
El futuro de la manufactura farmacéutica · envasados asépticamente, durables, a prueba de...
76
Más: La administración de EEUU pelea con el financiamiento regulatorio ¿Es momento de revisar el ICH Q9? La USP señala las actividades para los estándares de impurezas Consideraciones para el proceso estéril Volumen 11, Número 1 Investigación Arbitrada • Una novedosa tecnología para tabletas de liberación modificada • Indexación del patrón de difracción de polvos con rayos X para aplicaciones farmacéuticas ANTI-FALSIFICACIÓN: El Rx-360 proporciona una actualización RESOLUCIÓN DE PROBLEMAS: Cómo optimizar los sistemas de limpieza AÑO EN REVISIÓN: Innovación en las grandes farmacéuticas SÍNTESIS Y MANUFACTURA DE APIs: Estrategias en el escalamiento SUBCONTRATACIÓN: Biomanufactura en el extranjero El futuro de la manufactura farmacéutica El ámbito cambiante de la ciencia regulatoria SEGURIDAD LABORAL Los resultados de la encuesta anual sobre el empleo muestran una mayor confianza
Transcript of El futuro de la manufactura farmacéutica · envasados asépticamente, durables, a prueba de...

Más:
La administración de EEUU pelea con el financiamiento regulatorio
¿Es momento de revisar el ICH Q9?
La USP señala las actividades para los estándares de impurezas
Consideraciones para el proceso estéril
Volumen 11, Número 1
Investigación Arbitrada• Una novedosa tecnología para tabletas de liberación modificada • Indexación del patrón de difracción de polvos con rayos X para aplicaciones farmacéuticas
ANTI-FALSIFICACIÓN: El Rx-360 proporciona una actualización
RESOLUCIÓN DE PROBLEMAS:Cómo optimizar los
sistemas de limpieza
AÑO EN REVISIÓN: Innovación en las
grandes farmacéuticas
SÍNTESIS Y MANUFACTURA DE APIs: Estrategias en el escalamiento
SUBCONTRATACIÓN: Biomanufactura en el extranjero
El futuro de la manufactura farmacéuticaEl ámbito cambiante de laciencia regulatoria
SEgURIDAD LABORAL Los resultados de la encuesta anual sobre el empleo muestran una mayor confianza

Solu
cion
es In
tegr
ales
par
a su
Pro
ceso
y A
nális
is
Visi
tano
s:Ab
ril 2
013,
Expo
Pack
WTC
Pla
nta
Baja
& S
tand
: 115
0Ju
nio
2013
, Ex
po F
arm
aCe
ntro
Ban
amex
& S
tand
391
4
turn
ing
scie
nce
in
to s
olu
tio
ns
Sart
oriu
s de
Méx
ico
S.A.
de
C.V.
Cto.
Cir
cunv
alac
ión
Pte.
No.
149
Ciud
ad S
atél
ite,
Nau
calp
an, E
do. d
e M
éx. 5
3100
Tel
: +52
-55-
5562
-110
2 F
ax: +
52-5
5-55
62-2
942
sart
oriu
s@sa
rtom
ex.c
om.m
x
Bala
nzas
de
Labo
rato
rio
Básc
ulas
y P
lata
form
as In
dust
rial
esPe
sas
de V
erif
icac
ión
y Aj
uste
Anal
izad
ores
de
Hum
edad
pH-M
etro
sCe
ldas
de
Carg
a y
Kits
de
Mon
taje
Veri
fica
dore
s de
Pes
o y
Dete
ctor
es d
e M
etal
Serv
icio
Téc
nico
Calib
raci
ón d
e Pe
sas
e In
stru
men
tos
Soft
war
e pa
ra e
l Pro
cesa
mie
nto
de M
edic
ione
sM
icro
biol
ogía
Sist
ema
de p
urif
icac
ión
de a
gua
para
Lab
orat
orio
Filt
raci
ón y
Ult
rafi
ltra
cció
n pa
ra L
abor
ator
ios
Man
ejo
de lí
quid
os:M
icro
pipe
tas,
punt
as, d
ispe
nsad
ores
�E�X
�P�O
�P�A
�C�K
� �a�u
�t�o�r�i�z�a
�d�o
�v�i�e
�r�n�e
�s�,� �0
�1� �d
�e� �m
�a�r�z
�o� �d
�e� �2
�0�1
�3� �1
�1�:�1
�3�:�0
�0� �a
�.�m�.
Marque en la tarjeta de servicio al lector el No. 5

modelo 624
Nutra sus cualidades más valiosas.En Weiler Engineering, nuestras máquinas de envasado Blow/Fill/Seal ASEP-TECH® producen productos envasados asépticamente, durables, a prueba de fragmentaciones, en un ambiente “sin contacto manual”, que elimina virtualmente los problemas de contaminación del llenado convencional de viales.
Qué más esperaria del sistema de envasado aséptico de líquidos más avanzado del mundo?
Nuestras máquinas de Soplado/Llenado/Sellado integran moldeado por soplado, llenado estéril y sellado hermético
en una operación ininterrumpida—un proceso de producción sin contacto humano que garantiza que los productos parenterales,
soluciones oftálmicas, fármacos respiratorios y otros medicamentos líquidos de su compañía puedan llegar al mercado de la manera más
rentable posible—en todas las ocasiones.
Por más de 40 años, hemos determinado el estándar industrial para el desarrollo del procesamiento estéril con un compromiso continuo
por la calidad e innovación por tecnología aséptica. Trabajaremos con su compañía para desarrollar una estrategia a medida para cada uno
de sus productos.
adentro es más seguro
©20
12 W
eile
r En
gine
erin
g, In
c.
Soluciones protectoras mediante envasado innovador
Para ver los sistemas
ASEP-TECH® en acción,
visite www.asep-tech.com/ptes
14004-47 WEIL PharmTech ES_Sept2012.indd 1 5/23/12 1:46 PM
Mar
que
en la
tarje
ta d
e se
rvici
o al
lect
or e
l No.
9

Pharmaceutical Technology en Español MARZO / ABRIL 20132
En
la P
ort
ad
aMARZO / ABRIL 2013 VOLUMEN 11, NÚMERO 1Pharmaceutical Technology en Español, proporciona información importante, confiable, y oportuna sobre todos los aspectos relacionados con Desarrollo e Investigación Aplicada; y con las Tecnologías de Proceso, Fabricación, Formulación, y Empaque para la Industria Farmacéutica Convencional y la de Biotecnología.
5 Seguridad laboralAmy Ritter
Los resultados de la encuesta anual sobre el empleo muestran mayor confianza en la industria farmacéutica.
InvestIgacIón arbItrada
FORMULACIÓN
32 Desarrollo y comercialización de una novedosa tecnología para tabletas de liberación modificadaKevin D. Altria y James Taylor
Los autores describen la ruta desde el desarrollo a la comercialización de una técnica de liberación modificada.
52 Indexación del patrón de difracción con rayos X para aplicaciones farmacéuticas Los autores discuten la valiosa información que pue-de obtenerse de la indexación, y sus aplicaciones en la selección y análisis rutinario de formas sólidas.
en eL terreno de juego
NOVEDADES Y ANÁLISIS
8 Reporte desde Corea del SurJane Wan
9 Atención: Terapia génicaStephanie Sutton
CONVERSACIÓN Y COMUNIDAD
51 Tomando el pulso de la industriaLos lectores piensan que…
aspectos
REPORTE ESPECIAL
26 Innovación farmacéuticaAngie Drakulich
un vistazo a los líderes del año en estrategias de innovación, incluyendo a las principales compañías bio/farmacéuticas y a los ganadores de los premios de la aaps, phrMa y cphl.
ANTIFALSIFICACIÓN
38 Colaboración para proteger la cadena de suministro de los falsificadores Adeline Siew
pharmtech habla con Lynne byers y brian johnson acerca de las iniciativas del rx-360 para proteger la seguridad del paciente.
SÍNTESIS Y MANUFACTURA DE APIS
49 Estrategias en el escalamiento de APIs Los químicos de procesos emplean una variedad de esquemas para mejorar el rendimiento, la pureza y la estereoselectividad.
POSTURA OFICIAL
19 GMPs del desarrollo inicial para la manufactura de productos farmacéuticos de moléculas pequeñas (Parte III) Richard Creekmore, Eleni Dokou, Amnon Eylath, Dennis Joiner, Michael Lovdahl, Jackson Pellett, Eric Schmitt, y John W. Skoug
Los representantes del consorcio IQ exploran y definen esquemas y prácticas comunes de la industria para la aplicación de las gMps en el desarrollo inicial.
PRIMERA PLANA
10 Un vistazo a futuros de la manufactura y la regulaciónLa Fda en el ámbito cambiante de la ciencia regulatoria
Illustración por dan Ward Imagenes: dan Ward/buena vista Images/getty Images

Pharmaceutical Technology en Español MARZO / ABRIL 2013 3
CONTENIDO
Pharmaceutical Technology es selectivamente extraida o indexada en:Biological Sciences Database (Cambridge Scientific Abstracts)Biotechnology and Bioengineering Database (Cambridge Scientific Abstracts)Business and Management Practices (RDSI)Chemical Abstracts (CAS)Current Packaging AbstractsDECHEMADerwent Biotechnology Abstracts (Derwent Information, Ltd.)Excerpta Medica (Elsevier)International Pharmaceutical Abstracts (ASHP)Science Citation Index (Thomson)Pharmaceutical Technology está orgullosa de ser miembro asociado de DCAT, IPEC y PDA.
5 Seguridad laboralAmy Ritter
Los resultados de la encuesta anual sobre el empleo muestran mayor confianza en la industria farmacéutica.
coLuMnas
VIGILANCIA REGULATORIA
41 Desafíos de la política de salud para la administración de ObamaJill Wechsler
La casa blanca y el congreso probablemente luchen por el financiamiento para la regulación bio/farmacéutica
SOLUCIONES ESTADÍSTICAS
43 Momento para revisar el ICH Q9 Lynn D. Torbeck
un cambio en la terminología podría enfatizar la protección del paciente.
RESOLUCIÓN DE PROBLEMAS
45 Un esquema de ciclo de vida para la optimización de los sistemas de limpieza Andrew Wong y Cody Shrader
Los sistemas de limpieza en el sitio deberían optimizarse durante el diseño y la puesta en marcha y después de la validación.
BIOFORO
56 La transformación para la organización centrada en el proceso La organización centrada en el proceso en la manufactura biofarmacéutica… ¿es un escalón o una piedra en el camino?
PERSPECTIVA DE LA SUBCONTRATACIÓN
59 Biomanufactura en el extranjero La subcontratación de biomanufactura internacional ¿se convertirá en la corriente principal en esta década?
reguLacIón Y cuMpLIMIento
VIGILANCIA REGULATORIA EN EEUU
61 Resumen• Controversia sobre el tratado de medicamentos falsificados
• La EMA busca acción sobre los faltantes
• Colaboración sobre las células madre
• Precios de fármacos de especialidades
• La FTC se opone a los cambios de formulación anti-genéricos
• Nuevas guías publicadas
62 La calidad de fármacos en la etapa central para la FDA y los fabricantes La escasez y el desastre cada vez mayor estimulan los esfuerzos para revisar la supervisión de la manufactura y estimular las mejoras en la industria.
DENTRO DE LA USP
64 Actividades de establecimiento de estándares para impurezas el centro de la usp en el 2013 involucra a los estándares que se relacionan con impurezas orgánicas, con la medición del adn residual y las proteínas de la célula huésped en pro-ductos biotecnológicos, y con las impurezas elementales.
RESOLUCIÓN DE PROBLEMAS DEL CMC
64 Requerimientos de validación para la eficacia de la desinfección La revalidación de la desinfección de cuartos limpios puede ser innecesaria.
PREGÚNTELE AL EXPERTO
58 La EMA y la FDA en la Validación de procesos siegfried schmitt, un consultor importante de pareXeL, discute la guía de la agencia europea de Medicamen-tos sobre la validación de procesos y cómo se compara con la guía de validación de procesos de la Fda.
seccIones67 ¿Qué hay de nuevo?
67 Calendario de eventos
68 Cápsulas Farmacéuticas
71 Directorio Clasificado
72 Directorio de anunciantes

Pharmaceutical Technology en Español MARZO / ABRIL 20134
James P. AgallocoPresident, Agalloco & Associates
Larry L. Augsburger, PhDProfessor, Department of Pharmaceutics, University of Maryland
David H. Bergstrom, PhDCOO, NovaDel Pharma Inc.
Phil BormanQbD Lead & Data Management & Analysis Manager GlaxoSmithKline
Rory BudihandojoDirector, Quality Systems Audit, Boehringer-Ingelheim Shanghai Pharmaceuticals Co. (China)
Todd L. CecilVice-PresidentCompendial ScienceUnited States Pharmacopeia
Metin Çelik, PhDPresident, Pharmaceutical Technologies International (PTI)
Zak T. Chowhan, PhDConsultant, Pharmaceutical Development
Suggy S. Chrai, PhDPresident and CEO,Chrai Associates, Inc.
Roger Dabbah, PhDPrincipal Consultant, Tri-Intersect Solutions
Tim FreemanManaging Director, FreemanTechnology
Sanjay Garg, PhDProfessor, Pharmaceutical Sciences, University of South Australia
R. Gary Hollenbeck, PhDChief Scientific Officer, UPM Pharmaceuticals
Ruey-ching (Richard) Hwang, PhDSenior Director, Pharmaceutical Sciences,Pfizer Global R&D
Mansoor A. Khan, PhDDirector, FDA/CDER/DPQR
Russell E. MadsenPresident, The Williamsburg Group, LLC
Heidi M. Mansour, PhDAssistant Professor,College of Pharmacy, University of Kentucky
Jim MillerPresident, PharmSource Information Services Bio/Pharmaceutical Outsourcing Report
Colin Minchom, PhDVice President Particle DesignHovione
Christine Moore, PhDDeputy Director for Science and Policy, Office of New Drug Quality Assessment, CDER, FDA
R. Christian Moreton, PhDVice-President, Pharmaceutical Sciences, Finnbrit Consulting
Fernando J. Muzzio, PhDDirector, NSF Engineering Research Center on Structured Organic Particulate Systems, Dept. of Chemical and Biochemical Engineering, Rutgers University
Moheb M. Nasr, PhDVice-President, CMC Regulatory Strategy, Global Regulatory Affairs, GlaxoSmithKline
Garnet E. Peck, PhDProfessor Emeritus of Industrial Pharmacy, Purdue University
James Polli, PhDProfessor, School of Pharmacy, University of Maryland
Wendy Saffell-ClemmerDirector, Research, BioPharma Solutions
Gurvinder Singh Rekhi, PhDDirector,Research and Development, Elan Drug Delivery Inc.
Susan J. SchnieppPharmaceutical Consultant, Schniepp & Associates, LLC
David R. SchonekerDirector of Global Regulatory Affairs, Colorcon
Eric B. Sheinin, PhDPresident, Sheinin and Associates
Charles A. Signorino, PhDCEO, Emerson Resources, Inc.
Aloka SrinivasanPrincipal Consultant, PAREXEL International
Heinz Sucker, PhDProfessor Emeritus,Pharmaceutical Institute, University of Bern
Scott Sutton, PhDMicrobiology Network
Lynn D. TorbeckStatistician, PharmStat Consulting
Pharmaceutical Technology en Español, V.11 No. 1 Marzo-Abril de 2013. Publicación bimestral editada por Revistas para la Industria, S.A. de C.V. Editor responsable: Ma. Antonieta Gue-rrero Paz. No. de Certificado de Reserva de Derechos al Uso Exclusivo otorgado por el Instituto Na-cional del Derecho al Autor No. 04-2011-010610533100-102. No. de Certificado de Licitud de Título y Contenido otorgado por la Secretaría de Gobernación No. 15794. Domicilio de la Publicación: Av. Insurgentes Sur 605, Desp. 404-D, Col. Nápoles, C.P. 03810, Deleg. Benito Juárez, México, D.F. Impreso en: Polymasters de México, S.A. de C.V. - Calle Dos No. 123-C, Col. Granjas San Antonio C.P. 09070, México, D. F. Distribuida por: Revistas para la Industria, S.A. de C.V. - Av. Insurgentes Sur 605, Desp. 404-D, Col. Nápoles, C.P. 03810, Deleg. Benito Juárez, México, D.F.
Toda la información y conceptos que aquí aparecen son responsabilidad exclusiva de cada uno de los autores y firmas comerciales.
Esta prohibida y será castigada la reproducción total o parcial de cualquiera de los materiales que aquí aparecen.

Pharmaceutical Technology en Español MARZO / ABRIL 2013 5
EncuEsta dE EmplEo
Seguridad laboralLos resultados de la encuesta anual de empleo muestran mayor confianza en la industria farmacéutica.
Amy Ritter
La ola de grandes fusiones que domi-naron las noticias hace unos cuantos años parece estar desacelerándose, pero las compañías bio/farmacéu-
ticas todavía enfrentan presiones para hacer funcionar negocios más esbeltos y para ver un mejor retorno de la inversión de sus divisiones de IyD. La economía global continúa cojean-do, y la farmacéutica enfrenta presiones de precios de los pagadores conscientes del costo y de países en desarrollo determinados a man-tener la línea sobre los precios de fármacos. La industria continúa adaptándose a su desafiante entorno de negocios, y estos retos no pueden ayudar pero afectan a los empleados farma-céuticos. Pharmaceutical Technology le pre-guntó a los lectores acerca de la situación en sus empleos: qué tan seguros se sienten en sus puestos, qué tan satisfechos están con sus tra-bajos, y cómo ven el futuro de sus compañías y de la industria global. Las siguientes páginas resaltan los resultados claves de la encuesta.
El nivel de inseguridad en el empleo está cayendo entre los empleados biofarmacéuti-cos. Año tras año, menos respondedores dicen que se sienten menos seguros en sus puestos que lo que sentían el año anterior- En 2010, el 53% dijo que se sentían menos seguros. Esto cayó a 41% en 2011 y en 2012, sólo el 34% se sentía menos seguro. Aunque esto parece estimulante, los lectores no dijeron que se sen-tían más seguros. A su vez, un porcentaje in-crementado (47% en 2012) dijo que se sentían más o menos igual que el año anterior. Parece, entonces, que los empleados farmacéuticos se están acostumbrando al nuevo entorno de negocios, más fluido. ¿Se está volviendo la inseguridad la nueva normal? Quizás, pero quienes respondieron sentían la confianza de que podrían encontrar un nuevo empleo si era necesario, y continúan obteniendo satisfac-ción de la estimulación intelectual y de los de-safiantes proyectos asociados con sus trabajos.
El difícil clima económico se está dejando sentir por aquéllos que están en la industria. Una tercera parte de los que respondieron, el 33%, indicaron que el negocio había declinado durante el año anterior. Aun así. Los que res-pondieron estaban optimistas acerca del futu-ro, tanto por sus propias compañías como por la industria como un todo. Sólo el 34% espera que el negocio decline en su propia compañía el próximo año, con el resto esperando ya sea ningún cambio (18%) o un incremento (48%). Cuando se les preguntó acerca de la industria como un todo, el 49% dijo que esperaban que el negocio mejorara. El próximo año traerá su propia serie de desafíos para la industria, pero tenemos la esperanza de que este optimismo esté bien fundamentado.
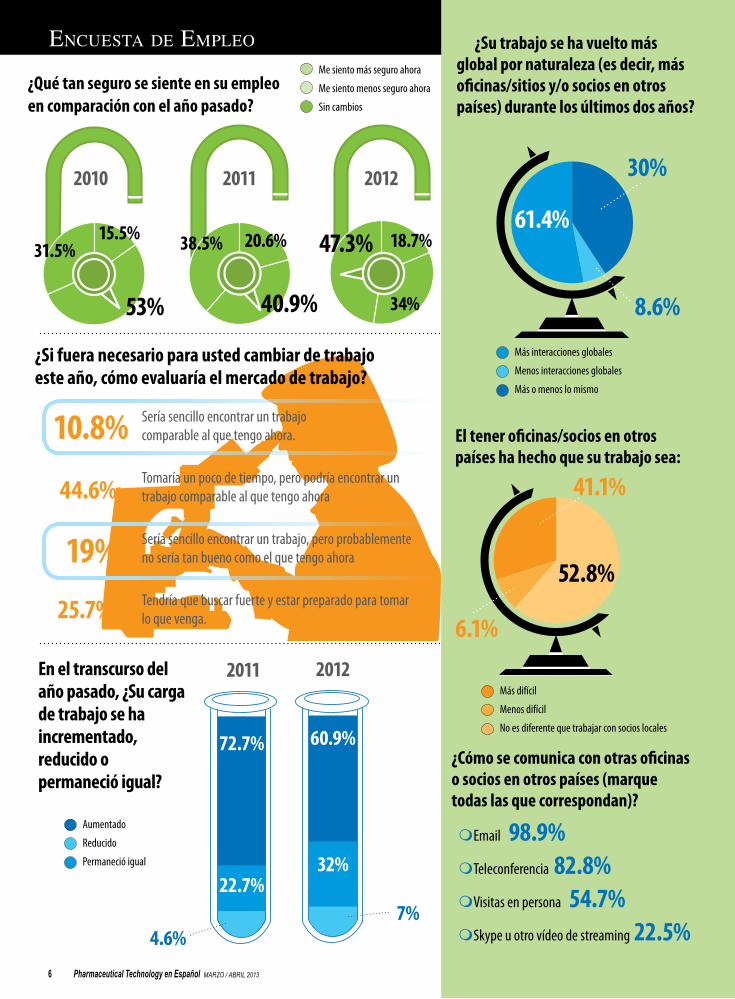
Pharmaceutical Technology en Español MARZO / ABRIL 20136
En el transcurso del año pasado, ¿Su carga de trabajo se ha incrementado, reducido o permaneció igual?
Sería sencillo encontrar un trabajo comparable al que tengo ahora.
Tomaría un poco de tiempo, pero podría encontrar un trabajo comparable al que tengo ahora
Sería sencillo encontrar un trabajo, pero probablemente no sería tan bueno como el que tengo ahora
Tendría que buscar fuerte y estar preparado para tomar lo que venga.
¿Si fuera necesario para usted cambiar de trabajo este año, cómo evaluaría el mercado de trabajo?
10.8%
¿Su trabajo se ha vuelto más global por naturaleza (es decir, más oficinas/sitios y/o socios en otros países) durante los últimos dos años?
¿Cómo se comunica con otras oficinas o socios en otros países (marque todas las que correspondan)?
mEmail 98.9%mTeleconferencia 82.8%mVisitas en persona 54.7%mSkype u otro vídeo de streaming 22.5%
61.4%
30%
8.6%
Más interacciones globales
Menos interacciones globales
Más o menos lo mismo
44.6%
25.7%
Aumentado
Reducido
Permaneció igual
19%
El tener oficinas/socios en otros países ha hecho que su trabajo sea:
Más difícil
Menos difícil
No es diferente que trabajar con socios locales
52.8%
41.1%
6.1%2011
72.7%
4.6%
22.7%
2012
60.9%
32%
7%
¿Qué tan seguro se siente en su empleo en comparación con el año pasado?
Me siento más seguro ahora
Me siento menos seguro ahora
Sin cambios
2011 20122010
31.5%15.5%
53%
38.5% 20.6%
40.9% 34%
18.7%47.3%
EncuEsta dE EmplEo

Pharmaceutical Technology en Español MARZO / ABRIL 2013 7
EEUU (USD) 105,000Canadá (CAD) 104,500 (104,544 USD)
Mediana de salarios
Proyectos Desafiantes
Estímulo Intelectual
Esta es la principal razón por la que vengo a trabajar
El negocio mejorará
El negocio declinará
No se espera cambio significativo
¿Cuál es su pronóstico para los prospectos de negocio de su compañía el próximo año?
Desde su punto de vista, ¿cuál es la perspectiva general para la industria bio/farmacéutica en el corto y largo plazo?
El negocio mejorará
El negocio declinará
El negocio mejorará en el extranjero, pero no domésticamente
El negocio mejorará domésticamente, pero no en el extranjero
El negocio declinará domésticamente, pero no en el extranjero
El negocio declinará en el extranjero, pero no domésticamente
No se espera ningún cambio significativo
9.6%18.7%
2.6%4.1%
0%15.9%
29.6%
31%29.2%
39.3%38%
En los dos últimos años ¿ha pasado por una fusión, compra, reducción
de personal o reestructuración?
SÍ NO2012 36.1% 63.9%2011 46.7% 53.3%
49.1%
47.6%
$
18.1%34.3%
Cambiaría de trabajo sólo por esto
Salario
Seguridad en el trabajoAvance profesional
*Debido al redondeo, algunos porcentajes pueden no sumar 100%. Algunas preguntas permitían respuestas múltiples.
Gr
ap
hic
s:
Me
lis
sa
Mc
ev
oy

Pharmaceutical Technology en Español MARZO / ABRIL 20138
Reporte desde: Corea del SurJane Wan
Cuatro compañías domésticas presentaron una demanda contra el Ministerio de Salud y Bienestar de Corea del Sur reclamando que los recientes recortes de precios hechos por el ministro habían afectado su negocio. En abril 1 del 2012, los precios de los fármacos se redujeron en un promedio de 17% y los recortes afectaron los precios de 6506 fármacos en todos los ámbitos. El pri-mer recorte, anunciado en 2011, redujo el precio de fármacos que habían per-dido la patente en 30%, con el precio del primer fármaco genérico establecido en 60% del precio del fármaco sin patente. Originalmente, los precios de los fármacos sin patente se redujeron en 20%, mientras que la primera versión ge-nérica se establecía en 68% del fármaco sin patente. El segundo recorte reduce el precio de los fármacos debido a descuentos ilegales. La combinación de los dos recortes significa que ciertos fármacos se someterán a una reducción doble del precio y la tasa del precio final puede ser hasta de 53.55%.
Las compañías farmacéuticas están desesperadas porque estos recortes tendrían un impacto directo sobre las utilidades del negocio aun cuando la agencia afirmó que este cambio aseguraría la sustentabilidad del mercado y erradicaría el problema de descuentos ilícitos. De hecho, una fuente de Sinhan Investment & Securities ha indicado que la mayoría de las compañías experi-mentaron una caída del 20% en utilidades desde que la política entró en vigor.
En respuesta a sus fortunas en declive, algunas compañías han optado por incrementar los precios de sus fármacos de libre venta (OTC) mientras que otras han incrementado su gasto en IyD. Las compañías domésticas tales como Dong-A Pharmaceutical y LG Life Sciences han comprometido el 22% y el 19% de las ventas netas en IyD, respectivamente.
Cher Boon Piang, un analista para Asia Pacífico Farmacéutico y de Salud del Monitoreo Internacional de Negocios (Asia), dice “Dados los recortes de precio, las compañías pueden sacar fármacos que no son lucrativos. Las com-pañías locales pueden incluso alejarse de los genéricos. Existe también un cambio hacia los biosimilares que abre la oportunidad para las compañías.” En octubre de 2011, Dong-A Pharmaceutical cerró tratos con Meiji Seika Phar-ma, de Tokio, para construir una planta de biosimilares en Songdo. Reciente-mente, las compañías locales Yuhan Corp y Teregen ETEX están colaborando para comercializar la provisión de servicios de genoma individuales.
El reciente acuerdo de libre comercio (FTA) también ha lastimado a los actores domésticos ya que contiene provisiones que protegen los derechos de
Las compañías domésticas están cambiando sus modelos de negocio en respuesta a los recientes recortes de precios de medicamentos.
Pharmaceutical Technology en Español MARZO / ABRIL 20138

Pharmaceutical Technology en Español MARZO / ABRIL 2013 9
propiedad intelectual de los desarrolladores originales del fár-maco. Por ejemplo, la agencia coreana tiene que informar a los fabricantes originales de que hay compañías que buscan producir versiones genéricas. A las compañías se les niega la aprobación para el mercado si existen objeciones por par-te de los fabricantes originales del fármaco y cuando existe una demanda de patente. También es obligatorio para el fa-bricante de genéricos proporcionar información de seguridad y eficacia para asegurar que la versión genérica no infringe la original.
Quizás un recorte gradual de los precios habría ayudado a aliviar las presiones a las que se enfrentan los actores de la industria en Corea del Sur. Cher dice, “En general, las compa-ñías enfrentaron problemas tanto financieros como de tiempo cuando se introdujeron dos recortes de precios en el 2011. Si la reducción de precios es gradual y se hace del conocimiento para las compañías en un marco de tiempo que las prepare para dichas reducciones, estas compañías pueden dar un giro e implementar estrategias para minimizar el impacto de la reducción de precios. Además, es más fácil tener soluciones al corto plazo contra las reducciones graduales de precios en comparación con una reducción mucho más grande.
Cher señala que los recortes de precios no pueden ser la única solución para contener los gastos crecientes del gobier-no y también es injusto hacer recaer la carga en las empresas. En lugar de esto, el gobierno debe buscar maneras de recortar el gasto otorgando sólo los subsidios necesarios y/o incre-mentar las primas. Estas estrategias pueden crear una impre-sión negativa, pero son esenciales si a un país no le está yendo bien económicamente. Adicionalmente, la carga de los costos de salud debe descansar a su vez sobre los individuos, agrega.
Claramente, los objetivos del gobierno y de los actores de la industria farmacéutica difieren grandemente. Por un lado, el gobierno busca reducir los costos de salud. Por otro lado, los actores de la industria están buscando maneras de maximizar las utilidades. Al preguntársele si se puede alcanzar el equili-brio entre ambas partes, Cher dice, “El nivel de compromiso depende de lo atractivo que sea el mercado. Típicamente, un mercado atractivo le permite al gobierno sacar adelante sus políticas ya que las compañías están dispuestas a renunciar a mayores márgenes de utilidad a cambio de un crecimiento sostenible durante un período de tiempo.”
El mercado de Corea del Sur se caracteriza por su pobla-ción anciana y una población mayor. El crecimiento potencial está limitado ya que ha evolucionado a un mercado desarro-llado, y los actores de la industria esperan tener regulacio-nes establecidas. Por lo tanto, tiene sentido que los actores de la industria estén pre-alertados de cualquier política en la agenda del gobierno. Por ejemplo, los actores de la industria fueron informados en 2010 de que la política de revelado de precios entrara en vigor en 2012. En Japón, los actores de la industria comprenden que es la práctica general que los recor-tes de precio ocurren una vez cada dos años.
A pesar de su mercado fragmentado y del revés en el re-corte de precios, Corea del Sur está clasificada entre los 12 principales del mundo con ventas anuales de $8,000 mdd.
Cher agrega, “A la larga, tiene los ingredientes necesarios para continuar con su exitosa industria farmacéutica, fuerte apoyo para la innovación, la voluntad del sector privado para explorar estas tecnologías innovadoras y el perfil demográfico también respalda el uso incrementado de fármacos. El con-flicto entre el gobierno y la industria surgirá definitivamente otra vez en el futuro, pero pensamos que las dos partes llega-rán a un compromiso.”
-Jane Wan es escritora independiente en Singapur
Los recortes de precios no pueden ser la única solución para contener los gas-tos crecientes del gobierno y también es injusto hacer recaer la carga en las empresas.
Atención: Terapia génica Primera terapia génica aprobada en EuropaLa Comisión Europea (CE) ha publicado la aprobación final para la primera terapia génica de Europa –un tratamiento para un raro trastorno genético que actualmente no tiene otras opciones de trata-miento. De acuerdo a una declaración de prensa, el tratamiento es la primera terapia génica en ser aprobada por las autoridades regulato-rias en el mundo Occidental.
El Glybera de uniQure (alipogene tiparvovec) está dirigido a paci-entes con deficiencia de lipoproteína lipasa (LPLD) que sufren de pan-creatitis aguda recurrente. El LPDL es un trastorno heredado que afec-ta a aproximadamente una a dos personas por millón. Los pacientes son incapaces de metabolizar las partículas de grasa en la sangre, llevando a inflamación del páncreas y, en algunos casos, la aparición temprana de diabetes y complicaciones cardiovasculares. Utilizando vectores de virus adeno-asociados como vehículo de entrega, Glybera añade copias de trabajo del gen de lipoproteína lipasa en las células musculares para habilitar la producción de enzimas.
“La aprobación final de Glybera de la CE marca un paso mayor adelante en hacer las terapias génicas disponibles no sólo para el LPLD sino también para un gran número de enfermedades raras con una muy alta necesidad médica sin cumplir,” dijo Jörn Aldag, CEO de uniQure en una declaración. La compañía está también planeando solicitar la aprobación regulatoria en EEUU, Canadá y otros mercados.
La Agencia Europea de Medicamentos (EMA) recomendó pri-mero Glybea para su aprobación en Julio de 2012, con el Comité para Productos Medicinales para Uso Humano (CHMP) del EMA que recomienda el otorgamiento de una autorización de comercialización bajo “circunstancias excepcionales”. uniQure sometió primero Glybera como tratamiento para LPLD al EMA en el 2009., pero recibió una opinión negativa. Sin embargo, al principio del 2012, la CE le pidió a la EMA que reevaluara la aplicación en un grupo restringido de pacientes con ataques de pancreatitis severas o múltiples. Se espera que el despliegue comercial del tratamiento comience en la segunda mitad del 2013.
—Stephanie Sutton

Pharmaceutical Technology en Español MARZO / ABRIL 201310
manufactura y rEgulación dE fármacos
Vistazo al futuro en manufactura y regulaciónAngie Drakulich
La FDA habla acerca del panorama cambiante de la ciencia regulatoria y su efecto en las revisiones de fármacos, las inspecciones al sitio y los enfoques generales.
La ciencia regulatoria sustenta la innovación y prácticas en la manufactura de fármacos. La definición más común de
ciencia regulatoria es una disciplina que crea nuevas herramientas, estándares, y esquemas para uso en la evaluación de la seguridad, efectividad, calidad y desem-peño de productos.
En octubre de 2010, la FDA publicó un marco de trabajo para avanzar en la ciencia regulatoria para la salud pública, el cual se enfocó en lo siguiente: entrega acelerada de nuevos tratamientos médi-cos; mejora de la salud pediátrica y de niños; protección contra enfermedades infecciosas emergentes y terrorismo; mejora de la seguridad y salud a través de la informática; protección del sumi-nistro de alimentos; modernización del análisis de seguridad; cumplimiento de desafíos para la regulación del tabaco; y establecer un marco de trabajo en cola-boración para la implementación.
Después de este marco de trabajo, en agosto 2011, la agencia publicó un plan estratégico para el avance de la ciencia regulatoria en la FDA, enfocado en gran medida en la toma de decisiones basada en la ciencia para mejorar la salud pú-blica. Parte de esta iniciativa identifica estándares específicos, métodos, y pre-guntas para que los revisores de fárma-
cos los usen en su proceso de revisión. El esfuerzo general se construye en la Iniciativa del Camino Crítico de la agen-cia, lanzado en el 2004, para dirigir la in-novación en procesos científicos, la cual incluye el reporte del 2011 que prioriza ocho áreas, las cuales incluyeron soporte para nuevos esquemas para mejora de la manufactura y calidad del producto.
La FDA ha lanzado varios Centros para la Excelencia en la Ciencia Regu-latoria y la Innovación (CERSI) para llevar a cabo estas prioridades (ver el recuadro sobre el papel de la acade-mia). La iniciativa de la agencia en la ciencia regulatoria también fue mejora-da con la aprobación del Acta de Segu-ridad e Innovación de la Administración de Alimentos y Fármacos (FDASIA) en Julio de 2012. La sección 1124 del FDASIA demanda la mejora de la toma de decisiones de productos médicos a través de documentos guía y la adop-ción de herramientas, métodos y proce-sos. La Secretaría de Servicios y Salud Humanos (HHS) de EEUU debe emitir reportes de desempeño sobre estos ob-jetivos para los años fiscales (FY) 2014 y 2016. Pharmaceutical Technology ha-bló con la FDA acerca de estos temas en una entrevista especial sobre el futuro de la regulación bio/farmacéutica.
Calidad de la manufactura de fármacosPharmTech: En agosto de 2011, La FDA publicó un plan estratégico, Avan-ce de la Ciencia Regulatoria en la FDA. La prioridad 3 de ese plan se enfoca en la manufactura y calidad del producto, que incluye permitir el desarrollo y eva-luación de nuevos y mejorados métodos de manufactura para mejorar la calidad del producto, esto es, a través de la ma-nufactura continua, nuevas tecnologías de manufactura, excipientes y comple-jas formas farmacéuticas, y los esque-mas de tecnología analítica de proceso (PAT) y calidad por diseño (QbD). ¿Qué es lo que la FDA y la industria esperan obtener concentrándose en estas áreas?
FDA: La FDA es responsable de la protección del consumidor a través de la disponibilidad de productos de cali-dad. Estos esquemas se concentran en la ciencia profunda, buena para lograr un producto de alta calidad. Aplicando la QbD y comprendiendo el papel de los excipientes y de las formas farmacéu-ticas complejas, se obtendrá un mayor nivel de comprensión del producto y del proceso. Los otros esquemas (PAT, ma-nufactura continua y nuevas tecnologías de manufactura) utilizan esa compren-sión para lograr procesos de manufac-tura más robustos y capaces, resultando en mayor calidad del producto. Con este mayor nivel de calidad del producto, se espera que caiga la tasa de fallas del producto, lo que a su vez incrementará la disponibilidad del producto y redu-cirá el costo del producto. Por lo tanto, enfocándose en estos esquemas basados en la ciencia, la FDA confía que la cali-dad general de los productos farmacéu-ticos continuará incrementándose.
Modernizando los esquemas de ma-nufactura para los farmacéuticos, como sería a través del QbD, la manufactura continua y/o el PAT, tiene el potencial de beneficiar a la industria, a los regu-ladores y a los pacientes. Ya ha habido múltiples reportes de calidad y benefi-cios en costo para los fabricantes a tra-vés del uso de los esquemas del QbD y/o del PAT. Para los pacientes, estos esquemas pueden llevar a un mayor ase-guramiento de calidad y disponibilidad del producto. Para los reguladores, esto significa potencialmente menos super-visión regulatoria post-aprobación de-

Pharmaceutical Technology en Español MARZO / ABRIL 2013 11Marque en la tarjeta de servicio al lector el No. 7

Pharmaceutical Technology en Español MARZO / ABRIL 201312
manufactura y rEgulación dE fármacosJu
pit
er
iMa
Ge
s/t
hin
ks
toc
k i
Ma
Ge
s
La FDA tiene dos Centros de Excelencia en Ciencia Regulatoria e Innovación (CERSI) relativamente nuevos. La FDA tiene Centros de Excelencia existentes con el Estado de Arkansas y otros institutos, pero los programas de la Universidad de Maryland (UMD) y Georgetown son los más recientemente establecidos, lanzados en octubre del 2011 a través
de una subvención anual de $2 mdd. Los programas del CERSI en general están pensados para ayudar a la FDA en su estrategia de ciencia regulatoria identificando estándares, métodos y preguntas para los revisores de fármacos.
El equipo de trabajo de la UMD está focalizado en la mejora de las evaluaciones preclínicas de seguridad, aprovechamiento de datos para mejorar los resultados de salud (es decir, “Big Data”) y asegurar que la FDA esté lista para evaluar tecnologías innovadoras y emergentes. El grupo de Georgetown está concentrado en aprovechamiento de datos así como estimular la innovación en evaluaciones clínicas y medicina personalizada para mejorar el desarrollo del producto y los resultados en el paciente. Pharmaceutical Technology habló con los directores del proyecto en el programa CERSI de la UMD, James Polli, PhD, y en el programa CERSI de la Universidad de Georgetown, Erin Wilhelm.
PharmTech: ¿Qué temas clave espera abordar su equipo del CERSI dentro de estas áreas prioritarias de investigación, y puede dar un ejemplo?
Polli (UMD): Con respecto a nuestro enfoque sobre “asegurar que esté listo”, las dos áreas en las que se está concentrando la UMD son imagenología hiperespectral (HSI) y estructuras de construcción de tejidos. El HSI representa una tecnología emergente con un rango de aplicaciones clínicas vigentes y potenciales, incluyendo oximetría de tejidos y detección temprana de cáncer. Sin embargo, hay actualmente una carencia de métodos estandarizados para asegurar la exactitud y consistencia de instrumentos de imagenología óptica. Hemos medido experimentalmente varias nanopartículas utilizando HSI. Se ha identificado una serie óptima de características para métodos de prueba basados en el blanco y en fantasmas. Evaluaremos además los beneficios e inconvenientes de utilizar características específicas de calidad de imagen, métricas, blancos/fantasmas e identificar los que parecen más apropiados para su incorporación en un protocolo de análisis estándar. Este estudio propuesto resultará en métodos de prueba con calidad de imagen validada para la FDA en la evaluación de sistemas de imagen y después la comprensión de los sistemas de imagenología espectral que se someten a la FDA.
El objetivo del proyecto de estructuras de construcción de tejidos es establecer relaciones entre la arquitectura de los andamios de construcción de tejidos y la respuesta resultante del tejido que puede ser adaptada a una estrategia de regeneración. Un aspecto crítico de este trabajo es la fabricación de andamios con arquitectura precisa y la alta exactitud de caracterización de estos andamios.
Wilhelm (Georgetown): ONuestro objetivo en Georgetown es ayudar a la agencia a poder responder las preguntas de ciencia regulatoria estratégica. El Centro de Innovación de Georgetown para Informática Biomédica (ICBI), por ejemplo, se asocia con la FDA para expandir la utilidad de las herramientas bioinformáticas existentes o para crear nuevas
herramientas. En las primeras etapas “beta”, estas herramientas le ayudan a la agencia a lograr su misión regulatoria. Finalmente, las herramientas pueden ser más desarrolladas para soportar la toma de decisiones clínica, pero todavía no estamos en esa etapa. Un equipo del ICBI está trabajando con la FDA en su sistema de reporte de eventos adversos de vacunas (VAERS) para buscar los potenciales vínculos entre las vacunas y los trastornos autoinmunes. Esta área es importante porque la FDA recibe reportes VAERS de cualquiera –pacientes, abogados, clínicos, miembros de la familia, u otros- de manera que los datos no han sido verificados, lo que indica que un reporte no significa que una vacuna haya causado realmente una respuesta autoinmune. Para ayudar a que los datos tengan sentido, el equipo de Georgetown cura la gran base de datos, la cual usa la FDA para identificar los verdaderos vínculos causales, si existe alguno. Dichos proyectos tienen implicaciones importantes para la salud pública.
PharmTech: La FDA ha identificado varias lagunas que ve en la ciencia regulatoria, y estas lagunas le han dado forma a su plan estratégico y a las áreas de prioridad de investigación del CERSI. ¿Hay algún hueco adicional que piense que debería abordarse a futuro?
Polli (UMD): Todos estamos familiarizados con la necesidad de tener mejores herramientas para un desarrollo más pronosticable. La investigación CERSI de la UMD está enfocada en ayudar a desarrollar la guía de la FDA en las áreas de fármacos y dispositivos. Son necesarios los mejores biomarcadores, por ejemplo. En un nivel más elevado del sistema, existen indudablemente áreas que pueden ser racionalizadas, como la armonización. Por el lado del CMC, el análisis para liberación en tiempo real y la implementación de la calidad por diseño (QbD) necesitan más avance. Algunos en la industria piensan que el tiempo es correcto para actualizar el escalamiento y los cambios post-aprobatorios (SUPACs) también. Se genera gran cantidad de datos de estabilidad, pero la mayoría de estos datos no son esenciales.
Wilhelm (Georgetown): Un área sería el concepto de la ciencia regulatoria en general. Existen muchas ideas falsas de lo que representa el término. Muchos seleccionan enfocarse en los aspectos ‘regulatorios’ y no comprenden que ‘ciencia regulatoria’ está enfocada en la ciencia que sustenta la regulación profunda. No es sólo acerca de leyes y regulaciones, sino más bien, acerca de la toma de decisiones y decisiones basadas en la ciencia. A lo largo de estas líneas, existen enormes brechas en los programas de educación y capacitación en ciencia regulatoria para la agencia regulatoria así como entre la industria y la academia. La promoción de la educación en ciencia regulatoria ayudará a tener científicos con una mentalidad activa de ciencia regulatoria en campos de vanguardia.
PharmTech: Los programas del CERSI sostienen sesiones mensuales del personal de capacitación con los miembros del personal de la FDA. ¿Qué está involucrado y pueden participar los miembros de la industria?
Polli (UMD): En la UMD, las conferencias mensuales han sido bien recibidas. Hemos trabajado con el personal de desarrollo profesional de la FDA para identificar las necesidades del revisor, y solicitamos a los miembros del personal de ciencias de la FDA que sugieran temas y ponentes. Los ponentes han incluido a la facultad de Maryland, académicos de otras universidades y ponentes de la industria. Los elementos subsiguientes del portafolio de capacitación incluyen intercambio de conferencias, un programa de educación personalizada para científicos de la FDA, y un grado de maestría en línea en ciencia
Papel de la academia en el futuro de la manufactura y regulación farmacéutica, por Angie Drakulich

