
Ejemplo de Ana lisis de Datos de Microarrays con R y Bioconductor · 2012-03-22 · 4.1.1 Matriz de...
17
Ejemplo de An´ alisis de Datos de Microarrays con R y Bioconductor Alex S´ anchez Statistics and Bioinformatics Research Group Departament d’Estad´ ıstica. Universitat de Barcelona April 25, 2009 Contents 1 Introducci´ on 1 1.1 El ejemplo ............................... 2 1.2 Directorios y opciones de trabajo ................. 2 2 Obtenci´ on y lectura de los datos 3 2.1 Los datos ............................... 3 2.2 Lectura de los datos ......................... 3 3 Exploraci´ on, Control de Calidad y Normalizaci´ on 4 3.1 Exploraci´ on y visualizaci´ on ..................... 4 3.2 Control de calidad .......................... 6 3.3 Normalizacion y Filtraje ....................... 7 4 Selecci´ on de genes diferencialmente expresados 8 4.1 An´ alisis basado en modelos lineales ................. 8 4.1.1 Matriz de dise˜ no ....................... 9 4.1.2 Contrastes ........................... 10 4.1.3 Estimaci´ on del modelo y selecci´ on de genes ........ 11 4.2 Anotaci´ on de resultados (1) ..................... 12 4.3 Comparaciones m´ ultiples ....................... 14 4.4 Visualizaci´ on de los perfiles de expresi´ on .............. 15 1 Introducci´on El an´ alisis de datos de microarrays suele proceder, en general secuencialmente, a trav´ es de una serie de etapas tal y como se muestra en la figura 1. El objetivo de este documento es ilustrar de forma breve pero completa como se hace un an´ alisis de microarrays con Bioconductor y R. Uno de los problemas en este tipo de estudios es que en cada etapa se puede proceder de varias formas lo que da lugar a un asfixiante n¸cumero de posibilidades especialmente para el ne¸cofito. Con el fin de evitar este problema de momento se describe una sola de las opciones posibles para cada paso, lo que constituye un proceso “al estilo Bioconductor”. 1
Transcript of Ejemplo de Ana lisis de Datos de Microarrays con R y Bioconductor · 2012-03-22 · 4.1.1 Matriz de...

Ejemplo de Analisis de Datos de Microarrays con
R y Bioconductor
Alex SanchezStatistics and Bioinformatics Research Group
Departament d’Estadıstica. Universitat de Barcelona
April 25, 2009
Contents
1 Introduccion 11.1 El ejemplo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21.2 Directorios y opciones de trabajo . . . . . . . . . . . . . . . . . 2
2 Obtencion y lectura de los datos 32.1 Los datos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32.2 Lectura de los datos . . . . . . . . . . . . . . . . . . . . . . . . . 3
3 Exploracion, Control de Calidad y Normalizacion 43.1 Exploracion y visualizacion . . . . . . . . . . . . . . . . . . . . . 43.2 Control de calidad . . . . . . . . . . . . . . . . . . . . . . . . . . 63.3 Normalizacion y Filtraje . . . . . . . . . . . . . . . . . . . . . . . 7
4 Seleccion de genes diferencialmente expresados 84.1 Analisis basado en modelos lineales . . . . . . . . . . . . . . . . . 8
4.1.1 Matriz de diseno . . . . . . . . . . . . . . . . . . . . . . . 94.1.2 Contrastes . . . . . . . . . . . . . . . . . . . . . . . . . . . 104.1.3 Estimacion del modelo y seleccion de genes . . . . . . . . 11
4.2 Anotacion de resultados (1) . . . . . . . . . . . . . . . . . . . . . 124.3 Comparaciones multiples . . . . . . . . . . . . . . . . . . . . . . . 144.4 Visualizacion de los perfiles de expresion . . . . . . . . . . . . . . 15
1 Introduccion
El analisis de datos de microarrays suele proceder, en general secuencialmente,a traves de una serie de etapas tal y como se muestra en la figura 1.
El objetivo de este documento es ilustrar de forma breve pero completa comose hace un analisis de microarrays con Bioconductor y R. Uno de los problemasen este tipo de estudios es que en cada etapa se puede proceder de varias formaslo que da lugar a un asfixiante ncumero de posibilidades especialmente para elnecofito. Con el fin de evitar este problema de momento se describe una solade las opciones posibles para cada paso, lo que constituye un proceso “al estiloBioconductor”.
1

Figure 1: The Microarray Analysis Process
1.1 El ejemplo
El ejemplo analizado consiste en unos datos disponibles en forma de paquete(estrogen) en Bioconductor. Se trata de un estudio en el que se estudia lainfluencia del tratamiento con estrogeno y del tiempo transcurrido desde eltratamiento. Esto se implementa en un diseno factorial de 2 factores (Estrogeno:Presente/Ausente, Tiempo 8h / 48h).
Se puede obtener mas detalles en la documentacion del paquete estrogen.
1.2 Directorios y opciones de trabajo
Para facilitar supondremos que trabajamos en un directorio escogido por nosotrosy cuya localizacion se asigna a la variable workingDir. Los datos se copiaran enun subdirectorio del anterior denominado “datos” que se almacenara en la vari-able dataDir y los resultados se almacenarcan en un directorio “results” cuyonombre completo se almacenara en la variable resultsDir.
Supondremos tambien concimientos basicos de Ry familiaridad con el proyectoBioconductor.
> workingDir <-getwd()
> dataDir <-file.path(workingDir, "datos")
> resultsDir <- file.path(workingDir,"results")
> setwd(workingDir)
> options(width=80)
> options(digits=5)
> memory.limit(4095)
NULL
2

2 Obtencion y lectura de los datos
Para un analisis de datos de microarrays de Affymetrix se necesitan los archivosde imagenes escaneadas (“.CEL”) y un archivo en el que se asigne una condicionexperimental a cada archivo.
2.1 Los datos
Una ventaja de este ejemplo es que los datos se encuentran disponibles tras in-stalar el paquete estrogen por lo que tan solo hace falta copiarlos a un directoriode trabajo. El directorio en que se encuentran los archivos .CEL es:
> library(estrogen)
> estrogenDir <- system.file("extdata", package = "estrogen")
> print(estrogenDir)
[1] "C:/ARCHIV~1/R/R-28~1.1/library/estrogen/extdata"
Para realizar el analisis es preciso copiar todos los archivos del directorio“extdata” a nuestro directorio de trabajo “datos”.
ATENCION: Esta operacioon que depende de la instalacicon y delsistema operativo no se indica aqui.
2.2 Lectura de los datos
El proceso de leer los datos puede parecer un poco extrano a primera vista perola idea es simple:
� En primer lugar creamos algunas estructuras de datos que contienen lainformacion sobre las variables y - opcionalmente - sobre las anotacionesy el experimento y
� A continuacion nos basamos en estas estructuras para leer los datos y crearlos objetos principales que se utilizaran para el analisis.
> require(Biobase)
> require(affy)
> sampleInfo <- read.AnnotatedDataFrame(file.path(dataDir,"targlimma.txt"),
+ header = TRUE, row.names = 1, sep="\t")
> fileNames <- pData(sampleInfo)$FileName
> rawData <- read.affybatch(filenames=file.path(dataDir,fileNames),
+ phenoData=sampleInfo)
Una forma alternativa de leer los datos consiste en acceder al directorio endonde se encuentran y escribir ReadAffy En este ejemplo se incluye un archivodefectuoso con fines didacticos, llamado “badcel”. Para ilustrar las diferenciasentre archivos correctos e incorrectos se puede leer en otro objeto.
> require(affy)
> setwd(dataDir)
> rawData.wrong <- ReadAffy()
> setwd(workingDir)
3

3 Exploracion, Control de Calidad y Normal-izacion
Tras leer los datos pasamos al preprocesado. Aunque puede interpretarse dedistintas formas esta fase suele consistir en
1. Realizar algunos graficos con los datos para hacerse una idea de como haresultado el experimento.
2. Realizar un control de calidad mas formal.
3. Normalizar y, en el caso de Affymetrix, resumir las expresiones
3.1 Exploracion y visualizacion
La exploracion previa puede hacerse paso a paso o en bloque si se utiliza algunpaquete como affycoretools. En la ayuda de este paquete se encuentra unadescripcion de los analisis basicos que permite realizar.
Un histograma permite hacerse una idea de si las distribuciones de los dis-tintos arrays son similares en forma y posicion.
4 6 8 10 12 14 16
0.0
0.2
0.4
0.6
0.8
Signal distribution
log intensity
dens
ity
neg10hneg10hest10hest10hneg48hneg48hest48hest48h
El grafico de degradacion –que no aparece en este caso, ya que daba proble-meas al representarlo– permite hacerse una idea de como ha sido el proceso dehibridacion de las muestras. Lineas paralelas sugieren una calidad similar.
low10A low10B hi10A hi10B low48A low48B hi48A hi48Bslope -0.103 -0.217 -0.129 -0.3810 -0.50400 -0.55300 -0.3510 -0.66700pvalue 0.550 0.194 0.394 0.0341 0.00482 0.00109 0.0464 0.00032
EL codigo para el grafico de degradacion serıa:
4

