
Elucidating the druggable interface of protein protein ... · interactions using fragment docking...
8
Elucidating the druggable interface of protein-protein interactions using fragment docking and coevolutionary analysis Fang Bai a , Faruck Morcos b,c,d , Ryan R. Cheng a , Hualiang Jiang e,1 , and José N. Onuchic a,f,g,h,1 a Center for Theoretical Biological Physics, Rice University, Houston, TX 77005; b Department of Biological Sciences, University of Texas at Dallas, Dallas, TX 75080; c Department of Bioengineering, University of Texas at Dallas, Dallas, TX 75080; d Center for Systems Biology, University of Texas at Dallas, Dallas, TX 75080; e Drug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China; f Department of Physics and Astronomy, Rice University, Houston, TX 77005; g Department of Chemistry, Rice University, Houston, TX 77005; and h Department of Biosciences, Rice University, Houston, TX 77005 Contributed by José N. Onuchic, September 28, 2016 (sent for review June 28, 2016; reviewed by Shi-Jie Chen, Shaomeng Wang, and Martin Weigt) Protein-protein interactions play a central role in cellular function. Improving the understanding of complex formation has many practical applications, including the rational design of new therapeu- tic agents and the mechanisms governing signal transduction net- works. The generally large, flat, and relatively featureless binding sites of protein complexes pose many challenges for drug design. Fragment docking and direct coupling analysis are used in an inte- grated computational method to estimate druggable protein-protein interfaces. (i ) This method explores the binding of fragment-sized molecular probes on the protein surface using a molecular docking- based screen. (ii ) The energetically favorable binding sites of the probes, called hot spots, are spatially clustered to map out candidate binding sites on the protein surface. (iii ) A coevolution-based interface interaction score is used to discriminate between different candidate binding sites, yielding potential interfacial targets for therapeutic drug design. This approach is validated for important, well-studied disease-related proteins with known pharmaceutical targets, and also identifies targets that have yet to be studied. Moreover, therapeutic agents are proposed by chemically connecting the fragments that are strongly bound to the hot spots. protein−protein interface | druggable surface | hot spots | direct coupling analysis | drug design P rotein−protein interactions (PPIs) mediate a wide range of important cellular functions, including signal transduction and enzymatic processes. The regulation of these interactions has drawn intense focus in physiology and pathology, where PPI interfaces have emerged as a new class of molecular targets for pharmacological intervention (1, 2). The design of drugs to tar- get PPIs, however, faces numerous challenges (3). Although over 41,000 unique human protein interactions have been experi- mentally discovered and reported on Human Protein Reference Database (4), only ∼2,500 nonredundant multiprotein complexes have experimentally determined structures available in the Pro- tein Data Bank (5). Therefore, the current experimental meth- ods cannot accommodate the high demand for structural details of these interactions. Even when the structural complexes are known, targeting PPIs with small molecules poses a significant challenge. Many protein interfaces have large, featureless sur- faces that lack obvious small-molecule binding pockets, making it difficult to design drugs with the ability to modulate (inhibit or stabilize) the PPIs with the necessary selectivity and potency (6). In the past two decades, two major classes of computational methods for protein−protein interface prediction have emerged (7): (i ) data-driven methods (8–13) and (ii ) molecular docking methods (14). Data-driven methods include homology modeling (8, 9), machine learning (10), and coevolution-based statistical models (11–13). These approaches make predictions using ho- mologous data as templates or by extracting interaction patterns from data using statistical models. On the other hand, molecular docking approaches (14) search for putative binding modes with favorable interaction energies and surface complementarity by using physics-based or geometric models. Although the data- driven methods are predictive, they are often limited by the amount of data that is available to train a statistical model. Docking methods can make structural predictions at atomic-level detail, but they are limited by the high-dimensional complexity of performing conformational searches as well as the accuracy of the force field being used. The predictive capabilities of docking approaches can be improved by incorporating relevant in- formation (e.g., amino acid coevolution) (15), which can be extracted from the existing wealth of sequence data. Hence, data-driven methods can be combined with physics-based dock- ing approaches to improve their predictive power. To overcome these challenges, an integrated computational approach called fragment docking and direct coupling analysis (Fd-DCA) is de- veloped by combining a docking methodology with coevolu- tionary signals from sequence data. This approach offers a distinct direction toward identifying PPI-mediated biological functions as well as the development of therapeutic agents that target protein interactions. Druggable Interface Identification Design Framework To identify druggable surfaces, one needs to find the residues or epitopes in the proteins that are responsible for the majority of Significance Protein-protein interfaces have become an emerging class of molecular targets for the design of therapeutic drugs. However, major challenges exist for the correct identification of binding sites on the protein surface as well as drug-like modulators of protein-protein interaction. An integrated approach using mo- lecular fragment docking and coevolutionary analysis is presented to face these challenges. This approach can accurately predict and characterize the binding sites for protein-protein interactions as well as provide clusters of bound, fragment-sized molecules on the druggable regions of the predicted binding site. These bound, molecular fragments can be chemically combined to create candidate drugs. Author contributions: F.B., H.J., and J.N.O. designed research; F.B., F.M., and R.R.C. per- formed research; F.B., F.M., and R.R.C. contributed new reagents/analytic tools; F.B., F.M., R.R.C., and J.N.O. analyzed data; and F.B., F.M., R.R.C., H.J., and J.N.O. wrote the paper. Reviewers: S.-J.C., University of Missouri–Columbia; S.W., University of Michigan; and M.W., Université Pierre et Marie Curie. The authors declare no conflict of interest. Freely available online through the PNAS open access option. 1 To whom correspondence may be addressed. Email: [email protected] or hljiang@mail. simm.ac.cn. This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10. 1073/pnas.1615932113/-/DCSupplemental. www.pnas.org/cgi/doi/10.1073/pnas.1615932113 PNAS | Published online November 29, 2016 | E8051–E8058 BIOPHYSICS AND COMPUTATIONAL BIOLOGY PNAS PLUS
-
Upload
nguyenkien -
Category
Documents
-
view
214 -
download
0
Transcript of Elucidating the druggable interface of protein protein ... · interactions using fragment docking...

