![David Carlson: Alpha Lipoic Acid - Silicon Valley Health Institute · 2016. 4. 17. · researching R-lipoic acid products. Although many of us say [inaudible 00:01:08] are familiar](https://static.fdocuments.in/doc/165x107/60a429feb9fbbf346950a043/david-carlson-alpha-lipoic-acid-silicon-valley-health-2016-4-17-researching.jpg)
Effect of Long-Term Administration of -Lipoic Acid on ... of Lipoic Acid... · Address...
36
Effect of Long-Term Administration of -Lipoic Acid on Retinal Capillary Cell Death and the Development of Retinopathy in Diabetic Rats Renu A. Kowluru and Sarah Odenbach Oxidative stress is increased in the retina in diabetes, and it is considered to play an important role in the develop- ment of retinopathy. -Lipoic acid, a thiol antioxidant, has been shown to have beneficial effects on polyneuropathy and on the parameters of oxidative stress in various tissues, including nerve, kidney, and retina. The purpose of this study was to examine the effect of -lipoic acid on retinal capillary cell apoptosis and the development of pathology in diabetes. Retina was used from streptozoto- cin-induced diabetic rats receiving diets supplemented with or without -lipoic acid (400 mg/kg) for 11 months of diabetes. Capillary cell apoptosis (by terminal trans- ferase-mediated dUTP nick-end labeling) and formation of acellular capillaries were investigated in the trypsin-di- gested retinal microvessels. The effect of -lipoic acid administration on retinal 8-hydroxy-2deoxyguanosine (8- OHdG) and nitrotyrosine levels was determined by enzyme-linked immunosorbent assay. -Lipoic acid admin- istration for the entire duration of diabetes inhibited capillary cell apoptosis and the number of acellular capil- laries in the retina, despite similar severity of hyperglyce- mia in the two diabetic groups (with and without -lipoic acid). Retinal 8-OHdG and nitrotyrosine levels were in- creased by over twofold and 70%, respectively, in diabe- tes, and -lipoic acid administration inhibited these increases. Our results demonstrate that the long-term administration of -lipoic acid has beneficial effects on the development of diabetic retinopathy via inhibition of ac- cumulation of oxidatively modified DNA and nitrotyrosine in the retina. -Lipoic acid supplementation represents an achievable adjunct therapy to help prevent vision loss in diabetic patients. Diabetes 53:3233–3238, 2004 D iabetic retinopathy is the leading cause of ac- quired blindness among young adults, and stud- ies have shown that hyperglycemia per se initiates its development (1). Diabetes increases oxidative stress, which plays a key regulatory role in the development of its complications (2– 4). Reactive oxygen species generated by high glucose are considered as a causal link between elevated glucose and the other meta- bolic abnormalities important in the development of dia- betic complications (5). In diabetes, mitochondria in the retina experience dysfunction (6), and the therapies that inhibit superoxide production in the retina also inhibit the development of diabetic retinopathy (7). However, the mechanism by which oxidative stress can contribute to the development of retinopathy in diabetes remains to be elucidated. In the pathogenesis of retinopathy in diabetes, retinal microvascular endothelial, Muller, and ganglion cells and pericytes are lost selectively via apoptosis before other histopathology is detectable, or loss of vision is evident (8,9). The detection of terminal transferase-mediated dUTP nick-end labeling (TUNEL)-positive cells in animal models of diabetic retinopathy is shown to serve as a surrogate end point to screen efficacy of interventions to inhibit the development of diabetic retinopathy. Administration of antioxidants to diabetic rats prevents the development of retinopathy and also retinal metabolic abnormalities postulated to be involved in the develop- ment of retinopathy (3,10). Vitamin E supplementation reduces the retinal hemodynamic abnormalities seen in diabetic patients (11), and pyridoxamine inhibits the for- mation of diabetes-induced retinal acellular strands in rats (12). By contrast, some studies have failed to show any effects of antioxidants on retinal vascular lesions (13), and the differences for such discrepancies are not clear. The antioxidant therapy that inhibits the development of reti- nopathy in diabetic rats inhibits the activation of nuclear transcriptional factor-B (NF-B) and the apoptosis exe- cution enzyme caspase-3 (14,15), suggesting that the ben- eficial effects of antioxidants on the development of diabetic retinopathy might involve inhibition of activation of NF-B and caspase-3. -Lipoic acid, a disulfide derivative of octanoic acid, can alter the redox status of cells and interact with thiols and other antioxidants (16). Administration of -lipoic acid to diabetic rats has been shown to inhibit parameters of oxidative stress in various organs of diabetic rats, includ- ing kidney, nerve, and retina (10). -Lipoic acid is also shown to inhibit diabetes-induced activation of a small– molecular weight G-protein in the retina (H-Ras), in- creased levels of vascular endothelial growth factor, and leukostaisis (10,17,18), and these abnormalities have been postulated to play roles in the pathogenesis of retinopathy in diabetes. However, the effects of -lipoic acid on retinal capillary cell apoptosis and the development of retinopa- thy are not known. From the Kresge Eye Institute, Wayne State University, Detroit, Michigan. Address correspondence and reprint requests to Renu A. Kowluru, PhD, Kresge Eye Institute, Wayne State University, 4717 St. Antoine, Detroit, MI 48201. E-mail: [email protected]. Received for publication 24 June 2004 and accepted in revised form 24 August 2004. ELISA, enzyme-linked immunosorbent assay; NF-B, nuclear transcrip- tional factor-B; 8-OHdG, 8-hydroxy-2deoxyguanosine; TUNEL, transferase- mediated dUTP nick-end labeling. © 2004 by the American Diabetes Association. DIABETES, VOL. 53, DECEMBER 2004 3233
Transcript of Effect of Long-Term Administration of -Lipoic Acid on ... of Lipoic Acid... · Address...

Effect of Long-Term Administration of �-Lipoic Acid onRetinal Capillary Cell Death and the Development ofRetinopathy in Diabetic RatsRenu A. Kowluru and Sarah Odenbach
Oxidative stress is increased in the retina in diabetes, andit is considered to play an important role in the develop-ment of retinopathy. �-Lipoic acid, a thiol antioxidant, hasbeen shown to have beneficial effects on polyneuropathyand on the parameters of oxidative stress in varioustissues, including nerve, kidney, and retina. The purposeof this study was to examine the effect of �-lipoic acid onretinal capillary cell apoptosis and the development ofpathology in diabetes. Retina was used from streptozoto-cin-induced diabetic rats receiving diets supplementedwith or without �-lipoic acid (400 mg/kg) for 11 months ofdiabetes. Capillary cell apoptosis (by terminal trans-ferase-mediated dUTP nick-end labeling) and formation ofacellular capillaries were investigated in the trypsin-di-gested retinal microvessels. The effect of �-lipoic acidadministration on retinal 8-hydroxy-2�deoxyguanosine (8-OHdG) and nitrotyrosine levels was determined byenzyme-linked immunosorbent assay. �-Lipoic acid admin-istration for the entire duration of diabetes inhibitedcapillary cell apoptosis and the number of acellular capil-laries in the retina, despite similar severity of hyperglyce-mia in the two diabetic groups (with and without �-lipoicacid). Retinal 8-OHdG and nitrotyrosine levels were in-creased by over twofold and 70%, respectively, in diabe-tes, and �-lipoic acid administration inhibited theseincreases. Our results demonstrate that the long-termadministration of �-lipoic acid has beneficial effects on thedevelopment of diabetic retinopathy via inhibition of ac-cumulation of oxidatively modified DNA and nitrotyrosinein the retina. �-Lipoic acid supplementation represents anachievable adjunct therapy to help prevent vision loss indiabetic patients. Diabetes 53:3233–3238, 2004
Diabetic retinopathy is the leading cause of ac-quired blindness among young adults, and stud-ies have shown that hyperglycemia per seinitiates its development (1). Diabetes increases
oxidative stress, which plays a key regulatory role in thedevelopment of its complications (2–4). Reactive oxygenspecies generated by high glucose are considered as acausal link between elevated glucose and the other meta-
bolic abnormalities important in the development of dia-betic complications (5). In diabetes, mitochondria in theretina experience dysfunction (6), and the therapies thatinhibit superoxide production in the retina also inhibit thedevelopment of diabetic retinopathy (7). However, themechanism by which oxidative stress can contribute to thedevelopment of retinopathy in diabetes remains to beelucidated.
In the pathogenesis of retinopathy in diabetes, retinalmicrovascular endothelial, Muller, and ganglion cells andpericytes are lost selectively via apoptosis before otherhistopathology is detectable, or loss of vision is evident(8,9). The detection of terminal transferase-mediateddUTP nick-end labeling (TUNEL)-positive cells in animalmodels of diabetic retinopathy is shown to serve as asurrogate end point to screen efficacy of interventions toinhibit the development of diabetic retinopathy.
Administration of antioxidants to diabetic rats preventsthe development of retinopathy and also retinal metabolicabnormalities postulated to be involved in the develop-ment of retinopathy (3,10). Vitamin E supplementationreduces the retinal hemodynamic abnormalities seen indiabetic patients (11), and pyridoxamine inhibits the for-mation of diabetes-induced retinal acellular strands in rats(12). By contrast, some studies have failed to show anyeffects of antioxidants on retinal vascular lesions (13), andthe differences for such discrepancies are not clear. Theantioxidant therapy that inhibits the development of reti-nopathy in diabetic rats inhibits the activation of nucleartranscriptional factor-�B (NF-�B) and the apoptosis exe-cution enzyme caspase-3 (14,15), suggesting that the ben-eficial effects of antioxidants on the development ofdiabetic retinopathy might involve inhibition of activationof NF-�B and caspase-3.
�-Lipoic acid, a disulfide derivative of octanoic acid, canalter the redox status of cells and interact with thiols andother antioxidants (16). Administration of �-lipoic acid todiabetic rats has been shown to inhibit parameters ofoxidative stress in various organs of diabetic rats, includ-ing kidney, nerve, and retina (10). �-Lipoic acid is alsoshown to inhibit diabetes-induced activation of a small–molecular weight G-protein in the retina (H-Ras), in-creased levels of vascular endothelial growth factor, andleukostaisis (10,17,18), and these abnormalities have beenpostulated to play roles in the pathogenesis of retinopathyin diabetes. However, the effects of �-lipoic acid on retinalcapillary cell apoptosis and the development of retinopa-thy are not known.
From the Kresge Eye Institute, Wayne State University, Detroit, Michigan.Address correspondence and reprint requests to Renu A. Kowluru, PhD,
Kresge Eye Institute, Wayne State University, 4717 St. Antoine, Detroit, MI48201. E-mail: [email protected].
Received for publication 24 June 2004 and accepted in revised form 24August 2004.
ELISA, enzyme-linked immunosorbent assay; NF-�B, nuclear transcrip-tional factor-�B; 8-OHdG, 8-hydroxy-2�deoxyguanosine; TUNEL, transferase-mediated dUTP nick-end labeling.
© 2004 by the American Diabetes Association.
DIABETES, VOL. 53, DECEMBER 2004 3233

In the present study, we investigated the effect oflong-term administration of �-lipoic acid on retinal capil-lary cell apoptosis and the development of retinal capillarylesions in the animal model of the early stages of diabeticretinopathy, the streptozotocin-induced diabetic rat. Tohelp interpret the effects of this dietary supplementationon retinal capillary cell apoptosis and histopathology, wehave investigated the effect of �-lipoic acid on oxidativelymodified DNA and nitrative stress in the retina.
RESEARCH DESIGN AND METHODS
Wistar rats (200–220 g, male) were made diabetic by intraperitoneal injectionof streptozotocin (55 mg/kg body wt). Insulin was administered to diabeticrats to allow slow weight gain while maintaining hyperglycemia (bloodglucose levels of 20–25 mmol/l). Age-matched normal rats served as control.Diabetic rats were divided into two groups: the rats in group 1 receivedpowder diet (Purina 5001) supplemented with �-lipoic acid (400 mg/kg), andin group 2 the diet was without any supplementation; these diets wereinitiated soon after establishment of diabetes (3–4 days after administration ofstreptozotocin). Each group had 12–15 rats, and the entire colony of rats(normal, diabetic, and diabetic with �-lipoic acid diet) received fresh powderdiet weekly. The rats were weighed two times a week, and their foodconsumption was measured once every week to calculate the amount of�-lipoic acid consumed. GHb was measured at 2 months of diabetes, and every3 months thereafter, using affinity columns (kit 442-B; Sigma Chemicals) (17).At the end of the experiment (11 months’ duration), the rats were killed byoverdose of pentobarbital, and the eyes were removed. The eyes were eithersuspended in 10% formalin to prepare trypsin-digested microvessels, or theretina was removed immediately and frozen in liquid nitrogen for biochemicalmeasurements. Treatment of the animals conformed to the National Instituteof Health principals of laboratory animal care, the Association for Research inVision and Ophthalmology resolution on the use of animals in research, andthe institutional guidelines.Capillary cell apoptosis and histopathology in retinal vessels. Theretinas were removed from the eyes, which were fixed in 10% formalin for 2–3days, and digested with 3% crude trypsin in Tris-HCl buffer (pH 7.8) containing0.2 mol/l sodium fluoride for 90 min to isolate the microvessels (3,8).
Apoptosis was determined by evaluating the trypsin-digested preparationsof retinal vessels for TUNEL-positive cells using a commercially available kit(in situ cell death kit; Roche Molecular Biochemicals, Indianapolis, IN) aspreviously reported by us (8,9). The slides containing retinal vessels wererehydrated in PBS and permeabilized with 0.25% Triton X-100 in PBS for 1 hat room temperature. The slides were mounted in Vectashield (VectorLaboratories, Burlingame, CA) after incubation with terminal deoxynucleoti-dyl transferase to add deoxynucleotide to the free 3�-OH end of DNA breaks,which is characteristic of apoptotic cell death. In each experiment the positivecontrol was run by exposing the retinal vessels to DNase (2,000 units/ml in 20mmol/l Tris-HCl, pH 7.5) for 10 min at room temperature before initiation ofthe TUNEL reaction. TUNEL-positive cells were identified in a maskedfashion; each trypsin digest was surveyed systematically under a ZeissAxiophot photomicroscope by scanning the specimen with downward andupward motion beginning at the upper left margin (8,9).
After TUNEL staining, the vessel preparations were stained with periodicacid-Schiff and hematoxylin for histologic evaluation. The number of acellularcapillaries was counted in multiple mid-retinal fields (one field adjacent toeach of the five to seven retinal arterioles radiating out from the optic disc)and expressed per millimeter squared of retinal area examined (3,9).
Oxidative stress. Oxidative stress was measured in the retina by quantifyingthe levels of oxidatively modified DNA (8-hydroxy-2�deoxyguanosine[8-OHdG]) and the levels of the intracellular antioxidant reduced glutathione.
8-OHdG levels were measured using an enzyme-linked immunosorbentassay (ELISA) kit from Oxis Research (Portland, OR), as described by uspreviously (19). To improve the accuracy and reproducibility of 8-OHdGmeasurement, DNA purified from the retina was digested with DNase (20).The 8-OHdG standard (0.5–40 ng/ml) or 15–20 �g DNA was incubated for 1 hwith monoclonal antibody against 8-OHdG in a microtiter plate precoated with8-OHdG. The final color was developed by the addition of 3,3�5,5�-tetrameth-ylbenzidine, and absorbance was measured at 450 nm (19).
Glutathione was measured using a glutathione assay kit from CaymanChemicals (Ann Arbor, MI) according to the manufacturer’s instructions (19).The retinal sample (50–75 �g) was deproteinized using phosphoric acid, andthe amount of 5-thio-2-nitrobenzoic acid produced was measured in thesupernatant.Nitrotyrosine. Nitrotyrosine levels were quantified by enzyme immunoassayusing a nitrotyrosine enzyme immunosorbent assay kit from Oxis Researchaccording to the manufacturer’s instructions. Nitrotyrosine standard or retinalhomogenates were incubated with nitrotyrosine antibody in the microplate for1 h; this was followed by incubation with streptavidin peroxidase for 1 h. Thesamples were incubated with tetramethylbenzidine substrate for 30 min, andthe reaction was stopped by 2.0 mol/l citric acid. The formation of yellowproduct was measured at 450 nm. The assay was sensitive as low as 0.05 pmolof nitrotyrosine.NF-�B. Activation of NF-�B was determined in the retina by ELISA, using anNF-�B kit from Active Motif (Carlsbad, CA) and following the manufacturer’sinstructions. The assay is based on the principle that only the active form ofNF-�B in the sample binds to oligonucleotide containing the NF-�B consensussite (5�-GGGACTTTCC-3�) that is immobilized on the microtiter plate. Theprimary antibody against the p65 subunit of NF-�B used in the assay systemis accessible only when NF-�B is activated and bound to its target DNA. Fora sensitive colorimetric readout, the secondary antibody used is conjugated tohorseradish peroxidase. Retina was homogenized in the lysis buffer (asprovided by the manufacturer) containing dithiothreitol and protease inhibi-tor, and after removing the cell debris, 8–10 �g protein was used for theELISA.
Experimental groups were compared statistically using the nonparametricKruskal-Wallis test followed by the Mann-Whitney test for multiple groupcomparisons. ANOVA with Fisher or Tukey group comparisons gave similarresults.
RESULTS
Effect of �-lipoic acid on capillary cell apoptosis and
histopathology. The number of TUNEL-positive cells inthe trypsin-digested retinal vessels (Fig. 1) was signifi-cantly higher in diabetic rats than that observed in thevessels from age-matched normal control rats (P � 0.02)(Table 1), thus suggesting increased apoptosis. The totalnumber of nuclei (including pericyte, endothelial cell, andnuclei with undetermined cellular attribution) positive forTUNEL staining was 4.1 � 2.2 in diabetes compared with1.6 � 1.0 in normal control retinal vessels (Table 1).Administration of �-lipoic acid for the entire duration ofdiabetes prevented an increase in TUNEL-positive nuclei(Table 1).
FIG. 1. TUNEL staining in retinal trypsin digest. Trypsin-digested microvessels were stained to detect TUNEL-positive cells using a kit from RocheMolecular Biochemicals. A: The photomicrograph of a retinal trypsin digest prepared from rat diabetic for 11 months. The arrows indicateTUNEL-positive capillary cells. B: A positive control where a trypsin digest from a normal rat retina was incubated with DNase before TUNELstaining.
DIABETIC RETINOPATHY AND �-LIPOIC ACID
3234 DIABETES, VOL. 53, DECEMBER 2004

Microvascular lesions consistent with the early stages ofdiabetic retinopathy were observed in the trypsin-digestedretinal preparation prepared from diabetic rats (Table 1);the number of acellular capillaries was significantlyincreased in diabetes compared with the age-matchednormal control eyes (P � 0.0002). �-Lipoic acid adminis-tration also reduced the number of acellular capillaries indiabetic rats; the number of acellular capillaries in theretina was decreased from 3.4 in the diabetic group to 1.2in the diabetes plus �-lipoic acid group, and the valuesobtained in the diabetes plus �-lipoic acid group were notstatistically different (P � 0.297) from those in the normalcontrol group (Table 1).Retinal oxidative stress. Oxidative stress, as deter-mined by the concentrations of oxidatively modified DNAand intracellular antioxidant, remained elevated in theretina of rats diabetic for 11 months. The levels of 8-OHdGwere elevated by over twofold and the concentration ofglutathione decreased by �40% in the retina of ratsdiabetic for 11 months as compared with those in theretina obtained from age-matched normal control rats(Fig. 2). Long-term administration of �-lipoic acid inhib-ited the diabetes-induced increase in retinal 8-OHdG lev-els; 8-OHdG values were not statistically different in the�-lipoic acid group and the normal control group (P �0.5101). Similarly, the decrease in glutathione levels in the
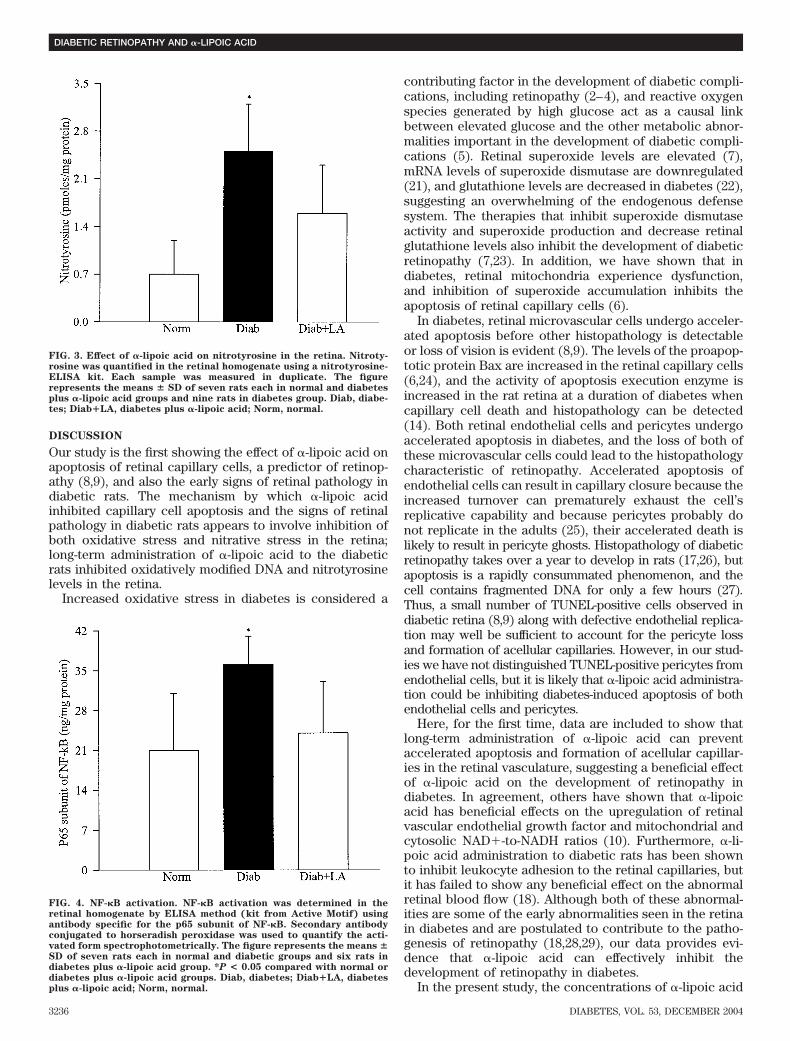
retina seen in diabetes was prevented by administration of�-lipoic acid (Fig. 2).Nitrative stress. In the retina obtained from the ratsdiabetic for 11 months, nitrotyrosine levels remain ele-vated by 70% as compared with those obtained fromage-matched normal rat retina (Fig. 3). In the same retina,NF-�B was also activated by 70% (Fig. 4). Diabetes-induced changes in retinal nitrative stress were inhibitedby administration of �-lipoic acid, and nitrotyrosine levelswere similar in the retina obtained from diabetic ratsreceiving �-lipoic acid and normal control rats. �-Lipoicacid administration also inhibited the activation of NF-�Bobserved in the retina of diabetic rats (Figs. 3 and 4).Severity of hyperglycemia. The severity of hyperglyce-mia, as measured by GHb, body weight, and 24-h urinevolume, was strikingly increased in the diabetic groupcompared with the normal control group. Administrationof �-lipoic acid did not ameliorate the severity of hyper-glycemia in diabetic rats: the values obtained for GHb,body weight, and 24-h urine volumes throughout theduration of the experiment were comparable between thetwo diabetic groups (diabetes and diabetes plus �-lipoicacid) and were significantly different (P � 0.001) from thenormal control group (Table 2).
TABLE 1Effect of administration of �-lipoic acid on capillary cell apoptosis and acellular capillaries in the retina
n TUNEL-positive capillary cells/retina Acellular capillaries/mm2retina
Normal 13 1.6 � 1.0 0.69 � 0.63Diabetes 9 4.1 � 2.2* 3.33 � 1.41*Diabetes plus �-lipoic acid 11 2.1 � 1.4 1.18 � 1.08
TUNEL-positive cells were identified in the trypsin-digested retinal microvessel in a masked fashion by scanning the specimen withdownward and upward motion beginning at the upper left margin. The same microvessel preparations after TUNEL staining were stainedwith periodic acid-Schiff and hematoxylin for histologic evaluation. The number of acellular capillaries was counted in multiple mid-retinalfields. *P � 0.05 vs. normal or diabetes plus �-lipoic acid groups.
FIG. 2. Effect of �-lipoic acid on diabetes-induced oxidative stress in the retina. Oxidative stress was measured by quantifying the levels of8-OHdG using the 8-OHdG ELISA kit and 10–15 �g DNA prepared from the retina. Each measurement was performed in duplicate, and the graphrepresents the means � SD of the values obtained from five normal, six diabetic, and eight diabetic plus �-lipoic acid rats. Glutathione (GSH)was measured in the deproteinized sample of the retina using colorimetric assay kit from Cayman Chemicals. The results are the means � SDobtained from 10 rats in the normal control group and 8 rats each in the diabetes and diabetes plus �-lipoic acid groups. *P < 0.05 compared withnormal or diabetes plus �-lipoic acid groups. Diab, diabetes; Diab�LA, diabetes plus �-lipoic acid; Norm, normal.
R.A. KOWLURU AND S. ODENBACH
DIABETES, VOL. 53, DECEMBER 2004 3235
DISCUSSION
Our study is the first showing the effect of �-lipoic acid onapoptosis of retinal capillary cells, a predictor of retinop-athy (8,9), and also the early signs of retinal pathology indiabetic rats. The mechanism by which �-lipoic acidinhibited capillary cell apoptosis and the signs of retinalpathology in diabetic rats appears to involve inhibition ofboth oxidative stress and nitrative stress in the retina;long-term administration of �-lipoic acid to the diabeticrats inhibited oxidatively modified DNA and nitrotyrosinelevels in the retina.
Increased oxidative stress in diabetes is considered a
contributing factor in the development of diabetic compli-cations, including retinopathy (2–4), and reactive oxygenspecies generated by high glucose act as a causal linkbetween elevated glucose and the other metabolic abnor-malities important in the development of diabetic compli-cations (5). Retinal superoxide levels are elevated (7),mRNA levels of superoxide dismutase are downregulated(21), and glutathione levels are decreased in diabetes (22),suggesting an overwhelming of the endogenous defensesystem. The therapies that inhibit superoxide dismutaseactivity and superoxide production and decrease retinalglutathione levels also inhibit the development of diabeticretinopathy (7,23). In addition, we have shown that indiabetes, retinal mitochondria experience dysfunction,and inhibition of superoxide accumulation inhibits theapoptosis of retinal capillary cells (6).
In diabetes, retinal microvascular cells undergo acceler-ated apoptosis before other histopathology is detectableor loss of vision is evident (8,9). The levels of the proapop-totic protein Bax are increased in the retinal capillary cells(6,24), and the activity of apoptosis execution enzyme isincreased in the rat retina at a duration of diabetes whencapillary cell death and histopathology can be detected(14). Both retinal endothelial cells and pericytes undergoaccelerated apoptosis in diabetes, and the loss of both ofthese microvascular cells could lead to the histopathologycharacteristic of retinopathy. Accelerated apoptosis ofendothelial cells can result in capillary closure because theincreased turnover can prematurely exhaust the cell’sreplicative capability and because pericytes probably donot replicate in the adults (25), their accelerated death islikely to result in pericyte ghosts. Histopathology of diabeticretinopathy takes over a year to develop in rats (17,26), butapoptosis is a rapidly consummated phenomenon, and thecell contains fragmented DNA for only a few hours (27).Thus, a small number of TUNEL-positive cells observed indiabetic retina (8,9) along with defective endothelial replica-tion may well be sufficient to account for the pericyte lossand formation of acellular capillaries. However, in our stud-ies we have not distinguished TUNEL-positive pericytes fromendothelial cells, but it is likely that �-lipoic acid administra-tion could be inhibiting diabetes-induced apoptosis of bothendothelial cells and pericytes.
Here, for the first time, data are included to show thatlong-term administration of �-lipoic acid can preventaccelerated apoptosis and formation of acellular capillar-ies in the retinal vasculature, suggesting a beneficial effectof �-lipoic acid on the development of retinopathy indiabetes. In agreement, others have shown that �-lipoicacid has beneficial effects on the upregulation of retinalvascular endothelial growth factor and mitochondrial andcytosolic NAD-to-NADH ratios (10). Furthermore, �-li-poic acid administration to diabetic rats has been shownto inhibit leukocyte adhesion to the retinal capillaries, butit has failed to show any beneficial effect on the abnormalretinal blood flow (18). Although both of these abnormal-ities are some of the early abnormalities seen in the retinain diabetes and are postulated to contribute to the patho-genesis of retinopathy (18,28,29), our data provides evi-dence that �-lipoic acid can effectively inhibit thedevelopment of retinopathy in diabetes.
In the present study, the concentrations of �-lipoic acid
FIG. 4. NF-�B activation. NF-�B activation was determined in theretinal homogenate by ELISA method (kit from Active Motif) usingantibody specific for the p65 subunit of NF-�B. Secondary antibodyconjugated to horseradish peroxidase was used to quantify the acti-vated form spectrophotometrically. The figure represents the means �SD of seven rats each in normal and diabetic groups and six rats indiabetes plus �-lipoic acid group. *P < 0.05 compared with normal ordiabetes plus �-lipoic acid groups. Diab, diabetes; Diab�LA, diabetesplus �-lipoic acid; Norm, normal.
FIG. 3. Effect of �-lipoic acid on nitrotyrosine in the retina. Nitroty-rosine was quantified in the retinal homogenate using a nitrotyrosine-ELISA kit. Each sample was measured in duplicate. The figurerepresents the means � SD of seven rats each in normal and diabetesplus �-lipoic acid groups and nine rats in diabetes group. Diab, diabe-tes; Diab�LA, diabetes plus �-lipoic acid; Norm, normal.
DIABETIC RETINOPATHY AND �-LIPOIC ACID
3236 DIABETES, VOL. 53, DECEMBER 2004