El diagrama de cajas muestra da, copmo el histograma, una idea de la dis-
tribucion de los datos.
neg1
0h
neg1
0h
est1
0h
est1
0h
neg4
8h
neg4
8h
est4
8h
est4
8h
6
8
10
12
14
Finalmente un cluster jerarquico seguido de un dendrograma nos puede ayu-dar a hacernos una idea de si las muestras se agrupan por condiciones experi-mentales.
Si lo hacen es bueno, pero si no, no es necesariamente indicador de problemas,puesto que es un grafico basado en todo los datos.
est1
0h
neg1
0h
neg1
0h
est1
0h
neg4
8h
neg4
8h
est4
8h
est4
8h
050
0000
1500
000
2500
000
Hierarchical clustering of samples
hclust (*, "average")dist(t(exprs(rawData)))
Hei
ght
5

3.2 Control de calidad
Las exploraciones anteriores nos proporcionan una idea de como son los datos.Se pueden realizar controles de calidad mas estrictos como:
� Los controles de calidad estandar de Affymetrix, descritos en el paquetesimpleaffy.
� Controles basados en modelos a nivel de sondas, descritos en el paqueteaffyPLM.
El paquete affyQCReport encapsula los analisis que pueden realizarse con elpaquete simpleaffy, de forma que con una instruccion se pueden realizar todoslos analisis y enviar la salida a un archivo.
> stopifnot(require(affyQCReport))
> QCReport(rawData,file=file.path(resultsDir,"QCReport.pdf"))
EL paquete affyPLM realiza un control de calidad basado en “probe-levelmodels” (PLM).
> stopifnot(require(affyPLM))
> computePLM <- F
> if(computePLM){
+ Pset<- fitPLM(rawData)
+ save(Pset, file=file.path(dataDir,"PLM.Rda"))
+ }else{
+ load (file=file.path(dataDir,"PLM.Rda"))
+ }
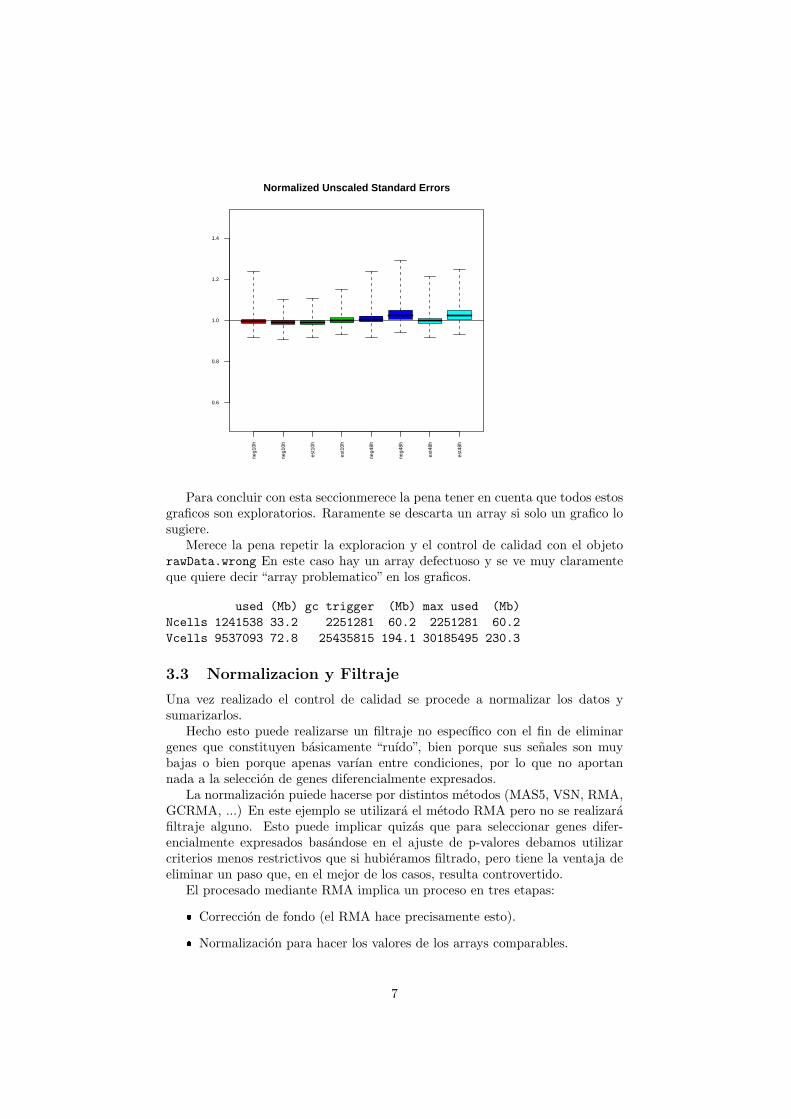
Como resultado del ajuste PLM se pueden obtener dos graficos, uno deexpresiones relativas y otro con errores estandarizados. Si los datos son decalidad ambos graficos deben ser centrados y relativamente simetricos. Cam-bios en esta situacion sugieren problemas en los arrays que no las verifiquen.
neg1
0h
neg1
0h
est1
0h
est1
0h
neg4
8h
neg4
8h
est4
8h
est4
8h
−4
−2
0
2
4
Relative Log Expression
6
neg1
0h
neg1
0h
est1
0h
est1
0h
neg4
8h
neg4
8h
est4
8h
est4
8h
0.6
0.8
1.0
1.2
1.4
Normalized Unscaled Standard Errors
Para concluir con esta seccionmerece la pena tener en cuenta que todos estosgraficos son exploratorios. Raramente se descarta un array si solo un grafico losugiere.
Merece la pena repetir la exploracion y el control de calidad con el objetorawData.wrong En este caso hay un array defectuoso y se ve muy claramenteque quiere decir “array problematico” en los graficos.
used (Mb) gc trigger (Mb) max used (Mb)Ncells 1241538 33.2 2251281 60.2 2251281 60.2Vcells 9537093 72.8 25435815 194.1 30185495 230.3
3.3 Normalizacion y Filtraje
Una vez realizado el control de calidad se procede a normalizar los datos ysumarizarlos.
Hecho esto puede realizarse un filtraje no especıfico con el fin de eliminargenes que constituyen basicamente “ruıdo”, bien porque sus senales son muybajas o bien porque apenas varıan entre condiciones, por lo que no aportannada a la seleccion de genes diferencialmente expresados.
La normalizacion puiede hacerse por distintos metodos (MAS5, VSN, RMA,GCRMA, ...) En este ejemplo se utilizara el metodo RMA pero no se realizarafiltraje alguno. Esto puede implicar quizas que para seleccionar genes difer-encialmente expresados basandose en el ajuste de p-valores debamos utilizarcriterios menos restrictivos que si hubieramos filtrado, pero tiene la ventaja deeliminar un paso que, en el mejor de los casos, resulta controvertido.
El procesado mediante RMA implica un proceso en tres etapas:
� Correccion de fondo (el RMA hace precisamente esto).
� Normalizacion para hacer los valores de los arrays comparables.
7

� Summarizacion de las diversas sondas asociadas a cada grupo de sondaspara dar un unico valor.
> stopifnot(require(affy))
> normalize <- F
> if(normalize){
+ eset_rma <- rma(rawData)
+ save(eset_rma, file=file.path(dataDir,"normalized.Rda"))
+ }else{
+ load (file=file.path(dataDir,"normalized.Rda"))
+ }
La normalizacion hace que los valores de los arrays sean comparables entreellos, aunque los distintos metodos situan los valores en escalas distintas, por loque lo que no resulta directamente comparable son los valores normalizados pordistintos metodos.
Un boxplot de los valores normalizados sugiere que los valores ya estan enuna escala en donde se pueden comparar.
> boxplot(eset_rma,main="RMA", names=sampleNames, cex.axis=0.7, col=info$grupo+1,las=2)
El codigo siguiente, que no se ejecuta muestra como se podrıan aplicar dis-tintos metodos para lego compararlos entre ellos.
> eset_mas5 <- mas5(rawData) # Uses expresso (MAS 5.0 method) much slower than RMA!
> stopifnot(require(gcrma))
> eset_gcrma <- gcrma(rawData) # The 'library(gcrma)' needs to be loaded first.
> stopifnot(require(plier))
> eset_plier <- justPlier(rawData, normalize=T) # The 'library(plier)' needs to be loaded first.
> compara <-data.frame(RMA=exprs(eset_rma)[,1], MAS5 =exprs(eset_mas5)[,1],
+ GCRMA=exprs(eset_gcrma)[,1], PLIER =exprs(eset_plier)[,1])
> pairs(compara)
4 Seleccion de genes diferencialmente expresa-dos
Como en las etapas anteriores la seleccion de genes diferencialmente expresados(GDE) puede basarse en distintas aproximaciones, desde la t de Student alprograma SAM pasando por multitud de variantes.
En este ejemplo se aplicara la aproximacion presentada por [?] basado enla utilizacion del modelo lineal general combinada con un metodo para obteneruna estimacion mejorada de la varianza.
4.1 Analisis basado en modelos lineales
La presentacion http://www.ub.es/stat/docencia/bioinformatica/microarrays/ADM/slides/4b -LinearModelsAndEmpiricalBayes.pdf o el manual del programa limma contienenexplicaciones detalladas sobre construir un modelo lineal para este problema ycomo utilizarlo para seleccionar genes diferencialmente expresados.
8