Elucidating the druggable interface of protein−proteininteractions using fragment docking andcoevolutionary analysisFang Baia, Faruck Morcosb,c,d, Ryan R. Chenga, Hualiang Jiange,1, and José N. Onuchica,f,g,h,1
aCenter for Theoretical Biological Physics, Rice University, Houston, TX 77005; bDepartment of Biological Sciences, University of Texas at Dallas, Dallas,TX 75080; cDepartment of Bioengineering, University of Texas at Dallas, Dallas, TX 75080; dCenter for Systems Biology, University of Texas at Dallas, Dallas,TX 75080; eDrug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences,Shanghai 201203, China; fDepartment of Physics and Astronomy, Rice University, Houston, TX 77005; gDepartment of Chemistry, Rice University, Houston,TX 77005; and hDepartment of Biosciences, Rice University, Houston, TX 77005
Contributed by José N. Onuchic, September 28, 2016 (sent for review June 28, 2016; reviewed by Shi-Jie Chen, Shaomeng Wang, and Martin Weigt)
Protein−protein interactions play a central role in cellular function.Improving the understanding of complex formation has manypractical applications, including the rational design of new therapeu-tic agents and the mechanisms governing signal transduction net-works. The generally large, flat, and relatively featureless bindingsites of protein complexes pose many challenges for drug design.Fragment docking and direct coupling analysis are used in an inte-grated computational method to estimate druggable protein−proteininterfaces. (i) This method explores the binding of fragment-sizedmolecular probes on the protein surface using a molecular docking-based screen. (ii) The energetically favorable binding sites of theprobes, called hot spots, are spatially clustered to map out candidatebinding sites on the protein surface. (iii) A coevolution-based interfaceinteraction score is used to discriminate between different candidatebinding sites, yielding potential interfacial targets for therapeuticdrug design. This approach is validated for important, well-studieddisease-related proteins with known pharmaceutical targets, and alsoidentifies targets that have yet to be studied. Moreover, therapeuticagents are proposed by chemically connecting the fragments that arestrongly bound to the hot spots.
protein−protein interface | druggable surface | hot spots | direct couplinganalysis | drug design
Protein−protein interactions (PPIs) mediate a wide range ofimportant cellular functions, including signal transduction
and enzymatic processes. The regulation of these interactionshas drawn intense focus in physiology and pathology, where PPIinterfaces have emerged as a new class of molecular targets forpharmacological intervention (1, 2). The design of drugs to tar-get PPIs, however, faces numerous challenges (3). Although over41,000 unique human protein interactions have been experi-mentally discovered and reported on Human Protein ReferenceDatabase (4), only ∼2,500 nonredundant multiprotein complexeshave experimentally determined structures available in the Pro-tein Data Bank (5). Therefore, the current experimental meth-ods cannot accommodate the high demand for structural detailsof these interactions. Even when the structural complexes areknown, targeting PPIs with small molecules poses a significantchallenge. Many protein interfaces have large, featureless sur-faces that lack obvious small-molecule binding pockets, making itdifficult to design drugs with the ability to modulate (inhibit orstabilize) the PPIs with the necessary selectivity and potency (6).In the past two decades, two major classes of computational
methods for protein−protein interface prediction have emerged(7): (i) data-driven methods (8–13) and (ii) molecular dockingmethods (14). Data-driven methods include homology modeling(8, 9), machine learning (10), and coevolution-based statisticalmodels (11–13). These approaches make predictions using ho-mologous data as templates or by extracting interaction patternsfrom data using statistical models. On the other hand, molecular
docking approaches (14) search for putative binding modes withfavorable interaction energies and surface complementarity byusing physics-based or geometric models. Although the data-driven methods are predictive, they are often limited by theamount of data that is available to train a statistical model.Docking methods can make structural predictions at atomic-leveldetail, but they are limited by the high-dimensional complexity ofperforming conformational searches as well as the accuracy ofthe force field being used. The predictive capabilities of dockingapproaches can be improved by incorporating relevant in-formation (e.g., amino acid coevolution) (15), which can beextracted from the existing wealth of sequence data. Hence,data-driven methods can be combined with physics-based dock-ing approaches to improve their predictive power. To overcomethese challenges, an integrated computational approach calledfragment docking and direct coupling analysis (Fd-DCA) is de-veloped by combining a docking methodology with coevolu-tionary signals from sequence data. This approach offers adistinct direction toward identifying PPI-mediated biologicalfunctions as well as the development of therapeutic agents thattarget protein interactions.
Druggable Interface Identification Design FrameworkTo identify druggable surfaces, one needs to find the residues orepitopes in the proteins that are responsible for the majority of
Significance
Protein−protein interfaces have become an emerging class ofmolecular targets for the design of therapeutic drugs. However,major challenges exist for the correct identification of bindingsites on the protein surface as well as drug-like modulators ofprotein−protein interaction. An integrated approach using mo-lecular fragment docking and coevolutionary analysis is presentedto face these challenges. This approach can accurately predict andcharacterize the binding sites for protein−protein interactions aswell as provide clusters of bound, fragment-sized molecules onthe druggable regions of the predicted binding site. These bound,molecular fragments can be chemically combined to createcandidate drugs.
Author contributions: F.B., H.J., and J.N.O. designed research; F.B., F.M., and R.R.C. per-formed research; F.B., F.M., and R.R.C. contributed new reagents/analytic tools; F.B., F.M.,R.R.C., and J.N.O. analyzed data; and F.B., F.M., R.R.C., H.J., and J.N.O. wrote the paper.
Reviewers: S.-J.C., University of Missouri–Columbia; S.W., University of Michigan; and M.W.,Université Pierre et Marie Curie.
The authors declare no conflict of interest.
