
Elucidating the phylodynamics of endemic rabies …Elucidating the phylodynamics of endemic rabies...
11
Elucidating the phylodynamics of endemic rabies virus in eastern Africa using whole-genome sequencing Kirstyn Brunker, 1,2,3, * Denise A Marston, 3 Daniel L Horton, 3,4 Sarah Cleaveland, 1,2 Anthony R Fooks, 3 Rudovick Kazwala, 5 Chanasa Ngeleja, 6 Tiziana Lembo, 1,2 Maganga Sambo, 7 Zacharia J Mtema, 7 Lwitiko Sikana, 7 Gavin Wilkie, 8 Roman Biek, 1,2,† and Katie Hampson 1,2,† 1 Institute of Biodiversity, Animal Health and Comparative Medicine, University of Glasgow, Glasgow G12 8QQ, UK, 2 The Boyd Orr Centre for Population and Ecosystem Health, University of Glasgow, Glasgow G12 8QQ, UK, 3 Animal and Plant Health Agency, Weybridge, Woodham Lane, KT15 3NB, UK, 4 School of Veterinary Medicine, University of Surrey, Guildford GU2 7XH, UK, 5 Department of Veterinary Medicine and Public Health, Sokoine University of Agriculture, Morogoro, United Republic of Tanzania, 6 Tanzania Veterinary Laboratory Agency, Dar es Salaam, United Republic of Tanzania, Temeke Veterinary, Mandela Road, P.O. BOX 9254, 7 Ifakara Health Institute, Ifakara, United Republic of Tanzania, P.O. Box 53 and 8 MRC Centre for Virus Research, University of Glasgow, Sir Michael Stoker Building, Garscube Campus, 464 Bearsden Road, Glasgow G61 1QH, UK *Corresponding author: E-mail: [email protected] † These authors contributed equally to this work. Abstract Many of the pathogens perceived to pose the greatest risk to humans are viral zoonoses, responsible for a range of emerging and endemic infectious diseases. Phylogeography is a useful tool to understand the processes that give rise to spatial patterns and drive dynamics in virus populations. Increasingly, whole-genome information is being used to uncover these patterns, but the limits of phylogenetic resolution that can be achieved with this are unclear. Here, whole-genome variation was used to uncover fine-scale population structure in endemic canine rabies virus circulating in Tanzania. This is the first whole-genome population study of rabies virus and the first comprehensive phylogenetic analysis of rabies virus in East Africa, providing important insights into rabies transmission in an endemic system. In addition, sub-continental scale patterns of population structure were identified using partial gene data and used to determine population structure at larger spatial scales in Africa. While rabies virus has a defined spatial structure at large scales, increasingly frequent levels of admixture were observed at regional and local levels. Discrete phylogeographic analysis revealed long-distance dispersal within Tanzania, which could be attributed to human-mediated movement, and we found evidence of multiple persistent, co-circulating lineages at a very local scale in a single district, despite on-going mass dog vaccination campaigns. This may reflect the wider endemic circulation of these lineages over several decades alongside increased admixture due to human- mediated introductions. These data indicate that successful rabies control in Tanzania could be established at a national level, since most dispersal appears to be restricted within the confines of country borders but some coordination with neighbouring countries may be required to limit transboundary movements. Evidence of complex patterns of rabies circula- tion within Tanzania necessitates the use of whole-genome sequencing to delineate finer scale population structure that V C The Author 2015. Published by Oxford University Press. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse, distribution, and reproduction in any medium, provided the original work is properly cited. 1 Virus Evolution, 2015, 1(1): 1–11 doi: 10.1093/ve/vev011 Research article
Transcript of Elucidating the phylodynamics of endemic rabies …Elucidating the phylodynamics of endemic rabies...

Elucidating the phylodynamics of endemic rabies virus
in eastern Africa using whole-genome sequencingKirstyn Brunker123 Denise A Marston3 Daniel L Horton34
Sarah Cleaveland12 Anthony R Fooks3 Rudovick Kazwala5
Chanasa Ngeleja6 Tiziana Lembo12 Maganga Sambo7 Zacharia J Mtema7
Lwitiko Sikana7 Gavin Wilkie8 Roman Biek12dagger and Katie Hampson12dagger
1Institute of Biodiversity Animal Health and Comparative Medicine University of Glasgow Glasgow G12 8QQUK 2The Boyd Orr Centre for Population and Ecosystem Health University of Glasgow Glasgow G12 8QQ UK3Animal and Plant Health Agency Weybridge Woodham Lane KT15 3NB UK 4School of Veterinary MedicineUniversity of Surrey Guildford GU2 7XH UK 5Department of Veterinary Medicine and Public Health SokoineUniversity of Agriculture Morogoro United Republic of Tanzania 6Tanzania Veterinary Laboratory AgencyDar es Salaam United Republic of Tanzania Temeke Veterinary Mandela Road PO BOX 9254 7IfakaraHealth Institute Ifakara United Republic of Tanzania PO Box 53 and 8MRC Centre for Virus ResearchUniversity of Glasgow Sir Michael Stoker Building Garscube Campus 464 Bearsden Road Glasgow G61 1QHUK
Corresponding author E-mail kbrunker1researchglaacukdaggerThese authors contributed equally to this work
Abstract
Many of the pathogens perceived to pose the greatest risk to humans are viral zoonoses responsible for a range of emergingand endemic infectious diseases Phylogeography is a useful tool to understand the processes that give rise to spatialpatterns and drive dynamics in virus populations Increasingly whole-genome information is being used to uncover thesepatterns but the limits of phylogenetic resolution that can be achieved with this are unclear Here whole-genome variationwas used to uncover fine-scale population structure in endemic canine rabies virus circulating in Tanzania This is the firstwhole-genome population study of rabies virus and the first comprehensive phylogenetic analysis of rabies virus in EastAfrica providing important insights into rabies transmission in an endemic system In addition sub-continental scalepatterns of population structure were identified using partial gene data and used to determine population structure at largerspatial scales in Africa While rabies virus has a defined spatial structure at large scales increasingly frequent levels ofadmixture were observed at regional and local levels Discrete phylogeographic analysis revealed long-distance dispersalwithin Tanzania which could be attributed to human-mediated movement and we found evidence of multiple persistentco-circulating lineages at a very local scale in a single district despite on-going mass dog vaccination campaigns This mayreflect the wider endemic circulation of these lineages over several decades alongside increased admixture due to human-mediated introductions These data indicate that successful rabies control in Tanzania could be established at a nationallevel since most dispersal appears to be restricted within the confines of country borders but some coordination withneighbouring countries may be required to limit transboundary movements Evidence of complex patterns of rabies circula-tion within Tanzania necessitates the use of whole-genome sequencing to delineate finer scale population structure that
VC The Author 2015 Published by Oxford University PressThis is an Open Access article distributed under the terms of the Creative Commons Attribution License (httpcreativecommonsorglicensesby40)which permits unrestricted reuse distribution and reproduction in any medium provided the original work is properly cited
1
Virus Evolution 2015 1(1) 1ndash11
doi 101093vevev011Research article
can that can guide interventions such as the spatial scale and design of dog vaccination campaigns and dog movementcontrols to achieve and maintain freedom from disease
Key words RNA virus phylodynamics zoonoses endemic rabies translocation
1 Introduction
The general trend of increasing incidence and expansion ofemerging or re-emerging zoonotic diseases (eg EbolaChikungunya and avian influenza) (Woolhouse 2002 Greger2007 Jones et al 2008) and persistence of established zoonosessuch as canine rabies highlights the ongoing challenges facedas we attempt to characterize and control them The processesthat drive the spread and persistence of infectious diseases arereflected in a genetic signature in pathogen genomes (Biek andReal 2010) Understanding the processes that give rise to spatialpopulation structure in pathogens can inform the managementand control of infectious diseases For example analyses of evo-lutionary epidemiological and ecological data have recentlydemonstrated that global live swine trade strongly predicts theglobal dissemination of influenza A viruses in swine (Nelsonet al 2015) and air travel has been revealed as a major factordriving the intra-continental spread of Dengue virus (Nuneset al 2014) Viral pathogens particularly fast-evolving RNA vi-ruses are model systems to explore pathogen populations asthey rapidly accumulate genetic diversity on a timescale similarto epidemiological processes (Drummond et al 2003 Biek et al2015) Statistical phylogeographic approaches are available(Lemey et al 2009 Bedford et al 2014 Bielejec et al 2014) to de-velop a quantitative understanding of the processes that giverise to spatial patterns in RNA viruses (Holmes and Grenfell2009) on epidemiological time scales Whole-genome sequenc-ing (WGS) is increasingly being used as a means to extract thesepatterns but it is unclear how much resolution can be gainedand at what temporal and spatial scale In this article caninerabies virus (RABV) is used as a model to determine the spatio-temporal patterns of an endemic zoonotic virus using whole-genome data to distinguish structure at an increasingly finescale
Rabies is a globally distributed zoonotic disease caused by asingle-stranded negative sense RNA virus from the Lyssavirusgenus Though capable of infecting any mammal given virusvariants are typically maintained in distinct species-specific cy-cles within the orders Carnivora and Chiroptera (RupprechtHanlon and Hemachudha 2002) The disease causes thousandsof human deaths every year predominantly in Asia and Africawhere the virus circulates endemically in domestic dogs (Canislupus familiaris) (Knobel et al 2005 Shwiff Hampson andAnderson 2013) The majority of these deaths (99) are causedby bites from rabid dogs instilling fear into the many communi-ties that live under continuous threat from a disease that is al-most invariably fatal but entirely preventable Although the roleof domestic dogs as key vectors of rabies is recognized muchless is known about the dog-associated RABV variant than wild-life variants such as raccoon or skunk RABVs circulating inNorth America (Brunker et al 2012) Moreover while epidemicexpansions of wildlife RABV have been well documented andstudied (eg Real et al 2005 Biek et al 2007 Kuzmina et al2013) we know little about the persistence and spread of rabiesin endemic landscapes
Characterizing the spatial scales of canine rabies dispersalis a critical step toward identifying the processes and factors
driving its dynamics and the scale at which control strategiesneed to be implemented On a global scale canine RABV ex-hibits a strong phylogeographic structure with the distribu-tion of seven distinct major clades reflecting the position ofmajor barriers such as oceans and mountain ranges or his-torical mass human colonizationmigration events (Davidet al 2007 Bourhy et al 2008) However it is unclear whetherthis genetic structure will persist in endemic scenarios or atsmaller scales and how much it is influenced by human-me-diated dispersal Indeed on a regional scale this landscapestructure becomes less distinct some landscape features forexample geopolitical boundaries can act as apparent barriersto movement as seen in North Africa (Talbi et al 2010)whilst contradictory patterns of synchronous cycles of RABVacross multiple countries (Hampson et al 2007) and repeatedcross-border incursions (Hayman et al 2011) have also beenobserved elsewhere in the continent
While there is a growing understanding of the epidemiologyof canine rabies in Africa (Hampson et al 2007 2009 Lemboet al 2008) effective rabies control is still hindered by limitedknowledge of some of the key drivers of viral transmission andspread Mass dog vaccination is the mainstay of successful ra-bies control but requires sustained coverage of at least 70 percent (WHO 2005 Townsend et al 2013) In addition spatial het-erogeneity may affect how vaccine is most effectively distrib-uted to interrupt key transmission corridors and target regionsseeding RABV dispersal An important aspect of this heteroge-neity is the impact of human factors on RABV transmissionwhich has direct implications for control including the designand scale of interventions necessary to interrupt transmissionand maintain freedom from disease For example movement ofpeople between urban and rural areas and the dog meat tradehave been postulated as means of spreading RABV through hu-man-mediated dog movements in rabies-endemic countries inAsia (Denduangboripant et al 2005 Tao et al 2009 Ahmed et al2015) Uncovering the viral population structure and dynamicsof RABV in Tanzania may identify similar patterns attributed tohuman-mediated movements that can aid the identification ofsources key to viral persistence
There have been few in-depth spatial epidemiology studies ofRABV in sub-Saharan Africa probably owing to the lack of re-sources for effective surveillance including sample collection (Nel2013) However recent studies provide intriguing insights intothe dynamics of rabies in certain parts of Africa (see Talbi et al2010 Mollentze et al 2014) indicating a degree of spatial struc-ture with evidence of long distance movements facilitated by hu-mans RABV spatiotemporal dynamics in East Africa in particularare poorly resolved and very few sequences are publically avail-able At present little is known about the genetic diversity orstructure of RABV in Tanzania other than coarse phylogeneticanalyses of partial or full nucleoprotein gene (N gene) sequenceslimited to well-studied regions (Kissi Tordo and Bourhy 1995Lembo et al 2007) Furthermore whole-genome population stud-ies of RABV have not yet been attempted despite their great po-tential to provide a better understanding of the processesdetermining rabies spread and persistence
2 | Virus Evolution 2015 Vol 1 No 1
In this study finely resolved space-time-genetic data in-cluding whole-genome sequences were used to determine thespatial and temporal dynamics of endemic RABV at both a re-gional and local scale in Tanzania The utility of partial genomedata was also demonstrated as a means to characterize large-scale phylogeographic patterns of RABV in Africa Specificallywe aimed to characterize the dynamics of rabies virus in an en-demic system including the role of human-mediated transport
2 Materials and methods21 Samples
For this study fifty-nine new whole-genome sequences were ob-tained from animal hosts (primarily domestic dogs) from nine re-gions in Tanzania sampled between 2003 and 2012 A previouslysequenced sample (RV2772 accession KF155002) (Marston et al2013) from Southern Tanzania was included in the dataset andused as a reference sequence (sample details in SupplementaryTable S1) The main study area encompassed the SerengetiDistrict in northwest Tanzania where approximately half thesamples (nfrac14 33) were obtained from active surveillance whichenabled the collection of brain material from suspect rabid ani-mals and GPS coordinates and dates recorded as described inHampson et al (2009) The remaining twenty-seven sampleswere obtained opportunistically from other regions in Tanzaniaas part of surveillance by the Tanzanian Ministry of Livestockand Fisheries Development and the Tanzanian VeterinaryLaboratory Agency All samples were sent to the Animal amp PlantHealth Agency (APHA) in Weybridge UK for processing
In addition fifty new partial N gene sequences (405 bp) fromTanzania were obtained via reverse transcription-polymerasechain reaction (RT-PCR) and Sanger sequencing using samplesarchived at APHA and submitted to GenBank with accessionnumbers KR534217ndashKR534266 (details in SupplementaryMaterial and Table S2)
22 RNA extraction and WGS
Total RNA was extracted at APHA directly from brain tissue us-ing TRIZOL according to manufacturerrsquos instructions(Invitrogen) Precipitated total RNA was re-suspended in molec-ular-grade water at a one in ten dilution and quantified using aNanoDrop spectrophotometer (Thermo Scientific) Sampleswere sequenced on a range of next generation sequencing plat-forms during NGS protocol optimization (see SupplementaryMaterial for details) The majority of samples (nfrac14 48) were se-quenced by the following method TRIzol-extracted viral RNAwas depleted of host genomic DNA using the on-column DNasetreatment in RNeasy plus mini kit (Qiagen) as per manufac-turerrsquos instructions with elution in 30 ml molecular grade waterThis was followed by host ribosomal RNA depletion usingTerminator 5rsquo-phosphate-dependent exonuclease (EpicentreBiotechnologies) as detailed in (Marston et al 2013) First- andsecond-strand cDNA was synthesized using a Roche cDNA syn-thesis system kit with random hexamers (Roche) ResultantcDNA was quantified using Picogreen dsDNA quantitation re-agent (Invitrogen) and approximately 1 ng of each sample usedin a lsquotagmentationrsquo reaction mix using a Nextera XT samplepreparation kit (Illumina) according to the manufacturerrsquos pro-tocol (minus the bead normalization step) DNA libraries foreach sample were quantified using a Quant-iT PicoGreendsDNA Assay Kit (Invitrogen) or a Qubit assay kit (Life technolo-gies) and average library size was measured with a high-
sensitivity DNA Bioanalyzer chip on a model 2100 Bioanalyzer(Agilent) Sample libraries were transported to the MRC Centrefor Virus Research at the University of Glasgow UK for the finalsteps of library preparation and sequencing Individual librarieswere pooled and normalized to equimolar concentrations at asuitable plexity (x24 for MiSeq runs) Libraries were sequencedas 150-bp paired-end reads on an Illumina MiSeq Additional se-quencing was conducted on a NextSeq 500 platform (GlasgowPolyomics at the University of Glasgow Glasgow UK) and readsmerged with MiSeq reads to increase coverage for poorly se-quenced samples (see Supplementary Material)
23 Bioinformatics and sequence analysis
Raw reads were assessed in FastQC (Andrews 2010) andTrimmomatic (Bolger Lohse and Usadel 2014) was used to trim3rsquo-ends remove adapter contamination and to filter based onquality with default parameters Filtered reads were mapped tothe previously sequenced genome of Tanzanian RABV sampleRV2772 (accession KF155002) with BWA mem version 0710(Li and Durbin 2009) and converted to bam file format usingSAMtools v 0118 (Li et al 2009)
A conservative single-nucleotide polymorphism (SNP) call-ing routine was implemented in GATK utilizing theUnifiedGenotyper tool to identify high confidence SNPs whichhad passed GATK filters on strand bias (FSgt 60 SORgt 4) map-ping quality (MQlt 40 MQRankSumlt125) read position(ReadPosRankSumlt8) and depth of coverage (DPlt 5) Indelswere filtered if FSgt 200 and ReadPosRankSumlt20 and furthermanually inspected for inclusion (eg dismissed if near a ho-mopolymer run) Consensus sequences were built using a cus-tom script in R which called filtered SNPs with a 75 per centconsensus rule (positions with lt75 consensus were given aIUPAC code for the corresponding ambiguous base call) and ge-nome positions with a depth of coverage less than two were la-belled lsquoNrsquo Potential SNP calls that failed only the depth filterthat is had a depthlt 5 but gt1 were passed if the same poly-morphism had been present as a high confidence SNP in at leasttwo other samples Otherwise the position was given an IUPACcode representing the population-level calls and the potentialSNP In addition a set of consensus sequences using a more re-laxed approach to SNP calling was produced which involvedstrict calls of all SNPs with depthgt 1 and gaps filled with themajority population consensus sequence These relaxed con-sensus sequences were used to produce initial starting trees forBEAST analyses (see Section 26)
Sequencing resulted in 93ndash100 per cent coverage of the ge-nome with gt99 per cent genome coverage achieved for 95 percent of samples and a median depth of coverage of seventy-seven (range 6ndash1871 see Supplementary Table S1) Nextera XT isa transposase-based method of library preparation and sequencereads typically miss the ends of the genome however as theends of lyssaviruses are highly conserved (Marston et al 2007Kuzmin et al 2008) it is unlikely that any informative variationwas missed We therefore consider our analyses to be based ongenome-wide variation and henceforth refer to our dataset aswhole-genome sequences Consensus sequences were alignedusing MAFFT v7149b (Katoh and Standley 2013) and submitted toGenBank (accession numbers KR906734ndashKR906792)
24 Phylogenetic reconstruction
Initial datasets of (1) partial N gene 405 bp (1317 sequences) and(2) full N gene 1350 bp (674 sequences) sequences isolated in
K Brunker et al | 3
Africa were constructed using sequences retrieved fromGenBank and including new Tanzanian isolates sequenced forthis study (59 new WGS samples and 50 new partial genome se-quences) Following maximum likelihood (ML) phylogenetic re-construction with the initial sequence datasets subset treeswere extracted for samples in the Africa 1b clade (430 samplesin the partial N dataset and 100 in the full N dataset)
Alignments for whole genome full N and partial N genewere created in MAFFT (Katoh and Standley 2013) and esti-mated phylogenetic relationships using both ML and Bayesianmethods ML phylogenies were estimated in RAxML(Stamatakis et al 2012) with a general time reversible (GTR) nu-cleotide substitution model and a gamma distribution model ofamong-site rate variation A Chinese dog RABV sequence fromGenBank (accession no FJ712193) was used as an outgroup andnode support was evaluated with 1000 bootstrap replicatesBayesian phylogenetic reconstruction was conducted in BEASTv181 (Drummond et al 2012) using a posterior distribution oftrees (without a molecular clock model) Phylogenies were visu-alized and annotated in R using the packages adegenet (Jombart2008) and APE (Paradis Claude and Strimmer 2004) and mapswere made in R with Maptools (Lewin-koh et al 2009) and sppackages (Pebesma and Bivand 2005 Bivand Pebesma andGomez-Rubio 2008) The degree of spatial admixture at largephylogeographic scales ie sub-continental and country levelwas quantified by an association index (AI) using BaTS softwarewith beast phylogenies (Parker et al 2008)
25 Selecting an evolutionary model
An initial nucleotide substitution model was chosen based onthe model selected by PartitionFinder (Lanfear et al 2012) Ourwhole-genome alignments were partitioned into sixteen sets ofnucleotides one for each codon position (CP) in each gene (fivegenes) and one for concatenated non-coding regions Modelscheme selection was based on the best AIC score from a greedysearch of substitution models which favoured a GTR modelwith partitioning into three CPs (CP123)
In addition model comparison based on marginal likelihoodestimates using path sampling (PS) and stepping stone (SS)sampling implemented in BEAST (100 path steps and a chainlength of 100000 steps) were used to test varying levels of com-plexity in the substitution model Non-coding sequence wasconcatenated and partitioned as a lsquogenersquo with its own evolution-ary model Specifically we tested the HKY model with (1) agene-specific nucleotide model with gene-specific rate varia-tion (2) a gene-linked CP partitioned model with among CP rateheterogeneity and homogeneous rates among genes and (3) agene-specific CP partitioned model with among CP and amonggene rate heterogeneity Model types 2 and 3 were also testedwith CP112 and CP123 partitioning schemes Following the re-sults for the best HKY model we did a final step comparing themost supported HKY model with the same structure but using aGTR model Results (Supplementary Table S2) strongly favouredGTR and CP models (CP123 was best supported) but there wasno support for partitions according to genes which all had simi-lar rates of substitution This significantly reduced the complex-ity of the model and is an important finding in the context ofanalysing RABV whole-genome sequence
26 Bayesian evolutionary analyses
Bayesian Markov-chain Monte Carlo analyses were performedusing BEAST v181 (Drummond et al 2012) and the BEAGLE
library (Ayres et al 2012) Based on model comparisons themost supported evolutionary model was a GTR model withdifferent substitution parameters for CPs one two and three(GTR123thornCP123thornC123) and homogeneous rates among geneswith a GTRthornG substitution model for non-coding sequenceA relaxed molecular clock with a lognormal distribution wasused to model rate variation among branches with aBayesian skyline model (Drummond et al 2005) with tengroups as a flexible tree prior A Bayesian skyline plot wasused to estimate the viral effective population size throughtime (Drummond et al 2005) To reconstruct the spatial dy-namics of RABV spread in Tanzania we implemented a dis-cretized diffusion process among nine regional samplinglocations formalized as an asymmetric continuous timeMarkov chain (CTMC) model (Lemey et al 2009) Three inde-pendent Markov-chain Monte Carlo chains with 50 millionstates and a sampling frequency of 50000 were combined inLogCombiner after discarding at least 10 per cent burnPosterior distributions were inspected in Tracer v16(Rambaut et al 2014) to ensure adequate mixing and conver-gence Initial analyses revealed issues with tree likelihoodconvergence Therefore a CTMC model using relaxed consen-sus sequences (see above) which contained fewer ambigui-ties was implemented first and the maximum cladecredibility (MCC) tree used as a starting tree for the finalCTMC models with conservative consensus sequences
To estimate the most significant pathways of viral dispersalbetween regions a stochastic search variable selection (BSSVS)procedure was implemented to identify the best supported dif-fusion rates through Bayes factor (BF) testing (Lemey et al 2009Bielejec et al 2011) For the per lineage rate of migration(Kuhnert Wu and Drummond 2011) a conditional referenceprior (Ferreira and Suchard 2008) is commonly used but for ourdata resulted in convergence problems with some of the BSSVSparameters Instead we used an exponential prior with a meanof 001 which gave the most robust results out of a range of val-ues tested (data not shown) The degree of spatial admixturewas scored using a modified Association Index (Wang et al2001 Lemey et al 2009) and quantified using the inferred num-ber of lineage migrationmovement events between locationsestimated by Markov jump counts (Minin and Suchard 2008)along the branches of the posterior tree distribution For BEASTmodels with Markov Jump counts implemented a conditionalreference prior on the per lineage rate of migration was chosenA summarized history of Markov jump counts was used to iden-tify movements between regions that occurred on very shortbranches and thus over unusually short time frames Dogsrarely move 1 km from their homestead (Hampson et al 2009Woodroffe and Donnelly 2011) and Hampson et al (2009) founda maximum distance of 20 km between linked RABV cases inthe Serengeti District Lineage migrations between regions rep-resenting distances gt100 km were therefore considered un-likely to be attributable to dog movement alone if they occurredover a period of 2 years or less and were instead interpreted asbeing the result of human-mediated movement In additionthe same form of discretized diffusion model was used to assessdiffusion at a broader scale that is between the North andSouth of Tanzania with Pemba Island classed as a third discretestate A BF test in the program SPREAD (Bielejec et al 2011) wasused to identify well-supported migration pathways (BFgt 3)Sampled trees were summarized as an MCC tree with mediannode heights using TreeAnnotator v181 and Figtree v142 wasused to visualize trees and the inferred ancestral locations forinternal branches
4 | Virus Evolution 2015 Vol 1 No 1
3 Results31 Geographic resolution partial versus full viralgenomes
Consistent with previous large-scale phylogenetic studies par-tial genome phylogenies indicated that RABV in sub-SaharanAfrica falls into several regional groups with viruses fromEastern Africa generally being genetically distinct from those inwestern central and southern parts of the continent(Supplementary Figs S1 and S2) Of the four major lineages ofRABV in Africa (David et al 2007 Bourhy et al 2008 Talbi et al2009) only the Cosmopolitan clade and more specifically theAfrica 1b lineage was detected in Tanzania as found previously(Lembo et al 2007) Within the Africa 1b lineage there was evi-dence of admixture between Tanzania and neighbouring coun-tries and occasional long-range admixture at a continental scale(ML trees in Fig 1 and Bayesian MCC trees in SupplementaryFig S3) Sequences clustering most closely with Tanzanian se-quences came from Kenya which shares a border to the northAlthough partial genome data were sufficient to identify suchlarge-scale spatial patterns these data did not provide adequateresolution to distinguish between samples at sub-national
scales within Tanzania The proportion of nodes with bootstrapsupport 80 per cent was only 011 (Bayesian posterior probabil-ity 90 018) for partial N and 027 (Bayesian 041) for full Ngene phylogenies Furthermore out of the sixty TanzanianWGS 60 per cent were identical at the partial N and 25 per centat the full N gene level In contrast ML and Bayesian trees basedon whole-genome sequences were fully resolved and well sup-ported (proportion of nodes with ML bootstrap support 80086 Bayesian posterior probability 90 089) even whensamples had been taken in close spatial and temporal proxim-ity For example RV2498 and RV2499 were sampled a day apartfrom the same village in Morogoro region both samples onlydiffered by a single SNP for the full N gene but were differenti-ated by twenty-five SNPs in the whole-genome alignment Thislarge divergence strongly suggests that the two samples are notfrom the same chain of transmission which might have beenthe conclusion based on partial genome data The median rawpairwise genetic distance between Tanzanian whole-genomesequences was 259 (range 0ndash608) nucleotides and between theSerengeti District samples was 120 (range 2ndash212) nucleotidesshowing considerable diversity at even a small spatiotemporalscale However much of this high divergence is due to the
(a) (b) (c)
Figure 1 ML trees derived from datasets of rabies virus sequences from the Africa 1b clade for increasing levels of genome coverage (a) 430 sequences from African
countries highlighted on the map for a 405 bp fragment of the nucleoprotein gene (b) 100 sequences of full 1350 bp nucleoprotein gene from the same countries (except
Botswana Ghana Kenya and Zimbabwe) and (c) sixty full or near-full genome sequences (range 11076ndash11923 bp) from Tanzania Trees are scaled by number of sub-
stitutions per site and node symbols indicate nodes with bootstrap support08 Historical samples from the Serengeti District (20 years old) are circled in partial ge-
nome trees
K Brunker et al | 5
presence of multiple lineages some of which are evident evenbased on partial genome data Using WGS we identified fivedistinct lineages with high posterior probability support (anno-tated in Fig 2)mdashmedian pairwise genetic distance withineach lineage is listed in Table 1 Bayesian phylogenetic recon-struction of WGS yielded a mean evolutionary rate of 144 104
substitutionssiteyear (95 highest posterior density578 107 to 319 104) similar to previous estimates for N
gene and G gene evolution (David et al 2007 Talbi et al 2009Ahmed et al 2015)
32 Phylogeography of RABV in Tanzania
Within Tanzania we found evidence of phylogeographic struc-ture (AIfrac14 070 Plt 0001) similar to other estimates of Africanintra-country AI values (Table 2) (Talbi et al 2009) Comparedwith the strong spatial structure between countries and largerspatial aggregations (Table 3) this indicates more fluid and dy-namic dispersal patterns within Tanzania as has been found inother African countries (Talbi et al 2010) Across the posteriordistribution of trees there were 24 (95 highest posterior den-sity 21ndash28) independent lineage movement events Using asummarized history of Markov Jump counts across the phylog-eny we found that 43 per cent of these migrations occurred inthe most recent ten years of the phylogeny (Fig 2c) A BSSVSprocedure in BEAST identified eighteen potential diffusionpathways to explain the observed phylogeographic patterns inthe posterior distribution However only four received substan-tial support based on BFgt 5 (Fig 2) Support was particularlystrong for dispersal from the Serengeti District to Morogoro
Figure 2 Regional phylogeography among sixty rabies virus whole-genome sequences sampled in Tanzania from 2003 to 2012 (a) an MCC tree with branches coloured
according to the most probable posterior location of its descendent node inferred by discrete-state phylogeographic reconstruction in BEAST Five major phylogenetic
groups (Tz1-5) are annotated on the tree and node symbols indicate node posterior support 09 (b) The four most significant dispersal pathways indicated by BF re-
sults from a BSSVS procedure in BEAST with the median number of transitions estimated by Markov jump counts indicated in cases where posterior support for a tran-
sition was gt07 (c) Markov jump densities for total number of transitions through time (d) Bayesian Skyline plot showing Nes the product of the effective population
size (Ne) and the generation time in years (s) through time
Table 1 Raw median genetic distance within each of the five mainrabies virus lineages identified in Tanzania
Genetic distance
RV lineage No of substitutions per site No of SNPs
Tz1 947E-03 110Tz2 227E-03 27Tz3 883E-03 95Tz4 417E-03 495Tz5 0 0
6 | Virus Evolution 2015 Vol 1 No 1
(BFfrac14 13530) and Markov jump counts estimated a median ofsix (range 3ndash10) migrations along this dispersal route with atleast one (range 1ndash3) occurring on a branch representing a pe-riod of less than 2 years Most lineages were sampled in morethan one region in Tanzania with some distributed across alarger geographic area than others (Fig 3) The largest lineageTz1 contains not only a cluster of Serengeti samples but alsoencompassed samples from a larger geographic extent and wasfound in eight out of the nine districts sampled The Bayesian
skyline plot revealed that the effective population size has re-mained fairly constant over the past 150 years (Fig 2d) Becauseof the much higher availability of samples from the SerengetiDistrict we also conducted a coarser phylogeographic analysisgrouping sequences into lsquoNorthrsquo lsquoSouthrsquo or lsquoPemba Islandrsquo Thisidentified a predominance of north to south dispersal (esti-mated thirteen independent movements compared with onemovement south to north) and evidence of dispersal back andforth between the North of mainland Tanzania and PembaIsland (Supplementary Fig S4)
4 Discussion
Using viral genetic data from a hierarchy of spatial scales andvarying levels of genome coverage we were able to demonstratethe advantages of whole-genome resolution to describe thespatio-temporal dynamics of endemically circulating caninerabies viruses
We found a clear phylogeographic structure between coun-tries in Africa which could be identified with partial genomedata This suggests that the majority of dispersal occurs at awithin-country scale and control programs would be most ap-propriate at a national scale with strategies to deal with poten-tial incursions from other countries However at regional andlocal scales within Tanzania the discriminatory power of par-tial genome data became too limited to reveal fine-scale popula-tion structure that could aid the effectiveness of controlinterventions In contrast WGS provided the resolution to ge-netically distinguish between all samples and produced a well-supported phylogeny Although sub-genomic information hasutility at broad phylogeographic scales (eg Horton et al 2015)this finding supports the application of WGS for studies aimingto discern population structure at a scale most relevant tocontrol
Although large-scale population structure according tosub-continental areas or country-specific lineages was appar-ent from sub-genomic data we also found incidences of oc-casional large-scale admixture (Supplementary Figs S1 andS2) We continued to find evidence at increasingly fine scalesTanzania and other countries within the Africa 1b cladeshowed signs of admixture particularly when countriesshared a border and within country movements of RABV fa-cilitated by humans were a feature of Tanzanian RABV Evenat a very local scale within the Serengeti District we foundmultiple co-circulating lineages This recurrent theme may bea characteristic of endemic RABV in Africa reflecting decadesof endemic circulation and human-mediated introductionsSimilar patterns have been observed in canine rabies-en-demic countries in South and Southeast Asia (Lumlertdachaet al 2006 Matsumoto et al 2013 Ahmed et al 2015) wherethe presence of co-circulating lineages was attributed to a
Figure 3 Spatial distribution of rabies virus lineages sampled from regions in
Tanzania between 2003 and 2012 with a colour gradient (yellow to red) indicat-
ing the total number of lineages (low to high) sampled in each region
Table 2 Degree of within-country rabies virus spatial admixture in three African countries measured using an Association Index (AI) whichranges from 0 indicating strong population subdivision to 1 indicating complete panmixis Results are shown for Tanzania (this study) andAlgeria and Morocco (Talbi et al 2010)
Country AI [95 BCI] P No of sequences No of locations Median distance (km) Min Max
Algeria 067 [062ndash073] lt0001 117 20 23308 2327 67462Morocco 055 [051ndash063] lt0001 133 28 32645 2862 92670Tanzania 070 [060-079] lt0001 60 9 43976 3905 108826
BCI Bayesian confidence interval
Table 3 Degree of spatial admixture between rabies virus samplesfrom Africa according to an Association Index (AI) AI values canrange from 0 indicating complete population subdivision to 1 indi-cating complete panmixis
Data Spatial clustering level No ofgroups
AI [95 BCI] P
N405 Sub-continent 3 004 [002005] lt001Country 14 006 [005008] lt001
N1350 Sub-continent 3 006 [004008] lt001Country 10 013 [009013] lt001
BCI Bayesian confidence interval Datasets of partial (N405) and full (N1350) nu-
cleoprotein sequences were tested at two levels of spatial aggregation (1) sub-
continent geographical partitions relative to Tanzania (three states Tanzania
neighbouring country and other African country) and (2) country of origin
K Brunker et al | 7
combination of historical introductions from neighbouringcountries and human-mediated movements
Much of the viral population structure we found withinTanzania is consistent with initial invasive waves that havepersisted endemically with structure eroding over time aidedby human-mediated movement Historical accounts describe arabies outbreak in southern regions of Tanzania in the mid-1950s which spread throughout the country and was recordedin the Serengeti District in the late 1970s (Magembe 1985Siongok and Karama 1985) This invasion perhaps shaped theinitial RABV population structure in Tanzania The timeline ofregional scale migrations (Fig 2c) indicates a strong rise in viraldispersal around this period possibly reflecting increasing hu-man connectivity Samples in Tz1 (Fig 2) share partial N geneidentity with samples from Kenya (Fig 1) indicating a sharedevolutionary history that may relate to an initial outbreakacross both countries Lineages Tz1 and Tz3 appear to have awide geographic distribution (Fig 3) and we found evidence of ageneral north to south dispersal pattern in Tanzania(Supplementary Fig S4) This highlights the potential for wide-spread dispersal should an incursion occur from Kenya and sug-gests a need for co-operative cross-border rabies managementonce a national control program is established Variation in thespread of different lineages across Tanzania may reflect the in-fluence of heterogeneous landscape features or dog populationstructure in impeding or facilitating RABV disseminationIdentifying and quantifying such features is the logical nextstep to provide additional information that can inform controlprogrammes such as identifying and strengthening pre-existing barriers Recent extensions of phylogeographic tech-niques have highlighted how this might be achieved using anintegrated approach combining evolutionary and ecologicalanalyses to quantify drivers of viral transmission (Lemey et al2014 Magee et al 2014)
The ancestry of lineages Tz4 and 5 (denoting lineages foundin the southern mainland and on the Island of Pemba respec-tively) points toward an introduction from the north ofTanzania (posterior probabilityfrac14 062) but the uncertainty ofthis ancestral location and the ancestral node itself (poste-riorfrac14 045) suggest these may be distinct historical lineageswith low-level persistence or undetected circulation elsewherein Tanzania Alternatively these clusters may represent in-stances where lineages from external sources have more re-cently invaded and resulted in sustained transmission inTanzaniamdashpartial genome phylogenies indicate Tz4 is relatedto clusters containing samples from countries south ofTanzania South Africa and Mozambique Furthermore Tz5shares a common ancestry with a Nigerian and several CentralAfrican Republic samples This again underlines the threat ofre-invasion or introduction from external sources and the po-tential value of whole-genome resolution to robustly and moreaccurately identify sources of new introductions To date themajority of RABV genetic data from Africa comes from partialgenome analyses however our data suggest that whole-genome characterization would be valuable and should be anaim for the future
Islands represent isolated landscapes with natural barriersto dispersal but incidents of RABV outbreaks instigated by hu-man-mediated introductions (eg to islands in Indonesia[Windiyaningsih et al 2004 Susilawathi et al 2012]) have oftenbeen recorded Sequences from the island of Pemba were sug-gestive of multiple introductions from various sources with evi-dence of dispersal to and from the mainland (SupplementaryFig S4) Samples from Pemba were scattered throughout the
phylogeny and the most divergent lineage Tz5 consisted of twosamples from Pemba The distribution of these lineages may re-flect earlier invasions of RABV into Pemba from elsewhere inTanzania (Tz1 and 2) and the African continent (Tz5) resultingfrom Pembarsquos location on an important trading route Howeversince these lineages were not resampled and no cases havebeen detected from Pemba in over a year (Lushasi pers comm)RABV may have been only transiently circulating Neverthelesslessons from Indonesia where lack of a swift and coordinatedresponse to a rabies incursion led to an epidemic (Townsendet al 2013) highlight the importance of active surveillance andrapid response to incursions