4.1.1 Matriz de diseno
El primer paso para el analisis es crear la matriz de diseno.La situacion discutida en este ejemplo se puede modelizar de dos formas, tal
como se discute en la presentacion citada mas arriba:
� Como un modelo de dos factores Estrogeno(Pres/Aus) Tiempo (10h/48h)con interaccion.
� Como un modelo de un factor con cuatro niveles (Pres.10h/Pres.48h/Aus.10h/Aus.48h).
Tal como se describe en el manual de limma la segunda parametrizacion re-sulta a menudo mas comoda, a pesar de parecer menos intuitiva, porque permiteformular con mas facilidad que la de dos factores las preguntas que tıpicamenteinteresan a los investigadores.
El modelo lineal para este estudio sera:
y1y2y3y4y5y6y7y8
=
1 0 0 01 0 0 00 1 0 00 1 0 00 0 1 00 0 1 00 0 0 10 0 0 1
︸ ︷︷ ︸Design Matrix,X
α1
α2
α3
α4
+
ε1ε2ε3ε4ε5ε6ε7ε8
Los parametros del modelos representan las cuatro combinaciones tiempo/estrogeno.
α1 = E(logAbs.10h),α2 = E(logPres.10h),α3 = E(logAbs.48h),α4 = E(logPres.48h).
La matriz de diseno puede definirse manualmente o a partir de un factorcreado especıficamente para ello.
Manualmente, seria:
> design.1<-matrix(
+ c(1,1,0,0,0,0,0,0,
+ 0,0,1,1,0,0,0,0,
+ 0,0,0,0,1,1,0,0,
+ 0,0,0,0,0,0,1,1),
+ nrow=8,
+ byrow=F)
> colnames(design.1)<-c("neg10h", "est10h", "neg48h", "est48h")
> rownames(design.1) <- c("low10A", "low10B", "hi10A" , "hi10B", "low48A", "low48B", "hi48A" , "hi48B")
> print(design.1)
neg10h est10h neg48h est48hlow10A 1 0 0 0
9

low10B 1 0 0 0hi10A 0 1 0 0hi10B 0 1 0 0low48A 0 0 1 0low48B 0 0 1 0hi48A 0 0 0 1hi48B 0 0 0 1
Alternativamente puede crearse la matriz de diseno a partir de la informacionsobre las condiciones contenida en el phenoData, siempre que exista un campoadecuado para ello.
En este caso la columna Target se ha creado para utilizarla con esta fi-nalidad. El objeto phenoData puede recrearse a partir del archivo original oextrayendolo del objeto ExpresionSet que contiene los datos y las covariables.
neg10h est10h neg48h est48hlow10A 1 0 0 0low10B 1 0 0 0hi10A 0 1 0 0hi10B 0 1 0 0low48A 0 0 1 0low48B 0 0 1 0hi48A 0 0 0 1hi48B 0 0 0 1attr(,"assign")[1] 1 1 1 1attr(,"contrasts")attr(,"contrasts")$lev[1] "contr.treatment"
Ambas matrices, design, design.1 resultan iguales.
4.1.2 Contrastes
Dado un modelo lineal definido a traves de una matriz de diseno pueden formu-larse las preguntas de interes como contrastes es decir comparaciones entre losparametros del modelo.
Cada parametrizacion distinta requerira de unos contrastes diferentes paralas mismas preguntas, por lo que habitualmente se utilizara la parametrizacionque permita formular de forma mas clara las comparaciones de interes.
En este caso interesa estudiar
� Efecto del estrogeno al inicio del tratamiento
� Efecto del estrogeno al cabo de un tiempo del tratamiento
� Efecto del tiempo en ausencia de estrogeno
Esto se puede formular facilmente con la parametrizacion adoptada.
β11 = α2 − α1, Efecto del estrogeno pasadas 10 horasβ1
2 = α4 − α3, Efecto del estrogeno pasadas 48 horasβ1
3 = α3 − α1, Efecto del tiempo en ausencia de Estrogeno
10

> cont.matrix <- makeContrasts (
+ Estro10=(est10h-neg10h),
+ Estro48=(est48h-neg48h),
+ Tiempo=(neg48h-neg10h),
+ levels=design)
> cont.matrix
ContrastsLevels Estro10 Estro48 Tiemponeg10h -1 0 -1est10h 1 0 0neg48h 0 -1 1est48h 0 1 0
4.1.3 Estimacion del modelo y seleccion de genes
Una vez definida la matriz de diseno y los contrastes podemos pasar a estimarel modelo, estimar los contrastes y realizar las pruebas de significacion que nosindiquen, para cada gen y cada comparacion, si puede considerarse diferencial-mente expresado.
El metodo implementado en limma amplıa el analisis tradicional utilizandomodelos de Bayes empıricos para combinar la informacion de toda la matriz dedatos y de cada gen individual y obtener estimaciones de error mejoradas.
El analisis proporciona los estadısticos de test habituales como Fold-changet-moderados o p-valores ajustados que se utilizan para ordenar los genes de masa menos diferencialmente expresados.
A fin de controlar el porcentaje de falsos positivos que puedan resultar delalto numero de contrastes realizados simultaneamente los p–valores se ajustande forma que tengamos control sobre la tasa de falsos positivos utilizando elmetodo de Benjamini y Hochberg ([1]).
La funcion topTable genera para cada contraste una lista de genes ordenadosde mas a menos diferencialmente expresados.
> topTabEstro10 <- topTable (fit.main, number=nrow(fit.main), coef="Estro10", adjust="fdr")
> topTabEstro48 <- topTable (fit.main, number=nrow(fit.main), coef="Estro48", adjust="fdr")
> topTabTiempo <- topTable (fit.main, number=nrow(fit.main) , coef="Tiempo", adjust="fdr")
Una forma de visualizar los resultados es mediante un volcano plot querepresenta en abscisas los cambios de expresion en escala logarıtmica y en or-denadas el “menos logaritmo” del p-valor o alternativamente el estadıstico B.
11