Pharmaceutical Technology en Español MARZO / ABRIL 2013 13
Mar
que
en la
tarje
ta d
e se
rvici
o al
lect
or e
l No.
4
regulatoria. Adicionalmente, estamos trabajando con la FDA para identificar un número de conferencias de un día que se realizarán en la UMD y serán abiertas al público. Estas conferencias de intercambio para la industria empezarán en febrero del 2013.
Wilhelm (Georgetown): Hemos trabajado con nuestros colegas de la UMD en su serie de conferencias para la FDA. Georgetown también ha promovido intercambios científicos entre los científicos que trabajan en la agencia y dentro de nuestros departamentos académicos para colaborar en proyectos. Varios de los proyectos en los que trabajan los equipos de la FDA y del ICBI de Georgetown surgieron de los talleres “Día de Investigación” que hacíamos en la FDA y que estamos planeando preparar futuros eventos para identificar otros proyectos donde la universidad y la FDA puedan trabajar juntos. El CERSI de Georgetown también ha trabajado para abrir un número de oportunidades de educación continua para los cientos de
clínicos que trabajan en la FDA y quieren mantener sus licencias médicas, de enfermería y otras licencias clínicas. Finalmente, Georgetown lanzó su maestría del programa de ciencias en ciencia regulatoria en otoño del 2012 con nuestro curso introductorio, Introducción a la Ciencia Regulatoria. La clase fue un enorme éxito. El programa completo de la maestría estará disponible en línea empezando el otoño de 2013. Como la ciencia regulatoria tiene aplicaciones para muchas disciplinas científicas, e impacta la salud pública en numerosas formas, pensamos que es importante asociarse con un amplio rango de interesados. El CERSI de Georgetown ha llegado a otros centros académicos, industria, gobierno, como el NIH, así como al público, para ayudar a darle forma a este campo en ciernes.
Para más información de los programas CERSI, contacte: [email protected] o visite http://regulatoryscience.georgetown.edu.
bido a la flexibilidad regulatoria (p.ej., espacio de diseño) sometida con la soli-citud. La FDA está actualmente tras de vías adicionales para facilitar cambios post-aprobatorios para procesos bien comprendidos y controlados bajo un sistema de calidad robusto.
La manufactura continua, aunque nueva para la mayoría de la manu-factura farmacéutica, es una tecnolo-
gía comúnmente usada en alimentos y procesos químicos. Conforme más compañías adquieren experiencia con la manufactura continua, emergen los beneficios, incluyendo casos de proble-mas reducidos en el escalamiento de la manufactura, uso reducido de material en el desarrollo y capacidades flexibles de manufactura. Debido a la cambian-te dinámica de nuevos fármacos hacia
productos más especializados, de me-nor volumen, la manufactura continua podría volverse un lugar común en los años por delante.
Finalmente, se está volviendo cada vez más importante comprender las in-teracciones entre fármacos, excipientes y componentes del dispositivo conforme las formas farmacéuticas se vuelven más complejas, tal como ingredientes múlti-

Pharmaceutical Technology en Español MARZO / ABRIL 201314
ples activos en un solo producto farma-céutico o en combinaciones de fármaco-dispositivo. El mayor nivel de compren-sión debe permitir productos más robus-tos que entreguen el desempeño que se
manufactura y rEgulación dE fármacos
pretende cuando se introduzcan cambios ya sea planeados o sin planear.
Métodos AnalíticosPharmTech: La prioridad 3 en el plan
estratégico también demanda el desa-rrollo de nuevos métodos analíticos, que incluyan aquéllos para determinar la ‘similitud’ entre productos de refe-rencia y biosimilares así como herra-
PharmTech: DDSM Pharmaceutical Products, la empresa de manufactura y tecnología a la medida de Royal DSM N.V., recibió un premio por la mejor CMO (Organización de Manufactura por Contrato) en el verano de 2012 del Segundo evento anual de Premios a la Industria de Asia Biofarmacéutica en Singapur por su suite de extremo a extremo de soluciones para el cliente. Innovaciones y estándares en la región. ¿Qué esquemas únicos ha tomado DSM
con respecto a este reconocimiento?Wessels: DSM está comprometido a proporcionar soluciones para
el futuro de la manufactura bio/farmacéutica mientras conduce la sustentabilidad para la producción de moléculas grandes y pequeñas. Pensamos que DSM tiene una de las más diversas CMO y la tecnología la posiciona en el espacio de la subcontratación farmacéutica, y con este increíble rango de tecnología y conocimiento, estamos concentrados en llevar el siguiente nivel de soluciones a nuestros clientes ya que enfrentan mayores retos para cumplir las demandas del mercado encontrando mientras tanto nuevos caminos en la manufactura de fármacos.
Por ejemplo, DSM ha mantenido una amplia base de experiencia y operaciones a través de las plataformas de la manufactura farmacéutica, que incluyen las áreas de mamíferos, microbiológicos, bio/químicos y formas farmacéuticas terminadas a través de las altas y bajas del mercado durante los pasados años. Estamos apalancando estos recursos para encontrar soluciones viables únicas para los clientes a quienes servimos. DSM le da servicio a nueve de las 10 principales compañías farmacéuticas, y de esta forma tenemos desafíos a diario en todas estas áreas. PharmTech: En su experiencia, ¿Qué capacidades se espera que tengan las CMOs hoy día que no tenían hace 5 o 10 años?
Wessels: DLa apuesta de DSM en el mercado de la subcontratación farmacéutica se basa en un portafolio global de recursos para servir continuamente a las necesidades cambiantes del cliente y darle valor real conforme la industria farmacéutica cambia modelos de negocio. Esto requiere amplitud y profundidad en la experiencia en las diversas áreas de la tecnología farmacéutica que pueden ser aplicadas a los retos del cliente para procesos cada vez más eficientes. Estamos incluyendo dicha innovación a través de nuestras plataformas de mamíferos, microbiológicas, químicas y de formas farmacéuticas terminadas. En el área de tecnología analítica de proceso (PAT), por ejemplo, DSM ha demostrado las eficiencias y seguridad de la tecnología de microrreactor para la producción a escala comercial de APIs como un ejemplo de la intensificación del proceso para lograr una producción comercial más eficiente, mejor manejo de los volúmenes de
manufactura, y baja del costo total. De esta forma podemos desafiar las tecnologías de proceso actuales con innovaciones en la intensificación del proceso para las soluciones de manufactura de la siguiente generación. PharmTech: ¿Cuál es su opinión sobre la consolidación de la industria que ha estado ocurriendo?
Wessels: Algunos sectores del mercado de la CMO experimentan más presión que otros; el crecimiento biofarmacéutico es todavía alto mientras que la consolidación en el sector de los APIs, particularmente en los países occidentales, es algo que hemos visto que sucede conforme las menores tasas de crecimiento para los fármacos innovadores se equilibran con los genéricos. El aumento de la competencia en este espacio es intenso tanto en la perspectiva del Oriente como en la del Occidente así como donde las CMOs pueden llevar soluciones rentables, sostenibles y confiables a la mesa de cualquier región del mercado. Yo creo que es importante señalar que la consolidación está siendo forzada no sólo desde una perspectiva de capacidad sustentable, sino también por razones de calidad y seguridad.
Nuestras cadenas de suministro global se están expandiendo y la seguridad de los ingredientes y de los fármacos terminados se ha vuelto crítica conforme los recursos de subcontratación, nuevos y viejos, son puestos a prueba. El incremento de las cartas de advertencia emitidas indica claramente una preocupación creciente desde una perspectiva regulatoria. En DSM, somos extremadamente sensibles a esta responsabilidad en la manufactura. PharmTech: ¿Qué nuevas tendencias y tópicos espera ver en el radar de la industria de los CMOs farmacéuticos en el año?
Wessels: Se ha vuelto críticamente claro que la calidad y confiabilidad son fundamentales para el desempeño del CMO y que llegará a tener en el equilibrio de los proveedores a través de las regiones globales en términos de consolidación y alianzas. DSM tiene una organización mundial de expertos en asuntos regulatorios. Estos especialistas siguen cambios en las regulaciones locales y mantienen una estrecha relación con las autoridades locales relevantes, asegurando que nuestros productos siempre cumplen con las regulaciones de la región. Esta organización global nos permite respaldar y aconsejar a nuestros clientes en un amplio rango de cuestiones regulatorias, asegurando por último la calidad y confiabilidad.
Adicionalmente, la sustentabilidad de los propios CMOs es un tema clave. En DSM, no nos limitamos a nosotros mismos al perfil tradicional del CMO, sino que nos convertimos en un amplio socio farmacéutico con un ojo en el futuro del mercado farmacéutico desde una perspectiva de cadena de suministro y la necesidad de modelos de manufactura sustentables para soportarlo. Nosotros estamos tomando un esquema híbrido fortaleciendo nuestro núcleo y las diversas actividades del CMO en biotecnología y en las tradicionales de APIs y de producción de formas farmacéuticas terminadas únicas, y equilibrando nuestro portafolio con actividades en biosimilares y genéricos, involucrando socios globalmente, para ayudarlos a manejar sus ciclos de vida del producto.
¿A dónde se dirigen las CMOs? Sesión de preguntas y respuestas con el CEO de DSM Pharmaceutical Products Alexander Wessels, por Angie Drakulich
DSM Pharmaceutical Products CEO Alexander Wessels

Pharmaceutical Technology en Español MARZO / ABRIL 2013 15
mientas para detectar propiedades físi-cas de formas farmacéuticas complejas. ¿Qué ganancia se espera obtener entre los científicos de la agencia en estas áreas analíticas?
FDA: Los avances en métodos analí-ticos para determinar la ‘similitud’ entre productos de referencia y biosimilares así como herramientas para detectar pro-piedades físicas de formas farmacéuticas complejas permitirá una mayor confian-za de que no haya diferencias estructu-rales entre productos o que cualquier diferencia observada sea menor. Para los biosimilares, esto reducirá la incertidum-bre y permitirá un programa de desarro-llo dirigido. Esto puede también facilitar el desarrollo y aprobación de genéricos con formas farmacéuticas complejas así como informar las estrategias de control para la calidad de los productos origina-dores complejos.
Contaminación microbianaPharmTech: La prioridad 3 del plan estratégico de agosto del 2011 deman-da la reducción de la contaminación microbiana de los productos médicos. ¿Qué objetivos específicos tiene la FDA en esta área?
FDA: La FDA ha intentado identifi-car y estudiar los huecos específicos en la manufactura farmacéutica. Los estu-dios están en curso en la filtración es-terilizante, efectos de las energías para esterilización terminal y métodos para la detección de microorganismos elusi-vos en componentes de fármacos y en entornos de manufactura.
Los estudios de filtración han in-cluido tasas de penetración microbio-lógica basadas en el tamaño de la cé-lula y la composición de la matriz del filtro así como la penetración debida al tiempo en varios filtros con dos tama-ños de microorganismos en desarrollo. El objetivo de estos estudios es mejorar los estudios de validación de filtros que previamente demostraron tener vul-nerabilidades. Los pasos futuros en la investigación buscarán condiciones de solución que estimulen la miniaturiza-ción de las células, neutralicen las cé-lulas o cargas en la superficie del filtro que mejoren la captura de células en las membranas del filtro.
Muchos productos son fabricados utilizando proceso aséptico, el cual puede ser menos efectivo que la este-rilización terminal. Algunos otros pro-ductos quirúrgicos no son fabricados para ser estériles del todo, y también poseen un riesgo. Cada una de estas
situaciones es el resultado de las eva-luaciones del riesgo que están basadas en la naturaleza del producto y su vul-nerabilidad a la energía esterilizante u otras condiciones. La FDA y el NIPTE [Instituto Nacional para la Tecnología Farmacéutica y la Educación] han ini-ciado estudios de productos (el enfoque inicial son los antisépticos quirúrgicos) para evaluar los efectos de la energía es-terilizante en productos seleccionados. El objetivo de estos estudios es definir condiciones que puedan permitir la ma-nufactura usando procesos destinados a producir un producto estéril, el cual es más seguro de usar en procedimientos quirúrgicos.
Los productos no estériles con fre-cuencia requieren una demostración de que los microorganismos, que normal-mente se espera que estén en el pro-ducto, no crecerán o serán eliminados. Se espera una demostración similar de los productos estériles que pueden ser usados varias veces (p.ej., viales mul-tidosis). Sin embargo, existen cepas microbiológicas que pueden resistir conservadores usados para controlar la contaminación, y algunas de estas cepas incluso crecen en presencia de los agen-tes antimicrobianos. Si se les permite crecer, un gran número de microorga-nismos puede alcanzar una dosis po-tencialmente infecciosa al exponerse a los pacientes. Entre estas especies están las bacterias comunes del agua que los CDC [Centros para el Control y Preven-ción de Enfermedades] han demostrado que son difíciles de detectar utilizando métodos farmacopeicos. En colabo-ración con el CDC y con la ayuda de
la Universidad de Michigan, estamos examinando métodos de cultivo para el recobro de estos organismos del agua farmacéutica. También se están consi-derando tecnologías alternativas para su detección. El objetivo de estos estudios es el desarrollo de métodos para detec-
tar confiablemente estos contaminantes potenciales.
Papel de los CERSIsPharmTech: Una manera de mejorar la ciencia regulatoria (desde un punto de vista de revisión del fármaco) es traba-jar con la industria directamente para identificar y llenar los huecos científicos y tecnológicos que existen a través del desarrollo y manufactura de fármacos. La FDA ha creado unos pocos CERSIs basados en subvenciones, específica-mente con la Universidad de George-town (UG), la Universidad de Maryland (UMD), y el Estado de Arkansas. ¿Es-tará abriendo la agencia algunos centros de excelencia adicionales?
FDA: Esperamos en el futuro fundar uno a dos CERSIs adicionales fuera de la región, pero el cómo y cuándo depen-derán de la capacidad de financiamiento.
PharmTech: ¿Puede comentar sobre los talleres de capacitación del personal realizados a la fecha con estos centros y su beneficio para los revisores de la agencia?
FDA: Los revisores de la agencia han tenido la oportunidad de participar en eventos de capacitación patrocina-dos tanto por los CERSIs de la UMD y de la UG. Estos eventos han propor-cionado educación médica continua al personal y oportunidades para enlazar-se con investigadores líderes de todo el país. Adicionalmente, los revisores de la agencia también han tenido la opor-tunidad de mantener sus habilidades científicas y médicas participando en el trabajo clínico, la investigación o en
Debido a la dinámica cambiante de los nuevos fárma-cos hacia productos más especializados, de menor volumen, la manufactura continua podría volverse un lugar más común en los años futuros.

Pharmaceutical Technology en Español MARZO / ABRIL 201316
manufactura y rEgulación dE fármacos
La Sociedad Internacional de Ingeniería Farmacéutica (ISPE) emitió un documento guía en septiembre de 2012 dirigido a establecer una línea base para el diseño de las instalaciones del
laboratorio de calidad. La Guía de Buenas Prácticas del ISPE: Instalaciones del Laboratorio de Calidad provee consideraciones paso a paso para producir un laboratorio de productos farmacéuticos de calidad. Aquí, la Presidente del ISPE, Nancy Berg, habla acerca de la guía.
PharmTech: ¿Cuáles fueron los objetivos primarios para emitir una guía sobre este tema? En otras palabras ¿Qué retos regulatorios y huecos de tecnología/ingeniería estuvieron enfrentando las industrias bio/farmacéuticas que necesitaban ser abordados?
Berg: Uno de los objetivos generales de ISPE es ayudar a la industria a crear puntos comunes de referencia y lenguaje común, de manera que a todos los niveles y funciones de trabajo se pueda trabajar en conjunto más fácilmente para abordar y resolver retos técnicos. En el espacio del laboratorio de calidad, identificamos que los propietarios e ingenieros del laboratorio de calidad necesitaban una herramienta para ayudar a definir los requerimientos para la renovación de los laboratorios existentes y/o desarrollar nuevas instalaciones de laboratorio. Ese fue el primer objetivo de la Guía de Buenas Prácticas del ISPE: Instalaciones del Laboratorio de Calidad, la cual presenta las guías de diseño enfocadas en los laboratorios de calidad farmacéutica dentro o como parte de un entorno regulado GxP.
El uso de la guía ayudará a eliminar los huecos de ingeniería creados por un equipo de ingeniería que se mueve hacia delante sin un alcance del trabajo apropiadamente definido. La guía aborda problemas tales como la definición de los requerimientos del laboratorio y finalmente realizando la evaluación de riesgo con el equipo completo antes de detallar el diseño. Enfocándose en el cliente para definir los requerimientos, esta guía es útil tanto para el propietario como para el arquitecto/ingeniero. Nuestro objetivo es que las empresas utilicen esta guía como un camino a seguir para sus clientes para ayudarlos a definir los entregables y eliminar la incertidumbre.
PharmTech: La guía la proporciona a la industria recomendaciones específicas para cumplir los requerimientos de las GMPs de regulaciones globales como el 21 CFR 210-211, la guía Q7 de la Conferencia Internacional de Armonización sobre GMPs para APIs, la Guía de GMPs de la Unión Europea y otras cGMPs. La realización de una evaluación de riesgo es un tema clave a través de estos requerimientos, y la guía del ISPE describe cómo aplicar dicha evaluación a una instalación de laboratorio de calidad.
PharmTech: ¿Puede ofrecer algunos puntos clave de la guía en esta área en términos de lo que la industria debe considerar que quizás no hayan considerado en el pasado con respecto al riesgo?
Berg: La guía provee orientación muy necesaria sobre cómo aplicar una evaluación de riesgo a una instalación de laboratorio de calidad e identifica elementos a ser considerados cuando se realiza la evaluación de riesgo. Básicamente, se adapta a muchos de los mismos conceptos de riesgo descritos en el ICH Q9 para este tipo de instalación. Por ejemplo, una evaluación de riesgo puede revelar que un espacio de laboratorio es insuficiente para el almacenamiento planeado o el montaje de muestras y reactivos para
análisis. Desde luego, este tipo de deficiencia puede llevar a varios problemas, incluyendo una mezcla de las muestras o reactivos.
La evaluación de riesgo permite que el cliente considere los requerimientos necesarios para cumplir las regulaciones. Muy a menudo, el grupo de soporte de arquitectos e ingenieros entrega una instalación técnicamente elaborada que está muy lejos de la necesidad del propietario para soportar el análisis. El propietario está entonces atrapado con la calibración y la validación en curso que no es necesaria para los protocolos de análisis. Por otro lado, si la guía se utiliza al máximo, el propietario del laboratorio puede definir aquellas áreas necesarias que están técnicamente elaboradas.
PharmTech: Los laboratorios de especialidades, tales como los que analizan y liberan la sustancia farmacéutica o producto farmacéutico que tiene riesgo biológico o producido en un proceso estéril, enfrentan desafíos únicos. La guía del ISPE incluye una sección dedicada a estos complejos laboratorios. ¿Cómo tienen que ajustar su esquema los equipos de calidad cuando trabajan con estos tipos de laboratorios e instalaciones?
Berg: Esta es un área muy interesante y muy compleja. La guía aborda varios tipos de laboratorios de especialidades, incluyendo laboratorios asépticos y para pruebas de esterilidad, laboratorios con riesgo biológico, y laboratorios de compuestos potentes. Cada uno de estos laboratorios tiene retos de diseño únicos y consideraciones regulatorias, y por lo tanto es difícil hacer generalizaciones acerca de esta sección.
Dependiendo del tamaño de la muestra, muchas de las condiciones especiales pueden lograrse con aisladores o biocampanas, especialmente si la muestra está cerrada y sólo se abre para el análisis en pequeñas cantidades. Otras áreas de preocupación son el manejo de solventes dentro del laboratorio y su remoción como productos de desecho del laboratorio. Los solventes juegan un gran papel en el análisis y es crítico asegurarse de que se mitiguen los riesgos de explosión así como los riesgos de fuego. La ley permite cantidades fijas de solventes dentro del laboratorio, por lo que el aspecto de diseño de esto es crítico para su operación continua. En general, sin embargo, he referido a los interesados en este tema al contenido de la guía, la cual contiene información mucho más detallada que la que puedo darles aquí.
PharmTech: La guía ISPE dedica una sección para registrar el manejo y recomienda que cada laboratorio desarrolle un manual de operaciones para identificar riesgos potenciales, junto con prácticas y procedimientos a ser seguidos para minimizar o eliminar esos riesgos. ¿Los equipos del laboratorio de calidad bio/farmacéutica de hoy día tienden a tener dichos manuales y piensa que estarán inclinados a tenerlos más adelante?
Berg: Aunque siempre es difícil predecir el futuro, creo que es seguro decir que la industria tendrá alguna forma de documentación basada en el riesgo, particularmente debido al ICH Q8, Q9 y Q10. Como el laboratorio de calidad maneja una variedad de productos, materias primas y solventes, existe una necesidad definida por los procedimientos estándar de operación. Existe también la necesidad de pruebas documentadas de que haya en el lugar un programa de capacitación para confirmar que los científicos experimentados y el personal de soporte estén completamente conscientes de los riesgos que rodean su operación y de que los dispositivos dentro del laboratorio se utilicen apropiadamente. El objetivo es minimizar el riesgo de estos peligros, tanto para el científico como para la muestra. Además de la capacitación y de los procedimientos de operación, el laboratorio de calidad debe estar equipado con un sistema de validación en el mantenimiento
El laboratorio de calidad futuro, de acuerdo al ISPE, por Angie Drakulich
ry
an
Mc
va
y/t
hin
ks
toc
k iM
aG
es

Pharmaceutical Technology en Español MARZO / ABRIL 2013 17
de registros del análisis realizado, lo cual es la base para la liberación del producto al mercado.
PharmTech: La guía incluye un apéndice para las diferencias clave a considerar en los laboratorios bio/farmacéuticos con base en Europa. ¿Estuvieron los funcionarios de la FDA o del EMA involucrados en la revisión de la guía antes de su publicación y éstos respaldan sus recomendaciones?
Berg: Afortunadamente para el ISPE, ambas agencias estuvieron involucradas en el proceso de revisión de la guía y su retroalimentación fue incorporada en el borrador final. No es típico para los cuerpos regulatorios, como la FDA y EMA, ratificar o respaldar tácitamente las guías para la industria
–y no es su papel hacerlo. Con esto, yo creo que es justo decir que teniendo su aportación durante el proceso de revisión ha ayudado a que la guía sea reflexiva del pensamiento regulatorio actual de ambas agencias a este respecto.
PharmTech: En general, basado en esta nueva guía, ¿Qué diferencias clave espera usted y el ISPE en el laboratorio de calidad bio/farmacéutica del futuro?
Berg: Finalmente, el ISPE espera que esta guía le de a las instalaciones del laboratorio de calidad con las necesarias herramientas basadas en el riesgo para respaldar la liberación de productos farmacéuticos de alta calidad para los pacientes.
oportunidades de enseñanza en el CER-SI de la UMD y de la UG.
Avances en la cienciaregulatoriaPharmTech: La sección 1124 del FDA-SIA establece que la Secretaría de HHS de EEUU debe tener una estrategia y un plan de implementación ‘para el avance de la ciencia regulatoria para productos médicos con el fin de promover la salud pública y el avance de la innovación en la toma de decisiones regulatoria’ para julio de 2013. ¿Cuál es el papel de la FDA?
FDA: La FDA está tomando el lide-razgo en el desarrollo de la Estrategia y Plan de Implementación para Productos Médicos, según se requiere en la sec-ción 1124 del FDASIA.
PharmTech: ¿Se esperan algunos objetivos adicionales mayores más allá del plan estratégico vigente de la FDA que rodeen a la ciencia regulatoria?
FDA: No, la FDA espera permane-cer dentro del marco de trabajo señala-do en el Plan Estratégico para la Ciencia Regulatoria. Las principales áreas de enfoque detalladas en el Plan Estratégi-co para la Ciencia Regulatoria identifi-can amplias áreas donde son necesarios los avances en la ciencia regulatoria y son por lo tanto muy amplias. Se espera que los reportes anuales de avance, los cuales seguirán en 2014 y 2016, inclu-yan información más detallada de los logros específicos relacionados con la ciencia regulatoria.
Inspecciones en el extranjero e importacionesPharmTech: Parte del FDASIA ayuda a financiar las inspecciones en el extran-jero de la FDA a través de nuevas tari-fas del usuario (es decir, las Enmiendas
a las Tarifas del Usuario de Fármacos Genéricos [GDUFA]). Empezando en el 2014, la inspecciones utilizarán me-todologías basadas en el riesgo. ¿Qué factores utilizará la agencia para deter-minar si y cuándo se inspecciona una planta en el extranjero?
FDA: Todavía tenemos que finalizar el esquema basado en el riesgo modifi-cado aunque el FDASIA Sección 705 detalla los factores relevantes para la evaluación del riesgo. Todas las instala-ciones serán inspeccionadas más pronto o más tarde. Las empresas nuevas para la FDA serán una prioridad como lo son las empresas que están buscando la apro-bación para un tipo de operación de ma-nufactura que es nueva para ese sitio. La FDA considerará el establecimiento de diferentes frecuencias para las plantas de producción de ingredientes activos y las plantas de formas farmacéuticas termi-nadas. Las instalaciones que hagan fár-macos estériles probablemente tendrán una mayor probabilidad que otros tipos de producción. La GDUFA espera que la frecuencia y rigor de las inspecciones a instalaciones extranjeras y domésticas sean equitativas, lo que tiene sentido para todos los fármacos, de manera que el modelo basado en el riesgo tendrá que asegurar que se logre esto.
PharmTech: El FDASIA requiere que la agencia emita una guía para julio de 2013 que defina cuándo puede negar-se o limitarse una inspección. ¿Cuál es el estatus?
FDA: De acuerdo a lo que requie-re la Sección 707del FDASIA, la FDA está actualmente trabajando en un pro-yecto de guía que abordará el alcance de las acciones o inacciones que cons-tituyan un retraso, una negación o una limitación de una inspección. La Sec-ción 707 considera un medicamento
mal etiquetado si se produce en una planta para la cual una inspección está retrasada, denegada, rehusada o limita-da. Esta provisión es importante porque crea un fuerte incentivo para todas las instalaciones, incluyendo instalaciones extranjeras, para permitir que la FDA realice inspecciones oportunas y com-pletas. Si una instalación doméstica retarda, niega, rehúsa o limita una ins-pección, la FDA tiene jurisdicción para obtener una orden de inspección. Antes del FDASIA, sin embargo, la FDA no tenía una herramienta efectiva para fa-cilitar la cooperación de instalaciones extranjeras con las inspecciones. Ac-tualmente, si una instalación extranjera retrasa, deniega, limita o rehúsa una inspección, la FDA puede rechazar la entrada para todos los productos farma-céuticos que se producen en esa instala-ción. Esto le ahorrará tiempo y recursos a la FDA y ayudará a asegurar la seguri-dad de nuestro suministro de fármacos.
PharmTech: El FDASIA permite que la FDA utilice información de las inspecciones de otros gobiernos o agen-cias. ¿Puede usted ampliar esto y com-partir otra información?
FDA: Los reportes de inspección de la FDA son generalmente descrip-ciones detalladas de cada inspección que incluyen el propósito, la descrip-ción de la amplitud y profundidad de la cobertura, los hallazgos y las discusio-nes significativas con el personal de la empresa. La FDA podría hacer uso de reportes comparablemente detallados de nuestras contrapartes y, bajo el FDA-SIA Sección 712, la FDA podría reco-nocer las inspecciones de un gobierno extranjero una vez que una revisión y auditoría de su sistema verifique sus inspecciones a satisfacción de la FDA. La FDA actualmente comparte repor-

Pharmaceutical Technology en Español MARZO / ABRIL 201318
tes de inspección, así como el acceso a sistemas de datos, con la contraparte de las agencias regulatorias nacionales que frecuentemente utilizan estos reportes o hallazgos de inspección en lugar de
su propia inspección. Afortunadamente, existe ahora una organización de ins-pectores farmacéuticos que armoniza las prácticas regulatorias de fármacos: el Esquema de Cooperación de la Ins-pección Farmacéutica (PIC/S). La FDA se convirtió en miembro del PIC/S hace aproximadamente dos años. La mem-bresía le da a la FDA una oportunidad de trabajar con autoridades colegas miembros del PIC/S para asegurar que los estándares de inspección sean ro-bustos y se sigan.
PharmTech: Con respecto a las im-portaciones, el FDASIA señala que los fabricantes extranjeros deben demostrar el estatus regulatorio de sus fármacos así como el cumplimiento de las cGMP y demostrar el registro de la planta den-tro de EEUU; de otra forma, la agencia tiene el derecho de destruir los fárma-cos importados que no cumplen, que tengan un valor inferior a $2500 dlls. Se estableció que estas reglas entren en vigor en julio de 2014 después de que sean desarrolladas más regulaciones. ¿Cómo diferirán los nuevos estándares de la importación actual y de los reque-rimientos de registro extranjero?
La GDUFA espera que la frecuencia y rigor de las inspecciones a planta domésticas y extranjeras sea equitativa.
FDA: El FDASIA le da a la FDA autoridad clara para requerir informa-ción acerca de los fármacos que se ofre-cen para importación y a rehusar su ad-misión si no se da esto, lo cual fortalece
la capacidad de la FDA para monitorear fármacos importados para ver si cum-plen los las leyes aplicables. De manera similar a muchos otros países, el FDA-SIA coloca la carga sobre el importador o propietario del producto para demos-trar que su fármaco cumple con los re-querimientos aplicables en EEUU. Es-tas autoridades de importación fortale-cen la capacidad de la FDA para evaluar que los fármacos importados cumplen con las leyes y regulaciones de EEUU. Las autoridades también posibilitan a la FDA para apuntar mejor a los productos de alto riesgo y evitar que entren a los Estados Unidos productos violatorios.
El FDASIA también le otorga a la FDA autoridad explícita para destruir los fármacos violatorios a los que se negó la admisión a los Estados Unidos si están valorados en $2500 dólares o menos. Antes de la promulgación del FDASIA, la FDA no tenía ninguna au-toridad de destrucción administrativa independiente. En lugar de esto, la FDA sólo podía emitir una ‘notificación de rechazo a la admisión’ por los fármacos que no cumplían ofrecidos para la ad-misión. Una vez que la FDA emitía la ‘notificación de rechazo a la admisión,
manufactura y rEgulación dE fármacos
la FDA tenía que apoyarse en la Pro-tección de Aduanas y Fronteras (CBP) para exportar o destruir la importación que no cumplía bajo el Acta Arancela-ria (19 U.S.C. 1595). Otorgándole a la FDA autoridad para la destrucción ad-ministrativa después de la notificación y la audiencia adecuadas, para fármacos violatorios valorados en $2500 o menos, estas nuevas autoridades de FDASIA no sólo crearán eficiencias y transparencia en el proceso de importación, sino que también fortalecerá la capacidad de la FDA para proteger la salud pública.
PharmTech: ¿Cómo afectarán estos nuevos requerimientos a la cadena de su-ministro y el desabasto de fármacos?
FDA: Las nuevas autoridades de seguridad de fármacos que el FDASIA le otorga a la FDA refleja un punto de vista más amplio y transparente de la cadena de suministro de fármacos. Por ejemplo, el FDASIA Sección 714 re-quiere que los importadores comercia-les se registren con la FDA, y la Sección 703 requiere que los listados de produc-tos farmacéuticos identifiquen la infor-mación de los excipientes. Mediante el otorgamiento a la FDA de nuevas auto-ridades que reflejan más exactamente el ciclo de vida completo del fármaco, el FDASIA equipa mejor a la agencia para proteger la salud pública y minimizar la exposición del consumidor a fármacos no seguros, inefectivos y de pobre ca-lidad en la cadena de suministro globa-lizada de hoy día. La FDA continuará todos los esfuerzos para abordar y evi-tar los desabastos de fármacos mientras implementa estos nuevos requerimien-tos, de los cuales se anticipa que fortale-cerán más los esfuerzos para garantizar la seguridad y calidad de los fármacos para los pacientes de EEUU. PT
Informes y Contrataciones:Tel: 52 (55) 5659-8880, 5536-2100, 5543-1486
E-mail: info@pharmatechespañol.com.mx

Pharmaceutical Technology en Español MARZO / ABRIL 2013 19
Los autores, parte del Consorcio Internacional sobre Innovación y Calidad en Desarrollo Farmacéutico (IQ Consortium), exploran y definen los esquemas y prácticas comunes de la industria cuando se aplican las GMPs en el desarrollo inicial. Un grupo de trabajo del consorcio se encarga de desarrollar una serie recomendaciones que pueden ayudar a la industria a identificar oportunidades para mejorar el tiempo de entrega para los primeros estudios en humanos y reducir los costos de desarrollo manteniendo mientras tanto los estándares de calidad requeridos y garantizando la seguridad del paciente. Este artículo es el tercero de una serie y se enfoca en la manufactura del producto farmacéutico.
Richard Creekmore está en desarrollo farmacéutico en AstraZeneca (Wilmington, DE); Eleni Dokou está en desarrollo farmacéutico en Vertex Pharmaceuticals (Cambridge, MA); Amnon Eylath está en calidad en ARIAD Pharmaceuticals (Cambridge, MA); Dennis Joiner está en ciencias farmacéu-ticas y suministros clínicos en Merck (Summit, NJ); Michael Lovdahl está en ciencias farmacéuticas en Pfizer (Groton, CT); Jackson Pellet está en ciencias farmacéuticas de moléculas pequeñas en Genentech (San Francisco Sur, CA); y Eric Schmitt y John W. Skoug* están en desarrollo de pro-ductos farmacéuticos en Abbott Laboratories (Abbott Park, IL).
*A quien debe dirigirse la correspondencia.
El Consorcio Internacional sobre Innovación y Calidad en Desarrollo Farmacéutico (IQ) se formó en 2010 y es una asociación de más de 25 compañías farmacéu-ticas y biotecnológicas con la misión de avanzar en
los estándares y regulaciones basadas en la ciencia y manejadas científicamente para productos farmacéuticos en todo el mun-do. En la emisión de junio del 2012 del Pharmaceutical Tech-nology, un artículo escrito por el Grupo de Trabajo de GMPs en el Desarrollo Inicial del IQ Consorcio describió el deseo y la justificación para recomendaciones más claras y consolidadas para las GMPs en el desarrollo inicial (de la Fase 1 a la Fase 2a) (1). Una consecuencia de la ausencia de claridad que rodea a las guías de GMPs en la fase inicial ha sido la variada interpre-tación y aplicación de las guías de GMPs existentes dentro de diferentes compañías de acuerdo a su propia cultura y toleran-cia al riesgo. Los debates internos con frecuencia resultan en interpretaciones conservadoras de “un tamaño se ajusta a todo” que se apoyan en las guías de la Conferencia Internacional de Armonización (ICH) que son mayormente relevantes para el desarrollo del producto comercial y no distinguen diferencias en los requerimientos entre el desarrollo inicial y la última eta-pa del desarrollo (Fase 2b y más allá). Un conductor clave de este grupo de trabajo (WG), por lo tanto, ha sido definir colecti-vamente las prácticas mínimas aceptables dentro de la industria con respecto a las expectativas de las GMP en el desarrollo inicial que permiten una flexibilidad añadida, son consistentes con la guía y los estatutos existentes y las cuales aseguran la calidad del producto y la seguridad del paciente (2 – 4).
El segundo artículo en esta serie abordó las recomenda-ciones para la validación del método analítico tanto para las sustancias farmacéuticas (DS) como para los productos farma-céuticos (DP) (5). En este tercer artículo de la serie, los autores abordan la aplicación de las GMPs en el desarrollo inicial en lo que se refiere a la manufactura del producto farmacéutico.
AntecedentesSegún se señaló anteriormente en la introducción a la serie de artículos, debido al alto desgaste en el desarrollo inicial, la mayoría de las compañías tratan de minimizar los recur-sos requeridos para avanzar en los compuestos en los estudios clínicos en humanos empleando formulaciones y procesos de manufactura simples (1). Por ejemplo, frecuentemente se usa la sustancia farmacéutica llenada en un frasco, el fármaco en la cápsula o simples mezclas de polvos llenados en cápsu-
Postura oficial: GMPs en fase teMPrana
GMPs del desarrollo inicial para la manufactura de productos farmacéuticos de moléculas pequeñasUna perspectiva de la industria (Parte III) Richard Creekmore, Eleni Dokou, Amnon Eylath, Dennis Joiner, Michael Lovdahl, Jackson Pellet,Eric Schmitt y John W. Skoug
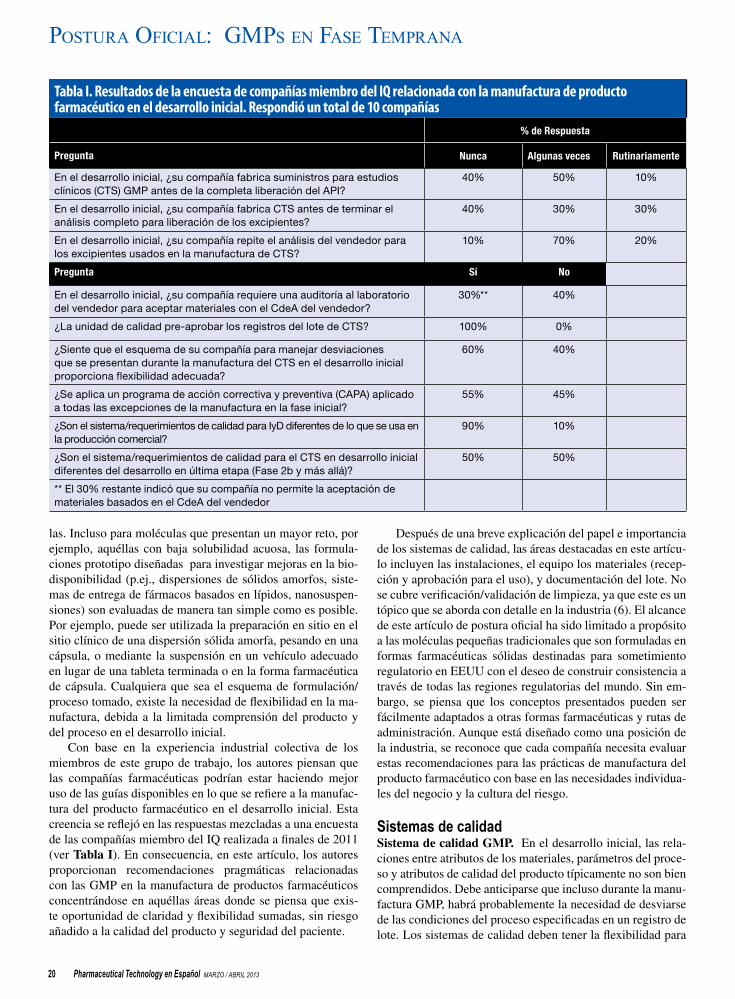
Pharmaceutical Technology en Español MARZO / ABRIL 201320
las. Incluso para moléculas que presentan un mayor reto, por ejemplo, aquéllas con baja solubilidad acuosa, las formula-ciones prototipo diseñadas para investigar mejoras en la bio-disponibilidad (p.ej., dispersiones de sólidos amorfos, siste-mas de entrega de fármacos basados en lípidos, nanosuspen-siones) son evaluadas de manera tan simple como es posible. Por ejemplo, puede ser utilizada la preparación en sitio en el sitio clínico de una dispersión sólida amorfa, pesando en una cápsula, o mediante la suspensión en un vehículo adecuado en lugar de una tableta terminada o en la forma farmacéutica de cápsula. Cualquiera que sea el esquema de formulación/proceso tomado, existe la necesidad de flexibilidad en la ma-nufactura, debida a la limitada comprensión del producto y del proceso en el desarrollo inicial.
Con base en la experiencia industrial colectiva de los miembros de este grupo de trabajo, los autores piensan que las compañías farmacéuticas podrían estar haciendo mejor uso de las guías disponibles en lo que se refiere a la manufac-tura del producto farmacéutico en el desarrollo inicial. Esta creencia se reflejó en las respuestas mezcladas a una encuesta de las compañías miembro del IQ realizada a finales de 2011 (ver Tabla I). En consecuencia, en este artículo, los autores proporcionan recomendaciones pragmáticas relacionadas con las GMP en la manufactura de productos farmacéuticos concentrándose en aquéllas áreas donde se piensa que exis-te oportunidad de claridad y flexibilidad sumadas, sin riesgo añadido a la calidad del producto y seguridad del paciente.
Después de una breve explicación del papel e importancia de los sistemas de calidad, las áreas destacadas en este artícu-lo incluyen las instalaciones, el equipo los materiales (recep-ción y aprobación para el uso), y documentación del lote. No se cubre verificación/validación de limpieza, ya que este es un tópico que se aborda con detalle en la industria (6). El alcance de este artículo de postura oficial ha sido limitado a propósito a las moléculas pequeñas tradicionales que son formuladas en formas farmacéuticas sólidas destinadas para sometimiento regulatorio en EEUU con el deseo de construir consistencia a través de todas las regiones regulatorias del mundo. Sin em-bargo, se piensa que los conceptos presentados pueden ser fácilmente adaptados a otras formas farmacéuticas y rutas de administración. Aunque está diseñado como una posición de la industria, se reconoce que cada compañía necesita evaluar estas recomendaciones para las prácticas de manufactura del producto farmacéutico con base en las necesidades individua-les del negocio y la cultura del riesgo.
Sistemas de calidadSistema de calidad GMP. En el desarrollo inicial, las rela-ciones entre atributos de los materiales, parámetros del proce-so y atributos de calidad del producto típicamente no son bien comprendidos. Debe anticiparse que incluso durante la manu-factura GMP, habrá probablemente la necesidad de desviarse de las condiciones del proceso especificadas en un registro de lote. Los sistemas de calidad deben tener la flexibilidad para
Tabla I. Resultados de la encuesta de compañías miembro del IQ relacionada con la manufactura de producto farmacéutico en el desarrollo inicial. Respondió un total de 10 compañías
% de Respuesta
Pregunta Nunca Algunas veces Rutinariamente
en el desarrollo inicial, ¿su compañía fabrica suministros para estudios clínicos (cts) GMp antes de la completa liberación del api?
40% 50% 10%
en el desarrollo inicial, ¿su compañía fabrica cts antes de terminar el análisis completo para liberación de los excipientes?
40% 30% 30%
en el desarrollo inicial, ¿su compañía repite el análisis del vendedor para los excipientes usados en la manufactura de cts?
10% 70% 20%
Pregunta Sí No
en el desarrollo inicial, ¿su compañía requiere una auditoría al laboratorio del vendedor para aceptar materiales con el cdea del vendedor?
30%** 40%
¿la unidad de calidad pre-aprobar los registros del lote de cts? 100% 0%
¿siente que el esquema de su compañía para manejar desviaciones que se presentan durante la manufactura del cts en el desarrollo inicial proporciona flexibilidad adecuada?
60% 40%
¿se aplica un programa de acción correctiva y preventiva (capa) aplicado a todas las excepciones de la manufactura en la fase inicial?
55% 45%
¿son el sistema/requerimientos de calidad para iyD diferentes de lo que se usa en la producción comercial?
90% 10%
¿son el sistema/requerimientos de calidad para el cts en desarrollo inicial diferentes del desarrollo en última etapa (Fase 2b y más allá)?
50% 50%
** el 30% restante indicó que su compañía no permite la aceptación de materiales basados en el cdea del vendedor
Postura oficial: GMPs en fase teMPrana