that inhibited microvascular apoptosis and pathology inthe retina in diabetes prevented an increase in oxidativestress in the retina. The levels of retinal cytosolic antiox-idant glutathione were similar in the diabetes plus �-lipoicacid and age-matched normal control groups. The concen-tration of retinal glutathione that we have obtained in thepresent study (including that in normal and diabetesgroups) is significantly lower than our previous reports(22,30); however, the results are consistent and show thatthe amount of glutathione is significantly decreased indiabetes. Although we cannot pinpoint the reasons forsuch discrepancies in the absolute numbers, here we haveused a colorimetric assay based on the enzymatic recy-cling that uses 5,5�-dithio-bis-2-nitrobenzoic acid, and theprevious measurements were performed fluorometricallyusing o-phthalaldehyde. Because �-lipoic acid is a power-ful free radical scavenger that can directly chelate metalions and regenerate cytosolic antioxidants, the beneficialeffects of �-lipoic acid on the apoptosis of retinal capillarycells and histopathology seen in this study could includeboth scavenging of free radicals and increasing glutathi-one. In support, others have reported beneficial effects of�-lipoic acid on oxidative stress in diabetes, including adecrease in mitochondrial and cytosolic NAD/NADH,malondialdehyde plus 4-hydroxyalkenal concentrations inthe retina, and glutathione levels in the kidney (10,31,32).
Increased levels of 8-OHdG are reported in the leukocytesof patients with idiopathic retinal inflammatory disease (33),and our data show that diabetic retinopathy, a disease thatshares many similarities with a chronic inflammatory disease(34,35), also has increased oxidatively modified DNA in theretina. Higher levels of 8-OHdG are observed in diabetes incardiomyocytes, kidney, and urine (19,36,37). Although �-li-poic acid is shown to terminate free radicals and chelatetransition metal ions (16,38), we believe this is the first reportshowing that �-lipoic acid can effectively inhibit the accumu-lation of oxidatively modified DNA; the diabetes-inducedincrease in retinal 8-OHdG was inhibited by �-lipoic acidadministration. This suggests that oxidative modification ofDNA might be playing an important role in the pathogenesisof retinopathy in diabetes.
Our studies and those of others have shown that theretina experiences increased nitrative stress in diabetes:the levels of peroxynitrite (formed by the reaction be-tween NO and superoxides) (39) are elevated and NF-�B isactivated in diabetes, and they remain elevated whencapillary cell apoptosis and pathology can be seen in theretina of diabetic rats (15,24,40). The activation of NF-�Bin the present study was demonstrated by using theantibody against its p65 subunit; this is because theexpression of p65 subunit is increased in the retinalmicrovasculature obtained from diabetic patients and in
the isolated retinal capillary cells incubated in high glu-cose medium (15,24). In addition, Podesta et al. (24) haveshown that in bovine retinal pericytes, only p65 antibody,but not p50 antibody, inhibits electrophoretic migration ofthe NF-�B complex, and this activation is consideredresponsible for the hyperglycemia-induced acceleratedloss of pericytes observed in diabetic retinopathy.
Nitrative modifications in retina that are formed early inthe course of development of retinopathy in diabetesappear to contribute to the progression of retinopathyafter reinstitution of good glycemic control (22). Thetherapies that inhibit the activation of apoptosis executionenzyme development of retinopathy in diabetic rats inhibitNO and nitrotyrosine levels and NF-�B activation in theretina (3,14,15). Here we provide data showing that theconcentration of �-lipoic acid that inhibits capillary cellapoptosis and pathology in the retina of diabetic rats alsoprevents the increase in diabetes-induced nitrative stressin the retina. Nitration of proteins is considered to play arole in the apoptosis of retinal cells; it can disrupt proteinassembly and functions, with possible pathological conse-quences, and results in oxidation of protein sulfhydryls(41–43). Our results show that �-lipoic acid inhibits thediabetes-induced increase in nitrotyrosine and capillarycell apoptosis in the retina. �-Lipoic acid treatment, bypreventing the formation of nitrotyrosine, has been shownto protect the endothelial NO system of the mesentericvasculature and the nitrergic innervation of corpus caver-nosum in diabetic rats (43). In addition, �-lipoic acidreduces the advanced glycation end product–induced ac-tivation of NF-�B by the reduction of oxidative stress inthe cell (44) via inhibition of the release and translocationof NF-�B from the cytoplasm into the nucleus.
�-Lipoic acid inhibits capillary cell apoptosis and pathol-ogy in the retina, despite similar severity of hyperglycemiain diabetic rats receiving diets supplemented with andwithout �-lipoic acid. Although �-lipoic acid has beenshown to stimulate glucose uptake into fat cells by acti-vating the insulin signaling pathway (45), our results showthat the beneficial effects of �-lipoic acid seen in thepresent study are not caused by amelioration of bloodglucose levels, because the levels of GHb, a parameter oflong-term blood glucose, were not different in diabetes anddiabetes plus �-lipoic acid groups.
In summary, long-term administration of �-lipoic acidcan inhibit the apoptosis of retinal capillary cells and thedevelopment of the early stages of diabetic retinopathy.The mechanism by which �-lipoic acid inhibits retinopathypossibly involves inhibition of both oxidative damage toDNA and nitrative stress in the retina. Thus, supplemen-tation with �-lipoic acid represents an achievable adjuncttherapy to help prevent loss of vision in diabetic patients.
TABLE 2Effect of �-lipoic acid on the severity of hyperglycemia in rats
GHb (%) Body weight (g) Urine volume (ml/24 h)
Normal 4.9 � 0.8 507 � 45 13 � 6Diabetes 12.7 � 1.2* 296 � 34* 122 � 25*Diabetes plus �-lipoic acid 11.5 � 2.1*† 274 � 27*† 95 � 21*†
Data are means � SD of seven rats each in normal group and eight rats each in diabetes and diabetes plus �-lipoic acid groups. GHb and24-h urine excretion (measured over 2–3 consecutive days) were quantified at 8 weeks of diabetes, and the process was repeated every 3months thereafter. *P � 0.02 vs. normal; †P 0.02 vs. diabetes.
R.A. KOWLURU AND S. ODENBACH
DIABETES, VOL. 53, DECEMBER 2004 3237

ACKNOWLEDGMENTS
This study was supported in part by grants from theJuvenile Diabetes Research Foundation, the ThomasFoundation, and Research to Prevent Blindness.
We thank Saiyeda Noor Abbas for technical assistance.
REFERENCES
1. Engerman RL, Kern TS: Experimental galactosemia produces diabetic-likeretinopathy. Diabetes 33:97–100, 1984
2. Baynes JW, Thrope SR: Role of oxidative stress in diabetic complications:a new perspective on an old paradigm. Diabetes 48:1–9, 1999
3. Kowluru RA, Tang J, Kern TS: Abnormalities of retinal metabolism indiabetes and experimental galactosemia. VII. Effect of long-term adminis-tration of antioxidants on the development of retinopathy. Diabetes
50:1938–1942, 20014. Haskins K, Bradley B, Powers K, Fadok V, Flores S, Ling X, Pugazhenthi S,
Reusch J, Kench J: Oxidative stress in type 1 diabetes. Ann N Y Acad Sci
1005:43–54, 20035. Brownlee M: Biochemistry and molecular cell biology of diabetic compli-
cations. Nature 414:813–820, 20016. Kowluru RA, Abbas SN: Diabetes-induced mitochondrial dysfunction in
the retina. Inves Ophthal Vis Sci 44:5327–5334, 20037. Du Y, Miller CM, Kern TS: Hyperglycemia increases mitochondrial super-
oxide in retina and retinal cells. Free Rad Biol Med 35:1491–1499, 20038. Mizutani M, Kern TS, Lorenzi M: Accelerated death of retinal microvascu-
lar cells in human and experimental diabetic retinopathy. J Clin Invest
97:2883–2890, 19969. Kern TS, Tang J, Mizutani M, Kowluru R, Nagraj R, Lorenzi M: Response of
capillary cell death to aminoguanidine predicts the development of reti-nopathy: comparison of diabetes and galactosemia. Invest Ophthalmol Vis
Sci 41:3972–3978, 200010. Obrosova IG, Minchenko AG, Marinescu V, Fathallah L, Kennedy A,
Stockert CM, Frank RN, Stevens MJ: Antioxidants attenuate early upregulation of retinal vascular endothelial growth factor in streptozotocin-diabetic rats. Diabetologia 44:1102–1110, 2001
11. Bursell SE, Clermont AC, Aiello LP, Aiello LM, Schlossman DK, Feener EP,Laffel L, King GL: High-dose vitamin E supplementation normalizes retinalblood flow and creatinine clearance in patients with type 1 diabetes.Diabetes Care 22:1245–1251, 1999
12. Stitt A, Gardiner TA, Anderson NL, Canning P, Frizzell N, Duffy N, Boyle C,Januszewski AS, Chachich M, Baynes JW, Thorpe SR: The AGE inhibitorpyridoxamine inhibits development of retinopathy in experimental diabe-tes. Diabetes 51:2826–2832, 2002, Erratum in: Diabetes 2003 52:223
13. Robison WG, Jacot JL, Katz ML, Glover JP: Retinal vascular changesinduced by the oxidative stress of alpha-tocopherol deficiency contrastedwith diabetic microangiopathy. J Ocul Pharmacol Ther 16:109–120, 2000
14. Kowluru RA, Koppolu P: Diabetes-induced activation of caspase-3 inretina: effect of antioxidant therapy. Free Radic Res 36:993–999, 2002
15. Kowluru RA, Koppolu P, Chakrabarti S, Chen S: Diabetes-induced activa-tion of nuclear transcriptional factor in the retina, and its inhibition byantioxidants. Free Radic Res 37:1169–1180, 2003
16. Packer L, Kraemer K, Rimbach G: Molecular aspects of lipoic acid in theprevention of diabetes complications. Nutrition 17:888–895, 2001
17. Kowluru RA, Kowluru A, Chakrabarti S, Khan Z: Potential contributoryrole of H-Ras, a small G-protein, in the development of retinopathy indiabetic rats. Diabetes 53:775–783, 2004
18. Abiko T, Abiko A, Clermont AC, Shoelson B, Horio N, Takahashi J, AdamisAP, King GL, Bursell SE: Characterization of retinal leukostasis andhemodynamics in insulin resistance and diabetes: role of oxidants andprotein kinase-C activation. Diabetes 52:829–837, 2003
19. Kowluru RA, Abbas SN, Odenbach S: Reversal of hyperglycemia anddiabetic nephropathy: effect of reinstitution of good metabolic control onoxidative stress in the kidney of diabetic rats. J Diabetes Complications
18:282–288, 200420. Huang X, Powell J, Mooney LA, Li C, Frenkel K: Importance of complete
DNA digestion in minimizing variability of 8-oxo-dG analyses. Free Rad
Biol Med 31:1341–1351, 200121. Li W, Yanoff M, Jian B, He Z: Altered mRNA levels of antioxidant enzymes
in pre-apoptotic pericytes from human diabetic retinas. Cell Mol Biol
45:59–66, 199922. Kowluru RA: Effect of reinstitution of good glycemic control on retinal
oxidative stress and nitrative stress in diabetic rats. Diabetes 52:818–823, 200323. Kowluru RA, Kern TS, Engerman RL: Abnormalities of retinal metabolism
in diabetes or experimental galactosemia. IV. Antioxidant defense system.Free Rad Biol Med 22:587–592, 1996
24. Podesta F, Romeo G, Liu WH, Krajewski S, Reed JC, Gerhardinger C,Lorenzi M: Bax is increased in the retina of diabetic subjects and isassociated with pericyte apoptosis in vivo and in vitro. Am J Pathol
156:1025–1032, 200025. Roth T, Podesta F, Stepp MA, Boeri D, Lorenzi M: Integrin overexpression
induced by high glucose and by human diabetes: potential pathway to celldysfunction in diabetic microangiopathy. Proc Natl Acad Sci U S A
90:9640–9644, 199326. Kern TS, Engerman RL: Comparison of retinal lesions in alloxan-diabetic
rats and galactose-fed rats. Curr Eye Res 13:863–867, 199427. Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD:
Programmed cell death and the control of cell survival: lessons from thenervous system. Science 262:695–700, 1993
28. Clermont AC, Brittis M, Shiba T, McGovern T, King GL, Bursell SE:Normalization of retinal blood flow in diabetic rats with primary interven-tion using insulin pumps. Inves Ophthal Vis Sci 35:981–990, 1994
29. Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP:Leukocyte-mediated endothelial cell injury and death in the diabetic retina.Am J Pathol 158:147–152, 2001
30. Kern TS, Kowluru RA, Engerman RL: Abnormalities of retinal metabolismin diabetes or galactosemia: ATPases and glutathione. Invest Ophthalmol
Vis Sci 35:2962–2967, 199431. Obrosova IG, Fathallah L, Greene DA: Early changes in lipid peroxidation
and antioxidative defense in diabetic rat retina: effect of DL-alpha-lipoicacid. Eur J Pharmacol 398:139–146, 2000
32. Obrosova IG, Fathallah L, Liu E, Nourooz-Zadeh J: Early oxidative stress inthe diabetic kidney: effect of DL-[alpha]-lipoic acid. Free Rad Biol Med
34:186–195, 200333. Rajesh M, Ramesh A, Ravi PE, Balakrishnamurthy P, Coral K, Punitham R,
Sulochana KN, Biswas J, Ramakrishnan S: Accumulation of 8-hy-droxydeoxyguanosine and its relationship with antioxidant parameters inpatients with Eales’ disease: implications for antioxidant therapy. Curr
Eye Res 27:103–110, 200334. Adamis AP: Is diabetic retinopathy an inflammatory disease? Br J Oph-
thalmol 86:363–365, 200235. Mohr S: Potential new strategies to prevent the development of diabetic
retinopathy. Expert Opin Investig Drugs 13:189–198, 200436. Farhangkhoee H, Khan ZA, Mukherjee S, Cukiernik M, Barbin YP,
Karmazyn M, Chakrabarti S: Heme oxygenase in diabetes-induced oxida-tive stress in the heart. J Mol Cell Cardiol 35:1439–1448, 2004
37. Nishikawa T, Sasahara T, Kiritoshi S, Sonoda K, Senokuchi T, Matsuo T,Kukidome D, Wake N, Matsumura T, Miyamura N, Sakakida M, KishikawaH, Araki E: Evaluation of urinary 8-hydroxydeoxy-guanosine as a novelbiomarker of macrovascular complications in type 2 diabetes. Diabetes
Care 26:1507–1512, 200338. Smith AR, Shenvi SV, Widlansky M, Suh JH, Hagen TM: Lipoic acid as a
potential therapy for chronic diseases associated with oxidative stress.Curr Med Chem 11:1135–1146, 2004
39. Behar-Cohen FF, Heydolph SFV, Droy-Lefaix MT, Courtois Y, Goureau O:Peroxynitrite cytotoxicity on bovine retinal pigmented epithelial cells inculture. Biochem Biophys Res Commun 226:842–849, 1996
40. Du Y, Smith MA, Miller CM, Kern TS: Diabetes-induced nitrative stress in theretina, and correction by aminoguanidine. J Neurochem 80:771–779, 2002
41. Sennlaub F, Courtois Y, Goureau O: Inducible nitric oxide synthasemediates retinal apoptosis in ischemic proliferative retinopathy. J Neuro-
sci 22:3987–3993, 200242. Halliwell B: What nitrates tyrosine? Is nitrotyrosine specific as a biomarker
of peroxynitrite formation in vivo? FEBS Lett 411:157–160, 199743. Gibson TM, Cotter MA, Cameron NE: Effects of alpha-lipoic acid on
impaired gastric fundus innervation in diabetic rats. Free Rad Biol Med
35:160–168, 200344. Bierhaus A, Chevion S, Chevion M, Hofmann M, Quehenberger P, Illmer T,
Luther T, Berentshtein E, Tritschler H, Muller M, Wahl P, Ziegler R,Nawroth PP: Advanced glycation end product-induced activation of NF-kappaB is suppressed by alpha-lipoic acid in cultured endothelial cells.Diabetes 46:1481–1490, 1997
45. Konrad D, Somwar R, Sweeney G, Yaworsky K, Hayashi M, Ramlal T, KlipA: The antihyperglycemic drug alpha-lipoic acid stimulates glucose uptakevia both GLUT4 translocation and GLUT4 activation: potential role of p38mitogen-activated protein kinase in GLUT4 activation. Diabetes 50:1464–1471, 2001
DIABETIC RETINOPATHY AND �-LIPOIC ACID
3238 DIABETES, VOL. 53, DECEMBER 2004

]>
Original Communication
Oral antioxidant therapy for marginal dry eye
KJ Blades1, S Patel2 and KE Aidoo3*
1Department of Vision Sciences, Glasgow Caledonian University, Glasgow, UK; 2West Coast Eye Research, Oban, Scotland; and3Division of Human Nutrition, School of Biological Sciences and Biomedical Sciences, Glasgow Caledonian University, Glasgow, UK
Objective: To assess the ef®cacy of an orally administered antioxidant dietary supplement for managing marginaldry eye.Design: A prospective, randomised, placebo controlled trial with cross-over.Setting: Eye Clinic, Department of Vision Sciences, Glasgow Caledonian University.Subjects: Forty marginal dry eye sufferers composed of 30 females and 10 males (median age 53 y; range 38 ±69 y).Interventions: Baseline assessments were made of tear volume suf®ciency (thread test), tear quality (stability),ocular surface status (conjunctival impression cytology) and dry eye symptoms (questionnaire). Each subject wasadministered courses of active treatment, placebo and no treatment, in random order for 1 month each and resultscompared to baseline.Results: Tear stability and ocular surface status were signi®cantly improved following active treatment (P< 0.05).No changes from baseline were detected following administration of placebo and no treatment (P> 0.05).Absolute increase in tear stability correlated with absolute change in goblet cell population density. Tear volumewas not improved following any treatment period and dry eye symptom responses were subject to placebo effect.Conclusions: Oral antioxidants improved both tear stability and conjunctival health, although it is not yetunderstood whether increased ocular surface health mediates increased tear stability or vice versa.Sponsors: This study was supported by a PhD scholarship funded by the Department of Vision Sciences, GlasgowCaledonian University, Scotland. Antioxidant supplements and placebos were kindly donated by Vitabiotics.Descriptors: marginal dry eye; tear stability; tear volume; conjunctiva; antioxidants; vitaminsEuropean Journal of Clinical Nutrition (2001) 55, 589±597
Introduction
The health and optical clarity of the cornea of the humaneye are of great importance as detriment may lead todegradation or loss of vision. Although exposed to theatmosphere between blinks, the anterior ocular structuresof the cornea and the conjunctiva are protected by a wellorganised ®lm of tears. The tear ®lm is essentially atrilaminar structure comprising a deep mucus layer pro-
duced primarily by the goblet cells of the conjunctivawhich increases ocular surface wettability; a substantialaqueous layer produced by the main and accessory lacri-mal glands and a super®cial layer of oil from theMeibomian glands in the eyelids which inhibits. Thedynamic tear ®lm structure is refreshed with the sweepingaction of the eyelids on each blink.
Under normal conditions, the tear ®lm is of suf®cientquantity and quality to establish a refractive surface of highquality for the cornea and to ensure the well-being of thecorneal and conjunctival epithelium (Holly, 1980). Abnorm-alities in the production, quality or replenishment of the tear®lm will result in various pathological states regarded as dryeye conditions. Such conditions can result in ocular surfacedamage, and may lead to eventual corneal damage whichcould impair corneal transparency and visual performance.The term `dry eye syndrome' describes a variety of condi-tions, of mixed aetiology but sharing common subjectivesymptoms and objective clinical signs, leading to a physicaland functional break-down of the tear ®lm (Lemp, 1995).
*Correspondence: KE Aidoo, Division of Human Nutrition, School ofBiological and Biomedical Sciences, Glasgow Caledonian University,Cowcaddens Road, Glasgow, G4 0BA, UK.Guarantor: S Patel.Contributors: KJB conducted the study and collected all data and analysedthem. He did the major work in writing the manuscript. SP randomised thesubjects' treatment orders and supervised the study. KEA advisedregarding the micronutrients of the antioxidants and co-supervised thestudy.Received 6 April 2000; revised 2 January 2001;accepted 10 January 2001
European Journal of Clinical Nutrition (2001) 55, 589±597ß 2001 Nature Publishing Group All rights reserved 0954±3007/01 $15.00
www.nature.com/ejcn

