
editorial board - Haematologicasupplements.haematologica.org/Haematologica_2004_S10.pdf · Mario...
196
Mensile – Sped. Abb. Post. – 45% art. 2, comma 20B, Legge 662/96 - Filiale di Pavia. Il mittente chiede la restituzione dei fascicoli non consegnati impegnandosi a pagare le tasse dovute journal of hematology ISSN 1592-8721 educational edition volume 89 supplement no. 10 published by the ferrata-storti foundation, pavia, italy XXXI Congresso Nazionale Associazione Italiana Ematologia Oncologia Pediatrica Stresa, 10-12 ottobre 2004 Guest Editors: Franca Fossati Bellani, Giuseppe Masera h haematologica Journal of Hematology s10
Transcript of editorial board - Haematologicasupplements.haematologica.org/Haematologica_2004_S10.pdf · Mario...

Men
sile
–Sp
ed.A
bb.P
ost.
–45
%ar
t.2,
com
ma
20B,
Legg
e66
2/96
-Fili
ale
diPa
via.
Ilm
itten
tech
iede
lare
stitu
zione
deif
asci
coli
non
cons
egna
tiim
pegn
ando
sia
paga
rele
tass
edo
vute
journal ofhematology
ISSN 1592-8721educational edition
volume 89supplement no. 10
published by theferrata-storti
foundation, pavia, italy
XXXI Congresso Nazionale Associazione Italiana Ematologia Oncologia Pediatrica
Stresa, 10-12 ottobre 2004Guest Editors: Franca Fossati Bellani, Giuseppe Masera
hhaematologica Jour
nal o
f H
emat
olog
y
s10

haem
atolog
ica–
Vol.
89 s
uppl
emen
t n.
10,
Oct
ober
200
4 -
pp. 1
-182

e d i t o r i a l b o a r d
editor-in-chiefMario Cazzola (Pavia)
deputy editorsJoan Bladé (Barcelona), Carlo Brugnara (Boston), Rosangela Invernizzi (Pavia), Francesco Lo Coco (Roma), Paolo Rebulla (Milano), Gilles Salles (Lyon), Jordi Sierra Gil (Barcelona), Vicente Vicente Garcia (Murcia)
assistant editorsGaetano Bergamaschi (Pavia), Luca Malcovati (Pavia), Vittorio Rosti (Pavia)
scientific societies committeeSergio Amadori (Roma Italian Society of Hematology), Maria Benedetta Donati (Campobasso, Italian Society forHemostasis and Thrombosis Research), Gianluca Gaidano (Novara, Italian Society of Experimental Hematology),Momcilo Jankovic (Monza, Italian Association of Pediatric Hematology/Oncology), Fernando Martínez Brotons(Barcelona, Spanish Society of Thrombosis and Hemostasis), Ciril Rozman (Barcelona, Spanish Association ofHematology and Hemotherapy)
consulting editorsAdriano Aguzzi (Zürich), Claudio Anasetti (Seattle), Justo Aznar Lucea (Valencia), Carlo L. Balduini (Pavia), Michele Baccarani (Bologna), Giovanni Barosi (Pavia), Yves Beguin (Liège), Javier Batlle Fonrodona (A Coruña),Marie Christine Béné (Vandoeuvre Les Nancy), Dina Ben-Yehuda (Jerusalem), Mario Boccadoro (Torino), David T. Bowen (Dundee), Juan A. Bueren (Madrid), Dario Campana (Memphis), Marco Cattaneo (Milano), Michele Cavo (Bologna), Thérèsa L. Coetzer (Johannesburg), Francesco Dazzi (London), Valerio De Stefano (Roma),Judith Dierlamm (Hamburg), Meletios A. Dimopoulos (Athens), Ginés Escolar Albadalejo (Barcelona), Elihu H. Estey(Houston), J. H. Frederik Falkenburg (Leiden), Felicetto Ferrara (Napoli), Lourdes Florensa (Barcelona), Jordi Fontcuberta Boj (Barcelona), Renzo Galanello (Cagliari), Paul L. Giangrande (Oxford), Paolo G. Gobbi (Pavia),Lawrence T. Goodnough (St. Louis), Sakari Knuutila (Helsinki), Yok-Lam Kwong (Hong Kong), Bernhard Laemmle(Bern), Mario Lazzarino (Pavia), Ihor R. Lemischka (Princeton), Franco Locatelli (Pavia), Gabriel Márquez (Madrid),Estella Matutes (London), Cristina Mecucci (Perugia), Giampaolo Merlini (Pavia), Charlotte Niemeyer (Freiburg),Ulrike Nowak-Göttl (Münster), Michael O’Dwyer (Galway, Ireland), Alberto Orfao (Salamanca), Antonio Páramo(Pamplona), Stefano A. Pileri (Bologna), Giovanni Pizzolo (Verona), Susana Raimondi (Memphis), AlessandroRambaldi (Bergamo), Vanderson Rocha (Paris), Francesco Rodeghiero (Vicenza), Guillermo F. Sanz (Valencia),Miguel Angel Sanz (Valencia), Jerry L. Spivak (Baltimore), Alvaro Urbano-Ispizua (Barcelona), Elliott P. Vichinsky(Oakland), Giuseppe Visani (Pesaro), Neal S. Young (Bethesda)
editorial officeLuca Arcaini, Matteo Giovanni Della Porta, Igor Ebuli Poletti, Marta Fossati, Michele Moscato, Lorella Ripari,Rachel Stenner
o f f i c i a l o r g a n o fAEHH (Spanish Association of Hematology and Hemotherapy) AIEOP (Italian Association of Pediatric Hematology/Oncology)SETH (Spanish Society of Thrombosis and Hemostasis)SIE (Italian Society of Hematology)SIES (Italian Society of Experimental Hematology)SISET (Italian Society for Hemostasis and Thrombosis Research)
Direttore responsabile: Prof. Edoardo Ascari; Autorizzazione del Tribunale di Pavia n. 63 del 5 marzo 1955. Editing: m Mikimos - Medical Editions, via A. Fogazzaro 5, Voghera, ItalyPrinting: Tipografia PI-ME, via Vigentina 136, Pavia, ItalyPrinted in October 2004
Haematologica is sponsored by educational grants from the following institutions and companies:
IRCCS Policlinico S. Matteo, Pavia, Italy University of Pavia, Italy
José Carreras International Leukemia Foundation
haema
tologica

i n f o r m a t i o n f o r a u t h o r s , r e a d e r s a n d s u b s c r i b e r s
Haematologica (print edition, ISSN 0390-6078) publishes peer-reviewed papers on all areas of experimental and clinical hema-tology. The journal is owned by a non-profit organization, the Ferrata Storti Foundation, and the way it serves the scientificcommunity is detailed online: http://www. haematologica. org/main. htm (journal’s policy).
Papers should be submitted online: http://www. haematologica. org/submission. For the time being the journal also considerspapers submitted via surface mail (Editorial Office, Haematologica, Strada Nuova 134, 27100 Pavia, Italy) or as attachments toemail messages (office@haematologica. org). However, these submission modalities are discouraged and will be abolishedshortly.
Haematologica publishes editorials, research papers, decision making & problem solving papers, review articles and scien-tific letters. Manuscripts should be prepared according to the Uniform Requirements for Manuscripts Submitted to Biomed-ical Journals, prepared by the International Committee of Medical Journal Editors (ICMJE) and fully available online(http://www. icmje. org). Additional information is available online: http://www. haematologica. org/instructions. htm (instruc-tions to authors).
Additional papers may be considered for the purely online journal (Haematologica on Internet, ISSN 1592-8721). Becausethere are no space constraints online, Haematologica on Internet will publish several items deemed by peer review to be scien-tifically sound and mainly useful as educational papers. These will include case reports, irreplaceable images, educational mate-rial from scientific meetings, meeting abstracts, and letters to the Editor.
Galley Proofs and Reprints. Galley proofs should be corrected and returned by email, fax or express delivery within 72 hours.Minor corrections or reasonable additions are permitted; however, excessive alterations will require editorial re-evaluation andwill be possibly charged to the authors. Papers accepted for publication will be printed without cost. The cost of printing colorfigures will be communicated upon request. Reprints may be ordered at cost by returning the appropriate form sent by thePublisher.
Transfer of Copyright and Permission to Reproduce Parts of Published Papers. Authors will grant copyright of their articlesto the Ferrata Storti Foundation. No formal permission will be required to reproduce parts (tables or illustrations) of publishedpapers, provided the source is quoted appropriately and reproduction has no commercial intent. Reproductions with commer-cial intent will require written permission and payment of royalties.
Haematologica is published in two printed editions: International (worldwide except Spain, Portugal and Latin America) andSpanish (in Spain, Portugal and Latin America). Detailed information about subscriptions is available online: http://www.haematologica. org/subscribe. htm (subscriptions). While access to the online journal is free, online access to additional itemsof the website http://www. haematologica. org/ will require either institutional or personal subscription. Rates of the International edition for the year 2004 are as following:
Institutional Personal
Print edition and full access to the online journal plus additional items of haematologica. org Euro 350 Euro 150Full access to the online journal plus additional items of haematologica. org Euro 350 Euro 75
To subscribe to the International edition, please visit our web site http://www. haematologica. org/subscribe. htm or contact:Haematologica Journal Office, Strada Nuova 134, 27100 Pavia, Italy (phone +39. 0382.531182, fax +39. 0382.27721, E-mailoffice@haematologica. org). To subscribe to the Spanish print edition, please contact: Ediciones Doyma SA, Travesera de Gra-cia, 17-21, 08021 Barcelona, Spain (phone +34. 3. 4145706, fax +34. 3. 414-4911, E-mail: doyma@doyma. es).
Advertisements. Contact the Advertising Manager, Haematologica Journal Office, Strada Nuova 134, 27100 Pavia, Italy (phone+39. 0382.531182, fax +39. 0382.27721, E-mail: mikimos@haematologica. org).
Disclaimer. Whilst every effort is made by the publishers and the editorial board to see that no inaccurate or misleading data,opinion or statement appears in this journal, they wish to make it clear that the data and opinions appearing in the articles oradvertisements herein are the responsibility of the contributor or advisor concerned. Accordingly, the publisher, the editorialboard and their respective employees, officers and agents accept no liability whatsoever for the consequences of any inaccu-rate or misleading data, opinion or statement. Whilst all due care is taken to ensure that drug doses and other quantities arepresented accurately, readers are advised that new methods and techniques involving drug usage, and described within thisjournal, should only be followed in conjunction with the drug manufacturer’s own published literature.
Associated with USPI, Unione Stampa Periodica Italiana. Premiato per l’alto valore culturale dal Ministero dei Beni Culturali ed Ambientali
hhaematologica

The origin and power of a name
αιµα [aima] = blood; αιµατος [aimatos] = of blood, λογος [logos]= reasoning
haematologicus (adjective) = related to blood
haematologica (adjective, plural and neuter,used as a noun) = hematological subjects
Journal of Hematology2003 JCR® Impact Factor = 3.453
Ancient Greek
Scientific Latin
Scientific Latin
Modern English
h
haema
tologica



Haematologica 2004; vol. 89; supplement no. 10 - October 2004(indexed by Current Contents/Life Sciences and in Faxon Finder and Faxon XPRESS, also available on diskette with abstracts)
http://www. haematologica. org/
RELAZIONI
Domenica 10 ottobreAIEOP Formazione Medica Permanente. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1I “guariti”: luci ed ombre. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12Lettura Magistrale. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
Lunedi 11 ottobreIncontra l’Esperto . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19Il laboratorio triennale medico-infermieristico. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33L’ormone della crescita in Oncoematologia Pediatrica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46Lettura Magistrale. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50Osteosarcoma . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
Martedi 12 ottobreIncontra l’Esperto . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64Malattie orfane in Ematologia ed Immunologia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69Nuovi Farmaci per la Terapia di Supporto . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79Nuovi Farmaci Antitumorali . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89Le leucemie negli Infants . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
ABSTRACTS
Abstracts | Comunicazioni e PosterEmatologia e Coagulazione . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101Leucemie e Linfomi. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 109Tumori Solidi . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 124Trapianto . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 134Altro . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 141
Abstracts | Dati per Letti. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 155
Abstracts | Sessioni Infermieristiche. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 175
Indice degli Autori. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . i
supplement 10, October 2004 Table of Contents
XXXI Congresso Nazionale Associazione Italiana Ematologia Oncologia Pediatrica
Stresa, 10-12 ottobre 2004Guest Editors: Franca Fossati Bellani, Giuseppe Masera

1
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
Come e quando sospettare unaneoplasia: presentazione di casi clinici
Maura MassiminoUnità Complessa di Oncologia Pediatrica,Istituto Nazionale Tumori, Milano
IntroduzioneI tumori dei bambini non sono quelli degli adulti: han-
no nomi diversi, storia naturale diversa, cura diversa, pro-gnosi diversa.
L'oncologia pediatrica si occupa dello studio e della curadelle neoplasie dell'età pediatrica. Il range di età teoricoè di 0-15 anni, tuttavia, nella realtà, il limite dei 15/21anni viene esteso indefinitamente per alcune neoplasietipiche del bambini che si presentano, sia pure eccezio-nalmente, in età adulta.
Lo scopo dell'oncologia pediatrica è perfezionare i pia-ni di cura, aumentando il numero dei sopravviventi, ridur-re il numero delle ricadute, migliorare le terapie di salva-taggio, riconoscere i fattori prognostici di tipo clinico ebiologico, fornire un adeguato e continuo follow-up, pro-grammare la terapia di riabilitazione, prevenire, ricono-scere e trattare le sequele iatrogene, fornire, se richiesta,una consulenza genetica.
Vi è un costante aumento dell'incidenza delle neopla-sie dell'infanzia che è stato rappresentato dalle leucemienegli anni '80: da 3. 8 a 4. 8/100000, mentre dal '75 al '98vi è stato un aumento continuo delle neoplasie del siste-ma nervoso centrale (da 2.3 a 3/100000).
I tumori dell'infanzia sono circa l'1-2% di tutte le neo-plasie. Prevalgono neoplasie del sistema emopoietico eforme embrionali che derivano dal mesenchima: blastomi,sarcomi.
Il cancro dei bambini non è un'entità singola, ma unospettro di neoplasie diverse per istologia, sede di origine,razza dell'ospite, sesso ed età (Tabella 1).
Vi è una prevalenza nei maschi e l'incidenza aumentaal di sotto dei 5 anni ed al di sopra dei 15, con una mag-giore frequenza nella razza bianca.
Tra 0 e 2 anni prevalgono neuroblastoma, nefroblasto-ma e retinoblastoma; tra 3 e 5 anni, leucemie acute e sar-comi delle parti molli; tra 5 e 9 anni tumori del sistemanervoso centrale e linfomi maligni; tra 10 e 15 anni linfo-ma di Hodgkin, tumori dell'osso, sarcomi delle parti mol-li, che si presentano inoltre per tutta l'adolescenza e nelgiovane adulto.
Nei vent'anni compresi tra il 1975 ed il 1994, vi è sta-
to un trend all'incremento della sopravvivenza che è diver-samente rappresentato a seconda delle neoplasie: in par-ticolare esso è nettamente più spiccato per le leucemieche per le neoplasie del sistema nervoso (Tabella 2).
Quando e quante le diagnosi?Mentre in un grande centro oncologico vengono effet-
INCIDENZA DELLE NEOPLASIE IN ETA’ PEDIATRICA
TREND DI INCIDENZA E MORTALITA’ DELLE LEUCEMIE
XXXI Congresso Nazionale Associazione Italiana Ematologia Oncologia Pediatrica
Programma Medico
AIEOP Formazione Medica Permanente

tuate circa 150-170 nuove diagnosi di neoplasia malignaogni anno, un pediatra di famiglia vede nella sua vita 2bambini affetti da tumore, e presso la pediatria di un ospe-dale generale ne afferiscono circa 4 in un anno.
Le emergenze
La diagnosi di tumore in età pediatrica rappresenta, diper sé, qualunque essa sia, un'emergenza, per il turba-mento profondo che comporta nella vita del bambino edella sua famiglia. Viene richiesto ai medici un immedia-to coinvolgimento nel riconoscere, affrontare e, quandopossibile, risolvere, il problema. Situazioni di emergenzasono ben note al pediatra oncologo e sono relative aglieffetti della neoplasia su organi, o sistemi, o funzioni checompromettono le possibilità di vita più che la neoplasiastessa in quel momento. La conoscenza di tali problema-tiche può consentire un intervento terapeutico appro-priato e richiede quasi sempre la collaborazione pluri-spe-cialistica.
Le possibilità di successo nel trattamento di questeemergenze sono perciò legate principalmente ad una pre-coce interpretazione dei sintomi di esordio, alla tempesti-vità ed alle corrette modalità di intervento che non pos-sono prescindere da un'intensa, fiduciosa, ed attiva colla-borazione tra tutti coloro che si occupano di un bambinocon tumore.
In oncologia sono emergenze cliniche relativamente fre-quenti gli effetti meccanici legati allo “spazio” occupatodalla neoplasia ed i danni metabolici secondari alla proli-ferazione ed alla lisi cellulare.
Nell'ambito della patologia addominale non è rara l'e-venienza dell'addome acuto. Una massa in rapido accre-scimento può sanguinare, o provocare volvolo, invagina-zione, perforazione di anse intestinali. (Verrà illustrata unaTC con enorme neoplasia renale, operata d'urgenza permacroematuria). Un esordio di questo tipo, nei paesi occi-dentali, è caratteristico del linfoma di Burkitt (verrà illu-strata la TC post-operatoria di un paziente operato perlinfoma di Burkitt della valvola ileo-cecale con dissemi-nazione omentale e peritoneale).
Una massa addominale può anche comprimere reni e vieurinarie determinando insufficienza renale ingravescentefino all'anuria. In queste circostanze può essere difficiledistinguere quanto è imputabile ad un effetto meccanicodel tumore da quanto è attribuibile al meccanismo dellacrescita cellulare. (Verranno mostrate due immagini TC:l'una relativa ad una grossa massa addomino-pelvica, l'al-tra ad una massa pelvica condizionante idronefrosi).
Altre volte la diagnosi di linfoma può mimare condizio-ni più comuni (artrite, traumi, osteomieliti) e le condizio-ni generali del paziente ne possono essere estremamentecompromesse (verrà mostrata la radiografia di un femoreprossimale con multiple lesioni litiche).
Nell'ambito della patologia toracica, la cosiddetta sin-drome della vena cava o sindrome mediastinica, si riferi-sce alla compressione della cava e/o della trachea condi-zionante ostruzione del deflusso venoso e respiratorio. Inoncologia pediatrica si tratta di un'evenienza rara (il 12%dei linfomi mediastinici esordisce in questo modo) ed è piùfrequentemente secondaria all'esordio dei linfomi non-Hodgkin (verranno mostrate lastre coerenti con questo
quadro). Solo in casi eccezionali è necessario il supportodel medico rianimatore per la ventilazione assistita quan-do i provvedimenti terapeutici specifici sarebbero troppotardivi rispetto alla gravità della situazione respiratoria. Ingenerale, infatti, una volta definita la diagnosi, il tratta-mento specifico può indurre una rapida riduzione dell'in-gombro mediastino e la ripresa della normale funzionerespiratoria. Quando l'anamnesi, l'esame clinico ed i para-metri biochimici, oltre alla radiografia del torace in dueproiezioni, siano suggestivi per la diagnosi di tumore,occorre una rapida conferma cito-istologica che può avve-nire attraverso l'analisi del versamento pleurico, se pre-sente, o con l'ausilio chirurgico attraverso il prelievo (bio-psia a cielo aperto o agobiopsia) di eventuali adenopatieperiferiche o della massa mediastinica.
L'emergenza è legata, otre che alla rapida evoluzionedella malattia, anche all'anatomia del mediastino delbambino con scarsa resistenza di trachea e bronchi chepossono essere compressi o ostruiti prima che una sinto-matologia prodromica (dispnea da sforzo, tosse, ortopnea)possa far sospettare la presenza di un processo neoplasti-co intratoracico.
Una massa toracica può inoltre invadere il canale rachi-deo, come può avvenire per qualunque neoformazione adinsorgenza vertebrale o para-vertebrale, e condizionare lasindrome da compressione midollare, spesso primo sinto-mo di neoplasia. In questo caso è indispensabile una col-laborazione immediata e continua tra pediatra-oncologo,neurochirurgo, radioterapista, ed ortopedico, che dovran-no, di volta in volta, decidere il primo approccio terapeu-tico come: (i) l'effettuazione della laminectomia, (ii) del-la terapia medica, o (iii) della radioterapia. (Verranno illu-strate TC relative a processi espansivi di questa natura). Leneoplasie che possono esordire con sintomi da compres-sione epidurale acuta sono soprattutto il neuroblastoma,il sarcoma di Ewing, e, più raramente, i linfomi. E' impor-tante ricordare che dalla tempestività di un intervento sulcanale rachideo può dipendere la qualità futura della vitadel paziente in relazione all'entità dei deficit già prodot-ti dalla compressione midollare (verrà mostrata unasequenza RM):
Gravi emergenze neurologiche sono rappresentate dal-l'esordio clinico delle lesioni espansive del sistema nervo-so centrale, con alterazione dello stato di coscienza, sin-tomi riferibili ad ipertensione endocranica, variazioni delritmo respiratorio, deficit di forza o sensibilità, convulsio-ni. (Verrà mostrata RM che documenta grossa neoforma-zione espansiva sovratentoriale).
Talvolta segni e sintomi di compromissione neurologi-ca sono presenti per periodi prolungati, ma sottovalutatio equivocati fino all'aggravamento acuto. (Verrà mostra-ta RM con neoplasia della fossa posteriore ed idrocefalotetraventricolare).
Diagnosi inaspettate
Altre volte le diagnosi sono completamente inaspetta-te, e, pertanto, ritardate fino ad una maggiore evidenzaclinica. (Verranno illustrate storie cliniche che possonoessere suggestive di quanto esposto).
Le diagnosi, infine, possono essere casuali, con il riscon-tro di processi neoplastici nell'ambito di accertamenti col-
2
haematologica vol. 89[supplement n. 10]:October 2004
Domenica 10 ottobre

laterali. In questo assetto clinico-diagnostico verrannoillustrate due storie cliniche che si ritengono rappresen-tative.
Attualmente le possibilità di guarigione dei bambiniaffetti da neoplasia sono ancora molto diverse a secondadella sede di origine e dell'istotipo del tumore.
Occorre inoltre ricordare che i sopravviventi da tumoredell'età pediatrica sono oltre 100000 in Europa, più che isopravvissuti alla poliomielite negli anni 50.
Il nostro costante investimento deve essere rivolto, oltreche all'aumento del numero delle guarigioni, alla riduzio-ne della sofferenza fisica e psicologica, che in qualsiasifase della malattia può accompagnare i tempi diagnosti-ci o terapeutici, o gli effetti a distanza della malattia e del-le cure specifiche.
Bibliografia
Pizzo P, Poplack D, editors. Principles & Practice of Pediatric Oncol-ogy, pub. Lippincott Williams & Wilkins (December 2001)http://www. seer. ims. nci. nih. govWu XC, Chen VW, Steele B, Roffers S, Klotz JB, Correa CN, et al.Cancer incidence in adolescents and young adults in the UnitedStates, 1992-1997. J Adolesc Health 2003; 32:405-15. Kramarova E, Stiller CA. The international classification of child-hood cancer. Int J Cancer 1996;68:759-65. Smith MA, Freidlin B, Ries LA, Simon R. Trends in reported incidenceof primary malignant brain tumors in children in the United States.J Natl Cancer Inst 1998; 90:1269-77Gatta G, Capocaccia R, Coleman MP, Ries LA, Berrino F. Childhoodcancer survival in Europe and the United States. Cancer 2002;95:1767-72.Rivista Italiana di Pediatria 2000;26:333-41.Terracini B, Coebergh JW, Gatta G, Magnani C, Stiller C, Verdec-chia A, et al. Childhood cancer survival in Europe: an overview. EurJ Cancer 2001;37:810-6.
La lettura dell'emocromo con le nuove appa-recchiature
Mauro Buttarello, Valeria TemporinDipartimento di Medicina di Laboratorio, Azienda Ospeda-liera- Azienda USL 16, Padova
IntroduzioneI moderni analizzatori ematologici oltre ai tradizionali
parametri dell'esame emocromocitometrico e della leu-cocitometria differenziale, sono in grado di produrrenumerose altre informazioni, sia qualitative che quanti-tative (Tabella 1).
Per alcuni parametri consolidati, quali i conteggi leu-cocitario, eritrocitario e piastrinico, la determinazione del-la concentrazione emoglobinica o del volume corpuscola-re medio, le performance analitiche sono generalmenteritenute di alta qualità, per altri, soprattutto per il con-teggio leucocitario differenziale o il conteggio dei retico-lociti sono meno soddisfacenti.
Accanto a queste considerazioni ne emergono altre con-nesse al miglior utilizzo clinico dei nuovi parametri ana-litici resisi disponibili soltanto grazie agli analizzatoriautomatici e perciò non del tutto esplorati nelle loropotenzialità. Fra questi vanno ricordati la frazione di reti-colociti immaturi (IRF) e gli indici reticolocitari come ilvolume reticolocitario medio (MCVr) e il contenuto reti-colocitario medio di emoglobina (CHr).
Per altri parametri quali l'indice di anisocitosi eritroci-taria (RDW) o gli indici piastrinici, come il volume piastri-nico medio (MPV) o l'indice di anisocitosi piastrinica(PDW), pur disponibili da parecchi anni, sono necessariealcune considerazioni sui limiti di utilizzo in quanto risul-tano tutt'ora poco standardizzati, e in alcuni casi, influen-zati da variabili preanalitiche quali il tempo trascorso dalmomento del prelievo, o dai principi analitici utilizzati dalsingolo analizzatore (metodo ad impedenza o a dispersio-ne di luce).
Basi tecnologicheGli analizzatori ematologici a flusso utilizzano in varia
associazione delle tecnologie riconducibili a due principifondamentali: il metodo ad impedenza e il metodo elet-tro-ottico o a dispersione di luce (Tabella 2). Le tecniche adimpedenza si basano sulla rilevazione dell'aumento dellaresistenza elettrica causato dal passaggio di una cellulaattraverso un foro di poche decine di micron di diametro.La variazione di resistenza è in prima approssimazionedirettamente correlata al volume della cellula. Nei meto-di elettro-ottici la quantità di luce dispersa a piccoli ango-li rispetto alla direzione del raggio incidente (0.5-3 gra-di), risulta correlata al volume della cellula. Se le cellulesono state preventivamente colorate utilizzando reazionicitochimiche selettive o rese fluorescenti con fluorocromiin grado di legarsi a specifici componenti cellulari è pos-sibile ottenere ulteriori informazioni che permettono diclassificare i diversi tipi di cellule. In ogni caso le celluleche siano allo stato nativo, o modificate nelle dimensionio nel colore, vengono sospese in un fluido di trasporto eintrodotte in successione attraverso l'apparato di rileva-
3
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

4
haematologica vol. 89[supplement n. 10]:October 2004
Domenica 10 ottobre
zione che permette di misurarne alcune caratteristichefisiche o chimiche atte ad identificarle, e per alcuni cito-tipi a fornire informazioni sulla dimensione, composizio-ne o sullo stato funzionale.
Le prestazioni analiticheLe prestazioni analitiche vengono tradizionalmente
valutate con l'imprecisione e l'inaccuratezza. L'imprecisione (o errore casuale) è importante nell'in-
terpretare i risultati ottenuti dai campioni dei pazientianche se non è direttamente percepita dai clinici poichéil risultato fornito risente sia della variabilità biologica
che di quella analitica. Perciò quando si giudica la preci-sione di un metodo è necessario conoscere la variabilitàbiologica in quanto miglioramenti oltre certi limiti nellaprecisione analitica per parametri con elevata variabilitàbiologica comportano soltanto minimi vantaggi all'usoclinico del test.
L'attuale stato dell'arte per l'imprecisione è riportatonella Tabella 3. 1
I risultati possono essere considerati buoni con l'ecce-zione di alcuni componenti della leucocitometria diffe-renziale (eosinofili e basofili) e dei reticolociti.
L'inaccuratezza (o errore sistematico) ha come conse-guenza la diversa collocazione del paziente rispetto ad unprestabilito valore soglia (limiti superiore o inferiore del-l'intervallo di riferimento o livelli decisionali utili dal pun-to di vista clinico). Le conseguenze sono una diminuzionedella sensibilità o della specificità del test a seconda chelo shift sia positivo o negativo.
La tabella 3 riporta i risultati disponibili relativamenteall'inaccuratezza strumentale ottenuta per confronto conmetodi di riferimento. 1,2 I risultati sono soddisfacenti perla gran parte dei parametri quali leucociti, eritrociti, emo-globina, granulociti neutrofili, e sugli strumenti più recen-ti, anche conteggio piastrinico. Sono accettabili per altricome il volume corpuscolare medio, il conteggio reticolo-citario o linfocitario o i granulociti eosinofili. Rimangonoancora lontani dall'ottimale per i monociti e i granulocitibasofili.
La frazione di reticolociti immaturi: utilità clinica e limita-zioni
Heilmeyer fu uno dei primi a proporre una classifica-zione maturativa dei reticolociti basata sulla quantità disostanza reticolofilamentosa evidenziabile al microscopiodopo colorazione con blu brillante di cresile. Nonostantela potenziale utilità di una classificazione basata sullamaturazione reticolocitaria come indice di attività eritro-poietica midollare, questa di fatto non ebbe applicazioneclinica in quanto i risultati non erano riproducibili. In tem-pi più recenti è stato dimostrato che il reticolo è compo-sto prevalentemente da proteine e RNA ribosomiale e l'in-
Tabella 1.Parametri analitici prodotti dai moderni analizzatori emato-logici.
Parametro Abbott ABX Bayer Beckman-Coulter Sysmex Cell Dyn 4000 Pentra 120 ADVIA 120 LH 750 XE-2100
EEssaammee EEmmooccrroommoocciittoommeettrriiccooLeucociti + + + + +Eritrociti + + + + +Emoglobina + + + + +Ematocrito + + + + +MCV + + + + +MCH + + + + +MCHC + + + + +RDW + + + + +Piastrine + + + + +MPV + + + + +PDW + + + + +
CCoonntteeggggiioo lleeuuccoocciittaarriioodifferenzialeNeutrofili + + + + +Linfociti + + + + +Monociti + + + + +Eosinofili + + + + +Basofili + + + + +Luc − − + − −Aly − − − + −Lic − − − + −
CCoonntteeggggiioo rreettiiccoolloocciittaarriiooReticolociti + + + + +IRF + + + + +MCVr − + + − −MRV − − − + −Ret-y (RET He) − − − − +
CHr − − + − −
AAllttrrii ccoonntteeggggiiEritroblasti + + − + +
HPC − − − − +
MCH: contenuto emoglobinico cellulare medio; MCHC concentrazione emoglobinica cellularemedia; Luc: grandi cellule non colorate; Aly: linfociti atipici; Lic: grandi cellule immature;MRV: volume reticolocitario medio; HPC: cellule staminali.
Tabella 3. Stato dell'arte per l'imprecisione e l'inaccuratezza in emo-citometria automatizzata.
Imprecisione (CV%) Inaccuratezza (bias %)
Leucociti 1.6 - 2.7 6. 6Eritrociti 0.5 - 1.3 1.6Emoglobina 0.6 - 1.2 1.5MCV 0.2 - 0.6 2.2Piastrine 1.9 - 3. 2 6. 5Neutrofili 1.7 - 2.9 5Linfociti 2.2 - 3. 8 12Monociti 4. 5 -10.0 26Eosinofili 10.1 - 18. 5 29Basofili 20.9 - 27. 7 42Reticolociti 1.0 - 13. 9 −

5
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
troduzione di metodi citometrici che utilizzano colorantiche si legano selettivamente all'RNA e quindi produconosegnali riproducibili proporzionali al contenuto in acidonucleico ha riproposto la classificazione maturativa deireticolociti.
Il termine frazione di reticolociti immaturi (IRF)3 è sta-to introdotto per indicare la frazione meno matura deireticolociti. Esistono tuttavia diverse modalità di espres-sione a seconda degli analizzatori utilizzati.
Alcuni dividono i reticolociti in 3 ed altri in 2 diversepopolazioni sulla base del contenuto in RNA; ne conse-guono intervalli di riferimento molto diversi e diventa pro-blematico qualsiasi confronto fra diverse tecnologie. 4
Indipendentemente dal modo in cui è prodotto, l'IRF èrisultato tuttavia un indice precoce e sensibile di attivitàeritropoietica. Il miglior uso clinico specialmente nellaclassificazione delle anemie basate sulla valutazione del-la risposta eritropoietica midollare può essere ottenutoriportando in una matrice bidimensionale l'IRF versus ilconteggio reticolocitario assoluto.
Una applicazione clinica particolare in corso di retico-locitopenia è la precoce identificazione della rigenerazio-ne midollare a seguito di trapianto di midollo o di che-mioterapia.
Questa condizione è caratterizzata dalla ricomparsa direticolociti ad alto contenuto in RNA e quindi da unaumento dell'IRF rispetto al valore prossimo allo zerocaratteristico delle condizioni di aplasia. I problemi tutto-ra aperti circa l'utilizzo generalizzato di questo indice sonolegati alla diversa sensibilità analitica dei vari strumenti,più elevata per gli analizzatori che utilizzano metodi dirilevazione in fluorescenza, e alla difficoltà nel confron-tare i risultati ottenuti con analizzatori ematologici diver-si per modello o Costruttore.
Gli indici reticolocitariL'ultima generazione di analizzatori ematologici forni-
sce altre informazioni sui reticolociti come gli indici reti-colocitari analoghi ai corrispondenti indici eritrocitari. Fraquesti, i più promettenti dal punto di vista clinico sono ilcontenuto reticolocitario medio di emoglobina (CHr) e ilvolume reticolocitario medio (MCVr). Il CHr, che riflettedirettamente la sintesi di emoglobina nei precursori midol-lari, è una misura dell'adeguatezza dei depositi di ferro.
Da un lato questo parametro è importante perché è ingrado di evidenziare una eritropoiesi sideropenica anchequando i marcatori biochimici come la ferritina e la tran-sferrina risultano inadeguati (ad esempio nelle infiamma-zioni o nell'anemia da malattia cronica), e dall'altro risul-ta particolarmente utile nel controllo precoce della rispo-sta alla terapia con ferro per via venosa. 5
Bassi valori di CHr sono risultati indicativi di eritropoiesisideropenica in pazienti dializzati. Il deficit funzionale diferro che compare in soggetti sani trattati con eritropoie-tina si presenta con una precoce riduzione del CHr rispet-to al valore di base. In questo caso, la somministrazionedi ferro per via venosa previene la formazione di reticolo-citi ipocromici, aumentando il valore del CHr.
Il CHr è considerato l'indice più utile di deficit di ferroe di anemia sideropenica anche nei pazienti pediatrici. 6
Un minor numero di studi risulta disponibile relativa-mente all'utilità clinica dell'MCVr. Nei soggetti con depo-
siti marziali depleti, questo indice aumenta rapidamentea seguito di terapia a base di ferro e diminuisce altrettantorapidamente all'instaurarsi dell'eritropoiesi ferrocarente.MCVr diminuisce e i reticolociti sono più piccoli degli eri-trociti circolanti nelle macrocitosi carenziali trattate conB12 e/o acido folico. E' stato anche visto che durante ilfollow-up dopo trapianto di midollo si ha l'inversione delrapporto tra MCVr e volume degli eritrociti maturi (MCV)(in condizioni normali MCVr/MCV >1) dovuta alla dimi-nuzione di MCVr. L'attecchimento del trapianto è indica-to precocemente dalla normalizzazione di questo rappor-to. 7 Si può pertanto notare come CHr e MCVr abbianomolte utilizzazioni cliniche in comune.
Attualmente i limiti principali all'utilizzo di questi indi-ci dipendono dalla ridotta disponibilità di questi parame-tri a livello strumentale.
Il CHr è infatti disponibile soltanto sugli analizzatoridella serie ADVIA 120 prodotti da Bayer e un indicesovrapponibile denominato reticulocyte hemoglobin equi-valent (RET-He) è ottenibile con gli analizzatori XE o XTprodotti da Sysmex. L'MCVr o parametri simili che sonoottenibili con più strumentazioni (ABX, Bayer, Beckman-Coulter) soffrono tuttavia di evidenti problemi di stan-dardizzazione che rendono difficilmente comparabili irisultati numerici ottenuti con strumenti di Costruttoridiversi.
L'indice quantitativo di anisocitosi eritrocitariaI moderni analizzatori ricavano dall'istogramma di
distribuzione dei volumi eritrocitari sia il valore medio(MCV) che il relativo indice di dispersione definito reddistribution width (RDW) quasi sempre espresso comecoefficiente di variazione percentuale e più raramentecome deviazione standard.
Fin dai tempi di Price-Jones l'anisocitosi ottenuta dal-la misura dei diametri eritrocitari venne riconosciuta diutilità clinica, tuttavia la notevole indaginosità ne ha limi-tato l'applicazione. La possibilità di disporre di un indicequantitativo e non soggettivo ha risvegliato l'interesse dimolti ricercatori nei confronti dell'anisocitosi eritrocitaria.Bessman nei primi anni 80, utilizzando un analizzatoreCoulter S Plus II studiò un'ampia casistica e dai risultatiottenuti propose una classificazione morfologica delleanemie basata sui due parametri MCV e RDW. 8 Questaclassificazione prevede che oltre alla differenziazione informe micro, normo e macrocitiche, queste possano esse-re ulteriormente classificate in “omogenee” (con RDWnormale) ed eterogenee (con RDW aumentato). Fra le pri-me sono comprese le anemie ipoproliferative, le anemieaplastiche e le eterozigoti talassemiche; tra le seconde leanemie da deficit nutrizionale di ferro, B12 e acido folicoe le anemie sideroblastiche. All'inizio ci furono numeroseconferme a questo inquadramento al punto che l'RDWentrò a far parte, in molti laboratori, del referto routina-rio. Tuttavia, accanto alle conferme cominciarono ad evi-denziarsi numerose eccezioni quali l'aumento dell'RDW inpazienti con anemia da infezioni croniche e in almenometà dei soggetti affetti da eterozigoti talassemica, o, alcontrario, valori normali in circa il 15-20 % di soggettiaffetti da anemia ferrocarenziale.
In conclusione esistono ampie dispersione dei valori diRDW all'interno della stessa patologia e ciò ne ha intac-

cato l'utilità nella diagnostica differenziale pur conser-vandola come generico indicatore di anormalità. 9 Unaulteriore complicazione deriva dal metodo di calcolo del-l'RDW, infatti sotto la medesima sigla si celano indiciespressi in modo del tutto differente: coefficiente di varia-zione nella maggioranza dei casi (Abbott, ABX, Bayer,Beckman-Coulter), ma anche misura diretta dell'ampiez-za della curva (Sysmex). Anche nel caso in cui RDW siaespresso allo stesso modo gli intervalli di riferimento cal-colati sui soggetti sani differiscono quando ottenuti conanalizzatori di Marchi diversi e talvolta anche con model-li diversi dello stesso Costruttore. 10 Questo è spiegabilecon l'adozione di diversi algoritmi per la troncatura delladistribuzione, indispensabile per eliminare i valori estre-mi spesso artefattuali. Pertanto ogni considerazione sul-l'uso clinico (diagnostico, diagnostico-differenziale o dicontrollo post terapeutico) dell'RDW deve essere ricon-dotta agli intervalli di riferimento stabiliti per ciascun tipodi analizzatore.
Gli indici piastriniciGli analizzatori automatici oltre al conteggio piastrini-
co forniscono anche il volume piastrinico medio (MPV) eil relativo coefficiente di variazione (PDW) come misuradella loro eterogeneità. Il volume delle piastrine è deter-minato durante la frammentazione dei megacariociti. Seb-bene il meccanismo sia tutt'ora non chiaro, la ploidia deimegacariociti appare strettamente correlata con il volu-me piastrinico. Modelli sperimentali mostrano che l'MPVè aumentato quando sia la produzione che la distruzionedelle piastrine sono stimolate dalla ipossia cronica o dal-la somministrazione di siero antipiastrine. Simili osserva-zioni sono state fatte in pazienti diabetici o in fumatoridove sembra esserci un aumento del turnover piastrinicomediato da citochine quali IL3, trombopoietina e IL6. Que-ste citochine, ed in particolare IL6, hanno influenza sullaploidia megacariocitaria portando alla produzione di pia-strine più grandi e funzionalmente più attive. 11 L'MPV èrisultato diminuito in pazienti trombocitopenici con ridot-ta attività midollare quali in corso di aplasia o con caren-za di B12 o acido folico.
Nei soggetti sani si ha una relazione diretta fra MPV ePDW, relazione mantenuta nella porpora trombocitopeni-ca idiopatica e nella leucemia mieloide cronica nelle qua-li aumentano entrambi. Ciò non avviene nelle anemie ipo-plastiche, nelle anemia megaloblastiche o durante che-mioterapia citotossica in cui si ha riduzione di MPV conaumento del PDW.
L'anticoagulante raccomandato per le determinazioniemocromocitometriche comprensive anche degli indicipiastrinici è l'EDTA bi o tripotassico. Quando il sangue vie-ne a contatto con l'EDTA le piastrine cambiano rapida-mente forma passando da dischi di 2-4 micron di diame-tro a sferoidi ricoperti di appendici filamentose. La sferi-cizzazione delle piastrine è inizialmente isovolumetricama nel giro di 1-2 ore il volume si modifica progressiva-mente fino a raggiungere una condizione di equilibrioanche se non definitivo. Nell'arco di questo periodo l'MPVrisulta aumentato mediamente di circa il 20% quandomisurato con analizzatori ad impedenza e diminuito dicirca il 10% quando ottenuto con metodi elettro-ottici(12). I vari tentativi di correggere matematicamente a
posteriori i risultati sono peraltro falliti per il comporta-mento non prevedibile dei singoli campioni.
Con l'uso dell'EDTA l'MPV rappresenta pertanto un para-metro poco attendibile13 e soltanto il conteggio delle pia-strine ha mantenuto la condizione di parametro con appli-cazioni cliniche certe.
ConclusioniL'evoluzione della tecnologia applicata agli analizzato-
ri ematologici ha fornito nuove opportunità, basti pensa-re agli indici reticolocitari, e ha sicuramente contribuitoa rendere più attendibili altri parametri quali il conteggioreticolocitario e quello piastrinico soprattutto alle basseconcentrazioni. E' inoltre già possibile contare accurata-mente gli eritroblasti e in un futuro prossimo avremo l'e-stensione della differenziazione leucocitaria ad ulterioricitotipi rispetto alle 5 popolazioni normali (ad esempio igranulociti immaturi). Non dobbiamo però dimenticareche la verifica microscopica dei campioni patologici ètutt'ora indispensabile come lo è l'interpretazione del datoda parte dello Specialista.
Bibliografia
1. Buttarello M. Quality specification in haematology: the automat-ed blood cell count. Clin Chim Acta 2004;346:45-54.
2. Groner W, Simson E. Pratical guide to modern hematology ana-lyzers. Chichester: Wiley. 1995.
3. Davis BH. Report on the ISLH-sponsored Immature reticulocyteFraction (IRF) workshop. Lab Hematol 1997;3:261-3.
4. Buttarello M, Bulian P, Farina G, Petris MG, Temporin V, Toffolo L.Five fully automated methods for performing Immature Reticulo-cyte Fraction. Comparison in diagnosis of bone marrow aplasia. AmJ Clin Pathol 2002;117:871-9.
5. Buttarello M, Temporin V, Ceravolo R, Farina G, Bulian P. The newreticulocyte parameter (Ret-Y) of the Sysmex XE 2100.Its use in thediagnosis and monitoring of posttreatment sideropenic anemia.Am J Clin Pathol 2004;121:489-95.
6. Brugnara C, Zurakowski D, DiCanzio J, Boyd T, Platt O. Reticulocytehemoglobin content to diagnose iron deficiency in children. JAMA1999;23:2225-30.
7. d'Onofrio G, Chirillo R, Zini G, Caenaro G, Tommasi M, Micciulli G.Simultaneous measurement of reticulocyte and red cell indices inhealthy subjects and patients with microcytic and macrocytic ane-mia. Blood 1995;85:818-23.
8. Bessman JD, Gilmer PR, Gardner FH. Improved classification ofanemias by MCV and RDW. Am J Clin Pathol 1983;80:322-6.
9. Simel DL. Is the RDW-MCV classification of anaemia useful? ClinLab Haematol 1987;9:349-59.
10. Buttarello M, Toffolo L, Bulian P, Temporin V, Farina G. Anisocitosiquantitativa (RDW) : comparabilità dei risultati ottenuti con le piùrecenti tecnologie. Riv Med Lab 2001;2:163[abstract].
11. Brown AS, Hong Y, De Belder A, Beacon H, Beeso J, Sherwood R,et al. Megakariocyte ploidy and platelet changes in human diabetesand atherosclerosis. Arterioscler Thromb Vasc Biol 1997;17:802-7.
12. Jackson SR, Carter JM. Platelet volume: laboratory measurementand clinical application. Blood Rev 1993;7:104-13.
13. Lippi U, Cappelletti P, Schinella M, Signori D. Mean platelet vol-umes: facts or artefacts ? Am J Clin Pathol 1985;84:11-3.
6
haematologica vol. 89[supplement n. 10]:October 2004
Domenica 10 ottobre

