
Emergencias Hematológicas
154
Emergencias Hematológicas Virgilio Salinas Rodríguez
description
Emergencias Hematológicas. Virgilio Salinas Rodríguez. Algunas Emergencias. Trombocitopenia. Hemorragia masiva. Coagulopatía por trauma. Hemofilia congénita y adquirida. Neutropenia febril. Anemia hemolítica autoinmune. Purpura trombocitopenica trombótica. Pancitopenia con CID. - PowerPoint PPT Presentation
Transcript of Emergencias Hematológicas

Emergencias Hematológicas
Virgilio Salinas Rodríguez

Algunas Emergencias
• Trombocitopenia.• Hemorragia masiva.• Coagulopatía por trauma.• Hemofilia congénita y adquirida.• Neutropenia febril.• Anemia hemolítica autoinmune.• Purpura trombocitopenica trombótica.• Pancitopenia con CID.

Algunas Emergencias
• Trombocitopenia.• Hemorragia masiva.• Coagulopatía por trauma.• Hemofilia congénita y adquirida.• Neutropenia febril.• Anemia hemolítica autoinmune.• Purpura trombocitopenica trombótica.• Pancitopenia con CID.

Dos Trombocitopenias
• Trombocitopenia por destrucción en sangre.
• Trombocitopenia por falla medular.

Terapia para sangrado por Trombocitopenia por destrucción
• Medicamentos:– Esteroides.– Inmunosupresores.
– Aprotinina, Acido tranexámico.– Plasmaferesis.– Inmunoglobulinas endovenosas.
– Agentes: eltrombopag, romiplostin– Factor VII recombinante activo– Trombopoyetina

Advertencias
• El uso de transfusión de plaquetas en Trombocitopenia por destrucción no esta justificado.
• Son ineficaces y pueden exacerbar la enfermedad.
• Hay terapias alternativas.

¿Cuáles Emplear en Emergencia?
• Inmunoglobulinas Endovenosas 1 gr/kilo/dosis– Acción demora 4 días.
• Dexametasona 40 mg diario por 4 días.– Actúa más rápido de prednisona.
• Inmunoglobulina Anti D 7.5 mg/kilo/día IM.– Acción al 4 día.
• Rituximab 375 mg/m2 EV.– Combinado con dexametasona aumenta eficacia.
• Acido tranexámico 10 mg/kilo/dosis cada 6 horas.– Reduce sangrado mientras actúan los otros.

Conclusiones
• La Trombocitopenia por falla medular:– La profilaxis tiene empleo dudoso pero
aumenta las necesidades trasfusionales.
– La terapia con plaquetas controlan el sangrado significativo.
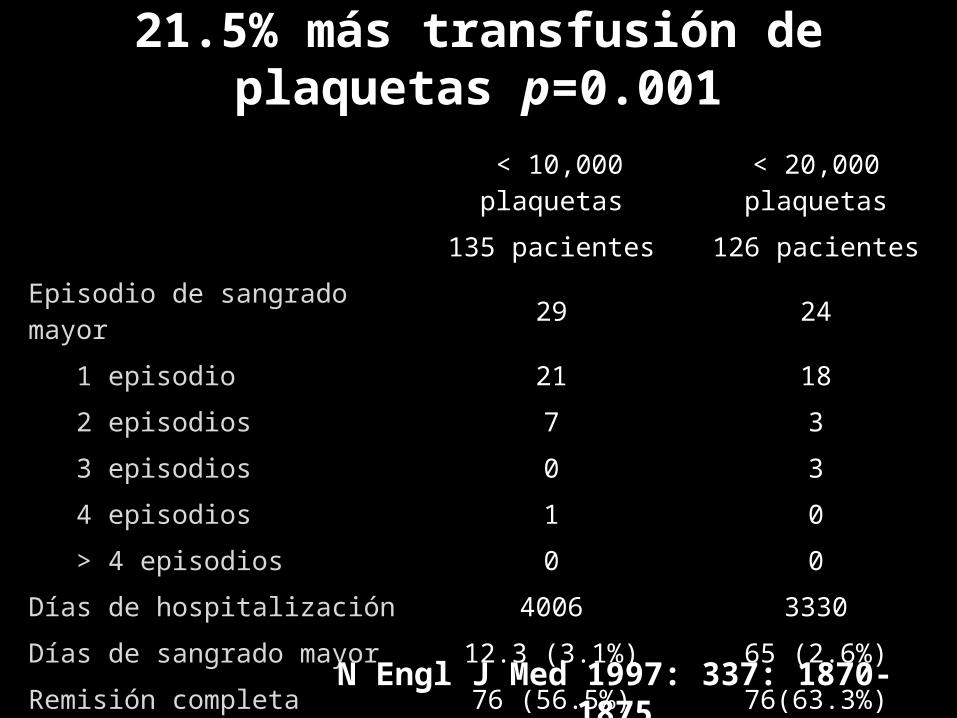
< 10,000 plaquetas < 20,000 plaquetas135 pacientes 126 pacientes
Episodio de sangrado mayor 29 24 1 episodio 21 18 2 episodios 7 3 3 episodios 0 3 4 episodios 1 0 > 4 episodios 0 0Días de hospitalización 4006 3330Días de sangrado mayor 12.3 (3.1%) 65 (2.6%)Remisión completa 76 (56.5%) 76(63.3%)Muertes 18 (13.3%) 9 (7.5%)
N Engl J Med 1997: 337: 1870-1875
21.5% más transfusión de plaquetas p=0.001
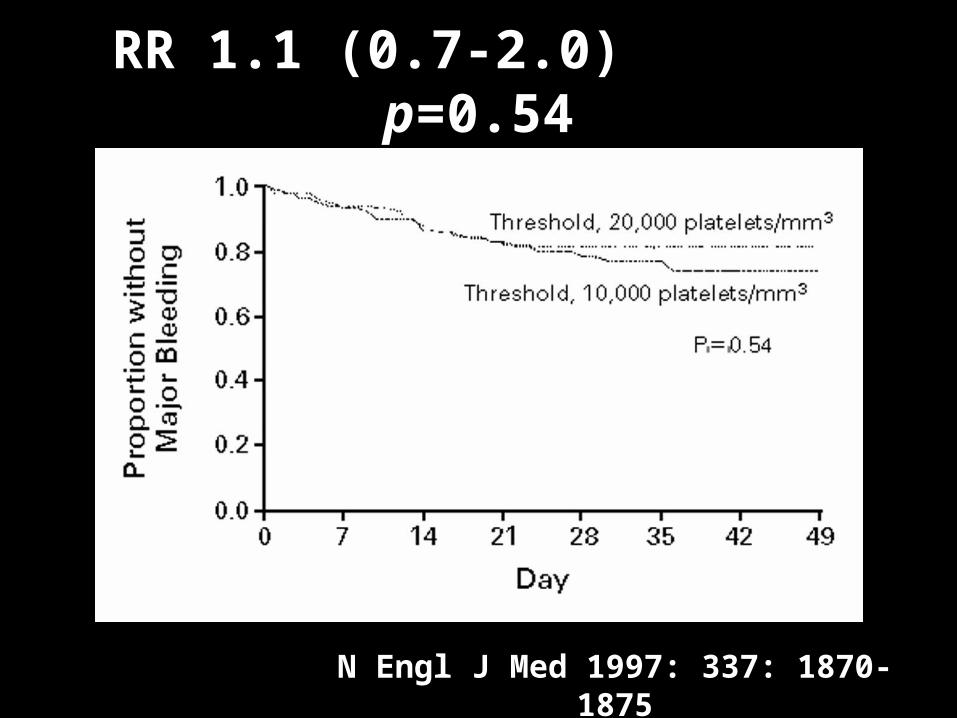
RR 1.1 (0.7-2.0) p=0.54
N Engl J Med 1997: 337: 1870-1875

¿quienes tienen más riesgo?
FACTORES DE SAGRADO OR (IC95%)
Uremia 1.64 (1.4-1.92)
Hipo-albuminemia 1.54 (1.33-1.79)
TMO reciente (<100días) 1.32 (1.22-1.43)
Reciente sangrado 6.72 (5.53-8.18)
Leucopenia 0.7 (0.6-0.79)
Transfusion Med Rev 2002: 16: 34-45
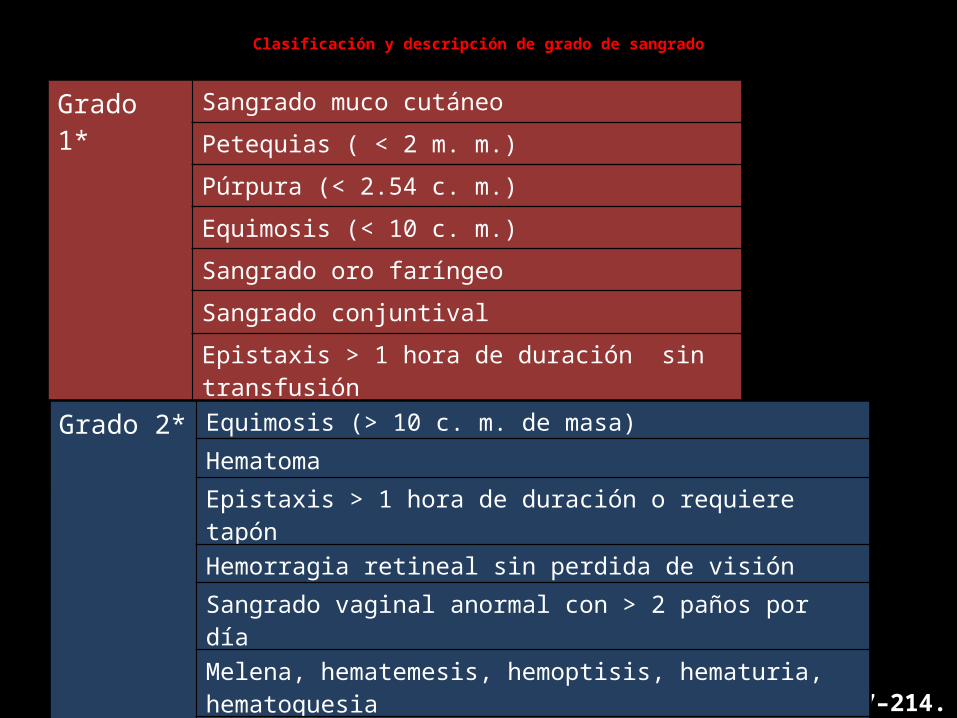
Clasificación y descripción de grado de sangrado
Grado 1* Sangrado muco cutáneoPetequias ( < 2 m. m.)Púrpura (< 2.54 c. m.)Equimosis (< 10 c. m.)Sangrado oro faríngeoSangrado conjuntivalEpistaxis > 1 hora de duración sin transfusiónSangrado vaginal anormales (< 2 paños por día)
* No requiere transfusión WHO Cancer. 1981;47:207–214.
Grado 2* Equimosis (> 10 c. m. de masa)HematomaEpistaxis > 1 hora de duración o requiere tapón
Hemorragia retineal sin perdida de visiónSangrado vaginal anormal con > 2 paños por díaMelena, hematemesis, hemoptisis, hematuria, hematoquesiaSangrado por sitios de puntura o muscular
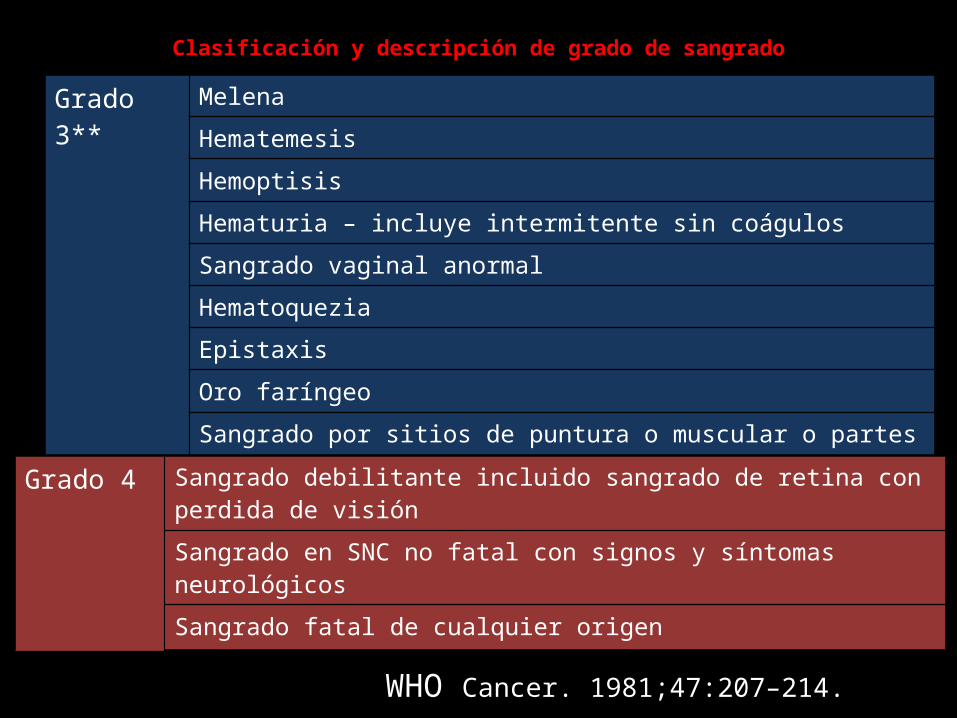
Clasificación y descripción de grado de sangrado
Grado 3** MelenaHematemesisHemoptisisHematuria – incluye intermitente sin coágulosSangrado vaginal anormalHematoqueziaEpistaxisOro faríngeoSangrado por sitios de puntura o muscular o partes blandas
** Requiere transfusión en especial para sangrado de más de 24 horas
Grado 4 Sangrado debilitante incluido sangrado de retina con perdida de visiónSangrado en SNC no fatal con signos y síntomas neurológicosSangrado fatal de cualquier origen
WHO Cancer. 1981;47:207–214.
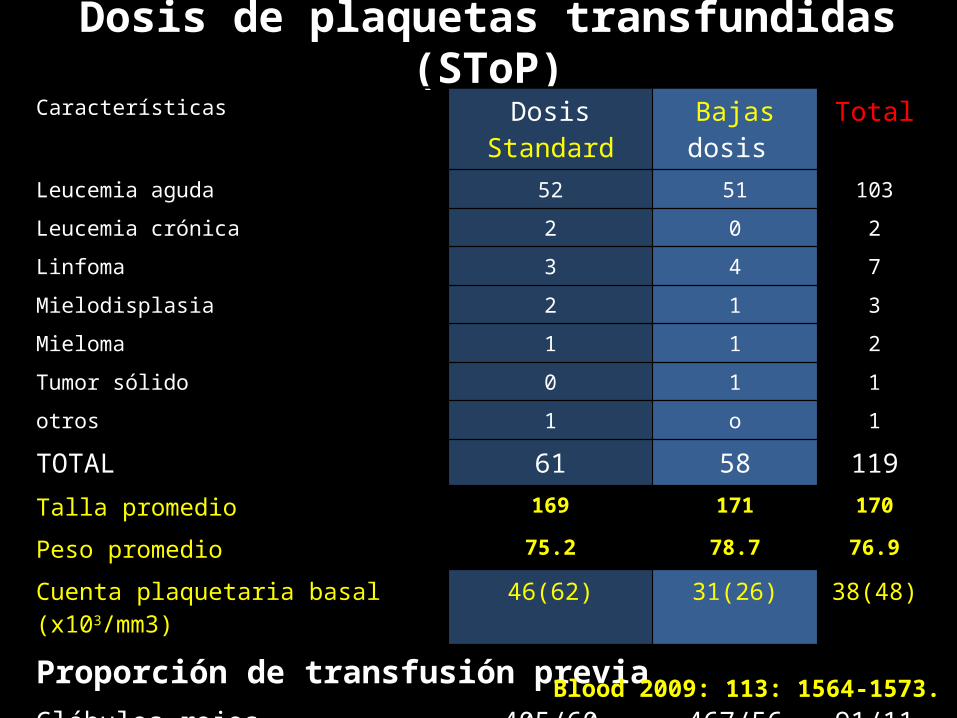
Dosis de plaquetas transfundidas (SToP)Características Dosis Standard Bajas dosis TotalLeucemia aguda 52 51 103Leucemia crónica 2 0 2Linfoma 3 4 7Mielodisplasia 2 1 3Mieloma 1 1 2Tumor sólido 0 1 1otros 1 o 1
TOTAL 61 58 119Talla promedio 169 171 170
Peso promedio 75.2 78.7 76.9
Cuenta plaquetaria basal (x103/mm3) 46(62) 31(26) 38(48)
Proporción de transfusión previaGlóbulos rojos 405/60 467/56 91/116plaquetas 33/61 35/56 68/117
Blood 2009: 113: 1564-1573.
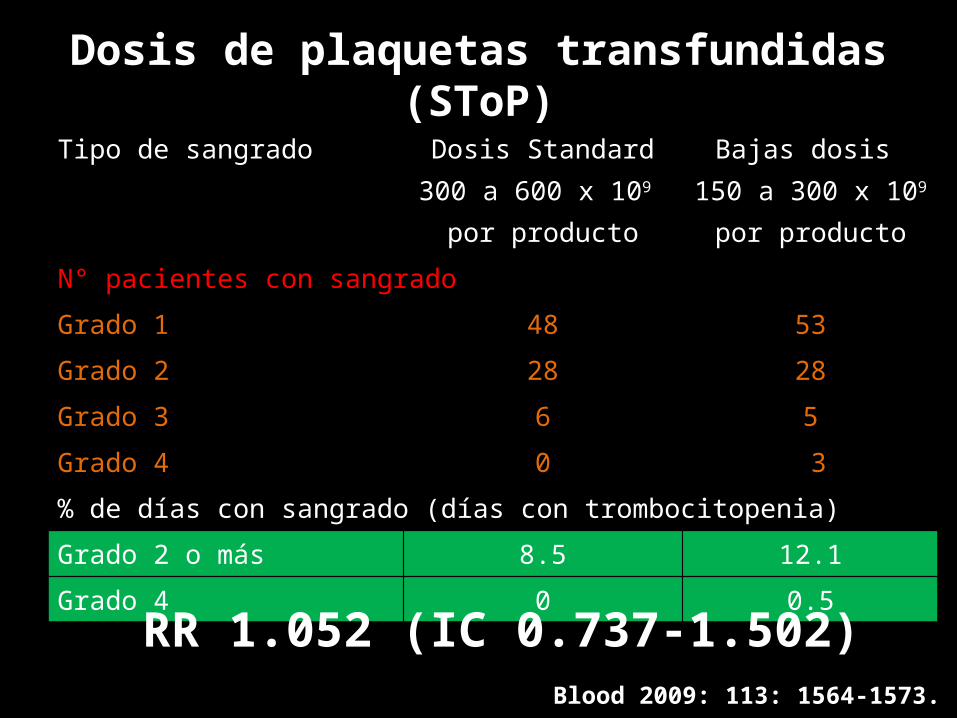
Dosis de plaquetas transfundidas (SToP)Tipo de sangrado Dosis Standard
300 a 600 x 109 por producto
Bajas dosis 150 a 300 x 109
por productoN° pacientes con sangradoGrado 1 48 53Grado 2 28 28Grado 3 6 5Grado 4 0 3% de días con sangrado (días con trombocitopenia)
Grado 2 o más 8.5 12.1Grado 4 0 0.5
Blood 2009: 113: 1564-1573.
RR 1.052 (IC 0.737-1.502)
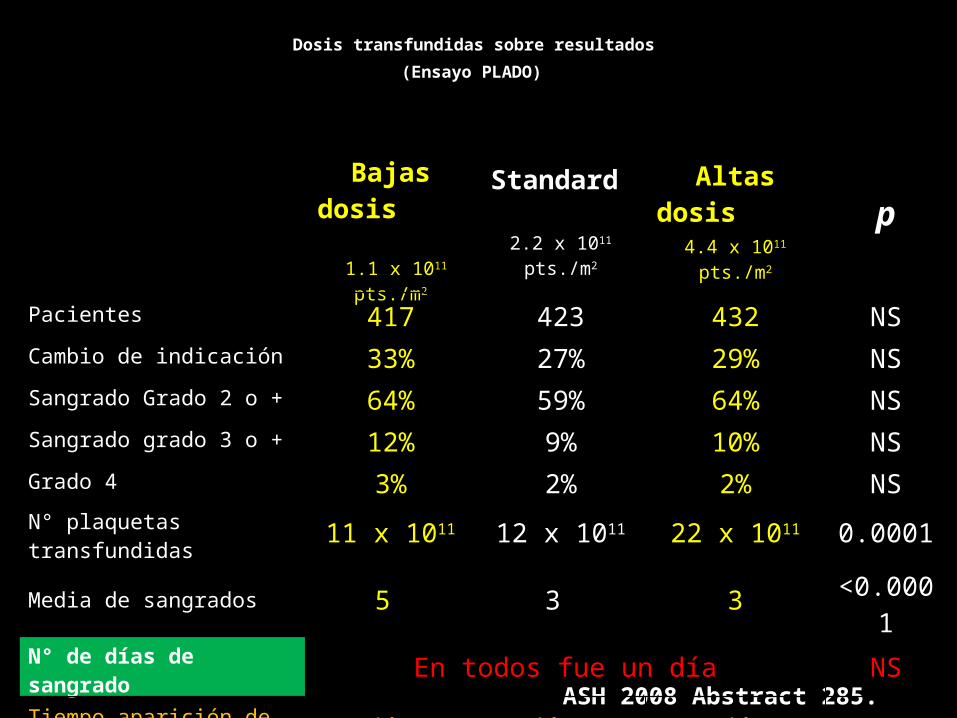
Dosis transfundidas sobre resultados
(Ensayo PLADO)
ASH 2008 Abstract 285.
Bajas dosis
1.1 x 1011 pts./m2
Standard
2.2 x 1011 pts./m2
Altas dosis
4.4 x 1011 pts./m2
p
Pacientes 417 423 432 NSCambio de indicación 33% 27% 29% NSSangrado Grado 2 o + 64% 59% 64% NSSangrado grado 3 o + 12% 9% 10% NSGrado 4 3% 2% 2% NSN° plaquetas transfundidas 11 x 1011 12 x 1011 22 x 1011 0.0001Media de sangrados 5 3 3 <0.0001N° de días de sangrado En todos fue un día NSTiempo aparición de sangrado grado 2 2 días 2 días 3 días NS
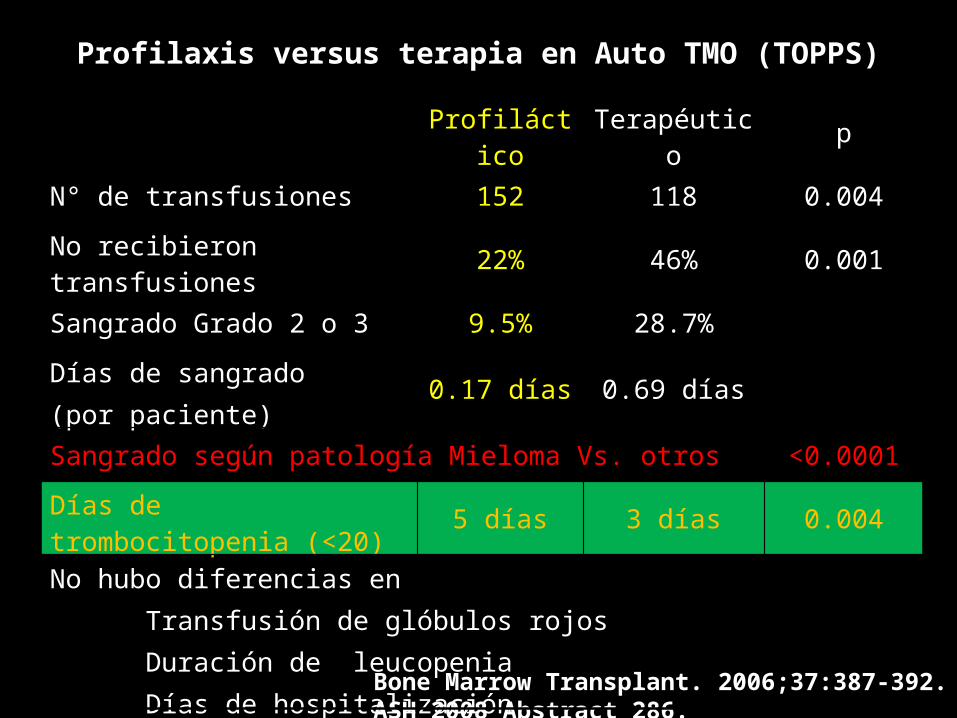
Profilaxis versus terapia en Auto TMO (TOPPS)
Bone Marrow Transplant. 2006;37:387-392.ASH 2008 Abstract 286.
Profiláctico Terapéutico p
N° de transfusiones 152 118 0.004
No recibieron transfusiones 22% 46% 0.001
Sangrado Grado 2 o 3 9.5% 28.7%
Días de sangrado (por paciente)
0.17 días 0.69 días
Sangrado según patología Mieloma Vs. otros <0.0001
Días de trombocitopenia (<20) 5 días 3 días 0.004
No hubo diferencias en Transfusión de glóbulos rojos Duración de leucopenia Días de hospitalización
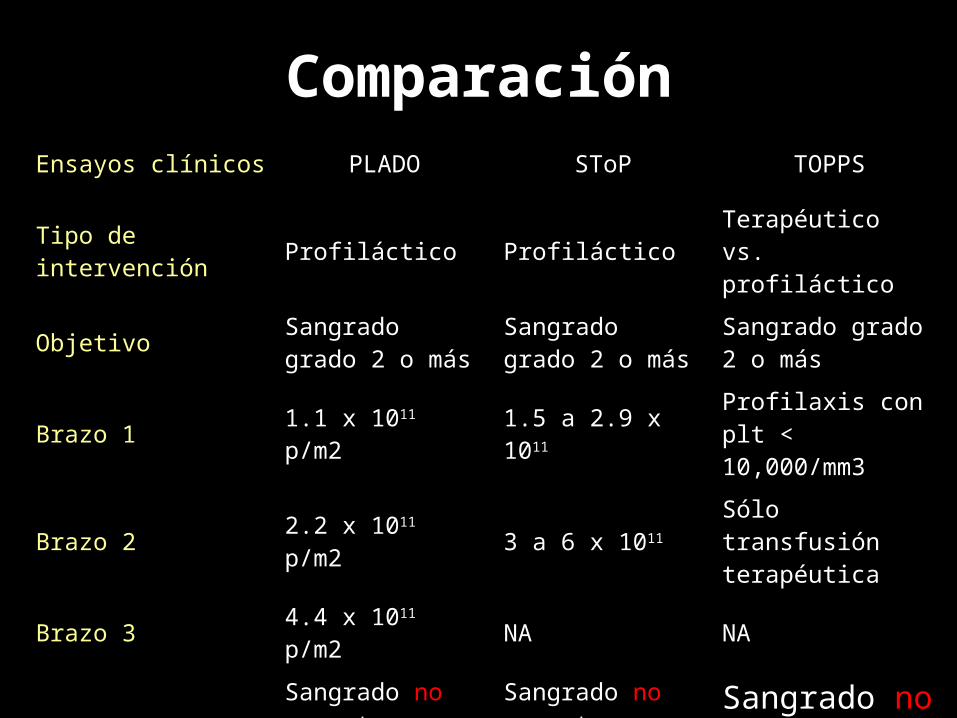
ComparaciónEnsayos clínicos PLADO SToP TOPPS
Tipo de intervención Profiláctico Profiláctico Terapéutico vs. profiláctico
Objetivo Sangrado grado 2 o más
Sangrado grado 2 o más
Sangrado grado 2 o más
Brazo 1 1.1 x 1011 p/m2 1.5 a 2.9 x 1011 Profilaxis con plt < 10,000/mm3
Brazo 2 2.2 x 1011 p/m2 3 a 6 x 1011 Sólo transfusión terapéutica
Brazo 3 4.4 x 1011 p/m2 NA NA
Conclusión
Sangrado no aumenta con menos dosis transfundida
Sangrado no aumento con menos dosis transfundida
Sangrado no aumenta sin profilaxis