Freely available online through the PNAS open access option.1To whom correspondence may be addressed. Email: [email protected] or [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1615932113/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1615932113 PNAS | Published online November 29, 2016 | E8051–E8058
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
PNASPL
US

the binding affinity between proteins or between a protein and aligand. These regions are commonly called “hot spots” (16). Thedesign of therapeutic agents that interact with hot spots hasproven to be a valid methodology for inferring PPI (1). Com-putationally differentiating hot spots from other regions on aprotein surface remains a difficult challenge (17). Overcomingthese difficulties is the main focus of this work.One computational framework that has successfully been ap-
plied to find hot spots on protein surfaces is called FTMap(18, 19). FTMap is a solvent mapping-based approach that ex-plores the binding of 16 small organic molecular probes to thesurface of a protein. This method first places the probes in adense grid around the protein. An empirical free energy is thenused as a scoring function to minimize each probe and findstrongly bound probe configurations on the protein surface.Regions that bind clusters of multiple probe types are recognizedas the hot spots of a protein. However, this process presentsseveral challenges. First, the current probes implemented in FTMapare small organic molecules, which are only appropriate for de-termining druggable sites for conventional drug design. Thesefragments may not be appropriate for identifying the binding sitesfor peptide drugs as well as nonligand-bound PPI interfaces. Sec-ond, FTMap relies on a two-step downhill energy minimization tolocate the binding mode for each probe, which may unexpectedlyneglect binding modes that involve conformational rearrangement.To overcome these challenges, a hot spot identification method is
introduced in this work. The binding of probes on a protein surfaceis now performed using 31 probe molecules, which include15 amino acid side chain residues in addition to 16 small organicmolecules (SI Appendix, Fig. S1). Not only can this method be usedto detect ligand−protein binding hot spots, but it also reveals protein/peptide−protein interacting regions (SI Appendix). Moreover, thishot spot identification method uses the in-house molecular dockingtool iFitDock (20), which features an accurate method for estimatingbinding affinity (i.e., Molecular Mechanics−Generalized Born SurfaceArea) as well as a robust binding conformation search algorithm.These improvements allow the method to globally search for ener-getically favorable binding positions to generate more reliable boundconformations of the probes (details in SI Appendix).To ensure that the hot spot predictions capture the true PPI-
forming surface of a protein, one can discriminate between dif-ferent hot spots based on the evolutionary signature of PPI foundin amino acid sequence data. The association of interactingproteins is often, in part, mediated by the unique recognitionpatterns on their complementary binding surfaces. These aminoacid patterns arise from the constraint to maintain importantamino acid complementarity over the course of natural selection(21), e.g., residues that form native contacts. Thus, coevolutionarypatterns in protein sequence data can be used to identify functionalcomplex interfaces. Extracting this rich information has been anarea of great focus (12, 13, 22), particularly with the advancement ofsequencing technologies over the recent decades (23).Recently, DCA (24–26) has allowed for the quantification of
amino acid coevolution from multiple sequence alignments ofprotein families or interacting protein families. DCA has success-fully been used to find highly coevolved pairs of residues, which areintraprotein (24, 27) and interprotein amino acid (12, 24, 25, 28, 29)contacts. Remarkably, DCA also captures the residue contacts thatstabilize functionally important conformational transitions (30).DCA has also been applied to study specificity in bacterial signalingsystems, quantifying the effect of mutations on phosphotransfer in asignaling network (29, 31).Preliminary results demonstrated the complementary role of
DCA in the prediction of drug targets for treating human breastcancer (22, 32). Earlier work used DCA and FTmap to predictthe binding interface between the nutrient-deprivation auto-phagy factor-1 (NAF-1) and the B-cell lymphoma-2 (Bcl-2), aswell as discover a NAF-1 small molecular binder, MAD-28,
which may disrupt NAF-1/Bcl-2 binding. Motivated by these find-ings, this current work introduces a generalized computational ap-proach that combines a hot spot identification method andcoevolutionary analysis using DCA to find binding sites for PPIthat are potential drug targets. The basic idea behind this integratedapproach consists of a molecular docking-based search of candidatebinding sites followed by a DCA-based discrimination of evolu-tionarily relevant candidates. The computational scheme consists ofthree steps: (i) The surface of a target protein is globally screenedfor the binding of 31 probe molecules using iFitDock (20) to findhot spots. (ii) The adjacent hot spots are spatially clustered toobtain candidate binding sites on the surface of a protein. (iii) Acoevolutionary interface interaction score (Eq. 1) is used todiscriminate between the candidate binding sites to find the mostpromising candidates, which demonstrate functional interactionsthat can potentially be targets for therapeutics.To demonstrate the power of this integrated approach in pre-
dicting functionally relevant protein−protein binding sites, six im-portant pharmaceutical protein−protein targets are analyzed in thisstudy. Furthermore, the bound molecular probes identified in thehot spot search are compared directly with known drugs for thosetargets, showing the potential for creating novel drugs by chemicallyconnecting the probes.
ResultsMost of the potential PPI targets for drug design do not have aknown complex structure. The power of the integrated approachintroduced in this work is that it does not require a known com-plex structure for the identification of the druggable interface of aprotein. To validate this integrated approach, the druggable in-terface is determined for six different, well-studied PPIs that arerelated to several diseases. For comparison purposes, this analysisis restricted to PPI systems for which the oligomeric structure isexperimentally solved and there exists sufficient amino acid se-quence diversity (>1,000 sequences for each family of proteins).The systems are the HIV-1 protease homodimer, human murinedouble minute 2 homolog (MDM2) homodimer, tumor necrosisfactor-alpha (TNF-α) homotrimer, cyclin-dependent kinase1(CDK1)−cyclin-dependent kinases regulatory subunit 1(CKS1),histone deacetylate 1(HDAC1)–metastasis-associated pro-tein MTA1 and B-cell lymphoma 2 (Bcl-2)−Bcl-2 homologousantagonist/killer (BAK) (described in SI Appendix).
Identification of a Druggable Interface for HIV-1 Protease Homodimer.HIV-1 protease is a symmetrical homodimer, which plays an essentialrole in the proper assembly and maturation of infectious virionsduring the HIV life cycle (33, 34). A common strategy for designingdrugs to block the spread of HIV is to target its dimerization in-terface, thereby preventing the assembly of the inactive monomersinto an active homodimer (34). In this case, however, there are twostructural interfaces: a terminal interface and an active site interface,which are both involved in HIV-1 protease dimerization (35).The molecular screening of the unliganded HIV-1 protease
monomer structure [Protein Data Bank (PDB) ID 3PHV] yieldedtwo relatively large, candidate binding sites, which are denoted assite 1 and site 2 (Fig. 1A, Middle). Site 1 covers a large portion ofthe protein surface, including the flap domain, active site, and theterminal domain (Fig. 1A). The smaller site 2 is mainly located onthe core domain of the protein. Accordingly, dimerization of HIV-1protease could occur between two monomers at different interfacialcombinations, e.g., site 1 to site 1, site 1 to site 2, or site 2 to site 2,respectively. Assessing the coevolutionary interface interactionscore, SInterfaceAB (Eq. 1), for each of the combinations of candidatebinding sites (Fig. 1A, Right) indicates that the strongest coevolutionoccurs in site 1 to site 1. This is in agreement with the crystalstructure of the HIV-1 protease homodimer (PDB ID 3R0Y). Theresidue composition of the site 1 is shown in SI Appendix, Table S1.Furthermore, homodimeric systems such as HIV-1 protease contain
E8052 | www.pnas.org/cgi/doi/10.1073/pnas.1615932113 Bai et al.

intramonomer as well as intermonomer residue pairs that coevolve.If one were to focus on coevolution exclusively between residuesthat lead to dimerization (i.e., exclude intramonomer coevolution),the contrast between the interaction score for site 1 to site 1 and theother combinations is more significant (as shown in SI Appendix,Fig. S2). Fig. 1B shows the predicted top 50 coevolving pairs ofintermonomer residues across the obtained binding sites of theHIV-1 protease homodimer, i.e., site 1 to site 1. The top coevolvingpairs of residues are consistent with the dimerization contacts ob-served in the homodimer of HIV-1 protease (PDB ID 3R0Y).These coevolving residue pairs include the flap domains, which arerelatively flexible and act as a couple of gatekeepers to controlsubstrate or ligand access to the active site.