Leucemie e manifestazioni orali in età pedia-trica: epidemiologia e diagnosi
Marco Baldoni, Ruggero PapagnaUniversità degli Studi di Milano-Bicocca, Clinica Odon-toiatrica, Corso di Laurea in Igiene Dentale, Milano
IntroduzioneLe leucemie, forme di proliferazione abnorme e atipica
di elementi immaturi della serie bianca del sangue, sonole più comuni forme di neoplasia maligna dell'età infan-tile. Esse vengono classificate secondo criteri clinici emorfocitologici, che comprendono il tipo di cellula, ilnumero di globuli bianchi nel sangue periferico e la rapi-dità del decorso clinico; le leucemie acute sono più fre-quenti nell'infanzia, mentre le leucemie croniche sono piùfrequenti nell'età adulta. 1,2
Nella fascia di età compresa tra 0 e 14 anni, secondo lestime del World Health Organisation Cancer MortalityDatabase, durante il periodo compreso tra il 1995 e il 1997negli Stati Uniti sono stati registrati 3315 decessi causa-ti dalle leucemie (corrispondenti a una mortalità di 1.9persone ogni 100000 appartenenti alla popolazione arischio); per la stessa fascia di età e nello stesso periodo,negli Stati Uniti sono stati registrati 9388 decessi causa-ti da tutte le forme neoplastiche (corrispondenti a unamortalità di 5. 4 persone ogni 100000 appartenenti allapopolazione a rischio). Sulla base di questi dati è possibi-le osservare che il 35. 3% dei decessi per neoplasia nellafascia di età compresa tra 0 e 14 anni è legato a formeleucemiche; i dati relativi alle fasce di età superiorimostrano una graduale diminuzione della percentualerelativa di decessi per leucemie, rappresentando il 3. 35%delle morti per neoplasia tra 25 e 64 anni e il 2.8% tra 60e 64 anni. Ogni anno in tutto il mondo si registrano circa250000 nuovi casi di leucemia e circa 195000 decessilegati alla malattia; i valori più elevati di incidenza e mor-talità si osservano a carico dei paesi più sviluppati. 3
Le leucemie sono caratterizzate da un'elevata inciden-za di complicanze orali che, seguendo uno schema validoanche per quelle sistemiche, possono essere suddivise intre gruppi. Le lesioni primarie sono indotte direttamentedall'infiltrazione delle strutture orali da parte delle cellu-le maligne proliferate (infiltrazione della gengiva, dellamucosa orale o delle strutture ossee della regione orale);le secondarie sono in gran parte il risultato della progres-siva tendenza mieloftisica che caratterizza il decorso del-la malattia (manifestazioni orali di anemia, diatesi emor-ragica, aumentata suscettibilità alle infezioni); le terzia-rie sono indotte dalla terapia antileucemica (modificazio-ni displastiche a carico degli epiteli legate a effetti cito-tossici, manifestazioni legate a mielosoppressione oimmunosoppressione). 4
Le lesioni orali possono rappresentare il primo segnodella malattia, fornendo un notevole contributo alla dia-gnosi di leucemia, anche se la diagnosi di certezza richie-de l'esame istologico del midollo;5 inoltre, la diagnosi pre-coce delle lesioni orali rappresenta un importante momen-to per la prevenzione e il trattamento delle complicanzeche possono insorgere durante la terapia,6 contribuendo
a migliorare la prognosi e a ridurre la morbilità e la mor-talità per questa malattia. 7 Anche dopo la remissione, ilgiovane paziente continua ad avere un'aumentata neces-sità di monitoraggio del cavo orale, a causa degli effettiritardati della malattia e della terapia (aumentata fre-quenza di carie, anomalie di sviluppo dentale e scheletri-co, recidiva della malattia). 8,9
Recenti evidenze scientifiche dimostrano che, se la reci-diva leucemica non si manifesta entro 2 anni dal trapian-to di midollo, la probabilità di guarigione è elevata; tut-tavia per molti anni dopo il trapianto il tasso di mortalitàrimane più elevato rispetto all'aspettativa della popola-zione generale, a causa della recidiva leucemica o di graft-versus-host disease. 10 È evidente che, per prevenire ecurare gli eventi tardivi che minacciano la sopravvivenzadel paziente, deve essere condotto un attento e costantemonitoraggio anche dei soggetti sopravvissuti.
Nel presente lavoro, dedicato al ruolo dello stomatolo-go nella gestione del giovane paziente affetto da leuce-mia, si intende chiarire alcuni aspetti di diagnosi, preven-zione e terapia delle manifestazioni del cavo orale duran-te la malattia leucemica, attraverso una sistematica revi-sione della letteratura associata all'analisi dell'approcciometodologico alla cura dei pazienti presso la Sezione diEmato-Oncologia Pediatrica della Clinica Odontostoma-tologica dell'Università degli Studi di Milano-Bicocca.
Razionale per un approccio sistematicoLa gestione del paziente affetto da leucemia può esse-
re distinta in tre fasi:1) approccio diagnostico,2) approccio preventivo,3) approccio terapeutico.
Le lesioni orali causate dalla patologia leucemica e dal-le sue terapie possono avere ripercussioni negative sullapsicologia del bambino, a causa dell'importanza assuntadal distretto orale nella percezione che l'individuo ha delproprio corpo;11-13 inoltre, l'aumento della sopravvivenzadei giovani pazienti deve necessariamente condurre a unmiglioramento della loro qualità di vita a guarigione avve-nuta. Attraverso un approccio globale al cavo orale delbambino leucemico sopravvissuto, lo stomatologo è ingrado di contribuire a tale miglioramento. 14,15
Approccio diagnosticoIl 38% dei soggetti affetti da leucemia riferisce una sin-
tomatologia relativa a lesioni del cavo orale, ma i segnioggettivi legati alla malattia sono osservabili nel 69% del-la popolazione affetta. 16-19 Al momento dell'ospedalizza-zione per la terapia della leucemia, l'incidenza di compli-canze orali aumenta fino a 89%. 20
L'incidenza delle manifestazioni orali primarie e secon-darie è strettamente dipendente dal tipo di leucemia,essendo più elevata nelle forme acute che in quelle cro-niche, così come in quelle monocitiche rispetto alle mie-locitiche o linfocitiche, ma non esiste alcuna correlazio-ne tra tipo e frequenza delle lesioni orali e stato emato-logico. 21,22
Tra le più frequenti manifestazioni osservate in corso dileucemia si possono elencare linfoadenopatia (71% nelleforme linfoidi acute (ALL), 41% nelle mieloidi acute
7
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

(AML)), pallore del volto (60%) e della mucosa (39%),dolore laringeo (53% nelle ALL, 37% nelle AML), eritema,ulcerazioni o sanguinamento della gengiva (43% nelleAML; 29% nelle ALL), delle labbra, della lingua e del pala-to, ulcerazioni orali e aumento di volume gengivale. 23,24
Le lesioni orali causate dalla terapia (lesioni terziarie),prevalentemente legate ad effetti citotossici, a mielosop-pressione e a immunosoppressione, sono rappresentateda fenomeni emorragici (77%), ulcerazioni da neutrope-nia (49%), infezioni (34%), infezioni da HHV-1 (39%)mucositi (16%). 25,26 Sembra che il 47% dei soggetti trat-tati per leucemia acuta sviluppi lesioni orali. 27 Tale valo-re è di poco più elevato rispetto a quello riportato in unostudio che ha valutato l'impatto delle complicanze oralidurante la terapia per non-head-and-neck malignancies,in cui si è osservato una frequenza di problematiche ora-li di circa il 40%. 28
InfezioniLe complicanze orali durante la terapia sono potenzia-
li fonti di infezioni sistemiche;29 il rischio di setticemia èparticolarmente elevato e sembra essere di origine oralein più del 50% dei casi;30 durante la fase neutropenicadella terapia con trapianto di midollo, le setticemie pro-vocate da Streptococcus alfa-emolitico proveniente dalcavo orale possono rappresentare il 41% del numero tota-le di setticemie. 31 L'Herpes virus e la Candida albicanssono i più frequenti patogeni opportunisti per i soggettiaffetti da patologia neoplastica e, in particolare, durantela chemioterapia; l'Herpes Simplex Virus è frequentemen-te causa delle ulcerazioni orali in corso di terapia anti-leu-cemica, specialmente dopo trapianto di midollo, e puòcausare lesioni multiple che possono provocare, in rareoccasioni, polmoniti e disseminazione dell'infezione. 32-34
Tra i pazienti affetti da leucemia, un numero significati-vo di decessi sono causati da setticemia da Candida e mol-to frequentemente tali setticemie sono legate a prece-denti infezioni orali. 35 Lo Staphylococcus aureus è causadel 7% delle batteriemie nei pazienti neutropenici affet-ti da neoplasia; sembra che la gravità delle mucositi e lafrequenza delle metastasi settiche in corso di leucemiasia legata alla presenza di una batteriemia da Staphylo-coccus aureus. 36 Le sepsi da Streptococcus alfa-emoliti-co sono in aumento nei pazienti sottoposti a terapia anti-neoplastica;37 le setticemie provocate da Streptococcus
alfa-emolitico sembrano essere legate alla profilassi diGraft Versus Host Disease con methotrexate e il conse-guente aumento di ulcerazioni orali. 38 In letteratura èstato riportato 1 caso di infezione da Stomatococcusmucilaginosus durante la fase neutropenica in seguito achemioterapia per AML: esso aveva provocato un quadrodi polmonite e setticemia; in letteratura sono stati ripor-tati soltanto 26 casi di infezione da Stomatococcus muci-laginosus in pazienti neutropenici affetti da neoplasieematiche. Le complicanze causate da questo normale abi-tante del cavo orale sono serie e possono essere fatali:shock settico, meningiti, acute respiratory distress syn-drome. 39
Numb Chin SyndromeRisale al 1986 uno dei primi report di neuropatia men-
tale bilaterale associata con la leucemia: un paziente cheè stato osservato per l'improvviso insorgere di dolore den-tale associato a una sintomatologia di formicolio del lab-bro inferiore, in assenza di altre compromissione orali, èstato sottoposto a una valutazione medica completa ed èstato rilevato un titolo anticorpale positivo a HTLV-IIIassociato ad una leucemia linfoblastica acuta. 40 Succes-sivamente, sono apparsi numerosi lavori sulla cosiddettaNumb Chin Syndrome, causata da una lesione infiltrativamaligna del distretto mandibolare che accompagna ildecorso di soggetti affetti da neoplasia (in particolare leu-cemia, carcinoma mammario e linfoma maligno). 41 Anchese la Numb Chin Syndrome è spesso considerata unamanifestazione tardiva di malattia neoplastica,42 la lette-ratura riporta che essa può condurre alla diagnosi preco-ce di neoplasia e contribuire alla definizione delle impli-cazioni prognostiche; la Numb Chin Syndrome può esse-re infatti il primo segno della patologia43 o della recidiva,potendo addirittura comparire nello stesso paziente inentrambi i momenti a distanza di tempo. 44
La Numb Chin Syndrome si presenta con i seguenti sin-tomi:45
- parestesia o ipoestesia nel territorio di distribuzionenel nervo mentoniero; dolore in corrispondenza delforame mentoniero;
- parestesia e dolore all'occlusione46 e alla percussione;diminuzione o scomparsa della sensibilità dentinale;mobilità dentale;
- open bite anteriore (probabilmente causato da estru-
8
haematologica vol. 89[supplement n. 10]:October 2004
Domenica 10 ottobre

sione bilaterale dei molari);- evidenze radiografiche di scomparsa bilaterale del
canale mandibolare, distruzione della cresta alveola-re, allargamento dello spazio parodontale e assotti-gliamento o perdita della lamina dura;
- raramente: linfoadenopatia sottomandibolare e a cari-co delle ghiandole salivari maggiori; paralisi del ner-vo ipoglosso; neuropatia del trigemino;47 sintomi dacompressione midollare. 48-50
Le ipotesi patogenetiche più accreditate sembrano esse-re la compressione del nervo da parte di infiltrati tumo-rali mandibolari, l'invasione diretta dei nervi cranici daparte delle cellule tumorali, metastasi trigeminali di tumo-ri delle meningi. 51
Graft-versus-Host diseaseUna complicanza particolarmente grave che si verifica
dopo trapianto di midollo osseo (BMT) è la Graft-Versus-Host Disease (GVHD), fenomeno immunologico che ponea rischio la sopravvivenza del paziente, causato dalla rea-zione del midollo trapiantato immunocompetente controi tessuti dell'ospite. 52 La condizione può affiggere più del45% dei soggetti che hanno subito il trapianto di midol-lo osseo e le lesioni acute e più aggressive si verificano nelcorso del primo mese dopo il trapianto. 53,54 La forma cro-nica risulta essere la più frequente causa di decesso nonlegata alla recidiva della malattia; il decesso può soprav-venire per diretta complicanza (per esempio obliterazio-ne bronchiolare) o per lo stato di immunodeficienza asso-ciato alla malattia, che aumenta significativamente la dia-tesi infettiva. 55-59 Gli organi bersaglio del fenomeno sonoil fegato, i polmoni, il tratto gastrointestinale le ghiando-le esocrine, la cute e la mucosa; la cavità orale è coinvol-ta in più dell'80% dei casi. 60 Segni e sintomi della cavitàorale sono simili a quelli riscontrati in altre sindromi opatologie caratterizzate da eziopatogenesi immunitaria: illupus erythematosus, il lichen planus, la sclerodermia e lasindrome di Sjögren. 61-63
La diagnosi di GVHD si basa sull'identificazione dellemanifestazioni cutanee e mucose e sull'identificazione dimanifestazioni di disfunzione epatica. I segni orali posso-no variare da un moderato fenomeno eritematoso a seve-re manifestazioni di mucosite, ulcerazioni, gengiviti,desquamazioni dei tessuti orali, xerostomia e infezioni.64–66 Chiaramente, lo stato di xerostomia predispone i sog-
getti ad aumentata incidenza di carie dentali e amplifica-zione in termini di estensione e severità di mucosità einfezioni orali. 67 Fenomeni osservati con minor frequen-za in soggetti affetti da GVHD consistono in alterazioniritentive delle ghiandole salivari, che si manifestano comevescicole ripiene di saliva, xantomi verrocosi e granulomipiogenici atipici elicitati da traumatismi. 68-71
La medicazione utilizzata più frequentemente per laprevenzione di GVHD è la ciclosporina, che si associa, inuna percentuale significativa di casi, ad aumento di volu-me dei tessuti gengivali; in rari casi si è osservata l'insor-genza di carcinomi orali, linfomi e sarcomi di Kaposi in sitisede di aumenti di volume gengivale causati da ciclospo-rina, probabilmente come risultato di una eccessiva immu-nosoppressione. 72-76
La diagnosi di certezza di GVHD deve essere eseguitacon l'esame istologico delle ghiandole salivari labiali (GSL),poiché l'esame istologico della mucosa buccale (MB) con-duce al 20% di falsi negativi, mentre per le GSL tale valo-re si riduce al 11%. 77,78 Nella Tabella 2 si riporta il valorediagnostico delle alterazioni orali cliniche e radiograficherilevate in corso di GVHD. 79
DiscussioneUn recente studio80 ha paragonato il tasso di sopravvi-
venza di pazienti sottoposti a trapianto di midollo osseoe affetti da manifestazioni orali recrudescenti di infezio-ne sostenuta da human herpes virus-1 con il tasso disopravvivenza di pazienti sottoposti a trapianto di midol-lo osseo e non manifestanti fenomeni legati ad infezioneda human herpes virus-1.
L'analisi a mostrato l'evidenza che un numero di pia-strine inferiore a 100000 cellule/mm3 100 giorni dopo iltrapianto e il fenomeno di recrudescenza dell'infezioneorale da HHV-1 sono variabili prognostiche indipendentinegative per il tasso di sopravvivenza dei soggetti sotto-posti a trapianto di midollo osseo nel corso dei 24 mesisuccessivi al trapianto.
In base ai dati riportati è evidente che il ruolo dello sto-matologo assume una rilevante importanza nell'intercet-tamento delle manifestazioni orali durante le varie fasi ditutta la storia naturale della leucemia. Il ruolo dello sto-matologo è particolarmente delicato, poiché l'adesionealle prescrizioni terapeutiche per il cavo orale negli ado-lescenti affetti da neoplasia non è influenzata da variabi-li demografiche o variabili terapeutiche. I soggetti che nonaderiscono adeguatamente al regime terapeutico possie-dono un concetto meno sviluppato di malattia, non per-cepiscono in modo significativo il senso di vulnerabilità permalattia e sono caratterizzati da elevati livelli di negazio-ne per difesa psicologica. In altri termini, i pazienti costrui-scono il proprio soggettivo punto di vista della malattia edel trattamento, che può avere notevoli implicazioni intermini di aderenza al regime terapeutico. 81
A tal propositi, si consideri anche che, nella popolazio-ne occidentale, fenomeni di neoplasia del cavo orale sonopiù frequentemente associati a soggetti immunosoppres-si, con condizioni predisponenti quali leucemia e infezio-ni associate a malnutrizione. 82
In tutte queste situazioni, una diagnosi precoce ed unprotocollo terapeutico razionale rappresentano il momen-to per condurre al miglioramento della prognosi delle
9
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

lesioni orali associate alla malattia e della leucemia stes-sa; infine, l'appicazione sistematica di un protocollo pre-ventivo riduce significativamente l'incidenza delle com-plicanze orali anche in questi pazienti. 83,84
Bibliografia
1. Devine SM, Larson RA. Acute leukemia in adults: Recent develop-ments in diagnosis and treatment. CA Cancer J Clin 1994;44:326-352
2. Robbins SL, Cotran R, Kumar V, Collins T. Pathologic Basis of Dis-ease. Saunders, 6th edition, 1999
3. GLOBOCAN 2000: Cancer Incidence, Mortality and PrevalenceWorldwide, Version 1.0.IARC CancerBase No. 5. Lyon, IARCPress,2001
4. Robbins SL, Cotran R, Kumar V, Collins T. Pathologic Basis of Dis-ease. Saunders, 6th edition, 1999
5. Devine SM, Larson RA. Acute leukemia in adults: Recent develop-ments in diagnosis and treatment. CA Cancer J Clin 1994;44:326-352
6. Guggenheimer J, Verbin RS, Appel BN, Schmutz J. Clinicopathologiceffects of cancer chemotherapeutic agents on human buccalmucosa. Oral Surg Oral Med Oral Pathol 1977 Jul;44(1):58-63
7. Vinckier F, Declerck D. [Oral manifestations in leukemic childrenand their diagnostic value]. Acta Stomatol Belg 1989Oct;86(3):219-26
8. Ayers KM, Colquhoun AN. Leukaemia in children. Part II--Dentalcare of the leukaemic child, includine management of oral sideeffects of cancer treatment. N Z Dent J 2000 Dec;96(426):141-6
9. Uderzo C, Fraschini D, Balduzzi A, et al. Long-term effects of bonemarrow transplantation on dental status in children withleukaemia. Bone Marrow Transplant 1997 Nov;20(10):865-9
10. Socié G, Veum Stone J, Wingard J R, et al. Long-Term Survival andLate Deaths after Allogeneic Bone Marrow Transplantation. N EnglJ Med 1999; 341(1):14-21
11. Bunetel L, Bonnaure-Mallet M. Oral pathoses caused by Candidaduring chemotherapy. Oral Surg Oral Med Oral Pathol Oral RadiolEndod 1996;82:161-5
12. De Beule F, Bercy P et al. The effectiveness of a preventive regi-men on the periodontal health of patients undergoing chemother-apy for leukemia and lymphoma. J Clin Periodontol 1991;18:346-7
13. Halton JM, Atkinson SA et al. Altered mineral metabolism andbone mass in children during treatment for acute lymphoblasticleukemia. J Bone Miner Res 1996 November;11(11):1774-83
14. Maguire A, Craft AW et al. The long-term effects of treatment onthe dental condition of children surviving malignant disease. Can-cer 1987;60:2570-75
15. Wright WE, Haller JM et al. An oral disease prevention program forpatients receiving radiation and chemotherapy. JADA1985;110:43-47
16. Williams MC, Lee GT. Childhood leukemia and dental considera-tions. J Clin Pediatr Dent 1991; 15:160-164
17. Barrett AP. Gingival lesions in leucemia. A classification. J Peri-odontol 1984;55:585-588
18. Bressman E, Decter JA, Chasens AI, Sackler RS. Acute myeloblas-tic leukemia with oral manifestations. Report of a case. Oral SurgOral Med Oral Pathol 1982;54:401-403
19. O'Sullivan EA, Duggal MS, Bailey CC. Changes in the oral health ofchildren during treatment for acute lymphoblastic leukaemia. IntJ Paediatr Dent 1994 Mar;4(1):31-4
20. Barrett AP. A long-term prospective clinical study of oral compli-cations during conventional chemotherapy for acute leukemia.Oral Surg Oral Med Oral Pathol 1987 Mar;63(3):313-6
21. Hou GL, Huang JS, Tsai CC. Analysis of oral manifestations ofleukemia: a retrospective study. Oral Dis 1997 Mar;3(1):31-8
22. Kleinheinz J, Meyer U, Buchner T, Kosters G, Weingart D, Joos U.[Oral manifestations of acute leukemia]. Mund Kiefer Gesichtschir1997 Feb;1(1):57-60
23. Theodorpoulos-Papadimitriou K, Donta AN, Kosmidi EB. [Incidenceof oral manifestations in children with acute leukemia]. Odon-tostomatol Proodos 1989 Aug;43(4):357-63
24. Hou GL, Huang JS, Tsai CC. Analysis of oral manifestations of
leukemia: a retrospective study. Oral Dis 1997 Mar;3(1):31-825. Barrett AP. A long-term prospective clinical study of oral compli-
cations during conventional chemotherapy for acute leukemia.Oral Surg Oral Med Oral Pathol 1987 Mar;63(3):313-6
26. Dreizen S, McCredie KB, Bodey GP, Keating MJ. Quantitative analy-sis of the oral complications of antileukemia chemotherapy. OralSurg Oral Med Oral Pathol 1986 Dec;62(6):650-3
27. Dreizen S, McCredie KB, Bodey GP, Keating MJ. Quantitative analy-sis of the oral complications of antileukemia chemotherapy. OralSurg Oral Med Oral Pathol 1986 Dec;62(6):650-3
28. Sonis S, Kunz A. Impact of improved dental services on the fre-quency of oral complications of cancer therapy for patients withnon-head-and-neck malignancies. Oral Surg Oral Med Oral Pathol1988 Jan;65(1):19-22
29. Childers NK, Stinnett EA, Wheeler P, Wright JT, Castleberry RP,Dasanayake AP. Oral complications in children with cancer. OralSurg Oral Med Oral Pathol 1993 Jan;75(1):41-7
30. Cousin GC. Oral manifestations of leukaemia. Dent Update 1997Mar;24(2):67-70
31. Heimdahl A, Mattsson T, Dahllof G, Lonnquist B, Ringden O. Theoral cavity as a port of entry for early infections in patients treat-ed with bone marrow transplantation. Oral Surg Oral Med OralPathol 1989 Dec;68(6):711-6
32. Saral R. Oral complications of cancer therapies. Management ofacute viral infections. NCI Monogr 1990;(9):107-10
33. Beiraghi S, Sanders B. Oral complications in a patient with acutelymphocytic leukemia: a report of case. Spec Care Dentist 1988Jan-Feb;8(1):13-5
34. Orbak R, Orbak Z. Oral condition of patients with leukemia andlymphoma. J Nihon Univ Sch Dent 1997 Jun;39(2):67-70
35. Stinnett EA, Childers NK, Wright JT, Rodu BK, Bradley EL Jr. Thedetection of oral Candida in pediatric leukemia patients. PediatrDent 1992 Jul-Aug;14(4):236-9
36. Gonzalez-Barca E, Carratala J, Mykietiuk A, Fernandez-Sevilla A,Gudiol F. Predisposing factors and outcome of Staphylococcusaureus bacteremia in neutropenic patients with cancer. Eur J ClinMicrobiol Infect Dis 2001 Feb;20(2):117-9
37. Barker GJ, Call SK, Gamis AS. Oral care with vancomycin paste forreduction in incidence of alpha-hemolytic streptococcal sepsis. JPediatr Hematol Oncol 1995 May;17(2):151-5
38. Heimdahl A, Mattsson T, Dahllof G, Lonnquist B, Ringden O. Theoral cavity as a port of entry for early infections in patients treat-ed with bone marrow transplantation. Oral Surg Oral Med OralPathol 1989 Dec;68(6):711-6
39. Gruson D, Hilbert G, Pigneux A, Vargas F, Guisset O, Texier J, BoironJM, Reiffers J, Gbikpi-Benissan G, Cardinaud JP. Severe infectioncaused by Stomatococcus mucilaginosus in a neutropenic patient:case report and review of the literature. Hematol Cell Ther 1998Aug;40(4):167-9
40. Milam SB, Rees TD, Leiman HI. An unusual cause of bilateral men-tal neuropathy in an AIDS patient. Report of a case. J Periodontol1986 Dec;57(12):753-5
41. Hiraki A, Nakamura S, Abe K, Takenoshita Y, Horinouchi Y, Shino-hara M, Shirasuna K. Numb chin syndrome as an initial symptomof acute lymphocytic leukemia: report of three cases. Oral SurgOral Med Oral Pathol Oral Radiol Endod 1997 May;83(5):555-61
42. Lossos A, Siegal T. Numb Chin Syndrome in cancer patients: etiol-ogy, response to treatment and prognostic significance. Neurolo-gy 1992;42(6):1181-4
43. Hiraki A, Nakamura S, Abe K, Takenoshita Y, Horinouchi Y, Shino-hara M, Shirasuna K. Numb chin syndrome as an initial symptomof acute lymphocytic leukemia: report of three cases. Oral SurgOral Med Oral Pathol Oral Radiol Endod 1997 May;83(5):555-61
44. Kuklok KB, Burton RG, Wilhelm ML. Numb chin syndrome leadingto a diagnosis of acute lymphoblastic leukemia: report of a case.J Oral Maxillofac Surg 1997 Dec;55(12):1483-5
45. Hiraki A, Nakamura S, Abe K, Takenoshita Y, Horinouchi Y, Shino-hara M, Shirasuna K. Numb chin syndrome as an initial symptomof acute lymphocytic leukemia: report of three cases. Oral SurgOral Med Oral Pathol Oral Radiol Endod 1997 May;83(5):555-61
46. Benito LJ, Simon R, Miera C. Numb Chin Syndrome as the initialmanifestation of HIV infection. Neurology 1998;50(2):511-2
47. Diago MP, Sebastian JVB, Giner AA, Orenga VE. Mental nerve neu-ropathy in systemic cancer. Oral Surg Oral Med Oral Pathol Oral
10
haematologica vol. 89[supplement n. 10]:October 2004
Domenica 10 ottobre

Radiol Endod 1990; 69:48-5148. Hiraki A, Nakamura S, Abe K, Takenoshita Y, Horinouchi Y, Shino-
hara M, Shirasuna K. Numb chin syndrome as an initial symptomof acute lymphocytic leukemia: report of three cases. Oral SurgOral Med Oral Pathol Oral Radiol Endod 1997 May;83(5):555-61
49. Kuklok KB, Burton RG, Wilhelm ML. Numb chin syndrome leadingto a diagnosis of acute lymphoblastic leukemia: report of a case.J Oral Maxillofac Surg 1997 Dec;55(12):1483-5
50. Benito LJ, Simon R, Miera C. Numb Chin Syndrome as the initialmanifestation of HIV infection. Neurology 1998;50(2):511-2
51. Hiraki A, Nakamura S, Abe K, Takenoshita Y, Horinouchi Y, Shino-hara M, Shirasuna K. Numb chin syndrome as an initial symptomof acute lymphocytic leukemia: report of three cases. Oral SurgOral Med Oral Pathol Oral Radiol Endod 1997 May;83(5):555-61
52. Dahllof G, Modeer T, Bolme P, Ringden O, Heimdahl A. Oral healthin children treated with bone marrow transplantation: A oneyearfollow-up. J Dent Child 1988;55:196-200
53. Heimdahl A, Mattsson T, Dahllof G, Lonnquist B, Ringden O. Theoral cavity as a port of entry for early infections in patients treat-ed with bone marrow transplantation. Oral Surg Oral Med OralPathol 1989;68:711-716
54. Curtis JW Jr, Caughman GB. An apparent unusual relationshipbetween rampant caries and graft-versus-host disease. Oral SurgOral Med Oral Pathol 1994;78:267-272
55. Socié G, Veum Stone J, Wingard J R, et al. Long-Term Survival andLate Deaths after Allogeneic Bone Marrow Transplantation. N EnglJ Med 1999; 341(1):14-21
56. Sullivan KM. Graft-versus-host disease. In: Forman SJ, Blume KG,Thomas ED, eds. Bone marrow transplantation. Cambridge, Mass.: Blackwell Scientific, 1994:339-62
57. Storek J, Gooley T, Witherspoon RP, Sullivan KM, Storb R. Infec-tious morbidity in long-term survivors of allogeneic marrow trans-plantation is associated with low CD4 T cell counts. Am J Hema-tol 1997;54:131-138
58. Hiroki A, Nakamura S et al. Significance of oral examination inchronic graft-versus-host disease. J Oral Pathol Med 1994;23:209-15
59. Nakamura et al. Oral involvement in chronic graft-versus-host dis-ease after allogenic bone transplantation. Oral Surg Oral Med OralPathol Oral Radiol Endod 1996; 82:556-63
60. Curtis JW Jr, Caughman GB. An apparent unusual relationshipbetween rampant caries and graft-versus-host disease. Oral SurgOral Med Oral Pathol 1994;78:267-272
61. Schubert MM, Izutsu KT. Iatrogenic causes of salivary gland dys-function. J Dent Res 1987;66(S):680-688
62. Dahllof G, Heimdahl A, Modeer T, Twetman S, Bolme P, Ringden O.Oral mucous membrane lesions in children treated with bone mar-row transplantation. Scand J Dent Res 1989;97:268-277
63. Hiroki A, Nakamura S, Shinohara M, Oka M. Significance of oralexamination in chronic graft-versus-host disease. J Oral PatholMed 1994;23:209-215
64. Rhodus NL, Little JW. Dental management of the bone marrowtransplant patient. Compendium Cont Educ Dent 1992;13:1042-1050
65. Wingard JR, Niehaus CS, Peterson DE, et al. Oral mucositis afterbone marrow transplantation. A marker of treatment toxicity andpredictor of hepatic veno-occlusive disease. Oral Surg Oral Med
Oral Pathol 1991;72:419-424. 66. Eggleston TI, Ziccardi VB, Lumerman H. Graft-versus-host disease.
Case report and discussion. Oral Surg Oral Med Oral Pathol OralRadiol Endod 1998 Dec;86(6):692-6
67. Curtis JW Jr, Caughman GB. An apparent unusual relationshipbetween rampant caries and graft-versus-host disease. Oral SurgOral Med Oral Pathol 1994;78:267-272
68. Rhodus NL, Little JW. Dental management of the bone marrowtransplant patient. Compendium Cont Educ Dent 1992;13:1042-1050
69. Allen CM, Kapoor N. Verruciform xanthoma in a bone marrowtransplant recipient. Oral Surg Oral Med Oral Pathol 1993;75: 591-594
70. Barrett AP. Graft-versus-host disease. A clinicopathologic review.Ann Dent 1987;46:7-11
71. Wandera A, Walker PO. Bilateral pyogenic granuloma of the tonguein graft-versus-host disease: Report of a case. J Dent Child1994;61:401-403
72. Little JW, Rhodus NL. Dental treatment of the liver transplantpatient. Oral Surg Oral Med Oral Pathol 1992;73:419-426
73. Maxymiw WG, Wood RE, Lee L. Primary, multi-focal, non-Hodgk-in's Iymphoma of the jaws presenting as periodontal disease in arenal transplant patient. Int J Oral Maxillofac Surg 1991;20:6970
74. 171.Regev E, Zeltser R, Lustmann J. Lip carcinoma in renal allograftrecipient with long-term immunosuppressive therapy. Oral SurgOral Med Oral Pathol 1992;73:412-414
75. 173. Varga E, Tyldesley WR. Carcinoma arising in cyclosporin-induced gingival hyperplasia. Br Dent J 1991;171:26 27
76. 172.Thomas DW, Seddon SV, Shepherd JP. Systemic immunosup-pression and oral malignancy: A report of a case and review of theliterature. Br J Oral Maxillofac Surg 1993;31:391-393
77. Hiroki A, Nakamura S et al. Significance of oral examination inchronic graft-versus-host disease. J Oral Pathol Med 1994;23:209-15
78. Nakamura et al. Oral involvement in chronic graft-versus-host dis-ease after allogenic bone transplantation. Oral Surg Oral Med OralPathol Oral Radiol Endod 1996; 82:556-63
79. Nakamura et al. Oral involvement in chronic graft-versus-host dis-ease after allogenic bone transplantation. Oral Surg Oral Med OralPathol Oral Radiol Endod 1996; 82:556-63
80. Gomez RS, Carneiro MA, Souza LN, Victoria JM, de Azevedo WM,De Marco L, Kalapothakis E. Oral recurrent human herpes virusinfection and bone marrow transplantation survival. Oral Surg OralMed Oral Pathol Oral Radiol Endod 2001 May;91(5):552-6
81. Tamaroff MH, Festa RS, Adesman AR, Walco GA. . II. Clinical andpsychologic correlates. J Pediatr 1992 May;120(5):812-7
82. Nash ES, Cheng LH, Smart K. Cancrum oris-like lesions. Br J OralMaxillofac Surg 1991 Feb;29(1):51-3
83. Sonis S, Kunz A. Impact of improved dental services on the fre-quency of oral complications of cancer therapy for patients withnon-head-and-neck malignancies. Oral Surg Oral Med Oral Pathol1988
84. Levy-Polack MP, Sebelli P, Polack NL. Incidence of oral complica-tions and application of a preventive protocol in children withacute leukemia. Spec Care Dentist 1998 Sep-Oct;18(5):189-93.
11
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

12
haematologica vol. 89[supplement n. 10]:October 2004
Domenica 10 ottobre
La dimensione del problema
Riccardo Haupt,* Maura Faraci,* M. G. Valsecchi,°Momcilo Jankovic°*Servizio di Epidemiologica e Biostatistica, Ematologia edOncologia Pediatrica, Istituto G. Gaslini Genova;°Clinica Pediatrica, Università di Milano-Bicocca, Monza
I tumori pediatrici sono un evento raro: in Italia, secon-do le stime del Registro Tumori Infantili (RTI) del Piemon-te, si ammalano ogni anno 147 bambini ogni 1.000.000,il ché corrisponde ad un numero di circa 1350 nuove dia-gnosi attese ogni anno in bambini di età compresa tra 0e 14 anni. 1 La loro incidenza si è mantenuta sostanzial-mente stabile negli ultimi 20 anni, pur con un apparentelieve aumento (stimato < all'1% annuo) negli ultimi anni;nello stesso periodo, invece, la mortalità per tumore pedia-trico è andata via via diminuendo, tanto che le stimeattuali riportano, considerando tutti tumori assieme, unaprobabilità di sopravvivenza a 5 anni superiore al 75%. 2,3
LuciNonostante la loro rarità, i tumori pediatrici sono un
evento sociale importante innanzitutto perché, pur essen-do possibile per molti di essi una completa guarigione, neipaesi industrializzati rappresentano purtroppo ancora laprima causa di morte in età pediatrica. L'altro aspetto,invece decisamente confortante, è legato al fatto chestanno aumentando sempre più tra la popolazione adultale persone che sono state curate per tumore in età pedia-trica. Stime attuali, applicate alla popolazione degli StatiUniti d'America, ove i progressi nel trattamento dei tumo-ri pediatrici sono iniziati relativamente prima che in Ita-lia, valutano che ai giorni nostri, 1 persona su 450 nellapopolazione generale è stata curata per tumore in etàpediatrica. 4
In Italia, la popolazione dei soggetti potenzialmenteguariti da tumore è censita attraverso il Registro Fuoriterapia (ROT) dell'Associazione Italiana di Oncologia edEmatologia Pediatrica (AIEOP). Nel ROT vengono raccolteinformazioni sui bambini affetti da tumore maligno che,indipendentemente dall'evoluzione successiva della malat-tia, hanno raggiunto in persistente remissione completa ilprogramma terapeutico per loro previsto. 5 Il registro èstato attivato nel 1980 ed inizialmente raccoglieva infor-mazioni solamente sui bambini ammalatisi di leucemiaacuta (linfoblastica o non linfoblastica), linfomi, tumore diWilms, malattia di Hodgkin, o neuroblastoma. Successiva-mente (nel 1985) sono stati inclusi nel ROT anche i bam-bini con tumori del sistema nervoso centrale (SNC) o consarcomi dei tessuti molli. Solamente a partire dal 1996,sono registrati tutti i bambini andati fuori terapia e segui-ti presso i centri AIEOP, indipendentemente dal tipo ditumore per cui sono stati curati. 6 La popolazione regi-strata nel ROT non rappresenta quindi la totalità delle per-sone che in Italia hanno superato positivamente una dia-gnosi di tumore in età pediatrica, ma ne è sicuramente uncampione molto rappresentativo. All'ultimo censimentodel ROT sono state registrate 11.226 persone, e la loro
probabilità di sopravvivenza a oltre 20 anni dalla sospen-sione delle cure è stata stimata essere superiore all'80%.7 Si può stimare quindi che oggi esistano in Italia più di 9.000 soggetti guariti da tumore contratto in età pediatri-ca. Poiché oltre il 70% dei bambini che si ammalno ditumore, se trattato con i moderni protocolli terapeutici,può raggiungere la sospensione elettiva delle cure ed esse-re vivo ed in remissione completa a più di 5 anni dalla dia-gnosi, si può affermare che, ogni anno, si possono poten-zialmente aggiungere al ROT circa 900 nuovi soggetti cherappresentano il 70% dei circa 1.400 nuovi casi di tumo-re mediamente annualmente diagnosticati in Italia. Adoggi (Figura 1), l'età mediana delle 8883 persone entratenel ROT entro il 1998 e tuttora sopravviventi è di 21 anni(range 6-51). Per quanto riguarda i tipi di tumore per cuiqueste persone sono state curate, le LLA rappresentano il37% della popolazione, seguite dai tumori di Wilms,malattia di Hodgkin e dai linfomi (10% ciascuna), dal neu-roblastoma (8%), e dai tumori del SNC (7%).
Man mano che ci si allontana nel tempo dalla data del-la diagnosi di tumore e dalla data di fine terapia, la com-prensibile paura che la malattia possa ripresentarsi tendea scomparire. Per certi versi, invece, aumenta la paura chepossano manifestarsi degli effetti tardivi prevalentemen-te legati alla somministrazione di trattamenti che interfe-riscono sulla crescita cellulare,8 in un periodo importantedella vita quale quello dell'accrescimento.
OmbreCol termine di effetto tardivo viene generalmente defi-
nito un problema clinico o psicologico che è comparsodurante il trattamento e persiste nel tempo, o che com-pare dopo più di 5 anni dalla diagnosi. Ovviamente l'in-tensità e le probabilità che si manifesti un effetto tardi-vo in un paziente dipendono dal tipo di tumore per cui èstato curato, dal tipo e dosaggio di chemio e/o radiotera-pia ricevute, dagli eventuali interventi chirurgici maggio-ri da lui subiti, oltre che dall'età al trattamento.
In generale, sequele delle neoplasie pediatriche posso-no interferire oltre che sull'aspetto psico-sociale, anchesulla crescita e lo sviluppo, sulla funzionalità di organivitali, sulla fertilità e riproduzione, così come sul rischiodi tumori secondari. Dai dati della letteratura e dall'espe-rienza personale si può dire che, sebbene si possa evi-denziare abbastanza frequentemente un qualche effettotardivo in persone curate per tumore, questi sono gene-
Figura 1.Distribuzione per età attaule (nel 2004) di 8883 soggetti fuoriterapia dopo diagnosi di tumore in età pediatrica e registarti nel ROTentro il 2001.
num
ero
di guariti
età attuale
I “guariti”: luci ed ombre

ralmente di entità molto lieve. 9,10 Sono da considerarsi amaggior rischio per effetti tardivi gravi in maniera tale dapoter interferire sulla qualità e quantità di vita, quellepersone trattate molto tempo fa con terapie oggi consi-derate obsolete, o quelle curate per neoplasie ad altorischio o che hanno sviluppato una o più recidive: situa-zioni che hanno necessariamente richiesto l'utilizzo difarmaci in dosi od intensità maggiori. 11 Il dato confor-tante, peraltro, è che la maggior parte dei “guariti” (oltreil 90%) riferisce di considerarsi in buona salute. In con-clusione, il fenomeno delle persone guarite da tumorepediatrico sta diventando una condizione sempre più fre-quente; si potrebbe stimare che mediamente ogni medi-co di base ha o avrà tra i suoi assistiti almeno 2 soggettiguariti da tumore in età pediatrica. La rilevanza di que-sto problema richiede quindi che anche la società di medi-cina generale acquisisca competenza su quelli che sono onon sono i problemi potenzialmente collegati ai tratta-menti ricevuti in età pediatrica, questo al fine di evitareinutili ed odiose discriminazioni, e al fine di instaurare, senecessari, programmi di prevenzione primaria o seconda-ria.
Bibliografia
1. Magnani C, Capocaccia R, Giordano L, Mosso ML, Pastore G, PratiS, et al. Stima del numero di casi incidenti di tumore maligno inetà pediatrica in Italia, per Regione. Riv Ital Ped 1992;18:203-7.
2. Robertson CM, Hawkins MM, Kingston JE. Late deaths and sur-vival after childhood cancer: Implication for cure. Br Med J 1994;309:162-6.
3. Magnani C, Pastore G. Survival of childhood cancer patients inItaly, 1978-1989. The ITACARE working group. Tumori 1997;83:426-89.
4. Meadows AT. Pediatric cancer survivors: past history and futurechallenges. Curr Probl Cancer 2003;27:112-6.
5. Haupt R, Jankovic M, Valsecchi MG, Fraschini D, Corbetta A,Manganini C, et al. I soggetti fuori terapia dopo tumore malig-no contratto in età pediatrica. Il registro italiano (OTR). Riv ItalPediatr 1995;21:621-5.
6. Pession A, Rondelli R, Locatelli F, Jankovic M, Haupt R, Basso G,et al. I Registri e l'assistenza al bambino con patologia neoplas-tica. Riv Ital Pediatr 2000;26:570-3.
7. Haupt R, Valsecchi MG, Silvestri D, De Lorenzo P, Napoli S,Masera G, et al. Early and late deaths after elective end of ther-apies for childhood cancer in Italy. The Italian Registry of the Off-Therapy patients (OTR). Int J Cancer 2000;86:393-8.
8. Hudson MM, Jones D, Boyett J, Sharp GB, Pui CH. Late mortali-ty of long-term survivors of childhood cancer. J Clin Oncol 1997;15:2205-13.
9. Seitzman RL, Glover DA, Meadows AT, Mills JL, Nicholson HS,Robison LL, et al. Self-concept in adult survivors of childhoodacute lymphoblastic leukemia: a cooperative Children's CancerGroup and National Institutes of Health study. Pediatr BloodCancer 2004; 42:230-40.
10. Garrè ML, Gandus S, Cesana B, Haupt R, De Bernardi B, ComelliA, et al. Health status of long term survivors after cancer in child-hood: results of an uni-institutional study in Italy. Am J PediatrHematol Oncol 1994;16:143-52.
11. Haupt R, Jankovic M, Massimo L. 95th course : survivors of child-hood cancer. “Follow-up” and emerging problems. Int J PediatrHematol Oncol 1999;6:209-11.
Come viene usata la parola “guariti” in Italia enel mondo
Momcilo Jankovic, Elena Viganò, Tiziana ColivaClinica Pediatrica Università di Milano-Bicocca, A. O. S.Gerardo di Monza
IntroduzioneOggi nel campo più strettamente oncologico, ma non solo,
la medicina ha fatto tanti e tali progressi che in percentua-li sempre crescenti si raggiunge la guarigione dalla malat-tia pregressa.
Secondo però le direttive dell'OMS (Organizzazione Mon-diale della Sanità) un soggetto viene considerato guarito see quando raggiunge un completo recupero fisico, psicologi-co e sociale. 1
Ciò comporta però una necessità di definizione ancor ogginon universalmente e scientificamente riconosciute: chi è difatto un soggetto guarito e pur riconoscerlo quanto l'OMSdice, quando è giusto considerarlo tale? E proprio i medicipossono avere maggiori difficoltà ad usare il termine di“guarito” per i soggetti che interrompono le cure per unpregresso tumore. I medici infatti sono particolarmenteattenti a quanto può accadere al paziente e alla famigliadopo che è stato completato con successo il piano di curae sono particolarmente attenti alle complicanze o effettitardivi che possono comparire successivamente.
Obiettivo dello studio e metodologiaCi siamo chiesti se e quando i pediatri-oncologi Europei
(e oltre oceano) usano il termine “guarito” nella loro prati-ca quotidiana. Nel 2003 abbiamo quindi distribuito nel-l'ambito dell'International BFM Study Group ad “esperiti”oncologi un questionario di 1 pagina (Figure 1). La raccoltadati computerizzata e l'analisi statistica è stata fatta daJulianne Byrne, PhD, Research Professor of Pediatrics al Chil-dren's National Medical Center di Washington e al BoyneResearch Intitute a Drogheda in Irlanda usando il sistemaEPI-INFO 6. 04d.
RisultatiSono stati inviati 300 questionari e abbiamo ricevuto
risposta da 110 medici. La Tabella 1 riporta i paesi di appar-tenenza dei medici che hanno risposto al questionario.
Essendo stato promosso dall'AIEOP (Associazione Italia-na di Ematologia e Oncologia Pediatrica) la percentuale dirisposte in Italia è stata la più alta. L'età media dei mediciche hanno risposto è stata di 49 anni (range 30-73) e nonabbiamo osservato differenza statisticamente significativanell'uso del termine ”guarito” tra medici di età <49 anni(57=86%) e >49 anni (40=93%) (p>0.06).
La distribuzione per sesso è stata di: maschi 67 (60.9%)e femmine 33 (39. 1%) senza differenza statisticamentesignificativa tra i 2 gruppi (p>0.05).
La Tabella 2 indica invece quanti medici oggi usano il ter-mine “guarito” in un soggetto trattato con successo pertumore (sì l'88% verso no il 12%).
Nella Tabella 3 viene riportato come viene usato oggi iltermine “guarito” nei diversi paesi Europei (non si è osser-vata variabilità statisticamente significativa).
Le Tabelle 4 e 5 riportano invece il criterio seguito dai
13
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

14
haematologica vol. 89[supplement n. 10]:October 2004
Domenica 10 ottobre
medici per considerare guarito un soggetto dopo tratta-mento per tumore: le curve di Kaplan-Meier e/o l'analisi delrischio.
Infine nella Tabella 6 riportiamo la percentuale dei medi-ci che hanno risposto di attendere almeno 5 anni dal com-pletamento delle cure prima di usare la parola “guarigione”.
I medici hanno poi espresso alcune considerazioni signi-ficative riguardo alle modalità dell'indagine che abbiamocondotto, come ad esempio:- Sfumare il concetto di guarigione sottolineando che non
c'è nulla di certo (100%) nella nostra vita anche se ègiusto parlare di guarigione.
Tabella 1.
Tabella 2.
Tabella 3.
Tabella 4.
Tabella 5.
Tabella 6.