Pharmaceutical Technology en Español MARZO / ABRIL 2013 21
permitir estos cambios a ser documentados en los registros de lote sin la aprobación formal previa del departamento de Calidad. Sin embargo, estos cambios deberán ser revisados por Calidad después de la producción, para evaluar el impacto potencial a la calidad del producto y a la seguridad del pa-ciente. Es crucial resumir y rastrear los cambios del proceso ya que mucha de esta información será usada para desarro-llar la comprensión del proceso y puede ser incluida en los reportes de la historia del desarrollo. De manera similar, las desviaciones no planeadas de los procedimientos de manufac-tura escritos deberán ser documentadas y justificadas, aunque pueden no necesitar abordarse en un sistema formal de acción correctiva y preventiva (CAPA), ya que el proceso está cam-biando continuamente como parte del proceso de desarrollo. Las desviaciones no planeadas que tienen la probabilidad de impactar la calidad del producto y la seguridad del paciente, como las fallas en la limpieza, las contaminaciones y ciertas fallas del equipo, deben ser investigadas y aplicar las acciones correctivas para evitar la recurrencia.
Manejo del riesgo en el desarrollo inicial. Para ayudar en la aplicación de la toma de decisiones basada en el riesgo en el desarrollo y manufactura del producto farmacéutico, los autores recomiendan que las compañías apliquen un esquema científico y basado en el riesgo, similar en principio al de la guía ICH Q9 Manejo del Riesgo de Calidad (7). El Anexo I de la guía describe varios métodos y herramientas que pueden ser útiles para determinar los riesgos relativos relacionados con los riesgos del sistema, tales como la instalación y la gen-te; las organizaciones (incluyendo los sistemas de calidad); los riesgos de proceso y los riesgos de producto (seguridad y eficacia). Existen muchas maneras de identificar, calificar y mitigar los riesgos operacionales y de calidad. Sin importar la metodología usada, una estrategia documentada y buenos re-gistros de las decisiones basadas en el riesgo son importantes para asegurar que se consideran los factores apropiados para la protección de los pacientes y la calidad del producto.
Instalaciones y equipoSin importar la escala de la manufactura, la instalación uti-lizada para fabricar los suministros de los estudios clínicos deben cumplir los requerimientos básicos de las GMPs se-gún se describe en las regulaciones y los documentos guía. Más adelante se dan tres escenarios del desarrollo inicial y las ventajas de cada uno con referencia al desarrollo inicial. El primero involucra una planta piloto diseñada y equipada para operaciones GMP de rutina. El segundo escenario apunta a establecer un área GMP dentro del entorno del laboratorio. El tercer ejemplo se enfoca en la realización de la manufactura GMP o el apalancamiento de la práctica de farmacia en estre-cha proximidad al sitio clínico.
Instalación GMP para la manufactura del produc-to farmacéutico. El esquema tradicional en la manufactura GMP del producto farmacéutico es usar una instalación dedi-cada (llamada con frecuencia planta piloto) para los estudios clínicos en fase inicial. Las ventajas de este esquema incluyen
que los sistemas de calidad para la instalación (es decir, man-tenimiento, calibración, limpieza, manejo de cambios, CAPA y documentación) están bien definidos, y que la capacitación y otras actividades requeridas para mantener el cumplimiento de las GMPs, están centralizadas. Otros conductores para usar una planta piloto en el desarrollo inicial pueden ser la necesi-dad de equipo especializado, o tamaños más grandes de lote en situaciones especiales.
Área GMP dentro de un entorno de laboratorio. En algunos casos, puede ser ventajoso establecer un área GMP dentro de un “entorno de laboratorio” (es decir, una instala-ción de desarrollo de fármacos no dedicada a la producción de suministros clínicos) para la manufactura de producto farmacéutico en desarrollo inicial. La justificación para este esquema podría ser evitar la significante inversión en la pre-paración de una instalación dedicada y crear sistemas más simples, más flexibles que cumplan los requisitos GMP pero están adaptados para la actividad específica prevista. Ejem-plos en donde puede considerarse este esquema incluyen la necesidad de contención especial no disponible en la planta piloto; la necesidad de trabajar con materiales radioactivos o peligrosos, el uso de sustancias controladas y la producción de producto “fabricado de una sola vez” usado para la prueba de concepto. La justificación del negocio debe estar docu-mentada y aprobada por los grupos de manufactura y Calidad. En tanto se mantengan los controles apropiados de las GMPs, especialmente los relacionados con la seguridad del opera-dor, la limpieza y la prevención de contaminación cruzada, no hay barrera de cumplimiento para usar instalaciones “tipo laboratorio” para la manufactura de lotes clínicos de etapa ini-cial. Antes de que se inicie la manufactura GMP, sin embargo, debe realizarse una evaluación de riesgo y documentarse. La inclusión de representantes de Calidad, analítica, manufactura clínica, desarrollo de producto, y salud ambiental y seguridad sería prudente. Cuando se selecciona/diseña una instalación de manufactura clínica en desarrollo inicial, debe darse con-sideración para la recepción almacenamiento, dispensado y movimiento de materiales. Los procesos de manufactura en el área no dedicada deben proteger el producto, el paciente y los operadores de manufactura.
Adicionalmente, las compañías deben considerar qué ele-mentos son apropiados para la manufactura. Por ejemplo, el uso de una campana de flujo laminar certificada puede ser una mejor elección para la manufactura que una campana de extracción, ya que la primera está diseñada para evitar con-taminación del producto, proteger al operador y el ambiente del laboratorio. Adicionalmente, con la limpieza apropiada, una campana de flujo laminar puede ser más fácilmente usada para múltiples productos. El equipo de pequeña escala/ma-nual o los procedimientos pueden ser el mejor esquema ya que probablemente el espacio sea limitado. Con un tamaño de lote pequeño, el uso de pequeña escala o equipo manual/procedimientos minimizará la pérdida de rendimiento. Medi-das adicionales a ser evaluadas incluyen el vestido apropiado y los dispositivos de protección personal del operador, mo-

Pharmaceutical Technology en Español MARZO / ABRIL 201322
nitoreo del área y del operador para exposición a fármacos potentes o radiomarcados, y así sucesivamente.
Se recomienda la documentación de la preparación de la instalación, la manufactura del producto, y el retorno de la instalación al estado previo, si es necesario. Esta documenta-ción debe describir la justificación para la manufactura en el área no dedicada, la evaluación de riesgo, la preparación del área, los procedimientos de limpieza, y la lista de personas responsables. Esta documentación puede hacer referencia a procedimientos existentes o a procedimientos normalizados de operación (PNOs) junto con los documentos asociados con las reuniones y preparación para la manufactura del lote. Los registros de lote y los registros de limpieza deben ser parte de la documentación y deben seguir la política de retención de datos de la compañía.
Preparación en el sitio clínico. La preparación de las for-mulaciones en el sitio, algunas veces conocidas como “prepa-ración extemporánea” o “combinado (compounding)” no es considerada como manufactura, aunque es un método efectivo de preparar suministros clínicos iniciales utilizando las leyes locales que gobiernan la práctica de la farmacia. Los procesos usados para la preparación en sitio pueden ser desde tan sim-ples como preparar y diluir soluciones o llenar cápsulas con el API hasta procesos más complicados tales como el mezclado y la compresión de tabletas. Un grupo de trabajo del IQ que aborda las prácticas actuales en la preparación extemporánea han realizado una encuesta a ser comunicada en una fecha posterior, la cual confirma que sólo unas cuantas compañías han adoptado prácticas para sacar ventaja de esquemas más avanzados de formulación en el sitio clínico (8). Esta es una oportunidad que se pierde, porque la preparación o las for-mulaciones en sitio tienen el potencial de acelerar dramática-mente la capacidad de la industria para responder cuestiones críticas relacionadas con los parámetros farmacocinéticos, la biodisponibilidad absoluta o relativa, la factibilidad de libera-ción extendida y los esquemas de mejora de la biodisponibi-lidad para moléculas difíciles de entregar. Actualmente, cada compañía debe desarrollar sus propias mejores prácticas para asegurar la calidad del producto y la seguridad del paciente; con frecuencia, estas prácticas están basadas en publicaciones compendiales o de asociaciones profesionales (9 – 11). Los autores piensan que este tema justifica más discusión entre la industria y las autoridades regulatorias para determinar cuán-do es apropiado la preparación de la dosis en el sitio clínico y el nivel de controles de calidad requeridos. Los resultados de dicha discusión podrían ayudar a incrementar la utilización de la preparación en sitio de materiales de estudios clínicos para facilitar respuestas rápidas a cuestiones críticas en el de-sarrollo inicial del fármaco y finalmente llevar los mejores productos posibles a los pacientes en el tiempo más corto.
Equipo. La mayoría del equipo usado para la manufactu-ra GMP inicial de producto farmacéutico debe manejarse bajo un programa de calificación, mantenimiento preventivo, y ca-libración para la instalación GMP. Sin embargo, en el desarro-llo inicial, puede haber ocasionalmente la necesidad de usar
equipo que no es parte de dicho programa. Más que llevar a cabo una amplia calificación para una pieza de equipo que no se espera que sea frecuentemente usada, una organización puede elegir calificarla para un solo paso o campaña. La do-cumentación de una calificación de la instalación/calificación operacional (IQ/OQ) y/o la verificación del desempeño en la condición de operación propuesta es suficiente. Por ejemplo, si la preparación de la solución necesita un mezclador con una velocidad de rotación de 75 rpm, entonces será suficien-te la documentación en el registro del lote de que se usó un tacómetro calibrado para verificar que el mezclador estaba operando a 75 rpm.
El uso de equipo o partes en contacto con el producto dedicadas o desechables puede ser preferible a seguir los pro-cedimientos de limpieza estándar para asegurar que el equipo esté limpio y sea aceptable para el uso. Sin embargo, no todo el equipo o partes del equipo son desechables o pueden tener un costo sustancial que haga de lo desechable algo prohibiti-vo. En este caso, las partes en contacto con el producto po-drían estar dedicadas a una sustancia farmacéutica específica para usar en la manufactura del producto farmacéutico. Las partes en contacto con el producto dedicadas a un compuesto pueden ser costosas y evitarse en algunos casos considerando cuidadosamente el cambio del producto y métodos de limpie-za efectiva cuando se compra equipo.
Otro elemento a considerar con respecto al equipo es que mientras más complicado sea el equipo para trabajarlo o mantenerlo, menos deseable puede ser para los primeros lotes GMP. En la mayoría de los casos, el equipo simple es ade-cuado y utilizará menos material y consumirá menos tiempo total para la preparación, operación y actividades de limpieza.
Materias primasEdificios e instalaciones. Las GMPs bajo el 21 Code of Fe-deral Regulations (CFR) Parte 211.42 establece que los edifi-cios o áreas usadas en la recepción, almacenamiento y manejo de materias primas deben ser de tamaño, construcción y ubi-cación adecuados para permitir la limpieza, mantenimiento y operación apropiados (7). El tema común para esta sección del CFR Partes 210 y 211 es la prevención de errores y conta-minación. En principio, los requerimientos para los edificios e instalaciones usadas en la fase inicial de manufactura no son significativamente diferentes de los de las últimas fases o aún de la producción comercial. Sin embargo, existen algunas áreas que son únicas para la manufactura clínica inicial.
Control de materiales. Las regulaciones del CFR bajo la parte 211.80 proporcionan una buena dirección con respecto a la identificación del lote, el inventario, la recepción, el al-macenamiento y la destrucción de materiales (7). El intento claro es garantizar la seguridad del paciente estableciendo controles para evitar errores o contaminación cruzada y ase-gurar la trazabilidad de los componentes desde la recepción hasta el uso clínico. En general, los requerimientos para el control de materiales son idénticos a través de todas las fases del desarrollo, por lo que es importante considerar estos re-
Postura oficial: GMPs en fase teMPrana

Pharmaceutical Technology en Español MARZO / ABRIL 2013 23
querimientos cuando se diseñe una instalación GMP dentro de un entorno de laboratorio.
Por ejemplo, a todos los materiales se les debe asignar un número de lote único y tener un etiquetado apropiado. Un sistema de inventarios debe proporcionar el rastreo de cada lote de cada componente con un registro para cada uso. En la recepción, cada lote debe ser examinado visualmente para el etiquetado apropiado y en busca de evidencia de violación o contaminación. Los materiales deben ser colocados en cua-rentena o en el área de aprobados o el área de rechazos con el etiquetado apropiado para identificar el material y evitar mez-clas con otros materiales en el área de almacenamiento. Debe hacerse una provisión para los materiales con requerimientos de almacenamiento especiales (p.ej., refrigeración, alta segu-ridad). El etiquetado del almacenamiento debe coincidir con las condiciones reales en que el material se está almacenando y debe incluir fechas de caducidad/reanálisis para los mate-riales aprobados. Aunque dicho etiquetado es inconveniente para los nuevos materiales, en donde puede cambiar la fecha de caducidad o la fecha de reanálisis conforme se conozca más información, esto le permite al personal poder determi-nar rápidamente si un lote particular de material está cerca o excede la fecha de caducidad o la fecha de reanálisis. Las fechas generales de caducidad/reanálisis para materiales co-munes deberán basarse en la recomendación del fabricante o en la literatura.
Finalmente, existen claros requerimientos regulatorios o ambientales para la destrucción de materiales caducos o re-chazados. Es importante observar los requerimientos regiona-les e internacionales con respecto al uso de materiales de ori-gen animal (12). Se recomienda usar materiales que no sean de origen animal y que exista certificación disponible por parte de los fabricantes de materia prima de que no contienen materiales de origen animal. Si deben usarse materiales de origen animal, entonces es necesaria la certificación de los fa-bricantes de la materia prima de que ésta se originó de fuentes certificadas y aprobadas (por los cuerpos regulatorios) para usarse en farmacéuticos para humanos, o que el material ha sido analizado al nivel requerido para su aceptación por parte de las agencias regulatorias (siguiendo las guías de EEUU, la UE o de Japón, según aplique).
Recepción y aprobaciónEspecificaciones. Es un requerimiento de las GMPs que to-das las materias primas para la manufactura de un produc-to farmacéutico tengan las especificaciones apropiadas para asegurar la calidad. Los requerimientos compendiales deben ser usados para establecer especificaciones siempre que el material esté listado en al menos un compendio farmacéuti-co (p.ej., Farmacopeas de EEUU, Europeas y Japonesas). Es importante que el uso de materiales que cumplan los reque-rimientos de un solo compendio sea aceptable para usar en la primera fase de los estudios clínicos realizados en EEUU, Europa y Japón. Por ejemplo, un material que cumpla los cri-terios de la USP y sea usado en la manufactura de un producto farmacéutico debe ser aceptable para usarse en los estudios
clínicos iniciales en la Unión Europea. En ausencia de una monografía de compendio farmacéutico, la especificación del vendedor y/o especificaciones compendiales alternativas ta-les como el Food Chemical Codex de la USP, deben guiar el establecimiento de la especificación. En cualquier caso, el pa-trocinador es el responsable del establecimiento de las espe-cificaciones apropiadas. Por lo tanto, es postura de los autores que la buena práctica es tener al menos una comprensión bási-ca de la manufactura, química y toxicología de los materiales para guiar el apropiado establecimiento de la especificación.
Análisis y evaluación del material. El mínimo análisis requerido para los materiales de entrada es la inspección vi-sual y la identificación. Sin embargo, como se mencionó an-teriormente, las pruebas apropiadas deben estar determinadas para el material con base en el conocimiento de la manufac-tura, química, y toxicología. Si el vendedor está calificado, entonces el certificado de análisis puede ser aceptable en con-junto con la inspección visual y el análisis de identificación (ver la sección “Calificación del vendedor” más adelante).
Aprobación para el uso. Idealmente, la manufactura de un producto farmacéutico a granel debe empezar con las es-pecificaciones del material aprobado y con los materiales que están completamente analizados y liberados. Sin embargo, existen circunstancias en donde puede ser no factible em-pezar la manufactura con especificaciones aprobadas y ma-teriales completamente analizados y liberados, incluyendo el API. La manufactura previa a la liberación final (algunas veces llamada manufactura “en riesgo”) puede ser aceptable, sin embargo, porque el sistema de calidad asegura que todas las especificaciones están aprobadas, los resultados de prueba están dentro de las especificaciones, y todos los documentos relevantes están en orden antes de que el producto sea libe-rado para administración a humanos. El “riesgo” debe estar completamente con el fabricante y no con el paciente.
Calificación del vendedor. Los vendedores que sumi-nistran excipientes, materias primas, o APIs deben estar ca-lificados por el patrocinador. La calificación apropiada debe depender de la etapa del desarrollo y de una evaluación del riesgo interno. Por ejemplo, si un vendedor tiene una historia de abastecimiento de la industria farmacéutica y el material va a ser usado en el desarrollo inicial, debe ser suficiente una evaluación en papel (p.ej., un cuestionario). Si un proveedor no tiene una historia de abastecimiento de la industria farma-céutica, debe realizarse una evaluación de riesgo y dependien-do del resultado puede requerirse una auditoría del sitio antes de aceptar el material para uso.
Idealmente, los vendedores deben ser calificados antes de usar las materias primas para la manufactura. Sin embargo, es aceptable para la calificación proceder en paralelo en tanto que la documentación/evaluaciones de riesgo estén disponibles an-tes de la liberación del producto y, como en la sección previa, todo el riesgo recae en el fabricante y no en el paciente.
Documentación y ejecución del lotePreparación de la documentación del registro del lote. La documentación de manufactura es un requerimiento bá-

Pharmaceutical Technology en Español MARZO / ABRIL 201324
sico para todas las fases del desarrollo clínico. El 21 CFR Partes 211.186 y 211.188 describen la producción maestra y los registros de producción del lote, respectivamente (7). El propósito establecido del registro de producción maestra es “asegurar la uniformidad de lote a lote.” Aunque el asegura-miento del registro es importante para un proceso de manu-factura comercial validado, no necesariamente aplica para los lotes de desarrollo clínico. Las propiedades del material, la escala de manufactura y el perfil del producto de calidad ob-jetivo cambian frecuentemente de lote a lote. Por lo tanto, los registros de producción del lote son la documentación apro-piada para los suministros del estudio clínico. Los registros de producción del lote para los materiales de la Fase 1 deben incluir como mínimo:
• Nombre, potencia y descripción de la forma farmacéutica• Una lista completa de ingredientes activos e inactivos,
incluyendo peso o medida por dosis unitaria y peso total o medida por unidad
• Tamaño teórico del lote (número de unidades)• Instrucciones de manufactura y control
Estos requerimientos mínimos son consistentes con la Guía para la Industria: cGMP para Fármacos de Investigación en Fase Inicial de la FDA, la cual requiere un registro de manufac-tura que detalle los materiales, equipo, procedimientos usados y cualquier problema encontrado durante la manufactura (2). Los registros deben permitir la replicación del proceso. Sobre esta base, existe flexibilidad en la manera para la cual la do-cumentación de las actividades del lote puede ocurrir, siempre que la documentación permita la revisión post-ejecución por la unidad de calidad y para la retención de estos registros.
Aprobaciones de la documentación del lote. Se requiere la revisión y aprobación de los registros del lote ejecutado por la unidad de Calidad de acuerdo al 21 CFR Parte 211.192 (7). Esta revisión y aprobación se requiere para todas las etapas de la manufactura clínica. Las pre-aprobaciones de los registros del lote deben estar gobernadas por procedimientos internos ya que no hay requerimientos en el CFR 21 de que la unidad de Calidad pre-apruebe el registro de lote (aunque esto es al-tamente recomendado con el fin de minimizar la posibilidad de errores). De hecho, la Tabla I muestra que la pre-aproba-ción de los registros de lote por la unidad de Calidad es prac-ticada por las 10 compañías que participaron en la encuesta de manufactura de productos de IQ Consortium relacionada con el desarrollo inicial. Los registros de lote deben ser retenidos durante al menos un año después de la caducidad del lote, de acuerdo al CFR Parte 211.180, pero muchas compañías mantienen sus registros de GMP archivados durante plazos más largos.
Despeje del cuarto. El 21 CFR Parte 211.130 requiere inspección de las instalaciones de acondicionado y etiquetado inmediatamente antes de usar para asegurar que todos los pro-ductos farmacéuticos de las operaciones anteriores hayan sido removidos. Esta inspección debe estar documentada y puede ser realizada por cualquier individuo calificado.
Aunque el despeje de línea para la manufactura a granel no está específicamente mencionado en el CFR, es de esperar que se realice el despeje del cuarto. Como mínimo, este des-peje debe ser realizado antes del inicio de un nuevo lote (es decir, antes de los materiales del lote que entran al cuarto de proceso).
Tiempo de permanencia. Durante las etapas iniciales del desarrollo, el análisis de liberación de la forma farmacéutica final confirma la calidad del producto y soporta el estableci-miento de tiempos de permanencia posteriormente en el desa-rrollo clínico. No existe requerimiento para establecer tiem-pos de permanencia para el trabajo en proceso en el desarrollo inicial. La formulación específica y la experiencia de esta-bilidad, la cual está generalmente limitada en esta etapa del desarrollo, deberían ser apalancadas para evaluar cualquier variación sustancial de los tiempos esperados de procesado del lote. Los datos colectados de estos lotes y el desarrollo posterior pueden ser usados para establecer tiempos de per-manencia para lotes futuros. (Las excepciones a este esquema pueden incluir preparaciones de solución o suspensión usadas en la manufactura de la forma farmacéutica sólida, donde los procedimientos típicamente gobiernan tiempos de permanen-cia permitidos para asegurar la ausencia de contaminación microbiana en el producto final).
Control de cambios. Los cambios en las materias primas, procesos y productos durante el desarrollo inicial son inevita-bles. No se requiere que estos cambios sean controlados por un sistema central sino más bien pueden ser apropiadamente documentados en reportes técnicos y en registros de manufac-tura del lote. Cualquier cambio en el proceso de manufactura a partir de un lote anterior debe ser capturado como parte de la documentación de registro del lote y comunicado a las áreas afectadas- La justificación para estos cambios también debe ser documentada ya que esto sirve como una fuente para los reportes de la historia del desarrollo y para la actualización de los sometimientos regulatorios. Los autores recomiendan que aquéllos cambios que pudieran afectar un sometimiento regulatorio sean capturados en un sistema formal.
Cambios de proceso. Los parámetros de proceso deben ser registrados pero no necesitan estar predeterminados por-que los procesos pueden no ser fijos o establecidos en el de-sarrollo inicial. Dada la limitada disponibilidad del API en el desarrollo inicial, un lote clínico es con frecuencia la primera vez que un producto se fabrica en una escala particular o que usa un tren de proceso particular. Por lo tanto, los cambios en el proceso deben ser esperados. Los trenes del proceso y los parámetros de operación deben estar documentados en el registro del lote pero los cambios no deben desencadenar un reporte de excepción o CAPA. Los cambios deben ser docu-mentados como una nota operacional o modificación al regis-tro del lote en tiempo real. Dichos cambios conducidos por observaciones técnicas no deben requerir aprobación previa por la unidad de Calidad, sino que deben tener la justificación
Postura oficial: GMPs en fase teMPrana

Pharmaceutical Technology en Español MARZO / ABRIL 2013 25
científica apropiada (vía formulador/científico) o la flexibili-dad apropiada construida en el registro del lote para permitir los cambios. Esta documentación debe estar disponible para la revisión de Calidad antes de la disposición del producto.
Cálculo del rendimiento. Los rendimientos reales deben ser calculados para pasos de proceso mayores para mayor comprensión del proceso y permitir la optimización de los procesos. Las tolerancias del rendimiento esperado no son siempre aplicables a la manufactura en el desarrollo inicial. En esta etapa del desarrollo inicial, cuando la formulación y el conocimiento del proceso están extremadamente limitados, puede no haber bases técnicas para establecer tolerancias del rendimiento y, por lo tanto, este rendimiento puede no ser un indicador de la calidad del producto final.
Controles en proceso y muestreo de IyD. Las pruebas y controles en proceso deben seguir los requerimientos básicos de las GMPs para documentar la consistencia del lote. Para productos en cápsula, estos requerimientos pueden incluir el peso de las cápsulas y la inspección física. Para la producción de tabletas, la fuerza de compresión o la dureza y peso de las tabletas deben ser monitoreadas junto con la apariencia. El muestreo de IyD, definido como las muestras tomadas para propósitos de comprender más el proceso pero no tomadas para las decisiones de disposición del lote, es una parte nor-mal de todas las fases de la manufactura clínica. En la manu-factura en el desarrollo inicial, se requiere un plan de mues-treo para las pruebas de control en proceso, pero no para las muestras de IyD. Sin embargo, para el propósito del conteo de material, el muestreo de IyD debe documentarse como parte de la ejecución del lote. Para estas muestras, los resultados del análisis pueden manejarse por separado y no son requeridos para incluirse en la documentación regulatoria.
ConclusiónLas organizaciones involucradas en la manufactura de sumi-nistros clínicos Fase 1 y 2a deben reconocer que la compren-sión del proceso es muy limitada en el desarrollo inicial. Los sistemas de calidad deben ser lo suficientemente robustos para asegurar la seguridad del paciente, pero deben ser también lo suficientemente flexibles para acomodarse a plazos acelera-dos y cambios de proceso en tiempo real. Las necesidades especiales de manufactura GMP en pequeña escala deben también ser consideradas cuando se diseñan instalaciones, se compra equipo y se selecciona el tipo de formas farmacéuti-cas para usar en los estudios clínicos iniciales. Las compañías están alentadas a aplicar los principios del manejo del riesgo de calidad para soportar estas decisiones basadas en el riesgo.
Los autores también piensan que los esquemas subutili-zados existen (p.ej., preparación en sitio de formas farmacéu-ticas más complejas) para responder rápida y eficientemente preguntas relacionadas con la formulación acerca de la bio-
disponibilidad, la farmacocinética y las tasas de liberación objetivo para formulaciones de liberación controlada. Los beneficios y riesgos potenciales de estos esquemas justifican más discusiones. Finalmente, la documentación de las ope-raciones de manufactura debe estar basada en el riesgo. Las instrucciones de manufactura en el desarrollo inicial no deben ser demasiado prescriptivas como para restringir los cambios en el proceso o desalentar el muestreo para la comprensión adicional del proceso. Los cambios deben esperarse y poder ser rápidamente revisados y aprobados por el departamento de Calidad o por un delegado calificado.
Para estimular más las discusiones sobre estos esquemas dentro de la industria y con las autoridades de salud en el mundo, el Grupo de Trabajo de GMPs en el Desarrollo Inicial del IQ Consortium está planeando realizar un taller en el futu-ro cercano para promover un debate robusto y discusión sobre estos esquemas recomendados para las GMPs en el desarrollo inicial. El grupo piensa fuertemente que dicho diálogo mejo-rará la relación entre desarrollo, aseguramiento de la calidad y los grupos regulatorios de química/ manufactura/controles (CMC) dentro de la industria farmacéutica. Adicionalmente, el acuerdo entre la industria y las autoridades regulatorias de fármacos con respecto a los esquemas aceptables para aplicar las GMPs en las fases iniciales del desarrollo de fármacos permitiría un enfoque más ágil y flexible en el desarrollo ini-cial, proporcionando mientras tanto controles apropiados para garantizar la seguridad del paciente.
Referencias1. A. Eylath et al., Pharm. Technol. 36 (6) 54–58 (2012).2. FDA, Guidance for Industry: CGMP for Phase 1 Investigational
Drugs (Rockville, MD, July 2008).3. FDA, “Current Good Manufacturing Practice and Investigational
New Drugs Intended for Use in Clinical Trials,” Fed. Reg. 73 (136), 40453–63 (2008).
4. EU, Guidelines to Good Manufacturing Practice: Medicinal Pro-ducts for Human and Veterinary Use, Annex 13: Investigational Medicinal Products (Feb. 2010).
5. D. Chambers, Pharm. Technol. 36 (7) 76–84 (2012).6. D.A. LeBlanc, in “Validated Cleaning Technologies for Pharmaceu-
tical Manufacturing,” (Interpharm/CRC, Washington, DC, 2000). 7. Code of Federal Regulations, Title 21, Food and Drugs (Government
Printing Office, Washington, DC).8. Personal communication with Mike Kress, Leader, IQ Consortium
Drug Product Working Group on Extemporaneous Formulations (May 2012).
9. USP 35–NF 30 General Chapter <795> Pharmaceutical Compoun-ding—Nonsterile Preparation, 344–350.
10. USP 35–NF 30 General Chapter <797> Pharmaceutical Compoun-ding—Sterile Preparations, 350–387.
11. L.V. Allen, The Art, Science and Technology of Pharmaceutical Compounding (American Pharmacists Association, 2008).
12. See, for example: the USP Excipient Supplier Qualification Pro-gram, Section 10 (V. 1.1, Sept. 2008), and references therein, for specific guidance. PT

Pharmaceutical Technology en Español MARZO / ABRIL 201326
Innovación Farmacéutica Angie Drakulich
Un vistazo a los líderes del año en la estrategia de innovación, incluyendo las principales
compañías bio/farmacéuticas y los que recibieron reconoci-mientos de AAPS, PhRMA y CPhI.
rEportE EspEcial: innovación
“Innovación”. Esta palabra está en el corazón del desarrollo bio/farmacéutico y la manufactura, ya sea que los inno-vadores le estén apuntando a una nueva línea celular o formulación, un objetivo para entrega de fármacos o un método, un esquema analítico o de empaque, o un sistema de calidad. Como dijo la Co-misionada de la FDA, Margaret Ham-burg en abril de 2012 en la conferencia de NEHI en Boston sobre “Salvando la Brecha de la Innovación”, la innovación “es un tema que es importante para to-dos nosotros, ya sea que pertenezcas a la industria, a la academia, a la práctica clínica o al gobierno.” Ella habló acer-ca de los retos que enfrentan IyD y los proyectos de productos, incluyendo las implicaciones de las patentes, y esta-bleciendo que, “Las líneas de tendencia para los productos innovadores con re-lación a las inversiones en investigación y desarrollo no son lo que a cualquiera de nosotros nos gustaría.”
Como ejemplo, Hamburg señaló la actual “era dorada” del descubrimien-to biomédico en el cual la industria ha secuenciado el genoma humano para revelar potenciales blancos de fármacos
y ha obtenido ganancias en separación de alto rendimiento y nanotecnología y todavía no necesariamente ha traducido estos desarrollos en terapias o curas. Hamburg propuso en su discurso que una solución potencial para esta brecha en la innovación, requiere una “estra-tegia amplia, integrada que acople el ‘eco-sistema’ completo.”
Dicha estrategia incluiría “inversio-nes nuevas y estratégicas en investiga-ción,” y “verdadera colaboración por parte de los interesados,” incluyendo un “fuerte liderazgo del negocio, de la aca-demia y del gobierno,” dijo.
Aquí, el Pharmaceutical Technology trata algunos de los esfuerzos de colabo-ración que tienen lugar entre estos titula-res para continuar el avance en la innova-ción en el desarrollo y la manufactura de fármacos. Las siguientes páginas desta-can los honores otorgados a estudiantes graduados y profesores así como a equi-pos de trabajo en IyD y manufactura bio/farmacéutica a lo largo del 2012. Como nos preparamos para entrar al año 2013, esperamos que estos individuos y su duro trabajo proporcionen la inspiración para más innovación farmacéutica.
ESTRATEGIAS DE INNOVACIÓN DE PRODUCTOS FARMACÉUTICOS ENTRE LAS GRANDES FARMACÉUTICAS
Cada año, nuestra publicación her-mana Pharmaceutical Executive pu-blica un reporte sobre las primeras 50 compañías farmacéuticas con base en las ventas globales de fármacos de pres-cripción. Estas compañías han logrado mantenerse por delante del juego con-forme la industria enfrenta múltiples caducidades de patentes y un paisaje cambiante que incluye nuevos modelos de subcontratación, capacidad de inter-cambio y estrategias de consolidación.
Viendo a futuro, Pharmaceutical Technology le pidió a las principales compañías en esta lista que hablaran acerca de sus estrategias de innovación, con un enfoque específico en el desa-rrollo y manufactura de fármacos. Estas compañías incluyen, en orden: Pfizer (Nueva York) con ventas globales de prescripciones en el 2011 de $57,700 mdd; Novartis (Suiza) con ventas de $54,000 mdd; Merck (Nueva Jersey) con ventas de $41,300 mdd; y Sanofi (Francia) con ventas de $37,000 mdd. A continuación están sus respuestas.
PfizerKevin Nepveux, vicepresidente, Servi-cios de Tecnología Global, Pfizer Sumi-nistro Global“La innovación es una mentalidad, no un proceso que puede encenderse y apagarse. Los estrategas organizaciona-les nos dicen que los comportamientos comen procesos para el desayuno, sin embargo persiste la tendencia para se-parar habilidades técnicas ‘duras’ de las habilidades comunicativas ‘más suaves’ que conducen la interacción de la fuerza laboral global. Las organizaciones ba-sadas en la técnica continúan poniendo gran valor en las habilidades ‘duras’. En vez de esto, necesitamos entender mejor cómo nuestros tecnólogos inte-ractúan entre sí, con nuestros negocios y sus retos ambientales para inspirar las innovaciones de todos los días así como sus avances.
“En 2010, Pfizer Suministro Global (PGS) re-examinó su esquema para la
iMa
Ge
n:
Da
n W
ar
D;
Bu
en
a v
ista
iMa
Ge
s/G
et
ty
iM
aG
es

Pharmaceutical Technology en Español MARZO / ABRIL 2013 27
innovación. Nosotros habíamos dise-ñado nuestras tácticas para inspirar el pensamiento creativo, pero nuestras métricas de seguimiento incluían perse-guir el valor monetario de los proyec-tos. Empezamos a ver la categorización de los proyectos como innovadora para cumplir la métrica fiscal, más que el desencadenamiento de una mentalidad que derivara en soluciones verdadera-mente creativas. Era tiempo de dar un nuevo impulso a nuestras metodologías de innovación.
“Realizamos un viaje que duró un año a nuestras ubicaciones de manu-factura y con nuestros grupos técnicos para escucharlos. Formamos un equipo de trabajo global diagonalmente jerár-quico para dirigir el desarrollo del nue-vo esquema, con patrocinio del grupo de trabajo ejecutivo y participación, un operador mecánico de manufactura, tecnólogos y los niveles organizaciona-les entre éstos –todos con los mismos sitios en la mesa de estrategias.
Hoy día, nuestra oficina de innova-ción en la sede, deliberadamente peque-ña, capacita a entrenadores en innova-ción en todo el mundo. Esta red global de más de 200 tiene la responsabilidad de construir ecosistemas locales de in-novación y de adaptar las herramientas de innovación a las necesidades y cultu-ras locales. Más que estar en el negocio de la innovación, la innovación es a su vez empleada en las necesidades del ne-gocio local.
“las ideas necesitan llegar de todos lados dentro de nuestra organización global, pero la innovación debe ser nu-trida desde arriba. Por supuesto, toda-vía necesitamos mostrar el valor fiscal de todo este esfuerzo. Sin embargo, hemos visto un retorno significativo en un tiempo corto desplazando nuestro enfoque al cambio en la mentalidad de innovación primero, y permitir que el valor surja como resultado de la nueva manera de pensar y de comportarse. Los tecnólogos de PGD del mañana no tienen el mismo molde que antes, y tampoco nuestro negocio.”
NovartisTimothy Wright, director global de de-sarrollo
“Existe un consenso en la industria far-macéutica de que necesitamos acelerar el ritmo del desarrollo de fármacos. Si fracasamos para hacerlo, no nos man-tendremos avanzando con los retos de salud cada vez más altos que enfrenta nuestra sociedad. Novartis ha hecho un buen progreso con algunos fuertes cambios en nuestro esquema para el desarrollo de fármacos. Un cambio es invertir en el desarrollo de múltiples indicaciones simultáneamente (más que en serie), en donde avanzamos con los compuestos en más de un escena-rio clínico en paralelo, incrementando así la probabilidad de que los fármacos lleguen al paciente más rápido. Una vez que comprendes la biología de la enfermedad y las vías moleculares in-volucradas, entonces cada compuesto puede ser probado simultáneamente en múltiples enfermedades que comparten la misma vía. Un ejemplo es nuestro inhibidor mTor everolimus, el cual está ahora aprobado en EEUU para cinco indicaciones de cáncer (bajo el nombre de marca Afinitor) y para trasplante re-nal (bajo el nombre de marca Zortress), y nuestro fármaco huérfano Ilaris, ori-ginalmente aprobado en una muy rara enfermedad genética y que ahora conti-núa siendo desarrollado en mayores po-blaciones de enfermedad, incluyendo la artritis gotosa y la enfermedad cardíaca.
“Otra solución para hacer frente a la sequía en la innovación es la colabora-ción y el intercambio de conocimientos. El costo de desarrollar un fármaco se ha elevado exponencialmente. Actualmen-te, se lleva un promedio de 10 años y los costos un promedio de $4,000 mdd para llevar una nueva terapia al mercado. Este modelo no es sustentable. Nece-sitamos trabajar juntos en la industria, la academia, y el gobierno para hacer que nuestros dólares en la innovación trabajen fuerte. Sólo trabajando juntos podemos asegurar tratamientos nuevos e innovadores que lleguen a los pacien-tes que los necesitan principalmente.”
MerckRich Tillyer, vicepresidente senior, Des-cubrimiento y Ciencias Preclínicas, Merck Laboratorios de Investigación“Las oportunidades para que Merck y la
industria farmacéutica impacten la salud humana son reales y numerosas dada la rápida evolución continua de las ciencias biomédicas, aunque esta rápida evolu-ción no se dará sin una inversión conti-nua en IyD y el énfasis en la innovación. La innovación en IyD es un factor crítico de éxito y Merck entiende que esto re-quiere un compromiso a largo plazo.
“En Merck, nuestro enfoque está en desarrollar medicamentos primero y los mejores en su clase que crearán una di-ferencia significativa para los pacientes. Para lograr este objetivo, necesitamos establecer una cultura de innovación alimentada por colaboraciones con so-cios internos y externos, y con un en-foque en el talento creciente. Nosotros continuaremos dirigiendo estas inicia-tivas en 2013 y más allá para asegurar que nuestros proyectos de candidatos permanezcan fuertes y continúen su avance hacia los pacientes que necesi-tan nuestros medicamentos.”
SanofiMarc Bonnefoi, director de Sanofi Nor-teamérica, IyD Central“Sanofi está evolucionando su esquema de IyD para poder tener acceso a las me-jores ideas, ciencia y gente en investiga-ción. Esto requiere una combinación de innovación tanto interna como externa y se implementa a través de la creación de centros de investigación integrados geográficamente localizados en los cua-les los científicos de Sanofi se asocian cada vez más con equipos de trabajo externos. Los centros de IyD han sido creados en Norteamérica con un énfasis especial en el área de Boston, así como en Alemania y Asia, y se está discutien-do un proyecto para implementar un centro en Francia.
“Durante muchos años, la industria farmacéutica diseño fármacos a partir de blancos biológicos que no estaban siempre bien validados y donde la vía para la prueba de concepto clínico y la validación última impacientes era in-cierta. Nosotros examinamos puntua-ciones de moléculas para ver si éstas te-nían algún efecto en diferentes modelos de enfermedad que con frecuencia esta-ban calificados de manera incompleta. Existían demasiados supuestos con res-

