diseases, and potentials for therapy. Annu Rev Biochem, 64, 403-434.
Quax, P.H., van Leeuwen, R.T., Verspaget, H.W. and Verheijen, J.H. (1990) Protein and messenger RNA levels of plasminogen activators and inhibitors analyzed in 22 human tumor cell lines. Cancer Res, 50, 1488-1494.
Rafii, S., Lyden, D., Benezra, R., Hattori, K. and Heissig, B. (2002) Vascular and haematopoietic stem cells: novel targets for anti-angiogenesis therapy? Nat Rev Cancer, 2, 826-835.
Rak, J., Yu, J.L., Kerbel, R.S. and Coomber, B.L. (2002) What do oncogenic mutations have to do with angiogenesis/vascular dependence of tumors? Cancer Res, 62, 1931-1934.
Read, T.A., Sorensen, D.R., Mahesparan, R., Enger, P.O., Timpl, R., Olsen, B.R., Hjelstuen, M.H., Haraldseth, O. and Bjerkvig, R. (2001) Local endostatin treatment of gliomas administered by microencapsulated producer cells. Nat Biotechnol, 19, 29-34.
Rehn, M., Veikkola, T., Kukk-Valdre, E., Nakamura, H., Ilmonen, M., Lombardo, C., Pihlajaniemi, T., Alitalo, K. and Vuori, K. (2001) Interaction of endostatin with integrins implicated in angiogenesis. Proc Natl Acad Sci U S A, 98, 1024-1029.
Reimer, C.L., Agata, N., Tammam, J.G., Bamberg, M., Dickerson, W.M., Kamphaus, G.D., Rook, S.L., Milhollen, M., Fram, R., Kalluri, R., Kufe, D. and Kharbanda, S. (2002) Antineoplastic effects of chemotherapeutic agents are potentiated by NM-3, an inhibitor of angiogenesis. Cancer Res, 62, 789-795.
Ren, X.D., Kiosses, W.B. and Schwartz, M.A. (1999) Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. Embo J, 18, 578-585.

69
Resnati, M., Guttinger, M., Valcamonica, S., Sidenius, N., Blasi, F. and Fazioli, F. (1996) Proteolytic cleavage of the urokinase receptor substitutes for the agonist-induced chemotactic effect. Embo J, 15, 1572-1582.
Resnati, M., Pallavicini, I., Wang, J.M., Oppenheim, J., Serhan, C.N., Romano, M. and Blasi, F. (2002) The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc Natl Acad Sci U S A, 99, 1359-1364.
Reynolds, L.E., Wyder, L., Lively, J.C., Taverna, D., Robinson, S.D., Huang, X., Sheppard, D., Hynes, R.O. and Hodivala-Dilke, K.M. (2002) Enhanced pathological angiogenesis in mice lacking β3 integrin or β3 and β5 integrins. Nat Med, 8, 27-34.
Ridley, A.J., Comoglio, P.M. and Hall, A. (1995) Regulation of scatter factor/hepatocyte growth factor responses by Ras, Rac, and Rho in MDCK cells. Mol Cell Biol, 15, 1110-1122.
Ridley, A.J. and Hall, A. (1992) The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell, 70, 389-399.
Ridley, A.J., Paterson, H.F., Johnston, C.L., Diekmann, D. and Hall, A. (1992) The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell, 70, 401-410.
Ridley, A.J., Self, A.J., Kasmi, F., Paterson, H.F., Hall, A., Marshall, C.J. and Ellis, C. (1993) rho family GTPase activating proteins p190, bcr and rhoGAP show distinct specificities in vitro and in vivo. Embo J, 12, 5151-5160.
Risau, W. (1997) Mechanisms of angiogenesis. Nature, 386, 671-674. Rousseau, S., Houle, F., Landry, J. and Huot, J. (1997) p38 MAP kinase activation by
vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene, 15, 2169-2177.
Ruoslahti, E. (1996) RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol, 12, 697-715.
Ruoslahti, E. (2002) Specialization of tumour vasculature. Nat Rev Cancer, 2, 83-90. Rønne, E., Behrendt, N., Ellis, V., Ploug, M., Danø, K. and Høyer-Hansen, G. (1991) Cell-
induced potentiation of the plasminogen activation system is abolished by a monoclonal antibody that recognizes the NH2-terminal domain of the urokinase receptor. FEBS Lett, 288, 233-236.