●●
●●
●
●
●
●
●
●
●
●
●●●● ●● ●● ●
●
●
●
●
●
●
●
●
●
●
●
●
●● ●
●●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●●●
●
●●
●
●
●
●●
●●●
●
●
●
●
●●●●
●
●
●
●●●●
●
●
●
●
●● ●●
●●
●
●
●●●
●
●
●
●
● ●
●
●
●
●
●
●●
●●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●● ●
●
●
●
●●
●
●
●
●●
●
●●
●
●
●
●●
●●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●●
●●
●
●
●
●●
●
●
●
●
●
●
●●
●
●●●
● ●●
●
●
●
●
●
●
●●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●●
●●
●
●●
●
●
●
●
●●
●
●
●●
●
●
●
●
●●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
● ●
●●●●
●
●
●
●
●
●
●●
●
●
●
●●
●
● ●
●●
●
● ●
●
●●
●
●
●
●
●●
●
●●
●●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●● ●●
● ●
●●●
●●
●●●
●
●●
●
●
●
●
●●
●
●●●
●
●●
●
●
●
●●
●
●●●
●
●
●
●
●●
●
●
●
●
●●
● ●
●
●
●
●●
●
●●
●
●
●●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●●
●
●
●
● ●● ●● ●
●
●●
●●●
●
●
●●●
●
●
●
●
●
●
●●
● ●
●
●
●
●
●●
●●●●
●
●
●●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●●●
●●
●
●
●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●●
●●
●
●●●●
●
●●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●● ●
●
●
●●
●
●●
●
●
●●●
●
●
●
●
●●
●
●
●
●●
●
●
●
●
● ●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●●
●●●●
●●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●●
●
●●●●
●
●
●
●
●
●
●
●●
●
●
●●● ●
●
●
●
●
●●●
● ●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●●●
●
●
●●
●
●
●●●
●
●●●
●
●
●
●
●●
●
●●●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●●●
●
●
●●
●
●
●
●●
● ●●●
●
●
●
●●
●● ●
●
●
●●
●
●
●
●
●
●
●
●●●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
●●●●
●
●●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●●
●●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●●● ●
●
●
●
●
●● ●
●
●
●
●
●
●
●
●
●●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●● ●
●
●
●
●
●
●
●
●
●
●
●●
●●●
●
●
●
●●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●●●
●
●
● ●
●
● ●●
●
●●● ●●
●
●
●
●
●●
●
●●
●
● ●●
●
●
●●●
●
●●
●
●
●
●
●
●
●
●●
●
●
●
● ●●●
●
●
●
●●
●
●
●
●
●
●
●
●
●●
●
●
●
●●
●
●●
●
● ●●
●●
●
●●●●
●
●●●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●●
●●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●●●
●●
●●
●
●
●
●
●
●
●
● ●●
●
●
●
●
●●
●●●
●
●
●
●
●
●
●
●●
●●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●●●
●
●
●
●
●
●●
●
●
● ●
●
●
●
●
●
●●
●●
●
●
●
●
● ●
●
●
●
●●
●
● ●●●
●●
●
●●
●●
●
●
●
●
●●
●
●
●●
●●
●
●
●
●
●
●●●
●
●
●●
●●●●●●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
● ●●
●●
●●
●
●
●
●
●
●
● ●●
●
●
●
●●● ●
●
●
●
●●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
● ●
●● ●
●
●
●●
●
●
●
●
●
●●
●
●●●
●●
●
●
●
●●●
●
●
●
●
●
●
●
●
● ●●●
●
●
●
●
●●
●
●
●●●
●
●●●●●
●●
● ●● ●
●
●
●●
●
●
●
●
●● ●●
●
● ●● ●
●
●
●
● ●
●
●● ●●
●
●
●
●●
●
●
●
●
● ●
●
●
●●● ●●
●
●●
●
●
●●●
●
● ●●
●●
●
●●
●
●
●●
●
●
● ●●
●
●
●●
●
●
●●●
●
●
●●●
● ●●
● ●
●
●
●
●
●
●●
●●
●
●●●●
●
●
●
●●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●●
●
●
●●
●
●
●
● ●● ●
●
●
●●
●
●
●
●
●
●●
●
●●
●
●
●●●●
● ● ●
●●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●●●● ●
●
●●
●
● ●●●
●
●●
●
●
●
●●
●
●
●
●
●●
●
●
●
●●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
● ●●
●●
●●
●●
●
●●
●●
●●
●
●
●
●
●
●
●
●
●
● ●●
●
●
●
●●
●● ●
●●
●
●●●●
●
●● ●
●●
●●●●●
●● ●
●
●
●
●●
●
●
●
●
●
●
●
●●
●
●
● ●
●
●●●
●
●
●●
●
●
●
●●
●
●
●●
● ●
●
●
●
●
●
●
●●●
●
●●●● ●
● ●
●
●
● ●●●
●
●
●
●
●●
●
●
●●●
●
●●●
● ●
●●
●
●
●● ●
●
●
●
●
●
●●
●
●●
●
●
●●
●
●
●
●●
●
●
● ●● ●
●
●
●
●
●●
●
●
●
●●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●●
●
●
●
●
●
●
●●●
●●●
●●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●●●
●●
●
●●
●
●
●
●
● ●
●●●
●
●
●
●
●
●●● ●
●
●
●
●
●
●
●
●●● ●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●● ●
●
●
●
●
●●● ●
●
● ●● ●
●
● ●●
●
●
●●
●
●
●
●●
●
●●●
●●
●
●
●
●
●●●●
●
●
●
●●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●
●●●
●●
●●
●
●
●●
●
●●●●
●●●
●
●
●
● ●
●
●●
●
●
●●●●
●
●
●
●
●●●
●●
●
●●
●
●
●● ●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●●
●
●
●●
●
●●●
●
●
●●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●●
●●
●
●
●●
●●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●
● ●●
●
●
●
●
●●
●
●
●
●
●
●●●
● ●
●
●
●● ●
●
●
●
●●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●●●
●●●
●
●
●
●
●
●●
●
●
●
●
●
●●●
●●
●
●
●
●
●
●
●
● ●
●
●
●
●● ●
●
●●
●●
●
●●
●
●
●
●●●● ●
●
●
●●●
●
● ●● ●
●
●●●●● ●●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●●●
●
●●●
●
●
●●
●
●
●
●●
●
●●
●●
●
●
●●
●
●●
●
●
●
●●
● ●●
●
●
●
●
●
●●
●
●
●
●●
●
●●●
●●
●
●●
●●●
●
●
●
●
●●
●
●
●
●
● ●
●
●
●
●
●
●●●
●
●
●●
●
●●
●●●
●
●
●
●
●●
●●●
●
●
●●●●
●●●●
●
●●
●
●
●
●
●
●
●●●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●●●
●●
●
●
●
●●
●
●
●●
●●
●
●
● ●
●
●
●
●●
●
●●
●
●
●
●
●
●●●●●
●●● ●● ●●
●
● ●● ●
●
●
●
●● ●
●●
●
●
● ●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●●
●
●
●●
●
●
●
●●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
● ● ●●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●●
●
●
●
●●
●
● ●
● ●
●●●
●
●
●
●
●
●
●●●
●●
●●
●
●●●
●●
●
●
●
●
●●
●
●
●●
●●
●
●
●●
●
●
●●
●
●●●●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●●
●
●●●
●
●
●●
●
●
●
●
●
● ●
●
●
● ●
●
●
●
● ●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●
●
● ●●
●
●
●●
●
●
●
●
●
● ●
●●●
●
●
●
●●
●
●
●●
● ●
●
●●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●●
●●●
●●
●●
●
●
●
●
●
●
●
●●
●
●
● ●
●
● ●●
●
●
●
●
●
●
●●
● ●●●●
●
●
●
●
●●
●●●
●
●
●●
●
●
●
●
●●
●
●●●●●●
●
●● ●●
●
●●
●
●●
●
● ●
●●
●
●●
●
●
●●
●
●
●●
●
●
●
●●
●
●
●
●
●●
●●●
●
●
●
●
●●
●
●
●
●●
●
●
●●●
●●
●
●
●
●
●●●
●●
●
●
●●
●
●
●
●
●
●● ●
●
●●
●●●
●
●●
●
● ●●●●●
●
●
●
●
●
●
●
●
●●●
●●●
●●●
●
● ●
●● ●
●
●
● ●
●
●●●
●
●●
●●
●
●
●
●
●
●●●
●
●●●
●
●● ● ●●●●●
●●●●
●
●● ●
●●
●
●
●
●
●
●
●
●●
●
●
●●●
●
●●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●●●
●●
●
●●
●
●
●
●
●●●
●
●
● ●
●●
●
●
●
●
●
●
●●●●
●
●
●
●
●
●
●●●
●
●●
●
●●
●
●
●
●
●
●
●●●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●●
●●
●
●
● ●●● ●●
●
●●
●
● ●
●
●
●●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
● ●
●
●●
●
●
●●●
●
●
●
●
●●
●
●
●●
●
●
●
●●
●
●
●
●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●●
● ●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●●
●
●
●
●●
●
●
●
●
●
●
●
●● ●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●●
●● ●
●
●
●
●
●●
●●●● ●
●
●
● ●●
●
●
●
●●●
●●
●●
●●●
●
●
●
●
●
●
●●● ●
●
●
●
●
●
●
●
●●●●
●● ●
●
●
●
●●●●
●
●
●
●
●●
●
●●
●●
●
●
●●
●●
● ●
●●
●
●
●
●●●
●
●●
●
●●●
●
●
●
●●
●
●
●●
●●
●●
●● ●●
●
●●●
●
●
●
●●● ●●
●
●
●
●
●● ●●
● ●
●
●
●
●● ●●
●
●
●
●
●●
●
●
● ●
●
●
●●
●
●●
●●
●
●
●
●
●
●●●
●
●
●
●
●
●●
●
●●
●
●
●
●
●●●●
●
●
●
●●● ●●
●
●
●
●
● ●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●●● ●
●●
●
●●
●
●●
●●●
●●
●
●
●
●●
●
●
●
●
●●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●●● ●●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●●
●●
●
●
●
● ●●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
● ●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●●
●●
●
●
●●
●●●
●
●
●
●
●●●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●●
●●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●●●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●●
●
●
●
●●
●●
●
●
●
●
●
●●●●●●
●
●●
●●
●
●
●●●
●
●●
●●
● ●
●●
●
●●●
●
●● ●
●●●
●
●
●
●●
●
●●
●
●
●
●●●●
●
●●
●●
●●
●
●
●
●
●●
●
●●
●●
●
●
●
●
●●
●●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●●
●
●
●● ●●
●
● ●●●
●
●
●
●●
●
●
●● ●●
●
●
●●
●
●
●●
●
●●
●
●
●●
●
●
●●●
●●● ●
●
●●
●
●●● ●●
●● ● ●●
●
●●●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
● ●
●●●
●● ●●●
●
●
●●
●
●
● ●●●
●
●
●
●●
●
●
●●
●
● ●
●
●●
●
●●
●
●●
●●
●●
●
●
●●
●
●●
●
●●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●●● ●
● ●
●●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●●●
●
●
●
●
●
●
●
●
●●●
●●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
● ●
●
●●
●
●
●●●
●
●●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●●
●
●
●
●
●
●●
●●
● ●●
●
●
●
●
●
●
●
●
●●●
●
●
●
●● ●
●
●
●
●●
●
●●
●● ●● ●
●
●●
●
●
● ●●●●
●●
●●
●
●
●●
●
●
●
●
●
●
●
●
●●
●
●
●
●●●●
●
● ●
●●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
●
● ●
●
●
●
●
●●●
●
●
●
●
●●
●●
●● ●●●●●●●
●
● ●
●
●
●
●
●
●●●
●
●●
●●
●
●
●
●
●● ●●
●
●
●
●
●●
●
●●●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
● ●
●● ●
●
●
●
●●
●
●
●
●
●
●●
● ●
●●●
●
●●● ●
●●
●
●
●
● ●●●
●
●
●
●
●
●
●●
●
● ●
●●
●
●●●
●
●●
●
●● ●
●
●●
●
●●
●
●
●●
●●
●●
●●
●
●●●
●●
●
●
●
●
●
●●
●●
●●●
●
●
●
●
●
●●
●
●
●
● ●
● ●
●
●
●
●
●●
●●● ●
●
●●● ●●
●●
●
●
●●
●
●● ●●
●
●
●
●
●
●
●
●●
●
●
●
●
●
● ●
●
●
●
●
●●
●
●●●●●
●
●
●● ●●
●
●●
●●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●●●
●●
●
●
●
●
●
●
●
●
●
● ●●
●
●
●
●
●
●
●●●
●
●●
●
●
●
●
●●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●●●
●
●●●
●●
●●
●
●
●
●
●
●●
●
●
●●●●
●●
●
●●
●●●
●●●●●
●
●
● ●●●●
●● ●
●
●●
●●
●
●●
●●
●
●
●
●
●●
●●
●
●
●
●
●
●
●●
●
●●
●
●
●●
●
●
●
●
●
●
●
●●
●●
●●
●
●
●
●●
●●●●
●
●●
●
●●
●
●
●
●
●●●
●
●
●
●
●●
●
●
●
●
●●
●
●●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
● ●●
●
●
●
●
●
●
● ● ●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●●●●
●●
●
●●●●●
●
●
●
●●
●
●●
●
●
●●
●●
●
●
●●
●
●
●
●●●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●●
●
● ●
●
●
●
●
●
●
●
●
●
● ●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●●●
●
●
●
● ●
●●●●●●
●
●
●●●●●
●
●
●
●
●
●
●●
●
●● ●●
●●
●
●
● ●
●
●●●
●
●
●
●
●
●
●
●
● ●
●
●●
●
● ●
●
● ●
●●● ●
●
●●●●
●
●
●
●●
●
●●●
●●
●●
●
●
●●
● ●
● ●●
●●
●
●
●
●
●●
●
●●●
●
●●
●
●
●
●
● ●
●
●●●
●
●
●
●
●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●
●
●● ●
●
●
●
●
●
●●
●●
●●
●
●
●
●
●●● ●●
●●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●●●
●
●
●
● ●●
●
●●
●●
●
●
●
●
● ●●●
●
●●
●
●
● ●●
●
●
●
●●
●
●
●
●● ●●
●
●
●
●
●
●
●
●●●
●
●●
●
●
●●●
●
●●●
●
●●
●●
●
●
●
●
●●
●
●
●
●
●
●●
●●
●
●
●
●
●
●●
●●●
●
●●●
●
●
●●
●
● ●
●
●
●
●●●
●
●
●●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●●●
●●
●●
●
●
●
●
●
●
●
● ●●
●
●● ●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●●
●
● ●
●
●●
●
●
●
●●
●
●
● ●● ●●●●
●●● ●
●
●●
● ●●
●
●
●●
●
●
●
●
●●
●
●
●
●
●●
●
●● ●
●
●●●●
●
●
●
●
●
●
●
●
●
●●
●●
●
●●
●
●●
●
●●
● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●●● ●●
●●
●
●
●
●
●
●
●●
●
●
●
●
●●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●●
●
●●
●
●
●
●
●● ●
●
●
●
●
●●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●●●●
●
●
●
●
●
●
●●●
●
●●●●●
●
●
●
●
●●
●
●
●
●
●
●●
●●
●
●
●●●●
●
●
●
●
●
●
●
●
● ●
●
●
●
●
●
●
●
●
●●●
●
●●
●
●●
●
●●
●●
●
●
●
●
●
●
●●
●
●●●
●
●
●
●
●
●
●
● ●●●
●●
●
●
●●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●●
●
●
●●●
●
●
●
●
●
●
● ●●●
●
●●
●●
●
●
●●
●●
●●
●
●●
●
●
●●●●
●●
●●
●●●●
●
●
●
●●●●
●
●
●● ●
●
●●●
●
●●
●
●●
●
●●
●●
●
●
●●
●●
●
●●
●●
●
●
●
●
●
●●● ●
●
●
●●●
●●
●
●
●●●●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●●●
●●●
●
●●●
●
●
●●
●
●
●
●●
●
●
●
●●
●
●
●
●
● ●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●●●●
●
●●
●●
●●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●●
●●
●
●
●●
●
●●
●
●
●
●
●●●
●●
●
●
● ●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●●●
●
●
●
●
●●
●●●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●●
●
●●
●
● ●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
● ●
●
●●●
●
●
●
●●●●
●
●●
●
●
●
●● ●● ●
●
●
●
●●●
●●●
●● ●
●●
●●
●
●
●
●●
●
●
●●●
●
●
●
●
●●
●
●●●●
●
●
● ●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
● ●
●
●
●
●
●●●
●
●
●●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●●
● ●●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●●
●● ●
●
● ●●●
●
●
●●
●●
●
●
●
●
●
●
●●●
●●
●●●
●●
●●
●●
●●
●●
●
●
●
●
●
●●
●
●●●● ●●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●● ●●●●
●
●
●●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●● ●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
● ●
●●
●
●●●
●
●●
●
●
●
●●●
●
●
●
●
●
●●
●●
●
●
● ●
●
● ●
●●
●●●
●
●●
●
●
●
●●
●
●
●
●
●
●●
●
●
● ●
●
●●●
●●
●●
● ●●●
●
●●●
●
●
●
●
●
●●●
●
●
●
●
●●
●
● ●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●●
●● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●●
●●●
●●
●●
●
●
●
●
●
● ●●
●
●
●
●
●
●
●
●●●●●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●●
●
●●
●● ●
●
●●
●
●
●
●●
●●
●
●
●
●
●
●
●
●
●
●●
● ●
●
●●
●
●
●
●
●
●
●
●
● ●● ●●
●
●
●
●
●
●
●
●
●
●
●●●●●●●
●
●
●●
●
●
●● ●
●●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●●
●●
●●
●●
●
●
●● ●●
●
●
●●● ●●
●
●●●●
●
●
●
●●
●
●●
●
●
●
●●●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
● ●
●●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●●
●●
●
● ●●
●●
●
●
●●●
●
●
●
●
●● ●● ●● ●
●
●●● ●
●
●
●
●
●●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●● ●● ●
●
●
●
●
●
●●●
●● ●● ●
●
●●●
●
●
●●
●●●
●
●
●
●
●
●●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●●
●
●
●●
●
●
●
●
●● ●●
●
●
●
●
●
●
●
●●
●
●●●
●●
●●
●●
●
●
●●●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●●●
●
●
●
●
● ●●
●
●
●
●
●
●
●
●
●
●●
●
●●●●
●
●●
● ●
● ●
●
●
●
●
●
●●●
●●
●
●
●
●●
●
●
●●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●●
●
●
● ●
●
●●
●
● ●
●
●●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●●
●●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●●
●
●●
●●
●
●
●
●● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●● ●
●
●
●
●
●●●
●
●
● ●● ●
● ●
●
●
●
●
●
● ●
●
● ●●●●
●
●
●
●
●
●
●
●●
●●
●
●●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●●
●
●
●
●●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
● ●● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
●●●●●●
●
●●
●●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●●
●●●
●
●
●●
●
●
●●
●
●
●●
●
●
●
●
●●
●
●
●
●●
●●
●
●
●
●
●
●
●
●●
●
●
●
●
●●●●●●
●
●
●
●
●
●●●
●●
●
●
●
●
●
●●
●●
●●
●
●
● ●
●
●
●
●●●
●
●●
●
●
●●
●●
●
●
●
●
●
●●
●
●
●
●●
●●
●
●
●
● ●●
●
●
●
●
● ●
●●
●
●
●
●
●
●
●● ●
●
●
●
●
●
● ●●●
●
●
●
●
●
●
●
●
●
●
● ●●●●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●●
●
●
●
●
●
● ●
● ●●
●
●
●
●
●
●
●●
● ●
●
●
●●●
●
●
●●
●
●
●
●
●●
●
●●
●●●
●
●
●
●●●
●●
●
●
●
●●● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
● ●●
●●
●
●
●
●
●
● ●● ●●
●●●
●
●●
●
●●
●
●
●●
●
●
●●
●
●
●●●●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●●
●●●
●●●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●●●
●
●●●●
●
●
●
●
●●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●●●●
●
●●
●
●
●
●●
●
●
●
●
●
●●●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●●●●
●
●●
●
●
●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●● ●●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●●
●●●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
●●●
●
●
●
●
●
●
●
●
●
●●
●●●
●●●●
●
● ●
●●
●
●●
●
●●
●
●
●●
●●
●
●
●
●●
●
●
●
●
●
●
●
● ●●●
●
●
●
● ●●
●
●●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
● ●●
●●● ●●●
●●
●
●
●
●
●
●●
●●
●●
●
●
●
●
●●●●● ●●
●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
● ●●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●●●
●
●
●●
●●
●
●
●
●
●
●
●●
●
●
●●●
●
●● ● ●
●
●
●●
●●
●
●
●
●
●
●
●
●
●
● ●
●●
●●
●
●
●
●
●
●
●●● ●●●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●● ●● ●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●●
● ●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●●●
●
●
●
● ●
●
●●●
●
●
●
●
● ●●
●
●
●
●
●
●●
●●●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●●
●●
●●
●
●●
●
●●
●
●
●●
●
●
●
●
●
●
●
● ●
●●●●
●●
●
●
●●
●
●
●
●
●●●
●
●
●
●●
●●
●
●
●
●
●
●
●●●
●●
●
●●
●●
●●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●● ●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
● ●●●●
●
●
●
●
●●●
●
●
●
●
●
●
●●
●
●
●
●
● ●
●●●
●
●●●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
● ●
●
●●
●
●
●●
●
●
●
●●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
● ●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●
●
●● ●
●
●
●
●●
●●
●●
●
●
●
●●
●
●
●
●
●
●● ●
●
●
●
●
●
●
●
●
●●
●●
●●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●●●
●●
●●● ●
●
●
●
●
●●
●
●
●
●
●
●●●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●●●● ●●
●
●
●
●
●●
●
●
●
● ●●
●
●
●
●●
●
●
●
●
●●
●
● ●●
●●
●
●
●●
●
●●
●
●●
●
●
●
●
●
●●
●
●●
●●●
●
●
●
●
●
●
●
● ●●
●
●
●
●
●
●
●
●●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●● ●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●●●● ●
●
●
●
●
●
●
●
●
● ●●
●●
●
●
●
●
● ●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
● ●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●● ●●
●
●
●
●
●
●
●
●●
●
●●
●
●
●●
●
●
●
●
●
● ●
●●
●
●●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●●●
●
●
●
●
●●
●
●
●
●
●●
●
●●
●●
●
●●
●
●
●
● ●
●
●● ●
●
●●
●
●● ●
●●
●
●
●●
●
●● ●● ●●
●●
●
●
●
●
●
●
●
●●●●
●
●
●●
●●●
●
● ●● ●●● ●●
●●
●
●
●●
●
●
●●
●●●
●
●
●
●
●
●●
●
●
●●
●● ●●
●
●
●
●●
●
●
●
●
●●
●
●
●
●●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
●●●
●
●
●
●
●
●
●
●● ●●●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●●
●●●●
●
●
●
●
●
●
●
●
● ●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●●●
●
●
●●
●●
●
●
●
●
●
●●
●●
●
●
●
●
●
●
●
● ●
●
●
●●●
●
●
●●
●
●
●
● ● ●
●
●
●
●
●●
●●
●
●
●
●
●●
●●
●
● ●●
●
●
● ●
●
●
●
●●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●●●● ●
●
●●
●
●
●
●
●●
●●
●
●●●●
●
●●
●●●
●
●
●
●
●
● ●●●●
●
●●●● ●
●
●●
●
●
●
●
●
●
● ●●●
●
●●●●
●●●
●●●
●
●
●
●
●
●
●
●●
●●●
●●
●●
●●
●
●
●●●
●
●
●
●●
●
●●
●
●●●
●
●
●●
●
●
● ●
●●
●
●
●
●●
●●●
●
●
●●
●●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●
●
●●
●
●
●
●
●
●●
●
●●
●
●
●
●
●●●
●
●●●
●
●●
●●
●
●
●
●
●
●●
●
●
●●●
●
●
● ●●● ●●●●
●
●●●●
●
●●
●
●
●●
●
●
●
●
●
● ●
●●
●●●
●
●
●
●
●●
● ●
●
●
●
●
● ●●
●
●●●
●
●
●
●
●
●●
●
●●●
●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
● ●● ●
●
●
●
●
●
●●
●
●
●
●
●
● ●
●
●
●
●
●●
●
● ●●
●
●
●
●
●
●
●●
●
●
●
● ●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●●●
●
●
●●
●
●
●
●
●●
●
●●
●●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●●●●
●
●
●
●
●
●
● ●●
●
●
●●
●
●● ●
●
●
● ●
●
●
●
●
●
●● ●●●
●●
●●
●
●● ●●
● ●●●
●●●●
●
●
●●
●
●
●●●●
●
●●●
●
●
●
●●
●
●
●
●
●
●●
●●●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●●
●●
●
●
● ●
●
●●●●●
●●
●
●
●●
●
●
●
●
●
●
●
●●
●●● ●
●●
● ●
●●
●●
●
●●
●
●
●●●
●
●
●
● ●●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●● ●
●
●
●●
●●
●
●
●●
●●
●
●●●●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●●
●●
●
●
● ●● ●
●
● ●●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●●●
●
●
●●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●●
●●●
●
●
●●
●
●
●
●
●●
●●●
●●
●
●
● ●●
●
●
●
●
●
●
●
●
●
●
●
● ●●●
●
●
●
●
●
●
●
●●
●●●
●
●●
●●
●●
●●
●
●
●
●●●
●
●
●
●
●
●
●
●●
●
●●●
●
●
●
●
● ●●●●●●
●
●
●
●
● ●
● ●●
●
●
●
●●
●
●
●
●●
●
●
●● ●
●
●
● ●●●
●
●
●●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●●
●
●
●
●
●●
●
●●
●
●
●
●●
●
●
●
●●● ●
●
●
●
●
●
●
●●
●
●
●
●
●
● ●
●
●
●●●
● ●
●
●
●
●
●●
●
●
●
●
●●●
●
●
●
●
●
●
● ●
●
●
●
●●
●
●
●●
●
●
●
●
●●
●
●
● ●● ●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●●
●
●●
●
●
●●
●
● ●
●
●●
● ●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●●
●●●
●
●
●
●
●●●
●
●
●
●
●
●
●●
●
●
●●
●
●●
●
●● ●
●
●
●
●
●●
●
●
●
●●
●
● ●
● ●
●
●
●●
●●
●
●●
●
●
●
●●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●●●
●
●
● ●
●
● ●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●●●
●
●
●
●
●●
●
● ●●
●
●●
●
● ●●
●●
●●
●●●
●
●
●
●
●●
●
●● ●●
●●
●
●
●
●
●●
●
●
●
●● ●●
●
●
●● ●
●
●
●●
●●●
●
●
●
●
●
●
●
●● ●
●●
●
●
●
●
●●●
●
● ●
●●
●
●
●
●
●●
●
●
●●●
●
●
●
●
●
●
●●
●
●●
●●
●
●
●●
●
●●●●
●●
●
●
●●●
●●
●
●
●
●●●
●
● ●
●
●
●
●●
●
●
●
●
●●
●
●
●
●●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
● ●
●
●
●
●●●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●●●●
●
●●●
●
●
●●
●
●●
●
●
●
●●● ●
●●
●
●
●
●
●● ●●●
●
●
●
●
●
●
●●
●●
●●
●● ●
●
●
●●
●
●●●
●
●●● ●
●
●
●
●●
●●●
●
●
●
●
●
● ●●
●●
●
●
●
●
●●●
● ●
●● ●
●
●
●
●●● ●● ●
●
●●
● ●
●●●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●●
●
●
●●●●●● ●
●●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●●●●
●
●
●
●●
●
●
●●
●
●●
●
●
●●
●
●
●
●●●●
●
●●
●
●
●
●
●
●●
●
●●
● ●●●
●●
●
●●
●●
●
●
●●
●
●●
●
●
● ●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●●●
●
●
●
●
●●●
●
●
●●
●
●
●
●
●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●●
●
●●●
●
●
●
●
●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●● ●
●
●●
●
●●●
●
●●●
●
●●
●
●
●
●●
●
●
●
●● ●
●
●●
●
●
●● ● ●
●
●
●
●
●
●
●●
●
● ●● ●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●●
● ●● ●
●●
●
●
●
●
●
●●
●
●
●
●
●
●● ●●
●
●
● ●
●
● ●●
●
●
●
●
●●
●
●●
●●
●
●
●
●●
●
●
●
●●
●
●
●●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●●
●
●
●●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
● ●●
●
●● ●●●●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
● ●
●
●
●
●
●
●
●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●●●
●
●
●
●●
●
●
●
●●●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●● ●
●
●
●
●
●
●
●
●
●
●
●
● ●●
●●
●
●
●●
●
●●
●●
●●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
● ●
●
●●●
●
●● ●
●
●●
●
●
●●
●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
● ●
●
●●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●● ●
●
●
●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
● ●
● ●
●
●●●
●
●
●●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●●
●
●●●
●
●
●
●
●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●●
●
●●
●
●●
●●
●
●
●
●
●●
● ●●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
● ●
●
●●●●
●
●
●
●
●●
●
●
●
●
●●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●●
●
●
●
●
●
●
● ●
●
●
●
●
● ●●
●
●● ●●●●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●●
●●●
●
●
●●●
●
●
●●
●●
●
●
●
●●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
● ●●
●
●
●●
●
●
●●
●
●
●●●●●●
●●
●
●
●
●
●●
●
●
●
●
●●
● ●
●
●●●
●
●
●●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●●
●
●
●●
●
●
●
●●
●
●
●
●
●
●
●●
●●
● ●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●
●
●
●
●●
●●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●●
●●
●
●●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●●●
●
●●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●
●
●●●
●
●
●
●
●
●
●
●
●●
●●●
●●
●
●
●
●●
●
●●●
●
●●●
●
●
●
●
●
●
●
●●
●
●
●●
●
●
●
●●
● ●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●●
●●●●
●
●
● ●
●
●●
●
●●
●
●
●
●
●
●
●
●●
●
●
●
●
●●●
●
●●
●
●
●
●
●
●●
●
●
●●●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●●
●●●
●
●
●
●
●●
●
●●
●●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●● ●●
●●
●●●
●
●●
●
●
●
●●
●
●
●
●
●
●
● ●●●
●
●
●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
● ●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
● ●●● ●
●●
●●●
●
●
●
●
●
●
●
●
●
●●
●●●●
●
●●
●
●
●
●
●
●●
●
●
●●
●
●●●
●
−2 −1 0 1 2 3
−5
05
10Differentially expressed genes
Estro10
Log Fold Change
Log
Odd
s
973512472
181411509102149539848922140 580
4.2 Anotacion de resultados (1)
La identificacion de los genes seleccionados puede resultar mas sencilla para elespecialista en un campo si se utilizan nombres estandar como el simbolo del geno “gene symbol”. Con esta finalidad cada paquete de anotaciones tiene tablasde correspondencia entre los distintos tipos de identificadores, principalmenteentre los del array y los de otras bases de datos.
Para saber que anotaciones estan disponibles debe cargarse el paquete yllamar la funcion del mismo nombre
> require(hgu95av2.db)
> hgu95av2()
Quality control information for hgu95av2:
This package has the following mappings:
hgu95av2ACCNUM has 12625 mapped keys (of 12625 keys)hgu95av2ALIAS2PROBE has 37258 mapped keys (of 37258 keys)hgu95av2CHR has 12019 mapped keys (of 12625 keys)hgu95av2CHRLENGTHS has 25 mapped keys (of 25 keys)hgu95av2CHRLOC has 11718 mapped keys (of 12625 keys)hgu95av2CHRLOCEND has 11718 mapped keys (of 12625 keys)hgu95av2ENSEMBL has 11417 mapped keys (of 12625 keys)hgu95av2ENSEMBL2PROBE has 8525 mapped keys (of 8525 keys)hgu95av2ENTREZID has 12024 mapped keys (of 12625 keys)hgu95av2ENZYME has 1975 mapped keys (of 12625 keys)hgu95av2ENZYME2PROBE has 722 mapped keys (of 722 keys)
12