Conclusiones
• La trombocitopenia por falla medular:– La profilaxis tiene empleo dudoso pero
aumenta las necesidades trasfusionales.
– La terapia con plaquetas controlan el sangrado significativo.

Algunas Emergencias
• Trombocitopenia.• Hemorragia masiva.• Coagulopatía por trauma.• Hemofilia congénita y adquirida.• Neutropenia febril.• Anemia hemolítica autoinmune.• Purpura trombocitopenica trombótica.• Pancitopenia con CID.
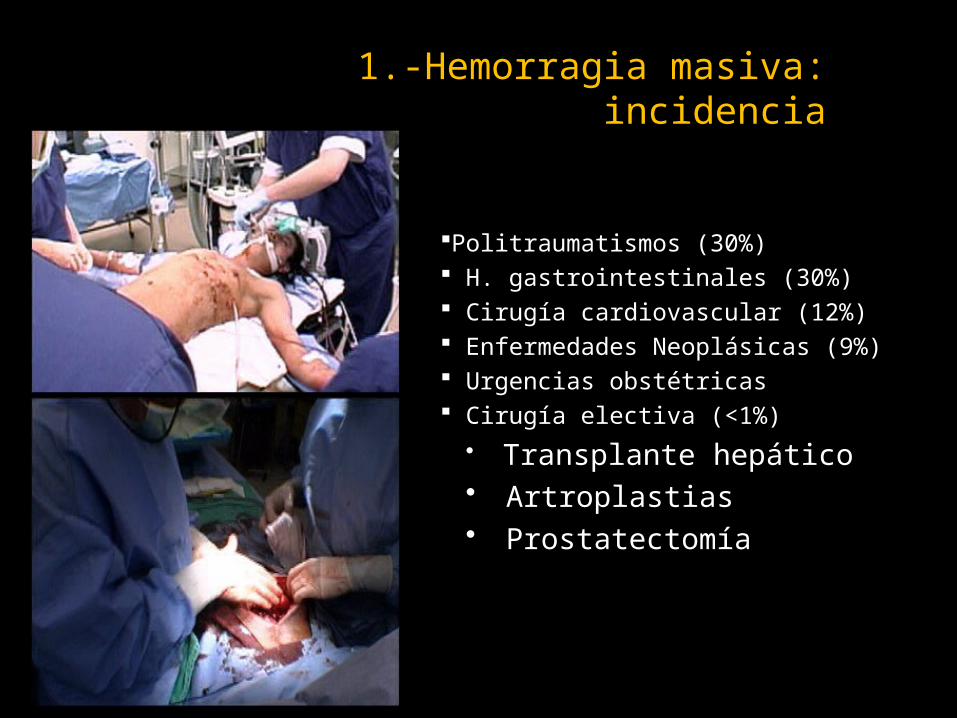
1.-Hemorragia masiva: incidencia
Politraumatismos (30%) H. gastrointestinales (30%) Cirugía cardiovascular (12%) Enfermedades Neoplásicas (9%) Urgencias obstétricas Cirugía electiva (<1%)
• Transplante hepático• Artroplastias• Prostatectomía
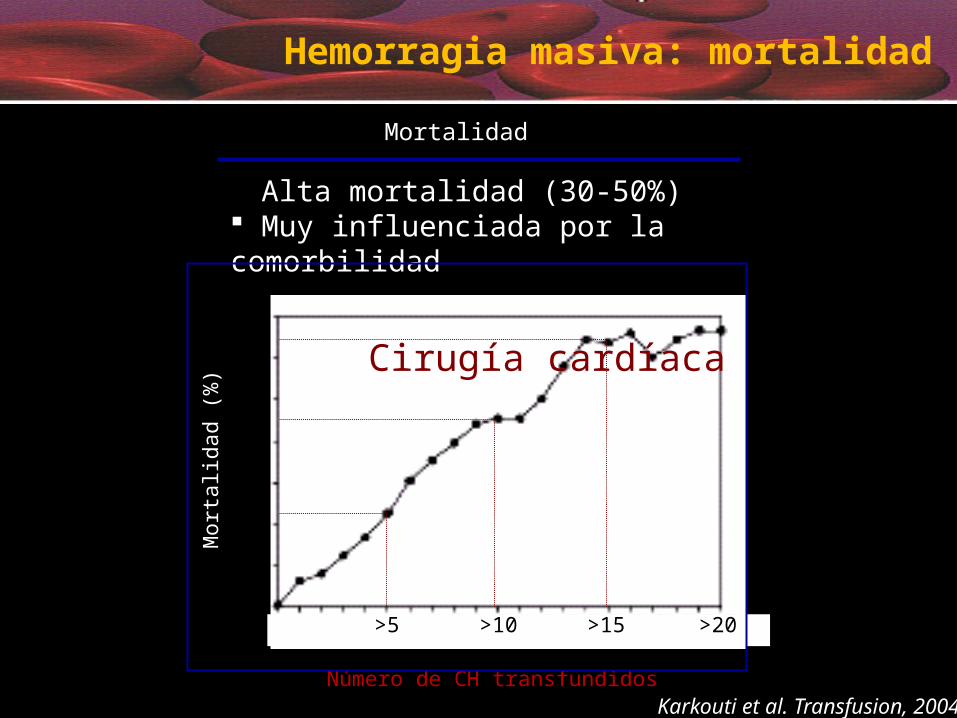
Hemorragia masiva: mortalidad
Mortalidad
Alta mortalidad (30-50%) Muy influenciada por la comorbilidad
35
25
15
>5
5
>10 >15 >20
Número de CH transfundidos
Mor
talid
ad (%
)
Cirugía cardíaca
Karkouti et al. Transfusion, 2004

• ¿Qué es una hemorragia grave y qué es una coagulopatía asociada?
• ¿Qué origen tiene?
• ¿Cómo la diagnosticamos?
• ¿Cómo la tratamos y/o podríamos tratar?
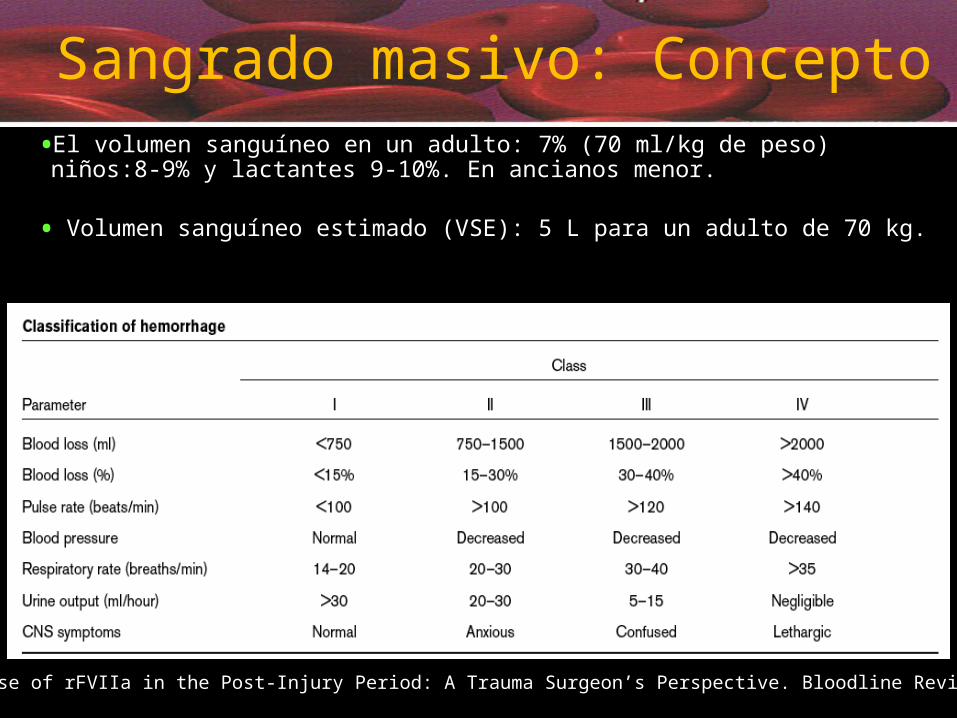
•El volumen sanguíneo en un adulto: 7% (70 ml/kg de peso) niños:8-9% y lactantes 9-10%. En ancianos menor.
• Volumen sanguíneo estimado (VSE): 5 L para un adulto de 70 kg.
Lynn M. Use of rFVIIa in the Post-Injury Period: A Trauma Surgeon’s Perspective. Bloodline Reviews 2001; 1.
Sangrado masivo: Concepto

Hemorragia masiva: definiciones
• La que precisa de la infusión de hemo - componentes en un volumen igual a una o más veces el volumen sanguíneo de un individuo en un tiempo inferior a las 24 horas.
• Transfusion de 4 unidades de CH en una hora con alta probabilidad de necesitar más unidades.
• Reemplazo de un 50% de volumen sanguíneo total en 3 horas.

• ¿Qué es una hemorragia grave y qué es una coagulopatía asociada?
• ¿Qué origen tiene?
• ¿Cómo la diagnosticamos?
• ¿Cómo la tratamos y/o podríamos tratar?
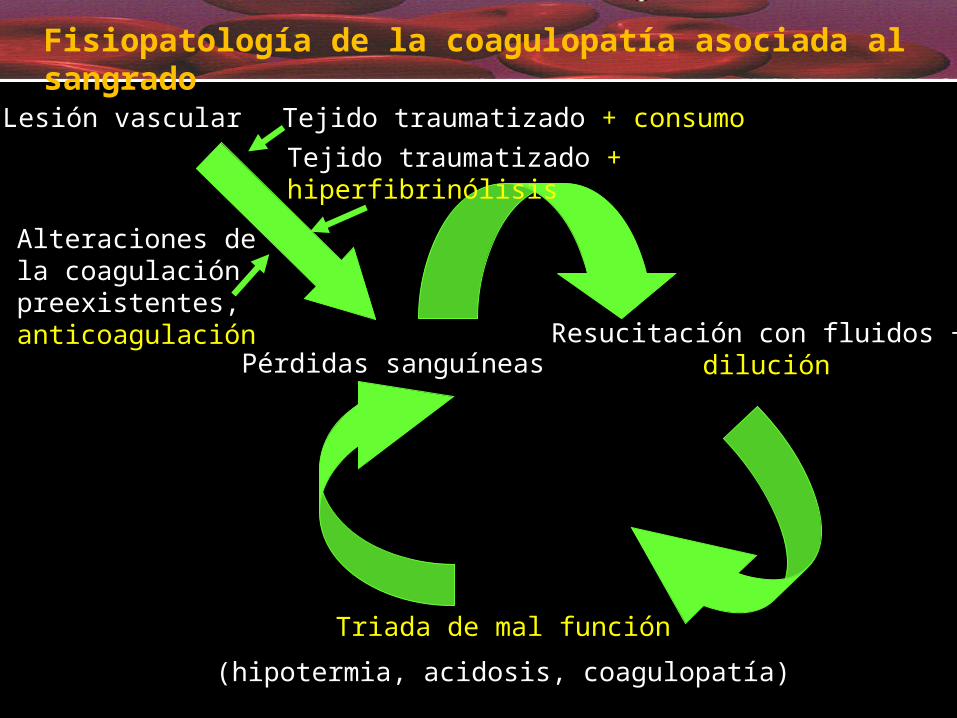
Resucitación con fluidos + diluciónPérdidas sanguíneas
Triada de mal función(hipotermia, acidosis, coagulopatía)
Fisiopatología de la coagulopatía asociada al sangrado
Tejido traumatizado + consumo Tejido traumatizado + hiperfibrinólisis
Lesión vascular
Alteraciones de la coagulación preexistentes, anticoagulación

Coagulación Intravascular Diseminada
•Defecto de la hemostasia sin manifestaciones clínicas
CID biológica •Elevación de D-dímeros (>500 mg/L) más
•1 criterio mayor de consumo de plaquetas o factores
•ó 2 criterios menores
•más hemorragia o isquemiaCID clínica •más sangrado microvascular y/o trombosis
• más compromiso de la funcionalidad orgánica o el pronóstico del paciente
CID complicada •más fallo orgánico simple o múltiple
Clasificación Definición Criterios diagnósticos
Criterios laboratorio Consumo de plaquetas Consumo de factores• Criterio mayores: Plaquetas: <50.000 /mL PT: INR >1.5• Criterios menores: Plaquetas: 50-100.000/mL PT: INR 1.2-1.5
SRLF. Lille, Francia, 2002
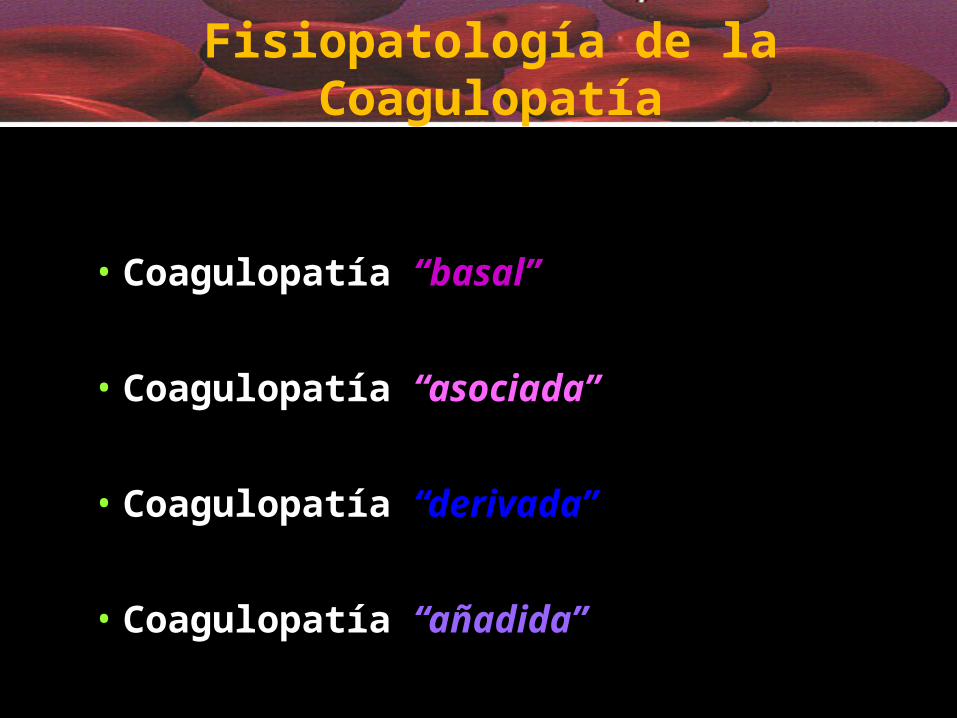
• Coagulopatía “basal”
• Coagulopatía “asociada”
• Coagulopatía “derivada”
• Coagulopatía “añadida”
Fisiopatología de la Coagulopatía

• Exposición de tromboplastina por la rotura vascular, que favorece la activación de la coagulación (coagulopatía de consumo)*
• Pérdida/consumo de factores y plaquetas (inhibición) por sangrado inicial descontrolado*
• Aumento de la actividad fibrinolítica -> hiperfibrinólisis*
Coagulopatía “basal”
*Ungerstedt JS et al. J Neurosurg Anesthesiol 2003; 15:13-18*Lynn M et al. Intensive Care Med 2002; 28 (Suppl 2):S241-247*Schreiber MA et al. J Trauma 2005; 58:475-480
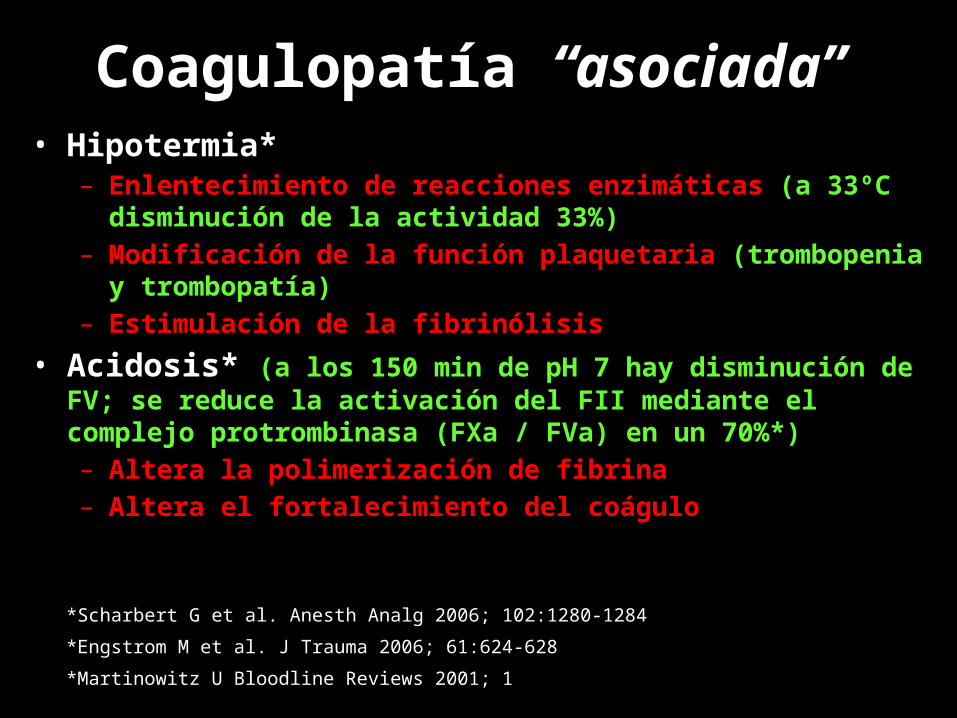
Coagulopatía “asociada”
• Hipotermia*– Enlentecimiento de reacciones enzimáticas (a 33ºC
disminución de la actividad 33%)– Modificación de la función plaquetaria (trombopenia y
trombopatía)– Estimulación de la fibrinólisis
• Acidosis* (a los 150 min de pH 7 hay disminución de FV; se reduce la activación del FII mediante el complejo protrombinasa (FXa / FVa) en un 70%*)– Altera la polimerización de fibrina– Altera el fortalecimiento del coágulo
*Scharbert G et al. Anesth Analg 2006; 102:1280-1284*Engstrom M et al. J Trauma 2006; 61:624-628*Martinowitz U Bloodline Reviews 2001; 1
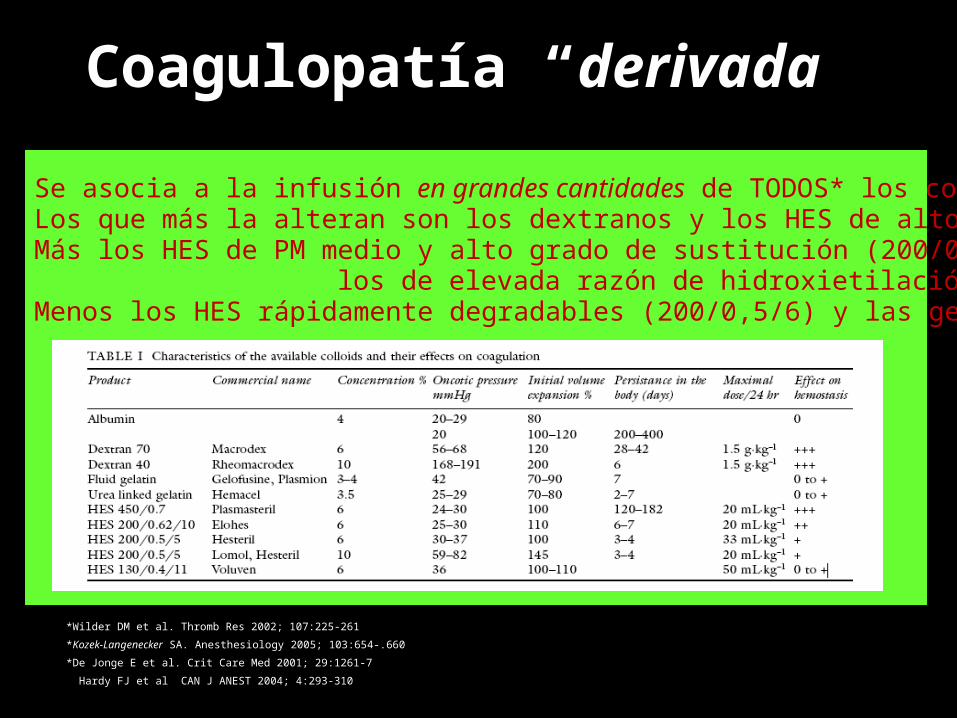
Coagulopatía “derivada”
• Transfusiones masivas (10-12 PRBC)– Hemodilución: Reposición de 1 volemia (35% plaquetas) y de
1,5 (Fbrg 1) – Consumo de factores y activación de la fibrinolisis
• Fluidoterapia– Efectos anticoagulantes y antiplaquetarios directos*– Grado de coagulopatía dilucional dependiente del fluido
utilizado*• Influyen en la función plaquetaria• Inhiben la polimerización de fibrina• Inducen un Sdme de v. Willebrand
Se asocia a la infusión en grandes cantidades de TODOS* los coloides.Los que más la alteran son los dextranos y los HES de alto PMMás los HES de PM medio y alto grado de sustitución (200/0,62) o los de elevada razón de hidroxietilación (200/0,5/13) Menos los HES rápidamente degradables (200/0,5/6) y las gelatinas.
*Wilder DM et al. Thromb Res 2002; 107:225-261*Kozek-Langenecker SA. Anesthesiology 2005; 103:654-.660*De Jonge E et al. Crit Care Med 2001; 29:1261-7 Hardy FJ et al CAN J ANEST 2004; 4:293-310