Saarela, J., Rehn, M., Oikarinen, A., Autio-Harmainen, H. and Pihlajaniemi, T. (1998a) The short and long forms of type XVIII collagen show clear tissue specificities in their expression and location in basement membrane zones in humans. Am J Pathol, 153, 611-626.
Saarela, J., Ylikärppä, R., Rehn, M., Purmonen, S. and Pihlajaniemi, T. (1998b) Complete primary structure of two variant forms of human type XVIII collagen and tissue-specific differences in the expression of the corresponding transcripts. Matrix Biol, 16, 319-328.
Sage, E.H., Reed, M., Funk, S.E., Truong, T., Steadele, M., Puolakkainen, P., Maurice, D.H. and Bassuk, J.A. (2003) Cleavage of the matricellular protein SPARC by matrix metalloproteinase 3 produces polypeptides that influence angiogenesis. J Biol Chem, 278, 37849-37857.
Saksela, O. and Rifkin, D.B. (1990) Release of basic fibroblast growth factor-heparan sulfate complexes from endothelial cells by plasminogen activator-mediated proteolytic activity. J Cell Biol, 110, 767-775.
Samad, F., Yamamoto, K. and Loskutoff, D.J. (1996) Distribution and regulation of plasminogen activator inhibitor-1 in murine adipose tissue in vivo. Induction by tumor necrosis factor-alpha and lipopolysaccharide. J Clin Invest, 97, 37-46.
Santos, M.F., McCormack, S.A., Guo, Z., Okolicany, J., Zheng, Y., Johnson, L.R. and Tigyi, G. (1997) Rho proteins play a critical role in cell migration during the early phase of mucosal restitution. J Clin Invest, 100, 216-225.
Sasaki, T., Fukai, N., Mann, K., Gohring, W., Olsen, B.R. and Timpl, R. (1998) Structure, function and tissue forms of the C-terminal globular domain of collagen XVIII containing the angiogenesis inhibitor endostatin. Embo J, 17, 4249-4256.
Sasaki, T., Hohenester, E. and Timpl, R. (2002) Structure and function of collagen-derived endostatin inhibitors of angiogenesis. IUBMB Life, 53, 77-84.
Sasaki, T., Larsson, H., Kreuger, J., Salmivirta, M., Claesson-Welsh, L., Lindahl, U., Hohenester, E. and Timpl, R. (1999) Structural basis and potential role of heparin/heparan sulfate binding to the angiogenesis inhibitor endostatin. Embo J, 18, 6240-6248.

70
Sastry, S.K. and Horwitz, A.F. (1993) Integrin cytoplasmic domains: mediators of cytoskeletal linkages and extra- and intracellular initiated transmembrane signaling. Curr Opin Cell Biol, 5, 819-831.
Sauter, B.V., Martinet, O., Zhang, W.J., Mandeli, J. and Woo, S.L. (2000) Adenovirus-mediated gene transfer of endostatin in vivo results in high level of transgene expression and inhibition of tumor growth and metastases. Proc Natl Acad Sci U S A, 97, 4802-4807.
Schoenwaelder, S.M. and Burridge, K. (1999) Bidirectional signaling between the cytoskeleton and integrins. Curr Opin Cell Biol, 11, 274-286.
Schuch, G., Heymach, J.V., Nomi, M., Machluf, M., Force, J., Atala, A., Eder, J.P., Jr., Folkman, J. and Soker, S. (2003) Endostatin inhibits the vascular endothelial growth factor-induced mobilization of endothelial progenitor cells. Cancer Res, 63, 8345-8350.
Schvartz, I., Seger, D. and Shaltiel, S. (1999) Vitronectin. Int J Biochem Cell Biol, 31, 539-544.
Schwartz, M.A. and Baron, V. (1999) Interactions between mitogenic stimuli, or, a thousand and one connections. Curr Opin Cell Biol, 11, 197-202.
Schwarzbauer, J.E. (1997) Cell migration: may the force be with you. Curr Biol, 7, R292-294.
Schwarzbauer, J.E. and Sechler, J.L. (1999) Fibronectin fibrillogenesis: a paradigm for extracellular matrix assembly. Curr Opin Cell Biol, 11, 622-627.
Seiffert, D., Ciambrone, G., Wagner, N.V., Binder, B.R. and Loskutoff, D.J. (1994) The somatomedin B domain of vitronectin. Structural requirements for the binding and stabilization of active type 1 plasminogen activator inhibitor. J Biol Chem, 269, 2659-2666.