A Strategy for Drug Design: HIV-1 Protease Homodimer. A combi-nation of the fragment (probe) binding and coevolutionary analysisindicates that site 1 is the best target for the design of inhibitory drugs.The small molecules in site 1 (i.e., “Probe” cluster 1) could potentiallybe used to create a drug molecule through chemical linking. To testthe plausibility of such a strategy, the probes in cluster 1 are comparedwith the known HIV-1 protease inhibitors (drugs) (SI Appendix, Fig.S3). So far, almost all of the inhibitors have been designed to targetagainst the formation of the active site interface (site 1/site 1). Fig. 1Cshows the binding of two example inhibitors on site 1: Tipranavir(from PDB ID 3SPK) and tripeptide (from PDB ID 1A30).Probe cluster 1 (site 1) contained the bound conformations
of the 30 molecular probe types with the exception of tyrosine
(Fig. 1A, Left). By superimposing the native binding conforma-tion of Tipranavir and tripeptide on Probe cluster 1 (Fig. 2), it isobserved that the bound probes are consistent with the moietiesof the drug molecules, i.e., benzene, phenylalanine, acetone, anddimethyl ether are found on the corresponding binding sites. Theresults imply that the integrated approach is sufficiently robust todetect druggable sites and identify appropriate molecular frag-ments that can bind to those sites. These fragments can be usedto assemble drugs according to combinatorial principles by linkingor decorating a given scaffold. It should be noted that, althoughthe probe types are consistent with the side chains of Tipranavirand tripeptide, they appear bound in several additional confor-mations not observed in the PDB structures of 3SPK and 1A30;this can be attributed to the lack of chemical bonds (e.g., abackbone) linking the fragments, which would act as an addi-tional constraint in the free energy minimization. Additionally,similar results are obtained from the superimposition of theHIV-1 protease substrate p2/NC on the Probe cluster 1 (SI Ap-pendix, Fig. S4). These results open the door for designing sub-strate-competitive peptide drugs by mutating the residues of thesubstrate to the ones that are predicted by fragment docking tohave stronger binding affinities.Although the binding sites of Tipranavir and a tripeptide are
located on site 1, additional hot spot regions that are adjacent onsite 1 may also be potential drug targets (Fig. 1C and SI Appendix,Fig. S3). These regions have not yet been targeted for drug design.One region is located on the terminal domain and comprises the
Fig. 1. Predicting the druggable interface for HIV-1 protease homodimer. (A) Two candidate binding sites (Middle, site 1 and site 2) are mapped out on thesurface of the HIV-1 protease monomer structure by spatially clustering the hot spots where multiple probe types (Left, dark cyan sticks) are located. Theinterface interaction score (Eq. 1) is calculated to find which combination of binding sites forms the homodimeric interface (i.e., site 1 to site 1, site 2 tosite 2, site 1 to site 2) (Right). Eq. 1 quantifies the amount of coevolutionary information (in nat units) between the residues in one predicted binding siteand residues in another predicted binding site. The site 1 to site 1 interface is found to exhibit the strongest evolutionary signal of PPIs according to Eq. 1,and therefore is predicted to form the homodimeric interface (Right). (B) The amount of coevolution between pairs of residues is quantified by DCA usingthe DI metric (Eq. 2). (Left) The top 50 coevolving residue pairs (orange connecting sticks) are shown across site 1 and site 1 of two monomers. (Right)These top 50 coevolving residues are also plotted on a contact map as orange dots, where light magenta circles and light blue circles are the experimentalintermonomer and intramonomer contacts, respectively. (C) The drugs, Tipranavir (magenta) and a tripeptide inhibitor (orange), target site 1 to interferein the homodimerization of HIV-1 protease. The areas on site 1 shown in red dashed circles are potential drug binding regions that have not yet beenexplored in experiment.
Bai et al. PNAS | Published online November 29, 2016 | E8053
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
PNASPL
US

residues of R8, P9, L24, L25, N25, and T26. The other region islocated on the flap domain and comprises the residues of I47, V54,and P79. Further analysis of these adjacent regions would shedlight on their potential as drug targets.
Identifying the Druggable Interface for MDM2 Homodimer and TNF-αHomotrimer. Similar results were obtained for proteins MDM2and TNF-α (Fig. 3). Here, concise interpretations of the resultsfor each are presented with detailed description in SI Appendix.The MDM2/MDMX complex has been considered as a potentialtarget for cancer therapies (36). The structure of the unligandedMDM2 was screened using the 31 molecular fragments, re-vealing two candidate binding sites (Fig. 3A). The highest in-terface interaction score, SInterfaceAB (Eq. 1), was observed betweenthe homodimerization interface formed by site 1 of two monomers(SI Appendix, Fig. S5), and the residues on site 1 is listed in SI Ap-pendix, Table S1. The predicted homodimeric interface is in agree-ment with the crystal structure of the homodimer (PDB ID 3VBG).Additionally, the molecular probes are found to bind to the smalldruggable cavity formed by residues of Q71, Q72, H73, and V93(Fig. 3B and SI Appendix, Fig. S6). This predicted druggable cavity isdistinct from the binding site of the inhibitors shown in Fig. 3B, thusproviding a future direction for MDM2 inhibitor optimization.Biological agents targeting TNF-α, to promote disassembly of
its homotrimerization, have been licensed for treating a variety ofinflammatory conditions (37). A monomer structure was isolatedfrom the homotrimer complex of TNF-α (PDB ID 1TNF) as thestarting structure for the binding site search. Two main candidatebinding sites were found during the molecular screening (Fig. 3C),
Fig. 2. Bound molecular probes are consistent with the side chains ofknown drugs at the binding site. (A) Tipranavir (magenta) overlaps with theprobe molecules (dark cyan) of benzene, phenylalanine, acetone, dimethylether, and dimethyl ether. (B) Tripeptide inhibitor (orange) overlaps with theamino acid side chains (dark cyan) of leucine, aspartic acid, and glutamicacid. Two low-energy binding poses are selected for each probe molecule.
Fig. 3. Inferring druggable sites on the interfaces of MDM2 homodimer and TNF-α homotrimer. (A) (Middle) Two candidate binding sites obtained on thesurface of the monomer structure (Left, dark cyan sticks are bound probes). The site 1 to site 1 interface is favored by the coevolutionary interface interactionscore given by Eq. 1 (SI Appendix, Fig. S5). (Right) Plotted on a contact map are the top 50 coevolving residue pairs for MDM2 given by DI (Eq. 2) (orangecircles) alongside the experimental (PDB ID 3VBG) intermonomer (light magenta circles) and intramonomer (light blue circles) contacts. (B) The known MDM2homodimer stabilizer RO-2443 targets the obtained site 1 (green and cyan stickball models) and the potential binding site discovered is highlighted with a reddashed circle. (C) Two candidate binding sites are found on the surface of monomer structure of TNF-α (Middle) based the hot spot search. The interfaceformed by site 1 in both monomers exhibits the highest amount of interfacial coevolution using Eq. 1 (SI Appendix, Fig. S5). The top 50 coevolving residuepairs given by DI (Eq. 2) are plotted on a contact map (orange dots) with the intermonomer (magenta circles) and intramonomer (light blue circles) contactsfrom the experimental structure (PDB ID 1TNF). (D) The experimentally known destabilizer of TNF-α targets the predicted site 1 to interfere in the homotrimerformation (green stick model is a small molecule destabilizer, and yellow cartoon model is the peptide destabilizer M21) is shown. An additional region on site1 that is predicted to have potential for drug design (circled with red dash lines) is also shown.