Although we found evidence of RABV spatial structurewithin Tanzania it was evident that dispersal was also fre-quent with at least one long distance migration occurringwithin a small temporal window (lt2 years) Lineage move-ments occurring on branches representing short evolutionarytimes such as those identified between Serengeti and Morogoro(gt750 km) indicate rates of dispersal much higher than thoserecorded for endemic wildlife rabies (Biek 2007) and imply hu-man mediated movements as seen in parts of North Africa(Talbi et al 2010) Movement of pastoralist and agro-pastoralistcommunities from the Lake zone in Northern Tanzania tosouthern regions for example Morogoro have been ongoingsince the 1950s (Walsh 2008) with a major influx reported from2003 to 2006 (PINGOrsquos Forum 2013) attributed to climate changeinduced drought and forced evictions from newly protectedareas (Kideghesho et al 2013) During these migrations pasto-ralists are accompanied by their dogs which may facilitate thelong distance movement of animals incubating the virus priorto transmission On further inspection we found that Morogorosamples indicated as instances of long-distance translocationcame from rural areas where pastoralists are likely to migrateto whereas other samples from urban Morogoro formed a dis-tinct cluster (Fig 2) We found three RABV lineages in Morogorofrom only ten samples (Fig 3) Quantifying networks of human-mediated movements (including livestock trade) would providea valuable proxy for connectivity for many zoonotic diseases af-fecting domesticated animals
Further to our findings of regional admixture in Tanzaniawe also observed considerable diversity at a very local scalethat is within the Serengeti District with several lineages co-circulating These lineages appear to have persisted for at least20 years (older Serengeti samples obtained from GenBank alsocluster within these lineages indicated in Fig 1) despite dogvaccination campaigns having been conducted in the districtsince 1996 While vaccination coverage has varied substantiallyacross years and between villages (Viana et al 2015) rabies inci-dence has at times significantly declined for example falling by97 per cent in the late 1990s (Cleaveland et al 2003) Yet our ge-netic data show that despite substantial declines in incidencethese lineages must have persisted at very low levels within thedistrict or subsequently reinvaded from neighbouring districtsWithout sampling the surrounding districts it is not possible todistinguish between these possibilities The skyline plot indi-cated a stable effective RABV population size (Ne) through time(Fig 3d) While this could be expected for an endemic pathogenit is worth noting that there was no evidence of viral populationsize reductions in response to vaccination campaigns Rabiescontrol efforts across much of Tanzania have been very limiteduntil recently and high turnover in the dog population(Hampson et al 2009) likely contributes to the stable persistenceof rabies in Tanzania It has also been noted that geographicstructure can obscure local fluctuations in subpopulations while
8 | Virus Evolution 2015 Vol 1 No 1
maintaining the appearance of a constant Ne in skyline plots(Carrington et al 2005)
This study represents a snapshot of RABV dynamics inTanzania indicating that human movements have dissemi-nated RABV out of locally endemic areas at scales relevant tocontrol that is administrative units such as regions or districtsThese frequent translocations have probably lead to the exis-tence of multiple co-circulating lineages (Fig 3) but relativelyfew introductions lead to sustained chains of transmission thatare detectable as lineage movement events However in diseasesystems closely associated with human activities the probabil-ity of successful translocation and establishment is likely to bemuch greater This suggests that without some level of regionalco-operation Tanzania will be unable to eliminate rabies andmaintain freedom from disease Human movements are oftenin response to social drivers which could be used as signals forincreased vigilance and surveillance in at risk areas
Our findings highlight the use of WGS to uncover fine scaletransmission patterns that can directly inform control effortsHowever sub-genomic approaches can still have utility at acoarser scale and are more easily obtained particularly whensample quality is an issue In particular they can be used to ini-tially identify admixture between countries which may indicatethe necessity of coordinated regional control programs and sur-veillance Co-circulation of multiple lineages and introductionsfacilitated by humans appear to be a feature of endemic rabies vi-rus and complicate the design of a sustainable control strategyHowever using whole-genome data we were able to identifysources of dispersal within Tanzania that could direct efforts to-ward surveillance and control The finding that humans play animportant role in the dynamics of RABV in Tanzania suggeststhat increasing awareness and dog vaccination in lsquohigh-riskrsquo com-munities such as pastoralists could help to reduce long-range dis-persal Moreover the design of enhanced surveillance andcontainment strategies to mitigate human-mediated incursionsand maintain disease freedom should be a priority once controlprograms are established and elimination is being targeted
Supplementary data
Supplementary data is available at VEVOLU Journal online
Acknowledgements
We acknowledge the Ministries of Livestock and FisheriesDevelopment and of Health and Social Welfare TanzaniaNational Parks Tanzania Wildlife Research InstituteNgorongoro Conservation Area Authority TanzaniaCommission for Science and Technology and NationalInstitute for Medical Research for permission and collabora-tion and the Arusha Mtwara and Mwanza VeterinaryInvestigation Centres Pemba Veterinary Office and theFrankfurt Zoological Society for logistical and technical sup-port We are grateful to the veterinary and livestock field of-ficers healthworkers and paravets throughout Tanzaniaand in particular to the following research assistants JoelChangalucha Zilpah Kaare Gurdeep Kour Ahmed LugeloKennedy Lushasi Mathias Magoto Khadija Said andRenatus Herman We thank Pelagia Muchuruza MatthewMaziku Geoffrey Mchau Eberhard Mbunda Francois-XavierMeslin and Bernadette Abela-Ridder for their collaborationsthrough the Bill amp Melinda GatesWHO rabies eliminationdemonstration project in Tanzania This work was
supported by the Wellcome Trust the Medical ResearchCouncil the UBS Optimus Foundation Lincoln Park Zooand the Research and Policy for Infectious DiseaseDynamics Program of the Science and TechnologyDirectorate Department of Homeland Security FogartyInternational Centre National Institute of Health
Conflict of interest None declared
ReferencesAhmed K et al (2015) lsquoMolecular Epidemiology of Rabies
Viruses Circulating in Two Rabies Endemic Provinces of Laos2011ndash2012 Regional Diversity in Southeast Asiarsquo PLoS NeglectedTropical Diseases 9 e0003645
Andrews S (2010) FastQC a quality control tool for highthroughput sequence datalthttpwwwbioinformaticsbbsrcacukprojectsfastqcgt accessed 4 Jun 2014
Ayres D L et al (2012) lsquoBEAGLE An Application ProgrammingInterface and High-Performance Computing Library forStatistical Phylogeneticsrsquo Systematic Biology 61 170ndash3
Bedford T et al (2014) lsquoIntegrating Influenza AntigenicDynamics with Molecular Evolutionrsquo eLife 2014 1ndash26
Biek R and Real L A (2010) lsquoThe Landscape Genetics of InfectiousDisease Emergence and Spreadrsquo Molecular Ecology 19 3515ndash31
mdashmdash et al (2007) lsquoA High-Resolution Genetic Signature ofDemographic and Spatial Expansion in Epizootic Rabies VirusrsquoProceedings of the National Academy of Sciences of the United Statesof America 104 7993ndash8
mdashmdash et al (2015) lsquoMeasurably Evolving Pathogens in the GenomicErarsquo Trends in Ecology amp Evolution 30 306ndash13
Bielejec F et al (2011) lsquoSPREAD Spatial PhylogeneticReconstruction of Evolutionary Dynamicsrsquo Bioinformatics(Oxford England) 27 2910ndash2
mdashmdash et al (2014) lsquoInferring Heterogeneous Evolutionary Processesthrough Time From Sequence Substitution toPhylogeographyrsquo Systematic Biology 63 493ndash504
Bivand R S Pebesma E J and Gomez-Rubio V (2008) AppliedSpatial Data Analysis with R New York Springer
Bolger A M Lohse M and Usadel B (2014) lsquoTrimmomatic AFlexible Trimmer for Illumina Sequence Datarsquo Bioinformatics30 2114ndash20
Bourhy H et al (2008) lsquoThe Origin and Phylogeography of DogRabies Virusrsquo The Journal of General Virology 89 2673ndash81
Brunker K et al (2012) lsquoIntegrating the Landscape Epidemiologyand Genetics of RNA Viruses Rabies in Domestic Dogs as aModelrsquo Parasitology 139 1899ndash913
Carrington C et al (2005) lsquoInvasion and Maintenance of DengueVirus Type 2 and Type 4 in the Americasrsquo Journal of Virology 7914680ndash7
Cleaveland S et al (2003) lsquoA Dog Rabies Vaccination Campaignin Rural Africa Impact on the Incidence of Dog Rabies andHuman Dog-Bite Injuriesrsquo Vaccine 21 1965ndash73
David D et al (2007) lsquoIdentification of Novel Canine Rabies VirusClades in the Middle East and North Africarsquo The Journal ofGeneral Virology 88 967ndash80
Denduangboripant J et al (2005) lsquoTransmission Dynamics ofRabies Virus in Thailand Implications for Disease ControlrsquoBMC Infectious Diseases 5 52
Drummond A J et al (2003) lsquoMeasurably Evolving PopulationsrsquoTrends in Ecology amp Evolution 18 481ndash8
mdashmdash et al (2005) lsquoBayesian Coalescent Inference of PastPopulation Dynamics from Molecular Sequencesrsquo MolecularBiology and Evolution 22 1185ndash92
K Brunker et al | 9
mdashmdash et al (2012) lsquoBayesian Phylogenetics with BEAUti and theBEAST 17rsquo Molecular Biology and Evolution 29 1969ndash73
Ferreira M A R and Suchard M A (2008) lsquoBayesian Analysis ofElapsed Times in Continuous-Time Markov Chainsrsquo CanadianJournal of Statistics 36 355ndash68
Greger M (2007) lsquoThe HumanAnimal Interface Emergence andResurgence of Zoonotic Infectious Diseasesrsquo Critical Reviews inMicrobiology 33 243ndash99
Hampson K et al (2007) lsquoSynchronous Cycles of Domestic DogRabies in Sub-Saharan Africa and the Impact of ControlEffortsrsquo Proceedings of the National Academy of Sciences of theUnited States of America 104 7717ndash22
mdashmdash et al (2009) lsquoTransmission Dynamics and Prospects for theElimination of Canine Rabiesrsquo PLoS Biology 7 e53
Hayman D T S et al (2011) lsquoEvolutionary History of Rabies inGhanarsquo PLoS Neglected Tropical Diseases 5 e1001
Holmes E C and Grenfell B T (2009) lsquoDiscovering the Phylodynamicsof RNA Virusesrsquo PLoS Computational Biology 5 e1000505
Horton D L et al (2015) lsquoComplex Epidemiology of a ZoonoticDisease in a Culturally Diverse Region Phylogeography ofRabies Virus in the Middle Eastrsquo PLOS Neglected TropicalDiseases 9 e0003569
Jombart T (2008) lsquoadegenet A R Package for the MultivariateAnalysis of Genetic Markersrsquo Bioinformatics (Oxford England)24 1403ndash5
Jones K E et al (2008) lsquoGlobal Trends in Emerging InfectiousDiseasesrsquo Nature 451 990ndash3
Katoh K and Standley D M (2013) lsquoMAFFT MultipleSequence Alignment Software Version 7 Improvements inPerformance and Usabilityrsquo Molecular Biology and Evolution30 772ndash80
Kideghesho J et al (2013) lsquoEmerging Issues and Challenges inConservation of Biodiversity in the Rangelands of TanzaniarsquoNature Conservation 6 1ndash29
Kissi B Tordo N and Bourhy H (1995) lsquoGenetic Polymorphismin the Rabies Virus Nucleoprotein Genersquo Virology 209 526ndash37
Knobel D L et al (2005) lsquoRe-Evaluating the Burden of Rabies inAfrica and Asiarsquo Bulletin of the World Health Organization 83360ndash8
Kuhnert D Wu C-H and Drummond A J (2011) lsquoPhylogeneticand Epidemic Modeling of Rapidly Evolving InfectiousDiseasesrsquo Infection Genetics and Evolution Journal of MolecularEpidemiology and Evolutionary Genetics in Infectious Diseases 111825ndash41
Kuzmin I V et al (2008) lsquoComplete Genomes of AravanKhujand Irkut and West Caucasian Bat Viruses with SpecialAttention to the Polymerase Gene and Non-Coding RegionsrsquoVirus Research 136 81ndash90
Kuzmina N A et al (2013) lsquoThe Phylogeography andSpatiotemporal Spread of South-Central Skunk Rabies VirusrsquoPloS One 8 e82348
Lanfear R et al (2012) lsquoPartitionfinder Combined Selectionof Partitioning Schemes and Substitution Models forPhylogenetic Analysesrsquo Molecular Biology and Evolution 291695ndash701
Lembo T et al (2007) lsquoMolecular Epidemiology Identifies Only aSingle Rabies Virus Variant Circulating in Complex CarnivoreCommunities of the Serengetirsquo Proceedings Biological SciencesThe Royal Society 274 2123ndash30
mdashmdash et al (2008) lsquoExploring Reservoir Dynamics A Case Study ofRabies in the Serengeti Ecosystemrsquo Journal of Applied Ecology45 1246ndash57
Lemey P et al (2009) lsquoBayesian Phylogeography Finds Its RootsrsquoPLoS Computational Biology 5 e1000520
mdashmdash et al (2014) lsquoUnifying Viral Genetics and HumanTransportation Data to Predict the Global TransmissionDynamics of Human Influenza H3N2rsquo PLoS Pathogens 10e1003932
Lewin-koh A N J et al (2009) Maptools Tools for Reading andHandling Spatial Objects R package version 08-23 lthttpcranr-projectorgwebpackagesmaptoolsindexhtmlgt accessed 1Sept 2014
Li H and Durbin R (2009) lsquoFast and Accurate Short ReadAlignment with Burrows-Wheeler Transformrsquo Bioinformatics(Oxford England) 25 1754ndash60
mdashmdash et al (2009) lsquoThe Sequence AlignmentMap Format andSAMtoolsrsquo Bioinformatics (Oxford England) 25 2078ndash9
Lumlertdacha B et al (2006) lsquoComplex Genetic Structure ofthe Rabies Virus in Bangkok and Its Surrounding ProvincesThailand Implications for Canine Rabies ControlrsquoTransactions of the Royal Society of Tropical Medicine andHygiene 100 276ndash81
Magee D et al (2014) lsquoCombining Phylogeography and SpatialEpidemiology to Uncover Predictors of H5N1 Influenza A VirusDiffusionrsquo Archives of Virology 160 215ndash24
Magembe S (1985) lsquoEpidemiology of Rabies in United Republic ofTanzaniarsquo in Kuwer E et al (eds) Rabies in the Tropics pp 392ndash8 New York Springer Berlin Heidelberg
Marston D A et al (2007) lsquoComparative Analysis of the FullGenome Sequence of European Bat Lyssavirus Type 1 andType 2 with Other Lyssaviruses and Evidence for a ConservedTranscription Termination and Polyadenylation Motif in theG-L 3rsquo Non-Translated Regionrsquo The Journal of General Virology88 1302ndash14
mdashmdash et al (2013) lsquoNext Generation Sequencing of Viral RNAGenomesrsquo BMC Genomics 14 444
Matsumoto T et al (2013) lsquoMolecular Epidemiology of HumanRabies Viruses in Sri Lankarsquo Infection Genetics and Evolution 18160ndash7
Minin V N and Suchard M A (2008) lsquoCounting LabeledTransitions in Continuous-Time Markov Models of EvolutionrsquoJournal of Mathematical Biology 56 391ndash412
Mollentze N et al (2014) lsquoA Bayesian Approach for Inferring theDynamics of Partially Observed Endemic Infectious Diseasesfrom Space-Time-Genetic Datarsquo Proceedings Biological SciencesThe Royal Society 281 20133251
Nel L H (2013) lsquoDiscrepancies in Data Reporting for RabiesAfricarsquo Emerging Infectious Diseases 19 529ndash33
Nelson M I et al (2015) lsquoGlobal Migration of Influenza A Virusesin Swinersquo Nature Communications 6 6696
Nunes M R T et al (2014) lsquoAir Travel is Associated withIntracontinental Spread of Dengue Virus Serotypes 1-3 inBrazilrsquo PLoS Neglected Tropical Diseases 8 e2769
Paradis E Claude J and Strimmer K (2004) lsquoAPE Analyses ofPhylogenetics and Evolution in R Languagersquo Bioinformatics 20289ndash90
Parker J Rambaut A and Pybus O G (2008) lsquoCorrelatingViral Phenotypes with Phylogeny Accounting forPhylogenetic Uncertaintyrsquo Infection Genetics and Evolution 8239ndash46
Pebesma E J and Bivand R S (2005) lsquoClasses and Methods forSpatial Data in Rrsquo R News 5 9ndash13
PINGOrsquos Forum (2013) Pastoralists Indigenous Non GovernmentalOrganizationsrsquo Forum Annual Report 2012-2013 lthttpwwwpin-gosforumortzgt accessed 23 Nov 2014
Rambaut A et al (2014) lsquoTracer v16rsquo Computer program anddocumentation distributed by the author lthttpbeastbioedacukTracergt accessed 4 Feb 2015
10 | Virus Evolution 2015 Vol 1 No 1
Real L A et al (2005) lsquoUnifying the Spatial Population Dynamicsand Molecular Evolution of Epidemic Rabies Virusrsquo Proceedingsof the National Academy of Sciences of the United States of America102 12107ndash11
Rupprecht C E Hanlon C A and Hemachudha T (2002)lsquoRabies Re-examinedrsquo The Lancet Infectious Diseases 2 327ndash43
Shwiff S Hampson K and Anderson A (2013) lsquoPotentialEconomic Benefits of Eliminating Canine Rabiesrsquo AntiviralResearch 98 352ndash6
Siongok T and Karama M (1985) lsquoEpidemiology of HumanRabies in Kenyarsquo in E Kuwer et al (eds) Rabies in the Tropicspp 445ndash50 New York Springer Berlin Heidelberg
Stamatakis A et al (2012) lsquoRAxML-Light A Tool for ComputingTerabyte Phylogeniesrsquo Bioinformatics (Oxford England) 28 2064ndash6
Susilawathi N M et al (2012) lsquoEpidemiological and ClinicalFeatures of Human Rabies Cases in Bali 2008-2010rsquo BMCInfectious Diseases 12 81
Talbi C et al (2009) lsquoEvolutionary History and Dynamics of DogRabies Virus in Western and Central Africarsquo The Journal ofGeneral Virology 90 783ndash91
mdashmdash et al (2010) lsquoPhylodynamics and Human-Mediated Dispersalof a Zoonotic Virusrsquo PLoS Pathogens 6 e1001166
Tao X Y et al (2009) lsquoMolecular Epidemiology of Rabies inSouthern Peoplersquos Republic of Chinarsquo Emerging InfectiousDiseases 15 1192ndash8
Townsend S E et al (2013) lsquoDesigning Programs for EliminatingCanine Rabies from Islands Bali Indonesia as a Case StudyrsquoPLoS Neglected Tropical Diseases 7 e2372
Viana M et al (2015) lsquoDynamics of a Morbillivirus at theDomesticndashWildlife Interface Canine Distemper Virus inDomestic Dogs and Lionsrsquo Proceedings of the National Academy ofSciences of the United States of America 112 1464ndash9
Walsh M (2008) Pastoralism and Policy Processes in TanzaniaMbarali Case Study Report to the Tanzania Natural ResourceForum Arusha
Wang T H et al (2001) lsquoIdentification of Shared Populations ofHuman Immunodeficiency Virus Type 1 Infecting Microgliaand Tissue Macrophages Outside the Central Nervous SystemrsquoJournal of Virology 75 11686ndash99
WHO (2005) lsquoWHO Expert Consultation on Rabiesrsquo World HealthOrganization technical report series 931 1ndash121
Windiyaningsih C et al (2004) lsquoThe Rabies Epidemic on FloresIsland Indonesia (1998-2003)rsquo Journal of the Medical Associationof Thailandfrac14Chotmaihet thangphaet 87 1389ndash93
Woodroffe R and Donnelly C A (2011) lsquoRisk of Contact be-tween Endangered African Wild Dogs Lycaon pictus andDomestic Dogs Opportunities for Pathogen TransmissionrsquoJournal of Applied Ecology 48 1345ndash54
Woolhouse M E J (2002) lsquoPopulation Biology of Emerging and Re-emerging Pathogensrsquo Trends in Microbiology 10(10 Suppl) S3ndash7
K Brunker et al | 11
- vev011-TF1
- vev011-TF2
-

can that can guide interventions such as the spatial scale and design of dog vaccination campaigns and dog movementcontrols to achieve and maintain freedom from disease
Key words RNA virus phylodynamics zoonoses endemic rabies translocation
1 Introduction
The general trend of increasing incidence and expansion ofemerging or re-emerging zoonotic diseases (eg EbolaChikungunya and avian influenza) (Woolhouse 2002 Greger2007 Jones et al 2008) and persistence of established zoonosessuch as canine rabies highlights the ongoing challenges facedas we attempt to characterize and control them The processesthat drive the spread and persistence of infectious diseases arereflected in a genetic signature in pathogen genomes (Biek andReal 2010) Understanding the processes that give rise to spatialpopulation structure in pathogens can inform the managementand control of infectious diseases For example analyses of evo-lutionary epidemiological and ecological data have recentlydemonstrated that global live swine trade strongly predicts theglobal dissemination of influenza A viruses in swine (Nelsonet al 2015) and air travel has been revealed as a major factordriving the intra-continental spread of Dengue virus (Nuneset al 2014) Viral pathogens particularly fast-evolving RNA vi-ruses are model systems to explore pathogen populations asthey rapidly accumulate genetic diversity on a timescale similarto epidemiological processes (Drummond et al 2003 Biek et al2015) Statistical phylogeographic approaches are available(Lemey et al 2009 Bedford et al 2014 Bielejec et al 2014) to de-velop a quantitative understanding of the processes that giverise to spatial patterns in RNA viruses (Holmes and Grenfell2009) on epidemiological time scales Whole-genome sequenc-ing (WGS) is increasingly being used as a means to extract thesepatterns but it is unclear how much resolution can be gainedand at what temporal and spatial scale In this article caninerabies virus (RABV) is used as a model to determine the spatio-temporal patterns of an endemic zoonotic virus using whole-genome data to distinguish structure at an increasingly finescale
Rabies is a globally distributed zoonotic disease caused by asingle-stranded negative sense RNA virus from the Lyssavirusgenus Though capable of infecting any mammal given virusvariants are typically maintained in distinct species-specific cy-cles within the orders Carnivora and Chiroptera (RupprechtHanlon and Hemachudha 2002) The disease causes thousandsof human deaths every year predominantly in Asia and Africawhere the virus circulates endemically in domestic dogs (Canislupus familiaris) (Knobel et al 2005 Shwiff Hampson andAnderson 2013) The majority of these deaths (99) are causedby bites from rabid dogs instilling fear into the many communi-ties that live under continuous threat from a disease that is al-most invariably fatal but entirely preventable Although the roleof domestic dogs as key vectors of rabies is recognized muchless is known about the dog-associated RABV variant than wild-life variants such as raccoon or skunk RABVs circulating inNorth America (Brunker et al 2012) Moreover while epidemicexpansions of wildlife RABV have been well documented andstudied (eg Real et al 2005 Biek et al 2007 Kuzmina et al2013) we know little about the persistence and spread of rabiesin endemic landscapes
Characterizing the spatial scales of canine rabies dispersalis a critical step toward identifying the processes and factors
driving its dynamics and the scale at which control strategiesneed to be implemented On a global scale canine RABV ex-hibits a strong phylogeographic structure with the distribu-tion of seven distinct major clades reflecting the position ofmajor barriers such as oceans and mountain ranges or his-torical mass human colonizationmigration events (Davidet al 2007 Bourhy et al 2008) However it is unclear whetherthis genetic structure will persist in endemic scenarios or atsmaller scales and how much it is influenced by human-me-diated dispersal Indeed on a regional scale this landscapestructure becomes less distinct some landscape features forexample geopolitical boundaries can act as apparent barriersto movement as seen in North Africa (Talbi et al 2010)whilst contradictory patterns of synchronous cycles of RABVacross multiple countries (Hampson et al 2007) and repeatedcross-border incursions (Hayman et al 2011) have also beenobserved elsewhere in the continent
While there is a growing understanding of the epidemiologyof canine rabies in Africa (Hampson et al 2007 2009 Lemboet al 2008) effective rabies control is still hindered by limitedknowledge of some of the key drivers of viral transmission andspread Mass dog vaccination is the mainstay of successful ra-bies control but requires sustained coverage of at least 70 percent (WHO 2005 Townsend et al 2013) In addition spatial het-erogeneity may affect how vaccine is most effectively distrib-uted to interrupt key transmission corridors and target regionsseeding RABV dispersal An important aspect of this heteroge-neity is the impact of human factors on RABV transmissionwhich has direct implications for control including the designand scale of interventions necessary to interrupt transmissionand maintain freedom from disease For example movement ofpeople between urban and rural areas and the dog meat tradehave been postulated as means of spreading RABV through hu-man-mediated dog movements in rabies-endemic countries inAsia (Denduangboripant et al 2005 Tao et al 2009 Ahmed et al2015) Uncovering the viral population structure and dynamicsof RABV in Tanzania may identify similar patterns attributed tohuman-mediated movements that can aid the identification ofsources key to viral persistence
There have been few in-depth spatial epidemiology studies ofRABV in sub-Saharan Africa probably owing to the lack of re-sources for effective surveillance including sample collection (Nel2013) However recent studies provide intriguing insights intothe dynamics of rabies in certain parts of Africa (see Talbi et al2010 Mollentze et al 2014) indicating a degree of spatial struc-ture with evidence of long distance movements facilitated by hu-mans RABV spatiotemporal dynamics in East Africa in particularare poorly resolved and very few sequences are publically avail-able At present little is known about the genetic diversity orstructure of RABV in Tanzania other than coarse phylogeneticanalyses of partial or full nucleoprotein gene (N gene) sequenceslimited to well-studied regions (Kissi Tordo and Bourhy 1995Lembo et al 2007) Furthermore whole-genome population stud-ies of RABV have not yet been attempted despite their great po-tential to provide a better understanding of the processesdetermining rabies spread and persistence
2 | Virus Evolution 2015 Vol 1 No 1
In this study finely resolved space-time-genetic data in-cluding whole-genome sequences were used to determine thespatial and temporal dynamics of endemic RABV at both a re-gional and local scale in Tanzania The utility of partial genomedata was also demonstrated as a means to characterize large-scale phylogeographic patterns of RABV in Africa Specificallywe aimed to characterize the dynamics of rabies virus in an en-demic system including the role of human-mediated transport
2 Materials and methods21 Samples
For this study fifty-nine new whole-genome sequences were ob-tained from animal hosts (primarily domestic dogs) from nine re-gions in Tanzania sampled between 2003 and 2012 A previouslysequenced sample (RV2772 accession KF155002) (Marston et al2013) from Southern Tanzania was included in the dataset andused as a reference sequence (sample details in SupplementaryTable S1) The main study area encompassed the SerengetiDistrict in northwest Tanzania where approximately half thesamples (nfrac14 33) were obtained from active surveillance whichenabled the collection of brain material from suspect rabid ani-mals and GPS coordinates and dates recorded as described inHampson et al (2009) The remaining twenty-seven sampleswere obtained opportunistically from other regions in Tanzaniaas part of surveillance by the Tanzanian Ministry of Livestockand Fisheries Development and the Tanzanian VeterinaryLaboratory Agency All samples were sent to the Animal amp PlantHealth Agency (APHA) in Weybridge UK for processing
In addition fifty new partial N gene sequences (405 bp) fromTanzania were obtained via reverse transcription-polymerasechain reaction (RT-PCR) and Sanger sequencing using samplesarchived at APHA and submitted to GenBank with accessionnumbers KR534217ndashKR534266 (details in SupplementaryMaterial and Table S2)
22 RNA extraction and WGS
Total RNA was extracted at APHA directly from brain tissue us-ing TRIZOL according to manufacturerrsquos instructions(Invitrogen) Precipitated total RNA was re-suspended in molec-ular-grade water at a one in ten dilution and quantified using aNanoDrop spectrophotometer (Thermo Scientific) Sampleswere sequenced on a range of next generation sequencing plat-forms during NGS protocol optimization (see SupplementaryMaterial for details) The majority of samples (nfrac14 48) were se-quenced by the following method TRIzol-extracted viral RNAwas depleted of host genomic DNA using the on-column DNasetreatment in RNeasy plus mini kit (Qiagen) as per manufac-turerrsquos instructions with elution in 30 ml molecular grade waterThis was followed by host ribosomal RNA depletion usingTerminator 5rsquo-phosphate-dependent exonuclease (EpicentreBiotechnologies) as detailed in (Marston et al 2013) First- andsecond-strand cDNA was synthesized using a Roche cDNA syn-thesis system kit with random hexamers (Roche) ResultantcDNA was quantified using Picogreen dsDNA quantitation re-agent (Invitrogen) and approximately 1 ng of each sample usedin a lsquotagmentationrsquo reaction mix using a Nextera XT samplepreparation kit (Illumina) according to the manufacturerrsquos pro-tocol (minus the bead normalization step) DNA libraries foreach sample were quantified using a Quant-iT PicoGreendsDNA Assay Kit (Invitrogen) or a Qubit assay kit (Life technolo-gies) and average library size was measured with a high-
sensitivity DNA Bioanalyzer chip on a model 2100 Bioanalyzer(Agilent) Sample libraries were transported to the MRC Centrefor Virus Research at the University of Glasgow UK for the finalsteps of library preparation and sequencing Individual librarieswere pooled and normalized to equimolar concentrations at asuitable plexity (x24 for MiSeq runs) Libraries were sequencedas 150-bp paired-end reads on an Illumina MiSeq Additional se-quencing was conducted on a NextSeq 500 platform (GlasgowPolyomics at the University of Glasgow Glasgow UK) and readsmerged with MiSeq reads to increase coverage for poorly se-quenced samples (see Supplementary Material)
23 Bioinformatics and sequence analysis
Raw reads were assessed in FastQC (Andrews 2010) andTrimmomatic (Bolger Lohse and Usadel 2014) was used to trim3rsquo-ends remove adapter contamination and to filter based onquality with default parameters Filtered reads were mapped tothe previously sequenced genome of Tanzanian RABV sampleRV2772 (accession KF155002) with BWA mem version 0710(Li and Durbin 2009) and converted to bam file format usingSAMtools v 0118 (Li et al 2009)
A conservative single-nucleotide polymorphism (SNP) call-ing routine was implemented in GATK utilizing theUnifiedGenotyper tool to identify high confidence SNPs whichhad passed GATK filters on strand bias (FSgt 60 SORgt 4) map-ping quality (MQlt 40 MQRankSumlt125) read position(ReadPosRankSumlt8) and depth of coverage (DPlt 5) Indelswere filtered if FSgt 200 and ReadPosRankSumlt20 and furthermanually inspected for inclusion (eg dismissed if near a ho-mopolymer run) Consensus sequences were built using a cus-tom script in R which called filtered SNPs with a 75 per centconsensus rule (positions with lt75 consensus were given aIUPAC code for the corresponding ambiguous base call) and ge-nome positions with a depth of coverage less than two were la-belled lsquoNrsquo Potential SNP calls that failed only the depth filterthat is had a depthlt 5 but gt1 were passed if the same poly-morphism had been present as a high confidence SNP in at leasttwo other samples Otherwise the position was given an IUPACcode representing the population-level calls and the potentialSNP In addition a set of consensus sequences using a more re-laxed approach to SNP calling was produced which involvedstrict calls of all SNPs with depthgt 1 and gaps filled with themajority population consensus sequence These relaxed con-sensus sequences were used to produce initial starting trees forBEAST analyses (see Section 26)
Sequencing resulted in 93ndash100 per cent coverage of the ge-nome with gt99 per cent genome coverage achieved for 95 percent of samples and a median depth of coverage of seventy-seven (range 6ndash1871 see Supplementary Table S1) Nextera XT isa transposase-based method of library preparation and sequencereads typically miss the ends of the genome however as theends of lyssaviruses are highly conserved (Marston et al 2007Kuzmin et al 2008) it is unlikely that any informative variationwas missed We therefore consider our analyses to be based ongenome-wide variation and henceforth refer to our dataset aswhole-genome sequences Consensus sequences were alignedusing MAFFT v7149b (Katoh and Standley 2013) and submitted toGenBank (accession numbers KR906734ndashKR906792)
24 Phylogenetic reconstruction
Initial datasets of (1) partial N gene 405 bp (1317 sequences) and(2) full N gene 1350 bp (674 sequences) sequences isolated in
K Brunker et al | 3
Africa were constructed using sequences retrieved fromGenBank and including new Tanzanian isolates sequenced forthis study (59 new WGS samples and 50 new partial genome se-quences) Following maximum likelihood (ML) phylogenetic re-construction with the initial sequence datasets subset treeswere extracted for samples in the Africa 1b clade (430 samplesin the partial N dataset and 100 in the full N dataset)
Alignments for whole genome full N and partial N genewere created in MAFFT (Katoh and Standley 2013) and esti-mated phylogenetic relationships using both ML and Bayesianmethods ML phylogenies were estimated in RAxML(Stamatakis et al 2012) with a general time reversible (GTR) nu-cleotide substitution model and a gamma distribution model ofamong-site rate variation A Chinese dog RABV sequence fromGenBank (accession no FJ712193) was used as an outgroup andnode support was evaluated with 1000 bootstrap replicatesBayesian phylogenetic reconstruction was conducted in BEASTv181 (Drummond et al 2012) using a posterior distribution oftrees (without a molecular clock model) Phylogenies were visu-alized and annotated in R using the packages adegenet (Jombart2008) and APE (Paradis Claude and Strimmer 2004) and mapswere made in R with Maptools (Lewin-koh et al 2009) and sppackages (Pebesma and Bivand 2005 Bivand Pebesma andGomez-Rubio 2008) The degree of spatial admixture at largephylogeographic scales ie sub-continental and country levelwas quantified by an association index (AI) using BaTS softwarewith beast phylogenies (Parker et al 2008)
25 Selecting an evolutionary model
An initial nucleotide substitution model was chosen based onthe model selected by PartitionFinder (Lanfear et al 2012) Ourwhole-genome alignments were partitioned into sixteen sets ofnucleotides one for each codon position (CP) in each gene (fivegenes) and one for concatenated non-coding regions Modelscheme selection was based on the best AIC score from a greedysearch of substitution models which favoured a GTR modelwith partitioning into three CPs (CP123)
In addition model comparison based on marginal likelihoodestimates using path sampling (PS) and stepping stone (SS)sampling implemented in BEAST (100 path steps and a chainlength of 100000 steps) were used to test varying levels of com-plexity in the substitution model Non-coding sequence wasconcatenated and partitioned as a lsquogenersquo with its own evolution-ary model Specifically we tested the HKY model with (1) agene-specific nucleotide model with gene-specific rate varia-tion (2) a gene-linked CP partitioned model with among CP rateheterogeneity and homogeneous rates among genes and (3) agene-specific CP partitioned model with among CP and amonggene rate heterogeneity Model types 2 and 3 were also testedwith CP112 and CP123 partitioning schemes Following the re-sults for the best HKY model we did a final step comparing themost supported HKY model with the same structure but using aGTR model Results (Supplementary Table S2) strongly favouredGTR and CP models (CP123 was best supported) but there wasno support for partitions according to genes which all had simi-lar rates of substitution This significantly reduced the complex-ity of the model and is an important finding in the context ofanalysing RABV whole-genome sequence
26 Bayesian evolutionary analyses
Bayesian Markov-chain Monte Carlo analyses were performedusing BEAST v181 (Drummond et al 2012) and the BEAGLE
library (Ayres et al 2012) Based on model comparisons themost supported evolutionary model was a GTR model withdifferent substitution parameters for CPs one two and three(GTR123thornCP123thornC123) and homogeneous rates among geneswith a GTRthornG substitution model for non-coding sequenceA relaxed molecular clock with a lognormal distribution wasused to model rate variation among branches with aBayesian skyline model (Drummond et al 2005) with tengroups as a flexible tree prior A Bayesian skyline plot wasused to estimate the viral effective population size throughtime (Drummond et al 2005) To reconstruct the spatial dy-namics of RABV spread in Tanzania we implemented a dis-cretized diffusion process among nine regional samplinglocations formalized as an asymmetric continuous timeMarkov chain (CTMC) model (Lemey et al 2009) Three inde-pendent Markov-chain Monte Carlo chains with 50 millionstates and a sampling frequency of 50000 were combined inLogCombiner after discarding at least 10 per cent burnPosterior distributions were inspected in Tracer v16(Rambaut et al 2014) to ensure adequate mixing and conver-gence Initial analyses revealed issues with tree likelihoodconvergence Therefore a CTMC model using relaxed consen-sus sequences (see above) which contained fewer ambigui-ties was implemented first and the maximum cladecredibility (MCC) tree used as a starting tree for the finalCTMC models with conservative consensus sequences
To estimate the most significant pathways of viral dispersalbetween regions a stochastic search variable selection (BSSVS)procedure was implemented to identify the best supported dif-fusion rates through Bayes factor (BF) testing (Lemey et al 2009Bielejec et al 2011) For the per lineage rate of migration(Kuhnert Wu and Drummond 2011) a conditional referenceprior (Ferreira and Suchard 2008) is commonly used but for ourdata resulted in convergence problems with some of the BSSVSparameters Instead we used an exponential prior with a meanof 001 which gave the most robust results out of a range of val-ues tested (data not shown) The degree of spatial admixturewas scored using a modified Association Index (Wang et al2001 Lemey et al 2009) and quantified using the inferred num-ber of lineage migrationmovement events between locationsestimated by Markov jump counts (Minin and Suchard 2008)along the branches of the posterior tree distribution For BEASTmodels with Markov Jump counts implemented a conditionalreference prior on the per lineage rate of migration was chosenA summarized history of Markov jump counts was used to iden-tify movements between regions that occurred on very shortbranches and thus over unusually short time frames Dogsrarely move 1 km from their homestead (Hampson et al 2009Woodroffe and Donnelly 2011) and Hampson et al (2009) founda maximum distance of 20 km between linked RABV cases inthe Serengeti District Lineage migrations between regions rep-resenting distances gt100 km were therefore considered un-likely to be attributable to dog movement alone if they occurredover a period of 2 years or less and were instead interpreted asbeing the result of human-mediated movement In additionthe same form of discretized diffusion model was used to assessdiffusion at a broader scale that is between the North andSouth of Tanzania with Pemba Island classed as a third discretestate A BF test in the program SPREAD (Bielejec et al 2011) wasused to identify well-supported migration pathways (BFgt 3)Sampled trees were summarized as an MCC tree with mediannode heights using TreeAnnotator v181 and Figtree v142 wasused to visualize trees and the inferred ancestral locations forinternal branches
4 | Virus Evolution 2015 Vol 1 No 1
3 Results31 Geographic resolution partial versus full viralgenomes
Consistent with previous large-scale phylogenetic studies par-tial genome phylogenies indicated that RABV in sub-SaharanAfrica falls into several regional groups with viruses fromEastern Africa generally being genetically distinct from those inwestern central and southern parts of the continent(Supplementary Figs S1 and S2) Of the four major lineages ofRABV in Africa (David et al 2007 Bourhy et al 2008 Talbi et al2009) only the Cosmopolitan clade and more specifically theAfrica 1b lineage was detected in Tanzania as found previously(Lembo et al 2007) Within the Africa 1b lineage there was evi-dence of admixture between Tanzania and neighbouring coun-tries and occasional long-range admixture at a continental scale(ML trees in Fig 1 and Bayesian MCC trees in SupplementaryFig S3) Sequences clustering most closely with Tanzanian se-quences came from Kenya which shares a border to the northAlthough partial genome data were sufficient to identify suchlarge-scale spatial patterns these data did not provide adequateresolution to distinguish between samples at sub-national
scales within Tanzania The proportion of nodes with bootstrapsupport 80 per cent was only 011 (Bayesian posterior probabil-ity 90 018) for partial N and 027 (Bayesian 041) for full Ngene phylogenies Furthermore out of the sixty TanzanianWGS 60 per cent were identical at the partial N and 25 per centat the full N gene level In contrast ML and Bayesian trees basedon whole-genome sequences were fully resolved and well sup-ported (proportion of nodes with ML bootstrap support 80086 Bayesian posterior probability 90 089) even whensamples had been taken in close spatial and temporal proxim-ity For example RV2498 and RV2499 were sampled a day apartfrom the same village in Morogoro region both samples onlydiffered by a single SNP for the full N gene but were differenti-ated by twenty-five SNPs in the whole-genome alignment Thislarge divergence strongly suggests that the two samples are notfrom the same chain of transmission which might have beenthe conclusion based on partial genome data The median rawpairwise genetic distance between Tanzanian whole-genomesequences was 259 (range 0ndash608) nucleotides and between theSerengeti District samples was 120 (range 2ndash212) nucleotidesshowing considerable diversity at even a small spatiotemporalscale However much of this high divergence is due to the
(a) (b) (c)
Figure 1 ML trees derived from datasets of rabies virus sequences from the Africa 1b clade for increasing levels of genome coverage (a) 430 sequences from African
countries highlighted on the map for a 405 bp fragment of the nucleoprotein gene (b) 100 sequences of full 1350 bp nucleoprotein gene from the same countries (except
Botswana Ghana Kenya and Zimbabwe) and (c) sixty full or near-full genome sequences (range 11076ndash11923 bp) from Tanzania Trees are scaled by number of sub-
stitutions per site and node symbols indicate nodes with bootstrap support08 Historical samples from the Serengeti District (20 years old) are circled in partial ge-
nome trees
K Brunker et al | 5
presence of multiple lineages some of which are evident evenbased on partial genome data Using WGS we identified fivedistinct lineages with high posterior probability support (anno-tated in Fig 2)mdashmedian pairwise genetic distance withineach lineage is listed in Table 1 Bayesian phylogenetic recon-struction of WGS yielded a mean evolutionary rate of 144 104
substitutionssiteyear (95 highest posterior density578 107 to 319 104) similar to previous estimates for N
gene and G gene evolution (David et al 2007 Talbi et al 2009Ahmed et al 2015)
32 Phylogeography of RABV in Tanzania
Within Tanzania we found evidence of phylogeographic struc-ture (AIfrac14 070 Plt 0001) similar to other estimates of Africanintra-country AI values (Table 2) (Talbi et al 2009) Comparedwith the strong spatial structure between countries and largerspatial aggregations (Table 3) this indicates more fluid and dy-namic dispersal patterns within Tanzania as has been found inother African countries (Talbi et al 2010) Across the posteriordistribution of trees there were 24 (95 highest posterior den-sity 21ndash28) independent lineage movement events Using asummarized history of Markov Jump counts across the phylog-eny we found that 43 per cent of these migrations occurred inthe most recent ten years of the phylogeny (Fig 2c) A BSSVSprocedure in BEAST identified eighteen potential diffusionpathways to explain the observed phylogeographic patterns inthe posterior distribution However only four received substan-tial support based on BFgt 5 (Fig 2) Support was particularlystrong for dispersal from the Serengeti District to Morogoro
Figure 2 Regional phylogeography among sixty rabies virus whole-genome sequences sampled in Tanzania from 2003 to 2012 (a) an MCC tree with branches coloured