• Alteraciones preexistentes*– Coagulopatías congénitas– Pacientes anticoagulados – Pacientes antiagregados– Anemización previa
Coagulopatía “añadida”
*DeLoughery TG. Crit Care Clin 2004; 20:13-24

•Controlado •Masivo e incontrolado•Sin retraso •Retraso variable
•Normovolemia•No shock
•Hipovolemia frecuente•Shock frecuente
•Normotermia •Hipotermia frecuente•Continuada•Posibilidad de anticipación
•Tardía•Resultados de las pruebas cuando ya hay coagulopatía
•Por dilución de factores
•Por coagulación intravascular diseminada
•Corrección de anemia•PFC y CCP según tests de laboratorio/clínica
•Corrección de hipoperfusión, hipotermia y anemia
•PFC y CCP según tests de laboratorio/clínica
Daño tisular
Inicio de la transfusión masiva
Volemia/shock
Temperatura
Monitorización de la hemostasia
Coagulopatía
Tratamiento de la coagulopatía
Cirugía electiva Trauma
Transfusión masiva:diferencias entre cirugía electiva y trauma

• ¿Qué es una hemorragia grave y qué es una coagulopatía asociada?
• ¿Qué origen tiene?
• ¿Cómo la diagnosticamos?
• ¿Cómo la tratamos y/o podríamos tratar?

• Recuento de plaquetas• Tiempo de protrombina (PT)• Tiempo de tromboplastina parcial activada
(aPTT) • Cuantificación del fibrinógeno• Dosificación de factores• Marcadores moleculares del sistema de
coagulación• Marcadores moleculares del sistema
fibrinolítico
Pruebas de rutina

Pruebas de rutina• Limitaciones severas (en rango al ingreso, tardan en alterarse,..)• No predicen de forma adecuada el sangrado• Patología dinámica (rapidez de variación,…)• Tardanza en los resultados (> 30 min lo que obliga a la
“transfusión empírica”)• Realizados en plasma “limpio”• Realizados a 37º• Díficil reproductibilidad del “modelo celular”• Dificultad en su validación por insuficiente estandarización
(sujetos a problemas metodológicos que incluyen variaciones en en los reactivos, entre los laboratorios e investigadores)
Dificultad en reproducir el “escenario sangrante”Detectan anormalidades “groseras” pero no las identifican
aPTT > 1,8 ó INR > 1,5-1,8 aumenta la mortalidad 35%TP es el marcador más sensible (cuando esta alterado)
Las plaquetas se muestran como predictores independientes

• No hay un método para medirla• El tiempo de sangrado no predice
el sangrado• Tests de 2ª línea• Analizadores de la función
plaquetaria• PFA-100 (Data)• Agregagometros de impedancia
Pruebas de función plaquetaria

• Tromboelastografía (TEG)
• Tromboelastometría de rotación (ROTEM)
Pruebas a la cabecera del enfermo
• Pruebas de viscosidad hechas en sangre completa
• Determinación de la viscoelasticidad de la sangre “de verdad” (no anticoagulada o coagulada con citrato)
• El patrón de modificaciones de la viscosidad que determinan refleja la cinética de todos los estadios de formación del trombo
• Tiempo de coagulación• Tiempo de formación del coágulo• Tiempo de la estabilización y firmeza del coágulo• Tiempo de firmeza máxima del coágulo • Tiempo de disolución del coágulo Permite el análisis cuantitativo inducido
por la hipotermia y acidosis.*valor añadido al déficit de fibrinógeno
Pruebas a la cabecera del enfermo

• Al ingreso (basal)• Cuando ocurra un sangrado relevante o
hemostasia quirúrgica incorrecta• Tras cada aporte de hemoderivados• Tras cada tratamiento procoagulante• Si se detecta una hipercoagulabilidad
postquirúrgica
Monitorización de la coagulación¿cuándo?
• ¿Qué es una hemorragia grave y qué es una coagulopatía asociada?
• ¿Qué origen tiene?
• ¿Cómo la diagnosticamos?
• ¿Cómo la tratamos y/o podríamos tratar?
Un paciente traumatizado “resucitado”es aquel en el que se ha controlado el
sangrado
“Trauma Rules”-BMJ Publications, UK (Hodgets, Ed), 1997

Hemorragia masiva: tratamiento
Reposición de la volemia Cristaloides y coloides
Optimización de la oxigenación tisular Reposición de hematíes ¿TAO?
Corrección de la hemostasia PFC y/o CCP Concentrado de plaquetas Fármacos hemostáticos
Los tres pilares básicos del tratamiento
Objetivos del tratamiento

• Si se mantiene, se tolera hasta 70% de pérdida
• Inicio precoz para asegurar aceptable perfusión vital (PAS 90 mmHg) -> mantiene la vasoconstricción compensatoria inicial, evita la desestabilización de los trombos y disminuye el riesgo de coagulopatía dilucional
• Coloides + cristaloides• Prevención de la hipotermia
Objetivos del tratamiento Reemplazo del volumen sanguíneo intravascular

• Disponer de una adecuada hemoglobina circulante transportadora de oxígeno (Hb >9 g/dl)
• Mantenimiento del gasto cardiaco (uso de inotrópicos)
• Favorecer la liberación del oxígeno y su captación por la célula (previniendo y tratando la acidosis metabólica)
Objetivos del tratamiento Mantenimiento de la oxigenación tisular

• Control del sangrado difuso por coagulopatía• Coagulopatía de consumo• Fibrinólisis excesiva• Coagulopatía dilucional• Disfunción plaquetaria• Hipotermia• Síndrome Politransfundido• Cambios metabólicos
• Control del sangrado de vasos • Quirúrgico• Endoscópico• Arterográfico
Objetivos del tratamiento Control de la hemostasia

Tratamiento “normalizado” • Medidas Coadyuvantes
• Corrección del equilibrio ácido-base• Obtención de la normotermia
• Medidas Quirúrgicas• Técnicas angiográficas de embolización• Cirugía/Cirugía de “control de daños”• Pegamentos biológicos
• Tratamiento sustitutivo• Concentrados de hematíes, plaquetas, PFC• Concentrados de fibrinógeno, crioprecipitados
control de la hemorragia y
de las alteraciones de la coagulación

Hemorragia masiva: Guía Europea Consenso
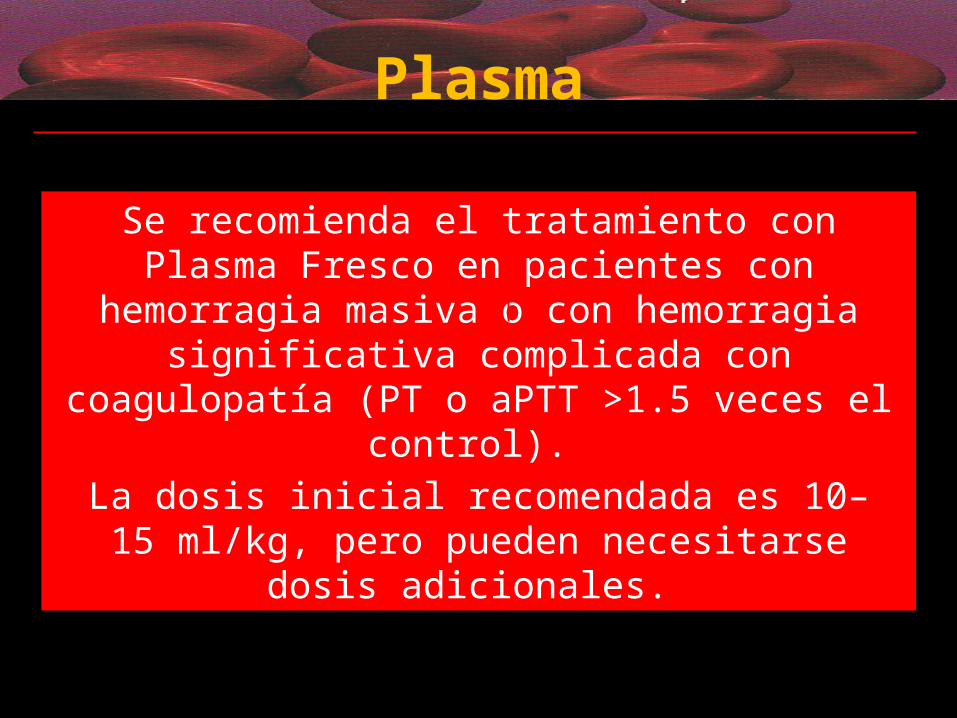
Plasma
Grado 1C
Se recomienda el tratamiento con Plasma Fresco en pacientes con hemorragia masiva o con hemorragia significativa complicada con coagulopatía (PT o aPTT
>1.5 veces el control). La dosis inicial recomendada es 10–15 ml/kg, pero
pueden necesitarse dosis adicionales.
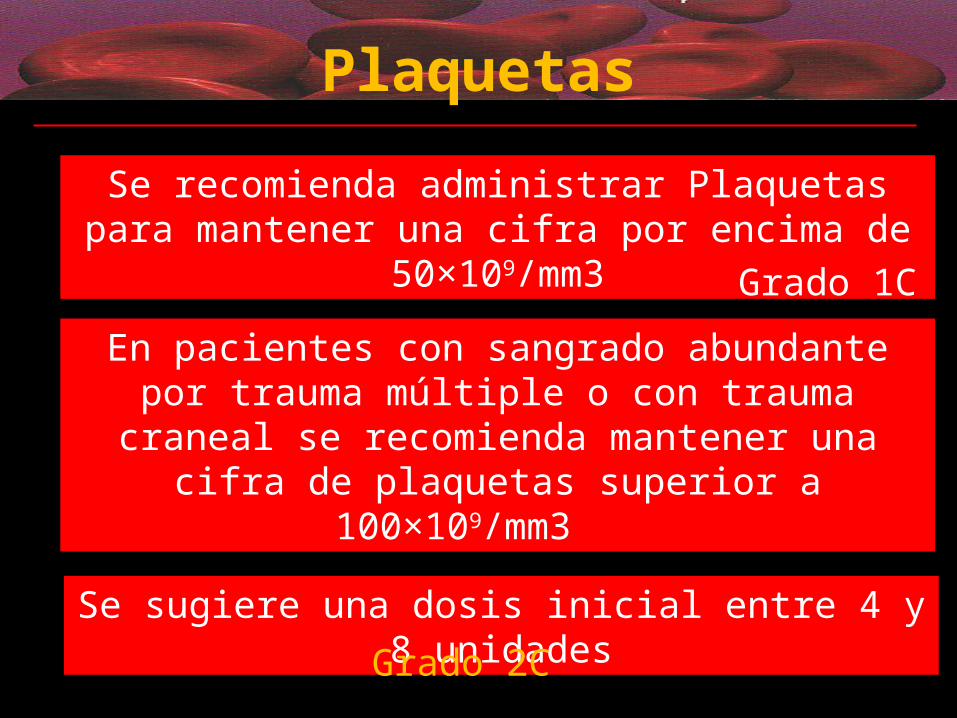
Plaquetas
Se sugiere una dosis inicial entre 4 y 8 unidades
Grado 2C
En pacientes con sangrado abundante por trauma múltiple o con trauma craneal se recomienda mantener
una cifra de plaquetas superior a 100×109/mm3
Se recomienda administrar Plaquetas para mantener una cifra por encima de 50×109/mm3
Grado 2C
Grado 1C

Fibrinógeno
Grado 1C
Se recomienda el tratamiento con concentrado de fibrinógeno o crioprecipitados en casos de hemorragia acompañada de niveles de fibrinógeno inferior a 1 g/l.
Se sugiere una dosis inicial de fibrinógeno de 3-4 gr o 50 mg/kg de crioprecipitados, equivalente a 15-20 unidades
en un adulto de 70 kg. La repetición de la dosis debe realizarse guiada por la valoración de los niveles posteriores de fibrinógeno.

Antifibrinolíticos
Grado 1C
Se sugiere que los agentes antifibrinolíticos sean considerados en el tratamiento del paciente traumatizados
sangrante. Las dosis sugeridas son: acido tranexámico 10–15 mg/kg seguidas de una perfusión
de 1–5 mg/kg/h;aprotinina 2 mill KIU seguidas inmediatamente por 500,000
KIU/h. Tratamiento antifibrinolítico debe suspenderse una vez que
la hemorragia ha sido controlada.
* A large body of evidence for the use of antifibrinolytic agents for the management of bleeding in elective surgery and cardiac surgery patients exists. For the purpose of these guidelines, we have assumed that these effects are transferable to trauma patients, and our recommendation is based upon this unproven assumption.

Factor VII activado
Grado 2C
Se sugiere que el uso de rFVIIa sea considerado en pacientes con trauma cerrado en los que persiste el
sangrado a pesar de los intentos estandard para controlar la hemorragia y el uso adecuado de hemoderivados.
La dosis inicial sugerida es de 200 µg/kg seguidos por dos dosis de 100 µg/kg administrados 1 y 3 horas despues de la
primera dosis.

Objetivos del tratamiento
• Corrección del sangrado• Prevenir el sangrado después de
procedimientos invasivos• No hay que optimizar los parámetros de
coagulación en ausencia de sangrado• Minimizar la transfusión indiscriminada• Minimizar la intervención hemostática
empírica
Naturaleza multifactorial de las alteraciones de la coagulación en la hemorragia grave
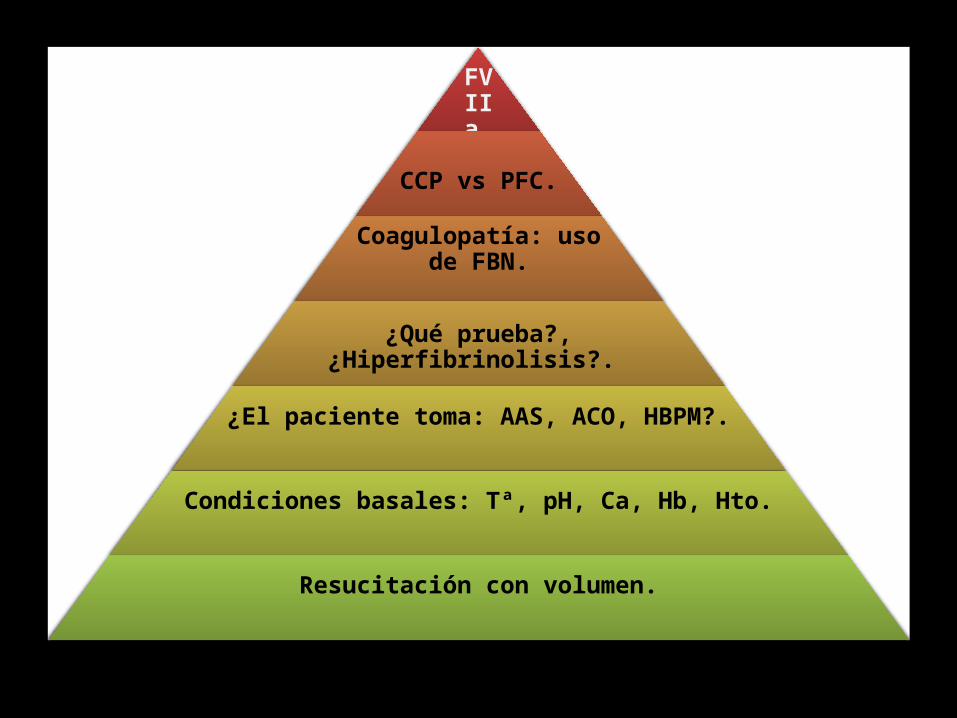
FVIIa.
CCP vs PFC.
Coagulopatía: uso de FBN.
¿Qué prueba?, ¿Hiperfibrinolisis?.
¿El paciente toma: AAS, ACO, HBPM?.
Condiciones basales: Tª, pH, Ca, Hb, Hto.
Resucitación con volumen.

INDICACIÓN DE TRANSFUSIÓN
Búsqueda de nuevos tratamientos
Guías Clínicas Equipo Trabajo
Control de calidad Sentido común

Algunas Emergencias
• Trombocitopenia.• Hemorragia masiva.• Coagulopatía por trauma.• Hemofilia congénita y adquirida.• Neutropenia febril.• Anemia hemolítica autoinmune.• Purpura trombocitopenica trombótica.• Pancitopenia con CID.

Coagulopatía del trauma
• Perdida de sangre con dilución.National Academy of Sciences: 1973:39-40.
J Trauma 2003;55:886-891.
Blood 1999; 94:199-207
J Clin Invest. 1997;100:2276-2285
Ann Surg. 2007;245:812-818
Ann Surg. 1969;169:455-482.
Contribución de la Guerra de Vietnam.
Consumo de factores de coagulación y plaquetas.
Disfunción plaquetaria por hipotermia.
Reducción de actividad de factores de coagulación por acidosis por hipoperfusión.
Activación de fibrinolisis.
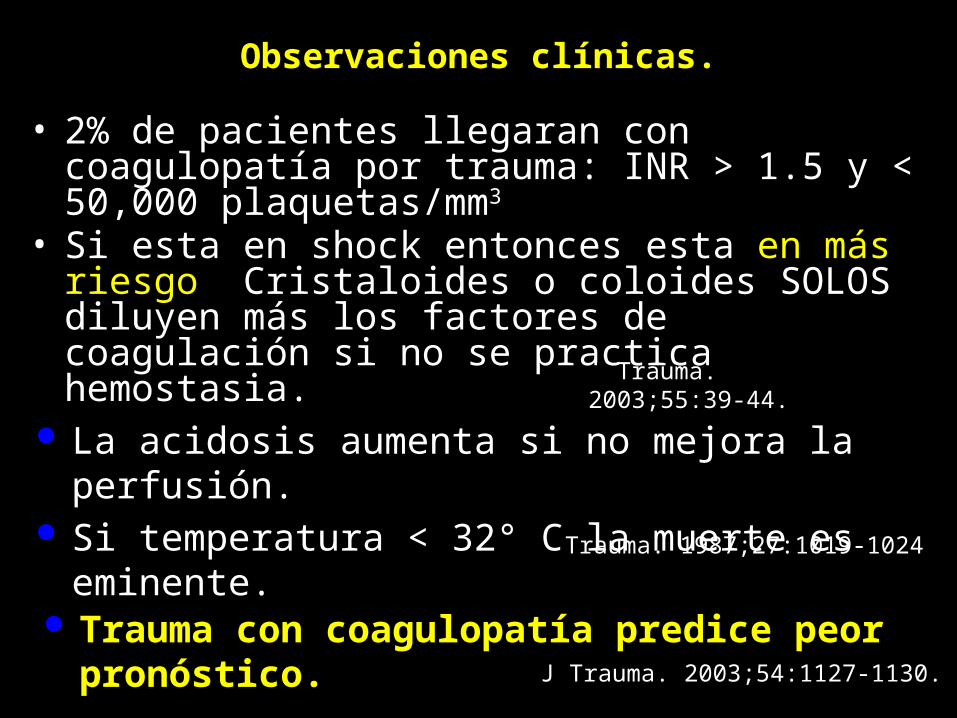
Observaciones clínicas.
• 2% de pacientes llegaran con coagulopatía por trauma: INR > 1.5 y < 50,000 plaquetas/mm3
• Si esta en shock entonces esta en más riesgo. Cristaloides o coloides SOLOS diluyen más los factores de coagulación si no se practica hemostasia.
J Trauma. 2003;55:39-44.
J Trauma. 1987;27:1019-1024
Trauma con coagulopatía predice peor pronóstico. J Trauma. 2003;54:1127-1130.
La acidosis aumenta si no mejora la perfusión. Si temperatura < 32° C la muerte es eminente.