E8054 | www.pnas.org/cgi/doi/10.1073/pnas.1615932113 Bai et al.

of which, coevolutionary analysis indicates that the TNF-α as-sembles via site 1 to site 1 (SI Appendix, Fig. S7). The residues onsite 1 are listed in SI Appendix, Table S1. Looking at where theprobes bind on TNF-α, it is observed that Probe cluster 1 corre-sponds to the binding target of both known inhibitors. However,these existing inhibitors only occupy approximately one-quarter ofthe identified druggable binding region on site 1 and prevent theformation of the trimer from the dimer (as shown in SI Appendix,Fig. S8). This suggests that more effective inhibitors may bedesigned that bridge the three discovered binding regions of site 1(Fig. 3D and SI Appendix, Fig. S9).
Elucidating a Druggable Interface for the CDK1−CKS1 Heterodimer.The CDK1–CKS1 complex is recruited to multiple coding re-gions in the genome and is necessary for the efficient expressionof a significant subset of genes (38). Recently, the CDK1–CKS1complex was crystallized with four CDK1–CKS1 heterodimers inthe asymmetric unit (39). This structure revealed that the C lobeof the CDK1 interacts with two CKS1 proteins at their N lobe.Overexpression of CDK1 or CKS1 is strongly associated withaggressive breast tumor, and studies suggest that inhibition ofCDK1 or CKS1 abrogation could be an effective treatmentstrategy for cancer (40, 41).Hot spot searches were separately performed on the isolated
structures of CDK1 and CKS1, obtained from the crystal struc-ture of the complex (PDB ID 4YC6; no monomeric structuresavailable). The results of these searches for CDK1 and CKS1 are
shown in Fig. 4A. Four different candidate binding sites withdiverse shapes and sizes were found on the surface of CDK1, andthree candidate binding sites were found on CKS1. The threecandidate binding sites on CKS1 almost span the entire surfaceof CKS1. Assessing the coevolutionary interface interactionscore, SInterfaceAB (Eq. 1), among the combinations of the candidatebinding sites between CDK1 and CKS1 reveals two highlycoevolving interfaces, i.e., site 1 of CDK1 to site 2 of CKS1 andsite 2 of CDK1 to site 1 of CKS1 (Fig. 4A, Right). These highlycoevolving interfaces suggest that CDK1 may interact with twoCKS1 at the two different binding sites (residues are shown in SIAppendix, Table S1), which is consistent with a previous struc-tural study (39). Remarkably, the interface interaction score forsite 2 of CDK1 to site 1 of CKS1 is much stronger than the onefor site 1 of CDK1 to site 2 of CKS1. The higher interactionscore suggests that the binding of CDK1 (site 2) to CKS1 (site 1)may be functionally more important. As shown in Fig. 4C, thecleft in site 2 of CDK1 is the ATP-binding site (active site), whichwas also shown to bind to the ATP competitive inhibitor (fromPDB ID 5HQ0). The top 50 coevolving interprotein residuepairs are plotted on a contact map for both interfaces (Fig. 4B),further illustrating the coevolution between residue pairs thatpreserve those interfaces.
Prediction of the Druggable Interface for the HDAC1–MTA1 Heterodimer.MTA1 has been regarded as a molecule that potentially plays animportant role in tumor invasion and metastasis by interacting with
Fig. 4. Identifying potential CDK1–CKS1 heterodimeric interfaces. (A) Four candidate binding sites are obtained on the surface of monomer structures ofCDK1 (Top Left) and three for CKS1 (Bottom Left). Two interfaces (site 2 of CDK1 to site 1 of CKS1 and site 1 of CDK1 to site 2 of CKS1) are highly coevolvingbased on the interface interaction score (Right). (B) Plotted on contact maps are the top 50 coevolving residue pairs between site 2 of CDK1 and site 1 of CKS1(dark red dots) against their native contacts (light blue dots) and the top 50 coevolving residue pairs between site 1 of CDK1 and site 2 of CKS1 (orange dots)against their native contacts (magenta dots) (Right). These contacts are drawn on the complex structure as dark red sticks and orange sticks (Left), re-spectively. (C) The known inhibitor targeted the obtained site 2 of CDK1 to interfere in the heterodimer formation (green stick model).
Bai et al. PNAS | Published online November 29, 2016 | E8055
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
PNASPL
US
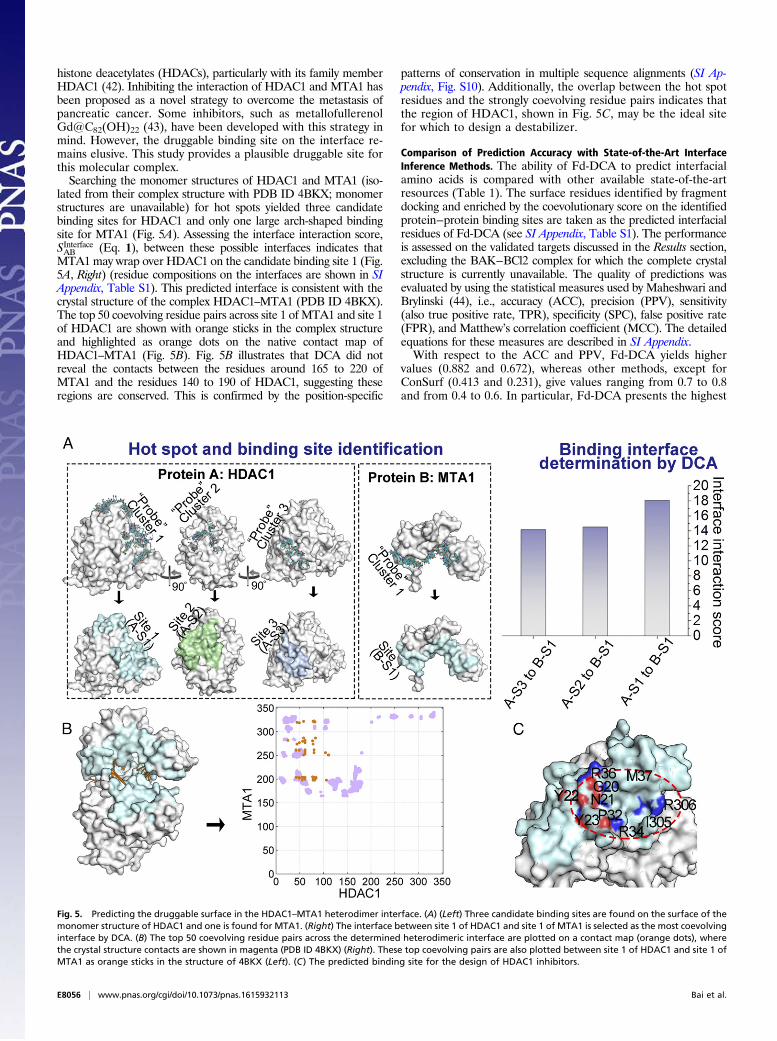
histone deacetylates (HDACs), particularly with its family memberHDAC1 (42). Inhibiting the interaction of HDAC1 and MTA1 hasbeen proposed as a novel strategy to overcome the metastasis ofpancreatic cancer. Some inhibitors, such as metallofullerenolGd@C82(OH)22 (43), have been developed with this strategy inmind. However, the druggable binding site on the interface re-mains elusive. This study provides a plausible druggable site forthis molecular complex.Searching the monomer structures of HDAC1 and MTA1 (iso-
lated from their complex structure with PDB ID 4BKX; monomerstructures are unavailable) for hot spots yielded three candidatebinding sites for HDAC1 and only one large arch-shaped bindingsite for MTA1 (Fig. 5A). Assessing the interface interaction score,SInterfaceAB (Eq. 1), between these possible interfaces indicates thatMTA1 may wrap over HDAC1 on the candidate binding site 1 (Fig.5A, Right) (residue compositions on the interfaces are shown in SIAppendix, Table S1). This predicted interface is consistent with thecrystal structure of the complex HDAC1–MTA1 (PDB ID 4BKX).The top 50 coevolving residue pairs across site 1 of MTA1 and site 1of HDAC1 are shown with orange sticks in the complex structureand highlighted as orange dots on the native contact map ofHDAC1–MTA1 (Fig. 5B). Fig. 5B illustrates that DCA did notreveal the contacts between the residues around 165 to 220 ofMTA1 and the residues 140 to 190 of HDAC1, suggesting theseregions are conserved. This is confirmed by the position-specific
patterns of conservation in multiple sequence alignments (SI Ap-pendix, Fig. S10). Additionally, the overlap between the hot spotresidues and the strongly coevolving residue pairs indicates thatthe region of HDAC1, shown in Fig. 5C, may be the ideal sitefor which to design a destabilizer.