Such tear ®lm disorders range in severity, from the border-line dry eye, which may only be apparent under conditionssuch as environmental challenge (McMonnies, 1986), to thesevere (pathological) dry eye (keratoconjunctivitis sicca,KCS), as often found in SjoÈgren's syndrome. Dry eyeproblems may be the result of low tear volume (aqueousde®ciency), or inadequate quality (lipid de®ciency or mucusde®ciency). Any factor or pathology which damages thesurface of the cornea or conjunctiva, or disrupts the structureand shape of the eyelids is also likely to promote dry eye.
The involvement of nutritional components in the aetiol-ogy of some dry eye states has been reported (Sommer &Muhialal, 1982; Sullivan et al, 1973). Vitamin A is impor-tant for the integrity and function of the corneal andconjunctival epithelial cells and its de®ciency may promoteabnormal conjunctival changes (van Agtmaal et al, 1988;Udomkesmalle et al, 1992). While vitamin A de®ciency isa rare condition in the Western world, it is found undersome conditions, such as chronic liver disease, cystic®brosis, regional enteritis and other causes of diet restric-tion or poor absorption (Petersen et al, 1968; Russell et al,1973; Sullivan et al, 1973; Smith et al, 1975).
Detrimental conjunctival changes are known to be anearly consequence of vitamin A de®ciency (Natadisastraet al, 1987) and conjunctival effects of nutritional de®-ciency have been shown to be reversible with systemicvitamin A therapy (Sommer, 1983).
Patel et al (1993) demonstrated a signi®cant increase intear ®lm quality (stability) following supplementary multi-vitamin intake by a normal Western population. In terms ofimproved tear stability they reported that the synergisticeffects of a multivitamin treatment were more predictablethan the effects of a single nutritional component (vitamin C)alone. Similarly, combined supplementation of zinc andvitamin A promotes a better reversal of conjunctival changesin non-xerophthalmic patients with suboptimal nutriture,than zinc or vitamin A alone (Udomkesmalle et al, 1992).These ®ndings imply that nutritional in¯uences on tear ®lmcomposition and physiology are complex. The ®ndings ofPatel et al (1993) are of particular importance because lowtear ®lm stability is a common sign and consequence ofmany dry eye conditions. If the effect noted in normalsextends also to dry eye sufferers, then nutritional supple-ments could be used as a convenient treatment.
Dry eye has previously been treated using arti®cial tearsubstitutes to address tear volume or quality issues(Foulkes, 1998). Such prophylactic treatments may offertransient relief of symptoms, but must be repeated fre-quently as required (Swanson, 1998). Others have attemptedto address tear insuf®ciency problems by occluding thepuncta, the channels which normally drain the tears fromthe surface of the eye into the naso-lacrimal system forclearance (Murube & Murube, 1996). While this is per-formed to increase the volume of tears present at the ocularsurface, there is evidence that the lacrimal system canreduce tear production in response to punctal occlusion,presumably through a feedback mechanism which normally
prevents epiphora in normals (Tomlinson et al, 1998).While punctal occlusion may bene®t aqueous de®cientpatients, it may not be a suitable treatment for patientswho are not aqueous de®cient, but suffer from another formof dry eye.
It has been proposed that prospective clinical trials shouldbe conducted to assess the ef®cacy of vitamin supplementsfor treating non-SjoÈgren's syndrome dry eye (Foulkes,1998). However, most of the published investigations haveused topical vitamin-containing eye drops (Holly, 1993).
The purpose of this study was to assess whether an orallyadministered antioxidant dietary supplement could improvethe objective clinical signs and alleviate the subjectivesymptoms of marginal dry eye in a Western population.
Experimental design
A prospective, cross-over, placebo-controlled, randomised,predominantly double-masked clinical trial design wasadopted, whereby each subject was evaluated at baseline(prior to any intervention) and again at monthly intervals,following each of three treatment periods. The treatmentperiods were: (i) no treatment given for 30 days; (ii) placebotreatment given for 30 days, and (iii) oral antioxidantsupplements given for 30 days. Treatments were assignedin random order, by the method of Latin squares (Fleiss,1986).
This trial was double-masked for the periods of placebotreatment and antioxidant supplements, but only single maskfor the period with no treatment as there was no way ofmasking the subjects to the lack of treatment in this period.
The treatment was an antioxidant complex product calledVisionACE1 (Vitabiotics, London, UK). This is a commer-
Table 1 Visionace1 (Vitabiotics, London, UK)
Constituent Average per two capsules
b-Carotene 6.mgVitamin E (natural source) 120.mgVitamin C 300.mgVitamin B6 30.mgVitamin D (200 IU) 5.mgThiamin (vitamin B1) 15.mgRibo¯avin (vitamin B2) 10.mgVitamin B12 9.mgFolacin (as folic acid) 500.mgVitamin K 200.mgPantothenic acid 20.mgMagnesium 100.mgZinc 15.mgIron 6.mgIodine 200.mgCopper 2.mgManganese 4.mgSelenium 200.mgChromium 100.mgCystine 40.mgMethionine 40.mgBio¯avinoids 30.mg
Oral antioxidant therapyKJ Blades et al
590
European Journal of Clinical Nutrition

cially available product, in the UK. The active ingredients ofVisionACE1 are given in Table 1. The placebos for the studywere of identical colour, texture, shape and size, howeverthey contained only starch powder. The placebo capsuleswere also manufactured and supplied by Vitabiotics.
Prior to administration to the subjects, the capsules werehygienically loaded into standard unmarked tablet bottles,labelled with a random ®ve digit identi®er code. Thesecodes were not broken until the completion of the study.
Experimental procedures
To assess the signs and symptoms of marginal dry eye,several tests were employed. Tests were included to assesstear volume and quality, health of the underlying depen-dent ocular tissues, and severity of dry eye symptomsexperienced.
The tests were performed in a standard order to preventany one procedure in¯uencing the results of subsequenttests. The order was Tear Thinning Time (TTT); GlasgowCaledonian University Thread (GCUT) test; Dry Eye Ques-tionnaire; Conjunctival Impression Cytology.
Subject recruitment
The required individuals were sufferers of marginal dry eye,complaining of ocular discomfort or having low tear stabi-lity. Only subjects not receiving any conventional treatmentfor the condition, such as arti®cial tears, were recruited. Thesubjects were recruited proactively, through the Glasgow
Caledonian University eye clinic, with a questionnairescreening to identify patients suffering dry eye symptoms.Additional marginal dry eye sufferers referred by optometricpractitioners and ophthalmologists were also accepted,provided they ful®lled the standard recruitment criteria andwere not receiving any dry eye treatment.
Subject selection criteria
The selection criteria were set to allow broad intake ofmarginal sufferers. Individuals were enrolled as subjects if:(i) they had a non-invasive TTT of under 10 s; and=or(ii) their McMonnies dry eye questionnaire responses atinitial contact were considered indicative of signi®cantprimary or secondary dry eye symptomology (Blades &Patel 1996b).
Tear quality (stability) Ð tear thinning time (TTT)
Tear stability tests investigate the ability of the tear ®lm toadequately cover the otherwise exposed anterior surface ofthe eye, for a suf®cient duration of time to prevent dryingand subsequent damage to the underlying tissues. The tear®lm is respread and reformed with each blink (van Haerin-gen, 1981). Spontaneous blinks occur every 4 ± 5 s innormal eyes (Patel et al, 1991; Zaman et al, 1998). Tear®lm stability is taken to be insuf®cient if break-up occurs inunder 10 s (Farrell et al, 1992). Tear ®lm stability assess-ment techniques can be considered as invasive or non-invasive, with non-invasive tests such as the HIRCAL gridconsidered to give more valid results (Craig & Blades,
Table 2 From McMonnies' dry eye questionnaire
Questions regarding symptoms of dry eye that are constantly experienced:2. Do your ever experience any of the following symptoms? (Please underline those that apply to you)
1. Soreness2. Scratchiness3. Dryness4. Grittiness5. Burning
3. How often do you have these symptoms? (Underline)NeverSometimesOftenConstantly
Questions regarding symptoms of dry eye that are experienced in response to provocative stimuli:4. Are you unusually sensitive to cigarette smoke, smog, air conditioning, or central heating?
YesNoSometimes
5. Do your eyes easily become very red and irritated when swimming?Not applicableYesNoSometimes
6. Are your eyes dry and irritated the day after drinking alcohol?Not applicableYesNoSometimes
Oral antioxidant therapyKJ Blades et al
591
European Journal of Clinical Nutrition

1997). The HIRCAL grid measures the time taken from ablink until the tear ®lm begins to show irregular thinning,which happens just before the tear ®lm ruptures, exposingthe underlying epithelium. The HIRCAL grid is a modi®edBausch and Lomb keratometer, with the original miresreplaced by a plate consisting of a white grid etched on ablack background. The illuminated pattern of horizontaland vertical white lines was projected onto the tear ®lmsurface. The image of this grid was observed following ablink, for the ®rst sign of a discontinuity or distortion of thewhite lines. At this point, the time was recorded as the TTT.This was repeated ®ve times and the average TTT wascalculated (Hirji et al, 1989).
Tear volume Ð the Glasgow Caledonian University threads(GCUT) phenol red thread wetting
The GCUT phenol red thread test uses a ®ne, pH-indicator-impregnated thread to measure the amount oftears absorbed from the lower lacrimal lake in unit time.The test was performed by dipping the end of a ®nethread over the lower eyelid into the tear meniscus, andleaving it place for 2 min. After this the extent of threadwetting was measured as the alkaline tears cause thewetted portion of the thread to change colour from lightyellow to red (Blades & Patel, 1996a). Previous workwith this particular thread test has shown that the extentof thread wetting is lower for a group of aqueousde®cient patients than for non-aqueous de®cient normals(Patel et al, 1998).
Dry eye symptom perception
Subjective symptom assessment was performed, usingMcMonnies' dry eye questionnaire (McMonnies & Ho,1987a,b). This contains several questions pertaining to theperception of the ocular symptoms of dry eye. In additionto providing a total questionnaire score, it was possible tocompute scores to describe the perception of constantlyexperienced dry eye symptoms and symptoms experiencedin response to provocative stimuli such as environmentalfactors (Blades & Patel, 1996b).
Ocular surface status Ð Squamous metaplasia assessmentand goblet cell density
Conjunctival impression cytology (CIC) is a method ofinvestigating surface damage in dry eye, at a cellularlevel (Lemp, 1995). The test involved pressing a smallMillipore# ®lter against the conjunctiva to collect a sampleof conjunctival epithelial cells which were ®xed, stainedand examined by light microscopy. By this technique, twoparameters were investigated: (i) the goblet cells thatproduce much of the basement mucus of the tear ®lmwere selectively visualised by the Periodic acid-Schiffreaction (PAS). The number of goblet cells per unit ®eldof view (0.6 mm2) was calculated (Nelson & Wright, 1984;
Rivas et al, 1993); and (ii) appearance of and degree ofsquamous metaplasia. Haematoxylin was used to visualisethe other epithelial cells collected. These were compared toa photographic scale of increasing ocular surface damage(Tseng, 1985).
Statistical analyses
Data which were of Gaussian distribution (Shapiro ± Wilktest, P> 0.05) were analysed using parametric analysis ofvariance (one-way ANOVA); and Student's t-test and wereexpressed as a bar graph. Non-Gaussian data were analysedusing distribution-free statistical tests and expressed as boxplots. In these, the medians are represented by horizontalbars: the 25 ± 75th percentiles are enclosed by boxes; the10 ± 90th percentiles are enclosed by vertical bars and theremaining 20% (extreme) data points are shown as hollowcircles. Factorial analysis (Student's t-test and the Mann ±Whitney test) was employed to compare the results.
Results
Forty marginal dry eye sufferers with poor tear stabilityand=or perceived dry eye symptomology were recruited forthis trial. This subject group was composed of 30 females and10 males, with a mean age of 53.4� 15.3. Initial diagnosticparameters were: (i) mean GCUT wetting was 16.4 mm witha standard deviation of 6.8; (ii) median TTT was 7 s with arange of 1.2 ± 20; (iii) median goblet cell population densitywas 7.7 per 0.6 mm2 with a range of 0 ± 66.7; (iv) medianTseng's squamous metaplasia scale score was 1.5 with arange of 0 ± 5; (v) median total McMonnies' dry eye ques-tionnaire score was 13 with a range of 4 ± 22; (vi) medianprovoked dry eye symptom score (derived from McMonnies'dry eye questionnaire responses) was 2.5 with a range of 0 ±5; and (vii) median provoked dry eye symptom score
Figure 1 The mean (� s.d.) G-CUT wetting prior to any treatment, andfollowing each of the treatment periods.
Oral antioxidant therapyKJ Blades et al
592
European Journal of Clinical Nutrition

Figure 2 `Box and Whiskers' plot representing the spread of tear thinning times (TTT) recorded following each treatment.
Figure 3 The distribution of goblet cell density data collected prior to any treatment and following each of the treatment periods.
Figure 4 The distribution of Tseng's metaplasia scale score data collected prior to any treatmdent and following each of the treatment periods.
Oral antioxidant therapyKJ Blades et al
593
European Journal of Clinical Nutrition
(derived from McMonnies' dry eye questionnaire responses)was 5 with a range of 0 ± 8.
Figures 1 ± 5 present the GCUT thread wetting, TTT,conjunctival status and subjective symptom data from base-line and following each of the treatment periods.
The effect of no treatment
After no treatment was given to the subjects for 1 month,there was no signi®cant difference in any of the objectiveor subjective parameters assessed (P> 0.05).
The effect of placebo treatment
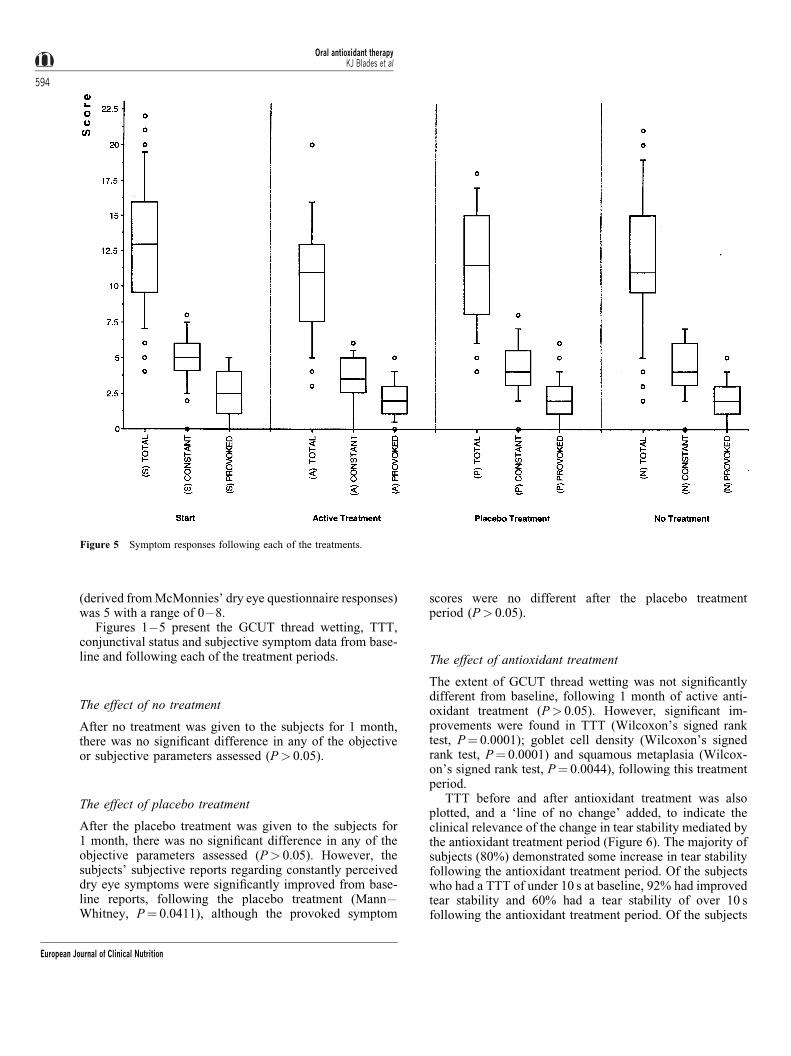
After the placebo treatment was given to the subjects for1 month, there was no signi®cant difference in any of theobjective parameters assessed (P> 0.05). However, thesubjects' subjective reports regarding constantly perceiveddry eye symptoms were signi®cantly improved from base-line reports, following the placebo treatment (Mann ±Whitney, P� 0.0411), although the provoked symptom
scores were no different after the placebo treatmentperiod (P> 0.05).
The effect of antioxidant treatment
The extent of GCUT thread wetting was not signi®cantlydifferent from baseline, following 1 month of active anti-oxidant treatment (P> 0.05). However, signi®cant im-provements were found in TTT (Wilcoxon's signed ranktest, P� 0.0001); goblet cell density (Wilcoxon's signedrank test, P� 0.0001) and squamous metaplasia (Wilcox-on's signed rank test, P� 0.0044), following this treatmentperiod.
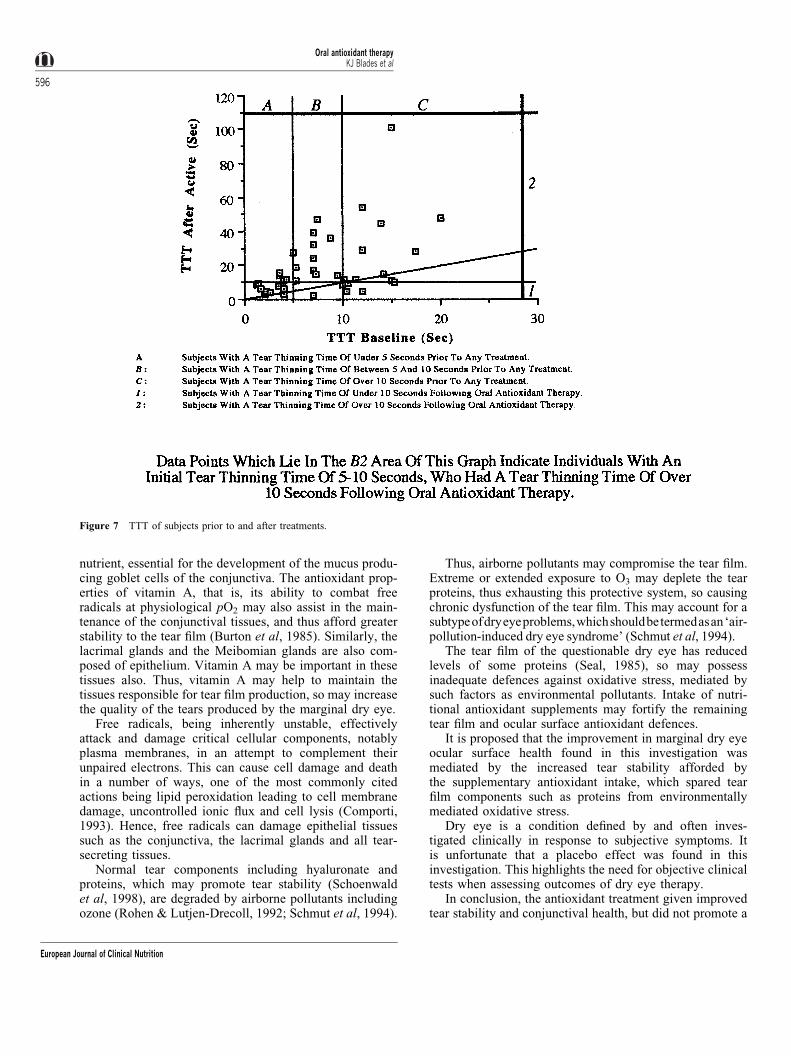
TTT before and after antioxidant treatment was alsoplotted, and a `line of no change' added, to indicate theclinical relevance of the change in tear stability mediated bythe antioxidant treatment period (Figure 6). The majority ofsubjects (80%) demonstrated some increase in tear stabilityfollowing the antioxidant treatment period. Of the subjectswho had a TTT of under 10 s at baseline, 92% had improvedtear stability and 60% had a tear stability of over 10 sfollowing the antioxidant treatment period. Of the subjects
Figure 5 Symptom responses following each of the treatments.
Oral antioxidant therapyKJ Blades et al
594
European Journal of Clinical Nutrition

who had a TTT of under 5 s at baseline, 84.6% had improvedtear stability and 30.8% had a tear stability of over 10 sfollowing the antioxidant treatment period. Of the subjectswho had a TTT of between 5 and 10 s at baseline, 91.7% hadimproved tear stability, and all 91.7% had a tear stability ofover 10 s following the antioxidant treatment period.
`Carry over effect'
To assess whether a `carry over effect' (sustained activityof the active treatment, without the active treatment period)had in¯uenced the results collected following the placebotreatment period, treatment orders were divided into thosewhere the active treatment was administered immediatelyprior to the placebo treatment, and those where it was eitheradministered after the placebo period or another treatmentseparating the active and placebo treatment periods. Thedata analysed using Mann ± Whitney test were: (i) TTT; (ii)conjunctival hyperaemia; (iii) subjective symptom percep-tion; (iv) conjunctival epithelial squamous metaplasia; and(v) conjunctival goblet cell population density. Data fromGCUT wetting were analysed using Student's t-test. The P-values of these analyses for all groups were over 0.05,indicating that no carry-over effects affected the parametersassessed following the placebo treatment period. The activ-ity of the treatment given therefore did not mediate anychanges which persisted for 1 month following cessation ofactive treatment.
A signi®cant correlation was found between the absolutechanges from baseline in TTT and goblet cell densitiesfollowing the antioxidant treatment period (Figure 7;Spearman's rank correlation coef®cient� 0.568;P< 0.002). The subjects' subjective reports regarding con-stantly perceived dry eye symptoms were again signi®-cantly improved from baseline reports following the activetreatment (Mann ± Whitney, P� 0.001), but provoked
symptom scores were no different following this treatmentperiod (P> 0.05).
Discussion
The results demonstrate that the orally administered anti-oxidant dietary supplements given improve the tear ®lmstability and the health of the conjunctival surface inmarginal dry eye sufferers. No net improvements in tearvolume were found. True improvement in subjectivelyreported symptoms was not found, because the subjectivesymptoms were improved when the placebo treatment wasgiven.
The tear ®lm=anterior eye is in a very dynamic relation-ship and Patel et al (1993) showed a signi®cant increasein tear ®lm stability with nutritional supplements for 10days.
The most consistent and clinically important improve-ment was the increase in tear stability in subjects who had aTTT of between 5 and 10 s at baseline. These subjectswould initially have been indicated as having poor tearstability (Farrell et al, 1992), but 91.7% of them had a tearstability of over 10 s, which would be classed as normal,following the antioxidant treatment period.
We detected a signi®cant correlation in the increasedtear stability and the increased conjunctival health. How-ever, we cannot determine if tear stability improved as adirect consequence of an increase in conjunctival healthand goblet cell count or vice versa.
Several mechanisms, could explain the clinical improve-ments mediated by the antioxidants, both individually orin concert. Both the nutritional and the antioxidant proper-ties of the dietary supplements given could explain theresults. For example, vitamin A is known to be involved inepithelial differentiation and as such is an important micro-
Figure 6 Absolute changes in TTT and goblet cells in the subjects.
Oral antioxidant therapyKJ Blades et al
595
European Journal of Clinical Nutrition
nutrient, essential for the development of the mucus produ-cing goblet cells of the conjunctiva. The antioxidant prop-erties of vitamin A, that is, its ability to combat freeradicals at physiological pO2 may also assist in the main-tenance of the conjunctival tissues, and thus afford greaterstability to the tear ®lm (Burton et al, 1985). Similarly, thelacrimal glands and the Meibomian glands are also com-posed of epithelium. Vitamin A may be important in thesetissues also. Thus, vitamin A may help to maintain thetissues responsible for tear ®lm production, so may increasethe quality of the tears produced by the marginal dry eye.
Free radicals, being inherently unstable, effectivelyattack and damage critical cellular components, notablyplasma membranes, in an attempt to complement theirunpaired electrons. This can cause cell damage and deathin a number of ways, one of the most commonly citedactions being lipid peroxidation leading to cell membranedamage, uncontrolled ionic ¯ux and cell lysis (Comporti,1993). Hence, free radicals can damage epithelial tissuessuch as the conjunctiva, the lacrimal glands and all tear-secreting tissues.
Normal tear components including hyaluronate andproteins, which may promote tear stability (Schoenwaldet al, 1998), are degraded by airborne pollutants includingozone (Rohen & Lutjen-Drecoll, 1992; Schmut et al, 1994).
Thus, airborne pollutants may compromise the tear ®lm.Extreme or extended exposure to O3 may deplete the tearproteins, thus exhausting this protective system, so causingchronic dysfunction of the tear ®lm. This may account for asubtypeofdryeyeproblems,whichshouldbetermedasan`air-pollution-induced dry eye syndrome' (Schmut et al, 1994).
The tear ®lm of the questionable dry eye has reducedlevels of some proteins (Seal, 1985), so may possessinadequate defences against oxidative stress, mediated bysuch factors as environmental pollutants. Intake of nutri-tional antioxidant supplements may fortify the remainingtear ®lm and ocular surface antioxidant defences.
It is proposed that the improvement in marginal dry eyeocular surface health found in this investigation wasmediated by the increased tear stability afforded bythe supplementary antioxidant intake, which spared tear®lm components such as proteins from environmentallymediated oxidative stress.
Dry eye is a condition de®ned by and often inves-tigated clinically in response to subjective symptoms. Itis unfortunate that a placebo effect was found in thisinvestigation. This highlights the need for objective clinicaltests when assessing outcomes of dry eye therapy.
In conclusion, the antioxidant treatment given improvedtear stability and conjunctival health, but did not promote a
Figure 7 TTT of subjects prior to and after treatments.
Oral antioxidant therapyKJ Blades et al
596
European Journal of Clinical Nutrition