according to the most probable posterior location of its descendent node inferred by discrete-state phylogeographic reconstruction in BEAST Five major phylogenetic
groups (Tz1-5) are annotated on the tree and node symbols indicate node posterior support 09 (b) The four most significant dispersal pathways indicated by BF re-
sults from a BSSVS procedure in BEAST with the median number of transitions estimated by Markov jump counts indicated in cases where posterior support for a tran-
sition was gt07 (c) Markov jump densities for total number of transitions through time (d) Bayesian Skyline plot showing Nes the product of the effective population
size (Ne) and the generation time in years (s) through time
Table 1 Raw median genetic distance within each of the five mainrabies virus lineages identified in Tanzania
Genetic distance
RV lineage No of substitutions per site No of SNPs
Tz1 947E-03 110Tz2 227E-03 27Tz3 883E-03 95Tz4 417E-03 495Tz5 0 0
6 | Virus Evolution 2015 Vol 1 No 1
(BFfrac14 13530) and Markov jump counts estimated a median ofsix (range 3ndash10) migrations along this dispersal route with atleast one (range 1ndash3) occurring on a branch representing a pe-riod of less than 2 years Most lineages were sampled in morethan one region in Tanzania with some distributed across alarger geographic area than others (Fig 3) The largest lineageTz1 contains not only a cluster of Serengeti samples but alsoencompassed samples from a larger geographic extent and wasfound in eight out of the nine districts sampled The Bayesian
skyline plot revealed that the effective population size has re-mained fairly constant over the past 150 years (Fig 2d) Becauseof the much higher availability of samples from the SerengetiDistrict we also conducted a coarser phylogeographic analysisgrouping sequences into lsquoNorthrsquo lsquoSouthrsquo or lsquoPemba Islandrsquo Thisidentified a predominance of north to south dispersal (esti-mated thirteen independent movements compared with onemovement south to north) and evidence of dispersal back andforth between the North of mainland Tanzania and PembaIsland (Supplementary Fig S4)
4 Discussion
Using viral genetic data from a hierarchy of spatial scales andvarying levels of genome coverage we were able to demonstratethe advantages of whole-genome resolution to describe thespatio-temporal dynamics of endemically circulating caninerabies viruses
We found a clear phylogeographic structure between coun-tries in Africa which could be identified with partial genomedata This suggests that the majority of dispersal occurs at awithin-country scale and control programs would be most ap-propriate at a national scale with strategies to deal with poten-tial incursions from other countries However at regional andlocal scales within Tanzania the discriminatory power of par-tial genome data became too limited to reveal fine-scale popula-tion structure that could aid the effectiveness of controlinterventions In contrast WGS provided the resolution to ge-netically distinguish between all samples and produced a well-supported phylogeny Although sub-genomic information hasutility at broad phylogeographic scales (eg Horton et al 2015)this finding supports the application of WGS for studies aimingto discern population structure at a scale most relevant tocontrol
Although large-scale population structure according tosub-continental areas or country-specific lineages was appar-ent from sub-genomic data we also found incidences of oc-casional large-scale admixture (Supplementary Figs S1 andS2) We continued to find evidence at increasingly fine scalesTanzania and other countries within the Africa 1b cladeshowed signs of admixture particularly when countriesshared a border and within country movements of RABV fa-cilitated by humans were a feature of Tanzanian RABV Evenat a very local scale within the Serengeti District we foundmultiple co-circulating lineages This recurrent theme may bea characteristic of endemic RABV in Africa reflecting decadesof endemic circulation and human-mediated introductionsSimilar patterns have been observed in canine rabies-en-demic countries in South and Southeast Asia (Lumlertdachaet al 2006 Matsumoto et al 2013 Ahmed et al 2015) wherethe presence of co-circulating lineages was attributed to a
Figure 3 Spatial distribution of rabies virus lineages sampled from regions in
Tanzania between 2003 and 2012 with a colour gradient (yellow to red) indicat-
ing the total number of lineages (low to high) sampled in each region
Table 2 Degree of within-country rabies virus spatial admixture in three African countries measured using an Association Index (AI) whichranges from 0 indicating strong population subdivision to 1 indicating complete panmixis Results are shown for Tanzania (this study) andAlgeria and Morocco (Talbi et al 2010)
Country AI [95 BCI] P No of sequences No of locations Median distance (km) Min Max
Algeria 067 [062ndash073] lt0001 117 20 23308 2327 67462Morocco 055 [051ndash063] lt0001 133 28 32645 2862 92670Tanzania 070 [060-079] lt0001 60 9 43976 3905 108826
BCI Bayesian confidence interval
Table 3 Degree of spatial admixture between rabies virus samplesfrom Africa according to an Association Index (AI) AI values canrange from 0 indicating complete population subdivision to 1 indi-cating complete panmixis
Data Spatial clustering level No ofgroups
AI [95 BCI] P
N405 Sub-continent 3 004 [002005] lt001Country 14 006 [005008] lt001
N1350 Sub-continent 3 006 [004008] lt001Country 10 013 [009013] lt001
BCI Bayesian confidence interval Datasets of partial (N405) and full (N1350) nu-
cleoprotein sequences were tested at two levels of spatial aggregation (1) sub-
continent geographical partitions relative to Tanzania (three states Tanzania
neighbouring country and other African country) and (2) country of origin
K Brunker et al | 7
combination of historical introductions from neighbouringcountries and human-mediated movements
Much of the viral population structure we found withinTanzania is consistent with initial invasive waves that havepersisted endemically with structure eroding over time aidedby human-mediated movement Historical accounts describe arabies outbreak in southern regions of Tanzania in the mid-1950s which spread throughout the country and was recordedin the Serengeti District in the late 1970s (Magembe 1985Siongok and Karama 1985) This invasion perhaps shaped theinitial RABV population structure in Tanzania The timeline ofregional scale migrations (Fig 2c) indicates a strong rise in viraldispersal around this period possibly reflecting increasing hu-man connectivity Samples in Tz1 (Fig 2) share partial N geneidentity with samples from Kenya (Fig 1) indicating a sharedevolutionary history that may relate to an initial outbreakacross both countries Lineages Tz1 and Tz3 appear to have awide geographic distribution (Fig 3) and we found evidence of ageneral north to south dispersal pattern in Tanzania(Supplementary Fig S4) This highlights the potential for wide-spread dispersal should an incursion occur from Kenya and sug-gests a need for co-operative cross-border rabies managementonce a national control program is established Variation in thespread of different lineages across Tanzania may reflect the in-fluence of heterogeneous landscape features or dog populationstructure in impeding or facilitating RABV disseminationIdentifying and quantifying such features is the logical nextstep to provide additional information that can inform controlprogrammes such as identifying and strengthening pre-existing barriers Recent extensions of phylogeographic tech-niques have highlighted how this might be achieved using anintegrated approach combining evolutionary and ecologicalanalyses to quantify drivers of viral transmission (Lemey et al2014 Magee et al 2014)
The ancestry of lineages Tz4 and 5 (denoting lineages foundin the southern mainland and on the Island of Pemba respec-tively) points toward an introduction from the north ofTanzania (posterior probabilityfrac14 062) but the uncertainty ofthis ancestral location and the ancestral node itself (poste-riorfrac14 045) suggest these may be distinct historical lineageswith low-level persistence or undetected circulation elsewherein Tanzania Alternatively these clusters may represent in-stances where lineages from external sources have more re-cently invaded and resulted in sustained transmission inTanzaniamdashpartial genome phylogenies indicate Tz4 is relatedto clusters containing samples from countries south ofTanzania South Africa and Mozambique Furthermore Tz5shares a common ancestry with a Nigerian and several CentralAfrican Republic samples This again underlines the threat ofre-invasion or introduction from external sources and the po-tential value of whole-genome resolution to robustly and moreaccurately identify sources of new introductions To date themajority of RABV genetic data from Africa comes from partialgenome analyses however our data suggest that whole-genome characterization would be valuable and should be anaim for the future
Islands represent isolated landscapes with natural barriersto dispersal but incidents of RABV outbreaks instigated by hu-man-mediated introductions (eg to islands in Indonesia[Windiyaningsih et al 2004 Susilawathi et al 2012]) have oftenbeen recorded Sequences from the island of Pemba were sug-gestive of multiple introductions from various sources with evi-dence of dispersal to and from the mainland (SupplementaryFig S4) Samples from Pemba were scattered throughout the
phylogeny and the most divergent lineage Tz5 consisted of twosamples from Pemba The distribution of these lineages may re-flect earlier invasions of RABV into Pemba from elsewhere inTanzania (Tz1 and 2) and the African continent (Tz5) resultingfrom Pembarsquos location on an important trading route Howeversince these lineages were not resampled and no cases havebeen detected from Pemba in over a year (Lushasi pers comm)RABV may have been only transiently circulating Neverthelesslessons from Indonesia where lack of a swift and coordinatedresponse to a rabies incursion led to an epidemic (Townsendet al 2013) highlight the importance of active surveillance andrapid response to incursions
Although we found evidence of RABV spatial structurewithin Tanzania it was evident that dispersal was also fre-quent with at least one long distance migration occurringwithin a small temporal window (lt2 years) Lineage move-ments occurring on branches representing short evolutionarytimes such as those identified between Serengeti and Morogoro(gt750 km) indicate rates of dispersal much higher than thoserecorded for endemic wildlife rabies (Biek 2007) and imply hu-man mediated movements as seen in parts of North Africa(Talbi et al 2010) Movement of pastoralist and agro-pastoralistcommunities from the Lake zone in Northern Tanzania tosouthern regions for example Morogoro have been ongoingsince the 1950s (Walsh 2008) with a major influx reported from2003 to 2006 (PINGOrsquos Forum 2013) attributed to climate changeinduced drought and forced evictions from newly protectedareas (Kideghesho et al 2013) During these migrations pasto-ralists are accompanied by their dogs which may facilitate thelong distance movement of animals incubating the virus priorto transmission On further inspection we found that Morogorosamples indicated as instances of long-distance translocationcame from rural areas where pastoralists are likely to migrateto whereas other samples from urban Morogoro formed a dis-tinct cluster (Fig 2) We found three RABV lineages in Morogorofrom only ten samples (Fig 3) Quantifying networks of human-mediated movements (including livestock trade) would providea valuable proxy for connectivity for many zoonotic diseases af-fecting domesticated animals
Further to our findings of regional admixture in Tanzaniawe also observed considerable diversity at a very local scalethat is within the Serengeti District with several lineages co-circulating These lineages appear to have persisted for at least20 years (older Serengeti samples obtained from GenBank alsocluster within these lineages indicated in Fig 1) despite dogvaccination campaigns having been conducted in the districtsince 1996 While vaccination coverage has varied substantiallyacross years and between villages (Viana et al 2015) rabies inci-dence has at times significantly declined for example falling by97 per cent in the late 1990s (Cleaveland et al 2003) Yet our ge-netic data show that despite substantial declines in incidencethese lineages must have persisted at very low levels within thedistrict or subsequently reinvaded from neighbouring districtsWithout sampling the surrounding districts it is not possible todistinguish between these possibilities The skyline plot indi-cated a stable effective RABV population size (Ne) through time(Fig 3d) While this could be expected for an endemic pathogenit is worth noting that there was no evidence of viral populationsize reductions in response to vaccination campaigns Rabiescontrol efforts across much of Tanzania have been very limiteduntil recently and high turnover in the dog population(Hampson et al 2009) likely contributes to the stable persistenceof rabies in Tanzania It has also been noted that geographicstructure can obscure local fluctuations in subpopulations while
8 | Virus Evolution 2015 Vol 1 No 1
maintaining the appearance of a constant Ne in skyline plots(Carrington et al 2005)
This study represents a snapshot of RABV dynamics inTanzania indicating that human movements have dissemi-nated RABV out of locally endemic areas at scales relevant tocontrol that is administrative units such as regions or districtsThese frequent translocations have probably lead to the exis-tence of multiple co-circulating lineages (Fig 3) but relativelyfew introductions lead to sustained chains of transmission thatare detectable as lineage movement events However in diseasesystems closely associated with human activities the probabil-ity of successful translocation and establishment is likely to bemuch greater This suggests that without some level of regionalco-operation Tanzania will be unable to eliminate rabies andmaintain freedom from disease Human movements are oftenin response to social drivers which could be used as signals forincreased vigilance and surveillance in at risk areas
Our findings highlight the use of WGS to uncover fine scaletransmission patterns that can directly inform control effortsHowever sub-genomic approaches can still have utility at acoarser scale and are more easily obtained particularly whensample quality is an issue In particular they can be used to ini-tially identify admixture between countries which may indicatethe necessity of coordinated regional control programs and sur-veillance Co-circulation of multiple lineages and introductionsfacilitated by humans appear to be a feature of endemic rabies vi-rus and complicate the design of a sustainable control strategyHowever using whole-genome data we were able to identifysources of dispersal within Tanzania that could direct efforts to-ward surveillance and control The finding that humans play animportant role in the dynamics of RABV in Tanzania suggeststhat increasing awareness and dog vaccination in lsquohigh-riskrsquo com-munities such as pastoralists could help to reduce long-range dis-persal Moreover the design of enhanced surveillance andcontainment strategies to mitigate human-mediated incursionsand maintain disease freedom should be a priority once controlprograms are established and elimination is being targeted
Supplementary data
Supplementary data is available at VEVOLU Journal online
Acknowledgements
We acknowledge the Ministries of Livestock and FisheriesDevelopment and of Health and Social Welfare TanzaniaNational Parks Tanzania Wildlife Research InstituteNgorongoro Conservation Area Authority TanzaniaCommission for Science and Technology and NationalInstitute for Medical Research for permission and collabora-tion and the Arusha Mtwara and Mwanza VeterinaryInvestigation Centres Pemba Veterinary Office and theFrankfurt Zoological Society for logistical and technical sup-port We are grateful to the veterinary and livestock field of-ficers healthworkers and paravets throughout Tanzaniaand in particular to the following research assistants JoelChangalucha Zilpah Kaare Gurdeep Kour Ahmed LugeloKennedy Lushasi Mathias Magoto Khadija Said andRenatus Herman We thank Pelagia Muchuruza MatthewMaziku Geoffrey Mchau Eberhard Mbunda Francois-XavierMeslin and Bernadette Abela-Ridder for their collaborationsthrough the Bill amp Melinda GatesWHO rabies eliminationdemonstration project in Tanzania This work was
supported by the Wellcome Trust the Medical ResearchCouncil the UBS Optimus Foundation Lincoln Park Zooand the Research and Policy for Infectious DiseaseDynamics Program of the Science and TechnologyDirectorate Department of Homeland Security FogartyInternational Centre National Institute of Health
Conflict of interest None declared
ReferencesAhmed K et al (2015) lsquoMolecular Epidemiology of Rabies
Viruses Circulating in Two Rabies Endemic Provinces of Laos2011ndash2012 Regional Diversity in Southeast Asiarsquo PLoS NeglectedTropical Diseases 9 e0003645
Andrews S (2010) FastQC a quality control tool for highthroughput sequence datalthttpwwwbioinformaticsbbsrcacukprojectsfastqcgt accessed 4 Jun 2014
Ayres D L et al (2012) lsquoBEAGLE An Application ProgrammingInterface and High-Performance Computing Library forStatistical Phylogeneticsrsquo Systematic Biology 61 170ndash3
Bedford T et al (2014) lsquoIntegrating Influenza AntigenicDynamics with Molecular Evolutionrsquo eLife 2014 1ndash26
Biek R and Real L A (2010) lsquoThe Landscape Genetics of InfectiousDisease Emergence and Spreadrsquo Molecular Ecology 19 3515ndash31
mdashmdash et al (2007) lsquoA High-Resolution Genetic Signature ofDemographic and Spatial Expansion in Epizootic Rabies VirusrsquoProceedings of the National Academy of Sciences of the United Statesof America 104 7993ndash8
mdashmdash et al (2015) lsquoMeasurably Evolving Pathogens in the GenomicErarsquo Trends in Ecology amp Evolution 30 306ndash13
Bielejec F et al (2011) lsquoSPREAD Spatial PhylogeneticReconstruction of Evolutionary Dynamicsrsquo Bioinformatics(Oxford England) 27 2910ndash2
mdashmdash et al (2014) lsquoInferring Heterogeneous Evolutionary Processesthrough Time From Sequence Substitution toPhylogeographyrsquo Systematic Biology 63 493ndash504
Bivand R S Pebesma E J and Gomez-Rubio V (2008) AppliedSpatial Data Analysis with R New York Springer
Bolger A M Lohse M and Usadel B (2014) lsquoTrimmomatic AFlexible Trimmer for Illumina Sequence Datarsquo Bioinformatics30 2114ndash20
Bourhy H et al (2008) lsquoThe Origin and Phylogeography of DogRabies Virusrsquo The Journal of General Virology 89 2673ndash81
Brunker K et al (2012) lsquoIntegrating the Landscape Epidemiologyand Genetics of RNA Viruses Rabies in Domestic Dogs as aModelrsquo Parasitology 139 1899ndash913
Carrington C et al (2005) lsquoInvasion and Maintenance of DengueVirus Type 2 and Type 4 in the Americasrsquo Journal of Virology 7914680ndash7
Cleaveland S et al (2003) lsquoA Dog Rabies Vaccination Campaignin Rural Africa Impact on the Incidence of Dog Rabies andHuman Dog-Bite Injuriesrsquo Vaccine 21 1965ndash73
David D et al (2007) lsquoIdentification of Novel Canine Rabies VirusClades in the Middle East and North Africarsquo The Journal ofGeneral Virology 88 967ndash80
Denduangboripant J et al (2005) lsquoTransmission Dynamics ofRabies Virus in Thailand Implications for Disease ControlrsquoBMC Infectious Diseases 5 52
Drummond A J et al (2003) lsquoMeasurably Evolving PopulationsrsquoTrends in Ecology amp Evolution 18 481ndash8
mdashmdash et al (2005) lsquoBayesian Coalescent Inference of PastPopulation Dynamics from Molecular Sequencesrsquo MolecularBiology and Evolution 22 1185ndash92
K Brunker et al | 9
mdashmdash et al (2012) lsquoBayesian Phylogenetics with BEAUti and theBEAST 17rsquo Molecular Biology and Evolution 29 1969ndash73
Ferreira M A R and Suchard M A (2008) lsquoBayesian Analysis ofElapsed Times in Continuous-Time Markov Chainsrsquo CanadianJournal of Statistics 36 355ndash68
Greger M (2007) lsquoThe HumanAnimal Interface Emergence andResurgence of Zoonotic Infectious Diseasesrsquo Critical Reviews inMicrobiology 33 243ndash99
Hampson K et al (2007) lsquoSynchronous Cycles of Domestic DogRabies in Sub-Saharan Africa and the Impact of ControlEffortsrsquo Proceedings of the National Academy of Sciences of theUnited States of America 104 7717ndash22
mdashmdash et al (2009) lsquoTransmission Dynamics and Prospects for theElimination of Canine Rabiesrsquo PLoS Biology 7 e53
Hayman D T S et al (2011) lsquoEvolutionary History of Rabies inGhanarsquo PLoS Neglected Tropical Diseases 5 e1001
Holmes E C and Grenfell B T (2009) lsquoDiscovering the Phylodynamicsof RNA Virusesrsquo PLoS Computational Biology 5 e1000505
Horton D L et al (2015) lsquoComplex Epidemiology of a ZoonoticDisease in a Culturally Diverse Region Phylogeography ofRabies Virus in the Middle Eastrsquo PLOS Neglected TropicalDiseases 9 e0003569
Jombart T (2008) lsquoadegenet A R Package for the MultivariateAnalysis of Genetic Markersrsquo Bioinformatics (Oxford England)24 1403ndash5
Jones K E et al (2008) lsquoGlobal Trends in Emerging InfectiousDiseasesrsquo Nature 451 990ndash3
Katoh K and Standley D M (2013) lsquoMAFFT MultipleSequence Alignment Software Version 7 Improvements inPerformance and Usabilityrsquo Molecular Biology and Evolution30 772ndash80
Kideghesho J et al (2013) lsquoEmerging Issues and Challenges inConservation of Biodiversity in the Rangelands of TanzaniarsquoNature Conservation 6 1ndash29
Kissi B Tordo N and Bourhy H (1995) lsquoGenetic Polymorphismin the Rabies Virus Nucleoprotein Genersquo Virology 209 526ndash37
Knobel D L et al (2005) lsquoRe-Evaluating the Burden of Rabies inAfrica and Asiarsquo Bulletin of the World Health Organization 83360ndash8
Kuhnert D Wu C-H and Drummond A J (2011) lsquoPhylogeneticand Epidemic Modeling of Rapidly Evolving InfectiousDiseasesrsquo Infection Genetics and Evolution Journal of MolecularEpidemiology and Evolutionary Genetics in Infectious Diseases 111825ndash41
Kuzmin I V et al (2008) lsquoComplete Genomes of AravanKhujand Irkut and West Caucasian Bat Viruses with SpecialAttention to the Polymerase Gene and Non-Coding RegionsrsquoVirus Research 136 81ndash90
Kuzmina N A et al (2013) lsquoThe Phylogeography andSpatiotemporal Spread of South-Central Skunk Rabies VirusrsquoPloS One 8 e82348
Lanfear R et al (2012) lsquoPartitionfinder Combined Selectionof Partitioning Schemes and Substitution Models forPhylogenetic Analysesrsquo Molecular Biology and Evolution 291695ndash701
Lembo T et al (2007) lsquoMolecular Epidemiology Identifies Only aSingle Rabies Virus Variant Circulating in Complex CarnivoreCommunities of the Serengetirsquo Proceedings Biological SciencesThe Royal Society 274 2123ndash30
mdashmdash et al (2008) lsquoExploring Reservoir Dynamics A Case Study ofRabies in the Serengeti Ecosystemrsquo Journal of Applied Ecology45 1246ndash57
Lemey P et al (2009) lsquoBayesian Phylogeography Finds Its RootsrsquoPLoS Computational Biology 5 e1000520
mdashmdash et al (2014) lsquoUnifying Viral Genetics and HumanTransportation Data to Predict the Global TransmissionDynamics of Human Influenza H3N2rsquo PLoS Pathogens 10e1003932
Lewin-koh A N J et al (2009) Maptools Tools for Reading andHandling Spatial Objects R package version 08-23 lthttpcranr-projectorgwebpackagesmaptoolsindexhtmlgt accessed 1Sept 2014
Li H and Durbin R (2009) lsquoFast and Accurate Short ReadAlignment with Burrows-Wheeler Transformrsquo Bioinformatics(Oxford England) 25 1754ndash60
mdashmdash et al (2009) lsquoThe Sequence AlignmentMap Format andSAMtoolsrsquo Bioinformatics (Oxford England) 25 2078ndash9
Lumlertdacha B et al (2006) lsquoComplex Genetic Structure ofthe Rabies Virus in Bangkok and Its Surrounding ProvincesThailand Implications for Canine Rabies ControlrsquoTransactions of the Royal Society of Tropical Medicine andHygiene 100 276ndash81
Magee D et al (2014) lsquoCombining Phylogeography and SpatialEpidemiology to Uncover Predictors of H5N1 Influenza A VirusDiffusionrsquo Archives of Virology 160 215ndash24
Magembe S (1985) lsquoEpidemiology of Rabies in United Republic ofTanzaniarsquo in Kuwer E et al (eds) Rabies in the Tropics pp 392ndash8 New York Springer Berlin Heidelberg
Marston D A et al (2007) lsquoComparative Analysis of the FullGenome Sequence of European Bat Lyssavirus Type 1 andType 2 with Other Lyssaviruses and Evidence for a ConservedTranscription Termination and Polyadenylation Motif in theG-L 3rsquo Non-Translated Regionrsquo The Journal of General Virology88 1302ndash14
mdashmdash et al (2013) lsquoNext Generation Sequencing of Viral RNAGenomesrsquo BMC Genomics 14 444
Matsumoto T et al (2013) lsquoMolecular Epidemiology of HumanRabies Viruses in Sri Lankarsquo Infection Genetics and Evolution 18160ndash7
Minin V N and Suchard M A (2008) lsquoCounting LabeledTransitions in Continuous-Time Markov Models of EvolutionrsquoJournal of Mathematical Biology 56 391ndash412
Mollentze N et al (2014) lsquoA Bayesian Approach for Inferring theDynamics of Partially Observed Endemic Infectious Diseasesfrom Space-Time-Genetic Datarsquo Proceedings Biological SciencesThe Royal Society 281 20133251
Nel L H (2013) lsquoDiscrepancies in Data Reporting for RabiesAfricarsquo Emerging Infectious Diseases 19 529ndash33
Nelson M I et al (2015) lsquoGlobal Migration of Influenza A Virusesin Swinersquo Nature Communications 6 6696
Nunes M R T et al (2014) lsquoAir Travel is Associated withIntracontinental Spread of Dengue Virus Serotypes 1-3 inBrazilrsquo PLoS Neglected Tropical Diseases 8 e2769
Paradis E Claude J and Strimmer K (2004) lsquoAPE Analyses ofPhylogenetics and Evolution in R Languagersquo Bioinformatics 20289ndash90
Parker J Rambaut A and Pybus O G (2008) lsquoCorrelatingViral Phenotypes with Phylogeny Accounting forPhylogenetic Uncertaintyrsquo Infection Genetics and Evolution 8239ndash46
Pebesma E J and Bivand R S (2005) lsquoClasses and Methods forSpatial Data in Rrsquo R News 5 9ndash13
PINGOrsquos Forum (2013) Pastoralists Indigenous Non GovernmentalOrganizationsrsquo Forum Annual Report 2012-2013 lthttpwwwpin-gosforumortzgt accessed 23 Nov 2014
Rambaut A et al (2014) lsquoTracer v16rsquo Computer program anddocumentation distributed by the author lthttpbeastbioedacukTracergt accessed 4 Feb 2015
10 | Virus Evolution 2015 Vol 1 No 1
Real L A et al (2005) lsquoUnifying the Spatial Population Dynamicsand Molecular Evolution of Epidemic Rabies Virusrsquo Proceedingsof the National Academy of Sciences of the United States of America102 12107ndash11
Rupprecht C E Hanlon C A and Hemachudha T (2002)lsquoRabies Re-examinedrsquo The Lancet Infectious Diseases 2 327ndash43
Shwiff S Hampson K and Anderson A (2013) lsquoPotentialEconomic Benefits of Eliminating Canine Rabiesrsquo AntiviralResearch 98 352ndash6
Siongok T and Karama M (1985) lsquoEpidemiology of HumanRabies in Kenyarsquo in E Kuwer et al (eds) Rabies in the Tropicspp 445ndash50 New York Springer Berlin Heidelberg
Stamatakis A et al (2012) lsquoRAxML-Light A Tool for ComputingTerabyte Phylogeniesrsquo Bioinformatics (Oxford England) 28 2064ndash6
Susilawathi N M et al (2012) lsquoEpidemiological and ClinicalFeatures of Human Rabies Cases in Bali 2008-2010rsquo BMCInfectious Diseases 12 81
Talbi C et al (2009) lsquoEvolutionary History and Dynamics of DogRabies Virus in Western and Central Africarsquo The Journal ofGeneral Virology 90 783ndash91
mdashmdash et al (2010) lsquoPhylodynamics and Human-Mediated Dispersalof a Zoonotic Virusrsquo PLoS Pathogens 6 e1001166
Tao X Y et al (2009) lsquoMolecular Epidemiology of Rabies inSouthern Peoplersquos Republic of Chinarsquo Emerging InfectiousDiseases 15 1192ndash8
Townsend S E et al (2013) lsquoDesigning Programs for EliminatingCanine Rabies from Islands Bali Indonesia as a Case StudyrsquoPLoS Neglected Tropical Diseases 7 e2372
Viana M et al (2015) lsquoDynamics of a Morbillivirus at theDomesticndashWildlife Interface Canine Distemper Virus inDomestic Dogs and Lionsrsquo Proceedings of the National Academy ofSciences of the United States of America 112 1464ndash9
Walsh M (2008) Pastoralism and Policy Processes in TanzaniaMbarali Case Study Report to the Tanzania Natural ResourceForum Arusha
Wang T H et al (2001) lsquoIdentification of Shared Populations ofHuman Immunodeficiency Virus Type 1 Infecting Microgliaand Tissue Macrophages Outside the Central Nervous SystemrsquoJournal of Virology 75 11686ndash99
WHO (2005) lsquoWHO Expert Consultation on Rabiesrsquo World HealthOrganization technical report series 931 1ndash121
Windiyaningsih C et al (2004) lsquoThe Rabies Epidemic on FloresIsland Indonesia (1998-2003)rsquo Journal of the Medical Associationof Thailandfrac14Chotmaihet thangphaet 87 1389ndash93
Woodroffe R and Donnelly C A (2011) lsquoRisk of Contact be-tween Endangered African Wild Dogs Lycaon pictus andDomestic Dogs Opportunities for Pathogen TransmissionrsquoJournal of Applied Ecology 48 1345ndash54
Woolhouse M E J (2002) lsquoPopulation Biology of Emerging and Re-emerging Pathogensrsquo Trends in Microbiology 10(10 Suppl) S3ndash7
K Brunker et al | 11
- vev011-TF1
- vev011-TF2
-

In this study finely resolved space-time-genetic data in-cluding whole-genome sequences were used to determine thespatial and temporal dynamics of endemic RABV at both a re-gional and local scale in Tanzania The utility of partial genomedata was also demonstrated as a means to characterize large-scale phylogeographic patterns of RABV in Africa Specificallywe aimed to characterize the dynamics of rabies virus in an en-demic system including the role of human-mediated transport
2 Materials and methods21 Samples
For this study fifty-nine new whole-genome sequences were ob-tained from animal hosts (primarily domestic dogs) from nine re-gions in Tanzania sampled between 2003 and 2012 A previouslysequenced sample (RV2772 accession KF155002) (Marston et al2013) from Southern Tanzania was included in the dataset andused as a reference sequence (sample details in SupplementaryTable S1) The main study area encompassed the SerengetiDistrict in northwest Tanzania where approximately half thesamples (nfrac14 33) were obtained from active surveillance whichenabled the collection of brain material from suspect rabid ani-mals and GPS coordinates and dates recorded as described inHampson et al (2009) The remaining twenty-seven sampleswere obtained opportunistically from other regions in Tanzaniaas part of surveillance by the Tanzanian Ministry of Livestockand Fisheries Development and the Tanzanian VeterinaryLaboratory Agency All samples were sent to the Animal amp PlantHealth Agency (APHA) in Weybridge UK for processing
In addition fifty new partial N gene sequences (405 bp) fromTanzania were obtained via reverse transcription-polymerasechain reaction (RT-PCR) and Sanger sequencing using samplesarchived at APHA and submitted to GenBank with accessionnumbers KR534217ndashKR534266 (details in SupplementaryMaterial and Table S2)
22 RNA extraction and WGS
Total RNA was extracted at APHA directly from brain tissue us-ing TRIZOL according to manufacturerrsquos instructions(Invitrogen) Precipitated total RNA was re-suspended in molec-ular-grade water at a one in ten dilution and quantified using aNanoDrop spectrophotometer (Thermo Scientific) Sampleswere sequenced on a range of next generation sequencing plat-forms during NGS protocol optimization (see SupplementaryMaterial for details) The majority of samples (nfrac14 48) were se-quenced by the following method TRIzol-extracted viral RNAwas depleted of host genomic DNA using the on-column DNasetreatment in RNeasy plus mini kit (Qiagen) as per manufac-turerrsquos instructions with elution in 30 ml molecular grade waterThis was followed by host ribosomal RNA depletion usingTerminator 5rsquo-phosphate-dependent exonuclease (EpicentreBiotechnologies) as detailed in (Marston et al 2013) First- andsecond-strand cDNA was synthesized using a Roche cDNA syn-thesis system kit with random hexamers (Roche) ResultantcDNA was quantified using Picogreen dsDNA quantitation re-agent (Invitrogen) and approximately 1 ng of each sample usedin a lsquotagmentationrsquo reaction mix using a Nextera XT samplepreparation kit (Illumina) according to the manufacturerrsquos pro-tocol (minus the bead normalization step) DNA libraries foreach sample were quantified using a Quant-iT PicoGreendsDNA Assay Kit (Invitrogen) or a Qubit assay kit (Life technolo-gies) and average library size was measured with a high-
sensitivity DNA Bioanalyzer chip on a model 2100 Bioanalyzer(Agilent) Sample libraries were transported to the MRC Centrefor Virus Research at the University of Glasgow UK for the finalsteps of library preparation and sequencing Individual librarieswere pooled and normalized to equimolar concentrations at asuitable plexity (x24 for MiSeq runs) Libraries were sequencedas 150-bp paired-end reads on an Illumina MiSeq Additional se-quencing was conducted on a NextSeq 500 platform (GlasgowPolyomics at the University of Glasgow Glasgow UK) and readsmerged with MiSeq reads to increase coverage for poorly se-quenced samples (see Supplementary Material)
23 Bioinformatics and sequence analysis
Raw reads were assessed in FastQC (Andrews 2010) andTrimmomatic (Bolger Lohse and Usadel 2014) was used to trim3rsquo-ends remove adapter contamination and to filter based onquality with default parameters Filtered reads were mapped tothe previously sequenced genome of Tanzanian RABV sampleRV2772 (accession KF155002) with BWA mem version 0710(Li and Durbin 2009) and converted to bam file format usingSAMtools v 0118 (Li et al 2009)
A conservative single-nucleotide polymorphism (SNP) call-ing routine was implemented in GATK utilizing theUnifiedGenotyper tool to identify high confidence SNPs whichhad passed GATK filters on strand bias (FSgt 60 SORgt 4) map-ping quality (MQlt 40 MQRankSumlt125) read position(ReadPosRankSumlt8) and depth of coverage (DPlt 5) Indelswere filtered if FSgt 200 and ReadPosRankSumlt20 and furthermanually inspected for inclusion (eg dismissed if near a ho-mopolymer run) Consensus sequences were built using a cus-tom script in R which called filtered SNPs with a 75 per centconsensus rule (positions with lt75 consensus were given aIUPAC code for the corresponding ambiguous base call) and ge-nome positions with a depth of coverage less than two were la-belled lsquoNrsquo Potential SNP calls that failed only the depth filterthat is had a depthlt 5 but gt1 were passed if the same poly-morphism had been present as a high confidence SNP in at leasttwo other samples Otherwise the position was given an IUPACcode representing the population-level calls and the potentialSNP In addition a set of consensus sequences using a more re-laxed approach to SNP calling was produced which involvedstrict calls of all SNPs with depthgt 1 and gaps filled with themajority population consensus sequence These relaxed con-sensus sequences were used to produce initial starting trees forBEAST analyses (see Section 26)
Sequencing resulted in 93ndash100 per cent coverage of the ge-nome with gt99 per cent genome coverage achieved for 95 percent of samples and a median depth of coverage of seventy-seven (range 6ndash1871 see Supplementary Table S1) Nextera XT isa transposase-based method of library preparation and sequencereads typically miss the ends of the genome however as theends of lyssaviruses are highly conserved (Marston et al 2007Kuzmin et al 2008) it is unlikely that any informative variationwas missed We therefore consider our analyses to be based ongenome-wide variation and henceforth refer to our dataset aswhole-genome sequences Consensus sequences were alignedusing MAFFT v7149b (Katoh and Standley 2013) and submitted toGenBank (accession numbers KR906734ndashKR906792)
24 Phylogenetic reconstruction
Initial datasets of (1) partial N gene 405 bp (1317 sequences) and(2) full N gene 1350 bp (674 sequences) sequences isolated in
K Brunker et al | 3
Africa were constructed using sequences retrieved fromGenBank and including new Tanzanian isolates sequenced forthis study (59 new WGS samples and 50 new partial genome se-quences) Following maximum likelihood (ML) phylogenetic re-construction with the initial sequence datasets subset treeswere extracted for samples in the Africa 1b clade (430 samplesin the partial N dataset and 100 in the full N dataset)
Alignments for whole genome full N and partial N genewere created in MAFFT (Katoh and Standley 2013) and esti-mated phylogenetic relationships using both ML and Bayesianmethods ML phylogenies were estimated in RAxML(Stamatakis et al 2012) with a general time reversible (GTR) nu-cleotide substitution model and a gamma distribution model ofamong-site rate variation A Chinese dog RABV sequence fromGenBank (accession no FJ712193) was used as an outgroup andnode support was evaluated with 1000 bootstrap replicatesBayesian phylogenetic reconstruction was conducted in BEASTv181 (Drummond et al 2012) using a posterior distribution oftrees (without a molecular clock model) Phylogenies were visu-alized and annotated in R using the packages adegenet (Jombart2008) and APE (Paradis Claude and Strimmer 2004) and mapswere made in R with Maptools (Lewin-koh et al 2009) and sppackages (Pebesma and Bivand 2005 Bivand Pebesma andGomez-Rubio 2008) The degree of spatial admixture at largephylogeographic scales ie sub-continental and country levelwas quantified by an association index (AI) using BaTS softwarewith beast phylogenies (Parker et al 2008)
25 Selecting an evolutionary model
An initial nucleotide substitution model was chosen based onthe model selected by PartitionFinder (Lanfear et al 2012) Ourwhole-genome alignments were partitioned into sixteen sets ofnucleotides one for each codon position (CP) in each gene (fivegenes) and one for concatenated non-coding regions Modelscheme selection was based on the best AIC score from a greedysearch of substitution models which favoured a GTR modelwith partitioning into three CPs (CP123)
In addition model comparison based on marginal likelihoodestimates using path sampling (PS) and stepping stone (SS)sampling implemented in BEAST (100 path steps and a chainlength of 100000 steps) were used to test varying levels of com-plexity in the substitution model Non-coding sequence wasconcatenated and partitioned as a lsquogenersquo with its own evolution-ary model Specifically we tested the HKY model with (1) agene-specific nucleotide model with gene-specific rate varia-tion (2) a gene-linked CP partitioned model with among CP rateheterogeneity and homogeneous rates among genes and (3) agene-specific CP partitioned model with among CP and amonggene rate heterogeneity Model types 2 and 3 were also testedwith CP112 and CP123 partitioning schemes Following the re-sults for the best HKY model we did a final step comparing themost supported HKY model with the same structure but using aGTR model Results (Supplementary Table S2) strongly favouredGTR and CP models (CP123 was best supported) but there wasno support for partitions according to genes which all had simi-lar rates of substitution This significantly reduced the complex-ity of the model and is an important finding in the context ofanalysing RABV whole-genome sequence
26 Bayesian evolutionary analyses
Bayesian Markov-chain Monte Carlo analyses were performedusing BEAST v181 (Drummond et al 2012) and the BEAGLE
library (Ayres et al 2012) Based on model comparisons themost supported evolutionary model was a GTR model withdifferent substitution parameters for CPs one two and three(GTR123thornCP123thornC123) and homogeneous rates among geneswith a GTRthornG substitution model for non-coding sequenceA relaxed molecular clock with a lognormal distribution wasused to model rate variation among branches with aBayesian skyline model (Drummond et al 2005) with tengroups as a flexible tree prior A Bayesian skyline plot wasused to estimate the viral effective population size throughtime (Drummond et al 2005) To reconstruct the spatial dy-namics of RABV spread in Tanzania we implemented a dis-cretized diffusion process among nine regional samplinglocations formalized as an asymmetric continuous timeMarkov chain (CTMC) model (Lemey et al 2009) Three inde-pendent Markov-chain Monte Carlo chains with 50 millionstates and a sampling frequency of 50000 were combined inLogCombiner after discarding at least 10 per cent burnPosterior distributions were inspected in Tracer v16(Rambaut et al 2014) to ensure adequate mixing and conver-gence Initial analyses revealed issues with tree likelihoodconvergence Therefore a CTMC model using relaxed consen-sus sequences (see above) which contained fewer ambigui-ties was implemented first and the maximum cladecredibility (MCC) tree used as a starting tree for the finalCTMC models with conservative consensus sequences
To estimate the most significant pathways of viral dispersalbetween regions a stochastic search variable selection (BSSVS)procedure was implemented to identify the best supported dif-fusion rates through Bayes factor (BF) testing (Lemey et al 2009Bielejec et al 2011) For the per lineage rate of migration(Kuhnert Wu and Drummond 2011) a conditional referenceprior (Ferreira and Suchard 2008) is commonly used but for ourdata resulted in convergence problems with some of the BSSVSparameters Instead we used an exponential prior with a meanof 001 which gave the most robust results out of a range of val-ues tested (data not shown) The degree of spatial admixturewas scored using a modified Association Index (Wang et al2001 Lemey et al 2009) and quantified using the inferred num-ber of lineage migrationmovement events between locationsestimated by Markov jump counts (Minin and Suchard 2008)along the branches of the posterior tree distribution For BEASTmodels with Markov Jump counts implemented a conditionalreference prior on the per lineage rate of migration was chosenA summarized history of Markov jump counts was used to iden-tify movements between regions that occurred on very shortbranches and thus over unusually short time frames Dogsrarely move 1 km from their homestead (Hampson et al 2009Woodroffe and Donnelly 2011) and Hampson et al (2009) founda maximum distance of 20 km between linked RABV cases inthe Serengeti District Lineage migrations between regions rep-resenting distances gt100 km were therefore considered un-likely to be attributable to dog movement alone if they occurredover a period of 2 years or less and were instead interpreted asbeing the result of human-mediated movement In additionthe same form of discretized diffusion model was used to assessdiffusion at a broader scale that is between the North andSouth of Tanzania with Pemba Island classed as a third discretestate A BF test in the program SPREAD (Bielejec et al 2011) wasused to identify well-supported migration pathways (BFgt 3)Sampled trees were summarized as an MCC tree with mediannode heights using TreeAnnotator v181 and Figtree v142 wasused to visualize trees and the inferred ancestral locations forinternal branches
4 | Virus Evolution 2015 Vol 1 No 1
3 Results31 Geographic resolution partial versus full viralgenomes
Consistent with previous large-scale phylogenetic studies par-tial genome phylogenies indicated that RABV in sub-SaharanAfrica falls into several regional groups with viruses fromEastern Africa generally being genetically distinct from those inwestern central and southern parts of the continent(Supplementary Figs S1 and S2) Of the four major lineages ofRABV in Africa (David et al 2007 Bourhy et al 2008 Talbi et al2009) only the Cosmopolitan clade and more specifically theAfrica 1b lineage was detected in Tanzania as found previously(Lembo et al 2007) Within the Africa 1b lineage there was evi-dence of admixture between Tanzania and neighbouring coun-tries and occasional long-range admixture at a continental scale(ML trees in Fig 1 and Bayesian MCC trees in SupplementaryFig S3) Sequences clustering most closely with Tanzanian se-quences came from Kenya which shares a border to the northAlthough partial genome data were sufficient to identify suchlarge-scale spatial patterns these data did not provide adequateresolution to distinguish between samples at sub-national