hgu95av2GENENAME has 12024 mapped keys (of 12625 keys)hgu95av2GO has 11540 mapped keys (of 12625 keys)hgu95av2GO2ALLPROBES has 8635 mapped keys (of 8635 keys)hgu95av2GO2PROBE has 6068 mapped keys (of 6068 keys)hgu95av2MAP has 11997 mapped keys (of 12625 keys)hgu95av2OMIM has 10397 mapped keys (of 12625 keys)hgu95av2PATH has 4529 mapped keys (of 12625 keys)hgu95av2PATH2PROBE has 210 mapped keys (of 210 keys)hgu95av2PFAM has 11955 mapped keys (of 12625 keys)hgu95av2PMID has 11990 mapped keys (of 12625 keys)hgu95av2PMID2PROBE has 192162 mapped keys (of 192162 keys)hgu95av2PROSITE has 11955 mapped keys (of 12625 keys)hgu95av2REFSEQ has 11927 mapped keys (of 12625 keys)hgu95av2SYMBOL has 12024 mapped keys (of 12625 keys)hgu95av2UNIGENE has 11996 mapped keys (of 12625 keys)hgu95av2UNIPROT has 11877 mapped keys (of 12625 keys)
Additional Information about this package:
DB schema: HUMANCHIP_DBDB schema version: 1.0Organism: Homo sapiensDate for NCBI data: 2008-Sep2Date for GO data: 200808Date for KEGG data: 2008-Sep2Date for Golden Path data: 2006-Apr14Date for IPI data: 2008-Sep02Date for Ensembl data: 2008-Jul23
Cafa tabla de asociacion puede consultarse de diversas formas,
� Con las funciones get o mget.
� Convirtiendola en una tabla y extrayendo valores
� En algunos casos utilizando funciones especıficas como getSYMBOL o getEG(por “Entrez Gene”) cuando exitan.
Por ejemplo si tomamos los cinco primeros genes seleccionados en la com-paracion “Estro10”
> top5 <-topTabEstro10$ID[1:5]
> cat("Usando mget\n")
Usando mget
> geneSymbol5.1 <- unlist(mget(top5, hgu95av2SYMBOL))
> geneSymbol5.1
39642_at 910_at 31798_at 41400_at 40117_at"ELOVL2" "TK1" "TFF1" "TK1" "MCM6"
13

