
Electro- and photo-chemistry of rhenium and rhodium ... · ide from carbon dioxide, rhenium...
14
134 ice | science Nanomaterials and Energy Volume 2 Issue NME3 Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini review Portenkirchner, Oppelt, Egbe, Knör and Sariçiftçi Pages 134–147 http://dx.doi.org/10.1680/nme.13.00004 Themed Issue Review Paper Received 12/02/2013 Accepted 05/04/2013 Published online 06/04/2013 Keywords: catalyst/energy conversion/photocatalysts/solar fuel generation ICE Publishing: All rights reserved 1. Introduction Hydrocarbons are currently considered as the most versatile energy carriers available for the growing fuel demand of mankind. Due to several advantageous properties such as high-energy density, trans- portation and storage properties, this situation will certainly also continue for the next decades. 1 However, the ongoing combustion of fossil fuel resources leads to many environmental impacts including the problem of global warming. 2–4 Moreover, the con- ventional hydrocarbon-based fuel production chain gradually faces difficulties to keep pace with rising demand and prices. 5 As an alternative approach, a sustainable production chain for fuels should be established, where combustible chemical compounds are directly generated by utilizing sunlight as the primary source of energy. In order to obtain synthetic hydrocarbons as solar fuels, the reduction of carbon dioxide and the supply of reducing equivalents based on water as an electron donor supplier are highly desirable starting points. 6–9 In this context, mixtures of carbon dioxide and hydrogen are very attractive, since they can be used in various reactors that are designed similar to the conversion of syngas (i.e. a mixture of carbon monoxide and hydrogen) into carbon-based fuels following the long-established Fischer-Tropsch process. 10–12 Besides hydrocarbons, the solar-driven production of methanol starting from carbon dioxide and water as abundant resources could provide a viable solution for solving the problem of diminishing fos- sil fuels. 13 An interesting achievement in this direction was reported recently by Delacourt et al., 14 who described syngas conversion into Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini review Engelbert Portenkirchner DI (FH)* Linz Institute for Organic Solar Cells (LIOS), Physical Chemistry, Johannes Kepler University Linz, Linz, Austria Kerstin Oppelt DI Institute of Inorganic Chemistry, Center for Nanobionics and Photochemical Sciences (CNPS), Johannes Kepler University Linz, Linz, Austria Daniel A. M. Egbe PD Dr.rer.nat. habil. Professor, Linz Institute for Organic Solar Cells (LIOS), Physical Chemistry, Johannes Kepler University Linz, Linz, Austria Günther Knör Univ.-Prof. Dr. Professor, Institute of Inorganic Chemistry, Center for Nanobionics and Photochemical Sciences (CNPS), Johannes Kepler University Linz, Linz, Austria Niyazi Serdar Sariçiftçi o.Univ. Prof. Mag. Dr. DDr. h.c. Professor, Linz Institute for Organic Solar Cells (LIOS), Physical Chemistry, Johannes Kepler University Linz, Linz, Austria Rhenium and rhodium complexes with bipyridyl ligands have been proven to be efficient homogeneous catalysts in the field of carbon dioxide and proton reduction. In this work, the authors provide several examples of these compounds with modified ligand structures and discuss their electro- and photo-catalytic capabilities toward carbon dioxide reduction and NAD + cofactor regeneration. The electrocatalysis is studied by cyclic voltammetry and controlled potential electrolysis for determining the over potentials, Faradaic efficiencies and reaction rate constants. In addition, the photophysics of these compounds is discussed based on UV-visible absorption, photoluminescence and infrared absorption spectroscopy. Results on comparing two different rhenium catalysts for homogeneous photocatalytic carbon dioxide reduction using a sacrificial electron donor are reported. 1 2 3 4 5 *Corresponding author e-mail address: [email protected] 1 2 3 4 5
Transcript of Electro- and photo-chemistry of rhenium and rhodium ... · ide from carbon dioxide, rhenium...

134
ice | science
Nanomaterials and EnergyVolume 2 Issue NME3
Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini reviewPortenkirchner, Oppelt, Egbe, Knör and Sariçiftçi
Pages 134–147 http://dx.doi.org/10.1680/nme.13.00004Themed Issue Review PaperReceived 12/02/2013 Accepted 05/04/2013Published online 06/04/2013Keywords: catalyst/energy conversion/photocatalysts/solar fuel generation
ICE Publishing: All rights reserved
1. IntroductionHydrocarbons are currently considered as the most versatile energy carriers available for the growing fuel demand of mankind. Due to several advantageous properties such as high-energy density, trans-portation and storage properties, this situation will certainly also continue for the next decades.1 However, the ongoing combustion of fossil fuel resources leads to many environmental impacts including the problem of global warming.2–4 Moreover, the con-ventional hydrocarbon-based fuel production chain gradually faces difficulties to keep pace with rising demand and prices.5
As an alternative approach, a sustainable production chain for fuels should be established, where combustible chemical compounds are directly generated by utilizing sunlight as the primary source of
energy. In order to obtain synthetic hydrocarbons as solar fuels, the reduction of carbon dioxide and the supply of reducing equivalents based on water as an electron donor supplier are highly desirable starting points.6–9 In this context, mixtures of carbon dioxide and hydrogen are very attractive, since they can be used in various reactors that are designed similar to the conversion of syngas (i.e. a mixture of carbon monoxide and hydrogen) into carbon-based fuels following the long-established Fischer-Tropsch process.10–12
Besides hydrocarbons, the solar-driven production of methanol starting from carbon dioxide and water as abundant resources could provide a viable solution for solving the problem of diminishing fos-sil fuels.13 An interesting achievement in this direction was reported recently by Delacourt et al.,14 who described syngas conversion into
Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini review
Engelbert Portenkirchner DI (FH)*Linz Institute for Organic Solar Cells (LIOS), Physical Chemistry, Johannes Kepler University Linz, Linz, Austria
Kerstin Oppelt DIInstitute of Inorganic Chemistry, Center for Nanobionics and Photochemical Sciences (CNPS), Johannes Kepler University Linz, Linz, Austria
Daniel A. M. Egbe PD Dr.rer.nat. habil.Professor, Linz Institute for Organic Solar Cells (LIOS), Physical Chemistry, Johannes Kepler University Linz, Linz, Austria
Günther Knör Univ.-Prof. Dr.Professor, Institute of Inorganic Chemistry, Center for Nanobionics and Photochemical Sciences (CNPS), Johannes Kepler University Linz, Linz, Austria
Niyazi Serdar Sariçiftçi o.Univ. Prof. Mag. Dr. DDr. h.c.Professor, Linz Institute for Organic Solar Cells (LIOS), Physical Chemistry, Johannes Kepler University Linz, Linz, Austria
Rhenium and rhodium complexes with bipyridyl ligands have been proven to be efficient homogeneous catalysts
in the field of carbon dioxide and proton reduction. In this work, the authors provide several examples of these
compounds with modified ligand structures and discuss their electro- and photo-catalytic capabilities toward carbon
dioxide reduction and NAD+ cofactor regeneration. The electrocatalysis is studied by cyclic voltammetry and controlled
potential electrolysis for determining the over potentials, Faradaic efficiencies and reaction rate constants. In addition,
the photophysics of these compounds is discussed based on UV-visible absorption, photoluminescence and infrared
absorption spectroscopy. Results on comparing two different rhenium catalysts for homogeneous photocatalytic
carbon dioxide reduction using a sacrificial electron donor are reported.
1
2
3
4
5
*Corresponding author e-mail address: [email protected]
1 2 3 4 5

Offprint provided courtesy of www.icevirtuallibrary.comAuthor copy for personal use, not for distribution
Nanomaterials and EnergyVolume 2 Issue NME3
Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini reviewPortenkirchner, Oppelt, Egbe, Knör and Sariçiftçi
135
methanol using an electrochemical cell similar to proton-exchange membrane fuel cells. This group achieved 45% energy efficiency at 10 mA/cm2, but unfortunately, the efficiency was decreasing with higher current densities (30% at 100 mA/cm2).
The homogeneous catalysis of carbon dioxide assimilating fuel generation based on water splitting represents another interesting approach in solar fuel research.15,16 These artificial photosynthetic systems are inspired by the overall function of the natural enzymatic reactions, where the industrially important role of hydrogen gas as a two-electron reductant for carbon dioxide fixation is replaced by organic redox cofactors, such as nicotinamide-adenine-dinucleotide (NADH), acting as hydride-transfer reagents. In all kinds of techni-cal scenarios following this direction, this leads to the fundamental problem of catalytic cofactor regeneration, that is, a reversible NAD+ to NADH conversion to supply electrons and protons for a multistep carbon dioxide conversion into energy-rich compounds.17–20
Despite of many current activities in this direction, the most challenging basic research need is to design new efficient and robust multielectron- and multiproton-transfer catalysts for the desired reactions. To lower the actual reduction potential of the carbon dioxide reduction process, suitable redox mediators are required.21–24 Concerning the generation of carbon monox-ide from carbon dioxide, rhenium complexes with bipyridine ligands are among the most frequently investigated candidates for homogeneous catalysis in terms of activities and lifetimes.23,25 Rhodium-based systems, on the other hand, have been success-fully established for catalyzing the recycling of redox cofactors such as nicotine amides. In this context, the combination of both rhenium and rhodium complexes for the catalytic production of syngas equivalents (carbon monoxide and hydrogen
or NADH)
powered by solar energy is an attractive new approach that we have started to follow recently.26,27 These energy-storing primary reactions could then be utilized to form hydrocarbons such as methane or methanol as more convenient types of solar fuels via the established processes.13
2. Catalyst materialsAt present most of the best studied catalysts are metal complexes with bipyridine ligands, where the catalyst center consists of transi-tion metals based on rhenium, rhodium or ruthenium. Despite their high-current efficiencies and high selectivity, problems in the field of artificial solar fuel production by these catalysts are manifold. Although these molecular catalyst compounds can be used to sta-bilize intermediate steps of the carbon dioxide reduction process and thus lower the required overpotential, achieving a simultaneous multiple electron and proton transfer is kinetically extremely diffi-cult to realize and over potentials of most reported catalyst systems are still significantly high. Furthermore, systems based on these catalyst materials, as reported up to now, suffer from low stability and low turnover frequency.