7 Estrategias para control.
• Mejorar la perfusión con mejora de la PAM.
• Vendajes hemostáticos.
• Empleo de Fibrinolíticos.
• Empleo de sangre fresca.
• Empleo de Factor rVIIa o CCP.
• Disponer plasma AB masivamente.
• Combinación de los anteriores.
N Engl J Med. 1994;331:1105-1109.
Crit Care. 2004;8:S57-S60.
Cochrane Database Syst Rev. 2011:CD004896.
J Trauma. 2006;61:181-184
J Trauma. 2004;57:709-719
J Trauma. 2007;62:307-310
Transfusion. 2006;46:685-686
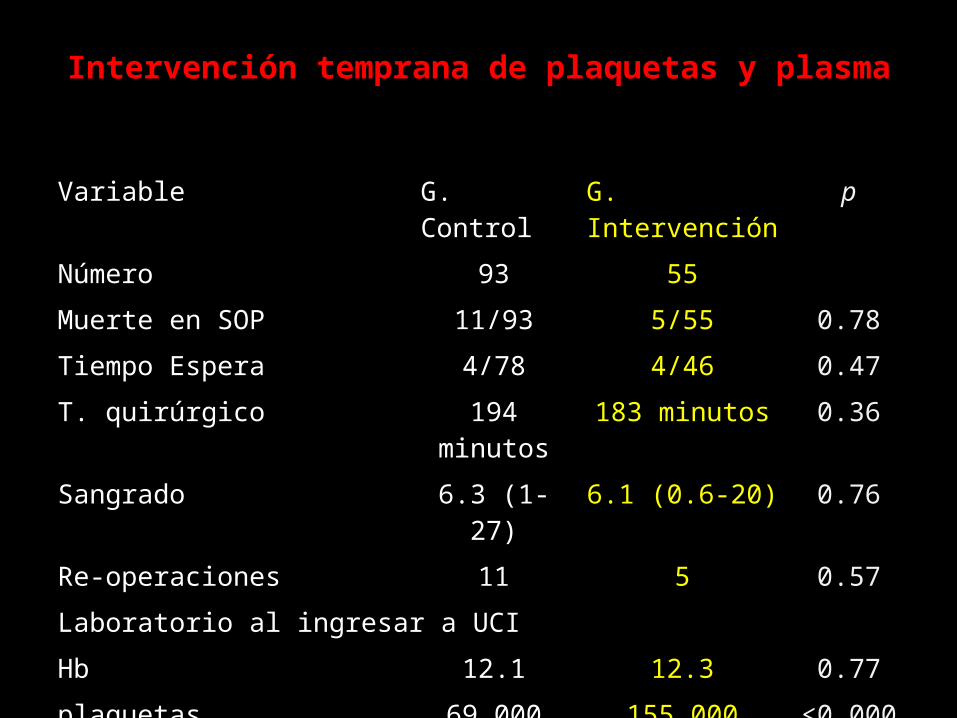
Intervención temprana de plaquetas y plasma
Variable G. Control G. Intervención pNúmero 93 55Muerte en SOP 11/93 5/55 0.78Tiempo Espera 4/78 4/46 0.47T. quirúrgico 194 minutos 183 minutos 0.36Sangrado 6.3 (1-27) 6.1 (0.6-20) 0.76Re-operaciones 11 5 0.57Laboratorio al ingresar a UCIHb 12.1 12.3 0.77plaquetas 69,000 155,000 <0.0001TPT 44” 39” >0.001Creatinina 1.7 1.3 0.99
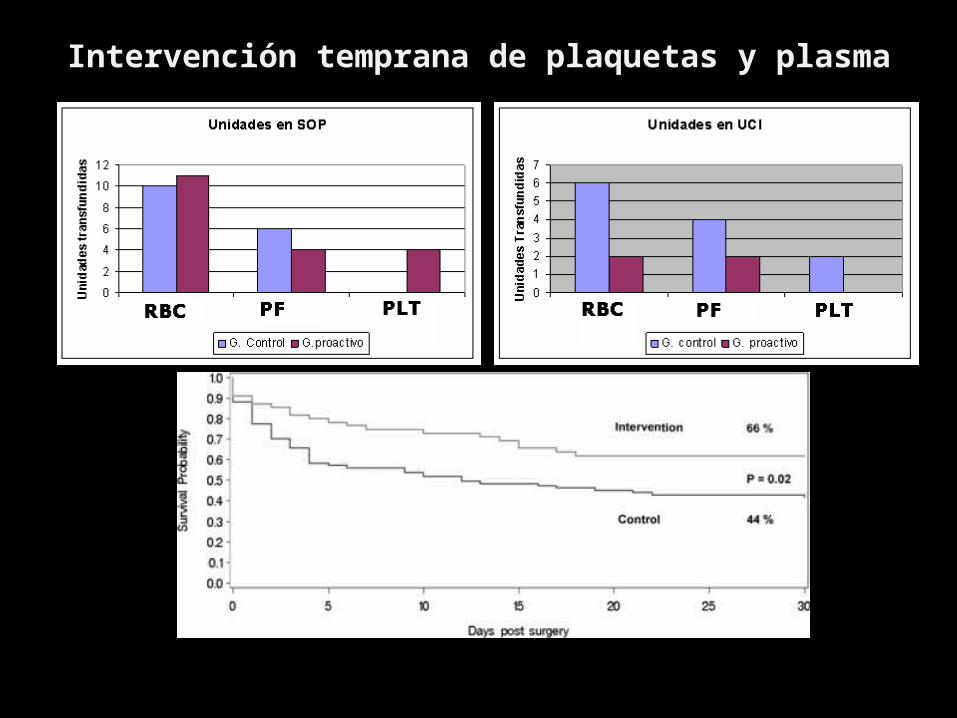
TRANSFUSION 2007;47:593-598
Intervención temprana de plaquetas y plasma
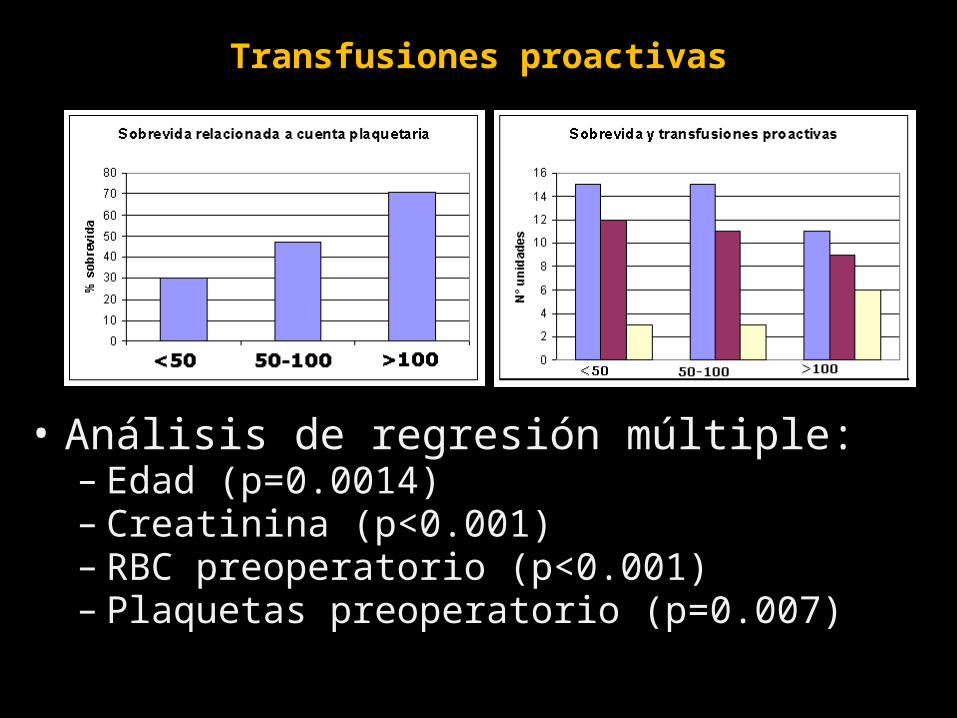
Transfusiones proactivas
• Análisis de regresión múltiple:– Edad (p=0.0014)– Creatinina (p<0.001)– RBC preoperatorio (p<0.001)– Plaquetas preoperatorio (p=0.007)
TRANSFUSION 2007;47:593-598

Conclusión
• Coagulopatía de Trauma esta asociado a traumas severos.
• Esta asociado a alta mortalidad.• Su diagnóstico es simple.• Su diagnóstico temprano impide emplear cristaloides
y usar hemoderivados.• Requiere mejorar abastecimiento de sangre.• Requiere mejorar control de hemorragia.

Tratamiento “de rescate”
• “Clásicos revisados” o Ácido tranexámico ( ®Transamin)o Ácido épsilon amino caproico (®Caproamin)o Desmopresina (®DDAVP)
• “Nuevas indicaciones”o Complejo Protrombínico (®Octaplex, ,®Beriplex, ®Protromplex)o Factor rVIIa (®Novoseven)o Concentrado de FXIII (®Fibrammin NH)o Fibrinógeno (®Haemocompletan)
Agentes prohemostáticosSustancias capaces de promover la hemostasis por formación de
fibrina o por bloqueo de la actividad fibrinolítica
24

Reversión de sobredosis de AVK
• Plasma fresco solo debe emplearse en caso de sangrado con INR > 9
Chest 2008;133: 123S–131S.¿quién es superior?
Plasma fresco congelado.Concentrado de complejo protrombínico.Factor VII activado recombinante.
Rapidez de reversión vs. Riesgo de trombosis.29

Dosis de plasma fresco• 121 pacientes con TP entre 13.1 y 17 segundos fueron
tratados con dos dosis. 12 ml/kilo y 33 ml/kilo. • Los pacientes que recibieron dosis bajas no alcanza a
corregir TP.
Plasma tiene riesgos: Enfermedades hemotrasmisibles. sobrecarga de líquidos Reacciones alérgicas Reacciones inmunológicas.
Profilaxis versus riesgos30
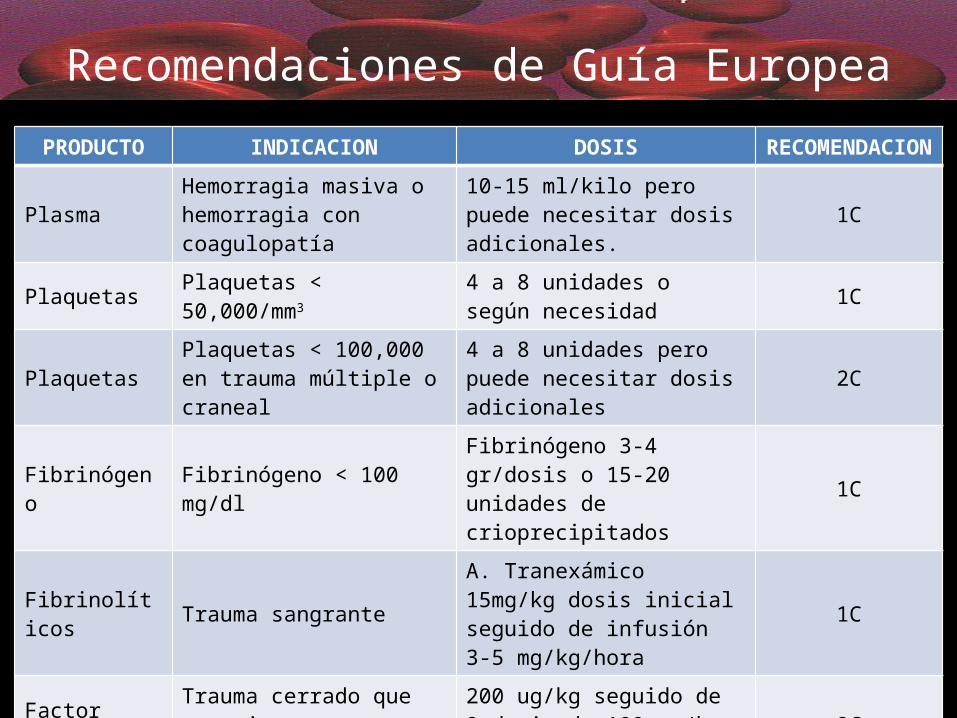
Recomendaciones de Guía EuropeaPRODUCTO INDICACION DOSIS RECOMENDACION
Plasma Hemorragia masiva o hemorragia con coagulopatía
10-15 ml/kilo pero puede necesitar dosis adicionales. 1C
Plaquetas Plaquetas < 50,000/mm3 4 a 8 unidades o según necesidad 1C
Plaquetas Plaquetas < 100,000 en trauma múltiple o craneal
4 a 8 unidades pero puede necesitar dosis adicionales 2C
Fibrinógeno Fibrinógeno < 100 mg/dl Fibrinógeno 3-4 gr/dosis o 15-20 unidades de crioprecipitados 1C
Fibrinolíticos Trauma sangranteA. Tranexámico 15mg/kg dosis inicial seguido de infusión 3-5 mg/kg/hora
1C
Factor rVIIa Trauma cerrado que no mejora con hemoderivados
200 ug/kg seguido de 2 dosis de 100 ug/kg 1 a 3 horas después 2C
Complejo Protrombínico
Revertir efecto de anticoagulantes
INR>5 = 30 UI/kgINR<5 = 15 UI/kg 1C
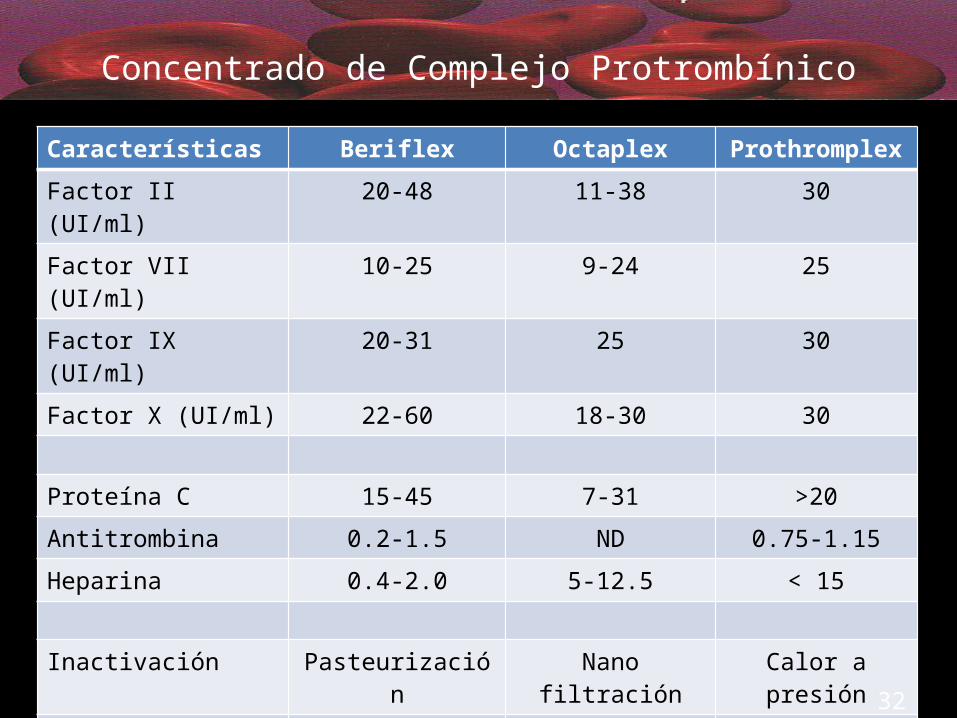
Concentrado de Complejo ProtrombínicoCaracterísticas Beriflex Octaplex Prothromplex
Factor II (UI/ml) 20-48 11-38 30
Factor VII (UI/ml) 10-25 9-24 25
Factor IX (UI/ml) 20-31 25 30
Factor X (UI/ml) 22-60 18-30 30
Proteína C 15-45 7-31 >20Antitrombina 0.2-1.5 ND 0.75-1.15Heparina 0.4-2.0 5-12.5 < 15
Inactivación Pasteurización Nano filtración Calor a presión
Velocidad de Infusión 8.4 ml/min 3 ml/min 1 ml/min
Conservación < 25º C < 25º C +2 a +8º C
Validez 3 años 2 años 3 años
32

Ensayos clínicos con Octaplex
Ensayos Clínicos
Número de pacientes
Razones de enrolamiento
Respuesta Clínica
Posibles efectos adversos
LEX-201 10 Deficiencia F VII= 4Deficiencia F IX = 6
96% excelente4% bueno 2 cefalea
LEX-202Lubetsky 2004 20 Sangrado mayor en AO
Cirugía o P. invasivo en AO 85% bueno15% moderado
Seroconversión parvoB19Anormal función hepática
LEX-203Riess 2007 60 Sangrado mayor por AO
Cirugía o P. invasivo en AOINR<1.4 en 93%Excelente control en 100% de casos
Seroconversión parvoB19Reacción en sitio de inyecciónHipertensión arterial
LEX-204Franken 2007 101
Deficiencia adquiridaSangrado agudoProfilaxis o sobredosis
84% muy buena15% moderada1% insuficiente
Ninguna
33
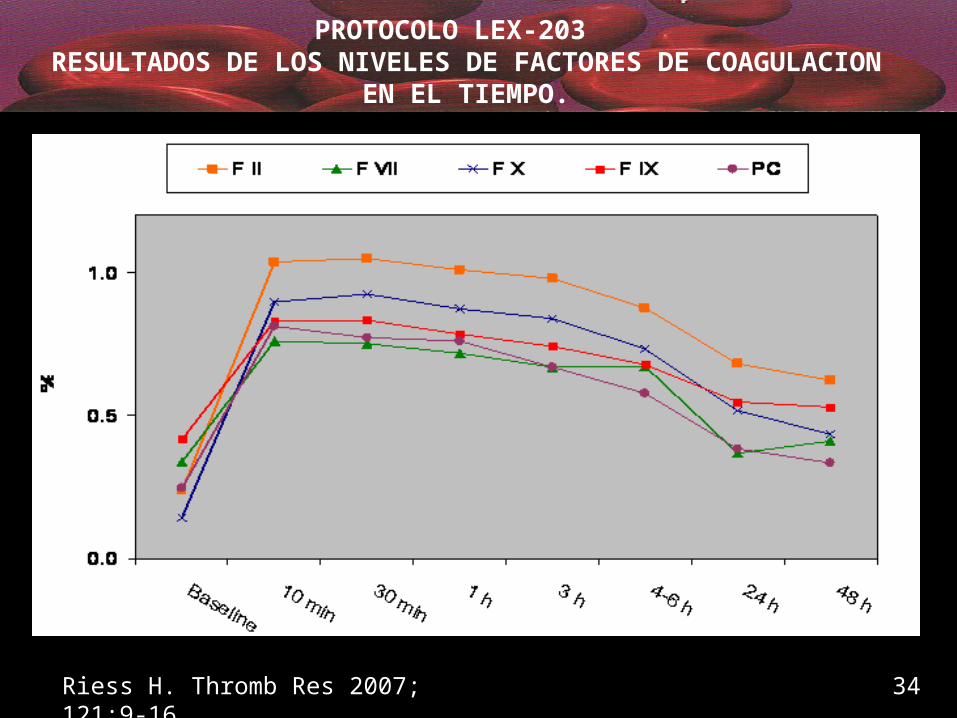
PROTOCOLO LEX-203 RESULTADOS DE LOS NIVELES DE FACTORES DE
COAGULACION EN EL TIEMPO.
Riess H. Thromb Res 2007; 121:9-16 34
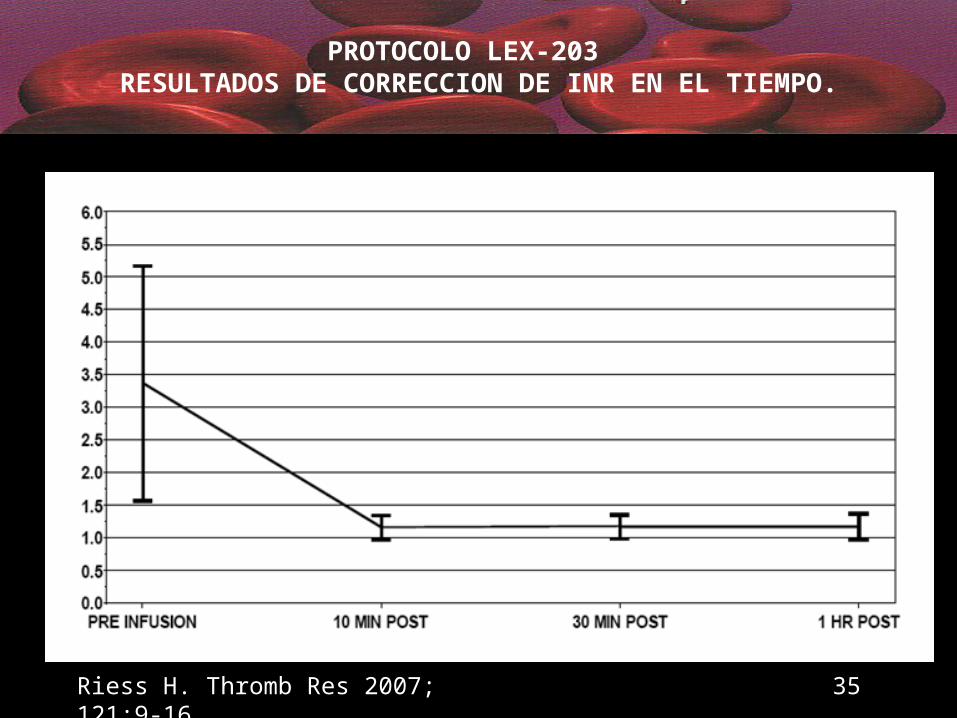
PROTOCOLO LEX-203 RESULTADOS DE CORRECCION DE INR EN EL TIEMPO.
Riess H. Thromb Res 2007; 121:9-16 35

USO DE COMPLEJO PROTROMBINICO
”Para los pacientes con sangrado en peligro de su vida o hemorragia intracraneal, se recomienda el uso de concentrados de complejo protrombínico o factor VIIa recombinante para anular inmediatamente el INR alterado”
Ansell J et al., The pharmacology and management of the vitamin K antagonists: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy, Chest 2004; 126:204S- 233S
36
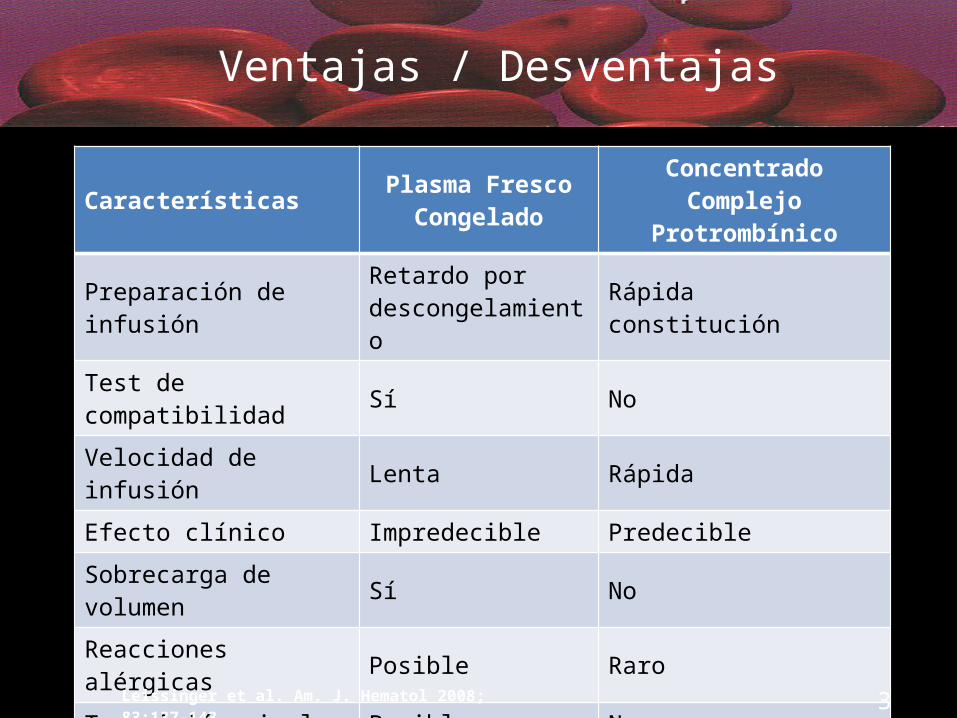
Ventajas / Desventajas
Características Plasma Fresco Congelado
Concentrado Complejo Protrombínico
Preparación de infusión Retardo por descongelamiento Rápida constitución
Test de compatibilidad Sí No
Velocidad de infusión Lenta Rápida
Efecto clínico Impredecible Predecible
Sobrecarga de volumen Sí No
Reacciones alérgicas Posible Raro
Trasmisión viral Posible No
Trombosis No 1.5% (primera generación)
Leissinger et al. Am. J. Hematol 2008; 83:137–143 37

Dosis y precios
Producto Dosis Supuesto Unidades Costos
Plasma fresco congelado 15 ml/kg Peso 70 kg
1050 ml 5 unidades 425 euros
Complejo protrombínico
INR>5=30UI/kgINR<5=15 UI/kg
Peso 70 kgINR<5 2 viales 420 euros
38

INDICACIONES DE USO DE CONCENTRADO PROTROMBINICO.
DEFICIENCIA ADQUIRIDA DE FACTORES DEL COMPLEJO DE PROTROMBINA:
– RAPIDA REVERSION DE LA TERAPIA ANTICOAGULANTE ORAL:• CIRUGIA EN PACIENTES ANTICOAGULADOS• SOBREDOSIS DE ANTICOAGULANTES
– RAPIDA CORRECCION DEL SANGRADO DEBIDO A LA DEFICIENCIA EN FACTORES DE COAGULACIÓN VITAMINA-K DEPENDIENTES:
• FALLA HEPÁTICA SEVERA• CIRUGÍA HEPATICA • TRANSFUSION MASIVA CON COAGULOPATIA DILUCIONAL • TRANSPLANTES
DEFICIENCIA HEREDITARIA DE FACTORES DEL COMPLEJO DE PROTROMBINA ?????
39

CONTRAINDICACIONES PARA EL USO DEL COMPLEJO PROTROMBINICO
• COAGULOPATIA DE CONSUMO
– POR SER UN PRODUCTO PROCOAGULANTE– SOLO USARLO SI NO HAY RESERVA DE PFC EN PRESENCIA
DE SANGRADO MASIVO
• TROMBOCITOPENIA INDUCIDA POR HEPARINA
– POR CONTENER HEPARINA PUEDE EXCERBAR EL FENOMENO TROMBOTICO
– LOS PRODUCTOS MODERNOS NO CONTIENEN HEPARINA
40

OTROS USOS DEL COMPLEJO PROTROMBINICO
• TRATAMIENTO STANDAR DE DEFICIENCIAS HEREDITARIAS DE LOS FACTORES DE LA COAGULACIÓN IX, II, VII Y/O X .
• PACIENTES QUIRURGICOS CON HEMORRAGIA MASIVA QUE PRESENTAN UNA IMPOSIBILIDAD DE ACABAR LA CIRUGÍA POR SANGRADO MASIVO DE CAUSA NO QUIRURGICA.
• PACIENTES PREQUIRURGICOS, CON IMPORTANTES ANORMALIDADES DE LA COAGULACIÓN O ALTO RIESGO DE SANGRADO INTRAOPERATORIO DE DIFICIL CONTROL.
• PACIENTES DE PROGRAMAS DE TRASPLANTE (HEPATICO).41

Algunas Emergencias
• Trombocitopenia.• Hemorragia masiva.• Coagulopatía por trauma.• Hemofilia congénita y adquirida.• Neutropenia febril.• Anemia hemolítica autoinmune.• Purpura trombocitopenica trombótica.• Pancitopenia con CID.