Pharmaceutical Technology en Español MARZO / ABRIL 201328
pecto a la biología de las enfermedades. Hoy día, buscamos empezar con una comprensión de la causa o las causas subyacentes de una enfermedad dada y trabajar para desarrollar una solución para interferir con ese proceso.
“Estamos tratando de integrar un es-quema transnacional para estos esfuer-zos, aplicando el conocimiento a partir de las poblaciones de pacientes y de la experiencia médica mucho más tempra-no en el proceso de IyD. Para hacer una realidad estos esfuerzos de investiga-ción transnacional, Sanofi está utilizan-do la capacidad de traducir a la situa-ción del paciente como un criterio para juzgar la calidad de los proyectos. Nada de esto puede ser hecho por Sanofi IyD solo; tenemos gente magnífica e ideas fantásticas, pero ninguna organización puede por sí sola hacer frente a la com-plejidad de las enfermedades humanas crónicas, las cuales permanecen más allá de nuestro alcance por el momento. Implementando efectivamente la inno-vación abierta y elevando el nivel para que los proyectos sean aceptados en desarrollo y alcanzar hitos adicionales a la inversión en el futuro, Sanofi espera crear uno de los mejores portafolios en la industria.”
INNOVACIÒN DE ESTUDIANTES A TRAVÉS DE LAS CIENCIAS FARMACÉUTICAS
En la reunión anual de 2012 de la Aso-ciación Estadounidense de Científicos Farmacéuticos (AAPS) en Chicago, los estudiantes graduados del país fueron honrados por su investigación y traba-jo en innovación bio/farmacéutica. Los premios del simposio fueron entregados a estudiantes graduados en las áreas que van desde el análisis y calidad farma-céutica hasta la entrega y disposición de fármacos oculares. Pharmaceutical Te-chnology tuvo la oportunidad de hablar con unos cuantos de los que recibieron el reconocimiento acerca de su trabajo.
El ganador de la categoría de bio-tecnología, David W. Woessner, señaló que su trabajo de equipo en la Univer-sidad de Utah sobre “Combinaciones Sintéticamente Letales para Leucemia Mieloide Crónica (CML) Terapia de
rEportE EspEcial: innovación
Alteración de la Dimerización de Bcr-Abl y Vías Específicas de Leucemia Secundaria” demuestra el primer desa-rrollo de una terapia basada en proteína/péptido para la CML.
Aunque las terapias actuales en la clínica (inhibidores de la tirosina cinasa [TKIs]) actúan sobre la porción ABL de la proteína, estamos demostrando eficacia al apuntar a la porción BCR, necesaria para la oligomerización y activación de la tirosina cinasa. Adicio-nalmente, este trabajo también aprove-cha una estrategia de terapia combinada al principio en el desarrollo,” dijo. La investigación futura en esta área ya se ha movido hacia esquemas de terapia combinada, explica Woessner. “Existen actualmente esencialmente cinco TKIs que tratan efectivamente la CML. Las combinaciones con otras moléculas pe-queñas que apuntan a las células madre leucémicas, pero no a las células madre hematopoyéticas normales/saludables son altamente valoradas. Continuare-mos haciendo ensayos para combina-ciones de fármacos,” agregó.
Mamta Kapoor de la Universidad de Connecticut también ganó en la catego-ría de biotecnología. Su investigación “Elucidación del Mecanismo de Cap-tación Celular de Nuevos Lipoplexos Aniónico-siRNA Ternarios”, orientada a obtener una comprensión sobre la in-teracción célula-formulación que pue-de facilitar la optimización más rápida para lograr una entrega eficiente. “Esta área de investigación es interesante por-que involucra un esquema mecanístico para comprender los pasos limitantes de la velocidad para la entrega intracelular competente de agentes terapéuticos,” explicó Kapoor. Los estudios mecanís-ticos “han ayudado en la comprensión de la contribución de los componentes de la formulación en el proceso de cap-tación y la bioactividad consecuente.” Mirando a futuro, el equipo de trabajo de Kapoor pretende repetir los estudios con otras nuevas formulaciones.
Lakshmi Prasanna Kolluru, quien ganó en la categoría de diseño de fár-macos e interfase de desarrollo, se con-centró en el “Diseño y Desarrollo de Nanopartículas Teragnósticas con base de Albúmina para la Entrega de Fár-
macos Dirigidos a Tumores”. Ubicado en la Universidad Mercer, el candidato a PhD se inspiró como un adolescente por una prima, una paciente con cáncer que sufría los efectos colaterales de la quimioterapia. “Ella se alteró psico-lógicamente cuando perdió su cabello debido a los efectos colaterales y esto alentó el interés en mí para concentrar-me en la investigación del cáncer. Así, cuando mi profesor me dio la libertad para diseñar un proyecto, opté por tra-bajar en el desarrollo de un sistema de entrega que dirigiera los fármacos para el cáncer al tumor y reducir los efectos colaterales de la quimioterapia”, dijo Kolluru. Viendo hacia delante, el estu-diante de grado dice que el equipo de la universidad está “planeando decorar este sistema de entrega con sondas dua-les dirigidas para examinar la eficacia de los ligandos in vitro e in vivo para la localización selectiva del fármaco en la región del tumor.”
Jiban Jyoti Panda del Centro Interna-cional para Ingeniería Genética y Biotec-nología en Nueva Delhi y de la Universi-dad FM de Balasore, Odisha, India, ganó en la categoría de diseño y desarrollo de la formulación para el artículo, “Nanotu-bos de Dipéptidos, Nanovesículas y Na-nogeles Auto-Ensamblados: Vehículos Potenciales para la Entrega de Fármacos Tumorales Dirigidos.” La investigación se enfocó en probar el potencial de la nanotecnología en la terapia de cáncer. “Se han investigado diferentes tipos de nanopartículas poliméricas, inorgánicas y metálicas por su potencial para la en-trega y terapia efectiva en tumores. Sin embargo, existen muchos factores que evitan el desarrollo de estas nanopartí-culas para un uso humano seguro,” ex-plicó Panda. “Tratamos de desarrollar nanopartículas basadas en péptidos, las cuales se espera que sean altamente bio-compatibles debido a su origen peptídi-co. Además, como el mayor problema con los fármacos o sistemas basados en péptidos es su baja estabilidad in vivo, tratamos de desarrollar nanopartículas a partir de péptidos pequeños diseña-dos con el aminoácido modificado a,β-dehidrofenilalanina (ΔPhe), residuo que es único y que proporcionaría las estruc-

Pharmaceutical Technology en Español MARZO / ABRIL 2013 29
Innovación para un objetivo líder de enfermedad: AlzheimerA principios del otoño pasado, la asociación de Investigación Farmacéutica y Fabricantes de América (PhRMA por sus siglas en inglés) honró a nueve personas por su investigación en la lucha contra la Enfermedad de Alzheimer (AD) como parte de los Premios a la Investigación y Esperanza de la asociación. Los premios se lanzaron en 2012, y reemplazan al programa del premio Descubridores de la organización. PharmTech fue uno de los medios de comunicación de los premios. Lo que es interesante acerca del grupo de ganadores es que los que recibieron el premio incluyen no sólo a científicos e investigadores, sino también a defensores de los pacientes y voluntarios. El presidente de PhRMA y CEO John J. Castellani se refiere a las cuatro categorías de premiación como el “ecosistema” requerido para tratar, prevenir y derrotar al AD de una vez por todas. “Cada uno de estos ganadores tiene una perspectiva que es necesaria para el éxito cuando estás tratando de combatir una enfermedad como el Alzheimer” dijo.
El Premio para la Investigación y Esperanza por la Investigación Académica en la AD fue para Bradley T. Hyman, DM, PhD, profesor de neurología en el Hospital General de Massachusetts/Escuela de Medicina de Harvard, y a David Holtzman, Profesor en Andrew B. Y Gretchen P. Jones y Director del Departamento de Neurología de la Escuela de Medicina de la Universidad de Washington.
Holtzman, quien ha estado trabajando con temas de la AD en varias capacidades durante aproximadamente 20 años, piensa que el área más prometedora en términos de buscar obtener un fármaco preventivo o un fármaco terapéutico que tengan un impacto a largo plazo en la enfermedad. “La genética, bioquímica, trabajo con modelos animales, datos clínicos/patológicos, y datos de biomarcadores todavía sugiere que el objetivo de la construcción y acumulación de amiloide-beta y tau en el cerebro siguen siendo los objetivos más prometedores.” Su laboratorio está trabajando en maneras de entender mejor estos hallazgos asociados con el ciclo de sueño-alerta que influye en el comienzo de la acumulación del amiloide-beta en el cerebro…y mejores formas para apuntar a la proteína tau.
Hyman agrega como área prometedora la “reciente capacidad para detectar cambios patológicos antes de los síntomas clínicos. Es esta capacidad para atajar la enfermedad al principio lo que yo creo que nos dará la ventaja para prevenir la enfermedad más que “curarla” dice.
El Premio para la Investigación y Esperanza para la Investigación en la Industria Farmacéutica en AD fue para el equipo de trabajo BACE de Merck, que incluye a Eric M. Parker, director ejecutivo y líder del sitio de neurociencias; Andrew W. Stamford, director de descubrimientos químicos; Matthew E. Kennedy, director asociado de neurociencias; Mark S. Forman, director de investigación clínica; y Julie A. Stone, directora científica senior.
El equipo de BACE ha estado evaluando la seguridad y eficacia de la enzima que rompe el sitio de la proteína precursora del beta-amiloide, o inhibidor del BACE, incluyendo la MK-8931, el cual es el compuesto principal del proyecto AD de Merck. Los resultados de los estudios clínicos hasta ahora asocian las dosis simples del MK-8931 con reducciones marcadas en los niveles de concentración del péptido Aβ.
“La hipótesis del amiloide sugiere que una de las maneras de alterar la causa molecular del AD es inhibir la actividad del BACE. Con los así llamados inhibidores del BACE, la producción de proteínas Aβ estaría estancada, impactando posiblemente el inicio del AD”, explica Parker. “A pesar de los diversos compuestos en investigación y de anticuerpos monoclonales que se han probado en la hipótesis amiloide con poco, si no es que con ningún éxito, pensamos que la inhibición del BACE reduce los niveles de los péptidos Aβ a un mayor grado que otros tratamientos que se han probado. Por lo tanto, los inhibidores BACE son un medio prometedor para probar la hipótesis del
amiloide. Sin embargo, estamos igualmente dedicados al hallazgo de esquemas no amiloides y terapias sintomáticas para la AD, y continuaremos nuestra investigación en esas áreas,” añade.
El Premio para la Investigación y Esperanza para la Defensa del Paciente fue para Kate Maslow en el Instituto de Medicina (IOM) del Centro Keck, en Maslow, una estudiante en residencia en la Academia Nacional de Ciencias del IOM, quien se ha enfocado en problemas que van unidos al cuidado de las personas con AD y otras demencias. Maslow tiene una perspectiva única de cómo la academia y la industria pueden asociarse mejor con los trabajadores de la salud y con aquéllos que están comprometidos en el día a día con el cuidado del paciente AD para mejorar los resultados y tratamientos del paciente. “Algunas veces, los investigadores de la academia y la industria están desconectados de la experiencia diaria de las personas con AD y de otras demencias y de sus familias. Aunque estos investigadores saben, al menos en teoría, cómo afectan las condiciones a la persona y a la familia y que pueden tener parientes que tienen las condiciones, la búsqueda de tratamientos biomédicos, biomarcadores y tecnologías de imagenología los pueden aislar del propósito último de esa búsqueda, dice. “Es posible que un mayor contacto e interacción entre las personas con estas condiciones, con las familias y con los investigadores académicos y de la industria, resultaría en una nueva comprensión e ideas que pudieran ayudar a concentrar el esfuerzo de investigación.”
Maslow insiste en añadir, sin embargo, que la comunicación va en dos vías. “Algunas personas con las condiciones, las familias y los defensores se beneficiarían de una comprensión más sofisticada del esfuerzo de investigación continuo, la complejidad de las condiciones, y la dificultad de aislar las causas y tratamientos efectivos. Mucha de la información disponible públicamente acerca de estos temas está sobresimplificada.”
El Premio para la Investigación y Esperanza para el Campeón Voluntario fue para Neja Chauhan en Adolescentes AFA para la Conciencia del AD. Chauhan ha sido voluntaria en la lucha contra la AD desde que tenía 15 años como fundadora de Adolescentes AFA, el cual es una rama activa de la AD Fundación de América, dedicada a la defensa de jóvenes. Hoy, dice Chauhan, los Adolescentes AFA tiene un consejo consultor de adolescentes en todo EEUU que proporciona conocimiento de cómo los adolescentes están afectados por la enfermedad. Adicionalmente, aproximadamente 1200 estudiantes solicitan entrar al concurso para la beca del ensayo de la organización cada año. Chauhan es actualmente estudiante de MBA en Stanford. “Espero que cualquiera que sea el camino que escoja ayudará a asegurar que la AD sea una memoria lejana algún día”, dice.
Viendo a futuro, cada receptor del premio tiene grandes esperanzas para la investigación futura y el cuidado de la AD. Maslow dice que le gustaría ver “el desarrollo y capacidad de los tratamientos biomédicos que pudieran prevenir y/o retrasar el inicio y avance de la enfermedad AD.” Las políticas gubernamentales le permiten a los sistemas de salud identificar y evaluar el deterioro cognitivo en pacientes/afiliados dando mientras tanto cuidado médico continuo y soporte están también en la lista de deseos de Maslow.
Holtzman concuerda en que la cantidad de asociaciones entre la academia y la industria necesitan incrementarse y señala que tales proyectos están a la alza, como es el caso de la iniciativa de neuroimagenología de la enfermedad AD así como de los estudios de prevención de la enfermedad AD que se hereda de manera dominante. “Ellos también se están asociando vía proyectos de investigación en el nivel de grupos individuales de laboratorios con compañías específicas,” dice.
Estas esperanzas para la investigación futura del AD y el cuidado del paciente son clave, especialmente considerando algunos de los contratiempos de los estudios clínicos que se han presentado durante 2012 (Johnson & Johnson, Pfizer y Eli Lilly

Pharmaceutical Technology en Español MARZO / ABRIL 201330
todos publicaron reportes de estudios clínicos fracasados dirigidos a la proteína beta-amiloide). Algunos reportes han establecido que las personas han perdido la esperanza así como el financiamiento para la investigación para la cura de la AD como resultado.
Parker responde: “Muchos de los fracasos en los estudios clínicos… pueden atribuirse a al menos dos factores. Primero, el tratamiento a ser probado tenía efectos colaterales significativos que limitaban las dosis que podían ser administradas, lo cual puede también haber limitado su efectividad. Segundo, estos estudios han reforzado una opinión colectiva en el campo de que podemos necesitar iniciar el tratamiento años antes de que los pacientes empiecen a mostrar síntomas. Sin embargo, eso significa que se debe tener la capacidad de identificar a la gente que está en riesgo de desarrollar AD en primer lugar, y esa investigación está todavía en curso.”
Hymann agrega: “el problema [del AD] es demasiado inmenso –con 6 millones de estadounidenses ya afectados y $200,000 mdd en costos de cuidado- para renunciar sólo porque los primeros pocos intentos no tuvieron éxito. No sabemos si los fármacos que se han ensayado se dieron demasiado tarde, en una dosis demasiado baja, o simplemente si debieron darse combinaciones más poderosas de fármacos. Nuestro conocimiento del proceso de la enfermedad ha llegado muy lejos para considerar la renuncia ahora como opción”.
Para escuchar la entrevista completa con Castellani, visite PharmTech.com/PharmTechTV. Para leer la entrevista completa con el equipo BACE, entre a PharmTech.com. El bios completo de los ganadores esta en www.phrma.org/awards.
—Angie Drakulich
turas con una conducta mejorada de ensamble y resistencia a la degradación enzimática, llevando a una mejor estabilidad.”
El siguiente objetivo de Panda es demostrar la eficacia de estos sistemas en otros modelos de tumor y demostrar su apli-cación potencial más amplia, como los nanosistemas basados en péptidos que pueden apuntar a la tuberculosis, SIDA, y otras enfermedades globales.
La estudiante de la Universidad de California en San Francisco, Rachel Jean Eclov ganó por su investigación en “Caracterización in vivo de Potenciadores ABCG2,” en la ca-tegoría de farmacocinética, farmacodinamia, metabolismo de fármacos, farmacología clínica e investigación traslacional. “La investigación previa sobre la funcionalidad del MXR ha sido principalmente dirigida a variantes funcionales (codifica-ción) del MXR. Mi investigación es única en que busca cómo otros tipos de variaciones genómicas pueden alterar la expre-sión del MXR y de esta forma la disposición del fármaco. Yo utilizo herramientas epigenéticas para desenmarañar cómo la expresión del MXR puede ser específica del tejido o incre-mentarse transitoriamente o reducirse,” explicó.
“Yo entonces considero cómo la variación genética en los elementos regulatorios identificados puede alterar las pro-piedades transcripcionales de ese elemento. Aunque se han publicado otras investigaciones epigenéticas, la mayoría de éstas son sobre las variaciones en los elementos regulatorios que causan defectos del desarrollo. Mi investigación utiliza epigenética para identificar variaciones genéticas que impac-tan el mundo más sutil de la disposición de fármacos en don-de los efectos de estas variaciones no son tan obvios aunque pueden ser severos.” En términos de cómo la investigación de Eclov puede afectar el trabajo futuro de la industria, dice, “Aun cuando mi investigación está sólo en los bordes inicia-les de la comprensión de la regulación genética de un gen ADME, puede usarse como una plantilla general para que otros investiguen la regulación genética de su propio gen de interés. Ésta muestra que existen muchas maneras en que una expresión del gen puede estar epigenéticamente regulada y de esta manera desregularse en cánceres o mediante tratamiento con fármacos. También es importante para los científicos dar-se cuenta de que las variaciones genéticas que no codifican,
Reconocimiento a la Innovación Farmacéutica en el CPhI Mundial-
Es crucial la innovación en tecnología que soporte el desarrollo de fármacos y la manufactura para cumplir la revolución de las necesidades y los productos farmacéuticos. Los Premios Farmacéuticos del CPhI reconocen dicha innovación y fueron entregados en el CPhI Mundial, la gran exposición de fabricantes por contrato y proveedores de ingredientes farmacéuticos y tecnología relacionada, organizada por UBM Live, y realizada de Oct. 9-11, 2012 en Madrid. Conocida anteriormente como los Premios a la Innovación, los renombrados Premios Farmacéuticos CPhI nombraron a los ganadores del Oro, la Plata y el Bronce por la Mejor Innovación Farmacéutica. Los premios fueron diseñados para estimular el ingreso en diversas áreas, incluyendo desarrollo de formulaciones, entrega de fármacos, manufactura química de APIs e intermedios, y biomanufactura.
El ganador del Premio Farmacéutico CPhI de Oro fue Haemopharm Healthcare, un fabricante con base en Milán de bolsas flexibles para soluciones estériles y contenedores y componentes relacionados. Haemopharm fue reconocido por su vial para inyección sin aguja (NIV), un sistema de cierre del vial sin aguja que puede ser usado en productos suministrados en un vial de vidrio o plástico, ya sea que esté en forma de líquido, gel o polvo. El sistema NIV mantiene la esterilidad y evita contaminación debido a un sello que se vuelve a cerrar herméticamente. El re-cierre evita el reflujo y la pérdida del contenido.
Merck Millipore, la división de ciencias de la vida de Merck KGaA, ganó el Premio Farmacéutico CPhI de Plata por su sílica bimodal como plataforma para entrega de fármacos, la cual está diseñada para facilitar la solubilidad de APIs escasamente solubles. La innovación de Merck Millipore saca ventaja de la estructura del poro plegado en dos de la sílica, lo cual ofrece una gran área de superficie y buenas propiedades de transporte para la entrega de fármacos. En octubre de 2012, Merck Millipore anunció la disponibilidad de un rango de productos para mejorar la biodisponibilidad de fármacos y abordar otros retos en la formulación farmacéutica. Estas ofertas incluyen productos que abordan la solubilidad y la modificación farmacocinética y farmacodinámica y la focalización, lo cual debe tomarse en cuenta para optimizar la biodisponibilidad del fármaco final.
rEportE EspEcial: innovación

Pharmaceutical Technology en Español MARZO / ABRIL 2013 31
encontradas en grandes pantallas, como un GWAS, podrían ser ‘aciertos’ muy relevantes y de que existen herramientas para ayudar a desarrollar modelos epigenéticos para la fun-ción de estas variantes.”
Otros estudiantes de la Universidad de Connecticut, Ek-neet K. Sahni, ganó en la categoría de ciencia de la manufac-tura e ingeniería por “Secado por Contacto en un Secador con Filtro Agitado: Experimentos y Simulaciones.” Dice Sahni acerca de la investigación “A pesar de los recientes avances
El Premio Farmacéutico CPhI de Bronce fue entregado a BioClin, una compañía farmacéutica localizada en Delft, Holanda, por su gel de remineralización Multi-Oral Remin, un producto que se aplica en la cavidad oral para tratar y evitar el sangrado de encías y la erosión de los dientes. La formulación está basada en un 2QR-complejo, un complejo acetilado de polimanosa, el cual bloquea la adhesión de bacterias, las neutraliza y combina esta actividad con el efecto de remineralización con propiedades anti-placa.
Aunque no estuvieron en los primeros tres, hubo otros dos finalistas para los Premios farmacéuticos CPhI. Los finalistas fueron Linhardt, un proveedor con base en Alemania de sistemas de empaque, por su tecnología de tubo colapsable Multiflex, la cual está diseñada para mejorar los tubos laminados tradicionales, y YaoPharma, una compañía de fármacos genéricos con base en Chonggin, China y una subsidiaria de Fosun Pharmaceutical, por el alprostadil, una emulsión liofilizada para inyección.
Los Premios Farmacéuticos CPhI también reconocieron los avances en la sustentabilidad, los cuales incluyeron un nuevo premio para el Mejor Empaque Sustentable, que reconoce una compañía para un material específico, maquinaria o proceso que facilita el empaque sustentable. El premio fue para MeadWestvaco Healthcare por su empaque para fármacos con mejor adherencia, que consiste en una cajilla exterior resistente a la rotura y reciclable y un blíster de deslizado fácil con un calendario integrado para ayudar a los pacientes en la administración de sus medicamentos.
Para información adicional sobre los ganadores del Premio Farmacéutico CPhI, escuche nuestros podcasts en www.PharmTech.com/PharmTechTV.
—Patricia Van Arnum
hechos hacia la comprensión del fenómeno de secado, las complejidades involucradas en el proceso, no sólo debido a la naturaleza acoplada del proceso que involucra calor, masa y momentum de transporte sino también de su dependencia de las propiedades del material y de las condiciones de secado, no permite predicciones cuantitativas con extrema exactitud. El modelo de penetración ha sido conocido desde hace mu-cho y se considera como el estándar industrial para el secado por contacto. No obstante, tiene desventajas en la considera-ción de la mecánica granular debido a su naturaleza continua. Hasta recientemente, la mayoría del trabajo de transferencia de masas basado en el DEM fue ya sea bi-dimensional o en lechos granulares estáticos.” Sahni explica que el trabajo ga-nador fue el primer estudio que empleó 3D-DEM “para com-prender mejor la conducta granular en un secador con filtro agitado investigando el efecto de las variables de proceso so-bre el desempeño del secado. Se ha dado más consideración al efecto de la velocidad, la cual no ha sido claramente com-prendida en la literatura.”
En términos de la relevancia del trabajo para estudios futu-ros, Sahni dice “Junto con la mejora de nuestra confianza para el uso del DEM como una herramienta emergente, se llena el vacío en la literatura para esquemas discretos. Los beneficios se ven también en los recursos reducidos para desarrollo y en los costos de manufactura sin algún retraso en la producción. Por lo tanto, el modelado del proceso basado en el desarrollo del producto puede reducir el tiempo requerido para llevar pro-ductos al mercado así como los costos de salud mediante el ahorro de recursos. Las conclusiones para los científicos son que se desarrolle una herramienta predictiva del desempeño a priori que pueda ayudar a predecir el resultado final incluso para los parámetros del proceso fuera del rango estudiado y para cualquier material el cual le dé un borde sobre otros dise-ños así como modelos que estén principalmente restringidos a los parámetros y/o al material estudiado.”
Breve presentación en Power Point de cada uno de los estudiantes graduados aparecen en PharmTech.com/AAPSstudents2012. PT
SU PRESENCIAEN LA REVISTA MÁS LEIDA POR LA INDUSTRIA FARMACÉUTICA
CONTRATE SU PAUTA PUBLICITARIA
AHORA
Mayores Informes y Contrataciones:Tel: 52 (55) 5659-8880, 5536-2100, 5543-1486 E-mail: [email protected]

Pharmaceutical Technology en Español MARZO / ABRIL 201332
GlaxoSmithKline recientemente desarrolló una novedosa tecnología para la formulación de tabletas de liberación modificada. Comercializada como Diff-CORE, la tecnología combina el uso de aperturas que están excavadas mecánicamente en tabletas con recubrimiento pelicular funcional, con matri-ces tradicionales de polímero que controlan el mecanismo de erosión del núcleo y la difusión. Los autores describen la ruta desde el desarrollo hasta la comercialización de este esquema de liberación modificada.
Kevin D. Altria es investigador científico senior, [email protected] y James Taylor* es investi-gador, [email protected], ambos con Desarrollo Farmacéutico, GSK R&D, New Frontiers Science Park, Third Avenue, Harlow, Essex, CM19 5AW, RU.
*A quien debe dirigirse la correspondencia.
Sometido: Feb. 15, 2010. Aceptado: Mar. 15, 2012
DedicatoriaEste artículo está dedicado a la memoria del Dr. John Hempenstall, quien respaldó enormemente el desarrollo y comercialización de esta tecnología. John fue un gran amigo y mentor para muchos colegas en GSK.
Existe un número de esquemas para desarrollar una formulación de liberación modificada (LM), la cual incluye matrices tradicionales de polímeros, como la matriz de hipromelosa (1), y matrices multicapas
más complejas con o sin excipientes funcionales adicionales (2). Hay beneficios y restricciones para cada esquema, los cuales incluyen requerimientos de tecnología y aplicabilidad para el tipo de fármaco/rango de concentración. Reciente-mente se ha desarrollado y comercializado un nuevo esquema de LM por parte de GlaxoSmithKline (GSK) bajo el nombre comercial DiffCORE. Este artículo describe el esquema de LM, su desempeño in vivo, in vitro y comercial industrial, y los beneficios potenciales del esquema.
¿Cuál es el respaldo para el esquema de LM y su desarrollo tecnológico?El esquema involucra el uso de excavación mecánica de table-tas con recubrimiento pelicular funcional (ver Figura 1) para formar aberturas de tamaño y posición conocidos en el recu-brimiento pelicular. La velocidad de liberación del fármaco puede ser modificada y controlada a través de la alteración del área de superficie expuesta y de la composición del núcleo de la tableta. La manufactura de las tabletas utiliza operaciones unitarias de manufactura estándar (es decir, mezclado, granu-lación y compresión). La formulación del núcleo y el proce-so de manufactura impactan el desempeño del producto final como lo haría en cualquier dosis sólida tradicional.
GSK adquirió una patente original en 1993, la cual uti-lizaba un recubrimiento pelicular basado en etilcelulosa. Aunque este recubrimiento pelicular era un recubrimiento semipermeable, cuando se usa en el proceso DiffCORE, se comporta esencialmente como una barrera impermeable. El recubrimiento retarda la liberación del API de la mayoría del área superficial del núcleo. Las aberturas en el recubrimien-to mantienen la velocidad de liberación del API del núcleo a lo largo del tracto gastrointestinal completo. Se encontró que este esquema es adecuado para compuestos que tienen alta solubilidad, principalmente ácidos débiles. Las bases débiles, que en ese tiempo formaban una gran parte del portafolio de LM, se encontró que exhibían liberación reducida al dejar el
forMulación
Desarrollo y comercialización de una novedosa tecnología para tabletas de liberación modificadaKevin D. Altria y James Taylor
isto
ck
ph
oto
/th
ink
sto
ck
iMa
Ge
s

Pharmaceutical Technology en Español MARZO / ABRIL 2013 33
usados de Alta Calidad!
Your source for
equipment!
p. 216.271.3500 • f. 216.271.5210 • [email protected] • 8200 Bessemer Avenue • Cleveland, Ohio, USA 44127
FEDERAL EQUIPMENT COMPANY ofrece equipos usadosen procesos Farmacéuticas y Enpaque de alta calidada una fracción del costo de equipos nuevos y disponibleinmediatamente. Por favor visite nuestra página webahora accesible via móvil para ver más de 15,000equipos diverso en inventario.
.atelpmoc atnalp o nóiccudorp aenil ,opiuqe nu
mercado asegura que recibirán ofertas valiosas por
En adición proporcionamos todos servicios auxiliaros
¡Visítenos Hoy!
Federal Equipment.pdf 1 08/03/13 16:28
entorno gástrico debido a la reducción de solubilidad produ-cida por el pH y por lo tanto en la disponibilidad.
Utilizando experimentos dirigidos, se desarrolló un nue-vo sistema de recubrimiento pelicular, el cual permitía que se usara la tecnología con bases débiles (3). El incremento en los potenciales productos que podían beneficiarse del nuevo sistema de recubrimiento pelicular provocó más inversión de tiempo y recursos para explorar completamente las capacida-des de esta tecnología.
El nuevo sistema de recubrimiento desarrollado fue un recubrimiento pelicular entérico basado en la investigación previa con diferentes agentes antiadherentes. Para las bases débiles, este recubrimiento inicialmente retarda la liberación del material activo a las aberturas excavadas mientras está dentro del entorno gástrico de alta solubilidad. Al llegar al mayor pH de los intestinos, el recubrimiento se disocia y se vuelve soluble. La disolución del recubrimiento incrementa el área de superficie expuesta del núcleo, lo cual incremen-ta la disponibilidad de la sustancia farmacéutica expuesta, compensando así la solubilidad reducida. Haciendo uso de técnicas de matriz de polímero establecidas, el núcleo está formulado para asegurar que se mantenga una velocidad de liberación controlada.
Se han hecho más mejoras para compuestos específicos, dependientes de la farmacodinamia del producto. El uso de un núcleo bi-capa permite la combinación de una capa de li-
beración inmediata (LI) y una capa de LM. La capa de LI reduce el tiempo para alcanzar la dosis mínima terapéutica para el paciente mientras que la capa LM proporciona una dosis de mantenimiento. Este esquema bi-capa puede obvia-mente extenderse a productos combinados aunque esto no se
Figura 1: Tabletas con recubrimiento pelicular con aperturas de tamaño conocido, de 2 mm (azul), 3 mm (café) y 4 mm (verde).
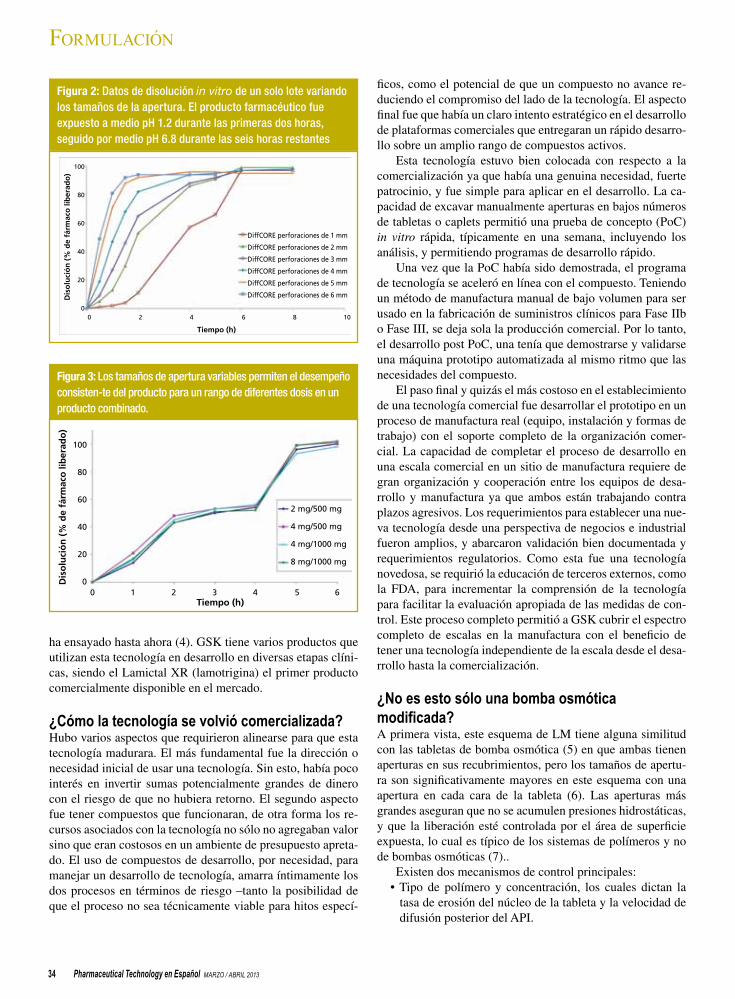
Pharmaceutical Technology en Español MARZO / ABRIL 201334
ha ensayado hasta ahora (4). GSK tiene varios productos que utilizan esta tecnología en desarrollo en diversas etapas clíni-cas, siendo el Lamictal XR (lamotrigina) el primer producto comercialmente disponible en el mercado.
¿Cómo la tecnología se volvió comercializada?Hubo varios aspectos que requirieron alinearse para que esta tecnología madurara. El más fundamental fue la dirección o necesidad inicial de usar una tecnología. Sin esto, había poco interés en invertir sumas potencialmente grandes de dinero con el riesgo de que no hubiera retorno. El segundo aspecto fue tener compuestos que funcionaran, de otra forma los re-cursos asociados con la tecnología no sólo no agregaban valor sino que eran costosos en un ambiente de presupuesto apreta-do. El uso de compuestos de desarrollo, por necesidad, para manejar un desarrollo de tecnología, amarra íntimamente los dos procesos en términos de riesgo –tanto la posibilidad de que el proceso no sea técnicamente viable para hitos especí-
forMulación
Figura 2: Datos de disolución in vitro de un solo lote variando los tamaños de la apertura. El producto farmacéutico fue expuesto a medio pH 1.2 durante las primeras dos horas, seguido por medio pH 6.8 durante las seis horas restantes
Figura 3: Los tamaños de apertura variables permiten el desempeño consisten-te del producto para un rango de diferentes dosis en un producto combinado.
100
80
60
40
20
00 2 4 6 8 10
DiffCORE perforaciones de 1 mm
DiffCORE perforaciones de 2 mm
DiffCORE perforaciones de 3 mm
DiffCORE perforaciones de 4 mm
DiffCORE perforaciones de 5 mm
DiffCORE perforaciones de 6 mmDis
olu
ció
n (
% d
e fá
rmac
o li
ber
ado
)
Tiempo (h)
100
80
60
40
20
0Dis
olu
ció
n (
% d
e fá
rmac
o li
ber
ado
)
0 1 2 3 4 5 6Tiempo (h)
2 mg/500 mg
4 mg/500 mg
4 mg/1000 mg
8 mg/1000 mg
ficos, como el potencial de que un compuesto no avance re-duciendo el compromiso del lado de la tecnología. El aspecto final fue que había un claro intento estratégico en el desarrollo de plataformas comerciales que entregaran un rápido desarro-llo sobre un amplio rango de compuestos activos.
Esta tecnología estuvo bien colocada con respecto a la comercialización ya que había una genuina necesidad, fuerte patrocinio, y fue simple para aplicar en el desarrollo. La ca-pacidad de excavar manualmente aperturas en bajos números de tabletas o caplets permitió una prueba de concepto (PoC) in vitro rápida, típicamente en una semana, incluyendo los análisis, y permitiendo programas de desarrollo rápido.
Una vez que la PoC había sido demostrada, el programa de tecnología se aceleró en línea con el compuesto. Teniendo un método de manufactura manual de bajo volumen para ser usado en la fabricación de suministros clínicos para Fase IIb o Fase III, se deja sola la producción comercial. Por lo tanto, el desarrollo post PoC, una tenía que demostrarse y validarse una máquina prototipo automatizada al mismo ritmo que las necesidades del compuesto.
El paso final y quizás el más costoso en el establecimiento de una tecnología comercial fue desarrollar el prototipo en un proceso de manufactura real (equipo, instalación y formas de trabajo) con el soporte completo de la organización comer-cial. La capacidad de completar el proceso de desarrollo en una escala comercial en un sitio de manufactura requiere de gran organización y cooperación entre los equipos de desa-rrollo y manufactura ya que ambos están trabajando contra plazos agresivos. Los requerimientos para establecer una nue-va tecnología desde una perspectiva de negocios e industrial fueron amplios, y abarcaron validación bien documentada y requerimientos regulatorios. Como esta fue una tecnología novedosa, se requirió la educación de terceros externos, como la FDA, para incrementar la comprensión de la tecnología para facilitar la evaluación apropiada de las medidas de con-trol. Este proceso completo permitió a GSK cubrir el espectro completo de escalas en la manufactura con el beneficio de tener una tecnología independiente de la escala desde el desa-rrollo hasta la comercialización.
¿No es esto sólo una bomba osmóticamodificada?A primera vista, este esquema de LM tiene alguna similitud con las tabletas de bomba osmótica (5) en que ambas tienen aperturas en sus recubrimientos, pero los tamaños de apertu-ra son significativamente mayores en este esquema con una apertura en cada cara de la tableta (6). Las aperturas más grandes aseguran que no se acumulen presiones hidrostáticas, y que la liberación esté controlada por el área de superficie expuesta, lo cual es típico de los sistemas de polímeros y no de bombas osmóticas (7)..
Existen dos mecanismos de control principales:• Tipo de polímero y concentración, los cuales dictan la
tasa de erosión del núcleo de la tableta y la velocidad de difusión posterior del API.

Pharmaceutical Technology en Español MARZO / ABRIL 2013 35
• El área de superficie del núcleo expuesto para la libera-ción del API, la cual está controlada por (a) el tamaño de la apertura en la región gástrica y por (b) la geometría del núcleo en la región intestinal.
¿Para qué tipos y dosis de APIs puede usarse el esquema?Debido a la naturaleza de esta tecnología, ha habido un nivel de riesgo percibido asociado con “ser el primero” de mane-ra que la captación inicial ha sido con compuestos que han tenido un número de retos o dificultades en el desarrollo de una forma farmacéutica de LM tradicional. Sin embargo, en resumen, se ha probado un número de compuestos a través del Sistema de Clasificación de Biofarmacéuticos (BCS) y ha demostrado gran éxito. Más recientemente, el lanzamiento de un producto comercial ha ayudado a reducir este riesgo perci-bido y ha incrustado la tecnología dentro de GSK.
Tres compuestos en fase tardía han demostrado la ampli-tud de la tecnología. Dos productos utilizaron dosis tan bajas como 2 mg y han demostrado ser consistentes y reproducibles en su entrega, mientras que un producto de dosis alta, de 1000 mg, se ha mantenido como una tableta diaria única para ayu-dar al cumplimiento del paciente sin tener que usar niveles innecesarios de polímeros.
¿Cómo se modifica y controla la velocidad de liberación?Esta novedosa tecnología utiliza la combinación de apertu-ras en recubrimientos peliculares funcionales con matrices de polímeros tradicionales. A diferencia de las matrices de polímeros típicos, estas formulaciones utilizan un polímero de baja viscosidad para controlar el mecanismo de erosión y difusión del núcleo. Los polímeros de baja viscosidad son más adecuados porque la hidratación del polímero está res-
tringida por el recubrimiento pelicular en el entorno gástrico de manera que ocurre poca erosión antes de la formación de la estructura de gel.
La capacidad para combinar estas matrices con un recu-brimiento pelicular funcional también proporciona mecanis-mos de control dual que confieren varias ventajas. Pueden lograrse velocidades de liberación difíciles o distintas, las cuales muestran poco efecto de alimento; el paciente está doblemente protegido por los mecanismos de control, redu-ciendo el riesgo de descarga de dosis; puede usarse un solo lote de tabletas para desarrollar una serie de velocidades de liberación simplemente modificando el área expuesta de la
Figura 4: Comportamiento de la disolución del producto inicial y después de 56 meses de almacenamiento a 30ºC / 65% de humedad relativa (HR).
100
80
60
40
20
0
Dis
olu
ció
n (
% d
e fá
rmac
o li
ber
ado
)
0 2 4 6 8 10 12 14 16
Producto inicial56 meses a 30ºC / 65% HR
Tiempo (h)
Tabla I: Número de formulaciones evaluadas en estudios clínicos con y sin DiffCORE (GlaxoSmithKline).
Molécula
Estudios clínicos pre DiffCORE
Número de formulaciones
Estudios clínicos DiffCORE
Número de formulaciones
Lamictal XR 0 NA 1 3
GSK1 5 20 3 11*
GSK2 4 27 3 15*
GSK3 0 NA 2 2
GSK4 /Metformin 0 NA 1 6
GSK5 0 NA 1 4*
*DiffCORE más otras formulaciones de liberación modificada.DiffCORE es tecnología de entrega de fármacos, de liberación modificada, propiedad de GlaxoSmithKline.NA = No aplica
Figura 5: Equipo de desarrollo con sistemas de inspección en línea.