For photocatalysis, it is important to extend the absorption of these compounds in the visible region. One way to address this is to cova-lently bind the catalyst to a photosensitizer with high absorption in the visible region. One of the best results concerning quantum yield and turnover numbers (TON) was achieved by bridging a rhenium-based catalyst with a ruthenium-based photosensitizer reported by Ishitani and coworkers.24 Another promising approach is to combine catalysts with inorganic semiconductor materials. In a recent work, Kubiak et al. showed light-assisted cogenera-tion of carbon monoxide and hydrogen from carbon dioxide and H
2O in an acetonitrile/water mixture, exhibiting high Faradic effi-
ciency at low homogeneous catalyst concentration.28 Bocarsly and coworkers29 reported selective reduction of carbon dioxide to meth-anol by combining p-GaP semiconductor electrode and pyridine as catalyst material. This attempt of a kinetically difficult 6e− aqueous photoreduction of carbon dioxide to methanol allows astonishingly low reduction potentials below the standard reduction potential of −0.52 V versus saturated calomel electrode.
The materials investigated in this study are rhenium and rhodium diimine complexes with different ligand systems. The molecular struc-tures are shown below. Figure 1 shows the schematics of four rhenium(I) tricarbonyl chloride complexes with different diimine ligand systems, that is, (2,2ʹ-bipyridyl)Re(CO)
3Cl (1), (4,4ʹ-dicarboxyl-2,2ʹ-bipyridyl)
Re(CO)3Cl (2), (5,5ʹ-bisphenylethinyl-2,2ʹ-bipyridyl)Re(CO)
3Cl
Figure 1. Schematic chemical structures of four different rhenium
compounds (2,2ʹ-bipyridyl)Re(CO)3Cl (1), (4,4ʹ-dicarboxyl-2,2ʹ-
bipyridyl)Re(CO)3Cl (2) (5,5ʹ-bisphenylethinyl-2,2ʹ-bipyridyl)Re(CO)3Cl
(3) and [5,5ʹ-bis ((2,6-bis-octyloxy-4-formyl)phenylethinyl)-2,2ʹ-
bipyridyl] Re(CO)3Cl (4) for carbon dioxide reduction.
H
H
O O
O
OOO
O
O O
O OO
O
NN
N N
NN
CICI
(1)
(3)
(4)
(2)
ReRe
CIRe
OO
OO
O O
HH15C8O
H
OO
N N
CIRe

Offprint provided courtesy of www.icevirtuallibrary.comAuthor copy for personal use, not for distribution
Nanomaterials and EnergyVolume 2 Issue NME3
Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini reviewPortenkirchner, Oppelt, Egbe, Knör and Sariçiftçi
136
(3) and [5,5ʹ-bis ((2,6-bis-octyloxy-4-formyl)phenylethinyl)-2,2ʹ-bipyridyl]Re(CO)
3Cl (4).
The (2,2ʹ-bipyridyl) and (4,4ʹ-dicarboxyl-2,2ʹ-bipyridyl) ligands were purchased from commercial suppliers (Fluka, Aldrich). The multichromophoric diimine ligand (5,5ʹ-bisphenylethinyl-2,2ʹ-bipyridyl) was synthesized from 2,2ʹ-bipyridine according to published routes through the formation of 5,5ʹ-dibromo-2,2ʹ-bipyridine and additional palladium-catalyzed Sonogashira coupling of the dibromo compound with phenylacetylene.26,30–32
[5,5ʹ-bis((2,6-bis-octyloxy-4-formyl)phenylethinyl)-2,2ʹ-bipyridyl] was obtained via coupling of 5,5ʹ-dibromobipyridine with 1,5-dioctyloxy-4-ethinyl-benzaldehyde. Synthesis of the rhe-nium complexes was then achieved by further metallation with Re(CO)
5Cl in toluene as described in the literature.33–36
These compounds (1–4) have been further investigated for their capability of electro- and photo-catalytical carbon dioxide reduction.
Figure 2 shows two different rhodium compounds, namely, [Cp*(bpy)RhCl]Cl (5) and [Cp*(5,5ʹ-bis(phenylethinyl)-2,2-bpy)RhCl]Cl (6). Synthesis of the rhodium complex 6 was first done by Oppelt et al.26 via palladium-catalyzed Sonogashira coupling of 5,5ʹ-dibromobipyridine with phenyl acetylene and subsequent metallation with [Cp*RhCl
2]
2 in methanol/toluene.
Compounds 5 and 6 have been further investigated for the elec-tro- and photo-catalytic reduction of NAD+ to NADH. A more detailed description of the synthesis and characterization of these compounds (1–6) can be found in the literature.26,27
3. Electrocatalytic studiesMethods for electrochemical activation of carbon dioxide have long been studied, and significant progress has been made over the
past decade).7,23,25,37 The electrochemical reduction of carbon diox-ide usually requires a high potential of about –1.9 V versus normal hydrogen electrode (NHE) for a one-electron activation process. This high potential is partly explained by the necessary large inner-shell rearrangements upon electron transfer and the corresponding bending of the molecule upon one-electron activation resulting in an O-C-O angle of 133°.38,39 By performing the carbon dioxide reduction in a multielectron process, however, this transformation can be facilitated. As a consequence of this multielectron and atom transfer, the required reduction potentials are lowered significantly. This is reflected by the change in the equilibrium redox potentials between reactions 1 and 2 in Table 1.40 The equilibrium potential of the carbon dioxide reduction progressively shifts to more positive values as the number of electrons and protons participating in the reduction process increase, comparing different net reactions 3–8 in Table 1.
Although the equilibrium reduction potentials for a proton coupled multielectron carbon dioxide reduction appear to be tolerably low; in reality however, the actual reduction potentials on bare metal electrodes are much higher than the Nernst potential due to bar-rier induced over potentials. In order to decrease the actual redox potential needed for the carbon dioxide reduction process, suitable catalysts are required.
Several desirable products such as methanol and others are cur-rently industrially synthesized from syngas, a mixture of carbon monoxide and hydrogen. The electro- and photo-catalytic for-mation of syngas is therefore a desired path for the efficient and selective synthesis of various hydrocarbon products.45,46 In addi-tion, carbon dioxide reduction by a cascade of enzymatic reactions where the cofactor NADH is consumed, has been applied to obtain methanol, while at the same time, NADH is oxidized to NAD+.47
Figure 2. Schematic chemical structures of two different rhodium
compounds [Cp*(bpy)RhCl]Cl (5) and [Cp*(5,5ʹ-bis(phenylethinyl)-
2,2ʹ-bpy)RhCl]Cl (6) for the electro- and photo-catalytic reduction of
NAD+ to NADH.
(5)
(6)
N NCI-
CIRh+
N NCI-
CIRh+
Redox reaction E°/V
1 CO2 + e– → CO–
2−1.90
2 2CO2 + e– → CO + CO2
3
–−0.65
3 CO2 + 2H+ + 2e– → HCOOH −0.61
4 CO2 + 2H+ + 2e– → CO + H2O −0.53
5 CO2 + 4H+ + 4e– → C + 2H2O −0.20
6 CO2 + 4H+ + 4e– → HCHO + H2O −0.48
7 CO2 + 6H+ + 6e– → CH3OH + H2O −0.38
8 CO2 + 8H+ + 8e– → CH4 + 2H2O −0.24
*Values are a collection from Refs. 41–44.
Table 1. Redox potentials versus normal hydrogen electrode for one-
and multi-electron reduction of carbon dioxide including proton-
dependent reduction pathways in aqueous solution at pH 7.
Offprint provided courtesy of www.icevirtuallibrary.comAuthor copy for personal use, not for distribution
Nanomaterials and EnergyVolume 2 Issue NME3
Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini reviewPortenkirchner, Oppelt, Egbe, Knör and Sariçiftçi
137
NAD+ regeneration to NADH according to reaction 9 requires a potential of about −0.32 V versus NHE at pH 7.48
9. NAD H 2e NADH+ + −+ + →
10. 2H O 2NAD 2NADH 2H O2 + → + ++ +2
When compared with hydrogen production via water splitting, thermodynamically NADH regeneration is slightly favorable. According to reaction 10 a potential of −1.14 V versus NHE (at pH 7, 25°C and an activity of NAD+ of 1) is required to convert NAD+ back to NADH by electrolysis in aqueous solu-tion. This potential is slightly less compared with water elec-trolysis with a standard redox potential of −1.23 V versus NHE. Practically, however, for both processes, high overpotentials are necessary.49–51
4. Cyclic voltammetryCyclic voltammetry is a useful technique to determine the redox properties of the soluble compounds 1–6. It allows indirect studies about the catalytic activity for carbon dioxide reduction to carbon monoxide and NADH regeneration. In the presented studies, a one-compartment cell was used for cyclic voltammetry experiments, either with a platinum or glassy carbon working electrode, a plati-num counter electrode and a Ag/AgCl quasi reference electrode calibrated with ferrocene/ferrocenium (Fc/Fc+) as an internal ref-erence. For carbon dioxide reduction experiments, the cyclic vol-tammogram of the metal complexes under nitrogen saturation was compared with to the spectrum in the presence of carbon dioxide. Electrochemical experiments for carbon dioxide reduction were performed in acetonitrile with 0.1-M tetrabutylammonium hex-afluorophosphate (TBAPF
6) as supporting electrolyte. It was found
that purging of the system with nitrogen or carbon dioxide respec-tively, for about 15 min under stirring, is sufficient to achieve gas saturation of the electrolyte solution.