net increase in tear volume in marginal dry eye sufferers.These clinical improvements may have been promoted bysparing tear ®lm components from oxidative stress. Themost consistent, clinically relevant improvements werenoted in marginal dry eye sufferers initially presentingwith TTT of between 5 and 10 s. Although dry eyesymptom perception is an important indicator of dry eye,it is not a reliable indicator of dry eye therapy ef®cacy. Fora more reliable clinical monitoring of the dry eye conditionand its progress tear stability should be assessed.
References
Blades K & Patel S (1996a): The dynamics of tear ¯ow within a phenol redimpregnated thread. Ophthal. Physiol. Opt. 16, 409 ± 415.
Blades K & Patel S (1996b): The incidence of dry eye symptoms in of®ceworkers. Optom. Visual Sci. 72, 40.
Burton GW, Foster DO, Perly B, Slater TF, Smith KP & Ingold KU(1985): Biological antioxidants. Phil. Trans. R. Soc. Lond. B 311, 565 ±578.
Comporti M (1993): Lipid peroxidation. An overview, In Free Radicals:from Basic Science to Medicine, eds. G Poli, E Albano & MU Dianzanipp 65 ± 79. Basel: Birkhauser, Switzerland.
Craig J & Blades K (1997): Preocular tear ®lm assessment. Optician 213,20 ± 27.
Farrell J, Grierson DJ, Patel S & Sturrock RD (1992): A classi®cation fordry eye following comparison of tear thinning time with Schirmer teartest. Arch. Ophthal. 70, 357 ± 360.
Fleiss JL (1986): The Design and Analysis of Clinical Experiments, pp281 ± 286. New York: Wiley.
Foulkes GN (1998): The now and future therapy of the non-SjoÈgren's dryeye, In Lacrimal Gland, Tear Film and Dry Eye Syndromes 2: BasicScience and Clinical Relevance, eds. DA Sullivan, DA Dart & MAMeneray, pp 959 ± 964. New York: Plenum Press.
Hirji N, Patel S & Callander M (1989): Human tear ®lm pre rupture time(TP-RPT), a non invasive technique for evaluating the pre corneal tear®lm using a novel keratometer mire. Ophthal. Physiol. Opt. 9, 139 ± 142.
Holly FJ (1980): Tear ®lm physiology. Am. J. Optom. Physiol. Opt. 57,252 ± 257.
Holly FJ (1993): Diagnostic methods and treatment modalities of dry eyeconditions. Int. Ophthal. 17, 113 ± 125.
Lemp MA (1995): Report of the National Eye Institute=Industry Work-shop on Clinical Trials in Dry Eyes. C.L.A.O. J. 21, 221 ± 232.
McMonnies CW (1986): Key questions in a dry eye history. J. Am. Optom.Assoc. 57, 512 ± 517.
McMonnies CW & Ho A (1987a): Patient history in screening for dry eyeconditions. J. Am. Optom. Assoc. 58, 296 ± 301.
McMonnies CW & Ho A (1987b): Responses to a dry eye questionnairefrom a normal population. J. Am. Optom. Assoc. 58, 588 ± 591.
Murube J & Murube E (1996): Treatment of dry eye by blocking thelacrimal canaliculi. Surv. Ophthal. 40, 463 ± 480.
Natadisastra G, Wittpen JR, West KP, Mukilal A & Sommer A (1987):Impression cytology for detection of vitamin A de®ciency. Arch.Ophthal. 105, 1224 ± 1228.
Nelson JD & Wright JC (1984): Conjunctival goblet cell densities inocular surface disease. Arch. Ophthal. 102, 1049 ± 1051.
Patel S, Henderson R, Bradley L, Galloway B & Hunter L (1991): Effectof visual display unit use on blink rate and tear stability. Optom. VisualSci. 68, 888 ± 892.
Patel S, Plaskow J & Ferrier C (1993): The in¯uence of vitamins and traceelement supplements on the stability of the pre-corneal tear ®lm. ActaOphthal. 71, 825 ± 829.
Patel S, Farrell J, Blades KJ & Grierson DJ (1998): The value of a phenolred impregnated thread for differentiating between the aqueous andnon-aqueous de®cient dry eye. Ophthal. Physiol. Opt. 18, 471 ± 476.
Petersen RA, Petersen VS & Robb RM (1968): Vitamin A de®ciency withxerophthalmia and night blindness in cystic ®brosis. Am. J. Dis. Child116, 662 ± 665.
Rivas L, Rodriguez JJ, Alvarez MI, Oroza MA & Murube del CastilloJ (1993): Correlation between impression cytology and tear functionparameters in SjoÈgren's syndrome. Acta Ophthal. 71, 353 ± 359.
Rohen JW & Lutjen-Drecoll E (1992): Functional morphology of theconjunctiva. In The Dry Eye, a Comprehensive Guide, eds. MA Lemp &R Marquardt, pp 35 ± 63. Berlin: Springer.
Russell RM, Smith VC, Multack R, Krill AE & Rosenberg IH (1973):Dark-adaptation testing for diagnosis of subclinical vitamin A de®-ciency and evaluation of therapy. Lancet ii, 1161 ± 1164.
Schmut O, Grubber E, El-Shabrawi M & Faulborn J (1994): Destruction ofhuman tear proteins by ozone. Free Rad. Biol. Med. 17, 165 ± 169.
Schoenwald RD, Vidvauns S, Wurster DE & Barfknecht CF (1998): Therole of tear proteins. In Tear Film Stability In The Dry Eye Patient AndIn The Rabbit; Lacrimal Gland, Tear Film, And Dry Eye Syndromes 2:Basic Science And Clinical Relevance, eds. DA Sullivan, DA Dartt &MA Meneray, pp 391 ± 400. New York: Plenum Press.
Seal DV (1985): The effect of ageing and disease on tear constituents.Trans. Ophthal. Soc UK 104, 355 ± 362.
Smith RS, Farrell T & Bailey T (1975): Keratomalacia. Sury. Ophthal. 20,213 ± 219.
Sommer A (1983): Effects of vitamin A de®ciency on the ocular surface.Ophthalmology 90, 592 ± 600.
Sommer A & Mukilal A (1982): Nutritional factors in corneal xerophthal-mia. Arch. Ophthal. 100, 399 ± 403.
Swanson (1998): Compliance with and typical usage of arti®cial tears indry eye conditions. J. Am. Optom. Assoc. 69, 649 ± 655.
Sullivan WR, McCulley JP & Dohlman CH (1973): Return of goblet cellsafter vitamin A therapy in xerosis of the conjunctiva. Am. J. Ophthal.75, 720 ± 725.
Tomlinson A, Craig JP & Lowether GE (1998): The biophysical role intear regulation. In Tear Film Stability in the Dry Eye Patient and in theRabbit; Lacrimal Gland, Tear Film, and Dry Eye Syndromes 2: BasicScience and Clinical Relevance, eds. DA Sullivan, DA Dartt &MA Meneray, pp 371 ± 380. New York: Plenum Press.
Tseng SCG (1985): Staging of squamous metaplasia by impressioncytology. Ophthalmology 92, 728 ± 733.
Udomkesmalle E, Dhanamitta S, Sirisinha S, Charoenkiatkul S,Tuntipopitat S, Banjong O, Roroongwasinkul N, Kramer TR & SmithJC (1992): Effect of vitamin A and zinc supplementation on the nurtureof children in northeast Thailand. Am. J. Clin. Nutr. 56, 50 ± 57.
van Agtmaal EJV, Bloem MW, Speek AJ, Saowakontha S, Schreurs WHP& Van Haeringen NJ (1988): The effects of vitamin A supplementationon tear ¯uid retinol levels of marginally nourished preschool children.Curr. Eye Res. 7, 43 ± 48.
van Haeringen NJ (1981): Clinical biochemistry of tears. Surv. Ophthal.26, 84 ± 96.
Zaman ML, Doughty MJ & Button NF (1998): The exposed ocular surfaceand its relationship to spontaneous eyeblink rate in elderly Caucasians.Exp. Eye Res. 67, 681 ± 686.
Oral antioxidant therapyKJ Blades et al
597
European Journal of Clinical Nutrition

Oxidized Omega-3 Fatty Acids Inhibit NF-�B Activation Viaa PPAR�-Dependent PathwayArchana Mishra, Ashok Chaudhary, Sanjeev Sethi
Objective—The aim of this study was to determine the effects of oxidized versus native omega-3 fatty acids on theendothelial expression of chemokines MCP-1 and IL-8, and, if effective in inhibiting chemokine expression, todetermine the mechanism for the inhibition of chemokine expression.
Methods and Results—Using enzyme-linked immunosorbent assays, we show that oxidized EPA and DHA but notunoxidized EPA or DHA inhibit cytokine-induced endothelial expression of monocyte chemoattractant protein (MCP)-1and, to a lesser extent, IL-8. In electrophoretic mobility shift assays, oxidized EPA but not unoxidized EPA potentlyinhibited cytokine-induced activation of endothelial nuclear factor-�B (NF-�B). Using Western blot analyses, we showthat the inhibition of NF-�B activation was not caused by prevention of phosphorylation of I�B� because oxidized EPAdid not inhibit cytokine-induced phosphorylation and ubiquination of I�B�. Furthermore, oxidized EPA inhibitedNF-�B activation in endothelial cells derived from wild-type mice but had no inhibitory effects on NF-�B activation inendothelial cells derived from peroxisome proliferator-activated receptor � (PPAR�)-deficient mice, indicating thatoxidized EPA requires PPAR� for its inhibitory effects on NF-�B.
Conclusions—These studies show that the antiinflammatory effects of fish oil may result from the inhibitory effects ofoxidized omega-3 fatty acids on NF-�B activation via a PPAR�-dependent pathway. (Arterioscler Thromb Vasc Biol.2004;24:1621-1627.)
Key Words: monocyte chemoattractant protein-1 � oxidized omega-3 fatty acids � oxidized eicosapentaenoic acid� nuclear factor-�B � PPARa
Consumption of marine fish oil has been reported toimprove the prognosis of several chronic inflammatory
diseases characterized by leukocyte accumulation andleukocyte-mediated tissue injury, including atherosclerosis,IgA nephropathy, inflammatory bowel disease, rheumatoidarthritis, etc.1–4 These beneficial effects of fish oil have beenassociated with the omega-3 polyunsaturated fatty acids(PUFAs), eicosapentaenoic acid (EPA), and docosahexaenoicacid (DHA), which are abundant in marine fish oil.
EPA and DHA are highly polyunsaturated and easilyundergo auto-oxidation.5,6 In fact, it is very difficult to avoidthe oxidation of these very labile fatty acids. More impor-tantly, in vivo, a large increase in tissue and plasma accumu-lation of both omega-3 fatty acids and fatty acid oxidationproducts is noted in subjects consuming fish oil, even afteraddition of antioxidant supplements to the diet.7–10 Thissuggests the possibility that oxidized omega-3 fatty acids maybe an important component of the observed antiinflammatoryeffects of fish oil. Indeed, our previous studies have shownthat oxidized EPA, and not unoxidized EPA, potently inhibitsleukocyte–endothelial interactions, both in vitro and in vivo,
through a peroxisome proliferator-activated receptor(PPAR)�-dependent mechanism.11,12
One of the early events in inflammation is the upregulationof endothelial chemokines, monocyte chemoattractantprotein-1 (MCP-1) and IL-8, in response to proinflammatorycytokines such as tumor necrosis factor (TNF)-� and IL-1.MCP-1 and IL-8 in turn promote leukocyte chemotaxis,adhesion, and transendothelial migration,13,14 and neutraliza-tion of MCP-1 has been shown to attenuate in vivo injuryarising from inflammatory mechanisms.15–17 The aim of thisstudy was to determine the effects of oxidized versus nativeomega-3 fatty acids on the endothelial expression of chemo-kines MCP-1 and IL-8, and, if effective in inhibiting chemo-kine expression, to determine the mechanism for the inhibi-tion of chemokine expression.
MethodsPreparation of Fatty AcidsEPA, DHA, and arachidonic acid were purchased from CaymanChemicals. They were relatively unoxidized as assessed by thiobar-bituric acid-reactive substances assay using malondialdehyde as a
Received March 10, 2004; revision accepted May 25, 2004.From the Department of Pathology, University of Iowa Hospitals and Clinics, Iowa City, Iowa.A.M. and A.C. contributed equally to this workConsulting Editor for this article was Peter Libby, MD, Brigham and Women’s Hospital, Boston, Mass.Correspondence to Dr Sanjeev Sethi, Department of Pathology, 5243 RCP, University of Iowa Hospitals and Clinics, 200 Hawkins Drive, Iowa City,
IA 52242. E-mail [email protected]© 2004 American Heart Association, Inc.
Arterioscler Thromb Vasc Biol. is available at http://www.atvbaha.org DOI: 10.1161/01.ATV.0000137191.02577.86
1621 by guest on January 14, 2016http://atvb.ahajournals.org/Downloaded from by guest on January 14, 2016http://atvb.ahajournals.org/Downloaded from by guest on January 14, 2016http://atvb.ahajournals.org/Downloaded from

standard; this assay is a measure of fatty acid oxidation.18 The fattyacids were stored as a 100-mmol/L stock in 100% ethanol undernitrogen to ensure minimal oxidation. For oxidation, 3.3 mmol/LEPA, DHA, or arachidonic acid was prepared in phosphate-bufferedsaline (PBS; pH 4.9) and the sample was incubated at 37°C for 16hours. Gas chromatography mass spectrometry of the samplesrevealed a single peak in the unoxidized (native) EPA sample, whichwas consistent with EPA, whereas oxidized EPA had �5% of theunoxidized EPA and a number of additional peaks that likelycorrespond to EPA oxidation products (data not shown). Usingthiobarbituric acid-reactive substances assay, the aldehyde content ofoxidized EPA was 1.78 �mol/L malondialdehye in 100 �mol/LEPA.11 For treatment of cells, oxidized or unoxidized EPA, DHA, orarachidonic acid was diluted in medium containing 20% fetal calfserum to final concentrations between 10 and 100 �mol/L. Thevehicle control was media containing PBS.
Enzyme-Linked Immunosorbent Assays forMCP-1 and IL-8Confluent human umbilical vein endothelial cells (HUVEC) andhuman microvessel endothelial cells (Clonetics) in 96-well plateswere incubated with vehicle, unoxidized, or oxidized EPA, DHA, orarachidonic acid at varying concentrations of 10, 25, 50, 75, and100 �mol/L for 1 hour. The cells were washed with PBS and freshmedium was added before stimulation with hIL-1� (10 U/mL) orhTNF� (10 ng/mL) for 5 hours. Aliquots of the medium wereremoved for analysis of MCP-1 and IL-8 by enzyme-linked immu-nosorbent assay (ELISA) using matched antibodies as previouslydescribed.19,20 Antibody binding was visualized with horseradishperoxidase-conjugated streptavidin and tetra methyl benzidine liquidsubstrate system.
Results are expressed as MCP-1 and IL-8 (pg/mL) in the culturemedium using values determined on a standard curve. TNF�-induced expression of MCP-1 and IL-8 was taken as 100%, andresults are also expressed as percent decrease compared withTNF�-induced expression of MCP-1 and IL-8.
RNAse Protection AssaysHUVEC were grown to confluence in 100-mm dishes and werepretreated for 1 hour with vehicle control or unoxidized(100 �mol/L) or oxidized EPA (100 �mol/L) in standard growthmedium. The HUVEC were washed and the cells were stimulatedwith hTNF� (10 ng/mL) for 3 hours. RNA extraction was performedby guanidine thiocyanate (RNAzol) method of Chomczynski andSacchi.21 Twenty �g of RNA was subjected to multiprobe RPAsystem using probes for multiple human chemokine related RNAmolecules, including MCP-1 and IL-8, and GAPDH as described inthe manufacturer protocols (Riboquant; Pharmingen).
Isolation and Culture of Murine AorticEndothelial CellsWe isolated endothelial cells from thoracic aortas of 4-week-old129SV wild-type and PPAR��/� mice.22 Briefly, thoracic aortaswere removed from anesthetized mice and rinsed 3times with PBSand placed in endothelial cell growth medium (M199, 20% FBS, 25�g/mL Gentamycin, 5 mg/50 mL endothelial mitogen, 50 �g/mL,heparin, 2 mmol/L L-glutamine 2 mmol/L). The periadventitial fataround the aortas was carefully removed. The aortas were cut openlongitudinally and cut into 4 to 6 pieces of 1- to 2-mm2 and placedendothelial cell side-down in a fibronectin-coated 100-mm cultureplate. Initially only a small amount of media was placed around theexplants to prevent them from floating. After 12 hours, 8 mL ofmedium was added. Cells began to expand from the edges of theexplant between days 2 to 5. Medium was changed every 2 to 3 days.On day 7, the explants were removed. After 2 to 4 days of explantremoval, the cells were confluent and were easily subcultured frompassage 2 to 5. These cells express cytokine-inducible mouseadhesion receptors such as vascular cell adhesion molecule-1 andintercellular adhesion molecule-1 (data not shown). Oxidized andunoxidized EPA had no effect on the viability of these cells as
determined by tetrazolium salt 3-[4,5-dimethyliazol-2-yl]-2-diphenyltetrazolium bromide (MTT) assays (data not shown). Forelectrophoresis mobility shift assays, the endothelial cells werepretreated for 1 hour with vehicle control or unoxidized(100 �mol/L) or oxidized EPA (100 �mol/L) before stimulation withmouse TNF� (10 ng/mL) for 2 hours. Nuclear extracts were thenprepared and used in electrophoretic mobility shift assays.
Preparation of Nuclear and Cytoplasmic ExtractsHUVEC were grown to confluence in gelatin-coated 100-mm dishes.HUVEC were pretreated for 1 hour with vehicle control or unoxi-dized (100 �mol/L) or oxidized EPA (100 �mol/L) in standardgrowth medium before stimulation with TNF� (10 ng/mL) for 0,15,and 60 minutes. Nuclear and cytoplasmic extracts were prepared.23
The cytoplasmic extract was used in Western blot analyses while thenuclear extract was used in the electrophoretic gel shift assays.Protein concentration was determined by Bio-Rad DC protein assay.
Electrophoretic Mobility Gel Shift AssaysNuclear extracts from HUVEC pretreated with vehicle control orunoxidized or oxidized EPA in standard growth medium beforestimulation with TNF� (10 ng/mL) for 60 minutes was used in thegel shift assays. The double-stranded oligonucleotide containing theconsensus sequence for the NF-�B (5�-AGT TGA GGG GAC TTTCCC AGG C-3�) and AP-1 (5�-TTC CGG CTG ACT CAT CAAGCG -3�) was end-labeled by incubating the oligonucleotide with�32P-labeled ATP and T4 polynucleotide kinase at 37°C for 10minutes according to standard protocols (Promega).24 For bindingreactions, nuclear extracts (10 �g) of HUVEC or mice endothelialcells were incubated in 10 to 15 �L of total reaction volume with 20�L of binding buffer and 32P-labeled NF-�B or AP-1 oligonucleo-tides for 20 minutes at room temperature. Samples were electropho-resed on a 5% nondenaturing acrylamide gel. The gels were driedand autoradiographed.
Polyclonal antisera against NF-�B subunit p65 was used todetermine the subunit composition of gel-shifted NF-�B complexes;1 �g of antibody was added to the binding reaction 20 minutesbefore addition of labeled oligonucleotide probe. Incubation of thenuclear extract from TNF�-treated cells with excess cold NF-�B orAP-1 oligonucleotide was used to confirm the specificity of AP-1binding activity.
Western Blot AnalysesCytoplasmic extracts were resolved by SDS-PAGE on 10% poly-acrylamide gels and transferred to polyvinylidene fluoride mem-branes (Bio-Rad) in 25 mmol/L Tris, pH 8.3, 192 mmol/L glycine,and 20% methanol. Membranes were blocked overnight at 4°C inblocking solution (PBS with 10% milk and 0.2% Tween-20). Afterblocking the membranes, the primary antibodies to IkB�, p50, p65,and �-actin (Santa Cruz Biotechnology) or phospho-IkB� (Cellsignaling) were applied according to the manufacturers directions for1 hour at room temperature.25 Horseradish peroxidase-conjugatedsecondary antibodies, goat antirabbit IgG, and goat antimouse IgG(Santa Cruz Biotechnology) were used. After each antibody appli-cation, blots were washed 3 times in TBS containing 0.2% Tween-20. Antibody complexes were visualized with the use ofchemiluminescence.
Statistical AnalysisData are presented as average�SEM. Statistical significance wasassessed by unpaired Student t test.
ResultsOxidized EPA Inhibits Cytokine-InducedEndothelial Expression of MCP-1We used ELISA methods to determine the effect of oxidizedand unoxidized EPA on cytokine-induced endothelial expres-sion of MCP-1 and IL-8. As expected, treatment of HUVECs
1622 Arterioscler Thromb Vasc Biol. September 2004
by guest on January 14, 2016http://atvb.ahajournals.org/Downloaded from

with TNF� and IL-1� results in a 6- to 8-fold increase inMCP-1 and IL-8 expression. Pretreatment of HUVECs withoxidized EPA for 1 hour significantly inhibited TNF� andIL-1�–induced expression of MCP-1 whereas incubationwith unoxidized EPA had little effect (Figure 1A). Theinhibition was dose-dependent; 75 and 100 �mol/L oxidizedEPA reduced MCP-1 expression to levels close to those seenfor unstimulated HUVECs. When HUVECs were pretreatedwith 50 and 25 �mol/L oxidized EPA, there was a 30% anda 15% decrease in MCP-1 expression when compared with
TNF�-treated cells, and a 31% and a 24% decrease whencompared with IL-1�–treated cells, respectively. The effectof oxidized EPA on cytokine-induced IL-8 expression wasless marked. There was a modest but significant decrease inTNF�-induced and IL-1�–induced endothelial expression ofIL-8 when the HUVECs were pretreated with 100 �mol/Loxidized EPA, whereas incubation with unoxidized EPA hadlittle effect (Figure 1B). At concentrations of 75 �mol/L andlower, oxidized EPA had no significant effect in inhibitingTNF�-induced and IL-1�–induced IL-8 expression. Instead,there was mild increase in the expression of IL-8 when HUVECwere pretreated with lower concentrations of oxidized EPA.
Similar results were also noted when human microvesselendothelial cells were used instead of HUVECs (Table). Wealso compared the effect of EPA, DHA, and arachidonic acidon TNF�-induced endothelial MCP-1 and IL-8 expression(Table I, available online at http://atvb.ahajournals.org).
Using RNAse protection assays, we determined whetheroxidized EPA inhibits cytokine-induced expression of endo-thelial MCP-1 and IL-8 at the transcriptional level. Pretreat-ment of HUVECs with oxidized EPA for 1 hour beforestimulation with TNF� resulted in almost complete absenceof the MCP-1 RNA transcripts and a milder decrease in IL-8mRNA. Unoxidized EPA had no effect in decreasing eitherMCP-1 or IL-8 mRNA levels (Figure 1C). When the expres-sion of MCP-1 and IL-8 was normalized to GAPDH expres-sion, oxidized EPA pretreatment resulted in a 60% decreasein MCP-1 expression whereas there was only marginaldecrease (10%) in IL-8 expression when compared withTNF�-treated endothelial cells.
Oxidized EPA Inhibits Cytokine-Induced NF-�BActivation but Has no Effect on AP-1The expression of MCP-1 and IL-8 is regulated throughtranscription factors NF-�B and AP-1.26,27 Studies haveshown that although NF-�B activation is required for optimalactivation of MCP-1, AP-1 activation can bypass the NF-�B–mediated IL-8 gene transcription.28
We studied the effect of oxidized versus unoxidized EPA oncytokine-induced activation of NF-�B by performing gel shiftassays on nuclear extracts of endothelial cells (Figure 2A).Treatment of HUVECs with TNF� for 60 minutes results inactivation of NF-�B as indicated by the gel shifts. Pretreatmentof HUVECs with oxidized EPA for 1 hour almost completelyinhibited TNF�-induced activation of NF-�B whereas incuba-tion with unoxidized EPA had little effect. The p65 componentof NF-�B was confirmed in supershift assays using antibodies tothe p65 subunit of NF-�B. Next, we studied the effect ofoxidized versus unoxidized EPA on TNF�-induced activation ofAP-1 (Figure 2B). Treatment of HUVECs with TNF� for 1 hourresults in activation of AP-1 as indicated by the gel shifts.Pretreatment of HUVECs with oxidized EPA or unoxidizedEPA for 1 hour had no significant inhibitory effect on TNF�-induced activation of AP-1. Incubation of nuclear extract fromTNF�-treated cells with excess cold AP-1 oligonucleotide wasused to confirm the specificity of AP-1 binding activity.
Figure 1. Oxidized EPA inhibits cytokine-induced endothelialMCP-1 and, to a lesser extent, IL-8 expression. HUVECs were pre-treated for 1 hour with vehicle (Veh) or unoxidized (100 �mol/L,UnoxEPA), or oxidized EPA (OxEPA) at indicated concentrationsbefore stimulation with TNF� (white) or IL-1� (black) for 5 hours.Aliquots of the culture medium were analyzed for (A) MCP-1 and(B) IL-8 by ELISA techniques (n�3). *P�0.0001 and ** P�0.005compared with EPA/TNF� and Veh/TNF�. C, HUVECs were pre-treated for 1 hour with vehicle, UnoxEPA (100 �mol/L), or OxEPA(100 �mol/L) before stimulation with TNF� for 3 hours. Total RNAwas extracted and mRNA for MCP-1 and IL-8 was assayed usingthe multiprobe RPA system.
Mishra et al Oxidized EPA Inhibits NF-�B Activation 1623
by guest on January 14, 2016http://atvb.ahajournals.org/Downloaded from