Pharmaceutical Technology en Español MARZO / ABRIL 201336
superficie del núcleo, como se muestra en la Figura 2.Durante las primeras pocas horas, cuando el recubrimien-
to funcional es insoluble en el medio ácido, el área de su-perficie expuesta es el mecanismo de control dominante, y la velocidad de liberación está controlada por el tamaño de las aperturas. Conforme se incrementa el área de superficie expuesta, la formulación del núcleo muestra una influencia cada vez mayor sobre la velocidad de liberación. Una vez que el tamaño de la apertura es lo suficientemente grande, la for-mulación del núcleo y las características del API se vuelven dominantes en el control de la liberación. En este ejemplo se utiliza un fármaco soluble de dosis alta para demostrar este efecto. Después de cuatro horas, cuando el producto far-macéutico se expone al medio de pH 6.8, el recubrimiento funcional se vuelve soluble, y las características del núcleo dirigen la velocidad de liberación.
A lo largo del proceso de desarrollo, con frecuencia nece-sitará desarrollarse un rango de dosis para identificar la dosis más apropiada para tratar grados variables de condición del paciente o simplemente para la titulación de la dosis. Cuan-do se diseñan productos combinados, los grandes números de combinaciones de dosis pueden hacer que los programas de desarrollo sean algo más complejos.
Los datos en la Figura 3 muestran un producto que abarca cuatro dosis a través de dos APIs. Utilizando los granulados de la plataforma y variando solamente los tamaños de apertu-ra, se diseñaron dosis para tener perfiles sobrepuestos.
¿Cómo se diseña una formulación para obtener un resultado clínico utilizando este esquema?La formulación se desarrolla con base en la definición de los datos fisiológicos, farmacodinámicos y farmacocinéticos (FC) como requerimiento clínico. Una vez que el perfil de solubilidad y el objetivo FC están claros con los requerimien-tos clave (es decir, área bajo la curva [ABC] y concentración máxima [C
máx]) puede establecerse la velocidad de liberación
apropiada y el perfil in vitro para el producto terminado. La capacidad para usar una selección de tipos de productos de-mostrados, por ejemplo DiffCORE bi-capa o perfiles de libe-ración múltiple a partir de un solo lote permite la rápida ma-nufactura del producto para el análisis clínico contra el(los) perfil(es) definido(s).Uno de los beneficios asociados con el DiffCORE ha sido la reducción en el número de estudios clínicos para la evalua-ción de la formulación. Cada estudio clínico es costoso por los recursos en términos de equipo, materiales, planeación, y reclutamiento de pacientes. Una revisión de seis compuestos que han usado la tecnología DiffCORE demostró que la mitad requería una sola visita a la clínica para definir la formula-ción para el desarrollo final (ver Tabla I). También mostró que la tecnología facilitaba el desarrollo de una formulación adecuada para compuestos que históricamente eran difíciles de desarrollar como productos de LM.
forMulación
¿Qué controles se necesitan sobre las aperturas?GSK identificó cuatro atributos de calidad potenciales a lo largo del ciclo de vida de desarrollo para asegurar un desem-peño consistente y robusto del producto: presencia, tamaño, posición y profundidad de las aberturas.
El efecto que estas tienen sobre el desempeño del pro-ducto es dependiente del API y de la formulación del núcleo. Aquéllos productos con mayor robustez se evidencian por una amplia tolerancia del tamaño de la apertura (± 1 mm) con poco impacto de la posición y profundidad de la apertura, lo cual proporciona una amplia estrategia de control de la manu-factura utilizando controles genéricos del proceso. Aquéllos productos que requieren controles de proceso más rigurosos alrededor de la formación de la apertura requerirán una receta del proceso única, por ejemplo, una alta sensibilidad para la profundidad de la apertura, ± 10 μm, requeriría parámetros específicos del movimiento de la broca para asegurar la for-mación precisa de la apertura. Los controles típicos de proce-so mantienen el tamaño de la apertura dentro de ± 0.2 mm, la posición dentro de 0.3 mm y la profundidad dentro de 50 μm del objetivo.
Los sistemas de inspección en línea, la imagenología vi-sual, y el desplazamiento con láser, miden cada apertura for-mada en términos de tamaño, posición y profundidad. Las especificaciones controladas por la receta se utilizan para categorizar el producto como aceptable o no aceptable. Los productos que no cumplen la especificación son automática-mente retirados del flujo de proceso con una confirmación de la remoción. Durante la producción de rutina, las eficiencias actuales del proceso se están midiendo en más de 99.9%.
¿El proceso de perforado afecta la uniformidad o la estabilidad?Para los productos desarrollados a la fecha, GSK no obser-vó típicamente ningún cambio en el perfil de liberación en la estabilidad para los productos perforados. La Figura 4 mues-tra el comportamiento de la disolución en la estabilidad de un producto durante 56 meses sin diferencias discernibles, lo que indica muy buena estabilidad y reproducibilidad. Como el pro-ceso de perforado sólo expone una pequeña área de superficie del producto mientras que el resto está cubierto con un recubri-miento pelicular entérico de permeabilidad reducida, la vida de anaquel no parece ser ni se espera que sea diferente a un producto con recubrimiento pelicular cosmético.
El desempolvado y la revisión de metal se utilizan como parte de la manufactura, similarmente para la compresión, re-duciendo la exposición potencial del operador y la contami-nación corriente abajo. Se investigó el potencial para daño del producto en el proceso corriente abajo, como es el caso del empacado, demostrando que, utilizando el equipo típico, el proceso fue optimizado para eliminar los defectos potenciales.

Pharmaceutical Technology en Español MARZO / ABRIL 2013 37
Figura 6: Equipo de manufactura comercial con sistemas de inspección en línea.
¿Cómo se escala e proceso para la producción comercial?Después de la PoC, se propuso cons-truir una máquina de desarrollo con el propósito fabricar estos productos (ver Figura 5)- Esta máquina fabrica-rá hasta 10,000 unidades por hora con inspección en línea de los atributos de calidad del producto que impactan el desempeño.
Para la producción comercial, GSK utiliza máquinas que están actualmente instaladas en su planta de manufactura global en Carolina del Norte (ver Figu-ra 6). Estas máquinas tienen la misma funcionalidad y parámetros del proceso que el equipo de pequeña escala, elimi-nando así el escalamiento e incremen-tando el rendimiento (aproximadamente 120,000 unidades por hora). El proceso es independiente de la escala, y el pro-ceso de desarrollo final es transferido directamente a las máquinas comercia-les. El proceso de manufactura actual muestra una uniformidad de contenido consistente con buena variabilidad (me-dia = 99%, desviación estándar = 0.82, con base en 14 lotes comerciales).
¿Cuáles son las ventajas y limitaciones de este esquema de LM en comparación con otros esquemas?El desarrollo de productos de libera-ción modificada puede ser una actividad costosa, la cual puede involucrar varios
estudios clínicos y actividades de formu-lación antes de que se obtenga un perfil FC deseado. Existen varias característi-cas de este esquema de LM que permiten minimizar estos costos:
Ventajas• Rápido desarrollo de múltiples velo-
cidades /perfiles de liberación• Reducción de costos asociados con
cambios en los requerimientos clínicos• Producción de perfiles de liberación
comunes dentro de un rango de productos• Se adapta a los productos combinados• Técnicas de manufactura simples• Eliminación de transferencia técnica
para el proceso de formación de la apertura• Inspección en línea completa de los
atributos de calidad en el punto de ma-nufactura.
Desventajas• Se requiere un proceso extra durante
la manufactura• Debe usarse equipo nuevo específico• La capacidad de manufactura es típi-
camente de aproximadamente 120,000 unidades por hora.
ConclusiónEl desarrollo de tecnología puede ser un proceso riesgoso para un negocio y parece que se le deja cada vez más a la academia demostrar los principios y el prototipo. El éxito de esta tecnología dentro de GSK muestra que esta necesi-dad no es siempre el caso. Una pequeña inversión en el trabajo dirigido puede lle-var a avances que benefician al negocio
así como al paciente, siempre y cuando el riesgo/beneficio relevante sea moni-toreado en todas las etapas del proceso.
Referencias1. C.L. Li et al., J. Pharm. Pharmacol. 57 (5)
533–546 (2005). 2. U. Conte et al., J. Control. Rel. 26 (1) 39–47
(1993). 3. US Patent Application 20110135695, “Oral
Dosage Form for Controlled Drug Re-lease,” filed Dec. 2010, Inventors: C.L. Li et al, Assignee: Glaxo Group Limited.
4. S.R. Vaithiyalingam and V.A. Sayeed, J. Pharm. 398 (1–2) 9–13 (2010).
5. F. Theeuwes, J. Pharm. Sci. 64 (12) 1987–1991 (1975).
6. J. Shokri, Eur. J. Pharm. Biopharm. 68 (2) 289–297 (2008).
7. D.R. Reynolds, S.A. Mitchell and K.M. Balwinski, Drug Dev. Ind. Pharm. 28 (4) 457–466 (2002). PT
Informes y Contrataciones:Tel: 52 (55) 5659-8880, 5536-2100,
5543-1486 E-mail: info@pharmatechespañol.com.mx

Pharmaceutical Technology en Español MARZO / ABRIL 201338
iMa
Ge
: h
eM
er
a,
th
ink
sto
ck
so
ur
ce
Lynne Byers es vicepresidente de calidad en Glaxosmithkline Brian Johnson es director senior de seguridad de la cadena de suministro en pfizer.
Colaboración para proteger la cadena de suministro de los falsificadoresAdeline Siew
rEportE EspEcial: anti-falsificación
PharmTech habla con Lynne Byers y Brian Johnson acerca de las iniciativas Rx-360 para proteger la seguridad del paciente.
Los medicamentos pirata pre-valecen en todo el mundo. Desde las especialidades far-macéuticas hasta los fárma-
cos genéricos, los falsificadores están no sólo apuntando a los fármacos de es-tilo de vida, tales como Viagra y Cialis, sino también a los tratamientos costo-sos para las condiciones que amenazan la vida, tales como el cáncer. Este año vio la aparición del Avastin pirata, el cual afectó 19 consultorios médicos en los Estados Unidos, demostrando que incluso países con sistemas regulatorios estrictos y de ejecución no están exen-tos del problema cada vez mayor de las falsificaciones (1). En esta era digital, el Internet proporciona un canal fácil para que los falsificadores entreguen sus falsificaciones. La compra de me-dicamentos en línea con sólo un click del ratón se está haciendo cada vez más prevalente debido a la conveniencia que ofrece, y de acuerdo con la Orga-nización Mundial de la Salud (OMS), constituye más del 50% de los casos de falsificación (2). En julio de 2011, la Comisión Europea publicó la Directiva
de Medicamentos Falsificados (FMD) 2011/62/EU en un esfuerzo de proteger mejor a los pacientes y consumidores. La nueva directiva, la cual está dirigida a evitar que entren medicamentos falsi-ficados a la cadena de suministro legal y llegue a los pacientes, entrará en vi-gor en enero de 2013. Mientras tanto, el Acta de Seguridad e Innovación de la FDA (FDASIA), aplicada en julio de 2012, ha hecho provisiones para garan-tizar la seguridad de los fármacos, por ejemplo mediante la implementación de nuevas penalizaciones a la falsificación, incluyendo una para el tráfico de medi-camentos falsificados.
Para examinar el avance hecho a la fecha en la guerra contra las falsifi-caciones, PharmTech habló con Lyn-ne Byers, vicepresidente de calidad en GlaxoSmithKline y Brian Johnson, di-rector senior de seguridad de la cadena de suministros en Pfizer. Ambos son re-presentantes y miembros del Rx-360, un consorcio internacional de la cadena de suministros farmacéuticos desarrollada por voluntarios de la industria farma-céutica y biotecnológica, con la misión de proteger la seguridad del paciente mejorando la seguridad de la cadena de suministro farmacéutico para asegurar la calidad y autenticidad de los productos y materiales que se mueven a través de la cadena de suministro.
PharmTech: Qué tan exitosos han sido los reguladores y la industria far-macéutica en la batalla contra la falsi-ficación?
Johnson: Hemos hecho un gran pro-greso como industria en la batalla contra los medicamentos falsificados y otras infracciones a la seguridad en la cadena de suministros; sin embargo, todavía hay mucho más por hacer. Algunos ejemplos de avance positivo desde una perspecti-va legislativa incluyen la adopción del FMD en Europa y el FDASIA en EEUU. La Operación Pangea es un ejemplo de los esfuerzos en colaboración para la aplicación, donde una campaña interna-cional de aplicación de la ley que invo-lucró 100 países que cerraron con éxito más de 18,000 farmacias en línea. El Rx-360 también ha hecho parte de un gran trabajo en el espacio de seguridad de la cadena de suministro, con varios documentos técnicos que ayudarán a

Pharmaceutical Technology en Español MARZO / ABRIL 2013 39
prevenir, detectar y responder a la falsifi-cación y otras amenazas de seguridad de la cadena de suministro. Existen muchos otros ejemplos y todos ellos contribuyen a nuestra lucha de diferentes maneras. No obstante, incluso con estas historias de éxito, todavía tenemos ejemplos de infracciones a la seguridad en la cadena de suministros global en las noticias de cada día. Debemos estar vigilantes.
PharmTech: ¿Qué identificarías como las provisiones clave en el FMD de la UE o en el FDASIA de EEUU en cuanto a la seguridad de la cadena de suministros? ¿Cómo pueden los regu-ladores armonizar mejor los esfuerzos globalmente?
Byers: En términos de mejorar la seguridad de la cadena de suministros, las provisiones clave del FMD son las siguientes. Todas las sustancias activas o APIs, importados a la UE, se requeri-rá que sean fabricados en cumplimiento con las GMPs equivalente a las de la UE para enero 2, 2013. La confirma-ción por escrito de cumplimiento con este requerimiento, el cual será emi-tido por una autoridad en el país que exporta, necesitará acompañar a todas las sustancias activas importadas a la UE a partir de julio 2 del 2013. Los países exportadores pueden aplicar para una exención de estas reglas, aunque a ningún país se le ha concedido una exención a la fecha. En circunstancias excepcionales, y cuando sea necesario asegurar la disponibilidad de productos farmacéuticos, si las instalaciones de manufactura del API tiene un certifica-do GMP de la UE, entonces un estado miembro puede ceder al requerimiento de la confirmación escrita durante un período que no exceda la validez del certificado de GMP. Los estados miem-bros que hagan uso de la posibilidad de dicha renuncia deberán comunicar esto a la Comisión Europea. Apenas se le ha otorgado la exención de los requeri-mientos de confirmación por escrito al primer país, Suiza. El proceso, que va a ser usado por la autoridad regulatoria de cada estado miembro, aún no ha sido comunicado. En términos de los regula-dores que trabajan más estrechamente, se alienta el reconocimiento mutuo de los reportes de inspección ya que esto ayudará a los reguladores a inspeccio-nar más plantas.
Johnson: Colectivamente, la re-ciente legislación en EEUU y la UE ayudará a mejorar la seguridad de la cadena de suministro, pero necesita-mos iniciativas legislativas similares en otras regiones del mundo para nivelar el campo de juego. Nuestras cadenas de suministro son complejas y globales, y como el débil vínculo es lo que ex-plotan los criminales, la armonización global es crítica. Estamos avanzando en la dirección correcta con las iniciativas de los reguladores a colaborar en la au-ditoría, compartir la información y el análisis de riesgo. Otros titulares están colaborando a través de organizaciones tales como Pharmaceutical Cargo Secu-rity Coalition (PCSC), Pharmaceutical Security Institute (PSI), y Rx-360. Es difícil individualizar provisiones es-pecíficas de legislación regulaciones o esfuerzos mediante organizaciones es-pecíficas debido a que todas ellas con-tribuyen en diferentes formas y atacan diferentes aspectos del problema. La clave será la colaboración continua y evitar trabajar en silos, lo que tendemos a hacer algunas veces.
PharmTech: ¿Podría proporcionar una actualización sobre las iniciativas y actividades del Rx-360 hasta el 2012 y su centro de actividad para el 2013?
Byers: Primeramente, como con-sorcio, estamos mejorando y facilitando las auditorías conjuntas y compartidas. Ambos programas de auditoría son operacionales después de un período de pruebas piloto. En segundo lugar, estamos también abordando un rango más amplio de tópicos de seguridad de la cadena de suministro. Se estableció un nuevo grupo de seguridad de la ca-dena de suministro y ha desarrollado y compartido documentos de mejores prácticas. En tercer lugar, estamos ac-tivamente articulando a los reguladores y educando a los legisladores. En térmi-nos de comunicaciones, hemos organi-zado seminarios virtuales alrededor de muchas de las iniciativas del Rx-360. También hemos producido 326 reportes flash de novedades y proceso para men-sajería con la industria, 90 resúmenes de regulaciones o legislaciones propuestas o aprobadas con un tiempo promedio para publicar entre 6-10 días, 16 boleti-nes y realizado 4 reuniones abiertas.
Para 2013, tenemos objetivos para
incrementar la membresía en todas las categorías y continuar trabajando en nuestras prioridades de programas de auditoría, seguridad y análisis de la ca-dena de suministro, y diseminación de información de seguridad de la cadena de suministro.
PharmTech: A la fecha ¿Cuáles son los tres logros más grandes alcan-zados por el Rx-360?
Byers: Yo diría que nuestros tres logros más grandes son: colaboración global entre fabricantes y proveedores dentro de la organización; una opinión positiva de la Comisión de Comercio Federal (FTC) de que los programas de auditoría, si se opera como se describe al FTC, es improbable que infrinjan las leyes anti-monopolio de EEUU, y nues-tra capacidad para reunir a expertos del mundo a trabajar en un problema dentro de un estrecho marco de tiempo (p.ej., después del tsunami en Japón).
PharmTech: ¿Qué mejores prácti-cas pueden ofrecer para que la industria monitoree sus materiales que llegan a través de las cadenas de suministro glo-bales, especialmente con productos que llegan de mercados emergentes, tales como China e India?
Byers: El Rx-360 desarrolla y com-parte las mejores prácticas y puntos a ser considerados en relación con la se-guridad de la cadena de suministro. Esta información está libremente disponible en el sitio web del Rx-360. Un ejemplo de un punto a considerar se relaciona con los sellos inviolables, los cuales se utilizan en los contenedores de materia prima (3).
Johnson: Las compañías necesitan tener programas amplios de gestión de calidad del proveedor para monitorear y manejar materiales y productos de fuen-tes externas, independientemente de la región de donde se originan. Esto em-pieza con la transparencia de la cadena de suministro y una buena comprensión de todos los “jugadores” en la cadena de suministro. Cuando la cadena de su-ministro es bien entendida, las compa-ñías pueden entonces usar procesos de evaluación de riesgos para determinar las estrategias apropiadas para mitigar el riesgo, incluyendo los riesgos asocia-dos con el abastecimiento de regiones con infraestructura regulatoria menos desarrollada. Tener requerimientos cla-

Pharmaceutical Technology en Español MARZO / ABRIL 201340
ros, buenos contratos y programas de supervisión robustos que aseguren que sus proveedores cumplan sus expectati-vas es fundamental.
PharmTech: Con respecto a la seguridad de la cadena de suministro ¿Cuáles son los retos comunes que se enfrentan en la prevención y detección de la adulteración intencional, la des-viación ilegal, y la falsificación de los productos y del empaque? ¿Qué pasos o esquema holístico está tomando el Rx-360 para abordar estos problemas con-siderando lo compleja que puede ser la cadena de suministros farmacéutica?
Johnson: Como industria, nuestro reto más grande y nuestra mayor opor-tunidad es hacer frente a las amenazas a la seguridad de la cadena de suminis-tro holísticamente. Hemos organizado históricamente amenazas individuales, tales como la falsificación o partes es-pecíficas de la cadena de suministro, tales como la manufactura por contrato. También, no siempre hemos balanceado la prevención, la detección y la respues-ta a estas amenazas. Ganar esta batalla requerirá de colaboración a través de la cadena de suministro y entre todos los interesados.
Lo que estamos tratando de hacer en el Rx-360 es promover un esquema holístico para la seguridad de la cade-na de suministro. Esto incluye definir la cadena de suministro ampliamente (de extremo a extremo), reconocer las ventajas de trabajar con las amenazas (falsificación, robo, desviación y adul-teración intencional) colectivamente, y balancear la prevención, detección y respuesta. Nuestro primer documento técnico sobre los programas amplios de seguridad de la cadena de suministro captura muchos de estos conceptos y es un buen mapa para el trabajo de se-guridad de la cadena de suministro que estaremos haciendo.
PharmTech: ¿Podría dar algunas bases de los objetivos y funciones de los diferentes grupos de trabajo de la cadena de suministro en el Rx-360 y qué ha logrado cada grupo de trabajo a la fecha?
Johnson: El Rx-360 aprobó la crea-ción del grupo de seguridad de la cade-na de suministro en octubre del 2011. El objetivo de este grupo de trabajo era promover un esquema holístico para la
seguridad de la cadena de suministro. Iniciamos cuatro equipos de trabajo ha-cia finales del 2011. Los objetivos para cada uno de los equipos fueron:
Sistemas de Manejo de la SCS – Puntos de referencia y documento técni-co que describe el programa extenso de seguridad de la cadena de suministro.
Manejo del Riesgo de Transporte – Puntos de referencia, desarrollo de un modelo de riesgo y documento técnico sobre el uso de esquemas basados en el riesgo para la seguridad del transporte.
Monitoreo del mercado – Puntos de referencia, documento técnico y marco de trabajo sobre buenas prácticas/herra-mientas-técnicas, tecnologías emergen-tes, para monitorear la actividad crimi-nal en el mercado.
Auditorías y Evaluaciones de Ter-ceros LSP’s – Puntos a considerar para auditorías/evaluaciones; metodología estándar de auditoría; herramientas de auditoría para LSP’s.
Se realizaron seminarios virtuales gratuitos para cada grupo de trabajo y los documentos técnicos están actual-mente incluidos en el sitio web del Rx-360. Todo esto fue logrado aproximada-mente en seis meses.
También recientemente iniciamos cuatro grupos de trabajo adicionales con los siguientes objetivos:
Manejo de incidentes – Puntos de referencia y desarrollo de un documen-to técnico que describe un proceso para responder a las transgresiones a la cade-na de suministro.
Desvío ilegal – Puntos de referencia y desarrollo de un documento técnico sobre las estrategias para evitar, detec-tar y responder a los desvíos ilegales.
Escasez de fármacos – Puntos de re-ferencia y desarrollo de mejores prácti-cas de lo que las compañías pueden ha-cer para evitar brechas en la seguridad de la cadena de suministro que resulten en escasez de fármacos.
Grupo de discusión de “Seriación” – Grupo de discusión para compartir in-formación sobre las estrategias y retos para la implementación de la seriación globalmente.
PharmTech: ¿Podría discutir bre-vemente las mejores prácticas cuando se usan para responder a las brechas de seguridad en la cadena de suministro?
rEportE EspEcial: anti-falsificación
Johnson: Es importante un proceso de manejo centralizado, efectivo del in-cidente para la adulteración intencional, robo, desviación ilegal y falsificacio-nes. Un proceso exhaustivo debe tener los siguientes atributos: reporte exacto y oportuno del incidente; acciones in-mediatas cuando sea apropiado (p.ej., notificación a los reguladores y ejecu-ción de la ley); evaluación del riesgo; identificación de la causa raíz si aplica; ejecución de acciones correctivas y pre-ventivas; análisis y revisión de tenden-cias; y mejora continua. Esto no es tan diferente a los procesos que usamos en nuestros sistemas de gestión de la ca-lidad.
PharmTech: El grupo de discusión para implementación de la seriación se enfoca a conseguir compañías que cola-boren en la implementación de diferen-tes requerimientos globalmente; ¿Po-dría discutir los retos involucrados en este proceso y los pasos que está dando el Rx-360?
Johnson: Primero, es importante señalar lo que el Rx-360 no está ha-ciendo con este grupo de discusión. No estamos abogando por las diversas legislaciones y/o regulaciones que se están desarrollando globalmente. Exis-ten otras organizaciones activamente comprometidas en las que participan nuestras compañías miembros, por lo que decidimos enfocarnos en una nece-sidad no cumplida, la cual fue crear un foro donde los titulares de las cadenas de suministro pudieran compartir infor-mación y estrategias que se estuvieran tomando para implementar soluciones y hacer inversiones (p.ej., técnicas e IT) en un mundo donde se están desa-rrollando modelos significativamente diferentes. Los retos son obvios cuando todos trabajamos en un mercado global.
Referencias1. FDA, “Counterfeit Version of Avastin in
US Distribution” (FDA website, 2012)., accessed Nov. 12, 2012.
2. WHO, “Medicines: spurious/falsely-labe-lled/falsified/counterfeit (SFFC) medici-nes” (WHO website, 2012), accessed Nov. 12, 2012.
3. Rx–360, “Rx–360 Points-to-Consider: Optimizing Pharmaceutical Raw Ma-terial Tamper Evidence” (Rx¬–360 we-bsite, 2012), accessed Nov. 12, 2012. PT

Pharmaceutical Technology en Español MARZO / ABRIL 2013 41
Retos de la política de salud para la administración de ObamaJill Wechsler
La Casa Blanca y el Congreso probablemente luchen por el financiamiento para la regulación bio/farmacéutica.
VIGILANCIA REGULATORIA
La calma después de la canden-te batalla por las elecciones este año ha sido breve debido a la presión en los legisladores
para abordar los abrumadores proble-mas del presupuesto. Con los republi-canos manteniendo un estrecho control sobre la Cámara de los Representantes, pero perdiendo el piso en el Senado, mucho depende de la capacidad del Presidente Obama para construir alguna clase de “arreglo” para el déficit enorme durante el fin de año “incapacitado” de la sesión del Congreso. La política de salud fue un punto clave de la disputa durante la campaña de elección, marca-da por promesas de mejor cobertura y predicciones de aumento de costos por ambos candidatos. Ahora, los recortes programados y los incrementos signifi-cativos de impuestos se espera que jue-guen un gran papel en la elaboración del programa de reformas.
La victoria de Obama terminó con las posibilidades de revocación abso-luta del Acta de Asistencia Asequible (ACA). Los Republicanos continuarán desafiando varios requerimientos de la legislación de salud, pero las provi-siones clave para las compañías farma-
pn
iko
la
i pu
nin
/Ge
tt
y iM
aG
es
Jill Wechsler es editora en Washington del Pharmaceutical Technology, 7715 rocton ave., chevy chase, MD 20815, tel. 301.656.4634, [email protected]. lea los blogs de Jill en pharmtech.com/wechsler.
céuticas, tales como los descuentos en fármacos para mayores en el hueco de cobertura de la Parte D y la autorización para biosimilares, es poco probable que cambien.
Más ampliamente, la prometida expansión de la cobertura para algo así como 30 millones estadounidenses previamente no asegurados seguirá ca-minando, aunque con los consumidores pagando aumento de las primas y costos compartidos para cubrir los costos siem-pre a la alza, de la salud. Eso prepara el escenario para un crecimiento significa-tivo en el mercado para los fármacos de marca. El Departamento de Salud y Ser-vicios Humanos (HHS) está trabajando fuerte para cumplir una serie de plazos y marcos de tiempo para establecer inter-cambios, definir beneficios y expandir el Medicaid, mucho de lo cual involucra estados que han sido reluctantes a com-prometerse con estos nuevos programas en un período de incertidumbre política.
Recortes de gastos por delanteLa nube negra que se cierne sobre todos estos programas es el “precipicio fiscal” de fin de año, con cerca de $500,000 mdd en incrementos de impuesto y re-cortes de gasto programados para empe-zar el 1º de enero de 2013 a menos que el Congreso actúe. Los ejecutivos en las compañías biofarmacéuticas están vigi-lando muy de cerca cómo los impuestos y las propuestas de presupuesto afecta-rán las tasas de impuestos corporativos y la inversión, así como el financiamien-to específico para la FDA, los Institutos
Nacionales de Salud (NIH) y otras acti-vidades importantes para la innovación biomédica y la cobertura de salud.
Todas las partes reconocen la nece-sidad crucial de reducir los desembolsos tanto públicos como privados para la sa-lud en EEUU, y los precios de los fárma-cos y el reembolso son un blanco prin-cipal, particularmente relacionado con los desembolsos para los programas de salud del gobierno federal y el Medicare Parte D. Los Demócratas han presionado por descuentos adicionales en fármacos adquiridos por los planes de fármacos de Medicare para mayores de bajos in-gresos “doblemente elegibles”, lo cual podría totalizar más de $100 mdd duran-te 10 años. También hay propuestas de presupuesto sobre la mesa para reducir el gasto federal en fármacos para emplea-dos del gobierno federal, así como para otros programas de salud del gobierno.
La victoria de Obama ofrece algo de estabilidad para la FDA, ya que la agen-cia continúa implementando el Acta de Seguridad e Innovación de la FDA y lucha para encontrar un terreno neutral entre acelerar los nuevos medicamen-tos no probados para los pacientes y proteger al público de un riesgo y daño indebidos. Aunque no habrá un cambio total en el liderazgo de la rama ejecu-tiva, muchos funcionarios principales de la administración probablemente se cambien a otras actividades, y los exten-sos recortes en el presupuesto de 2013 podrían socavar muchos proyectos de la FDA. Un recorte de 8.2% en el pre-supuesto de la FDA, según se propuso

Pharmaceutical Technology en Español MARZO / ABRIL 201342
en el proceso de embargo, reduciría el presupuesto del 2013 de la FDA en $320 mdd y provocaría que la agencia despida aproximadamente 1000 empleados, de acuerdo al consultor Steven Grossman, editor de FDA Matters. Incluso sin di-cho recorte en todos los ámbitos, que podría poner en peligro la capacidad de la FDA de colectar tarifas de usua-rio de los fabricantes de farmacéuticos y dispositivos médicos, el presupuesto de la FDA permanecerá vulnerable a las presiones para reducir el gasto federal durante algunos de los años por venir.
Las severas reducciones en el fi-nanciamiento del NIH, además, arries-garían el ritmo del descubrimiento de nuevos fármacos y biotecnológicos y el soporte para la investigación clínica que es clave para estimular la innova-ción necesaria para llenar los reducidos proyectos de nuevos fármacos. La co-munidad de investigación biomédica
VIGILANCIA REGULATORIA
Otro empujón para rastreo y seguimiento Existe un renovado interés en establecer un sistema nacional más seguro de distribución de fármacos que pueda identificar los productos falsificados y adulterados que se mueven a través de la cadena de suministro. El Congreso no pudo lograr un acuerdo sobre dicha política para la inclusión en el Acta de Seguridad e Innovación de la FDA (FDASIA), pero los Senadores Michael Bennet (D-CO) y Richard Burr (R-NC) son autores de un proyecto de compromiso, con un ojo en la promulgación a principios de 2013. Los fabricantes prefieren estándares nacionales al parchado del pedigrí estatal y las políticas de rastreo, y quieren un acuerdo antes de que tengan que implementar los extensos requisitos de rastreo de California en 2015.
Un “proyecto de discusión” del Senado de 117 páginas establece un estándar nacional para el rastreo de productos farmacéuticos, empezando a nivel de lote y moviéndose hacia un sistema futuro de rastreo a nivel unitario electrónico interoperable. Los fabricantes identificarían productos con números de serie y códigos de fármaco, y habría otorgamiento de licencias de la FDA para los mayoristas y terceros distribuidores.
Marcando 1962 enmiendasLos funcionarios de la FDA y la comunidad legal de fármacos marcaron el 50º aniversario de las 1962
enmiendas Kefauver-Harris al Acta de Alimentos, Fármacos y Cosméticos en octubre de 2012, señalando cómo esa ley transformó radicalmente el desarrollo y la regulación de fármacos. La Comisionada de la FDA, Margaret Hamburg, recordó cómo el desastre de la talidomida alentó la aprobación de la legislación que requería preaprobación de la FDA de nuevos fármacos con base en los datos de seguridad y eficacia. Esto, a su vez, creó un nuevo mundo de estudios clínicos controlados aleatorizados, protecciones al sujeto humano, estándares de buenas prácticas de manufactura, supervisión de publicidad de fármacos y reporte de eventos adversos de fármacos.
Además, estos reglamentos también llevaron a un desarrollo de fármacos y proceso de análisis mucho más largo y más costoso, un problema para Peter Barton Hutt, abogado senior en Covington & Burling y abogado de respaldo de la FDA en 1962. Hutt discutió con Bob Temple, director suplente para la ciencia clínica del Centro para Evaluación e Investigación de Fármacos (CDER), en una recepción conmemorativa patrocinada por el Instituto de Leyes de Alimentos y Fármacos; Temple insistió en que la FDA es muy flexible en aceptar un rango de datos para soportar aprobaciones de nuevos fármacos, mientras que Hutt se quejaba de que el proceso de revisión es demasiado largo y engorroso. No obstante estuvieron de acuerdo que los cambios
comparables en la FD&CA probablemente no sean en el corto plazo.
Extendiendo las fechas de caducidadLos críticos de las compañías farmacéuticas están buscando extender las fechas de caducidad etiquetadas como una manera de reducir el gasto en los medicamentos de prescripción y de libre venta (OTC). Tres consumidores de Missouri recientemente presentaron una demanda de que las limitaciones de vida de anaquel muy corta ayudan solamente a los fabricantes a impulsar las ventas. Aunque los autores reconocieron que la caducidad de los fármacos está basada en pruebas de estabilidad requeridas, proponen que los análisis de la compañía deberían extender esas fechas –no mantenerlas tan cortas como sea posible. De manera similar, una carta en los Anales de Medicina Interna en octubre cita la investigación que demuestra que muchos medicamentos OTC para el dolor que caducaron hace tiempo permanecen efectivos. El estudio probó 14 fármacos y encontró que 12 tenían concentraciones al menos del 90% del etiquetado. La FDA y el Departamento de Defensa tienen un Programa de Extensión de Vida de Anaquel (SLEP) para soportar el uso más prolongado de fármacos en los inventarios de medicamentos y que podrían Proveer una base para la extensión de las fechas de caducidad más ampliamente.
Resumen de temas candentes
está destacando la importancia tanto de la FDA como del NIH en la protección de la salud pública para pacientes en el país y alrededor del mundo.
El Congreso aborda las farma-cias de recetas magistralesMientras tanto, las muertes ocasionadas por inyectables de esteroides contami-nados hechos por una farmacia de ma-gistrales en Massachusetts está centran-do la atención en la necesidad de una autoridad legal más amplia de la FDA en esta y otras áreas. La necesidad de reautorizar las tarifas de usuario de fár-macos para animales en 2013 se espera que provea un vehículo para la legisla-ción que asegurará mejor la cadena de suministro de fármacos de prescripción y también abordará la supervisión de combinados de fármacos (Para más información de este asunto, ver “Com-
pounding and FDA Regulation” en www.pharmtech.com/pharmtechtv.)
El brote de meningitis fúngica ha en-fermado a más de 425 individuos y ha causado alrededor de 30 muertes, desde principios de noviembre. El representan-te Edward Markey (D-MA) ha propuesto legislar para mejorar la supervisión de la FDA de las farmacias de recetas magis-trales, y dirigir audiencias de comités de la Cámara y el Senado justo después del día de la elección para dirigir la respues-ta de la FDA y los reguladores estatales y para analizar acciones por parte del far-macéutico infractor, el Centro de Com-binados de Nueva Inglaterra (NECC). El proyecto de Markey clarifica el derecho de la FDAS a inspeccionar y regular a los grandes preparadores de combina-
“Vigilancia Regulatoria”continúa en la pág. 44

Pharmaceutical Technology en Español MARZO / ABRIL 2013 43
Momento para modificar el ICH Q9Lynn D. Torbeck
Un cambio en la terminología podría acentuar la protección al paciente.
SOLUCIONES ESTADÍSTICAS
La guía Q9 vigente, de la Con-ferencia Internacional de Ar-monización (ICH), Manejo del Riesgo de Calidad, fue
recomendada para adopción por los tres cuerpos reguladores de los Estados Unidos, Japón y la Unión Europea, el 9 de noviembre de 2005 (1). El Grupo de Trabajo de Expertos (EWG por sus siglas en inglés) de la guía es digno de elogio por el contenido y calidad de la versión actual.
El éxito del EWG queda evidencia-do por la rápida adopción por las com-pañías farmacéuticas, la industria en ge-neral y por las muchas presentaciones públicas, cursos de capacitación y artí-culos publicados. Casi todas las compa-ñías farmacéuticas tienen un programa de manejo del riesgo de calidad (QRM por sus siglas en inglés) en alguna eta-pa del desarrollo. La cercana relación del ICH Q9 con el ICH Q8 Desarrollo Farmacéutico, y el ICH Q10 Sistema de Calidad Farmacéutica ha solidificado más el amplio uso difundido del QRM en aplicaciones prácticas.
ph
oto
Dis
c/G
et
ty
iMa
Ge
s
Lynn D. Torbeck es estadístico en pharmstat consulting, 2000 Dempster, evanston, il 60202, tel. 847.424.1314, [email protected], www.pharmstat.com.
QRM y protección del pacienteA pesar del éxito del ICH Q90, esta guía podría beneficiarse por modificaciones que pusieran un mayor énfasis en la mi-nimización del riesgo para el paciente más que en el manejo del riesgo de ca-lidad. El Q9 actual fue tomado extensa-mente de la Guía 73:2002 del ISO/IEC y ha incorporado mucho del formato y conceptos en la Guía 73:2002 del ISO/IEC (2). El QRM, por implicación, está concentrado internamente, pero existe una necesidad de agregar un enfoque externo para minimizar más el riesgo para los pacientes.
El ICH Q9 sí tiene observaciones sobre el papel del paciente en la estruc-tura del programa QRM, pero ese con-texto está limitado. Se implica, aunque realmente no se dice, que QRM signi-fica manejo del riesgo para el paciente. La palabra paciente aparece sólo siete veces en el documento; dos veces se hace referencia al paciente como de-positario, dos veces con respecto a la protección del paciente, una vez como parte interesada, una vez con respecto a la necesidad del paciente y una vez con respecto a los fármacos de alta calidad. Paciente y riesgo no se mencionan jun-tos. En contraste, la frase “Manejo del Riesgo de Calidad” aparece 81 veces.
Se asume que el EWG pretendía que el QRM redujera el riesgo para el paciente, pero la mayoría de los lectores
de la industria pueden no reconocer di-cho supuesto y pueden implementar el Q9 con énfasis en el manejo más que en el riesgo para el paciente; obviamente, dos diferentes objetivos.
El Q9 necesita ser más explícito en su objetivo de minimización del riesgo para el paciente (PRM por sus siglas en inglés). Esto puede lograrse sim-plemente reemplazando las palabras “manejo del riesgo de calidad” donde aparecen en la versión actual con “mi-nimización del riesgo para el paciente”. El cambio de calidad para el paciente y de manejo a minimización reenfocaría el programa de calidad completo de in-terno a externo y además soportaría el concepto de ciclo de vida de validación.
El riesgo para la calidad sería toda-vía manejado mediante la minimización del riesgo para el paciente. Por ejemplo, una frase en el Q9 dice: “…la protec-ción del paciente mediante el manejo del riesgo para la calidad debe conside-rarse de primera importancia” (1).
Considere ahora el centro de este nuevo enunciado: “El mejoramiento de la calidad mediante la minimización del riesgo para el paciente debe considerar-se como el objetivo principal.” Observe cómo este cambio modifica el enfoque del manejo del riesgo a la minimización del riesgo. Este enunciado modificado tiene el beneficio adicional de implicar la mejora continua y no sólo de mante-ner el status quo.
continúa en la pág. 44