15
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
FIG. 1 ELTEC Survey of ‘CURE’Version 2
3 Dec, 2003 Page 1
YOUR NAME _____ __________________________________________________No. |___|___|___|___|
ADDRESS ___________________________________________________________________________
COUNTRY ___________________________________________________________________
YOUR INSTITUTION: _________________________________________________________________
SEX: M |__| F |__| YOUR YEAR OF BIRTH |_1_|_9_|_ __|___|
TODAY’S DATE: |__|__| - |__|__| - |_2_|_0_|_0_|____|
DAY MONTH YEAR
QUESTIONS
1. Do you use the word “CURE” when speaking to your oncology patients
at the end of therapy, or during follow-up YES |__| (go to 2)
NO |__| (go to 6)
2. If YES, do you mean …
|__| ‘Cured’ from their primary oncologic disease only, or
|__| ‘Cured’ in a total, general sense, or
|__| ‘Cured’ from oncologic disease, but with a risk of late effects or second malignancies, or
|__| ‘Cured’ in another sense, please specify:
3. If YES, when do you tell patients that they are cured?Please check each cancer type below that applies to your patients?
Time since end of therapy, in years, in CCR*Cancer Type
At end oftherapy 1 2 3 4 5 6 7 8 9 - 10
Leukemia & Lymphomaa. B type
b. Non-B type
c. Brain Tumor
Lymphomad. Hodgkins
e. Soft Tissue Sarcoma
f. Bone Cancers
Neuroblastoma,
g. Stage 1 & 2
h. Stage 3 & 4
i. Wilms Tumor
j. Germ Cell Tumors
k. Retinoblastoma
l. Hepatoblastoma
m. Other, specify

16
haematologica vol. 89[supplement n. 10]:October 2004
Domenica 10 ottobre
FIG. 1 ELTEC Survey of ‘CURE’Version 2
3 Dec, 2003 Page 2
4. Are there other possibilities for the correct time to tell patients that they are cured?
Please write in your answer
5. What criteria do you use to decide when a patient is cured? Check answer.
a. Based on Kaplan-Meier curves in the relevant (e.g., BFM) study groupYES |__|
NO |__|
b. Based on the time when the risk of relapse is the same as the risk of thegeneral population of developing a first cancer
YES |__|
NO |__|
c. Other, specify
6. If NO, do you think that
|__| In life nothing is 100% certain, and former cancer patients still have a slight risk of recurrence
|__| The risk for recurrences or for second cancers is still higher than expected
|__| Other late complications (neurological, cognitive, etc) make the use of the word ‘cure’ difficult
|__| Other, please specify
If you have other comments about this topic please write them here.
Thank you very much for your assistance,
Yours sincerely,
Momcilo Jankovic MDfor the ELTEC Committee of the I-BFM-SGMonza, Italy
3 December, 2003

- E' corretto e dovrebbe essere “obbligatorio” parlare di“guarigione” anche se ci sono rischi di ricadute tardiveo di comparsa tardiva di gravi effetti tossici.
- E' importante pur con queste “attenzioni” (caveat!) par-lare di guarigione: ha un effetto psicologico significati-vo.
- Forse è troppo rigido considerare un soggetto guarito sìo no. Dovrebbe esserci una maggiore gradualità a con-siderare “guarito” un soggetto per pregresso tumore eper pregressa terapia.
- Più passa il tempo maggiore è la certezza di considera-ta il soggetto veramente guarito.
CommentoQuesto studio eseguito con i Pediatri Oncologi Europei su
un campione sufficientemente ampio dimostra che tuttosommato c'è una buona confidenza (>80%) nell'uso del ter-mine “guarito” per i bambini trattati quando si parla alle lorofamiglie. Rimane aperto e sollevato dalla maggior parte deglioncologi pediatri il problema “sentito” del rischio delle reci-dive tardive e della possibile comparsa di gravi effetti tar-divi (es. secondi tumori indotti delle terapie precedenti). Quiperò occorre fare una considerazione importante o megliouna distinzione non filosofica: quando si parla di guarigio-ne si intende scomparsa di un tumore adeguatamente trat-tato, e ciò non può e non deve includere gli effetti tossicitardivi e/o la comparsa di un secondo tumore. 2-6 Ciò puòaccadere ma va a compromettere non la guarigione ma lostato di salute generale di un soggetto che giustifica pie-namente la necessità, in questi soggetti, di lunghi (per sem-pre?) follow-up clinici e strumentali. Ci teniamo quindi atenere ben distinti i due concetti di: a) guarigione b) stato
di salute globale. 2, 6 E' bello veder sorrider un bambino (eanche un adulto) specie se sono usciti trionfanti da un tun-nel insidioso e complesso come quello di una malattia tumo-rale. La guarigione è una grande vittoria ma racchiude in sel'insidia di una minata “qualità di vita” che pertanto va pro-tetta e garantita per poter parlare di “vero successo” e di“vera guarigione”.
Prospettiva futuraE' in corso un analogo censimento con i più grandi grup-
pi cooperativi internazionali anche oltre oceano per far siche a livello di comunità scientifica mondiale possa essereelaborato un documento chiaro ed esauriente sul concettodi guarigione indispensabile per far si che il concetto di gua-rigione anche sociale possa finalmente essere ottenuto sen-za limitazioni e compromessi.
Bilbiografia
1. Jankovic M, Fraschini D, Amici A et al. Outcome after cessation oftherapy in childhood acute lymphoblastic leukemia. Eur J Cancer,1993; 29A, 13:1839-1843
2. Leplege A, Hunt S. The problem of quality of life in medicine. JAMA,1997; 278:47-50
3. Haupt R, Jankovic M, Valsecchi MG et al. I soggetti fuori terapiadopo tumore maligno contratto in età pediatrica. Il Registro Italiano(OTR). Riv Ital Pediatr, 1995; 21:621-5.
4. Reaman GH, Haase GM. Quality of life research in childhood cancer.Cancer, 1996;78:1330-2.
5. Bradiyn AS, Ritchey AK, Harris CV et al. Quality of life research inpediatric oncology. Cancer, 1996;78:1333-9.
6. Palmer RH. Quality of care. JAMA, 1997; 277:1896-7.
17
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

Chi decide in pediatria
Sandro SpinsantiIstituto Giano, Roma
Il Comitato nazionale per la bioetica nel documentoInformazione e consenso all'atto medico (1992) invita anon considerare il consenso informato in pediatria comeun capitolo a parte - o un caso eccezionale - del consen-so informato in generale; propone invece la cultura pedia-trica in quanto capace di “dare un contributo alla cultu-ra del consenso informato”. Accogliamo l'invito a ripen-sare quest'ultimo a partire da quanto ci insegna il rapportocon bambini e adolescenti malati. Il punto di partenza nonpuò essere che la consapevolezza della complessità deirapporti che si stringono attorno a un bambino. Il bambi-no vive immerso in un tessuto di relazioni interpersonali,delle quali ha una immediata conoscenza intuitiva. Il sape-re relazionale del bambino è enormemente sviluppato.Sfrutta al massimo la comunicazione non verbale e sa rac-cogliere informazioni per vie che gli adulti non immagi-nano. Allo stesso modo, il consenso informato ai tratta-menti sanitari prende vita dalle relazioni.
Sinteticamente, si è soliti designare la trama di rappor-ti che si stringono intorno a una persona malata comealleanza terapeutica. L'alleanza è tanto più efficace quan-to più rispetta tutti i protagonisti nella loro specificità.Quando ha a che fare con il minore, il medico può sba-gliare inclinando in due direzioni opposte: può dare o unpeso eccessivo o nessun peso al ruolo che sono chiamatia svolgere i familiari. Nel primo caso, il medico delega ifamiliari di essere gli interlocutori con il minore, coloro chespiegano e fanno accettare le decisioni mediche. Il medi-co diventa così per il minore una figura distante, chiusanell'ambito della professionalità. Nel secondo caso i sani-tari tendono a escludere i genitori - o per risparmiare loroil peso emotivo delle decisioni, o perché li ritengono unelemento di disturbo - e a rivolgersi direttamente al mino-re, scavalcando le sue figure di riferimento. L'obiettivo acui deve tendere l'alleanza terapeutica non è di divideree contrapporre le figure coinvolte, ma di tenerle insieme.Non diversa dovrebbe essere la strategia anche nel casodi un consenso informato che ha come protagonisti degliadulti.
Un secondo elemento qualificante del consenso infor-mato pediatrico è che non può limitarsi a un atto isolato,avulso da tutto il processo comunicativo. È la relazione checonta, e questa si costruisce con il tempo, mediante pic-coli gesti e comportamenti apparentemente irrilevanti.Anche una piccola bugia o una reticenza di scarso rilievopossono compromettere la relazione con un bambino. E larelazione - con il bambino, non meno di quella con l'adulto- comincia con l'ascolto, prima che con l'informazione.
Il clinico impara soprattutto con il bambino che percomunicare efficacemente (con tutto il groviglio di pro-blemi che gli si pongono: che cosa dire e che cosa omet-tere; come spiegare; come trovare un accordo su quantointende proporre) deve innanzi tutto ascoltare la persona
alla quale vuole trasmettere le informazioni. In questoprocesso è artificiale distinguere le piccole decisioni dal-le grandi: il processo comunicativo è unitario; può essereincrementato o compromesso anche da aspetti perifericirispetto a quelle scelte di grande rilievo che si ritiene esi-gano il consenso informato a sé stante. Il paradigma ope-rativo che presuppone di considerare la comunicazionecome un processo unitario è anche l'approccio più correttoper il consenso informato con gli adulti.
In terzo luogo l'ambito pediatrico rende manifesto comesia facile scambiare il consenso con l'assenso. La comuni-cazione complementare (quella che si esprime nella rela-zione one up/one down) è quasi connaturata alla minoreetà e soprattutto all'infanzia. Finché si mantiene questotipo di relazione, è abbastanza agevole indurre chi sta inposizione subordinata a fare ciò che ha deciso - per il suobene - chi occupa la posizione di potere sovrastante. Conle buone (seduzione, lusinghe, ricatti affettivi) o con lecattive. La tentazione autoritaria è presente in medicina,così come in tutto il vasto ambito dell'educazione. Mal'assenso così ottenuto è ben lontano dal consenso inte-so come partecipazione attiva alle decisioni.
Nel documento Informazione e consenso all'atto medi-co troviamo un'opportuna differenziazione della capacitàdi arrivare a un consenso in base alle fasi di sviluppocognitivo. Prima dei 6-7 anni non possiamo parlare di unconsenso autonomo:
Il consenso è in qualche modo concepibile tra 7 e 10-12 anni, ma sempre non del tutto autonomo e da consi-derare insieme con quello dei genitori. Solo entrando nel-l'età adolescenziale si può pensare che il consenso diven-ti progressivamente autonomo.
Di conseguenza dopo i 7 anni va ricercato il consensodel bambino e dei genitori, e dopo i 14 è prioritario il con-senso dell'adolescente. Dobbiamo considerare un falli-mento clamoroso della gestione della decisione consen-suale con un giovane se prevale la logica giuridica (a 18anni meno un giorno non è autorizzato a prendere deci-sioni su di sé, mentre può farlo a compimento formaledella maggiore età. . . ), a danno dell'alleanza terapeuti-ca, dell'insieme unitario del processo comunicativo, dellaconquista comune di un punto di accordo negoziato conil minore stesso.
18
haematologica vol. 89[supplement n. 10]:October 2004
Domenica 10 ottobre
Lettura Magistrale

19
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
Trattamento dell'Ipertensione Polmonare nel-le anemie emolitiche croniche
Gian Luca Forni, Giorgio Derchi°Centro della Microcitemia e °S. C. di Cardiologia, Ospeda-le Galliera, Genova
L'Ipertensione Polmonare (IP) è caratterizzata da unaumento delle resistenze del letto vascolare polmonareche porta alla disfunzione cardiaca destra ed alla morte.Ha una eziopatogenesi multifattoriale con una prevalen-za totale che va da 30 a 50/milione, può essere primitivao associata a malattie del connettivo, a malformazionicongenite cardiache, infezioni HIV, tromboembolismo pol-monare etc. 1 Nelle anemie emolitiche croniche è descrit-ta una incidenza che va dal 10% della Thalassemia Majoral 60% della Sickle cell anemia, ma è stata descritta anchenella Sferocitosi e nella Emoglobinuria Parossistica Not-turna. 2-4 Queste cifre hanno fatto sì che si parlasse di unasindrome di “hemolysis-associated pulmonary hyperten-sion” e che l'ipertensione polmonare venisse indicata comeprincipale causa di morte nella Sickle cell anemia. 4
Le opzioni terapeutiche sono limitate ed includono ilcalcio antagonisti, il Bosentan antagonista degli endothe-lin-receptor e la terapia anticoaugulante a lungo termi-ne. I calcio-antagonisti, che sono sono efficaci solo nel30% dei casi, sono di difficile gestione in pazienti nor-malmente ipotesi come i thalassemici. Il Bosentan fre-quentemente causa aumento delle transaminasi e questorappresenta un problema in pazienti epatopatici qualisono la grande maggioranza dei pazienti thalassemici. IlSildenafil citrato è un potente e selettivo inibitore dellefosfodiesterasi cGMP specifiche che promuovono il rilasciodella muscolatura liscia del letto vascolare polmonare edè stato utilizzato con successo nel trattamento dell'IP pri-mitiva e secondaria. 5
Abbiamo arruolato 7 pazienti affetti da emoglobino-patie in un protocollo che prevede l'impiego di Sildenafilnel trattamento delIa IP severa che non ha risposto ai trat-tamenti convenzionali. IP è stato definita come Tricuspidgradient (TG) ≥45 mmHg al EcoDoppler CW. Il disegnodello studio prevedeva come end-point primario il gradodi cambiamento della ipertensione polmonare. End-pointsecondario includeva il cambiamento di classe funziona-le NYHA. Il protocollo è stato approvato dal Comitato dibioetica del nostro ospedale ed i pazienti hanno dato illoro consenso informato. Il periodo di trattamento, tutto-ra in corso, varia da 2 a 50 mesi.
A 8 settimane in tutti i pazienti abbiamo ottenuto unasignificativa riduzione dell'ipertensione polmonare misu-rata come gradiente ticuspidale sistolico ed il migliora-mento della classe funzionale NYHA senza osservare effet-ti avversi. Nel follow up questo trend si è mantenuto.
In conclusione i dati preliminari di questo studio dimo-strano che il Sildenafil Citrato è efficace e senza effettiavversi nei pazienti con emoglobinopatie affetti da iper-tensione polmonare non trattabile con altre terapie orali.
Bibliografia
1) Rubin LJ. Primary pulmonary hypertension N Engl J Med1997;336:111-17
2) Derchi G, Fonti A, Forni GL, Cappellini MD, Turati F. Pulmonaryhypertension in patients with thalassemia major. Am Heart J. 1999Aug;138(2Pt 1):384
3) Aessopos A, Farmakis D, Karagiorga M, Voskaridou E, Loutradi A,Hatziliami A, Joussef J, Rombos J, Loukopoulos D. Cardiac involve-ment in thalassemia intermedia: a multicenter study. Blood 2001Jun 1;97(11):3411-6
4) Gladwin M. T. , Sachdev V. , Jison M. L. , et al Pulmonary hyper-tension as a risk factor for death in patients with sickle cell dis-ease N Engl J Med 2004;350:886-95
5) Littera R, La Nasa G, Derchi G, Cappellini MD, Chang CY, Contu L.Long-term treatment with sildenafil in a thalassemic patient withpulmonary hypertension. Blood 2002 Aug 15;100(4):151.
Coagulazione intravascolare disseminata
Giuseppe AvvisatiEmatologia, Università Campus Bio-MeCIDo, Roma
Con il termine di CID (Coagulazione intravascolare dif-fusa) si inCIDa una condizione morbosa caratterizzata daun'attivazione generalizzata della coagulazione in vivo.L'attivazione in vivo della coagulazione è responsabile del-la presenza di trombina circolante con formazione con-temporanea in vivo di fibrina e microaggregati piastriniciche insieme danno origine a microtrombi circolanti. Que-sti ultimi, arrestandosi nei piccoli vasi riducono l'afflussodi sangue con conseguente grave compromissione dinumerosi organi per scompenso metabolico ed emodina-mico. A tutto ciò l'organismo cerca di rispondere conun'attivazione secondaria della fibrinolisi responsabile,insieme alla riduzione del numero di piastrine circolantiper la formazione di microaggregati in vivo, della com-parsa di una grave sintomatologia emorragica. A secondadella gravità della sintomatologia clinica tromboemorra-gica possiamo distinguere una forma acuta ed una croni-ca di CID cui alcuni autori aggiungono anche una formasubacuta.
EziologiaLe cause responsabili dello scatenamento di una CID
sono numerose e quindi, la CID di per sé non è una malat-tia ad eziologia unica, ma una sindrome dovuta a causeapparentemente differenti tra loro. Le forme morbose piùfrequentemente responsabili di una CID sono: alcunepatologie ostetriche, le sepsi, alcune neoplasie maligne,soprattutto se metastatizzate, le emopatie acute, le con-dizioni morbose in cui è presente un' iperemolisi acuta, lacirrosi epatica, gli interventi chirurgici, i traumi e le ustio-ni gravi, gli emangiomi giganti.
PatogenesiIn condizioni fisiologiche, il sistema vascolare resta per-
vio e il sangue non coagula grazie al normale flusso ema-tico, alla presenza di inibitori naturali della coagulazione(come l'antitrombina III e la proteina C) e all'attivazionedella fibrinolisi, con formazione di plasmina, in manierabilanciata rispetto all'attivazione della cascata coagula-tiva. Questo equilibrio fisiologico nella CID viene alterato
Incontra l’Esperto

perché si realizza uno stimolo marcato o in senso coagu-lativo o in senso fibrinolitico o in ambedue i sensi. La CIDrappresenta comunque il risultato finale di una comples-sa sequenza di eventi correlati tra loro in maniera cosìstretta da essere ricostruita con difficoltà.
Quale sono i fattori capaci di scatenare una CID?Le osservazioni cliniche e sperimentali suggeriscono che
vi sono fattori che agiscono con meccanismo diretto oindiretto. Il meccanismo diretto è in causa quando i fat-tori scatenanti, come nel caso degli estratti tessutali conazione tromboplastino-simile e degli enzimi proteolitici(tipo i veleni di serpenti), sono capaci di attivare diretta-mente la cascata coagulativa in punti diversi provocandola formazione d trombina e successiva deposizione di fibri-na, consumo di piastrine e conseguente danno tessutale.Attraverso un meccanismo indiretto agiscono invece leendotossine e i complessi antigene-anticorpo che, provo-cando un danno tessutale, attivano contemporaneamen-te la cascata coagulativa e il sistema fibrinolitico per mez-zo della contatto-attivazione che trasforma il fattore XIIin XIIa.
Quali sono i fattori che favoriscono la genesi di una CID?I più importanti sono i seguenti:a) shock, ipossiemia, acidosi;b) blocco del sistema istiocito-macrofagico;c) presenza di lipidi plasmatici elevati,d) inibizione del sistema fibrinolitico. Alcuni autori ritengono che una CID sia presente in tut-
te le forme di shock e che rappresenti la caratteristicaprincipale di tutti gli shock irreversibili. Lo shock svolge unruolo fisiopatologico importante anche se non ancoradefinito; infatti la ridotta perfusione, con conseguenteipossiemia e acidosi, produce uno stato di ipercoagulabi-lità e favorisce l'aggregazione intravascolare di piastrine.Inoltre l' ipoperfusione splancnica riduce l'efficienza delsistema istiocito-macrofagico e la funzione epatica diclearance, mantenendo più a lungo in circolo i fattori atti-vati della coagulazione con conseguente attivazione del-la cascata coagulativa.
FisiopatologiaDopo l'attivazione del sistema coagulativo con com-
parsa in circolo della trombina e della plasmina, la fisio-patologia della CID è relativamente costante ed indipen-dentemente dalla eziologia.
Qual è il ruolo della trombina in una CID?Una volta formatasi, la trombina comincia a staccare
dalla molecola di fibrinogeno prima il fibrinopeptide A(FpA) e successivamente il fibrinopeptide B (FpB). In que-sto modo vengono lasciati liberi i monomeri di fibrina chesuccessivamente sotto l'azione del fattore XIII, polimeriz-zano dando luogo alla formazione di microtrombi di fibri-na, nei quali vengono catturate le piastrine con conse-quente piastrinopenia. Altre azioni svolte dalla trombinasono l'inizio dell'aggregazione piastrinica e il potenzia-mento della cascata coagulativa, aumentando l'attivitàcoagulativa dei fattori V e VIII; tuttavia la sua azione con-tinua conduce alla degradazione di questi fattori per mez-zo della proteina C. Quindi la trombina è responsabile del-
la diminuzione del fibrinogeno, delle piastrine e dei fat-tori II, V, VIII, XIII, ma alla diminuzione di questi fattoriconcorre anche la plasmina con la sua azione proteoliti-ca. Tuttavia alcune volte le concentrazioni dei fattori V eVIII in corso di CID possono essere normali o addiritturaelevate; ciò si verifica quando si ha una minore azioneproteolitica della trombina, così che l'attivazione di que-sti fattori avviene senza che vi sia una corrispondenteinattivazione.
Qual è il ruolo della plasmina in una CID? Anche la plasmina circola liberamente e comincia a
staccare dalla molecola del fibrinogeno i frammenti X, Y,D ed E, che danno luogo ai prodotti di degradazione delfibrinogeno (FDP). Inoltre la plasmina, prima ancora distaccare i frammenti X,Y, D ed E, rilascia dalla molecola delfibrinogeno anche peptidi specifici tra cui il peptide B-1-42 ed il frammento B-15-42.
Prima della polimerizzazione in fibrina gli FDP possonocombinarsi ai monomeri di fibrina impedendone la poli-merizzazione e dando luogo ai monomeri solubili di fibri-na. Questi, sono responsabili della positività dei test diparacoagulazione, in quanto l'etanolo o il solfato di pro-tamina staccano gli FDP dal monomero di fibrina permet-tendo la polimerizzazione con formazione di maglie difibrina. L'interferenza degli FDP con la polimerizzazionedei monomeri di fibrina altera profondamente l'emostasi,favorendo l'emorragia. Inoltre, gli FDP hanno un'alta affi-nità per la membrana piastrinica, rivestendo le piastrinee impedendone la normale funzionalità. Inoltre, attivan-do le frazioni C1 e C3 del complemento, la plasmina atti-va il sistema del complemento fino a C8 e C9 con conse-guente lisi cellulare e piastrinica, liberazione di ADP efosfolipidi e quindi presenza in circolo di una maggiorequantità di materiale dotato di proprietà procoagulanti.
L'attivazione del sistema del complemento aumenta lapermeabilità vascolare favorendo l'ipotensione e lo shockche sono aggravati anche dalla contemporanea attivazio-ne del sistema delle chinine attraverso l'attivazione delfattore XII (mediata dal danno tessutale provocato dallaipotensione e dallo shock). Il fattore XIIa è infatti capacedi attivare la precallicreina in callicreina, che a sua voltaattiva il chininogeno ad alto peso molecolare in bra-CIDhinina.
Una volta che il processo della CID è stato scatenatocontinuerà ad automantenersi fino a che non interverrà unevento in grado di interrompere questo circolo vizioso.
Solo la conoscenza del meccanismo fisiopatologico, ciconsente di capire facilmente perché la maggior parte deipazienti con CID presenta, contemporaneamente o in tem-pi successivi, fenomeni trombotici ed emorragici.
Manifestazioni clinicheLa manifestazione clinica più evidente della CID è la
presenza di emorragie, spesso gravi, che insorgono ina-spettate ed interessano contemporaneamente più sedi.Alle manifestazioni emorragiche si associano spesso feno-meni trombotici, specialmente del microcircolo, che con-tribuiscono ad aggravare il quadro clinico. I fenomenitrombotici del microcircolo quasi sempre sono responsa-bili di una insufficienza renale con i segni di ridotta per-fusione vascolare aggravati anche da uno stato di shock
20
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre

di entità variabile. Molto comuni sono i sintomi di dannoepatico (alterazione degli enzimi epatici), renale (aumen-to della creatinina), polmonare (ipossia) e del SNC (obnu-bilamento del sensorio fino al coma). Queste manifesta-zioni possono essere dovute alla CID, ma possono ancheessere in rapporto con la malattia di base. D'altra parte lafisiopatologia della CID è così complessa che è spessoimpossibile stabilire con certezza la genesi della sintoma-tologia clinica e delle alterazioni dei dati di laboratorio.Altro segno importante di CID può essere uno sviluppoinspiegato di una sindrome clinica da shock, senza emor-ragie manifeste, dovuta al sequestro di sangue e plasmanei tessuti danneggiati e nelle cavità sierose, secondariaall'attivazione del sistema delle chinine. Infine, dal puntodi vista clinico possiamo distinguere una CID acuta ed unaCID cronica e la possibilità di forme con caratteristicheintermedie.
CID acutaE' la consequenza di uno stimolo intenso, continuo e
prolungato nel tempo. In questo caso, in molti pazientipredominano le gravi manifestazioni emorragiche conecchimosi generalizzate, petecchie ed emorragie nelle sedidei prelievi venosi, degli agoaspirati e delle manovre biop-tiche, di traumi o di interventi chirurgici. Comuni sono legengivorragie, le epistassi, le emorragie gastrointestinali,le emorragie polmonari e l'ematuria; possono esservi ver-samenti ematici nelle cavità sierose. Questa forma di CIDè molto più frequente nelle neoplasie ematologiche ed inparticolare nelle leucemie acute.
CID cronicaAlcune forme di CID sono causate da uno stimolo debo-
le o intermittente. In questi pazienti si raggiunge un equi-librio tra distruzione dei fattori della coagulazione e del-le piastrine. Clinicamente, oltre a emorragie di minoreintensità, sono presenti anche processi tromboflebitici otromboembolici ricorrenti; tuttavia col progredire dellamalattia di base possono svilupparsi manifestazioni emor-ragiche più gravi. Forme croniche di CID sono statedescritte in molti casi di tumori metastatizzati. La fisio-patologia di questa CID compensata, cronica o subacuta,è fondamentalmente la stessa di quella acuta. Ciò nono-stante è necessario fare una distinzione in quanto il qua-dro clinico e i dati di laboratorio nella forma cronica sonopiù variabili che nella forma acuta e spesso possono crea-re problemi dal punto di vista diagnostico. La CID cronicapresenta parametri clinici e di laboratorio che assomi-gliano a uno stadio intermedio fra uno stato di ipercoa-gulabilità e la CID acuta. E' perciò possibile che tutti glistati di ipercoagulabilità non siano altro che lenti proces-si di coagulazione intravascolare. Inoltre è attualmentechiaro che la sindrome di Trousseau (attivazione della coa-gulazione associata a tromboflebiti migranti e recidivan-ti in corso di neoplasie) in molti casi non è altro che unaforma di CID cronica.
DiagnosiPur rappresentando la CID conclamata una sindrome di
non frequente riscontro, deve essere sospettata e diagno-sticata rapidamente per la possibile gravità del quadromorboso. Il primo e più importante passo per una corret-
ta diagnosi di CID è quello di sospettare che le manife-stazioni cliniche di natura emorragica o tromboemorragi-ca presentate da un paziente possano essere dovute aduna coagulazione intravascolare disseminata. Infatti, trop-po spesso questo riconoscimento è tardivo e ciò può cau-sare danni irreversibili. Una volta sospettata la presenzadi una CID, l'uso di alcune indagini di laboratorio di faci-le e rapida esecuzione ci consentirà di confermare o menoil nostro sospetto. Tuttavia, i risultati ottenuti con le pro-ve di laboratorio vanno attentamente valutati tenendopresente anche la sintomatologia clinica, in modo da potergiungere ad una corretta decisione terapeutica e tra lenumerose prove a nostra disposizione è opportuno sce-gliere quegli esami che sono in grado di rispondere alnostro quesito diagnostico nel minor tempo possibile.
Sistema a punti per la diagnosi di CID (Tabella 1)Alcuni ricercatori, al fine di semplificare il lavoro dei
clinici, hanno proposto di utilizzare in tutti quei casi in cuiil paziente ha una patologia che può associarsi ad unaCID, un sistema a punti che si basa sui risultati di alcuniesami di laboratorio di semplice esecuzione come: PT, FDPo XDP, Fibrinogeno e Numero di Piastrine, per conferma-re o escludere la presenza di una CID. Questo sistema, inbase al punteggio finale, consente di suddividere i pazien-ti in due categorie:
(i). Pazienti con un punteggio ≥5, compatibile con unadiagnosi di CID;
(ii). Pazienti con un punteggio < 5, suggestivo ma noncompatibile con una diagnosi di CID;
I pazienti con punteggio 5 devono esser trattati secon-do le linee terapeutiche consigliate per una CID; al con-trario, nei pazienti con punteggio <5 è necessario con-trollare gli esami di laboratorio giornalmente ed attende-re che il punteggio diventi 5, prima di iniziare il tratta-mento.
Bisogna ricordarsi però che questo sistema a punti èvalido solo per quei pazienti affetti da una patologia capa-ce di scatenare una CID.
21
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
Tabella 1.Sistema a punti per la diagnosi di CID.
(i). Il paziente ha una patologia che può associarsi ad una CID?SI = procedi ; NO = non usare questo sistema
(ii). Richiedi Screening Coagulativo ( PT, Fibrinogeno,FDP/XDP, Conta Piastrine)
(iii). Dai un punteggio ai risultati:
Aumento del PT (<3 sec = 0 ; >3 sec =1 ; >6 sec = 2) w
Fibrinogeno (>100mg/dL = 0; <100 mg/dL =1) w
FDP/XDP* (N = 0; A =1; AAA = 2) w
Piastrine x 103/ºL (>100 = 0; <100 =1; <50 = 2) w
(iv). Calcola il Punteggio w5 compatibile con CID, ripetere giornalmente
< 5 suggestivo, non affermativo di CID, ripetere giornalmente
* N: Normale ; A: Aumentati ; AAA: Molto aumentati

TerapiaNel caso si sospetti una CID, vi sono delle misure tera-
peutiche di carattere generale che vanno prontamenteattuate senza attendere i risultati di laboratorio. Questemisure terapeutiche generali sono:
(i). terapia della malattia di base (diversa a seconda del-la etiologia della CID),
(ii). eventuale terapia contro lo Shock;Inoltre, se il paziente presenta delle emorragie in atto,
o deve essere sottoposto a manovre invasive e si ha ilsospetto che ci si trovi in presenza di un paziente con CID,è necessaria:
(iii). La trasfusione di concentrati piastrinici (alla dosedi 1 concentrato ogni 10kg di peso corporeo)
(iv). ) La trasfusione di plasma fresco congelato in infu-sione continua (alla dose di 15-20 ml /kg di peso corpo-reo).
Se invece il paziente non presenta una sintomatologiaemorragica o non deve essere sottoposto a manovre inva-sive, non è necessario utilizzare i concentrati piastrinici oil plasma fresco congelato.
Generalmente queste misure terapeutiche, se pronta-mente attuate, sono in grado di risolvere la maggior par-te delle CID, tuttavia, nei casi in cui prevalgono i fenome-ni trombotici, è necessario attuare anche una terapia confarmaci anticoagulanti.
Terapia con farmaci anticoagulantiIl farmaco anticoagulante più comunemente usato,
anche se fino ad ora non è stato mai stato dimostrato unsuo effetto benefico in studi cinici controllati, è l'eparina.Considerando la complessa fisiopatologia della CID è chia-ro che la somministrazione di eparina è in grado di inter-rompere solo uno dei due momenti fisiopatologici. Tutta-via, quando necessaria la somministrazione di eparina puòesser fatta con modalità diverse ma sempre a dosaggirelativamente bassi. In particolare, in presenza di insuffi-cienza renale è inCIData la somministrazione di eparina ininfusione continua con dosaggi variabili che dipendono dalpeso del paziente. Invece, in assenza di insufficienza rena-le è utile la somministrazione sottocutanea di dosi tera-peutiche di eparine a basso peso molecolare (EBPM)secondo gli schemi utilizzati per il trattamento dellamalattia tromboembolica per le diverse EBPM.
Terapia con Inibitori fisiologici della coagulazione Poiché l'antitrombina III (ATIII) è uno dei più potenti
inibitori della coagulazione, il suo uso nei pazienti conCID è stato oggetto di numerosi studi, anche se la mag-gior parte degli studi randomizzati hanno riguardatopazienti con Sepsi o Shock settico. Al momento attuale,però, sulla base di numerosi studi clinici non vi è inCIDa-zione all'utilizzo dei concentrati di ATIII in corso di CID conl'esclusione di quei pazienti in trattamento eparinico egravi deficit di ATIII. Per quanto riguarda invece l'utilizzodei concentrati di proteina C attivata è ancora troppo pre-sto consigliarne l'uso al di fuori di studi clinici prospetti-ci controllati.
Terapia con farmaci antifibrinoliticiLa somministrazione di questi farmaci in corso di CID
dovrebbe essere presa in considerazione solo in queipazienti nei quali è presente un'eccessiva risposta fibri-nolitica secondaria all'attivazione della coagulazioneoppure un' iperfibrinolisi primaria. Anche questi farmacivanno somministrati per infusione continua alla dose di40-100 mg /kg/die per l'acido tranexamico.
ConclusioniI pazienti con CID presentano problemi non solo dia-
gnostici, ma anche terapeutici, per la possibile concomi-tante presenza di complicanze emorragiche e trombotiche.Fondamentale è comunque in questi casi identificare etrattare in modo corretto la condizione morbosa di basee l'eventuale condizione di shock che spesso si associaalla CID. Inoltre, un ruolo di primaria importanza è svoltoanche dal corretto supporto trasfusionale. Infine, deveessere tenuto ben presente che la somministrazione dieparina richiede un'attenta valutazione clinica e non puòin alcun modo essere considerato, come avveniva una vol-ta, l'unico trattamento specifico per questa patologia.
Bibliografia
Levi M, ten Cate H. Disseminated Intravascular Coagulation. NEngl J Med 1999;341:586-92.Levi M, de Jonge E, Meijers J. The diagnosis of disseminatedintravascular coagulation. Blood Rev 2002;16:217-23.
22
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre

Epidemiologia descrittiva dei tumori maligniin età pediatrica. Il contributo dei RegistriTumori di popolazione: dalla registrazionealla valutazione della qualità degli interventi
Guido Pastore, Paola Dalmasso,* Milena Maule,Maria Luisa Mosso, Silvia Viscomi, Luisa Zuccolo, Corrado Magnani°Registro dei Tumori Infantili del Piemonte, CPO-Piemonte,Università degli Studi di Torino, *Dipartimento di SanitàPubblica, Università di Torino, °Dipartimento di Scienzemediche, Università del Piemonte Orientale, Novara
Tradizionalmente lo scopo principale dei registri tumo-ri (RT) di popolazione è stato quello di fornire indicazionisulla frequenza delle patologie neoplastiche in un conte-sto geografico definito. Le misure di frequenza comune-mente utilizzate fanno riferimento ad una popolazioneresidente (denominatore) in un area geografica circo-scritta e in un periodo temporale definito e hanno alnumeratore (i) il numero di nuovi casi diagnosticati nellapopolazione in studio (tassi di incidenza); (ii) il numero dipersone che muoiono a causa del tumore tra i residentinell'area sotto osservazione (tassi di mortalità/letalità e(iii) il numero di persone che vivono dopo che la diagno-si di tumore è stata posta in una popolazione e in un ambi-to temporale definito (tassi di prevalenza).
Inoltre i RT provvedono a calcolare percentuali cumu-lative di sopravivvenza delle persone affette da tumorediagnosticate in periodi di tempo differenti in popolazio-ni definite. 1
Accanto a queste statistiche routinarie, principalmentedi interesse di salute pubblica in quanto utili per la piani-ficazione degli interventi assistenziali, i RT possono forni-re informazioni su possibili fattori di rischio, valutazionedei programmi di diagnosi precoce/screening e, più recen-temente, anche indicazioni sulla qualità delle cure chevengono offerte ai malati, sempre con una visione dipopolazione. I RT di popolazione forniscono quindi unaprospettiva più completa di molti studi sulla valutazionedella qualità dell'assistenza e il conseguente sviluppo dilinee guida, per lo più basati su casistiche ospedaliere, chesono selezionate e non rappresentative dell'insieme deimalati per il diverso case-mix e referral pattern delle isti-tuzioni di ricovero e cura. 2
I RT di popolazione si suddividono in 2 categorie: quel-li generali che raccolgono informazioni su tutti casi ditumore maligno diagnosticati durante l'intero arco dellavita e quelli specifici per sede tumorale o per fascia di etàcome quelli pediatrici. A partire dagli anni 1970, 63 RTeuropei operano nell'ambito del programma AutomatedChildhood Cancer Information System (ACCIS) in 19 nazio-ni con procedure standardizzate di raccolta dati, codificadelle informazioni cliniche e sono caratterizzati da similecompletezza della registrazione. ACCIS svolge la sua atti-vità sotto l'egida dell'Agenzia Internazionale per la Ricer-ca sul Cancro (IARC). Circa il 50% della popolazione euro-pea (0-14 anni), ma solo il 20% della popolazione italia-na, risiede in aree coperte da RT pediatrici. Il Registro deiTumori Infantili del Piemonte (RTIP) è il più vecchio RT
dell'Europa meridionale e uno dei più grandi. Gli altri RTpediatrici italiani operano nelle Marche e in provincia diRagusa. Sono anche disponibili stime di incidenza da par-te di altre popolazioni italiane servite da un RT non spe-cifico, che si riferiscono circa il 15% della popolazioneitaliana. 3,4
I tumori maligni in età pediatrica (0-14 anni) costitui-scono circa 1-2% di tutti tumori maligni che vengonodiagnosticati nel corso della vita. 5 I tassi di incidenza nel-le nazioni a standard di vita occidentale sono compresi tra140-180 casi per 106 di bambini/anno solare. I tassi diincidenza tra i maschi sono solo leggermente più elevatiche nelle femmine, con l'eccezione dei linfomi nonHodgkin (LNH) e nei linfomi di Hodgkin (HD) dove si osser-va un rapporto M:F di 2:1 e dei tumori delle cellule ger-minali e dei tumori di Wilms (TW) dove predominano lebambine. Nel primo anno di vita i neuroblastomi (NB)costituiscono il più frequente tipo di tumore seguiti dalleleucemie e dai TW, nella classe 1-4 anni le leucemie lin-fatiche acute (LLA) rappresentano il 35-40% dei casiseguiti dai tumori cerebrali (TC), nella classe 5-9 anni i TCe le leucemie sono rispettivamente il 25 e il 30% dei casie nella classe 10-14 anni predominano i linfomi, i TC, leleucemie e sarcomi ossei. Tra gli adolescenti (15-19 anni)sono rari i tumori peculiari dell'età pediatrica (WT, NB,retinoblastoma e LLA), mentre prevalgono i linfomi e i car-cinomi analogamente a quanto si osserva nell'età adulta.Nei paesi economicamente più avvantaggiati, a partiredagli anni '60-'70, è comparso un picco di incidenza perle LLA nel 2°-4° anno di vita che, nei decenni successivi,è progressivamente aumentato in ampiezza e ristretto allafascia di età 3-4 anni. Questo picco si è evidenziato neipaesi dell'Europa orientale a partire dagli anni '80.4,6
Nel corso degli 3 decenni, i tassi di incidenza per tutti itumori, nel loro complesso, e per alcuni tipi specifici han-no mostrato una lieve ma percettibile (intorno a 1-2 % peranno) tendenza all'aumento. Questo trend, osservato siain Europa sia negli USA, è in gran parte da mettere in rela-zione con i miglioramenti delle tecniche diagnostiche perquanto riguarda i TC e i tumori non altrimenti specificati.Anche l'incremento dei NB, particolarmente evidente neibambini di età inferiore all'anno, è da attribuirsi all'au-mento di tumori addominali, per lo più localizzati e a buo-na prognosi, rilevati occasionalmente in corso di ecogra-fie eseguite durante la gravidanza e nei primi mesi di vita.Nelle nazioni (Giappone, Canada e Germania), dove sonostati avviati programmi di screening, si è osservato unaumento di casi localizzati con andamento clinico favo-revole, senza per altro una riduzione dei casi metastaticiad evoluzione fatale. Questo trend è stata interpretatocome una sovradiagnosi di casi di NB che altrimenti nonsarebbero diventati clinicamente evidenti, infatti in con-comitanza con l'aumento dell'incidenza dei NB nei primianni di vita, non si osservato una riduzione dei tassi diincidenza nelle classi di età seguenti né una variazione deitassi di mortalità. Per contro, l'incremento dei tassi di inci-denza delle LLA, specialmente limitato alla classe di età 1-4 anni e per le forme pre-B, sembra costituire un fatto rea-le, connesso con mutamenti ambientali, almeno in alcu-ne popolazioni dove il fenomeno è stato analizzato conadeguate tecniche statistiche. 4,7
Se l'aumento di alcuni tipi di tumori può essere spia
23
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

della presenza di fattori di rischio ambientali, la diminu-zione dei tassi di mortalità per i principali tumori pedia-trici è indice dei successi dell'oncologia pediatrica. 8 Neipaesi occidentali, i tumori sono la causa di morte più fre-quente dopo le malformazioni congenite e gli incidenti. Itassi di mortalità per tutti i tumori in Piemonte sono pas-sati da 90/106 di bambini negli anni '70 a circa 50 alla finedegli anni '90, con un trend simile a quelli degli altri pae-si occidentali. La diminuzione per le LLA è stata ancora piùmarcata: da 30 /106 negli anni '70 a 10 alla fine degli anni'90 (9). I tassi di mortalità sono influenzati dalla frequen-za (e dall'incidenza) delle neoplasie e dalla sopravvivenzadei pazienti, l'effetto del miglioramento delle cure si river-bera sui tassi di mortalità più lentamente di quanto nonavvenga per le percentuali di sopravvivenza, che per altronecessitano una maggior supporto organizzativo per esse-re calcolati (contatto periodico con gli uffici anagrafici diresidenza del paziente, ricerca dei pazienti che si trasfe-riscono presso altri comuni, acquisizione dei documenti distato in vita). 2,8
A partire dagli anni '70 le percentuali di sopravvivenzaa 5 anni dalla diagnosi, sono aumentate in modo costan-te: da circa il 40% fino al circa il 75% negli anni '90.Inparticolare il miglioramento della prognosi è stato piùmarcato per i bambini affetti da LLA, da TW e da HD, peri quali le percentuali di sopravvivenza a 5 anni dalla dia-gnosi si sono avvicinate al 90% negli anni più recenti. Peri TC, i NB, le leucemie non linfatiche acute e i sarcomidelle parti molli e i sarcomi ossei i risultati mostranomiglioramenti più contenuti. Il miglioramento prognosti-co è stato osservato dapprima negli USA e poi in Europa.La situazione europea mostra differenze rilevanti tra i pae-si occidentali e quelli orientali. I dati italiani si confron-tano favorevolmente con quelli degli altri paesi industria-lizzati. 10-13 I risultati osservati dal sistema di registrazio-ne implementato dall'AIEOP Mod 101, sebbene non esau-stivo perchè su base ospedaliero, fornisce dati di soprav-vivenza per alcune patologie, dove le segnalazioni dei cen-tri AIEOP si avvicinano alla totalità dei casi diagnosticatiin Italia (ad es LLA, NB), assai simili a quelle dei RT dipopolazione e confermano l'evoluzione della prognosi deibambini con tumori maligni in Italia, specialmente se trat-tati in accordo con protocolli terapeutici definiti (vedi rela-zione del Dr. Pession). In modo concorde i dati sulla soprav-vivenza dopo una diagnosi di tumore hanno evidenziatoil progressivo aumento dei casi guariti a livello regiona-le,10 a livello nazionale11 e sovranazionale. 12,13 L'aumentodella prevalenza dei bambini con tumori che si affaccia-no alla vita adulta è evidente e deve essere ben presenteper chi opera nel campo della pianificazione delle strut-ture sanitarie, perché queste persone richiedono maggio-ri interventi da parte del sistema sanitario nazionale. InPiemonte, come in altre parti del mondo occidentale,almeno una persona di età 20-29 anni ogni 1000 è unsopravvissuto/o un guarito dopo un tumore in età pedia-trica, questa osservazione sollecita l'attenzione nei con-fronti dei numerosi aspetti della loro qualità di vita: dalraggiungimento delle mete scolastiche, all'inserimento nelmondo del lavoro, alla capacità di instaurare duraturi lega-mi affettivi, alla capacità di procreare, al possibile svilup-po di una seconda neoplasia maligna. 2,14-17
Le analisi di sopravvivenza, prodotte dai RT, permetto-
no di dare una misura reale dell'impatto che interventimedici sofisticati, messi a punto in istituzioni altamentespecializzate, hanno sullo stato di salute della popolazio-ne in generale. Ad esempio uno studio recente del RTIP hadimostrato come le percentuali di sopravvivenza dei bam-bini affetti da LLA residenti in Piemonte siano assoluta-mente analoghe a quelle rilevati dagli studi collaboratividell'AIEOP. 16 Analogamente, i fattori che possono influi-re sulla sopravvivenza subito dopo la diagnosi (età, tipo ditumore, centro dove il bambino è stato trattato nei primigiorni dopo la diagnosi, ecc. . . ), possono essere meglioanalizzati tramite i dati dei RT che dagli studi clinici. Infat-ti, è possibile che i pazienti che muoiono rapidamentepossono essere esclusi dalle casistiche ospedaliere. 15 Altrifattori non biologici che influiscono sulla prognosi (esse-re trattatati preso istituzioni specializzate dove strategiemultidisciplinari più facilmente sono implementate e dovevengono trattatati numerosi pazienti in accordo a proto-colli terapeutici definiti, accessibilità ai servizi sanitari)possono essere meglio valutati attraverso le analisi disopravvivenza condotte dai RT. 2 Un limite dei RT neglistudi di sopravvivenza su base di popolazione rispetto aquelli ospedalieri sta nel numero di informazioni clinicheraccolte. E' auspicabile che i RT espandano i loro datasetincludendo maggiori dati di interesse clinico (ad es. esten-sione della malattia alla diagnosi, indicazioni sulle proce-dure usate per valutare l'estensione della malattia alladiagnosi, la comparsa di metastasi e dati sulla co-morbi-dità) possibilmente interagendo in maniera complemen-tare con i sistemi di registrazione ospedalieri (come l'ar-chivio AIEOP Mod 1.01 i cui risultati verranno presentatidal Dr. Pession e l'archivio AIEOP ROT che sarà illustratodal Dr Haupt). Considerando l'aumento della mole di lavo-ro connesso e i benefici derivanti dall'estensione degliarchivi dei RT, sono stati proposti studi di popolazionemirati a tumori specifici (EUROCARE high-resolutionstudy). 2 L'evoluzione dei RT da registri di casi a sistemi perla valutazione della qualità delle cure è auspicabile: infat-ti è necessaria una attenta pianificazione delle limitaterisorse economiche per ottenere elevati standard di qua-lità nell'assistenza di malattie rare, come i tumori pedia-trici.
L'attività del RTIP è finanziata dalla Regione Piemonte,dall'Associazione Italiana per le Ricerche sul Cancro (AIRC)e dal progetto Oncologia Compagnia San Paolo. Si ringra-ziano i prof Benedetto Terracini e Franco Merletti per il lorocontinuo e costruttivo supporto e Marinella Nonnato,Daniela Alessi, Elisa Dama Vanda Macerata e AssuntaRasulo per il loro impegno nella raccolta dei dati e nella lorogestione.
Bibliografia
1. Parkin DM, Stiller CA, Draper GJ, Bieber CA, Terracini B, Young JL.International Incidence of Childhood Cancer. IARC Scientific Pub-lications No. 87. Lyon: International Agency for Research on Can-cer; 1988
2. Sankila R, Coebergh JW. Cancer registries contribute to qualityimprovements in clinical care for all European cancer patients EurJ Cancer 2004;40:635-7.
3. Steliarova-Foucher E, Stiller C, Kaatsch P, Berrino F, Coebergh JW,Lacour B, Parkin M. Geographical patterns and time trends of can-cer incidence and survival among children and adolescents inEurope since 1970s: the ACCIS project. Lancet, in press 2004.
24
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre

4. Kramárová E, Stiller CA. The International Classification of Child-hood Cancer. Int J Cancer 1996;68:759-65.
5. Little J. Epidemiology of childhood cancer. IARC Scientific Publi-cation No. 149.
6. Ries LAG, Eisner MP, Kosary CL, Hankey BF, Miller BA, Clegg L,Mariotto A, Feuer EJ, Edwards BK, editors. SEER Cancer StatisticsReview, 1975-2001, National Cancer Institute: Bethesda, MD. USA;http://seer. cancer. gov/csr/1975_2001/. 2004.
7. Magnani C, Dalmasso P, Pastore G, Terracini B, Martuzzi M, MossoML et al. Increasing incidence of childhood leukaemia in North-west Italy, 1975-98. Int J Cancer 2003;105:552-7.
8. Levi F, La Vecchia C, Negri E, Lucchini F. Childhood cancer mortal-ity in Europe, 1955-1995. Eur J Cancer 2001;37:785-809.
9. Zuccolo L, Pastore G, Maule M, Gregori D, Terracini B, Merletti F,et al. Time trends of childhood cancer mortality rates: a reportfrom the childhood cancer registry of piedmont, Italy, 1971-98.Pediatr Blood and Cancer in press, 2004.
10. Viscomi S, Pastore G, Mosso ML, Terracini B, Madon E, Merletti F,et al. Population-based survival after childhood cancer during1970-98: A Report from the Childhood Cancer Registry of Pied-mont (Italy) Haematologica 2003; 88:974-82
11. Pannelli F, Mosciatti P, Felici L, Magnani C, Pascucci C, Pastore G.Survival trends of childhood cancer during the period 1978-1994in Italy: a first report from the Italian Cancer Registries. Epidemi-ol Prev 2001;25:354-75.
12. Capocaccia R, Gatta G, Magnani C, Stiller CA, Coebergh JW (eds.) Childhood Cancer Survival in Europe 1978-92: the EUROCAREstudy. Eur J Cancer, 37 Special issue; 2001.
13. Gatta G, Corazziari I, Magnani C, Peris-Bonet R, Roazzi P, Stiller C.Childhood cancer survival in Europe. The EUROCARE WorkingGroup. Ann Oncol 2003;14:119-27.
14. Magnani C Population-based survival after childhood lym-phoblastic leukaemia in time periods corresponding to specificclinical trials from 1979 to 1998 - a report from the ChildhoodCancer Registry of Piedmont (Italy). Eur J Cancer 2003;39:952-60.
15. Pastore G, Viscomi S, Mosso ML, Maule M, Terracini B, Magnani C,et al. Early deaths from childhood cancer a report from the Child-hood Cancer Registry of Piedmont, Italy, 1967-98 Eur J Pediatr2004;163:313-9.
16. Pastore G, Viscomi S, Gerov G, Terracini B, Madon E, Magnani C.et al. Incidence of second primary malignancies after a malignanttumor in childhood: a population survey in Piedmont (Italy). Int JCancer 1996;67:6-10.
17. Pastore G, Magnani C, Mosso ML, Viscomi S, Terracini B, MerlettiF. Marriage and offspring in adult long-term survivors of childhoodcancer: a study from the Childhood Cancer Registry of the Pied-mont region (Italy). Ital J Pediatr 2002;28:121-7.
Il Mod. 1.01: risultati e prospettive
Andrea PessionClinica Pediatrica Università di Bologna
IntroduzioneDal 1989 é stato adottato un sistema di rilevazione di
tutti i bambini affetti da tumore maligno diagnosticati e/ovisti per la prima volta nei centri AIEOP al fine di quantifi-care il numero di casi diagnosticati e trattati nei diversicentri, il rapporto tra casi osservati e casi attesi, l'adesioneo meno ai protocolli diagnostico-terapeutici promossi dalgruppo e la migrazione interna italiana. Questo strumen-to, noto come Mod. 1.01, ha preso l'avvio dapprima in ver-sione di scheda cartacea, sostituita nel corso del 2000 dauna scheda elettronica fruibile attraverso il portale AIEOPdai centri autorizzati. La possibilità di usufruire di un siste-ma comune per la condivisione in rete di dati provenientida ricerche multicentriche, ha consentito di acquisire inmaniera interattiva l'aggiornamento del follow-up deipazienti arruolati in protocolli AIEOP e di richiedere l'ag-giornamento mirato del follow-up per i casi trattati conprotocolli diversi da quelli ufficiali. Ciò ha reso possibileanalizzare la sopravvivenza di tutti i casi registrati, sia perpatologia sia per periodo di diagnosi, nonché verificarequanto essa possa essere influenzata dall'essere stati omeno arruolati in una sperimentazione clinica.
MetodiLa scheda Mod. 1.01 si compone di 3 parti per un tota-
le di 31 voci, delle quali 5 a risposta obbligatoria. Nella pri-ma parte, oltre all'identificazione del centro di diagnosi e/otrattamento, sono richiesti i dati anagrafici del paziente(data e comune di nascita, comune di residenza e/o didomicilio) e i principali dati riguardanti la diagnosi. I tipiistologici dei tumori sono codificati secondo Birch J. M.1.Nella seconda sezione sono richieste 4 informazioni sup-plementari miranti a definire per i tumori solidi e i linfo-mi: lo stadio, i criteri di stadiazione, la presenza o menodi metastasi, e la sede di insorgenza, codificata secondola classificazione proposta da Donaldson S. S. 2, mentreper le leucemie le informazioni sui gruppi di rischio sonoinvece desunte dal tipo di protocollo terapeutico utilizza-to. Nella ultima parte sono infine richieste informazioni sulprotocollo terapeutico di prima scelta adottato per il bam-bino, nonché la/le causa/e che hanno motivato l'eventua-le mancato reclutamento del paziente in uno dei proto-colli ufficiali AIEOP.
L'esigenza di aumentare la tempestività, correttezza equalità della segnalazione dei casi attraverso una regi-strazione computerizzata per via telematica demandata aicentri ha portato alla realizzazione di una banca dati (BD)del Mod. 1.01 “web based” attivata, secondo la metodo-logia AMR (Advanced Multicenter Research), grazie allacollaborazione del Centro Interuniversitario del Nord-Estitaliano di Calcolo Automatico (CINECA) di Bologna egestita dal Centro Operativo (CO) AIEOP. L'accesso al pro-getto di registrazione dei casi avviene attraverso il sitoInternet AIEOP (www. aieop. org), tramite la digitazionedi un codice identificativo del centro e di una password,diversi per ciascun centro.
25
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

A differenza della versione originale, la versione elet-tronica del Mod. 1.01 risulta composta di due sole parti,la prima riservata ai dati anagrafici del paziente sui qua-li il sistema opera il controllo automatico delle omonimie,ed una seconda riservata alle informazioni relative alladiagnosi e all'eventuale successivo trattamento.
Nell'ambito della BD Mod. 1.01 ai centri sono consen-tite, oltre all'inserimento dei nuovi casi e alla modifica deidati dei pazienti del proprio centro già registrati o ad essotrasferiti da altro centro, le seguenti funzioni: a) aggior-namento follow-up dei pazienti del proprio centro o adesso trasferiti da altro centro; b) trasferimento di un pro-prio paziente ad altro centro; c) consultazione report dimonitoraggio relativi al proprio centro; d) consultazionereport di monitoraggio relativi ai dati globali aggregati. IlCO AIEOP mantiene invece un ruolo di coordinamento sul-l'intero sistema, con possibilità di modifica dei dati egestione delle abilitazioni degli utenti.
Risultati
Al 31 dicembre 2003, dopo 15 anni dalla sua attivazio-ne, 57/61 (93%) centri AIEOP hanno registrato con Mod.1.01 18891 casi affetti da tumore maligno, dei quali17422 (92%) di età alla diagnosi compresa tra 0 e 14 anni,1096 (6%) tra 15-17 anni e 373 (2%) > 18 anni.
Il rapporto tra casi osservati e casi attesi (O/A), calco-lato sui casi registrati residenti in Italia e diagnosticatinel periodo 1989-1998, sulla base dei tassi d'incidenzaetà-specifici del RTI del Piemonte, è risultato complessi-vamente pari a 0.82, con valori superiori a 0.95 per i disor-dini mielo-linfoproliferativi (DMLP) e compresi tra 0.85 e0.45 per i tumori solidi (TS) (vedi Figura 1).
Il 66% dei casi registrati è stato trattato con protocol-li AIEOP (80% DMLP, 50% TS), con un lieve, ma progres-sivo, incremento nel corso degli anni, passando dal 60%nel 1989-1993 al 70% nel 1999-2003.
Il fenomeno migratorio extraregionale, valutato attra-verso il rapporto tra i casi residenti trattati in centri AIEOPdella regione ed i casi residenti, comunque trattati in uncentro AIEOP, interessa circa il 20% dei casi (15% DMLP,30% TS), variando da valori inferiori al 5% per la Liguriaad oltre il 65% per Abruzzo e Calabria. Confrontando l'an-damento di questo fenomeno nel tempo, si assiste nelcomplesso ad una sua riduzione significativa, mediamen-te nell'ordine del 4% (p <. 01), passando dal 27. 6% nelperiodo 1989-1994 al 23. 6% nel 1995-2000.Tale ridu-zione, che risulta particolarmente significativa in Tosca-na (14%, p<. 05), Sardegna (15%, p<. 05) e Calabria (14%,p<. 01), si osserva sia nel caso dei DMLP (19. 3% vs 14.9%, p<. 05) sia per i TS (37. 0% vs 33. 0%, p=n. s. ). Nelcaso dei DMLP, la massima riduzione del fenomeno migra-torio nel tempo si osserva in Calabria con il 17% (p<. 05),mentre permane costantemente elevata in Abruzzo, inte-ressando il 40% dei DMLP residenti. Una significativa ridu-zione delle migrazioni dei TS si registra in Sardegna, conoltre il 17% in meno nel periodo 1995-2000 rispetto alprecedente (p<. 05), mentre il fenomeno permane anco-ra molto elevato in Abruzzo e Calabria. La sopravvivenza(SUR) a 5 anni dalla diagnosi calcolata su un campione di10791 casi registrati e diagnosticati tra il 1989 ed il 1998con età < 15 anni è risultata pari al 71.3% proiettandosial 67. 3% e 65. 8% a 10 e 15 anni rispettivamente (Figu-ra 2). La sopravvivenza è stata stimata complessivamen-te e per le principali categorie diagnostiche, includendocome variabili di stratificazione il sesso (Figura 3), l'età alladiagnosi (Figura 4), il periodo di diagnosi (Figura 5) e latipologia di protocollo clinico in cui è stato inserito ilpaziente.
Discussione
Il rapporto O/A per tutti i tumori conferma la leadershipdell'AIEOP, come punto di riferimento delle istituzioni ita-liane coinvolte nel trattamento dei tumori pediatrici e talerisultato è sovrapponibile a quello ottenuto in altri paesioccidentali. Per la maggioranza dei tumori il numero deicasi osservati è sovrapponibile a quello dei casi attesi,mentre per i tumori del sistema nervoso centrale, il nume-ro dei casi registrati è significativamente inferiore all'at-teso (O/A = 0.47, p <. 001). Come osservato anche in altri
26
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre
Figura 2.95% CI: intervallo di confidenza al 95%.
Figura 1.O/A: rapporto casi osservati su casi attesi, LAL: leucemiaacuta linfoblastica, LAM: leucemia acuta mieloblastica, Altre L. : altreleucemie, SNC: sistema nervoso centrale, SNS: sistema nervoso sim-patico, LH: linfoma di Hodgkin, LnH: linfomi non Hodgkin, STM: sarco-mi dei tessuti molli, RTB: retinoblastoma, Altri T. : altri tumori.

27
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
paesi, i bambini affetti da questi tumori afferiscono in
prevalenza a centri di neurochirurgia, la maggior parte deiquali non aderisce all'AIEOP.
Il rapporto tra casi trattati con protocolli ufficiali AIEOP
e casi registrati, pari complessivamente al 66%, ha fattorilevare una grande variabilità per patologia. Infatti, sel'arruolamento nei protocolli AIEOP per i DLMP può dirsinel complesso soddisfacente, è da rilevare il più bassoreclutamento dei protocolli per i TS, e soprattutto per itumori del sistema nervoso centrale, di cui per altro unaquota richiede solo terapia chirurgica e/o radioterapica equindi può non afferire a centri di oncologia pediatrica.
Anche se da oltre vent'anni la politica di assistenza sani-taria perseguita dall'AIEOP è quella di avvicinare il più pos-sibile le strutture di diagnosi e terapia al luogo di residen-za dei bambini affetti da neoplasia, è ancora oggi presen-te un fenomeno migratorio che interessa mediamente lametà dei pazienti residenti in alcune regioni centro-meri-dionali. Le cause principali che ancora spingono le lorofamiglie ad intraprendere i cosiddetti “viaggi della speran-za” sono forse radicate nella tradizione di queste aree geo-grafiche, ed in parte dovute alla crescente tecnologianecessaria per assicurare un ottimale percorso diagnosticoterapeutico, nonché a carenze strutturali di assistenza-offerta ancora presenti in alcune regioni. Sebbene la spin-ta migratoria si sia ridotta nel tempo, l'AIEOP dovrà pro-durre ancora più sforzi tendenti non tanto ad invertire que-sto fenomeno, che si traduce molte volte in una riduzionedella qualità della vita sia del paziente sia dei suoi familia-ri, quanto a monitorarne le cause cercando di ridurre ladurata di questa permanenza lontano da casa.
L'analisi della sopravvivenza ha dimostrato come com-plessivamente la prognosi dei soggetti affetti da tumoremaligno in Italia sia in linea con quanto è possibile otte-nere anche negli altri Paesi occidentali. Un miglioramentonel tempo è documentabile per la maggior parte della cate-gorie diagnostiche. Le femmine vanno mediamente megliorispetto ai maschi e la prognosi peggiora all'aumentare del-l'età anche se rilevanti sono le differenze che si registranoper singola patologia. I vantaggi in termine di prognosi rile-vati per i casi arruolati in protocolli AIEOP sono evidenti esignificative per le patologie più frequenti.
La creazione di una banca dati web-based per il Mod.1.01 ha costituito il prototipo per la successiva trasfor-mazione di altre banche dati AIEOP malattia specifiche odi registri e, a partire dal 2002, la stessa metodologia èstata adottata per la raccolta e gestione dei dati relativiai casi con immunodeficienze trattati nei centri parteci-panti ai protocolli AIEOP. La tecnologia utilizzata consen-te, attraverso l'interattività del processo, il controllo auto-matico e la sicurezza dell'informazione registrata, nelrispetto delle norme di privacy, la piena fruibilità del-l'informazione da esse derivata a tutte le diverse compo-nenti interessate.
Bibliografia
1. Birch JM, Marsden HB. A classification scheme for childhood can-cer. Int J Cancer 1987;40:620-4.
2. Donaldson SS, Draper GJ, Flamant F, Gerard-Marchant R,Mouriesse H, Newton WA, et al. Topography of Childhood Tumors:Pediatric Coding System. Pediatric Hematology and Oncology1986;3:249-58.
3. Pession A, Rondelli R, Haupt R, Magnani C, Pastore G, Terracini B,et al. Sistema di rilevazione dei casi di tumore maligno in età pedi-atrica in Italia su base ospedaliera. Rivista Italiana di Pediatria2000;26:333-41.
Figura 4.
Figura 5.
Figura 3.

Il nuovo censimento del Registro Fuori Terapia
Riccardo Haupt,1 Daniela Silvestri,2 Silvia Caruso,1Stefano Parodi,1 Franca Fossati Bellani,3 AlessandroSandri,4 Corrado Magnani,5 Modesto Carli,6Andrea Pession,7 Francesca Fioredda,1 Marta Pillon,6Angelo Rosolen,6 Guia Hanau,1 Momcilo Jankovic2
1Servizio di Epidemiologica e Biostatistica - Ematologia edOncologia Pediatrica. Istituto G. Gaslini Genova; 2ClinicaPediatrica, Università di Milano-Bicocca, Monza; 3Divisionedi Pediatria, Ist. Nazionale dei Tumori, Milano; 4ClinicaPediatrica, Università di Torino; 5Registro dei Tumori Infan-tili del Piemonte, Dipartimento di Scienze Mediche, Univer-sità Amodeo Avogadro del Piemonte Orientale, Novara; 6Cli-nica Pediatrica, Università di Padova; 7Clinica Pediatrica,Università di Bologna
In Italia, così come nella maggior parte dei paesi occi-dentali, sta aumentando sempre più il numero dei sogget-ti che hanno superato con successo il percorso terapeuti-co successivo a una diagnosi di tumore contratto in etàpediatrica. 1-3 Molte di queste persone sono ormai entratenell'età adulta, ma poco si sa ancora se, e in che misura, itrattamenti ricevuti abbiano interferito o interferiranno conil loro stato di salute e con la loro spettanza di vita. 4-7
Nel nostro paese, la popolazione potenzialmente gua-rita da tumore è censita sin dal 1981, tramite il RegistroOff-Therapy (ROT) dell'Associazione Italiana di Ematolo-gia ed Oncologia Pediatrica (AIEOP). Il ROT raccoglie infor-mazioni sui bambini affetti da tumore maligno che, indi-pendentemente dall'evoluzione successiva della malattia,hanno concluso in persistente remissione completa il pro-gramma terapeutico per loro previsto. 8
All'ultimo aggiornamento del ROT sono stati registratioltre 11.000 soggetti e stime recenti permettono di direche oltre l'80% dei bambini con tumore che ha concluso inpersistente remissione completa i trattamenti previsti saràvivo a più di 20 anni dopo la sospensione delle cure. 7
Obiettivo del ROT è quello di raccogliere informazionisull'evoluzione clinica della malattia dopo la sospensioneelettiva del cure e su eventuali effetti tardivi che posso-no comparire anche a distanza di molti anni. Il ROT vieneaggiornato periodicamente con l'inserimento di nuovi casi,grazie all'interazione col il Modello 1.01 che da informa-zioni su tutti i casi diagnosticati annualmente in centriAIEOP. 9,10 Il follow-up dei soggetti già registrati nel ROTviene invece effettuato ad intervalli più lunghi (4-5 anni)tramite richiesta di informazioni ai centri curanti e/o alleanagrafe dei comuni di nascita o residenza. Tramite que-sti aggiornamenti si raccolgono informazioni su eventua-li effetti tardivi, ma allo stato attuale, è ancora scarsa laqualità dell'informazione disponibile circa le dosi cumu-lative di chemio e /o radioterapia ricevuta da ogni sog-getto inserito nel ROT. Questo articolo descrive un pro-getto recentemente attivato che ha l'obiettivo di integra-re le informazioni già disponibili nel ROT, sintetizzando perogni soggetto la sua storia di malattia con specificazionesugli interventi chirurgici subiti, sulle dosi e sedi di even-tuali trattamenti radioterapici, sulle dosi cumulative difarmaci potenzialmente tossici, oltre a note su possibilicomplicazioni avvenute durante le cure.
Materiali e MetodiAssegnazione di un identificatore univoco: Inizialmente
si è assegnato un codice univoco identificativo (UPN) adogni soggetto inserito nel ROT. Poiché sin dal 1989 è atti-vo il registro AIEOP dei nuovi casi diagnosticati in tutti icentri italiani (Mod. 101)9, si è deciso che, per i pazientidiagnosticati dopo il 1989, tale codice identificativo fos-se quello (a 7 cifre) assegnato dal Mod. 1.01.Per i casi condiagnosi antecedente al 1989, è stato invece attribuitoun nuovo UPN anch'esso di 7 cifre, ottenuto dall'associa-zione del prefisso '777' al codice identificativo a 4 cifre giàpresente nel database ROT. In seguito, sono stati inviati airesponsabili dei singoli protocolli terapeutici di malattia(ad es. LLA, tumore di Wilms, Neuroblastoma, linfomi nonHodgkin, ecc) gli elenchi dei pazienti con quella neopla-sia conosciuti al ROT. I responsabili dei protocolli hannoquindi inserito l'UPN anche nei loro data base in modo dapermettere il trasferimento delle informazioni eventual-mente disponibili sui trattamenti (dai data base dei pro-tocolli di malattia al ROT) o su effetti a distanza (dal ROTai data base dei protocolli di malattia).
Creazione della scheda raccolta dati: Dopo aver concor-dato con i responsabili dei protocolli di terapia, ed aver ana-lizzato esperienze simili effettuate in altri studi cooperati-vi internazionali, sono state identificate le informazioni chesi è ritenuto necessario raccogliere per ogni paziente circale dosi cumulative di chemio e radioterapia ricevute, oltreche su eventuali interventi chirurgici subiti durante il decor-so di malattia. E' stata quindi predisposta una scheda siacartacea che elettronica per la raccolta dati che permetteràdi acquisire informazioni specifiche per ogni soggetto sul-l'avvenuta esposizione ad un determinato farmaco e sulladose cumulativa (mg/m2) somministrata. Per i trattamentiradianti verranno riportate indicazioni sull'area irradiata,sulla dose massima ricevuta oltre che sul tipo di apparec-chiatura utilizzata. Infine, con la scheda raccolta dati ver-ranno riportate anche informazioni sul tipo di eventualiinterventi chirurgici subiti.
Trasferimento elettronico delle informazioni sui tratta-menti ricevuti: Una volta completata l'operazione UPN, uti-lizzando le banche dati dei protocolli terapeutici che ave-vano raccolto tali informazioni, si è proceduto al trasferi-mento elettronico dei dati sulle dosi cumulative effettiva-mente ricevute da ogni soggetto.
Estrazione dati direttamente dalle cartelle cliniche: Per ipazienti per i quali non è stato possibile raccogliere com-pletamente le informazioni sui trattamenti ricevuti, taliinformazioni dovranno essere raccolte direttamente dallecartelle cliniche. A questo scopo è stato effettuato un cor-so per tutti i monitor dei centri AIEOP al fine di garantireun'omogenea metodologia nella raccolta dati.
Calcolo dosi attese: In previsione del fatto che per alcu-ni pazienti, specialmente quelli trattati molto tempo fa, laqualità dei dati ottenibili dalle cartelle cliniche non saràottimale, si è deciso comunque di raccogliere dati sulle dosicumulative attese secondo lo schema originale propostodai vari protocolli terapeutici utilizzati nel corso degli anni.
Controllo di qualità: Per i soggetti inseriti in protocolli distudio, la qualità dei dati verrà valutata confrontando ledosi somministrate con quelle attese secondo il disegno ori-ginale del protocollo. Deviazioni importanti, in eccesso odin difetto, verranno ricontrollate tramite ricerca diretta sul-
28
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre

la documentazione clinica Si estrarrà invece un campionedei casi le cui informazioni saranno state raccolte dai moni-tor e, tramite una seconda revisione della cartella fatta dapersonale indipendente, se ne controllerà la corrisponden-za con quelli raccolti.
Aggiornamento follow-up: Contestualmente alla raccol-ta di informazioni sui trattamenti ricevuti, si aggiorneràanche il follow-up di ogni soggetto ROT. In particolare, verràaggiornato lo stato in vita, e le informazioni sulla compar-sa di eventuali eventi maggiori (recidiva di malattia, secon-do tumore, matrimonio, figli, ecc).
Inoltre, in via sperimentale, e per il momento solamenteper quanto riguarda i soggetti residenti nella regione Pie-monte, è stata messa a punto una procedura per l'appaia-mento (linkage) tra records con dati nominativi. Con talesistema si prevede di appaiare i nominativi dei soggetti ROTcon l'archivio delle dimissioni ospedaliere permettendoquindi l'individuazione delle principali patologie prevalen-ti ed incidenti in tale popolazione.
Risultati attesiDal punto di vista statistico ed epidemiologico la popo-
lazione Italiana già censita con ROT è tra le più grandi diquelle descritte in letteratura, ed è numericamente suffi-ciente per poter dare informazioni significative sui proble-mi di interesse.
Una volta conclusa questa operazione di aggiornamentosarà possibile ottenere un profilo personale per ogni sog-getto inserito nel ROT contenente informazioni sui dati cli-nici della malattia di base e sulla sua storia di malattia (tipoe stadio del tumore, tipo e dosaggio cumulativo dei tratta-menti chemio e radioterapici ricevuti, chirurgie maggiori, edeventuali complicazioni). Questo profilo, in forma cartaceaod elettronica, sarà parte integrante del ROT e disponibilesu richiesta per tutti i soggetti guariti o per i loro medicicuranti. In tal modo, sarà possibile avere una documenta-zione esaustiva di quei trattamenti ricevuti in età pediatri-ca che potrebbero avere un ruolo nella determinazione distati clinici che potrebbero comparire anche in età adulta.11
Tramite le routinarie attività del ROT e dei vari archiviche concorrono a questo progetto, sarà inoltre possibileottenere informazioni cliniche aggiornate su specifici end-points di interesse e quantificare quindi l'eventuale aumen-to nel rischio di sviluppare una determinata condizionemorbosa.
Una volta conclusa questa operazione, il nostro Registroavrà raccolto una quantità di dati rilevante e qualitativa-mente molto buona tale da poter permettere confronti conaltri registri simili presenti a livello internazionale quale ilChildhood Cancer Survivors Study (CCSS) americano o iregistri su base di popolazione attivi in Gran Bretagna e neipaesi Scandinavi.
Infine, la diffusione dei risultati ottenuti, in termini distima del rischio per determinati eventi sanitari avversi saràutile per le autorità competenti per decidere l'attivazione dieventuali programmi di prevenzione primaria e secondariain soggetti a rischio.
Bibliografia
1. Gatta G, Corazziari I, Magnani C. The EUROCARE Working Group.Childhood cancer survival in Europe. Ann Oncol 2003;14 Suppl5:V119-V127.
2. Pastore G, Viscomi S, Gerov GL, Terracini B, Madon E, Magnani C.Population-based survival after childhood lymphoblastic leukaemiain time periods corresponding to specific clinical trials from 1979to 1998--a report from the Childhood Cancer Registry of Piedmont(Italy). Eur J Cancer 2003;39:952-60.
3. McNeil DE, Cote TR, Clegg L, Mauer A. SEER update of incidenceand trends in pediatric malignancies: acute lymphoblasticleukemia. Med Pediatr Oncol 2002;39:554-7.
4. Pui CH, Cheng C, Leung W, Rai SN, Rivera GK, Sandlund JT, et al.Extended follow-up of long-term survivors of childhood acute lym-phoblastic leukemia. N Engl J Med 2003;349:640.
5. Robison LL, Bhatia S. Late-effects among survivors of leukaemiaand lymphoma during childhood and adolescence. Br J Haematol2003;122:345-59.
6. Hudson MM, Jones D, Boyett J, Sharp GB, Pui CH. Late mortalityof long-term survivors of childhood cancer. J Clin Oncol 1997;15:2205-13.
7. Haupt R, Valsecchi MG, Silvestri D, De Lorenzo P, Napoli S, MaseraG, et al. Early and late deaths after elective end of therapies forchildhood cancer in Italy. Int J Cancer 200;86:393-8.
8. Zurlo MG, Pastore G, Masera G, Terracini B, Burgio R, Ceci A, et al.Italian Registry of patients off-therapy after childhood acute lym-phoblastic leukemia. Cancer 1986;57:1052-5.
9. Pession A, Rondelli R, Haupt R, et al. Il sistema di rilevazione deicasi di tumore maligno in eta' pediatrica in italia su baseospedaliera. Riv Ital Pediatr 2000;26:333-341
10. Pession A, Rondelli R, Locatelli F, et al. I Registri e l'assistenza albambino con patologia neoplastica. Riv Ital Pediatr 2000;26:570-573
11. Masera G, Chesler M, Jankovic M, et al. SIOP Working Committeeon Psychosocial issues in pediatric oncology: guidelines for care oflong-term survivors. Guidelines for Care of Long-Term Survivors.Med Pediatr Oncol 1996; 27:1-2
29
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

AIEOP clinical data warehouse
M. De Rosa,* F. Ronchetti,* G. Greco,* A. Pession,°R. Rondelli°*CINECA, Casalecchio di Reno (BO); °Clinica di Oncologia edEmatologia Pediatrica, Policlinico S. Orsola-Malpighi,Bologna
L'impatto delle nuove tecnologie informatiche e tele-matiche ha dato un enorme impulso alla ricerca in ambi-to biomedico e sanitario grazie alla possibilità di racco-gliere e condividere le informazioni in modo semplice erapido. L'avvento di Internet ha influito radicalmente sul-le metodologie di ricerca favorendo l'approccio coopera-tivo tra gli istituti di ricerca ed ottimizzando tempi e costinel complesso iter delle sperimentazioni multicentriche.
L'AIEOP (Associazione Italiana di Ematologia ed Onco-logia Pediatrica, www. aieop. org) è stato tra i primi grup-pi in Italia a cogliere la portata della trasformazione inatto. Nel 1985 ha istituito, avvalendosi della partnershiptecnologica del CINECA, un servizio per il monitoraggiodella ricerca sulle patologie oncologiche-ematologiche in63 centri dislocati su tutto il territorio nazionale. Il CINE-CA (www. cineca. it), fondato per Decreto Presidenziale nel1969, è un consorzio no-profit composto da 23 UniversitàItaliane (Ancona, Bari, Bologna, Camerino, Catania, Ferra-ra, Firenze, Insubria, Macerata, Milano, Modena, Messina,Padova, Parma, Pavia, Pisa, Salerno, Siena, Trento, Trieste,Udine, Urbino e Venezia), dal CNR (Centro NazionaleRicerche) e da una rappresentanza del MIUR. E' il piùimportante centro di calcolo italiano e uno dei più impor-tanti ed avanzati a livello europeo, attivo ad ampio rag-gio in tutti i settori dell'Information Technology. La suacompetenza è testimoniata dalla presenza nei primi 50posti della classifica mondiale delle 500 migliori organiz-zazioni di supercomputing (www. top500.org).
Nel 1985 l'AIEOP ha iniziato con il Cineca una collabo-razione per la gestione e il monitoraggio dei dati relativiai protocolli di ricerca del gruppo. Utilizzando la metodo-logia AMR (Advanced Multicenter Research), è stato orga-nizzato un database orientato al paziente che raccoglietutte le informazioni relative ai pazienti e ai protocolliAIEOP.
L'AMR è una metodologia realizzata dal Cineca per laraccolta, la condivisione e l'analisi di informazioni prove-nienti da sperimentazioni cliniche a carattere multicen-trico che consente la gestione dell'intero flusso informa-tivo, dalla pianificazione dello studio all'analisi finale deidati.
Il sistema è completamente web-based: tutto il flussoinformativo, dall'inserimento all'analisi dei dati, avvienedirettamente in Internet. I servizi web-based hanno il van-taggio di non avere bisogno di applicazioni di software alivello locale e di avere un controllo centrale della ricercanel suo insieme.
Nel sistema l'accesso alle informazioni da parte dei sin-goli centri partecipanti (in fase di input o di consultazio-ne e analisi dei dati) avviene in modo sicuro. L'AIEOP, tito-lare del trattamento dei dati contenuti in BD ha nomina-to il Cineca responsabile del trattamento dei dati. Il Cine-ca per ottemperare alle normative vigenti nazionali e
internazionali relative alla sicurezza e alla privacy dei datiha sviluppato tecnologie e sistemi con i più elevati stan-dard di sicurezza e riservatezza.
Nel 2003 è stato implementato nel sistema il ClinicalData Warehouse: un insieme di strutture dati e strumen-ti di analisi necessari a fornire un supporto decisionale. IlClinical Data Warehouse contiene informazioni prove-nienti da distinti database operazionali arricchite da datiaggregati. Il suo obiettivo è offrire una serie di dati clini-ci e di strumenti per facilitare il processo di ricerca e ditrattamento.
Il CDW AIEOP è composto da un registro (RegistroModello 1.01), che raccoglie un dataset minimo di infor-mazioni relative ad anagrafica e diagnosi di tutti i soggettiche hanno ricevuto una diagnosi in ambito oncologico-ematologico presso uno dei centri AIEOP.
Quando i pazienti registrati nel Registro Mod. 1.01accedono ai protocolli di ricerca AIEOP, l'AMR provvede aregistrare i loro dati in banche dati patologie e protocol-lo specifiche che consentono una maggiore complessità e
30
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre
Figura 1.www. aieop. org.
Figura 2.