> cat("Usando toTable\n")
Usando toTable
> genesTable<- toTable(hgu95av2SYMBOL)
> rownames(genesTable) <- genesTable$probe_id
> genesTable[top5, 2]
[1] "ELOVL2" "TK1" "TFF1" "TK1" "MCM6"
> cat("Usando getSYMBOL\n")
Usando getSYMBOL
> require(annotate)
> geneSymbol5.3 <- getSYMBOL(top5, "hgu95av2.db")
> geneSymbol5.3
39642_at 910_at 31798_at 41400_at 40117_at"ELOVL2" "TK1" "TFF1" "TK1" "MCM6"
4.3 Comparaciones multiples
Cuando se realizan varias comparaciones a la vez puede resultar importante verque genes cambian simultaneamente en mas de una comparacion. Si el numerode comparaciones es alto tambien puede ser necesario realizar un ajuste de p-valores entre las comparaciones, distinto del realizado entre genes.
La funcion decidetests permite realizar ambas cosas. En este ejemplo nose ajustaran los p–valores entre comparaciones. Tan solo se seleccionaran losgenes que cambian en una o mas condiciones.
EL resultado del analisis es una tabla res que para cada gen y cada com-paracion contiene un 1 (si el gen esta sobre-expresado o “up” en esta condicion),un 0 (si no hay cambio significativo) o un -1 (si esta “down”-regulado).
Para resumir dicho analisis podemos contar que filas tienen como mınimouna celda distinta de cero:
> sum.res.rows<-apply(abs(res),1,sum)
> res.selected<-res[sum.res.rows!=0,]
> print(summary(res))
Estro10 Estro48 Tiempo-1 23 99 270 12512 12375 125911 90 151 7
Un diagrama de Venn permite visualizar la tabla anterior sin diferenciarentre genes “up” o “down” regulados.
14