Pharmaceutical Technology en Español MARZO / ABRIL 201344
El cambio de manejo estático de la calidad a reducción continua en el ries-go para el paciente, sugiere la adición de herramientas y conceptos estadísti-camente orientados. Una nueva versión se beneficiaría grandemente con estas inclusiones estadísticas:
• Definiciones de probabilidad para frecuentistas y teoría Bayesiana
• Una revisión corta de cómo difiere el análisis frecuentista y el Bayesiano
• Una nota de que en el campo de la estadística, el riesgo es con frecuencia sólo un valor de probabilidad
SOLUCIONES ESTADÍSTICAS
• Una corta revisión de los riesgos de la decisión según están defini dos por los errores Tipo I y Tipo II de la hipótesis
• Una breve revisión de los riesgos del muestreo según están definidos por el límite de calidad aceptable (AQL) y por la calidad limitante (LQ).
Observe que en el campo de la es-tadística, el error Tipo II y el LQ están asociados con el concepto de riesgo del paciente o del consumidor. La minimi-zación de estos riesgos es inherente en el buen diseño de datos, la recolección y el análisis.
ConclusiónEs un momento oportuno para afilar el enfoque sobre el riesgo para el paciente ahora que se ha alcanzado una medida del éxito con el manejo del riesgo de calidad. Reemplazando el término “ma-nejo del riesgo de calidad” con “mini-mización del riesgo para el paciente” es una manera de moverse al siguiente nivel de protección al paciente mientras se construye sobre la fuerte base actual del ICH Q9.
Referencias1. ICH, Q9, Quality Risk Management (2005). 2. ISO/IEC Guide 73:2002. PT
dos que califiquen como fabricantes de fármacos. Las farmacias pequeñas de mezclado continuarían operando con la licencia del estado, y la FDA podría emitir exenciones a los operadores que respondan a la escasez de fármacos y a las crisis de salud pública. Los fármacos combinados tienen que etiquetarse di-ciendo que no han sido analizados para los estándares de seguridad y eficacia de la FDA, y la FDA tiene que publicar una lista de fármacos inseguros o inefectivos no adecuados para combinarse.
La regulación de la FDA de los que preparan combinaciones ha sido un asunto espinoso por décadas, ya que los esfuerzos previos de la agencia para im-poner reglas más estrictas con los que preparan combinados han sido revoca-dos por las cortes. Pero la reciente crisis ha reabierto el debate sobre la conve-niencia de la regulación estatal contra la federal de las farmacias y cuando el combinado califica como manufactura de fármaco.
Los esfuerzos de la FDA y de los reguladores de Massachusetts para ce-rrar NECC y su compañía hermana,
Ameridose, destacan los vínculos entre los desabastos de fármacos y el combi-nado. La Comisionada de la FDA Mar-garet Hamburg señaló que la agencia está trabajando duro para minimizar los desabastos en importantes productos de Ameridose usados en cirugía y para pre-venir la insuficiencia cardíaca congesti-va. El ex-funcionario de la FDA Scott Gottlieb también comentó que las regu-laciones tan cerradas de la FDA han lle-vado a escasez de fármacos inyectables de bajo costo, impulsando a los hospita-les y pacientes a buscar alternativas con los que preparan combinados. El muy bajo reembolso para los inyectables genéricos también puede limitar el in-terés de la industria farmacéutica en la producción de estas terapias, llevando a desabastos y mayor dependencia de los que preparan combinados.
KV Pharma comentó que el caso de NECC ilustra el error de la FDA en per-mitir que los combinadores continúen produciendo hidroxiprogesterona para evitar nacimientos prematuros después de la aprobación del Makena de KV. Las agencias de salud del estado y las
aseguradoras han estado optando por la versión combinada menos costosa, pero ahora puede cambiar al producto de KV para evitar la exposición a me-dicamentos combinados posiblemente inseguros.
Habrá más debates sobre cuanta au-toridad legal añadida necesita la FDA para lidiar con los preparadores de com-binados. Algunos críticos de la agencia se quejaron de que la FDA no hacia un uso completo de su autoridad legal existente para regular a NECC después de las inspecciones iniciales insatis-factorias. Los Republicanos general-mente se oponen dándole a la agencia poderes más fuertes, y las farmacias de combinados afirman que están suficien-temente reguladas por los consejos de licencias del estado. Los farmacéuti-cos objetan que el proyecto de ley de Markey para imponer la regulación de la FDA de los combinados podría blo-quear el acceso del paciente a los me-dicamentos necesarios y además exigir mayores contribuciones a la FDA. Esto es un asunto de costo contra seguridad y un reto para encontrar un compromiso que sea aprobado. PT
“Vigilancia Regulatoria”continuación de la pág. 42

Pharmaceutical Technology en Español MARZO / ABRIL 2013 45
Glo
WiM
aG
es
/Ge
tt
y iM
aG
es
Equipo y Proceso
Un esquema de ciclo de vida para la optimización de los sistemas de limpiezaAndrew Wong y Cody Shrader
Los sistemas de limpieza en el sitio deben optimizarse durante el diseño y la puesta en marcha y después de la validación.
RESOLUCIÓN DE PROBLEMAS
En los 90’s, las instalaciones de manufactura farmacéuti-ca empezaron a adoptar las tecnologías de limpieza en
el sitio (CIP) para mejorar los pro-cesos de limpieza e incrementar el tiempo de actividad crítica del equi-po. Aunque estos sistemas iniciales proporcionaron beneficios significa-tivos sobre la limpieza manual, eran armados antes de tener una guía más moderna sobre la construcción y op-timización. Sus diseños se han pro-pagado posteriormente a otras insta-laciones de producción sin una ree-valuación significativa. Como tal, los ciclos de limpieza son con frecuencia una idea de último momento durante el diseño del proceso actual y de los esfuerzos de desarrollo, resultando en ciclos que son pobremente conce-bidos, laboriosamente largos o inne-cesariamente despilfarradores.
Enfocarse en el diseño del sistema de limpieza a lo largo del ciclo de vida puede producir significativos ahorros en costo y tiempo para una organización. Al inicio de un proyecto, el equipo y la tubería deben ser revisados para el dise-ño sanitario que facilite la metodología de la CIP. Después del diseño y cons-trucción, los ciclos de limpieza deben
Andrew Wong y Cody Shrader son ingenieros senior en hyde engineering + consulting, Boulder, co, [email protected].
ser puestos en marcha apropiadamente a través del ensayo y el análisis. Con frecuencia, los sistemas de limpieza y los ciclos son calificados y validados como fueron entregados, imponien-do así barreras al control de cambios para realizar la optimización del ciclo de limpieza. Aunque la modificación de los ciclos de limpieza después de la validación es más compleja, existe un camino para mejoras medidas y con-troladas a través del diseño mecánico o desarrollo de la automatización. Este camino requiere balancear los bene-ficios y los resultados deseados de la optimización con los costos y recursos disponibles para el diseño y la imple-mentación. El siguiente es un breve vistazo a las técnicas para optimizar los ciclos de limpieza a lo largo del ciclo de vida del equipo.
Diseño del equipoUn eficiente ciclo de limpieza empieza con el equipo diseñado para asegurar una limpieza exitosa. El diseño de los tanques y tubería debe ser revisado para su facilidad de limpieza sanitaria, según se describe en la sección SD-3.1 del es-tándar de Equipo de Bioproceso de la Sociedad Americana de Ingenieros Me-cánicos (1). Este diseño puede incluir la minimización de piernas muertas; verifi-cación de que la tubería tenga pendiente hacia un drenado; verificación del punto
bajo de los drenados, conexiones sanita-rias, y válvulas; y la verificación de que todas las superficies de contacto con el producto sean accesibles a las soluciones de limpieza.
El siguiente paso en revisión de la capacidad de limpieza es crear un diseño preliminar de vías de flujo para los cir-cuitos del CIP. Un ejemplo se muestra en la Figura 1. Los segmentos del equipo y la tubería deben esta apropiadamente separados y/o combinados en diferentes circuitos de limpieza como parte de un diseño preliminar. Las consideraciones importantes incluyen requerimientos del proceso y del programa, residuos poten-ciales y diseño de la tubería.
Proceso y programa. El conoci-miento del uso del equipo puede pro-porcionar una idea sobre los tiempos de permanencia o transferencia del pro-ceso. Las líneas de transferencia y los tanques pueden necesitar encadenarse en un solo circuito de CIP para el cam-bio rápido del equipo que cumpla estas demandas. Los tiempos de permanencia del equipo limpio y sucio también pue-den afectar la programación del equipo y los requerimientos de limpieza.
Residuos. La caracterización de los residuos a través de los estudios de lim-pieza y la identificación asociada con las superficies en contacto con el producto ayudan en el desarrollo de los paráme-tros. Ciertos residuos pueden requerir di-ferentes soluciones de limpieza, concen-

Pharmaceutical Technology en Español MARZO / ABRIL 201346
traciones y temperaturas para que ocurra una limpieza adecuada. Este análisis puede ayudar a organizar circuitos me-diante parámetros comunes de limpieza.
Diseño de la tubería. Las conexio-nes disponibles del panel de transfe-rencia pueden limitar la combinación de ciertas líneas de transferencia y tan-ques. El usuario debe tomar en cuenta los tamaños de línea y las longitudes ya que las caídas de presión pueden redu-cir el flujo y la turbulencia dentro de la tubería. Pueden requerirse dentro del sistema bombas adicionales y otras pie-zas del carrete. Debe tenerse precaución en estos casos para minimizar los pa-sos de configuración manual y reducir el riesgo de errores en la preparación. Finalmente, el usuario debe considerar la disponibilidad de drenados por gra-vedad en los puntos bajos a lo largo del circuito del CIP. Los drenados por gravedad siguen siendo cruciales para ciclos eficientes del CIP.
Diseño del sistema de automatizaciónEl diseño de automatización de la lim-pieza también debe ser revisado para sus características de limpieza eficien-te. El desarrollo de ciclos y secuencias que complementan un sistema parti-cular del control de la automatización reduce grandemente los costos de ope-ración a largo plazo. Por ejemplo, un controlador lógico de proceso (PLC) de respuesta rápida, de acción directa, pue-den minimizar los tiempos de enjuague y consumo de agua alternando a través de cada vía auxiliar en un biorreactor complejo lo suficientemente rápido en paralelo con las bolas de atomización aunque no se extiendan las duraciones de la prueba de cobertura de la bola de atomización. En contraste, el tiempo de transición por la vía dentro de un sis-tema de control distribuido (DCS) de-pende de su estilo de programación y puede requerir varias capas del módulo del equipo (EM) y comandos sub-EM antes de alcanzar finalmente los mó-dulos de control (CMs) objetivo. Sólo después de esperar las confirmaciones de la válvula y el estado puede empezar el siguiente paso. Aquí, la creación de un ciclo que combine múltiples transi-
Figura 1: Ejemplo de un diseño del trayecto del flujo
Bola deatomización
V-04
V-02
V-22
V-12
V-10
Desdeel CIP
Bomba CIPS
Tanque de buffer 1
TP-01
PuertoA
PuertoCIPR
Al CIP
Bomba CIPR
V-19V-11
M-4M-3
M-2
M-1
Líneade alivio
Loop #1 – Línea de alivioLoop #2 – Tubo JLoop #3 – Línea de venteoLoop #4 – Bola de atomizado
RESOLUCIÓN DE PROBLEMAS
ciones dentro de un solo agrupamiento, resultará en el ciclo más corto y más rentable posible. La combinación de acciones de limpieza (p.ej., enjuague, drenado y soplado con aire) dentro de las fases también reduce la duración del ciclo. En contraste, una estrategia de uso de fases individuales más modula-res, puede alargar el ciclo.
Varios métodos que reducen costos y tiempo deben estar balanceados con la capacidad del equipo. Por ejemplo, uno puede ser capaz de sacar ventaja de las capacidades del equipo con PLC integrado (p.ej., centrífugas basadas en PLC proporcionadas por el vendedor). Estas consideraciones del diseño deben estar identificadas al principio del pro-yecto para asegurar que la calidad y los procedimientos de validación pueden ser desarrollados para abordar el mues-treo, la instrumentación y los requisitos de verificación que se están construyen-do dentro del CIP y sistemas receptores.
Para el propio ciclo del CIP, la se-cuencia del paso automatizado, los cri-terios de transición del paso y los valo-res de los parámetros deben estar bien definidos y documentados para optimi-zar el uso de la utilidad proporcionando mientras tanto un control suficiente del proceso.
Secuencia. Típicamente, un ciclo de limpieza debe iniciar con enjuagues de agua seguidos por limpieza con de-tergente y enjuagues después del de-tergente. Entre cualquier enjuague o lavada con detergente, el sistema debe ser drenado completamente para evi-tar dilución o reacción química con el siguiente paso del ciclo. Una paso de soplado con aire, colocado antes del drenado, puede reducir en gran medida el tiempo de drenado por gravedad y así reducir el tiempo del ciclo global.
Criterios de transición. La defi-nición de los criterios de transición del paso proporciona una manera de con-trolar los parámetros críticos del ciclo de limpieza. Por ejemplo, la duración del lavado químico, el punto de ajuste mínimo de la temperatura, y la concen-tración objetivo, pueden todos ajustarse de acuerdo a los requisitos antes de las transiciones del paso de lavado al si-guiente paso.
Valores de los parámetros. Los es-tudios de limpieza de residuos a escala de laboratorio pueden proporcionar un excelente punto de inicio para los pa-rámetros del ciclo CIP. Los atributos escalables, tales como concentración del agente de limpieza, temperatura del proceso, tiempo de exposición y energía externa, pueden ser explorados dentro del espacio de diseño de limpie-za para aislar el(los) parámetro(s) más crítico(s). Combinado con una evalua-ción del agente de limpieza más efecti-vo y la identificación de los residuos del proceso en el peor caso, estos esfuerzos a escala de laboratorio pueden reducir dramáticamente el número de iteracio-nes del ciclo que deben realizarse du-rante la puesta en marcha y permiten un enfoque en la mejora de la eficiencia.
Puesta en marchaDurante la fase de puesta en marcha, el ciclo de la CIP puede optimizarse más con las pruebas prácticas de desarro-llo. Se requiere el conocimiento de la secuencia del paso CIP y los criterios de transición para establecer apropia-damente parámetros significativos y eficientes para las recetas del CIP. La verificación en campo puede incluir el monitoreo de los puntos de drenado, la visualización de los niveles del tanque durante la recirculación o el registro de las lecturas de presión a lo largo de la línea de suministro.
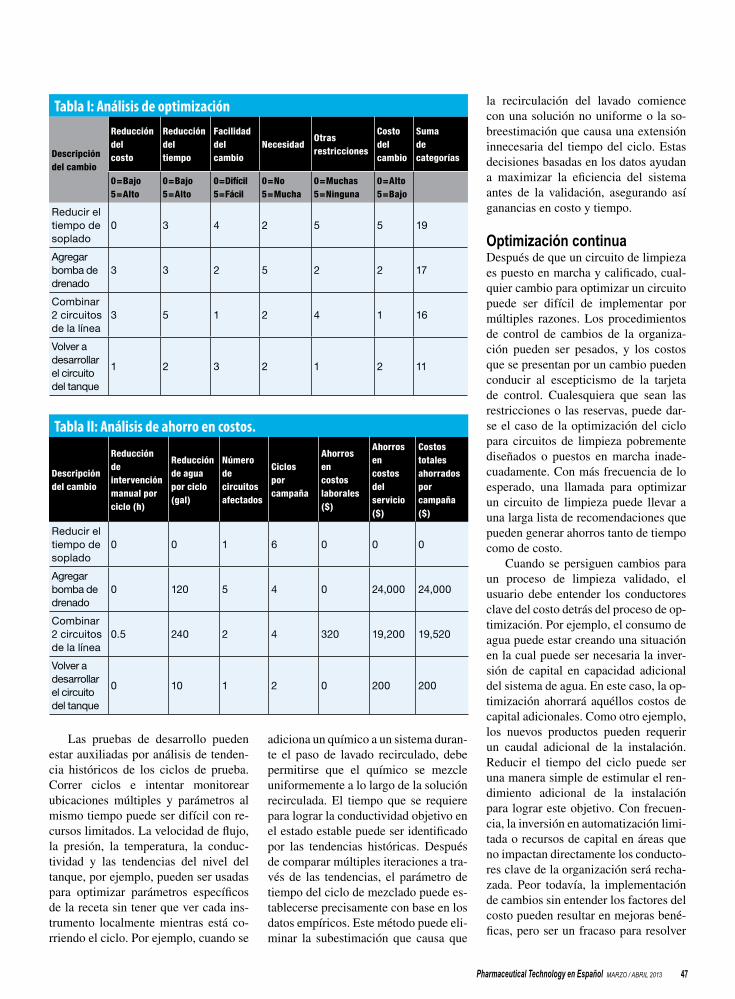
Pharmaceutical Technology en Español MARZO / ABRIL 2013 47
Las pruebas de desarrollo pueden estar auxiliadas por análisis de tenden-cia históricos de los ciclos de prueba. Correr ciclos e intentar monitorear ubicaciones múltiples y parámetros al mismo tiempo puede ser difícil con re-cursos limitados. La velocidad de flujo, la presión, la temperatura, la conduc-tividad y las tendencias del nivel del tanque, por ejemplo, pueden ser usadas para optimizar parámetros específicos de la receta sin tener que ver cada ins-trumento localmente mientras está co-rriendo el ciclo. Por ejemplo, cuando se
Tabla I: Análisis de optimización
Descripción del cambio
Reducción del costo
Reducción del tiempo
Facilidad del cambio
NecesidadOtras restricciones
Costo del cambio
Sumade categorías
0=Bajo5=Alto
0=Bajo5=Alto
0=Difícil5=Fácil
0=No5=Mucha
0=Muchas5=Ninguna
0=Alto5=Bajo
reducir el tiempo de soplado
0 3 4 2 5 5 19
agregar bomba de drenado
3 3 2 5 2 2 17
combinar 2 circuitos de la línea
3 5 1 2 4 1 16
volver a desarrollar el circuito del tanque
1 2 3 2 1 2 11
Tabla II: Análisis de ahorro en costos.
Descripción del cambio
Reducción de intervención manual por ciclo (h)
Reducción de agua por ciclo (gal)
Número de circuitos afectados
Ciclospor campaña
Ahorrosencostos laborales($)
Ahorros en costos del servicio ($)
Costos totales ahorrados por campaña ($)
reducir el tiempo de soplado
0 0 1 6 0 0 0
agregar bomba de drenado
0 120 5 4 0 24,000 24,000
combinar 2 circuitos de la línea
0.5 240 2 4 320 19,200 19,520
volver a desarrollar el circuito del tanque
0 10 1 2 0 200 200
adiciona un químico a un sistema duran-te el paso de lavado recirculado, debe permitirse que el químico se mezcle uniformemente a lo largo de la solución recirculada. El tiempo que se requiere para lograr la conductividad objetivo en el estado estable puede ser identificado por las tendencias históricas. Después de comparar múltiples iteraciones a tra-vés de las tendencias, el parámetro de tiempo del ciclo de mezclado puede es-tablecerse precisamente con base en los datos empíricos. Este método puede eli-minar la subestimación que causa que
la recirculación del lavado comience con una solución no uniforme o la so-breestimación que causa una extensión innecesaria del tiempo del ciclo. Estas decisiones basadas en los datos ayudan a maximizar la eficiencia del sistema antes de la validación, asegurando así ganancias en costo y tiempo.
Optimización continuaDespués de que un circuito de limpieza es puesto en marcha y calificado, cual-quier cambio para optimizar un circuito puede ser difícil de implementar por múltiples razones. Los procedimientos de control de cambios de la organiza-ción pueden ser pesados, y los costos que se presentan por un cambio pueden conducir al escepticismo de la tarjeta de control. Cualesquiera que sean las restricciones o las reservas, puede dar-se el caso de la optimización del ciclo para circuitos de limpieza pobremente diseñados o puestos en marcha inade-cuadamente. Con más frecuencia de lo esperado, una llamada para optimizar un circuito de limpieza puede llevar a una larga lista de recomendaciones que pueden generar ahorros tanto de tiempo como de costo.
Cuando se persiguen cambios para un proceso de limpieza validado, el usuario debe entender los conductores clave del costo detrás del proceso de op-timización. Por ejemplo, el consumo de agua puede estar creando una situación en la cual puede ser necesaria la inver-sión de capital en capacidad adicional del sistema de agua. En este caso, la op-timización ahorrará aquéllos costos de capital adicionales. Como otro ejemplo, los nuevos productos pueden requerir un caudal adicional de la instalación. Reducir el tiempo del ciclo puede ser una manera simple de estimular el ren-dimiento adicional de la instalación para lograr este objetivo. Con frecuen-cia, la inversión en automatización limi-tada o recursos de capital en áreas que no impactan directamente los conducto-res clave de la organización será recha-zada. Peor todavía, la implementación de cambios sin entender los factores del costo pueden resultar en mejoras bené-ficas, pero ser un fracaso para resolver
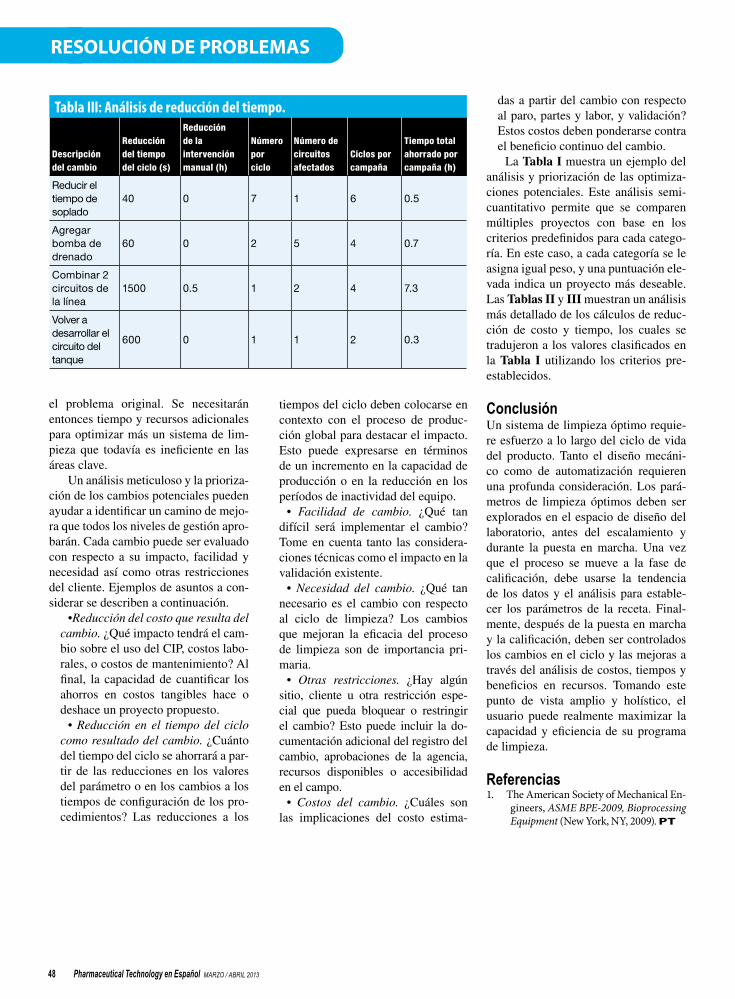
Pharmaceutical Technology en Español MARZO / ABRIL 201348
RESOLUCIÓN DE PROBLEMAS
el problema original. Se necesitarán entonces tiempo y recursos adicionales para optimizar más un sistema de lim-pieza que todavía es ineficiente en las áreas clave.
Un análisis meticuloso y la prioriza-ción de los cambios potenciales pueden ayudar a identificar un camino de mejo-ra que todos los niveles de gestión apro-barán. Cada cambio puede ser evaluado con respecto a su impacto, facilidad y necesidad así como otras restricciones del cliente. Ejemplos de asuntos a con-siderar se describen a continuación.
•Reducción del costo que resulta del cambio. ¿Qué impacto tendrá el cam-bio sobre el uso del CIP, costos labo-rales, o costos de mantenimiento? Al final, la capacidad de cuantificar los ahorros en costos tangibles hace o deshace un proyecto propuesto.
• Reducción en el tiempo del ciclo como resultado del cambio. ¿Cuánto del tiempo del ciclo se ahorrará a par-tir de las reducciones en los valores del parámetro o en los cambios a los tiempos de configuración de los pro-cedimientos? Las reducciones a los
Tabla III: Análisis de reducción del tiempo.
Descripción del cambio
Reducción del tiempo del ciclo (s)
Reducción de la intervención manual (h)
Número por ciclo
Número de circuitos afectados
Ciclos por campaña
Tiempo total ahorrado por campaña (h)
reducir el tiempo de soplado
40 0 7 1 6 0.5
agregar bomba de drenado
60 0 2 5 4 0.7
combinar 2 circuitos de la línea
1500 0.5 1 2 4 7.3
volver a desarrollar el circuito del tanque
600 0 1 1 2 0.3
tiempos del ciclo deben colocarse en contexto con el proceso de produc-ción global para destacar el impacto. Esto puede expresarse en términos de un incremento en la capacidad de producción o en la reducción en los períodos de inactividad del equipo.
• Facilidad de cambio. ¿Qué tan difícil será implementar el cambio? Tome en cuenta tanto las considera-ciones técnicas como el impacto en la validación existente.
• Necesidad del cambio. ¿Qué tan necesario es el cambio con respecto al ciclo de limpieza? Los cambios que mejoran la eficacia del proceso de limpieza son de importancia pri-maria.
• Otras restricciones. ¿Hay algún sitio, cliente u otra restricción espe-cial que pueda bloquear o restringir el cambio? Esto puede incluir la do-cumentación adicional del registro del cambio, aprobaciones de la agencia, recursos disponibles o accesibilidad en el campo.
• Costos del cambio. ¿Cuáles son las implicaciones del costo estima-
das a partir del cambio con respecto al paro, partes y labor, y validación? Estos costos deben ponderarse contra el beneficio continuo del cambio.
La Tabla I muestra un ejemplo del análisis y priorización de las optimiza-ciones potenciales. Este análisis semi-cuantitativo permite que se comparen múltiples proyectos con base en los criterios predefinidos para cada catego-ría. En este caso, a cada categoría se le asigna igual peso, y una puntuación ele-vada indica un proyecto más deseable. Las Tablas II y III muestran un análisis más detallado de los cálculos de reduc-ción de costo y tiempo, los cuales se tradujeron a los valores clasificados en la Tabla I utilizando los criterios pre-establecidos.
ConclusiónUn sistema de limpieza óptimo requie-re esfuerzo a lo largo del ciclo de vida del producto. Tanto el diseño mecáni-co como de automatización requieren una profunda consideración. Los pará-metros de limpieza óptimos deben ser explorados en el espacio de diseño del laboratorio, antes del escalamiento y durante la puesta en marcha. Una vez que el proceso se mueve a la fase de calificación, debe usarse la tendencia de los datos y el análisis para estable-cer los parámetros de la receta. Final-mente, después de la puesta en marcha y la calificación, deben ser controlados los cambios en el ciclo y las mejoras a través del análisis de costos, tiempos y beneficios en recursos. Tomando este punto de vista amplio y holístico, el usuario puede realmente maximizar la capacidad y eficiencia de su programa de limpieza.
Referencias1. The American Society of Mechanical En-
gineers, ASME BPE-2009, Bioprocessing Equipment (New York, NY, 2009). PT

Pharmaceutical Technology en Español MARZO / ABRIL 2013 49
Patricia Van Arnum es editor ejecutivo del pharmaceutical technology, 485 route one south, edif. F, primer piso, iselin, nJ 08830, tel. 732.346.3072, [email protected]@pharmtech/arnum
Estrategias en elescalamiento de APIsPatricia Van Arnum
síntEsis y manufactura dE apis
Los químicos de proceso emplean una variedad de esquemas para mejorar el rendimiento, la pureza y la estereoselectividad.
Los químicos de síntesis orgá-nica en las industrias farma-céuticas y de químicos finos buscan avances para ayudar
a mejorar el rendimiento, la pureza y la estereoselectividad de intermedios y APIs. Este objetivo toma el rumbo en una multitud de maneras desde los avances generales en química orgánica hasta beneficios específicos de la reac-ción. Los avances en síntesis asimétri-ca son de particular importancia dada la prevalencia de compuestos quirales como candidatos potenciales a fárma-cos y el consiguiente interés en produ-cir enantiómeros simples. Este objetivo
tiene que ser cumplido con la capacidad de escalamiento de cualquier síntesis estereoselectiva dada para manufac-tura comercial, y en esta área se han hecho varios desarrollos interesantes. La economía de la producción también es una consideración así como en el escalamiento de reacciones a niveles comerciales. Una ruta rentable para el fármaco antimalaria artemisinina, y una ruta biocatalítica para reacciones de nitración aromática son algunos de los desarrollos recientes.
Síntesis asimétricaImino-cianación enantioselectiva/reacción de Strecker
Los investigadores del Instituto de Ciencias Químicas e Ingeniería, un ins-tituto nacional de investigación bajo el auspicio de la Agencia para la Ciencia, Tecnología e Investigación de Singapur, reportó recientemente sobre un nuevo catalizador robusto que genera selec-tivamente precursores de aminoácidos
a partir del cianuro a temperatura am-biente a escala industrial. Específica-mente, ellos reportaron sobre un nuevo catalizador más dócil al escalamiento en la reacción asimétrica de Strecker, un catalizador en racimo de titanio qui-ral (SCTC) auto-soportado heterogéneo en la imino-cianación enantioselectiva/reacción de Strecker usada tanto en lote como en procesos continuos (1, 2). Una limitación en la reacción asimétrica de Strecker es la carencia de disponibilidad de catalizadores heterogéneos eficientes y reutilizables que trabajen a tempera-tura ambiente. Los investigadores saca-ron ventaja de la naturaleza hidrolizable del alcóxido de titanio para sintetizar un catalizador SCTC mediante la hidrólisis controlada de un complejo quiral pre-formado de titanio-alcóxido (2). Los investigadores reportaron que los cata-lizadores SCTC aislados eran estables y mostraron hasta 98% de enantioselec-tividad con conversión completa de la imina en un lapso de dos horas para una amplia variedad de iminas a temperatu-ra ambiente. Además, los catalizadores heterogéneos fueron reciclables más de 10 veces sin ninguna pérdida en activi-dad o selectividad (2).
Los investigadores reportaron que el catalizador fue usado en un reactor de lecho empacado para llevar a cabo la cianación bajo flujo continuo hasta con 97% de enantioselectividad bajo condi-ciones de flujo optimizado a temperatura ambiente en el caso de la benzhidrili- mina. También se realizó una reacción de Strecker de tres componentes bajo flujo continuo usando los correspondientes al-dehídos e iminas en lugar de las iminas pre-formadas bajo el cual se obtuvo una buena distribución del producto para la formación de amino nitrilos con valores de enantioselectividad de hasta 98% (2).
Centros estereogénicos todos de carbón cuaternario en sistemas ací-clicos. Un equipo de investigadores en-cabezados por Ilan Marek, profesor en el Instituto Technion-Israel de Tecnolo-gía en Haifa, describieron su trabajo en la formación de centros estereogénicos todos de carbón cuaternario en sistemas acíclicos de alquinos (3). La construc-ción de centros quirales todos de carbón cuaternario había estado limitada en metodologías previas basadas en aldol, de acuerdo a un comunicado de prensa

Pharmaceutical Technology en Español MARZO / ABRIL 201350
del Instituto Technion-Israel de Tecno-logía. Los investigadores usaron una operación de un solo recipiente, empe-zando desde los hidrocarburos clásicos, para formar productos de aldol conte-niendo el estereocentro todo de carbón cuaternario deseado a través de la for-mación concomitante de tres nuevos enlaces, de acuerdo al comunicado. Un reto en la formación de estereocentros todos de carbón cuaternario en sistemas acíclicos había sido la preparación de estereocentros todos de carbón cuater-nario en aductos de aldol (3). El prin-cipal problema que limita la formación de estos estereocentros es la ausencia de un método eficiente de preparación de enolatos tri-sustituidos estereodefinidos en sistemas acíclicos. Los investigado-res desarrollaron un esquema diferente que involucra la formación de dos nue-vos centros estereogénicos, incluyendo
síntEsis y manufactura dE apis
el todo de carbón cuaternario, a través de una reacción de carbometalación-oxidación combinada de un organocu-prato para dar un enolato tri-sustituido estereodefinido (3). Los investigadores utilizaron este método para generar una serie de productos de aldol y Mannich de inamidas. Para este trabajo en sínte-sis orgánica, Marek recibió el Premio de la Real Sociedad de Química Orga-nometálico en el 2011 y el Premio Jans-sen Pharmaceutica por Creatividad en Síntesis Orgánica en 2012.
Funcionalización de carbón asimé-trico-hidrógeno. Los investigadores de la Escuela Politécnica Federal de Lau-sanne, Escuela de Ciencias Básicas, Ins-tituto de Ciencias Químicas e Ingeniería, Laboratorio de Catálisis Asimétrica y Síntesis, en Lausanne, Suiza, reportaron recientemente acerca del desarrollo de ligandos ciclopentadienil quirales como
elementos de control en la funcionali-zación de carbón-hidrógeno asimétrico. Los investigadores buscaban abordar una limitación en la aplicación de com-plejos metálicos coordinada por un solo ligando ciclopentadienil como catali-zador en reacciones asimétricas (4). La dificultad, reportaron, ha sido diseñar los sustituyentes ciclopentadienil que des-víen la esfera de coordinación. Los in-vestigadores desarrollaron una clase de derivados ciclopentadienil C2
-simétricos que controlan el arreglo espacial de los reactantes coordinados transitoriamente alrededor del átomo metálico central, desarrollando complejos de rodio (III) con estos ligandos como catalizadores altamente enantioselectivos para las funcionalizaciones dirigidas del enlace carbón-hidrógeno de derivados del ácido hidroxámico (4).
Enfoque del desarrollo de fármacos: fármacos huérfanos
Las mayores presiones del mercado han obligado a las compañías farmacéuticas a reconsiderar sus estrategias de desarrollo de producto. La innovación de productos en medio de un mercado cada vez más competitivo, que incluye la incursión de fármacos genéricos en áreas terapéuticas, hace que el desarrollo de nuevos fármacos sea aún más desafiante. Los fármacos huérfanos, tradicionalmente perseguidos por las compañías biofarmacéuticas más pequeñas, se está volviendo una opción atractiva para las compañías biofarmacéuticas de tamaño medio a grande. Con frecuencia soportados por precios favorables desde la perspectiva de la compañía farmacéutica y llenando la demanda del paciente por fármacos para tratar necesidades médicas no cumplidas, los fármacos huérfanos ofrecen un buen potencial de mercado.
Un reciente análisis de Thomson Reuters del caso de economía e inversión para el desarrollo y comercialización de fármacos huérfanos muestra que aunque los fármacos huérfanos apuntan a poblaciones de pacientes mucho más pequeñas que los fármacos tradicionales dominantes, los precios favorables para dichos productos y otros factores tales como los incentivos del gobierno, los estudios clínicos más pequeños y más cortos, la extensión de la exclusividad y las altas tasas de aprobación regulatoria, hacen de los fármacos huérfanos principales una alternativa viable para los fármacos dominantes. Parte de esa oportunidad también proviene de la abundancia de áreas terapéuticas para el posible desarrollo de fármacos. El reporte de Thomson Reuters ofrece estimados de los Institutos Nacionales de Salud y de la Organización Europea para Enfermedades Raras que indican que existen aproximadamente 7000 enfermedades raras en el mundo y el número reconocido de dichas enfermedades se incrementa en una tasa de 250 enfermedades por año.
El mercado de los fármacos huérfanos fue valorado en aproximadamente $50,000 mdd globalmente a finales del 2011, según el análisis de Thomson Reuters. El gasto en fármacos huérfanos actualmente suma aproximadamente 6% del total de ventas de farmacéuticos, asumiendo un total del mercado farmacéutico de $880,000 mdd. La tasa de crecimiento anual compuesto (CAGR) del mercado de fármacos huérfanos entre el 2001 y el 2010 fue de 25.8% en comparación con un
CAGR de sólo 20.1% para un grupo control equiparable de fármacos no huérfanos, de acuerdo al análisis de Thomson Reuters. Dadas estas tasas comparativas de crecimiento y con las expectativas de que las tasas de aprobación de fármacos huérfanos continuarán siendo robustas, los indicadores positivos para los fármacos huérfanos continuarán probablemente a largo plazo. Este crecimiento es en parte debido al alto número de fármacos huérfanos basados en biológicos, los cuales son menos vulnerables a la incursión de fármacos genéricos en comparación con los fármacos de molécula pequeña, de acuerdo al análisis de Thomson Reuters.
El análisis de Thomson Reuters evaluó las ganancias actuales y futuras de los fármacos que generan ganancias de alto ingreso durante el ciclo de vida de un producto. El valor presente promedio, basado en estimados del 2010 de los fármacos huérfanos fue de $637 mdd, el cual estuvo a la par con el valor promedio presente de fármacos no huérfanos de $638 mdd. Viendo el valor de los fármacos durante el período de vida del producto, se encontró que el Lipitor (atorvastatina), el fármaco anti-colesterol de Pfizer, es el fármaco valorado más alto. Éste generó un estimado de $197,000 mdd durante su período de vida medido a través del descuento del ingreso potencial. El descuento del ingreso potencial es el valor pico descontinuado de la ganancia del fármaco durante su período de vida. Es un estimado basado en el valor de todos los ingresos históricos y de los ingresos futuros proyectados. El fármaco anticáncer Rituxan (rituximab), de Roche/Genentech, es el segundo fármaco clasificado más alto en términos del descuento del ingreso potencial y es el único fármaco entre los primeros diez fármacos que tiene ventas significativas que surgen de las indicaciones de fármaco huérfano, de acuerdo al análisis de Thomson Reuters. Aunque los fármacos huérfanos están asociados con nichos de áreas terapéuticas, su éxito en el mercado ha sido bastante robusto. De los 86 fármacos huérfanos incluidos en el análisis de Thomson Reuters, 25 fueron productos estrella, representando el 29% de los fármacos huérfanos. Los productos estrella son fármacos definidos con ventas de más de $1,000 mdd. Ese 29% fue comparable con el 29%, o con 83 de 291 fármacos no huérfanos que fueron considerados productos estrella.

Pharmaceutical Technology en Español MARZO / ABRIL 2013 51
Escalamiento de la artemisininaUna síntesis a gran escala, rentable, para el producto natural artemisinina, un tra-tamiento anti-malaria, ha sido esquiva, pero los investigadores de la Universidad de Indiana (UI) desarrollaron un nuevo esquema que se dirige hacia ese objetivo. La Organización Mundial de la Salud ha establecido un costo objetivo por gra-mo para la artemisinina de 25 centavos o menos, pero el costo actual es de al-rededor de $2.40 dólares por gramo, de acuerdo a un comunicado de prensa de la UI el 12 de septiembre de 2012. “En el 2005, la OMS afirmaba que la estructura de la artemisinina era demasiado com-pleja para una síntesis rentable,” dijo el profesor Silas Cook, profesor de quími-ca del Colegio Bloomington de Artes y Ciencias de la UI en el comunicado de prensa de la universidad.
Silas y otros investigadores de la UI buscaron un esquema rentable para la artemisinina que utiliza ciclohexenona como materia inicial clave y bases para el desarrollo de cascadas de reacción en la producción de artemisinina en una secuencia de cinco recipientes en una escala de gramos (5). “La clave para el éxito final de la artemisinina sinté-tica será la producción a gran escala del fármaco,” dijo Cook en el comu-nicado de prensa. “Como tal, tuvimos
que repensar en los materiales de inicio calificados como adecuados para esta síntesis e inventar una nueva química.” El siguiente reto será movernos desde la escala de gramos a la producción en escala de kilogramos.
Reacciones de nitraciónaromáticaLa biocatálisis puede jugar un im-portante papel en la química sintética proporcionando transformaciones quí-micas más eficientes y mejorando las condiciones del proceso. Tal es el caso de los químicos de la Universidad de Warwick en el Reino Unido, quienes en colaboración con investigadores de la Universidad de Cornell en Nueva York, reportaron recientemente sobre el des-cubrimiento de una enzima en la bac-teria Streptomyces scabies que cataliza una reacción de nitración aromática (6).
Los investigadores afirman que el TxtE, una enzima del citocromo P450, es el primer ejemplo de una enzima que evolucionó específicamente para catali-zar una reacción de nitración aromática, de acuerdo a un comunicado de prensa de la Universidad Warwick el 21 de sep-tiembre del 2012. Esto lo hace utilizan-do los gases de monóxido de nitrógeno, generados a partir del aminoácido argi-nina mediante la enzima óxido nítrico
sintasa txtD y dioxígeno. La construc-ción de la enzima TxtE puede permitir que se desarrolle como un biocataliza-dor de nitración para la producción de químicos industriales sin la necesidad de usar ácido nítrico.
Las enzimas del citocromo P450 están diseminadas en los organismos vivos. Generalmente, éstas son respon-sables de la hidroxilación y otras reac-ciones oxidativas, pero esta es la prime-ra vez que se ha reportado que catalizan una reacción de nitración, de acuerdo a los investigadores. La TxtE cataliza la introducción de un grupo nitro en una posición específica del anillo aromáti-co del aminoácido triptófano. Este es el primer paso clave en la biosíntesis del thaxtomin A.
Referencias1. “Catalyst: Putting Cyanide to Work”, A*Star
Research, online, www.research.a-star.edu.sg/research/6566, Oct. 10, 2012.
2. A.M. Seayad et al., Chemistry–A European Journal 18 (18), 5693–5700 (2012).
3. I. Marek et al, Nature 490 (7421), 522–526 (2012).
4. B.E. and N. Cramer, Science 338 (6106), 504–506 (2012).
5. C. Zhu and S.P. Cook. J. Am. Chem. Soc. 134 (33), 13577–13579 (2012).
6. G. Challis, Nature Chem. Biol. 8 (10), 814–816 (2012). PT
CONVERSACIÓN FARMACÉUTICA Y COMUNIDAD
LOS LECTORES PIENSAN QUE…
En comparación con el 2011, ¿Su empre-sa ha asistido a más o menos exposiciones industriales en 2012?