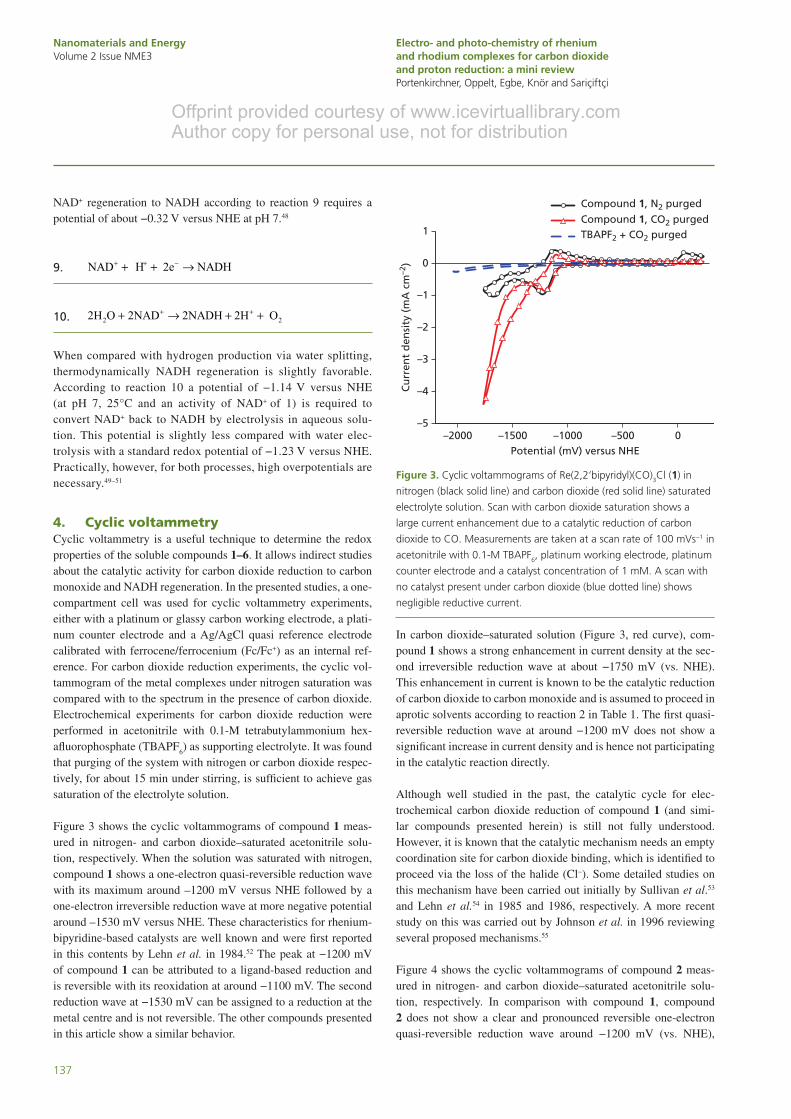
Figure 3 shows the cyclic voltammograms of compound 1 meas-ured in nitrogen- and carbon dioxide–saturated acetonitrile solu-tion, respectively. When the solution was saturated with nitrogen, compound 1 shows a one-electron quasi-reversible reduction wave with its maximum around –1200 mV versus NHE followed by a one-electron irreversible reduction wave at more negative potential around –1530 mV versus NHE. These characteristics for rhenium-bipyridine-based catalysts are well known and were first reported in this contents by Lehn et al. in 1984.52 The peak at −1200 mV of compound 1 can be attributed to a ligand-based reduction and is reversible with its reoxidation at around −1100 mV. The second reduction wave at −1530 mV can be assigned to a reduction at the metal centre and is not reversible. The other compounds presented in this article show a similar behavior.
In carbon dioxide–saturated solution (Figure 3, red curve), com-pound 1 shows a strong enhancement in current density at the sec-ond irreversible reduction wave at about −1750 mV (vs. NHE). This enhancement in current is known to be the catalytic reduction of carbon dioxide to carbon monoxide and is assumed to proceed in aprotic solvents according to reaction 2 in Table 1. The first quasi-reversible reduction wave at around −1200 mV does not show a significant increase in current density and is hence not participating in the catalytic reaction directly.
Although well studied in the past, the catalytic cycle for elec-trochemical carbon dioxide reduction of compound 1 (and simi-lar compounds presented herein) is still not fully understood. However, it is known that the catalytic mechanism needs an empty coordination site for carbon dioxide binding, which is identified to proceed via the loss of the halide (Cl−). Some detailed studies on this mechanism have been carried out initially by Sullivan et al.53 and Lehn et al.54 in 1985 and 1986, respectively. A more recent study on this was carried out by Johnson et al. in 1996 reviewing several proposed mechanisms.55
Figure 4 shows the cyclic voltammograms of compound 2 meas-ured in nitrogen- and carbon dioxide–saturated acetonitrile solu-tion, respectively. In comparison with compound 1, compound 2 does not show a clear and pronounced reversible one-electron quasi-reversible reduction wave around −1200 mV (vs. NHE),
Figure 3. Cyclic voltammograms of Re(2,2ʹbipyridyl)(CO)3Cl (1) in
nitrogen (black solid line) and carbon dioxide (red solid line) saturated
electrolyte solution. Scan with carbon dioxide saturation shows a
large current enhancement due to a catalytic reduction of carbon
dioxide to CO. Measurements are taken at a scan rate of 100 mVs−1 in
acetonitrile with 0.1-M TBAPF6, platinum working electrode, platinum
counter electrode and a catalyst concentration of 1 mM. A scan with
no catalyst present under carbon dioxide (blue dotted line) shows
negligible reductive current.
1
0
–1
–2
–3
–4
–5–2000 –1500 –1000
Potential (mV) versus NHE–500 0
Cu
rren
t d
ensi
ty (
mA
cm
–2)
Compound 1, N2 purgedCompound 1, CO2 purgedTBAPF2 + CO2 purged

Offprint provided courtesy of www.icevirtuallibrary.comAuthor copy for personal use, not for distribution
Nanomaterials and EnergyVolume 2 Issue NME3
Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini reviewPortenkirchner, Oppelt, Egbe, Knör and Sariçiftçi
138
although the onset is still observable. A nonreversible wave can be observed at around –1500 mV (vs. NHE), which can be attrib-uted to the additional carboxyl groups on the bipyridyl ligand. In contrast to previously published data,56 the modified compound 2 showed some catalytic behavior toward carbon dioxide reduction when the electrolyte solution was saturated with carbon dioxide, as can be seen in Figure 4, red curve. However, carbon dioxide poten-tiostatic bulk electrolysis at –2100 mV (vs. NHE) revealed that the compound seems to be unstable and loses its catalytic activity within several minutes of electrolysis time.
Figure 5 shows the cyclic voltammograms of compound 3 recorded under N2
− and carbon dioxide–saturated acetonitrile solution. In contrast to compound 1, compound 3 does not show a clear separation between different reductive waves. In addition, the onset of the reductive current occurred at a potential around −750 mV (vs. NHE) which is about 330 mV more positive com-pared with compound 1 (Figure 3). This significant differences between compound 1 and 3 can be attributed to the addition of the bisphenylethinyl groups at the 5,5ʹ position. When compound 3 was scanned repeatedly under N2
− saturated conditions to very negative potentials (i.e. –1600 mV vs. NHE), a violet film formed on the platinum working electrode. The origin of this film forma-tion and its potential toward carbon dioxide reduction is currently under investigation.
When the acetonitrile solution was saturated with carbon dioxide, compound 3 showed a strong enhancement in the second reduction wave current density. In relative comparison, a 6.5-fold increase at the second irreversible reduction wave under carbon dioxide at −1750 mV (vs. NHE; Figure 5) was observed. Comparing this with compound 1, the relative increase in current density is higher for compound 3. A more detailed study on compound 3 has been published elsewhere.27
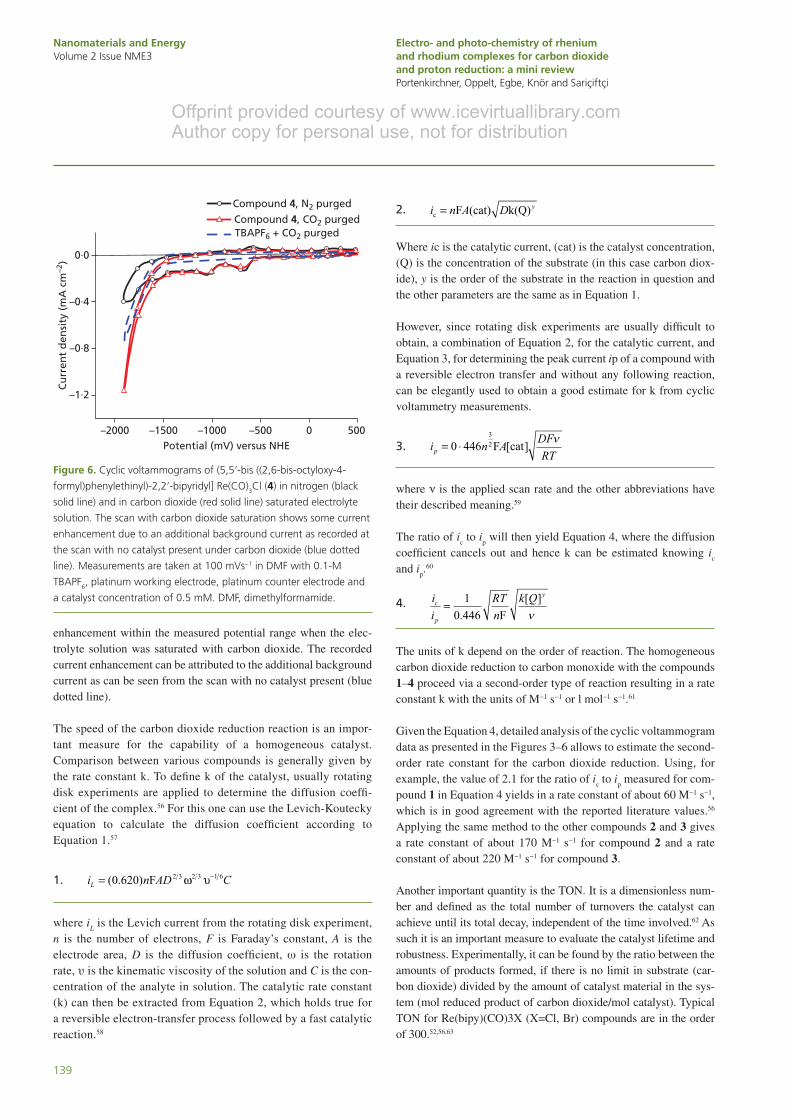
Different to the compound materials 1–3, compound 4 was poorly soluble in acetonitrile. Therefore, cyclic voltammograms were measured in dimethylformamide (DMF). Figure 6 shows the cor-responding cyclic voltammograms of compound 4 recorded in N2
− and carbon dioxide–saturated DMF solution. Due to the extended ligand of this compound, the redox characteristics are much more diverse compared with above presented measurements of the more simple compounds 1–3. Within the potential of 0 to −1800 mV (vs. NHE), four distinct reversible reduction peaks can be found with their half-wave potential (E
1/2) at −635, −1020, −1250 and −1500
mV (vs. NHE). Since it is generally known for rhenium compounds with bipyridine ligands that a metal-center-based reduction shows an irreversible behavior, these peaks might be attributed to a ligand-based reduction. Further studies would be necessary to fully clarify their true nature. Different to the other compounds 1–3, the cyclic voltammogram of compound 4 did not reveal catalytic current
Figure 4. Cyclic voltammograms of (4,4ʹ-dicarboxyl-2,2ʹ-bipyridyl)
Re(CO)3Cl (2) in nitrogen (black solid line) and carbon dioxide (red
solid line) saturated electrolyte solution. Scan with carbon dioxide
saturation shows a large current enhancement due to the catalytic
reduction of carbon dioxide to carbon monoxide. Measurements are
taken at a scan rate of 50 mVs-1 in acetonitrile with 0.1-M TBAPF6,
platinum working electrode, platinum counter electrode and a catalyst
concentration of 2 mM. A scan without catalyst under carbon dioxide
(blue dotted line) shows negligible reductive current.