Oxidized EPA Promotes Cytoplasmic Retention ofp50 and p65 Subunits of NF-�B AfterCytokine-Induced ActivationProinflammatory cytokines activate I�B kinase complex thatphosphorylate NF-�B inhibitor, I�B�, resulting in its conju-gation with ubiquitin and subsequent degradation. The free
p50/p65 subunits of the NF-�B complex then translocate tothe nucleus and induce target genes.29
To determine whether oxidized EPA prevented the nucleartranslocation of p50 and p65 subunits of NF-�B, we per-formed Western blot analyses using antibodies to p50 (Figure3A) and p65 (Figure 3B) on the cytosolic extracts ofendothelial cells. Baseline levels of p50 and p65 are presentin the cytoplasm before stimulation of HUVECs with TNF�.Stimulation with TNF� for 15 minutes results in almostcomplete disappearance of p50 and p65 subunits from thecytoplasm. At 60 minutes, both p50 and p65 reappear in thecytoplasm. After pretreatment of HUVECs with oxidizedEPA, p50 and p65 are present at 15 and 60 minutes,indicating cytoplasmic retention of the subunits at both timepoints after stimulation with TNF�. Unoxidized EPA had noeffect in preventing the nuclear translocation of p50 and p65subunits, and at 15 minutes of treatment with TNF� virtuallyno cytoplasmic p50 or p65 was identified.
Oxidized EPA Does not Inhibit Cytokine-InducedPhosphorylation and Ubiquination of I�BaTo determine whether the mechanism of oxidized EPA-mediated inhibition of NF-�B was via inhibition of phosphor-ylation of I�B�, we performed Western blot analyses oncytoplasmic extracts of HUVECs using antibodies to I�B�(Figure 4A) and phosphorylated I�B� (Figure 4B). At base-line, I�B� was noted in the cytoplasm although no phosphor-
Effect of Unoxidized and Oxidized EPA on Expression of Cytokine-Induced Human Microvessel Endothelial Cell MCP-1 and IL-8
Veh Veh�TNF�EPA
(100 mM)�TNF�Ox EPA
(100 mM)�TNF�Ox EPA
(75 mM)�TNF�Ox EPA
(50 mM)�TNFaOx EPA
(25 mM)�TNF�Ox EPA
(10 mM)�TNF�
MCP-1 (pg/mL) 1358 (�13) 3482 (�45) 3357 (�42) 1056* (�41) 1088 (�15) 1409 (�41) 2465 (�30) 3332 (�64)
IL-8 (pg/mL) 911 (�15) 2716 (�60) 2940 (�143) 747* (�14) 1273 (�31) 3425 (�30) 3992 (�52) 3804 (�74)
Human microvessel endothelial cells were pretreated for 1 hour with vehicle (Veh) or unoxidized (EPA) or oxidized EPA (OxEPA) at indicated concentrations beforestimulation with TNF� for 5 hours.
Aliquots of the culture medium were analyzed for MCP-1 and IL-8 by ELISA techniques, and the results are expressed as pg/mL in the culture medium using valuesdetermined on a standard curve (�SEM).
*P�0.0001 compared to vehicle�TNF� and unoxidized EPA�TNF� (n�3).
Figure 2. Oxidized EPA inhibits TNF�-induced activation ofNF-�B but has no significant inhibitory effect on AP-1 activation.Nuclear extracts were prepared from HUVEC-treated with vehi-cle or unoxidized (UnoxEPA, 100 �mol/L) or oxidized EPA(OxEPA, 100 �mol/L) before stimulation with TNF� for 1 hour.The nuclear extracts were coincubated with 32P-labeled (A)NF-�B oligonucleotide or (B) AP-1 oligonucleotide, and electro-phoretic mobility shift assays were performed. In (A), nuclearextracts were preincubated with antibodies to p65 or excessunlabelled NF-�B (cold) as indicated. In (B) (lane 5), nuclearextracts were preincubated with excess unlabelled AP-1 beforeincubation with 32P-labeled AP-1 oligonucleotide (n�3).
Figure 3. Oxidized EPA inhibits TNF�-induced nuclear translo-cation of p50 and p65. HUVEC were treated with vehicle orunoxidized (UnoxEPA, 100 �mol/L) or oxidized EPA (OxEPA,100 �mol/L) for 1 hour before stimulation with TNF� for varyingtime points as indicated. Cytoplasmic extracts were preparedand the proteins were separated by SDS-PAGE, transferred topolyvinylidene fluoride membrane, and probed with antibodiesto p50, p65, and �-actin.
1624 Arterioscler Thromb Vasc Biol. September 2004
by guest on January 14, 2016http://atvb.ahajournals.org/Downloaded from

ylated I�B� was detected. After stimulation with TNF� for15 minutes, minimal I�B� was detectable in the cytoplasmwhereas a significant amount of phosphorylated I�B� wasnoted. After 60 minutes, I�B� reappeared in the cytoplasmeven though phosphorylated I�B� was still present. Similarresults were noted in HUVECs pretreated with oxidized EPA;after 15 minutes of stimulation with TNF�, there wascomplete absence of I�B� in the cytoplasm. This correlatedwith appearance of phosphorylated I�B�. At 60 minutes,even though phosphorylated I�B� was still present, I�B�reappeared in the cytoplasm (although the expression wasslightly decreased). These findings are similar to HUVECswith no oxidized EPA pretreatment, indicating that oxidizedEPA does not prevent phosphorylation and ubiquination ofI�B�, suggesting a different mechanism for the inhibition ofnuclear translocation of p50/p65 subunits.
Oxidized EPA Does not Inhibit Cytokine-InducedActivation of NF-�B in PPAR�-DeficientEndothelial CellsOur previous studies have shown that oxidized EPA is apotent activator of PPAR�.11 To determine whether PPAR�is required for the inhibitory effects of oxidized EPA onNF-�B activation, we isolated and grew endothelial cellsfrom wild-type and PPAR��/� mice thoracic aortas. Treat-ment of wild-type endothelial cells with TNF� for 2 hoursresulted in activation of NF-�B as indicated by the gel shifts.Pretreatment of wild-type endothelial cells with oxidizedEPA for 1 hour almost completely inhibited TNF�-inducedactivation of NF-�B, whereas incubation with unoxidizedEPA had little effect (Figure 5A). Endothelial cells fromPPAR��/� mice showed TNF�-induced activation of NF-�B.However, oxidized EPA pretreatment had no significantinhibitory effect on TNF� induced activation of NF-�B inPPAR��/� endothelial cells (Figure 5B).
DiscussionOmega-3 fatty acids in fish oil has been reported to improvethe prognosis of several chronic inflammatory diseases char-
acterized by leukocyte accumulation, including atherosclero-sis, inflammatory bowel disease, rheumatoid arthritis, psori-asis, etc.1–4
Omega-3 fatty acids, such as EPA and DHA, are highlypolyunsaturated and readily undergo oxidation at ambient andsubambient temperatures, even in the absence of exogenousoxidizing reagents.5,6 In view of the ease with which omega-3PUFA spontaneously oxidize and in vivo data suggestingextensive accumulation of oxidation products after fish oilconsumption, we investigated the possibility that oxidizedomega-3 fatty acids may be an important component of theobserved antiinflammatory effects of fish oil. In our previousstudies, we showed that oxidized EPA and not unoxidizedEPA pretreatment of HUVEC inhibits leukocyte adhesion tocytokine-stimulated HUVEC, and this effect is mediatedthrough a PPAR�-dependent pathway.11,12 In our presentstudies, we extend these observations and show that oxidizedEPA is also effective in inhibiting cytokine-induced endothe-lial chemokine expression, particularly MCP-1, and propose amechanism for these antiinflammatory effects.
The fact that oxidation of the omega-3 fatty acids isrequired for the aforementioned antiinflammatory effects isalso pointed out by other studies. Nohe et al and De Caterinaet al have shown that prolonged incubation of endothelialcells with native omega-3 fatty acids (26 hours of totalexposure, 6 hours before and 20 hours after TNF� stimula-tion) results in inhibition of cytokine and adhesion molecule
Figure 4. Oxidized EPA does not inhibit TNF�-induced phos-phorylation and ubiquination of I�B�. HUVEC were treated withvehicle or unoxidized (UnoxEPA, 100 �mol/L) or oxidized EPA(OxEPA, 100 �mol/L) for 1 hour before stimulation with TNF� forvarying time points as indicated. Cytoplasmic extracts were pre-pared and the proteins were separated by SDS-PAGE, trans-ferred to polyvinylidene fluoride membrane, and probed withantibodies to (A) I�B� and (B) phosphorylated-I�B�.
Figure 5. Oxidized EPA inhibits TNF�-induced activation ofNF-�B wild-type endothelial cells but has no inhibitory effect inendothelial cells derived from PPAR��/� mice. Nuclear extractswere prepared from thoracic aortic endothelial cells derivedfrom (A) wild-type and (B) PPAR��/� mice treated with vehicle orunoxidized EPA (EPA, 100 �mol/L) or oxidized EPA (OxEPA,100 �mol/L) before stimulation with TNF� for 2 hours. Nuclearextracts were preincubated with antibodies to p65 as indicated.The nuclear extracts were coincubated with 32P-labeled NF-�Boligonucleotide, and electrophoresis mobility shift assays wereperformed (n�3).
Mishra et al Oxidized EPA Inhibits NF-�B Activation 1625
by guest on January 14, 2016http://atvb.ahajournals.org/Downloaded from

expression, whereas shorter incubation periods (6 hours) hasno effect.30–32 This suggests that oxidation of omega-3 fattyacids takes place during the long incubation period, and thatthe oxidation product(s) and not the native omega-3 fattyacids are most likely responsible for the inhibition of leuko-cyte–endothelial interactions.
To ascertain that the inhibition of chemokine expressionwas not caused by the cytotoxic effects of oxidized EPA, weperformed MTT assays, which showed that oxidized EPA inthe doses used for our experiments had no effect on theviability of endothelial cells (data not shown). Also, oxidizedEPA did not have any effect on the constitutively expressedsurface proteins such as von Willebrand factor, endoglin, andhuman leukocyte antigen class I molecules.11,12
The expression of MCP-1 and IL-8 is regulated throughactivation of NF-�B. NF-�B activation appears to be neces-sary for the induction of chemokine genes, and deletion ofNF-�B binding sites results in an inability to induce thesegenes.26 In these studies, we show that oxidized EPA and notunoxidized EPA inhibits cytokine-induced activation ofNF-�B and promotes cytosolic retention of the p50 and p65subunits. Hence, the oxidized EPA-mediated inhibition ofMCP-1 and IL-8 in cytokine-stimulated endothelial cellscould be explained by an inhibitory effect of oxidized EPA onNF-�B activity.
The difference in the extent of inhibition of MCP-1 andIL-8 expression by oxidized EPA is most likely caused by thedifferential effects of oxidized EPA on endothelial NF-�Band AP-1 activation. Oxidized EPA almost completely inhib-its endothelial NF-�B activation, whereas it had minimaleffects in preventing AP-1 activation. Because AP-1 activa-tion alone can result in IL-8 expression,28 oxidized EPA hadonly a mild inhibitory effect on IL-8 expression.
What is the mechanism for the oxidized EPA-mediatedinhibition of cytokine-induced NF-�B activation? We firsthypothesized that oxidized EPA inhibits phosphorylation ofI�B� by inhibiting the IKK (kinase) complex. However, it isunlikely that oxidized EPA inhibits IKK (kinase) activitybecause phosphorylation and ubiquination of IkB� is noted inoxidized EPA-treated endothelial cells. In fact, when normal-ized to actin controls, oxidized EPA pretreatment beforeTNF� stimulation for 60 minutes resulted in 30% morep-IkB� when compared with TNF�-treated cells. Also, itunlikely that oxidized EPA induces IkB� expression becauseafter 15 minutes of cytokine stimulation, no IkB� is noted inthe cytoplasm of cells pretreated with oxidized EPA. After 60minutes, I�B� starts to appear in the cytoplasm of oxidizedEPA pretreated cells and its concentration is, in fact, some-what less than the TNF�-treated cells. Thus, it is likely thatoxidized EPA does not prevent NF-�B activation by increas-ing the expression of I�B�.
In our previous studies we noted that oxidized EPA is apotent activator of PPAR� and that PPAR� is needed for theinhibitory effects of oxidized EPA on leukocyte–endothelialinteractions,12 which lead us to hypothesize that oxidizedEPA might inhibit cytokine-induced NF-�B activationthrough a PPAR�-dependent pathway. Here, we show thatalthough oxidized EPA inhibits cytokine-induced NF-�Bactivation in wild-type cells, it has no inhibitory effects in
PPAR�-deficient endothelial cells, suggesting that oxidizedEPA mediates its inhibitory effects on NF-�B throughPPAR�. The PPAR�-mediated inhibitory effects of oxidizedEPA on NF-�B activation are possibly through direct inter-actions of PPAR� with the p50/p65 subunits. Delerive et al(1999) have shown, using glutathione S-transferase pull-down experiments, that after fibrate (PPAR� agonist) treat-ment, PPAR� physically interacts with p65 in vitro.33
Taken together with our previous studies,11,12 we show thatauto-oxidation of omega-3 fatty acids results in the genera-tion of oxidized compounds with potent antiinflammatoryproperties that inhibit proinflammatory responses such asleukocyte adhesion receptor and chemokine expression. Thecentral theme for the antiinflammatory effects of oxidizedomega-3 fatty acids is likely through inhibition of NF-�B viaa PPAR�-dependent pathway. The oxidation of omega-3fatty acids is likely to occur in areas of active inflammationcaused by increased expression of oxidative enzymes (eg,NADPH oxidase, myeloperoxidase, cyclooxygenase, lipoxy-genase) and the generation of reactive oxygen species in theseareas, which are capable of oxidizing PUFAs. The identifi-cation of these products could result in potent, low-toxicity,proinflammatory response inhibitors with potent PPAR�agonist and anti-NF-�B properties for the treatment of in-flammatory diseases.
AcknowledgmentsWe thank Dr. Lynn Stoll for assistance with ELISA assays and JoelCarl for assistance with the figures. We acknowledge support(Scientist Development Grant) from the American HeartAssociation (S.S.)
References1. Kromhout D, Bosschieter EB, de Lezenne Coulander C. The inverse
relation between fish consumption and 20-year mortality from coronaryheart disease. N Engl J Med. 1985;312:1205–1209.
2. Belluzzi A, Brignola C, Campieri M, Pera A, Boschi S, Miglioli M. Effectof and enteric coated fish oil preparation on relapses in Crohn’s disease.N Engl J Med. 1996;334:1557–1560.
3. Donadio JV Jr., Bergstralh EJ, Offord KP, Spencer DC, Holley KE. Acontrolled trial of fish oil in IgA nephropathy. N Engl J Med. 1994;331:1194–1199.
4. Volker D, Fitzgerald P, Major G, Garg M. Efficacy of fish oil concentratein the treatment of rheumatoid arthritis. J Rheumatol. 2000;27:2343–2346.
5. Chan HWS. The mechanism of autoxidation. Autoxidation of unsaturatedlipids. Academic Press: UK; 1987.
6. Porter NA, Caldwell SE, Mills KA. Mechanisms of free radical oxidationof unsaturated lipids. Lipids. 1995;30:277–290.
7. Gonzalez MJ, Gray JI, Schemmel RA, Dugan L Jr, Welsch CW. Lipidperoxidation products are elevated in fish oil diets even in the presence ofadded antioxidants. J Nutr. 1992;122:2190–2195.
8. Wander RC, Du S-H. Oxidation of plasma proteins is not increased aftersupplementation with eicosapentaenoic and docosahexaenoic acids. Am JClin Nutr. 2000;72:731–737.
9. Meydani M, Natiello F, Goldin B, Free N, Woods M, Schaefer E,Blumberg JB, Gorbach SL. Effect of long-term fish oil supplementationon vitamin E status and lipid peroxidation in women. J Nutr. 1991;121:484–91.
10. Higdon JV, Liu J, Du SH, Morrow JD, Ames BN, Wander RC. Supple-mentation of postmenopausal women with fish oil rich in eicosapenta-enoic acid and docosahexaenoic acid is not associated with greater in vivolipid peroxidation compared with oils rich in oleate and linoleate asassessed by plasma malondialdehyde and F2-isoprostanes Am J Clin Nutr.2000;72:714–722.
1626 Arterioscler Thromb Vasc Biol. September 2004
by guest on January 14, 2016http://atvb.ahajournals.org/Downloaded from

11. Sethi S, Eastman AY, Eaton JW. Inhibition of phagocyte-endotheliuminteractions by oxidized fatty acids: a natural anti-inflammatorymechanism? J Lab Clin Med. 1996;128:27–38.
12. Sethi S, Ziouzenkova O, Ni H, Wagner DD, Plutzky J, Mayadas TN.Oxidized omega-3 fatty acids in fish oil inhibit leukocyte-endothelialinteractions through activation of PPAR� Blood. 2002;100:1340–1346.
13. Gerszten RE, Garcia-Zepeda EA, Lim YC, Yoshida M, Ding HA,Gimbrone MA Jr, Luster AD, Luscinskas FW, Rosenzweig A. MCP-1and IL-8 trigger firm adhesion of monocytes to vascular endotheliumunder flow conditions. Nature. 1999;398:718–723.
14. Maus U, Henning S, Wenschuh H, Mayer K, Seeger W, Lohmeyer J. Roleof endothelial MCP-1 in monocyte adhesion to inflamed human endo-thelium under physiological flow Am J Physiol. 2002;283:H2584–H2591.
15. Shimizu H, Maruyama S, Yuzawa Y, Kato T, Miki Y, Suzuki S, Sato W,Morita Y, Maruyama H, Egashira K, Matsuo S. Anti-monocyte chemoat-tractant protein-1 gene therapy attenuates renal injury induced by protein-overload proteinuria. J Am Soc Nephrol. 2003;14:1496–505.
16. Egashira K. Molecular mechanisms mediating inflammation in vasculardisease: special reference to monocyte chemoattractant protein-1. Hyper-tension. 2003;41:834–41.
17. Dawson J, Miltz W, Mir AK, Wiessner C. Targeting monocyte chemoat-tractant protein-1 signalling in disease. Exper Opin Ther Targets. 2003;7:35–48.
18. Halliwell B, Gutteridge JM. Free radicals in biology and medicine, 3rded. New York, NY: Oxford University Press; 1993.
19. Denning GM, Wollenweber LA, Railsback MA, Cox CD, Stoll LL,Britigan BE. Pseudomonas Pyocyanin increases interleukin-8 expressionby human airway epithelial cells. Infect Immun. 1998;66:5777–5784.
20. Rice JB, Stoll LL, Li W-G, Denning GM, Weydert J, Charipar E,Richenbacher WE, Miller FJ Jr, Weintraub NL. Low-Level EndotoxinInduces Potent Inflammatory Activation of Human Blood Vessels: Inhi-bition by Statins. Arterioscler Thromb Vasc Biol. 2003;23:1576–1582.
21. Chomczynski P, Sacchi N. Single-step method of RNA isolation by acidguanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem.1987;162:156–9.
22. Huang H, McIntosh J, Hoyt DG. An efficient, nonenzymatic method forisolation and culture of murine aortic endothelial cells and their responseto inflammatory stimuli. In Vitro Cell Dev Biol Anim. 2003;39:43–50.
23. Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiationby RNA polymerase II in a soluble extract from isolated mammaliannuclei. Nucleic Acids Res. 1983;11:1475–1489.
24. Canty TG Jr., Boyle EM Jr, Farr A, Morgan EN, Verrier ED, PohlmanTH. Oxidative stress induces NF-kappaB nuclear translocation withoutdegradation of IkappaB�. Circulation. 1999;100:361–364.
25. Zandi E, Chen Y, Karin M. Direct phosphorylation of I�B by IKK� andIKK�: discrimination between free and NF-�B-bound substrate. Science.1998;281:1360–1363.
26. Martin T, Cardarelli PM, Parry GC, Felts KA, Cobb RR. Cytokineinduction of monocyte chemoattractant protein-1 gene expression inhuman endothelial cells depends on the cooperative action of NF-kappa Band AP-1. Eur J Immunol. 1997;27:1091–1097.
27. Roebuck KA. Regulation of Interleukin-8 gene expression. J InterferonCytokine Res. 1999;19:429–438.
28. Hipp MS, Urbich C, Mayer P, Wischhusen J, Weller M, Kracht M,Spyridopoulos I. Proteosome inhibition leads to NF-kB-independent IL-8transactivation in human endothelial cells through induction of AP-1. EurJ Immunol. 2002;32:2208–2217.
29. Lenard MJ, Baltimore D. NF-kB: a pleiotropic mediator of inducible andtissue specific gene control. Cell. 1988;58:227–229.
30. Nohe B, Johannes T, Dieterich HJ, Mayer K, Merfels M, Grimminger F,Seeger W. Antiinflammatory effects of omega-3 fatty acids vary atdifferent stages of inflammation. Am J Physiol. 2003;285:H2248–H2250.
31. De Caterina R, Bernini W, Carluccio MA, Liao JK, Libby P. Structuralrequirements for inhibition of cytokine-induced endothelial activation byunsaturated fatty acids. J Lipid Res. 1998;39:1062–1070.
32. De Caterina R CM, Clinton SK, Gimbrone MA Jr, Libby P. The omega-3fatty acid docosahexaenoate reduces cytokine-induced expression ofproatherogenic and proinflammatory proteins in human endothelial cells.Arterioscler Thromb Vasc Biol. 1994;14:1829–1836.
33. Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM,Gonzalez FJ, Fruchart J-C, Tedgui A, Haegeman G, Staels B. Peroxisomeproliferator-activated receptor alpha negatively regulates the vascularinflammatory gene response by negative cross-talk with transcriptionfactors NF-kappa B and AP-1. J Biol Chem. 1999;274:32048–32054.
Mishra et al Oxidized EPA Inhibits NF-�B Activation 1627
by guest on January 14, 2016http://atvb.ahajournals.org/Downloaded from
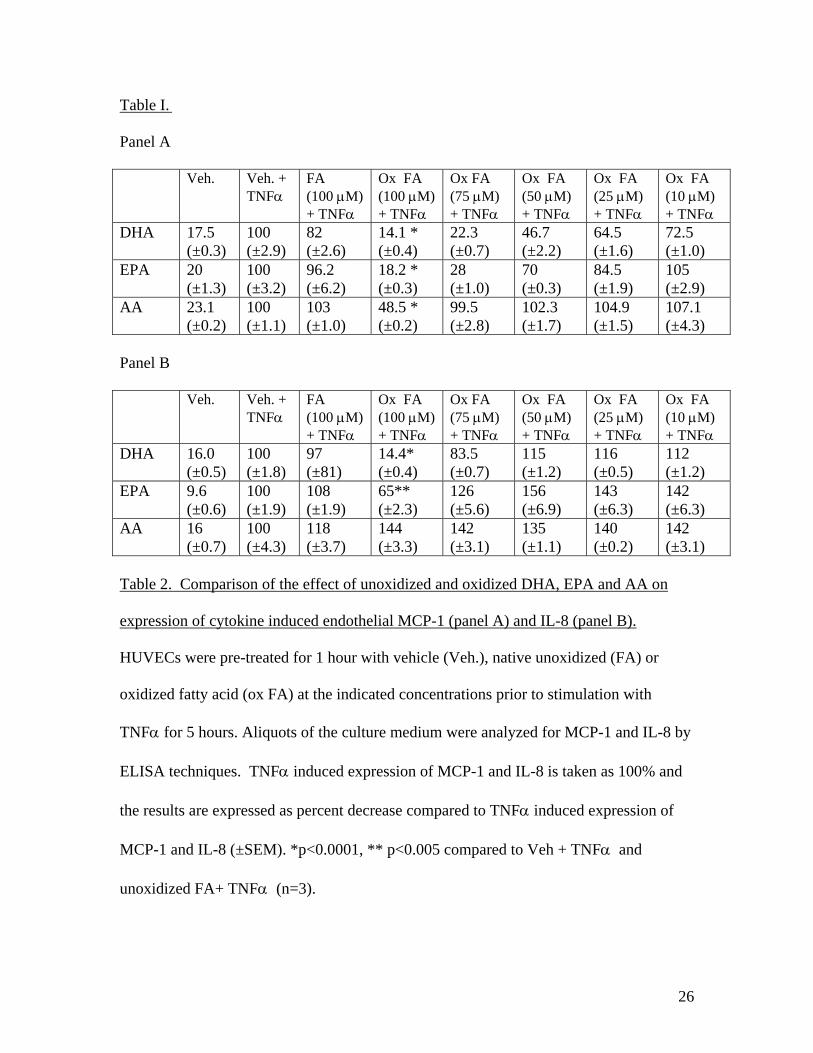
Table I.
Panel A
Veh. Veh. + TNFα
FA (100 µM) + TNFα
Ox FA (100 µM) + TNFα
Ox FA (75 µM) + TNFα
Ox FA (50 µM) + TNFα
Ox FA (25 µM) + TNFα
Ox FA (10 µM) + TNFα
DHA 17.5 (±0.3)
100 (±2.9)
82 (±2.6)
14.1 * (±0.4)
22.3 (±0.7)
46.7 (±2.2)
64.5 (±1.6)
72.5 (±1.0)
EPA 20 (±1.3)
100 (±3.2)
96.2 (±6.2)
18.2 * (±0.3)
28 (±1.0)
70 (±0.3)
84.5 (±1.9)
105 (±2.9)
AA 23.1 (±0.2)
100 (±1.1)
103 (±1.0)
48.5 * (±0.2)
99.5 (±2.8)
102.3 (±1.7)
104.9 (±1.5)
107.1 (±4.3)
Panel B Veh. Veh. +
TNFα FA (100 µM) + TNFα
Ox FA (100 µM) + TNFα
Ox FA (75 µM) + TNFα
Ox FA (50 µM) + TNFα
Ox FA (25 µM) + TNFα
Ox FA (10 µM) + TNFα
DHA 16.0 (±0.5)
100 (±1.8)
97 (±81)
14.4* (±0.4)
83.5 (±0.7)
115 (±1.2)
116 (±0.5)
112 (±1.2)
EPA 9.6 (±0.6)
100 (±1.9)
108 (±1.9)
65** (±2.3)
126 (±5.6)
156 (±6.9)
143 (±6.3)
142 (±6.3)
AA 16 (±0.7)
100 (±4.3)
118 (±3.7)
144 (±3.3)
142 (±3.1)
135 (±1.1)
140 (±0.2)
142 (±3.1)
Table 2. Comparison of the effect of unoxidized and oxidized DHA, EPA and AA on
expression of cytokine induced endothelial MCP-1 (panel A) and IL-8 (panel B).
HUVECs were pre-treated for 1 hour with vehicle (Veh.), native unoxidized (FA) or
oxidized fatty acid (ox FA) at the indicated concentrations prior to stimulation with
TNFα for 5 hours. Aliquots of the culture medium were analyzed for MCP-1 and IL-8 by
ELISA techniques. TNFα induced expression of MCP-1 and IL-8 is taken as 100% and
the results are expressed as percent decrease compared to TNFα induced expression of
MCP-1 and IL-8 (±SEM). *p<0.0001, ** p<0.005 compared to Veh + TNFα and
unoxidized FA+ TNFα (n=3).
26