Pharmaceutical Technology en Español MARZO / ABRIL 201352
La indexación es el proceso de determinar el tamaño, la forma y la simetría de la celda unitaria cristalográfica para un componente cristalino responsable de una serie de picos en un patrón de difracción en polvos con rayos X (XRPD). Los autores discuten la valiosa información que puede obtenerse de la indexación y sus aplicaciones en la seleccióny análisis rutinario de formas sólidas.
Richard B. McClurg*, PhD, es becario de investigación, [email protected], y Jared P. Smith, PhD, es investigador, ambos en SSCI, una división de Aptuit LLC, 3065 Kent Ave., West Lafayette, IN, USA 47906.
*A quien debe dirigirse la correspondencia.
Sometido: Mayo 2, 2012. Aceptado: Septiembre 18, 2012.
La determinación de estructuras cristalinas utilizan-do difracción con rayos X de cristales individuales es una técnica bien desarrollada (1). Una estructura cristalina individual proporciona la caracterización
estructural completa de la forma a escala atómica. El princi-pal inconveniente para la difracción cristalina individual es la necesidad de hacer crecer un cristal suficientemente grande y sin defectos para el análisis, lo cual no es práctico para todas las formas cristalinas. Además, un solo cristal no es siempre representativo del material policristalino del cual se obtuvo. La difracción en polvo con rayos x (XRPD) es un conjunto de análisis que generalmente es representativo de un material en polvo, y utiliza muestras de polvo que con frecuencia son más fáciles de producir que los cristales individuales. Por estas razones, la XRPD se utiliza rutinariamente para la caracteri-zación de sólidos cristalinos.
El patrón de la XRPD de una forma cristalina en un punto dado del estado termodinámico sirve como huella digital para la forma bajo las condiciones dadas. La información codifica-da en el patrón del XRPD incluye si un material es una sola fase o una mezcla de fases; el tamaño, forma y simetría de la celda unitaria; la posición de las moléculas en la celda uni-taria; y la cepa cristalina, entre otra información. A pesar de la riqueza de la información disponible, la interpretación de rutina de los patrones de XRPD está con frecuencia limitada a una comparación visual cualitativa que, a lo más, subutiliza la información disponible y, en el peor de los casos, lleva a conclusiones incorrectas. La extracción de información de un patrón de XRPD más allá de la interpretación visual agrega un valor significativo y mejora en gran medida la compren-sión de las formas cristalinas. La indexación del XRPD es un método que puede ser usado para extraer información y ayudar a la interpretación de los patrones de XRPD.
La indexación XRPD es el proceso de determinar el tama-ño, la forma y la simetría de la celda unitaria cristalográfica para un componente cristalino responsable de una serie de pi-cos en un patrón de XRPD. La indexación obtiene su nombre de la asignación de las etiquetas del índice de Miller a cada
Indexación del patrón de difracción de polvos con rayos X para aplicaciones farmacéuticasRichard B. McClurg y Jared P. Smit
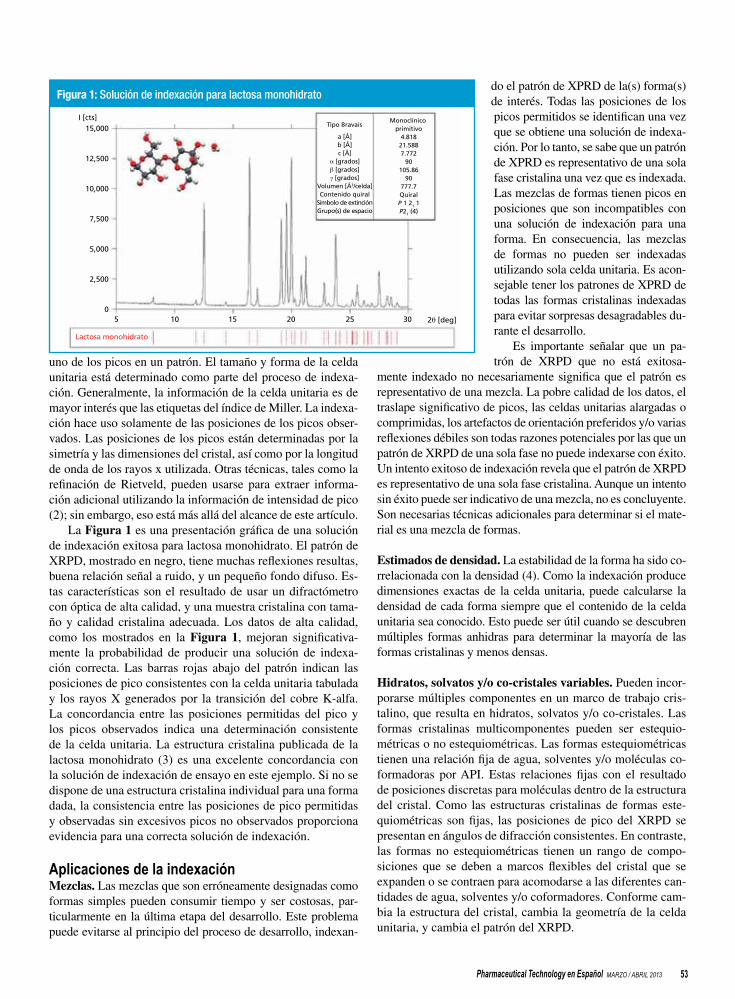
Pharmaceutical Technology en Español MARZO / ABRIL 2013 53
uno de los picos en un patrón. El tamaño y forma de la celda unitaria está determinado como parte del proceso de indexa-ción. Generalmente, la información de la celda unitaria es de mayor interés que las etiquetas del índice de Miller. La indexa-ción hace uso solamente de las posiciones de los picos obser-vados. Las posiciones de los picos están determinadas por la simetría y las dimensiones del cristal, así como por la longitud de onda de los rayos x utilizada. Otras técnicas, tales como la refinación de Rietveld, pueden usarse para extraer informa-ción adicional utilizando la información de intensidad de pico (2); sin embargo, eso está más allá del alcance de este artículo.
La Figura 1 es una presentación gráfica de una solución de indexación exitosa para lactosa monohidrato. El patrón de XRPD, mostrado en negro, tiene muchas reflexiones resultas, buena relación señal a ruido, y un pequeño fondo difuso. Es-tas características son el resultado de usar un difractómetro con óptica de alta calidad, y una muestra cristalina con tama-ño y calidad cristalina adecuada. Los datos de alta calidad, como los mostrados en la Figura 1, mejoran significativa-mente la probabilidad de producir una solución de indexa-ción correcta. Las barras rojas abajo del patrón indican las posiciones de pico consistentes con la celda unitaria tabulada y los rayos X generados por la transición del cobre K-alfa. La concordancia entre las posiciones permitidas del pico y los picos observados indica una determinación consistente de la celda unitaria. La estructura cristalina publicada de la lactosa monohidrato (3) es una excelente concordancia con la solución de indexación de ensayo en este ejemplo. Si no se dispone de una estructura cristalina individual para una forma dada, la consistencia entre las posiciones de pico permitidas y observadas sin excesivos picos no observados proporciona evidencia para una correcta solución de indexación.
Aplicaciones de la indexaciónMezclas. Las mezclas que son erróneamente designadas como formas simples pueden consumir tiempo y ser costosas, par-ticularmente en la última etapa del desarrollo. Este problema puede evitarse al principio del proceso de desarrollo, indexan-
Figura 1: Solución de indexación para lactosa monohidrato
15,000
12,500
10,000
7,500
5,000
2,500
0
I [cts]
Lactosa monohidrato
2θ [deg]5 10 20 3015 25
Tipo Bravais
a [Å]b [Å]c [Å]
α [grados]β [grados]γ [grados]
Volumen [Å3/celda]Contenido quiral
Símbolo de extinciónGrupo(s) de espacio
Monoclínico primitivo
4.81821.5887.772
90105.86
90777.7QuiralP 1 21 1P21 (4)
do el patrón de XPRD de la(s) forma(s) de interés. Todas las posiciones de los picos permitidos se identifican una vez que se obtiene una solución de indexa-ción. Por lo tanto, se sabe que un patrón de XPRD es representativo de una sola fase cristalina una vez que es indexada. Las mezclas de formas tienen picos en posiciones que son incompatibles con una solución de indexación para una forma. En consecuencia, las mezclas de formas no pueden ser indexadas utilizando sola celda unitaria. Es acon-sejable tener los patrones de XPRD de todas las formas cristalinas indexadas para evitar sorpresas desagradables du-rante el desarrollo.
Es importante señalar que un pa-trón de XRPD que no está exitosa-
mente indexado no necesariamente significa que el patrón es representativo de una mezcla. La pobre calidad de los datos, el traslape significativo de picos, las celdas unitarias alargadas o comprimidas, los artefactos de orientación preferidos y/o varias reflexiones débiles son todas razones potenciales por las que un patrón de XRPD de una sola fase no puede indexarse con éxito. Un intento exitoso de indexación revela que el patrón de XRPD es representativo de una sola fase cristalina. Aunque un intento sin éxito puede ser indicativo de una mezcla, no es concluyente. Son necesarias técnicas adicionales para determinar si el mate-rial es una mezcla de formas.
Estimados de densidad. La estabilidad de la forma ha sido co-rrelacionada con la densidad (4). Como la indexación produce dimensiones exactas de la celda unitaria, puede calcularse la densidad de cada forma siempre que el contenido de la celda unitaria sea conocido. Esto puede ser útil cuando se descubren múltiples formas anhidras para determinar la mayoría de las formas cristalinas y menos densas.
Hidratos, solvatos y/o co-cristales variables. Pueden incor-porarse múltiples componentes en un marco de trabajo cris-talino, que resulta en hidratos, solvatos y/o co-cristales. Las formas cristalinas multicomponentes pueden ser estequio-métricas o no estequiométricas. Las formas estequiométricas tienen una relación fija de agua, solventes y/o moléculas co-formadoras por API. Estas relaciones fijas con el resultado de posiciones discretas para moléculas dentro de la estructura del cristal. Como las estructuras cristalinas de formas este-quiométricas son fijas, las posiciones de pico del XRPD se presentan en ángulos de difracción consistentes. En contraste, las formas no estequiométricas tienen un rango de compo-siciones que se deben a marcos flexibles del cristal que se expanden o se contraen para acomodarse a las diferentes can-tidades de agua, solventes y/o coformadores. Conforme cam-bia la estructura del cristal, cambia la geometría de la celda unitaria, y cambia el patrón del XRPD.

Pharmaceutical Technology en Español MARZO / ABRIL 201354
Los cocristales estequiométricos de ácido p-coumárico y de nicotinamida son ejemplos para ilustrar esto. La Ta-bla I presenta la información de la cel-da unitaria para el ácido p-coumárico (5), nicotinamida (6), y dos cocristales que contienen diferentes estequiome-trías de los mismos dos componentes (7). Las soluciones de indexación para los componentes se tomaron de la Base de Datos Estructurales de Cambridge (CSD) (5, 6). Los patrones de XRPD para cada uno de los cristales fueron indexados con éxito. El volumen por unidad asimétrica para el cocristal 1:1 (345 Å3) concuerda con la suma del volumen por unidad asimétrica para los componentes (339 Å3). La estequiome-tría 1:1 fue confirmada utilizando la determinación de la estructura del cris-tal individual. El volumen por unidad asimétrica para el cocristal 2:1 (538 Å3) está en concordancia con la suma ponderada de sus componentes (533 Å3). La estequiometría 2:1 fue confir-mada utilizando solución 1H RMN. En este ejemplo, cada uno de los compo-nentes y los cocristales comparten el mismo grupo espacial (P2
1/c, no. 14).
Esta coincidencia no es necesaria para la estimación de la estequiometría.
Las posiciones del pico de XRPD de hidrato, solvatos y/o cocristales va-riables pueden aparecer ligeramente cambiadas debido a cambios menores de la celda unitaria, o pueden aparecer en ángulos de difracción significativa-mente diferentes si los cambios en la celda unitaria son grandes y/o anisotró-picos. Los patrones del XRPD para una forma de composición variable pueden parecer que son diferentes y, por lo tan-to, pueden ser erróneamente designados como formas distintas cuando en reali-dad representan puntos del estado en una forma con propiedades variables. Loa cambios anisotrópicos de la celda pueden causar picos traslapados que se resuelven conforme la composición de la celda cambia, y el “nuevo” pico puede ser incorrectamente vis-to como evidencia de una nueva forma. La indexación de una serie de patrones para una forma variable puede usarse para mostrar que un material mantiene su simetría y tiene cambios continuos en la geometría de la celda unitaria. Esta continuidad es evidencia para una forma variable. En contraste, los cambios en la simetría de la celda y/o los cambios discretos en los pará-metros de la celda unitaria son evidencia de múltiples formas.
Comparaciones con los datos de un solo cristalLas celdas unitarias generalmente se expanden con el aumento de temperatura. La indexación puede ser usada para conside-rar la expansión térmica cuando se compara un patrón XRPD calculado que fue generado a partir de una colección de datos de un cristal individual a baja temperatura para un patrón de XRPD experimental que fue colectado a temperatura ambiente. Los datos de un cristal individual son con frecuencia colectados a bajas temperaturas para mejorar la calidad de la estructura (1). La expansión térmica anisotrópica puede causar un patrón
Figure 2: del patrón de difracción en polvo con rayos X (XRPD) a temperatura ambiente (rojo) y el patrón de XRPD calculado desde la estructura del cristal individual a 120 K (azul) para el cocristal 1:1 de ácido p-coumárico y nicotinamida. La intensidad máxima de ambos patrones está escalada a 10,000 conteos para facilidad de comparación.
10,000
7,500
5,000
2,500
0
Inte
nsid
ad (c
onte
os)
2θ
5 10 20 3015 25
Tabla I: Determinación de la estequiometría del cristalInformación de la celda unitaria
Àcido p-coumárico
Nicotinamida Cocristal 1:1 Cocristal 2:1
Grupo espacial
P21/c P21/c P21/c P21/c
a (Å) 8.707 3.975 15.694 9.601
b (Å) 5.256 15.632 6.358 6.984
c (Å) 17.207 9.422 14.465 32.131
a 90 90 90 90
β 99.67 99.03 106.97 91.60
γ 90 90 90 90
volumen/celda (Å3) 776.27 578.20 1380.5 2153.7
volumen/unidad asimétrica (Å3)
194.07 144.55 345.1 538.4
referencia (5) (6) (7) (7)
invEstigación arbitrada
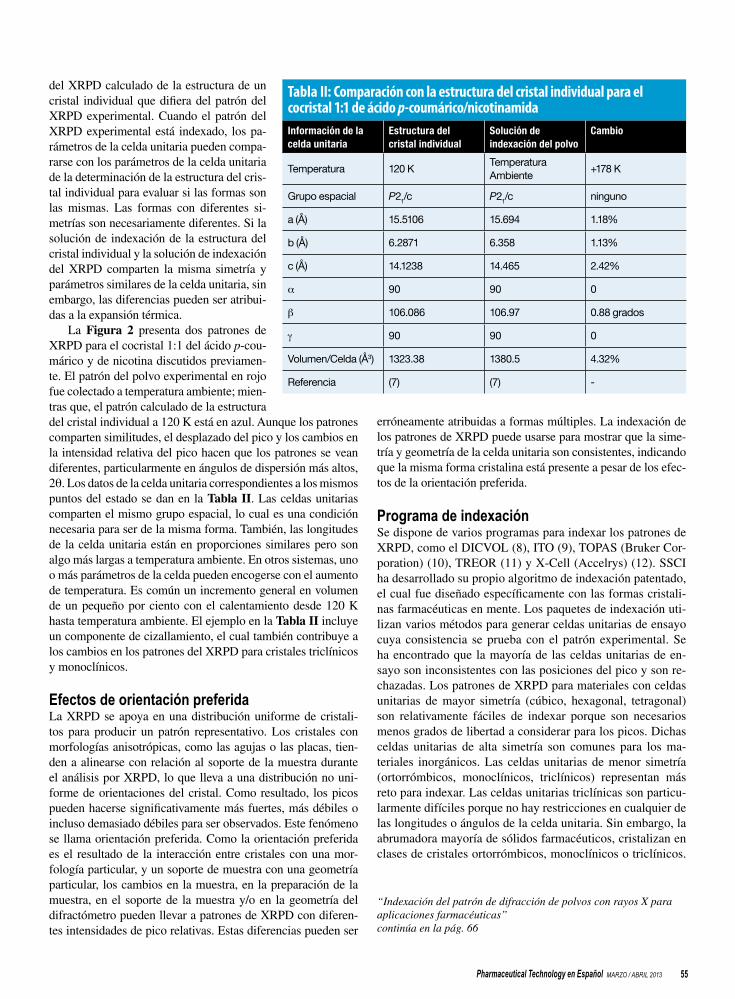
Pharmaceutical Technology en Español MARZO / ABRIL 2013 55
del XRPD calculado de la estructura de un cristal individual que difiera del patrón del XRPD experimental. Cuando el patrón del XRPD experimental está indexado, los pa-rámetros de la celda unitaria pueden compa-rarse con los parámetros de la celda unitaria de la determinación de la estructura del cris-tal individual para evaluar si las formas son las mismas. Las formas con diferentes si-metrías son necesariamente diferentes. Si la solución de indexación de la estructura del cristal individual y la solución de indexación del XRPD comparten la misma simetría y parámetros similares de la celda unitaria, sin embargo, las diferencias pueden ser atribui-das a la expansión térmica.
La Figura 2 presenta dos patrones de XRPD para el cocristal 1:1 del ácido p-cou-márico y de nicotina discutidos previamen-te. El patrón del polvo experimental en rojo fue colectado a temperatura ambiente; mien-tras que, el patrón calculado de la estructura del cristal individual a 120 K está en azul. Aunque los patrones comparten similitudes, el desplazado del pico y los cambios en la intensidad relativa del pico hacen que los patrones se vean diferentes, particularmente en ángulos de dispersión más altos, 2θ. Los datos de la celda unitaria correspondientes a los mismos puntos del estado se dan en la Tabla II. Las celdas unitarias comparten el mismo grupo espacial, lo cual es una condición necesaria para ser de la misma forma. También, las longitudes de la celda unitaria están en proporciones similares pero son algo más largas a temperatura ambiente. En otros sistemas, uno o más parámetros de la celda pueden encogerse con el aumento de temperatura. Es común un incremento general en volumen de un pequeño por ciento con el calentamiento desde 120 K hasta temperatura ambiente. El ejemplo en la Tabla II incluye un componente de cizallamiento, el cual también contribuye a los cambios en los patrones del XRPD para cristales triclínicos y monoclínicos.
Efectos de orientación preferidaLa XRPD se apoya en una distribución uniforme de cristali-tos para producir un patrón representativo. Los cristales con morfologías anisotrópicas, como las agujas o las placas, tien-den a alinearse con relación al soporte de la muestra durante el análisis por XRPD, lo que lleva a una distribución no uni-forme de orientaciones del cristal. Como resultado, los picos pueden hacerse significativamente más fuertes, más débiles o incluso demasiado débiles para ser observados. Este fenómeno se llama orientación preferida. Como la orientación preferida es el resultado de la interacción entre cristales con una mor-fología particular, y un soporte de muestra con una geometría particular, los cambios en la muestra, en la preparación de la muestra, en el soporte de la muestra y/o en la geometría del difractómetro pueden llevar a patrones de XRPD con diferen-tes intensidades de pico relativas. Estas diferencias pueden ser
Tabla II: Comparación con la estructura del cristal individual para el cocristal 1:1 de ácido p-coumárico/nicotinamidaInformación de la celda unitaria
Estructura del cristal individual
Solución de indexación del polvo
Cambio
temperatura 120 ktemperaturaambiente
+178 k
Grupo espacial P21/c P21/c ninguno
a (Å) 15.5106 15.694 1.18%
b (Å) 6.2871 6.358 1.13%
c (Å) 14.1238 14.465 2.42%
a 90 90 0
β 106.086 106.97 0.88 grados
γ 90 90 0
volumen/celda (Å3) 1323.38 1380.5 4.32%
referencia (7) (7) -
erróneamente atribuidas a formas múltiples. La indexación de los patrones de XRPD puede usarse para mostrar que la sime-tría y geometría de la celda unitaria son consistentes, indicando que la misma forma cristalina está presente a pesar de los efec-tos de la orientación preferida.
Programa de indexaciónSe dispone de varios programas para indexar los patrones de XRPD, como el DICVOL (8), ITO (9), TOPAS (Bruker Cor-poration) (10), TREOR (11) y X-Cell (Accelrys) (12). SSCI ha desarrollado su propio algoritmo de indexación patentado, el cual fue diseñado específicamente con las formas cristali-nas farmacéuticas en mente. Los paquetes de indexación uti-lizan varios métodos para generar celdas unitarias de ensayo cuya consistencia se prueba con el patrón experimental. Se ha encontrado que la mayoría de las celdas unitarias de en-sayo son inconsistentes con las posiciones del pico y son re-chazadas. Los patrones de XRPD para materiales con celdas unitarias de mayor simetría (cúbico, hexagonal, tetragonal) son relativamente fáciles de indexar porque son necesarios menos grados de libertad a considerar para los picos. Dichas celdas unitarias de alta simetría son comunes para los ma-teriales inorgánicos. Las celdas unitarias de menor simetría (ortorrómbicos, monoclínicos, triclínicos) representan más reto para indexar. Las celdas unitarias triclínicas son particu-larmente difíciles porque no hay restricciones en cualquier de las longitudes o ángulos de la celda unitaria. Sin embargo, la abrumadora mayoría de sólidos farmacéuticos, cristalizan en clases de cristales ortorrómbicos, monoclínicos o triclínicos.
“Indexación del patrón de difracción de polvos con rayos X para aplicaciones farmacéuticas”continúa en la pág. 66

Pharmaceutical Technology en Español MARZO / ABRIL 201356
iMa
Ge
n:
sto
ck
By
te
/Ge
tt
y iM
aG
es
BIOFORO
La transformación para la organización centrada en el proceso
Simon Chalk
Los retos científicos y de inge-niería para la producción de productos biofarmacéuticos a escala industrial, histórica-
mente, han requerido que la experiencia profunda en la materia sea desplegada eficientemente y con responsabilidad directa para el resultado. Este modus operandi ha permanecido intacto por-que la mayoría de las instalaciones eran para un solo producto (es decir, alto volumen, baja variedad) y sufrían pequeños cambios. Sin embargo, para competir en el mercado actual, las ins-talaciones farmacéuticas necesitan ser cada vez más multiproducto, flexibles, de cambios rápidos, y más centradas en el cliente (es decir., bajo volumen, alta variedad). El modelo tradicional de co-mando y control con frecuencia no es lo suficientemente responsivo o ágil para cumplir este reto.
Las estructuras de organización centrada en el proceso (PCO) han esta-do de moda durante los pasados años. Las estructuras PCO realinean los blo-ques de construcción de la organización con los procesos de valor agregado en el
¿Es la organización centrada en el proceso en manufactura biofarmacéutica un trampolín o un obstáculo?
negocio. En lugar de que las jerarquías funcionales sean la estructura dominan-te, la gente está organizada en equipos multidisciplinarios, cuyos objetivos están enfocados en la administración de las actividades de extremo a extre-mo que entregan valor a su cliente. El tamaño del equipo puede extenderse a muchas decenas de miembros.
Nada llama más la atención que una revisión en la organización que altera potencialmente el status quo.
Este concepto se ha vuelto la mejor práctica en muchas industrias donde los flujos del material y la información esbeltos, rápidos son cruciales para sobrevivir. La remoción de límites de funciones cruzadas y un elemento de auto-gestión y empoderamiento son fundamentales. Cuando trabaja, el flujo de valor se puede ejecutar con menos capas de gestión; se incrementan las eficiencias de la plantilla; y la toma de decisiones puede delegarse más efecti-vamente. Lo que es más importante, las vías de comunicación pueden facilitarse y los hoyos negros encontrados en las interfaces de la organización son elimi-nados con frecuencia.
Ejemplos de organizacióncentrada en el procesoVarias compañías farmacéuticas han hecho con éxito la transición de una es-tructura de organización tradicional a un nuevo modelo progresivo. El resultado, en algunos casos, ha sido la diferencia entre la sobrevivencia y el eventual cierre de la planta. En otros casos, el cambio ha sido un fracaso y finalmente ha sido ne-cesario un cambio en la dirección.
En un ejemplo innovador, un equipo de proceso interno de logística de mate-riales se formó de funciones separadas de programación de materiales, ope-raciones de almacenamiento, asegura-miento de calidad y control de calidad. Los equipos fueron colocados en una oficina adyacente al flujo de materiales interno y se les dio autoridad para traba-jar juntos para cumplir objetivos comu-nes. Un período de mapeo del flujo de valores, análisis y mejora llevó al redi-seño de la manera de trabajar, a ajustes en la asignación de responsabilidades, y capacitación cruzada para aumentar el rango de habilidades en el equipo. Previo al cambio, de la recepción a la liberación le podía tomar a cualquiera de 7 a más de 40 días. Los inventarios del material de seguridad reflejaban la longitud y variabilidad de estos tiem-pos de ciclo. Después del cambio, los tiempos de ciclo se lograron consisten-temente en el rango de uno a tres días. El equipo tomó con entusiasmo el reto
Simon Chalk es director del Grupo BioPhorum Operations, [email protected]

Pharmaceutical Technology en Español MARZO / ABRIL 2013 57
de empoderamiento y una cultura de “auto-dirección” surgida del esquema. Se dio soporte para asegurar que estu-vieran en el sitio las habilidades inter-personales y de trabajo en equipo para los individuos. El proyecto se convirtió en el punto de inicio para un rediseño más demandante en las áreas de manu-factura en donde se tomó un esquema de abajo hacia arriba para transformar la organización operacional más am-plia. En este caso, la estructura siguió a la estrategia de una manera que soporta-ba el cambio cultural irreversible.
Otro caso de estudio igualmente informativo mostró como la estructura de la organización en una planta era re-diseñada de manera radical con menos énfasis en la mejora del desempeño, maneras de trabajar, agrandamiento de las habilidades y más énfasis en geren-tes que se convirtieron en propietarios del flujo del proceso dentro de un arre-glo de equipo auto-dirigido. Las líneas en el organigrama fueron el centro de atención. A su debido tiempo, se en-contró que el cambio era demasiado radical con un enfoque insuficiente en la mejora del desempeño del negocio. La estructura no siguió a la estrategia, y la organización se cambió de regreso a una estructura lineal clásica. Después de un período de reflexión, las lecciones aprendidas llevaron a la adopción de un esquema más efectivo.
Un escalón común para el PCO es usar más la estructura de tipo matriz donde los equipos por disciplina están vinculados a los equipos de proceso de la producción mediante puntos de con-tacto simples. Estas líneas punteadas construyen el compromiso y la propie-dad para los objetivos del equipo de
proceso sin debilitar a los miembros del equipo funcional.
El papel de la alta direcciónCuando se trata de hacer una diferencia, la alta dirección realmente tiene pocas palancas que pueda jalar. Si tienen que hacer un cambio rápidamente, las op-ciones se reducen más. El único cam-bio que siempre permanece abierto es el cambio en la organización. Muchos altos directivos escogen jalar esta pa-lanca como primera opción, particular-mente si están bajo presión para mejo-rar el desempeño, reducir los costos, o simplemente para que se vea que hacen algo. Nada llama más la atención que una revisión de la organización que potencialmente altera el status quo. El problema es que el cambio en la orga-nización puede ser difícil y no siempre lleva al resultado esperado.
¿Así que los nuevos modelos orga-nizacionales y teorías de las que se ha-bla deben tomarse en cuenta? ¿Qué tan relevantes son éstas para un ambiente estrechamente regulado como la ma-nufactura farmacéutica? Y, si se adopta ¿Cómo deben ser implementadas?
El dilema es que desde una perspec-tiva cultural, la industria farmacéutica está enraizada en la mentalidad de “co-mando y control” en donde la fuerte su-pervisión, las políticas claras, y los pro-cedimientos meticulosamente definidos dirigen la conducta. El requerimiento es que el empleado aprenda cómo se realiza una tarea y ejecutar consisten-temente esa tarea una y otra vez (y no necesariamente pregunta por qué). Los gerentes y supervisores están ahí para definir la tarea, para asegurar que se hace correctamente cada vez por gente
adecuadamente entrenada, y para resol-ver problemas cuando se presentan. El cumplimiento es clave. Los expertos tales como los ingenieros, los profesio-nales de calidad y el personal de respal-do son también (altamente) capacitados para enfocarse en sus propias áreas de especialidad.
No hay duda de que los cambios donde a los operadores de producción se les da más responsabilidad para sim-ples rutinas de mantenimiento, control de calidad y programación de lotes, por ejemplo, confrontan paradigmas exis-tentes. Puede verse que esta realidad es controversial. Sin embargo, el profesio-nalismo y la capacidad de la gente que supervisa los sitios de manufactura de la industria son más que adecuadas para asegurar que estos cambios evoluciones de manera que se mantenga la calidad del producto y la seguridad del paciente.
Para los altos directivos que buscan tener un impacto, con frecuencia les hace sentido dejar la palanca del cam-bio de la organización en la última posi-ción en la lista. Se vuelve entonces una herramienta efectiva con la cual hacer mejoras al negocio y cambios culturales permanentes.
La comprensión de la diferencia en-tre el éxito y el fracaso yace no en la teoría de la organización, aun cuando es importante tomar en cuenta los factores de la gestión del cambio. Yace en una filosofía de “regreso a lo básico” donde los caminos materiales se simplifican y acortan, el conocimiento fluye a donde es necesario, a la gente se le da una va-riedad más amplia de capacitación y las actividades adicionales sin valor, mal-gastadas, son eliminadas. PT
SU PRESENCIAEN LA REVISTA MÁS LEIDA POR LA INDUSTRIA FARMACÉUTICA
CONTRATE SU PAUTA PUBLICITARIA
AHORA
Mayores Informes y Contrataciones:Tel: 52 (55) 5659-8880, 5536-2100, 5543-1486 E-mail: [email protected]

Pharmaceutical Technology en Español MARZO / ABRIL 201358
PREGÚNTELE AL EXPERTO
La EMA y la FDA en la validación de procesos
P.Desde que la FDA publicó su Guía para la Validación de Procesos de la Industria: Principios Generales y Prácti-
cas en el 2011, La Agencia Europea de Medicamentos (EMA) ha publicado su proyecto Guía sobre la Validación de Proce-sos. ¿Están estas dos guías alineadas?
R.Aunque hay diferencias en la terminología, los princi-pios que subyacen en ésta son los mismos. El principio
general es el esquema de ciclo de vida para la validación. Una vez que se ha establecido el conocimiento suficiente acerca del producto y del proceso (es decir, la fase de calificación del proceso), pueden fabricarse los lotes de validación. Lo que es nuevo aquí es que todos los siguientes lotes (comer-ciales) sean considerados lotes de verificación. Por lo tanto, la validación es un proceso continuo hasta la suspensión del producto.
P.¿De manera que todavía están bien tres lotes para demos-trar la validación inicial?
R.En Europa esto es poco más o menos el estándar acep-tado. A la FDA, por el otro lado, realmente le gustaría
ver una justificación para el número de lotes. Esto no quiere decir que tres lotes no sean aceptables; pero debe proporcio-narse el razonamiento científico o al menos los datos históri-cos (de campañas de validación previas).
P.¿Puede decirnos más acerca del conocimiento y com-prensión del proceso?
R.Durante las pasadas una o dos décadas, se alentaba a las compañías a someter solamente la apenas mínima
información en sus solicitudes. Como resultado, la compren-sión del proceso y del producto que podía haber ayudado con las mejoras de calidad en el proceso y el producto no era ex-plorada. Actualmente, las agencias han cambiado su postura al respecto y no sólo alientan, sino que realmente esperan que la industria establezca, mantenga y mejore continuamente su base de conocimientos. Además, la expectativa es también someter mucha de esta comprensión en los sometimientos.
Al obtener una comprensión del proceso más amplia y más profunda, la industria puede lograr mayor flexibilidad con sus procesos de manufactura, ya que los rangos de trabajo pueden expandirse en consecuencia. Dicha flexibilidad mejo-rada resulta típicamente en una reducción significativa en las
Sigfredo Schmitt, director consultor en PAREXEL, discute la guía de la Agencia Europea de Medicamentos sobre la validación de procesos y cómo se comparan con la guía de validación de procesos de la FDA.
desviaciones del proceso registrado y/o en menores cambios en el registro (variaciones en la UE).
P. ¿No es la verificación del proceso continuo (CPV) algo que ya está haciendo la industria?
R. Las compañías tienen que preparar los reportes anuales de calidad del producto (APQ), los cuales proporcionan
detalles acerca del número de lotes producidos en ese perío-do, eventos anormales, tales como desviaciones y recupera-ciones de producto del mercado, y el estatus de validación de los procesos y métodos analíticos. La información para estos reportes se colecta retrospectivamente al final del año; para algunos lotes casi un año después de su fecha de manufactura. Esto es lo que la industria ha estado haciendo, pero eso no es lo que los reguladores intentan con respecto al CPV. La veri-ficación continua requiere datos en tiempo real o datos “cer-canos al evento” y análisis de la información. En principio, la verificación del estado validado debe ser realizada antes de la manufactura de cualquier nuevo lote. Dicho esquema requie-re, en primer lugar, datos/información fácilmente disponible, y en segundo lugar, métodos estadísticos para su análisis. La disponibilidad de los datos sólo puede ser razonablemente lo-grada cuando éstos se proporcionan en formato electrónico. Imagine que tiene que trasladar datos manualmente del regis-tro de lote a una hoja de cálculo o similar; eso no sería factible para la mayoría de los entornos de la manufactura.
P. Mencionó el análisis estadístico, ¿podría detallar?
R. Aunque los datos han sido siempre analizados para de-terminar si cumplen sus especificaciones, con frecuen-
cia dichos datos no se investigaron en busca de tendencias. Obviamente, si no tengo datos en tiempo real u omito realizar análisis de tendencia cercano al evento, hay poco valor en ha-cerlo, y se habrán perdido las oportunidades para acciones correctivas. Las expectativas de las agencias regulatorias son que la industria ahora realice tendencias de datos utilizando reglas estadísticas ampliamente usadas y métodos, los cuales permitirán la detección y el análisis de eventos fuera de ten-dencia. El propósito es evitar que ocurran eventos fuera de especificaciones. Dichos métodos estadísticos llevan mucho tiempo; sin embargo, su uso en este contexto no está todavía diseminado. PT

Pharmaceutical Technology en Español MARZO / ABRIL 2013 59
PERSPECTIVA DE LA SUBCONTRATACIÓN
Jon
at
ha
n e
va
ns
/ph
oto
Dis
c/G
et
ty
iMa
Ge
s
Biomanufactura en el extranjeroEric Langer
Los biofarmacéuticos constitu-yen una industria global, y los biofabricantes e innovadores de fármacos han reconocido
desde hace mucho la necesidad de una estrategia global de producto. Confor-me crecen las economías emergentes, ellos están empezando a demandar bio-lógicos y vacunas producidos domésti-camente para cumplir las necesidades de salud pública de sus crecientes po-blaciones, lo cual a su vez, ha abierto oportunidades estratégicas en las prin-cipales compañías biofarmacéuticas. Los centros de biomanufactura fuera de los Estados Unidos y de Europa Oc-cidental están proporcionando nuevas opciones para las operaciones globales. En el pasado, los ahorros potenciales en costos manejaron muchas decisiones de subcontratación, pero hoy, las decisio-nes son más complejas conforme los in-novadores de fármacos integran las de-mandas de un mercado global creciente.
BioPlan Associates rastreó estas tendencias en su 9º. Reporte Anual y Encuesta de Fabricantes Biofarma-céuticos (1). Conforme las compañías de biológicos contratan proyectos más sofisticados, la subcontratación global se ha movido desde una decisión táctica a una estratégica para muchas compa-ñías. El cálculo de la decisión que rodea la elección de un CMO global refleja muchos de estos cambios. Por ejem-plo, las percepciones del extranjero están cambiando, y los innovadores de fármacos se han vuelto mucho menos
¿La subcontratación para biomanufactura internacional se volverá dominante en esta década?
Eric Langer es presidente de Bioplan associates, tel. 301.921.5979, [email protected], y un contribuyente periódico de Perspectivas de la Subcontratación
preocupados acerca de si su CMO está basado localmente. De los 19 atributos del CMO identificados en el estudio de BioPlan, “ser local” fue el último en términos de importancia con sólo el 6.3% de los 302 respondedores que identificaron este atributo como “muy importante” (1). En 2005, 10.4% de los que respondieron citaron una ubica-ción del CMO como “muy importante”, pero sólo el 6.3% lo hizo en 2012. Este cambio refleja mayor aceptación de las actividades de subcontratación en el ex-tranjero debido a la globalización de la industria, más proyectos de producción en el extranjero con éxito, y una expe-riencia mejorada de los patrocinadores en la gestión de la subcontratación en el extranjero.
Futuro de la deslocalizaciónLa encuesta de BioPlan le pregun-
tó a los encuestados que reflexionaran sobre su subcontratación internacional a futuro de sus instalaciones (es decir, deslocalización) durante los próximos
cinco años. Dependiendo de la opera-ción de subcontratación, los encuesta-dos indicaron que entre 35 – 42% de sus operaciones de manufactura (desarrollo de proceso, operaciones de biomanu-factura y otras operaciones) podrían ser deslocalizadas en alguna medida (ver Figura 1), en cinco años, y el 46% de las operaciones de manufactura para es-tudios clínicos podría ser deslocalizada en algún grado.
Un gran porcentaje de biofabricantes en 2012 –tantos como 36%- esperan des-localizar algo de la manufactura de sus plantas durante los próximos cinco años (ver Figura 1) aunque el porcentaje que espera deslocalizar más de la mitad de sus operaciones calificó sólo en un solo dígito. Este hallazgo sugiere que puede ser demasiado pronto para afirmar que la deslocalización se convertirá en una actividad dominante aunque hay razo-nes para esperar que la deslocalización se vuelva más aceptada, especialmente conforme las operaciones regionales fuera de EEUU y Europa empiecen a
Figura 1: Porcentaje de biofabricantes en las encuestas de 2012 y 2011 de BioPlan Associates que respondieron que deslocalizarían al menos alguna biomanufactura durante los próximos cinco años (1).
35.7%% de desarrollode proceso parabiomanufactura
% deoperaciones de
biomanufactura
% de otrasoperaciones
22.40%
40.5%
37.60%
41.79%
Porcentaje de encuestados
29.60%
2012
2011
FiG
ur
a 1
es
co
rt
es
ia D
el
au
tor

Pharmaceutical Technology en Español MARZO / ABRIL 201360
cumplir los estándares regulatorios y conforme los dispositivos para bioproce-so de un solo uso entren a la manufactu-ra a escala comercial. Estas instalaciones integradas, completamente desechables, permitirán la manufactura en regiones más desarrolladas, ya que muchos de los procesos de manejo de calidad requeri-dos por las instalaciones tradicionales pueden estar “integrados” en los dispo-sitivos desechables.
Al comparar los resultados de la encuesta en 2012 y 2011, la encuesta mostró un incremento en todos los ám-bitos en el porcentaje de los encuesta-dos, quienes dijeron que esperan la des-localización de al menos alguna de sus operaciones durante los próximos cinco años (ver Figura 1). Específicamente, la encuesta mostró que:
• 35.7% dijo que deslocalizaría al menos algún desarrollo de proce-so para biomanufactura, más que el 22.4% en 2011.
• 40.5% indicó que deslocalizaría al menos algunas operaciones de bio-manufactura, más que el 37.6% en 2011.
• 41.7% espera subcontratar inter-nacionalmente otras operaciones, más que el 29.6% en 2011.
Viendo a futuroDe acuerdo al Índice de las 1000 Prin-cipales Instalaciones Biofarmacéuticas Globales de BioPlan, el 37.5% de la ca-pacidad de manufactura biofarmacéuti-ca global existe fuera de Norteamérica y Europa (2). Participando respectiva-mente con 8.5% y 8.1% de la capaci-dad de biomanufactura global, China y
la India están surgiendo en el escenario global, seguidos por Japón y otros paí-ses asiáticos, los cuales tienen 9.6% de la capacidad de biomanufactura global y Latinoamérica en 6.5%.
No es de sorprender que países tales como China e India, los cuales están de-sarrollando rápidamente clusters de bio-manufactura, aparezcan en posiciones al-tas en la lista de probables destinos para la subcontratación internacional. Entre todos los encuestados de la industria para el estudio anual de BioPlan, China cla-sificó más alto como destino potencial para subcontratación internacional con 26.2% de los encuestados (por arriba del 17% en 2011) indicando que había una “probabilidad” o “fuerte probabilidad” de que subcontrataran la producción a las instalaciones en los próximos cinco años. El siguiente en la lista fue India, citado por el 18.4% de los encuestados, por arriba del 13.2% en 2011.
En alguna medida, las tendencias de la subcontratación geográfica tien-den a reflejar la disponibilidad (perci-bida o real) de CMOs acreditados en un país dado. Las nuevas instalaciones que vienen en línea y el limitado nú-mero de CMOs conocidos pueden ser razones más importantes que los facto-res culturales o políticos mientras que los problemas de patentes y propiedad intelectual también juegan claramente un papel. No obstante, China es cla-ramente el destino potencial principal para la subcontratación internacional futura (incluso cuando se segregó a los encuestados de EEUU y de Europa), e India está creciendo a favor, particular-mente entre los encuestados de EEUU.
Es valioso tener en mente que Chi-na fue un iniciador tardío en el área de la manufactura por contrato. Los pocos CMOs internacionales con base en Chi-na están trabajando fuerte para estable-cer una presencia global, y los aspectos legales y regulatorios continúan difi-cultando la manufactura por contrato a escala comercial. No obstante, continúa habiendo un exceso de capacidad de producción farmacéutica en China, de manera que la subcontratación puede permitir mejoras conforme la capaci-dad libre de muchas instalaciones de producción es abordada. Sin embargo, puede tomar años para que los CMOs chinos cumplan los estándares comple-tos de las cGMPs y que se les permita operar a escala comercial. Los datos de la encuesta de BioPlan muestran que para el 2017, entre un tercio y casi la mitad de la industria estarán subcon-tratando alguna forma de operación in-ternacionalmente, con China como un componente clave de los planes futuros. Asumiendo que la aceptación de la des-localización continúe incrementándose y que los destinos emergentes clave, como China, alcancen los estándares regulatorios necesarios, es razonable suponer que para finales de esta década, la deslocalización se habrá convertido en una actividad dominante.
Referencias1. BioPlan Associates, 9th Annual Report and
Survey of Biopharmaceutical Manufactu-rers (Rockville, MD, 2012).
2. BioPlan Associates, Top 1000 Global Biopharmaceutical Facilities Index (Rock-ville, MD). PT
PERSPECTIVA DE LA SUBCONTRATACIÓN
Informes y Contrataciones:Tel: 52 (55) 5659-8880, 5536-2100,
5543-1486 E-mail: info@pharmatechespañol.com.mx

Pharmaceutical Technology en Español MARZO / ABRIL 2013 61
RE
GU
LA
CIÓ
N Y
CU
MP
LIM
IEN
TO
resuMen REGULATORIO
Controversia sobre el tratado de fármacos falsificadosLos representantes de 65 naciones que pertenecen a la Orga-nización Mundial de la Salud (OMS) se reunieron en Argen-tina en noviembre del 2012 para discutir un tratado interna-cional para combatir el comercio de fármacos falsificados –o “productos médicos Subestándar/Espúreos/Falsamente eti-quetados/Falsificados/Piratas” (SSFFCs) en el lenguaje de la OMS. Esta primera reunión del “mecanismo” de los SSFFC establecido por la OMS en mayo del 2011 produjo un plan de trabajo y un acuerdo para obtener información más precisa sobre la “naturaleza y magnitud” de la falsificación. El avance puede ser lento porque algunas naciones emergentes consi-deran las iniciativas anti-falsificación de fármacos como una táctica de las grandes empresas farmacéuticas para eliminar la competencia de genéricos. Los representantes de la India bloquearon la participación en la reunión de la OMS de los académicos que autorizaron un artículo en el British Medical Journal sobre la importancia de un tratado global para dete-ner el comercio de medicamentos falsos. Dicho tratado, ideal-mente, llevaría a estándares de autenticación comunes para identificar y desalentar la falsificación de fármacos, además de la cooperación para permitir que los reguladores desafíen a los falsificadores a través de las fronteras.
La EMA busca acciones para los desabastosUn “artículo de reflexión” de la Agencia Europea de Medica-mentos (EMA) describe un rango de iniciativas para los regu-ladores y fabricantes para tratar los desabastos de fármacos que surgen de las dificultades en la manufactura. La EMA quiere una notificación temprana de los problemas potencia-les de la industria, de manera que pueda coordinar mejor la respuesta a los desabastos por parte de los estados miembros. La EMA también propone la “planeación de continuidad del negocio” por los fabricantes para asegurar el acceso del paciente a los medicamentos esenciales, más el análisis de riesgo de las debilidades en las plantas de producción, que probablemente crean las situaciones de desabasto. El reporte está disponible en el sitio del EMA.
Colaboración con las células madreLas compañías farmacéuticas líder y los académicos están creando una colección mayor de 1500 líneas celulares madre pluripotentes inducidas (IPS) para usar en análisis y desarro-
llo de fármacos. Roche y otras nueve empresas están respal-dando el proyecto StemBANCC, el cual será manejado por la Universidad de Oxford. La iniciativa de Medicamentos Inno-vadores de Europa (IMI) respalda esta iniciativa de $73 mdd como una manera de mejorar el desarrollo de fármacos.
Precios de fármacos especialesUn gran brinco (23%) en el gasto en fármacos especiales en 2012 parece ser el responsable de una notable elevación en precios para fármacos de marca en general. El gerente de be-neficios de farmacia (PBM por sus siglas en inglés) Express Scripts rastrea un grupo de medicamentos de marca usados ampliamente y encontró que sus precios aumentaron 13.3% desde septiembre de 2011 a septiembre de 2012, sobrepa-sando por mucho el 2% de inflación. Al mismo tiempo, los precios de los fármacos genéricos cayeron cerca del 22%. Los fabricantes objetaron que la cifra de inflación del precio está sesgada por unos cuantos medicamentos de especialidad caros que tratan condiciones graves, como hepatitis C, escle-rosis múltiple, y cáncer. Mientras tanto, el crecimiento en el mercado de especialidades farmacéuticas está provocando a los PBMs y a las aseguradoras a acaparar estas empresas de servicio mientras que los empleadores y los pagadores están vigilando más de cerca las estrategias para coordinar el cui-dado y monitoreando la prescripción a los pacientes que usan medicamentos de alto costo (ver “La brecha de precios se am-plía entre los fármacos de marca y los genéricos” en www.express-scripts.com).
La FTC se opone a los cambios de formulación
anti-genéricosLa Comisión de Comercio Federal está respaldando al fabri-cante de fármacos genéricos Mylan Pharmaceuticals en un li-tigio que desafía las modificaciones del innovador a la forma farmacéutica, potencia y etiquetado diseñados para frustrar la competencia del genérico. Mylan afirma que Warner Chilcott buscó retrasar los genéricos a través de cambios menores, no terapéuticos, en su fármaco antibacteriano Doryx (doxiciclina hiclato). Un Amicus Brief de la FTC en noviembre de 2012 se opone a tales tácticas de “salto de producto” por parte de las empresas de marca como tácticas para manipular el pro-ceso regulatorio de la FDA y socavar las leyes federales y del estado que estimulan la competencia de los genéricos.
1 Un Amicus Brief es un documento sometido a la corte por alguien que no está relacionado directamente con el caso en consideración (N. de la T.)