Comparison of Prediction Accuracy with State-of-the-Art InterfaceInference Methods. The ability of Fd-DCA to predict interfacialamino acids is compared with other available state-of-the-artresources (Table 1). The surface residues identified by fragmentdocking and enriched by the coevolutionary score on the identifiedprotein−protein binding sites are taken as the predicted interfacialresidues of Fd-DCA (see SI Appendix, Table S1). The performanceis assessed on the validated targets discussed in the Results section,excluding the BAK−BCl2 complex for which the complete crystalstructure is currently unavailable. The quality of predictions wasevaluated by using the statistical measures used by Maheshwari andBrylinski (44), i.e., accuracy (ACC), precision (PPV), sensitivity(also true positive rate, TPR), specificity (SPC), false positive rate(FPR), and Matthew’s correlation coefficient (MCC). The detailedequations for these measures are described in SI Appendix.With respect to the ACC and PPV, Fd-DCA yields higher
values (0.882 and 0.672), whereas other methods, except forConSurf (0.413 and 0.231), give values ranging from 0.7 to 0.8and from 0.4 to 0.6. In particular, Fd-DCA presents the highest
Fig. 5. Predicting the druggable surface in the HDAC1–MTA1 heterodimer interface. (A) (Left) Three candidate binding sites are found on the surface of themonomer structure of HDAC1 and one is found for MTA1. (Right) The interface between site 1 of HDAC1 and site 1 of MTA1 is selected as the most coevolvinginterface by DCA. (B) The top 50 coevolving residue pairs across the determined heterodimeric interface are plotted on a contact map (orange dots), wherethe crystal structure contacts are shown in magenta (PDB ID 4BKX) (Right). These top coevolving pairs are also plotted between site 1 of HDAC1 and site 1 ofMTA1 as orange sticks in the structure of 4BKX (Left). (C) The predicted binding site for the design of HDAC1 inhibitors.
E8056 | www.pnas.org/cgi/doi/10.1073/pnas.1615932113 Bai et al.

sensitivity, with a TPR of 0.823. It should be noted that Fd-DCApresents a slightly inferior SPC and FPR compared with cons-PPISP, PIER, VORFFIR, eFindSitePPI, and InterProSurf. How-ever, PPV, TPR, SPC, and FPR only measure partial aspects ofperformance and do not provide a global picture of predictionperformance (45). To this end, MCC is a more balanced, com-prehensive measure that considers true/false positive and true/falsenegative aspects together, for which Fd-DCA largely outperformsall other methods, with a value of 0.838 versus 0.087 to 0.324. Insummary, the performance of Fd-DCA in predicting interfacialresidues compares favorably to other related approaches.
DiscussionFd-DCA is introduced to surmount the computational difficul-ties of finding druggable protein−protein interfaces. It combinesan accurate hot spot prediction methodology with clues fromevolutionary signatures of PPIs (i.e., coevolution) to discriminatebetween different hot spots. Hot spots that are found to coevolveare deduced to form the PPI interface, which are the most likelytargets for drug design. The incorporation of DCA is a key factorleading to the success of the proposed method, establishing a linkbetween candidate binding sites and PPI interfaces. It also showsthat identifying candidate binding sites by using fragment dockingis essential to discriminate true positive coevolutionary signalsfrom false positive ones (SI Appendix, Fig. S13), therefore im-proving the estimation of protein−protein interfacial residuepairs. Because DCA targets covariant regions, then, in some cases,this methodology could miss very highly conserved regions. Thepresent methodology is seen as a relevant complement to thetypical and straightforward analysis of conservation. This studyhighlights the importance of such coevolving regions, because theyare not picked up by methods that assume site independence.However, it would be valuable and straightforward to include a
metric of conservation in our score. Otherwise, additional clues,such as shape complementarity of the pair of sites, could also behelpful for binding site selection. Additionally, an important featureof this approach is that no structural information about the proteincomplex is necessary for the identification of a druggable interface.This is especially relevant because structural data only exist for asmall fraction of known PPIs.Fd-DCA can also be used to characterize the types of small mo-
lecular fragments and their binding orientations that can mimic thebinding interaction between the protein and its binding partners.Therefore, Fd-DCA can potentially be an important tool for the de-velopment of small-molecule or peptide agents for competitive bind-ing, complementing the experimental approaches to drug design. Inparticular, it is now possible to perform de novo drug design andcreate small molecules or peptides by following the fragment-baseddrug discovery concept (54); this entails expanding or linking thesefragment-sized probes with chemical bonds for small molecules, orpeptide bonds and alanine for peptides (SI Appendix, Fig. S14). Hence,the bound probes can act as core moieties to generate new drug leads.