> vennDiagram (res.selected[,1:3], main="Genes in common #1", cex=0.9)
Genes in common #1
Estro10 Estro48
Tiempo 0
19
169
9
38
3
69
3
Figure 2: Numero de genes seleccionado en cada comparacion
4.4 Visualizacion de los perfiles de expresion
Tras seleccionar los genes diferencialmente expresados podemos visualizar lasexpresiones de cada gen agrupandolas para destacar los genes que se encuentranup o down regulados simultaneamente constituyendo perfiles de expresion.
Hay distintas formas de visualizacion pero aquı tan solo se presenta el usode mapas de color o Heatmaps.
En primer lugar seleccionamos los genes avisualizar: Se toman todos aquellosque han resultado diferencialmente expresados en alguna de las tres compara-ciones.
> probeNames<-rownames(res)
> probeNames.selected<-probeNames[sum.res.rows!=0]
> exprs2cluster <-exprs(eset_rma)[probeNames.selected,]
Para representar el Heatmap tan solo necesitamos la matriz de datos resul-tante.
> color.map <- function(horas) { if (horas< 20) "yellow" else "red" }
> grupColors <- unlist(lapply(pData(eset_rma)$time.h, color.map))
> heatmap(exprs2cluster, col=rainbow(100), ColSideColors=grupColors, cexCol=0.9)
15