Pharmaceutical Technology en Español MARZO / ABRIL 201362
Jill Wechsler es editora en Washington del Pharmaceutical
Technology, tel. 301.656.4634, jwechsler@advanstar.
com. Lea los blogs de Jill en PharmTech.com/wechsler
RE
GU
LA
CIÓ
N Y
CU
MP
LIM
IEN
TOobservancIa REGULATORIA
La escasez de fármacos y el desastre que la acompaña alienta los esfuerzos para revisar la supervisión de la manufactura y estimular las mejoras en la industria.
calidad de los fármacos en el centro del escenario para la Fda y los fabricantes
El esfuerzo de una década para modernizar la producción bio-farmacéutica y las políticas re-gulatorias ha estado a la altura
de sus objetivos y está llevando a una reevaluación de los esfuerzos regulato-rios de la FDA y de la respuesta de la in-dustria. El cambio en el abastecimiento de fármacos y la producción a sitios en el extranjero aumenta la demanda por una supervisión más eficiente, como lo hacen las provisiones en el Acta de Seguridad e Innovación de la FDA (FDASIA) que respaldan el desarrollo acelerado y la aprobación de terapias avanzadas para mejorar el cuidado del paciente.
En consecuencia, Janet Woodcock, director del Centro para la Evaluación e Investigación de Fármacos (CDER), le está dando una nueva mirada a los programas de la FDA diseñados para asegurar que los requerimientos de la calidad de manufactura promuevan la innovación biomédica que lleve a pro-ductos seguros y efectivos. La iniciati-va “Calidad Farmacéutica para el Siglo XXI” del CDER se lanzó en el 2002 para estimular la adopción del fabrican-te de sistemas de gestión de calidad mo-derna, tecnología analítica de proceso y métodos de producción automatizada. Este programa ha dado muchos logros, “aunque todavía no estamos ahí”, de-claró Woodcock en el simposio de Di-ciembre patrocinado por el Consorcio Internacional para la Innovación y Cali-dad en el Desarrollo Farmacéutico (IQ).
Woodcock hizo observaciones si-milares acerca de la inversión del fa-bricante en esquemas de calidad por diseño (QbD) en junio de 2012 en la conferencia de la FDA/ISPE en Bal-timore. A pesar de años de discusión
acerca de la QbD y los sistemas mo-dernos de manufactura, la FDA todavía ve altas tasas de rechazo y defectos de producto extendidos, señala Woodcock. La continua escasez de fármacos, la re-ciente crisis de mezclado de fármacos y las frecuentes recuperaciones de pro-ducto del mercado han intensificado la atención de la FDA en estrategias para mejorar la producción de fármacos de calidad. Estas fallas se suman al costo de la producción de fármacos, el cual reduce los recursos de IyD de una com-pañía y con frecuencia ocasiona precios más elevados de los fármacos.
A su vez, los ejecutivos Biofarma-céuticos deberían considerar la produc-ción de fármacos de calidad como un medio para alcanzar una clara ventaja competitiva sobre sus competidores y como una “competencia básica” para la administración –una actitud que Wood-cock encuentra rara. Este esquema po-dría involucrar el desarrollo de métricas de calidad cuantificables, algo que se hace en otras industrias como la auto-motriz o en las tasas de reparación de televisiones o en el porcentaje de sali-das en tiempo de las aerolíneas.
Para las compañías farmacéuticas, las métricas podrían ser públicas, como las recuperaciones de producto del mer-cado o las citaciones de inspección. Las medidas más internas podrían incluir retrasos en el ciclo de producción, ín-dice en el cumplimiento de especifica-ciones, o tiempos de ciclo teóricos con-tra reales. Dichas evaluaciones pueden documentar cómo la producción más eficiente reduce el desperdicio y aho-rra dinero evitando también las recu-peraciones de producto del mercado y los problemas de seguridad que dañan reputaciones corporativas. Y los aho-
rros en la arena de la manufactura que soportan precios más bajos para nuevas terapias serán particularmente impor-tantes conforme surjan más tratamien-tos personalizados. La administración farmacéutica podría estar más dispuesta a invertir en tecnología moderna de pro-ducción si pudiera reducir el costo de la producción, respaldar aprobaciones más rápidas, evitar quemar el tiempo de cobertura de la patente, y facilitar el reembolso de los pagadores.
Aprobaciones más rápidasGarantizar la producción de fármacos de calidad se ha vuelto más urgente conforme la FDA enfrenta más presión para facilitar el acceso del paciente a nuevos fármacos “de avanzada”, un objetivo que podría ser indeterminado por los retrasos en el escalamiento de la manufactura. La nueva Designación de Terapia de Avanzada, la cual puede aplicar la FDA al principio en el desa-rrollo clínico según sea autorizado por el FDASIA, “parece un cambio de jue-go” para el cáncer, enfermedades gené-ticas y otras condiciones serias, señala Woodcock. Sin embargo, si la FDA está preparada para aprobar un fármaco prioritario mientras el patrocinador está todavía en la escala de producción clíni-ca, la aprobación final podría retrasarse si los revisores encuentran que el pro-ducto no cumple los criterios de “listo para la manufactura”.
“Esto ha sucedido”, afirma Wood-cock, “y va a empeorar” conforme la FDA encuentre terapias para el cáncer más dirigidas que demuestren una res-puesta completa en la Fase I o II, pero fabricantes que carecen de la capacidad para suministrar el producto rápida-mente. En una conferencia de noviem-

Pharmaceutical Technology en Español MARZO / ABRIL 2013 63
bre del 2012, sobre la investigación del cáncer, patrocinada por Amigos de la Investigación del Cáncer y la Institución Brookings, Woodcock destacó los beneficios potenciales de la nueva política para las terapias de avanzada, advirtiendo al mismo tiempo que la manufactura del fármaco podría ser un “paso limitante” en el desarrollo. Para evitar dichos obs-táculos, ella recomendó reuniones iniciales, por separado, de la FDA con los patrocinadores sobre los problemas de manu-factura para discutir el plan de escalamiento para un fármaco potencialmente crítico.
Estas fallas se suman al costo de producción del fármaco.
Tales discusiones podrían resaltar los beneficios de la adopción de los procesos de manufactura continuos para fa-cilitar una transferencia eficiente desde el desarrollo hasta la producción comercial. Los fabricantes que adopten la QbD al principio del proceso de desarrollo también pueden experi-mentar “menos sorpresas desagradables” en producción, me-nos desecho y más inspecciones positivas de la planta en el camino., señala Woodcock.
Cambios regulatoriosEl impacto potencial de los sistemas de manufactura de cali-dad sobre la innovación y el acceso a fármacos proporciona una fuerte plataforma para el cambio en la regulación de cali-dad, dice Woodcock. Las iniciativas de calidad farmacéutica del CDER han dado muchos éxitos, como se ha visto en el monitoreo en línea expandido en las líneas de producción; el desarrollo de software y otras tecnologías para monitorear los procesos de manufactura y detectar errores; la armonización de los conceptos de manejo del riesgo de calidad a través de la Conferencia Internacional de Armonización; y la implemen-tación de los esquemas de la QbD en muchas compañías.
La FDA ha sido menos productiva con los esfuerzos para proveer a la industria con la ayuda sobre los suplementos de manufactura y para construir un cuerpo de inspectores farma-céuticos con mayor experiencia para evaluar los sistemas de producción de calidad innovadores. Woodcock está esperan-zada en que los recursos añadidos proporcionados por el Acta de Tarifas de Usuarios de Fármacos Genéricos (GDUFA) re-vivirá el programa de los inspectores y dará más paridad a las visitas a los sitios extranjeros y domésticos.
El CDER también está respondiendo a estos desarrollos es-tableciendo una Oficina de Calidad Farmacéutica (OPQ), otra “súper oficina” que coordinará la revisión de los datos de quí-mica, manufactura y controles (CMC) en las solicitudes con es-fuerzos de cumplimiento para asegurar la adhesión a las GMPs en los sitios de las plantas. La OPQ reemplazará a la oficina de Ciencia Farmacéutica (OPS), cuya directora desde hace mucho tiempo, Helen Winkle, se está retirando de la agencia.
Este cambio reunirá a todos los revisores del CMC para fármacos genéricos, fármacos convencionales y biológicos. Adicionalmente, la Oficina de Manufactura y Calidad de Pro-ducto (OMPQ) en la Oficina de Cumplimiento del CDER se unirá a la nueva entidad. Mientras tanto, la Oficina de Fár-macos Genéricos (OGD) se está convirtiendo en una súper oficina separada del CDER y operará más como Oficina de Nuevos Fármacos del CDER, con responsabilidad para la re-visión de bioequivalencia, microbiología y datos clínicos y hará las aclaraciones de las etiquetas de los fármacos. Durante los próximos seis meses surgirán más detalles acerca de la reorganización y del OPQ.
Las regulaciones para los sometimientos del CMC y los GMPs cubren mucho del mismo territorio, así que tiene sen-tido reunir a los miembros del personal que las evalúan, co-mentó Jon Clark, director asociado de la OPS para políticas regulatorias, en el simposio del IQ. Una ventaja de la nueva estructura es que estimulará la discusión de las GMPs mucho más temprano en el proceso de desarrollo y es de esperar que evite las clases de retrasos de producción que podrían frustrar la aprobación para el mercado. El nombramiento de Howard Sklamberg, suplente comisionado adjunto para asuntos regu-latorios, encabezará la oficina de cumplimiento del CDER y es también alguien familiarizado con las operaciones de la fuerza en campo de la FDA para ayudar a darles forma a los nuevos modelos de inspección.
Woodcock está esperanzada en que esta reorganización y otras iniciativas promoverán mejores maneras de asegurar la calidad de los fármacos estimulando mientras tanto la inno-vación. El sistema actual para el monitoreo de las actividades de manufactura es muy intenso en recursos, sin embargo, no puede dirigir la supervisión a situaciones de mayor riesgo. “No estamos sólo cambiando cajas”, insiste, sino buscando enfo-carnos más en la vigilancia de los datos y la métrica, en vez de las inspecciones en sitio, para proveer una revisión más rápida de las solicitudes además de procedimientos mejorados para asegurar la manufactura de calidad alrededor del mundo. PT
SU PRESENCIAEN LA REVISTA MÁS LEIDA POR LA INDUSTRIA FARMACÉUTICA
CONTRATE SU PAUTA PUBLICITARIA
AHORA
Mayores Informes y Contrataciones:Tel: 52 (55) 5659-8880, 5536-2100, 5543-1486 E-mail: [email protected]

Pharmaceutical Technology en Español MARZO / ABRIL 201364
RE
GU
LA
CIÓ
N Y
CU
MP
LIM
IEN
TO
dentro de LA USP
El foco de atención de la USP en 2013 involucra estándares relacionados con impurezas orgánicas, medición de ADN residual y proteínas de la célula huésped en productos biotecnológicos, e impurezas elementales.
actividades para el establecimiento de estándares de impurezas
Conforme han ido evolucionando los productos far-macéuticos a través de las décadas, así también los procesos para su manufactura y las técnicas ana-líticas para comprender su composición. Por cada
medicamento tomado por un paciente, es importante para el que hace el fármaco poder identificar las impurezas de cual-quier significancia que puedan ser resultado de su proceso de manufactura y tomar las medidas para controlar las impurezas indeseadas, especialmente porque éstas pueden afectar la se-guridad y eficacia de un producto.
Una impureza es una sustancia que se presenta en un pro-ducto farmacéutico terminado que no juega ningún papel fun-cional y no es un componente inherente de la forma pura de los ingredientes usados intencionalmente. Puede ser el resul-tado de un proceso de manufactura, puede ser agregada inten-cionalmente como catalizador durante la síntesis del fármaco, o puede ser un subproducto de degradación que surge durante el ciclo de vida de un fármaco.
Empezando en los 90’s, la Conferencia Internacional de Armonización (ICH) empezó el desarrollo de guías relacio-
requisitos de validación para la eficacia de la desinfecciónLa revalidación de la desinfección de los cuartos limpios puede ser innecesaria.
La revalidación periódica de un
desinfectante no sirve a ningún propósito
científico útil.
John S. Kent, PhDDirector, Consulto de Desarrollo Farmacéutico San Mateo, CA 94403
resoLucIón de PROBLEMAS DEL CMC
P.¿Es útil de alguna manera la re-evaluación periódica de un desin-
fectante? Se me ha pedido que repita estos estudios aun cuando ya se hicie-ron en el pasado. No ha habido ningún cambio a los materiales o métodos des-de entonces.
R.En la industria de la manufactu-ra farmacéutica, que opera bajo
las GMPs, la revalidación periódica de la eficacia de la desinfección no está científicamente justificada o requeri-da siempre que el desinfectante usado siga siendo el mismo, la dilución del desinfectante es todavía la misma, los organismos de desafío (incluyendo los aislados del ambiente) son todavía los mismos, las superficies del ambiente de manufactura son todavía las mismas, y el vendedor del desinfectante es toda-vía el mismo. Hasta donde yo sé, sólo
cuando uno de estos cinco elementos con respecto a la desinfección del cuar-to limpio cambia, se le requiere que re-pita la validación de la prueba de efica-cia de la desinfección. La revalidación periódica de un desinfectante no sirve a ningún propósito científico útil, y no se de alguna regulación de la FDA que es-tablezca que uno debe revalidar periódi-camente los desinfectantes. Asegúrese de ejecutar la validación de la eficacia de la desinfección apropiadamente y tenga la documentación completa. PT
John S. Kent, PhD, Director, consulto de Desarrollo Farmacéutico san Mateo, ca 94403

Pharmaceutical Technology en Español MARZO / ABRIL 2013 65
RE
GU
LA
CIÓ
N Y
CU
MP
LIM
IEN
TO
nadas con las impurezas en los productos farmacéuticos. Las guías del ICH proporcionan un marco de trabajo para los sometimientos regulatorios para nuevos fármacos. Este cuer-po de trabajo sobre impurezas llevó a lo que ahora está bien reconocido como la serie Q3 del ICH, la cual incluye guías (niveles permitidos) para impurezas orgánicas e inorgánicas así como para solventes residuales. En el desarrollo de sus propios estándares para impurezas, la Convención de la Far-macopea de EEUU (USP) ha trabajado para y armonizarse con el ICH, en lo posible, manteniendo al mismo tiempo sus objetivos de modernización y creación de estándares que re-flejen las prácticas actuales en la industria que ayudan a ase-gurar la calidad del fármaco.
Como organización científica no lucrativa, la USP desa-rrolla estándares para la identidad, potencia, calidad y pureza de los medicamentos y sus ingredientes. Adicionalmente, los estándares de la USP incluyen aquéllos para impurezas espe-cíficas asociadas con sustancias farmacéuticas y productos far-macéuticos. Los estándares documentales en el compendio de la USP –Farmacopea de los Estados Unidos y el Formulario Nacional (USP-NF)- son publicados como monografías indi-viduales y capítulos generales así como en Noticias Generales.
Con respecto a las impurezas orgánicas, la USP estableció en años recientes estándares para solventes residuales en pro-ductos farmacéuticos, sustancias farmacéuticas y excipientes. Adicionalmente, la USP ha iniciado una revisión de otros capítulos generales relevantes para las impurezas orgánicas en general y ha desarrollado estándares modificados para las impurezas elementales. En 2013, la USP fue el anfitrión de un taller en otro grupo de impurezas –proteínas y ADN residua-les de células huésped en productos biotecnológicos.
Impurezas orgánicasLas impurezas orgánicas cumplen un amplio espectro de compuestos que tienen estructuras, conducta y características variables. Éstas pueden ser resultado del proceso de manu-factura, de las condiciones de almacenamiento o incluso de degradación. Algunas pueden ser volátiles y pueden ser muy propensas a cambiar como resultado de la exposición al calor, la luz u otros factores externos.
En junio de 2013, la USP será anfitrión de un taller enfocado a la medición de ADN y proteínas residuales de la célula huésped en productos biotecnológicos.
Un panel de expertos de la USP ha estado a cargo para ha-cer las recomendaciones sobre las modificaciones al Capítulo General <1086> Impurezas en Sustancias Farmacéuticas y Productos Farmacéuticos, el Capítulo General <466> - Impu-
rezas ordinarias y Noticias Generales 5.60. La composición del Panel de Expertos consultor incluye representantes de fa-bricantes de productos farmacéuticos y de libre venta (OTC) y sus grupos de marca (es decir, la Asociación de Productos para la Salud del Consumidor, Investigación Farmacéutica y Fabricantes de América, y otros), miembros de los Comités de Expertos de la USP sobre Análisis Físico, Moléculas Peque-ñas y Toxicología, y enlaces de la FDA.
El Capítulo General <1086> incluye algunas definiciones clave asociadas con impurezas que están alineadas con las es-tablecidas en el ICH, otras farmacopeas y la guía actual de la FDA. El Capítulo General <466> aplica un método relativa-mente sencillo, aunque antiguo –la cromatografía en capa del-gada (CCD)- para la detección de impurezas. Aunque la CCD le permite al usuario comparar un artículo de prueba contra un estándar conocido y confirmar la presencia de ese artículo en una muestra, está limitada en su capacidad para distinguir entre APIs, excipientes o impurezas y cuantificar exactamente las impurezas dentro de una muestra dada.
La revisión del Panel de Expertos tomará en cuenta las impurezas asociadas con cientos de monografías que apare-cen en la USP-NF, las cuales incluyen fármacos OTC y diver-sas formas farmacéuticas asociadas con un solo OTC. Como parte de su trabajo, el Panel de Expertos está evaluando la factibilidad de categorizar diferentes tipos de impurezas or-gánicas (p.ej., compuestos heterocíclicos) y, con base en esas categorizaciones, explorará métodos relevantes para la detec-ción y la medición que puedan ser aplicadas a múltiples mo-nografías dentro de un solo grupo.
Proteínas residuales de la célula huésped y ADNAunque los fármacos tradicionales de moléculas pequeñas se hacen a través de rutas de síntesis química, muchos productos biológicos y biotecnológicos se hacen utilizando tecnologías recombinantes en células huésped diseñadas para producir una proteína terapéutica construida genéticamente. Estos productos recombinantes son con frecuencia transcritos de secuencias del ADN humano que están colocadas dentro de células huésped como las líneas celulares de ratón, células de levaduras, o Escherichia coli. Después que las células produ-cen las proteínas, éstas son purificadas y caracterizadas. Los fabricantes de estos productos de proteína deben ser capaces de demostrar que sus productos finales contienen niveles muy bajos del ADN y las proteínas de la célula huésped porque po-dría haber un riesgo de tumorigenicidad o inmunogenicidad, respectivamente, de estos materiales cuando se administren al paciente. Actualmente, la USP está desarrollando un ca-pítulo sobre el ADN residual, y al menos dos estándares de referencia estarán disponibles para respaldar el capítulo. La USP también está desarrollando un capítulo de información general que contiene las mejores prácticas para el desarrollo y caracterización de reactivos críticos así como el desarrollo, validación y uso de los procedimientos de medición de proteí-nas de la célula huésped.

Pharmaceutical Technology en Español MARZO / ABRIL 201366
RE
GU
LA
CIÓ
N Y
CU
MP
LIM
IEN
TO En junio de 2013, la USP en colaboración con la Asocia-ción de Mejores Prácticas Emergentes de los BioFarmacéuti-cos (BEBPA) será anfitriona de un taller enfocado en la me-dición de las proteínas residuales de la célula huésped y del ADN en productos de biotecnología. La reunión presentará prácticas actuales de la industria y proporcionará perspectivas de la comunidad regulatoria. Para la descripción del taller y la información del registro, ver http://uspgo.to/host-cell-protein.
Impurezas elementalesCon respecto a las impurezas elementales, la USP ha de-
sarrollado dos capítulos generales relevantes -<232> Impu-rezas elementales – Límites y <233> Impurezas elementales – Procedimientos. El Capítulo General <232> especifica lí-mites para impurezas elementales seleccionadas, incluyendo
límites para mercurio, cadmio, arsénico y plomo –elementos tóxicos comúnmente encontrados en el ambiente. En el Ca-pítulo General <233>, se describen procedimientos para la identificación de impurezas utilizando tecnología de plasma acoplado inductivamente. Adicionalmente, el Capítulo Ge-neral <233> proporciona criterios de validación que el fa-bricante seleccionaría para usar procedimientos diferentes a los descritos en los nuevos Capítulos Generales. Como es el caso de los solventes residuales, los capítulos de impurezas elementales serán aplicados a los artículos reconocidos en la USP-NF por medio de una provisión de Noticias Generales, que se espera que se proponga en enero del 2013 en el Foro Farmacéutico y que entre en vigor el 1º. De Mayo de 2014. Para información actualizada acerca del estatus actual de los estándares de la USP para impurezas elementales, ver http//usogo.to/elemental-impurities. PT
Más del 96% de las estructuras cristalinas de moléculas far-macéuticamente relevantes resueltas en el SSCI adoptan una de estas tres clases de cristal. Por lo tanto, los programas de indexación que se enfocan en las clases de cristal de menor simetría son los recomendados para farmacéuticos.
Las rutinas de indexación deben ser usadas por individuos con experiencia en difracción y cristalografía. La mayoría de los paquetes de indexación darán salida a una mejor solución para cualquier patrón de entrada, pero depende del usuario determinar si la solución propuesta es consistente con el pa-trón de entrada. Como las celdas unitarias no son únicas, un problema común es reportar la celda unitaria correcta en una forma no estándar. Otro problema común es que una celda unitaria de ensayo, la cual es una fracción de la celda correcta, sea reportada, y no se reconoce una mayor simetría. Alterna-tivamente, una simetría demasiado alta puede ser reportada. Los patrones de polvo de mezclas no son indexables en un principio, pero los programas de indexación generalmente producen una solución de ensayo no sensible para los patro-nes de mezcla que deberían ser rechazados por el usuario. Al-gunos paquetes de indexación, sin embargo, pueden indexar exitosamente un material cristalino en presencia de una im-pureza cristalina. El conocimiento del volumen molecular, el grado de hidratación y/o solvatación, la quiralidad del com-puesto, y los grupos espaciales comunes pueden auxiliar a un experto en la evaluación de soluciones de indexación de ensayo. Por ejemplo, aunque hay 230 grupos espaciales, las frecuencias a las que se presentan están lejos de ser iguales. Trabajar en SSCI en moléculas farmacéuticamente relevantes ha demostrado que sobre el 90% de las moléculas quirales adoptan uno de cuatro grupos espaciales (P21
, P212
12
1, C2, ó
P1) y más del 90% de moléculas no quirales adoptan uno de cuatro grupos espaciales también (P2
1/c, P-1, C2/c, ó Pbca).
“Indexación del patrón de difracción de polvos con rayos X para aplicaciones farmacéuticas”continuación de la pág. 55
Aunque las soluciones de indexación han sido determinadas para APIs en otros grupos espaciales, éstas son menos co-munes. Las soluciones de indexación propuestas fuera de los grupos espaciales comunes deben ser examinadas.
ConclusiónLa indexación de XRPD puede ser usada para extraer infor-mación de patrones de XRPD de alta calidad y añadir valor a su interpretación. Puede ser usada para identificar patrones que representan fases simples así como para determinar si los patrones representan la misma o diferentes formas. La in-dexación de patrones de XRPD para formas cristalinas debe ser una parte rutinaria de la selección y análisis de formas sólidas.
Referencias1. W. Massa, Crystal Structure Determination (Springer, Berlin,
Germany, 2nd ed. 2004).2. R. A. Young, “Introduction to the Rietveld method,” in The
Rietveld Method, R. A. Young, Ed., (Oxford University Press, New York, NY, 1993), pp. 1–38.
3. C. A. Beevers and H. N. Hansen, Acta Cryst., B27, 1323–1325 (1971).
4. A. Burger and R. Ramberger, Mikrochim. Acta, 72 (3–4) 273–316 (1979).
5. R. F. Bryan and P. G. Forcier, Mol. Cryst. Liq. Cryst., 60 (3) 157–165 (1980).
6. Y. Miwa et al., Acta Cryst., B55, 78–84 (1999). 7. Results from research at SSCI, a division of Aptuit, West La-
fayette, IN, USA, not previously published.8. A. Boultif and D. Louër, J. Appl. Cryst., 37 (5) 724–731 (2004).9. J. W. Visser, J. Appl. Cryst., 2 (3) 89–95 (1969).10. A. A. Coelho, J. Appl. Cryst., 36 (1) 86–95 (2003).11. P. E. Werner, L. Erikson, and M. Westdahl, J. Appl. Cryst., 18
(5) 367–370 (1985).12. M. A. Neumann, J. Appl. Cryst., 36 (2) 356–365 (2003). PT

Pharmaceutical Technology en Español MARZO / ABRIL 2013 67
CALENDARIO DE EVENTOS
Cuando usted contacte a alguno de los organizadores de éstos eventos, por favor mencione que vio su evento en
MARZO 12 – 15PROPAK AFRICA 2013. The Packaging, Food Processing, Printing, Plastics & Labelling Exhibition. Lugar: Expo Centre, Nasrec, JHB, South Africa. Teléfonos: + 27(11) 8351565, Contacto / Info: [email protected], Página Web: www.propakafrica.co.za
14 - 15II Seminario Internacional de Innovación en Tecnologías del Agua. Lugar: Hotel Hacienda Jurica, Qro. Contacto / Info: L.R.C. Eidy Arzola Franco, Tel. 01 55 57529600, E-mail: [email protected], Página web: www.helguera.com.mx
17 – 21 PITTCON 2013. Lugar: Pennsylvania Convention Center Philadelphia, PA USA. Contacto / Info: [email protected], Teléfonos: 18008253221, Página Web: http://pittcon.org/
20 – 22CPhI South East Asia (CPhI SEA), Lugar: Jakarta international Expo, Jakarta, Indonesia. Contacto / Info: [email protected], Teléfonos: +622126645000, Página Web: www.cphi-sea.com
18 – 21BIO-PHARMA ASIA 2013. Lugar: Singapur, Asia - Resorts World Sentosa. Contacto / Info: [email protected], Teléfonos: + (65) 6222 8550, Página Web: www.terrapinn.com/exhibition/biopharma-asia/index.stm
Lugar: Hotel Hyatt Regency, ciudad de México, Teléfonos: (55) 5601 3082 (55)5601 3083, E-mail: [email protected], dirección@vector_pharma.com, Página Web: www.vector-pharma.com
Junio 4, 5 y 6Ciudad de México
13 – 16Achemasia 2013. Lugar: Beijing, PRC China National Convention Center. Contacto / Info: [email protected], Teléfonos: + (49) 6975 64152, Página Web: http://achema-content.dechema.de/
14 – 16XXI COLAMIQC. Lugar: São Paulo – SP BRASIL. Contacto / Info: [email protected], Teléfonos: +551150445466, Página Web: www.colamiqc2013.com.br
ABRIL17 – 19PHARMINTECH 2013. Exhibition for the Pharmaceutical, nutraceutical and personal care industry. Lugar: Bologna Italia. Contacto / Info: [email protected], Teléfonos: (39) 02 3191091, Página Web: www.pharmintech.it/ita/home
17 – 19EXPOFARMA 2013. Lugar: México D.F., Word Trade Center, Ciudad de México. Contacto / Info: [email protected], Teléfonos: 52 (55) 9183 2060, 01800 0020 236, Página Web: www.afmac.org.mx/actividades/expofarma
23 – 25 INTERPHEX NY 2013. Es la feria mundial que engloba las últimas tecnologías, educación, fuente de productos y servicios para el progreso y desarrollo del sector de las ciencias de la vida. Lugar: New York, Estados Unidos, Javits Convention Center. Contacto / Info: [email protected], Teléfonos: 1 (2)12 2162000, Fax: +1 (2)12 2162588, Página Web: www.interphex.com
24 – 26CPhI/ICSE/P-MEC/BioPh Japan. Lugar: Tokyo, Japan. Contacto / Info: [email protected], Teléfonos: 31 203 632 616, Página Web: www.cphijapan.com
MAYO8 – 10BIOtech Japón. Lugar: Tokyo Big Sight. Contacto / Info: [email protected], Teléfonos: 81-3-3349-8518, Página Web: www.bio-t.jp/en

Pharmaceutical Technology en Español MARZO / ABRIL 201368
Principales tendencias que se deben tener en cuenta contra la falsificación de
medicamentos
Los bienes falsificados representan casi un 10% del comercio global y su valor estimado ronda los 500 mil millones de dólares. Entre los bienes que más se falsi-fican en el mundo están los productos de lujo como el calzado, los relojes, la ropa, etc. y los productos farma-céuticos. Es indiscutible que la falsificación de produc-tos en todos estos ramos causa pérdidas irreparables a las marcas que los comercializan a escala mundial. Sin embargo es la venta de medicamentos falsificados, que alcanzó un valor de 75 mil millones de dólares en el año 2010, la que en última instancia provoca la pérdida de vidas humanas.
Aunque tanto la OMS como las compañías farma-céuticas están haciendo mucho para concienciar a la
gente sobre el problema de los medicamentos falsifica-dos y de los riesgos que comportan, los falsificadores están recurriendo cada vez más a tácticas fraudulen-tas para engañar al público e inducirle a que compre medicamentos baratos pero letales. Comúnmente, la falsificación de medicamentos en la actualidad res-ponde a las cinco pautas siguientes. Cerca del 43% de medicamentos falsificados no contienen compo-nentes activos, mientras que un 21% contienen bajos niveles de componente activo. Otro 24% de medica-mentos seguros se reemplazan por medicamentos de baja calidad, el etiquetado y empaquetado fraudulento representa un 7% de la falsificación de medicamentos a nivel global.
Cada vez más y más compañías farmacéuticasestán adoptando diversas y múltiples estrategias en
sus soluciones contra la falsificación de medicamentos.En este artículo trataremos las tendencias másrecientes y hablaremos sobre su importancia.
Principales tendenciascontínua en pág. 70.

Pharmaceutical Technology en Español MARZO / ABRIL 2013 69
ACG Pam Mexico.pdf 1 05/09/12 11:42
Mar
que
en la
tarje
ta d
e se
rvici
o al
lect
or e
l No.
2

Pharmaceutical Technology en Español MARZO / ABRIL 20137070
inglés) y permite disponer de información directa del consumidor, particularmente en países con un alto por-centaje de uso de móviles.
Si combinamos esta función con la serialización, contamos con una estructura casi perfecta para erra-dicar los medicamentos falsificados del mercado. Con la serialización, cada caja lleva impreso un número de serie único que se registra en la base de datos durante el proceso de producción; dicho número puede servir para identificar productos con el mismo número de se-rie. También puede ayudar a resolver el problema de la desviación de productos originales para comercializar en el mercado negro.
Junto a los métodos de seguimiento y rastreo, y a la serialización, las técnicas de autenticación – visibles u ocultas – sirven como otra capa de protección con-tra la falsificación. En el empaquetado del producto se utilizan técnicas visibles, como hologramas o tintas de color cambiante para permitir una rápida identificación de la marca por parte del consumidor, mientras que técnicas ocultas tales como pigmentos ultravioletas o rayos infrarrojos sólo son detectables con equipos es-pecializados.
La falsificación de medicamentos en general, y de algunos que permiten salvar vidas, ha causado la pérdida de muchas vidas humanas a nivel mundial y continúa poniendo en riesgo la vida de muchas otras personas. Mediante la implementación de múltiples es-trategias y medidas contra la falsificación, las compa-ñías farmacéuticas no sólo pueden recuperar millones de dólares, sino también seguir haciendo lo que mejor saben hacer – salvar vidas.
Para contrarrestar esta amenaza, muchas compa-ñías farmacéuticas han optado por asociarse con em-presas que ponen sobre la mesa su conocimiento sobre etiquetado y empaquetado para garantizar la seguridad del producto, dificultar la falsificación e incrementar el valor de la marca. Mientras que las compañías farma-céuticas tratan de mantener su política de reducción de costes, las empresas de empaquetado consideran que la inversión en innovación e investigación y desarrollo es la única manera de estar por delante de la compe-tencia y, lo que es más importante, de los falsificadores.
Enfrentarse a los falsificadores exige un enfoque de múltiples niveles. En otras palabras, un falsificador puede ser capaz de reproducir una réplica exacta de un holograma, pero puede que no sea capaz de eludir las etiquetas RFID o métodos de seguimiento y rastreo. En consecuencia, las compañías farmacéuticas tienen que utilizar lo mejor de las tecnologías visibles y ocultas en el empaquetado para garantizar una óptima seguridad y minimizar en lo posible el riesgo que pueda conllevar la falsificación de sus productos.
Uno de los mejores métodos para evitar la falsifica-ción de medicamentos, es implementar una solución de seguimiento y rastreo en la que se controla el proceso completo desde la fase de fabricación, hasta la fase de empaquetado del medicamento. No sirve únicamente como una herramienta adicional de protección para la seguridad de la cadena de suministros, sino que tam-bién confiere al usuario final la capacidad de verificar la autenticidad del medicamento en el mismo punto de venta a través de un simple SMS. Esta solución para la prevención de la falsificación asegura la rentabilidad a largo plazo del capital invertido (ROI, por sus siglas en
Shabbir BadamiACG WorldwidePara más información, por favor, envíe su email a [email protected] o visítenos en www.acg-world.com
Informes y Contrataciones:Tel: 52 (55) 5659-8880, 5536-2100, 5543-1486
E-mail: info@pharmatechespañol.com.mx

Pharmaceutical Technology en Español MARZO / ABRIL 2013 71
MAQUINARIA DE ACONDICIONAMIENTO / LLENADORAS Y TAPONADORAS
MAQUINARIA DE ACONDICIONAMIENTO / BLISTERAS Y ENCARTONADORAS
Directorio Clasificado
INST. Y EQUIPO PARA INSPECCIÓNCONTROL Y MONITOREO DE PROCESOS
MAQUINARIA DE ACONDICIONAMIENTO / AGRUPADORAS Y ENVOLVEDORAS
VARIOS / CHAT MASCOTAS
El lugar para los amantes de sus mascotas
Síguenos en...
desdeRecibe consejos e información para el cuidado de tu mascota y Consulta a Expertos que responderán a tus principales inquietudes sobre: • Comportamiento / Entrenamiento• Alimentación• Higiene• Control de plagas• Enfermedades / padecimientos comunes• y más Comparte experiencias con otros dueños de mascotas Conoce a otras mascotas como la tuya Encuentra pareja para tu mascota Compra regalos para tu mascota
Encuentra, Participa y Aprende
MAQUINARIA DE ACONDICIONAMIENTO / LLENADORAS Y TAPONADORAS

Pharmaceutical Technology en Español MARZO / ABRIL 201372
Cuando usted contacte a alguno de estos anunciantes, por favor m
encione que vió su anuncio en
ÍND
ICE
DE
AN
UN
CIA
NT
ES
PRO
DU
CTO
/SERVIC
IOEM
PRESA
No. PÁ
GIN
AN
O. PA
RA TA
RJETA D
E SER
VICIO
AL LEC
TOR
Cápsulas de Gelatina Blanda
Construcción y Sistemas Críticos / Cuartos Am
bientales
Equipo de Proceso
Equipo de Proceso / Tableteadoras y Encapsuladras
Inst. y Equipo para Inspección. Control y Monitoreo de Procesos
Instrumentos y Equipos Analíticos - Biotecnología
Maquinaria de Acondicionam
iento / Agrupadoras y Envolvedoras
Maquinaria de Acondicionam
iento / Blisteras y Encartonadoras
Maquinaria de Acondicionam
iento / Llenadoras y Taponadoras
Maquinaria de Acondicionam
iento / Llenadoras y Taponadoras
Maquinaria de Acondicionam
iento / Llenadoras y Taponadoras
Maquinaria de Acondicionam
iento / Soplado, Llenado, Sellado
Materiales y Accesorios para Laboratorio / Purificadores de Agua para Laboratorio
Sistemas de Aire Acondicionado
Varios / Chat mascotas
Varios / Estetica para Mascotas
CATALEN
T
GRU
PO ID
EA
FEDERA
L EQU
IPMEN
T
ACG -pam
- PACK & PRO
CESS
OPTREL - PACK &
PROCESS
SARTO
RIUS
POLY PACK - PACK &
PROCESS
FABRIM
A - PACK &
PROCESS
MA
R - PACK & PRO
CESS
ROTA
- PACK & PRO
CESS
TGM
- PACK & PRO
CESS
WEILER EN
GIN
EERING
, INC.
VEOLIA
WATER SYSTEM
S MEXICO
S.A. D
E C.V.
EOLIS A
MERICA
LATINA
S.A. D
E C.V.
PETFACEBOO
K
MA
S QU
E PERRUQ
UERIA
4ta. Forros
1133
6971
2da. Forros
71717171671
3ra. Forros
137167
1 7 8 2
10
5
11
12
14
13
159 6 4 3
16
ww
w.catalent.com
/ Email: consultas@
catalent.com
ww
w.grupoidea.com
.mx / e-m
ail: [email protected]
.mx
ww
w.fedequip.com
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.sartorius.com
/ Email: sartorius-stedim
@sartom
ex.com.m
x
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.pack-process.com
/ E-mail: info@
pack-process.com
ww
w.w
eilerengineering.com / e-m
ail: solutions@w
eilerengineering.com
ww
w.purelabflex.com
/ Email: elga.m
exico@veoliaw
ater.com
ww
w.eolis.com
.mx y w
ww
.comsaem
te.com
ww
w.petfacebook.com
.mx
facebook Mas Q
Perruqueria twitter @
MA
SQU
EPERRUQ
UERIA
DIR
ECC
IÓN
WEB
/ CO
RR
EO ELEC
TRÓ
NIC
O

Mar
que
en la
tarje
ta d
e se
rvici
o al
lect
or e
l No.
6

Marque en la tarjeta de servicio al lector el No. 1


