specificità di elaborazione delle informazioni raccolte. Oltre al Registro Mod. 1.01 il database AIEOP contiene
il Registro TMO che raccoglie informazioni relative a tut-ti i trapianti di midollo osseo effettuati nei centri AIEOPabilitati.
Il CDW mette a disposizione la navigazione trasversaledelle informazioni su pazienti, protocolli e patologie. Que-sto sistema consente di visualizzare l'informazione a par-tire dal massimo livello di aggregazione (ad esempio tut-ti i protocolli di una patologia) fino alla singola scheda diraccolta dati del paziente.
Attraverso questo strumento si accede a report prede-finiti sui singoli protocolli di ricerca, sulle patologie e suiRegistri AIEOP, si accede a un Protocol status overviewche mostra lo stato di avanzamento del protocollo nelcorso del tempo e fornisce in tempo reale lo stato di arruo-lamento, i centri partecipanti e i risultati conseguiti (pub-blicazioni, ecc. ).
L'accesso è consentito a diversi profili utente (Centropartecipante, Coordinatore del Comitato Scientifico diStudio, Coordinatore del protocollo e Centro Operativo)con differenti visioni di analisi: il Centro Operativo haaccesso a tutta la banca dati AIEOP, i Coordinatori posso-no visualizzare i dati relativi ai propri protocolli o allapatologia, mentre i Centri hanno accesso ai dati relativi aipropri pazienti ed ad una visione aggregata dell'interodatabase.
Rischio di leucemia infantile ed esposizioneambientale a campi elettromagnetici adesposizione estremamente bassa
Corrado MagnaniDipartimento di Scienze Mediche dell'Università del Pie-monte Orientale e Servizio di Epidemiologia dei Tumori delCentro di Riferimento per l'Epidemiologia e la PrevenzioneOncologica - CPO - Piemonte, Novara
Viene presentato lo stato dell'arte delle indagini sul-l'associazione tra campi magnetici a frequenza ELF etumori. L'esposizione a campi magnetici ELF è stata valu-tata dalla IARC come possibile cancerogeno per l' uomo -gruppo 2B. Le conclusioni sono state tratte in base ai risul-tati degli studi sulle leucemie infantili e sulle leucemielinfatiche croniche associate alla esposizione lavorativa.
IntroduzioneI possibili rischi per la salute umana conseguenti alla
esposizione della popolazione a campi elettromagnetici afrequenza di rete ed a radiofrequenza costituiscono unargomento di diffusa preoccupazione, talora anche ineccesso rispetto alla evidenza scientifica disponibile.
In questa nota presento una sintesi della evidenza epi-demiologica rifacendomi quando possibile alle indicazio-ni prodotte da gruppi di lavoro di organismi scientifici.L'argomento è trattato limitatamente alle frequenze direte ed al rischio di leucemia infantile.
Campi elettrici e magnetici a frequenza estremamentebassa (ELF) ed insorgenza di neoplasie
Come campi a frequenza estremamente bassa o ELFintendiamo quelli compresi in un intervallo di frequenzatra 3 e 3000 Hz: dal punto di vista pratico le frequenze dimaggiore interesse sono quelle di 50 e 60Hz, corrispon-denti alla frequenza della corrente alternata rispettiva-mente della rete elettrica europea e della rete americanaed indicate anche come frequenza di rete. L'intervallo ELFcomprende anche le loro armoniche.
La prima ipotesi di una associazione tra campi elettricie magnetici ELF è stata avanzata da uno studio epide-miologico pubblicato nel 1979 da due ricercatori statuni-tensi: N. Wertheimer e E. Leeper. Le ricerche successive sisono progressivamente concentrate sull'associazione conle neoplasie del sistema linfoemopoietico in conseguenzadi esposizione ambientale e lavorativa a campi elettrici emagnetici ELF. L'elenco dei lavori scientifici pubblicati suquesto argomento in riviste internazionali è assai nume-roso ed è opportuno non limitarsi alla lettura di uno o piùlavori ma riferirsi a meta-analisi.
Senza entrare nel dettaglio di argomenti meglio svilup-pati dai colleghi fisici, occorre ricordare che il campo elet-trico è schermato dalle pareti e che in presenza di ogget-ti a superficie isolante (come il corpo umano) viene modi-ficato rispetto al campo teorico che si avrebbe in uno spa-zio vuoto. Questo non avviene invece per il campo magne-tico, che non è schermato o influenzato dai comuni mate-riali. Dal punto di vista pratico questo porta ad un mag-giore interesse per il campo magnetico per la sua minorevariabilità. Inoltre, ad ulteriore supporto dell'interesse pre-
31
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
Tabella 1.Dati aggiornati al 20 luglio 2004.
Registro Mod. 1.01 ppaazziieennttii ttoottaallii: 25. 108;17. 000 dei quali - pari al 67,7%- hanno eseguito
un protocollo AIEOP
Registro BMT 4. 309 pazienti, 5. 093 trapianti
Data Base protocolli AIEOP AMR 5 patologie (Leucemia acuta linfoblastica,Leucemia acuta non linfoblastica,
Malattia di Hodgkin, Sarcoma di Ewing ed Immunodeficienze)
53 studi, di cui:- 3 in fase di start up
- 7 aperti- 11 chiusi all'accrual
- 29 terminati

valente verso il campo magnetico, i risultati degli studisull'esposizione ambientale condotti con la stima del cam-po magnetico e del campo elettrico hanno indicato un'as-sociazione prevalentemente con il campo magnetico.
Relativamente all'esposizione ambientale ed alle leu-cemie infantili, le analisi di maggior interesse sono stateprodotte da Ahlbom et al. (2000) e da Greenland et al.(2000). Entrambi hanno usato il metodo del 'pool' dei datidall'insieme degli studi epidemiologici disponibili, per untotale di oltre 4000 casi di leucemia infantile ed oltre10000 controlli.
Lo studio di Ahlbom è stato limitato alle indagini epi-demiologiche in cui il campo magnetico era stato misu-rato (Linet et al. 1997; Michaelis et al. 1998; Dockerty et al.1999; UKCCS 1999; McBride et al. 1999) o calcolato (Fei-chting e Ahlbom 1993; Olsen et al. 1993; Verkasalo et al.1993 Tynes et al. 1997). In sintesi: Non è stato osservatoun aumento del rischio per i bambini nelle cui abitazioniil campo magnetico era inferiore a 0.4 microtesla mentreper campi magnetici superiori si osserva un aumento sta-tisticamente significativo del rischio di leucemia: RR=2,00(I. C. 95%: 1,27-3,13) (Figura 1). La stima del rischio nonvaria in modo rilevante considerando possibili fattori
confondenti (stato socio economico, mobilità residenzia-le, residenza urbana o rurale, tipo di abitazione, traffico)o limitandola alle leucemie linfatiche acute. Il numero dicontrolli con esposizione superiore a 0.4 microtesla era di62 su 10400 (0.6% del totale).
Lo studio di Greenland et al. (2000) ha incluso ancheindagini che non avevano misure del campo magnetico masolo la classificazione wire-code (Wertheimer e Leeper,1978; Fulton et al. 1980; Savitz et al. 1988; London et al.1991; Fajardo-Gutierrez et al. 1997) o misure condotte perun tempo inferiore a 24 ore (Tomenius, 1986; Coghill et al.1996). Sono stati invece esclusi i dati degli studi di Greenet al, 1999 ed UKCCS, 1999, perché non pervenuti in tem-po utile. Greenland et al. (2000) presentano la stima delrischio per esposizioni superiori a 0.3 microtesla, osser-vando un aumento statisticamente significativo (RRMH:1,68; IC 95%: 1,23-2,31) (Figura 2). La stima del rischioaumenta includendo nelle analisi un indicatore di statosocio economico (non sono stati considerati altri possibi-li confondenti). Sono esposti a oltre 0,3 microtesla 130controlli su 7084 (1,8%) mentre lo sono a oltre 0,4 micro-tesla 74 controlli (1,0%).
In entrambi gli studi, la rarità dell'esposizione ai livelliassociati con un aumento del rischio, impedisce di scio-gliere l'incertezza legata al possibile effetto di bias neldeterminare il risultato finale.
Un contributo per ridurre l'incertezza potrà venire daglistudi attualmente in corso, quale lo studio italiano SETILsull'eziologia di leucemia, linfoma e neuroblastoma nelbambino tra 0 e 10 anni, i cui primi risultati saranno dispo-nibili nel 2004. Lo studio ha incluso 980 casi e1200 con-trolli. Al convegno saranno presentati indicatori di pro-cesso.
ConclusioniL'Agenzia Internazionale per la Ricerca sul Cancro -
IARC (2002) ha ritenuto di classificare i campi magneticicome 'Possibili Cancerogeni per l'Uomo - gruppo IARC 2B'.Le conclusioni sono state tratte in base ai risultati deglistudi sulle leucemie infantili ed agli studi sulle leucemielinfatiche croniche associate alla esposizione lavorativanell'adulto.
In questa situazione di incertezza paiono condivisibili leraccomandazioni presenti in forma abbastanza similedocumenti prodotti in Svezia (The Swedish NationalBoards, 1996), USA (NIESH, 1999) ed in Italia (Lagorio etal. , 1998) che suggeriscono una politica di riduzione del-le esposizioni più elevate e di adozione di obbiettivi diqualità a livelli inferiori per le nuove costruzioni. Questalinea è stata adottata dal Ministero dell'Ambiente nel1998 relativamente alla normativa sulle radiofrequenze.
Bibliografia
Feychting M, Ahlbom A. Magnetic fields and cancer in children residingnear Swedish high-voltage power lines. Am J Epidemiol1993;138:467-81.
Fulton JP, Coob S, Preble L, Leone L, Forman E. Electrical wiring config-urations and childhood leukaemia in Rhode Island. Am J Epidemi-ol 1980; 111:292-6.
IARC. IARC monographs on the evaluation of carcinogenic risks tohumans- volume 80 - Non-Ionizing Radiation, Part 1: Static andExtremely Low-Frequency (ELF) Electric and Magnetic Fields. IARC,Lyon. 2002.
32
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre
Figura 1.Rischio di leucemia infantile e campi magnetici. Analisi pooleddi Ahlbom et al. , 2000.
Figura 2.Rischio di leucemia infantile e campi magnetici. Analisi pooleddi Grenland et al.
<= 0,1 0,1 <= 0,2 0,2 <= 0,4 > 0,4
<= 0,1 0,1 <= 0,2 0,2 <= 0,3 > 0,3microtestla
4
3
2
1
0
4
3
2
1
0

33
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
Lagorio S, Comba P, Iavarone I, Zapponi GA. Tumori e malattie neurode-generative in relazione all'esposizione a campi elettrici e mag-netici a 50-60 Hz: rassegna degli studi epidemiologici. RapportoISTISAN 98/31.Roma: ISS. 1998
Linet MS, Hatch EE, Kleinerman RA, Robison LL, Kaune WT, Friedman DR,et al. Residential exposure to magnetic fields and acute lym-phoblastic leukemia in children. N Engl J Med 1997;337:1-7.
London SJ, Thomas DC, Bowman JD, et al. Exposure to residential elec-tric fields and risk of childhood leukemia. Am J Epidemiol 1991;134:923-37.
Mc Bride ML, Gallagher RP, Theriault G, Armstrong BG, Tamaro S, Spinel-li JJ, et al. Power frequency electric and magnetic fields and riskof childhood leukemia in Canada. Am J Epidemiol 1999; 149:831-42.
Michaelis J, Schuz J, Meinert R, Menger M, Grigat JP, Kaatsch P, et al.Childhood leukemia and electromagnetic fields: results of a pop-ulation-based case-control study in Germany. Cancer Causes Con-trol 1997;8:167-74.
Michaelis J, Schuz J, Meinert R, Zemann E, Grigat JP, Kaatsch P, et al.Combined risk estimates for two German population-based case-control studies on residential magnetic fields and childhood acuteleukemia. Epidemiol 1998;9:92-4.
Myers A, Clayden AD, Cartwright RA, Cartwright SC. Childhood cancerand overhead powerlines: a case-control study. Br Med J1998;307:891-5.
Olsen JH, Nielsen A, Shulgen G. Residence near high voltage facilities andrisk of cancer in children. Br Med J 1993;137:318-30.
National Institute for Environmental Health Sciences - National Insti-tute of Health (NIESH). Report on Health effects from exposure toPower line Frequency Electric and Magnetic fields. NIH1999;99:4493.
Savitz DA, Wachtel H, Barnes FA, John EM, Tvrdik JG. Case-control studyof childhood cancer and exposure to 60-Hz magnetic fields. Am JEpidemiol 1988;128:21-38.
Severson RK, Stevens RG, Kaune WT, Thomas DB, Heuser L, Davis S, etal. Acute nonlymphocytic leukemia and residential exposure topower frequency magnetic fields. Am J Epidemiol 1998;128:10-20.
Tynes T, Haldorsen T. Electromagnetic fields and cancer in children resid-ing near Norvegian high-voltage power lines. Am J Epidemiol1997;145:219-26.
Verkasalo PK, Pukkula E, Hongisto MY, Valjus JE, Jarvinen PJ, Heikkila KV,et al. Risk of cancer in Finnish children living close to power lines.Br Med J 1993; 307:895-9.
Wertheimer N, Leeper E. Electrical wiring configurations and childhoodcancer. Am J Epidemiol 1979;109:273-84.
The Swedish National board of Occupational Safety and health, Nation-al board of Housing, Building and planning, National Electricitysafety board, National Board of Health and Welfare, RadiationProtection Institute. Low frequency electrical and magnetic fields.The precautionary principle for national authorities, guidance fordecision makers. Stockholm, 1996
Un po' di storia
Giorgio Perilongo, Donatella Bastianello, LoredanaBeltrame, Elisa Berto, Michela Bonetto, Patrizia Gavin,Cristina Salgarello, Cinzia Cereda, Laura SainatiClinica di Onco-ematologia Pediatrica, Dipartimento Assi-stenziale Integrato di Pediatria, Azienda Ospedaliera,Padova
Il 21 e 22 settembre 2000 si è tenuto ad Abano Terme(Padova) un convegno sul tema “L'alleanza tra infermieree medico in onco-ematologia pediatrica”. L'idea di orga-nizzare delle giornate di studio su questo argomentonasceva dall'esigenza, fortemente sentita, di una maggiorintegrazione culturale, professionale ed organizzativa traqueste due figure mediche. Il presupposto da cui si erapartiti era che “l'alleanza” tra medici ed infermieri esiste-va già, di fatto, ma che essa doveva tuttavia essere raffi-nata e migliorata. La riunione di “Abano 2000” non è sta-to un congresso scientifico vero e proprio ma piuttostoun'occasione per giudicare realtà e situazioni obsolete,capire ed analizzare i bisogni emergenti derivanti dallemoderne realtà assistenziali ed individuare le possibilestrategie applicative per migliorare il rapporto professio-nale tra medico ed infermiere. Così facendo si completòun itinerario di studio e ricerca che fu di aiuto nel defini-re innanzitutto la situazione attuale per quel che riguar-da l'integrazione, come già anticipato nell'identificare ibisogni, i fattori che ostacolano una fattiva collaborazio-ne ed infine, quelli che la rendono possibile.
Il risultato principale raggiunto è stato l'elaborazione diun così detto “laboratorio triennale di studio, formazionee intervento” centrato proprio sul tema dell'Integrazioneinfermiere e medico in onco-ematologia pediatrica”; ilsottotitolo del progetto era: “condizione indispensabili peruna assistenza qualificata”. Il principio ispiratore è statoquello di passare dalle parole a fatti, magari piccoli fatti,ma potenzialmente alla lunga molto efficaci. Detto diver-samente l'idea ispiratrice è stata quella di attivare un“laboratorio”, ossia dar vita ad occasioni concrete, reali peresperimentare nei fatti un lavoro sinergico. Le occasionipreviste sono state dei momenti residenziali, di incontro,durante i quali medici ed infermieri potevano confrontar-si apertamente, sgravati delle sovrastrutture che la corsiadi un ospedale necessariamente impone, e quindi dei pro-getti operativi concreti di lavoro integrato da realizzareuna volta tornati nel proprio ambiente di lavoro. L'altroelemento fondante di questa proposta è stato il convin-cimento che l'integrazione non può essere il frutto di buo-na volontà ma bensì il risultato di un processo educativoculturale e dell'adozione, nella prassi lavorative quotidia-na, di processi organizzativi e procedure operative speci-ficatamente concepite per favorire l'integrazione tra figu-re professionali diverse. Lasciando ad altri di entrare piùnel merito del progetto, e parlando di prospettive storiche,si anticipa che il successo di questa iniziativa è stata taleche quest'anno la branca Europea della SIOP in coopera-zione con la “European Oncology Nursing Society” adot-terà, elaborandolo, un progetto del tutto simile al presen-
Il laboratorio triennale medico-infermieristico

te, in un programma che vedrà coinvolte molte nazionidell'Europa occidentale ed orientale. Il gruppo italiano haun ruolo leader in questa iniziativa. Non vi è dubbio che iprogressi in campo oncologico pediatrico passano ancheattraverso questo lavoro, forse meno visibili, ma certa-mente altrettanto efficace.
Questo progetto nato a Padova si è tuttavia sviluppatograzie al contributo fondamentale del dott. Roberto Cot-ta che della prof. Paola di Giulio e di tutti i colleghi medi-ci ed infermieri che si sono lasciati coinvolgere attiva-mente.
Il disegno del progetto
Paola Di GiulioUniversità degli Studi di Torino
Gli obiettivi del Laboratorio di studio e formazione Gli obiettivi generali del Laboratorio sono stati i seguen-
ti: (i) provare a descrivere e misurare il livello di integra-zione in una o più aree dell'assistenza: programmazione,organizzazione, controllo e valutazione delle attività (inte-grazione organizzativa); (ii) migliorare l'integrazione inalcune aree dell'assistenza: programmazione, organizza-zione, gestione e valutazione dell'assistenza clinica (inte-grazione tecnico-specialistica), (iii) favorire lo scambio el'arricchimento delle conoscenze culturali e tecnico-pro-fessionali tra gli operatori (medici ed infermieri) dello stes-so contesto (integrazione socio-culturale).
I requisitiPer raggiungere gli obiettivi di questa iniziativa di for-
mazione intervento era indispensabile la partecipazionediretta, il protagonismo attivo, la motivazione degli ope-ratori direttamente coinvolti nel processo di cambiamen-to. I processi di integrazione risultano autentici ed effica-ci se riescono a coniugare sviluppo organizzativo, svilup-po professionale e crescita individuale.
Per questi motivi la partecipazione al seminario è statariservata alle equipe di reparto, in particolare alle due figu-re che svolgono un ruolo di primaria importanza: il medi-co e l'infermiere, che dovevano anche essere nella posi-zione di prendere decisioni per realizzare i progetti o icambiamenti proposti (ad esempio il direttore di unità ela caposala). Poiché si trattava di un percorso triennale,dove i momenti sono tra di loro concatenati, si dovevacercare di garantire un minimo di continuità nelle pre-senze.
I seminari avevano anche l'obiettivo di consolidare unamodalità di lavoro assieme, e favorire il costante con-fronto reciproco, nel rispetto delle diversità e specificità.L'effettuazione dei progetti sul campo implica necessa-riamente un forte livello di integrazione e di condivisionedei modelli e degli schemi di riferimento.
Medici ed infermieri di 16 centri si sono incontrati aMilano nel novembre del 2001
I tre laboratori organizzati su base annuale (a Milano,Roma e Napoli) hanno scandito le tappe del progetto: a.selezione del problema e articolazione del progetto; b.attivazione del progetto, raccolta dati e modalità di valu-tazione; c. valutazione dei risultati e definizione dellemodalità di consolidamento delle esperienze. Gli incontrisono stati anche l'occasione di valutazione e confronto trai gruppi, che hanno analizzato criticamente i progetti,offerto suggerimenti, consigli, ma anche condiviso pro-blemi e difficoltà.
Il percorsoLe fasi del percorso possono essere così sintetizzate:1.Un iniziale momento di aula, a carattere intensivo e
con una didattica attiva, con esercitazioni basate sul pro-prio contesto operativo, per metabolizzare i contenutitrattati;
34
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre

2.Un secondo momento, all'interno del proprio conte-sto di lavoro, in cui i partecipanti cercano di applicare allapropria realtà le metodologie e gli strumenti di analisi eriprogettazione appresi durante il momento formativo;
3. Una tutorship a distanza per monitorare lo svolgi-mento dei lavori sul campo;
4. La presentazione ufficiale dei lavori svolti dai singo-li gruppi nel successivo seminario, con valutazione ediscussione- sia da parte del gruppo che degli esperti, deiprogressi ottenuti, delle difficoltà incontrate e delle solu-zioni da adottare. Tra una fase e l'altra è sempre stataprevista una tutorship a distanza.
5. Presentazione finale dei lavori e discussione dellemodalità di consolidamento delle esperienze fatte.
I lavori svolti hanno rappresentato, infatti, il punto dipartenza per un ulteriore salto nello sviluppo delle cono-scenze.
Il primo incontro: scelta ed analisi del problema. Milano22-24 Novembre 2001
Nel primo incontro sono stati forniti alcuni contenutiorganizzativi e metodologici essenziali per analizzare iproblemi ed il fabbisogno di integrazione tra infermieri emedici nelle unità di oncoematologia pediatrica, è statapresentata la metodologia per fare un'analisi organizza-tiva dei problemi e si è discusso su come lavorare perobiettivi. Sono stati via via forniti i contenuti teoricinecessari a sostenere il lavoro dei gruppi sia in aula che ilsuccessivo lavoro sul campo. Obiettivo del seminario erainfatti di offrire ai medici e agli infermieri l'opportunità diconoscere e di cominciare a sperimentare all'interno delproprio contesto lavorativo alcuni strumenti di integra-zione.
Tenendo presente che ogni singola realtà ha caratteri-stiche, consuetudini, culture, problemi e livelli di parten-za diversi, le esperienze da realizzare potevano esserediverse.
Ciascun gruppo ha selezionato un problema che ha ana-lizzato prima in aula e successivamente, al rientro nellapropria sede di lavoro, secondo il seguente schema:
(i) analisi organizzativa del proprio contesto, (ii) indivi-duazione dei fattori di criticità in rapporto al fabbisognodi integrazione, (iii) analisi delle cause e (iv) individuazio-ne dei possibili interventi.
E' stata fornita una consulenza on-line, sia sull'analisidel problema che sulla definizione degli obiettivi e dellestrategie di risoluzione.
Per consolidare l'apprendimento è importante docu-mentare sia il processo di progettazione e realizzazionedelle esperienze che gli esiti raggiunti, in modo da potervalutare, attraverso la scelta di indicatori predefiniti, irisultati ottenuti sia sugli operatori che, dove possibile, irisultati sul paziente, pertanto è stato chiesto ai gruppi dipresentare, nell'incontro successivo, i problemi analizzatied i progetti per risolverli, condividere le difficoltà, i pro-blemi incontrati.
Secondo incontro. Il progetto e la sua realizzazione Roma21-23 Novembre 2002
Obiettivo del seminario era di sviluppare competenzeprogettuali, in stretta sintonia con le esperienze concretesviluppate dai singoli gruppi nei loro contesti.
L'incontro si è basato fondamentalmente sulle presen-tazione dei lavori prodotti e loro supervisione. Dato che igruppi dovevano attivare il progetto presentato nelle pro-prie realtà, sono stati forniti contenuti teorici sugli stru-menti di integrazione, su come gestire una riunione dicoordinamento. Sono state definite le modalità di raccol-ta dati, discusse metodologie e strumenti, definiti gli indi-catori di valutazione.
Naturalmente - anche se poco utilizzata, è stata forni-ta consulenza on line.
Terzo incontro. I risultati ed il consolidamento. Napoli 11-13 Dicembre 2003
Il terzo seminario aveva l'obiettivo di (i) consolidare le competenze progettuali maturate,(ii) verificare la qualità e la tenuta del percorso com-
piuto, sia in rapporto agli aspetti metodologici che allaqualità delle relazioni inter professionali,
(iii) sviluppare un processo di apprendimento delle sin-gole equipe, anche attraverso una rilettura critica delleproprie esperienze,
(iv) acquisire chiara consapevolezza dell'importanza deivincoli di risorse e di contesto nello sviluppo di un concretoprogetto di miglioramento,
(v) sviluppare competenze nella gestione dei processi dicambiamento.
I gruppi hanno presentato i progetti con particolareattenzione a tutto ciò che ne ha frenato od ostacolatol'attuazione. I problemi presentati hanno costituito occa-sione di riflessione e formazione. E' stato dato spazio allestrategie da utilizzare -diversificate in base ai contesti, altipo di problema ed alle risorse disponibili, per consolida-re il processo di cambiamento attivato (sia a livello orga-nizzativo che relazionale)
35
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

VEDI ALTRO FILE AIEOP_36
36
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre

Progetto di analisi e qualificazionedell'assistenza nel reparto dionco-ematologia pediatrica di Brescia
Stefania Dotti,* Daniela Brontesi,*Carmelita D'Ippolito,+ Chiara Caldani,§ Fulvio Porta#
*Infermiera; +pediatra; §specializzando in Pediatria;#responsabile DH onco-ematologia Pediatrica, Brescia
Il dolore nei pazienti oncologici pediatrici è un argo-mento molto rilevante ma spesso non ancora adeguata-mente affrontato. Nel nostro reparto non esistevanomodalità omogenee né di valutazione del dolore né ditrattamento; inoltre esistevano diversità di gestione traDH e Reparto.
Il nostro lavoro aveva i seguenti obiettivi: - uniformare la valutazione del dolore; - uniformare il trattamento; - rendere il paziente partecipe alla gestione di un sin-
tomo cosi invalidante. Dal dicembre 2001 al settembre 2002 sono state ela-
borate le scale di rilevazione del dolore: comportamenta-le (< 3 anni), analogico affettiva ( 3 - 7anni) e numerica oanalogico visiva (> 7 anni); si è tenuto un corso di forma-zione medico-infermieristico, e, in collaborazione con laRianimazione Pediatrica si è redatto un opuscolo infor-mativo sul dolore e la relativa terapia, infine sono stateintrodotte delle schede di rilevazione e di trattamento deldolore relativo a procedure invasive, alla mucosite o adaltri stati dolorosi (pazienti terminali).
Le schede venivano redatte dai pazienti o dai genitoriin collaborazione con il personale medico o infermieristi-co. Per la scala analogico-affettiva abbiamo consideratopunteggio critico >2, mentre per quella analogico visi-va>3.
Lo studio è stato affrontato nel periodo da settembre2002 a marzo 2004; le schede valutabili sono quelle rela-tive alle procedure dolorose.
Sono state raccolte 199 schede di punture lombari (33bambini) e 82 di puntati midollari (20 bambini). Per le pri-me il punteggio critico è stato superato in 25 schede su199 (12,5%) relative a 10 bambini, 5 di essi hanno ricon-fermato il dato, per 4 di loro si è passati ad una sedazio-ne profonda nelle successive procedure.
Per gli aspirati midollari il punteggio critico è statosuperato in 12 schede su 84 (14,6%) relative a 7 bambi-ni, 5 dei quali hanno riconfermato il dato, per uno solo èstato possibile passare a sedazione profonda.
Questo progetto prosegue e viene rutinariamente appli-cato su tutti i bambini seguiti nel nostro Centro.
Grazie alle schede di rilevazione e trattamento siamoriusciti ad obiettivare ed uniformare la gestione del dolo-re tra DH e Reparto; il coinvolgimento dei pazienti e geni-tori li ha resi partecipi e più fiduciosi; l'analisi delle sche-de ha permesso di isolare un numero limitato di pazientiper i quali è stato possibile procedere alla sedazioneprofonda coinvolgendo la Rianimazione Pediatrica che hamostrato un progressivo interesse.
Assistenza domiciliare integrata nel bambinooncologico
Edvige GombachResponsabile Infermieristica Dipartimento Chirurgico,IRCCS Burlo Garofolo, Trieste
L'assistenza domiciliare integrata Vale a dire l'insieme di prestazioni mediche, infermieristi-
che, riabilitative, socio-assistenziali e psicologiche, fornite adomicilio a pazienti cronici in forma coordinata ed integratasecondo piani assistenziali individuali definiti e programma-ti da una equipé multidisciplinare:
è un Diritto del bambino malatoL'articolo 3 della Carta dei Diritti del Bambino in Ospeda-
le, enunciando il diritto a ricevere il miglior livello di cura eassistenza, specifica che il ricorso all'ospedalizzazione deveessere limitato “alle situazioni in cui non sia possibile far fron-te alle esigenze assistenziali…in altro modo” e che “ vengo-no favoriti day hospital e assistenza domiciliare…”.
è suggerita dal Piano Sanitario Nazionale 2002 - 2004Nella parte seconda: “La salute del neonato, del bambino
e dell'adolescente” tra gli obiettivi vi è quello di incremen-tare l'adozione di strutture socio-sanitarie alternative (qua-li l'ospedalizzazione a domicilio).
è prevista nella Proposta di progetto obiettivo materno-infantile e dell'età evolutiva della Regione FVG. Il DGR n.2588, del 18. 7. 2002 sottolinea la necessità di definire leintegrazioni con il Distretto sanitario per assicurare la conti-nuità assistenziale.
Sino ad ora esperienze d'assistenza domiciliare in emato-oncologia pediatrica riguardavano prevalentemente terapiepalliative in pazienti terminali (assistenza in case d'acco-glienza prossime all'ospedale (Gaslini); assistenza in ambitometropolitano (Meyer, Bambino Gesù).
L'U. O. di Emato-oncologia pediatrica dell'IRCCS BurloGarofolo di Trieste ha sentito la necessità di avviare un pro-getto per trasferire alcune prestazioni assistenziali a casadei propri assistiti, cercando d'integrarsi con le strutture sani-tarie presenti nel territorio regionale che costituisce il suobacino d'utenza.
Il progetto, oltre a soddisfare un diritto fondamentale delbambino, offre anche vantaggi all'insieme paziente/famiglia,(minor rischio infettivo, migliore continuità assistenziale,riduzione del costo sociale secondario all'ospedalizzazione).
ObiettivoCreare attorno al bambino oncologico una rete assisten-
ziale in grado di garantirgli a domicilio: (i) prelievi(ii) gestione presidi (iii) piccola chirurgia (iv) trattamenti chemioterapici a minore intensità (v) trattamenti antibiotici parenterali(vi) monitoraggio dei parametri vitali(vii) supporto nutrizionale (enterale o parenterale)(viii) gestione effetti collaterali dei farmaci
43
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

(ix) gestione di eventuali malattie intercorrenti(x) supporto psicologico e pedagogico(xi) educazione al self care(xii) terapia palliativa (nel bambino terminale)
erogati omogeneamente, mantenendo standard di eccel-lenza, senza aumento dei costi umani o economici per il SSN.
PercorsoIl progetto è stato suddiviso nelle seguenti fasi:
Analisi e definizione interna della domandanumero di nuovi casi attesipazienti eleggibiliprestazioni trasferibilipeculiarità di gestione (leadership del Centro, rapidità di trasmissione delle informazioni, urgenza della refertazione, gestione “dinamica”)
Verifica dell'offerta numero, formazione, esperienza in ambito pediatrico degli operatori sanitari del territoriotipo di assistenza medica ricevuta (MMG o PLS)modalità di attivazione e di gestione dei servizi territoriali; disponibilità di presidi.
Accordi interaziendalicon ognuna delle 6 ASS della Regione vengono definite:(i) competenze di ciascuna figura implicata (chi fa, cosa, quando, come)(ii) procedura di attivazione dell'assistenza domiciliare(iii) linee guida comuni all'assistenza del bambino onco-
logico(iv) modulistica da utilizzare(v) fabbisogno formativo del personale territoriale.
Il risultato di questi accordi costituiscono la sostanza di con-venzioni sottoscritte dalle rispettive D. S.
Aggiornamento del personale sanitario del territorioPer Infermieri e Assistenti Sanitari:
stage individuale presso il Centro;
corso ECM su problematiche domiciliari del bambino onco-logico quali:
(i) profilassi delle infezioni(ii) igiene del paziente neutropenico (iii) gestione del catetere venoso centrale(iv) esecuzione prelievi da vena centrale (v) gestione di sistemi di infusione(vi) preparazione, somministrazione e smaltimento
dei farmaci antiblastici(vii)terapia palliativa o antidolorifica(viii) la vita di relazione (scuola, sport,
associazionismo) (ix) la comunicazione con il bambino e la sua famiglia(x) il bambino come cittadino utente
(etica, diritti, legislazione).
Per PLS e Pediatri ospedalieri:corso ECM su:
i) effetti collaterali della terapia antiblastica(riconoscimento e gestione)
ii) prevenzione e gestione delle complicanzeinfettive, metaboliche
iii) terapia antalgica iv) terapia palliativa nel bambino terminalev) la vita di relazione (scuola, sport, associazionismo) vi) la comunicazione con il bambino e la sua famigliavii)il bambino come cittadino utente
(etica, diritti, legislazione).
Sviluppo della rete di comunicazione(i) trasmissione delle informazioni in tempo reale
attraverso un programma sperimentale di e-govermentsviluppato all'interno del portale dalla ASS 1 e accessibi-le ai soli operatori abilitati
(ii) archiviazione delle informazioni nel server del portale(cartella informatica individuale)
FinanziamentoIl nostro IRCCS ha presentato il progetto come Ricerca di
rete oncologica che il Ministero della Salute ha approvatofinanziandolo con 55. 000 euro.
Risultatii) Stipulata convenzione con 3 delle 6 ASS della Regioneii) 30 infermieri di 8 distretti hanno frequentato nel perio-
do gennaio 2003 - marzo 2004 lo stage formativo pres-so il Centro per un totale di 210 ore
iii) sono stati assistiti 14 pazienti su 18 eleggibili (in parti-colare tutte le leucemie e i linfomi non Hodgkin)
iv) Le prestazioni più frequentemente erogate nell'ambitodella convenzione sono state:
prelievi 25%medicazioni del cvc 20%sostituzione tappo 20%rilevazione parametri vitali 12%sedute di logopedia, fisioterapia 12%terapie parenterali 10%(antibiotici, fattori di crescita. . ) altro 1%(i)La media di accessi ospedalieri risparmiati dall'assistenza
domiciliare per ogni singolo paziente è stata del 30% cir-ca. Per l'ospedale la conseguente diminuzione di ricoveri
impropri ha dato la possibilità di aumentare i tempi di assi-stenza ai pazienti ospedalizzati.
Risultati dell'esperienza e prospettive future
A. GaraventaGruppo di lavoro AIEOP per l'integrazione medico-infer-mieristica
Il rapido svilupparsi ed evolvere delle conoscenze bio-mediche e dei modelli assistenziali in emato-oncologiapediatrica, e la costante evoluzione dei bisogni e delleaspettative dei pazienti e delle famiglie da un lato e lasempre più limitata disponibilità di risorse dall'altro, spin-gono costantemente medici e infermieri a ripensare il pro-prio modo di lavorare in una duplice direzione:
44
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre

- accrescere le proprie competenze sia nel campo spe-cialistico che sul fronte comunicativo e relazionale;
- sviluppare modelli organizzativi e competenze rela-zionali indispensabili per soddisfare il fabbisogno diintegrazione esistente tra le differenti figure profes-sionali.
L'integrazione tra infermiere e medico è condizioneindispensabile per una assistenza qualificata ed un cor-retto uso delle risorse. La collaborazione interdisciplinaredelle professioni sanitarie è ampiamente discussa in let-teratura - quasi 200 negli ultimi 20 anni sono le vocibibliografiche sulla collaborazione medico-infermieristica.
Lavorare in modo integrato implica una progettualitàcomune (obiettivi condivisi) che si traduce nella costru-zione di strumenti d'integrazione (protocolli, progetti, per-corsi, strumenti del sistema informativo) che garantisca-no una presa in carico complessiva ed integrata delpaziente in tutte le fasi del processo assistenziale.
Il laboratorio AIEOP d'integrazione medico-infermieri-stica si e' sviluppato nell'arco di 4 anni come percorso for-mativo con l'obiettivo di sviluppare competenze metodo-logiche e organizzative per favorire l'integrazione fra lediverse professioni sanitarie in oncoematologia pediatri-ca.
L'effettiva acquisizione di abilità in questo camporichiede necessariamente un coinvolgimento forte dei sog-getti direttamente interessati e una reciproca disponibi-lità a confrontarsi.
Queste conoscenze e abilità di taglio organizzativo emetodologico si possono imparare solo facendo diretta-mente e quindi attivandosi in prima persona, possibil-mente in un contesto guidato e protetto.
Per questo motivo tutto il percorso di formazione e' sta-to progettato integrando momenti d'aula a momenti dilavoro sul campo, nel proprio contesto lavorativo, con tut-ti i vincoli della quotidianità e della scarsità delle risorse.
Il punto di partenza di questo percorso sono state leproposte di linee guida per l'attività' integrata tra infer-mieri e medici di ematologia oncologica pediatrica pub-blicate sulla RIP nel 1999 e su Med Ped Oncol nel 2000.
Questo documento voleva descrivere la realtà idealenell'ambito della quale si vorrebbe lavorare e si propone-va come materiale di lavoro per elaborare nel quotidianosoluzioni che meglio si adottino alla realtà specifica diogni centro AIEOP.
A tutti era evidente che si trattava di una realtà, unametodologia di lavoro da conquistare e consolidare con lasperimentazione sul campo per cui nel primo corso tenu-tosi a Milano (organizzato dalla Clinica Pediatrica di Mon-za e dall'Oncologia Pediatrica dell' Istituto Tumori) ognicentro individuo al suo interno un progetto-area critica daaffrontare come percorso formativo:
Centri partecipanti e Progetti♦ Dip. Biomedicina Eta' Evolutiva - Clinica Pediatrica- Bari
Progetto: Gestione domiciliare del catetere venoso cen-trale
♦ Clinica Pediatrica - Ospedale Civile - BresciaProgetto: Affronto del dolore da procedura
♦ Divisione di Ematologia - Ospedale A. Pugliese - Catan-zaro
Progetto: Dimissione integrata medico-infermieristica♦ Ospedale casa Sollievo Sofferenza - S. Giovanni Roton-
do - FoggiaProgetto: Gestione della febbre
♦ Dipartimento di Ematologia/Oncologia - Istituto G. Gasli-ni - GenovaProgetto: Riorganizzazione del Day Hospital
♦ Divisione di Oncologia Pediatrica - Ist. Naz. Studio e CuraTumori - MilanoProgetto: Organizzazione di un servizio di "call center"
♦ Clinica Pediatrica Univ. di Milano Bicocca - AziendaOspedaliera S. Gerardo - MonzaProgetto: Organizzazione e programmazione dei ricoveri
♦ Serv. di Oncologia Pediatrica - Dip. di Pediatria - II Ate-neo di NapoliProgetto: Organizzazione di un servizio di "call center"
♦ Dipartimento Pediatria Universita' di Padova II - Catte-dra Clinica Pediatrica PadovaProgetto: Assistenza e gestione del bambino terminale
♦ U. O. di Oncologia - Ospedale Pediatrico Bambin Gesu' -RomaProgetto: Inserimento del nuovo personaleinfermieristico in reparto di Oncologia Pediatrica
♦ Clinica Pediatrica - Ospedale Infantile Burlo Garofalo -TriesteProgetto: Assistenza domiciliare integrata con i serviziterritoriali
Nel secondo corso a Roma (organizzato dalla Divisionedi Oncologia pediatrica dell'Istituto Bambin Gesu'), ven-nero presentati e discussi i diversi progetti elaborati l'af-fronto di tali problematiche e nel terzo corso a Napoli(organizzato dal Servizio di Oncologia Pediatrica dellaSeconda Università degli Studi di Napoli), sono stati pre-sentati e discussi i risultati preliminari dei progetti porta-ti avanti dalle singole unità.
Le singole èquipe hanno discusso l' esperienza fatta,sviluppando un confronto tra le diverse esperienze in unaanalisi critica delle difficoltà incontrate e degli errori fat-ti nello sviluppo dei progetti e hanno poi rielaborato ilproprio lavoro facendone oggetto di una presentazioneorganica che verranno poi sottomessi per pubblicazione.
Certamente lo scopo di questo percorso non era la pub-blicazione di elaborati ma la ricerca di modalità per edu-carsi a lavorare in collaborazione per lavorare meglio. Altriargomenti possono essere affrontati o gli stessi in centridiversi. La conoscenza teorica e superficiale di una deter-minata metodologia non rappresenta però una garanziaper una effettiva applicazione nel concreto. Le compe-tenze metodologiche si possono consolidare solamentecon la sperimentazione in situazione ricche e diversifica-te e con il confronto con gli altri.
Per questa ragione abbiamo pensato a sviluppare unasequenza di scambi on line che aumenti il livello di inte-rattivita' tra i partecipanti e lo staff docenti ed abbiamosviluppato un progetto di ricerca europeo. Indicazioni e limiti del trattamento con RHGH nei bambi-ni con tumore cerebrale
45
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

Indicazioni e limiti nel trattamento con rhGhnei bambini con tumore cerebrale
Carlo de Sanctis, Patrizia Chiabotto, Jaele Bellone,Andrea CorriasDipartimento Specialistico Endocrinologia Pediatrica,Ospedale Infantile Regina Margherita, Torino
Nei pazienti con tumori cerebrali le strategie terapeu-tiche necessariamente aggressive possono comportarecomplicanze che coinvolgono diversi apparati e tra que-sti il sistema endocrino.
Il difetto secretorio di GH rappresenta la complicanzaendocrina di più frequente osservazione. Nei soggetti conneoplasia della regione ipotalamo - ipofisaria (craniofa-ringioma, germinoma, glioma opto-chiasmatico) il deficitè legato alla neoplasia, all'intervento chirurgico, alla radio-terapia ed, in minor misura, alla chemioterapia. Neipazienti con neoplasia che origina in altra sede cerebrale(medulloblastoma, ependimoma, glioma, pinealoblasto-ma), il deficit di GH è soprattutto conseguente alla radio-terapia cranica. 3,4
La radioterapia, frequentemente impiegata in moltitumori cerebrali, occupa un ruolo rilevante nel ridurre lasecrezione di GH. Il difetto di GH ha un tempo di latenzavariabile, e' strettamente legato alla dose radiante ed allasua modalità di erogazione nel senso che quanto più èelevata la dose e più breve è il periodo di erogazione tan-to più grave è il danno secretorio. 11,13,16,17
La diagnosi di deficit di GH richiede un'attenta valuta-zione clinica e auxologica che comporta il controllo del-l'altezza, della statura vertice - della statura ischiatica,della velocità di crescita, del peso e dello sviluppo pube-rale. Particolare attenzione va data alla velocità di cresci-ta che se < -2 DS in un anno o < -1.5 DS per 2 anni con-secutivi6 renderà necessaria la valutazione della secrezio-ne di GH. Tuttavia, è da tenere presente che la velocità dicrescita, pur in presenza di ridotta secrezione di GH, puòessere normale nei soggetti che si presentano obesi e/ocon anticipo puberale.
Sotto il profilo biochimico, la diagnosi di deficit di GHsi avvale dell'assetto secretorio di GH dopo test provoca-tivi classici e dello studio della secrezione spontanea diGH. Tra i test provocativi classici (insulina, clonidina, L-dopa, arginina, glucagone) quelli più usati sono il test coninsulina in soggetti di età > 2 anni e quello con glucago-ne in soggetti di età < 2 anni. La valutazione della secre-zione spontanea di GH è stata presa in considerazionesoprattutto in soggetti con tumore cerebrale distante dal-la regione ipotalamo -ipofisaria, irradiati in regione cra-nica, allo scopo di individuare deficit neurosecretori apatogenesi ipotalamica, caratterizzati da una ridottasecrezione spontanea di GH in presenza di una normalerisposta ai test provocativi.
Trattamento con GHIndicazioni. L'impiego del GH nei soggetti GHD gene-
ralmente viene proposto 2 anni dopo la terapia antitu-
morale quando le probabilità di recidiva sono ridotte. Inalcuni Centri il trattamento viene iniziato più precoce-mente, 1 anno dopo la fine della terapia antitumorale.
L'rhGH è somministrato a dosi 0.1-0.25 mg/kg/settimanao a dosi di 4-6.5 mg/m2/settimana, ripartite in 6 sommini-strazioni effettuate la sera per via sottocutanea. General-mente vi è la tendenza a somministrare la dose di rhGH chemantenga livelli di IGF1 nella fascia superiore di normalitàonde evitare che valori soprafisiologici di IGF1 possanoaumentare il rischio di recidiva o di secondo tumore. 9
Nei pazienti pediatrici e negli adolescenti con tumorecerebrale e GHD, l'rhGH trova indicazione allo scopo di: -promuovere il recupero della crescita staturale - favorireil trofismo e la forza muscolare; - indurre la riduzione delgrasso corporeo; - migliorare la mineralizzazione ossea.
Uno studio recente riporta 25 anni di osservazione e lealtezze finali raggiunte da 58 bambini sopravvissuti atumori cerebrali che avevano ricevuto trattamento conrhGH. L'altezza finale - l'altezza media parentale è risulta-ta di -3.8 cm. nei soggetti irradiati a livello cranico e di -11.8 cm. quando era stata impiegata anche la chemiotera-pia; di -11.5 cm. in quelli irradiati a livello cranio-spinalee di -18.9 cm. quando era stata impiegata anche la che-mioterapia. 7 In 11 soggetti della stessa casistica che pre-sentavano anticipo puberale è stata utilizzata l'associazio-ne rhGH piu' GnRH con risultati migliori nei 4 pazienti cheavevano ricevuto la sola irradiazione cranica.
In una nostra casistica di 21 soggetti con medullobla-stoma e deficit di GH che hanno raggiunto l'altezza fina-le, solo 4 sono stati trattati con rhGH. L'altezza finale -l'altezza media parentale è risultata di -2.3±0.9 DS nelgruppo dei non trattati mentre quella dei 4 soggetti trat-tati è risultata, rispettivamente, di -0.1, -1, -0.4 e -3.2 DS.
In sintesi le esperienze riportate in letteratura eviden-ziano risultati soddisfacenti. Alle dosi di rhGH prima indi-cate, vengono raggiunte altezze finali (corrette per lealtezze medie parentali) migliori nei casi in cui la situa-zione auxologica prima della terapia risulti poco compro-messa, quando è più breve l'intervallo di tempo tra la finedella terapia tumorale e l'inizio della terapia con rhGH, neisoggetti con pubertà ritardata ed in quelli irradiati solo inregione cranica rispetto a quelli irradiati anche in regio-ne spinale. 4,7
La terapia con rhGH puo' trovare indicazione per i suoieffetti metabolici sul ricambio proteico e glicolipidico.Consente, difatti, la riduzione del contenuto lipidico del-le cellule adipose, favorisce la redistribuzione dell'adipe eaumenta la sintesi proteica. A proposito sono riportatescarse esperienze in pazienti pediatrici; nell'adulto vieneriferita la riduzione della massa grassa del 10-12% e l'au-mento della massa magra del 5-10% (1,5).
Un'altra indicazione al trattamento con rhGH puo' esse-re rappresentata dalla correzione della osteopenia docu-mentata in questi soggetti. 8,17 L'impiego dell'rhGH sottoil profilo biochimico favorisce a livello renale il processodi idrossilazione della vitamina D, con conseguente mag-gior assorbimento di calcio e fosforo.
Pur con i limiti rappresentati dalla metodica attual-mente impiegata e dall'interpretazione dei dati ottenuti,nella nostra casistica di 15 soggetti trattati per tumorecerebrale in epoca prepuberale e con deficit di GH, è sta-ta documentata con la DEXA una migliore mineralizza-
46
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre
L’ormone della crescita in Oncoematologia Pediatrica