scales within Tanzania The proportion of nodes with bootstrapsupport 80 per cent was only 011 (Bayesian posterior probabil-ity 90 018) for partial N and 027 (Bayesian 041) for full Ngene phylogenies Furthermore out of the sixty TanzanianWGS 60 per cent were identical at the partial N and 25 per centat the full N gene level In contrast ML and Bayesian trees basedon whole-genome sequences were fully resolved and well sup-ported (proportion of nodes with ML bootstrap support 80086 Bayesian posterior probability 90 089) even whensamples had been taken in close spatial and temporal proxim-ity For example RV2498 and RV2499 were sampled a day apartfrom the same village in Morogoro region both samples onlydiffered by a single SNP for the full N gene but were differenti-ated by twenty-five SNPs in the whole-genome alignment Thislarge divergence strongly suggests that the two samples are notfrom the same chain of transmission which might have beenthe conclusion based on partial genome data The median rawpairwise genetic distance between Tanzanian whole-genomesequences was 259 (range 0ndash608) nucleotides and between theSerengeti District samples was 120 (range 2ndash212) nucleotidesshowing considerable diversity at even a small spatiotemporalscale However much of this high divergence is due to the
(a) (b) (c)
Figure 1 ML trees derived from datasets of rabies virus sequences from the Africa 1b clade for increasing levels of genome coverage (a) 430 sequences from African
countries highlighted on the map for a 405 bp fragment of the nucleoprotein gene (b) 100 sequences of full 1350 bp nucleoprotein gene from the same countries (except
Botswana Ghana Kenya and Zimbabwe) and (c) sixty full or near-full genome sequences (range 11076ndash11923 bp) from Tanzania Trees are scaled by number of sub-
stitutions per site and node symbols indicate nodes with bootstrap support08 Historical samples from the Serengeti District (20 years old) are circled in partial ge-
nome trees
K Brunker et al | 5
presence of multiple lineages some of which are evident evenbased on partial genome data Using WGS we identified fivedistinct lineages with high posterior probability support (anno-tated in Fig 2)mdashmedian pairwise genetic distance withineach lineage is listed in Table 1 Bayesian phylogenetic recon-struction of WGS yielded a mean evolutionary rate of 144 104
substitutionssiteyear (95 highest posterior density578 107 to 319 104) similar to previous estimates for N
gene and G gene evolution (David et al 2007 Talbi et al 2009Ahmed et al 2015)
32 Phylogeography of RABV in Tanzania
Within Tanzania we found evidence of phylogeographic struc-ture (AIfrac14 070 Plt 0001) similar to other estimates of Africanintra-country AI values (Table 2) (Talbi et al 2009) Comparedwith the strong spatial structure between countries and largerspatial aggregations (Table 3) this indicates more fluid and dy-namic dispersal patterns within Tanzania as has been found inother African countries (Talbi et al 2010) Across the posteriordistribution of trees there were 24 (95 highest posterior den-sity 21ndash28) independent lineage movement events Using asummarized history of Markov Jump counts across the phylog-eny we found that 43 per cent of these migrations occurred inthe most recent ten years of the phylogeny (Fig 2c) A BSSVSprocedure in BEAST identified eighteen potential diffusionpathways to explain the observed phylogeographic patterns inthe posterior distribution However only four received substan-tial support based on BFgt 5 (Fig 2) Support was particularlystrong for dispersal from the Serengeti District to Morogoro
Figure 2 Regional phylogeography among sixty rabies virus whole-genome sequences sampled in Tanzania from 2003 to 2012 (a) an MCC tree with branches coloured
according to the most probable posterior location of its descendent node inferred by discrete-state phylogeographic reconstruction in BEAST Five major phylogenetic
groups (Tz1-5) are annotated on the tree and node symbols indicate node posterior support 09 (b) The four most significant dispersal pathways indicated by BF re-
sults from a BSSVS procedure in BEAST with the median number of transitions estimated by Markov jump counts indicated in cases where posterior support for a tran-
sition was gt07 (c) Markov jump densities for total number of transitions through time (d) Bayesian Skyline plot showing Nes the product of the effective population
size (Ne) and the generation time in years (s) through time
Table 1 Raw median genetic distance within each of the five mainrabies virus lineages identified in Tanzania
Genetic distance
RV lineage No of substitutions per site No of SNPs
Tz1 947E-03 110Tz2 227E-03 27Tz3 883E-03 95Tz4 417E-03 495Tz5 0 0
6 | Virus Evolution 2015 Vol 1 No 1
(BFfrac14 13530) and Markov jump counts estimated a median ofsix (range 3ndash10) migrations along this dispersal route with atleast one (range 1ndash3) occurring on a branch representing a pe-riod of less than 2 years Most lineages were sampled in morethan one region in Tanzania with some distributed across alarger geographic area than others (Fig 3) The largest lineageTz1 contains not only a cluster of Serengeti samples but alsoencompassed samples from a larger geographic extent and wasfound in eight out of the nine districts sampled The Bayesian
skyline plot revealed that the effective population size has re-mained fairly constant over the past 150 years (Fig 2d) Becauseof the much higher availability of samples from the SerengetiDistrict we also conducted a coarser phylogeographic analysisgrouping sequences into lsquoNorthrsquo lsquoSouthrsquo or lsquoPemba Islandrsquo Thisidentified a predominance of north to south dispersal (esti-mated thirteen independent movements compared with onemovement south to north) and evidence of dispersal back andforth between the North of mainland Tanzania and PembaIsland (Supplementary Fig S4)
4 Discussion
Using viral genetic data from a hierarchy of spatial scales andvarying levels of genome coverage we were able to demonstratethe advantages of whole-genome resolution to describe thespatio-temporal dynamics of endemically circulating caninerabies viruses
We found a clear phylogeographic structure between coun-tries in Africa which could be identified with partial genomedata This suggests that the majority of dispersal occurs at awithin-country scale and control programs would be most ap-propriate at a national scale with strategies to deal with poten-tial incursions from other countries However at regional andlocal scales within Tanzania the discriminatory power of par-tial genome data became too limited to reveal fine-scale popula-tion structure that could aid the effectiveness of controlinterventions In contrast WGS provided the resolution to ge-netically distinguish between all samples and produced a well-supported phylogeny Although sub-genomic information hasutility at broad phylogeographic scales (eg Horton et al 2015)this finding supports the application of WGS for studies aimingto discern population structure at a scale most relevant tocontrol
Although large-scale population structure according tosub-continental areas or country-specific lineages was appar-ent from sub-genomic data we also found incidences of oc-casional large-scale admixture (Supplementary Figs S1 andS2) We continued to find evidence at increasingly fine scalesTanzania and other countries within the Africa 1b cladeshowed signs of admixture particularly when countriesshared a border and within country movements of RABV fa-cilitated by humans were a feature of Tanzanian RABV Evenat a very local scale within the Serengeti District we foundmultiple co-circulating lineages This recurrent theme may bea characteristic of endemic RABV in Africa reflecting decadesof endemic circulation and human-mediated introductionsSimilar patterns have been observed in canine rabies-en-demic countries in South and Southeast Asia (Lumlertdachaet al 2006 Matsumoto et al 2013 Ahmed et al 2015) wherethe presence of co-circulating lineages was attributed to a
Figure 3 Spatial distribution of rabies virus lineages sampled from regions in
Tanzania between 2003 and 2012 with a colour gradient (yellow to red) indicat-
ing the total number of lineages (low to high) sampled in each region
Table 2 Degree of within-country rabies virus spatial admixture in three African countries measured using an Association Index (AI) whichranges from 0 indicating strong population subdivision to 1 indicating complete panmixis Results are shown for Tanzania (this study) andAlgeria and Morocco (Talbi et al 2010)
Country AI [95 BCI] P No of sequences No of locations Median distance (km) Min Max
Algeria 067 [062ndash073] lt0001 117 20 23308 2327 67462Morocco 055 [051ndash063] lt0001 133 28 32645 2862 92670Tanzania 070 [060-079] lt0001 60 9 43976 3905 108826
BCI Bayesian confidence interval
Table 3 Degree of spatial admixture between rabies virus samplesfrom Africa according to an Association Index (AI) AI values canrange from 0 indicating complete population subdivision to 1 indi-cating complete panmixis
Data Spatial clustering level No ofgroups
AI [95 BCI] P
N405 Sub-continent 3 004 [002005] lt001Country 14 006 [005008] lt001
N1350 Sub-continent 3 006 [004008] lt001Country 10 013 [009013] lt001
BCI Bayesian confidence interval Datasets of partial (N405) and full (N1350) nu-
cleoprotein sequences were tested at two levels of spatial aggregation (1) sub-
continent geographical partitions relative to Tanzania (three states Tanzania
neighbouring country and other African country) and (2) country of origin
K Brunker et al | 7
combination of historical introductions from neighbouringcountries and human-mediated movements
Much of the viral population structure we found withinTanzania is consistent with initial invasive waves that havepersisted endemically with structure eroding over time aidedby human-mediated movement Historical accounts describe arabies outbreak in southern regions of Tanzania in the mid-1950s which spread throughout the country and was recordedin the Serengeti District in the late 1970s (Magembe 1985Siongok and Karama 1985) This invasion perhaps shaped theinitial RABV population structure in Tanzania The timeline ofregional scale migrations (Fig 2c) indicates a strong rise in viraldispersal around this period possibly reflecting increasing hu-man connectivity Samples in Tz1 (Fig 2) share partial N geneidentity with samples from Kenya (Fig 1) indicating a sharedevolutionary history that may relate to an initial outbreakacross both countries Lineages Tz1 and Tz3 appear to have awide geographic distribution (Fig 3) and we found evidence of ageneral north to south dispersal pattern in Tanzania(Supplementary Fig S4) This highlights the potential for wide-spread dispersal should an incursion occur from Kenya and sug-gests a need for co-operative cross-border rabies managementonce a national control program is established Variation in thespread of different lineages across Tanzania may reflect the in-fluence of heterogeneous landscape features or dog populationstructure in impeding or facilitating RABV disseminationIdentifying and quantifying such features is the logical nextstep to provide additional information that can inform controlprogrammes such as identifying and strengthening pre-existing barriers Recent extensions of phylogeographic tech-niques have highlighted how this might be achieved using anintegrated approach combining evolutionary and ecologicalanalyses to quantify drivers of viral transmission (Lemey et al2014 Magee et al 2014)
The ancestry of lineages Tz4 and 5 (denoting lineages foundin the southern mainland and on the Island of Pemba respec-tively) points toward an introduction from the north ofTanzania (posterior probabilityfrac14 062) but the uncertainty ofthis ancestral location and the ancestral node itself (poste-riorfrac14 045) suggest these may be distinct historical lineageswith low-level persistence or undetected circulation elsewherein Tanzania Alternatively these clusters may represent in-stances where lineages from external sources have more re-cently invaded and resulted in sustained transmission inTanzaniamdashpartial genome phylogenies indicate Tz4 is relatedto clusters containing samples from countries south ofTanzania South Africa and Mozambique Furthermore Tz5shares a common ancestry with a Nigerian and several CentralAfrican Republic samples This again underlines the threat ofre-invasion or introduction from external sources and the po-tential value of whole-genome resolution to robustly and moreaccurately identify sources of new introductions To date themajority of RABV genetic data from Africa comes from partialgenome analyses however our data suggest that whole-genome characterization would be valuable and should be anaim for the future
Islands represent isolated landscapes with natural barriersto dispersal but incidents of RABV outbreaks instigated by hu-man-mediated introductions (eg to islands in Indonesia[Windiyaningsih et al 2004 Susilawathi et al 2012]) have oftenbeen recorded Sequences from the island of Pemba were sug-gestive of multiple introductions from various sources with evi-dence of dispersal to and from the mainland (SupplementaryFig S4) Samples from Pemba were scattered throughout the
phylogeny and the most divergent lineage Tz5 consisted of twosamples from Pemba The distribution of these lineages may re-flect earlier invasions of RABV into Pemba from elsewhere inTanzania (Tz1 and 2) and the African continent (Tz5) resultingfrom Pembarsquos location on an important trading route Howeversince these lineages were not resampled and no cases havebeen detected from Pemba in over a year (Lushasi pers comm)RABV may have been only transiently circulating Neverthelesslessons from Indonesia where lack of a swift and coordinatedresponse to a rabies incursion led to an epidemic (Townsendet al 2013) highlight the importance of active surveillance andrapid response to incursions
Although we found evidence of RABV spatial structurewithin Tanzania it was evident that dispersal was also fre-quent with at least one long distance migration occurringwithin a small temporal window (lt2 years) Lineage move-ments occurring on branches representing short evolutionarytimes such as those identified between Serengeti and Morogoro(gt750 km) indicate rates of dispersal much higher than thoserecorded for endemic wildlife rabies (Biek 2007) and imply hu-man mediated movements as seen in parts of North Africa(Talbi et al 2010) Movement of pastoralist and agro-pastoralistcommunities from the Lake zone in Northern Tanzania tosouthern regions for example Morogoro have been ongoingsince the 1950s (Walsh 2008) with a major influx reported from2003 to 2006 (PINGOrsquos Forum 2013) attributed to climate changeinduced drought and forced evictions from newly protectedareas (Kideghesho et al 2013) During these migrations pasto-ralists are accompanied by their dogs which may facilitate thelong distance movement of animals incubating the virus priorto transmission On further inspection we found that Morogorosamples indicated as instances of long-distance translocationcame from rural areas where pastoralists are likely to migrateto whereas other samples from urban Morogoro formed a dis-tinct cluster (Fig 2) We found three RABV lineages in Morogorofrom only ten samples (Fig 3) Quantifying networks of human-mediated movements (including livestock trade) would providea valuable proxy for connectivity for many zoonotic diseases af-fecting domesticated animals
Further to our findings of regional admixture in Tanzaniawe also observed considerable diversity at a very local scalethat is within the Serengeti District with several lineages co-circulating These lineages appear to have persisted for at least20 years (older Serengeti samples obtained from GenBank alsocluster within these lineages indicated in Fig 1) despite dogvaccination campaigns having been conducted in the districtsince 1996 While vaccination coverage has varied substantiallyacross years and between villages (Viana et al 2015) rabies inci-dence has at times significantly declined for example falling by97 per cent in the late 1990s (Cleaveland et al 2003) Yet our ge-netic data show that despite substantial declines in incidencethese lineages must have persisted at very low levels within thedistrict or subsequently reinvaded from neighbouring districtsWithout sampling the surrounding districts it is not possible todistinguish between these possibilities The skyline plot indi-cated a stable effective RABV population size (Ne) through time(Fig 3d) While this could be expected for an endemic pathogenit is worth noting that there was no evidence of viral populationsize reductions in response to vaccination campaigns Rabiescontrol efforts across much of Tanzania have been very limiteduntil recently and high turnover in the dog population(Hampson et al 2009) likely contributes to the stable persistenceof rabies in Tanzania It has also been noted that geographicstructure can obscure local fluctuations in subpopulations while
8 | Virus Evolution 2015 Vol 1 No 1
maintaining the appearance of a constant Ne in skyline plots(Carrington et al 2005)
This study represents a snapshot of RABV dynamics inTanzania indicating that human movements have dissemi-nated RABV out of locally endemic areas at scales relevant tocontrol that is administrative units such as regions or districtsThese frequent translocations have probably lead to the exis-tence of multiple co-circulating lineages (Fig 3) but relativelyfew introductions lead to sustained chains of transmission thatare detectable as lineage movement events However in diseasesystems closely associated with human activities the probabil-ity of successful translocation and establishment is likely to bemuch greater This suggests that without some level of regionalco-operation Tanzania will be unable to eliminate rabies andmaintain freedom from disease Human movements are oftenin response to social drivers which could be used as signals forincreased vigilance and surveillance in at risk areas
Our findings highlight the use of WGS to uncover fine scaletransmission patterns that can directly inform control effortsHowever sub-genomic approaches can still have utility at acoarser scale and are more easily obtained particularly whensample quality is an issue In particular they can be used to ini-tially identify admixture between countries which may indicatethe necessity of coordinated regional control programs and sur-veillance Co-circulation of multiple lineages and introductionsfacilitated by humans appear to be a feature of endemic rabies vi-rus and complicate the design of a sustainable control strategyHowever using whole-genome data we were able to identifysources of dispersal within Tanzania that could direct efforts to-ward surveillance and control The finding that humans play animportant role in the dynamics of RABV in Tanzania suggeststhat increasing awareness and dog vaccination in lsquohigh-riskrsquo com-munities such as pastoralists could help to reduce long-range dis-persal Moreover the design of enhanced surveillance andcontainment strategies to mitigate human-mediated incursionsand maintain disease freedom should be a priority once controlprograms are established and elimination is being targeted
Supplementary data
Supplementary data is available at VEVOLU Journal online
Acknowledgements
We acknowledge the Ministries of Livestock and FisheriesDevelopment and of Health and Social Welfare TanzaniaNational Parks Tanzania Wildlife Research InstituteNgorongoro Conservation Area Authority TanzaniaCommission for Science and Technology and NationalInstitute for Medical Research for permission and collabora-tion and the Arusha Mtwara and Mwanza VeterinaryInvestigation Centres Pemba Veterinary Office and theFrankfurt Zoological Society for logistical and technical sup-port We are grateful to the veterinary and livestock field of-ficers healthworkers and paravets throughout Tanzaniaand in particular to the following research assistants JoelChangalucha Zilpah Kaare Gurdeep Kour Ahmed LugeloKennedy Lushasi Mathias Magoto Khadija Said andRenatus Herman We thank Pelagia Muchuruza MatthewMaziku Geoffrey Mchau Eberhard Mbunda Francois-XavierMeslin and Bernadette Abela-Ridder for their collaborationsthrough the Bill amp Melinda GatesWHO rabies eliminationdemonstration project in Tanzania This work was
supported by the Wellcome Trust the Medical ResearchCouncil the UBS Optimus Foundation Lincoln Park Zooand the Research and Policy for Infectious DiseaseDynamics Program of the Science and TechnologyDirectorate Department of Homeland Security FogartyInternational Centre National Institute of Health
Conflict of interest None declared
ReferencesAhmed K et al (2015) lsquoMolecular Epidemiology of Rabies
Viruses Circulating in Two Rabies Endemic Provinces of Laos2011ndash2012 Regional Diversity in Southeast Asiarsquo PLoS NeglectedTropical Diseases 9 e0003645
Andrews S (2010) FastQC a quality control tool for highthroughput sequence datalthttpwwwbioinformaticsbbsrcacukprojectsfastqcgt accessed 4 Jun 2014
Ayres D L et al (2012) lsquoBEAGLE An Application ProgrammingInterface and High-Performance Computing Library forStatistical Phylogeneticsrsquo Systematic Biology 61 170ndash3
Bedford T et al (2014) lsquoIntegrating Influenza AntigenicDynamics with Molecular Evolutionrsquo eLife 2014 1ndash26
Biek R and Real L A (2010) lsquoThe Landscape Genetics of InfectiousDisease Emergence and Spreadrsquo Molecular Ecology 19 3515ndash31
mdashmdash et al (2007) lsquoA High-Resolution Genetic Signature ofDemographic and Spatial Expansion in Epizootic Rabies VirusrsquoProceedings of the National Academy of Sciences of the United Statesof America 104 7993ndash8
mdashmdash et al (2015) lsquoMeasurably Evolving Pathogens in the GenomicErarsquo Trends in Ecology amp Evolution 30 306ndash13
Bielejec F et al (2011) lsquoSPREAD Spatial PhylogeneticReconstruction of Evolutionary Dynamicsrsquo Bioinformatics(Oxford England) 27 2910ndash2
mdashmdash et al (2014) lsquoInferring Heterogeneous Evolutionary Processesthrough Time From Sequence Substitution toPhylogeographyrsquo Systematic Biology 63 493ndash504
Bivand R S Pebesma E J and Gomez-Rubio V (2008) AppliedSpatial Data Analysis with R New York Springer
Bolger A M Lohse M and Usadel B (2014) lsquoTrimmomatic AFlexible Trimmer for Illumina Sequence Datarsquo Bioinformatics30 2114ndash20
Bourhy H et al (2008) lsquoThe Origin and Phylogeography of DogRabies Virusrsquo The Journal of General Virology 89 2673ndash81
Brunker K et al (2012) lsquoIntegrating the Landscape Epidemiologyand Genetics of RNA Viruses Rabies in Domestic Dogs as aModelrsquo Parasitology 139 1899ndash913
Carrington C et al (2005) lsquoInvasion and Maintenance of DengueVirus Type 2 and Type 4 in the Americasrsquo Journal of Virology 7914680ndash7
Cleaveland S et al (2003) lsquoA Dog Rabies Vaccination Campaignin Rural Africa Impact on the Incidence of Dog Rabies andHuman Dog-Bite Injuriesrsquo Vaccine 21 1965ndash73
David D et al (2007) lsquoIdentification of Novel Canine Rabies VirusClades in the Middle East and North Africarsquo The Journal ofGeneral Virology 88 967ndash80
Denduangboripant J et al (2005) lsquoTransmission Dynamics ofRabies Virus in Thailand Implications for Disease ControlrsquoBMC Infectious Diseases 5 52
Drummond A J et al (2003) lsquoMeasurably Evolving PopulationsrsquoTrends in Ecology amp Evolution 18 481ndash8
mdashmdash et al (2005) lsquoBayesian Coalescent Inference of PastPopulation Dynamics from Molecular Sequencesrsquo MolecularBiology and Evolution 22 1185ndash92
K Brunker et al | 9
mdashmdash et al (2012) lsquoBayesian Phylogenetics with BEAUti and theBEAST 17rsquo Molecular Biology and Evolution 29 1969ndash73
Ferreira M A R and Suchard M A (2008) lsquoBayesian Analysis ofElapsed Times in Continuous-Time Markov Chainsrsquo CanadianJournal of Statistics 36 355ndash68
Greger M (2007) lsquoThe HumanAnimal Interface Emergence andResurgence of Zoonotic Infectious Diseasesrsquo Critical Reviews inMicrobiology 33 243ndash99
Hampson K et al (2007) lsquoSynchronous Cycles of Domestic DogRabies in Sub-Saharan Africa and the Impact of ControlEffortsrsquo Proceedings of the National Academy of Sciences of theUnited States of America 104 7717ndash22
mdashmdash et al (2009) lsquoTransmission Dynamics and Prospects for theElimination of Canine Rabiesrsquo PLoS Biology 7 e53
Hayman D T S et al (2011) lsquoEvolutionary History of Rabies inGhanarsquo PLoS Neglected Tropical Diseases 5 e1001
Holmes E C and Grenfell B T (2009) lsquoDiscovering the Phylodynamicsof RNA Virusesrsquo PLoS Computational Biology 5 e1000505
Horton D L et al (2015) lsquoComplex Epidemiology of a ZoonoticDisease in a Culturally Diverse Region Phylogeography ofRabies Virus in the Middle Eastrsquo PLOS Neglected TropicalDiseases 9 e0003569
Jombart T (2008) lsquoadegenet A R Package for the MultivariateAnalysis of Genetic Markersrsquo Bioinformatics (Oxford England)24 1403ndash5
Jones K E et al (2008) lsquoGlobal Trends in Emerging InfectiousDiseasesrsquo Nature 451 990ndash3
Katoh K and Standley D M (2013) lsquoMAFFT MultipleSequence Alignment Software Version 7 Improvements inPerformance and Usabilityrsquo Molecular Biology and Evolution30 772ndash80
Kideghesho J et al (2013) lsquoEmerging Issues and Challenges inConservation of Biodiversity in the Rangelands of TanzaniarsquoNature Conservation 6 1ndash29
Kissi B Tordo N and Bourhy H (1995) lsquoGenetic Polymorphismin the Rabies Virus Nucleoprotein Genersquo Virology 209 526ndash37
Knobel D L et al (2005) lsquoRe-Evaluating the Burden of Rabies inAfrica and Asiarsquo Bulletin of the World Health Organization 83360ndash8
Kuhnert D Wu C-H and Drummond A J (2011) lsquoPhylogeneticand Epidemic Modeling of Rapidly Evolving InfectiousDiseasesrsquo Infection Genetics and Evolution Journal of MolecularEpidemiology and Evolutionary Genetics in Infectious Diseases 111825ndash41
Kuzmin I V et al (2008) lsquoComplete Genomes of AravanKhujand Irkut and West Caucasian Bat Viruses with SpecialAttention to the Polymerase Gene and Non-Coding RegionsrsquoVirus Research 136 81ndash90
Kuzmina N A et al (2013) lsquoThe Phylogeography andSpatiotemporal Spread of South-Central Skunk Rabies VirusrsquoPloS One 8 e82348
Lanfear R et al (2012) lsquoPartitionfinder Combined Selectionof Partitioning Schemes and Substitution Models forPhylogenetic Analysesrsquo Molecular Biology and Evolution 291695ndash701
Lembo T et al (2007) lsquoMolecular Epidemiology Identifies Only aSingle Rabies Virus Variant Circulating in Complex CarnivoreCommunities of the Serengetirsquo Proceedings Biological SciencesThe Royal Society 274 2123ndash30
mdashmdash et al (2008) lsquoExploring Reservoir Dynamics A Case Study ofRabies in the Serengeti Ecosystemrsquo Journal of Applied Ecology45 1246ndash57
Lemey P et al (2009) lsquoBayesian Phylogeography Finds Its RootsrsquoPLoS Computational Biology 5 e1000520
mdashmdash et al (2014) lsquoUnifying Viral Genetics and HumanTransportation Data to Predict the Global TransmissionDynamics of Human Influenza H3N2rsquo PLoS Pathogens 10e1003932
Lewin-koh A N J et al (2009) Maptools Tools for Reading andHandling Spatial Objects R package version 08-23 lthttpcranr-projectorgwebpackagesmaptoolsindexhtmlgt accessed 1Sept 2014
Li H and Durbin R (2009) lsquoFast and Accurate Short ReadAlignment with Burrows-Wheeler Transformrsquo Bioinformatics(Oxford England) 25 1754ndash60
mdashmdash et al (2009) lsquoThe Sequence AlignmentMap Format andSAMtoolsrsquo Bioinformatics (Oxford England) 25 2078ndash9
Lumlertdacha B et al (2006) lsquoComplex Genetic Structure ofthe Rabies Virus in Bangkok and Its Surrounding ProvincesThailand Implications for Canine Rabies ControlrsquoTransactions of the Royal Society of Tropical Medicine andHygiene 100 276ndash81
Magee D et al (2014) lsquoCombining Phylogeography and SpatialEpidemiology to Uncover Predictors of H5N1 Influenza A VirusDiffusionrsquo Archives of Virology 160 215ndash24
Magembe S (1985) lsquoEpidemiology of Rabies in United Republic ofTanzaniarsquo in Kuwer E et al (eds) Rabies in the Tropics pp 392ndash8 New York Springer Berlin Heidelberg
Marston D A et al (2007) lsquoComparative Analysis of the FullGenome Sequence of European Bat Lyssavirus Type 1 andType 2 with Other Lyssaviruses and Evidence for a ConservedTranscription Termination and Polyadenylation Motif in theG-L 3rsquo Non-Translated Regionrsquo The Journal of General Virology88 1302ndash14
mdashmdash et al (2013) lsquoNext Generation Sequencing of Viral RNAGenomesrsquo BMC Genomics 14 444
Matsumoto T et al (2013) lsquoMolecular Epidemiology of HumanRabies Viruses in Sri Lankarsquo Infection Genetics and Evolution 18160ndash7
Minin V N and Suchard M A (2008) lsquoCounting LabeledTransitions in Continuous-Time Markov Models of EvolutionrsquoJournal of Mathematical Biology 56 391ndash412
Mollentze N et al (2014) lsquoA Bayesian Approach for Inferring theDynamics of Partially Observed Endemic Infectious Diseasesfrom Space-Time-Genetic Datarsquo Proceedings Biological SciencesThe Royal Society 281 20133251
Nel L H (2013) lsquoDiscrepancies in Data Reporting for RabiesAfricarsquo Emerging Infectious Diseases 19 529ndash33
Nelson M I et al (2015) lsquoGlobal Migration of Influenza A Virusesin Swinersquo Nature Communications 6 6696
Nunes M R T et al (2014) lsquoAir Travel is Associated withIntracontinental Spread of Dengue Virus Serotypes 1-3 inBrazilrsquo PLoS Neglected Tropical Diseases 8 e2769
Paradis E Claude J and Strimmer K (2004) lsquoAPE Analyses ofPhylogenetics and Evolution in R Languagersquo Bioinformatics 20289ndash90
Parker J Rambaut A and Pybus O G (2008) lsquoCorrelatingViral Phenotypes with Phylogeny Accounting forPhylogenetic Uncertaintyrsquo Infection Genetics and Evolution 8239ndash46
Pebesma E J and Bivand R S (2005) lsquoClasses and Methods forSpatial Data in Rrsquo R News 5 9ndash13
PINGOrsquos Forum (2013) Pastoralists Indigenous Non GovernmentalOrganizationsrsquo Forum Annual Report 2012-2013 lthttpwwwpin-gosforumortzgt accessed 23 Nov 2014
Rambaut A et al (2014) lsquoTracer v16rsquo Computer program anddocumentation distributed by the author lthttpbeastbioedacukTracergt accessed 4 Feb 2015
10 | Virus Evolution 2015 Vol 1 No 1
Real L A et al (2005) lsquoUnifying the Spatial Population Dynamicsand Molecular Evolution of Epidemic Rabies Virusrsquo Proceedingsof the National Academy of Sciences of the United States of America102 12107ndash11
Rupprecht C E Hanlon C A and Hemachudha T (2002)lsquoRabies Re-examinedrsquo The Lancet Infectious Diseases 2 327ndash43
Shwiff S Hampson K and Anderson A (2013) lsquoPotentialEconomic Benefits of Eliminating Canine Rabiesrsquo AntiviralResearch 98 352ndash6
Siongok T and Karama M (1985) lsquoEpidemiology of HumanRabies in Kenyarsquo in E Kuwer et al (eds) Rabies in the Tropicspp 445ndash50 New York Springer Berlin Heidelberg
Stamatakis A et al (2012) lsquoRAxML-Light A Tool for ComputingTerabyte Phylogeniesrsquo Bioinformatics (Oxford England) 28 2064ndash6
Susilawathi N M et al (2012) lsquoEpidemiological and ClinicalFeatures of Human Rabies Cases in Bali 2008-2010rsquo BMCInfectious Diseases 12 81
Talbi C et al (2009) lsquoEvolutionary History and Dynamics of DogRabies Virus in Western and Central Africarsquo The Journal ofGeneral Virology 90 783ndash91
mdashmdash et al (2010) lsquoPhylodynamics and Human-Mediated Dispersalof a Zoonotic Virusrsquo PLoS Pathogens 6 e1001166
Tao X Y et al (2009) lsquoMolecular Epidemiology of Rabies inSouthern Peoplersquos Republic of Chinarsquo Emerging InfectiousDiseases 15 1192ndash8
Townsend S E et al (2013) lsquoDesigning Programs for EliminatingCanine Rabies from Islands Bali Indonesia as a Case StudyrsquoPLoS Neglected Tropical Diseases 7 e2372
Viana M et al (2015) lsquoDynamics of a Morbillivirus at theDomesticndashWildlife Interface Canine Distemper Virus inDomestic Dogs and Lionsrsquo Proceedings of the National Academy ofSciences of the United States of America 112 1464ndash9
Walsh M (2008) Pastoralism and Policy Processes in TanzaniaMbarali Case Study Report to the Tanzania Natural ResourceForum Arusha
Wang T H et al (2001) lsquoIdentification of Shared Populations ofHuman Immunodeficiency Virus Type 1 Infecting Microgliaand Tissue Macrophages Outside the Central Nervous SystemrsquoJournal of Virology 75 11686ndash99
WHO (2005) lsquoWHO Expert Consultation on Rabiesrsquo World HealthOrganization technical report series 931 1ndash121
Windiyaningsih C et al (2004) lsquoThe Rabies Epidemic on FloresIsland Indonesia (1998-2003)rsquo Journal of the Medical Associationof Thailandfrac14Chotmaihet thangphaet 87 1389ndash93
Woodroffe R and Donnelly C A (2011) lsquoRisk of Contact be-tween Endangered African Wild Dogs Lycaon pictus andDomestic Dogs Opportunities for Pathogen TransmissionrsquoJournal of Applied Ecology 48 1345ndash54
Woolhouse M E J (2002) lsquoPopulation Biology of Emerging and Re-emerging Pathogensrsquo Trends in Microbiology 10(10 Suppl) S3ndash7
K Brunker et al | 11
- vev011-TF1
- vev011-TF2
-

Africa were constructed using sequences retrieved fromGenBank and including new Tanzanian isolates sequenced forthis study (59 new WGS samples and 50 new partial genome se-quences) Following maximum likelihood (ML) phylogenetic re-construction with the initial sequence datasets subset treeswere extracted for samples in the Africa 1b clade (430 samplesin the partial N dataset and 100 in the full N dataset)
Alignments for whole genome full N and partial N genewere created in MAFFT (Katoh and Standley 2013) and esti-mated phylogenetic relationships using both ML and Bayesianmethods ML phylogenies were estimated in RAxML(Stamatakis et al 2012) with a general time reversible (GTR) nu-cleotide substitution model and a gamma distribution model ofamong-site rate variation A Chinese dog RABV sequence fromGenBank (accession no FJ712193) was used as an outgroup andnode support was evaluated with 1000 bootstrap replicatesBayesian phylogenetic reconstruction was conducted in BEASTv181 (Drummond et al 2012) using a posterior distribution oftrees (without a molecular clock model) Phylogenies were visu-alized and annotated in R using the packages adegenet (Jombart2008) and APE (Paradis Claude and Strimmer 2004) and mapswere made in R with Maptools (Lewin-koh et al 2009) and sppackages (Pebesma and Bivand 2005 Bivand Pebesma andGomez-Rubio 2008) The degree of spatial admixture at largephylogeographic scales ie sub-continental and country levelwas quantified by an association index (AI) using BaTS softwarewith beast phylogenies (Parker et al 2008)
25 Selecting an evolutionary model
An initial nucleotide substitution model was chosen based onthe model selected by PartitionFinder (Lanfear et al 2012) Ourwhole-genome alignments were partitioned into sixteen sets ofnucleotides one for each codon position (CP) in each gene (fivegenes) and one for concatenated non-coding regions Modelscheme selection was based on the best AIC score from a greedysearch of substitution models which favoured a GTR modelwith partitioning into three CPs (CP123)
In addition model comparison based on marginal likelihoodestimates using path sampling (PS) and stepping stone (SS)sampling implemented in BEAST (100 path steps and a chainlength of 100000 steps) were used to test varying levels of com-plexity in the substitution model Non-coding sequence wasconcatenated and partitioned as a lsquogenersquo with its own evolution-ary model Specifically we tested the HKY model with (1) agene-specific nucleotide model with gene-specific rate varia-tion (2) a gene-linked CP partitioned model with among CP rateheterogeneity and homogeneous rates among genes and (3) agene-specific CP partitioned model with among CP and amonggene rate heterogeneity Model types 2 and 3 were also testedwith CP112 and CP123 partitioning schemes Following the re-sults for the best HKY model we did a final step comparing themost supported HKY model with the same structure but using aGTR model Results (Supplementary Table S2) strongly favouredGTR and CP models (CP123 was best supported) but there wasno support for partitions according to genes which all had simi-lar rates of substitution This significantly reduced the complex-ity of the model and is an important finding in the context ofanalysing RABV whole-genome sequence
26 Bayesian evolutionary analyses
Bayesian Markov-chain Monte Carlo analyses were performedusing BEAST v181 (Drummond et al 2012) and the BEAGLE
library (Ayres et al 2012) Based on model comparisons themost supported evolutionary model was a GTR model withdifferent substitution parameters for CPs one two and three(GTR123thornCP123thornC123) and homogeneous rates among geneswith a GTRthornG substitution model for non-coding sequenceA relaxed molecular clock with a lognormal distribution wasused to model rate variation among branches with aBayesian skyline model (Drummond et al 2005) with tengroups as a flexible tree prior A Bayesian skyline plot wasused to estimate the viral effective population size throughtime (Drummond et al 2005) To reconstruct the spatial dy-namics of RABV spread in Tanzania we implemented a dis-cretized diffusion process among nine regional samplinglocations formalized as an asymmetric continuous timeMarkov chain (CTMC) model (Lemey et al 2009) Three inde-pendent Markov-chain Monte Carlo chains with 50 millionstates and a sampling frequency of 50000 were combined inLogCombiner after discarding at least 10 per cent burnPosterior distributions were inspected in Tracer v16(Rambaut et al 2014) to ensure adequate mixing and conver-gence Initial analyses revealed issues with tree likelihoodconvergence Therefore a CTMC model using relaxed consen-sus sequences (see above) which contained fewer ambigui-ties was implemented first and the maximum cladecredibility (MCC) tree used as a starting tree for the finalCTMC models with conservative consensus sequences
To estimate the most significant pathways of viral dispersalbetween regions a stochastic search variable selection (BSSVS)procedure was implemented to identify the best supported dif-fusion rates through Bayes factor (BF) testing (Lemey et al 2009Bielejec et al 2011) For the per lineage rate of migration(Kuhnert Wu and Drummond 2011) a conditional referenceprior (Ferreira and Suchard 2008) is commonly used but for ourdata resulted in convergence problems with some of the BSSVSparameters Instead we used an exponential prior with a meanof 001 which gave the most robust results out of a range of val-ues tested (data not shown) The degree of spatial admixturewas scored using a modified Association Index (Wang et al2001 Lemey et al 2009) and quantified using the inferred num-ber of lineage migrationmovement events between locationsestimated by Markov jump counts (Minin and Suchard 2008)along the branches of the posterior tree distribution For BEASTmodels with Markov Jump counts implemented a conditionalreference prior on the per lineage rate of migration was chosenA summarized history of Markov jump counts was used to iden-tify movements between regions that occurred on very shortbranches and thus over unusually short time frames Dogsrarely move 1 km from their homestead (Hampson et al 2009Woodroffe and Donnelly 2011) and Hampson et al (2009) founda maximum distance of 20 km between linked RABV cases inthe Serengeti District Lineage migrations between regions rep-resenting distances gt100 km were therefore considered un-likely to be attributable to dog movement alone if they occurredover a period of 2 years or less and were instead interpreted asbeing the result of human-mediated movement In additionthe same form of discretized diffusion model was used to assessdiffusion at a broader scale that is between the North andSouth of Tanzania with Pemba Island classed as a third discretestate A BF test in the program SPREAD (Bielejec et al 2011) wasused to identify well-supported migration pathways (BFgt 3)Sampled trees were summarized as an MCC tree with mediannode heights using TreeAnnotator v181 and Figtree v142 wasused to visualize trees and the inferred ancestral locations forinternal branches
4 | Virus Evolution 2015 Vol 1 No 1
3 Results31 Geographic resolution partial versus full viralgenomes
Consistent with previous large-scale phylogenetic studies par-tial genome phylogenies indicated that RABV in sub-SaharanAfrica falls into several regional groups with viruses fromEastern Africa generally being genetically distinct from those inwestern central and southern parts of the continent(Supplementary Figs S1 and S2) Of the four major lineages ofRABV in Africa (David et al 2007 Bourhy et al 2008 Talbi et al2009) only the Cosmopolitan clade and more specifically theAfrica 1b lineage was detected in Tanzania as found previously(Lembo et al 2007) Within the Africa 1b lineage there was evi-dence of admixture between Tanzania and neighbouring coun-tries and occasional long-range admixture at a continental scale(ML trees in Fig 1 and Bayesian MCC trees in SupplementaryFig S3) Sequences clustering most closely with Tanzanian se-quences came from Kenya which shares a border to the northAlthough partial genome data were sufficient to identify suchlarge-scale spatial patterns these data did not provide adequateresolution to distinguish between samples at sub-national
scales within Tanzania The proportion of nodes with bootstrapsupport 80 per cent was only 011 (Bayesian posterior probabil-ity 90 018) for partial N and 027 (Bayesian 041) for full Ngene phylogenies Furthermore out of the sixty TanzanianWGS 60 per cent were identical at the partial N and 25 per centat the full N gene level In contrast ML and Bayesian trees basedon whole-genome sequences were fully resolved and well sup-ported (proportion of nodes with ML bootstrap support 80086 Bayesian posterior probability 90 089) even whensamples had been taken in close spatial and temporal proxim-ity For example RV2498 and RV2499 were sampled a day apartfrom the same village in Morogoro region both samples onlydiffered by a single SNP for the full N gene but were differenti-ated by twenty-five SNPs in the whole-genome alignment Thislarge divergence strongly suggests that the two samples are notfrom the same chain of transmission which might have beenthe conclusion based on partial genome data The median rawpairwise genetic distance between Tanzanian whole-genomesequences was 259 (range 0ndash608) nucleotides and between theSerengeti District samples was 120 (range 2ndash212) nucleotidesshowing considerable diversity at even a small spatiotemporalscale However much of this high divergence is due to the
(a) (b) (c)
Figure 1 ML trees derived from datasets of rabies virus sequences from the Africa 1b clade for increasing levels of genome coverage (a) 430 sequences from African
countries highlighted on the map for a 405 bp fragment of the nucleoprotein gene (b) 100 sequences of full 1350 bp nucleoprotein gene from the same countries (except
Botswana Ghana Kenya and Zimbabwe) and (c) sixty full or near-full genome sequences (range 11076ndash11923 bp) from Tanzania Trees are scaled by number of sub-
stitutions per site and node symbols indicate nodes with bootstrap support08 Historical samples from the Serengeti District (20 years old) are circled in partial ge-
nome trees
K Brunker et al | 5
presence of multiple lineages some of which are evident evenbased on partial genome data Using WGS we identified fivedistinct lineages with high posterior probability support (anno-tated in Fig 2)mdashmedian pairwise genetic distance withineach lineage is listed in Table 1 Bayesian phylogenetic recon-struction of WGS yielded a mean evolutionary rate of 144 104