Sangrado en HemofiliaHemofilia severa:– Recidiva de sangrado en
articulaciones :• Sinovitis articular• Artropatía hemofílica.
– Consecuencia: • Necesidad de reemplazo
articular• En promedio, 20 a 30 años
después.
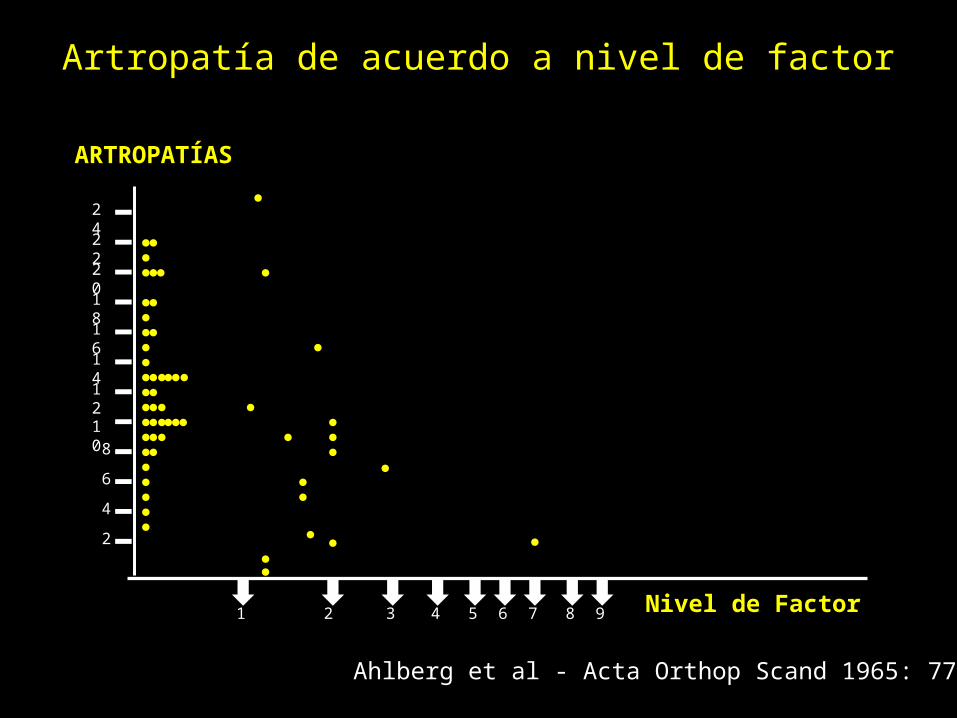
Artropatía de acuerdo a nivel de factor
2
4
6
810
12
14
16
18
20
22
24
1 2 3 4 5 6 7 8 9
Ahlberg et al - Acta Orthop Scand 1965: 77, 5-99
Nivel de Factor
ARTROPATÍAS
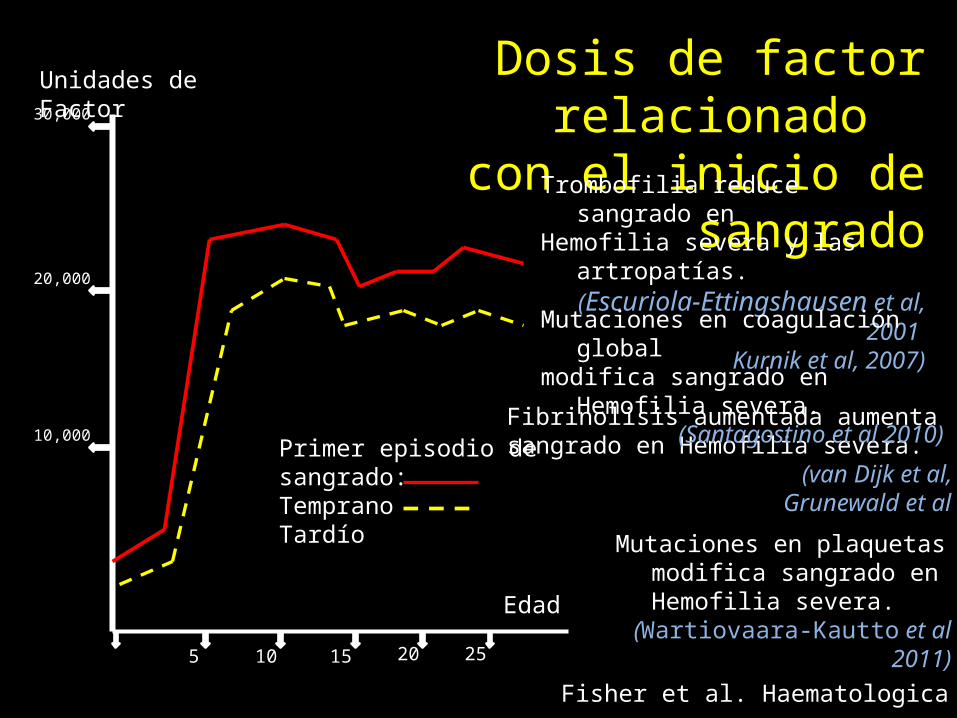
25
Edad
5 10 15 20
10,000
20,000
30,000
Unidades de Factor
Primer episodio de sangrado:TempranoTardío
Dosis de factor relacionado con el inicio de sangrado
Fisher et al. Haematologica 2011
Trombofilia reduce sangrado en Hemofilia severa y las artropatías.(Escuriola-Ettingshausen et al, 2001
Kurnik et al, 2007)
Fibrinolisis aumentada aumenta sangrado en Hemofilia severa.
(van Dijk et al, 2007, Grunewald et al 2002)
Mutaciones en coagulación global modifica sangrado en Hemofilia severa.
(Santagostino et al 2010)
Mutaciones en plaquetas modifica sangrado en Hemofilia severa.
(Wartiovaara-Kautto et al 2011)

Formas de atención.
• A Demanda.– Atención LUEGO de sangrado articular.– Detener el sangrado.
• Profilaxis.– Atención para PREVENIR sangrado articular.– Preservar la función articular.
• TRES TIPOS.– Primaria o continua.– Secundaria o limitada– Peri-operatoria
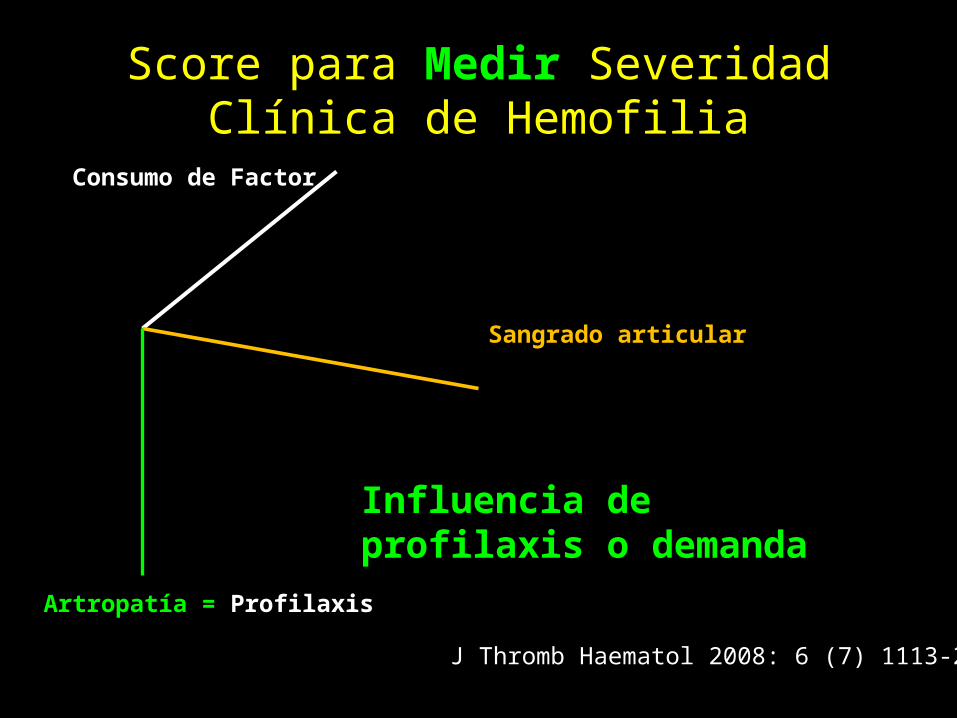
Score para Medir Severidad Clínica de Hemofilia
Consumo de Factor
Sangrado articular
Artropatía = Profilaxis
J Thromb Haematol 2008: 6 (7) 1113-21
Influencia de profilaxis o demanda
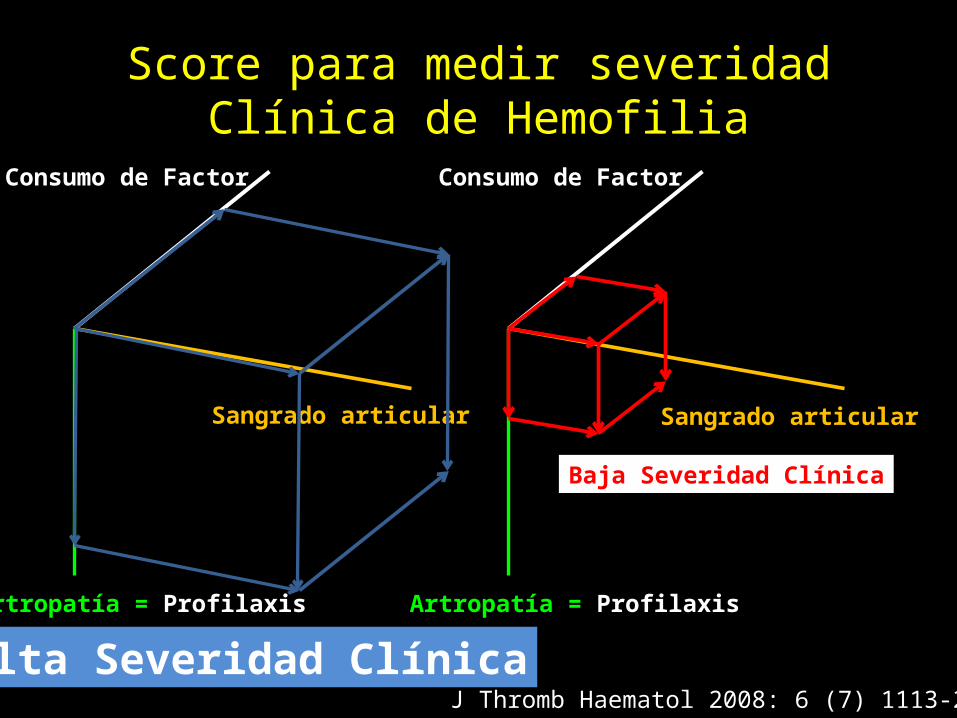
Score para medir severidad Clínica de Hemofilia
Consumo de Factor
Sangrado articular
Artropatía = Profilaxis
J Thromb Haematol 2008: 6 (7) 1113-21Alta Severidad Clínica
Consumo de Factor
Sangrado articular
Artropatía = Profilaxis
Baja Severidad Clínica
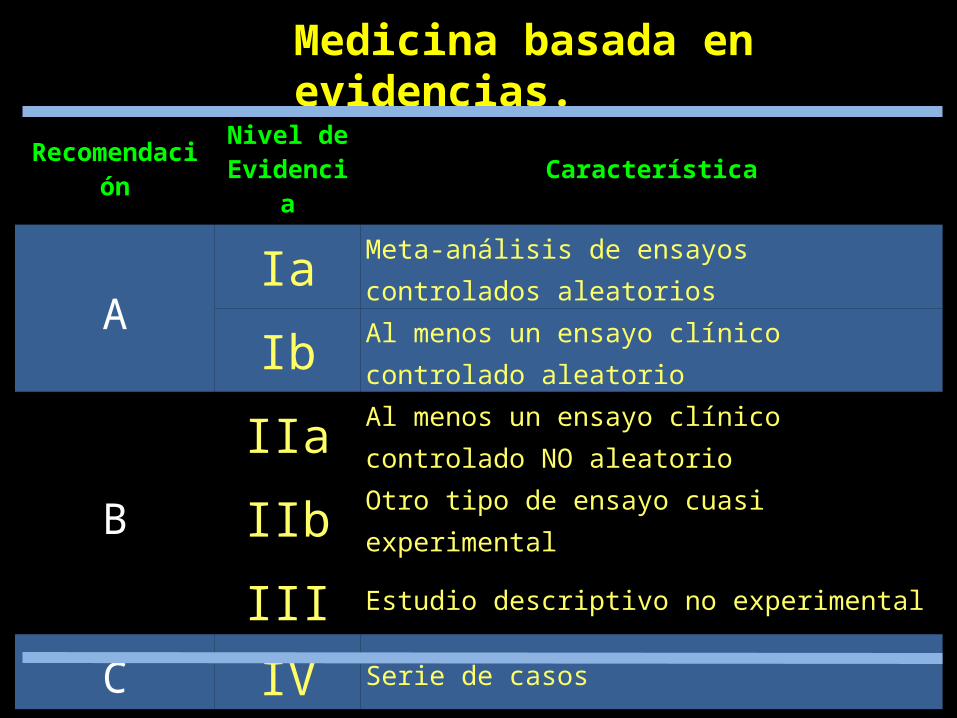
Recomendación Nivel de Evidencia Característica
A Ia Meta-análisis de ensayos controlados aleatorios
Ib Al menos un ensayo clínico controlado aleatorio
BIIa Al menos un ensayo clínico controlado NO aleatorio
IIb Otro tipo de ensayo cuasi experimental
III Estudio descriptivo no experimental
C IV Serie de casos
D V Informes de comité de expertos
Medicina basada en evidencias.

¿Es útil la prevención de hemorragias en los pacientes con hemofilia A o B?
• Búsqueda de base de datos en MEDLINE, EMBASE y en Cochrane Central, hasta abril 2011.
• Selección: Ensayos clínicos que incluye adultos y niños con hemofilia A o B. Comparan PROFILAXIS contra PLACEBO o tratamiento a DEMANDA.
• Objetivos: – Medida Primaria: Tasa de hemorragias.– Medidas Secundarias:
• Daño radiológico• Calidad de Vida• Efectos adversos• Costos.
• Inclusión: SEIS ENSAYOS, con 142 participantesCochrane Database of Systematic Reviews 2011; 9: CD003429
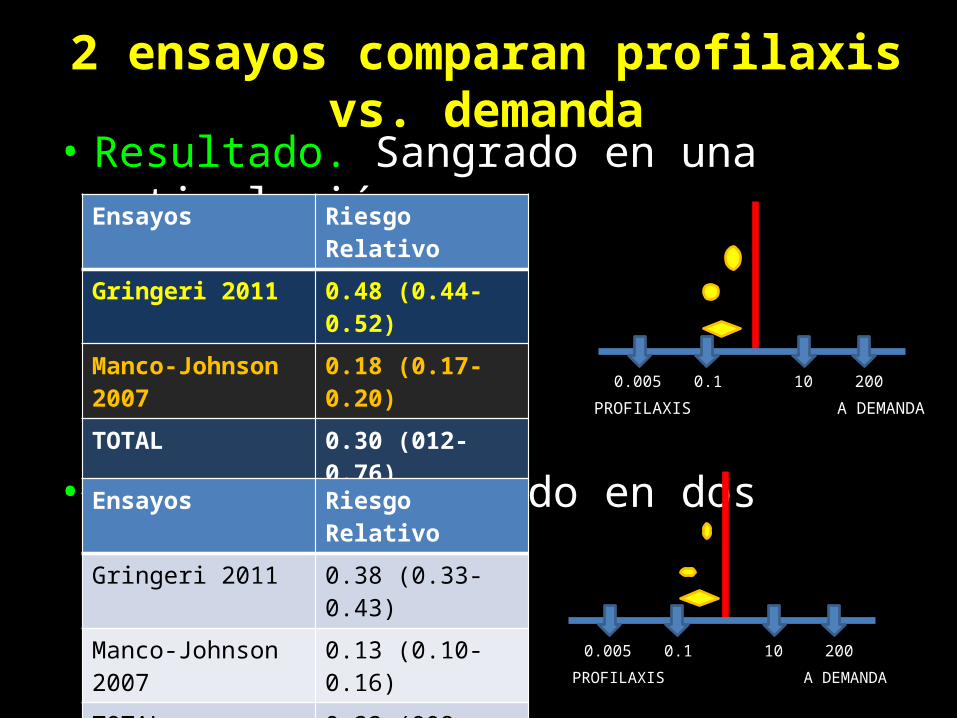
• Resultado. Sangrado en una articulación
• Resultado. Sangrado en dos articulaciones
Ensayos Riesgo Relativo
Gringeri 2011 0.48 (0.44-0.52)
Manco-Johnson 2007 0.18 (0.17-0.20)
TOTAL 0.30 (012-0.76)
Ensayos Riesgo Relativo
Gringeri 2011 0.38 (0.33-0.43)
Manco-Johnson 2007 0.13 (0.10-0.16)
TOTAL 0.22 (008-0.63)
PROFILAXIS A DEMANDA10 2000.10.005
PROFILAXIS A DEMANDA10 2000.10.005
2 ensayos comparan profilaxis vs. demanda

• Resultado. Calidad de vida
• Resultado. Uso de Factor VIII
Ensayos Riesgo Relativo
Gringeri 2011 32.73(22.3-43.16)
A DEMANDA PROFILAXIS10 2000.10.005
Ensayos Cociente
Gringeri 2011 4.87 (2.95-6.79)
Manco-Johnson 2007 5.44 (4.20-6.69)
TOTAL 5.27 (4.23-6.32)
A DEMANDA PROFILAXIS2 4-2-4
2 ensayos comparan profilaxis vs. demanda
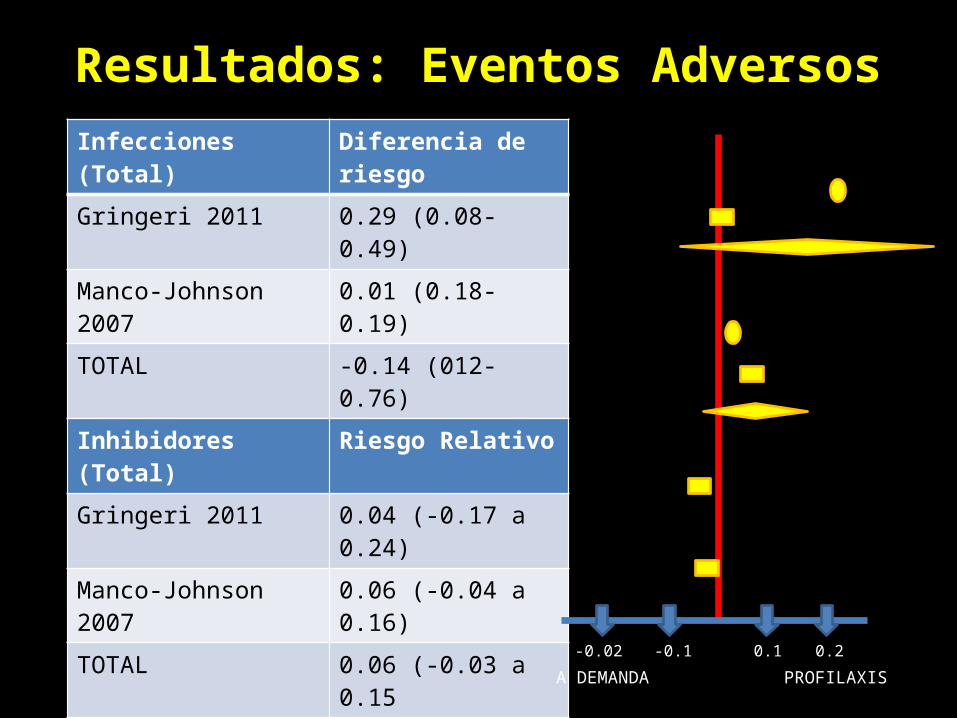
Resultados: Eventos AdversosInfecciones (Total) Diferencia de riesgo
Gringeri 2011 0.29 (0.08-0.49)
Manco-Johnson 2007 0.01 (0.18-0.19)
TOTAL -0.14 (012-0.76)
Inhibidores (Total) Riesgo Relativo
Gringeri 2011 0.04 (-0.17 a 0.24)
Manco-Johnson 2007 0.06 (-0.04 a 0.16)
TOTAL 0.06 (-0.03 a 0.15
Infección (en CVC) Riesgo Relativo
Manco-Johnson 2007 -0.03 (-0.26 a 0.19)
Inhibidores (en CVC) Riesgo Relativo
Manco-Johnson 2007 -0.01 (-0.11 a 0.10)
A DEMANDA PROFILAXIS0.1 0.2-0.1-0.02
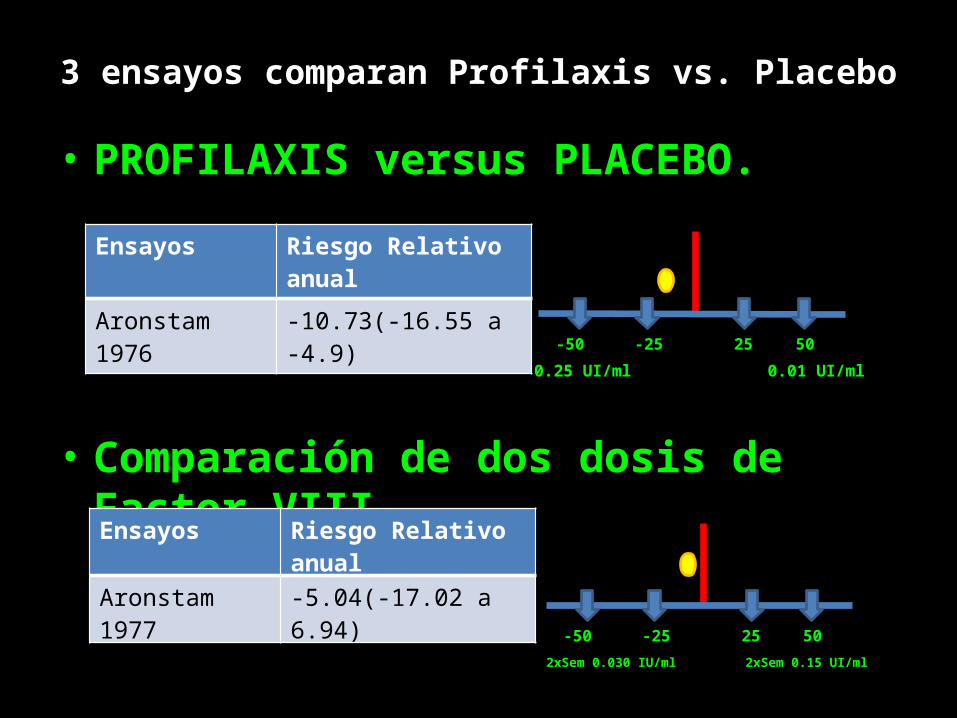
3 ensayos comparan Profilaxis vs. Placebo
• PROFILAXIS versus PLACEBO.
• Comparación de dos dosis de Factor VIII
Ensayos Riesgo Relativo anual
Aronstam 1976 -10.73(-16.55 a -4.9)
0.25 UI/ml 0.01 UI/ml25 50-25-50
Ensayos Riesgo Relativo anual
Aronstam 1977 -5.04(-17.02 a 6.94)
2xSem 0.030 IU/ml 2xSem 0.15 UI/ml25 50-25-50