zione ossea nel gruppo dei pazienti trattati con rhGH. Limiti. I principali effetti indesiderati dell'impiego dell'rh-GH sono rappresentati da: ridotta tolleranza glicidica aseguito dell'azione nota sul ricambio glicidico; cefalea,convulsioni, edemi, ipertensione in relazione all'effettosodioritentivo ; artralgie in conseguenza di edemi periar-ticolari; sviluppo di recidive e di un secondo tumore.
Tra questi effetti il rischio di recidiva tumorale puo' rap-presentare il vero fattore limitante.
Allo stato attuale, nonostante il GH sia un ormone mito-geno, non vi è evidenza di un aumento di recidive deltumore o di insorgenza di un secondo tumore. 2,10,12,14,15,18
In una nostra casistica di 12 pazienti con medullobla-stoma trattati con rhGH nessuno di questi ha presentatorecidiva tumorale o secondo tumore anche a distanza di7 anni dalla fine del trattamento del tumore.
L'impiego dell'rhGH è assolutamente controindicato neicasi in cui il tumore sia ancora in fase attiva. E', pertan-to, importante che l'inizio della terapia venga deciso congli oncologi pediatri che potranno meglio fornire indica-zioni anche sulla prognosi e sul rischio di recidiva.
Bibliografia
1. Chrisoulidou A, Beshya SA, Rutherford O, Spinks TJ, Mayet J, KydP, et al. Effects of 7 years of growth hormone replacement thera-py in hypopituitary adults. J Clin Endocrinol Metab 2000;85:3762-9
2. Corrias A, Picco P, Einaudi S, de Sanctis L, Besenzon L, Garre ML,et al. Growth hormone treatment in irradiated children with braintumours. J Pediatr Endocrinol Metab 1997;10:41-9.
3. de Sanctis C, Corrias A. Disordini endocrinologici in soggetti contumori cerebrali. Quaderni di Endocrinologia Pediatrica 1999;9:41-63
4. De Vile CJ, Grant DB, Hayward RD, Stanhope R. Growth and seque-lae of craniopharyngioma. Arch Dis Child 1996;75:108-14.
5. Fernholm R, Bramnert M, Hagg E, Hilding A, Baylink DJ, Mohan S,et al. Growth hormone replacement therapy improves body com-position and increases bone metabolism in elderly patients withpituitary disease. J Clin Endocrinol Metab 2000;85:4104-12.
6. GH Research Society. Consensus Guidelines for the Diagnosis andTreatment of Growth Hormone (GH) Deficiency in childhood andAdolescence: Summary Statement of the GH Research Society. JClin Endocrinol Metab 2000;85:3990-3.
7. Gleeson HK, Stoter R, Ogilvy-Stuart AL, Gattamaneni HR, BrennaBM, Shalet SM. Improvements in final height over 25 years ingrowth hormone (GH)-deficient childhood survivors of braintumours receiving GH replacement. J Clin Endocrinol Metab 2003;88:3682-9.
8. Krishnamoorthy P, Freeman C, Bernstein ML, Lawrence S, Rodd C.Osteopenia in children who have undergone posterior fossa cran-iospinal irradiation for brain tumors. Arch Pediatr Adolesc Med2004;158:491-6.
9. Juul A, Bernasconi S, Clayton P, Kiess W, DeMuinck-Keizer Schar-ma SMPF. European audit of current practise in diagnosis andtreatment of childhood growth hormone deficiency. Horm Res2002;58:233-41.
10. Maneatis T, Baptista J, Connelly K, Blethen S. Growth hormonesafety update from the National Cooperative Growth Study. J.Pediatr Endocrinol Metab 2000;13:1035-44.
11. Mars M, Beck JD, Grabenbauer CG, Dorr HG. Spontaneous noctur-nal growth hormone secretion in children after medulloblastomatherapy. Med Pediatr Oncol 2001;36:494-6.
12. Moshang T, Chen Rundle C, Graves D, Nickas J, Johanson A, Mead-ows A. Brain tumor recurrence in children treated with growthhormones: the National Cooperative Growth Study experience. J.Pediatr 1996;128:S4-7.
13. Ogilvy-Stuart AL, Shalet SM. Growth and puberty after growthhormone treatment after irradiation for brain tumours. Arch Dis
Child 1995;73:141-6. 14. Packer R, Bayett J, Janss A, Stavrou T, Kun L, Wisoff J, et al. Growth
hormone replacement therapy in children with medulloblastoma:use and effect on tumor control. J Clin Oncol 2001;19:480-7.
15. Sklar C, Merterns A, Mitby P, Occhiogrosso G, Qin J, Heller G, et al.Risk of disease recurrence and second neoplasms in survivors ofchildhood cancer treated with growth hormone: a report from thechildhood Cancer Survivor Study. J Clin Endocrinol Metab 2002;87:3136-41.
16. Shalet SM. Growth in irradiated children. In : Two decades of expe-rience in growth. Edited by Pombo and Rosenfield. Serono Sym-posia Pubblications for Raven Press; New York: 1993. p. 227-37.
17. Spoudeas HA. Growth and endocrine function after chemiothera-py and radiotherapy in childhood. Eur J Cancer 2002;38:1748-59.
18. Swerdlow A, Reddingius R, Higgins C, Spoudeas H, Phipps K, QiaoZ, et al. Growth hormone treatment of children with brain tumorsand risk of tumor recurrence. J Clin Endocrinol Metab 2000;85:4444-9.
La terapia con ormone somatotropo ricombi-nante umano (rHu-GH) nei pazienti sottopostia trapianto di cellule staminali emopoietiche
Giovanna Giorgiani, Alessandra De Cesare, Anna DehòOncoematologia Pediatrica, IRCCS Policlinico San Matteo,Pavia.
Il trapianto di cellule staminali emopoietiche (TCSE),autologo o allogenico, è stato sempre più largamente usa-to nel corso degli ultimi anni per il trattamento di nume-rose emopatie maligne o non neoplastiche, di errori con-geniti dell'immunità e per selezionati errori congeniti delmetabolismo. Con l'aumentare del numero dei soggettilungo-sopravviventi dopo TCSE, si è prestata sempre mag-giore attenzione al monitoraggio dei possibili effetti col-laterali associati alla procedura trapiantologica. Infatti,numerosi sono i possibili effetti a distanza che possonoessere diagnosticati nei pazienti sottoposti a TCSE in etàpediatrica. Fra essi, indubbiamente, per frequenza di dia-gnosi e per impatto sulla qualità di vita spicca il possibi-le rallentamento dello sviluppo staturale.
Diversi fattori, tra loro talvolta interagenti, possono con-tribuire al rallentamento dello sviluppo staturale. Infatti,una riduzione della velocità di crescita dopo TCSE puòessere osservata a causa di un danno diretto della terapiacitostatica e/o radiante sulle cartilagini di coniugazione,nel contesto di una graft versus host disease (GVHD) cro-nica, magari richiedente prolungato trattamento steroideoe per una ridotta produzione di ormone somatotropo (GH),motivata da un danno a carico dell'asse ipotalamo-ipofi-sario. Quest'ultimo fattore è stato largamente chiamato incausa per spiegare il ridotto accrescimento dei pazientisottoposti a TCSE e dei vari elementi che contribuisconoad una alterazione della velocità di crescita dopo trapiantoè l'unico suscettibile di correzione attraverso l'uso diormone somatotropo umano ricombinante (rHu-GH).
Dagli studi finora pubblicati, così come dai dati deriva-ti dalla nostra personale esperienza, è emerso in manieraincontrovertibile che il trattamento radiante (irradiazio-ne totale corporea o total body irradiation, TBI) è gravatoda una maggior incidenza di ritardato accrescimento sta-turale. In particolare, l'uso di TBI in dose singola, oppureimpiegata in pazienti precedentemente sottoposti a irra-
47
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

diazione craniale o cranio-spinale, risulta particolarmen-te deleterio per i pazienti. Poiché il busulfano, rispetto allaTBI, è gravato da una minor incidenza di effetti collatera-li, soprattutto di tipo neurologico e inerenti ad un ritardonell'accrescimento staturale, il suo uso, a parità di effica-cia rispetto alla radioterapia, deve essere raccomandato inmaniera particolare, nei soggetti al di sotto dei 2 anni dietà. Non stupisce, quindi, che, con l'eccezione della leu-cemia linfatica acuta (ove l'impiego della TBI offre, con-solidatamente, un vantaggio in termini di outcome fina-le) la radioterapia a dose convenzionale (990-1200 cGy)venga sempre meno frequentemente utilizzata. Deve esse-re, comunque, ricordato che neanche il busulfano è com-pletamente scevro da effetti deleteri sulla velocità diaccrescimento, potendosi osservare ritardi nell'accresci-mento post-trapianto in bambini che hanno ricevuto unregime di condizionamento al trapianto basato sull'im-piego di questo farmaco alchilante.
Come precedentemente ricordato, una larga parte deldanno all'accrescimento staturale nei pazienti sottopostia TCSE è dovuto a una ridotta produzione di GH. Sia l'i-potalamo, sia l'ipofisi possono risentire sfavorevolmentedella terapia pre-trapianto, in particolare di quella radian-te. Sarà, dunque, di utilità per un corretto inquadramen-to diagnostico del paziente effettuare in maniera tempe-stiva valutazioni delle produzione di GH dopo stimolo far-macologico (insulina, L-dopa, arginina, sleep test) in pre-senza di riduzione della velocità di crescita (sotto il 25°percentile o inferiore a 2 deviazioni standard rispetto alvalore pre-trapianto), associata a ritardo nella matura-zione scheletrica, documentabile attraverso una radio-grafia del carpo. Di minore interesse pratico, pur se con-cettualmente tutt'altro che irrilevante, sarà stabilire attra-verso il test al GHRH la sede (ipotalamica o ipofisaria) deldanno conseguente la terapia di condizionamento al tra-pianto.
Una volta diagnosticata una riduzione della velocità dicrescita motivata da una ridotta produzione di GH, si potràutilmente considerare l'inizio di una terapia sostitutivacon rHu-GH. Numerose evidenze indicano che i miglioririsultati in termini di miglioramento della velocità di cre-scita e, al meglio, raggiungimento del target staturalegenetico si hanno quando la terapia sostitutiva viene ini-ziata precocemente. Preliminare e necessaria all'inizio deltrattamento con rHu-GH sarà la dimostrazione di norma-le metabolismo glucidico nel paziente candidato alla tera-pia sostitutiva. Il rHu-GH verrà impiegato alla dose di 0,7UI/Kg (oppure 0,2 mg/Kg) alla settimana, suddivisa in dosirefratte che coprano almeno 5-6 giorni. Una volta intra-preso, il trattamento andrà sicuramente continuato finoa dimostrata saldatura delle cartilagini di coniugazione.Non mancano, tuttavia assertori dell'opportunità di man-tenere una terapia con rHu-GH anche in soggetti adulti,seppure a dosi ridotte, per il favorevole effetto svolto daltrattamento sul trofismo muscolare e più in generale sulmetabolismo.
Nel corso del trattamento andrà periodicamente moni-torato il miglioramento della velocità di crescita e l'evo-luzione dell'età ossea del paziente. Quasi superfluo ricor-dare come l'eventuale presenza di insufficiente produzio-ne ormonale tiroidea, GVHD cronica o ridotta produzioneepatica di somatomedine potranno sfavorevolmente con-
dizionare i risultati ottenibili con la terapia sostitutivabasata sull'uso di rHu-GH
Soprattutto in passato, sono state sollevate potenzialiriserve sull'uso di rHu-GH in pazienti sottoposti a TCSE,magari a causa di una pregressa emopatia maligna, per ilpotenziale rischio che questo trattamento basato sull'im-piego di una sostanza ad azione pro-mitogenica potessefavorire lo sviluppo di neoplasie secondarie o la compar-sa di una recidiva di malattia. Sebbene alcuni articoli, pro-venienti in particolare da autori giapponesi, abbiano por-tato supporto a questa ipotesi, più attente ed esaustiveanalisi hanno di molto, per non dire completamente, ridi-mensionato questo rischio e, oggi, numerosi gruppi, tra cuiil nostro, impiegano largamente la terapia con rHu-GHper ottimizzare la crescita del paziente trapiantato e l'ot-tenimento di una statura definitiva quanto più possibilevicino al target genetico.
Bibliografia
Brauner R, Adan L, Souberbielle JC, et al. Contribution of growth hormonedeficiency to the growth failure that follows bone marrow trans-plantation. J Pediatr 1997; 130: 785-92.
Brennan B, Shalet SM. Endocrine late effects after bone marrow trans-plant. Br J Haematol. 2002; 118: 58-66.
Clayton PE, Shalet SM. Dose dependency of time of onset of radiation-induced growth hormone deficiency. J. Pediatr 1991; 118: 226-228.
Clement-De Boers A, Oostadijk W, van Weel-Sipman MH, et al. Finalheight and hormonal function after bone marrow transplantationin children. J Pediatr 1996; 129: 544-550.
Davies S, Ramsay NK, Klein JP, et al. Comparison of preparative regimensin transplants for children with acute lymphoblastic leukemia. JClin Oncol 2000; 18:340-7.
Giorgiani G, Bozzola M, Locatelli F, et al: Role of busulfan and total bodyirradiation on growth of pre-pubertal children receiving bone mar-row transplantation and results of treatment with recombinanthuman growth hormone. Blood 1995; 86:825-831.
Leiper AD, Stanhope R, Lau T, et al. The effects of total body irradiationand bone marrow transplantation during childhood and adoles-cence on growth and endocrine function. Br J Haematol. 1987; 67:419-426.
Locatelli F, Burgio GR. Transplant of haematopoietic stem cells in child-hood.: where we are and where we are going. Haematologica1998; 83: 550-563.
Locatelli F, Labopin M, Ortega J, et al. Factors influencing outcome andincidence of long-term complications in children who underwentautologous stem cell transplantation for acute myeloid leukemiain first complete remission. Blood 2003; 101: 1611-1619.
Michel G, Gluckman E, Esperou-Bourdeau H, et al. Allogeneic bone mar-row transplantation for children with acute myeloblastic leukemiain first complete remission: impact of conditioning regimen with-out total-body irradiation-a report from the Societe Francaise deGreffe de Moelle. J Clin Oncol 1994; 12:1217-22.
Ogilvy-Stuart AL, Clark DJ, Wallace W.HB, et al. Endocrine deficit afterfractionated total body irradiation. Arch Dis Child 1992; 67: 1107-110.
Sanders J, Pritchard S, Mahoney P, et al. Growth and development fol-lowing marrow transplantation for leukaemia. Blood 1986; 68:1129-34.
Zecca M, Locatelli F. Management of graft-versus-host disease in pae-diatric bone marrow transplant recipients. Pediatr Drugs 2000; 2:29-55.
Zecca M, Prete A, Rondelli R, et al. Chronic graft-versus-host disease inchildren: incidence, risk factors, and impact on outcome. Blood2002;100 1192-1200.
Zilla Z, Boulad F, Black P., et al. Growth in children after bone marrowtransplantation for acute leukaemia. Blood 1995; 88: 819-24.
48
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre

49
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
Efficacia della terapia con ormone della cre-scita ricombinante (recGH) nei thalassemicimajor politrasfusi di bassa statura: valutazione degli effetti sull'altezza finale
L. Cavallo,1 V. De Sanctis,2 M. Cisternino,3 M. CarusoNicoletti,4 M. C. Galati,5 A. Acquafredda,1 C. Zecchino,1M. Delvecchio1
1U. O. di Pediatria Generale e Specialistica “B. Trambusti”,Dipartmento di Biomedicina dell'Età Evolutiva, Universitàdi Bari; 2U. O. di Pediatria dell'Adolescenza, A. O. Arcispe-dale Sant'Anna, Ferrara; 3U. O. di Pediatria, Università diPavia, Pavia; 4Istituto di Clinica Pediatrica, Università diCatania; 5U. O. di Ematoncologia Pediatrica, A. O. PuglieseCiaccio, Catanzaro
I thalassemici major politrasfusi (Th) mostrano un pat-tern di crescita caratterizzato da un deficit staturale chesi instaura all'età di 5-6 anni e si accentua all'epoca incui fisiologicamente si dovrebbe realizzare lo sviluppopuberale (ritardato o assente in questi pazienti). 1-3 Lacurva staturale presenta un tardivo recupero nella tardaadolescenza con il raggiungimento di un'altezza defini-tiva il cui deficit (espresso in SDS) corrisponde a quellopresente nella fase prepuberale. 1,3,4 Il deficit staturalesembra vere una genesi multifattoriale. 5 Benché il fre-quentemente rilevato deficit dell'asse GHRF-GH-IGFI nonsembra svolgere un ruolo decisivo nel determinare il defi-cit staturale6-8, tuttavia sono stati effettuati alcuni ten-tativi di migliorare la crescita con la somministrazionedi recGH. In una percentuale variabile ma significativa dipazienti la terapia effettuata per 1 anno ha determina-to un miglioramento della crescita, mentre risultatidiscordanti sono presenti in letteratura per trattamentipiù prolungati (2-3 anni). L'altezza finale (FH) cui per-vengono i Th trattati per lunghi periodi con recGH è sta-ta valutata in una piccola casistica di pazienti. 8
Materiali e MetodiIn questo studio abbiamo valutato la FH di 25 (18 M)
Th di bassa statura e con ridotta riserva di GH (picco altest con insulina ed al test con clonidina <10 ng/mL)trattati per 3. 3±1.2 (1.3-5. 2) anni con recGH (Saizen,Serono Laboratories; 0.033 mg/kg/die 6 sere/settimanasc). I dati dell'altezza sono stati espressi in SDS. All'ini-zio della terapia (start) tutti i pazienti erano clinica-mente prepuberi. La loro età cronologica (CA) ed ossea(BA) era, rispettivamente, di 13. 6±2.0 e 11.4±1.6 anni.Il 50% dei maschi e 5/7 femmine hanno presentato ini-zio di sviluppo puberale spontaneo durante il tratta-mento.
RisultatiA fine terapia (end): l'altezza calcolata per la CA
(HxCA) non presentava alcun incremento significativo,mentre l'altezza calcolata per la BA (HxBA) risultavasignificativamente diminuita (p=0.004). L'incrementodella BA (3. 29±1.65 anni) era significativamente mag-giore (p<0.001) dell'incremento dell'età staturale(2.16±0.98 anni). Sia la HxCA che la HxBA erano positi-vamente correlate con la BA rilevata allo start (rispetti-
vamente r=0.50 e 0.42, p=0.012 e 0.034). La FHxCA era significativamente superiore (p=0.001)
rispetto sia allo start che allo end, mentre la FHxBA dimi-nuiva significativamente (p<0.001) rispetto allo start. LaFH era positivamente correlata con la CA (r=0.63,p=0.001), la BA (r=0.68, p<0.001) e la HxBA (r=0.59,p=0.002) valutate allo start e con la HxCA e la HxBA(r=0.82 and 0.74, rispettivamente, p<0.001) valutate alloend. Una correlazione negativa fu rilevata tra la FH e ladurata del trattamento (r=-0.56, p=0.004).
DiscussioneI nostri dati sembrano escludere che la prolungata
terapia con recGH alla dose di 0.2 mg/kg/settimana pos-sa migliorare l'altezza finale in questi pazienti; al con-trario si può ipotizzare che la terapia possa determina-re un effetto negativo.
I nostri dati non escludono la possibilità che questipazienti possano beneficiare della terapia con recGH adosi più elevate, ma considerato che in precedenti studil'incremento della dose non ha modificato i risultati inmaniera significativa,7,9 che il rapporto costo/beneficio ditale incremento è elevato e che questi pazienti sono adalto rischio di iperinsulinemia ed alterato metabolismoglucidico anche in assenza di terapia con recGH non rite-niamo che vi siano le indicazioni per consigliare unaumento della dose di recGH. Tenuto conto che i nostripazienti all'inizio della terapia presentavano un'etàmedia relativamente avanzata (8. 5-14 anni), non pos-siamo escludere, infine, che l'inizio più precoce dellaterapia possa essere più efficace nel determinare unincremento significativo dell'altezza definitive. Tuttaviaalcuni dati ricavabili dal nostro studio (correlazioni posi-tive tra la età ossea allo start e sia l'altezza allo end chel'altezza finale), mostrando che l'efficacia è maggiorenei pazienti che presentano l'età ossea più avanazataallo start, rendono tale ipotesi improbabile.
ConclusioniIn base ai nostri dati suggeriamo che i Th di bassa sta-
tura dovrebbero ricevere terapia con recGH per 1 anno eche trattamenti più prolungati dovrebbero essere riserva-ti ad adolescenti selezionati che presentino problemi psi-cologici determinati dal deficit staturale per i quali l'ac-celerazione della crescita rappresenti lo scopo principaledella terapia, anche in assenza di un effetto significativosull'incremento dell'altezza definitiva.
Bibliografia
1. Cavallo L, De Mattia D, Giobbe T. Growth in homozygous β-tha-lassemia patients. In: Cavallo L, Job JC, New MI, eds. Growth Dis-orders: the State of the art. Serono Symposia No. 81.Rome: RavenPress; 1991.p. 47-54.
2. Garcia-Mayor RVG, Andrade Olivie A, Fernandez Catalina P, Cas-tro M, Rego Iraeta A, Reparaz A. Linear growth in thalassemic chil-dren treated with intensive chelation therapy. A longitudinal study.Horm Res 1993;40:189-93.
3. Rodda CP, Reid ED, Johnson S, Doery J, Matthews R, Bowden DKShort stature in homozygous β-thalassemia is due to dispropor-tionate truncal shortening. Clin Endocrinol 1995;42:587-92.
4. Perignon F, Brauner R, Souberbielle JC, de Montalembert M, GirotR. Croissance et fonction endocrine dans la thalassemie majeure.Arch Franç Pediatr 1993;50:657-63.
5. De Sanctis V, Vullo C, Bagni B. Talassemia, chelazione ed accresci-

mento staturale. In: Tatò L, Boghen M, Piemonte G, editors. LeIndicazioni Non Convenzionali dell'ormone biosintetico dellacrescita. Padova: San Marco Editrice; 1990.p. 78-86.
6. Cavallo L, Gurrado R, Gallo F, Zecchino C, De Mattia D, Tatò L.Growth deficiency in polytransfused β-thalassaemia patients isnot growth hormone dependent. Clin Endocrinol 1997;46:701-6.
7. Low LCK, Postel-Vinay MC, Kwan EYW, Cheung PT. Serum growthhormone (GH) binding protein, IGF-I and IGFBP-3 in patients withβ-thalassemia major and the effect of GH treatment. ClinEndocrinol 1998;48:641-6.
8. Theodoridis C, Ladis V, Papatheodorou A, Berdousi H, Palamidou F,Evagelopoulou C, et al. Growth and management of short staturein thalasseamia major. J Pediatr Endocrinol Metab 1998;11:835-44.
9. Low LCK, Kwan EYM, Lim YJ, Lee ACW, Tam CF, Lam KSL. Growthhormone treatment of short Chinese children with β-thalassemiamajor without GH deficiency. Clin Endocrinol 1995;42:359-63.
Quale futuro per l’adroterapia?
Ugo AmaldiUniversità di Milano Bicocca e Fondazione TERA
IntroduzioneGli acceleratori lineari di elettroni (linac) sono gli stru-
menti più usati dai radioterapisti: oggi circa 8000 macchi-ne acceleratrici di questo tipo sono funzionanti nel mon-do. La dose inevitabilmente somministrata ai tessuti sani èsempre il fattore limitante. Quindi meglio si riesce a'conformare' la dose al bersaglio, più la dose al tumore puòessere aumentata. I fasci di fotoni prodotti dai linac (disolito chiamati 'fasci di raggi X' dai medici) sono caratte-rizzati da un assorbimento esponenziale dopo un massimoche, per fasci di energia massima pari a 10 MeV, è rag-giunto a una profondità di circa 3 cm. La conformità delladose, difficile da ottenere per via dell'assorbimento espo-nenziale, è quindi la principale finalità di tutti i recenti svi-luppi della radioteleterapia del cancro. La IMRT (IntensityModulated Radio-Therapy) fa uso di 6-10 fasci di raggi Xincrociati.
Adroterapia è un termine generale che include molte tec-niche di radioterapia oncologica diverse tra loro, ma tuttebasata sull'uso di fasci di particelle non elementari velocicomposte di quarks: protoni e ioni carbonio sono gli adro-ni più usati per controllare localmente diversi tipi di tumo-re. La curva della dose in profondità di protoni e ioni leg-geri (come il carbonio) è completamente diversa da quelladei fotoni poiché queste particelle cariche rilasciano il mas-simo della densità di dose alla fine del proprio percorso,nel cosiddetto 'picco di Bragg'. Protoni e ioni leggeri, pri-vati dei loro elettroni, sono vantaggiosi nelle terapie IMHT(Intensity Modulated Hadron Therapy) per tre proprietà fisi-che. Innanzitutto, come detto, il massimo della densità dienergia depositata si ha alla fine del percorso di questeparticelle, nel picco di Bragg. In secondo luogo, questi adro-ni carichi penetrano il corpo dei pazienti praticamente sen-za diffusione. In terzo luogo queste particelle, essendo cari-che, possono essere canalizzate con dipoli e quadrupolimagnetici in modo da formare fasci sottili di penetrazionevariabile; muovendo il fascetto nelle due direzioni trasver-sali mediante due magneti è possibile irradiare qualunqueparte di un bersaglio tumorale in maniera rapida ed accu-rata (distribuzione attiva della dose). Fasci di adroni con-sentono quindi un trattamento molto conforme di tumorisituati in profondità con precisione millimetrica e con unadose minima ai tessuti traversati e limitrofi.
Per raggiungere tessuti molli a profondità superiori ai 25cm, com'è necessario per trattare tumori profondi, i fascidi protoni e di ioni di carbonio devono avere un'energiainiziale di non meno di 200 MeV e 4'800 MeV rispettiva-mente (cioè 400 MeV/nucleone).
I radioterapisti, che usano linac rotanti per trattarepazienti con fasci di raggi X, vorrebbero avere la stessa pos-sibilità usando fasci di protoni e ioni. La rigidità magneti-ca di protoni da 200 MeV è però tale che il campo magne-tico in grado di farlo ha un raggio tipico di 2-3 m, e unotre volte maggiore negli ioni carbonio. Per questo motivo
50
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre
Lettura

soltanto fasci di protoni fissi, soprattutto nel piano oriz-zontale, sono stati usati in tutto il mondo fino al 1992,quando il primo centro di trattamento adroterapico inambiente ospedaliero è divenuto operativo a Los Angelespresso il Loma Linda Medical Centre. Questo, e i nuovi cen-tri di protonterapia costruiti da allora, sono dotati in gene-re di grandi strutture meccaniche (10 metri di diametro)dette “testate rotanti” che fanno da supporto rigido ainecessari magneti di. Nel 1997 presso il PSI (Villigen-Swit-zerland) divenne operativa la prima struttura ruotante conun fascio di protoni da 250 MeV. Qui è stato introdotto unnuovo sistema 'attivo' per la distribuzione della dose: il ber-saglio è suddiviso in molte migliaia di unità elementari det-te voxels e ciascun elemento è irradiato in passi successi-vi da un fascio di protoni che ha una sezione di circa 5 mme un'energia tale di raggiungere la profondità desiderata.All'inizio del 2004 al PSI erano stati trattati con questatecnica circa duecento pazienti.
Per il melanoma dell'occhio, così come per il trattamen-to della degenerazione maculare, sono sufficienti fasci diprotoni orizzontali con energie nell'intervallo 60-70 MeV eil trattamento passivo è adeguato. Gli acceleratori usatisono per lo più ciclotroni relativamente piccoli. Il primocentro italiano di protonterapia è stato creato dall'INFNpresso i Laboratori Nazionali del Sud di Catania, ove fun-ziona da anni un ciclotrone superconduttore dal quale èestratto, per la terapia dell'occhio, un fascio di protoni da62 MeV. I trattamenti sono iniziati nel 2002.All'inizio del2004 erano stati trattati circa 80 pazienti.
Numero di pazientiA tutto oggi circa 35'000 pazienti sono stati sottoposti
a protonterapia e, per quanto riguarda i tumori profondi,ottimi risultati sono stati ottenuti nel trattamento deitumori del capo-collo, della spina dorsale e della prostata.
Riassumendo una lunga catena di argomenti scientificie clinici, la commissione adroterapia dell'Associazione Ita-liana di Adroterapia Oncologica (AIRO) è giunta nel 2003alla conclusione che la protonterapia dovrebbe assoluta-mente essere utilizzata al posto della radioterapia conven-zionale per l'1% circa dei pazienti. Poiché nei paesi occi-dentali sono irradiati con raggi X circa 20.000 pazienti/annoogni 10 milioni di abitanti, su questa popolazione la pro-tonterapia dovrebbe essere utilizzata in priorità quindi percirca 200 pazienti l'anno (circa mille in tutta Italia). Inol-tre, per circa il 12% dei trattamenti usuali, cioè per circa2400 pazienti l'anno su 10 milioni di abitanti, la protonte-rapia assicurerebbe un miglior controllo di molti tipi ditumore dei raggi X. Un maggior numero di dati clinici èperò necessario per quantificare con precisione i vantaggi.Ciò corrisponde a circa 14000 pazienti italiani l'anno.
Fasci di ioni carbonio di 4800 MeV penetrano in tessutobiologico fino a 27 cm di profondità, come i protoni da 200MeV, e sono indicati per il trattamento di quei tumoriprofondi che sono radioresistenti sia ai raggi X che ai pro-toni. Gli argomenti radiobiologici sono molti e complessi edè necessario rimandare alla letteratura specializzata. Qua-litativamente si può dire che ciascuno di questi ioni lasciain media, in ogni cellula attraversata, 24 volte più energiadi un protone dello stesso percorso. Questa energia produ-ce una 'colonna' di ionizzazioni che, in media, è 24 volte piùdensa e che, quando intercetta il DNA di una cellula, vi
causa rotture doppie (Double Strand Breaks) e danni mul-tipli (Multiple Damaged Sites). Per questo gli effetti sullacellula sono qualitativamente diversi da quelli prodotti dal-le radiazioni ionizzanti, che cedono energia in maniera piùdiffusa, come i raggi X e i protoni, che causano per lo piùrotture di una sola delle due eliche del DNA. A causa dellafrazione molto più grande di effetti 'diretti', gli ioni legge-ri hanno una efficacia radiobiologica (Radio BiologicalEffectiveness, RBE), che è fino a tre volte maggiore di quel-la dei raggi X e dei protoni. Gli ioni leggeri sono quindiadatti per quelle situazioni cliniche nelle quali la radiore-sistenza - dovuta a ipossia o ad altre ragioni - è un pro-blema difficile da superare sia con la terapia convenziona-le che con i protoni.
Per quanto riguarda il numero di pazienti che devonoessere trattati con ioni carbonio, va innanzitutto sottoli-neato che sino all'inizio del 2004 erano stati trattati circa2000 pazienti con ioni carbonio in tutto il mondo. In effet-ti, la maggior parte dei dati clinici disponibili nascono dal-l'attività, iniziata nel 1994 in Giappone, dell'HIMAC (HeavyIon Medical Accelerator Centre, Chiba) — dove circa 1800pazienti sono stati irradiati — e al laboratorio GSI di Darm-stadt.
Sono stati recentemente pubblicati dai medici di HIMAC(Giappone) lavori che riportano i risultati molto interes-santi ottenuti nel controllo di tumori del capo-collo, delpolmone e del fegato. Gli ottimi risultati di sopravvivenzaa tre anni (più del 70%) dei pazienti portatori di tumori pol-monari non microcitomi hanno fatto comprendere a moltile potenzialità di questa nuova radioterapia, che porta consé un ulteriore vantaggio. Proprio per il fatto che gli ionicarbonio hanno un'interazione con le cellule che è quali-tativamente diversa da quella dei raggi X e dei protoni, iricercatori di HIMAC hanno provato clinicamente quelloche la radiobiologia aveva già indicato: poiché la ripara-zione cellulare è praticamente assente non è utile suddivi-dere la dose in 30 sedute — come per i raggi X e i proto-ni — e, anzi, è possibile dare la dose necessaria a control-lare il tumore, che globalmente è minore, su soltanto 4-9sedute. L'accorciamento del trattamento costituisce unvantaggio psicologico e finanziario, ma la mancanza diriparazione cellulare può anche indurre recidive nei tessu-ti sani e richiede grande cura nel modellare l'efficacia bio-logica relativa di un campo di irradiamento complesso.
Sulla base dei più recenti risultati ottenuti sui 1800pazienti irradiati allo HIMAC di Chiba e al GSI, l'AIRO ègiunta alla conclusione che circa il 3% dei malati trattaticon raggi X si avvantaggerebbero, sia in termini di soprav-vivenza che di qualità di vita, se potessero essere irradiaticon fasci di ioni carbonio. Ciò corrisponde a 600 malatil'anno per 10 milioni di abitanti e, in Italia, a un totale dicirca 3500 pazienti l'anno. L'AIRO ritiene quindi necessa-ria la costruzione in Italia di un Centro nazionale per il trat-tamento con ioni carbonio.
Centri per la protonterapia profondaLa maggior parte dei centri di protonterapia profonda
esistenti al mondo fanno uso di fasci di particelle prodottida acceleratori di particelle usati per la ricerca di base (sianucleare sia in particelle elementari) che sono stati suc-cessivamente adattati alla protonterapia. La distribuzionedella dose è di tipo 'passivo', in quanto il fascio è disperso
51
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

52
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre
angolarmente facendogli attraversare un opportuno diffu-sore-assorbitore e, a valle, sagomato trasversalmente concollimatori. Questi centri sono operativi da molto tempo inEurolandia, Giappone, Russia ed USA. I 'centri ospedalieri'in funzione si trovano in California (Loma Linda e Northea-st Proton Therapy Center di Boston costruito dalla dittabelga IBA) e in Giappone: Kashiwa, Hyogo, Tsukuba eWasaka Bay.
A partire dal 2004-2005 in USA vi saranno cinque cen-tri 'ospedalieri' dedicati al trattamento di tumori profondisaranno operativi negli Stati Uniti. Due ospedali privaticinesi hanno ordinato all'IBA, per la fine del 2004, ciclotronie strutture rotanti. L'IBA ha ottenuto l'ordine di un centroin Corea.
In Europa, alla fine del 2000 è stato lanciato un nuovoprogetto, detto PROSCAN', dal Paul Scherrer Institute. Perla realizzazione di questo centro è stato ordinato alla dit-ta ACCEL un protonciclotrone superconduttore. La dittaACCEL ha poi venduto un secondo ciclotrone supercondut-tore a Prohealth AG vicino Monaco. Il Rinecker Proton The-rapy Centre (RPTC) avrà una sala di trattamento multiusoe quattro testate rotanti.
In conclusione, la protonterapia è in pieno sviluppo tan-to che quattro ditte offrono sistemi chiavi in mano per costidell'ordine di 50-60 milioni di euro per un centro con duetestate isocentriche. In Italia l'AIRO è giunta alla conclu-sione che l'Italia ha bisogno di 4-5 centri ospedalieri diprotonterapia e la Provincia di Trento ha già finanziato ilprimo.
Il futuro: il Cyclinac, combinazione di un ciclotrone ed unLinac
Nel 1993 la Fondazione TERA ha iniziato - in collabora-zione con l'ENEA (Frascati), e l'Istituto Superiore di Sanità(Roma) - lo studio di un nuovo acceleratore di adroni, il“cyclinac”, che è ora il principale programma di ricerca esviluppo di TERA (Figura 1).
Un cyclinac è la combinazione di un ciclotrone ad altacorrente e bassa energia con un linac ad alto gradiente diaccelerazione. Per realizzare un cyclinac per protoni, TERA
ha progettato e costruito, in collaborazione con il CERN el'INFN, un modulo lungo 1,25 metri di un acceleratorelineare a 3 GHZ battezzato “LIBO”, cioè LInac Booster. Nel2003 il modulo ha accelerato protoni presso i LaboratoriNazionali del Sud dell'INFN, che sono localizzati a Catania.
Con un grant della Fondazione Monzino (Milano), dall'i-nizio del 2004 TERA sta costruendo il LIBO 30-40, cioè ilprimo modulo definitivo di un cyclinac che porterà i pro-toni a 210 MeV.
Centri stranieri con fasci di ioni carbonioA Tokyo funziona dal 1992 il centro HIMAC, che ha aper-
to la strada all'utilizzo degli ioni carbonio nella terapia deitumori, particolarmente di quelli radioresistenti, con fascisia orizzontali che verticali,. Dal 1997 presso il laboratorio
Figura 1
Figura 2

GSI (Darmstadt) il prof. G. Kraft, il Dr. J. Debus e i loro col-laboratori trattano pazienti con un fascio orizzontale diioni di carbonio e un sistema attivo tridimensionale didistribuzione della dose. Presso il Centro HIBMC di Hyogoil primo paziente è stato trattato con protoni nel maggio2001.Questo centro ha tre sale per protoni (due di questecon testate rotanti) e due sale per ioni dotate di fasci ver-ticali e fasci inclinati.
In entrambi i centri giapponesi non si sono quindicostruite testate rotanti. Diversa è la scelta fatta per il nuo-vo centro che è in costruzione da parte del GSI presso la Cli-nica Oncologica dell'Università di Heidelberg mostrato nel-la figura 2 (Figura 2).
La costruzione è iniziata nel 2001 e si prevede di irradiareil primo paziente alla fine del 2006. Il costo previsto è di 75milioni di Euro, metà dei quali sono stati dati dal governofederale; il resto è coperto da un prestito bancario che sifonda su un rimborso, da parte delle assicurazioni e del ser-vizio sanitario nazionale, di circa 1000 Euro per seduta.
Il Proton Ion Medical Machine StudyAlla fine del 1995 l'autore di questo contributo - con
l'appoggio dei promotori del progetto Med-AUSTRON -convinse la Direzione del CERN dell'opportunità di proget-tare, a livello europeo, un sincrotrone ottimizzato per laterapia con ioni carbonio. PIMMS (= Proton and Ion Medi-cal Machine Study) è il nome della collaborazione che allo-ra si formò tra il CERN, Med-AUSTRON (Austria), Oncology2000 (Repubblica Ceca) e TERA (Italia). TERA, Med-AUSTRON e Oncology 2000 hanno investito 25, 10 e 3uomo ◊ anno rispettivamente. IL CERN ha partecipato conmolti esperti e disegnatori; alcuni esperti del GSI hannocontribuito con la loro competenza a molte delle riunionidel Project Advisor Committee che si sono tenute negli anniche vanno dal 1996 al 2000.
Nell'adroterapia profonda con distribuzione attiva delladose è necessario che il fascio di adroni sia uniforme neltempo; ciò è automatico nei ciclotroni ma non nei sincro-troni, i cui fasci estratti, soprattutto per le inevitabili insta-bilità delle correnti dei magneti di macchina, hanno unastruttura temporale caratterizzata dalla presenza di picchiirregolari di corrente su tempi inferiori al millisecondo Que-sta struttura temporale rende difficile la misura accuratadella dose durante la distribuzione attiva dei fasci clinici. Ilsincrotrone del PIMMS è stato quindi disegnato partendo
dal fascio clinico e procedendo andando 'indietro'. In par-ticolare, per ottenere un fascio uniforme l'estrazione èbasata su un'ottica magnetica speciale.
L'idea di partenza del PIMMS è che i gruppi europei inte-ressati alla costruzione di un proprio centro di terapia conioni possono adattare le proposte fatte nello studio alleloro esigenze e ai loro fondi. Per questo al gruppo di studionon furono posti limiti di costo e dimensioni.
Il Centro Nazionale ed il progetto PIMMS/TERAScopo principale della Fondazione TERA, creata nel 1992
e riconosciuta dal Ministero della Salute nel 1994, è la rea-lizzazione del Centro Nazionale di Adroterapia Oncologica(CNAO), una struttura equipaggiata con fasci di protoni edi ioni carbonio per il trattamento medico dei tumori, inparticolare dei tumori radioresistenti, e per la ricerca clini-ca. Una prima proposta fu presentata alle autorità italianenel 1995 e una seconda nel 1997. Negli anni 1998-2000 siè poi lavorato a un nuovo disegno basato su un sincrotro-ne di tipo PIMMS. Per ridurre i costi si sono progettate (nel-la fase 1) solo tre sale di trattamento, uno stesso linac iniet-tore è stato utilizzato tanto per i protoni che per gli ioni, lesorgenti di ioni e l'iniettore sono stati sistemati all'internodel sincrotrone, ed è stata scelta una configurazione mol-to compatta delle linee di fascio di modo che la superficiecoperta del bunker sotterraneo è stata ridotta a 3500m2.Come iniettore è stato adottato il disegno del GSI.
Dal 2001, responsabile della costruzione e della futurautilizzazione del Centro (Figura 3) è la Fondazione CNAO(Centro Nazionale di Adroterapia Oncologica) creata con lalegge finanziaria del 2001 e finanziata dal Ministero dellaSalute - sugli anni 2001-2005 - con 35,3 milioni di Euro.Altri finanziamenti sono stati dati da Fondazioni private ealtri enti, di modo che nel 2003 erano coperti il 60% deicosti totali, che ammontano a circa 70 milioni di Euro.
Nel luglio 2002 le Fondazioni CNAO e TERA firmaronouna convenzione sulla base della quale TERA ha prodottoil progetto definitivo del Centro, cioè 2900 pagine di dise-gni tecnici e delle specifiche dei circa duecento compo-nenti diversi del sincrotrone da 400 MeV/u, delle linee ditrasporto e dei quattro sistemi di distribuzione della doseche serviranno le tre sale di trattamento. Alla progettazio-ne hanno anche contribuito l'INFN, e in particolare i Labo-ratori Nazionali di Frascati, e il CERN di Ginevra.
Nel 2004 la ditta Calvi-Tekne, scelta dalla CNAO, ha com-
53
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
Figura 3 Figura 4

pletato il progetto degli edifici. La facciata del CNAO èmostrata in Figura 4.
Alla fine del 2003 TERA ha passato alla CNAO tutto il per-sonale che ha lavorato al Centro Nazionale per tutti que-sti anni: 15 persone con contratti a tempo pieno e 9 con-sulenti con contratti a tempo parziale.
Il progetto PIMMS/TERA è alla base anche di altri progettieuropei. Nel 1998 l'Università Claude Bernard di Lionerichiese a TERA un disegno preliminare di un centro dota-to di ioni di carbonio e nella primavera del 2001 la propo-sta descritta nel documento sottoposto al Governo erabasata precisamente sul disegno PIMMS/TERA. Nel 1999scienziati dell'Istituto e dell'Ospedale Karolinska di Stoc-colma e di TERA decisero di preparare insieme una propo-sta, basata su PIMMS/TERA, per un centro con ioni legge-ri da costruire molto vicino al dipartimento di radioterapiadell'Ospedale Karolinska.
Infine, nel 2003, dopo aver confrontato criticamente ilsincrotrone del GSI con il progetto PIMMS/TERA, il comi-tato tecnico di Med-AUSTRON ha scelto questo secondodisegno come cuore del progetto austriaco, che ha comescopo la realizzazione di un centro per protoni e ioni car-bonio a Wiener Neustadt nel sud dell'Austria. Nel 2004Med-AUSTRON ha firmato un accordo di collaborazionecon la Fondazione CNAO.
Dalla metà del 2002 i cinque progetti europei (Heidel-berg, Pavia, Wiener Neustadt, Lione e Stoccolma) fannoparte di un network europeo detto ENLIGHT (EuropeanNetwork fot Light Ion Therapy) insieme all'ESTRO (la Societàeuropea di radioterapia e oncologia), all'EORTC (l'organiz-zazione europea per la ricerca clinica sul cancro), al CERNe al GSI.
Il futuro: un Cyclinac per ioniPer accelerare ioni carbonio a energia di 4500-4800 MeV
sono oggi necessari o sincrotroni oppure ciclotroni super-conduttori. Entrambe sono macchine complicate e costo-se. La Fondazione TERA, datasi come compito lo sviluppo dinuovi acceleratori in grado di ridurre dimensioni e investi-menti dei centri di adroterapia, ha preso un brevetto di unanuova struttura lineare accelerante di tipo CH che produ-ce un gradiente 2-3 volte maggiore dalla struttura del LIBO.Ciò permetterà la realizzazione di un cyclinac per ioni car-bonio che sarà lungo 25-30 metri, cioè all'incirca il diame-tro del sincrotrone PIMMS/TERA.
Con un finanziamento della Fondazione Monzino (Mila-no), nel 2004 è stato costruito il prototipo della strutturaCLUSTER, che è mostrato nella figura 5.
54
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre
Figura 5

haematologica vol. 89[supplement n. 10]:October 2004
Il ruolo della chirurgia nell'osteosarcomapediatrico
M. Mercuri, M. Manfrini°°Istituto Ortopedico Rizzoli, Bologna
Il trattamento dell'osteosarcoma ha subito una straor-dinaria trasformazione a partire dalla settima decade delsecolo appena passato. La rarità della malattia, la cui inci-denza resta stabile peraltro nelle varie aree geografiche,e la sua peculiarità nel colpire prevalentemente gli arti disoggetti in età di accrescimento, hanno determinato uncrescente interesse, incrementato dall'importante miglio-ramento prognostico ottenuto quando all'approccio chi-rurgico si è associato il trattamento chemioterapico. Ini-zialmente la chemioterapia consisteva in un unico far-maco somministrato ciclicamente dopo l'amputazione.Attualmente, eseguita la diagnosi, il paziente viene sot-toposto ad alcuni cicli di polichemioterapia dopo i qualiviene rivalutata l'estensione loco-regionale del tumore eviene affrontata la rimozione chirurgica del segmentoscheletrico e la sua ricostruzione. Il significato oncologi-co della chirurgia si è quindi modificato: dall'unica tera-pia possibile ad un tassello, seppur ancora indispensabile,nel trattamento polichemioterapico che dopo la chirurgiaprosegue, con modalità basate sempre più frequente-mente sulla analisi istologica del segmento asportato. L'in-cremento delle possibilità chirurgiche conservative ed ilsensibile aumento della popolazione lungo sopravviventeha inoltre focalizzato l'attenzione del chirurgo verso l'a-nalisi delle varie tecniche chirurgiche ed in particolare deirisultati funzionali, di affidabilità e di durata.
MaterialiDal 1976 al 2000 furono trattati presso l'Istituto Orto-
pedico Rizzoli, 407 pazienti di età inferiore a 15 anni,affetti da osteosarcoma scheletrico, ad alto grado, loca-lizzato. Nello stesso periodo altri 80 soggetti della stessaetà presentarono la stessa diagnosi associata a metasta-si, includendo in questa definizione sia le localizzazionilocoregionali (skip metastasi e linfonodi), sia le metasta-si a distanza (polmoni, ossa).
Tutti i pazienti furono sottoposti a trattamento che-mioterapico in relazione con i protocolli adottati nel perio-do di osservazione.
In 394 pazienti con malattia localizzata fu eseguito unintervento chirurgico con l'obiettivo di asportare in bloc-co la lesione tumorale; si trattò di un intervento demoli-tivo in 131 casi (33%), mentre la conservazione dell'artofu realizzata in 263 casi (67%). In 13 casi l'intervento pro-posto fu invece rifiutato. Nei casi metastatici, 46 sogget-ti ebbero un trattamento chirurgico tendente a rimuove-re tutte le lesioni presenti all'esordio.
Nel corso dello stesso periodo l'approccio chirurgico fupiù volte modificato. Negli anni 1976-80 le amputazionirappresentarono il 90% dei trattamenti chirurgici. Talepercentuale si ridusse al 58% nel quinquennio 1981-85,quindi al 25% nel periodo 1986-90, per assestarsi al 12%
in entrambi i lustri dell'ultima decade. Negli ultimi 15anni, tra le amputazioni è stato incluso anche l'interven-to di giroplastica che rappresenta un'innovazione, conparticolari vantaggi funzionali, rispetto al concetto tradi-zionale di amputazione.
RisultatiLa percentuale di recidiva locale nei casi localizzati è
stata complessivamente del 5% (21/394) : tra le amputa-zioni classiche il rischio si è verificato nel 4% dei casi(4/100), nelle giroplastiche nel 3% (1/31) mentre negliinterventi conservativi ha raggiunto il 6% (16/263).
Ad un follow-up medio di 120 mesi (40-280), l'analisidei risultati oncologici tra i casi trattati chirurgicamenteè così schematizzabile: nei casi localizzati, il 52% non hapiù presentato segni di malattia ed il 9% è in remissionea distanza di tempo dopo il trattamento chirurgico di unao più ricadute; nei 46 casi metastatici sottoposti a trat-tamento chirurgico, 10 pazienti (22%) sono restati con-tinuativamente liberi da malattia, mentre altri 3 (7%) sipresentano in remissione dopo il trattamento chirurgicodi una ricaduta.
Le amputazioni secondarie ad interventi ricostruttivi persuccessive complicanze sono passate dal 14% dei primianni, al 10% dei periodi successivi e attualmente sonoattestate attorno al 6%.
Ma oltre a questi risultati l'innovazione principale é rap-presentata dalla possibilità di eseguire interventi rico-struttivi che permettono di minimizzare i tempi di ricu-pero, riducendo i deficit funzionali. Nella ricostruzionedegli arti è stata superata la fase in cui l'unica alternati-va chirurgica conservativa era l'artrodesi e possiamo oraeseguire interventi ricostruttivi con protesi, anche "allun-gabili", o con innesti osteoarticolari opportunamentesagomati o con protesi "composite". I vantaggi consen-titi dalla diagnostica radiologica sofisticata permettonoinoltre di ridurre il margine oncologico dal tessuto neo-plastico con resezioni guidate che consentono di salvarele strutture articolari e migliorare il risultato funzionale,anche a lungo termine.
55XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
Osteosarcoma