MethodsThe computational method, Fd-DCA, developed in this study consists of threesteps (details in SI Appendix). (i) The in-house molecular docking method iFitDock(20) is used to globally explore the protein surface using 31 small-fragment-sizedprobe molecules (SI Appendix, Fig. S1). Hot spots are identified by spatiallyclustering the probes bound to the surface of the protein using the algorithmcalled Density-Based Spatial Clustering of Applications with Noise (DBSCAN)(55). (ii) Candidate binding sites on the surface of a protein are obtained byclustering hot spots using DBSCAN. (iii) A coevolutionary interface interactionscore SInterfaceAB (Eq. 1) is calculated for each of the pairwise combinations of thebinding sites between the two different proteins to find the evolutionarilyconserved binding interface(s). Considering two proteins that contain bindingsites A and B, respectively, SInterfaceAB is calculated as
SInterfaceAB =X
i∈Protein 1
Xj∈Protein 2
DIijδði∈AÞδðj∈BÞ, [1]
where
DIij =Xqk=1
Xql=1
PðDCAÞij ðk, lÞln
"PðDCAÞij ðk, lÞPiðkÞPjðlÞ
#. [2]
DIij is the Direct Information (DI) metric (24), which quantifies the amount ofcoevolutionary information (in nats) in the inferred DCA pair distributionPðDCAÞij between residues i and j. Eq. 1 sums the DI terms for all pairs of res-
idues, i and j, where i belongs to binding site A [i.e., δði∈AÞ] and j belongs tobinding site B [i.e., δði∈BÞ]. Furthermore, each position in a protein, i or j,has q = 21 possible states, representing the 20 amino acids and multiplesequence alignment gap.
ACKNOWLEDGMENTS. Work at the Center for Theoretical Biological Physicswas sponsored by the National Science Foundation (Grants PHY-1427654,CHE-1614101, and MCB-1241332) and by the Cancer Prevention and Re-search Institute of Texas (Grant R1110). F.B. was partially supported by WelchFoundation Grant C-1792. H.J. was supported by the National Basic ResearchProgram of China (Grant 2015CB910304) and the National Natural ScienceFoundation of China (Grants 21210003, 81230076, and 91313000).
1. Jubb H, Higueruelo AP, Winter A, Blundell TL (2012) Structural biology and drug
discovery for protein−protein interactions. Trends Pharmacol Sci 33(5):241–248.2. Ivanov AA, Khuri FR, Fu H (2013) Targeting protein−protein interactions as an anti-
cancer strategy. Trends Pharmacol Sci 34(7):393–400.3. Zinzalla G, Thurston DE (2009) Targeting protein−protein interactions for therapeutic
intervention: A challenge for the future. Future Med Chem 1(1):65–93.4. Keshava Prasad TS, et al. (2009) Human Protein Reference Database—2009 update.
Nucleic Acids Res 37(Database issue):D767–D772.5. Watkins AM, Arora PS (2015) Structure-based inhibition of protein−protein interac-
tions. Eur J Med Chem 94:480–488.6. Jin L, Wang W, Fang G (2014) Targeting protein-protein interaction by small mole-
cules. Annu Rev Pharmacol Toxicol 54(1):435–456.7. Xue LC, Dobbs D, Bonvin AMJJ, Honavar V (2015) Computational prediction of protein
interfaces: A review of data driven methods. FEBS Lett 589(23):3516–3526.8. Jordan RA, El-Manzalawy Y, Dobbs D, Honavar V (2012) Predicting protein−protein in-
terface residues using local surface structural similarity. BMC Bioinformatics 13(1):41.
9. Shoemaker BA, et al. (2010) Inferred Biomolecular Interaction Server—A web server
to analyze and predict protein interacting partners and binding sites. Nucleic Acids
Res 38(Database issue):D518–D524.10. Ezkurdia I, et al. (2009) Progress and challenges in predicting protein−protein in-
teraction sites. Brief Bioinform 10(3):233–246.11. Halabi N, Rivoire O, Leibler S, Ranganathan R (2009) Protein sectors: Evolutionary
units of three-dimensional structure. Cell 138(4):774–786.12. dos Santos RN, Morcos F, Jana B, Andricopulo AD, Onuchic JN (2015) Dimeric interactions
and complex formation using direct coevolutionary couplings. Sci Rep 5:13652.13. Hopf TA, et al. (2014) Sequence co-evolution gives 3D contacts and structures of
protein complexes. eLife 3:e03430.14. Vakser IA (2014) Protein-protein docking: From interaction to interactome. Biophys J
107(8):1785–1793.15. Noel J, Morcos F, Onuchic J (2016) Sequence co-evolutionary information is a natural
partner to minimally-frustrated models of biomolecular dynamics. F1000Res 5(F1000
Faculty Rev):106.
Table 1. Comparison of the performance of Fd-DCA with eightweb servers for the prediction of protein interface residues
Method ACC PPV TPR SPC FPR MCC
cons-PPISP (46) 0.794 0.493 0.269 0.929 0.071 0.253ConSurf (47) 0.413 0.231 0.807 0.312 0.688 0.087PIER (48) 0.796 0.500 0.092 0.976 0.024 0.145PredUs (49) 0.728 0.402 0.683 0.739 0.261 0.324SPPIDER (50) 0.721 0.359 0.466 0.787 0.213 0.207VORFFIR (51) 0.783 0.451 0.297 0.907 0.093 0.236eFindSitePPI (52) 0.775 0.415 0.245 0.911 0.089 0.187InterProSurf (53) 0.802 0.561 0.149 0.970 0.030 0.214Fd-DCA 0.882 0.672 0.823 0.897 0.103 0.838
cons-PPISP, consensus protein–protein interaction site predictor; ConSurf,conservation surface-mapping; InterProSurf, protein–protein interaction server;PIER, protein interface recognition for structural proteomics; SPPIDER, solventaccessibility-based protein–protein interaction sites identification and recogni-tion; VORFFIR, Voronoi random forest feedback interface predictor.
Bai et al. PNAS | Published online November 29, 2016 | E8057
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
PNASPL
US

16. Morrow JK, Zhang S (2012) Computational prediction of protein hot spot residues.Curr Pharm Des 18(9):1255–1265.
17. Moreira IS, Fernandes PA, Ramos MJ (2007) Hot spots—A review of the protein−proteininterface determinant amino-acid residues. Proteins 68(4):803–812.
18. Kozakov D, et al. (2011) Structural conservation of druggable hot spots in protein−proteininterfaces. Proc Natl Acad Sci USA 108(33):13528–13533.
19. Dennis S, Kortvelyesi T, Vajda S (2002) Computational mapping identifies the bindingsites of organic solvents on proteins. Proc Natl Acad Sci USA 99(7):4290–4295.
20. Bai F, et al. (2013) Free energy landscape for the binding process of Huperzine A toacetylcholinesterase. Proc Natl Acad Sci USA 110(11):4273–4278.
21. Lovell SC, Robertson DL (2010) An integrated view of molecular coevolution in pro-tein−protein interactions. Mol Biol Evol 27(11):2567–2575.
22. Tamir S, et al. (2014) Integrated strategy reveals the protein interface between cancertargets Bcl-2 and NAF-1. Proc Natl Acad Sci USA 111(14):5177–5182.