substitutionssiteyear (95 highest posterior density578 107 to 319 104) similar to previous estimates for N
gene and G gene evolution (David et al 2007 Talbi et al 2009Ahmed et al 2015)
32 Phylogeography of RABV in Tanzania
Within Tanzania we found evidence of phylogeographic struc-ture (AIfrac14 070 Plt 0001) similar to other estimates of Africanintra-country AI values (Table 2) (Talbi et al 2009) Comparedwith the strong spatial structure between countries and largerspatial aggregations (Table 3) this indicates more fluid and dy-namic dispersal patterns within Tanzania as has been found inother African countries (Talbi et al 2010) Across the posteriordistribution of trees there were 24 (95 highest posterior den-sity 21ndash28) independent lineage movement events Using asummarized history of Markov Jump counts across the phylog-eny we found that 43 per cent of these migrations occurred inthe most recent ten years of the phylogeny (Fig 2c) A BSSVSprocedure in BEAST identified eighteen potential diffusionpathways to explain the observed phylogeographic patterns inthe posterior distribution However only four received substan-tial support based on BFgt 5 (Fig 2) Support was particularlystrong for dispersal from the Serengeti District to Morogoro
Figure 2 Regional phylogeography among sixty rabies virus whole-genome sequences sampled in Tanzania from 2003 to 2012 (a) an MCC tree with branches coloured
according to the most probable posterior location of its descendent node inferred by discrete-state phylogeographic reconstruction in BEAST Five major phylogenetic
groups (Tz1-5) are annotated on the tree and node symbols indicate node posterior support 09 (b) The four most significant dispersal pathways indicated by BF re-
sults from a BSSVS procedure in BEAST with the median number of transitions estimated by Markov jump counts indicated in cases where posterior support for a tran-
sition was gt07 (c) Markov jump densities for total number of transitions through time (d) Bayesian Skyline plot showing Nes the product of the effective population
size (Ne) and the generation time in years (s) through time
Table 1 Raw median genetic distance within each of the five mainrabies virus lineages identified in Tanzania
Genetic distance
RV lineage No of substitutions per site No of SNPs
Tz1 947E-03 110Tz2 227E-03 27Tz3 883E-03 95Tz4 417E-03 495Tz5 0 0
6 | Virus Evolution 2015 Vol 1 No 1
(BFfrac14 13530) and Markov jump counts estimated a median ofsix (range 3ndash10) migrations along this dispersal route with atleast one (range 1ndash3) occurring on a branch representing a pe-riod of less than 2 years Most lineages were sampled in morethan one region in Tanzania with some distributed across alarger geographic area than others (Fig 3) The largest lineageTz1 contains not only a cluster of Serengeti samples but alsoencompassed samples from a larger geographic extent and wasfound in eight out of the nine districts sampled The Bayesian
skyline plot revealed that the effective population size has re-mained fairly constant over the past 150 years (Fig 2d) Becauseof the much higher availability of samples from the SerengetiDistrict we also conducted a coarser phylogeographic analysisgrouping sequences into lsquoNorthrsquo lsquoSouthrsquo or lsquoPemba Islandrsquo Thisidentified a predominance of north to south dispersal (esti-mated thirteen independent movements compared with onemovement south to north) and evidence of dispersal back andforth between the North of mainland Tanzania and PembaIsland (Supplementary Fig S4)
4 Discussion
Using viral genetic data from a hierarchy of spatial scales andvarying levels of genome coverage we were able to demonstratethe advantages of whole-genome resolution to describe thespatio-temporal dynamics of endemically circulating caninerabies viruses
We found a clear phylogeographic structure between coun-tries in Africa which could be identified with partial genomedata This suggests that the majority of dispersal occurs at awithin-country scale and control programs would be most ap-propriate at a national scale with strategies to deal with poten-tial incursions from other countries However at regional andlocal scales within Tanzania the discriminatory power of par-tial genome data became too limited to reveal fine-scale popula-tion structure that could aid the effectiveness of controlinterventions In contrast WGS provided the resolution to ge-netically distinguish between all samples and produced a well-supported phylogeny Although sub-genomic information hasutility at broad phylogeographic scales (eg Horton et al 2015)this finding supports the application of WGS for studies aimingto discern population structure at a scale most relevant tocontrol
Although large-scale population structure according tosub-continental areas or country-specific lineages was appar-ent from sub-genomic data we also found incidences of oc-casional large-scale admixture (Supplementary Figs S1 andS2) We continued to find evidence at increasingly fine scalesTanzania and other countries within the Africa 1b cladeshowed signs of admixture particularly when countriesshared a border and within country movements of RABV fa-cilitated by humans were a feature of Tanzanian RABV Evenat a very local scale within the Serengeti District we foundmultiple co-circulating lineages This recurrent theme may bea characteristic of endemic RABV in Africa reflecting decadesof endemic circulation and human-mediated introductionsSimilar patterns have been observed in canine rabies-en-demic countries in South and Southeast Asia (Lumlertdachaet al 2006 Matsumoto et al 2013 Ahmed et al 2015) wherethe presence of co-circulating lineages was attributed to a
Figure 3 Spatial distribution of rabies virus lineages sampled from regions in
Tanzania between 2003 and 2012 with a colour gradient (yellow to red) indicat-
ing the total number of lineages (low to high) sampled in each region
Table 2 Degree of within-country rabies virus spatial admixture in three African countries measured using an Association Index (AI) whichranges from 0 indicating strong population subdivision to 1 indicating complete panmixis Results are shown for Tanzania (this study) andAlgeria and Morocco (Talbi et al 2010)
Country AI [95 BCI] P No of sequences No of locations Median distance (km) Min Max
Algeria 067 [062ndash073] lt0001 117 20 23308 2327 67462Morocco 055 [051ndash063] lt0001 133 28 32645 2862 92670Tanzania 070 [060-079] lt0001 60 9 43976 3905 108826
BCI Bayesian confidence interval
Table 3 Degree of spatial admixture between rabies virus samplesfrom Africa according to an Association Index (AI) AI values canrange from 0 indicating complete population subdivision to 1 indi-cating complete panmixis
Data Spatial clustering level No ofgroups
AI [95 BCI] P
N405 Sub-continent 3 004 [002005] lt001Country 14 006 [005008] lt001
N1350 Sub-continent 3 006 [004008] lt001Country 10 013 [009013] lt001
BCI Bayesian confidence interval Datasets of partial (N405) and full (N1350) nu-
cleoprotein sequences were tested at two levels of spatial aggregation (1) sub-
continent geographical partitions relative to Tanzania (three states Tanzania
neighbouring country and other African country) and (2) country of origin
K Brunker et al | 7
combination of historical introductions from neighbouringcountries and human-mediated movements
Much of the viral population structure we found withinTanzania is consistent with initial invasive waves that havepersisted endemically with structure eroding over time aidedby human-mediated movement Historical accounts describe arabies outbreak in southern regions of Tanzania in the mid-1950s which spread throughout the country and was recordedin the Serengeti District in the late 1970s (Magembe 1985Siongok and Karama 1985) This invasion perhaps shaped theinitial RABV population structure in Tanzania The timeline ofregional scale migrations (Fig 2c) indicates a strong rise in viraldispersal around this period possibly reflecting increasing hu-man connectivity Samples in Tz1 (Fig 2) share partial N geneidentity with samples from Kenya (Fig 1) indicating a sharedevolutionary history that may relate to an initial outbreakacross both countries Lineages Tz1 and Tz3 appear to have awide geographic distribution (Fig 3) and we found evidence of ageneral north to south dispersal pattern in Tanzania(Supplementary Fig S4) This highlights the potential for wide-spread dispersal should an incursion occur from Kenya and sug-gests a need for co-operative cross-border rabies managementonce a national control program is established Variation in thespread of different lineages across Tanzania may reflect the in-fluence of heterogeneous landscape features or dog populationstructure in impeding or facilitating RABV disseminationIdentifying and quantifying such features is the logical nextstep to provide additional information that can inform controlprogrammes such as identifying and strengthening pre-existing barriers Recent extensions of phylogeographic tech-niques have highlighted how this might be achieved using anintegrated approach combining evolutionary and ecologicalanalyses to quantify drivers of viral transmission (Lemey et al2014 Magee et al 2014)
The ancestry of lineages Tz4 and 5 (denoting lineages foundin the southern mainland and on the Island of Pemba respec-tively) points toward an introduction from the north ofTanzania (posterior probabilityfrac14 062) but the uncertainty ofthis ancestral location and the ancestral node itself (poste-riorfrac14 045) suggest these may be distinct historical lineageswith low-level persistence or undetected circulation elsewherein Tanzania Alternatively these clusters may represent in-stances where lineages from external sources have more re-cently invaded and resulted in sustained transmission inTanzaniamdashpartial genome phylogenies indicate Tz4 is relatedto clusters containing samples from countries south ofTanzania South Africa and Mozambique Furthermore Tz5shares a common ancestry with a Nigerian and several CentralAfrican Republic samples This again underlines the threat ofre-invasion or introduction from external sources and the po-tential value of whole-genome resolution to robustly and moreaccurately identify sources of new introductions To date themajority of RABV genetic data from Africa comes from partialgenome analyses however our data suggest that whole-genome characterization would be valuable and should be anaim for the future
Islands represent isolated landscapes with natural barriersto dispersal but incidents of RABV outbreaks instigated by hu-man-mediated introductions (eg to islands in Indonesia[Windiyaningsih et al 2004 Susilawathi et al 2012]) have oftenbeen recorded Sequences from the island of Pemba were sug-gestive of multiple introductions from various sources with evi-dence of dispersal to and from the mainland (SupplementaryFig S4) Samples from Pemba were scattered throughout the
phylogeny and the most divergent lineage Tz5 consisted of twosamples from Pemba The distribution of these lineages may re-flect earlier invasions of RABV into Pemba from elsewhere inTanzania (Tz1 and 2) and the African continent (Tz5) resultingfrom Pembarsquos location on an important trading route Howeversince these lineages were not resampled and no cases havebeen detected from Pemba in over a year (Lushasi pers comm)RABV may have been only transiently circulating Neverthelesslessons from Indonesia where lack of a swift and coordinatedresponse to a rabies incursion led to an epidemic (Townsendet al 2013) highlight the importance of active surveillance andrapid response to incursions
Although we found evidence of RABV spatial structurewithin Tanzania it was evident that dispersal was also fre-quent with at least one long distance migration occurringwithin a small temporal window (lt2 years) Lineage move-ments occurring on branches representing short evolutionarytimes such as those identified between Serengeti and Morogoro(gt750 km) indicate rates of dispersal much higher than thoserecorded for endemic wildlife rabies (Biek 2007) and imply hu-man mediated movements as seen in parts of North Africa(Talbi et al 2010) Movement of pastoralist and agro-pastoralistcommunities from the Lake zone in Northern Tanzania tosouthern regions for example Morogoro have been ongoingsince the 1950s (Walsh 2008) with a major influx reported from2003 to 2006 (PINGOrsquos Forum 2013) attributed to climate changeinduced drought and forced evictions from newly protectedareas (Kideghesho et al 2013) During these migrations pasto-ralists are accompanied by their dogs which may facilitate thelong distance movement of animals incubating the virus priorto transmission On further inspection we found that Morogorosamples indicated as instances of long-distance translocationcame from rural areas where pastoralists are likely to migrateto whereas other samples from urban Morogoro formed a dis-tinct cluster (Fig 2) We found three RABV lineages in Morogorofrom only ten samples (Fig 3) Quantifying networks of human-mediated movements (including livestock trade) would providea valuable proxy for connectivity for many zoonotic diseases af-fecting domesticated animals
Further to our findings of regional admixture in Tanzaniawe also observed considerable diversity at a very local scalethat is within the Serengeti District with several lineages co-circulating These lineages appear to have persisted for at least20 years (older Serengeti samples obtained from GenBank alsocluster within these lineages indicated in Fig 1) despite dogvaccination campaigns having been conducted in the districtsince 1996 While vaccination coverage has varied substantiallyacross years and between villages (Viana et al 2015) rabies inci-dence has at times significantly declined for example falling by97 per cent in the late 1990s (Cleaveland et al 2003) Yet our ge-netic data show that despite substantial declines in incidencethese lineages must have persisted at very low levels within thedistrict or subsequently reinvaded from neighbouring districtsWithout sampling the surrounding districts it is not possible todistinguish between these possibilities The skyline plot indi-cated a stable effective RABV population size (Ne) through time(Fig 3d) While this could be expected for an endemic pathogenit is worth noting that there was no evidence of viral populationsize reductions in response to vaccination campaigns Rabiescontrol efforts across much of Tanzania have been very limiteduntil recently and high turnover in the dog population(Hampson et al 2009) likely contributes to the stable persistenceof rabies in Tanzania It has also been noted that geographicstructure can obscure local fluctuations in subpopulations while
8 | Virus Evolution 2015 Vol 1 No 1
maintaining the appearance of a constant Ne in skyline plots(Carrington et al 2005)
This study represents a snapshot of RABV dynamics inTanzania indicating that human movements have dissemi-nated RABV out of locally endemic areas at scales relevant tocontrol that is administrative units such as regions or districtsThese frequent translocations have probably lead to the exis-tence of multiple co-circulating lineages (Fig 3) but relativelyfew introductions lead to sustained chains of transmission thatare detectable as lineage movement events However in diseasesystems closely associated with human activities the probabil-ity of successful translocation and establishment is likely to bemuch greater This suggests that without some level of regionalco-operation Tanzania will be unable to eliminate rabies andmaintain freedom from disease Human movements are oftenin response to social drivers which could be used as signals forincreased vigilance and surveillance in at risk areas
Our findings highlight the use of WGS to uncover fine scaletransmission patterns that can directly inform control effortsHowever sub-genomic approaches can still have utility at acoarser scale and are more easily obtained particularly whensample quality is an issue In particular they can be used to ini-tially identify admixture between countries which may indicatethe necessity of coordinated regional control programs and sur-veillance Co-circulation of multiple lineages and introductionsfacilitated by humans appear to be a feature of endemic rabies vi-rus and complicate the design of a sustainable control strategyHowever using whole-genome data we were able to identifysources of dispersal within Tanzania that could direct efforts to-ward surveillance and control The finding that humans play animportant role in the dynamics of RABV in Tanzania suggeststhat increasing awareness and dog vaccination in lsquohigh-riskrsquo com-munities such as pastoralists could help to reduce long-range dis-persal Moreover the design of enhanced surveillance andcontainment strategies to mitigate human-mediated incursionsand maintain disease freedom should be a priority once controlprograms are established and elimination is being targeted
Supplementary data
Supplementary data is available at VEVOLU Journal online
Acknowledgements
We acknowledge the Ministries of Livestock and FisheriesDevelopment and of Health and Social Welfare TanzaniaNational Parks Tanzania Wildlife Research InstituteNgorongoro Conservation Area Authority TanzaniaCommission for Science and Technology and NationalInstitute for Medical Research for permission and collabora-tion and the Arusha Mtwara and Mwanza VeterinaryInvestigation Centres Pemba Veterinary Office and theFrankfurt Zoological Society for logistical and technical sup-port We are grateful to the veterinary and livestock field of-ficers healthworkers and paravets throughout Tanzaniaand in particular to the following research assistants JoelChangalucha Zilpah Kaare Gurdeep Kour Ahmed LugeloKennedy Lushasi Mathias Magoto Khadija Said andRenatus Herman We thank Pelagia Muchuruza MatthewMaziku Geoffrey Mchau Eberhard Mbunda Francois-XavierMeslin and Bernadette Abela-Ridder for their collaborationsthrough the Bill amp Melinda GatesWHO rabies eliminationdemonstration project in Tanzania This work was
supported by the Wellcome Trust the Medical ResearchCouncil the UBS Optimus Foundation Lincoln Park Zooand the Research and Policy for Infectious DiseaseDynamics Program of the Science and TechnologyDirectorate Department of Homeland Security FogartyInternational Centre National Institute of Health
Conflict of interest None declared
ReferencesAhmed K et al (2015) lsquoMolecular Epidemiology of Rabies
Viruses Circulating in Two Rabies Endemic Provinces of Laos2011ndash2012 Regional Diversity in Southeast Asiarsquo PLoS NeglectedTropical Diseases 9 e0003645
Andrews S (2010) FastQC a quality control tool for highthroughput sequence datalthttpwwwbioinformaticsbbsrcacukprojectsfastqcgt accessed 4 Jun 2014
Ayres D L et al (2012) lsquoBEAGLE An Application ProgrammingInterface and High-Performance Computing Library forStatistical Phylogeneticsrsquo Systematic Biology 61 170ndash3
Bedford T et al (2014) lsquoIntegrating Influenza AntigenicDynamics with Molecular Evolutionrsquo eLife 2014 1ndash26
Biek R and Real L A (2010) lsquoThe Landscape Genetics of InfectiousDisease Emergence and Spreadrsquo Molecular Ecology 19 3515ndash31
mdashmdash et al (2007) lsquoA High-Resolution Genetic Signature ofDemographic and Spatial Expansion in Epizootic Rabies VirusrsquoProceedings of the National Academy of Sciences of the United Statesof America 104 7993ndash8
mdashmdash et al (2015) lsquoMeasurably Evolving Pathogens in the GenomicErarsquo Trends in Ecology amp Evolution 30 306ndash13
Bielejec F et al (2011) lsquoSPREAD Spatial PhylogeneticReconstruction of Evolutionary Dynamicsrsquo Bioinformatics(Oxford England) 27 2910ndash2
mdashmdash et al (2014) lsquoInferring Heterogeneous Evolutionary Processesthrough Time From Sequence Substitution toPhylogeographyrsquo Systematic Biology 63 493ndash504
Bivand R S Pebesma E J and Gomez-Rubio V (2008) AppliedSpatial Data Analysis with R New York Springer
Bolger A M Lohse M and Usadel B (2014) lsquoTrimmomatic AFlexible Trimmer for Illumina Sequence Datarsquo Bioinformatics30 2114ndash20
Bourhy H et al (2008) lsquoThe Origin and Phylogeography of DogRabies Virusrsquo The Journal of General Virology 89 2673ndash81
Brunker K et al (2012) lsquoIntegrating the Landscape Epidemiologyand Genetics of RNA Viruses Rabies in Domestic Dogs as aModelrsquo Parasitology 139 1899ndash913
Carrington C et al (2005) lsquoInvasion and Maintenance of DengueVirus Type 2 and Type 4 in the Americasrsquo Journal of Virology 7914680ndash7
Cleaveland S et al (2003) lsquoA Dog Rabies Vaccination Campaignin Rural Africa Impact on the Incidence of Dog Rabies andHuman Dog-Bite Injuriesrsquo Vaccine 21 1965ndash73
David D et al (2007) lsquoIdentification of Novel Canine Rabies VirusClades in the Middle East and North Africarsquo The Journal ofGeneral Virology 88 967ndash80
Denduangboripant J et al (2005) lsquoTransmission Dynamics ofRabies Virus in Thailand Implications for Disease ControlrsquoBMC Infectious Diseases 5 52
Drummond A J et al (2003) lsquoMeasurably Evolving PopulationsrsquoTrends in Ecology amp Evolution 18 481ndash8
mdashmdash et al (2005) lsquoBayesian Coalescent Inference of PastPopulation Dynamics from Molecular Sequencesrsquo MolecularBiology and Evolution 22 1185ndash92
K Brunker et al | 9
mdashmdash et al (2012) lsquoBayesian Phylogenetics with BEAUti and theBEAST 17rsquo Molecular Biology and Evolution 29 1969ndash73
Ferreira M A R and Suchard M A (2008) lsquoBayesian Analysis ofElapsed Times in Continuous-Time Markov Chainsrsquo CanadianJournal of Statistics 36 355ndash68
Greger M (2007) lsquoThe HumanAnimal Interface Emergence andResurgence of Zoonotic Infectious Diseasesrsquo Critical Reviews inMicrobiology 33 243ndash99
Hampson K et al (2007) lsquoSynchronous Cycles of Domestic DogRabies in Sub-Saharan Africa and the Impact of ControlEffortsrsquo Proceedings of the National Academy of Sciences of theUnited States of America 104 7717ndash22
mdashmdash et al (2009) lsquoTransmission Dynamics and Prospects for theElimination of Canine Rabiesrsquo PLoS Biology 7 e53
Hayman D T S et al (2011) lsquoEvolutionary History of Rabies inGhanarsquo PLoS Neglected Tropical Diseases 5 e1001
Holmes E C and Grenfell B T (2009) lsquoDiscovering the Phylodynamicsof RNA Virusesrsquo PLoS Computational Biology 5 e1000505
Horton D L et al (2015) lsquoComplex Epidemiology of a ZoonoticDisease in a Culturally Diverse Region Phylogeography ofRabies Virus in the Middle Eastrsquo PLOS Neglected TropicalDiseases 9 e0003569
Jombart T (2008) lsquoadegenet A R Package for the MultivariateAnalysis of Genetic Markersrsquo Bioinformatics (Oxford England)24 1403ndash5
Jones K E et al (2008) lsquoGlobal Trends in Emerging InfectiousDiseasesrsquo Nature 451 990ndash3
Katoh K and Standley D M (2013) lsquoMAFFT MultipleSequence Alignment Software Version 7 Improvements inPerformance and Usabilityrsquo Molecular Biology and Evolution30 772ndash80
Kideghesho J et al (2013) lsquoEmerging Issues and Challenges inConservation of Biodiversity in the Rangelands of TanzaniarsquoNature Conservation 6 1ndash29
Kissi B Tordo N and Bourhy H (1995) lsquoGenetic Polymorphismin the Rabies Virus Nucleoprotein Genersquo Virology 209 526ndash37
Knobel D L et al (2005) lsquoRe-Evaluating the Burden of Rabies inAfrica and Asiarsquo Bulletin of the World Health Organization 83360ndash8
Kuhnert D Wu C-H and Drummond A J (2011) lsquoPhylogeneticand Epidemic Modeling of Rapidly Evolving InfectiousDiseasesrsquo Infection Genetics and Evolution Journal of MolecularEpidemiology and Evolutionary Genetics in Infectious Diseases 111825ndash41
Kuzmin I V et al (2008) lsquoComplete Genomes of AravanKhujand Irkut and West Caucasian Bat Viruses with SpecialAttention to the Polymerase Gene and Non-Coding RegionsrsquoVirus Research 136 81ndash90
Kuzmina N A et al (2013) lsquoThe Phylogeography andSpatiotemporal Spread of South-Central Skunk Rabies VirusrsquoPloS One 8 e82348
Lanfear R et al (2012) lsquoPartitionfinder Combined Selectionof Partitioning Schemes and Substitution Models forPhylogenetic Analysesrsquo Molecular Biology and Evolution 291695ndash701
Lembo T et al (2007) lsquoMolecular Epidemiology Identifies Only aSingle Rabies Virus Variant Circulating in Complex CarnivoreCommunities of the Serengetirsquo Proceedings Biological SciencesThe Royal Society 274 2123ndash30
mdashmdash et al (2008) lsquoExploring Reservoir Dynamics A Case Study ofRabies in the Serengeti Ecosystemrsquo Journal of Applied Ecology45 1246ndash57
Lemey P et al (2009) lsquoBayesian Phylogeography Finds Its RootsrsquoPLoS Computational Biology 5 e1000520
mdashmdash et al (2014) lsquoUnifying Viral Genetics and HumanTransportation Data to Predict the Global TransmissionDynamics of Human Influenza H3N2rsquo PLoS Pathogens 10e1003932
Lewin-koh A N J et al (2009) Maptools Tools for Reading andHandling Spatial Objects R package version 08-23 lthttpcranr-projectorgwebpackagesmaptoolsindexhtmlgt accessed 1Sept 2014
Li H and Durbin R (2009) lsquoFast and Accurate Short ReadAlignment with Burrows-Wheeler Transformrsquo Bioinformatics(Oxford England) 25 1754ndash60
mdashmdash et al (2009) lsquoThe Sequence AlignmentMap Format andSAMtoolsrsquo Bioinformatics (Oxford England) 25 2078ndash9
Lumlertdacha B et al (2006) lsquoComplex Genetic Structure ofthe Rabies Virus in Bangkok and Its Surrounding ProvincesThailand Implications for Canine Rabies ControlrsquoTransactions of the Royal Society of Tropical Medicine andHygiene 100 276ndash81
Magee D et al (2014) lsquoCombining Phylogeography and SpatialEpidemiology to Uncover Predictors of H5N1 Influenza A VirusDiffusionrsquo Archives of Virology 160 215ndash24
Magembe S (1985) lsquoEpidemiology of Rabies in United Republic ofTanzaniarsquo in Kuwer E et al (eds) Rabies in the Tropics pp 392ndash8 New York Springer Berlin Heidelberg
Marston D A et al (2007) lsquoComparative Analysis of the FullGenome Sequence of European Bat Lyssavirus Type 1 andType 2 with Other Lyssaviruses and Evidence for a ConservedTranscription Termination and Polyadenylation Motif in theG-L 3rsquo Non-Translated Regionrsquo The Journal of General Virology88 1302ndash14
mdashmdash et al (2013) lsquoNext Generation Sequencing of Viral RNAGenomesrsquo BMC Genomics 14 444
Matsumoto T et al (2013) lsquoMolecular Epidemiology of HumanRabies Viruses in Sri Lankarsquo Infection Genetics and Evolution 18160ndash7
Minin V N and Suchard M A (2008) lsquoCounting LabeledTransitions in Continuous-Time Markov Models of EvolutionrsquoJournal of Mathematical Biology 56 391ndash412
Mollentze N et al (2014) lsquoA Bayesian Approach for Inferring theDynamics of Partially Observed Endemic Infectious Diseasesfrom Space-Time-Genetic Datarsquo Proceedings Biological SciencesThe Royal Society 281 20133251
Nel L H (2013) lsquoDiscrepancies in Data Reporting for RabiesAfricarsquo Emerging Infectious Diseases 19 529ndash33
Nelson M I et al (2015) lsquoGlobal Migration of Influenza A Virusesin Swinersquo Nature Communications 6 6696
Nunes M R T et al (2014) lsquoAir Travel is Associated withIntracontinental Spread of Dengue Virus Serotypes 1-3 inBrazilrsquo PLoS Neglected Tropical Diseases 8 e2769
Paradis E Claude J and Strimmer K (2004) lsquoAPE Analyses ofPhylogenetics and Evolution in R Languagersquo Bioinformatics 20289ndash90
Parker J Rambaut A and Pybus O G (2008) lsquoCorrelatingViral Phenotypes with Phylogeny Accounting forPhylogenetic Uncertaintyrsquo Infection Genetics and Evolution 8239ndash46
Pebesma E J and Bivand R S (2005) lsquoClasses and Methods forSpatial Data in Rrsquo R News 5 9ndash13
PINGOrsquos Forum (2013) Pastoralists Indigenous Non GovernmentalOrganizationsrsquo Forum Annual Report 2012-2013 lthttpwwwpin-gosforumortzgt accessed 23 Nov 2014
Rambaut A et al (2014) lsquoTracer v16rsquo Computer program anddocumentation distributed by the author lthttpbeastbioedacukTracergt accessed 4 Feb 2015
10 | Virus Evolution 2015 Vol 1 No 1
Real L A et al (2005) lsquoUnifying the Spatial Population Dynamicsand Molecular Evolution of Epidemic Rabies Virusrsquo Proceedingsof the National Academy of Sciences of the United States of America102 12107ndash11
Rupprecht C E Hanlon C A and Hemachudha T (2002)lsquoRabies Re-examinedrsquo The Lancet Infectious Diseases 2 327ndash43
Shwiff S Hampson K and Anderson A (2013) lsquoPotentialEconomic Benefits of Eliminating Canine Rabiesrsquo AntiviralResearch 98 352ndash6
Siongok T and Karama M (1985) lsquoEpidemiology of HumanRabies in Kenyarsquo in E Kuwer et al (eds) Rabies in the Tropicspp 445ndash50 New York Springer Berlin Heidelberg
Stamatakis A et al (2012) lsquoRAxML-Light A Tool for ComputingTerabyte Phylogeniesrsquo Bioinformatics (Oxford England) 28 2064ndash6
Susilawathi N M et al (2012) lsquoEpidemiological and ClinicalFeatures of Human Rabies Cases in Bali 2008-2010rsquo BMCInfectious Diseases 12 81
Talbi C et al (2009) lsquoEvolutionary History and Dynamics of DogRabies Virus in Western and Central Africarsquo The Journal ofGeneral Virology 90 783ndash91
mdashmdash et al (2010) lsquoPhylodynamics and Human-Mediated Dispersalof a Zoonotic Virusrsquo PLoS Pathogens 6 e1001166
Tao X Y et al (2009) lsquoMolecular Epidemiology of Rabies inSouthern Peoplersquos Republic of Chinarsquo Emerging InfectiousDiseases 15 1192ndash8
Townsend S E et al (2013) lsquoDesigning Programs for EliminatingCanine Rabies from Islands Bali Indonesia as a Case StudyrsquoPLoS Neglected Tropical Diseases 7 e2372
Viana M et al (2015) lsquoDynamics of a Morbillivirus at theDomesticndashWildlife Interface Canine Distemper Virus inDomestic Dogs and Lionsrsquo Proceedings of the National Academy ofSciences of the United States of America 112 1464ndash9
Walsh M (2008) Pastoralism and Policy Processes in TanzaniaMbarali Case Study Report to the Tanzania Natural ResourceForum Arusha
Wang T H et al (2001) lsquoIdentification of Shared Populations ofHuman Immunodeficiency Virus Type 1 Infecting Microgliaand Tissue Macrophages Outside the Central Nervous SystemrsquoJournal of Virology 75 11686ndash99
WHO (2005) lsquoWHO Expert Consultation on Rabiesrsquo World HealthOrganization technical report series 931 1ndash121
Windiyaningsih C et al (2004) lsquoThe Rabies Epidemic on FloresIsland Indonesia (1998-2003)rsquo Journal of the Medical Associationof Thailandfrac14Chotmaihet thangphaet 87 1389ndash93
Woodroffe R and Donnelly C A (2011) lsquoRisk of Contact be-tween Endangered African Wild Dogs Lycaon pictus andDomestic Dogs Opportunities for Pathogen TransmissionrsquoJournal of Applied Ecology 48 1345ndash54
Woolhouse M E J (2002) lsquoPopulation Biology of Emerging and Re-emerging Pathogensrsquo Trends in Microbiology 10(10 Suppl) S3ndash7
K Brunker et al | 11
- vev011-TF1
- vev011-TF2
-
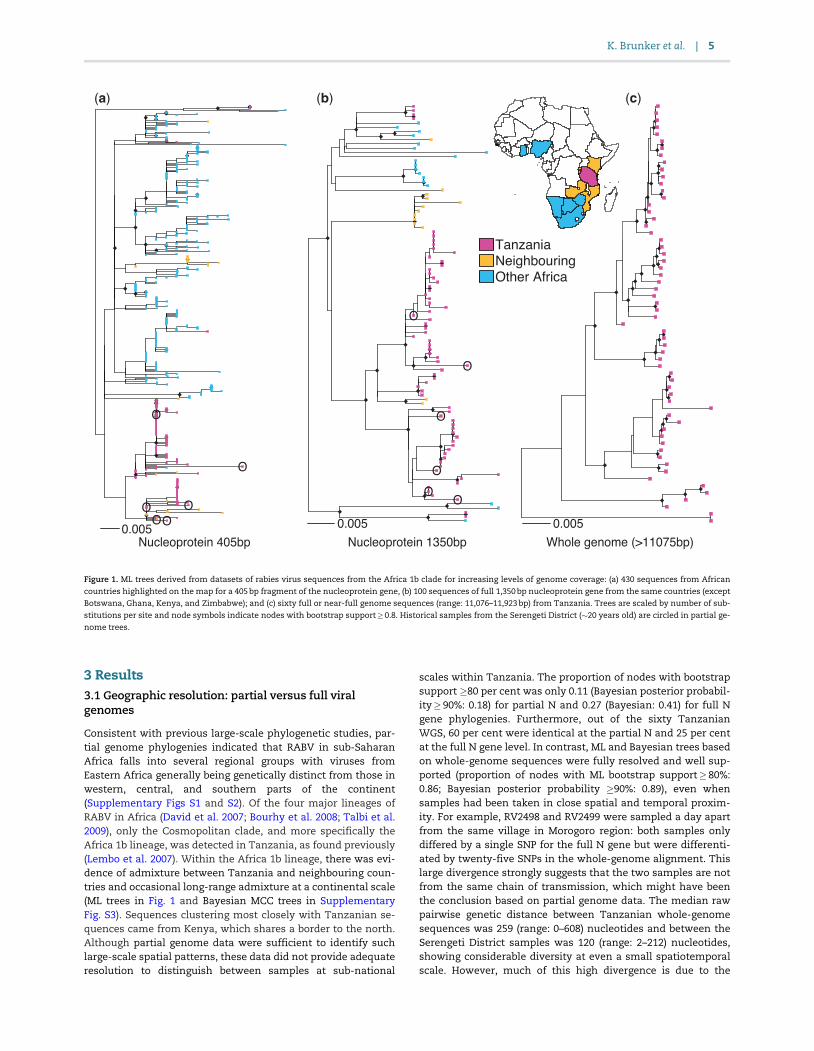
3 Results31 Geographic resolution partial versus full viralgenomes
Consistent with previous large-scale phylogenetic studies par-tial genome phylogenies indicated that RABV in sub-SaharanAfrica falls into several regional groups with viruses fromEastern Africa generally being genetically distinct from those inwestern central and southern parts of the continent(Supplementary Figs S1 and S2) Of the four major lineages ofRABV in Africa (David et al 2007 Bourhy et al 2008 Talbi et al2009) only the Cosmopolitan clade and more specifically theAfrica 1b lineage was detected in Tanzania as found previously(Lembo et al 2007) Within the Africa 1b lineage there was evi-dence of admixture between Tanzania and neighbouring coun-tries and occasional long-range admixture at a continental scale(ML trees in Fig 1 and Bayesian MCC trees in SupplementaryFig S3) Sequences clustering most closely with Tanzanian se-quences came from Kenya which shares a border to the northAlthough partial genome data were sufficient to identify suchlarge-scale spatial patterns these data did not provide adequateresolution to distinguish between samples at sub-national
scales within Tanzania The proportion of nodes with bootstrapsupport 80 per cent was only 011 (Bayesian posterior probabil-ity 90 018) for partial N and 027 (Bayesian 041) for full Ngene phylogenies Furthermore out of the sixty TanzanianWGS 60 per cent were identical at the partial N and 25 per centat the full N gene level In contrast ML and Bayesian trees basedon whole-genome sequences were fully resolved and well sup-ported (proportion of nodes with ML bootstrap support 80086 Bayesian posterior probability 90 089) even whensamples had been taken in close spatial and temporal proxim-ity For example RV2498 and RV2499 were sampled a day apartfrom the same village in Morogoro region both samples onlydiffered by a single SNP for the full N gene but were differenti-ated by twenty-five SNPs in the whole-genome alignment Thislarge divergence strongly suggests that the two samples are notfrom the same chain of transmission which might have beenthe conclusion based on partial genome data The median rawpairwise genetic distance between Tanzanian whole-genomesequences was 259 (range 0ndash608) nucleotides and between theSerengeti District samples was 120 (range 2ndash212) nucleotidesshowing considerable diversity at even a small spatiotemporalscale However much of this high divergence is due to the
(a) (b) (c)
Figure 1 ML trees derived from datasets of rabies virus sequences from the Africa 1b clade for increasing levels of genome coverage (a) 430 sequences from African
countries highlighted on the map for a 405 bp fragment of the nucleoprotein gene (b) 100 sequences of full 1350 bp nucleoprotein gene from the same countries (except
Botswana Ghana Kenya and Zimbabwe) and (c) sixty full or near-full genome sequences (range 11076ndash11923 bp) from Tanzania Trees are scaled by number of sub-
stitutions per site and node symbols indicate nodes with bootstrap support08 Historical samples from the Serengeti District (20 years old) are circled in partial ge-
nome trees
K Brunker et al | 5
presence of multiple lineages some of which are evident evenbased on partial genome data Using WGS we identified fivedistinct lineages with high posterior probability support (anno-tated in Fig 2)mdashmedian pairwise genetic distance withineach lineage is listed in Table 1 Bayesian phylogenetic recon-struction of WGS yielded a mean evolutionary rate of 144 104
substitutionssiteyear (95 highest posterior density578 107 to 319 104) similar to previous estimates for N
gene and G gene evolution (David et al 2007 Talbi et al 2009Ahmed et al 2015)
32 Phylogeography of RABV in Tanzania
Within Tanzania we found evidence of phylogeographic struc-ture (AIfrac14 070 Plt 0001) similar to other estimates of Africanintra-country AI values (Table 2) (Talbi et al 2009) Comparedwith the strong spatial structure between countries and largerspatial aggregations (Table 3) this indicates more fluid and dy-namic dispersal patterns within Tanzania as has been found inother African countries (Talbi et al 2010) Across the posteriordistribution of trees there were 24 (95 highest posterior den-sity 21ndash28) independent lineage movement events Using asummarized history of Markov Jump counts across the phylog-eny we found that 43 per cent of these migrations occurred inthe most recent ten years of the phylogeny (Fig 2c) A BSSVSprocedure in BEAST identified eighteen potential diffusionpathways to explain the observed phylogeographic patterns inthe posterior distribution However only four received substan-tial support based on BFgt 5 (Fig 2) Support was particularlystrong for dispersal from the Serengeti District to Morogoro
Figure 2 Regional phylogeography among sixty rabies virus whole-genome sequences sampled in Tanzania from 2003 to 2012 (a) an MCC tree with branches coloured
according to the most probable posterior location of its descendent node inferred by discrete-state phylogeographic reconstruction in BEAST Five major phylogenetic
groups (Tz1-5) are annotated on the tree and node symbols indicate node posterior support 09 (b) The four most significant dispersal pathways indicated by BF re-
sults from a BSSVS procedure in BEAST with the median number of transitions estimated by Markov jump counts indicated in cases where posterior support for a tran-
sition was gt07 (c) Markov jump densities for total number of transitions through time (d) Bayesian Skyline plot showing Nes the product of the effective population
size (Ne) and the generation time in years (s) through time
Table 1 Raw median genetic distance within each of the five mainrabies virus lineages identified in Tanzania
Genetic distance
RV lineage No of substitutions per site No of SNPs
Tz1 947E-03 110Tz2 227E-03 27Tz3 883E-03 95Tz4 417E-03 495Tz5 0 0
6 | Virus Evolution 2015 Vol 1 No 1
(BFfrac14 13530) and Markov jump counts estimated a median ofsix (range 3ndash10) migrations along this dispersal route with atleast one (range 1ndash3) occurring on a branch representing a pe-riod of less than 2 years Most lineages were sampled in morethan one region in Tanzania with some distributed across alarger geographic area than others (Fig 3) The largest lineageTz1 contains not only a cluster of Serengeti samples but alsoencompassed samples from a larger geographic extent and wasfound in eight out of the nine districts sampled The Bayesian
skyline plot revealed that the effective population size has re-mained fairly constant over the past 150 years (Fig 2d) Becauseof the much higher availability of samples from the SerengetiDistrict we also conducted a coarser phylogeographic analysisgrouping sequences into lsquoNorthrsquo lsquoSouthrsquo or lsquoPemba Islandrsquo Thisidentified a predominance of north to south dispersal (esti-mated thirteen independent movements compared with onemovement south to north) and evidence of dispersal back andforth between the North of mainland Tanzania and PembaIsland (Supplementary Fig S4)
4 Discussion
Using viral genetic data from a hierarchy of spatial scales andvarying levels of genome coverage we were able to demonstratethe advantages of whole-genome resolution to describe thespatio-temporal dynamics of endemically circulating caninerabies viruses
We found a clear phylogeographic structure between coun-tries in Africa which could be identified with partial genomedata This suggests that the majority of dispersal occurs at awithin-country scale and control programs would be most ap-propriate at a national scale with strategies to deal with poten-tial incursions from other countries However at regional andlocal scales within Tanzania the discriminatory power of par-tial genome data became too limited to reveal fine-scale popula-tion structure that could aid the effectiveness of controlinterventions In contrast WGS provided the resolution to ge-netically distinguish between all samples and produced a well-supported phylogeny Although sub-genomic information hasutility at broad phylogeographic scales (eg Horton et al 2015)this finding supports the application of WGS for studies aimingto discern population structure at a scale most relevant tocontrol
Although large-scale population structure according tosub-continental areas or country-specific lineages was appar-ent from sub-genomic data we also found incidences of oc-casional large-scale admixture (Supplementary Figs S1 andS2) We continued to find evidence at increasingly fine scalesTanzania and other countries within the Africa 1b cladeshowed signs of admixture particularly when countriesshared a border and within country movements of RABV fa-cilitated by humans were a feature of Tanzanian RABV Evenat a very local scale within the Serengeti District we foundmultiple co-circulating lineages This recurrent theme may bea characteristic of endemic RABV in Africa reflecting decadesof endemic circulation and human-mediated introductionsSimilar patterns have been observed in canine rabies-en-demic countries in South and Southeast Asia (Lumlertdachaet al 2006 Matsumoto et al 2013 Ahmed et al 2015) wherethe presence of co-circulating lineages was attributed to a
Figure 3 Spatial distribution of rabies virus lineages sampled from regions in
Tanzania between 2003 and 2012 with a colour gradient (yellow to red) indicat-
ing the total number of lineages (low to high) sampled in each region
Table 2 Degree of within-country rabies virus spatial admixture in three African countries measured using an Association Index (AI) whichranges from 0 indicating strong population subdivision to 1 indicating complete panmixis Results are shown for Tanzania (this study) andAlgeria and Morocco (Talbi et al 2010)
Country AI [95 BCI] P No of sequences No of locations Median distance (km) Min Max
Algeria 067 [062ndash073] lt0001 117 20 23308 2327 67462Morocco 055 [051ndash063] lt0001 133 28 32645 2862 92670Tanzania 070 [060-079] lt0001 60 9 43976 3905 108826
BCI Bayesian confidence interval
Table 3 Degree of spatial admixture between rabies virus samplesfrom Africa according to an Association Index (AI) AI values canrange from 0 indicating complete population subdivision to 1 indi-cating complete panmixis
Data Spatial clustering level No ofgroups
AI [95 BCI] P
N405 Sub-continent 3 004 [002005] lt001Country 14 006 [005008] lt001
N1350 Sub-continent 3 006 [004008] lt001Country 10 013 [009013] lt001
BCI Bayesian confidence interval Datasets of partial (N405) and full (N1350) nu-
cleoprotein sequences were tested at two levels of spatial aggregation (1) sub-
continent geographical partitions relative to Tanzania (three states Tanzania
neighbouring country and other African country) and (2) country of origin
K Brunker et al | 7
combination of historical introductions from neighbouringcountries and human-mediated movements
Much of the viral population structure we found withinTanzania is consistent with initial invasive waves that havepersisted endemically with structure eroding over time aidedby human-mediated movement Historical accounts describe arabies outbreak in southern regions of Tanzania in the mid-1950s which spread throughout the country and was recordedin the Serengeti District in the late 1970s (Magembe 1985Siongok and Karama 1985) This invasion perhaps shaped theinitial RABV population structure in Tanzania The timeline ofregional scale migrations (Fig 2c) indicates a strong rise in viraldispersal around this period possibly reflecting increasing hu-man connectivity Samples in Tz1 (Fig 2) share partial N geneidentity with samples from Kenya (Fig 1) indicating a sharedevolutionary history that may relate to an initial outbreakacross both countries Lineages Tz1 and Tz3 appear to have awide geographic distribution (Fig 3) and we found evidence of ageneral north to south dispersal pattern in Tanzania(Supplementary Fig S4) This highlights the potential for wide-spread dispersal should an incursion occur from Kenya and sug-gests a need for co-operative cross-border rabies managementonce a national control program is established Variation in thespread of different lineages across Tanzania may reflect the in-fluence of heterogeneous landscape features or dog populationstructure in impeding or facilitating RABV disseminationIdentifying and quantifying such features is the logical nextstep to provide additional information that can inform controlprogrammes such as identifying and strengthening pre-existing barriers Recent extensions of phylogeographic tech-niques have highlighted how this might be achieved using anintegrated approach combining evolutionary and ecologicalanalyses to quantify drivers of viral transmission (Lemey et al2014 Magee et al 2014)