56
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre
Ruolo della chemioterapia: forme localizzate
Gaetano BacciIstituti Ortopedici Rizzoli, Bologna
Terapia associata dell'osteosarcoma non-mentastaticodelle estremita': l'esperienza dell'Italian Sarcoma Groupnegli ultimi 25 anni
Fra il Marzo 1972 ed il Marzo 2.000 sono stati trattaticon chemioterapia adjuvante o neoadjuvante 1148 casi diosteosarcoma delle estremità*. La chemioterapia è stataattuata secondo 4 diversi protocolli adjuvanti (248 pazien-ti trattati fra il Marzo del 1972 ed il Febbraio 1983) e 6diversi protocolli neoadjuvanti (900 pazienti trattati fra ilMarzo 1983 ed il Marzo 2000).
La chirurgia fu limb salvage nel 71% dei casi, amputa-zione nel 26% e giroplastica nel 3% Nonostante i margi-ni chirurgici siano risultati inadeguati in 106 pazienti (9%)vi furono solo 61 recidive locali (5%). Queste, tranne duecasi, si associarono sempre ad una diffusione sistemicadella malattia. L'incidenza di recidive locali risultò invecesignificativamente correlata con i margini chirurgici (3.6% per margini adeguati vs 24% per margini inadeguati,p < 0.0001) e, nei casi trattati con chemioterapia neoadju-vante al grado di necrosi chemio-indotta (3. 9% per ipazienti good responders vs 8. 4% per i pazienti poorrespondersi).
Ad un follow-up medio di 15 anni (5-28), 635 pazientirimasero continuamente liberi da malattia, 488 ebberometastasi e/o recidiva locale, 18 morirono per tossicità e7 per ragioni non correlabili né al tumore né al tratta-mento (2 suicidi, 2 embolie polmonari, 1 complicanza delCVC, 1 incidente stradale, 1 secondo tumore).
La sopravvivenza libera da malatia a 5 anni (5SLM) fudel 57% e all'analisi univariata risultò significativamentecorrelato con il sesso (59% per le femmine vs 54% per imaschi, p < 0.007), l'istotipo (82% per gli osteosarcomifibroblastici vs 52% per le altre varianti, p < 0.0001), ilivelli sierici delle fosfatasi alcaline (67% per i pazienticon valori normali vs il 41% per i pazienti con valori ele-vati, p < 0.0001). Per quanto riguarda le variabili connes-se con il trattamento la sopravvivenza libera da eventi a5 anni risultò correlata al tipo di chirurgia (61% per ipazienti trattati con limb salvage vs il 47 per i pazientitrattati con amputazione, p < 0.0001), il tipo di chemio-terapia (61% per i casi trattati con neodjuvante vs 43%per i pazienti trattati con chemioterapia neoadjuvante, p< 0.0001) e la risposta istologica alla chemioterapia (67%per i good responders vs 48% per i poor responders, p <0.0001). All'analisi multivariata solo i valori sierici dellefosfatasi alcaline, il tipo di chemioterapia e la rispostaistologica al trattamento chemioterapico mantennero illoro indipendente significato prognostico.
Con l'eccezione del primo (SLE = 49%) nei restanti 5protocolli neoadjuvanti la sopravvivenza libera da malat-tia a 5 anni risultò essenzialmente sovrapponibile (65%,59%, 62%, 65%). 18 pazienti morirono per tossicità lega-ta alla chemioterapia (10 per cardiopatia da ADM, 6 persepsi durante leucopenia, uno per insufficienza renale daritardata eliminazione di MTX, uno per malattia veno-occlusiva epatica dopo MTX).
Oltre ai casi deceduti, si ebbero altri 9 severe cardio-tossicità da ADM, una delle quali comparsa a 12 anni dal-la fine del trattamento. Di questi pazienti 5 morirono peril tumore e 4 sono in vita, liberi da malattia ed in buonecondizioni. Tre di questi pazienti ebbero un trapianto car-diaco. In 11 pazienti si ebbe un secondo tumore diagno-sticato a 2-11 anni dall'inizio del trattamento. Tali tumo-ri furono 4 leucemie linfoblastiche acute, 4 carcinomi del-la mammella, 2 tumori polmonari. Un'ultima pazienteebbe un carcinoma della mammella dopo 5 anni trattatocon successo con mastectomia e radioterapia. Dopo altri3 anni ebbe un ca. ovarico che la condusse a morte.
Per quanto riguarda la chirurgia ci furono 298 compli-canze maggiori che interessarono 278 pazienti (24%). 20pazienti ebbero più di una complicanza e la complicanzapiù tardiva fu osservata a 6 anni dall'intervento. 135pazienti (12%) dovettero subire una seconda operazioneche in 11 casi fu un'amputazione in casi originariamentetrattati con limb salvage. I risultati funzionali nei pazien-ti trattati con limb salvage furono eccellenti nel 7% deicasi, buoni nel 66% e scarsi nel 27% . Per i pazienti trat-tati con amputazione , buoni nel 33% dei casi e scarsi nel67%.
Limiti degli studi multicentrici nell'osteosarcoma Se non vi sono dubbi che i trials multicentrici siano mol-
to importanti in tumori a larga diffusione, più discutibileè il loro impiego in tumori molto rari come l'osteosarco-ma, specialmente se nella terapia di tali tumori è previstoil coinvolgimento di specialisti di branche diverse. La que-stione che mi sono posto ed alla quale ho cercato dirispondere con una revisione della recente letteratura è laseguente : “I pazienti con un'osteosarcoma delle estre-mità trattati in una singola istituzione o trattati in trialsmulticentrici hanno le stesse probabilità di guarigione edi conservazione dei loro arti ?”.
Questa domanda sorge perché mentre i protocolli monoistituzionali sono condotti in centri che, soprattutto perquanto riguarda la chirurgia, hanno una larga esperienzanel trattamento delle neoplasie muscolo-scheletriche, neitrials multicentrici i pazienti vengono spesso trattati incentri che solo molto sporadicamente hanno occasione divedere tali neoplasie. Ad esempio nei 3 primi studi coo-perativi tedeschi della COSS vennero trattati, in 10 anni,412 pazienti, ossia 41 casi per anno. I centri che parteci-parono a questi trials furono 54, il ché significa che inmedia ogni centro trattò meno di un paziente per anno.Questo sul piano teorico. Sul piano reale il numero fu assaiminore. Infatti due dei centri aderenti agli studi COSS,Vienna e Munster, trattarono, rispettivamente 120 e 110pazienti. Ne deriva che le restanti 52 istituzioni si prese-ro cura in 10 anni di solo 182 pazienti, ossia, in media, 1paziente ogni 3 anni!
Questi numeri non sono molto diversi in altri studi mul-
(*) per quanto riguarda la chemioterapia, i pazienti sonostati trattati al Rizzoli, alla Clinica Pediatrica dell'Univer-sità di Torino, al “Meyer “ di Firenze, all'Oncologia Medicadi Ravenna e Torino. Per quanto riguarda la chirurgia alRizzoli ed al CTO di Firenze.

57
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
ticentrici relativi al trattamento neoadjuvante dell'osteo-sarcoma. I pazienti trattati in media ogni anno da un sin-golo centro sono stati 1 ogni 2 anni nel trial del PediatricOncology Group e nel 1° trial dello Scandinavian Sarco-ma Group; ed 1 paziente ogni anno nei due studi dell'Eu-ropean Osteosarcoma Group , nel secondo studio SSG enello studio del Cancer Children Group. Occorre sottoli-neare che questi numeri i riferiscono a medie, ma in cia-scuno di questi trials la distribuzione dei casi trattati daivari centri non è stata bilanciata. Ad esempio, nel primostudio dello SSG, Oslo ed Helsinki hanno trattato in 8 anni64 pazienti, mentre nellstesso periodo Bergen e Trondheimhanno trattato 1 solo caso! Da quanto detto scaturisceuna prima considerazione: nei trial multicentrici relativiall'osteosarcoma oltre il 50% dei pazienti viene curato inistituzioni con scarsa esperienza in questo tumore.
La seconda domanda che mi sono posto è stata quindila seguente: “Un oncologo pediatra ed un chirurgo orto-pedico che trattano un'osteosarcoma ogni 4-5 anni han-no realmente una competenza tale da garantire al pazien-te la migliore terapia?”
Per tentare di rispondere a questo quesito ho fatto unasorta di meta-analisi relativa ai pazienti trattati con che-mioterapia neoadjuvante fra il 1978 ed il 1989 in studimulticentrici ed in studi monoistituzionali. Sono stati cosìconfrontati i risultati ottenuti in 1548 pazienti trattati in9 diversi studi multicentrici (COSS-80, COSS-82, COSS-86,EOI-1, EOI-2, SSG-1, SSG-2, CCG-782, POG -8651) con irisultati ottenuti in 1020 pazienti trattati in 12 studimono-istituzionali condotti in 5 diversi ospedali (Rizzoli,Anderson Hospital, Sloan Kettering, G. Roussy, DebréHospital). Negli studi mono-istituzionali ed in quelli mul-tocentrici le percentuale di amputazioni eseguite e disopravvivenza libera da malattia (SLM) anni è stata rispet-tivamente del 27% vs il 45% (p < 0.0001)e del 63% vs il52% (p < 0.0001). In linea teorica queste differenze pote-vano essere dovute alla diversa efficacia dei protocollichemioterapici usati nei due tipi di studio. Per verificarequesta possibilità ho confrontato i risultati ottenuti neidiversi centri aderenti allo stesso studio multi-istiuziona-li. Tale confronto mi è stato possibile solo per gli studiCOSS in quanto i coordinatori dello studio e i chirurghiortopedici di due Istituzioni hanno pubblicato i loro datisu riviste rispettivamente di Oncologia e di Ortopedia. Nei3 studi COSS considerati, relativi a 412 pazienti, la per-centuale di amputazioni è stata del 50% a Vienna ed aMunster e dell'82% nelle restanti 52 Istituzioni (P <0.0001). E' difficile ipotizzare che tutti i casi “più impe-gnativi” siano capitati nelle istituzioni più piccole. Analo-gamente la sopravivenza libera da malattia a 5 anni è sta-ta del 69% a Munster e Vienna e del 44% nei restanti 52centri (p < 0.0001). Un altro dato è abbastanza preoccu-pante, ossia l'incidenza delle recidive locali. Questa fudell'11% nei pazienti operati presso le 52 istituzioni conmeno pratica nei tumori muscolo-scheletrici e di solo il2% nei pazienti operati a Vienna e a Munster. Riassu-mendo, se i pazienti curati nelle 52 istituzioni minori fos-sero stati trattati a Vienna o Munster, le loro probabilitàdi conservare l'arto e di guarire dal tumore sarebbero sta-te rispettivamente superiori del 30% e del 15% .
Il protocollo T-10 di Rosen è stata una pietra miliare nelmigliorare la prognosi dell'osteosarcoma. Tale protocollo
fu quindi adottato, oltre che dallo Sloan kettering , anchein diversi altri studi sia monoistituzionali (Gustavo Rous-sy, Debré Hospital) che multiistituzionali (EOI-2, SSG-2).Confrontando questi due tipi di studio, relativi a pazientitrattati con lo stesso protocollo chemioterapico (T-10 diRosen) la percentuale di amputazioni fu del 30% nei 3studi monoistituzionali e del 66% nei due studi multi-centrici (p < 0.0001). Analogamente, nei due gruppi dipazienti la sopravvivenza libera da malattia nei 2 gruppifu rispettivamente del 59% vs il 47% (p < 0.02).
Concludendo, questi dati sembrano indicare che nell'o-steosarcoma delle estremità i pazienti trattati presso isti-tuzioni che hanno esperienza nella terapia dei tumorimuscoloscheletrici hanno una prognosi quod valitudineme quod vitam nettamente migliore rispetto a quelli trat-tati, nell'ambito di studi multicentrici, in centri con pocaesperienza in tal senso. Questo dovrebbe indurre a far siche i futuri protocolli anche se multi-istituzionali si rivol-gessero solo a poche istituzioni specificamente preparatein questo tumore. Questo è quanto fatto nell'ultimo stu-dio dell'Italian Sarcoma Group in cui la chirurgia è attua-ta in 3 centri e la chemioterapia in 6. Purtroppo questonon è quanto si sta facendo in altri studi sull'osteosarco-ma. I centri che hanno cooperato all'ultimo studio COSSsono stati infatti 133 e più di 250, di 3 diversi continen-ti, quelli in cui verranno trattati i pazienti dell' EURAMOS-, attivato recentemente.
Se non vi è dubbio che per la soluzione scientifica diproblemi inerenti il trattamento dei tumori i numeri, e diconseguenza i trials multicentrici, sono molto importantiritengo che ancora più importante sia il salvaguardare ildiritto del paziente di ricevere il miglior trattamento pos-sibile.

58
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre
Ruolo della chemioterapia ed approcciterapeutici innovativi: forme metastatiche
Franca Fagioli*, A. Palmero*, S. Ferrari°*Dipartimento di Scienze Pediatriche e dell'Adolescenza,Università di Torino; °Istituti Ortopedici Rizzoli, Bologna
La prognosi dei pazienti affetti da osteosarcoma ad altogrado di malignità è notevolmente migliorata negli ultimi25 anni, con un aumento della sopravvivenza globale dal15% al 70%. Questo miglioramento è da attribuire, oltre alperfezionamento delle tecniche chirurgiche, a: 1) l'effettodella chemioterapia preoperatoria;1-4 2) l'introduzione diuna chemioterapia aggressiva con combinazioni differentidi alte dosi di Methotrexate (MTX), Adriamicina (ADM),Cisplatino (CDDP) ed Ifosfamide (IFO);5-8 3) l'identificazio-ne di una correlazione dose-risposta tra MTX, ADM e CDDPe cellule di osteosarcoma;9-10 4) l'identificazione della rispo-sta istologica alla chemioterapia preoperatoria come il prin-cipale fattore prognostico in base al quale modulare la che-mioterapia postoperatoria. 11-12
La prognosi dei pazienti con osteosarcoma in recidiva èperò tuttora molto severa, con una sopravvivenza globaledel 0-50% dopo metastasectomia e chemioterapia diseconda linea. 13-17 Solo il 5% dei pazienti ha una recidivalocale, mentre la maggior parte dei casi ricade per meta-stasi polmonari e molto raramente per metastasi ossee. 18
Come dimostrato da Ferrari et al. con un'analisi retrospet-tiva su 162 pazienti in recidiva, i fattori prognostici piùimportanti sono: 1) la sede delle metastasi (sopravvivenzaa 5 anni del 44% in caso di metastasi polmonari e del 19%per le altre sedi); 2) l'intervallo libero da malattia (soprav-vivenza a 5 anni del 8% in pazienti con intervallo libero damalattia inferiore all'anno, 23% se di 2 anni, 50% se di 3anni e 74% per un intervallo maggiore); 3) l'asportazionecompleta delle metastasi (sopravvivenza a 5 anni del 44%);4) il numero delle metastasi, con una prognosi significati-vamente maggiore per i pazienti con una o due metastasipolmonari. È pertanto possibile dividere i pazienti in grup-pi di rischio a seconda di questi fattori: prognosi moltobuona (sopravvivenza del 72% a 5 anni) per i pazienti conintervallo libero da malattia maggiore di 24 mesi e 1 o 2metastasi polmonari; prognosi intermedia per i pazienti con1 o 2 metastasi polmonari ma intervallo breve (41% a 5anni) o intervallo lungo ma 3 o più noduli (33% a 5 anni);prognosi severa (5% a 5 anni) per i pazienti con intervallobreve e 3 o più metastasi.19
Poiché la rarità della malattia e l'eterogeneità della dif-fusione metastatica rendono uno studio randomizzato dif-ficilmente realizzabile, sono presenti in letteratura solostudi clinici non controllati; numerosissime problematicherestano pertanto aperte. Un certo numero di regimi dichemioterapia con diverse combinazioni di farmaci sonostati utilizzati, con risultati difficili da analizzare (Tabella1). Nuove strategie terapeutiche si sono pertanto rivelatenecessarie. La più esplorata è stata quella dell'intensifica-zione delle dosi di chemioterapia con supporto di cellulestaminali autologhe (Tabella 2), che ha permesso di ridur-re il tempo di engraftment e la tossicità del ciclo tera-peutico, permettendo così di incrementare le dosi dei far-maci. Nell'esperienza del European Bone Marrow Tran-
splantation registry (EBMT), 149 pazienti pediatrici sonostati sottoposti a megaterapia: la sopravvivenza a 5 anniè stata del 33% per i pazienti trattati con alte dosi duran-te il primo trattamento contro il 14% per quelli trattatidopo la recidiva (p = 0.197, non significativa); con un sin-golo ciclo è stata del 26% contro il 18% con più cicli ripe-tuti (p = 0.842, non significativa).
Il protocollo di fase II (ISG-SSG-I) delineato dall'Italiane Scandinavian Sarcoma Group, consistente in due cicli dialte dosi di chemioterapia con carboplatino (CBDCA)(1500mg/m2) ed Etoposide (1800 mg/m2) (VP16) seguiti dareinfusione di cellule staminali periferiche autologhe, èstato recentemente terminato. 20 Il CBDCA e il VP16 era-no stati scelti per la loro scarsa tossicità, per le evidenzedi attività del VP16 nei sarcomi dell'osso e dei tessuti mol-li e per la dimostrata attività del CBDCA nell'osteosarco-ma. Con questo schema terapeutico, associato a chirurgia,25 pazienti in recidiva su 32 hanno ottenuto una remis-sione completa, associata, però, ad una elevata percen-tuale di recidiva-progressione (84,4% ad una mediana di8 mesi dall'ingresso in studio). La sopravvivenza globale ela sopravvivenza libera da malattia a 3 anni sono state
Tabella 1.Protocolli chemioterapici utilizzati nei pazienti affetti daosteosarcoma in recidiva.
Autore Referenze Numero pazienti Farmaci Sopravvivenzatrattati utilizzati
Saeter Cancer, 199543 60 MTX, CDDP, VP16, IFO SG a 5 anni: 24-50%Michelagnoli Br J Cancer, 199944 13 IFO, VP16, MTX SG a 5 anni: 25% Saylors J Clin Oncol, 200145 18 CTX + TP Qualche RPRodriguez-Galindo J Ped Hemat/Oncol, 200246 14 CBDCA + VP16 R = 28,5%
Methotrexate: MTX; Cisplatino: CDDP; Etoposide: VP16; Ifosfamide: IFO; Carboplatino: CBDCA;Ciclofosfamide : CTX; Topotecan: TP; Sopravvivenza globale: SG; Remissione parziale: RP; R:risposta (completa + parziale).
Tabella 2.Protocolli di alte dosi di chemioterapia seguite da reinfusionedi cellule staminali autologhe in pazienti con osteosarcoma in recidi-va.
Autore Referenze Numero pazienti Farmaci Beneficiotrattati utilizzati
Graham JCO, 199247 18 Bu + CTX ±Colombat Bone Marrow Transplant, 199448 7 L-PAM + CBDCA
(o L-PAM + IFO o CDDP + VP16 o BU)
Chan Bone Marrow Transplant, 199749 2 TT, L-PAM ±Miniero Bone Marrow Transplant, 199850 6 CBDCA + VP16 siLucidarme Bone Marrow Transplant, 199851 7 TT siSauerbrey Bone Marrow Transplant, 200152 15 L-PAM + VP16 no
(± CBDCA, TT, CTX, BU) Fagioli JCO, 200220 32 CBDCA + VP16 si
Carboplatino: BDCA; Ciclofosfamide: CTX; Cisplatino: CDDP; Busulfano : BU; Etoposide : VP16;Ifosfamide: IFO; Melphalan: L-PAM; Thiotepa: TT.

rispettivamente del 20% e del 12%. Questo protocollo hamostrato pertanto la fattibilità delle alte dosi di chemio-terapia nei pazienti con osteosarcoma in recidiva e la pos-sibilità di ottenere remissioni in un significativo, anche sepiccolo, numero di pazienti con una prognosi molto seve-ra, ma ha sottolineato la necessità di ulteriori nuove stra-tegie per mantenere lo stato di remissione. Per questaragione l'Italian Sarcoma Group ha recentemente deli-neato un nuovo protocollo terapeutico diviso in 4 fasi: 1)due cicli di chemioterapia con Ciclofosfamide (CTX) e VP16per mobilizzare le cellule staminali ed indurre lo stato diremissione; 2) un ciclo di alte dosi di CBDCA e VP16 conreinfusione di cellule staminali periferiche autologhe perconsolidare la remissione; 3) trattamento con Samarium(seguito da reinfusione di cellule staminali autologhe) peri pazienti con lesioni captanti; 4) una fase di immunote-rapia con trapianto non mieloablativo di cellule stamina-li da fratello HLA-identico o utilizzo dell'IL-2 nei pazien-ti senza donatore, per mantenere la remissione dellamalattia.
Il razionale per l'uso del trapianto non mieloablativo dicellule staminali allogeniche nei pazienti affetti da tumo-ri solidi si basa sulla generazione di una risposta immuneT-mediata verso le cellule tumorali. 21 Sono recentemen-te emersi dati che confermano l'esistenza di un effettotrapianto contro tumore simile all'effetto graft versusleukemia. Pertanto dal 1997 Childs e colleghi hanno ini-ziato a studiare i trapianti allogenici non mieloablativi neitumori renali metastatici e nel melanoma metastatico,con evidenza di attecchimento del trapianto e di succes-siva regressione delle metastasi. 22 La procedura trapian-tologica è utilizzata come base per l'induzione di uno sta-to di tolleranza tra donatore e ricevente; un regime dicondizionamento immunosoppressivo ma non mieloabla-tivo è sufficiente per ottenere l'attecchimento del tra-pianto, permette di ottimizzare la ricostituzione immune,di creare uno stato di chimerismo misto e di ridurre lamortalità correlata al regime, rendendo il trapianto allo-genico più sicuro ed estendibile anche a pazienti piùanziani o debilitati (23). A tale scopo vengono utilizzateper il condizionamento basse dosi di Fludarabina (FLU) (30mg/m2/die per 2 giorni), CTX (30 mg/Kg/die per 2 giorni)e Thiotepa (TT) (5 mg/Kg/die per 1 giorno), con cui si ottie-ne una potente immunosoppressione ma una modestamielosoppressione. 24 L'equilibrio tra le cellule emato-poietiche del donatore e del ricevente nel chimerismomisto può essere modificato aumentando o diminuendo ildosaggio della Ciclosporina o somministrando linfociti deldonatore (DLI). 25
Un effetto simil graft-versus-tumour può anche essereottenuto con innovativi protocolli di immunoterapia concellule dendritiche e linfociti T specificatamente attivati.L'identificazione di antigeni tumorali ed il loro riconosci-mento da parte di linfociti tumore-specifici ha permessolo sviluppo di strategie di immunoterapia in tumori soli-di, soprattutto nel melanoma; recentemente l'espressio-ne di antigeni della famiglia MAGE, SSX e SART3 è stataidentificata in linee cellulari di osteosarcoma e in tessutidi osteosarcoma freschi,26-28 permettendo lo sviluppo distrategie immunoterapeutiche anche in pazienti affettida osteosarcoma. Presso il Laboratorio del Centro Tra-pianti di Midollo Osseo di Torino è stato pertanto ideato
un progetto per espandere cloni linfocitari T citotossiciutilizzando cellule dendritiche specificatamente attivatecon peptidi di MAGE, SSX e SART3 o lisati tumorali di lineedi osteosarcoma e di valutare la loro specificità e la loroefficacia in vitro e in vivo.
Una certa efficacia dell'IL-2, un mediatore dalla rispo-sta immune cellulo-mediata, la cui azione si esplica attra-verso l'attivazione dei linfociti T e delle cellule NK, è sta-ta evidenziata con studi, tra cui quelli condotti presso l'I-stituto Nazionale Tumori di Milano, effettuati su pazien-ti affetti da tumori solidi e in particolare sull'osteosarco-ma (29). La chemioterapia intensiva associata non inibi-sce la possibilità di amplificare una risposta immunologi-ca IL-2-indotta anche in tempi molto ravvicinati rispettoalla somministrazione degli antiblastici.
Il Samarium-153 ethylene diamine tetramethylene pho-sphonate (153-Sm-EDTMP) è un radiofarmaco in gradodi legarsi all'osso fornendo irradiazione terapeutica allemetastasi ossee. I primi report sulle alte dosi di Samariumnel trattamento dei pazienti con metastasi ossee di osteo-sarcoma suggeriscono una sua efficacia ed un possibileincremento d'azione da parte di farmaci radiosensibiliz-zanti. Poiché la tossicità dose limitante è la trombocito-penia, Anderson et al hanno condotto un trial a dosi sca-lari con supporto con cellule staminali periferiche o midol-lari: con 30 mCi di 153-Sm-EDTMP/Kg sono necessariepiù di 2×106 CD34+/kg per superare la tossicità ematolo-gia. Gli effetti collaterali non ematologici sono risultatiminimi e tutti i pazienti hanno ridotto od eliminato glioppiacei. 30
Numerosi nuovi agenti chemioterapici, diversi da quel-li usati per il trattamento di prima linea, sono attualmen-te in studio per cercare di migliorare la prognosi deipazienti in recidiva. In Italia è stato recentemente attiva-to uno studio di fase II per valutare l'efficacia della Vino-relbina, un alcaloide della vinca semisintetico, nei pazien-ti in fase avanzata di malattia; questo farmaco ha datodelle ottime risposte (43-47% di percentuale di risposta)in studi di fase I e II su angiosarcoma, rabdomiosarcomae sarcoma di Kaposi31 e in 5 pazienti affetti da osteosar-coma trattati presso l'Istituto Nazionale Tumori di Mila-no (1 risposta completa, 2 malattie stabili e 2 progressio-ni di malattia).
Un altro farmaco in studio è l'Ecteinascidin-743 (ET-743), un alcaloide di origine marina che in vitro ha mostra-to una promettente attività nei confronti di linee cellula-ri di sarcomi. In uno studio di Delaloge et al l'ET-743 è sta-to utilizzato in 3 pazienti affetti da osteosarcoma avan-zato; si sono ottenute due remissioni parziali ed una pro-gressione di malattia. 32 L'ET-743 è stato utilizzato anchecome trattamento di terza linea in uno studio dell'ItalianSarcoma Group su 11 pazienti affetti da osteosarcomacon malattia in progressione o in recidiva; non sono sta-te osservate risposte obiettive. 33
Il Topotecan, un inibitore della topoisomerasi I, non hamostrato alcuna utilità quando utilizzato come unico far-maco,34 mentre, in associazione con il CTX, ha prodottouna risposta parziale in un paziente. 35
Altro inibitore della topoisomerasi I in studio nei tumo-ri solidi è l'Irinotecan; i risultati ottenuti al Memorial SloanKettering Cancer Center in 7 pazienti affetti da osteosar-coma hanno però evidenziato che il farmaco non è attivo
59
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

in questo tumore. 35
L'evidenza che l'Insulin-like Growth Factor-1 (IGF-1) èun potente mitogeno per le cellule di osteosarcoma hasuggerito che analoghi della Sandostatina (che parzial-mente riduce IGF-1), possibilmente in associazione conantagonisti degli ormoni rilascianti l'ormone della cresci-ta, possano essere utilizzati nella terapia dell'osteosarco-ma. 36 Tuttavia, uno studio di fase I condotto dal NationalCancer Institute in pazienti in recidiva e non chirurgica-mente trattabili non ha dimostrato un'attività antituma-rale, nonostante la documentata riduzione dei valori sie-rici di IGF-1 nei pazienti trattati. 37
Sono inoltre in studio farmaci per cercare di superare laresistenza alla chemioterapia determinata dall'alta espres-sione di glicoproteina-p 170: uno studio di fase I/II è incorso a Cleveland per valutare la Dexverapamile comereversore della resistenza alla Doxorubicina negli osteo-sarcomi in recidiva. 38
Particolare interesse ha suscitato un nuovo farmaco,inibitore selettivo dell'enzima tirosinokinasi tumorale (Gli-vec), che ha mostrato attività in neoplasie ematologichee nei tumori stromali gatroenterici. Il Children's OncologyGroup sta pertando conducendo uno studio in pazienticon sarcomi dei tessuti molli e dell'osso. 39
Relativamente all'uso di agenti in grado di modulare larisposta immune, particolare interesse è stato sollevatodagli studi sul Muramil Tripeptide liposomiale (L-MTP-PE)suggerendo una possibile sinergia con l'IFO. 40
Due protocolli di studio, l'uno coordinato dal Children'sOncology Group e l'altro dal Memorial Sloan Kettering,hanno come razionale il trattamento selettivo delle cel-lule tumorali utilizzando un anticorpo monoclonale spe-cifico per HER2 (Trastuzumab), sovraespresso soprattuttonei pazienti a peggior prognosi. 41
Altra frontiera per il trattamento dell'osteosarcoma è laterapia genica: è in corso uno studio che utilizza un vet-tore retrovirale (Ad-OC-E1a) che dovrebbe penetrareselettivamente nelle cellule tumorali e regolare la produ-zione intracellulare di una proteina virale che porterebbealla lisi delle cellule tumorali. 42
Bibliografia
1. Bacci G, Picci P, Ruggieri P, Mercuri M, Avella M, Capanna R, et al.Primary chemotherapy and delayed surgery (neoadjuvantchemotherapy) for osteosarcoma of the extremities. The IstitutoRizzoli's experience in 127 patients treated preoperatively withintravenous methotrexate (high versus moderate doses) andintraarterial cisplatin. Cancer 1990;65:2539-53.
2. Bacci G, Picci P, Ferrari S, Ruggieri P, Casadei R, Tienghi A, et al.Primary chemotherapy and delayed surgery for non metastaticosteosarcoma of the extremities: results in 164 patients pre-oper-atively treated with high doses of methotrexate followed by Cis-platin and Doxorubicin. Cancer 1993;72:3227-8.
3. Meyers PA, Gorlick R, Heller G, Casper E, Lane J, Huvos AG, et al.Intensification of preoperative chemotherapy for osteogenic sar-coma: results of the Memorial Sloan-Kettering (T12) Protocol. JClin Oncol 1998;16:2452-8.
4. Saeter G, Alvegard TA, Elomaa I, Stenwig AE, Holmstrom T, SolheimOP. Treatment of osteosarcoma of the extremities with the T-10protocol, with emphasis on the effects of pre-operativechemotherapy with single agent high-dose methotrexate. A Scan-dinavian Sarcoma Group study. J Clin Oncol 1991;9:1766-75.
5. Bacci G, Picci P, Avella M, Ferrari S, Casadei R, Ruggieri P, et al.Effect of intraarterial versus intravenosus cisplatin in addition tosystemic adriamycin and high-dose methotrexate on histologic
tumor response of osteosarcoma of the estremities. J Chemother1992;4:189-95.
6. Lewis IJ, Weeden S, Machin D, Stark D, Craft AW. Received doseand dose-intensity of chemotherapy and outcome in non-metastatic extremity osteosarcoma. European Osteosarcoma Inter-group. J Clin Oncol 2000;18:4028-37.
7. Saeter G. Treatment of osteosarcoma with high-dose methotrex-ate-containing neoadjuvant chemotherapy. Scandinavian Sarco-ma Group data. Med Ped Oncol 1996;27:263[abstact].
8. Saeter G, Wiebe T, Wiklund T, Monge O, Wahlqvist Y, Engstrom K,et al. Chemotherapy in osteosarcoma. The Scandinavian SarcomaGroup experience. Acta Orthopaedica Scandinavia 1999;70:S74-S82.
9. Smith MA, Ungerleider RS, Horowitz ME, Simon R. Influence ofdoxorubicin dose-intensity on response and outcome for patientswith osteogenic sarcoma and Ewing's sarcoma. J Natl Cancer Inst1991; 83:1460-70.
10. Bacci G, Picci P, Avella M, Dallari D, Ferrari S, Prasad R, et al. Theimportance of dose-intensity in neoadjuvant chemotherapy ofosteosarcoma: a retrospective analysis of high dose methotrexate,cisplatinum and adriamycin used preoperatively. J Chemother1990;2:127-35.
11. Bacci G, Ferrari S, Delepine N, Bertoni F, Picci P, Mercuri M, et al.Predictive factors of histologic response to primary chemothera-py in osteosarcoma of the extremity: study of 272 patients pre-operatively treated with high-dose methotrexate, doxorubicin, andcisplatin. J Clin Oncol 1998;16:658-63.
12. Picci P, Sangiorgi L, Rougraff BT, Neff JR, Casadei R, CampanacciM. Relationship of chemotherapy-induced necrosis and surgicalmargins to local recurrence in osteosarcoma. J Clin Oncol 1994;12: 2699-2705.
13. Saeter G, Hoie J, Stenwig AE, Johansson AK, Hannisdal E, SolheimOP. Systemic relapse of patients with osteogenic sarcoma. Prog-nostic factors for long term survival. Cancer 1995; 75: 1084-1093.
14. Saeter G. Treatment strategies and outcome in metastatic(relapsed) osteogenic sarcoma. The Scandinavian Sarcoma Group(SSG) Experience. Med Ped Oncol 1996;27:264[abstact].
15. Bacci G, Briccoli A, Picci P. Osteosarcoma of the extremitiesmetastatic at presentation: results obtained with primarychemotherapy-followed by simultaneous resection of the prima-ry and metastatic lesions. Cancer J 1990;3:213-8.
16. Marina NM, Pratt CB, Rao BN, Shema SJ, Meyer WH. Improvedprognosis of children with osteosarcoma metastatic to the lung(s)at the time of diagnosis. Cancer 1992;70:2722-7.
17. Meyers PA, Heller G, Healey JH, Huvos A, Applewhite A, Sun M, etal. Osteogenic sarcoma with clinically detectable metastasis atinitial presentation. J Clin Oncol 1993;11:449-53.
18. Bacci G, Ferrari S, Longhi A, Perin S, Forni C, Fabbri N, et al. Pat-tern of relapse in patients with osteosarcoma of the extremitiestreated with neoadjuvant chemotherapy. Eur J Cancer 2001;37:32-8.
19. Ferrari S, Briccoli A, Mercuri M, Bertoni F, Picci P, Tienghi A, et al.Postrelapse survival in osteosarcoma of the extremities: prognos-tic factors for long-term survival. J Clin Oncol 2003; 21:710-5.
20. Fagioli F, Aglietta M, Tienghi A, Ferrari S, Brach del Prever A, Vas-sallo E, et al. High dose chemotherapy in the treatment of relapsedosteosarcoma: an Italian Sarcoma Group study. J Clin Oncol 2002;20:2150-6.
21. Slavin S. New strategies for bone marrow transplantation. CurrOpin Immunol 2000;12:542-51
22. Childs R, Drachenberg D. Allogeneic stem cell transplantation forrenal cell carcinoma. Curr Opin Urol 2001;11:495-502.
23. Storb RF, Champlin R, Riddell SR, Murata M, Bryant S, Warren EH.Non-myeloablative transplants for malignant disease. Hematology(Am Soc Hematol Educ Program). 2001:375-91.
24. Corradini P, Tarella C, Olivieri A, Gianni AM, Voena C, Zallio F, etal. Reduced-intensity conditioning followed by allografting ofhematopoietic cells can produce clinical and molecular remissionsin patients with poor-risk hematologic malignancies. Blood 2002;99:75-82.
25. Lokhorst HM, Schattenberg A, Cornelissen JJ, Thomas LL, Verdon-ck LF. Donor leukocyte infusions are effective in relapsed multiplemyeloma afterallogeneic bone marrow transplantation. Blood.1997;90:4206-11.
60
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre

61
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
26. Sudo T, Kuramoto T, Komiya S, Inoue A, Itoh K. Expression of MAGEgenes in osteosarcoma. J Orthop Res 1997;15:128.
27. Naka N, Araki N, Nakanishi H, Itoh K, Mano M, Ishiguro S, et al.Expression of SSX genes in human osteosarcomas. Int J Cancer.2002;98:640.
28. Tsuda N, Murayama K, Ishida H, Matsunaga K, Komiya S, Itoh K,Yamada A. Expression of a newly defined tumor-rejection antigenSART3 in musculoskeletal tumors and induction of HLA class I-restricted cytotoxic T lymphocytes by SART3-derived peptides. JOrthop Res 2001;19:346-351.
29. Toren A, Nagler A, Rozenfeld-Granot G, Levanon M, Davidson J,Bielorai B, et al. Amplification of immunological functions by sub-cutaneous injection of intermediate-high dose interleukin-2 for 2years after autologous stem celltransplantation in children withstage IV neuroblastoma. Transplantation 2000;70:1100-4
30. Anderson PM, Wiseman,G. A. , Dispenzieri A, Arndt CA, HartmannLC, Smithson WA, et al. High-dose samarium-153 ethylenediamine tetramethylene phosphonate: low toxicity of skeletal irra-diation in patients with osteosarcoma and bone metastases. J. ClinOncol 2002;20:189-96.
31. Casanova M, Ferrari A, Spreafico F, Terenziani M, Massimino M,Luksch R, et al. Vinorelbine in previously treated advanced child-hood sarcomas: evidence of activity in rhabdomyosarcoma. Can-cer 2002;94:3263-8
32. Delaloge S, Yovine A, Taamma A, Riofrio M, Brain E, Raymond E,et al. Ecteinascidin-743: a marine-derived compound in advanced,pretreated sarcoma patients - preliminary evidence of activity. JClin Oncol 2001;19:1248-55.
33. Casanova M, Casali PG, Sessa C, Bacci G, Aglietta M, Riccardi R etal. Ecteinascidin-743 in infusione di tre ore in pazienti affetti dasarcoma pretrattato in progressione o alla recidiva: studio di faseII. Riv Ital Pediatr 2002;1 Suppl 2:150.
34. Blaney SM, Needle MN, Gillespie A, Sato JK, Reaman GH, Berg SL,et al. Phase II trial of topotecan administered as 72-hour contin-uous infusion in children with refractory solid tumors: a collabo-rative Pediatric Branch, National Cancer Institute, and Children'sCancer Group Study. Clin Cancer Res 1998;4:357-60
35. Saylors RL, Stewart CF, Zamboni, Wall DA, Bell B, Stine KC, et al.Phase I study of topotecan in combination with cyclophosphamidein pediatric patients with malignant solid tumors: a PediatricOncology Group Study. J Clin Oncol 1998;16:945-52
36. Cosetti M, Wexler LH, Calleja E, Trippett T, LaQuaglia M, Huvos AG,et al. Irinotecan for pediatric solid tumors: the Memorial Sloan-Kettering experience. J Pediatr Hematol Oncol 2002;24:101-5.
37. Mansky PJ, Liewehr DJ, Steinberg SM, Chrousos GP, Avila NA, Long,et al. Treatment of metastatic osteosarcoma with the somato-statin analog OncoLar: significant reduction of insulin-like growthfactor-1 serum levels. J Pediatr Hematol Oncol 2002;24:440-6
38. Pollak M, Sem AW, Richard M, Tetenes E, Bell R. Inhibition ofmetastatic behavior of murine osteosarcoma by hypophysectomy.J Natl Cancer Inst 1992;84:966-71.
39. Matsuyama S, Iwadate M, Kondo M, Saitoh M, Hanyu A, ShimizuK, et al. SB-431542 and Gleevec inhibit transforming growth fac-tor-β-induced proliferation of human osteosarcoma. cells. CancerRes 2003;63:7791-8.
40. Kleinerman ES, Gano JB, Johnston, Benjamin RS, Jaffe N. Efficacyof liposomal muramyl tripeptide (CGP 19835A) in the treatmentof relapsed osteosarcoma. Am J Clin Oncol. 1995;18:93-9.
41. Anninga JK, van de Vijver MJ, Cleton-Jansen AM, Kristel PM,Taminiau AH, Nooij M, et al. Overexpression of the HER-2 onco-gene does not play a role in high-grade osteosarcomas. Eur J Can-cer 2004;40:963-70.
42. Benjamin R, Helman L, Meyers P, Reaman G. A phase I/II dose esca-lation and activity study of intravenous injections of OCaP1 forsubjects with refractory osteosarcoma metastatic to lung. HumGene Ther 2001;12:1591-3.
43. Bramwell VH. The role of chemotherapy in the management ofnon-metastatic operable extremity osteosarcoma. Semin Oncol1997;24:561-71.
44. Saeter G, Høie J, Stenwig AE, Johansson AK, Hannisdal E, SolheimOP. Systemic relapse of patients with osteogenic sarcoma. Prog-nostic factors for long term survival. Cancer 1995;75:1084-93.
45. Michelagnoli MP, Lewis IJ, Gattamaneni HR, Bailey CC, Lashford LS.Ifosfamide/etoposide alternating with high-dose methotrexate:
evaluation of a chemotherapy regimen for poor-risk osteosarco-ma. Br J Cancer 1999;79:1174-8.
46. Saylors RL, Stine KC, Sullivan J, Kepner JL, Wall DA, Bernstein ML,et al. Cyclophosphamide plus topotecan in children with recurrentor refractory solid tumors: a Pediatric Oncology Group phase IIstudy. J Clin Oncol 2001;19:3463-9.
47. Rodriguez-Galindo C, Daw NC, Kaste SC, Meyer WH, Dome JS,Pappo AS, et al. Treatment of refractory osteosarcoma with frac-tionated cyclophosphamide and etoposide. J. Pediatr Hematol.Oncol 2002;24:250-5.
48. Graham ML, Yeager AM, Leventhal BG, Wiley JM, Civin CI, StraussLC, et al. Treatment of recurrent and refractory pediatric solidtumors with high-dose busulfan and cyclophosphamide followedby autologous bone marrow rescue. J Clin Oncol 1992;10:1857-64.
49. Colombat PH, Biron P, Coze C, Rosti G, Siimes MA, Blay JY, et al.Failure of high-dose alkylating agents in osteosarcoma. Bone Mar-row Transplant 1994;14:665-6.
50. Chan KW, Petropoulos D, Choroszy M, Herzog C, Jaffe N, Ater J, etal. High-dose sequential chemotherapy and autologous stem cellreinfusion in advanced pediatric solid tumors. Bone Marrow Trans-plant 1997;20:1039-43.
51. Miniero R, Brach del Prever A, Vassallo E, Nesi F, Busca A, FagioliF, et al. Feasibility of high-dose chemotherapy and autologousperipheral blood stem cell transplantation in children with highgrade osteosarcoma. Bone Marrow Transplant 1998;22:S37-S40.
52. Lucidarme N, Valteau-Couanet D, Oberlin O, Couanet D, Kalifa C,Beaujean F, et al. Phase II study of high-dose thiotepa andhematopoietic stem cell transplantation in children with solidtumors. Bone Marrow Transplant 1998;22:535-40.
53. Sauerbrey A, Bielack S, Kempf-Bielack B, Zoubek A, Paulussen M,Zintl F. High-dose chemotherapy (HDC) and autologoushematopoietic stem cell transplantation (ASCT) as salvage thera-py for relapsed osteosarcoma. Bone Marrow Transplant 2001;27:933-7.