Sexto Ensayo• En hemofílicos B comparan profilaxis semanal
(15 UI/kg) versus bisemanal (7.5UI/kg)
Ensayos Riesgo Relativo anual
Morfini 1976 -3.30(-5.5 a -1.1)
Bi-semanal Semanal5 10-5-10
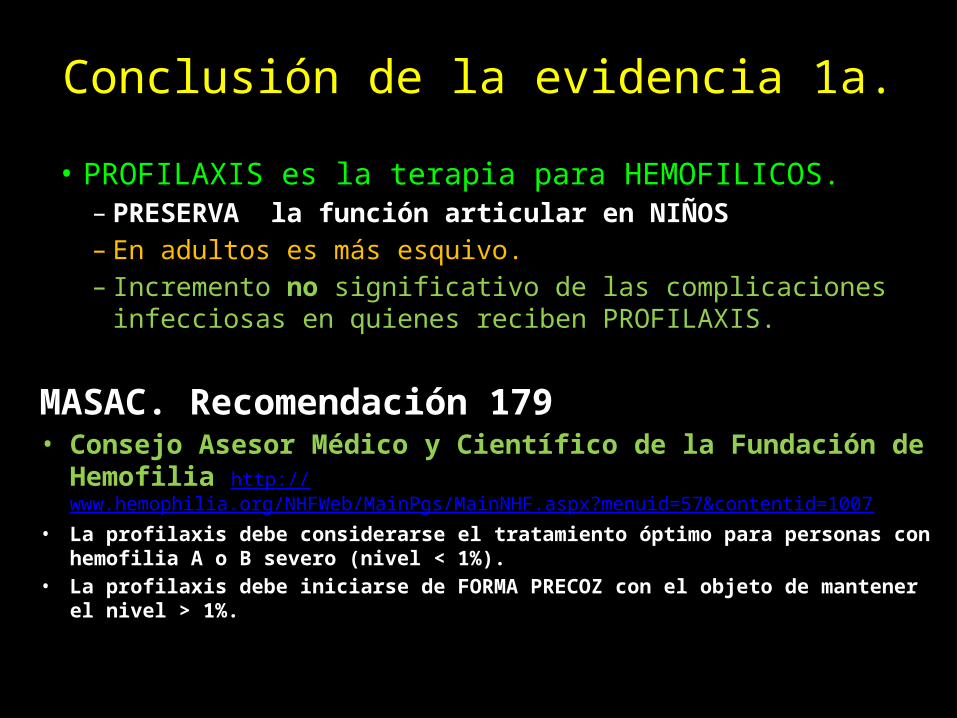
Conclusión de la evidencia 1a.
• PROFILAXIS es la terapia para HEMOFILICOS.– PRESERVA la función articular en NIÑOS– En adultos es más esquivo.– Incremento no significativo de las complicaciones infecciosas en
quienes reciben PROFILAXIS.
MASAC. Recomendación 179• Consejo Asesor Médico y Científico de la Fundación de Hemofilia
http://www.hemophilia.org/NHFWeb/MainPgs/MainNHF.aspx?menuid=57&contentid=1007• La profilaxis debe considerarse el tratamiento óptimo para personas con hemofilia A o
B severo (nivel < 1%).• La profilaxis debe iniciarse de FORMA PRECOZ con el objeto de mantener el nivel > 1%.
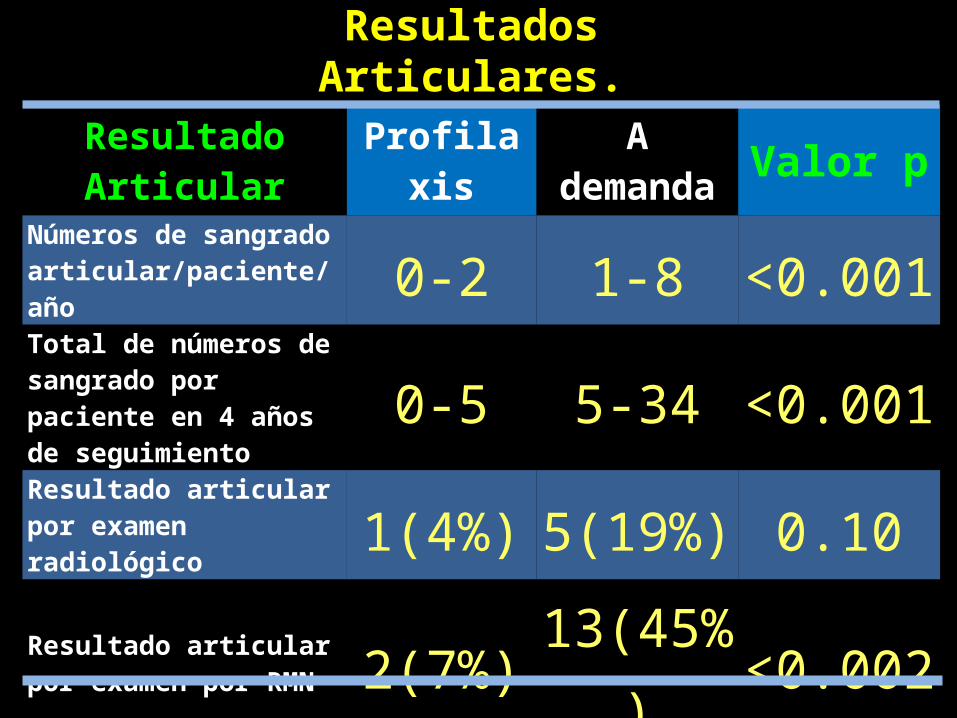
Resultado Articular Profilaxis A demanda Valor p
Números de sangrado articular/paciente/año 0-2 1-8 <0.001Total de números de sangrado por paciente en 4 años de seguimiento 0-5 5-34 <0.001
Resultado articular por examen radiológico 1(4%) 5(19%) 0.10Resultado articular por examen por RMN 2(7%) 13(45%) <0.002Número de articulaciones dañadas 2 16 <0.002
Resultados Articulares.
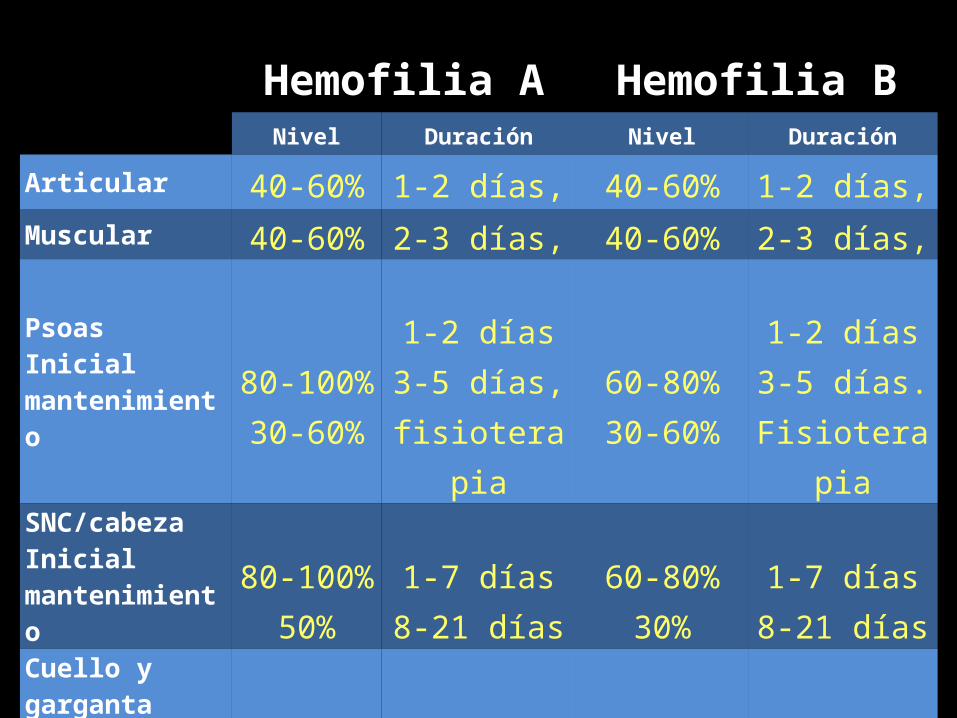
Hemofilia A Hemofilia BNivel Duración Nivel Duración
Articular 40-60% 1-2 días, 40-60% 1-2 días, Muscular 40-60% 2-3 días, 40-60% 2-3 días,
PsoasInicialmantenimiento
80-100%30-60%
1-2 días3-5 días,
fisioterapia
60-80%30-60%
1-2 días3-5 días.
Fisioterapia
SNC/cabezaInicialmantenimiento
80-100%50%
1-7 días8-21 días
60-80%30%
1-7 días8-21 días
Cuello y gargantaInicialmantenimiento
80-100%50%
1-7 días8-14 días
60-80%30%
1-7 días8-14 días

Hemofilia A Hemofilia BNivel Duración Nivel Duración
GastrointestinalInicialmantenimiento
80-100%50%
1-6 días8-14 días
60-80%30%
1-6 días8-14 días
Renal 50% 3-5 días 40% 3-5 días
Cirugía mayorPreoperatorioPostoperatorio
80-100%60-80%40-60%
1-3 días4-6 días
7-14 días
60-80%40-60%30-50%
1-3 días4-6 días
7-14 días

Procedimiento Hemofilia A Hemofilia B Objetivo
Angiografía Coronaria Bolo de 40U/k FVIII seguido de 20 U/k FVIII
Bolo de 80 U/k F IX seguido de 30 U/k F IX
Nivel > 80 U/dL
Colocar Stent de metal
Antes de procedimiento
Bolo de 40U/k FVIII seguido de 20 U/k FVIII
Bolo de 80 U/k F IX seguido de 30 U/k F IX Nivel > 80 U/dL
Durante terapia con aspirina y clopidogrel
50U/K F VIII cada dos días
60-70U/K F IX 2-3/ semana Nivel >30U/dL
Durante terapia con aspirina
25-40 U/k FVIII cada dos días
25-50U/k FIX 2-3/semana Nivel >5U/dL
Fibrinolisis Bolo de 40 U/K FVIII seguido de infusión
continúa (3-4U/K/h FVIII )
Bolo de 80 U/K F IX seguido de infusión
continúa (3-4 U/k/h F IX)Nivel de 80-100 U/dL. Seguido de >50 U/dL.
Terapia para síndrome coronario agudo
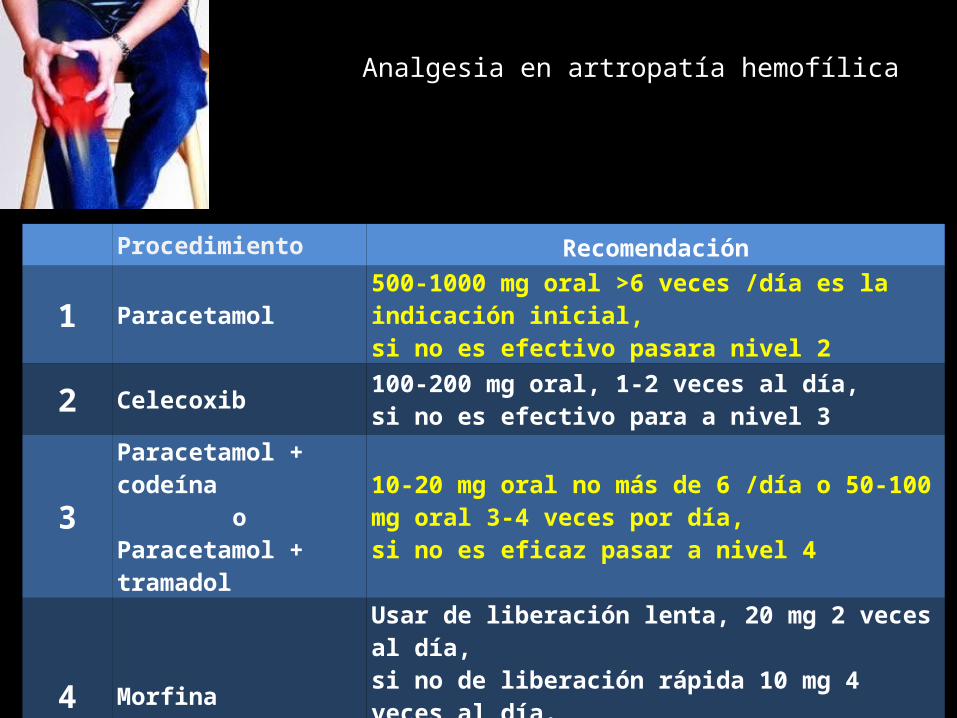
Procedimiento Recomendación
1 Paracetamol 500-1000 mg oral >6 veces /día es la indicación inicial, si no es efectivo pasara nivel 2
2 Celecoxib 100-200 mg oral, 1-2 veces al día, si no es efectivo para a nivel 3
3Paracetamol + codeína
oParacetamol + tramadol
10-20 mg oral no más de 6 /día o 50-100 mg oral 3-4 veces por día, si no es eficaz pasar a nivel 4
4 MorfinaUsar de liberación lenta, 20 mg 2 veces al día, si no de liberación rápida 10 mg 4 veces al día. Pasar a liberación rápida si no responde a liberación lenta
Analgesia en artropatía hemofílica

Algunas Emergencias
• Trombocitopenia.• Hemorragia masiva.• Coagulopatía por trauma.• Hemofilia congénita y adquirida.• Neutropenia febril.• Anemia hemolítica autoinmune.• Purpura trombocitopenica trombótica.• Pancitopenia con CID.
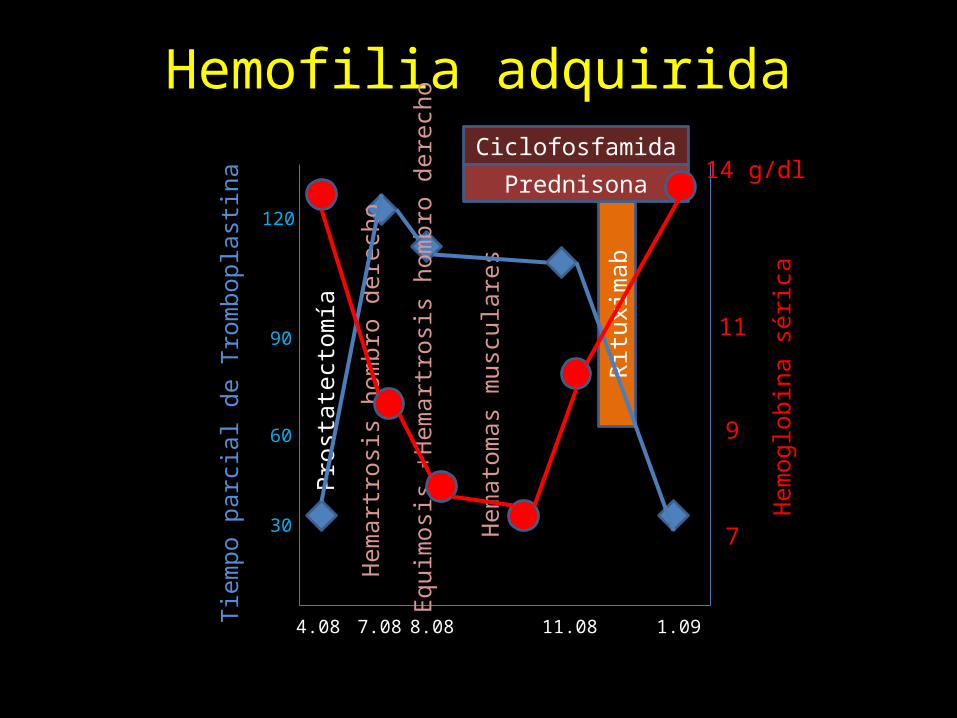
Hemofilia adquirida
4.08 7.08 8.08
CiclofosfamidaPrednisona
11.08Ri
tuxi
mab
1.09
Tiem
po p
arci
al d
e Tr
ombo
plas
tina
30
60
90
120
Pros
tate
ctom
ía
Hem
artr
osis
hom
bro
dere
cho
Equi
mos
is +H
emar
tros
is ho
mbr
o de
rech
o
Hem
atom
as m
uscu
lare
s
7
14 g/dl
9
11
Hem
oglo
bina
séric
a
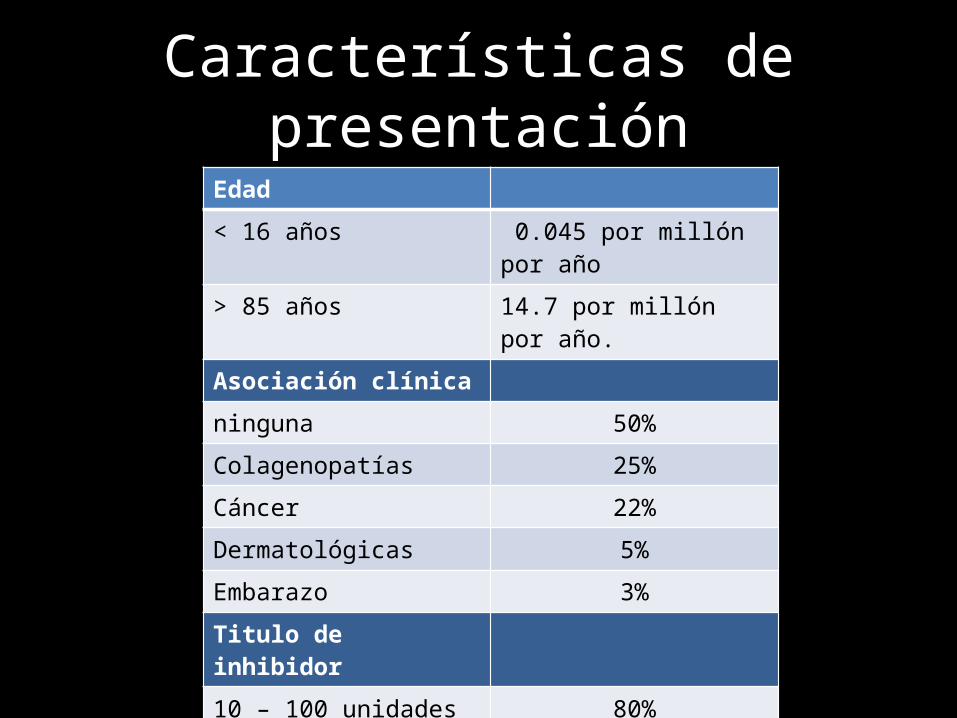
Características de presentaciónEdad
< 16 años 0.045 por millón por año
> 85 años 14.7 por millón por año.
Asociación clínica
ninguna 50%
Colagenopatías 25%
Cáncer 22%
Dermatológicas 5%
Embarazo 3%
Titulo de inhibidor
10 – 100 unidades 80%
> 100 unidades 10%
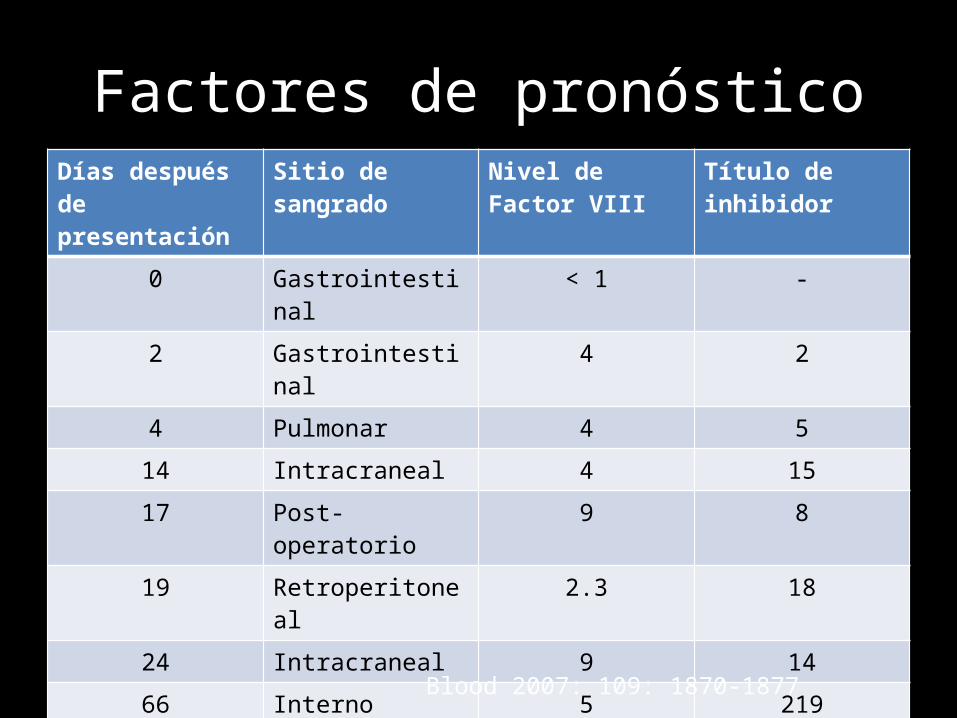
Factores de pronósticoDías después de presentación
Sitio de sangrado Nivel de Factor VIII Título de inhibidor
0 Gastrointestinal < 1 -
2 Gastrointestinal 4 2
4 Pulmonar 4 5
14 Intracraneal 4 15
17 Post-operatorio 9 8
19 Retroperitoneal 2.3 18
24 Intracraneal 9 14
66 Interno 5 219
106 Intracraneal 4 6
136 Gastrointestinal < 1 109
146 intracraneal 2 14
Blood 2007: 109: 1870-1877