Semenza, G.L. (2001) Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med, 7, 345-350.
Sertie, A.L., Sossi, V., Camargo, A.A., Zatz, M., Brahe, C. and Passos-Bueno, M.R. (2000) Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome). Hum Mol Genet, 9, 2051-2058.
Shaarawy, M. and El-Sharkawy, S.A. (2001) Biomarkers of intrinsic angiogenic and anti-angiogenic activity in patients with endometrial hyperplasia and endometrial cancer. Acta Oncol,40, 513-518.
Shichiri, M. and Hirata, Y. (2001) Antiangiogenesis signals by endostatin. Faseb J, 15,1044-1053.
Silletti, S., Kessler, T., Goldberg, J., Boger, D.L. and Cheresh, D.A. (2001) Disruption of matrix metalloproteinase 2 binding to integrin αvβ3 by an organic molecule inhibits angiogenesis and tumor growth in vivo. Proc Natl Acad Sci U S A, 98, 119-124.
Simon, D.I., Wei, Y., Zhang, L., Rao, N.K., Xu, H., Chen, Z., Liu, Q., Rosenberg, S. and Chapman, H.A. (2000) Identification of a urokinase receptor-integrin interaction site. Promiscuous regulator of integrin function. J Biol Chem, 275, 10228-10234.
Simon, K.O., Nutt, E.M., Abraham, D.G., Rodan, G.A. and Duong, L.T. (1997) The αvβ3
integrin regulates α5β1-mediated cell migration toward fibronectin. J Biol Chem, 272, 29380-29389.
Simons, K. and Ikonen, E. (1997) Functional rafts in cell membranes. Nature, 387, 569-572. Simons, K. and Toomre, D. (2000) Lipid rafts and signal transduction. Nat Rev Mol Cell
Biol, 1, 31-39. Small, J.V., Stradal, T., Vignal, E. and Rottner, K. (2002) The lamellipodium: where motility
begins. Trends Cell Biol, 12, 112-120. Solberg, H., Ploug, M., Høyer-Hansen, G., Nielsen, B.S. and Lund, L.R. (2001) The murine
receptor for urokinase-type plasminogen activator is primarily expressed in tissues actively undergoing remodeling. J Histochem Cytochem, 49, 237-246.
Standker, L., Schrader, M., Kanse, S.M., Jurgens, M., Forssmann, W.G. and Preissner, K.T. (1997) Isolation and characterization of the circulating form of human endostatin. FEBS Lett, 420,129-133.
Stefansson, S. and Lawrence, D.A. (1996) The serpin PAI-1 inhibits cell migration by blocking integrin αvβ3 binding to vitronectin. Nature, 383, 441-443.

71
Stefansson, S. and Lawrence, D.A. (2003) Old dogs and new tricks: proteases, inhibitors, and cell migration. Sci STKE, 2003, pe24.
Stoppelli, M.P., Tacchetti, C., Cubellis, M.V., Corti, A., Hearing, V.J., Cassani, G., Appella, E. and Blasi, F. (1986) Autocrine saturation of pro-urokinase receptors on human A431 cells. Cell,45, 675-684.
Stubbs, M.T., Renatus, M. and Bode, W. (1998) An active zymogen: unravelling the mystery of tissue-type plasminogen activator. Biol Chem, 379, 95-103.
Stupack, D.G., Puente, X.S., Boutsaboualoy, S., Storgard, C.M. and Cheresh, D.A. (2001) Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J Cell Biol, 155,459-470.
Sudhakar, A., Sugimoto, H., Yang, C., Lively, J., Zeisberg, M. and Kalluri, R. (2003) Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by αvβ3 and α5β1 integrins. Proc Natl Acad Sci U S A, 100, 4766-4771.
Suzuki, M., Iizasa, T., Ko, E., Baba, M., Saitoh, Y., Shibuya, K., Sekine, Y., Yoshida, S., Hiroshima, K. and Fujisawa, T. (2002) Serum endostatin correlates with progression and prognosis of non-small cell lung cancer. Lung Cancer, 35, 29-34.
Taddei, L., Chiarugi, P., Brogelli, L., Cirri, P., Magnelli, L., Raugei, G., Ziche, M., Granger, H.J., Chiarugi, V. and Ramponi, G. (1999) Inhibitory effect of full-length human endostatin on in vitro angiogenesis. Biochem Biophys Res Commun, 263, 340-345.