Archana Mishra, Ashok Chaudhary and Sanjeev SethiPathway
-DependentαB Activation Via a PPARκOxidized Omega-3 Fatty Acids Inhibit NF-
Print ISSN: 1079-5642. Online ISSN: 1524-4636 Copyright © 2004 American Heart Association, Inc. All rights reserved.
Greenville Avenue, Dallas, TX 75231is published by the American Heart Association, 7272Arteriosclerosis, Thrombosis, and Vascular Biology
doi: 10.1161/01.ATV.0000137191.02577.862004;24:1621-1627; originally published online July 1, 2004;Arterioscler Thromb Vasc Biol.
http://atvb.ahajournals.org/content/24/9/1621World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://atvb.ahajournals.org/content/suppl/2004/09/10/24.9.1621.DC1.htmlData Supplement (unedited) at:
http://atvb.ahajournals.org//subscriptions/
at: is onlineArteriosclerosis, Thrombosis, and Vascular Biology Information about subscribing to Subscriptions:
http://www.lww.com/reprints
Information about reprints can be found online at: Reprints:
document. Question and AnswerPermissions and Rightspage under Services. Further information about this process is available in the
which permission is being requested is located, click Request Permissions in the middle column of the WebCopyright Clearance Center, not the Editorial Office. Once the online version of the published article for
can be obtained via RightsLink, a service of theArteriosclerosis, Thrombosis, and Vascular Biologyin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on January 14, 2016http://atvb.ahajournals.org/Downloaded from

CLINICAL SCIENCE
Protective role of oral antioxidant supplementation inocular surface of diabetic patientsV Peponis, M Papathanasiou, A Kapranou, C Magkou, A Tyligada, A Melidonis,T Drosos, N M Sitaras. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Br J Ophthalmol 2002;86:1369–1373
Aim: To investigate the effect of vitamin C and E supplementation in the levels of nitrite, nitric oxide(NO) related metabolite, and ocular surface parameters in diabetic patients.Methods: 50 patients with non-insulin dependent diabetes mellitus were given vitamin C (1000mg/day) and vitamin E (400 IU/day) supplementation for 10 days. Nitrite levels in tears weremeasured by photometric determination before and after vitamin supplementation. Tear functionparameters (Schirmer test I, BUT, ocular ferning test) and brush cytology analysis of the conjunctivalepithelium were also evaluated.Results: Nitrite levels were found to be significantly reduced (p<0.05) after 10 days of vitamin C andE supplementation. Improved values for Schirmer test, BUT test, and ocular ferning test were also found.Goblet cell density and grading of squamous metaplasia showed a significant improvement.Conclusions: Oxidative stress and free radical production are elevated in diabetes mellitus. Antioxi-dants, such as vitamin C and vitamin E, probably have an important role in reducing the oxidativedamage produced by nitric oxide and other free radicals and improving the ocular surface milieu.
Diabetes mellitus is associated with a number of ocularcomplications which can lead even to blindness. Thestudy of ocular surface manifestations during the course
of diabetes mellitus has increased in recent years. Forexample, 47%–67% of diabetic patients have primary corneallesions during their lifetime.1 In addition many diabeticpatients complain of dry eye symptoms, indicating a clear rolefor tear film abnormalities.2
The ocular surface is relatively unprotected and constantlyexposed to radiation, atmospheric oxygen, environmentalchemicals and physical insults, resulting in the generation ofreactive oxygen species (ROS) which are thought to contributeto ocular damage.3 Vitamin C (ascorbic acid) is found in highconcentration in the eye and is thought to be a primarysubstrate in ocular protection.4 Moreover, most studies havefound that people with diabetes mellitus have circulatingascorbic acid concentrations at least 30% lower than peoplewithout diabetes mellitus.5
Nitric oxide (NO) serves a wide variety of functions in vari-ous biological systems, both in intracellular compartments asa second messenger that responds to activation of plasmamembrane receptors and in extracellular compartments as aparacrine factor that carries information between cells.6
Nitric oxide is synthesised from L-arginine by three distinctisoforms of nitric oxide synthase (NOS) which are products ofthree different genes.7 The three isoforms of NOS are classifiedas follows: neuronal NOS (NOS1), inducible NOS (NOS2), andendothelial NOS (NOS3).8 Nitric oxide is constantly producedby NOS1 and NOS3, which are activated via the calcium/calmodulin complex. By contrast, the activity of NOS2 is inde-pendent of calcium and capable of generating large amountsof NO in the presence of immunological and inflammatorystimuli.8
Nitric oxide has been implicated in various physiologicaland pathological processes in the eye. It contributes to theregulation of aqueous humour dynamics, retinal neurotrans-mission, phototransduction, regulation of retinal vasculartone, ocular inflammatory diseases (uveitis, retinitis), degen-erative diseases (glaucoma, retinal degeneration), allergic eyedisease, and diabetic retinopathy.6–8
Induction of iNOS (inducible NOS) by cytokines leads to
the production of large amounts of NO which mediate the
destructive responses in ocular inflammation.6 Peroxynitrite
(ONOO−) formed by the reaction between nitric oxide and
superoxide (O2
−) is a powerful oxidant capable of causing tis-
sue injury.9
There are observations that high glucose levels lead to
increased NOS expression and increased NOS activity.10 This
could probably be attributed to increased oxidative stress that
is evident in diabetes mellitus.9
The above observations raise the question of whether anti-
oxidant supplementation could modify NO levels as well as
tear film characteristics in diabetes mellitus.
In this study the potential effect of oral vitamins C and E on
NO levels and various clinical and cytological parameters of
tear film and ocular surfaces in diabetic patients was investi-
gated.
PATIENTS AND METHODSNinety seven eyes of 50 patients diagnosed with non-insulin
dependent diabetes mellitus (NIDDM) were enrolled in this
study from the department of ophthalmology and the diabetes
centre of the general hospital of Piraeus. Patients characteris-
tics are summarised in Table 1.
Table 1 Patient characteristics
Age (years) 63.64 (10.33)*Sex (M/F) 17/33Duration of diabetes (year) 10.85 (9.08)*Therapy (hypogl/insul) 35/15Smoke (yes/no) 7/43HBA1C (%) 8.56 (1.58)*Neuropathy (no/yes) 26/24Retinopathy (0/1/2/3)† 14/18/12/6
*Mean (SD)†0 = no diabetic retinopathy, 1 = background retinopathy 2 =non-proliferative retinopathy, 3 = proliferative retinopathy.
See end of article forauthors’ affiliations. . . . . . . . . . . . . . . . . . . . . . .
Correspondence to:Nikolaos M Sitaras, MD,PhD, Department ofPharmacology, MedicalSchool, University ofAthens, 75 Mikras AsiasStreet, Athens 11527,Greece; [email protected]
Accepted for publication2 July 2002. . . . . . . . . . . . . . . . . . . . . . .
1369
www.bjophthalmol.com
group.bmj.com on January 14, 2016 - Published by http://bjo.bmj.com/Downloaded from

Exclusion criteria from the study were (a) any systemic dis-ease other than NIDDM, (b) medications other than hypogly-caemic drugs or insulin that could interfere with tears andocular surface parameters, (c) topical eye medications withinthe past 6 months, (d) history of ophthalmic surgical or laserprocedures, (e) eye diseases other than dry eye and/or diabeticretinopathy, (f) history of taking vitamin supplements. Thediagnosis of peripheral neuropathy was based on abnormalnerve conduction velocity test results and the presence ofsymptoms and signs of diabetic polyneuropathy such as neu-ropathic ulcers, dysaesthesias, paraesthesias, and abnormaldeep tendon reflexes. The status of diabetic retinopathy wasassessed by fundus examination and, if necessary, by fluores-cein angiography.
Patients entered in the study had a careful slit lamp exam-ination and a detailed ocular and systemic medical history.The dietary intakes of vitamin C and E were assessed carefully.
Immediately after accrual patients were given a combina-tion of vitamin C (1000 mg/day) and E (400 IU/day)(Nutrition Headquarters, IL, USA) for a period of 10 days.Vitamin C can enhance the activity of vitamin E by reducingthe tocopheroxyl radical and thereby restoring the radicalscavenging activity of vitamin E.11 All the measurements weredone on days 1 and 11. Any adverse reaction was immediatelyreported.
The study was carried out in accordance with the principlesof the Declaration of Helsinki. The subjects entered the studyafter informed consent was given, following a full explanationdescribing the procedures and the nature of the study.
MethodsAll measurements were done by the same investigator in a
quiet, dimly lit examination room of relatively constant
temperature and humidity.
Schirmer testA conventional Schirmer I test without anaesthesia was
performed by placing the short portion of a folded Schirmer
test strip (Ciba-Vision, Switzerland) over the lower lid margin
and measuring the amount of wetting in mm after 5 minutes.
Values of less than 10 mm of wetting are considered abnormal.
Fluorescein tear break up time (BUT)The standard tear film BUT measurement was performed
using moistened fluorescein strips (Chauvin Pharmaceuticals
Ltd, Essex, UK). The time interval between the last complete
blink and the appearance of the first black spot in the preocu-
lar tear film was measured three times and the mean value of
the measurements was calculated. A BUT value of less than 10
seconds was considered abnormal.
Ocular ferning testA glass capillary tube was used to collect the tear fluid from
the lateral part of the inferior tear meniscus. The sample was
dropped onto a light microscopy slide, allowed to dry at room
temperature for 5–10 minutes, and then observed by phase
contrast light microscopy at a magnification of ×40–100. On
the basis of its average appearance each sample was assigned
to one of the four types in the Rolando classification.12
Ronaldo classified tear film crystallisation into four types(grade I–IV): grade I consists of large, homogeneous ferns anduniform closely branching arborisation; in grade II ferns aresmaller and sparsely distributed; in grade III arborisation isscarcely present with many empty spaces, and in grade IVferning is totally absent and clusters of mucus may be present.
Types III and IV are considered abnormal. All the ferningspecimens were evaluated by two investigators in a maskedfashion.
Conjunctival brush cytologyA disposable brush (Accelon R Multi Biosampler, Medscand,
Malmö, Sweden) was used. After topical installation of anaes-
thetic the temporal bulbar conjunctiva was scraped by gentle
rotations of the brush under slit lamp observation. Care was
taken not to touch any part of the surface except that being
examined. The collected material was smeared on slides by
rotations of the brush and then fixed with Cytospray. The
brush samples were stained with periodic acid Schiff (PAS)
and Papanicolaou stain and were evaluated for the number of
goblet cells and the presence of squamous metaplasia of con-
junctival epithelial cells with a light microscope (Zeiss,
Germany) at a magnification of ×10–40 according to a grading
system originally described by Nelson13 and slightly modified
by Rojas et al.14 All cytology specimens were evaluated by two
investigators in a masked fashion.
Nitric oxide measurementsA volume of 100 µl water for injection was installed in the
lower conjunctival fornix and after three forced blinkings lav-
age fluid was collected using a micropipette and avoiding any
contact with the ocular surface. Nitrite concentration, the
stable end product of NO, was measured using the Griess
reaction.15 Briefly, the samples were mixed with the same vol-
ume of Griess reagent (mixture of one part of 0.1% naphthyl-
ethylenediamine dihydrochloride in water and one part of 1%
sulfanilamide in 5% hydrochloric acid) and allowed to react at
room temperature for 30 minutes. The concentrations of
nitrite were then determined photochemically by measuring
absorbance at 540 nm.
The order of performing the tests was as follows. Lavage
fluid for nitrite determination was taken first in order to avoid
any ocular irritation that could interfere with subsequent
measurements; ocular ferning test; BUT test; Schirmer test;
brush cytology. An adequate time interval (15–20 minutes)
was left between the tests to prevent one procedure interfering
with the results of subsequent tests.
Statistical analysisIn order to compare the results before and after treatment
parametric Student’s t tests were used for data which followed
normal distribution and distribution free statistical tests
(Wilcoxon sign rank test) for non-normal data. Regression
analysis was used to test the effects of various factors
(baseline characteristics such as age, duration of diabetes,
smoking habits, status of peripheral neuropathy, severity of
retinopathy) on the continuous variables. Spearman’s correla-
tion analysis was used to explore correlations between the
vitamin induced changes in the parameters that were
evaluated. A probability level less than 5% was considered sta-
tistically significant.
RESULTSSchirmer testThe mean Schirmer test value in the diabetic patients before
vitamin supplementation was 12.94 (SD 6.81) mm and after
treatment was 15.86 (6.44) mm. The difference was statisti-
cally significant (p<0.001). Seventy eight per cent (95% con-
fidence interval (95% CI): 64 to 88) of patients who received
treatment showed an improvement in Schirmer test values;
77% of patients with values less than 10 mm before treatment
and 78% of patients with values more than 10 mm before
treatment showed improvement suggesting an equal effect in
both groups.
A mild but statistically significant correlation was found
between the differences in Schirmer test values before and
after supplementation and the differences in BUT values
(Spearman’s correlation coefficient, rs=0.384; p=0.006). An-
other significant correlation was observed between the differ-
ences in Schirmer test values and the differences in nitrite
levels (rs=0.388; p=0.008).
1370 Peponis, Papathanasiou, Kapranou, et al
www.bjophthalmol.com
group.bmj.com on January 14, 2016 - Published by http://bjo.bmj.com/Downloaded from

BUT testThe median BUT test value in the diabetic patients before
vitamin supplementation was 10.25 seconds (range 5.5–25
seconds) and after treatment was 12.75 seconds (range 8–27
seconds). The difference was statistically significant
(p=0.001). All the patients (n=19) with BUT less than 10 sec-
onds showed improvement compared to 60% of patients with
BUT of more than 10 seconds.
A mild but statistically significant correlation was found
between the differences in BUT test values before and after
supplementation and the differences in ferning test values
(rs=0.438; p=0.002). Another significant correlation was
found between the differences in BUT test values and the dif-
ferences in goblet cells densities (rs=0.459; p=0.001).
Ocular ferning testThe average grade of the ferning test was 2.43 (SD 0.85) before
supplementation and 1.96 (0.77) after supplementation (p
<0.001), which shows an improvement. A total of 64% (95%
CI: 49 to 77) of patients showed improvement (defined as
grade before >grade after supplementation) (see also Fig 1).
Patients with duration of diabetes >15 years showed less
improvement (mean value 0.06 (0.77)) compared to patients
with duration of diabetes <15 years (mean value 0.64 (0.63));
p value = 0.018. Additionally, patients >65 years old showed
less improvement (mean value 0.19 (0.76)) compared to
patients <65 years old (mean value 0.7 (0.6)); p value = 0.008.
A mild but statistically significant correlation was found
between the differences in ferning test values before and after
supplementation and the differences in goblet cell densities.
(rs=0.458; p=0.001).
Nitrite concentration measurementsThe mean nitrite value in the diabetic patients before vitamin
supplementation was 3.24 (SD 1.23) µM and 2.21 (0.85) µM
after treatment. The difference was statistically significant
(p<0.001); 78% (95% CI: 63 to 89) of treated patients showed
a reduction in nitrite values after supplementation.
Using univariate regression analysis we showed that the
reduction in nitrite was greater in patients with proliferative
diabetic retinopathy compared to patients without diabetic
retinopathy (p=0.001).
Brush cytology analysisGoblet cell densityThe average goblet cell densities were 49 cells/per unit field
(range 19–259) before vitamin supplementation and 58 cells/
per unit field (range 18–178) after supplementation with the
difference being statistically significant (p=0.003); 76% of
patients showed an increase in goblet cell densities (95% CI:
61 to 86). We also found that patients exhibiting signs of dia-
betic neuropathy showed less increase in goblet cell densities
(mean value 1.2 (18.7) cells/per unit field) compared to
patients without diabetic neuropathy (mean value 15.11
(34.43) cells/per unit field); p=0.012.
Using multivariate regression analysis we showed that the
increase in goblet cell densities was adversely affected by age
(p=0.027) and duration of diabetes (p=0.035).
Squamous metaplasiaThe average grade of squamous metaplasia was 1.1 (0.42)
before supplementation and 0.9 (0.49) after supplementation
(p=0.01) which also shows a significant improvement; 44%
(95% CI: 30 to 58) of patients showed improvement (defined
as grade before >grade after supplementation).
We also found that diabetic neuropathy adversely affects the
improvement in squamous metaplasia stage (Pearson’s χ2,
p=0.042).
DISCUSSIONAlthough the Diabetes Control and Complications Trial has
identified hyperglycaemia as a significant risk factor for the
development of diabetic complications,16 the full spectrum of
the pathophysiological mechanisms of chronic diabetic
complications hasn’t been thoroughly elucidated. Some
equally tenable hypotheses for the origin of complications are
oxidative stress damage,17 18 advanced glycation end products
(AGE) hypothesis,19 20 aldose reductase pathway,21 reductive
stress (pseudohypoxia),22 true hypoxia,23 carbonyl stress,24
altered lipoprotein metabolism,25 increased protein kinase C
activity,26 and altered growth factors,27 and cytokine28 activi-
ties.
The various hypotheses overlap and intersect with one
another: AGE formation and altered polyol pathway activity
may lead to oxidative stress, oxidative stress may accelerate
AGE formation, reductive stress may lead to activation of pro-
tein kinase C activity, AGEs may induce growth factors and
cytokines production, and so on.29
Oxidative stress has been defined as a disturbance in the
balance between the production of reactive oxygen species
(especially free radicals) and the antioxidant defence status
which may lead to tissue injury.30 Oxidative stress is an
acknowledged pathogenetic mechanism in diabetic complica-
tions. Beyond that it appears that a number of antioxidant
defence systems are compromised in diabetic individuals.31
These include decrease in plasma ascorbic acid levels,32 33
intracellular deficiency of ascorbic acid (cellular scurvy),34 and
decrease in cellular vitamin E levels.35 Moreover, ocular tissues
probably have a higher free radical activity than any other
organ, mainly because of ultraviolet exposure.36
Higher levels of NO were found in the aqueous humour of
diabetic patients and this may induce inflammatory reactions
that cause cell damage.37 Biswas et al have reported that in the
resting state the levels of NO were higher in diabetic compared
to normal polymorphonuclear leucocytes.38 Macrophages have
been shown to increase NO production in diabetes.39 The large
amounts of NO which are produced may occur during iNOS
mediated NO release.40 ROS mediate signal transduction,
including activation of the transcription factor NF-kB
(nuclear factor kB), which is crucial for the inducible expres-
sion of genes involved in proinflammatory cytokines produc-
tion (IL-1, IL-6, TNF-α).41 The cytokines could then activate
DNA directed mRNA synthesis that induces synthesis of
iNOS.42 Additionally, the reaction of nitric oxide with superox-
ide (O2
−) leads to the formation of peroxynitrite, a potent oxi-
dant which contributes to ocular inflammation.43
Our results show that vitamin C and E supplementation
decreases NO levels in the lavage fluid from the ocular surface
of diabetic patients towards the levels that we have found in
normal healthy subjects (2.11 (0.93) µM, range 1.13–2.97 µM;
unpublished data). We suggest that the antioxidant activity of
these compounds results in a decrease in the oxidative burden
Figure 1 Ocular ferning grading (evaluated according toRolando12) in diabetic patients before and after vitamin C and E sup-plementation.
Oral antioxidant supplementation in ocular surface of diabetic patients 1371
www.bjophthalmol.com
group.bmj.com on January 14, 2016 - Published by http://bjo.bmj.com/Downloaded from