0
–1
–2
–3
–4
–5
–6
–2000 –1500 –1000Potential (mV) versus NHE
–500 0
Cu
rren
t d
ensi
ty (
mA
cm
–2)
Compound 2, N2 purgedCompound 2, CO2 purgedTBAPF6 + CO2 purged
Figure 5. Cyclic voltammograms of Re(5,5ʹ-bisphenylethinyl-
2,2ʹbipyridyl)(CO)3Cl (3) in nitrogen (black solid line) and carbon
dioxide (red solid line) saturated electrolyte solution. The scan with
carbon dioxide saturation shows a large current enhancement due
to a catalytic reduction of carbon dioxide to carbon monoxide.
Measurements are taken at 100 mVs-1 in acetonitrile with 0.1-M
TBAPF6, platinum working electrode, platinum counter electrode
and a catalyst concentration of 1 mM. A scan without catalyst under
carbon dioxide (blue dotted line) shows negligible reductive current.
0·5
–3·0
–2·5
–2·0
–1·5
–1·0
–0·5
0·0
–2000 –1500 –1000Potential (mV) versus NHE
–500 0C
urr
ent
den
sity
(m
A c
m–2
)
Compound 3, N2 purged
Compound 3, CO2 purged
TBAPF6 + CO2 purged
Offprint provided courtesy of www.icevirtuallibrary.comAuthor copy for personal use, not for distribution
Nanomaterials and EnergyVolume 2 Issue NME3
Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini reviewPortenkirchner, Oppelt, Egbe, Knör and Sariçiftçi
139
enhancement within the measured potential range when the elec-trolyte solution was saturated with carbon dioxide. The recorded current enhancement can be attributed to the additional background current as can be seen from the scan with no catalyst present (blue dotted line).
The speed of the carbon dioxide reduction reaction is an impor-tant measure for the capability of a homogeneous catalyst. Comparison between various compounds is generally given by the rate constant k. To define k of the catalyst, usually rotating disk experiments are applied to determine the diffusion coeffi-cient of the complex.56 For this one can use the Levich-Koutecky equation to calculate the diffusion coefficient according to Equation 1.57
1. i n CL = −( . )0 620 2 3 2 3 1 6FAD ω υ
where iL is the Levich current from the rotating disk experiment,
n is the number of electrons, F is Faraday’s constant, A is the electrode area, D is the diffusion coefficient, ω is the rotation rate, υ is the kinematic viscosity of the solution and C is the con-centration of the analyte in solution. The catalytic rate constant (k) can then be extracted from Equation 2, which holds true for a reversible electron-transfer process followed by a fast catalytic reaction.58
2. i n D yc F (cat) k(Q)= A
Where ic is the catalytic current, (cat) is the catalyst concentration, (Q) is the concentration of the substrate (in this case carbon diox-ide), y is the order of the substrate in the reaction in question and the other parameters are the same as in Equation 1.
However, since rotating disk experiments are usually difficult to obtain, a combination of Equation 2, for the catalytic current, and Equation 3, for determining the peak current ip of a compound with a reversible electron transfer and without any following reaction, can be elegantly used to obtain a good estimate for k from cyclic voltammetry measurements.
3. i n A DFRTp = ⋅0 446
32F cat[ ] ν
where ν is the applied scan rate and the other abbreviations have their described meaning.59
The ratio of ic to i
p will then yield Equation 4, where the diffusion
coefficient cancels out and hence k can be estimated knowing ic
and ip.60
4.
ii
RTn
k Qc
p
y
= 10 446.
[ ]F ν
The units of k depend on the order of reaction. The homogeneous carbon dioxide reduction to carbon monoxide with the compounds 1–4 proceed via a second-order type of reaction resulting in a rate constant k with the units of M−1 s−1 or l mol−1 s−1.61
Given the Equation 4, detailed analysis of the cyclic voltammogram data as presented in the Figures 3–6 allows to estimate the second-order rate constant for the carbon dioxide reduction. Using, for example, the value of 2.1 for the ratio of i
c to i
p measured for com-
pound 1 in Equation 4 yields in a rate constant of about 60 M−1 s−1, which is in good agreement with the reported literature values.56 Applying the same method to the other compounds 2 and 3 gives a rate constant of about 170 M−1 s−1 for compound 2 and a rate constant of about 220 M−1 s−1 for compound 3.
Another important quantity is the TON. It is a dimensionless num-ber and defined as the total number of turnovers the catalyst can achieve until its total decay, independent of the time involved.62 As such it is an important measure to evaluate the catalyst lifetime and robustness. Experimentally, it can be found by the ratio between the amounts of products formed, if there is no limit in substrate (car-bon dioxide) divided by the amount of catalyst material in the sys-tem (mol reduced product of carbon dioxide/mol catalyst). Typical TON for Re(bipy)(CO)3X (X=Cl, Br) compounds are in the order of 300.52,56,63
Figure 6. Cyclic voltammograms of (5,5ʹ-bis ((2,6-bis-octyloxy-4-
formyl)phenylethinyl)-2,2ʹ-bipyridyl] Re(CO)3Cl (4) in nitrogen (black
solid line) and in carbon dioxide (red solid line) saturated electrolyte
solution. The scan with carbon dioxide saturation shows some current
enhancement due to an additional background current as recorded at
the scan with no catalyst present under carbon dioxide (blue dotted
line). Measurements are taken at 100 mVs−1 in DMF with 0.1-M
TBAPF6, platinum working electrode, platinum counter electrode and
a catalyst concentration of 0.5 mM. DMF, dimethylformamide.
–1·2
–0·8
–0·4
0·0
–2000 –1500 –1000Potential (mV) versus NHE
–500 0 500
Cu
rren
t d
ensi
ty (
mA
cm
–2)
Compound 4, N2 purgedCompound 4, CO2 purgedTBAPF6 + CO2 purged

Offprint provided courtesy of www.icevirtuallibrary.comAuthor copy for personal use, not for distribution
Nanomaterials and EnergyVolume 2 Issue NME3
Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini reviewPortenkirchner, Oppelt, Egbe, Knör and Sariçiftçi
140
Figure 7 shows a comparison of cyclic voltammograms and UV-vis absorption spectra between compounds 1 and 4. These measure-ments indicate the strong dependence of these compounds on the ligand system. For compound 4 the extended conjugated ligand results in a clear shift for the first reduction wave to a more posi-tive potential. As a consequence, also the UV-vis absorption maxi-mum (a metal-to-ligand charge-transfer [MLCT] band) is shifted to a longer wavelength as expected. This behavior is important to tune the properties of these metal-organic compounds to improve their capability for electro- and photo-catalytic carbon dioxide reduction.27,42,56
Figure 8 shows the comparison between cyclic voltammogram measurements of [Cp*(5,5ʹ-bis(phenylethinyl)-2,2ʹ-bpy)RhCl]Cl (6) with (red solid line) and without (black solid line) NAD+ pre-sent. When the systems contains NAD+, a large reductive current enhancement can be observed, starting at about –200 mV versus NHE with its peak maximum at about –350 mV versus NHE. This increase in current density is attributed to the catalytic reduction of NAD+ to NADH by the catalyst compound 6. The reduction of NAD+ is assumed to proceed according to reaction 9, which would require a potential of about –0.32 V versus NHE at pH 7. This is considerably close to the observed potential peak of about –350 mV versus NHE.48
5. Controlled potential electrolysisControlled potential electrolysis experiments were generally performed in a gas-tight one-compartment or H-cell with a platinum
working electrode, a platinum counter electrode and a Ag/AgCl quasi reference electrode. Although a one-compartment cell is easier to handle in terms of setup building and sealing, it has the big disadvan-tage that reduction products formed at the working electrode might
Figure 8. Cyclic voltammetry of Rh compound 6 with (black solid line)
and without (red solid line) NAD+ present.The scan with NAD+ in the
system shows a large current enhancement attributed to the catalytic
reduction of NAD+ to NADH. Voltammograms are taken at 50 mVs−1
in 50-mM potassium phosphate buffer at pH 7, glassy carbon working
electrode, Ag/AgCl/3-M NaCl reference electrode and a platinum wire
as counter electrode.