Terapia
• Control de Sangrado
• Erradicación de inhibidor
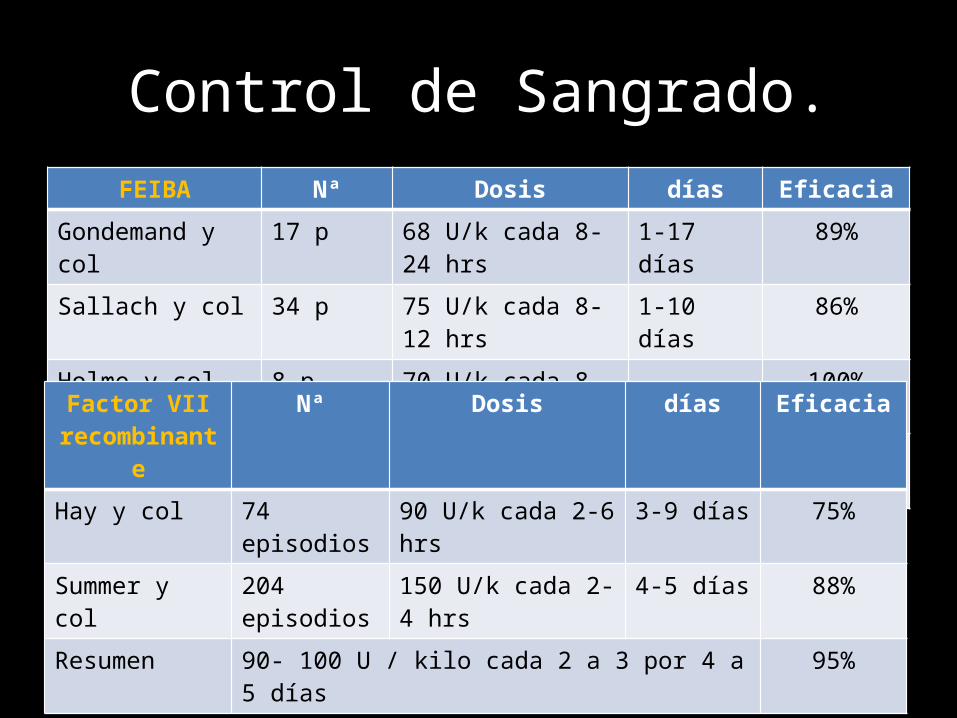
Control de Sangrado.FEIBA Nª Dosis días Eficacia
Gondemand y col 17 p 68 U/k cada 8-24 hrs 1-17 días 89%
Sallach y col 34 p 75 U/k cada 8-12 hrs 1-10 días 86%
Holme y col 8 p 70 U/k cada 8 hrs - 100%
Resumen 50-100 U / kilo cada 8 a 12 horas (<200U/k) 80%
Factor VII recombinante
Nª Dosis días Eficacia
Hay y col 74 episodios 90 U/k cada 2-6 hrs 3-9 días 75%
Summer y col 204 episodios 150 U/k cada 2-4 hrs 4-5 días 88%
Resumen 90- 100 U / kilo cada 2 a 3 por 4 a 5 días 95%

Seguridad de FEIBA y Factor VII rRiesgos FEIBA Factor VII recombinante
Trombosis (100,000 infusiones) 8.24 24.6Tipo de trombosis Infarto miocardio Cerebro-vascularEventos en hemofilia adquirida 8/67 7/16
Modalidad Nº pacientes Nº de pacientes con un solo tratamiento
Nada 51 (34.2%) -FEIBA 49 (32.9%) 25 (16.8%)Factor VII activado 47 (31.5%) 21 (14.1%)Factor VIII 38 (25.5%) 15 (10.1%)Desmopresina 7 (4.7%) 2 (1.3%)

Erradicación del inhibidorRemisión Completa
Nivel de Factor normal
Inhibidor no detectable
Título < 1 unidad Bethesda
Remisión Parcial
Nivel de factor VIII > 25%
Declina el inhibidor > 50%
No sangrado
Medicamento Dosis Eficacia Documento
Inmunoglobulinas endovenosas
2 gr/kilo diario por 2 días
12 a 25 % 66% si se suma prednisona
Tolerancia inmunológica
Protocolo Budapest 90% Factor VIII más ciclofosfamida 20 mg diario + prednisona 100 mg diario por 30 días
Protocolo Bonn-Malmö
50% Factor VIII + Ig EV + ciclofosfamida + prednisona.
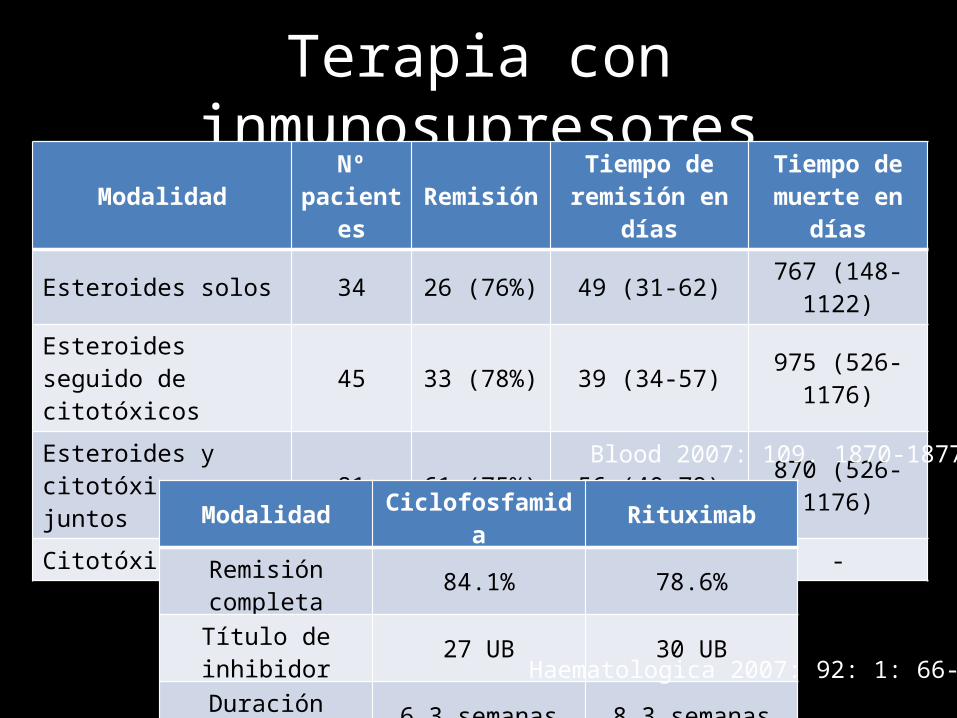
Terapia con inmunosupresoresModalidad Nº
pacientes Remisión Tiempo de remisión en días
Tiempo de muerte en días
Esteroides solos 34 26 (76%) 49 (31-62) 767 (148-1122)Esteroides seguido de citotóxicos 45 33 (78%) 39 (34-57) 975 (526-1176)
Esteroides y citotóxicos juntos 81 61 (75%) 56 (40-79) 870 (526-1176)
Citotóxicos 7 4(67%) - -
Blood 2007: 109. 1870-1877
Modalidad Ciclofosfamida Rituximab
Remisión completa 84.1% 78.6%
Título de inhibidor 27 UB 30 UB
Duración media 6.3 semanas 8.3 semanas
Haematologica 2007: 92: 1: 66-71
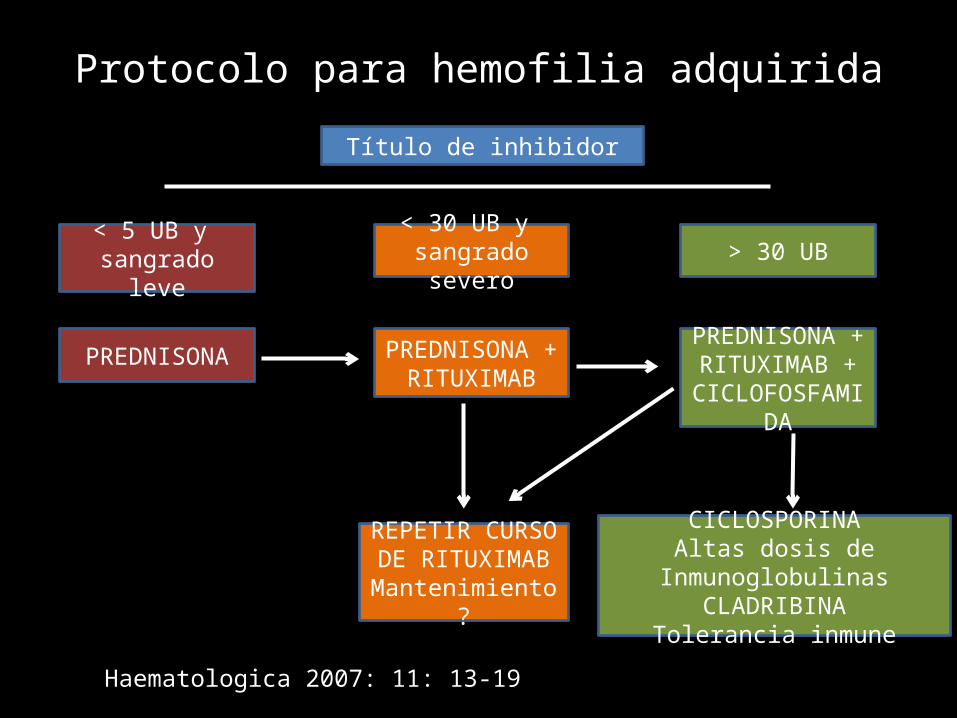
Protocolo para hemofilia adquiridaTítulo de inhibidor
< 5 UB y sangrado leve
< 30 UB y sangrado severo > 30 UB
PREDNISONA PREDNISONA +RITUXIMAB
PREDNISONA +RITUXIMAB +
CICLOFOSFAMIDA
REPETIR CURSO DE RITUXIMAB
Mantenimiento?
CICLOSPORINAAltas dosis de Inmunoglobulinas
CLADRIBINATolerancia inmune
Haematologica 2007: 11: 13-19

Prueba anormalRespuesta a mezcla con plasma normal (P+N)
No corrige Si corrige
Déficit de factoresInhibidores de factores
Prueba Valor de Corte
T. Protrombina TP p+n < TPp + TPn 2
T. Parcial de Tromboplastina TPT (p+n) + TPT n x100TPT p
T. Trombina TT p+n < TT n/2

Algunas Emergencias
• Trombocitopenia.• Hemorragia masiva.• Coagulopatía por trauma.• Hemofilia congénita y adquirida.• Neutropenia febril.• Anemia hemolítica autoinmune.• Purpura trombocitopenica trombótica.• Pancitopenia con CID.

Neutropenia Febril• Temperatura oral
– > 38.3º C en una sola toma– 38º C por una hora.
• Cuenta con tendencia a >500 neutrófilos/mm3
• Emergencia Médica. • Actuar antes de cumplir UNA HORA.• Cultivos necesarios.• Radiografía de Tórax.• Inicio de antibióticos parenterales. STAT

Indicaciones
• Ante Temperatura mayor a 38.3º C entonces antes de una hora:– 2 hemocultivos de 2 venas diferentes.– Metamisol 20mg/kilo/dosis EV en 45 minutos.– Cefepime 2g cada 8 horas EV

Medidas de Aislamiento.• Éxito depende de ENFERMERIA.• Profilaxis de antibióticos y antimicóticos.• Estudio microbiológico permanente.• Acción oportuna de Farmacia.• Compromiso Administrativo – Gerencial.• Participación de personal de limpieza y dieta.• Participación de FAMILIARES.

Niveles de Atención enNeoplasias Hematológicas

Niveles de atención.
• Determinantes:– Complejidad del tratamiento.– Duración de la neutropenia esperada después de la
quimioterapia.– El tipo de la enfermedad.
• Niveles de Atención:– Nivel 1– Nivel 2A– Nivel 2B
–Nivel 3
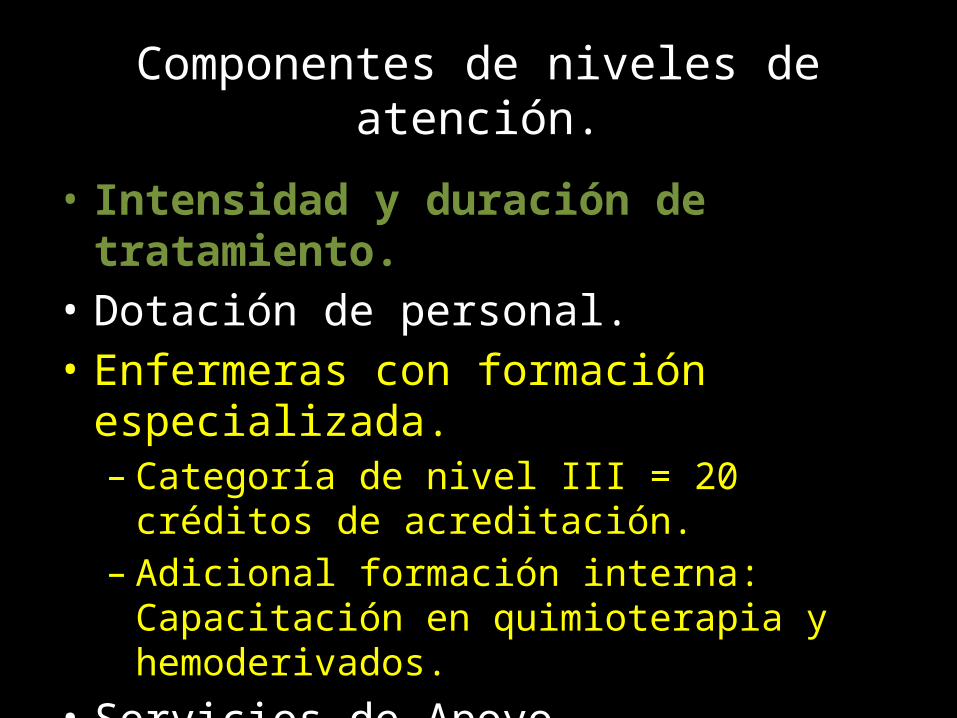
Componentes de niveles de atención.
• Intensidad y duración de tratamiento.• Dotación de personal.• Enfermeras con formación especializada.
– Categoría de nivel III = 20 créditos de acreditación.– Adicional formación interna: Capacitación en
quimioterapia y hemoderivados.• Servicios de Apoyo.• Disposiciones para atención en Emergencia.
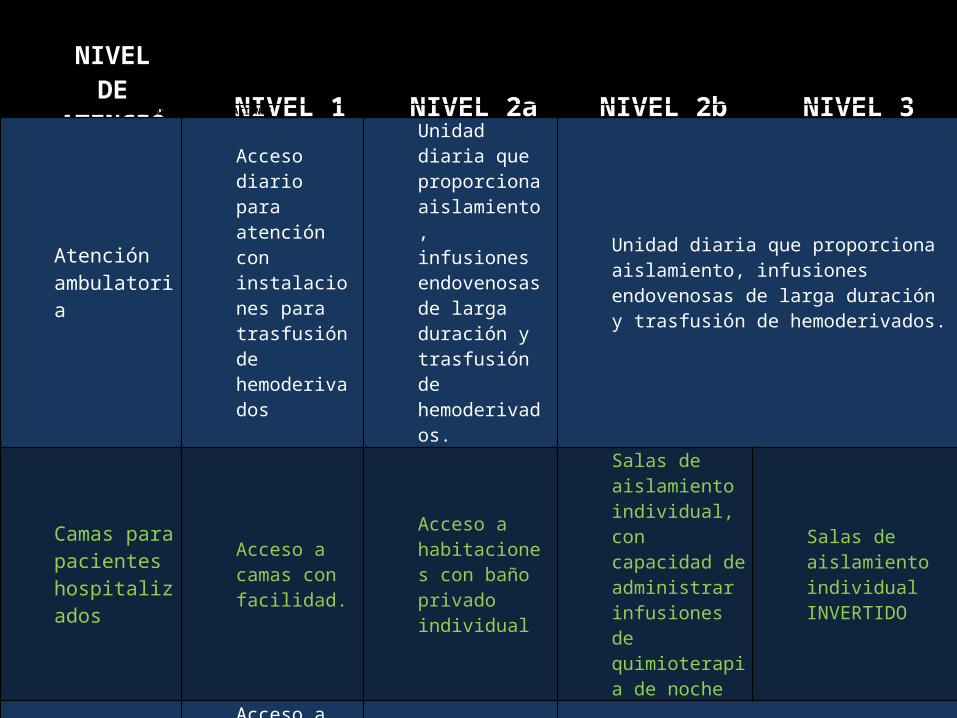
NIVEL DE ATENCIÓN NIVEL 1 NIVEL 2a NIVEL 2b NIVEL 3
ESPECIALIDADES DE APOYO E INSTALACIONES
Atención ambulatoria
Acceso diario para atención con instalaciones para trasfusión de hemoderivados
Unidad diaria que proporciona aislamiento, infusiones endovenosas de larga duración y trasfusión de hemoderivados.
Unidad diaria que proporciona aislamiento, infusiones endovenosas de larga duración y trasfusión de hemoderivados.
Camas para pacientes hospitalizados
Acceso a camas con facilidad.
Acceso a habitaciones con baño privado individual
Salas de aislamiento individual, con capacidad de administrar infusiones de quimioterapia de noche
Salas de aislamiento individual INVERTIDO
Acceso a camas
Acceso a sala de emergencia con empleo de guías acordadas para manejo de complicaciones de la quimioterapia.
Admisión a emergencia con empleo de guías acordadas para manejo de complicaciones de la quimioterapia.
Directo acceso a sala de hospitalización de hematología.

Algunas Emergencias
• Trombocitopenia.• Hemorragia masiva.• Coagulopatía por trauma.• Hemofilia congénita y adquirida.• Neutropenia febril.• Anemia hemolítica autoinmune.• Purpura trombocitopenica trombótica.• Pancitopenia con CID.

Anemia Hemolítica Autoinmune
• ¿Cuándo Trasfundir?• Terapia previa:
– Inmunoglobulinas endovenosas 1 gr/kilo/día.– Dexametasona 40 mg diario por 4 días.– Ciclofosfamida 500 mg/m2 dosis inicial.– Rituximab 375 mg/m2 dosis inicial. Opcional.
• Trasfusiones luego de 4 día.– Todo en caliente cuando son anticuerpos fríos.– Bajo vigilancia médica por si hay reacciones.

Algunas Emergencias
• Trombocitopenia.• Hemorragia masiva.• Coagulopatía por trauma.• Hemofilia congénita y adquirida.• Neutropenia febril.• Anemia hemolítica autoinmune.• Purpura trombocitopenica trombótica.• Pancitopenia con CID.
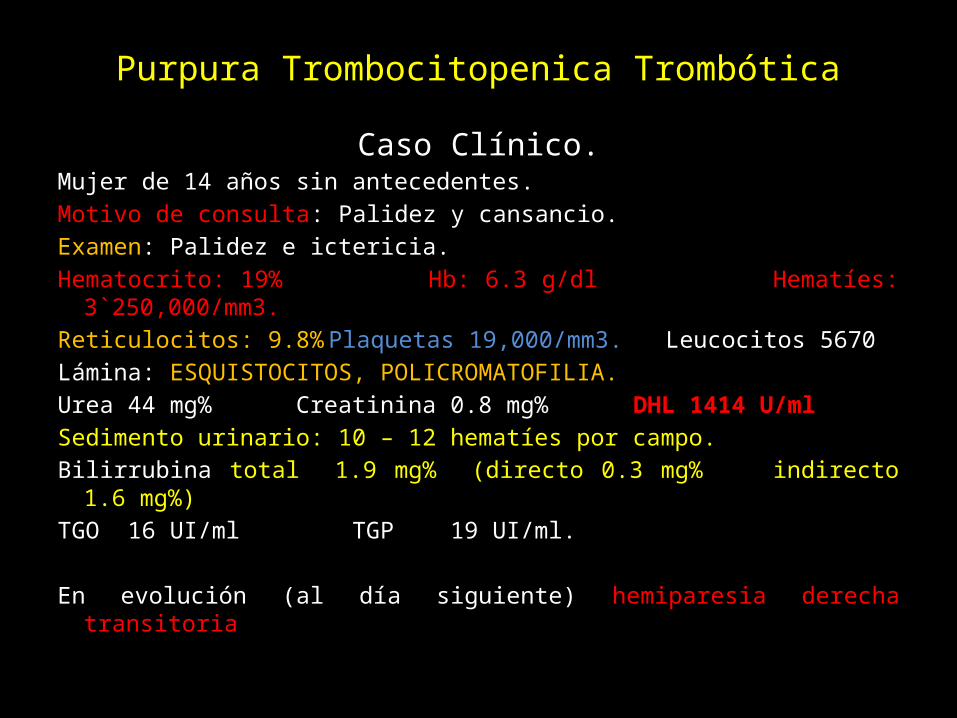
Purpura Trombocitopenica TrombóticaCaso Clínico.
Mujer de 14 años sin antecedentes.Motivo de consulta: Palidez y cansancio.Examen: Palidez e ictericia. Hematocrito: 19% Hb: 6.3 g/dl Hematíes: 3`250,000/mm3.Reticulocitos: 9.8% Plaquetas 19,000/mm3. Leucocitos 5670Lámina: ESQUISTOCITOS, POLICROMATOFILIA.Urea 44 mg% Creatinina 0.8 mg% DHL 1414 U/mlSedimento urinario: 10 – 12 hematíes por campo. Bilirrubina total 1.9 mg% (directo 0.3 mg% indirecto 1.6 mg%)TGO 16 UI/ml TGP 19 UI/ml.
En evolución (al día siguiente) hemiparesia derecha transitoria

Descripción
• La púrpura trombocitopénica trombótica es una microangiopatía trombótica microvascular severa: – agregación plaquetaria con isquemia tisular– trombocitopenica – fragmentación de los hematíes.
• Descrita por Moschcowitz en 1924• Incidencia 3.7 a 11 por 1`000,000 habitantes por
año.• Incidencia máxima entre 30 y 40 años.
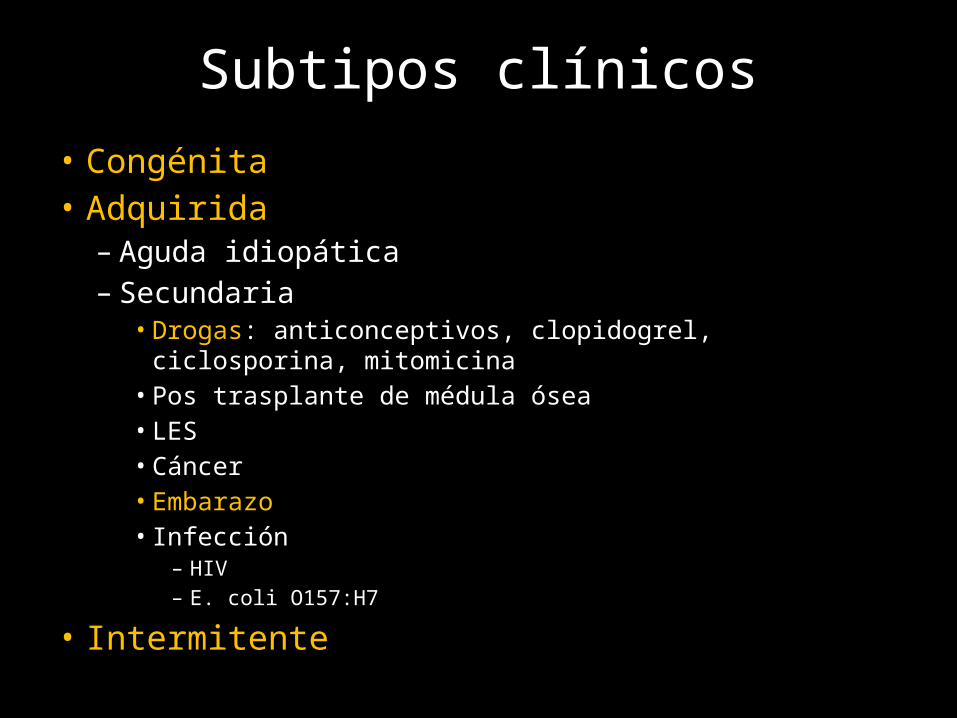
Subtipos clínicos• Congénita• Adquirida
– Aguda idiopática– Secundaria
• Drogas: anticonceptivos, clopidogrel, ciclosporina, mitomicina• Pos trasplante de médula ósea• LES• Cáncer• Embarazo• Infección
– HIV– E. coli O157:H7
• Intermitente

Manifestaciones clínicas
• Criterios:– Anemia hemolítica microangiopática– Trombocitopenia 75%– Síntomas neurológicos 40%– Fiebre– Disfunción renal.
• Menos común: dolor abdominal y stress respiratorio.• 10-40% infección de vías aéreas superiores o
síndrome gripal semanas previas.