in the ocular surface that could lead to elimination of NO andits cytotoxic effects. We suggest that this could be achievedthrough downregulation of the cytokine induced activation ofiNOS. Interestingly, patients with proliferative diabetic retin-opathy showed the greater reduction in nitrite levels. Thesepatients had the greatest pretreatment values of nitrite andbenefit more from vitamin supplementation. Recently Yilmazet al found elevated NO levels in the vitreous of diabeticpatients with proliferative diabetic retinopathy compared tocontrols.44 We haven’t found significant differences in NO lev-els between smokers and non-smokers although smoking is awell known oxidative factor.
Our results also demonstrate that the orally administeredantioxidant supplements improve the tear film stability, tearsecretion, and health of the ocular surface. These results are inaccordance with previous reports.45 46
There are several mechanisms that could explain ourfindings. The antioxidant properties of vitamins C and E couldprotect the ocular surface from free radical attack and preservethe integrity of the ocular epithelium. The above mentionedreduction of NO and probably peroxynitrite could abolish thecytotoxic effects of these compounds. Additionally, vitaminsA, C, and E are needed for cell differentiation, development,and maintenance.47 Deficiencies of these vitamins in animalscaused loss of goblet cells in the conjunctiva and abnormalchromatin distribution in the nucleus of epithelial cells.47
Tseng et al speculated that inflammation and loss of vasculari-sation are two possible mechanisms for loss of goblet cells invarious ocular surface disorders.48 On the other hand vitaminC could have an endogenous anti-inflammatory role in theeye.49 Human tears are rich in vitamin C which actsprotectively for the ocular tissues.50 51 Certainly the nutritionalinfluences on tear film composition and physiology arecomplex.
We also found that patients with diabetic neuropathy areless likely to benefit from vitamin supplementation regardingthe improvement in goblet cell counts and squamousmetaplasia grade. These patients had probably decreased cor-neal sensitivity as a manifestation of their diabeticneuropathy.52 The subsequent decrease of neurotrophic effectsof the trigeminal sensory nerve on the cornea and theconjunctiva may be responsible for these ocular surfacechanges.53 As a consequence these patients had lower initialvalues for goblet cells and squamous metaplasia and lessimprovement compared to the patients without neuropathy.Age and duration of diabetes, which are also correlated withdecreased corneal sensitivity, adversely affected the changes ingoblet cell counts.
The improved BUT values, which suggest increased tear filmstability, are clearly correlated with the number of goblet cells.However, because tear film stability is essential for the healthof the ocular surface we couldn’t determine if BUT valuesimproved as a result of increased mucin production from gob-let cells48 or vice versa.
Schirmer test values were found to be significantlyincreased after the treatment period. Although this test isconsidered a rough screening test for the detection of tearproduction when performed as a standardised procedure withthe same investigator, as in our study it could provide valuableinformation. Shreeve et al54 proposed that lacrimal glandsecretion is promoted by micronutrients (zinc, magnesium)and vitamins (C, B6, and niacin). The apparent improvementin Schirmer test values and the correlation with BUTimprovement could also reflect the water retentive propertiesof mucins,55 resulting in an increased precorneal residencetime,55 increased corneal wettability, and reduced tearevaporation from the ocular surface.56
Tear ferning patterns depend on the interaction betweenelectrolytes, protein, and mucin macromolecules.57 We found amild correlation between the changes in goblet cells densitiesand ferning test values. The increased number of goblet cells
with subsequent production of mucin probably accounts forthe improvement in ocular ferning grade after vitaminsupplementation. This could also account for the minorchanges in ferning observed in patients >65 years old andwith a duration of diabetes >15 years.
We concluded in this study that oral vitamin C and Esupplementation could have a protective role in the ocularsurface of diabetic patients, leading to improvement in variousclinical and cytological parameters as well as a significantreduction in potentially hazardous nitric oxide levels. Al-though the effect of vitamins was beneficial, the shortduration of supplementation does not allow us to drawconclusions for the effects of prolonged vitamin administra-tion. This is a subject of further investigation.
. . . . . . . . . . . . . . . . . . . . .Authors’ affiliationsV Peponis, M Papathanasiou, Department of Ophthalmology, GeneralHospital of Piraeus “Tzaneion”, GreeceV Peponis, C Magkou, A Tyligada, N M Sitaras, Department ofPharmacology, Medical School, University of Athens, GreeceA Kapranou, Department of Pathology, General Hospital of Piraeus“Tzaneion”, GreeceA Melidonis, Diabetes Center, General Hospital of Piraeus “Tzaneion”,GreeceT Drosos, “Pammakaristos” General Hospital, Athens, Greece
REFERENCES1 Schultz RO, Van Horn DL, Peters MA, et al. Diabetic keratopathy. Trans
Am Ophthalmol Soc 1981;79:180–99.2 Goebbles M. Tear secretion and tear film function in insulin dependent
diabetics. Br J Ophthalmol 2000;84:19–21.3 Knight JA. Reactive oxygen species and the neurodegenerative
disorders. Ann Clin Lab Sci 1997;27:11–25.4 Rose RC, Richer SP, Bode AM. Ocular oxidants and antioxidant
protection. Proc Soc Exp Biol Med 1998;217:397–407.5 Will JC, Byers T. Does diabetes mellitus increase the requirement for
vitamin C? Nutrition Rev 1996;54:193–202.6 Becquet F, Courtois Y, Goureau O. Nitric oxide in the eye: multifaceted
roles and diverse outcomes. Surv Ophthalmol 1997;42:71–82.7 Goldstein I, Ostwald P, Roth S. Minireview. Nitric oxide: a review of its
role in retinal function and disease. Vis Res 1996;36:2979–94.8 Schmetterer L, Polak K. Role of nitric oxide in the control of ocular
blood flow. Prog Retin Eye Res 2001;20:823–47.9 Rösen P, Nawroth PP, King G, et al. The role of oxidative stress in the
onset and progression of diabetes and its complications: a summary of aCongress Series sponsored by UNESCO-MCBN, the American DiabetesAssociation and the German Diabetes Society. Diabetes Metabol Res Rev2001;17:189–212.
10 Cosentino F, Hishikawa K, Katusic ZS, et al. High glucose increasesnitric oxide synthase expression and superoxide anion generation inhuman aortic endothelial cells. Circulation 1997;96:25–8.
11 Packer L. Vitamin C and redox cycling antioxidants. In: Packer L, FuchsJ, eds. Vitamin C in health and disease. New York: Marcel Dekker,1997:95–121.
12 Rolando M. Tear mucus ferning test in normal and keratoconjunctivitissicca eyes. Chibert Int J Ophthalmol 1984;2:32–41.
13 Nelson DJ. Cellulose acetate impressions of the ocular surface. Dry eyestates. Arch Ophthalmol 1983;101:1869–72.
14 Rojas MV, Rodriguez MT, Blanco JA, et al. Impression cytology inpatients with keratoconjuctivitis sicca. Cytopathology 1993;4:347–55.
15 Green LC, Warner DA, Glogowski J, et al. Analysis of nitrate, nitrite and(15N) nitrate in biological fluids. Anal Biochem 1982;126:131–8.
16 The DCCT Research Group. The effect of intensive diabetes treatmenton the development and progression of long-term complications ininsulin-dependent diabetes mellitus: the Diabetes Control andComplications Trial. N Engl J Med 1993;329:977–86.
17 Giugliano D, Cerriello A, Paolisso G. Oxidative stress and diabeticvascular complications. Diabetes Care 1996;19:257–67.
18 Baynes JW, Thorpe SR. The role of oxidative stress in diabeticcomplications. Curr Opin Endocrinol 1997;3:277–84.
19 Brownlee M. Advanced protein glycosylation in diabetes and aging.Ann Rev Med. 1996;46:223–34.
20 Vlassara H. Recent progress in advanced glycation end products anddiabetic complications. Diabetes 1997;46(Suppl 2):S19–25.
21 Hotta N. New approaches for treatment in diabetes: aldose reductaseinhibitors. Biomed Pharmacother 1995;49:232–42.
22 Ido Y, Kilo C, Williamson JR. Cytosolic NADH/NAD+, free radical andvascular dysfunction in early diabetes mellitus. Diabetologia1997;40(Suppl 2):S115–17.
23 Cameron NE, Cotter MA. Metabolic and vascular factors in thepathogenesis of diabetic neuropathy. Diabetes 1997;46 (Suppl2):S31–7.
24 Lyons TJ, Jenkins AJ. Glycation, oxidation and lipoxidation in thedevelopment of the complications of diabetes: a carbonyl stresshypothesis. Diabetes Rev 1997;5:365–91.
1372 Peponis, Papathanasiou, Kapranou, et al
www.bjophthalmol.com
group.bmj.com on January 14, 2016 - Published by http://bjo.bmj.com/Downloaded from

25 Witztum JL. Role of modified lipoproteins in diabetic macroangiopathy.Diabetes 1997;46 (Suppl 2):S112–14.
26 Ishii H, Daissuke K, King GL. Protein kinase C activation and its role inthe development of vascular complications in diabetes mellitus. J MolMed 1998;76:21–31.
27 Pfeiffer A, Schatz H. Diabetic microvascular complications and growthfactors. Exp Clin Endocrinol Diabetes 1994;103:7–14.
28 Sharma K, Ziyadeh FN. Biochemical events and cytokine interactionslinking glucose metabolism to the development of diabetic nephropathy.Sem Nephrol 1997;17:80–92.
29 Baynes JW, Thorpe SR. Role of oxidative stress in diabeticcomplications. A new perspective on an old paradigm. Diabetes1999;48:1–9.
30 Halliwell B. Free radicals, antioxidants and human disease: curiosity,cause or consequence. Lancet 1994;344:721–4.
31 Hunt JV. Ascorbic acid and diabetes mellitus. In: Harris RJ, ed.Subcellular biochemistry. Vol 25. Ascorbic acid: biochemistry andbiomedical cell biology. New York: Plennum Press, 1996:369–405.
32 Jennings PE, Chirico S, Jones AF, et al. Vitamin C metabolites andmicroangiopathy in diabetes mellitus. Diabetes Res 1987;6:151–4.
33 Som S, Basu D, Mukherjee S, et al. Ascorbic acid metabolism indiabetes mellitus. Metabolism 1981;30:572–7.
34 Welch RW, Bergsten P, Butler JD, et al. Ascorbic acid accumulation andtransport in human fibroblasts. Biochem J 1993;294:505–10.
35 Karpen C, Cataland S, Orisio TM, et al. Interaction of platelet vitamin Eand thromboxane synthesis in type 1 diabetes mellitus. Diabetes1984;33:239–43.
36 Florence TM. The role of free radicals in disease. Aust NZ J Ophthalmol1995;23:3–7.
37 Chiou SH, Chang CJ, Chou CK, et al. Increased nitric oxide levels inaqueous humor of diabetic patients with neovascular glaucoma. DiabetesCare 1999;22:861–2.
38 Biswas SK, Bhelwa AP, Upadhyay AU, et al. Status of nitric oxide freeradicals in diabetic neutrophils: effect of diabetic serum factor on thegeneration of these species in normal neutrophils and their relation tolysosomal degranulation. Indian J Biochem Biophys 1993;30:293–6.
39 Wu G. Nitric oxide synthesis and the effect of aminoguanidine andNG-monomethyl- L-arginine on the onset of diabetes in the spontaneouslydiabetic BB rat. Diabetes 1995;44:360–4.
40 Honing MLH, Morrison PJ, Banga JD, et al. Nitric oxide availability indiabetes mellitus. Diabetes Metab Rev 1998;14:241–9.
41 Baeuerle PA, Henkel T. Function and activation of NF-kB in the immunesystem. Annu Rev Immunol 1994;12:141–79.
42 McCann SM, Licinio M, Wong L, et al. The nitric oxide hypothesis ofaging. Exp Gerontol 1998;33:813–26.
43 Allen JB, Keng T, Privalle CH. Nitric oxide and peroxynitrite productionin ocular inflammation. Environ Health Perspect 1998;106 (Suppl5):1145–9.
44 Yilmaz G, Esser P, Kociek N, et al. Elevated vitreous nitric oxide levels inpatients with proliferative diabetic retinopathy. Am J Ophthalmol2000;130:87–90.
45 Patel S, Farrell J, Blades KJ, et al. The influence of vitamins and traceelement supplements on the stability of the precorneal tear film. ActaOphthalmol 1993;71:825–9.
46 Blades KJ, Patel S, Aidoo KE. Oral antioxidant therapy for marginal dryeye. Eur J Clin Nutr 2001;55:589–97.
47 Amemiya TS. The eye and nutrition. Jpn J Ophthalmol 2000;44:320.48 Tseng SCG, Hirst LW, Maumenee AE, et al. Possible mechanisms for the
loss of goblet cells in mucin-deficient disorders. Ophthalmology1984;91:545–52.
49 Paterson AP, O’Rourke MC. Vitamin C levels in human tears. ArchOphthalmol 1987;105:376–7.
50 Choy CKM, Benzie IFF, Cho P. Ascorbic acid concentration and totalantioxidant activity of human tear fluid measured using the FRASC assay.Invest Ophthalmol Vis Sci 2000;41:3293–8.
51 Gogia R, Richer SP, Rose RC. Tear fluid content of electrochemicallyactive components including water soluble antioxidants. Curr Eye Res1998;17:257–63.
52 Dogru M, Katakami C, Inoue M. Tear function and ocular surfacechanges in noninsulin-dependent diabetes mellitus. Ophthalmology2001;108:586–92.
53 Marfurt CF, Echtenkamp SF. The effect of diabetes on neuropeptidecontent in the rat cornea and iris. Invest Ophthalmol Vis Sci1995;36:1100–6.
54 Shreeve C. Treating the dry eye. Ophthalmic Optician 1982;25:650–1.55 Nakamura M, Hikida M, Nakano T, et al. Characterization of water
retentive properties of hyaluronan. Cornea 1993;12:433–6.56 Tsubota K, Yamada M. Tear evaporation from the ocular surface. Invest
Ophthalmol Vis Sci 1992;33:2942–50.57 Golding TR, Baker AT, Rechberger J, et al. X-ray and scanning electron
microscopic analysis of the structural composition of tear ferns. Cornea1994;13:58–66.
Oral antioxidant supplementation in ocular surface of diabetic patients 1373
www.bjophthalmol.com
group.bmj.com on January 14, 2016 - Published by http://bjo.bmj.com/Downloaded from

patientssupplementation in ocular surface of diabetic Protective role of oral antioxidant
Melidonis, T Drosos and N M SitarasV Peponis, M Papathanasiou, A Kapranou, C Magkou, A Tyligada, A
doi: 10.1136/bjo.86.12.13692002 86: 1369-1373 Br J Ophthalmol
http://bjo.bmj.com/content/86/12/1369Updated information and services can be found at:
These include:
References #BIBLhttp://bjo.bmj.com/content/86/12/1369
This article cites 44 articles, 11 of which you can access for free at:
serviceEmail alerting
box at the top right corner of the online article. Receive free email alerts when new articles cite this article. Sign up in the
Notes
http://group.bmj.com/group/rights-licensing/permissionsTo request permissions go to:
http://journals.bmj.com/cgi/reprintformTo order reprints go to:
http://group.bmj.com/subscribe/To subscribe to BMJ go to:
group.bmj.com on January 14, 2016 - Published by http://bjo.bmj.com/Downloaded from

C Basic & Clinical Pharmacology & Toxicology 2006, 98, 68–73.Printed in Denmark . All rights reserved
Copyright C
ISSN 1742-7835
a-Tocopherol and a-Lipoic Acid Enhance the ErythrocyteAntioxidant Defence in Cyclosporine A-Treated Rats
Louise A. Lexis1,3, Robert G. Fassett2,3 and Jeff S. Coombes2,3
1Physiology Laboratory, School of Community Health, Charles Sturt University, Albury, 2640, 2Renal Research Unit,Launceston General Hospital, Launceston, Tasmania, 7250, and 3Exercise and Oxidative Stress Research Group,
School of Human Movement Studies, University of QLD, St Lucia, 4072, Australia
(Received May 3, 2005; Accepted August 2, 2005)
Abstract: The aim of this study was to determine the effects of dietary antioxidant supplementation with a-tocopheroland a-lipoic acid on cyclosporine A (cyclosporine)-induced alterations to erythrocyte and plasma redox balance. Ratswere randomly assigned to either control, antioxidant (a-tocopherol 1000 IU/kg diet and a-lipoic acid 1.6 g/kg diet),cyclosporine (25 mg/kg/day), or cyclosporine π antioxidant treatments. Cyclosporine was administered for 7 days after an8 week feeding period. Plasma was analysed for a-tocopherol, total antioxidant capacity, malondialdehyde, and creatinine.Erythrocytes were analysed for glutathione, methaemoglobin, superoxide dismutase, catalase, glutathione peroxidase, glu-cose-6-phosphate dehydrogenase, a-tocopherol and malondialdehye. Cyclosporine administration caused a significant de-crease in superoxide dismutase activity (P�0.05 control versus cyclosporine) and this was improved by antioxidant supple-mentation (P�0.05 cyclosporine versus cyclosporine π antioxidant; P�0.05 control versus cyclosporine π antioxidant).Animals receiving cyclosporine and antioxidants showed significantly increased (P�0.05) catalase activity compared toboth groups not receiving cyclosporine. Cyclosporine administration induced significant increases in plasma malondial-dehyde and creatinine concentration (P�0.05 control versus cyclosporine). Antioxidant supplementation prevented thecyclosporine induced increase in plasma creatinine (P�0.05 cyclosporine versus cyclosporine π antioxidant; P�0.05 con-trol versus cyclosporine π antioxidant), however, supplementation did not alter the cyclosporine induced increase inplasma malondialdehyde concentration (P�0.05 cyclosporine versus cyclosporine π antioxidant). Antioxidant supple-mentation resulted in significant increases (P�0.05) in plasma and erythrocyte a-tocopherol in both of the supplementedgroups compared to non-supplemented groups. In conclusion, dietary supplementation with a-tocopherol and a-lipoicacid enhanced the erythrocyte antioxidant defence and reduced nephrotoxicity in cyclosporine treated animals.
A recent animal study from our group showed that 7 daysadministration of the commonly used immunosuppressantcyclosporine (25 mg/kg/day) induced changes to erythrocyteand plasma redox balance (Lexis et al. 2005). These resultsindicate that cyclosporine may contribute to the alterederythrocyte and plasma redox balance observed in trans-plant recipients (Cristol et al. 1996; Hussein et al. 1997).This is important since cyclosporine-induced oxidativestress is thought to play a role in the premature cardiovascu-lar morbidity observed in transplant recipients (Morris et
al. 2000; Heitzer et al. 2001). Furthermore, inverse associ-ations have been found between cardiovascular disease andantioxidant levels in plasma and erythrocytes (Bonithon-Kopp et al. 1997; Nyyssonen et al. 1997). It is thereforeof interest to determine if antioxidant supplementation canenhance antioxidant defences and reduce cyclosporine-in-duced oxidative stress in the plasma and erythrocytes ofcyclosporine-treated animals.
It is widely believed that an optimum antioxidant supple-ment contains more than one nutrient (Upston et al. 2003),and the combination of a-tocopherol and a-lipoic acid has
Author for correspondence: Louise A. Lexis, School of Commun-ity Health, Faculty of Health Studies, Charles Sturt University.PO Box 789, Albury, 2640. Australia (fax π61 2 60516727, e-mailllexis/csu.edu.au).
generated scientific interest (Haramaki et al. 1995; Coombeset al. 2000). In a study by Coombes et al. (2000) the dietarycombination of a-tocopherol and a-lipoic acid reducedmyocardial lipid peroxidation after ischaemia in aged rats.Haramaki et al. (1995) showed that a-tocopherol and dihy-drolipoic acid exerted separate and synergistic protective ef-fects against the hypoxic-reoxygenated rat heart.
In the circulation, a-tocopherol is the major lipid solubleantioxidant in plasma lipoproteins and the erythrocytemembrane (Constantinescu et al. 1993; Frei 1999). In ani-mal studies, the cyclosporine-induced rise in renal malondi-aldehyde was prevented by a-tocopherol supplementation(Wang & Salahudeen 1995; Parra et al. 1998). It is thereforepossible that a-tocopherol may also prevent the cyclospor-ine-induced rise in plasma malondialdehyde.
a-Lipoic acid is a naturally occurring cofactor within py-ruvate dehydrogenase and a-keto-glutarate dehydrogenaseand is soluble in both lipid and aqueous phases (Wollin &Jones 2003). Although very little free a-lipoic acid isthought to occur in unsupplemented conditions, free a-lipo-ic acid has the ability to scavenge superoxide, hydrogen per-oxide, hydroxyl radicals, and peroxynitrite, and can also re-cycle glutathione (GSH), a-tocopherol, and ascorbic acid(Wollin & Jones 2003). In vitro, a-lipoic acid decreasedplasma susceptibility to oxidation (Marangon et al. 1999),was protective against haemolysis of human erythrocytes

69CYCLOSPORINE AND ERYTHROCYTE ANTIOXIDANT DEFENCE
induced by peroxyl radicals (Constantinescu et al. 1994),and increased GSH synthesis in isolated human erythro-cytes (Han et al. 1997). In a study by Bhatti et al. (2005)a-lipoic acid attenuated superoxide generation and kidneyexpression of NADPH oxidase in diabetic rats, and it wasconcluded that a-lipoic acid improves pathology in diabetesby reducing oxidative stress.
Therefore, the present study was conducted to determineif a dietary combination of a-tocopherol and a-lipoic acidcould attenuate the cyclosporine-induced alterations toerythrocyte and plasma redox balance that we have shownpreviously after 7 days of cyclosporine administration at adose of 25 mg/kg/day (Lexis et al. 2005).
Materials and Methods
Animals and experimental design. After approval was given fromCharles Sturt University’s Animal Care and Ethics Committee, thefollowing experiments were conducted. Forty-eight male SpragueDawley rats (3 weeks old) weighing 91.5∫8.7 g (mean∫S.D.) wererandomly assigned (nΩ12 per group) to one of four experimentalgroups: 1) control, 2) antioxidant, 3) cyclosporine, or 4) cyclospor-ine and antioxidant. All rats were fed rat chow and water ad libitumand maintained on a 12 hr light/dark photo-period with a roomtemperature of 21∫2 æ. The rat chow given to groups 2) and 4) wassupplemented with d-a-tocopheryl acid succinate (1000 IU/kg diet)and a-lipoic acid (1.6 g/kg diet). The supplemented diet was con-tinued until sacrifice. After 8 weeks of antioxidant supplementation,cyclosporine-treated animals were administered with 25 mg/kg ofthe drug (30 mg/ml dissolved in extra light olive oil and 4% ethanol)via intraperitoneal injection for 7 days. Control and antioxidantgroups were injected daily for 7 days with the same volume of thevehicle. Animal weights were recorded daily and cyclosporine dosesadjusted to changes in body mass. All injections were performed atapproximately the same time of the day. The concentration of a-tocopherol (1000 IU/kg diet) in the supplemented rat chow and thesupplementation period (8 weeks) was based on a study reportingsignificant increases in a-tocopherol concentration in the plasmaand erythrocytes of rats after 8 weeks of a-tocopherol supplementa-tion (Machlin & Gabriel 1982). The concentration of a-lipoic acid(1.6 g/kg diet) in the supplemented rat chow was the same as astudy by Coombes et al. (2000) in aged rats. Food intake was notmonitored in the present study because we have shown previouslythat food intake is similar in rats consuming standard rat chow andthose consuming antioxidant-supplemented chow (Coombes et al.2000). It has also been shown that weight loss occurred in cyclo-sporine-treated rats despite similar food intake to that of rats notreceiving cyclosporine (Wang & Salahudeen 1995).
Blood collection. On the day of sacrifice, blood was collected 3 hr afterdrug administration as it has previously been shown that cyclospor-ine reaches peak levels approximately 3 hr after intraperitoneal injec-tion in the rat (Ibarra et al. 1996). Just prior to exanguination ratswere administered with 90 mg/kg sodium pentobarbital via intraper-itoneal injection. After reaching a surgical plane of anaesthesia, thechest cavity was opened and rats were exanguinated via intracardiacpuncture. Approximately 5 ml of blood was collected in glass EDTAvacutainer tubes and aliquots of whole blood removed for analysisof cyclosporine concentrations and methaemoglobin. The remainingwhole blood was centrifuged at 4 æ for 10 min. at 800¿g. The plasmawas then removed, divided into aliquots, and stored at ª80 æ until bio-chemical analysis of total antioxidant capacity, a-tocopherol, malon-dialdehyde and creatinine. The buffy coat was then removed and dis-carded. Erythrocytes were subsequently washed three times withphosphate-buffered saline. An aliquot of washed cell suspension wasused to determine the concentration of erythrocyte GSH. Erythro-
cyte aliquots were stored at ª80 æ until biochemical analysis of a-toc-opherol and malondialdehyde, and the activities of superoxide dismu-tase, catalase, glucose-6-phosphate dehydrogenase (G6PD) and GSHperoxidase. Because plasma contains comparatively little superoxidedismutase, catalase and GSH peroxidase compared to the erythrocyte(Halliwell & Gutteridge 1986), these measurements were made onlyin the erythrocyte.
Cyclosporine A. Peak blood cyclosporine concentrations were deter-mined at the Hunter Area Pathology Service (Hunter Hospital,NSW, Australia) using the EMIT 2000 Specific Assay (DadeBehring – Syva, Deerfield, ILL, USA). The method was based ona homogenous enzyme immunoassay and analysis was carried outon a Roche Cobas Mira chemistry system (Roche, Basel, Switzer-land).
Erythrocyte glutathione. Erythrocyte glutathione was determined bythe method of Beutler et al. (1963). This method is based on theprinciple that GSH reduction of 5,5-dithiobis (2-nitrobenzoic acid)forms a yellow coloured anion which can be measured spectropho-tometrically at 412 nm.
Methaemoglobin. Methaemoglobin was determined by the methodof Evelyn & Malloy (1938). Methaemoglobin has an absorptionpeak at 630 nm, which disappears upon addition of potassium cy-anide. The difference in absorbance is therefore proportional to theconcentration of methaemoglobin. Total haemoglobin was meas-ured by conversion to cyanmethaemoglobin by the addition of pot-assium cyanide and potassium ferricyanide.
Erythrocyte antioxidant enzymes. G6PD activity was determinedusing a modification of the Sigma Diagnostics kit method (pro-cedure .345-UV) which is based on the spectrophotometric methodof Lohr & Waller (1974). G6PD catalyses the first step in the pen-tose phosphate pathway, oxidising glucose-6-phosphate to 6-phos-phogluconate and reducing NADP to NADPH. The rate of forma-tion of NADPH is proportional to G6PD activity and was meas-ured spectrophotometrically as an increase in absorbance at 340nm. One unit of G6PD activity is defined as 1mM NADPH pro-duced per minute.
Catalase activity was measured using a modification of themethod of Slaughter & O’Brien (2000). This method is based onthe competition of catalase with peroxidase for H2O2 produced bythe action of uricase on uric acid. Peroxidase uses available H2O2
to catalyse the formation of a coloured quinoneimine, howevercatalase inhibits this reaction by eliminating H2O2. The procedurewas carried out on an automated spectrophotometer (Cobas, Mira,Roche Diagnostics, Switzerland) and one unit of catalase activitydefined as the concentration of catalase that inhibits quinoneiminecolour formation from H2O2 by 50%.
GSH peroxidase activity was determined using a modification ofthe method of Andersen et al. (1997) which measures the oxidationof NADPH to NADP. The procedure was carried out on an auto-mated spectrophotometer (Cobas, Mira, Roche Diagnostics, Switz-erland) and one unit of GSH peroxidase activity defined as 1 mmolNADPH oxidised per min.
Superoxide dismutase activity was determined based on themethod of Madesh & Balasubramanian (1998) which measures theability of the enzyme to inhibit reduction of the tetrazolium dye, 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide (MTT)by superoxide. The procedure was carried out on a microplatereader (Titertek Multiskan MCC340, Flow Laboratories, NorthRyde, Australia) and one unit of superoxide dismutase defined asthe amount of ennzyme required to inhibit MTT reduction by 50%.All erythrocyte enzyme activities were normalised to haemoglobinconcentration.
Malondialdehyde. High performance liquid chromatography(HPLC) was used to determine plasma and erythrocyte malondial-