–0·2
–0·1
0·0
0·2
–1000 –800 –600Potential (mV) versus NHE
–200 0–400 200
0·1
Cu
rren
t d
ensi
ty (
mA
cm
–2)
Rh complex 6
Rh complex 6 + NAD+
Blank
Figure 7. Comparison of Re(2,2ʹbipyridyl)(CO)3Cl (1) and (5,5ʹ-bis
((2,6-bis-octyloxy-4-formyl)phenylethinyl)-2,2ʹ-bipyridyl] Re(CO)3Cl
(4) in (a) cyclic voltammograms and (b) UV-vis absorption spectra.
Voltammograms are taken at 100 mVs-1 in dimethylformamide
with 0.1-M TBAPF6, platinum working electrode, platinum counter
electrode and a catalyst concentration of 0.5 mM. UV-vis absorption
spectra are recorded in methanol.
–0·4
–0·3
–0·2
–0·1
0·0
0·1
0·2
–2000 –1500 –1000Potential (mV) versus NHE
–500 0 500
Cu
rren
t d
ensi
ty (
mA
cm
–2)
Compound 1, N2 purged(a) (b)
Compound 4, N2 purged
300 400 500Wavelength (nm)
600 700A
bso
rpti
on
(ar
bit
rary
un
it) Compound 1
Compound 4

Offprint provided courtesy of www.icevirtuallibrary.comAuthor copy for personal use, not for distribution
Nanomaterials and EnergyVolume 2 Issue NME3
Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini reviewPortenkirchner, Oppelt, Egbe, Knör and Sariçiftçi
141
be reoxidized on the counter electrode. This problem can be avoided using an H-cell. However, since the expected product with this type of catalysts is carbon monoxide and the solubility of carbon monoxide in a given solvent is typically in the order off 100 times less than for carbon dioxide, it is expected that little to no carbon monoxide will remain in solution. In fact, these experiments did not show any differ-ence concerning carbon monoxide yield between the one-compart-ment cell electrolysis experiment and the H-cell experiment.
Figure 9 shows a typical current density versus time plot for carbon dioxide electrolysis experiment with compound 3 as performed in a one-compartment cell. It can be seen that the reductive cur-rent initially dropped significantly and afterwards stayed constant with a small decay reflecting the declining concentration of carbon dioxide substrate in solution over the electrolysis time. In addition, a plot of current density versus 1/time1/2, as shown in Figure 9b, can give useful information on the kinetics of the process. For exam-ple, Fick’s second law of diffusion shows that a linearity in the current versus 1/time1/2 plot suggests both, a fast electron-transfer rate and a time-independent surface concentration of the reactant (in this case carbon dioxide) within the electrolysis time, compare Equation 5.59
5.
j n D c c
t
s
=
−F≠
12 0
12
Where j is the current density, n is the number of electrons, F is the Faraday constant, D is the diffusion coefficient, c0 is the bulk concentration, c
s is the electrode surface concentration and t is the
measurement time.
The slope of the line also allows calculating the diffusion coefficient D, if the bulk and electrode surface concentrations are known. The equation cannot be applied for very short time scales, since the initial concentration gradient is very high and the current as such is not limited by diffusion, but by the electron-transfer process, compare Figure 9(a).43
As a direct proof of the catalytic carbon dioxide reduction capability of the rhenium catalysts, headspace gas samples are taken and analyzed with regard to the carbon monoxide concentra-tion using gas chromatography (GC) and fourier transform infrared spectroscopy (FTIR) measurements. The application of transmis-sion IR gas measurements, compared with standard GC analysis, has several advantages. IR measurement has a very short measure-ment time, high reproducibility, works at ambient temperatures and pressures, shows no vulnerability to interact with a mobile or sta-tionary phase, and the gas sample will not come in contact with the detector system. Details to this method used for carbon monoxide measurements have been reported in a previous articles.27
The Faradaic efficiency (ηF) can then be calculated according to
Equation 6.
6. ηF
COgas COsol=× +( )2 n n
ne
Where nCO gas
is the number of carbon monoxide molecules in the gas phase, n
CO sol is the number of carbon monoxide molecules dis-
solved in solution and ne is the number of electrons put into the
system during the electrolysis experiment.
The number of molecules of carbon monoxide in the gas phase was obtained by GC and FTIR analysis, while the number of molecules
Figure 9. Typical current density versus time plot for potential static
carbon dioxide electrolysis experiment of Re(5,5ʹ-bisphenylethinyl-
2,2ʹbipyridyl)(CO)3Cl (3) at constant −1950 mV versus NHE, performed
in acetonitrile solution saturated with carbon dioxide and an
electrolysis time of 3000 s (a) and same data plotted as current density
versus 1/time1/2 (b). NHE, normal hydrogen electrode.
–0·4
–0·0
0·0 0·1
(b)
(a)
0·21/t1/2
0·40·3 0·5
–0·3–0·2–0·1
–0·5
Cu
rren
t d
ensi
ty (
mA
cm
–2)
–0·4
–0·0
0 500 1000Time (s)
20001500 2500 3000
–0·3–0·2–0·1
–0·5
Figure 10. Schematic energy diagram of the lowest-lying excited
states of complexes 1–4. Data taken from Ref. 66.
AbsorptionPhosphorescence
1MLCT*3MLCT*
IC
ICISC
1ππ*
3ππ*
G.S.

Offprint provided courtesy of www.icevirtuallibrary.comAuthor copy for personal use, not for distribution
Nanomaterials and EnergyVolume 2 Issue NME3
Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini reviewPortenkirchner, Oppelt, Egbe, Knör and Sariçiftçi
142
of carbon monoxide dissolved in the electrolyte solution was esti-mated using Henry’s law following Equation 7.
7. p = ⋅k cH
The Henry constant kH is taken with 2507 atm mol solvent per
mol carbon monoxide, derived from data of Castillo et al.,64 p is the partial pressure of the solute carbon monoxide and c is the concentration of carbon monoxide in solution. The number of electrons consumed in the carbon dioxide electrolysis was deter-mined by integration of the current–time curve of the electrolysis experiment.
With this approach, a Faradaic efficiency for the reduction of car-bon dioxide to carbon monoxide by the compound 3 of about 43% was calculated and ranks equally with the efficiencies that have been measured for compound 1.27 Literature values of reported Faradaic efficiencies of similar or modified compounds reach val-ues up to 100%.44,56,65 A control experiment with a pure platinum plate electrode under otherwise identical conditions did not yield detectable amounts of carbon monoxide.
It has been shown that under these conditions (ACN:TBAPF6) also
small amounts of formate and oxalate can be formed, however, with typical Faradaic efficiencies below 1%.44
6. Photophysics and photocatalysisThe photocatalytic efficiency of different diimino rhenium carbonyl complexes for the reduction of carbon dioxide to carbon monoxide is strongly dependent on the electronic structure and the redox properties of these compounds. As stated by Takeda et al.66 the low-est electronic excited states of bipyridine-based rhenium diimime
carbonyl complexes, mainly of 3MLCT* and 3ππ* character, are frequently quite close in energy and may also be mixed with each other. Focusing on photochemical carbon dioxide reduction, almost all applicable catalysts have in common, that their lowest excited state is of the 3MLCT* character with a relatively long lifetime in the 10–100 ns range.66 This state can be quenched reductively by suitable sacrificial electron donors such as triethanolamine (TEOA or TEA) to generate a one-electron-reduced (OER) species. These OER molecules can form adducts with carbon dioxide, the struc-ture of which is not yet fully clear. There are various propositions for this intermediate by different research groups.53,67,68 In most of the suggested catalytic pathways, one of the OER-carbon dioxide adducts reacts with a second OER radical in order to obtain carbon monoxide as a reaction product and retrieve the initial catalyst.66
The nature of the bipyridyl ligand influences the energetic levels of the possible electronic transitions upon excitation with UV and visible light. A simplified scheme of the most common electronic structure for similar rhenium carbonyl complexes is shown in Figure 10.66 The prototype compound Re(bpy)(CO)
3Cl (1) is taken
as a basic template for the following descriptions. Its UV-visible absorbance spectrum in toluene is shown in Figure 12 (black trace). It shows two distinct maxima at 299 and 400 nm, and the room temperature photoluminescence (red trace) at an excitation wave-length of 380 nm shows a broad emission from 450 to 750 nm with a maximum at 550 nm.
The absorbance spectra of the rhenium bipydridyl complexes 1–4 exhibit strong electronic 1ππ* intraligand transitions of the diimine ligands in the higher energetic region, usually at wavelengths shorter than 330 nm and MLCT signatures at lower energies.26,69,70 Additional weaker UV bands of IL origin can be observed for com-pounds 3 and 4 in the 300–320-nm spectral region. These bands
Compound SolventAbsorbance
λmax (nm)Emission λmax (nm)
1 Re(2,2ʹ-bpy)(CO)3Cl Toluene 299, 403 550
1 Re(2,2ʹ-bpy)(CO)3Cl Methanol 369
2 Re(4,4ʹ-dicarboxy-2,2ʹ-bpy)(CO)3Cl Methanol 394
3 Re[5,5ʹ-bis(phenylethinyl)-2,2ʹ-bpy](CO)3Cl Dichloromethane 245, 290, 390
442, 689
3 Re[5,5ʹ-bis(phenylethinyl)-2,2ʹ-bpy](CO)3Cl Acetonitrile 360
4 Re[5,5ʹ-bis((2,6-bis-octyloxy-4-formyl)phenylethinyl)-2,2ʹ-bpy](CO)3Cl Toluene 430 428, 695
4 Re[5,5ʹ-bis((2,6-bis-octyloxy-4-formyl)phenylethinyl)-2,2ʹ-bpy](CO)3Cl Dichloromethane 427
4 Re[5,5ʹ-bis((2,6-bis-octyloxy-4-formyl)phenylethinyl)-2,2ʹ-bpy](CO)3Cl Methanol 411
ACN, acetonitrile; DCM, dichloromethane; MeOH, methanol.