The ancestry of lineages Tz4 and 5 (denoting lineages foundin the southern mainland and on the Island of Pemba respec-tively) points toward an introduction from the north ofTanzania (posterior probabilityfrac14 062) but the uncertainty ofthis ancestral location and the ancestral node itself (poste-riorfrac14 045) suggest these may be distinct historical lineageswith low-level persistence or undetected circulation elsewherein Tanzania Alternatively these clusters may represent in-stances where lineages from external sources have more re-cently invaded and resulted in sustained transmission inTanzaniamdashpartial genome phylogenies indicate Tz4 is relatedto clusters containing samples from countries south ofTanzania South Africa and Mozambique Furthermore Tz5shares a common ancestry with a Nigerian and several CentralAfrican Republic samples This again underlines the threat ofre-invasion or introduction from external sources and the po-tential value of whole-genome resolution to robustly and moreaccurately identify sources of new introductions To date themajority of RABV genetic data from Africa comes from partialgenome analyses however our data suggest that whole-genome characterization would be valuable and should be anaim for the future
Islands represent isolated landscapes with natural barriersto dispersal but incidents of RABV outbreaks instigated by hu-man-mediated introductions (eg to islands in Indonesia[Windiyaningsih et al 2004 Susilawathi et al 2012]) have oftenbeen recorded Sequences from the island of Pemba were sug-gestive of multiple introductions from various sources with evi-dence of dispersal to and from the mainland (SupplementaryFig S4) Samples from Pemba were scattered throughout the
phylogeny and the most divergent lineage Tz5 consisted of twosamples from Pemba The distribution of these lineages may re-flect earlier invasions of RABV into Pemba from elsewhere inTanzania (Tz1 and 2) and the African continent (Tz5) resultingfrom Pembarsquos location on an important trading route Howeversince these lineages were not resampled and no cases havebeen detected from Pemba in over a year (Lushasi pers comm)RABV may have been only transiently circulating Neverthelesslessons from Indonesia where lack of a swift and coordinatedresponse to a rabies incursion led to an epidemic (Townsendet al 2013) highlight the importance of active surveillance andrapid response to incursions
Although we found evidence of RABV spatial structurewithin Tanzania it was evident that dispersal was also fre-quent with at least one long distance migration occurringwithin a small temporal window (lt2 years) Lineage move-ments occurring on branches representing short evolutionarytimes such as those identified between Serengeti and Morogoro(gt750 km) indicate rates of dispersal much higher than thoserecorded for endemic wildlife rabies (Biek 2007) and imply hu-man mediated movements as seen in parts of North Africa(Talbi et al 2010) Movement of pastoralist and agro-pastoralistcommunities from the Lake zone in Northern Tanzania tosouthern regions for example Morogoro have been ongoingsince the 1950s (Walsh 2008) with a major influx reported from2003 to 2006 (PINGOrsquos Forum 2013) attributed to climate changeinduced drought and forced evictions from newly protectedareas (Kideghesho et al 2013) During these migrations pasto-ralists are accompanied by their dogs which may facilitate thelong distance movement of animals incubating the virus priorto transmission On further inspection we found that Morogorosamples indicated as instances of long-distance translocationcame from rural areas where pastoralists are likely to migrateto whereas other samples from urban Morogoro formed a dis-tinct cluster (Fig 2) We found three RABV lineages in Morogorofrom only ten samples (Fig 3) Quantifying networks of human-mediated movements (including livestock trade) would providea valuable proxy for connectivity for many zoonotic diseases af-fecting domesticated animals
Further to our findings of regional admixture in Tanzaniawe also observed considerable diversity at a very local scalethat is within the Serengeti District with several lineages co-circulating These lineages appear to have persisted for at least20 years (older Serengeti samples obtained from GenBank alsocluster within these lineages indicated in Fig 1) despite dogvaccination campaigns having been conducted in the districtsince 1996 While vaccination coverage has varied substantiallyacross years and between villages (Viana et al 2015) rabies inci-dence has at times significantly declined for example falling by97 per cent in the late 1990s (Cleaveland et al 2003) Yet our ge-netic data show that despite substantial declines in incidencethese lineages must have persisted at very low levels within thedistrict or subsequently reinvaded from neighbouring districtsWithout sampling the surrounding districts it is not possible todistinguish between these possibilities The skyline plot indi-cated a stable effective RABV population size (Ne) through time(Fig 3d) While this could be expected for an endemic pathogenit is worth noting that there was no evidence of viral populationsize reductions in response to vaccination campaigns Rabiescontrol efforts across much of Tanzania have been very limiteduntil recently and high turnover in the dog population(Hampson et al 2009) likely contributes to the stable persistenceof rabies in Tanzania It has also been noted that geographicstructure can obscure local fluctuations in subpopulations while
8 | Virus Evolution 2015 Vol 1 No 1
maintaining the appearance of a constant Ne in skyline plots(Carrington et al 2005)
This study represents a snapshot of RABV dynamics inTanzania indicating that human movements have dissemi-nated RABV out of locally endemic areas at scales relevant tocontrol that is administrative units such as regions or districtsThese frequent translocations have probably lead to the exis-tence of multiple co-circulating lineages (Fig 3) but relativelyfew introductions lead to sustained chains of transmission thatare detectable as lineage movement events However in diseasesystems closely associated with human activities the probabil-ity of successful translocation and establishment is likely to bemuch greater This suggests that without some level of regionalco-operation Tanzania will be unable to eliminate rabies andmaintain freedom from disease Human movements are oftenin response to social drivers which could be used as signals forincreased vigilance and surveillance in at risk areas
Our findings highlight the use of WGS to uncover fine scaletransmission patterns that can directly inform control effortsHowever sub-genomic approaches can still have utility at acoarser scale and are more easily obtained particularly whensample quality is an issue In particular they can be used to ini-tially identify admixture between countries which may indicatethe necessity of coordinated regional control programs and sur-veillance Co-circulation of multiple lineages and introductionsfacilitated by humans appear to be a feature of endemic rabies vi-rus and complicate the design of a sustainable control strategyHowever using whole-genome data we were able to identifysources of dispersal within Tanzania that could direct efforts to-ward surveillance and control The finding that humans play animportant role in the dynamics of RABV in Tanzania suggeststhat increasing awareness and dog vaccination in lsquohigh-riskrsquo com-munities such as pastoralists could help to reduce long-range dis-persal Moreover the design of enhanced surveillance andcontainment strategies to mitigate human-mediated incursionsand maintain disease freedom should be a priority once controlprograms are established and elimination is being targeted
Supplementary data
Supplementary data is available at VEVOLU Journal online
Acknowledgements
We acknowledge the Ministries of Livestock and FisheriesDevelopment and of Health and Social Welfare TanzaniaNational Parks Tanzania Wildlife Research InstituteNgorongoro Conservation Area Authority TanzaniaCommission for Science and Technology and NationalInstitute for Medical Research for permission and collabora-tion and the Arusha Mtwara and Mwanza VeterinaryInvestigation Centres Pemba Veterinary Office and theFrankfurt Zoological Society for logistical and technical sup-port We are grateful to the veterinary and livestock field of-ficers healthworkers and paravets throughout Tanzaniaand in particular to the following research assistants JoelChangalucha Zilpah Kaare Gurdeep Kour Ahmed LugeloKennedy Lushasi Mathias Magoto Khadija Said andRenatus Herman We thank Pelagia Muchuruza MatthewMaziku Geoffrey Mchau Eberhard Mbunda Francois-XavierMeslin and Bernadette Abela-Ridder for their collaborationsthrough the Bill amp Melinda GatesWHO rabies eliminationdemonstration project in Tanzania This work was
supported by the Wellcome Trust the Medical ResearchCouncil the UBS Optimus Foundation Lincoln Park Zooand the Research and Policy for Infectious DiseaseDynamics Program of the Science and TechnologyDirectorate Department of Homeland Security FogartyInternational Centre National Institute of Health
Conflict of interest None declared
ReferencesAhmed K et al (2015) lsquoMolecular Epidemiology of Rabies
Viruses Circulating in Two Rabies Endemic Provinces of Laos2011ndash2012 Regional Diversity in Southeast Asiarsquo PLoS NeglectedTropical Diseases 9 e0003645
Andrews S (2010) FastQC a quality control tool for highthroughput sequence datalthttpwwwbioinformaticsbbsrcacukprojectsfastqcgt accessed 4 Jun 2014
Ayres D L et al (2012) lsquoBEAGLE An Application ProgrammingInterface and High-Performance Computing Library forStatistical Phylogeneticsrsquo Systematic Biology 61 170ndash3
Bedford T et al (2014) lsquoIntegrating Influenza AntigenicDynamics with Molecular Evolutionrsquo eLife 2014 1ndash26
Biek R and Real L A (2010) lsquoThe Landscape Genetics of InfectiousDisease Emergence and Spreadrsquo Molecular Ecology 19 3515ndash31
mdashmdash et al (2007) lsquoA High-Resolution Genetic Signature ofDemographic and Spatial Expansion in Epizootic Rabies VirusrsquoProceedings of the National Academy of Sciences of the United Statesof America 104 7993ndash8
mdashmdash et al (2015) lsquoMeasurably Evolving Pathogens in the GenomicErarsquo Trends in Ecology amp Evolution 30 306ndash13
Bielejec F et al (2011) lsquoSPREAD Spatial PhylogeneticReconstruction of Evolutionary Dynamicsrsquo Bioinformatics(Oxford England) 27 2910ndash2
mdashmdash et al (2014) lsquoInferring Heterogeneous Evolutionary Processesthrough Time From Sequence Substitution toPhylogeographyrsquo Systematic Biology 63 493ndash504
Bivand R S Pebesma E J and Gomez-Rubio V (2008) AppliedSpatial Data Analysis with R New York Springer
Bolger A M Lohse M and Usadel B (2014) lsquoTrimmomatic AFlexible Trimmer for Illumina Sequence Datarsquo Bioinformatics30 2114ndash20
Bourhy H et al (2008) lsquoThe Origin and Phylogeography of DogRabies Virusrsquo The Journal of General Virology 89 2673ndash81
Brunker K et al (2012) lsquoIntegrating the Landscape Epidemiologyand Genetics of RNA Viruses Rabies in Domestic Dogs as aModelrsquo Parasitology 139 1899ndash913
Carrington C et al (2005) lsquoInvasion and Maintenance of DengueVirus Type 2 and Type 4 in the Americasrsquo Journal of Virology 7914680ndash7
Cleaveland S et al (2003) lsquoA Dog Rabies Vaccination Campaignin Rural Africa Impact on the Incidence of Dog Rabies andHuman Dog-Bite Injuriesrsquo Vaccine 21 1965ndash73
David D et al (2007) lsquoIdentification of Novel Canine Rabies VirusClades in the Middle East and North Africarsquo The Journal ofGeneral Virology 88 967ndash80
Denduangboripant J et al (2005) lsquoTransmission Dynamics ofRabies Virus in Thailand Implications for Disease ControlrsquoBMC Infectious Diseases 5 52
Drummond A J et al (2003) lsquoMeasurably Evolving PopulationsrsquoTrends in Ecology amp Evolution 18 481ndash8
mdashmdash et al (2005) lsquoBayesian Coalescent Inference of PastPopulation Dynamics from Molecular Sequencesrsquo MolecularBiology and Evolution 22 1185ndash92
K Brunker et al | 9
mdashmdash et al (2012) lsquoBayesian Phylogenetics with BEAUti and theBEAST 17rsquo Molecular Biology and Evolution 29 1969ndash73
Ferreira M A R and Suchard M A (2008) lsquoBayesian Analysis ofElapsed Times in Continuous-Time Markov Chainsrsquo CanadianJournal of Statistics 36 355ndash68
Greger M (2007) lsquoThe HumanAnimal Interface Emergence andResurgence of Zoonotic Infectious Diseasesrsquo Critical Reviews inMicrobiology 33 243ndash99
Hampson K et al (2007) lsquoSynchronous Cycles of Domestic DogRabies in Sub-Saharan Africa and the Impact of ControlEffortsrsquo Proceedings of the National Academy of Sciences of theUnited States of America 104 7717ndash22
mdashmdash et al (2009) lsquoTransmission Dynamics and Prospects for theElimination of Canine Rabiesrsquo PLoS Biology 7 e53
Hayman D T S et al (2011) lsquoEvolutionary History of Rabies inGhanarsquo PLoS Neglected Tropical Diseases 5 e1001
Holmes E C and Grenfell B T (2009) lsquoDiscovering the Phylodynamicsof RNA Virusesrsquo PLoS Computational Biology 5 e1000505
Horton D L et al (2015) lsquoComplex Epidemiology of a ZoonoticDisease in a Culturally Diverse Region Phylogeography ofRabies Virus in the Middle Eastrsquo PLOS Neglected TropicalDiseases 9 e0003569
Jombart T (2008) lsquoadegenet A R Package for the MultivariateAnalysis of Genetic Markersrsquo Bioinformatics (Oxford England)24 1403ndash5
Jones K E et al (2008) lsquoGlobal Trends in Emerging InfectiousDiseasesrsquo Nature 451 990ndash3
Katoh K and Standley D M (2013) lsquoMAFFT MultipleSequence Alignment Software Version 7 Improvements inPerformance and Usabilityrsquo Molecular Biology and Evolution30 772ndash80
Kideghesho J et al (2013) lsquoEmerging Issues and Challenges inConservation of Biodiversity in the Rangelands of TanzaniarsquoNature Conservation 6 1ndash29
Kissi B Tordo N and Bourhy H (1995) lsquoGenetic Polymorphismin the Rabies Virus Nucleoprotein Genersquo Virology 209 526ndash37
Knobel D L et al (2005) lsquoRe-Evaluating the Burden of Rabies inAfrica and Asiarsquo Bulletin of the World Health Organization 83360ndash8
Kuhnert D Wu C-H and Drummond A J (2011) lsquoPhylogeneticand Epidemic Modeling of Rapidly Evolving InfectiousDiseasesrsquo Infection Genetics and Evolution Journal of MolecularEpidemiology and Evolutionary Genetics in Infectious Diseases 111825ndash41
Kuzmin I V et al (2008) lsquoComplete Genomes of AravanKhujand Irkut and West Caucasian Bat Viruses with SpecialAttention to the Polymerase Gene and Non-Coding RegionsrsquoVirus Research 136 81ndash90
Kuzmina N A et al (2013) lsquoThe Phylogeography andSpatiotemporal Spread of South-Central Skunk Rabies VirusrsquoPloS One 8 e82348
Lanfear R et al (2012) lsquoPartitionfinder Combined Selectionof Partitioning Schemes and Substitution Models forPhylogenetic Analysesrsquo Molecular Biology and Evolution 291695ndash701
Lembo T et al (2007) lsquoMolecular Epidemiology Identifies Only aSingle Rabies Virus Variant Circulating in Complex CarnivoreCommunities of the Serengetirsquo Proceedings Biological SciencesThe Royal Society 274 2123ndash30
mdashmdash et al (2008) lsquoExploring Reservoir Dynamics A Case Study ofRabies in the Serengeti Ecosystemrsquo Journal of Applied Ecology45 1246ndash57
Lemey P et al (2009) lsquoBayesian Phylogeography Finds Its RootsrsquoPLoS Computational Biology 5 e1000520
mdashmdash et al (2014) lsquoUnifying Viral Genetics and HumanTransportation Data to Predict the Global TransmissionDynamics of Human Influenza H3N2rsquo PLoS Pathogens 10e1003932
Lewin-koh A N J et al (2009) Maptools Tools for Reading andHandling Spatial Objects R package version 08-23 lthttpcranr-projectorgwebpackagesmaptoolsindexhtmlgt accessed 1Sept 2014
Li H and Durbin R (2009) lsquoFast and Accurate Short ReadAlignment with Burrows-Wheeler Transformrsquo Bioinformatics(Oxford England) 25 1754ndash60
mdashmdash et al (2009) lsquoThe Sequence AlignmentMap Format andSAMtoolsrsquo Bioinformatics (Oxford England) 25 2078ndash9
Lumlertdacha B et al (2006) lsquoComplex Genetic Structure ofthe Rabies Virus in Bangkok and Its Surrounding ProvincesThailand Implications for Canine Rabies ControlrsquoTransactions of the Royal Society of Tropical Medicine andHygiene 100 276ndash81
Magee D et al (2014) lsquoCombining Phylogeography and SpatialEpidemiology to Uncover Predictors of H5N1 Influenza A VirusDiffusionrsquo Archives of Virology 160 215ndash24
Magembe S (1985) lsquoEpidemiology of Rabies in United Republic ofTanzaniarsquo in Kuwer E et al (eds) Rabies in the Tropics pp 392ndash8 New York Springer Berlin Heidelberg
Marston D A et al (2007) lsquoComparative Analysis of the FullGenome Sequence of European Bat Lyssavirus Type 1 andType 2 with Other Lyssaviruses and Evidence for a ConservedTranscription Termination and Polyadenylation Motif in theG-L 3rsquo Non-Translated Regionrsquo The Journal of General Virology88 1302ndash14
mdashmdash et al (2013) lsquoNext Generation Sequencing of Viral RNAGenomesrsquo BMC Genomics 14 444
Matsumoto T et al (2013) lsquoMolecular Epidemiology of HumanRabies Viruses in Sri Lankarsquo Infection Genetics and Evolution 18160ndash7
Minin V N and Suchard M A (2008) lsquoCounting LabeledTransitions in Continuous-Time Markov Models of EvolutionrsquoJournal of Mathematical Biology 56 391ndash412
Mollentze N et al (2014) lsquoA Bayesian Approach for Inferring theDynamics of Partially Observed Endemic Infectious Diseasesfrom Space-Time-Genetic Datarsquo Proceedings Biological SciencesThe Royal Society 281 20133251
Nel L H (2013) lsquoDiscrepancies in Data Reporting for RabiesAfricarsquo Emerging Infectious Diseases 19 529ndash33
Nelson M I et al (2015) lsquoGlobal Migration of Influenza A Virusesin Swinersquo Nature Communications 6 6696
Nunes M R T et al (2014) lsquoAir Travel is Associated withIntracontinental Spread of Dengue Virus Serotypes 1-3 inBrazilrsquo PLoS Neglected Tropical Diseases 8 e2769
Paradis E Claude J and Strimmer K (2004) lsquoAPE Analyses ofPhylogenetics and Evolution in R Languagersquo Bioinformatics 20289ndash90
Parker J Rambaut A and Pybus O G (2008) lsquoCorrelatingViral Phenotypes with Phylogeny Accounting forPhylogenetic Uncertaintyrsquo Infection Genetics and Evolution 8239ndash46
Pebesma E J and Bivand R S (2005) lsquoClasses and Methods forSpatial Data in Rrsquo R News 5 9ndash13
PINGOrsquos Forum (2013) Pastoralists Indigenous Non GovernmentalOrganizationsrsquo Forum Annual Report 2012-2013 lthttpwwwpin-gosforumortzgt accessed 23 Nov 2014
Rambaut A et al (2014) lsquoTracer v16rsquo Computer program anddocumentation distributed by the author lthttpbeastbioedacukTracergt accessed 4 Feb 2015
10 | Virus Evolution 2015 Vol 1 No 1
Real L A et al (2005) lsquoUnifying the Spatial Population Dynamicsand Molecular Evolution of Epidemic Rabies Virusrsquo Proceedingsof the National Academy of Sciences of the United States of America102 12107ndash11
Rupprecht C E Hanlon C A and Hemachudha T (2002)lsquoRabies Re-examinedrsquo The Lancet Infectious Diseases 2 327ndash43
Shwiff S Hampson K and Anderson A (2013) lsquoPotentialEconomic Benefits of Eliminating Canine Rabiesrsquo AntiviralResearch 98 352ndash6
Siongok T and Karama M (1985) lsquoEpidemiology of HumanRabies in Kenyarsquo in E Kuwer et al (eds) Rabies in the Tropicspp 445ndash50 New York Springer Berlin Heidelberg
Stamatakis A et al (2012) lsquoRAxML-Light A Tool for ComputingTerabyte Phylogeniesrsquo Bioinformatics (Oxford England) 28 2064ndash6
Susilawathi N M et al (2012) lsquoEpidemiological and ClinicalFeatures of Human Rabies Cases in Bali 2008-2010rsquo BMCInfectious Diseases 12 81
Talbi C et al (2009) lsquoEvolutionary History and Dynamics of DogRabies Virus in Western and Central Africarsquo The Journal ofGeneral Virology 90 783ndash91
mdashmdash et al (2010) lsquoPhylodynamics and Human-Mediated Dispersalof a Zoonotic Virusrsquo PLoS Pathogens 6 e1001166
Tao X Y et al (2009) lsquoMolecular Epidemiology of Rabies inSouthern Peoplersquos Republic of Chinarsquo Emerging InfectiousDiseases 15 1192ndash8
Townsend S E et al (2013) lsquoDesigning Programs for EliminatingCanine Rabies from Islands Bali Indonesia as a Case StudyrsquoPLoS Neglected Tropical Diseases 7 e2372
Viana M et al (2015) lsquoDynamics of a Morbillivirus at theDomesticndashWildlife Interface Canine Distemper Virus inDomestic Dogs and Lionsrsquo Proceedings of the National Academy ofSciences of the United States of America 112 1464ndash9
Walsh M (2008) Pastoralism and Policy Processes in TanzaniaMbarali Case Study Report to the Tanzania Natural ResourceForum Arusha
Wang T H et al (2001) lsquoIdentification of Shared Populations ofHuman Immunodeficiency Virus Type 1 Infecting Microgliaand Tissue Macrophages Outside the Central Nervous SystemrsquoJournal of Virology 75 11686ndash99
WHO (2005) lsquoWHO Expert Consultation on Rabiesrsquo World HealthOrganization technical report series 931 1ndash121
Windiyaningsih C et al (2004) lsquoThe Rabies Epidemic on FloresIsland Indonesia (1998-2003)rsquo Journal of the Medical Associationof Thailandfrac14Chotmaihet thangphaet 87 1389ndash93
Woodroffe R and Donnelly C A (2011) lsquoRisk of Contact be-tween Endangered African Wild Dogs Lycaon pictus andDomestic Dogs Opportunities for Pathogen TransmissionrsquoJournal of Applied Ecology 48 1345ndash54
Woolhouse M E J (2002) lsquoPopulation Biology of Emerging and Re-emerging Pathogensrsquo Trends in Microbiology 10(10 Suppl) S3ndash7
K Brunker et al | 11
- vev011-TF1
- vev011-TF2
-
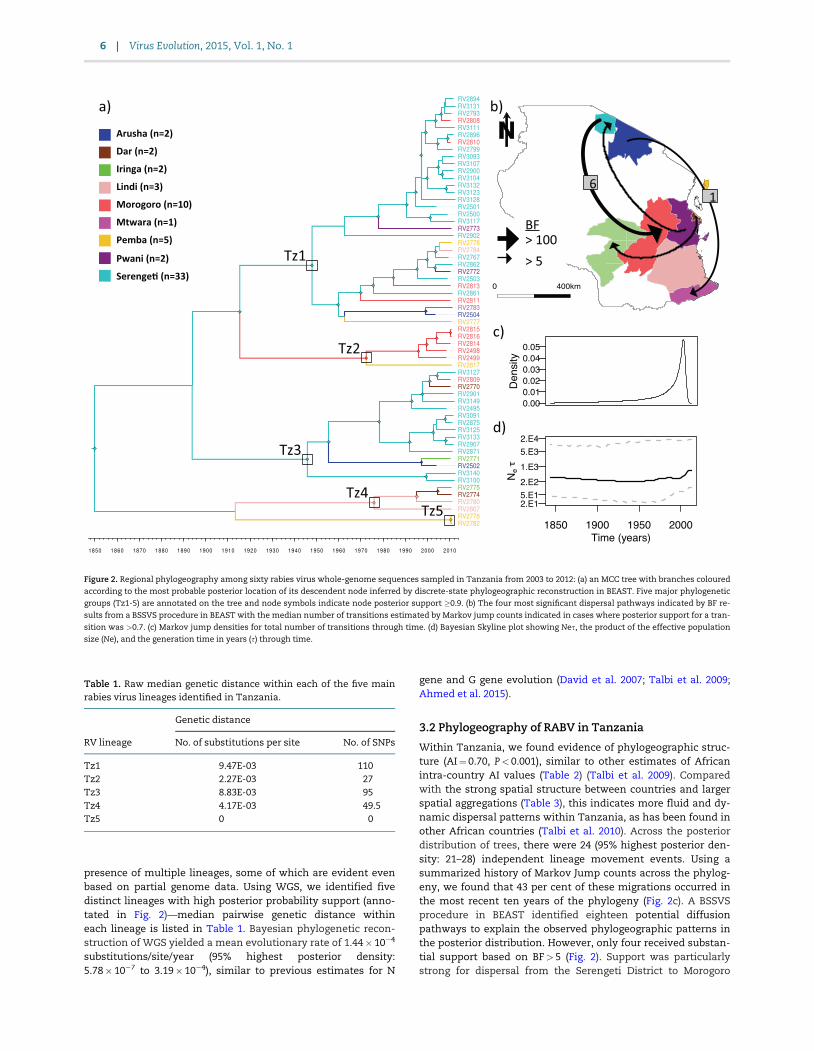
presence of multiple lineages some of which are evident evenbased on partial genome data Using WGS we identified fivedistinct lineages with high posterior probability support (anno-tated in Fig 2)mdashmedian pairwise genetic distance withineach lineage is listed in Table 1 Bayesian phylogenetic recon-struction of WGS yielded a mean evolutionary rate of 144 104
substitutionssiteyear (95 highest posterior density578 107 to 319 104) similar to previous estimates for N
gene and G gene evolution (David et al 2007 Talbi et al 2009Ahmed et al 2015)
32 Phylogeography of RABV in Tanzania
Within Tanzania we found evidence of phylogeographic struc-ture (AIfrac14 070 Plt 0001) similar to other estimates of Africanintra-country AI values (Table 2) (Talbi et al 2009) Comparedwith the strong spatial structure between countries and largerspatial aggregations (Table 3) this indicates more fluid and dy-namic dispersal patterns within Tanzania as has been found inother African countries (Talbi et al 2010) Across the posteriordistribution of trees there were 24 (95 highest posterior den-sity 21ndash28) independent lineage movement events Using asummarized history of Markov Jump counts across the phylog-eny we found that 43 per cent of these migrations occurred inthe most recent ten years of the phylogeny (Fig 2c) A BSSVSprocedure in BEAST identified eighteen potential diffusionpathways to explain the observed phylogeographic patterns inthe posterior distribution However only four received substan-tial support based on BFgt 5 (Fig 2) Support was particularlystrong for dispersal from the Serengeti District to Morogoro
Figure 2 Regional phylogeography among sixty rabies virus whole-genome sequences sampled in Tanzania from 2003 to 2012 (a) an MCC tree with branches coloured
according to the most probable posterior location of its descendent node inferred by discrete-state phylogeographic reconstruction in BEAST Five major phylogenetic
groups (Tz1-5) are annotated on the tree and node symbols indicate node posterior support 09 (b) The four most significant dispersal pathways indicated by BF re-
sults from a BSSVS procedure in BEAST with the median number of transitions estimated by Markov jump counts indicated in cases where posterior support for a tran-
sition was gt07 (c) Markov jump densities for total number of transitions through time (d) Bayesian Skyline plot showing Nes the product of the effective population
size (Ne) and the generation time in years (s) through time
Table 1 Raw median genetic distance within each of the five mainrabies virus lineages identified in Tanzania
Genetic distance
RV lineage No of substitutions per site No of SNPs
Tz1 947E-03 110Tz2 227E-03 27Tz3 883E-03 95Tz4 417E-03 495Tz5 0 0
6 | Virus Evolution 2015 Vol 1 No 1
(BFfrac14 13530) and Markov jump counts estimated a median ofsix (range 3ndash10) migrations along this dispersal route with atleast one (range 1ndash3) occurring on a branch representing a pe-riod of less than 2 years Most lineages were sampled in morethan one region in Tanzania with some distributed across alarger geographic area than others (Fig 3) The largest lineageTz1 contains not only a cluster of Serengeti samples but alsoencompassed samples from a larger geographic extent and wasfound in eight out of the nine districts sampled The Bayesian
skyline plot revealed that the effective population size has re-mained fairly constant over the past 150 years (Fig 2d) Becauseof the much higher availability of samples from the SerengetiDistrict we also conducted a coarser phylogeographic analysisgrouping sequences into lsquoNorthrsquo lsquoSouthrsquo or lsquoPemba Islandrsquo Thisidentified a predominance of north to south dispersal (esti-mated thirteen independent movements compared with onemovement south to north) and evidence of dispersal back andforth between the North of mainland Tanzania and PembaIsland (Supplementary Fig S4)
4 Discussion
Using viral genetic data from a hierarchy of spatial scales andvarying levels of genome coverage we were able to demonstratethe advantages of whole-genome resolution to describe thespatio-temporal dynamics of endemically circulating caninerabies viruses
We found a clear phylogeographic structure between coun-tries in Africa which could be identified with partial genomedata This suggests that the majority of dispersal occurs at awithin-country scale and control programs would be most ap-propriate at a national scale with strategies to deal with poten-tial incursions from other countries However at regional andlocal scales within Tanzania the discriminatory power of par-tial genome data became too limited to reveal fine-scale popula-tion structure that could aid the effectiveness of controlinterventions In contrast WGS provided the resolution to ge-netically distinguish between all samples and produced a well-supported phylogeny Although sub-genomic information hasutility at broad phylogeographic scales (eg Horton et al 2015)this finding supports the application of WGS for studies aimingto discern population structure at a scale most relevant tocontrol
Although large-scale population structure according tosub-continental areas or country-specific lineages was appar-ent from sub-genomic data we also found incidences of oc-casional large-scale admixture (Supplementary Figs S1 andS2) We continued to find evidence at increasingly fine scalesTanzania and other countries within the Africa 1b cladeshowed signs of admixture particularly when countriesshared a border and within country movements of RABV fa-cilitated by humans were a feature of Tanzanian RABV Evenat a very local scale within the Serengeti District we foundmultiple co-circulating lineages This recurrent theme may bea characteristic of endemic RABV in Africa reflecting decadesof endemic circulation and human-mediated introductionsSimilar patterns have been observed in canine rabies-en-demic countries in South and Southeast Asia (Lumlertdachaet al 2006 Matsumoto et al 2013 Ahmed et al 2015) wherethe presence of co-circulating lineages was attributed to a
Figure 3 Spatial distribution of rabies virus lineages sampled from regions in
Tanzania between 2003 and 2012 with a colour gradient (yellow to red) indicat-
ing the total number of lineages (low to high) sampled in each region
Table 2 Degree of within-country rabies virus spatial admixture in three African countries measured using an Association Index (AI) whichranges from 0 indicating strong population subdivision to 1 indicating complete panmixis Results are shown for Tanzania (this study) andAlgeria and Morocco (Talbi et al 2010)
Country AI [95 BCI] P No of sequences No of locations Median distance (km) Min Max
Algeria 067 [062ndash073] lt0001 117 20 23308 2327 67462Morocco 055 [051ndash063] lt0001 133 28 32645 2862 92670Tanzania 070 [060-079] lt0001 60 9 43976 3905 108826
BCI Bayesian confidence interval
Table 3 Degree of spatial admixture between rabies virus samplesfrom Africa according to an Association Index (AI) AI values canrange from 0 indicating complete population subdivision to 1 indi-cating complete panmixis
Data Spatial clustering level No ofgroups
AI [95 BCI] P
N405 Sub-continent 3 004 [002005] lt001Country 14 006 [005008] lt001
N1350 Sub-continent 3 006 [004008] lt001Country 10 013 [009013] lt001
BCI Bayesian confidence interval Datasets of partial (N405) and full (N1350) nu-
cleoprotein sequences were tested at two levels of spatial aggregation (1) sub-
continent geographical partitions relative to Tanzania (three states Tanzania
neighbouring country and other African country) and (2) country of origin
K Brunker et al | 7
combination of historical introductions from neighbouringcountries and human-mediated movements
Much of the viral population structure we found withinTanzania is consistent with initial invasive waves that havepersisted endemically with structure eroding over time aidedby human-mediated movement Historical accounts describe arabies outbreak in southern regions of Tanzania in the mid-1950s which spread throughout the country and was recordedin the Serengeti District in the late 1970s (Magembe 1985Siongok and Karama 1985) This invasion perhaps shaped theinitial RABV population structure in Tanzania The timeline ofregional scale migrations (Fig 2c) indicates a strong rise in viraldispersal around this period possibly reflecting increasing hu-man connectivity Samples in Tz1 (Fig 2) share partial N geneidentity with samples from Kenya (Fig 1) indicating a sharedevolutionary history that may relate to an initial outbreakacross both countries Lineages Tz1 and Tz3 appear to have awide geographic distribution (Fig 3) and we found evidence of ageneral north to south dispersal pattern in Tanzania(Supplementary Fig S4) This highlights the potential for wide-spread dispersal should an incursion occur from Kenya and sug-gests a need for co-operative cross-border rabies managementonce a national control program is established Variation in thespread of different lineages across Tanzania may reflect the in-fluence of heterogeneous landscape features or dog populationstructure in impeding or facilitating RABV disseminationIdentifying and quantifying such features is the logical nextstep to provide additional information that can inform controlprogrammes such as identifying and strengthening pre-existing barriers Recent extensions of phylogeographic tech-niques have highlighted how this might be achieved using anintegrated approach combining evolutionary and ecologicalanalyses to quantify drivers of viral transmission (Lemey et al2014 Magee et al 2014)
The ancestry of lineages Tz4 and 5 (denoting lineages foundin the southern mainland and on the Island of Pemba respec-tively) points toward an introduction from the north ofTanzania (posterior probabilityfrac14 062) but the uncertainty ofthis ancestral location and the ancestral node itself (poste-riorfrac14 045) suggest these may be distinct historical lineageswith low-level persistence or undetected circulation elsewherein Tanzania Alternatively these clusters may represent in-stances where lineages from external sources have more re-cently invaded and resulted in sustained transmission inTanzaniamdashpartial genome phylogenies indicate Tz4 is relatedto clusters containing samples from countries south ofTanzania South Africa and Mozambique Furthermore Tz5shares a common ancestry with a Nigerian and several CentralAfrican Republic samples This again underlines the threat ofre-invasion or introduction from external sources and the po-tential value of whole-genome resolution to robustly and moreaccurately identify sources of new introductions To date themajority of RABV genetic data from Africa comes from partialgenome analyses however our data suggest that whole-genome characterization would be valuable and should be anaim for the future
Islands represent isolated landscapes with natural barriersto dispersal but incidents of RABV outbreaks instigated by hu-man-mediated introductions (eg to islands in Indonesia[Windiyaningsih et al 2004 Susilawathi et al 2012]) have oftenbeen recorded Sequences from the island of Pemba were sug-gestive of multiple introductions from various sources with evi-dence of dispersal to and from the mainland (SupplementaryFig S4) Samples from Pemba were scattered throughout the
phylogeny and the most divergent lineage Tz5 consisted of twosamples from Pemba The distribution of these lineages may re-flect earlier invasions of RABV into Pemba from elsewhere inTanzania (Tz1 and 2) and the African continent (Tz5) resultingfrom Pembarsquos location on an important trading route Howeversince these lineages were not resampled and no cases havebeen detected from Pemba in over a year (Lushasi pers comm)RABV may have been only transiently circulating Neverthelesslessons from Indonesia where lack of a swift and coordinatedresponse to a rabies incursion led to an epidemic (Townsendet al 2013) highlight the importance of active surveillance andrapid response to incursions
Although we found evidence of RABV spatial structurewithin Tanzania it was evident that dispersal was also fre-quent with at least one long distance migration occurringwithin a small temporal window (lt2 years) Lineage move-ments occurring on branches representing short evolutionarytimes such as those identified between Serengeti and Morogoro(gt750 km) indicate rates of dispersal much higher than thoserecorded for endemic wildlife rabies (Biek 2007) and imply hu-man mediated movements as seen in parts of North Africa(Talbi et al 2010) Movement of pastoralist and agro-pastoralistcommunities from the Lake zone in Northern Tanzania tosouthern regions for example Morogoro have been ongoingsince the 1950s (Walsh 2008) with a major influx reported from2003 to 2006 (PINGOrsquos Forum 2013) attributed to climate changeinduced drought and forced evictions from newly protectedareas (Kideghesho et al 2013) During these migrations pasto-ralists are accompanied by their dogs which may facilitate thelong distance movement of animals incubating the virus priorto transmission On further inspection we found that Morogorosamples indicated as instances of long-distance translocationcame from rural areas where pastoralists are likely to migrateto whereas other samples from urban Morogoro formed a dis-tinct cluster (Fig 2) We found three RABV lineages in Morogorofrom only ten samples (Fig 3) Quantifying networks of human-mediated movements (including livestock trade) would providea valuable proxy for connectivity for many zoonotic diseases af-fecting domesticated animals
Further to our findings of regional admixture in Tanzaniawe also observed considerable diversity at a very local scalethat is within the Serengeti District with several lineages co-circulating These lineages appear to have persisted for at least20 years (older Serengeti samples obtained from GenBank alsocluster within these lineages indicated in Fig 1) despite dogvaccination campaigns having been conducted in the districtsince 1996 While vaccination coverage has varied substantiallyacross years and between villages (Viana et al 2015) rabies inci-dence has at times significantly declined for example falling by97 per cent in the late 1990s (Cleaveland et al 2003) Yet our ge-netic data show that despite substantial declines in incidencethese lineages must have persisted at very low levels within thedistrict or subsequently reinvaded from neighbouring districtsWithout sampling the surrounding districts it is not possible todistinguish between these possibilities The skyline plot indi-cated a stable effective RABV population size (Ne) through time(Fig 3d) While this could be expected for an endemic pathogenit is worth noting that there was no evidence of viral populationsize reductions in response to vaccination campaigns Rabiescontrol efforts across much of Tanzania have been very limiteduntil recently and high turnover in the dog population(Hampson et al 2009) likely contributes to the stable persistenceof rabies in Tanzania It has also been noted that geographicstructure can obscure local fluctuations in subpopulations while
8 | Virus Evolution 2015 Vol 1 No 1
maintaining the appearance of a constant Ne in skyline plots(Carrington et al 2005)
This study represents a snapshot of RABV dynamics inTanzania indicating that human movements have dissemi-nated RABV out of locally endemic areas at scales relevant tocontrol that is administrative units such as regions or districtsThese frequent translocations have probably lead to the exis-tence of multiple co-circulating lineages (Fig 3) but relativelyfew introductions lead to sustained chains of transmission thatare detectable as lineage movement events However in diseasesystems closely associated with human activities the probabil-ity of successful translocation and establishment is likely to bemuch greater This suggests that without some level of regionalco-operation Tanzania will be unable to eliminate rabies andmaintain freedom from disease Human movements are oftenin response to social drivers which could be used as signals forincreased vigilance and surveillance in at risk areas
Our findings highlight the use of WGS to uncover fine scaletransmission patterns that can directly inform control effortsHowever sub-genomic approaches can still have utility at acoarser scale and are more easily obtained particularly whensample quality is an issue In particular they can be used to ini-tially identify admixture between countries which may indicatethe necessity of coordinated regional control programs and sur-veillance Co-circulation of multiple lineages and introductionsfacilitated by humans appear to be a feature of endemic rabies vi-rus and complicate the design of a sustainable control strategyHowever using whole-genome data we were able to identifysources of dispersal within Tanzania that could direct efforts to-ward surveillance and control The finding that humans play animportant role in the dynamics of RABV in Tanzania suggeststhat increasing awareness and dog vaccination in lsquohigh-riskrsquo com-munities such as pastoralists could help to reduce long-range dis-persal Moreover the design of enhanced surveillance andcontainment strategies to mitigate human-mediated incursionsand maintain disease freedom should be a priority once controlprograms are established and elimination is being targeted
Supplementary data
Supplementary data is available at VEVOLU Journal online
Acknowledgements
We acknowledge the Ministries of Livestock and FisheriesDevelopment and of Health and Social Welfare TanzaniaNational Parks Tanzania Wildlife Research InstituteNgorongoro Conservation Area Authority TanzaniaCommission for Science and Technology and NationalInstitute for Medical Research for permission and collabora-tion and the Arusha Mtwara and Mwanza VeterinaryInvestigation Centres Pemba Veterinary Office and theFrankfurt Zoological Society for logistical and technical sup-port We are grateful to the veterinary and livestock field of-ficers healthworkers and paravets throughout Tanzaniaand in particular to the following research assistants JoelChangalucha Zilpah Kaare Gurdeep Kour Ahmed LugeloKennedy Lushasi Mathias Magoto Khadija Said andRenatus Herman We thank Pelagia Muchuruza MatthewMaziku Geoffrey Mchau Eberhard Mbunda Francois-XavierMeslin and Bernadette Abela-Ridder for their collaborationsthrough the Bill amp Melinda GatesWHO rabies eliminationdemonstration project in Tanzania This work was
supported by the Wellcome Trust the Medical ResearchCouncil the UBS Optimus Foundation Lincoln Park Zooand the Research and Policy for Infectious DiseaseDynamics Program of the Science and TechnologyDirectorate Department of Homeland Security FogartyInternational Centre National Institute of Health
Conflict of interest None declared
ReferencesAhmed K et al (2015) lsquoMolecular Epidemiology of Rabies
Viruses Circulating in Two Rabies Endemic Provinces of Laos2011ndash2012 Regional Diversity in Southeast Asiarsquo PLoS NeglectedTropical Diseases 9 e0003645
Andrews S (2010) FastQC a quality control tool for highthroughput sequence datalthttpwwwbioinformaticsbbsrcacukprojectsfastqcgt accessed 4 Jun 2014
Ayres D L et al (2012) lsquoBEAGLE An Application ProgrammingInterface and High-Performance Computing Library forStatistical Phylogeneticsrsquo Systematic Biology 61 170ndash3
Bedford T et al (2014) lsquoIntegrating Influenza AntigenicDynamics with Molecular Evolutionrsquo eLife 2014 1ndash26
Biek R and Real L A (2010) lsquoThe Landscape Genetics of InfectiousDisease Emergence and Spreadrsquo Molecular Ecology 19 3515ndash31
mdashmdash et al (2007) lsquoA High-Resolution Genetic Signature ofDemographic and Spatial Expansion in Epizootic Rabies VirusrsquoProceedings of the National Academy of Sciences of the United Statesof America 104 7993ndash8
mdashmdash et al (2015) lsquoMeasurably Evolving Pathogens in the GenomicErarsquo Trends in Ecology amp Evolution 30 306ndash13
Bielejec F et al (2011) lsquoSPREAD Spatial PhylogeneticReconstruction of Evolutionary Dynamicsrsquo Bioinformatics(Oxford England) 27 2910ndash2
mdashmdash et al (2014) lsquoInferring Heterogeneous Evolutionary Processesthrough Time From Sequence Substitution toPhylogeographyrsquo Systematic Biology 63 493ndash504
Bivand R S Pebesma E J and Gomez-Rubio V (2008) AppliedSpatial Data Analysis with R New York Springer
Bolger A M Lohse M and Usadel B (2014) lsquoTrimmomatic AFlexible Trimmer for Illumina Sequence Datarsquo Bioinformatics30 2114ndash20
Bourhy H et al (2008) lsquoThe Origin and Phylogeography of DogRabies Virusrsquo The Journal of General Virology 89 2673ndash81
Brunker K et al (2012) lsquoIntegrating the Landscape Epidemiologyand Genetics of RNA Viruses Rabies in Domestic Dogs as aModelrsquo Parasitology 139 1899ndash913
Carrington C et al (2005) lsquoInvasion and Maintenance of DengueVirus Type 2 and Type 4 in the Americasrsquo Journal of Virology 7914680ndash7
Cleaveland S et al (2003) lsquoA Dog Rabies Vaccination Campaignin Rural Africa Impact on the Incidence of Dog Rabies andHuman Dog-Bite Injuriesrsquo Vaccine 21 1965ndash73
David D et al (2007) lsquoIdentification of Novel Canine Rabies VirusClades in the Middle East and North Africarsquo The Journal ofGeneral Virology 88 967ndash80
Denduangboripant J et al (2005) lsquoTransmission Dynamics ofRabies Virus in Thailand Implications for Disease ControlrsquoBMC Infectious Diseases 5 52
Drummond A J et al (2003) lsquoMeasurably Evolving PopulationsrsquoTrends in Ecology amp Evolution 18 481ndash8
mdashmdash et al (2005) lsquoBayesian Coalescent Inference of PastPopulation Dynamics from Molecular Sequencesrsquo MolecularBiology and Evolution 22 1185ndash92
K Brunker et al | 9
mdashmdash et al (2012) lsquoBayesian Phylogenetics with BEAUti and theBEAST 17rsquo Molecular Biology and Evolution 29 1969ndash73
Ferreira M A R and Suchard M A (2008) lsquoBayesian Analysis ofElapsed Times in Continuous-Time Markov Chainsrsquo CanadianJournal of Statistics 36 355ndash68
Greger M (2007) lsquoThe HumanAnimal Interface Emergence andResurgence of Zoonotic Infectious Diseasesrsquo Critical Reviews inMicrobiology 33 243ndash99
Hampson K et al (2007) lsquoSynchronous Cycles of Domestic DogRabies in Sub-Saharan Africa and the Impact of ControlEffortsrsquo Proceedings of the National Academy of Sciences of theUnited States of America 104 7717ndash22
mdashmdash et al (2009) lsquoTransmission Dynamics and Prospects for theElimination of Canine Rabiesrsquo PLoS Biology 7 e53
Hayman D T S et al (2011) lsquoEvolutionary History of Rabies inGhanarsquo PLoS Neglected Tropical Diseases 5 e1001
Holmes E C and Grenfell B T (2009) lsquoDiscovering the Phylodynamicsof RNA Virusesrsquo PLoS Computational Biology 5 e1000505
Horton D L et al (2015) lsquoComplex Epidemiology of a ZoonoticDisease in a Culturally Diverse Region Phylogeography ofRabies Virus in the Middle Eastrsquo PLOS Neglected TropicalDiseases 9 e0003569
Jombart T (2008) lsquoadegenet A R Package for the MultivariateAnalysis of Genetic Markersrsquo Bioinformatics (Oxford England)24 1403ndash5