LOUISE A. LEXIS ET AL.70
dehyde using the method of Sim et al. (2003). The principle of thismethod is that malondialdehyde contained in plasma or erythro-cytes is derivatised with 2,4-di-nitrophenylhydrazine which formsstable hydrazones that can be easily separated by HPLC using diodearray detection (Shimadzu, Kyoto, Japan).
a-Tocopherol. HPLC was used to determine erythrocyte a-tocoph-erol concentrations using a modification of the method of Hatam &Kayden (1979). Proteins were precipitated from haemolysate withHPLC grade methanol and a-tocopherol extracted from the super-natant using HPLC grade hexane. Plasma a-tocopherol concen-trations were determined using a modification of the method ofTaibi & Nicotra (2002). Proteins were precipitated from solutionwith HPLC grade ethanol and a-tocopherol subsequently extractedusing HPLC grade hexane. a-Tocopherol extracted from erythro-cytes and plasma was measured using fluorometric detection (Shim-adzu, Kyoto, Japan). a-Lipoic acid was not assayed in the presentstudy as it has been shown to have a short elimination half-life andthere is a wide intersubject variation in time to peak concentrationin plasma after oral administration (Teichert et al. 1998). This issupported by unpublished observations in rats from our laboratory.
Plasma total antioxidant capacity. Total antioxidant capacity wasdetermined by the method of Miller et al. (1993) which is based onthe inhibition by antioxidants of the absorbance of the radical cat-ion 2,2ø-azinobis (3-ethylbenzthiazoline-6-sulfonate) (ABTSπ). TheABTS radical cation is formed by the interaction of ABTS withthe ferrylmyoglobin radical species, generated by the activation ofmetmyoglobin with hydrogen peroxide. Antioxidant compoundssuppress the absorbance of the ABTS radical cation to an extentand on a time scale dependent on the antioxidant capacity of theplasma. The assay was carried out on an automated spectro-photometer (Cobas, Mira, Roche Diagnostics, Switzerland).
Plasma creatinine. Plasma creatinine was determined using the JaffeReaction method. Absorbance was measured at 520 nm using anautomated spectrophotometer (Cobas, Mira, Roche Diagnostics,Switzerland).
Statistical analysis. Comparison between groups for cyclosporineconcentrations after the dosing period was made using an indepen-dent t-test. Comparison between groups for animal body weightdata, and for all markers of antioxidant status and oxidative stresswas made by one way ANOVA. If significance was determined, aTamhane’s (antioxidant status/oxidative stress data) or Tukey HSD(body weight data) post hoc test was used. All biochemical data isshown as the mean∫S.D. Significance was established at the 95%confidence level (P�0.05).
Results
Changes in body mass throughout the 7 day cyclosporineadministration period were significantly different (P�0.05)between cyclosporine-treated and placebo groups. Placebo-
Table 1.
Measures of antioxidant status, oxidative stress, and renal function in the plasma after 8 weeks antioxidant supplementation and 7 dayscyclosporine/placebo administration. Values are means∫S.D. Statistical difference at the level of P�0.05 using a Tamhane’s post hoc test:cΩdifferent from control, aoΩdifferent from antioxidant, csaΩdifferent from cyclosporine.
Cyclosporine πControl Antioxidant Cyclosporine antioxidant
a-Tocopherol (mM) 22.1∫7.3 39.6∫7.8c, csa 25.3∫3.7 37.4∫6.7c, csa
Total antioxidant capacity (mM) 1.59∫0.04 1.64∫0.14 1.59∫0.06 1.7∫0.14Malondialdehyde (mM) 23.9∫2 22.8∫2 32.5∫5c, ao 29.5∫4.9c, ao
Creatinine (mM) 53.5∫1.4 48.6∫3.2 68.3∫5.2c, ao 60.6∫6.6ao, csa
treated animals continued to gain weight (2.9% increase incontrol group, 4.7% increase in antioxidant group) at thesame rate (P�0.05 control versus antioxidant) during thecyclosporine administration period. Cyclosporine-treatedanimals failed to gain weight (0.7% decrease cyclosporine,0.4% decrease cyclosporine π antioxidant) during the drugtreatment period. Mean peak blood cyclosporine concen-trations of the cyclosporine treated, and cyclosporine πantioxidant treated animals at sacrifice were not significant-ly different (P�0.05) between groups, and were 3899∫1465mg/l and 3127∫1292 mg/l respectively.
Markers of plasma oxidative stress and antioxidant statusare presented in table 1. Antioxidant supplementation re-sulted in a significant increase in a-tocopherol concen-tration in both of the antioxidant supplemented groups(P�0.05 control versus antioxidant; control versus cyclo-sporine π antioxidant). Cyclosporine administrationcaused a significant increase in malondialdehyde concen-tration in both of the cyclosporine-treated groups (P�0.05control versus cyclosporine; control versus cyclosporine πantioxidant). Antioxidant supplementation did not signifi-cantly alter the cyclosporine-induced increase in plasmamalondialdehyde concentration (P�0.05 cyclosporine ver-sus cyclosporine π antioxidant). Plasma creatinine, meas-ured as an assessment of renal function at sacrifice, wassignificantly increased in the cyclosporine-treated animals(P�0.05 control versus cyclosporine). The mean creatinineconcentrations of cyclosporine π antioxidant-treated ani-mals were not significantly increased compared to controls(P�0.05), and were significantly lower than cyclosporinetreated animals (P�0.05 cyclosporine versus cyclosporineπ antioxidant).
Markers of erythrocyte antioxidant status and oxidativestress are presented in table 2. Cyclosporine administrationresulted in a significant decrease in superoxide dismutaseactivity (P�0.05 control versus cyclosporine). Antioxidantsupplementation attenuated the cyclosporine-induced de-crease in superoxide dismutase activity (P�0.05 cylosporineversus cyclosporineπantioxidant), however, activity re-mained significantly less than controls (P�0.05 control ver-sus cyclosporineπantioxidant). The combination of antioxi-dant supplementation and cyclosporine administration re-sulted in a significant increase in erythrocyte catalaseactivity when compared to both groups not receiving cyclo-sporine (P�0.05 control versus cyclosporine π antioxidant;

71CYCLOSPORINE AND ERYTHROCYTE ANTIOXIDANT DEFENCE
Table 2.
Measures of erythrocyte antioxidant status and oxidative stress after 8 weeks antioxidant supplementation and 7 days cyclosporine/placeboadministration. Values are means∫S.D. Statistical difference at the level of P�0.05 using a Tamhane’s post hoc test: cΩdifferent from control,aoΩdifferent from antioxidant, csaΩdifferent from cyclosporine.
Cyclosporine πControl Antioxidant Cyclosporine antioxidant
Superoxide dismutase (U/gHb) 6035∫443 3858∫528c 3053∫906c 4450∫383c, ao, csa
Catalase (U/gHb) 4855∫931 4981∫403 5232∫860 6045∫598c, ao
Glutathione peroxidase (U/gHb) 167.2∫28.9 182.9∫18.5 167.3∫18.1 177.7∫19.7Glucose-6-phosphate dehydrogenase (U/gHb) 22.9∫2 24.7∫2.1 23∫2 22.5∫4.4Glutathione (mg/100 ml RBC) 107.6∫11.1 123.9∫15.9 103∫14.8 119.6∫40Met Hb (% total Hb) 0.12∫0.14 0.19∫0.13 0.11∫0.15 0.07∫0.12a-Tocopherol (mg/ml packed cells) 2.82∫0.47 4.86∫0.35c 3.17∫0.28ao 5.09∫1.22c, csa
Malondialdehyde (mM) 16.9∫4 17.3∫1.5 20.4∫2.1 17.8∫1.7
antioxidant versus cyclosporine π antioxidant). Antioxi-dant supplementation resulted in a significant increase ina-tocopherol in both of the supplemented groups (P�0.05control versus antioxidant; control versus cyclosporine πantioxidant). Antioxidant supplementation alone caused asignificant decrease in superoxide dismutase activity(P�0.05 control versus antioxidant).
Discussion
These experiments showed that dietary supplementationwith a-tocopherol and a-lipoic acid enhanced the erythro-cyte antioxidant defence and reduced nephrotoxicity incyclosporine-treated rats. Antioxidant supplementation at-tenuated the cyclosporine-induced decrease in superoxidedismutase activity, and the combination of antioxidanttreatment and cyclosporine administration resulted in anincreased erythrocyte catalase activity. Antioxidant supple-mentation prevented the cyclosporine-induced increase inplasma creatinine, however, it did not alter the cyclosporine-induced increase in plasma malondialdehyde concentration.
In the present study, antioxidant supplementation attenu-ated the cyclosporine-induced decrease in erythrocyte su-peroxide dismutase activity, indicating an improved abilityof the erythrocyte to remove superoxide enzymatically. It ishypothesised that a-lipoic acid provided an additionalmechanism for the direct removal of superoxide, thus spar-ing superoxide dismutase under conditions of elevated reac-tive oxygen species. A limitation of the present study is thefailure to measure reactive oxygen species and future studiesdesigned to test this hypothesis via the direct measurementof superoxide are warranted.
Animals receiving both cyclosporine and antioxidantshad higher erythrocyte catalase activity compared to ani-mals not receiving cyclosporine, indicating an enhancedability to remove hydrogen peroxide from the circulation.We hypothesise that the increased activity is due to in-creased oxidative stress in conjunction with antioxidant sup-plementation. A potential source of plasma hydrogen per-oxide is the endothelium (Griendling et al. 2000) and Lon-goni et al. (2001) reported an increased production ofcellular oxidants after incubation of human endothelial cells
with cyclosporine. It has been shown that human erythro-cyte catalase contains tightly bound NADPH which is ef-fective in preventing and reversing inactivation of the en-zyme (Kirkman et al. 1987). It is possible in the presentstudy that antioxidant supplementation indirectly caused anincreased availability of erythrocyte NADPH leading to in-creased catalase activity. A limitation of the present studyis the failure to measure hydrogen peroxide levels. Futurestudies designed to test this hypothesis via the direct meas-urement of hydrogen peroxide and NADPH concentrationsare warranted.
Supplementation with a-tocopherol and a-lipoic acidprevented the cyclosporine-induced rise in plasma creati-nine. This supports the findings of previous studies showingthat a-tocopherol treatment prevented the cyclosporine-in-duced reduction in creatinine clearance in rats (Wang & Sal-ahudeen 1995; Parra et al. 1998). These results are not sur-prising given that kidney dysfunction is a major compli-cation of cyclosporine treatment, and available evidencesuggests a therapeutic role for antioxidants in the preven-tion of cyclosporine-induced nephrotoxicity (Parra Cid et
al. 2003). It has been shown that a-tocopherol inhibited thesynthesis of renal reactive oxygen species and improved re-nal function and histological alteration in cyclosporine-treated animals (Parra Cid et al. 2003).
Supplementation with a-tocopherol and a-lipoic acidfailed to attenuate the cyclosporine-induced increase inplasma malondialdehyde, indicating that either the dose orantioxidant combination was insufficient to prevent lipidperoxidation. In contrast, Naidu et al. (1999) showed thatco-administration of lacidipine (a calcium channel blockerwith an antioxidant effect) and cyclosporine to rats pre-vented the drug induced rise in plasma malondialdehyde.Similarly, Kumar et al. (1999) showed that co-administra-tion of melatonin and cyclosporine had the same effect. Po-tential sources of enhanced reactive oxygen species forma-tion after cyclosporine administration include the vascula-ture (Griendling et al. 2000) and the superoxide producingP450 enzyme systems in the kidney and liver (Yoshimura et
al. 1993). Future studies should be designed to determinethe relationship between reactive oxygen species in the circu-lation and lipid peroxidation products.

LOUISE A. LEXIS ET AL.72
In the present study, antioxidant supplementation re-sulted in significant increases in plasma and erythrocyte a-tocopherol concentration. These results support a previousstudy showing increases in a-tocopherol concentration inthe plasma and erythrocytes of rats after a similar supple-mentation protocol (Machlin & Gabriel 1982). This findingmay prove to be important for transplant recipients, as epi-demiologic studies have shown inverse associations betweenboth plasma and erythrocyte a-tocopherol concentration,and the incidence of cardiovascular disease (Gey & Puska1989; Bonithon-Kopp et al. 1997). Indeed, it has been re-ported that transplant recipients show decreased concen-trations of erythrocyte a-tocopherol (Cristol et al. 1996).Furthermore, a primary prevention study of 40 cardiactransplant recipients 0–2 years after transplantation showedthat a combination of a-tocopherol and vitamin C supple-mentation for 1 year retarded the progression of coronaryartery arteriosclerosis (Fang et al. 2002).
The cyclosporine-induced alterations to erythrocyteredox balance shown in the present study differ from a pre-vious study from our group despite the use of the samecyclosporine administration protocol (Lexis et al. 2005). Inour previous study, cyclosporine administration for 7 daysresulted in increased methaemoglobin, increased superoxidedismutase and catalase activities, and decreased GSH con-centration and G6PD activity (Lexis et al. 2005). The meanpeak cyclosporine levels (5726 mg/l) were significantly higher(P�0.05) than the levels of the present study (3899 mg/l and3127 mg/l for the cyclosporine, and cyclosporineπanti-oxidant-treated groups, respectively). The source of cyclo-sporine A in our previous study (Lexis et al. 2005) (Re-search Biology Institute (RBI) product .C224) differedfrom that of the present study (Fluka product .30024), andalthough unexpected, may explain the significantly differentmean blood concentrations. It is possible that the lowerconcentrations of cyclosporine in the present study resultedin decreased concentrations of reactive oxygen species in thecirculation and more moderate alterations to erythrocyteredox balance. A potential source of vascular superoxide isthe endothelium (Griendling et al. 2000) and Lopez-Ongilet al. (1998) reported a cyclosporine-induced dose-depend-ent increase of reactive oxygen species synthesis in bovinecultured aortic endothelial cells. To our knowledge, themechanism to explain alterations to erythrocyte antioxidantenzyme activity in the absence of cellular protein synthesisis not known, however, it is hypothesised that it is relatedto changing levels of oxidative stress within the erythrocyte.This may explain the controversial results between studiesand is an exciting area for future research.
In summary, these experiments show that dietary supple-mentation with a-tocopherol and a-lipoic acid enhanced theerythrocyte antioxidant defence and reduced nephrotoxicityin cyclosporine-treated rats. Antioxidant supplementationattenuated the cyclosporine-induced decrease in superoxidedismutase activity, and the combination of antioxidanttreatment and cyclosporine administration resulted in anincreased erythrocyte catalase activity. Antioxidant supple-
mentation prevented the cyclosporine-induced increase inplasma creatinine, however, it did not alter the cyclosporine-induced increase in plasma malondialdehyde. Future studiesshould measure reactive oxygen species concentrations as aresult of cyclosporine administration to test the hypothesisthat a cyclosporine-induced increase in vascular reactiveoxygen species alters erythrocyte redox balance. Additionalstudies should also determine the individual effects of die-tary supplementation with a-lipoic acid and a-tocopherolon cyclosporine-induced oxidative stress in the erythrocytebecause the mechanism by which a-lipoic acid exerts itsantioxidant effects are not completely understood. Al-though the relevance of these findings to the clinical settingis yet to be established, the results suggest that antioxidantsupplementation with a-tocopherol and a-lipoic acid maybe beneficial in the management of cyclosporine-dependentpatients.
AcknowledgementsThis work was supported by a grant from the Clifford
Craig Medical Research Trust. Technical assistance was pro-vided by Gary Wilson at the University of Queensland andBev de Jong at Charles Sturt University.
References
Andersen, H. R., J. B. Nielsen, F Nielsen & P. Grandjean: Antioxi-dative enzyme activities in human erythrocytes. Clin. Chem. 1997,43, 562–568.
Bhatti, F., R. W. Mankhey, L. Asico, M. T. Quinn, W. J. Welch &C. Maric: Mechanisms of antioxidant and pro-oxidant effects ofalpha-lipoic acid in the diabetic and nondiabetic kidney. KidneyInt. 2005, 67, 1371–1380.
Beutler, E., O. Duron & B. M. Kelly: Improved method for thedetermination of blood glutathione. J. Lab. Clin. Med. 1963, 61,882–888.
Bonithon-Kopp, C., C. Coudray, C. Berr, P. J. Touboul, J. M. Feve,A. Favier & P. Ducimetiere: Combined effects of lipid peroxi-dation and antioxidant status on carotid atherosclerosis in apopulation aged 59–71 y: The EVA study. Etude sur le Vieillise-ment Arteriel. Amer. J. Clin. Nutr. 1997, 65, 121–127.
Constantinescu, A., D. Han & L. Packer: Vitamin E recycling inhuman erythrocyte membranes. J. Biol. Chem. 1993, 268, 10906–10913.
Constantinescu, A., H. Tritschler & L. Packer: Alpha-lipoic acidprotects against hemolysis of human erythrocytes induced byperoxyl radicals. Biochem. Mol. Biol. Int. 1994, 33, 669–679.
Coombes, J. S., S. K. Powers, K. L. Hamilton, H. A. Demirel, R.A. Shanely, M. A. Zergeroglu, C. K. Sen, L. Packer & L. L. Ji:Improved cardiac performance after ischemia in aged rats supple-mented with vitamin E and alpha-lipoic acid. Amer. J. Physiol. –Regul. Integr. Comp. Physiol. 2000, 279, R2149–2155.
Cristol, J. P., M. F. Maggi, C. Vela, B. Descomps & G. Mourad:Lipid metabolism and oxidative stress in renal transplantation:implications for chronic rejection. Transplant. Proc. 1996, 28,2820–2821.
Evelyn, K. & H. Malloy: Microdetermination of oxyhemoglobin,methemoglobin and sulfhemoglobin in a single sample of blood.J. Biol. Chem. 1938, 126, 655–662.
Fang, J. C., S. Kinlay, J. Beltrame, H. Hikiti, M. Wainstein, D.Behrendt, J. Suh, B. Frei, G. H. Mudge, A. P. Selwyn & P. Ganz:Effect of vitamins C and E on progression of transplant-associ-ated arteriosclerosis: a randomised trial. Lancet 2002, 359, 1108–1113.

73CYCLOSPORINE AND ERYTHROCYTE ANTIOXIDANT DEFENCE
Lexis, L. A., R. S. Richards, R. G. Fassett & J. S. Coombes: Cyclo-sporine A induced changes to erythrocyte redox balance is timecourse dependent. Basic & Clinical Pharmacology & Toxicology2005, 97, 135–140.
Frei, B.: On the role of vitamin C and other antioxidants in athero-genesis and vascular dysfunction. Proc. Soc. Exp. Biol. Med.1999, 222, 196–204.
Gey, K. F. & P. Puska: Plasma vitamins E and A inversely correlatedto mortality from ischemic heart disease in cross-cultural epi-demiology. Ann. N.Y. Acad. Sci. 1989, 570, 268–282.
Griendling, K. K., D. Sorescu & M. Ushio-Fukai: NAD(P)H oxi-dase: role in cardiovascular biology and disease. Circ. Res. 2000,86, 494–501.
Halliwell, B. & J. M. Gutteridge: Oxygen free radicals and iron inrelation to biology and medicine: some problems and concepts.Arch. Biochem. Biophys. 1986, 246, 501–514.
Han, D., G. Handelman, L. Marcocci, C. K. Sen, S. Roy, H. Kobu-chi, H. J. Tritschler, L. Flohe & L. Packer: Lipoic acid increasesde novo synthesis of cellular glutathione by improving cystineutilization. BioFactors. 1997, 6, 321–338.
Haramaki, N., H. Assadnazari, G. Zimmer, V. Schepkin & L.Packer: The influence of vitamin E and dihydrolipoic acid oncardiac energy and glutathione status under hypoxia-reoxygen-ation. Biochem. Mol. Biol. Int. 1995, 37, 591–597.
Hatam, L. J. & H. J. Kayden: A high-performance liquid chromato-graphic method for the determination of tocopherol in plasmaand cellular elements of the blood. J. Lipid Res. 1979, 20, 639–645.
Heitzer, T., T. Schlinzig, K. Krohn, T. Meinertz & T. Munzel: Endo-thelial dysfunction, oxidative stress, and risk of cardiovascularevents in patients with coronary artery disease. Circulation 2001,104, 2673–2678.
Hussein, O., M. Rosenblat, G. Refael & M. Aviram: Dietary sel-enium increases cellular glutathione peroxidase activity and re-duces the enhanced susceptibility to lipid peroxidation of plasmaand low-density lipoprotein in kidney transplant recipients.Transplantation 1997, 63, 679–685.
Ibarra, A., G. Guizar-Sahagun, D. Correa, R. Kretschmer, I. Gri-jalva, F. J. Flores-Murrieta, G. Castaneda-Hernandez, A. Odor,R. M. Lopez, R. Franco-Bourland, A. L. Espitia, H. Salgado-Ceballos & I. Madrazo: Alteration of cyclosporin-A pharmaco-kinetics after experimental spinal cord injury. J. Neurotrauma1996, 13, 267–272.
Kirkman, H. N., S. Galiano & G. F. Gaetani: The function of cata-lase-bound NADPH. J. Biol. Chem. 1987, 262, 660–666.
Kumar, K. V., M. U. Naidu, A. A. Shifow, A. Prayag & K. S. Rat-nakar: Melatonin: an antioxidant protects against cyclosporine-induced nephrotoxicity. Transplantation 1999, 67, 1065–1068.
Lohr, G. W. & H. D. Waller: Glucose-6-phosphate dehydrogenase.In: Methods of enzymatic analysis. Ed.: H. U. Bergmeyer. Aca-demic Press, New York, USA, 1974, pp. 636–643.
Longoni, B., E. Boschi, G. C. Demontis, G. M. Ratto & F. Mosca:Apoptosis and adaptive responses to oxidative stress in humanendothelial cells exposed to cyclosporin A correlate with bcl-2expression levels. FASEB J. 2001, 15, 731–740.
Lopez-Ongil, S., O. Hernandez-Perera, J. Navarro-Antolin, G. Perezde Lema, M. Rodriguez-Puyol, S. Lamas & D. Rodriguez-Puyol:Role of reactive oxygen species in the signalling cascade of cyclo-sporine A-mediated up-regulation of eNOS in vascular endo-thelial cells. Brit. J. Pharmacol. 1998, 124, 447–454.
Machlin, L. J. & E. Gabriel: Kinetics of tissue alpha-tocopheroluptake and depletion following administration of high levels ofvitamin E. Ann N. Y. Acad. Sci. 1982, 393, 48–60.
Madesh, M. & K. A. Balasubramanian: Microtiter plate assay forsuperoxide dismutase using MTT reduction by superoxide. IndianJ. Biochem. Biophys. 1998, 35, 184–188.
Marangon, K., S. Devaraj, O. Tirosh, L. Packer & I. Jialal: Com-parison of the effect of alpha-lipoic acid and alpha-tocopherolsupplementation on measures of oxidative stress. Free Radic.Biol. Med. 1999, 27, 1114–1121.
Miller, N. J., C. Rice-Evans, M. J. Davies, V. Gopinathan & A.Milner: A novel method for measuring antioxidant capacity andits application to monitoring the antioxidant status in prematureneonates. Clin. Sci. 1993, 84, 407–412.
Morris, S. T., J. J. McMurray, R. S. Rodger, R. Farmer & A. G.Jardine: Endothelial dysfunction in renal transplant recipientsmaintained on cyclosporine. Kidney Int. 2000, 57, 1100–1106.
Naidu, M. U., K. V. Kumar, A. A. Shifow, A. Prayag & K. S. Rat-nakar: Lacidipine protects against cyclosporine-induced nephro-toxicity in rats. Nephron 1999, 81, 60–66.
Nyyssonen, K., M. T. Parviainen, R. Salonen, J. Tuomilehto & J. T.Salonen: Vitamin C deficiency and risk of myocardial infarction:prospective population study of men from Eastern Finland. Brit.Med. J. 1997, 314, 634–638.
Parra Cid, T., J. R. Conejo Garcia, F. Carballo Alvarez & G. deArriba: Antioxidant nutrients protect against cyclosporine Anephrotoxicity. Toxicology 2003, 189, 99–111.
Parra, T., G. de Arriba, J. R. Conejo, M. Cantero, I. Arribas, D.Rodriguez-Puyol, M. Rodriguez-Puyol & F. Carballo: Cyclospor-ine increases local glomerular synthesis of reactive oxygen speciesin rats: effect of vitamin E on cyclosporine nephrotoxicity. Trans-plantation 1998, 66, 1325–1329.
Sim, A. S., C. Salonikas, D. Naidoo & D. E. Wilcken: Improvedmethod for plasma malondialdehyde measurement by high-per-formance liquid chromatography using methyl malondialdehydeas an internal standard. J. Chromatogr. B: Analyt Technol. Biom-ed. Life Sci. 2003, 785, 337–344.
Slaughter, M. R. & P. J. O’Brien: Fully-automated spectrophoto-metric method for measurement of antioxidant activity of cata-lase. Clin. Biochem. 2000, 33, 525–534.
Taibi, G. & C. M. Nicotra: Development and validation of a fastand sensitive chromatographic assay for all-trans-retinol and toc-opherols in human serum and plasma using liquid-liquid extrac-tion. J. Chromatogr. B: Analyt. Technol. Biomed. Life Sci. 2002,780, 261–267.
Teichert, J., J. Kern, H. J. Tritschler, H. Ulrich & R. Preiss: Investi-gations on the pharmacokinetics of alpha-lipoic acid in healthyvolunteers. Int. J. Clin. Pharmacol. Therap. 1998, 36, 625–628.
Upston, J. M., L. Kritharides & R. Stocker: The role of vitamin Ein atherosclerosis. Prog. Lipid Res. 2003, 42, 405–422.
Wang, C. & A. K. Salahudeen: Lipid peroxidation accompaniescyclosporine nephrotoxicity: effects of vitamin E. Kidney Int.1995, 47, 927–934.
Wollin, S.D. & P. J. H. Jones: Alpha-lipoic acid and cardiovasculardisease. J. Nutr. 2003, 133, 3327–3330.
Yoshimura, R., N. Yoshimura, T. Nakatani, E. Kusunose, T. Yama-guchi, T. Oka & T. Kishimoto: The effects of cyclosporin on renalmicrosomal cytochrome P-450 systems. Clin. Nephrol. 1993, 40,339–345.


