23. Pareek CS, Smoczynski R, Tretyn A (2011) Sequencing technologies and genome se-quencing. J Appl Genet 52(4):413–435.
24. Morcos F, et al. (2011) Direct-coupling analysis of residue coevolution captures nativecontacts across many protein families. Proc Natl Acad Sci USA 108(49):E1293–E1301.
25. Weigt M, White RA, Szurmant H, Hoch JA, Hwa T (2009) Identification of direct res-idue contacts in protein−protein interaction by message passing. Proc Natl Acad SciUSA 106(1):67–72.
26. Ekeberg M, Lövkvist C, Lan Y, Weigt M, Aurell E (2013) Improved contact prediction inproteins: Using pseudolikelihoods to infer Potts models. Phys Rev E Stat Nonlin SoftMatter Phys 87(1):012707.
27. Sułkowska JI, Morcos F, Weigt M, Hwa T, Onuchic JN (2012) Genomics-aided structureprediction. Proc Natl Acad Sci USA 109(26):10340–10345.
28. Schug A, Weigt M, Onuchic JN, Hwa T, Szurmant H (2009) High-resolution proteincomplexes from integrating genomic information with molecular simulation. ProcNatl Acad Sci USA 106(52):22124–22129.
29. Cheng RR, Morcos F, Levine H, Onuchic JN (2014) Toward rationally redesigningbacterial two-component signaling systems using coevolutionary information. ProcNatl Acad Sci USA 111(5):E563–E571.
30. Morcos F, Jana B, Hwa T, Onuchic JN (2013) Coevolutionary signals across proteinlineages help capture multiple protein conformations. Proc Natl Acad Sci USA 110(51):20533–20538.
31. Cheng RR, et al. (2016) Connecting the sequence-space of bacterial signaling proteinsto phenotypes using coevolutionary landscapes. Mol Biol Evol 33(12):3054–3064.
32. Bai F, et al. (2015) The Fe-S cluster-containing NEET proteins mitoNEET and NAF-1 aschemotherapeutic targets in breast cancer. Proc Natl Acad Sci USA 112(12):3698–3703.
33. Brik A, Wong C-H (2003) HIV-1 protease: Mechanism and drug discovery. Org BiomolChem 1(1):5–14.
34. Boggetto N, Reboud-Ravaux M (2002) Dimerization inhibitors of HIV-1 protease. BiolChem 383(9):1321–1324.
35. Hayashi H, et al. (2014) Dimerization of HIV-1 protease occurs through two stepsrelating to the mechanism of protease dimerization inhibition by Darunavir. Proc NatlAcad Sci USA 111(33):12234–12239.
36. Graves B, et al. (2012) Activation of the p53 pathway by small-molecule-induced
MDM2 and MDMX dimerization. Proc Natl Acad Sci USA 109(29):11788–11793.37. Brazil M (2006) TNFR superfamily trimers. Nat Rev Drug Discov 5(1):20.38. Yu VPCC, Baskerville C, Grünenfelder B, Reed SI (2005) A kinase-independent function
of Cks1 and Cdk1 in regulation of transcription. Mol Cell 17(1):145–151.39. Brown NR, et al. (2015) CDK1 structures reveal conserved and unique features of the
essential cell cycle CDK. Nat Commun 6:6769.40. Westbrook L, et al. (2007) Cks1 regulates cdk1 expression: A novel role during mitotic
entry in breast cancer cells. Cancer Res 67(Suppl 23):11393–11401.41. Goga A, Yang D, Tward AD, Morgan DO, Bishop JM (2007) Inhibition of CDK1 as a
potential therapy for tumors over-expressing MYC. Nat Med 13(7):820–827.42. Santoro F, et al. (2013) A dual role for Hdac1: Oncosuppressor in tumorigenesis, on-
cogene in tumor maintenance. Blood 121(17):3459–3468.43. Pan Y, et al. (2015) Gd-metallofullerenol nanomaterial suppresses pancreatic cancer
metastasis by inhibiting the interaction of histone deacetylase 1 and metastasis-associated
protein 1. ACS Nano 9(7):6826–6836.44. Maheshwari S, Brylinski M (2015) Predicting protein interface residues using easily
accessible on-line resources. Brief Bioinform 16(6):1025–1034.45. Vihinen M (2012) How to evaluate performance of prediction methods? Measures
and their interpretation in variation effect analysis. BMC Genomics 13(Suppl 4):S2.46. Chen H, Zhou H-X (2005) Prediction of interface residues in protein−protein com-
plexes by a consensus neural network method: Test against NMR data. Proteins 61(1):
21–35.47. Ashkenazy H, et al. (2016) ConSurf 2016: An improved methodology to estimate and
visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44(W1):
W344–W350.48. Kufareva I, Budagyan L, Raush E, Totrov M, Abagyan R (2007) PIER: Protein Interface
Recognition for structural proteomics. Proteins 67(2):400–417.49. Zhang QC, et al. (2011) PredUs: A web server for predicting protein interfaces using
structural neighbors. Nucleic Acids Res 39(Web Server issue):W283–W287.50. Porollo A, Meller J (2007) Prediction-based fingerprints of protein−protein interac-
tions. Proteins 66(3):630–645.51. Segura J, Jones PF, Fernandez-Fuentes N (2011) Improving the prediction of protein
binding sites by combining heterogeneous data and Voronoi diagrams. BMC
Bioinformatics 12(1):352.52. Maheshwari S, Brylinski M (2015) Prediction of protein−protein interaction sites from
weakly homologous template structures using meta-threading and machine learning.
J Mol Recognit 28(1):35–48.53. Negi SS, Schein CH, Oezguen N, Power TD, Braun W (2007) InterProSurf: A web server
for predicting interacting sites on protein surfaces. Bioinformatics 23(24):3397–3399.54. Scott DE, Coyne AG, Hudson SA, Abell C (2012) Fragment-based approaches in drug
discovery and chemical biology. Biochemistry 51(25):4990–5003.55. Ester M, Kriegel H-p, Jörg S, Xu X (1996) A density-based algorithm for discovering
clusters in large spatial databases with noise. KDD 96(34):226–231.
E8058 | www.pnas.org/cgi/doi/10.1073/pnas.1615932113 Bai et al.











![Improving Protein Docking Using Sustainable Genetic …jianjunh/paper/autodockx.pdfImproving Protein Docking Using Sustainable Genetic Algorithms [40]. However, it does not guarantee](https://static.fdocuments.in/doc/165x107/5f0497177e708231d40eb93d/improving-protein-docking-using-sustainable-genetic-jianjunhpaper-improving-protein.jpg)






![Docking interactions in protein kinase and phosphatase ...interacting protein–protein motifs for MAP kinases and tyrosine phosphatases [12,13]. Docking interactions in protein phosphatases](https://static.fdocuments.in/doc/165x107/60ee63efe2bdd8639d7712a5/docking-interactions-in-protein-kinase-and-phosphatase-interacting-proteinaprotein.jpg)