Jones K E et al (2008) lsquoGlobal Trends in Emerging InfectiousDiseasesrsquo Nature 451 990ndash3
Katoh K and Standley D M (2013) lsquoMAFFT MultipleSequence Alignment Software Version 7 Improvements inPerformance and Usabilityrsquo Molecular Biology and Evolution30 772ndash80
Kideghesho J et al (2013) lsquoEmerging Issues and Challenges inConservation of Biodiversity in the Rangelands of TanzaniarsquoNature Conservation 6 1ndash29
Kissi B Tordo N and Bourhy H (1995) lsquoGenetic Polymorphismin the Rabies Virus Nucleoprotein Genersquo Virology 209 526ndash37
Knobel D L et al (2005) lsquoRe-Evaluating the Burden of Rabies inAfrica and Asiarsquo Bulletin of the World Health Organization 83360ndash8
Kuhnert D Wu C-H and Drummond A J (2011) lsquoPhylogeneticand Epidemic Modeling of Rapidly Evolving InfectiousDiseasesrsquo Infection Genetics and Evolution Journal of MolecularEpidemiology and Evolutionary Genetics in Infectious Diseases 111825ndash41
Kuzmin I V et al (2008) lsquoComplete Genomes of AravanKhujand Irkut and West Caucasian Bat Viruses with SpecialAttention to the Polymerase Gene and Non-Coding RegionsrsquoVirus Research 136 81ndash90
Kuzmina N A et al (2013) lsquoThe Phylogeography andSpatiotemporal Spread of South-Central Skunk Rabies VirusrsquoPloS One 8 e82348
Lanfear R et al (2012) lsquoPartitionfinder Combined Selectionof Partitioning Schemes and Substitution Models forPhylogenetic Analysesrsquo Molecular Biology and Evolution 291695ndash701
Lembo T et al (2007) lsquoMolecular Epidemiology Identifies Only aSingle Rabies Virus Variant Circulating in Complex CarnivoreCommunities of the Serengetirsquo Proceedings Biological SciencesThe Royal Society 274 2123ndash30
mdashmdash et al (2008) lsquoExploring Reservoir Dynamics A Case Study ofRabies in the Serengeti Ecosystemrsquo Journal of Applied Ecology45 1246ndash57
Lemey P et al (2009) lsquoBayesian Phylogeography Finds Its RootsrsquoPLoS Computational Biology 5 e1000520
mdashmdash et al (2014) lsquoUnifying Viral Genetics and HumanTransportation Data to Predict the Global TransmissionDynamics of Human Influenza H3N2rsquo PLoS Pathogens 10e1003932
Lewin-koh A N J et al (2009) Maptools Tools for Reading andHandling Spatial Objects R package version 08-23 lthttpcranr-projectorgwebpackagesmaptoolsindexhtmlgt accessed 1Sept 2014
Li H and Durbin R (2009) lsquoFast and Accurate Short ReadAlignment with Burrows-Wheeler Transformrsquo Bioinformatics(Oxford England) 25 1754ndash60
mdashmdash et al (2009) lsquoThe Sequence AlignmentMap Format andSAMtoolsrsquo Bioinformatics (Oxford England) 25 2078ndash9
Lumlertdacha B et al (2006) lsquoComplex Genetic Structure ofthe Rabies Virus in Bangkok and Its Surrounding ProvincesThailand Implications for Canine Rabies ControlrsquoTransactions of the Royal Society of Tropical Medicine andHygiene 100 276ndash81
Magee D et al (2014) lsquoCombining Phylogeography and SpatialEpidemiology to Uncover Predictors of H5N1 Influenza A VirusDiffusionrsquo Archives of Virology 160 215ndash24
Magembe S (1985) lsquoEpidemiology of Rabies in United Republic ofTanzaniarsquo in Kuwer E et al (eds) Rabies in the Tropics pp 392ndash8 New York Springer Berlin Heidelberg
Marston D A et al (2007) lsquoComparative Analysis of the FullGenome Sequence of European Bat Lyssavirus Type 1 andType 2 with Other Lyssaviruses and Evidence for a ConservedTranscription Termination and Polyadenylation Motif in theG-L 3rsquo Non-Translated Regionrsquo The Journal of General Virology88 1302ndash14
mdashmdash et al (2013) lsquoNext Generation Sequencing of Viral RNAGenomesrsquo BMC Genomics 14 444
Matsumoto T et al (2013) lsquoMolecular Epidemiology of HumanRabies Viruses in Sri Lankarsquo Infection Genetics and Evolution 18160ndash7
Minin V N and Suchard M A (2008) lsquoCounting LabeledTransitions in Continuous-Time Markov Models of EvolutionrsquoJournal of Mathematical Biology 56 391ndash412
Mollentze N et al (2014) lsquoA Bayesian Approach for Inferring theDynamics of Partially Observed Endemic Infectious Diseasesfrom Space-Time-Genetic Datarsquo Proceedings Biological SciencesThe Royal Society 281 20133251
Nel L H (2013) lsquoDiscrepancies in Data Reporting for RabiesAfricarsquo Emerging Infectious Diseases 19 529ndash33
Nelson M I et al (2015) lsquoGlobal Migration of Influenza A Virusesin Swinersquo Nature Communications 6 6696
Nunes M R T et al (2014) lsquoAir Travel is Associated withIntracontinental Spread of Dengue Virus Serotypes 1-3 inBrazilrsquo PLoS Neglected Tropical Diseases 8 e2769
Paradis E Claude J and Strimmer K (2004) lsquoAPE Analyses ofPhylogenetics and Evolution in R Languagersquo Bioinformatics 20289ndash90
Parker J Rambaut A and Pybus O G (2008) lsquoCorrelatingViral Phenotypes with Phylogeny Accounting forPhylogenetic Uncertaintyrsquo Infection Genetics and Evolution 8239ndash46
Pebesma E J and Bivand R S (2005) lsquoClasses and Methods forSpatial Data in Rrsquo R News 5 9ndash13
PINGOrsquos Forum (2013) Pastoralists Indigenous Non GovernmentalOrganizationsrsquo Forum Annual Report 2012-2013 lthttpwwwpin-gosforumortzgt accessed 23 Nov 2014
Rambaut A et al (2014) lsquoTracer v16rsquo Computer program anddocumentation distributed by the author lthttpbeastbioedacukTracergt accessed 4 Feb 2015
10 | Virus Evolution 2015 Vol 1 No 1
Real L A et al (2005) lsquoUnifying the Spatial Population Dynamicsand Molecular Evolution of Epidemic Rabies Virusrsquo Proceedingsof the National Academy of Sciences of the United States of America102 12107ndash11
Rupprecht C E Hanlon C A and Hemachudha T (2002)lsquoRabies Re-examinedrsquo The Lancet Infectious Diseases 2 327ndash43
Shwiff S Hampson K and Anderson A (2013) lsquoPotentialEconomic Benefits of Eliminating Canine Rabiesrsquo AntiviralResearch 98 352ndash6
Siongok T and Karama M (1985) lsquoEpidemiology of HumanRabies in Kenyarsquo in E Kuwer et al (eds) Rabies in the Tropicspp 445ndash50 New York Springer Berlin Heidelberg
Stamatakis A et al (2012) lsquoRAxML-Light A Tool for ComputingTerabyte Phylogeniesrsquo Bioinformatics (Oxford England) 28 2064ndash6
Susilawathi N M et al (2012) lsquoEpidemiological and ClinicalFeatures of Human Rabies Cases in Bali 2008-2010rsquo BMCInfectious Diseases 12 81
Talbi C et al (2009) lsquoEvolutionary History and Dynamics of DogRabies Virus in Western and Central Africarsquo The Journal ofGeneral Virology 90 783ndash91
mdashmdash et al (2010) lsquoPhylodynamics and Human-Mediated Dispersalof a Zoonotic Virusrsquo PLoS Pathogens 6 e1001166
Tao X Y et al (2009) lsquoMolecular Epidemiology of Rabies inSouthern Peoplersquos Republic of Chinarsquo Emerging InfectiousDiseases 15 1192ndash8
Townsend S E et al (2013) lsquoDesigning Programs for EliminatingCanine Rabies from Islands Bali Indonesia as a Case StudyrsquoPLoS Neglected Tropical Diseases 7 e2372
Viana M et al (2015) lsquoDynamics of a Morbillivirus at theDomesticndashWildlife Interface Canine Distemper Virus inDomestic Dogs and Lionsrsquo Proceedings of the National Academy ofSciences of the United States of America 112 1464ndash9
Walsh M (2008) Pastoralism and Policy Processes in TanzaniaMbarali Case Study Report to the Tanzania Natural ResourceForum Arusha
Wang T H et al (2001) lsquoIdentification of Shared Populations ofHuman Immunodeficiency Virus Type 1 Infecting Microgliaand Tissue Macrophages Outside the Central Nervous SystemrsquoJournal of Virology 75 11686ndash99
WHO (2005) lsquoWHO Expert Consultation on Rabiesrsquo World HealthOrganization technical report series 931 1ndash121
Windiyaningsih C et al (2004) lsquoThe Rabies Epidemic on FloresIsland Indonesia (1998-2003)rsquo Journal of the Medical Associationof Thailandfrac14Chotmaihet thangphaet 87 1389ndash93
Woodroffe R and Donnelly C A (2011) lsquoRisk of Contact be-tween Endangered African Wild Dogs Lycaon pictus andDomestic Dogs Opportunities for Pathogen TransmissionrsquoJournal of Applied Ecology 48 1345ndash54
Woolhouse M E J (2002) lsquoPopulation Biology of Emerging and Re-emerging Pathogensrsquo Trends in Microbiology 10(10 Suppl) S3ndash7
K Brunker et al | 11
- vev011-TF1
- vev011-TF2
-

(BFfrac14 13530) and Markov jump counts estimated a median ofsix (range 3ndash10) migrations along this dispersal route with atleast one (range 1ndash3) occurring on a branch representing a pe-riod of less than 2 years Most lineages were sampled in morethan one region in Tanzania with some distributed across alarger geographic area than others (Fig 3) The largest lineageTz1 contains not only a cluster of Serengeti samples but alsoencompassed samples from a larger geographic extent and wasfound in eight out of the nine districts sampled The Bayesian
skyline plot revealed that the effective population size has re-mained fairly constant over the past 150 years (Fig 2d) Becauseof the much higher availability of samples from the SerengetiDistrict we also conducted a coarser phylogeographic analysisgrouping sequences into lsquoNorthrsquo lsquoSouthrsquo or lsquoPemba Islandrsquo Thisidentified a predominance of north to south dispersal (esti-mated thirteen independent movements compared with onemovement south to north) and evidence of dispersal back andforth between the North of mainland Tanzania and PembaIsland (Supplementary Fig S4)
4 Discussion
Using viral genetic data from a hierarchy of spatial scales andvarying levels of genome coverage we were able to demonstratethe advantages of whole-genome resolution to describe thespatio-temporal dynamics of endemically circulating caninerabies viruses
We found a clear phylogeographic structure between coun-tries in Africa which could be identified with partial genomedata This suggests that the majority of dispersal occurs at awithin-country scale and control programs would be most ap-propriate at a national scale with strategies to deal with poten-tial incursions from other countries However at regional andlocal scales within Tanzania the discriminatory power of par-tial genome data became too limited to reveal fine-scale popula-tion structure that could aid the effectiveness of controlinterventions In contrast WGS provided the resolution to ge-netically distinguish between all samples and produced a well-supported phylogeny Although sub-genomic information hasutility at broad phylogeographic scales (eg Horton et al 2015)this finding supports the application of WGS for studies aimingto discern population structure at a scale most relevant tocontrol
Although large-scale population structure according tosub-continental areas or country-specific lineages was appar-ent from sub-genomic data we also found incidences of oc-casional large-scale admixture (Supplementary Figs S1 andS2) We continued to find evidence at increasingly fine scalesTanzania and other countries within the Africa 1b cladeshowed signs of admixture particularly when countriesshared a border and within country movements of RABV fa-cilitated by humans were a feature of Tanzanian RABV Evenat a very local scale within the Serengeti District we foundmultiple co-circulating lineages This recurrent theme may bea characteristic of endemic RABV in Africa reflecting decadesof endemic circulation and human-mediated introductionsSimilar patterns have been observed in canine rabies-en-demic countries in South and Southeast Asia (Lumlertdachaet al 2006 Matsumoto et al 2013 Ahmed et al 2015) wherethe presence of co-circulating lineages was attributed to a
Figure 3 Spatial distribution of rabies virus lineages sampled from regions in
Tanzania between 2003 and 2012 with a colour gradient (yellow to red) indicat-
ing the total number of lineages (low to high) sampled in each region
Table 2 Degree of within-country rabies virus spatial admixture in three African countries measured using an Association Index (AI) whichranges from 0 indicating strong population subdivision to 1 indicating complete panmixis Results are shown for Tanzania (this study) andAlgeria and Morocco (Talbi et al 2010)
Country AI [95 BCI] P No of sequences No of locations Median distance (km) Min Max
Algeria 067 [062ndash073] lt0001 117 20 23308 2327 67462Morocco 055 [051ndash063] lt0001 133 28 32645 2862 92670Tanzania 070 [060-079] lt0001 60 9 43976 3905 108826
BCI Bayesian confidence interval
Table 3 Degree of spatial admixture between rabies virus samplesfrom Africa according to an Association Index (AI) AI values canrange from 0 indicating complete population subdivision to 1 indi-cating complete panmixis
Data Spatial clustering level No ofgroups
AI [95 BCI] P
N405 Sub-continent 3 004 [002005] lt001Country 14 006 [005008] lt001
N1350 Sub-continent 3 006 [004008] lt001Country 10 013 [009013] lt001
BCI Bayesian confidence interval Datasets of partial (N405) and full (N1350) nu-
cleoprotein sequences were tested at two levels of spatial aggregation (1) sub-
continent geographical partitions relative to Tanzania (three states Tanzania
neighbouring country and other African country) and (2) country of origin
K Brunker et al | 7
combination of historical introductions from neighbouringcountries and human-mediated movements
Much of the viral population structure we found withinTanzania is consistent with initial invasive waves that havepersisted endemically with structure eroding over time aidedby human-mediated movement Historical accounts describe arabies outbreak in southern regions of Tanzania in the mid-1950s which spread throughout the country and was recordedin the Serengeti District in the late 1970s (Magembe 1985Siongok and Karama 1985) This invasion perhaps shaped theinitial RABV population structure in Tanzania The timeline ofregional scale migrations (Fig 2c) indicates a strong rise in viraldispersal around this period possibly reflecting increasing hu-man connectivity Samples in Tz1 (Fig 2) share partial N geneidentity with samples from Kenya (Fig 1) indicating a sharedevolutionary history that may relate to an initial outbreakacross both countries Lineages Tz1 and Tz3 appear to have awide geographic distribution (Fig 3) and we found evidence of ageneral north to south dispersal pattern in Tanzania(Supplementary Fig S4) This highlights the potential for wide-spread dispersal should an incursion occur from Kenya and sug-gests a need for co-operative cross-border rabies managementonce a national control program is established Variation in thespread of different lineages across Tanzania may reflect the in-fluence of heterogeneous landscape features or dog populationstructure in impeding or facilitating RABV disseminationIdentifying and quantifying such features is the logical nextstep to provide additional information that can inform controlprogrammes such as identifying and strengthening pre-existing barriers Recent extensions of phylogeographic tech-niques have highlighted how this might be achieved using anintegrated approach combining evolutionary and ecologicalanalyses to quantify drivers of viral transmission (Lemey et al2014 Magee et al 2014)
The ancestry of lineages Tz4 and 5 (denoting lineages foundin the southern mainland and on the Island of Pemba respec-tively) points toward an introduction from the north ofTanzania (posterior probabilityfrac14 062) but the uncertainty ofthis ancestral location and the ancestral node itself (poste-riorfrac14 045) suggest these may be distinct historical lineageswith low-level persistence or undetected circulation elsewherein Tanzania Alternatively these clusters may represent in-stances where lineages from external sources have more re-cently invaded and resulted in sustained transmission inTanzaniamdashpartial genome phylogenies indicate Tz4 is relatedto clusters containing samples from countries south ofTanzania South Africa and Mozambique Furthermore Tz5shares a common ancestry with a Nigerian and several CentralAfrican Republic samples This again underlines the threat ofre-invasion or introduction from external sources and the po-tential value of whole-genome resolution to robustly and moreaccurately identify sources of new introductions To date themajority of RABV genetic data from Africa comes from partialgenome analyses however our data suggest that whole-genome characterization would be valuable and should be anaim for the future
Islands represent isolated landscapes with natural barriersto dispersal but incidents of RABV outbreaks instigated by hu-man-mediated introductions (eg to islands in Indonesia[Windiyaningsih et al 2004 Susilawathi et al 2012]) have oftenbeen recorded Sequences from the island of Pemba were sug-gestive of multiple introductions from various sources with evi-dence of dispersal to and from the mainland (SupplementaryFig S4) Samples from Pemba were scattered throughout the
phylogeny and the most divergent lineage Tz5 consisted of twosamples from Pemba The distribution of these lineages may re-flect earlier invasions of RABV into Pemba from elsewhere inTanzania (Tz1 and 2) and the African continent (Tz5) resultingfrom Pembarsquos location on an important trading route Howeversince these lineages were not resampled and no cases havebeen detected from Pemba in over a year (Lushasi pers comm)RABV may have been only transiently circulating Neverthelesslessons from Indonesia where lack of a swift and coordinatedresponse to a rabies incursion led to an epidemic (Townsendet al 2013) highlight the importance of active surveillance andrapid response to incursions
Although we found evidence of RABV spatial structurewithin Tanzania it was evident that dispersal was also fre-quent with at least one long distance migration occurringwithin a small temporal window (lt2 years) Lineage move-ments occurring on branches representing short evolutionarytimes such as those identified between Serengeti and Morogoro(gt750 km) indicate rates of dispersal much higher than thoserecorded for endemic wildlife rabies (Biek 2007) and imply hu-man mediated movements as seen in parts of North Africa(Talbi et al 2010) Movement of pastoralist and agro-pastoralistcommunities from the Lake zone in Northern Tanzania tosouthern regions for example Morogoro have been ongoingsince the 1950s (Walsh 2008) with a major influx reported from2003 to 2006 (PINGOrsquos Forum 2013) attributed to climate changeinduced drought and forced evictions from newly protectedareas (Kideghesho et al 2013) During these migrations pasto-ralists are accompanied by their dogs which may facilitate thelong distance movement of animals incubating the virus priorto transmission On further inspection we found that Morogorosamples indicated as instances of long-distance translocationcame from rural areas where pastoralists are likely to migrateto whereas other samples from urban Morogoro formed a dis-tinct cluster (Fig 2) We found three RABV lineages in Morogorofrom only ten samples (Fig 3) Quantifying networks of human-mediated movements (including livestock trade) would providea valuable proxy for connectivity for many zoonotic diseases af-fecting domesticated animals
Further to our findings of regional admixture in Tanzaniawe also observed considerable diversity at a very local scalethat is within the Serengeti District with several lineages co-circulating These lineages appear to have persisted for at least20 years (older Serengeti samples obtained from GenBank alsocluster within these lineages indicated in Fig 1) despite dogvaccination campaigns having been conducted in the districtsince 1996 While vaccination coverage has varied substantiallyacross years and between villages (Viana et al 2015) rabies inci-dence has at times significantly declined for example falling by97 per cent in the late 1990s (Cleaveland et al 2003) Yet our ge-netic data show that despite substantial declines in incidencethese lineages must have persisted at very low levels within thedistrict or subsequently reinvaded from neighbouring districtsWithout sampling the surrounding districts it is not possible todistinguish between these possibilities The skyline plot indi-cated a stable effective RABV population size (Ne) through time(Fig 3d) While this could be expected for an endemic pathogenit is worth noting that there was no evidence of viral populationsize reductions in response to vaccination campaigns Rabiescontrol efforts across much of Tanzania have been very limiteduntil recently and high turnover in the dog population(Hampson et al 2009) likely contributes to the stable persistenceof rabies in Tanzania It has also been noted that geographicstructure can obscure local fluctuations in subpopulations while
8 | Virus Evolution 2015 Vol 1 No 1
maintaining the appearance of a constant Ne in skyline plots(Carrington et al 2005)
This study represents a snapshot of RABV dynamics inTanzania indicating that human movements have dissemi-nated RABV out of locally endemic areas at scales relevant tocontrol that is administrative units such as regions or districtsThese frequent translocations have probably lead to the exis-tence of multiple co-circulating lineages (Fig 3) but relativelyfew introductions lead to sustained chains of transmission thatare detectable as lineage movement events However in diseasesystems closely associated with human activities the probabil-ity of successful translocation and establishment is likely to bemuch greater This suggests that without some level of regionalco-operation Tanzania will be unable to eliminate rabies andmaintain freedom from disease Human movements are oftenin response to social drivers which could be used as signals forincreased vigilance and surveillance in at risk areas
Our findings highlight the use of WGS to uncover fine scaletransmission patterns that can directly inform control effortsHowever sub-genomic approaches can still have utility at acoarser scale and are more easily obtained particularly whensample quality is an issue In particular they can be used to ini-tially identify admixture between countries which may indicatethe necessity of coordinated regional control programs and sur-veillance Co-circulation of multiple lineages and introductionsfacilitated by humans appear to be a feature of endemic rabies vi-rus and complicate the design of a sustainable control strategyHowever using whole-genome data we were able to identifysources of dispersal within Tanzania that could direct efforts to-ward surveillance and control The finding that humans play animportant role in the dynamics of RABV in Tanzania suggeststhat increasing awareness and dog vaccination in lsquohigh-riskrsquo com-munities such as pastoralists could help to reduce long-range dis-persal Moreover the design of enhanced surveillance andcontainment strategies to mitigate human-mediated incursionsand maintain disease freedom should be a priority once controlprograms are established and elimination is being targeted
Supplementary data
Supplementary data is available at VEVOLU Journal online
Acknowledgements
We acknowledge the Ministries of Livestock and FisheriesDevelopment and of Health and Social Welfare TanzaniaNational Parks Tanzania Wildlife Research InstituteNgorongoro Conservation Area Authority TanzaniaCommission for Science and Technology and NationalInstitute for Medical Research for permission and collabora-tion and the Arusha Mtwara and Mwanza VeterinaryInvestigation Centres Pemba Veterinary Office and theFrankfurt Zoological Society for logistical and technical sup-port We are grateful to the veterinary and livestock field of-ficers healthworkers and paravets throughout Tanzaniaand in particular to the following research assistants JoelChangalucha Zilpah Kaare Gurdeep Kour Ahmed LugeloKennedy Lushasi Mathias Magoto Khadija Said andRenatus Herman We thank Pelagia Muchuruza MatthewMaziku Geoffrey Mchau Eberhard Mbunda Francois-XavierMeslin and Bernadette Abela-Ridder for their collaborationsthrough the Bill amp Melinda GatesWHO rabies eliminationdemonstration project in Tanzania This work was
supported by the Wellcome Trust the Medical ResearchCouncil the UBS Optimus Foundation Lincoln Park Zooand the Research and Policy for Infectious DiseaseDynamics Program of the Science and TechnologyDirectorate Department of Homeland Security FogartyInternational Centre National Institute of Health
Conflict of interest None declared
ReferencesAhmed K et al (2015) lsquoMolecular Epidemiology of Rabies
Viruses Circulating in Two Rabies Endemic Provinces of Laos2011ndash2012 Regional Diversity in Southeast Asiarsquo PLoS NeglectedTropical Diseases 9 e0003645
Andrews S (2010) FastQC a quality control tool for highthroughput sequence datalthttpwwwbioinformaticsbbsrcacukprojectsfastqcgt accessed 4 Jun 2014
Ayres D L et al (2012) lsquoBEAGLE An Application ProgrammingInterface and High-Performance Computing Library forStatistical Phylogeneticsrsquo Systematic Biology 61 170ndash3
Bedford T et al (2014) lsquoIntegrating Influenza AntigenicDynamics with Molecular Evolutionrsquo eLife 2014 1ndash26
Biek R and Real L A (2010) lsquoThe Landscape Genetics of InfectiousDisease Emergence and Spreadrsquo Molecular Ecology 19 3515ndash31
mdashmdash et al (2007) lsquoA High-Resolution Genetic Signature ofDemographic and Spatial Expansion in Epizootic Rabies VirusrsquoProceedings of the National Academy of Sciences of the United Statesof America 104 7993ndash8
mdashmdash et al (2015) lsquoMeasurably Evolving Pathogens in the GenomicErarsquo Trends in Ecology amp Evolution 30 306ndash13
Bielejec F et al (2011) lsquoSPREAD Spatial PhylogeneticReconstruction of Evolutionary Dynamicsrsquo Bioinformatics(Oxford England) 27 2910ndash2
mdashmdash et al (2014) lsquoInferring Heterogeneous Evolutionary Processesthrough Time From Sequence Substitution toPhylogeographyrsquo Systematic Biology 63 493ndash504
Bivand R S Pebesma E J and Gomez-Rubio V (2008) AppliedSpatial Data Analysis with R New York Springer
Bolger A M Lohse M and Usadel B (2014) lsquoTrimmomatic AFlexible Trimmer for Illumina Sequence Datarsquo Bioinformatics30 2114ndash20
Bourhy H et al (2008) lsquoThe Origin and Phylogeography of DogRabies Virusrsquo The Journal of General Virology 89 2673ndash81
Brunker K et al (2012) lsquoIntegrating the Landscape Epidemiologyand Genetics of RNA Viruses Rabies in Domestic Dogs as aModelrsquo Parasitology 139 1899ndash913
Carrington C et al (2005) lsquoInvasion and Maintenance of DengueVirus Type 2 and Type 4 in the Americasrsquo Journal of Virology 7914680ndash7
Cleaveland S et al (2003) lsquoA Dog Rabies Vaccination Campaignin Rural Africa Impact on the Incidence of Dog Rabies andHuman Dog-Bite Injuriesrsquo Vaccine 21 1965ndash73
David D et al (2007) lsquoIdentification of Novel Canine Rabies VirusClades in the Middle East and North Africarsquo The Journal ofGeneral Virology 88 967ndash80
Denduangboripant J et al (2005) lsquoTransmission Dynamics ofRabies Virus in Thailand Implications for Disease ControlrsquoBMC Infectious Diseases 5 52
Drummond A J et al (2003) lsquoMeasurably Evolving PopulationsrsquoTrends in Ecology amp Evolution 18 481ndash8
mdashmdash et al (2005) lsquoBayesian Coalescent Inference of PastPopulation Dynamics from Molecular Sequencesrsquo MolecularBiology and Evolution 22 1185ndash92
K Brunker et al | 9
mdashmdash et al (2012) lsquoBayesian Phylogenetics with BEAUti and theBEAST 17rsquo Molecular Biology and Evolution 29 1969ndash73
Ferreira M A R and Suchard M A (2008) lsquoBayesian Analysis ofElapsed Times in Continuous-Time Markov Chainsrsquo CanadianJournal of Statistics 36 355ndash68
Greger M (2007) lsquoThe HumanAnimal Interface Emergence andResurgence of Zoonotic Infectious Diseasesrsquo Critical Reviews inMicrobiology 33 243ndash99
Hampson K et al (2007) lsquoSynchronous Cycles of Domestic DogRabies in Sub-Saharan Africa and the Impact of ControlEffortsrsquo Proceedings of the National Academy of Sciences of theUnited States of America 104 7717ndash22
mdashmdash et al (2009) lsquoTransmission Dynamics and Prospects for theElimination of Canine Rabiesrsquo PLoS Biology 7 e53
Hayman D T S et al (2011) lsquoEvolutionary History of Rabies inGhanarsquo PLoS Neglected Tropical Diseases 5 e1001
Holmes E C and Grenfell B T (2009) lsquoDiscovering the Phylodynamicsof RNA Virusesrsquo PLoS Computational Biology 5 e1000505
Horton D L et al (2015) lsquoComplex Epidemiology of a ZoonoticDisease in a Culturally Diverse Region Phylogeography ofRabies Virus in the Middle Eastrsquo PLOS Neglected TropicalDiseases 9 e0003569
Jombart T (2008) lsquoadegenet A R Package for the MultivariateAnalysis of Genetic Markersrsquo Bioinformatics (Oxford England)24 1403ndash5
Jones K E et al (2008) lsquoGlobal Trends in Emerging InfectiousDiseasesrsquo Nature 451 990ndash3
Katoh K and Standley D M (2013) lsquoMAFFT MultipleSequence Alignment Software Version 7 Improvements inPerformance and Usabilityrsquo Molecular Biology and Evolution30 772ndash80
Kideghesho J et al (2013) lsquoEmerging Issues and Challenges inConservation of Biodiversity in the Rangelands of TanzaniarsquoNature Conservation 6 1ndash29
Kissi B Tordo N and Bourhy H (1995) lsquoGenetic Polymorphismin the Rabies Virus Nucleoprotein Genersquo Virology 209 526ndash37
Knobel D L et al (2005) lsquoRe-Evaluating the Burden of Rabies inAfrica and Asiarsquo Bulletin of the World Health Organization 83360ndash8
Kuhnert D Wu C-H and Drummond A J (2011) lsquoPhylogeneticand Epidemic Modeling of Rapidly Evolving InfectiousDiseasesrsquo Infection Genetics and Evolution Journal of MolecularEpidemiology and Evolutionary Genetics in Infectious Diseases 111825ndash41
Kuzmin I V et al (2008) lsquoComplete Genomes of AravanKhujand Irkut and West Caucasian Bat Viruses with SpecialAttention to the Polymerase Gene and Non-Coding RegionsrsquoVirus Research 136 81ndash90
Kuzmina N A et al (2013) lsquoThe Phylogeography andSpatiotemporal Spread of South-Central Skunk Rabies VirusrsquoPloS One 8 e82348
Lanfear R et al (2012) lsquoPartitionfinder Combined Selectionof Partitioning Schemes and Substitution Models forPhylogenetic Analysesrsquo Molecular Biology and Evolution 291695ndash701
Lembo T et al (2007) lsquoMolecular Epidemiology Identifies Only aSingle Rabies Virus Variant Circulating in Complex CarnivoreCommunities of the Serengetirsquo Proceedings Biological SciencesThe Royal Society 274 2123ndash30
mdashmdash et al (2008) lsquoExploring Reservoir Dynamics A Case Study ofRabies in the Serengeti Ecosystemrsquo Journal of Applied Ecology45 1246ndash57
Lemey P et al (2009) lsquoBayesian Phylogeography Finds Its RootsrsquoPLoS Computational Biology 5 e1000520
mdashmdash et al (2014) lsquoUnifying Viral Genetics and HumanTransportation Data to Predict the Global TransmissionDynamics of Human Influenza H3N2rsquo PLoS Pathogens 10e1003932
Lewin-koh A N J et al (2009) Maptools Tools for Reading andHandling Spatial Objects R package version 08-23 lthttpcranr-projectorgwebpackagesmaptoolsindexhtmlgt accessed 1Sept 2014
Li H and Durbin R (2009) lsquoFast and Accurate Short ReadAlignment with Burrows-Wheeler Transformrsquo Bioinformatics(Oxford England) 25 1754ndash60
mdashmdash et al (2009) lsquoThe Sequence AlignmentMap Format andSAMtoolsrsquo Bioinformatics (Oxford England) 25 2078ndash9
Lumlertdacha B et al (2006) lsquoComplex Genetic Structure ofthe Rabies Virus in Bangkok and Its Surrounding ProvincesThailand Implications for Canine Rabies ControlrsquoTransactions of the Royal Society of Tropical Medicine andHygiene 100 276ndash81
Magee D et al (2014) lsquoCombining Phylogeography and SpatialEpidemiology to Uncover Predictors of H5N1 Influenza A VirusDiffusionrsquo Archives of Virology 160 215ndash24
Magembe S (1985) lsquoEpidemiology of Rabies in United Republic ofTanzaniarsquo in Kuwer E et al (eds) Rabies in the Tropics pp 392ndash8 New York Springer Berlin Heidelberg
Marston D A et al (2007) lsquoComparative Analysis of the FullGenome Sequence of European Bat Lyssavirus Type 1 andType 2 with Other Lyssaviruses and Evidence for a ConservedTranscription Termination and Polyadenylation Motif in theG-L 3rsquo Non-Translated Regionrsquo The Journal of General Virology88 1302ndash14
mdashmdash et al (2013) lsquoNext Generation Sequencing of Viral RNAGenomesrsquo BMC Genomics 14 444
Matsumoto T et al (2013) lsquoMolecular Epidemiology of HumanRabies Viruses in Sri Lankarsquo Infection Genetics and Evolution 18160ndash7
Minin V N and Suchard M A (2008) lsquoCounting LabeledTransitions in Continuous-Time Markov Models of EvolutionrsquoJournal of Mathematical Biology 56 391ndash412
Mollentze N et al (2014) lsquoA Bayesian Approach for Inferring theDynamics of Partially Observed Endemic Infectious Diseasesfrom Space-Time-Genetic Datarsquo Proceedings Biological SciencesThe Royal Society 281 20133251
Nel L H (2013) lsquoDiscrepancies in Data Reporting for RabiesAfricarsquo Emerging Infectious Diseases 19 529ndash33
Nelson M I et al (2015) lsquoGlobal Migration of Influenza A Virusesin Swinersquo Nature Communications 6 6696
Nunes M R T et al (2014) lsquoAir Travel is Associated withIntracontinental Spread of Dengue Virus Serotypes 1-3 inBrazilrsquo PLoS Neglected Tropical Diseases 8 e2769
Paradis E Claude J and Strimmer K (2004) lsquoAPE Analyses ofPhylogenetics and Evolution in R Languagersquo Bioinformatics 20289ndash90
Parker J Rambaut A and Pybus O G (2008) lsquoCorrelatingViral Phenotypes with Phylogeny Accounting forPhylogenetic Uncertaintyrsquo Infection Genetics and Evolution 8239ndash46
Pebesma E J and Bivand R S (2005) lsquoClasses and Methods forSpatial Data in Rrsquo R News 5 9ndash13
PINGOrsquos Forum (2013) Pastoralists Indigenous Non GovernmentalOrganizationsrsquo Forum Annual Report 2012-2013 lthttpwwwpin-gosforumortzgt accessed 23 Nov 2014
Rambaut A et al (2014) lsquoTracer v16rsquo Computer program anddocumentation distributed by the author lthttpbeastbioedacukTracergt accessed 4 Feb 2015
10 | Virus Evolution 2015 Vol 1 No 1
Real L A et al (2005) lsquoUnifying the Spatial Population Dynamicsand Molecular Evolution of Epidemic Rabies Virusrsquo Proceedingsof the National Academy of Sciences of the United States of America102 12107ndash11
Rupprecht C E Hanlon C A and Hemachudha T (2002)lsquoRabies Re-examinedrsquo The Lancet Infectious Diseases 2 327ndash43
Shwiff S Hampson K and Anderson A (2013) lsquoPotentialEconomic Benefits of Eliminating Canine Rabiesrsquo AntiviralResearch 98 352ndash6
Siongok T and Karama M (1985) lsquoEpidemiology of HumanRabies in Kenyarsquo in E Kuwer et al (eds) Rabies in the Tropicspp 445ndash50 New York Springer Berlin Heidelberg
Stamatakis A et al (2012) lsquoRAxML-Light A Tool for ComputingTerabyte Phylogeniesrsquo Bioinformatics (Oxford England) 28 2064ndash6
Susilawathi N M et al (2012) lsquoEpidemiological and ClinicalFeatures of Human Rabies Cases in Bali 2008-2010rsquo BMCInfectious Diseases 12 81
Talbi C et al (2009) lsquoEvolutionary History and Dynamics of DogRabies Virus in Western and Central Africarsquo The Journal ofGeneral Virology 90 783ndash91
mdashmdash et al (2010) lsquoPhylodynamics and Human-Mediated Dispersalof a Zoonotic Virusrsquo PLoS Pathogens 6 e1001166
Tao X Y et al (2009) lsquoMolecular Epidemiology of Rabies inSouthern Peoplersquos Republic of Chinarsquo Emerging InfectiousDiseases 15 1192ndash8
Townsend S E et al (2013) lsquoDesigning Programs for EliminatingCanine Rabies from Islands Bali Indonesia as a Case StudyrsquoPLoS Neglected Tropical Diseases 7 e2372
Viana M et al (2015) lsquoDynamics of a Morbillivirus at theDomesticndashWildlife Interface Canine Distemper Virus inDomestic Dogs and Lionsrsquo Proceedings of the National Academy ofSciences of the United States of America 112 1464ndash9
Walsh M (2008) Pastoralism and Policy Processes in TanzaniaMbarali Case Study Report to the Tanzania Natural ResourceForum Arusha
Wang T H et al (2001) lsquoIdentification of Shared Populations ofHuman Immunodeficiency Virus Type 1 Infecting Microgliaand Tissue Macrophages Outside the Central Nervous SystemrsquoJournal of Virology 75 11686ndash99
WHO (2005) lsquoWHO Expert Consultation on Rabiesrsquo World HealthOrganization technical report series 931 1ndash121
Windiyaningsih C et al (2004) lsquoThe Rabies Epidemic on FloresIsland Indonesia (1998-2003)rsquo Journal of the Medical Associationof Thailandfrac14Chotmaihet thangphaet 87 1389ndash93
Woodroffe R and Donnelly C A (2011) lsquoRisk of Contact be-tween Endangered African Wild Dogs Lycaon pictus andDomestic Dogs Opportunities for Pathogen TransmissionrsquoJournal of Applied Ecology 48 1345ndash54
Woolhouse M E J (2002) lsquoPopulation Biology of Emerging and Re-emerging Pathogensrsquo Trends in Microbiology 10(10 Suppl) S3ndash7
K Brunker et al | 11
- vev011-TF1
- vev011-TF2
-

combination of historical introductions from neighbouringcountries and human-mediated movements
Much of the viral population structure we found withinTanzania is consistent with initial invasive waves that havepersisted endemically with structure eroding over time aidedby human-mediated movement Historical accounts describe arabies outbreak in southern regions of Tanzania in the mid-1950s which spread throughout the country and was recordedin the Serengeti District in the late 1970s (Magembe 1985Siongok and Karama 1985) This invasion perhaps shaped theinitial RABV population structure in Tanzania The timeline ofregional scale migrations (Fig 2c) indicates a strong rise in viraldispersal around this period possibly reflecting increasing hu-man connectivity Samples in Tz1 (Fig 2) share partial N geneidentity with samples from Kenya (Fig 1) indicating a sharedevolutionary history that may relate to an initial outbreakacross both countries Lineages Tz1 and Tz3 appear to have awide geographic distribution (Fig 3) and we found evidence of ageneral north to south dispersal pattern in Tanzania(Supplementary Fig S4) This highlights the potential for wide-spread dispersal should an incursion occur from Kenya and sug-gests a need for co-operative cross-border rabies managementonce a national control program is established Variation in thespread of different lineages across Tanzania may reflect the in-fluence of heterogeneous landscape features or dog populationstructure in impeding or facilitating RABV disseminationIdentifying and quantifying such features is the logical nextstep to provide additional information that can inform controlprogrammes such as identifying and strengthening pre-existing barriers Recent extensions of phylogeographic tech-niques have highlighted how this might be achieved using anintegrated approach combining evolutionary and ecologicalanalyses to quantify drivers of viral transmission (Lemey et al2014 Magee et al 2014)
The ancestry of lineages Tz4 and 5 (denoting lineages foundin the southern mainland and on the Island of Pemba respec-tively) points toward an introduction from the north ofTanzania (posterior probabilityfrac14 062) but the uncertainty ofthis ancestral location and the ancestral node itself (poste-riorfrac14 045) suggest these may be distinct historical lineageswith low-level persistence or undetected circulation elsewherein Tanzania Alternatively these clusters may represent in-stances where lineages from external sources have more re-cently invaded and resulted in sustained transmission inTanzaniamdashpartial genome phylogenies indicate Tz4 is relatedto clusters containing samples from countries south ofTanzania South Africa and Mozambique Furthermore Tz5shares a common ancestry with a Nigerian and several CentralAfrican Republic samples This again underlines the threat ofre-invasion or introduction from external sources and the po-tential value of whole-genome resolution to robustly and moreaccurately identify sources of new introductions To date themajority of RABV genetic data from Africa comes from partialgenome analyses however our data suggest that whole-genome characterization would be valuable and should be anaim for the future
Islands represent isolated landscapes with natural barriersto dispersal but incidents of RABV outbreaks instigated by hu-man-mediated introductions (eg to islands in Indonesia[Windiyaningsih et al 2004 Susilawathi et al 2012]) have oftenbeen recorded Sequences from the island of Pemba were sug-gestive of multiple introductions from various sources with evi-dence of dispersal to and from the mainland (SupplementaryFig S4) Samples from Pemba were scattered throughout the
phylogeny and the most divergent lineage Tz5 consisted of twosamples from Pemba The distribution of these lineages may re-flect earlier invasions of RABV into Pemba from elsewhere inTanzania (Tz1 and 2) and the African continent (Tz5) resultingfrom Pembarsquos location on an important trading route Howeversince these lineages were not resampled and no cases havebeen detected from Pemba in over a year (Lushasi pers comm)RABV may have been only transiently circulating Neverthelesslessons from Indonesia where lack of a swift and coordinatedresponse to a rabies incursion led to an epidemic (Townsendet al 2013) highlight the importance of active surveillance andrapid response to incursions
Although we found evidence of RABV spatial structurewithin Tanzania it was evident that dispersal was also fre-quent with at least one long distance migration occurringwithin a small temporal window (lt2 years) Lineage move-ments occurring on branches representing short evolutionarytimes such as those identified between Serengeti and Morogoro(gt750 km) indicate rates of dispersal much higher than thoserecorded for endemic wildlife rabies (Biek 2007) and imply hu-man mediated movements as seen in parts of North Africa(Talbi et al 2010) Movement of pastoralist and agro-pastoralistcommunities from the Lake zone in Northern Tanzania tosouthern regions for example Morogoro have been ongoingsince the 1950s (Walsh 2008) with a major influx reported from2003 to 2006 (PINGOrsquos Forum 2013) attributed to climate changeinduced drought and forced evictions from newly protectedareas (Kideghesho et al 2013) During these migrations pasto-ralists are accompanied by their dogs which may facilitate thelong distance movement of animals incubating the virus priorto transmission On further inspection we found that Morogorosamples indicated as instances of long-distance translocationcame from rural areas where pastoralists are likely to migrateto whereas other samples from urban Morogoro formed a dis-tinct cluster (Fig 2) We found three RABV lineages in Morogorofrom only ten samples (Fig 3) Quantifying networks of human-mediated movements (including livestock trade) would providea valuable proxy for connectivity for many zoonotic diseases af-fecting domesticated animals
Further to our findings of regional admixture in Tanzaniawe also observed considerable diversity at a very local scalethat is within the Serengeti District with several lineages co-circulating These lineages appear to have persisted for at least20 years (older Serengeti samples obtained from GenBank alsocluster within these lineages indicated in Fig 1) despite dogvaccination campaigns having been conducted in the districtsince 1996 While vaccination coverage has varied substantiallyacross years and between villages (Viana et al 2015) rabies inci-dence has at times significantly declined for example falling by97 per cent in the late 1990s (Cleaveland et al 2003) Yet our ge-netic data show that despite substantial declines in incidencethese lineages must have persisted at very low levels within thedistrict or subsequently reinvaded from neighbouring districtsWithout sampling the surrounding districts it is not possible todistinguish between these possibilities The skyline plot indi-cated a stable effective RABV population size (Ne) through time(Fig 3d) While this could be expected for an endemic pathogenit is worth noting that there was no evidence of viral populationsize reductions in response to vaccination campaigns Rabiescontrol efforts across much of Tanzania have been very limiteduntil recently and high turnover in the dog population(Hampson et al 2009) likely contributes to the stable persistenceof rabies in Tanzania It has also been noted that geographicstructure can obscure local fluctuations in subpopulations while
8 | Virus Evolution 2015 Vol 1 No 1
maintaining the appearance of a constant Ne in skyline plots(Carrington et al 2005)