Table 2. Summary of photophysical data of rhenium tetracarbonyldiimino complexes 1–4.

Offprint provided courtesy of www.icevirtuallibrary.comAuthor copy for personal use, not for distribution
Nanomaterials and EnergyVolume 2 Issue NME3
Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini reviewPortenkirchner, Oppelt, Egbe, Knör and Sariçiftçi
143
suggest energetic splitting of approximately 2200 cm−1 due to coupling to phenylethinyl C^C vibrational modes.26 The absorb-ance and emission data in various solvents for compounds 1–4 are summarized in Table 2.
Besides substituent effects on the spectra, what is most remarkable and also well known about the absorbance of the described com-pounds is the fact that the 1MLCT transition is strongly dependent on solvent effects;33,34,36 this can also be seen in Figure 11 compar-ing the spectra of compound 3 in dichloromethane to the spectrum recorded in acetonitrile. In the more polar solvents such as metha-nol or acetonitrile, the lowest lying-1MLCT absorbance is shifted to
the red by 30 nm compared with less polar solvents such as toluene or dichloromethane.
Furthermore, the carboxyl substituted compound 2 shows a strong red shift of the MLCT of about 2770 cm−1 as compared with rhe-nium complex 1, due to the electron withdrawing effect of the carboxyl groups.
The photophysics of the rhenium complex 3 is dominated by the presence of the reducing rhenium(I) tricarbonyl chloride donor fragment, which leads to a luminescent lowest-energy triplet excited state of the MLCT type.26
Figure 11. UV-visible absorption spectra of the rheniumcarbonyl-
complexes 1–4 in different solvents. In more polar solvents, such as
methanol or ACN, the lowest-lying 1MLCT absorbance is shifted to the
red by 30 nm compared with less polar solvents such as toluene or
DCM. ACN, acetonitrile; DCM, dichloromethane.
300 400 500 600
Ab
sorp
tio
n (
arb
itia
ry u
nit
)
Wavelength (nm)
Compound 4, in MeOHCompound 3, in ACN
Compound 1, in MeOHCompound 2, in MeOHCompound 3, in DCM
Figure 12. UV-visible absorption(black) with two maxima at 299 and
403 nm and photoluminescence (red) spectra of rhenium carbonyl
complex 1 in toluene at room temperature and λexc. = 380 nm.
300 400 500Wavelength (nm)
700600
Ab
sorp
tio
n (
arb
itia
ry u
nit
)
Lum
ines
cen
ce (
arb
itia
ry u
nit
)
Figure 13. UV-visible absorption (black) and emission spectra
at λexc. = 388 nm (red) of rhenium carbonyl complex 3 in
dichloromethane at room temperature. The 3MLCT emission spectra
are indicated by a maximum at approximately 650 nm and additional
shoulders at 590 and 700 nm.
300 400 500Wavelength (nm)
700600
Ab
sorp
tio
n (
arb
itia
ry u
nit
)
Lum
ines
cen
ce (
arb
itia
ry u
nit
)
Figure 14. UV-visible absorption and photoluminescence spectra
upon excitation at 380 nm of rhenium carbonyl complex 4 in toluene
at room temperature. The 1MLCT absorbance lies at a wavelength of
430 nm; the emission spectrum shows two peaks at 428 nm and a
broad 3MLCT emission from 600 nm on with a maximum at 695 nm.
400 500 600Wavelength (nm)
700
Ab
sorp
tio
n (
arb
itia
ry u
nit
)
Lum
ines
cen
ce (
arb
itia
ry u
nit
)

Offprint provided courtesy of www.icevirtuallibrary.comAuthor copy for personal use, not for distribution
Nanomaterials and EnergyVolume 2 Issue NME3
Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini reviewPortenkirchner, Oppelt, Egbe, Knör and Sariçiftçi
144
In some cases, such as with compounds 3 and 4, the emission quan-tum yield is very low (Φ < 0.001). The broad 3MLCT emission of 3 in dichloromethane solution is also covering a typical wide spectral region including orange and red light exhibiting a maxi-mum at around 650 nm and showing additional shoulders at 590 and 700 nm (Figure 13). Interestingly, upon UV-light excitation at the absorption peak maximum of 3, the compound also shows an additional structured blue green intraligand emission with max-ima at 443 and 465 nm, which is not completely quenched by the lower-lying MLCT states (Figure 13).26 This is not the case for the nonsubstituted parent compound (2,2ʹ-bipyridyl)Re(CO)
3Cl (1)
(Figure 12) and was found to be caused by the additional pheny-lethinyl groups of the diimine ligand of 3.
Comparable dual luminescence behavior has also been reported for similar multichromophore systems investigated by Schanze and coworkers.70 In addition, compound 4, the absorbance and lumines-cence spectra of which are shown in Figure 14, exhibits similar spec-troscopic properties as catalyst 3. This is not surprising, considering that 3 and 4 are structurally related. The IL luminescent transition of 4 lies in the range between 400 and 500 nm in this case, whereas the 3MLCT emission is observed in the region higher than 600 nm.
Comparing now photocatalytic carbon dioxide reduction experi-ments applying compounds 1 and 3, it was found that the reduc-tion in DMF/TEOA(5:1/v:v) similar to previous works of Koike and coworkers67 shows big differences in reaction efficiency. As has been reported by other groups before, compound 1 showed a good photocatalytic activity with a quantum yield in the range of of φ = 0.1.67 In later experiments with the new catalyst 3, a significantly lower performance in the generation of carbon monoxide was reported under similar conditions.27 These findings can be inter-preted by analyzing the above described spectrophotometric meas-urements,27 considering the assumption that a long lived 3MLCT* state is a necessary requirement for the successful application of the catalyst in a photochemical system for the reduction on carbon dioxide to carbon dioxide.
7. Conclusion and outlookIn the present work, several tricarbonylchlororhenium(I)pyridyl complexes (1–4) were reviewed regarding to their potential as cata-lysts for homogeneous electrochemical and photochemical reduc-tion of carbon dioxide to carbon monoxide. In addition, rhodium catalysts (5,6) for electrochemical and photochemical NAD+ cofac-tor regeneration and proton reduction were investigated.
It was found that the carbon dioxide reduction potential, determined by cyclic voltammetry, can be positively influenced by a modified ligand system. A comparison between the catalyst material 3 with added phenylethinyl groups at the 5,5ʹ position to the nonmodi-fied catalyst 1 showed a shift in the onset carbon dioxide reduction potential by about 300 mV (vs. NHE) to more positive values. Bulk
carbon dioxide electrolysis experiments showed Faraday efficien-cies around 45–50% for the formation of carbon monoxide. The best estimated rate constants according to cyclic voltammetry data are in the order of 200 M−1 s−1.
NAD+ cofactor regeneration using organometallic catalysts was also investigated by cyclic voltammetry measurements. First experiments show a large reductive current enhancement attributed to NAD+ reduction to NADH at potentials of about −350 mV (vs. NHE).
Furthermore, the compounds with an extended ligand system showed significantly higher optical absorption in the visible range, which is basically a clear benefit for photocatalytic applications. However, photocatalytic experiments of compounds 1 and 3 indi-cate that the a modified ligand system can lead to an inversion of the lowest-lying excited state properties of such compounds from a MLCT character present in 1 to an intraligand situation present in 3. Following this argumentation it can be concluded that certain ligand-based modifications are to be avoided since it is generally assumed that the long lived 3MLCT state is necessary for the suc-cessful application of the catalyst in a photochemical system for the direct reduction of carbon dioxide to carbon monoxide.
As a next step new and improved electro- and photo-catalysts for carbon dioxide reduction and NADH regeneration have to be devel-oped and characterized. The modification of the attached ligand system is a suitable way for systematic tuning of the excited state properties of such materials and hence their electro- and photo-catalytic abilities. A clear demonstration of this was shown by the comparison of compounds 1 and 3. Following this idea a modifica-tion where the phenylethinyl groups are attached to the 4,4ʹ posi-tion might be highly interesting, assuming a better conjugation to the metal center of the complex.
Although further catalyst improvements are crucial, finally, the two systems capable of catalytic carbon dioxide reduction and H
2O
splitting could be combined in a photo- or photoelectron-chemical cell. Such a system would be capable of solar-powered production of syngas and its equivalents (carbon monoxide and hydrogen or NADH).
AcknowledgementsFinancial support by the Austrian Science Foundation (FWF) within the Wittgenstein Prize as well as the projects P21045: “Bio-inspired Multielectron Transfer Photosensitizers” and P25038: “Functional Light-Responsive Carbonyl Systems” is gratefully acknowledged.
REFERENCES
1. Dresselhaus, M. S.; Thomas, I. L. Alternative energy technologies. Nature 2001, 414(6861), 332–337.

Offprint provided courtesy of www.icevirtuallibrary.comAuthor copy for personal use, not for distribution
Nanomaterials and EnergyVolume 2 Issue NME3
Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini reviewPortenkirchner, Oppelt, Egbe, Knör and Sariçiftçi
145
2. Kerr, R. A. Even oil optimists expect energy demand to outstrip supply. Science 2007, 317, 437.
3. Friedlingstein, P. A steep road to climate stabilization. Nature 2008, 451(7176), 297–298.
4. Richardson, K.; Steffen, W.; Hans, S. J.; Alcamo, J.; Barker, T.;
Kammen, D. M.; Leemans, R.; Liverman, D.; Munasinghe, M.;
Osman-Elasha, B.; Stern, L. N.; Wæver, O. Synthesis Report Climate Change – Global Risks, Challenges & Decisions, 2nd edn. Copenhagen: University of Copenhagen, 2009, 1–36.