low
48B
low
48A
low
10B
low
10A
hi48
B
hi48
A
hi10
B
hi10
A
AFFX−CreX−5_atAFFX−CreX−3_at31798_at39755_at36165_at39781_at32272_at33322_i_at40891_f_at32843_s_at319_g_at956_at769_s_at286_at36780_at31444_s_atAFFX−BioDn−3_at32609_at39967_at38254_at33308_at35828_at2047_s_at35276_at37043_at40898_at37383_f_at1395_at35485_at425_at34780_at39542_at31830_s_at41386_i_at1035_g_at36958_at39420_at1034_at1700_at37669_s_at38700_at39738_at1358_s_at34374_g_at38551_at36681_at1890_at38882_r_at2031_s_at38966_at37238_s_at893_at38720_at947_at239_at40215_at32174_at32186_at894_g_at39059_at38729_at1651_at33821_at39756_g_at32590_at1470_at1521_at41485_at1737_s_at40874_at33266_at41696_at40195_at1973_s_at34678_at37585_at40690_at673_at40619_at39073_at32536_at33371_s_at40078_at34851_at1158_s_at39631_at480_at592_at36123_at38827_at1544_at38875_r_at37485_at1126_s_at37576_at40726_at1536_at35249_at160043_at37899_at32441_at32027_at2036_s_at34717_s_at904_s_at418_at37263_at38501_s_at1474_s_at33324_s_at34363_at36396_at39637_at37302_at1610_s_at975_at1178_at39269_at39581_at2004_at35936_g_at35059_at1125_s_at861_g_at1993_s_at35407_at35803_at40855_at32239_at881_at892_at37821_at35224_at38116_at910_at41400_at35312_at40117_at1516_g_at41583_at38863_at37913_at1824_s_at33901_at37228_at32702_at430_at35141_at1462_s_at38350_f_at38414_at39109_at40412_at35723_at37739_at348_at38158_at651_at1782_s_at36499_at38368_at35995_at38065_at39337_at40407_at37242_at39092_at1854_at1884_s_at39353_at674_g_at37985_at37686_s_at37347_at1775_at1505_at1515_at1376_at36813_at36833_at1803_atAFFX−BioB−3_atAFFX−BioB−5_atAFFX−BioDn−5_atAFFX−BioC−3_atAFFX−BioB−M_atAFFX−BioC−5_at38814_at39397_at33899_at32043_at37014_at32597_at1197_at40071_at40767_at846_s_at37294_at35977_at40621_at40079_at33730_at37026_at32448_at35361_at757_at38064_at40314_at38584_at1005_at36890_at38881_i_at32001_s_at266_s_at32786_at40425_at39837_s_at634_atAFFX−HUMISGF3A/M97935_MB_at34512_at38735_at35597_at36634_at41439_at33878_at37331_g_at36617_at38653_at1687_s_at33900_at33218_at40249_at37028_at40841_at1842_at2049_s_at31792_at40759_at1913_at41742_s_at37287_at36927_at40290_f_at32355_at39248_at36767_at38261_at35057_at1087_at34624_at33377_at38944_at1433_g_at34863_s_at32901_s_at33802_at39677_at40145_at32263_at1599_at1943_at38422_s_at349_g_at32589_at36922_at653_at262_at41719_i_at34790_at40348_s_at36837_at36374_at34238_at35437_at35227_at35314_at543_g_at1738_at1670_at39715_at37646_at967_g_at34852_g_at1592_at34715_at40697_at1476_s_at685_f_at38131_at39651_at39642_at33252_at1823_g_at1053_at982_at32791_at981_at41490_at41480_at34563_at37157_at34736_at1945_at36134_at1801_at2042_s_at40533_at37458_at33255_at41569_at
Si se desea realizar mapas de color mas sofisticados puede utilizarse el pa-quete gplots que implementa una version mejorada en la funcion heatmap.2
> color.map <- function(horas) { if (horas< 20) "yellow" else "red" }
> grupColors <- unlist(lapply(pData(eset_rma)$time.h, color.map))
> require("gplots")
> heatmap.2(exprs2cluster,
+ col=bluered(75), scale="row",
+ ColSideColors=grupColors, key=TRUE, symkey=FALSE,
+ density.info="none", trace="none", cexCol=1)
16
low
48B
low
48A
low
10B
low
10A
hi48
B
hi48
A
hi10
B
hi10
A
AFFX−CreX−5_atAFFX−CreX−3_at31798_at39755_at36165_at39781_at32272_at33322_i_at40891_f_at32843_s_at319_g_at956_at769_s_at286_at36780_at31444_s_atAFFX−BioDn−3_at32609_at39967_at38254_at33308_at35828_at2047_s_at35276_at37043_at40898_at37383_f_at1395_at35485_at425_at34780_at39542_at31830_s_at41386_i_at1035_g_at36958_at39420_at1034_at1700_at37669_s_at38700_at39738_at1358_s_at34374_g_at38551_at36681_at1890_at38882_r_at2031_s_at38966_at37238_s_at893_at38720_at947_at239_at40215_at32174_at32186_at894_g_at39059_at38729_at1651_at33821_at39756_g_at32590_at1470_at1521_at41485_at1737_s_at40874_at33266_at41696_at40195_at1973_s_at34678_at37585_at40690_at673_at40619_at39073_at32536_at33371_s_at40078_at34851_at1158_s_at39631_at480_at592_at36123_at38827_at1544_at38875_r_at37485_at1126_s_at37576_at40726_at1536_at35249_at160043_at37899_at32441_at32027_at2036_s_at34717_s_at904_s_at418_at37263_at38501_s_at1474_s_at33324_s_at34363_at36396_at39637_at37302_at1610_s_at975_at1178_at39269_at39581_at2004_at35936_g_at35059_at1125_s_at861_g_at1993_s_at35407_at35803_at40855_at32239_at881_at892_at37821_at35224_at38116_at910_at41400_at35312_at40117_at1516_g_at41583_at38863_at37913_at1824_s_at33901_at37228_at32702_at430_at35141_at1462_s_at38350_f_at38414_at39109_at40412_at35723_at37739_at348_at38158_at651_at1782_s_at36499_at38368_at35995_at38065_at39337_at40407_at37242_at39092_at1854_at1884_s_at39353_at674_g_at37985_at37686_s_at37347_at1775_at1505_at1515_at1376_at36813_at36833_at1803_atAFFX−BioB−3_atAFFX−BioB−5_atAFFX−BioDn−5_atAFFX−BioC−3_atAFFX−BioB−M_atAFFX−BioC−5_at38814_at39397_at33899_at32043_at37014_at32597_at1197_at40071_at40767_at846_s_at37294_at35977_at40621_at40079_at33730_at37026_at32448_at35361_at757_at38064_at40314_at38584_at1005_at36890_at38881_i_at32001_s_at266_s_at32786_at40425_at39837_s_at634_atAFFX−HUMISGF3A/M97935_MB_at34512_at38735_at35597_at36634_at41439_at33878_at37331_g_at36617_at38653_at1687_s_at33900_at33218_at40249_at37028_at40841_at1842_at2049_s_at31792_at40759_at1913_at41742_s_at37287_at36927_at40290_f_at32355_at39248_at36767_at38261_at35057_at1087_at34624_at33377_at38944_at1433_g_at34863_s_at32901_s_at33802_at39677_at40145_at32263_at1599_at1943_at38422_s_at349_g_at32589_at36922_at653_at262_at41719_i_at34790_at40348_s_at36837_at36374_at34238_at35437_at35227_at35314_at543_g_at1738_at1670_at39715_at37646_at967_g_at34852_g_at1592_at34715_at40697_at1476_s_at685_f_at38131_at39651_at39642_at33252_at1823_g_at1053_at982_at32791_at981_at41490_at41480_at34563_at37157_at34736_at1945_at36134_at1801_at2042_s_at40533_at37458_at33255_at41569_at
−1 0 1Row Z−Score
Color Key
References
[1] Y. Benjamini and Y. Hochberg. Controlling the false discovery rate: Apractical and powerful approach to multiple testing. Journal of the RoyalStatistical Society B, 57:289–300, 1995.
17














![Matriz de Leopol[1]](https://static.fdocuments.in/doc/165x107/5695d1261a28ab9b02955a40/matriz-de-leopol1.jpg)