Taipale, J. and Keski-Oja, J. (1997) Growth factors in the extracellular matrix. Faseb J, 11,51-59.
Taipale, J., Koli, K. and Keski-Oja, J. (1992) Release of transforming growth factor-β1 from the pericellular matrix of cultured fibroblasts and fibrosarcoma cells by plasmin and thrombin. JBiol Chem, 267, 25378-25384.
Thomas, S.M. and Brugge, J.S. (1997) Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol, 13, 513-609.
Timpl, R. (1996) Macromolecular organization of basement membranes. Curr Opin Cell Biol, 8, 618-624.
Troyanovsky, B., Levchenko, T., Mansson, G., Matvijenko, O. and Holmgren, L. (2001) Angiomotin: an angiostatin binding protein that regulates endothelial cell migration and tube formation. J Cell Biol, 152, 1247-1254.
Uchiyama, T., Kurabayashi, M., Ohyama, Y., Utsugi, T., Akuzawa, N., Sato, M., Tomono, S., Kawazu, S. and Nagai, R. (2000) Hypoxia induces transcription of the plasminogen activator inhibitor-1 gene through genistein-sensitive tyrosine kinase pathways in vascular endothelial cells. Arterioscler Thromb Vasc Biol, 20, 1155-1161.
Urbich, C., Reissner, A., Chavakis, E., Dernbach, E., Haendeler, J., Fleming, I., Zeiher, A.M., Kaszkin, M. and Dimmeler, S. (2002) Dephosphorylation of endothelial nitric oxide synthase contributes to the anti-angiogenic effects of endostatin. Faseb J, 16, 706-708.
Wagner, S.N., Atkinson, M.J., Wagner, C., Hofler, H., Schmitt, M. and Wilhelm, O. (1996) Sites of urokinase-type plasminogen activator expression and distribution of its receptor in the normal human kidney. Histochem Cell Biol, 105, 53-60.
Waltz, D.A. and Chapman, H.A. (1994) Reversible cellular adhesion to vitronectin linked to urokinase receptor occupancy. J Biol Chem, 269, 14746-14750.
Watanabe, K., Yamada, H. and Yamaguchi, Y. (1995) K-glypican: a novel GPI-anchored heparan sulfate proteoglycan that is highly expressed in developing brain and kidney. J Cell Biol,130, 1207-1218.
Webb, D.J., Thomas, K.S. and Gonias, S.L. (2001) Plasminogen activator inhibitor 1 functions as a urokinase response modifier at the level of cell signaling and thereby promotes MCF-7 cell growth. J Cell Biol, 152, 741-752.
Wei, Y., Eble, J.A., Wang, Z., Kreidberg, J.A. and Chapman, H.A. (2001) Urokinase receptors promote beta1 integrin function through interactions with integrin α3β1. Mol Biol Cell,12, 2975-2986.
Wei, Y., Lukashev, M., Simon, D.I., Bodary, S.C., Rosenberg, S., Doyle, M.V. and Chapman, H.A. (1996) Regulation of integrin function by the urokinase receptor. Science, 273,1551-1555.

72
Wei, Y., Yang, X., Liu, Q., Wilkins, J.A. and Chapman, H.A. (1999) A role for caveolin and the urokinase receptor in integrin-mediated adhesion and signaling. J Cell Biol, 144, 1285-1294.
Wen, W., Moses, M.A., Wiederschain, D., Arbiser, J.L. and Folkman, J. (1999) The generation of endostatin is mediated by elastase. Cancer Res, 59, 6052-6056.
Werb, Z. (1997) ECM and cell surface proteolysis: regulating cellular ecology. Cell, 91, 439-442.
Werb, Z., Vu, T.H., Rinkenberger, J.L. and Coussens, L.M. (1999) Matrix-degrading proteases and angiogenesis during development and tumor formation. Apmis, 107, 11-18.
Wilcox, S.A., Reho, T., Higgins, P.J., Tominna-Sebald, E. and McKeown-Longo, P.J. (1996) Localization of urokinase to focal adhesions by human fibrosarcoma cells synthesizing recombinant vitronectin. Biochem Cell Biol, 74, 899-910.
Wind, T., Hansen, M., Jensen, J.K. and Andreasen, P.A. (2002) The molecular basis for anti-proteolytic and non-proteolytic functions of plasminogen activator inhibitor type-1: roles of the reactive centre loop, the shutter region, the flexible joint region and the small serpin fragment. BiolChem, 383, 21-36.