5. Murray, J.; King, D. Oil’s tipping point has passed. Nature 2009, 481, 433–435.
6. Peters, M.; Köhler, B.; Kuckshinrichs, W.; Leitner, W.;
Markewitz, P.; Müller, T. E. Chemical technologies for exploiting and recycling carbon dioxide into the value chain. ChemSusChem 4(9), 1216–1240.
7. Kumar, B.; Llorente, M.; Froehlich, J.; Dang, T.; Sathrum, A.;
Kubiak, C. P. Photochemical and photoelectrochemical reduc-tion of CO
2. Annual Review of Physical Chemistry 2012, 63,
541–569. 8. Kanan, M. W.; Nocera, D. G. In situ formation of an oxygen-
evolving catalyst in neutral water containing phosphate and CO2+. Science 2008, 321(5892), 1072–105.
9. Kanan, M. W.; Surendranath, Y.; Nocera, D. G. Cobalt–phosphate oxygen-evolving compound. Chemical Society Reviews 2009, 38, 109–114.
10. Shukla, S.; Halligudi, S. B.; Taqui Khan, M. M. Reduction of CO
2 by molecular hydrogen to formic acid and formal-
dehyde and their decomposition to CO and H2O. Journal of
Molecular Catalysis 1989, 57, 47–60.11. Xiaoding, X.; Moulijn, J. A. Mitigation of CO
2 by chemi-
cal conversion : plausible chemical reactions and promising products. Energy & Fuels 1996, 10, 305–325.
12. Schulz, H. Short history and present trends of Fischer–Tropsch synthesis. Applied Catalysis A: General 1999, 186(1–2), 3–12.
13. Olah, G.; Goeppert, A.; Prakash Surya, G. K. Beyond oil and gas: the methanol economy. Los Angeles: Wiley-VCH, 2006.
14. Delacourt, C.; Ridgway, P. L.; Kerr, J. B.; Newman, J. Design of an electrochemical cell making syngas (CO + H
2) from
CO2 and H
2O reduction at room temperature. Journal of The
Electrochemical Society 2008, 155(1), 42–49.15. Knör, G.; Monkowius, U. W. E. Photosensitization and photo-
catalysis in bioinorganic, bio-organometallic and biomimetic systems. Advanced Inorganic Chemistry 2011, 63, 235–289.
16. Concepcion, J. J.; House, R. L.; Papanikolas, J. M.;
Meyer, T. J. Chemical approaches to artificial photosynthesis. Proceedings of the National Academy of Sciences of the United States of America 2012, 109(39), 15560–15564.
17. Obert, R.; Dave, B. C. Enzymatic conversion of carbon dioxide to methanol: enhanced methanol production in silica sol−gel matrices. Journal of the American Chemical Society 1999, 121(51), 12192–12193.
18. El-zahab, B.; Donnelly, D.; Wang, P. Particle-tethered NADH for production of methanol from CO
2 catalyzed by coimmo-
bilized enzymes. Biotechnology and Bioengineering 2008, 99(3), 508–514.
19. Lu, Y.; Jiang, Z.; Xu, S.; Wu, H. Efficient conversion of CO2
to formic acid by formate dehydrogenase immobilized in a novel alginate–silica hybrid gel. Catalysis Today 2006, 115(1–4), 263–268.
20. Sun, Q.; Jiang, Y.; Jiang, Z.; Zhang, L.; Sun, X.; Li, J. Green and efficient conversion of CO
2 to methanol by biomimetic
coimmobilization of three dehydrogenases in protamine-tem-plated titania. Industrial & Engineering Chemistry Research 2009, 48(9), 4210–4215.
21. Arakawa, H.; Aresta, M.; Armor, J. N.; Barteau, M. A.;
Beckman, E. J.; Bell, A. T.; Bercaw, J. E.; Creutz, C.; Dinjus, E.;
Dixon, D. A.; Domen, K.; DuBois, D. L.; Eckert, J.; Fujita, E.;
Gibson, D. H.; Goddard, W. A.; Goodman, D. W.; Keller, J.;
Kubas, G. J.; Kung, H. H.; Lyons, J. E.; Manzer, L. E.;
Marks, T. J.; Morokuma, K.; Nicholas, K. M.; Periana, R.;
Que, L.; Rostrup-Nielson, J.; Sachtler, W. M. H.; Schmidt, L. D.;
Sen, A.; Somorjai, G. A.; Stair, P. C.; Stults, B. R.;
Tumas, W. Catalysis research of relevance to carbon man-agement: progress, challenges, and opportunities. Chemical Reviews 2001, 101(4), 953–996.
22. Balzani, V.; Credi, A.; Venturi, M. Photochemical conversion of solar energy. ChemSusChem 2008, 1, 26–58.
23. Roy, S. C.; Varghese, O. K.; Paulose, M.; Grimes, C. A. Toward solar fuels: photocatalytic conversion of carbon dioxide to hydrocarbons. American Chemical Society Nano 2010, 4(3), 1259–1278.
24. Yui, T.; Tamaki, Y.; Sekizawa, K.; Ishitani, O. Photocatalytic reduction of CO
2 : from molecules to semiconductors. Topics
in Current Chemistry 2011, 303(April), 151–184.25. Benson, E. E.; Kubiak, C. P.; Sathrum, A. J.;
Smieja, J. M. Electrocatalytic and homogeneous approaches to conversion of CO
2 to liquid fuels. Chemical Society
Reviews 2009, 38(1), 89–99.26. Oppelt, K.; Egbe, D. A. M.; Monkowius, U,.; List, M.;
Zabel, M.; Sariciftci, N. S. Luminescence and spectroscopic studies of organometallic rhodium and rhenium multi-chromophore systems carrying polypyridyl acceptor sites and phenylethynyl antenna subunits. Journal of Organometallic Chemistry 2011, 696(10), 2252–2258.
27. Portenkirchner, E.; Oppelt, K.; Ulbricht, C.; Egbe, D. A. M.;
Neugebauer, H.; Knör, G.; Sariciftci, N. S. Electrocatalytic and photocatalytic reduction of carbon dioxide to carbon monoxide using the alkynyl-substituted rhenium(I) complex (5ʹ,5ʹ-bisphenylethynyl-2,2-bipyridyl)Re(CO)
3Cl. Journal of
Organometallic Chemistry 2012, 716, 19–25.28. Kumar, B.; Smieja, J. M.; Sasayama, A. F.;
Kubiak, C. P. Tunable, light-assisted co-generation of CO and H
2 from CO
2 and H
2O by Re(bipy-tbu)(CO)
3Cl and p-Si

Offprint provided courtesy of www.icevirtuallibrary.comAuthor copy for personal use, not for distribution
Nanomaterials and EnergyVolume 2 Issue NME3
Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini reviewPortenkirchner, Oppelt, Egbe, Knör and Sariçiftçi
146
in non-aqueous medium. Chemical Communications 2012, 48(2), 272–274.
29. Barton, E. E.; Rampulla, D. M.; Bocarsly, A. B. Selective solar-driven reduction of CO
2 to methanol using a catalyzed
p-GaP based photoelectrochemical cell. Journal of Amercian Chemical Society 2008, 130, 6342–6344.
30. Romero, F. M.; Ziessel, R.; Strasbourg, C. A straightforward synthesis of 5-bromo and 5,5ʹ-dibromo-2,2ʹ-bipyridines. Tetrahedron Letters 1995, 36(36), 6471–6474.
31. Sanechika, K.; Yamamoto, T.; Yamamoto, A. Palladium catalyzed C-C coupling for synthesis of pi-conjugated poly-mers composed of arylene and ethynylene units. Bulletin of the Chemical Society of Japan 1984, 57(3), 752–755.
32. Egbe, D. A. M.; Klemm, E. Preparation of new rigid back-bone conjugated organic polymers with large fluorescence quantum yields. Macromolecular Chemistry and Physics 1998, 199(12), 2683–2688.
33. Knör, G.; Leirer, M.; Vogler, A. Synthesis, char-acterization and spectroscopic properties of 1,2-diiminetricarbonylrhenium(I)chloride complexes with o-benzoquinone diimines as ligands. Journal of Organometallic Chemistry 2000, 610(1–2), 16–19.
34. Monkowius, U.; Ritter, S.; König, B.; Zabel, M.;
Yersin, H. Synthesis, characterisation and ligand proper-ties of novel Bi-1,2,3-triazole ligands. European Journal of Inorganic Chemistry 2007, 2007(29), 4597–4606.
35. Monkowius, U.; Svartsov, Y. N.; Fischer, T.; Zabel, M.;
Yersin, H. Synthesis, crystal structures, and electronic spectra of (1,8-naphthyridine)ReI(CO)
3Cl and [(1,8-naphthyridine)
CuI(DPEPhos)]PF6. Inorganic Chemistry Communications 2007, 10(12), 1473–1477.
36. Leirer, M.; Knör, G.; Vogler, A. Electronic spectra of 1,2-diiminetricarbonylrhenium(I)chloride complexes with imidazole derivatives as ligands. Inorganica Chimica Acta 1999, 288(2), 150–153.
37. Abe, T.; Kaneko, M. Reduction catalysis by metal complexes confined in a polymer matrix. Progress in Polymer Science 2003, 28(10), 1441–1488.
38. Taylor, P.; Reddy, M. V. V. S.; Lingam, K. V.; Rao, T. K. G. Studies of radicals in oxalate systems. Molecular Physics 1980, 41(6), 1493–1500.