Criterio diagnóstico
• Debe hacerse en todo paciente con– Anemia hemolítica microangiopática– Trombocitopenia– Ausencia de otra causa identificable:
• Perfil de coagulación normal. • Test Coombs negativo.• Test de Ham negativo.• LES catastrófico. Vasculitis generalizada.• Cáncer diseminado.• Hipertensión maligna.

Fisiopatología
Evidencia de Hemolisis Evidencia de hemolisis Intravascular
Evidencia de microangiopatía trombótica
AnemiaAumento de reticulocitosLeucocitosisESQUISTOCITOSAumento de DHL
HemoglobinuriaHemosiderinuriaReducción de haptoglobinaAumento de bilirrubina indirecta
Trombocitopenia Perfil de coagulación NORMALPDF normalesFactores I, V, VIII normales o aumentados.

Anatomía Patológica
• Compromiso de arteriolas terminales y capilares.• Trombo compuesto de plaquetas, factor von
Willebrand con escasa fibrina.• Depósitos hialinos en el subendotelio de
capilares y capa muscular de arteriolas.• Ausencia de inflamación vascular y perivascular.• Órganos afectados: Cerebro, páncreas, corazón,
riñón, bazo, glándulas adrenales.

Mecanismo Molecular
• Falla de PROTEASA que degrada al Factor Von Willebrand.
• Miembro de la familia ADAMTS (a disintegrin and metalloproteinase with thrombospondin type-I: ADAMTS13.
• ADAMTS 13 es una glicoproteína codificada en el cromosoma 9, sintetizada en el hígado.
• También en endotelio vascular y podocitos glomerulares.
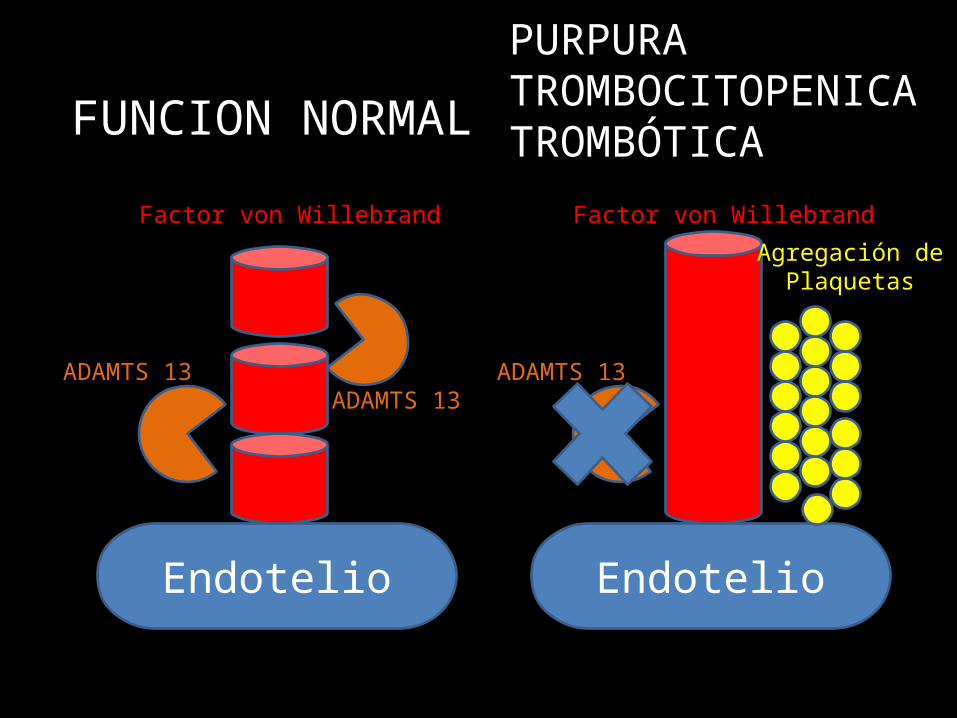
• z
Endotelio
Factor von Willebrand
ADAMTS 13ADAMTS 13
FUNCION NORMAL
Endotelio
Factor von Willebrand
ADAMTS 13
Agregación dePlaquetas
PURPURATROMBOCITOPENICA TROMBÓTICA

ADAMTS 13
• Valor normal en plasma 50% a 178%• Nivel reducido:
– Enfermedad hepática– CID– Uremia– Neoplasias diseminadas– Enfermedades metabólicas– Embarazo – parto
• PTT congénito e idiopático: < 5%

PTT CONGENITO
• Herencia autosómica recesiva.• PTT familiar Mutaciones de ADAMTS 13.• Actividad de ADAMTS 13 = < 5%.• Se presenta en infancia o adolescencia
recurrente a intervalos regulares (21-28 días)• Variantes menos severas se presenta a edades
más tardías con episodios intermitentes. Asociado a cuadro febril.

PTT ADQUIRIDA IDIOPATICA
• Ausencia o disminución severa de ADAMTS 13 en episodio inicial o recurrente.
• Auto-anticuerpos IgG inhiben a ADAMTS 13 en 44 a 94% en el episodio agudo. Defecto transitorio, intermitente o recurrente de regulación inmune.
• Pacientes sin anticuerpos anti-ADAMTS 13: defecto de producción o sensibilidad limitada de tests.

Tratamiento PTT IDIOPÁTICO.• PLASMAFERESIS
– Disminuye la MORTALIDAD de 90% a 20%– Debe ser instituido dentro de las 24 horas del
diagnóstico.– 1 a 1.5 volemias por día– Frecuencia diaria hasta por los menos 2 días
posteriores a la obtención de Remisión completa:• Ausencia de síntomas neurológicos• Plaquetas > 150,000 por mm3.• DHL normal• Hemoglobina en ascenso.

Tratamiento de PTT congénito
• Infusión profiláctica de:– plasma fresco congelado, – Plasma reducido en crioprecipitado– Concentrado de complejo protrombínico.
• Cada 3 a 4 semanas.

Tratamiento de PTT secundaria
• No hay deficiencia de ADAMTS 13 • Raramente responde a plasmaferesis.• Excepciones:
– Enfermedades autoinmunes.– Embarazo. Puede asociarse congénito o adquirido.– Ticlopidina. Induce anti – ADAMTS 13
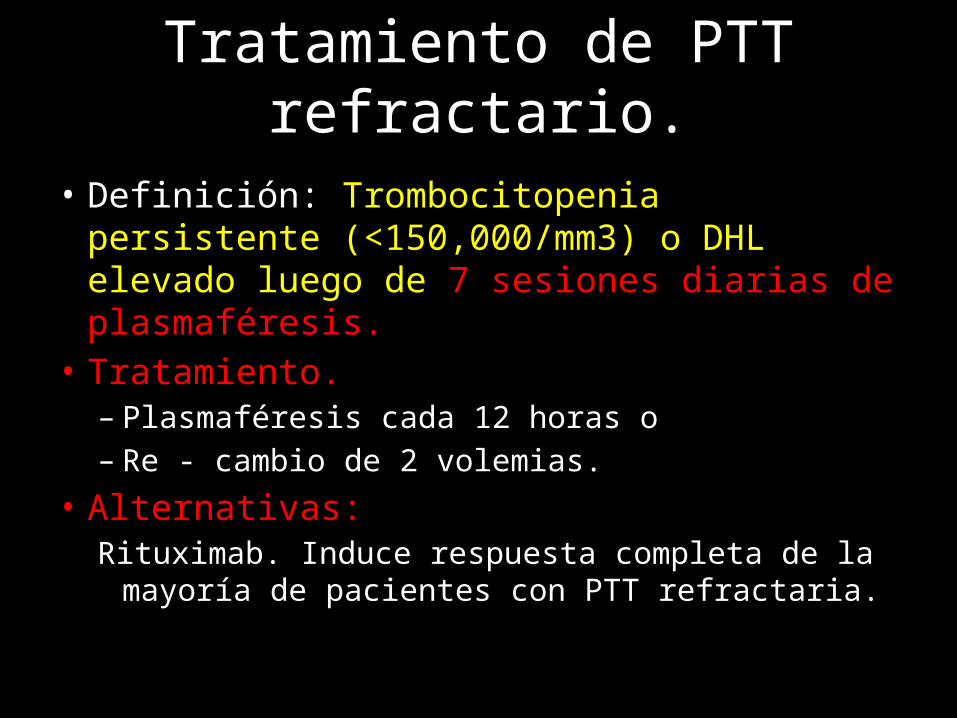
Tratamiento de PTT refractario.
• Definición: Trombocitopenia persistente (<150,000/mm3) o DHL elevado luego de 7 sesiones diarias de plasmaféresis.
• Tratamiento.– Plasmaféresis cada 12 horas o – Re - cambio de 2 volemias.
• Alternativas:Rituximab. Induce respuesta completa de la mayoría
de pacientes con PTT refractaria.

Recaída
• 36 % de los pacientes recaen en próximos 10 años.
• Hasta el momento es identificar pacientes en RIESGO. UGFVW elevado en periodos de remisión asociado a enfermedad autoinmune.
• Esplenectomía puede reducir el riesgo de RECAIDA.

Conclusiones
• Es una urgencia hematológica.• Síndrome clínico con múltiples etiologías,
mecanismos y manifestaciones clínicas.• Diagnóstico diferencial debe PENSARSE en
anemia hemolítica microangiopática con trombocitopenia.
• Sobre todo en embarazo, parto, sepsis y neoplasias.

Algunas Emergencias
• Trombocitopenia.• Hemorragia masiva.• Coagulopatía por trauma.• Hemofilia congénita y adquirida.• Neutropenia febril.• Anemia hemolítica autoinmune.• Purpura trombocitopenica trombótica.• Pancitopenia con CID.

Pancitopenia con CID.
Sepsis con Falla multi-orgánica
VasculitisCatastrófico
NeoplasiasHematológicas
Leucemia Promielocítica
Aguda
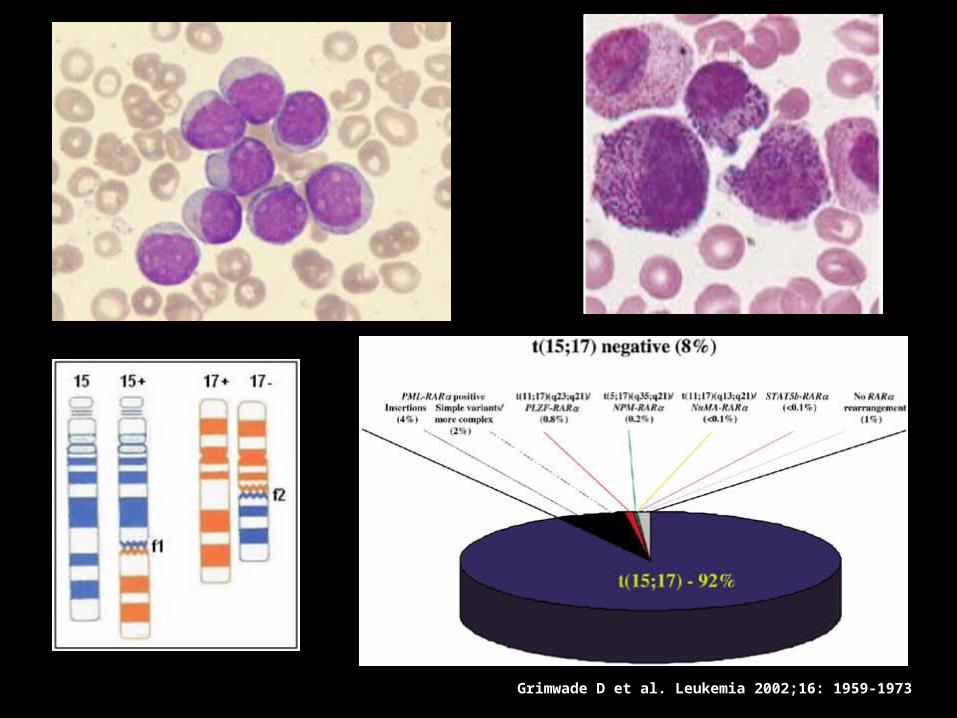
Grimwade D et al. Leukemia 2002;16: 1959-1973
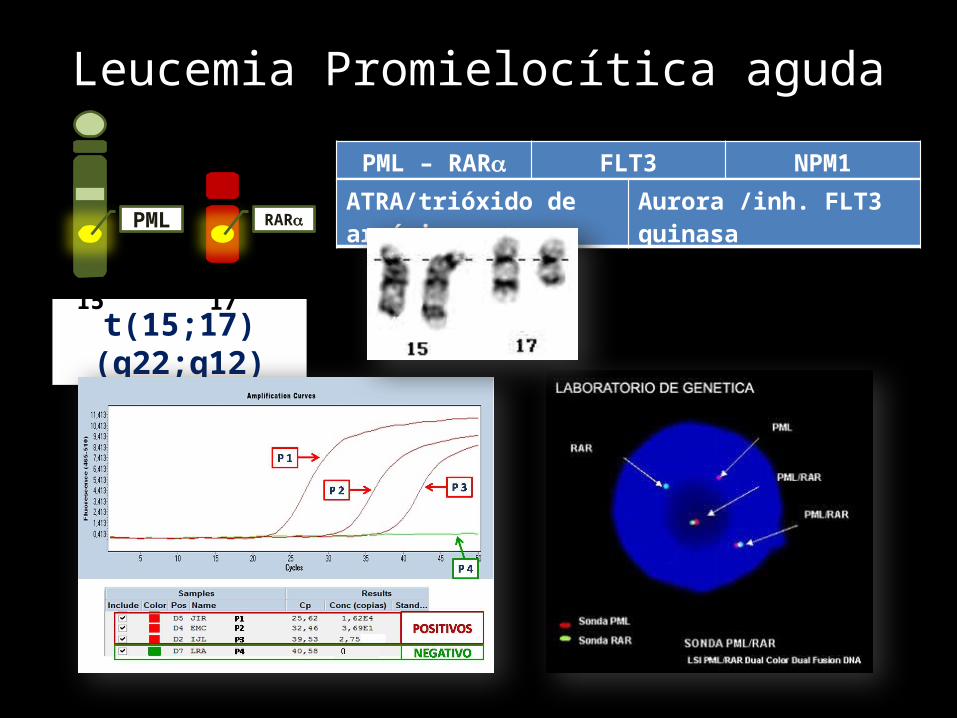
Leucemia Promielocítica aguda
t(15;17)(q22;q12)
X
X
PML
15
RAR
17
PML – RAR FLT3 NPM1ATRA/trióxido de arsénico Aurora /inh. FLT3 quinasa
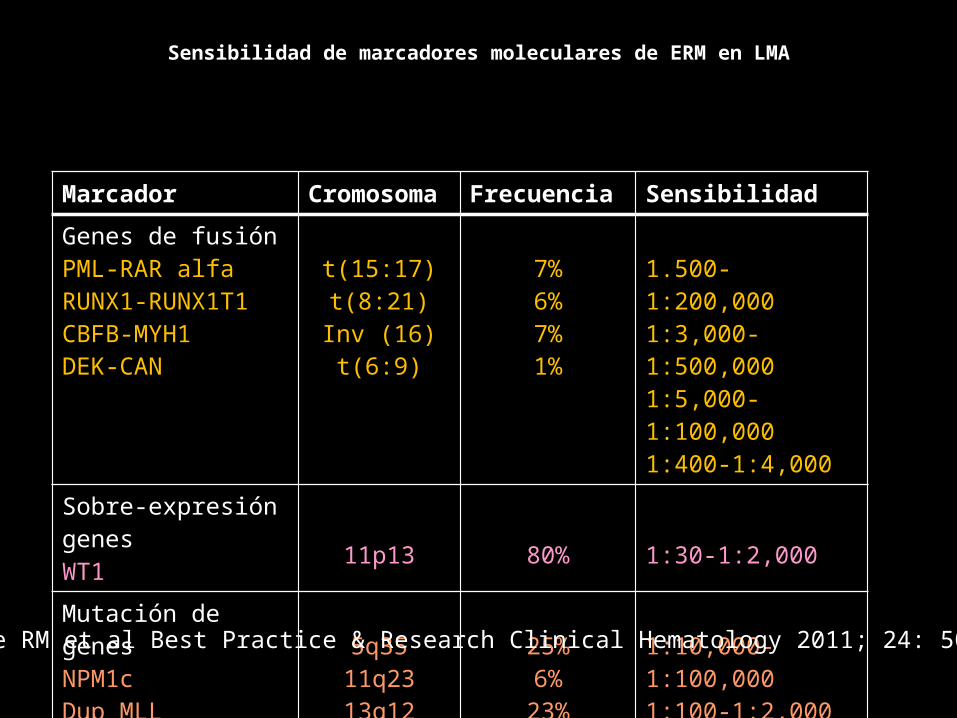
Marcador Cromosoma Frecuencia Sensibilidad
Genes de fusiónPML-RAR alfaRUNX1-RUNX1T1CBFB-MYH1DEK-CAN
t(15:17)t(8:21)Inv (16)t(6:9)
7%6%7%1%
1.500-1:200,0001:3,000-1:500,0001:5,000-1:100,0001:400-1:4,000
Sobre-expresión genesWT1 11p13 80% 1:30-1:2,000Mutación de genesNPM1cDup MLLFLT3-ITD
5q3511q2313q12
25%6%
23%
1:10,000-1:100,0001:100-1:2,0001:10,000
Sensibilidad de marcadores moleculares de ERM en LMA
Stone RM et al Best Practice & Research Clinical Hematology 2011; 24: 509-514.

Características clínicas• Pancitopenia.• Trastorno de coagulación con Dimero D elevado.• Hipofibrinogenemia• Sangrado muco - cutáneo.
• Tratamiento URGENTE. • Acido transretinoico 45 mg/m2 diario.• Plasma fresco congelado o concentrado de
complejo protrombínico.• Crioprecipitado.-

Cuadro clínicoAlta Tasa de proliferación celular.
Leucocitosis.
Sensibilidad a la terapia.
DHL elevado.
Co-morbilidades: Uremia,hiperuricemia,disminución de flujo urinario,orina ácida,Deshidratación,falla renal.

CriteriosDefinición de criterios de laboratorio de Cairo – Bishop Desde 3 días antes hasta 7 día después de iniciar quimioterapia.
Acido úrico >4.76 mg/ml o > 25% de incremento de basal
Potasio > 6 mEq / Lt o >25% de incremento de basal
Fósforo > 2.1 mg/lt (niños) o >1.45 mg/lt (adultos) o 25% de incremento de basal
Calcio < 1.75 mg/lt o 25% de reducción de basal.
Definición de criterios clínicos de Cairo – BishopDesde 3 días antes hasta 7 día después de iniciar quimioterapia.
Creatinina > 1.5 veces más para edad y sexo.
Arritmia cardiaca o muerte súbita.
Convulsiones.
Br J Haematol. 2004;127:3-11.

Conducta en alto riesgo. • Monitoreo:
– Cardiaco.– Bioquímico cada 6 a 8 horas.
• Terapia:– Rasburicasa 4.5 mg EV
• 0.2 mg/kilo en 50 ml de Solución salina en 30 minutos.• Repetir 2 dosis si ácido úrico >7.5 mg% ente 2 y 5 día• Dosis 12 mg EV en caso de Obesidad (> 87 kilos/m2)• Excepto en deficiencia de G6FD: Alopurinol.
– Hidratación 3 litros/m2 por día.

Conducta
• Riesgo Intermedio.– Vigilancia.– Aumentar hidratación.– Alopurinol 100 a 300 mg cada 8 horas oral
• Bajo riesgo.– Hidratación normal.– Alopurinol en caso de enfermedad voluminosa o
cambios metabólicos.
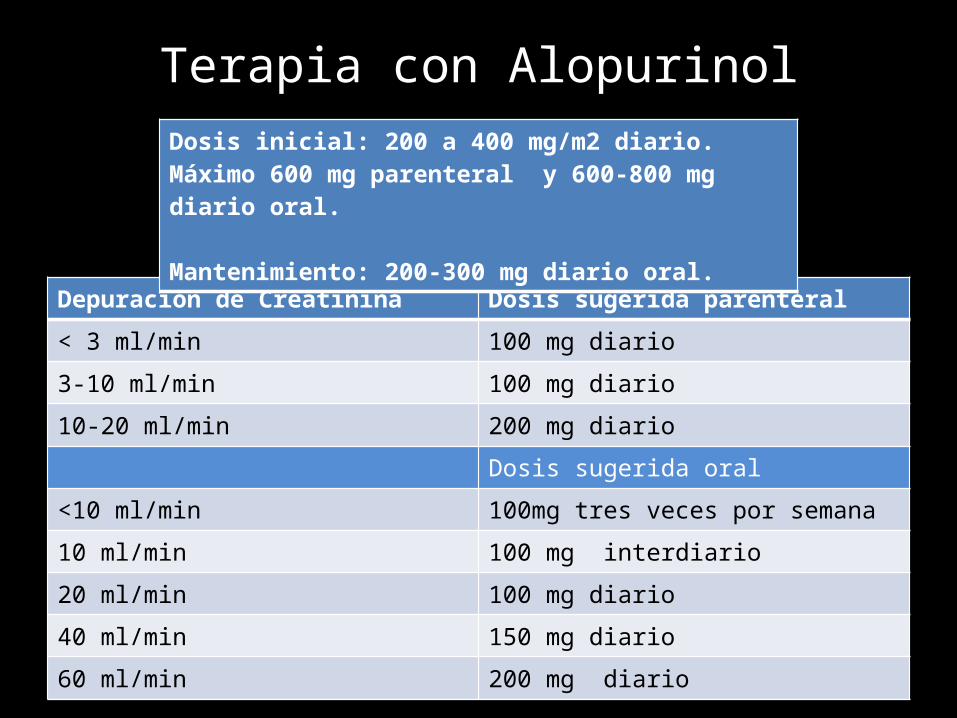
Terapia con Alopurinol
Depuración de Creatinina Dosis sugerida parenteral
< 3 ml/min 100 mg diario
3-10 ml/min 100 mg diario
10-20 ml/min 200 mg diario
Dosis sugerida oral
<10 ml/min 100mg tres veces por semana
10 ml/min 100 mg interdiario
20 ml/min 100 mg diario
40 ml/min 150 mg diario
60 ml/min 200 mg diario
Dosis inicial: 200 a 400 mg/m2 diario. Máximo 600 mg parenteral y 600-800 mg diario oral.
Mantenimiento: 200-300 mg diario oral.

Hiperfosfatemia.• 50% de falla renal, con 55% de mortalidad.• Moderado (< 6.5 mg/dl)
– Hidratación adecuada– Quelante:
• hidróxido de aluminio, • carbonato de calcio
• Grave (>8 mg/dl)– Hemodiálisis.– Diálisis peritoneal.



