Wohlwend, A., Belin, D. and Vassalli, J.D. (1987) Plasminogen activator-specific inhibitors produced by human monocytes/macrophages. J Exp Med, 165, 320-339.
Volberg, T., Romer, L., Zamir, E. and Geiger, B. (2001) pp60(c-src) and related tyrosine kinases: a role in the assembly and reorganization of matrix adhesions. J Cell Sci, 114, 2279-2289.
Worthylake, R.A. and Burridge, K. (2003) RhoA and ROCK promote migration by limiting membrane protrusions. J Biol Chem, 278, 13578-13584.
Wu, C. (1997) Roles of integrins in fibronectin matrix assembly. Histol Histopathol, 12, 233-240.
Xu, J., Rodriguez, D., Petitclerc, E., Kim, J.J., Hangai, M., Moon, Y.S., Davis, G.E., Brooks, P.C. and Yuen, S.M. (2001) Proteolytic exposure of a cryptic site within collagen type IV is required for angiogenesis and tumor growth in vivo. J Cell Biol, 154, 1069-1079.
Yamaguchi, N., Anand-Apte, B., Lee, M., Sasaki, T., Fukai, N., Shapiro, R., Que, I., Lowik, C., Timpl, R. and Olsen, B.R. (1999) Endostatin inhibits VEGF-induced endothelial cell migration and tumor growth independently of zinc binding. Embo J, 18, 4414-4423.
Yancopoulos, G.D., Davis, S., Gale, N.W., Rudge, J.S., Wiegand, S.J. and Holash, J. (2000) Vascular-specific growth factors and blood vessel formation. Nature, 407, 242-248.
Yang, J.T., Rayburn, H. and Hynes, R.O. (1993) Embryonic mesodermal defects in α5integrin-deficient mice. Development, 119, 1093-1105.
Yebra, M., Parry, G.C.N., Stromblad, S., Mackman, N., Rosenberg, S., Mueller, B.M. and Cheresh, D.A. (1996) Requirement of receptor-bound urokinase-type plasminogen activator for integrin αvβ5-directed cell migration. J Biol Chem, 271, 29393-29399.
Yin, G., Liu, W., An, P., Li, P., Ding, I., Planelles, V., Schwarz, E.M. and Min, W. (2002) Endostatin gene transfer inhibits joint angiogenesis and pannus formation in inflammatory arthritis. Mol Ther, 5, 547-554.
Ylikärppä, R., Eklund, L., Sormunen, R., Kontiola, A.I., Utriainen, A., Määttä, M., Fukai, N., Olsen, B.R. and Pihlajaniemi, T. (2003a) Lack of type XVIII collagen results in anterior ocular defects. Faseb J.
Ylikärppä, R., Eklund, L., Sormunen, R., Muona, A., Fukai, N., Olsen, B.R. and Pihlajaniemi, T. (2003b) Double knockout mice reveal a lack of major functional compensation between collagens XV and XVIII. Matrix Biol, 22, 443-448.
Yokoyama, Y., Green, J.E., Sukhatme, V.P. and Ramakrishnan, S. (2000) Effect of endostatin on spontaneous tumorigenesis of mammary adenocarcinoma in a transgenic mouse model. Cancer Res, 60, 4362-4365.
Yoon, S.S., Eto, H., Lin, C.M., Nakamura, H., Pawlik, T.M., Song, S.U. and Tanabe, K.K. (1999) Mouse endostatin inhibits the formation of lung and liver metastases. Cancer Res, 59,6251-6256.
Yu, J.L. and Rak, J.W. (2003) Host microenvironment in breast cancer development: inflammatory and immune cells in tumour angiogenesis and arteriogenesis. Breast Cancer Res, 5,83-88.

73
Zhou, Z., Apte, S.S., Soininen, R., Cao, R., Baaklini, G.Y., Rauser, R.W., Wang, J., Cao, Y. and Tryggvason, K. (2000) Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc Natl Acad Sci U S A, 97, 4052-4057.
Zorick, T.S., Mustacchi, Z., Bando, S.Y., Zatz, M., Moreira-Filho, C.A., Olsen, B. and Passos-Bueno, M.R. (2001) High serum endostatin levels in Down syndrome: implications for improved treatment and prevention of solid tumours. Eur J Hum Genet, 9, 811-814.


