39. Schwarz, H. A.; Creutz, C.; Sutin, N. Cobalt(I) polypyridine complexes. Redox and substitutional kinetics and thermody-namics in the aqueous 2,2ʹ-bipyridine and 4,4ʹ-dimethyl-2,2ʹ-bipyridine series studied by the pulse-radiolysis technique. Inorganic Chemistry 1985, 24, 433–439.
40. Schwarz, H. A.; Dodson, W. Reduction potentials of CO2- and the alcohol radicals. Journal of Physical Chemistry 1989, 93, 409–414.
41. Sullivan, B. P. Electrochemical and Electrocatalytic Reactions of Carbon Dioxide. Amsterdam: Elsevier, 1993, 14.
42. Takeda, H.; Ishitani, O. Development of efficient photo-catalytic systems for CO
2 reduction using mononuclear and
multinuclear metal complexes based on mechanistic studies. Coordination Chemistry Reviews 2010, 254(3–4), 346–54.
43. Hamann, C.; Hamnett, A.; Vielstich, W. Electrochemistry. Weinheim: WILEY-VCH, 1998.
44. Cosnier, S.; Deronzier, A.; Moutet, J.-C. Electrochemical coating of a platinum electrode by a poly(pyrrole) film containing the fac-Re(2,2ʹ-bipyridine)(CO)
3CI system.
Journal of Electroanalytical Chemistry 1986, 207, 315–321.45. Olah, G. A.; Goeppert, A.; Prakash, G. K. S. Chemical
recycling of carbon dioxide to methanol and dimethyl ether : from greenhouse gas to renewable, environmentally carbon neutral fuels and synthetic hydrocarbons. Journal of Organic Chemistry 2009, 74(74), 487–498.
46. Yu, K. M. K.; Curcic, I.; Gabriel, J.; Tsang, S. C. E. Recent advances in CO
2 capture and utilization. ChemSusChem 2008,
1(11), 893–899.47. Dibenedetto, A.; Stufano, P.; Macyk, W.; Baran, T.;
Fragale, C.; Costa, M.; Aresta, M. Hybrid technologies for an enhanced carbon recycling based on the enzymatic reduction of CO
2 to methanol in water: chemical and pho-
tochemical NADH regeneration. ChemSusChem 2012, 5(2), 373–378.
48. Davies, K. J. A.; Doroshowo, J. H. Redox cycling of anthra-cyclines by cardiac mitochondria. Journal of Biological Chemistry 1986, 261(7), 3060–3067.
49. Hollmann, F.; Witholt, B.; Schmid, A. [Cp*Rh(bpy)(H2O)]2+:
a versatile tool for efficient and non-enzymatic regeneration of nicotinamide and flavin coenzymes. Journal of Molecular Catalysis B 2003, 20, 167–176.
50. Azem, A.; Man, F.; Omanovic, S. Direct regeneration of NADH on a ruthenium modified glassy carbon electrode. Journal of Molecular Catalysis A: Chemical 2004, 219(2), 283–299.
51. Damian, A.; Omanovic, S. Electrochemical reduction of NAD+ on a polycrystalline gold electrode. Journal of Molecular Catalysis A: Chemical 2006, 253(1–2), 222–233.
52. Hawecker, J.; Lehn, J.; Ziessel, R. Electrocatalytic reduc-tion of carbon dioxide mediated by Re(bipy)(CO)
3Cl
(bipy = 2,2ʹ-bipyridine). Journal of Chemical Society, Chemical Communication 1984, 6, 328–330.
53. Sullivan, B. P.; Bolinger, C. M.; Conrad, D.; Vining, W.
J.; Meyer, T. J. One- and two-electron pathways in the electrocatalytic reduction of CO
2 by fac-Re(bpy)(CO)
3Cl
(bpy = 2,2ʹ-bipyridine). Journal of Chemical Society, Chemical Communication 1985, 20, 1414–1416.
54. Hawecker, J.; Lehn, J.; Ziessel, R. Photochemical and elec-trochemical reduction of carbon dioxide to carbon monoxide mediated by (2,2ʹ-bipyridine) tricarbonylchlororhenium (I) and related complexes as homogeneous catalysts). Helvetica Chimica Acta 1986, 69, 1990–2012.
55. Johnson, F. P. A.; George, M. W.; Hartl, F.;
Turner, J. J. Electrocatalytic reduction of CO2 using the com-
plexes [Re (bpy)(CO)3L] n (n)+1, L) P(OEt)3, CH3CN; n) 0,

Offprint provided courtesy of www.icevirtuallibrary.comAuthor copy for personal use, not for distribution
Nanomaterials and EnergyVolume 2 Issue NME3
Electro- and photo-chemistry of rhenium and rhodium complexes for carbon dioxide and proton reduction: a mini reviewPortenkirchner, Oppelt, Egbe, Knör and Sariçiftçi
147
catalyst precursors : infrared spectroelectrochemical investi-gation. Organometallics 1996, 15, 3374–3387.
56. Smieja, J. M.; Kubiak, C. P. Re(bipy-tBu)(CO)(3)Cl-improved catalytic activity for reduction of carbon dioxide: IR-spectroelectrochemical and mechanistic studies. Inorganic Chemistry 49(20), 9283–9289.
57. Bard, A. J.; Faulkner, C. J. Electrochemical Methods: Fundamentals and Applications, 2nd edn. New York: Wiley, 2001.
58. Saveant, J. M.; Vianello, E. Potential-sweep chronoamper-ometry theory of kinetic currents in the case of a first order chemical reaction preceding the electron-transfer process. Electrochimica Acta 1963, 8, 905–923.
59. Bard, A. J.; Faulkner, C. J. Electrochemical Methods. New York: John Wiley, 1980, 171–172, 218.
60. Dubois, D. L.; Miedanerlt, A.; Haltiwangert, R. C. Electrochemical reduction of CO
2 catalyzed by [Pd (triphosphine)
(solvent)](BF 4)2 complexes : synthetic and mechanistic studies. Journal of Amercian Chemical Society 1991, 113, 8753–8764.
61. Atkins, P. W. Physikalische Chemie, 3rd edn. Weinheim: WILEY-VCH, 2001, 823.
62. Kozuch, S.; Martin, J. M. L. “Turning over” definitions in catalytic cycles. ACS Catalysis 2012, 2(12), 2787–2794.
63. Yoshida, T.; Tsutsumida, K.; Teratani, S.; Yasufuku, K.;
Kaneko, M. Electrocatalytic reduction of CO2 in water by
[Re(bpy)(C0)3Br] and [Re(terpy)(CO)3Br] complexes incor-porated into coated nafion membrane (bpy = 2,2ʹ-bipyridine;terpy = 2,2ʹ : 6ʹ,2ʺ-terpyridine). Journal of Chemical Society, Chemical Communication 1993, 7, 631–633.
64. Lopez-Castillo, Z. K.; Aki, S. N. V. K.; Stadtherr, M. A.;
Brennecke, J. F. Enhanced solubility of oxygen and
carbon monoxide in CO2 – expanded liquids. Industrial &
Engineering Chemistry Research 2006, 45, 5351–5360.65. Hawecker, J.; Lehn, J.; Ziessel, R. Photochemical and elec-
trochemical reduction of carbon dioxide to carbon monoxide mediated by (2,2ʹ-bipyridine) tricarbonylchlororhenium (I) and related complexes as homogeneous catalysts. Helvetica Chimica Acta 1986, 69, 1990–2012.
66. Takeda, H.; Koike, K.; Morimoto, T. Photochemistry and photocatalysis of rhenium (I) diimine complexes. Elsevier 2011, 63(I), 137–186.
67. Koike, K.; Hori, H.; Ishizuka, M.; Westwellm, J. R.;
Takeuchi, K.; Ibusuki, T.; Enjouji, K.; Konno, H.;
Sakamoto, K.; Ishitani, O. Key process of the photo-catalytic reduction of CO
2 using [Re(4,4ʹ-X2-bipyridine)
(CO)3PR3]+(X = CH3, H, CF3; PR3 = phosphorus ligands): dark reaction of the one-electron-reduced complexes with CO
2. Organometallics 1997 16(26), 5724–5729.
68. Scheiring, T.; Klein, A.; Kaim, W. EPR study of paramagnetic rhenium(I) complexes (bpy>>.>>->>)Re(CO)3X relevant to the mechanism of electrocatalytic CO
2 reduction. Journal
of the Chemical Society, Perkin Transactions 2 1997, 12, 2569–2572.
69. Walters, K. A.; Premvardhan, L. L.; Liu, Y.; Peteanu, L. A.;
Schanze, K. S. Metal-to-ligand charge transfer absorption in a rhenium (I) complex that contains a conjugated bipyri-dine acceptor ligand. Chemical Physics Letters 2001, 339, 255–262.
70. Liu, Y.; Li, Y.; Schanze, K. S. Photophysics of π-conjugated oligomers and polymers that contain transition metal com-plexes. Journal of Photochemistry and Photobiology C: Photochemistry Reviews 2002, 3(1), 1–23.
WHAT DO YOU THINK?
To discuss this paper, please email up to 500 words to the managing editor at [email protected]
Your contribution will be forwarded to the author(s) for a reply and, if considered appropriate by the editor-in-chief, will be published as a discussion in a future issue of the journal.
ICE Science journals rely entirely on contributions sent in by professionals, academics and students coming from the field of materials science and engineering. Articles should be within 5000-7000 words long (short communications and opinion articles should be within 2000 words long), with adequate illustrations and references. To access our author guidelines and how to submit your paper, please refer to the journal website at www.icevirtuallibrary.com/nme









![Lability and Basicity of Bipyridine-Carboxylate ...sabrash/seminar/Outside Speaker... · ] (bpH 2 cH = 6′-phosphono-[2,2′-bipyridine]-6-carboxylic acid, L = 4-picoline or isoquinoline).](https://static.fdocuments.in/doc/165x107/61062003ba91955d9f7906a7/lability-and-basicity-of-bipyridine-carboxylate-sabrashseminaroutside-speaker.jpg)