This study represents a snapshot of RABV dynamics inTanzania indicating that human movements have dissemi-nated RABV out of locally endemic areas at scales relevant tocontrol that is administrative units such as regions or districtsThese frequent translocations have probably lead to the exis-tence of multiple co-circulating lineages (Fig 3) but relativelyfew introductions lead to sustained chains of transmission thatare detectable as lineage movement events However in diseasesystems closely associated with human activities the probabil-ity of successful translocation and establishment is likely to bemuch greater This suggests that without some level of regionalco-operation Tanzania will be unable to eliminate rabies andmaintain freedom from disease Human movements are oftenin response to social drivers which could be used as signals forincreased vigilance and surveillance in at risk areas
Our findings highlight the use of WGS to uncover fine scaletransmission patterns that can directly inform control effortsHowever sub-genomic approaches can still have utility at acoarser scale and are more easily obtained particularly whensample quality is an issue In particular they can be used to ini-tially identify admixture between countries which may indicatethe necessity of coordinated regional control programs and sur-veillance Co-circulation of multiple lineages and introductionsfacilitated by humans appear to be a feature of endemic rabies vi-rus and complicate the design of a sustainable control strategyHowever using whole-genome data we were able to identifysources of dispersal within Tanzania that could direct efforts to-ward surveillance and control The finding that humans play animportant role in the dynamics of RABV in Tanzania suggeststhat increasing awareness and dog vaccination in lsquohigh-riskrsquo com-munities such as pastoralists could help to reduce long-range dis-persal Moreover the design of enhanced surveillance andcontainment strategies to mitigate human-mediated incursionsand maintain disease freedom should be a priority once controlprograms are established and elimination is being targeted
Supplementary data
Supplementary data is available at VEVOLU Journal online
Acknowledgements
We acknowledge the Ministries of Livestock and FisheriesDevelopment and of Health and Social Welfare TanzaniaNational Parks Tanzania Wildlife Research InstituteNgorongoro Conservation Area Authority TanzaniaCommission for Science and Technology and NationalInstitute for Medical Research for permission and collabora-tion and the Arusha Mtwara and Mwanza VeterinaryInvestigation Centres Pemba Veterinary Office and theFrankfurt Zoological Society for logistical and technical sup-port We are grateful to the veterinary and livestock field of-ficers healthworkers and paravets throughout Tanzaniaand in particular to the following research assistants JoelChangalucha Zilpah Kaare Gurdeep Kour Ahmed LugeloKennedy Lushasi Mathias Magoto Khadija Said andRenatus Herman We thank Pelagia Muchuruza MatthewMaziku Geoffrey Mchau Eberhard Mbunda Francois-XavierMeslin and Bernadette Abela-Ridder for their collaborationsthrough the Bill amp Melinda GatesWHO rabies eliminationdemonstration project in Tanzania This work was
supported by the Wellcome Trust the Medical ResearchCouncil the UBS Optimus Foundation Lincoln Park Zooand the Research and Policy for Infectious DiseaseDynamics Program of the Science and TechnologyDirectorate Department of Homeland Security FogartyInternational Centre National Institute of Health
Conflict of interest None declared
ReferencesAhmed K et al (2015) lsquoMolecular Epidemiology of Rabies
Viruses Circulating in Two Rabies Endemic Provinces of Laos2011ndash2012 Regional Diversity in Southeast Asiarsquo PLoS NeglectedTropical Diseases 9 e0003645
Andrews S (2010) FastQC a quality control tool for highthroughput sequence datalthttpwwwbioinformaticsbbsrcacukprojectsfastqcgt accessed 4 Jun 2014
Ayres D L et al (2012) lsquoBEAGLE An Application ProgrammingInterface and High-Performance Computing Library forStatistical Phylogeneticsrsquo Systematic Biology 61 170ndash3
Bedford T et al (2014) lsquoIntegrating Influenza AntigenicDynamics with Molecular Evolutionrsquo eLife 2014 1ndash26
Biek R and Real L A (2010) lsquoThe Landscape Genetics of InfectiousDisease Emergence and Spreadrsquo Molecular Ecology 19 3515ndash31
mdashmdash et al (2007) lsquoA High-Resolution Genetic Signature ofDemographic and Spatial Expansion in Epizootic Rabies VirusrsquoProceedings of the National Academy of Sciences of the United Statesof America 104 7993ndash8
mdashmdash et al (2015) lsquoMeasurably Evolving Pathogens in the GenomicErarsquo Trends in Ecology amp Evolution 30 306ndash13
Bielejec F et al (2011) lsquoSPREAD Spatial PhylogeneticReconstruction of Evolutionary Dynamicsrsquo Bioinformatics(Oxford England) 27 2910ndash2
mdashmdash et al (2014) lsquoInferring Heterogeneous Evolutionary Processesthrough Time From Sequence Substitution toPhylogeographyrsquo Systematic Biology 63 493ndash504
Bivand R S Pebesma E J and Gomez-Rubio V (2008) AppliedSpatial Data Analysis with R New York Springer
Bolger A M Lohse M and Usadel B (2014) lsquoTrimmomatic AFlexible Trimmer for Illumina Sequence Datarsquo Bioinformatics30 2114ndash20
Bourhy H et al (2008) lsquoThe Origin and Phylogeography of DogRabies Virusrsquo The Journal of General Virology 89 2673ndash81
Brunker K et al (2012) lsquoIntegrating the Landscape Epidemiologyand Genetics of RNA Viruses Rabies in Domestic Dogs as aModelrsquo Parasitology 139 1899ndash913
Carrington C et al (2005) lsquoInvasion and Maintenance of DengueVirus Type 2 and Type 4 in the Americasrsquo Journal of Virology 7914680ndash7
Cleaveland S et al (2003) lsquoA Dog Rabies Vaccination Campaignin Rural Africa Impact on the Incidence of Dog Rabies andHuman Dog-Bite Injuriesrsquo Vaccine 21 1965ndash73
David D et al (2007) lsquoIdentification of Novel Canine Rabies VirusClades in the Middle East and North Africarsquo The Journal ofGeneral Virology 88 967ndash80
Denduangboripant J et al (2005) lsquoTransmission Dynamics ofRabies Virus in Thailand Implications for Disease ControlrsquoBMC Infectious Diseases 5 52
Drummond A J et al (2003) lsquoMeasurably Evolving PopulationsrsquoTrends in Ecology amp Evolution 18 481ndash8
mdashmdash et al (2005) lsquoBayesian Coalescent Inference of PastPopulation Dynamics from Molecular Sequencesrsquo MolecularBiology and Evolution 22 1185ndash92
K Brunker et al | 9
mdashmdash et al (2012) lsquoBayesian Phylogenetics with BEAUti and theBEAST 17rsquo Molecular Biology and Evolution 29 1969ndash73
Ferreira M A R and Suchard M A (2008) lsquoBayesian Analysis ofElapsed Times in Continuous-Time Markov Chainsrsquo CanadianJournal of Statistics 36 355ndash68
Greger M (2007) lsquoThe HumanAnimal Interface Emergence andResurgence of Zoonotic Infectious Diseasesrsquo Critical Reviews inMicrobiology 33 243ndash99
Hampson K et al (2007) lsquoSynchronous Cycles of Domestic DogRabies in Sub-Saharan Africa and the Impact of ControlEffortsrsquo Proceedings of the National Academy of Sciences of theUnited States of America 104 7717ndash22
mdashmdash et al (2009) lsquoTransmission Dynamics and Prospects for theElimination of Canine Rabiesrsquo PLoS Biology 7 e53
Hayman D T S et al (2011) lsquoEvolutionary History of Rabies inGhanarsquo PLoS Neglected Tropical Diseases 5 e1001
Holmes E C and Grenfell B T (2009) lsquoDiscovering the Phylodynamicsof RNA Virusesrsquo PLoS Computational Biology 5 e1000505
Horton D L et al (2015) lsquoComplex Epidemiology of a ZoonoticDisease in a Culturally Diverse Region Phylogeography ofRabies Virus in the Middle Eastrsquo PLOS Neglected TropicalDiseases 9 e0003569
Jombart T (2008) lsquoadegenet A R Package for the MultivariateAnalysis of Genetic Markersrsquo Bioinformatics (Oxford England)24 1403ndash5
Jones K E et al (2008) lsquoGlobal Trends in Emerging InfectiousDiseasesrsquo Nature 451 990ndash3
Katoh K and Standley D M (2013) lsquoMAFFT MultipleSequence Alignment Software Version 7 Improvements inPerformance and Usabilityrsquo Molecular Biology and Evolution30 772ndash80
Kideghesho J et al (2013) lsquoEmerging Issues and Challenges inConservation of Biodiversity in the Rangelands of TanzaniarsquoNature Conservation 6 1ndash29
Kissi B Tordo N and Bourhy H (1995) lsquoGenetic Polymorphismin the Rabies Virus Nucleoprotein Genersquo Virology 209 526ndash37
Knobel D L et al (2005) lsquoRe-Evaluating the Burden of Rabies inAfrica and Asiarsquo Bulletin of the World Health Organization 83360ndash8
Kuhnert D Wu C-H and Drummond A J (2011) lsquoPhylogeneticand Epidemic Modeling of Rapidly Evolving InfectiousDiseasesrsquo Infection Genetics and Evolution Journal of MolecularEpidemiology and Evolutionary Genetics in Infectious Diseases 111825ndash41
Kuzmin I V et al (2008) lsquoComplete Genomes of AravanKhujand Irkut and West Caucasian Bat Viruses with SpecialAttention to the Polymerase Gene and Non-Coding RegionsrsquoVirus Research 136 81ndash90
Kuzmina N A et al (2013) lsquoThe Phylogeography andSpatiotemporal Spread of South-Central Skunk Rabies VirusrsquoPloS One 8 e82348
Lanfear R et al (2012) lsquoPartitionfinder Combined Selectionof Partitioning Schemes and Substitution Models forPhylogenetic Analysesrsquo Molecular Biology and Evolution 291695ndash701
Lembo T et al (2007) lsquoMolecular Epidemiology Identifies Only aSingle Rabies Virus Variant Circulating in Complex CarnivoreCommunities of the Serengetirsquo Proceedings Biological SciencesThe Royal Society 274 2123ndash30
mdashmdash et al (2008) lsquoExploring Reservoir Dynamics A Case Study ofRabies in the Serengeti Ecosystemrsquo Journal of Applied Ecology45 1246ndash57
Lemey P et al (2009) lsquoBayesian Phylogeography Finds Its RootsrsquoPLoS Computational Biology 5 e1000520
mdashmdash et al (2014) lsquoUnifying Viral Genetics and HumanTransportation Data to Predict the Global TransmissionDynamics of Human Influenza H3N2rsquo PLoS Pathogens 10e1003932
Lewin-koh A N J et al (2009) Maptools Tools for Reading andHandling Spatial Objects R package version 08-23 lthttpcranr-projectorgwebpackagesmaptoolsindexhtmlgt accessed 1Sept 2014
Li H and Durbin R (2009) lsquoFast and Accurate Short ReadAlignment with Burrows-Wheeler Transformrsquo Bioinformatics(Oxford England) 25 1754ndash60
mdashmdash et al (2009) lsquoThe Sequence AlignmentMap Format andSAMtoolsrsquo Bioinformatics (Oxford England) 25 2078ndash9
Lumlertdacha B et al (2006) lsquoComplex Genetic Structure ofthe Rabies Virus in Bangkok and Its Surrounding ProvincesThailand Implications for Canine Rabies ControlrsquoTransactions of the Royal Society of Tropical Medicine andHygiene 100 276ndash81
Magee D et al (2014) lsquoCombining Phylogeography and SpatialEpidemiology to Uncover Predictors of H5N1 Influenza A VirusDiffusionrsquo Archives of Virology 160 215ndash24
Magembe S (1985) lsquoEpidemiology of Rabies in United Republic ofTanzaniarsquo in Kuwer E et al (eds) Rabies in the Tropics pp 392ndash8 New York Springer Berlin Heidelberg
Marston D A et al (2007) lsquoComparative Analysis of the FullGenome Sequence of European Bat Lyssavirus Type 1 andType 2 with Other Lyssaviruses and Evidence for a ConservedTranscription Termination and Polyadenylation Motif in theG-L 3rsquo Non-Translated Regionrsquo The Journal of General Virology88 1302ndash14
mdashmdash et al (2013) lsquoNext Generation Sequencing of Viral RNAGenomesrsquo BMC Genomics 14 444
Matsumoto T et al (2013) lsquoMolecular Epidemiology of HumanRabies Viruses in Sri Lankarsquo Infection Genetics and Evolution 18160ndash7
Minin V N and Suchard M A (2008) lsquoCounting LabeledTransitions in Continuous-Time Markov Models of EvolutionrsquoJournal of Mathematical Biology 56 391ndash412
Mollentze N et al (2014) lsquoA Bayesian Approach for Inferring theDynamics of Partially Observed Endemic Infectious Diseasesfrom Space-Time-Genetic Datarsquo Proceedings Biological SciencesThe Royal Society 281 20133251
Nel L H (2013) lsquoDiscrepancies in Data Reporting for RabiesAfricarsquo Emerging Infectious Diseases 19 529ndash33
Nelson M I et al (2015) lsquoGlobal Migration of Influenza A Virusesin Swinersquo Nature Communications 6 6696
Nunes M R T et al (2014) lsquoAir Travel is Associated withIntracontinental Spread of Dengue Virus Serotypes 1-3 inBrazilrsquo PLoS Neglected Tropical Diseases 8 e2769
Paradis E Claude J and Strimmer K (2004) lsquoAPE Analyses ofPhylogenetics and Evolution in R Languagersquo Bioinformatics 20289ndash90
Parker J Rambaut A and Pybus O G (2008) lsquoCorrelatingViral Phenotypes with Phylogeny Accounting forPhylogenetic Uncertaintyrsquo Infection Genetics and Evolution 8239ndash46
Pebesma E J and Bivand R S (2005) lsquoClasses and Methods forSpatial Data in Rrsquo R News 5 9ndash13
PINGOrsquos Forum (2013) Pastoralists Indigenous Non GovernmentalOrganizationsrsquo Forum Annual Report 2012-2013 lthttpwwwpin-gosforumortzgt accessed 23 Nov 2014
Rambaut A et al (2014) lsquoTracer v16rsquo Computer program anddocumentation distributed by the author lthttpbeastbioedacukTracergt accessed 4 Feb 2015
10 | Virus Evolution 2015 Vol 1 No 1
Real L A et al (2005) lsquoUnifying the Spatial Population Dynamicsand Molecular Evolution of Epidemic Rabies Virusrsquo Proceedingsof the National Academy of Sciences of the United States of America102 12107ndash11
Rupprecht C E Hanlon C A and Hemachudha T (2002)lsquoRabies Re-examinedrsquo The Lancet Infectious Diseases 2 327ndash43
Shwiff S Hampson K and Anderson A (2013) lsquoPotentialEconomic Benefits of Eliminating Canine Rabiesrsquo AntiviralResearch 98 352ndash6
Siongok T and Karama M (1985) lsquoEpidemiology of HumanRabies in Kenyarsquo in E Kuwer et al (eds) Rabies in the Tropicspp 445ndash50 New York Springer Berlin Heidelberg
Stamatakis A et al (2012) lsquoRAxML-Light A Tool for ComputingTerabyte Phylogeniesrsquo Bioinformatics (Oxford England) 28 2064ndash6
Susilawathi N M et al (2012) lsquoEpidemiological and ClinicalFeatures of Human Rabies Cases in Bali 2008-2010rsquo BMCInfectious Diseases 12 81
Talbi C et al (2009) lsquoEvolutionary History and Dynamics of DogRabies Virus in Western and Central Africarsquo The Journal ofGeneral Virology 90 783ndash91
mdashmdash et al (2010) lsquoPhylodynamics and Human-Mediated Dispersalof a Zoonotic Virusrsquo PLoS Pathogens 6 e1001166
Tao X Y et al (2009) lsquoMolecular Epidemiology of Rabies inSouthern Peoplersquos Republic of Chinarsquo Emerging InfectiousDiseases 15 1192ndash8
Townsend S E et al (2013) lsquoDesigning Programs for EliminatingCanine Rabies from Islands Bali Indonesia as a Case StudyrsquoPLoS Neglected Tropical Diseases 7 e2372
Viana M et al (2015) lsquoDynamics of a Morbillivirus at theDomesticndashWildlife Interface Canine Distemper Virus inDomestic Dogs and Lionsrsquo Proceedings of the National Academy ofSciences of the United States of America 112 1464ndash9
Walsh M (2008) Pastoralism and Policy Processes in TanzaniaMbarali Case Study Report to the Tanzania Natural ResourceForum Arusha
Wang T H et al (2001) lsquoIdentification of Shared Populations ofHuman Immunodeficiency Virus Type 1 Infecting Microgliaand Tissue Macrophages Outside the Central Nervous SystemrsquoJournal of Virology 75 11686ndash99
WHO (2005) lsquoWHO Expert Consultation on Rabiesrsquo World HealthOrganization technical report series 931 1ndash121
Windiyaningsih C et al (2004) lsquoThe Rabies Epidemic on FloresIsland Indonesia (1998-2003)rsquo Journal of the Medical Associationof Thailandfrac14Chotmaihet thangphaet 87 1389ndash93
Woodroffe R and Donnelly C A (2011) lsquoRisk of Contact be-tween Endangered African Wild Dogs Lycaon pictus andDomestic Dogs Opportunities for Pathogen TransmissionrsquoJournal of Applied Ecology 48 1345ndash54
Woolhouse M E J (2002) lsquoPopulation Biology of Emerging and Re-emerging Pathogensrsquo Trends in Microbiology 10(10 Suppl) S3ndash7
K Brunker et al | 11
- vev011-TF1
- vev011-TF2
-

maintaining the appearance of a constant Ne in skyline plots(Carrington et al 2005)
This study represents a snapshot of RABV dynamics inTanzania indicating that human movements have dissemi-nated RABV out of locally endemic areas at scales relevant tocontrol that is administrative units such as regions or districtsThese frequent translocations have probably lead to the exis-tence of multiple co-circulating lineages (Fig 3) but relativelyfew introductions lead to sustained chains of transmission thatare detectable as lineage movement events However in diseasesystems closely associated with human activities the probabil-ity of successful translocation and establishment is likely to bemuch greater This suggests that without some level of regionalco-operation Tanzania will be unable to eliminate rabies andmaintain freedom from disease Human movements are oftenin response to social drivers which could be used as signals forincreased vigilance and surveillance in at risk areas
Our findings highlight the use of WGS to uncover fine scaletransmission patterns that can directly inform control effortsHowever sub-genomic approaches can still have utility at acoarser scale and are more easily obtained particularly whensample quality is an issue In particular they can be used to ini-tially identify admixture between countries which may indicatethe necessity of coordinated regional control programs and sur-veillance Co-circulation of multiple lineages and introductionsfacilitated by humans appear to be a feature of endemic rabies vi-rus and complicate the design of a sustainable control strategyHowever using whole-genome data we were able to identifysources of dispersal within Tanzania that could direct efforts to-ward surveillance and control The finding that humans play animportant role in the dynamics of RABV in Tanzania suggeststhat increasing awareness and dog vaccination in lsquohigh-riskrsquo com-munities such as pastoralists could help to reduce long-range dis-persal Moreover the design of enhanced surveillance andcontainment strategies to mitigate human-mediated incursionsand maintain disease freedom should be a priority once controlprograms are established and elimination is being targeted
Supplementary data
Supplementary data is available at VEVOLU Journal online
Acknowledgements
We acknowledge the Ministries of Livestock and FisheriesDevelopment and of Health and Social Welfare TanzaniaNational Parks Tanzania Wildlife Research InstituteNgorongoro Conservation Area Authority TanzaniaCommission for Science and Technology and NationalInstitute for Medical Research for permission and collabora-tion and the Arusha Mtwara and Mwanza VeterinaryInvestigation Centres Pemba Veterinary Office and theFrankfurt Zoological Society for logistical and technical sup-port We are grateful to the veterinary and livestock field of-ficers healthworkers and paravets throughout Tanzaniaand in particular to the following research assistants JoelChangalucha Zilpah Kaare Gurdeep Kour Ahmed LugeloKennedy Lushasi Mathias Magoto Khadija Said andRenatus Herman We thank Pelagia Muchuruza MatthewMaziku Geoffrey Mchau Eberhard Mbunda Francois-XavierMeslin and Bernadette Abela-Ridder for their collaborationsthrough the Bill amp Melinda GatesWHO rabies eliminationdemonstration project in Tanzania This work was
supported by the Wellcome Trust the Medical ResearchCouncil the UBS Optimus Foundation Lincoln Park Zooand the Research and Policy for Infectious DiseaseDynamics Program of the Science and TechnologyDirectorate Department of Homeland Security FogartyInternational Centre National Institute of Health
Conflict of interest None declared
ReferencesAhmed K et al (2015) lsquoMolecular Epidemiology of Rabies
Viruses Circulating in Two Rabies Endemic Provinces of Laos2011ndash2012 Regional Diversity in Southeast Asiarsquo PLoS NeglectedTropical Diseases 9 e0003645
Andrews S (2010) FastQC a quality control tool for highthroughput sequence datalthttpwwwbioinformaticsbbsrcacukprojectsfastqcgt accessed 4 Jun 2014
Ayres D L et al (2012) lsquoBEAGLE An Application ProgrammingInterface and High-Performance Computing Library forStatistical Phylogeneticsrsquo Systematic Biology 61 170ndash3
Bedford T et al (2014) lsquoIntegrating Influenza AntigenicDynamics with Molecular Evolutionrsquo eLife 2014 1ndash26
Biek R and Real L A (2010) lsquoThe Landscape Genetics of InfectiousDisease Emergence and Spreadrsquo Molecular Ecology 19 3515ndash31
mdashmdash et al (2007) lsquoA High-Resolution Genetic Signature ofDemographic and Spatial Expansion in Epizootic Rabies VirusrsquoProceedings of the National Academy of Sciences of the United Statesof America 104 7993ndash8
mdashmdash et al (2015) lsquoMeasurably Evolving Pathogens in the GenomicErarsquo Trends in Ecology amp Evolution 30 306ndash13
Bielejec F et al (2011) lsquoSPREAD Spatial PhylogeneticReconstruction of Evolutionary Dynamicsrsquo Bioinformatics(Oxford England) 27 2910ndash2
mdashmdash et al (2014) lsquoInferring Heterogeneous Evolutionary Processesthrough Time From Sequence Substitution toPhylogeographyrsquo Systematic Biology 63 493ndash504
Bivand R S Pebesma E J and Gomez-Rubio V (2008) AppliedSpatial Data Analysis with R New York Springer
Bolger A M Lohse M and Usadel B (2014) lsquoTrimmomatic AFlexible Trimmer for Illumina Sequence Datarsquo Bioinformatics30 2114ndash20
Bourhy H et al (2008) lsquoThe Origin and Phylogeography of DogRabies Virusrsquo The Journal of General Virology 89 2673ndash81
Brunker K et al (2012) lsquoIntegrating the Landscape Epidemiologyand Genetics of RNA Viruses Rabies in Domestic Dogs as aModelrsquo Parasitology 139 1899ndash913
Carrington C et al (2005) lsquoInvasion and Maintenance of DengueVirus Type 2 and Type 4 in the Americasrsquo Journal of Virology 7914680ndash7
Cleaveland S et al (2003) lsquoA Dog Rabies Vaccination Campaignin Rural Africa Impact on the Incidence of Dog Rabies andHuman Dog-Bite Injuriesrsquo Vaccine 21 1965ndash73
David D et al (2007) lsquoIdentification of Novel Canine Rabies VirusClades in the Middle East and North Africarsquo The Journal ofGeneral Virology 88 967ndash80
Denduangboripant J et al (2005) lsquoTransmission Dynamics ofRabies Virus in Thailand Implications for Disease ControlrsquoBMC Infectious Diseases 5 52
Drummond A J et al (2003) lsquoMeasurably Evolving PopulationsrsquoTrends in Ecology amp Evolution 18 481ndash8
mdashmdash et al (2005) lsquoBayesian Coalescent Inference of PastPopulation Dynamics from Molecular Sequencesrsquo MolecularBiology and Evolution 22 1185ndash92
K Brunker et al | 9
mdashmdash et al (2012) lsquoBayesian Phylogenetics with BEAUti and theBEAST 17rsquo Molecular Biology and Evolution 29 1969ndash73
Ferreira M A R and Suchard M A (2008) lsquoBayesian Analysis ofElapsed Times in Continuous-Time Markov Chainsrsquo CanadianJournal of Statistics 36 355ndash68
Greger M (2007) lsquoThe HumanAnimal Interface Emergence andResurgence of Zoonotic Infectious Diseasesrsquo Critical Reviews inMicrobiology 33 243ndash99
Hampson K et al (2007) lsquoSynchronous Cycles of Domestic DogRabies in Sub-Saharan Africa and the Impact of ControlEffortsrsquo Proceedings of the National Academy of Sciences of theUnited States of America 104 7717ndash22
mdashmdash et al (2009) lsquoTransmission Dynamics and Prospects for theElimination of Canine Rabiesrsquo PLoS Biology 7 e53
Hayman D T S et al (2011) lsquoEvolutionary History of Rabies inGhanarsquo PLoS Neglected Tropical Diseases 5 e1001
Holmes E C and Grenfell B T (2009) lsquoDiscovering the Phylodynamicsof RNA Virusesrsquo PLoS Computational Biology 5 e1000505
Horton D L et al (2015) lsquoComplex Epidemiology of a ZoonoticDisease in a Culturally Diverse Region Phylogeography ofRabies Virus in the Middle Eastrsquo PLOS Neglected TropicalDiseases 9 e0003569
Jombart T (2008) lsquoadegenet A R Package for the MultivariateAnalysis of Genetic Markersrsquo Bioinformatics (Oxford England)24 1403ndash5
Jones K E et al (2008) lsquoGlobal Trends in Emerging InfectiousDiseasesrsquo Nature 451 990ndash3
Katoh K and Standley D M (2013) lsquoMAFFT MultipleSequence Alignment Software Version 7 Improvements inPerformance and Usabilityrsquo Molecular Biology and Evolution30 772ndash80
Kideghesho J et al (2013) lsquoEmerging Issues and Challenges inConservation of Biodiversity in the Rangelands of TanzaniarsquoNature Conservation 6 1ndash29
Kissi B Tordo N and Bourhy H (1995) lsquoGenetic Polymorphismin the Rabies Virus Nucleoprotein Genersquo Virology 209 526ndash37
Knobel D L et al (2005) lsquoRe-Evaluating the Burden of Rabies inAfrica and Asiarsquo Bulletin of the World Health Organization 83360ndash8
Kuhnert D Wu C-H and Drummond A J (2011) lsquoPhylogeneticand Epidemic Modeling of Rapidly Evolving InfectiousDiseasesrsquo Infection Genetics and Evolution Journal of MolecularEpidemiology and Evolutionary Genetics in Infectious Diseases 111825ndash41
Kuzmin I V et al (2008) lsquoComplete Genomes of AravanKhujand Irkut and West Caucasian Bat Viruses with SpecialAttention to the Polymerase Gene and Non-Coding RegionsrsquoVirus Research 136 81ndash90
Kuzmina N A et al (2013) lsquoThe Phylogeography andSpatiotemporal Spread of South-Central Skunk Rabies VirusrsquoPloS One 8 e82348
Lanfear R et al (2012) lsquoPartitionfinder Combined Selectionof Partitioning Schemes and Substitution Models forPhylogenetic Analysesrsquo Molecular Biology and Evolution 291695ndash701
Lembo T et al (2007) lsquoMolecular Epidemiology Identifies Only aSingle Rabies Virus Variant Circulating in Complex CarnivoreCommunities of the Serengetirsquo Proceedings Biological SciencesThe Royal Society 274 2123ndash30
mdashmdash et al (2008) lsquoExploring Reservoir Dynamics A Case Study ofRabies in the Serengeti Ecosystemrsquo Journal of Applied Ecology45 1246ndash57
Lemey P et al (2009) lsquoBayesian Phylogeography Finds Its RootsrsquoPLoS Computational Biology 5 e1000520
mdashmdash et al (2014) lsquoUnifying Viral Genetics and HumanTransportation Data to Predict the Global TransmissionDynamics of Human Influenza H3N2rsquo PLoS Pathogens 10e1003932
Lewin-koh A N J et al (2009) Maptools Tools for Reading andHandling Spatial Objects R package version 08-23 lthttpcranr-projectorgwebpackagesmaptoolsindexhtmlgt accessed 1Sept 2014
Li H and Durbin R (2009) lsquoFast and Accurate Short ReadAlignment with Burrows-Wheeler Transformrsquo Bioinformatics(Oxford England) 25 1754ndash60
mdashmdash et al (2009) lsquoThe Sequence AlignmentMap Format andSAMtoolsrsquo Bioinformatics (Oxford England) 25 2078ndash9
Lumlertdacha B et al (2006) lsquoComplex Genetic Structure ofthe Rabies Virus in Bangkok and Its Surrounding ProvincesThailand Implications for Canine Rabies ControlrsquoTransactions of the Royal Society of Tropical Medicine andHygiene 100 276ndash81
Magee D et al (2014) lsquoCombining Phylogeography and SpatialEpidemiology to Uncover Predictors of H5N1 Influenza A VirusDiffusionrsquo Archives of Virology 160 215ndash24
Magembe S (1985) lsquoEpidemiology of Rabies in United Republic ofTanzaniarsquo in Kuwer E et al (eds) Rabies in the Tropics pp 392ndash8 New York Springer Berlin Heidelberg
Marston D A et al (2007) lsquoComparative Analysis of the FullGenome Sequence of European Bat Lyssavirus Type 1 andType 2 with Other Lyssaviruses and Evidence for a ConservedTranscription Termination and Polyadenylation Motif in theG-L 3rsquo Non-Translated Regionrsquo The Journal of General Virology88 1302ndash14
mdashmdash et al (2013) lsquoNext Generation Sequencing of Viral RNAGenomesrsquo BMC Genomics 14 444
Matsumoto T et al (2013) lsquoMolecular Epidemiology of HumanRabies Viruses in Sri Lankarsquo Infection Genetics and Evolution 18160ndash7
Minin V N and Suchard M A (2008) lsquoCounting LabeledTransitions in Continuous-Time Markov Models of EvolutionrsquoJournal of Mathematical Biology 56 391ndash412
Mollentze N et al (2014) lsquoA Bayesian Approach for Inferring theDynamics of Partially Observed Endemic Infectious Diseasesfrom Space-Time-Genetic Datarsquo Proceedings Biological SciencesThe Royal Society 281 20133251
Nel L H (2013) lsquoDiscrepancies in Data Reporting for RabiesAfricarsquo Emerging Infectious Diseases 19 529ndash33
Nelson M I et al (2015) lsquoGlobal Migration of Influenza A Virusesin Swinersquo Nature Communications 6 6696
Nunes M R T et al (2014) lsquoAir Travel is Associated withIntracontinental Spread of Dengue Virus Serotypes 1-3 inBrazilrsquo PLoS Neglected Tropical Diseases 8 e2769
Paradis E Claude J and Strimmer K (2004) lsquoAPE Analyses ofPhylogenetics and Evolution in R Languagersquo Bioinformatics 20289ndash90
Parker J Rambaut A and Pybus O G (2008) lsquoCorrelatingViral Phenotypes with Phylogeny Accounting forPhylogenetic Uncertaintyrsquo Infection Genetics and Evolution 8239ndash46
Pebesma E J and Bivand R S (2005) lsquoClasses and Methods forSpatial Data in Rrsquo R News 5 9ndash13
PINGOrsquos Forum (2013) Pastoralists Indigenous Non GovernmentalOrganizationsrsquo Forum Annual Report 2012-2013 lthttpwwwpin-gosforumortzgt accessed 23 Nov 2014
Rambaut A et al (2014) lsquoTracer v16rsquo Computer program anddocumentation distributed by the author lthttpbeastbioedacukTracergt accessed 4 Feb 2015
10 | Virus Evolution 2015 Vol 1 No 1
Real L A et al (2005) lsquoUnifying the Spatial Population Dynamicsand Molecular Evolution of Epidemic Rabies Virusrsquo Proceedingsof the National Academy of Sciences of the United States of America102 12107ndash11
Rupprecht C E Hanlon C A and Hemachudha T (2002)lsquoRabies Re-examinedrsquo The Lancet Infectious Diseases 2 327ndash43
Shwiff S Hampson K and Anderson A (2013) lsquoPotentialEconomic Benefits of Eliminating Canine Rabiesrsquo AntiviralResearch 98 352ndash6
Siongok T and Karama M (1985) lsquoEpidemiology of HumanRabies in Kenyarsquo in E Kuwer et al (eds) Rabies in the Tropicspp 445ndash50 New York Springer Berlin Heidelberg
Stamatakis A et al (2012) lsquoRAxML-Light A Tool for ComputingTerabyte Phylogeniesrsquo Bioinformatics (Oxford England) 28 2064ndash6
Susilawathi N M et al (2012) lsquoEpidemiological and ClinicalFeatures of Human Rabies Cases in Bali 2008-2010rsquo BMCInfectious Diseases 12 81
Talbi C et al (2009) lsquoEvolutionary History and Dynamics of DogRabies Virus in Western and Central Africarsquo The Journal ofGeneral Virology 90 783ndash91
mdashmdash et al (2010) lsquoPhylodynamics and Human-Mediated Dispersalof a Zoonotic Virusrsquo PLoS Pathogens 6 e1001166
Tao X Y et al (2009) lsquoMolecular Epidemiology of Rabies inSouthern Peoplersquos Republic of Chinarsquo Emerging InfectiousDiseases 15 1192ndash8
Townsend S E et al (2013) lsquoDesigning Programs for EliminatingCanine Rabies from Islands Bali Indonesia as a Case StudyrsquoPLoS Neglected Tropical Diseases 7 e2372
Viana M et al (2015) lsquoDynamics of a Morbillivirus at theDomesticndashWildlife Interface Canine Distemper Virus inDomestic Dogs and Lionsrsquo Proceedings of the National Academy ofSciences of the United States of America 112 1464ndash9
Walsh M (2008) Pastoralism and Policy Processes in TanzaniaMbarali Case Study Report to the Tanzania Natural ResourceForum Arusha
Wang T H et al (2001) lsquoIdentification of Shared Populations ofHuman Immunodeficiency Virus Type 1 Infecting Microgliaand Tissue Macrophages Outside the Central Nervous SystemrsquoJournal of Virology 75 11686ndash99
WHO (2005) lsquoWHO Expert Consultation on Rabiesrsquo World HealthOrganization technical report series 931 1ndash121
Windiyaningsih C et al (2004) lsquoThe Rabies Epidemic on FloresIsland Indonesia (1998-2003)rsquo Journal of the Medical Associationof Thailandfrac14Chotmaihet thangphaet 87 1389ndash93
Woodroffe R and Donnelly C A (2011) lsquoRisk of Contact be-tween Endangered African Wild Dogs Lycaon pictus andDomestic Dogs Opportunities for Pathogen TransmissionrsquoJournal of Applied Ecology 48 1345ndash54
Woolhouse M E J (2002) lsquoPopulation Biology of Emerging and Re-emerging Pathogensrsquo Trends in Microbiology 10(10 Suppl) S3ndash7
K Brunker et al | 11
- vev011-TF1
- vev011-TF2
-

mdashmdash et al (2012) lsquoBayesian Phylogenetics with BEAUti and theBEAST 17rsquo Molecular Biology and Evolution 29 1969ndash73
Ferreira M A R and Suchard M A (2008) lsquoBayesian Analysis ofElapsed Times in Continuous-Time Markov Chainsrsquo CanadianJournal of Statistics 36 355ndash68
Greger M (2007) lsquoThe HumanAnimal Interface Emergence andResurgence of Zoonotic Infectious Diseasesrsquo Critical Reviews inMicrobiology 33 243ndash99
Hampson K et al (2007) lsquoSynchronous Cycles of Domestic DogRabies in Sub-Saharan Africa and the Impact of ControlEffortsrsquo Proceedings of the National Academy of Sciences of theUnited States of America 104 7717ndash22
mdashmdash et al (2009) lsquoTransmission Dynamics and Prospects for theElimination of Canine Rabiesrsquo PLoS Biology 7 e53
Hayman D T S et al (2011) lsquoEvolutionary History of Rabies inGhanarsquo PLoS Neglected Tropical Diseases 5 e1001
Holmes E C and Grenfell B T (2009) lsquoDiscovering the Phylodynamicsof RNA Virusesrsquo PLoS Computational Biology 5 e1000505
Horton D L et al (2015) lsquoComplex Epidemiology of a ZoonoticDisease in a Culturally Diverse Region Phylogeography ofRabies Virus in the Middle Eastrsquo PLOS Neglected TropicalDiseases 9 e0003569
Jombart T (2008) lsquoadegenet A R Package for the MultivariateAnalysis of Genetic Markersrsquo Bioinformatics (Oxford England)24 1403ndash5
Jones K E et al (2008) lsquoGlobal Trends in Emerging InfectiousDiseasesrsquo Nature 451 990ndash3
Katoh K and Standley D M (2013) lsquoMAFFT MultipleSequence Alignment Software Version 7 Improvements inPerformance and Usabilityrsquo Molecular Biology and Evolution30 772ndash80
Kideghesho J et al (2013) lsquoEmerging Issues and Challenges inConservation of Biodiversity in the Rangelands of TanzaniarsquoNature Conservation 6 1ndash29
Kissi B Tordo N and Bourhy H (1995) lsquoGenetic Polymorphismin the Rabies Virus Nucleoprotein Genersquo Virology 209 526ndash37
Knobel D L et al (2005) lsquoRe-Evaluating the Burden of Rabies inAfrica and Asiarsquo Bulletin of the World Health Organization 83360ndash8
Kuhnert D Wu C-H and Drummond A J (2011) lsquoPhylogeneticand Epidemic Modeling of Rapidly Evolving InfectiousDiseasesrsquo Infection Genetics and Evolution Journal of MolecularEpidemiology and Evolutionary Genetics in Infectious Diseases 111825ndash41
Kuzmin I V et al (2008) lsquoComplete Genomes of AravanKhujand Irkut and West Caucasian Bat Viruses with SpecialAttention to the Polymerase Gene and Non-Coding RegionsrsquoVirus Research 136 81ndash90
Kuzmina N A et al (2013) lsquoThe Phylogeography andSpatiotemporal Spread of South-Central Skunk Rabies VirusrsquoPloS One 8 e82348
Lanfear R et al (2012) lsquoPartitionfinder Combined Selectionof Partitioning Schemes and Substitution Models forPhylogenetic Analysesrsquo Molecular Biology and Evolution 291695ndash701
Lembo T et al (2007) lsquoMolecular Epidemiology Identifies Only aSingle Rabies Virus Variant Circulating in Complex CarnivoreCommunities of the Serengetirsquo Proceedings Biological SciencesThe Royal Society 274 2123ndash30
mdashmdash et al (2008) lsquoExploring Reservoir Dynamics A Case Study ofRabies in the Serengeti Ecosystemrsquo Journal of Applied Ecology45 1246ndash57
Lemey P et al (2009) lsquoBayesian Phylogeography Finds Its RootsrsquoPLoS Computational Biology 5 e1000520
mdashmdash et al (2014) lsquoUnifying Viral Genetics and HumanTransportation Data to Predict the Global TransmissionDynamics of Human Influenza H3N2rsquo PLoS Pathogens 10e1003932
Lewin-koh A N J et al (2009) Maptools Tools for Reading andHandling Spatial Objects R package version 08-23 lthttpcranr-projectorgwebpackagesmaptoolsindexhtmlgt accessed 1Sept 2014
Li H and Durbin R (2009) lsquoFast and Accurate Short ReadAlignment with Burrows-Wheeler Transformrsquo Bioinformatics(Oxford England) 25 1754ndash60
mdashmdash et al (2009) lsquoThe Sequence AlignmentMap Format andSAMtoolsrsquo Bioinformatics (Oxford England) 25 2078ndash9
Lumlertdacha B et al (2006) lsquoComplex Genetic Structure ofthe Rabies Virus in Bangkok and Its Surrounding ProvincesThailand Implications for Canine Rabies ControlrsquoTransactions of the Royal Society of Tropical Medicine andHygiene 100 276ndash81
Magee D et al (2014) lsquoCombining Phylogeography and SpatialEpidemiology to Uncover Predictors of H5N1 Influenza A VirusDiffusionrsquo Archives of Virology 160 215ndash24
Magembe S (1985) lsquoEpidemiology of Rabies in United Republic ofTanzaniarsquo in Kuwer E et al (eds) Rabies in the Tropics pp 392ndash8 New York Springer Berlin Heidelberg
Marston D A et al (2007) lsquoComparative Analysis of the FullGenome Sequence of European Bat Lyssavirus Type 1 andType 2 with Other Lyssaviruses and Evidence for a ConservedTranscription Termination and Polyadenylation Motif in theG-L 3rsquo Non-Translated Regionrsquo The Journal of General Virology88 1302ndash14
mdashmdash et al (2013) lsquoNext Generation Sequencing of Viral RNAGenomesrsquo BMC Genomics 14 444
Matsumoto T et al (2013) lsquoMolecular Epidemiology of HumanRabies Viruses in Sri Lankarsquo Infection Genetics and Evolution 18160ndash7
Minin V N and Suchard M A (2008) lsquoCounting LabeledTransitions in Continuous-Time Markov Models of EvolutionrsquoJournal of Mathematical Biology 56 391ndash412
Mollentze N et al (2014) lsquoA Bayesian Approach for Inferring theDynamics of Partially Observed Endemic Infectious Diseasesfrom Space-Time-Genetic Datarsquo Proceedings Biological SciencesThe Royal Society 281 20133251
Nel L H (2013) lsquoDiscrepancies in Data Reporting for RabiesAfricarsquo Emerging Infectious Diseases 19 529ndash33
Nelson M I et al (2015) lsquoGlobal Migration of Influenza A Virusesin Swinersquo Nature Communications 6 6696
Nunes M R T et al (2014) lsquoAir Travel is Associated withIntracontinental Spread of Dengue Virus Serotypes 1-3 inBrazilrsquo PLoS Neglected Tropical Diseases 8 e2769
Paradis E Claude J and Strimmer K (2004) lsquoAPE Analyses ofPhylogenetics and Evolution in R Languagersquo Bioinformatics 20289ndash90
Parker J Rambaut A and Pybus O G (2008) lsquoCorrelatingViral Phenotypes with Phylogeny Accounting forPhylogenetic Uncertaintyrsquo Infection Genetics and Evolution 8239ndash46
Pebesma E J and Bivand R S (2005) lsquoClasses and Methods forSpatial Data in Rrsquo R News 5 9ndash13
PINGOrsquos Forum (2013) Pastoralists Indigenous Non GovernmentalOrganizationsrsquo Forum Annual Report 2012-2013 lthttpwwwpin-gosforumortzgt accessed 23 Nov 2014
Rambaut A et al (2014) lsquoTracer v16rsquo Computer program anddocumentation distributed by the author lthttpbeastbioedacukTracergt accessed 4 Feb 2015
10 | Virus Evolution 2015 Vol 1 No 1
Real L A et al (2005) lsquoUnifying the Spatial Population Dynamicsand Molecular Evolution of Epidemic Rabies Virusrsquo Proceedingsof the National Academy of Sciences of the United States of America102 12107ndash11
Rupprecht C E Hanlon C A and Hemachudha T (2002)lsquoRabies Re-examinedrsquo The Lancet Infectious Diseases 2 327ndash43
Shwiff S Hampson K and Anderson A (2013) lsquoPotentialEconomic Benefits of Eliminating Canine Rabiesrsquo AntiviralResearch 98 352ndash6
Siongok T and Karama M (1985) lsquoEpidemiology of HumanRabies in Kenyarsquo in E Kuwer et al (eds) Rabies in the Tropicspp 445ndash50 New York Springer Berlin Heidelberg
Stamatakis A et al (2012) lsquoRAxML-Light A Tool for ComputingTerabyte Phylogeniesrsquo Bioinformatics (Oxford England) 28 2064ndash6
Susilawathi N M et al (2012) lsquoEpidemiological and ClinicalFeatures of Human Rabies Cases in Bali 2008-2010rsquo BMCInfectious Diseases 12 81
Talbi C et al (2009) lsquoEvolutionary History and Dynamics of DogRabies Virus in Western and Central Africarsquo The Journal ofGeneral Virology 90 783ndash91
mdashmdash et al (2010) lsquoPhylodynamics and Human-Mediated Dispersalof a Zoonotic Virusrsquo PLoS Pathogens 6 e1001166
Tao X Y et al (2009) lsquoMolecular Epidemiology of Rabies inSouthern Peoplersquos Republic of Chinarsquo Emerging InfectiousDiseases 15 1192ndash8
Townsend S E et al (2013) lsquoDesigning Programs for EliminatingCanine Rabies from Islands Bali Indonesia as a Case StudyrsquoPLoS Neglected Tropical Diseases 7 e2372
Viana M et al (2015) lsquoDynamics of a Morbillivirus at theDomesticndashWildlife Interface Canine Distemper Virus inDomestic Dogs and Lionsrsquo Proceedings of the National Academy ofSciences of the United States of America 112 1464ndash9
Walsh M (2008) Pastoralism and Policy Processes in TanzaniaMbarali Case Study Report to the Tanzania Natural ResourceForum Arusha
Wang T H et al (2001) lsquoIdentification of Shared Populations ofHuman Immunodeficiency Virus Type 1 Infecting Microgliaand Tissue Macrophages Outside the Central Nervous SystemrsquoJournal of Virology 75 11686ndash99
WHO (2005) lsquoWHO Expert Consultation on Rabiesrsquo World HealthOrganization technical report series 931 1ndash121
Windiyaningsih C et al (2004) lsquoThe Rabies Epidemic on FloresIsland Indonesia (1998-2003)rsquo Journal of the Medical Associationof Thailandfrac14Chotmaihet thangphaet 87 1389ndash93
Woodroffe R and Donnelly C A (2011) lsquoRisk of Contact be-tween Endangered African Wild Dogs Lycaon pictus andDomestic Dogs Opportunities for Pathogen TransmissionrsquoJournal of Applied Ecology 48 1345ndash54
Woolhouse M E J (2002) lsquoPopulation Biology of Emerging and Re-emerging Pathogensrsquo Trends in Microbiology 10(10 Suppl) S3ndash7
K Brunker et al | 11
- vev011-TF1
- vev011-TF2
-

Real L A et al (2005) lsquoUnifying the Spatial Population Dynamicsand Molecular Evolution of Epidemic Rabies Virusrsquo Proceedingsof the National Academy of Sciences of the United States of America102 12107ndash11
Rupprecht C E Hanlon C A and Hemachudha T (2002)lsquoRabies Re-examinedrsquo The Lancet Infectious Diseases 2 327ndash43
Shwiff S Hampson K and Anderson A (2013) lsquoPotentialEconomic Benefits of Eliminating Canine Rabiesrsquo AntiviralResearch 98 352ndash6
Siongok T and Karama M (1985) lsquoEpidemiology of HumanRabies in Kenyarsquo in E Kuwer et al (eds) Rabies in the Tropicspp 445ndash50 New York Springer Berlin Heidelberg
Stamatakis A et al (2012) lsquoRAxML-Light A Tool for ComputingTerabyte Phylogeniesrsquo Bioinformatics (Oxford England) 28 2064ndash6
Susilawathi N M et al (2012) lsquoEpidemiological and ClinicalFeatures of Human Rabies Cases in Bali 2008-2010rsquo BMCInfectious Diseases 12 81
Talbi C et al (2009) lsquoEvolutionary History and Dynamics of DogRabies Virus in Western and Central Africarsquo The Journal ofGeneral Virology 90 783ndash91
mdashmdash et al (2010) lsquoPhylodynamics and Human-Mediated Dispersalof a Zoonotic Virusrsquo PLoS Pathogens 6 e1001166
Tao X Y et al (2009) lsquoMolecular Epidemiology of Rabies inSouthern Peoplersquos Republic of Chinarsquo Emerging InfectiousDiseases 15 1192ndash8
Townsend S E et al (2013) lsquoDesigning Programs for EliminatingCanine Rabies from Islands Bali Indonesia as a Case StudyrsquoPLoS Neglected Tropical Diseases 7 e2372
Viana M et al (2015) lsquoDynamics of a Morbillivirus at theDomesticndashWildlife Interface Canine Distemper Virus inDomestic Dogs and Lionsrsquo Proceedings of the National Academy ofSciences of the United States of America 112 1464ndash9
Walsh M (2008) Pastoralism and Policy Processes in TanzaniaMbarali Case Study Report to the Tanzania Natural ResourceForum Arusha
Wang T H et al (2001) lsquoIdentification of Shared Populations ofHuman Immunodeficiency Virus Type 1 Infecting Microgliaand Tissue Macrophages Outside the Central Nervous SystemrsquoJournal of Virology 75 11686ndash99
WHO (2005) lsquoWHO Expert Consultation on Rabiesrsquo World HealthOrganization technical report series 931 1ndash121
Windiyaningsih C et al (2004) lsquoThe Rabies Epidemic on FloresIsland Indonesia (1998-2003)rsquo Journal of the Medical Associationof Thailandfrac14Chotmaihet thangphaet 87 1389ndash93
Woodroffe R and Donnelly C A (2011) lsquoRisk of Contact be-tween Endangered African Wild Dogs Lycaon pictus andDomestic Dogs Opportunities for Pathogen TransmissionrsquoJournal of Applied Ecology 48 1345ndash54
Woolhouse M E J (2002) lsquoPopulation Biology of Emerging and Re-emerging Pathogensrsquo Trends in Microbiology 10(10 Suppl) S3ndash7
K Brunker et al | 11
- vev011-TF1
- vev011-TF2
-


