Aspetti biologici dell’osteosarcoma
Piero PicciLaboratorio di Ricerca Oncologica, Istituti Ortopedici Riz-zoli, Bologna, Italy
L'osteosarcoma (OS), il più frequente tumore malignoprimitivo dell'osso, è caratterizzato da un decorsoestremamente aggressivo con sviluppo di metastasi pol-monari in circa il 40-50% dei pazienti.1,2 Nonostante l'u-so della chemioterapia adiuvante o neoadiuvante ne abbiasignificativamente migliorato la prognosi, permane tutto-ra un numero consistente di pazienti che sviluppa farma-coresistenza e va incontro ad un decorso clinico sfa-vorevole. 3
Gli attuali protocolli chemioterapici per l'OS ad alto gra-do sono basati sull'impiego di doxorubicina (DX), alte dosidi methotrexate (MTX), e cisplatino (CDDP), ai quali vieneeventualmente aggiunta l'ifosfamide nella fase post-oper-atoria. Studi clinici hanno dimostrato che la DX ed il MTXsono sicuramente i due farmaci più importanti per lachemioterapia dell'OS e che la chemioresistenza intrinse-ca o acquisita verso questi antiblastici rappresenta il mag-gior fattore limitante per l'efficacia del trattamento. 1,2,4,5
Pertanto, trattamenti con agenti in grado superare i mec-canismi legati alla farmacoresistenza presenti nelle celluledi OS potrebbero costituire la base nuovi protocolli ter-apeutici.
Nuovi farmaci in grado di superare i principali meccanismidi farmacoresistenza presenti nelle cellule di osteosarcoma
Nell'OS ad alto grado, la mancata risposta al tratta-mento è soprattutto determinata dalla sovraespressionedella P-glicoproteina, una proteina di membrana codifi-cata dal gene MDR1, che è responsabile della resistenzaalla DX ed è risultata essere il più importante fattore prog-nostico negativo nell'OS. 6-9 Studi clinici preliminari confarmaci capaci di superare la resistenza alla DX attraver-so l'inibizione della funzionalità della P-glicoproteina (inparticolare alcuni chemiorevertanti, quali ad esempio ilVerapamil) hanno mostrato solo parziali successi, rag-giungendo la massima efficacia solamente a dosi nontollerabili dal paziente a causa della grave tossicità col-laterale.
Una possibile strategia alternativa è rappresentata dal-l'utilizzazione di nuovi agenti antitumorali quali le alchi-cicline, una nuova classe di farmaci che sono risultatiessere efficaci in diversi tumori farmacoresistenti, ivi com-presi quelli sovraesprimenti la P-glicoproteina.10 La mol-ecola più efficace di questa nuova categoria di antiblas-tici è la 4-dimetossi-3'-diamino-3'aziridinil-4'metilsul-fonil-daunorubicin (comunemente identificata con la siglaPNU-159548), che è risultata essere attiva non solo con-tro cellule tumorali che sovraesprimono P-glicoproteina,ma anche contro cellule neoplastiche resistenti ad agen-ti alchilanti come cisplatino e ciclofosfamide. 11 Inoltre, ilPNU-159548 risulta essere una molecola di particolareinteresse per una possibile utilizzazione nella chemioter-apia dell'OS ad alto grado in quanto studi in vivo hannodimostrato che questo nuovo agente antitumorale ha unacardiotossicità significativamente inferiore alla DX, uno
dei fattori più fortemente limitanti l'utilizzazione della DXin schemi di trattamento polichemioterapici. 10
Ulteriori strategie terapeutiche possono essere prese inconsiderazione per superare la resistenza al MTX. In par-ticolare, il trimetrexate (TMTX) é stato indicato come unapossibile alternativa in quei tumori che non sono respon-sivi al MTX. 12 Come il MTX, il TMTX è un potente inibitoredell'enzima diidrofolato-riduttasi (DHFR), che svolge unruolo chiave nel metabolismo intracellulare dei folati edè indispensabile per la sintesi del DNA e la riproduzionecellulare. D'altra parte, il TMTX non è interessato dai mec-canismi di resistenza al MTX mediati da un insufficientetrasporto trans-membrana,13 meccanismo che è statodimostrato essere il fattore più importante per la manca-ta risposta clinica al MTX in pazienti con OS. 5
Nonostante PNU-159548 e TMTX siano stati studiati, siain vitro che in vivo, in diversi tumori, fino ad ora non sonostati riportati studi riguardo la loro efficacia su cellule diOS e sulle loro interazioni con gli altri farmaci comune-mente impiegati nel trattamento dell'OS ad alto grado. Lostudio qui presentato è stato quindi effettuato con l'in-tento di valutare l'efficacia in vitro di questi due farmaci
62
haematologica vol. 89[supplement n. 10]:October 2004
Lunedi 11 ottobre
Tabella 1.Linee cellulari di osteosarcoma umano utilizzate per la val-utazione in vitro dell'efficacia di PNU-159548 e TMTX.
Linea Origine della Livello di resistenzacellulare linea cellulare rispetto la corrispondente
linea parentale(numero di volte)
U-2OS OS umano −Saos-2 OS umano −IOR/OS9 OS umano −IOR/OS10 OS umano −IOR/OS14 OS umano −IOR/OS15 OS umano −IOR/OS18 OS umano −IOR/MOS OS umano −SARG OS umano −U-2OS/DX30 Variante DX-resistente di U-2OS 15U-2OS/DX580 Variante DX-resistente di U-2OS 330Saos-2/DX30 Variante DX-resistente di Saos-2 72Saos-2/DX580 Variante DX-resistente di Saos-2 338U-2OS/MTX3 Variante MTX-resistente di U-2OS 3U-2OS/MTX30 Variante MTX-resistente di U-2OS 21U-2OS/MTX100 Variante MTX-resistente di U-2OS 69U-2OS/MTX300 Variante MTX-resistente di U-2OS 135Saos-2/MTX30 Variante MTX-resistente di Saos-2 15Saos-2/MTX100 Variante MTX-resistente di Saos-2 24Saos-2/MTX300 Variante MTX-resistente di Saos-2 109Saos-2/MTX1µg Variante MTX-resistente di Saos-2 281
U-2OS/CDDP300 Variante CDDP-resistente di U-2OS 4U-2OS/CDDP1µg Variante CDDP-resistente di U-2OS 10
Saos-2/CDDP300 Variante CDDP-resistente di Saos-2 3Saos-2/CDDP1µg Variante CDDP-resistente di Saos-2 17

su cellule di OS umano, sia farmacosensibili che farma-coresistenti.
Materiali e MetodiL'efficacia in vitro per ciascun agente è stata valutata
utilizzando un pannello di 9 linee cellulari di OS umano,ottenute da campioni chirurgici di pazienti con OS ad altogrado, e 16 varianti farmacoresistenti selezionate attra-verso l'esposizione in vitro a dosi progressivamente cres-centi di DX, MTX o CDDP (Tabella 1). Tutte queste lineesono state stabilizzate e caratterizzate presso il Labora-torio di Ricerca Oncologica (Istituti Ortopedici Rizzoli,Bologna).
La sensibilità in vitro a PNU-159548 e TMTX è statadeterminata mediante la valutazione dell'inibizione dellacrescita cellulare dopo esposizione al farmaco per 96 oreed espressa come rapporto IC50 (concentrazione del far-maco capace di inibire al 50% la crescita cellulare dellecellule trattate rispetto ai rispettivi controlli).
Per valutare le interazioni in vitro tra il PNU-159548 oil TMTX con i farmaci convenzionali usati nella chemioter-apia dell'OS, le stesse linee di OS umano sono stateesposte a vari trattamenti combinati. In una prima seriedi esperimenti, le cellule sono state esposte a schemi ditrattamento basati sulla somministrazione simultanea diPNU-159548 o TMTX con DX, MTX o CDDP. L'entità del-l'inibizione della crescita cellulare è stata poi confronta-ta con quella ottenuta negli esperimenti con trattamen-to a singolo farmaco per identificare il tipo di interazione(additività, sinergismo, o antagonismo) di PNU-159548 oTMTX con i farmaci tradizionali.
In una seconda serie di esperimenti le cellule sono stateesposte in sequenza a PNU-159548 o a TMTX e poi, doporimozione del primo farmaco, a DX, MTX o CDDP. Lo stes-so schema di trattamento è stato poi ripetuto utilizzandoi farmaci in sequenza inversa. Gli effetti della combi-nazione dei farmaci in termini di additività, sinergismo, oantagonismo sono stati calcolati col metodo del “medianeffect plot”. Per ciascuna combinazione di farmaci, il coef-ficiente di interazione (CI) è stato determinato con la for-mula:
CI = (D)1 /(Dx)1 + (D)2/(Dx)2
dove (Dx)1 e (Dx)2 sono, rispettivamente, le dosi del far-maco 1 e farmaco 2 che produce una x% inibizione dellacrescita cellulare quando sono somministrati singolar-mente, mentre (D)1 e (D)2 sono, rispettivamente, la dose delfarmaco 1 e del farmaco 2 necessarie per produrre la stes-sa x% di inibizione della crescita cellulare quando sonosomministrati in combinazione. Quando CI=1 l'interazioneè considerata additiva; quando CI<1 l'interazione è con-siderata sinergica; quando CI>1 l'interazione è consider-ata antagonista.
RisultatiL'analisi di efficacia in vitro del PNU-159548 ha
mostrato come le celllule di OS siano, generalmente,abbastanza sensibili a questo farmaco. Inoltre, il PNU-159548 è risultato essere attivo anche in cellule consovraespressione della P-glicoproteina e resistenti allaDX, in cellule resistenti al MTX e, anche se in misura
minore, in cellule resistenti al CDDP. L'esposizione simultanea al PNU-159548 ed a DX, MTX
o CDDP ha prodotto soprattutto un'interazione additivanelle linee cellulari farmacosensibili e nelle lineeresistenti alla DX, mentre nelle linee resistenti a MTX oCDDP l'interazione era principalmente antagonista. Gliesperimenti di trattamento combinato con farmaci som-ministrati sequenzialmente hanno mostrato che la mag-gior efficacia del trattamento in vitro viene raggiuntaquando il PNU-159548 viene usato come primo farma-co, seguito immediatamente dal trattamento con DX,MTX o CDDP, anche se è stata riscontrata un'ampia vari-abilità da linea a linea.
L'analisi di efficacia in vitro del TMTX ha mostratocome le celllule di OS siano, generalmente, abbastanzasensibili anche a questo farmaco. Inoltre, il TMTX è risul-tato essere attivo anche in cellule resistenti al MTX, conla sola eccezione per quelle cellule che devono la lororesistenza al MTX ad un aumento del livello citoplas-matico di DHFR. Il TMTX è inoltre risultato essere efficaceanche in cellule resistenti al CDDP e, in misura legger-mente inferiore, in quelle resistenti alla DX.
La simultanea esposizione al TMTX ed alla DX o CDDPha prodotto soprattutto interazioni antagoniste, sia incellule di OS farmacosensibili che farmacoresistenti. Lesomministrazioni sequenziali hanno dimostrato come lamaggior efficacia del trattamento in vitro si raggiungaquando il TMTX viene usato come primo farmaco, segui-to immediatamente da DX o CDDP.
ConclusioniNonostante l'era post-genomica sia già sicuramente
iniziata con la speranza che ciò possa portare in futuroa trattamenti individualizzati, basati sulle caratteristichegenetiche di ogni singolo tumore, al presente è ancoradi grande attualità la messa a punto di nuovi farmaci ingrado di superare i limiti di efficacia e di utlizzazione deichemioterapici convenzionali.
In questo contesto, sia il PNU-159548 che il TMTX sipropongono come dei possibili e promettenti candidatiper un'utilizzazione clinica in nuovi protocolli terapeu-tici per pazienti con OS ad alto grado non responsivi aitrattamenti convenzionali.
Bibliografia
1. Bruland ØS, Pihl A. On the current management of osteosar-coma. A critical evaluation and a proposal for a modified treat-ment strategy. Eur J Cancer 1997;33:1725-1731.
2. Bacci G et al. Long-term outcome for patients with non-metastatic osteosarcoma of the extremity treated at the Isti-tuto Ortopedico Rizzoli according to the istituto ortopedicorizzoli/osteosarcoma-2 protocol: an updated report. J ClinOncol 2000;18:4016-4027.
3. Bramwell VH. The role of chemotherapy in the management ofnon-metastatic operable extremity osteosarcoma. Semin Oncol1997;24:561-571.
4. Delepine N et al. Influence of methotrexate dose intensity onoutcome of patients with high grade osteogenic osteosarco-ma. Analysis of the literature. Cancer 1996;78:2127-2135.
5. Guo W et al. Mechanisms of methotrexate resistance inosteosarcoma. Clin Cancer Res 1999;5:621-627.
6. Baldini N et al. Expression of P-glycoprotein in high-gradeosteosarcomas in relation to clinical outcome. N Eng J Med1995;333:1380-1385.
7. Chan HS et al: P-glycoprotein expression: critical determinant
63
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

64
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre
in the response to osteosarcoma chemotherapy. J Natl CancerInst 1997;89:1706-1715.
8. Hornicek FJ, et al. P-glycoprotein levels predict poor outcomein patients with osteosarcoma. Clin Orthop 2000;373:11-7.
9. Serra M, Scotlandi K, Reverter-Branchat G, Ferrari S, ManaraMC, Benini S, et al. Value of P-glycoprotein and clinicopatho-logic factors as basis for new treatment strategies in high-grade osteosarcoma of the extremities. J Clin Oncol2003;21:536-42.
10. Geroni C, Ripamonti M, Arrigoni C, Fiorentini F, Capolongo L,Moneta D, et al. Pharmacological and toxicological aspects of4-demethoxy-3'-deamino-3'-aziridinyl-4'-methylsulphonyl-daunorubicin (PNU-159548): a novel antineoplastic agent.Cancer Res 2001;61:1983-1990.
11. Marchini S, Damia G, Broggini M, Pennella G, Ripamonti M,Marsiglio A, et al. 4-demethoxy-3'-deamino-3'aziridinyl-4'methylsulphonyl-DNR (PNU-159548), a novel anticanceragent active against tumor cell lines with different resistancemechanisms. Cancer Res 2001;61:1991-5.
12. Takimoto CH. New antifolates: pharmacology and clinicalapplications. Oncologist 1996;1: 68-81.
13. Takemura Y, Kobayashi H, Miyachi H. Cellular and molecularmechanisms of resistance to antifolate drugs: new analoguesand approaches to overcome the resistance. Int J Hematol1997; 66:459-77.
Problemi riabilitativi in oncologia
Enrico CastelliDirettore U. O. di Neuroriabilitazione, IRCCS EugenioMedea, Bosisio Parini, Lecco
I tumori primitivi del sistema nervoso centrale (SNC)sono i più frequenti tumori solidi nel bambino, secondisolo alla leucemia, con una incidenza di 250-300 nuovicasi ogni anno in Italia per pazienti di età inferiore ai 15anni. Di conseguenza, i tumori cerebrali rappresentanouna causa significativa di mortalità e morbilità nell'in-fanzia. Inoltre, studi recenti indicano che l'incidenza ditumori cerebrali nel bambino e nell'adulto è in aumento.I gliomi, soprattutto gli astrocitomi, sono il tipo istologi-co più frequente in età infantile, seguiti dal medullobla-stoma. La fossa cranica posteriore è la sede più frequen-te di localizzazione: oltre la metà delle neoplasie del SNCinsorge nel cervelletto, nel tronco encefalico e nel IV° ven-tricolo. Il trattamento delle neoplasie del SNC prevedel'impiego integrato di neurochirurgia, radioterapia e che-mioterapia. Negli ultimi decenni, i miglioramenti nel trat-tamento di tali tumori hanno determinato un aumentodella sopravvivenza a 5 anni dalla diagnosi, passata dal50% all'80% per gli istotipi a basso grado di malignità. Peri gliomi maligni, in particolare per il glioblastoma mul-tiforme, e per le neoplasie intrinseche del tronco cerebra-le, i risultati sono meno incoraggianti. Tuttavia la soprav-vivenza si associa, in pazienti lungosopravviventi, a defi-cit complessi in ambito motorio, sensoriale, endocrino,cognitivo e psicologico, che compromettono significati-vamente la loro qualità di vita. Nei bambini trattati pertumore cerebrale il SNC può essere danneggiato, oltre chedal danno tissutale diretto causato dalla neoplasia e dal-l'intervento neurochirurgico, anche dall'effetto tossicodella chemioterapia e/o della radioterapia. In particolaregli effetti patologici della terapia radiante sono ben docu-mentati. L'aumentata vulnerabilità dei pazienti in etàpediatrica, quando più intensi sono i processi maturativi,è ben documentata e gli esiti legati alla radioterapia pos-sono manifestarsi anche molti anni dopo la sua esecuzio-ne. I bambini più piccoli sono poi particolarmente a rischiodi comparsa di disturbi neuropsicologici poiché la mieli-nizzazione e lo sviluppo del SNC, in particolare delle areecorticali associative determinanti per l'acquisizione dellecapacità cognitive superiori, non sono ancora completa-ti. I deficit neuropsicologici più frequenti sono a caricodelle funzioni attentive e mnestiche, che possono benefi-ciare di una riabilitazione specifica volta all'acquisizionedi strategie compensative ed alla creazione di abilitàdeterminanti per il reinserimento sociale. Le principalivariabili che incidono sulla comparsa di queste problema-tiche sono: l'età alla diagnosi, il tipo e la localizzazione deltumore, le complicanze perioperatorie, i protocolli tera-peutici impiegati. Dobbiamo anche considerare che in etàevolutiva lo sviluppo dell'intelligenza necessita di unamaturazione progressiva dei circuiti anatomici e di unadifferenziazione strutturale. La maturità funzionale del-l'encefalo si completa nell'arco di anni: è quindi evidente
Incontra l’Esperto

che le conseguenze di una lesione cerebrale saranno diver-se secondo l'età del paziente alla diagnosi. Le lesioniacquisite dell'encefalo nelle fasce più basse di età inter-feriscono con l'organizzazione neuronale di base, con lamaturazione dei circuiti neuroanatomici, con la differen-ziazione strutturale e determinano un outcome peggiorerispetto a lesioni analoghe in soggetti adulti. Le lesionicerebrali precoci compromettono la registrazione diffe-renziale e l'analisi dettagliata degli stimoli sensitiviambientali, la formazione di nessi associativi con prece-denti analoghe rappresentazioni recuperate nella memo-ria a lungo termine e con i segnali che rappresentano l'e-laborazione corrente. Tali funzioni complesse necessitanodell'attività di differenti lobi cerebrali e di un'intricataserie di interconnessioni anatomiche che sostanzialmen-te mettono in relazione la corteccia prefrontale con l'ip-pocampo dei lobi temporali, la corteccia parietale poste-riore ed il neocervelletto e più in generale zone differen-ti della corteccia cerebrale a livello locale, interregionaleed interemisferico. Proprio queste aree cerebrali e le lorointerconnessioni anatomiche possono essere sono dan-neggiate dalle lesioni causate dal tumore, sia con un mec-canismo focale che diffuso (a carico prevalentemente del-la sostanza bianca come risultato secondario ed indesi-derato della chemio e della radioterapia).
In età infantile i sintomi psicologici possono esserevariamente rappresentati da reazioni depressive, reazionid'ansia o miste, reazioni con aspetti emozionali o condisturbi della condotta. Gli adolescenti presentano soprat-tutto disturbi comportamentali, con conseguente isola-mento. Trattandosi di turbe secondarie a neoplasie cere-brali vanno considerati i disturbi psichiatrici su base orga-nica, che hanno una prevalenza compresa tra il 5% ed il40%. Anche le reazioni dei familiari alla malattia del figliosono elementi che possono influenzare la situazione cli-nica globale del bambino.
L'impatto del tumore cerebrale sulle relazioni familiariè significativamente correlato con la percezione di sinto-mi comportamentali ed affettivi e con le modificazionidella qualità di vita. Va posta particolare enfasi ai distur-bi comportamentali o emozionali, in modo che i familiaripossano comprenderne la natura e relazionarsi meglio conil paziente. Generalmente i disturbi emotivi sono la mag-gior fonte di stress per i parenti, poiché tali problemi ten-dono a persistere più a lungo delle disabilità fisiche.Durante le fasi precoci i familiari spesso rispondono conshock e negazione; questa prima reazione evolve attra-verso una serie di cambiamenti che variano dalla nega-zione e dall'attesa di un completo recupero, fino alladepressione, quando si rende evidente che i deficit sonodi lunga durata o permanenti. Vi sono delle importantidifferenze nelle reazioni psicologiche fra un caregiver chesi occupi di un adulto o di un bambino; mentre il caregi-ver di un paziente adulto ha delle preoccupazioni di ordi-ne emotivo, sessuale, finanziario, di cambiamento dellavita familiare, quello di un bambino spesso si trova a fron-teggiare sentimenti di perdita delle speranze e delle aspet-tative future. Nell'immediato periodo post-diagnosi lanegazione dei problemi del paziente può rappresentareuna normale reazione ad una situazione intollerabile,mentre l'auto-negazione può costituire un serio ostacoloalla riorganizzazione familiare se non viene correttamen-
te indirizzato durante la fase riabilitativa. Questi caregi-vers spesso sviluppano delle alterate interazioni familiarie sociali, focalizzando esclusivamente la loro attenzionesui bisogni del bambino, isolandolo dal contesto sociale.Quando persistono, tali strategie di relazione patologichepossono compromettere le interazioni e la salute menta-le dei vari membri della famiglia. La terapia familiare puòspesso aiutare queste famiglie in crisi, fornendo ancheinformazioni utili, strutture e supporto.
La valutazione multidisciplinareLa valutazione clinica, il programma riabilitativo e la sti-
ma delle sequele richiedono l'uso e la coordinazione di mol-teplici competenze. La valutazione di un tumore del SNC nelbambino è più complicata rispetto all'adulto sia per lo svi-luppo contemporaneo delle funzioni cognitive e psicologi-che che per le necessità di organizzazione della persona-lità. La valutazione e la gestione del processo riabilitativodevono essere multidisciplinari, con il coinvolgimento deivari specialisti competenti per problemi specifici. L'attivitàdi un team multidisciplinare non è di per sé sufficiente, maogni specialista coinvolto non può lavorare in maniera iso-lata ma deve integrare il proprio contributo col resto delgruppo al fine di garantire un trattamento omnicompren-sivo delle problematiche del bambino e della sua famiglia.L'approccio multidisciplinare permette una valutazione glo-bale delle disabilità presenti, facilita l'identificazione dellearee a rischio per deficit futuri, consente l'identificazionedei principali obiettivi riabilitativi e semplifica la gestionedel programma riabilitativo. Come conseguenza questamodalità di approccio, promuovendo il miglioramento fun-zionale e facilitando il recupero di una dimensione socia-le, riduce anche l'impatto economico a lungo termine diquesta patologia. Il team riabilitativo dovrebbe disporre inparticolare delle seguenti competenze: neurologia, fisia-tria, neuropsichiatria infantile, pediatria, psicologia, neu-ropsicologia, pedagogia, servizio sociale, terapisti della ria-bilitazione dei settori di fisioterapia, logopedia, neuropsi-cologia, psicomotricità e terapia occupazionale. La valuta-zione viene suddivisa in differenti parti, dalla raccolta deidati anamnestici all'analisi delle problematiche internisti-che, neurologiche, fisiatriche, oculistiche, ORL e foniatriche.Oltre alle visite specialistiche i pazienti vengono inseriti neisettori di trattamento riabilitativo che effettuano in unaprima fase un'osservazione delle disabilità presenti cheintegra e completa il bilancio clinico. La valutazione deveessere rapportata alla severità e complessità dei deficit pre-senti, senza la necessità di sottoporre sempre il bambino aduna valutazione estensiva. La valutazione dei deficit neu-ropsicologici si basa sull'osservazione clinica e sull'utilizzodi test psicometrici standardizzati che devono consentire lacomparazione delle prestazioni a differenti età, lo studiodelle relazioni tra alterazione del test e specifiche aree cere-brali con la verifica delle strategie di elaborazione delleinformazioni. I possibili problemi comportamentali, le dif-ficoltà familiari e sociali possono essere definiti mediantecolloqui clinici, osservazioni di gioco libero ed utilizzo ditest standardizzati. La valutazione multidisciplinare con-sente un completo bilancio clinico dei deficit del pazienteed è seguita dall'identificazione dei principali obiettivi ria-bilitativi che vengono perseguiti nei differenti settori ditrattamento.
65
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

Il trattamento riabilitativoNon esiste un unico approccio che possa prendersi cura
delle necessità di tutti i pazienti e delle loro famiglie, madeve essere proposto un approccio razionale che conside-ri la natura delle lesioni cerebrali e le loro conseguenze,l'entità del problema, la metodologia di trattamento, ilpersonale ed i servizi disponibili. L'essenza del trattamen-to del bambino post traumatico consiste nel fornire le cir-costanze ottimali che favoriscano il recupero dei dannicerebrali presenti, evitino la comparsa o l'accentuazionedel danno secondario da complicazioni, facilitino lo svi-luppo motorio, cognitivo, psicologico e gli apprendimen-ti, consentendo pertanto il massimo recupero funzionalecompatibile con le disabilità presenti. Essenziale appare,a fianco dei trattamenti riabilitativi, l'apporto dei familia-ri. Essi devono essere guidati e sostenuti nel processo diprogressiva presa di coscienza delle problematiche e del-le esigenze del loro figlio, anche rispetto alle loro reazio-ni psicologiche ed aspettative, rendendoli elementi attivied essenziali del progetto riabilitativo.
Dalla valutazione multidisciplinare effettuata all'in-gresso deriva la scelta degli obiettivi prioritari dell'inter-vento riabilitativo intensivo, impostati in base al bilancioclinico. Appare indispensabile l'impiego di strumenti vol-ti alla verifica dell'efficacia dell'intervento riabilitativo, daapplicare sia in ingresso che in uscita dai diversi settori ditrattamento.
Tutti i membri del team riabilitativo e di assistenza deb-bono avere una lunga esperienza di lavoro con i bambini.Anche l'ambiente di degenza e riabilitativo dovrebbe esse-re organizzato ed arredato in accordo con le necessità, igusti e gli interessi dei bambini, al fine di ridurne l'ansiae facilitarne la collaborazione.
Inizialmente il bambino viene trattato a letto ma, appe-na le condizioni cliniche di base si sono stabilizzate, vie-ne condotto fuori dalla stanza e continua l'intervento ria-bilitativo in idonei ambienti. Il principale obiettivo riabi-litativo è il raggiungimento del massimo livello di recu-pero, compatibilmente con i limiti imposti dalla severitàdelle lesioni, e di un benessere generale del bambino, favo-rendo il successivo reinserimento scolastico e sociale.
Le tecniche riabilitative utilizzate ed i loro principaliobiettivi sono riportate di seguito:
• fisioterapia per raggiungere il massimo recuperomotorio e l'autonomia personale
• la stimolazione multisensoriale, soprattutto con unospecifico training per i disturbi neurovisivi e l'eventualeipovisione, mentre le lesioni traumatiche dell'VIII nervocranico richiedono un training per l'ipoacusia ed i distur-bi dell'equilibrio
• gli obiettivi dell'intervento logopedico variano dalrecupero dell'alimentazione per bocca in fase post acutaprecoce, con masticazione e deglutizione, all'articolazio-ne delle parole, al training dell'eventuale comunicazioneextraverbale per gli anartrici, al trattamento dei disturbiafasici, della lettura, della scrittura e della comunicazio-ne indipendente
• il training delle funzioni cognitive per bambini di etàinferiore a quattro anni o con una compromissione seve-ra del livello intellettivo può essere effettuato più effica-cemente utilizzando tecniche psicomotorie volte al recu-pero sensomotorio, dell'organizzazione spazio-temporale
di base, dello schema corporeo, delle sequenze, dei rap-porti di causa-effetto, sino al gioco simbolico; i bambinicon età o competenze maggiori necessitano di una spe-cifica riabilitazione neuropsicologica che dal training del-le funzioni di base (attenzione, memoria, nessi logici, pro-blem solving, etc) giunge sino al loro uso integrato e quin-di indipendente. Questi obiettivi possono essere raggiun-ti anche utilizzando specifici software orientati sia allafacilitazione di eventuali deficit motori, sensoriali o comu-nicativi che al training delle funzioni neuropsicologiche.
• la terapia occupazionale è volta soprattutto al recu-pero dei livelli di autonomia personale per le attività del-la vita quotidiana, con supporto per l'eventuale adatta-mento della casa e della scuola.
• l'intervento cognitivo comportamentale dall'inizialerichiesta di contenimento di un eventuale stato di agita-zione o di comportamento inadeguato si rivolge quindialla comprensione ed al superamento delle problematichepsicologiche, per quanto concesso dall'età del bambino,prima con un intervento individuale e quindi in piccologruppo di coetanei con simili disabilità. Il gruppo vienegestito da educatori specializzati supervisionati da unopsicologo con competenze di tipo cognitivo-comporta-mentale. Questo ambiente stimolante facilita il recupero,l'integrazione e la creatività delle funzioni, riduce i disor-dini del comportamento e delle relazioni interpersonali,favorisce ed incrementa l'autostima del bambino.
• è molto importante effettuare un'attività di supportoe counseling ai familiari aiutandoli nell'elaborazione diquanto accaduto, riducendo il rischio di tensioni e conflitti,coinvolgendoli nel processo riabilitativo.
Il rientro a scuola viene preparato con molta attenzio-ne poiché i problemi presentati da questi bambini nonpossono essere assimilati a quelli tipici del paziente conritardo mentale, con problemi di apprendimento o condisturbi del comportamento. D'accordo con i familiari, pri-ma della dimissione, è necessario contattare gli insegnanti,al fine di renderli consapevoli delle problematiche delpaziente e di fornire indicazioni per un adeguato inter-vento didattico specializzato. Prima della dimissione ilServizio Sociale deve organizzare, con le strutture sanita-rie del territorio di residenza del paziente, l'eventuale pro-secuzione degli interventi.
In età evolutiva la presa in carico riabilitativa ed il fol-low up devono durare a lungo, spesso per tutta la fase disviluppo. Tutti i pazienti devono essere periodicamenterivalutati al fine di controllarne gli aspetti clinico-funzio-nali, di aggiornare il programma riabilitativo, di verifica-re l'eventuale insorgenza di complicazioni.
Bibliografia
Anderson NE. Late complications in childhood central nervous systemtumour survivors. Curr Opin Neurol 2003;16:677-683.
Anderson DM, Rennie KM, Ziegler RS, Neglia JP, Robison LR, Gurney JG.Medical and Neurocognitive late effects among survivors of child-hood central nervous system tumors. Cancer 2001;92: 2709-19.
Carpentieri SC, Mulhern RK, Douglas S, Hanna S, Fairclough DL. Behav-ioral resiliency among surviving brain tumors: a longitudinal study.Journal of Clinical Child Psychology 1993;2:236-46.
Douglas Ris M, packer R, Golwein J, Wallace D, Boyett JM. Intellectualoutcome after reduced-dose radiation therapy plus adjuvantchemotherapy for medulloblastoma: a children' cancer group
66
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre

67
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
study. J Clin Oncol 2001;19:3470-6. Fossen A, Abrahamsen TG, Storm-Mathisen I. Psychological outcome in
children treated for brain tumor. Pediatr Hematol Oncol1998;15:479-88.
Grill J, Keifper Renaux V, Bulteau C, Viguiler D, Levy-Piebois M, et al.Long-term intellectual outcome in children with posterior fossatumors according to radiation doses and volumes. Int J RadiatOncol Biol Phys 1999;45:137-45.
Hoppe-hirsch E, Brunet L, Laroussinie F, Cinalli G, Pierre-Kahn A, RenierD, et al. Intellectual outcome in children with malignant tumorsof the posterior fossa: influence of the field of irradiation andquality of surgery. Child's Nerv Syst 1995;11:340-6.
Kieran MW. Advances in pediatric neuro-oncology. Curr Opin Neurol2000;13:627-34.
Karatekin C, Lazareff JA, Asarnow RF. Relevance of the cerebellar hemi-spheres for executive functions. Pediatr Neurol 2000;22:106-12.
Levisohn L, Cronin-Golomb A, Schmahmann J D. Neuropsychologicalconsequences of cerebellar tumors resection in children. Brain.2000;123:1041-50.
Mulhern R M, Carpentieri S, Shema S, Stone P, Fairclough D. Factorsassociated with social and behavioral problems among childrenrecently diagnosed with brain tumor. J Pediatr Psychol
Packer RJ, Goldwein J, Nicholson HS, Vezina LG, Allen JC, Ris MD, et al.Treatment of children with medulloblastomas with reduced-dosecraniospinal radiation therapy and adjuvant chemotherapy: a chil-dren's cancer group study. J Clin Oncol 1999;17:2127-36.
Palmer SL, Goloubeva O, Reddick WE, Glass JO, Gajjar A, Kun L, et al. Pat-terns of intellectual development among survivors of pediatricmedulloblastoma: a longitudianal Analysis. J Clin Oncol2001;19:2302-8.
Radcliffe J, Bennett D, Kazak A E, Foley B, Phillips PC. Adjustment inchildhood brain tumor survival: child, mother, and teacher report.J Pediat Psychol 1996;21:529-39.
Reimers TS, Ehrenfels S, Mortensen EL, Schmiegelow M, Sonderkaer S,Carstensen H, et al. Cognitive deficits in long-term survivors ofchildhood brain tumors: identificatio of predictive factors. MedPediatr Oncol 2003;40:26-34.
Riva D, Giorgi C. The cerebellum contributes to higher functions duringdevelopment: evidence from a series of children surgically treat-ed for posterior fossa tumors. Brain 2000;123:1051-61.
La proteomica
Maria Monti, Marianna Caterino, Daniela Pagnozzi,Stefania Orrù, Piero PucciCEINGE Biotecnologie Avanzate SCARL, Napoli e Diparti-mento di Chimica Organica e Biochimica, Università diNapoli Federico II, Napoli, Italy
Il termine proteoma è utilizzato oggigiorno per indicarelo studio e la caratterizzazione di un elevato numero diproteine in miscele molto complesse provenienti daorganelli cellulari, intere cellule o addirittura (micro)-organismi. I moderni studi sul proteoma possono esseredivisi essenzialmente in due settori principali, il proteomadi espressione che tende alla definizione qualitativa equantitativa dell'aumento e/o diminuzione dei livelli diproteine, e il proteoma funzionale che tenta di identificarecomponenti di compartimenti cellulari, complessi multi-proteici e vie di trasduzione del segnale. Entrambi questiapprocci si basano sul frazionamento delle miscele di pro-teine mediante elettroforesi bidimensionale su gel di poli-acrilamide (2D-gel) e sulla successiva identificazione dellebande proteiche mediante tecniche di spettrometria dimassa. Questa procedura, infatti, è ottimale per l'analisiglobale di proteine in quanto si è dimostrata in grado diseparare, quantificare ed identificare migliaia di proteinepresenti in un singolo gel.
L'identificazione di bande proteiche da 2D-gel si ottienemediante costruzione della mappa peptidica di massa uti-lizzando essenzialmente la spettrometria di massa MAL-DI-TOF (Matrix-Assisted Laser Desorption Ionization-TimeOf Flight). La banda proteica è escissa dal gel, digerita insitu con opportuni enzimi proteolitici e la miscela pep-tidica risultante è direttamente analizzata mediante MAL-DI-MS. L'identificazione delle proteine viene effettuatautilizzando i valori di massa accurati dei peptidi determi-nati mediante MALDI-MS . Questi valori, infatti, insiemead altri parametri quali il tipo di proteasi usata per l'idrolisio il peso molecolare della proteina presunto dal gel di SDS,vengono introdotti in alcuni programmi di ricerca disponi-bili in rete (ProFound, Mascot, MS-Fit, etc. ). I valori dimassa registrati sugli spettri sono paragonati con quelliprovenienti dalla digestione teorica di tutte le proteinedella banca dati, consentendo l'identificazione delle pro-teine. Quando questa procedura non fornisce dati defini-tivi, vengono utilizzate metodologie di spettrometria dimassa tandem basate sulla ionizzazione ad electrospray(ES-MS/MS) per ottenere informazioni anche parziali disequenza di uno o più frammenti proteolitici. è statodimostrato che le informazioni ottenute dalla sequenzaparziale di un solo peptide sono spesso sufficienti ad iden-tificare la proteina in banca dati.
In genere gli studi di proteomica di espressione sonoindirizzati all'analisi delle differenze del pattern di espres-sione in cellule anormali (i. e. tumorali, stimolate da trat-tamenti farmacologici, etc. ) in paragone con quelle nor-mali, attraverso l'identificazione di bande proteiche checompaiono (o scompaiono) o che sono presenti a livelliquantitativi diversi. Nelle applicazioni biomediche, questoapproccio comparativo viene di solito utilizzato per iden-tificare proteine il cui livello di espressione aumenta o

diminuisce in seguito all'insorgere di una patologia e chepossono quindi essere utilizzate come marker specifici perla diagnosi o in qualità di target a scopo terapeutico. Inquesti studi quindi è particolarmente importante potercontare su una affidabile analisi delle variazioni quanti-tative delle singole bande. Questa analisi era di solitoottenuta dalla valutazione dell'intensità del colore dellesingole bande, una procedura laboriosa e niente affattoaccurata. Di recente, nelle procedure di proteomica diespressione sono stati introdotti metodi di quantizzazionebasati sulla modifica delle proteine mediante uso di reat-tivi marcati con isotopi stabili che consentono di deter-minare le variazioni relative nell'ammontare delle pro-teine. Il principio alla base di questi metodi consiste nel-l'incorporazione di un reattivo contenente un isotopo sta-bile nelle proteine provenienti da uno dei due stati che sivuole esaminare. Il rapporto quantitativo di una singolaproteina nei due stati può quindi essere valutato accu-ratamente dalla misura del rapporto dei due segnali cor-rispondenti al campione derivatizzato e non derivatizza-to. Infine, lo studio del proteoma di espressione necessitadi metodi in grado di provvedere all'analisi simultanea diun enorme numero di proteine. Recenti progressi nel cam-po dell'automazione sia per quanto riguarda il trattamentodei campioni utilizzando sistemi robotici che l'acquisizionee l'interpretazione degli spettri di massa mediante oppor-tuni programmi contribuiranno a velocizzare il processo diidentificazione delle proteine.
Proteoma funzionaleUna delle più importanti questioni nella moderna ricer-
ca biologica riguarda la comprensione a livello molecolaredelle funzioni di una cellula vivente. Poiché ormai è notal'intera sequenza del genoma di vari organismi, il numerodi proteine la cui funzione è ancora sconosciuta è cresci-uto enormemente negli ultimi tempi. Nel tentativo diottenere informazioni sull'attività biologica di singole pro-teine, si è sviluppato l'analisi del cosiddetto proteoma fun-zionale che potrebbe essere definito come lo studio dellefluttuazioni spaziali e temporali dei componenti e dei pro-cessi molecolari che avvengono in una cellula vivente.Infatti, in una cellula molti processi non vengono con-trollati soltanto dall'abbondanza relativa delle varie pro-teine, ma anche dalla regolazione transiente della loroattività mediante ad esempio modificazioni covalentireversibili, dalla loro localizzazione cellulare e dall'associ-azione con altri componenti. Gli studi di proteomica fun-zionale sono quindi indirizzati verso due obiettivi princi-pali, la comprensione della funzione biologica di proteinesconosciute e la definizione dei meccanismi molecolaridei più importanti processi cellulari.
Entrambi questi obiettivi possono essere perseguitimediante lo studio delle interazioni proteina-proteina invivo. E’ oggi chiaro, infatti, che un gran numero di proteinesvolge la sua funzione nella cellula associandosi sotto for-ma di complessi multiproteici; ne consegue che la com-prensione delle funzioni biologiche di queste proteine e ladelucidazione dei meccanismi molecolari attraverso cuiqueste funzioni sono svolte è legata all'identificazione deiloro partners molecolari. Procedure basate sull'analisi pro-teomica possono fornire un contributo basilare all'identi-ficazione dei componenti dei complessi multiproteici e
rappresentano oggigiorno un efficace metodo alternativoalle metodologie di biologia molecolare basato sulla tec-nica del doppio ibrido. La filosofia del metodo comune atutte le strategie sviluppate consiste nella possibilità diesprimere la proteina di interesse (esca) in forma ricom-binante modificata con uno specifica marcatura (tag); ilcomplesso dei partners molecolari della proteina esca puòquindi essere purificato dall'intero estratto cellulare medi-ante tecniche basate sulla cromatografia di affinità uti-lizzando l'opportuno ligando (anti-tag) immobilizzato suun supporto insolubile.
Negli ultimi anni sono state sviluppate diverse strate-gie che si basano su questo semplice concetto. Utilizzan-do sistemi di espressione di proteine commercialmentedisponibili, la proteina esca può essere espressa come ibri-do di fusione con la Glutatione-S-transferasi (GST-fusedprotein) o modificata con vari epitopi (FLAG, HA, Myc) ocon una coda di istidine (poli-His). In tutti i casi, l'esca puòessere immobilizzata su un supporto solido sfruttando l'in-terazione specifica del tag con un opportuno ligando. Adesempio, si possono utilizzare particelle di agarosio deriva-tizzate con glutatione per legare le proteine ibride conGST, o con opportuni anticorpi anti-epiropo (FLAG, HA oMyc) od infine le resine ad ioni Nichel che interagisconospecificamente con la coda di istidine. Una volta immobi-lizzata, l'esca viene incubata con l'intero estratto cellulareo, quando è il caso, con estratti da organelli specifici alloscopo di pescare i suoi specifici partners molecolari, di quiil nome dato a questa procedura Fishing for Partners. Laproteina esca, infatti, stabilisce interazioni stabili, noncovalenti con i suoi partners specifici presenti nell'estrat-to cellulare che saranno quindi trattenuti, mentre le pro-teine non riconosciute dall'esca saranno eluite durante illavaggio. I componenti proteici trattenuti sono quindieluiti e frazionati mediante SDS-PAGE. Le bande proteichevisualizzate sul gel sono digerite proteoliticamente in situe le miscele di peptidi risultanti sono analizzate mediantespettrometria di massa MALDI (MALDI-MS) o conmetodologie di cromatografia liquida accoppiata allaspettrometria di massa tandem (LC-MS/MS), consenten-do l'identificazione delle proteine secondo la proceduradescritta in precedenza.
La stessa strategia può anche essere applicata all'iden-tificazione di proteine che interagiscono con il DNA o l'R-NA utilizzando uno specifico oligonucleotide legato a par-ticelle di agarosio come esca.
Una strategia alternativa prevede che la proteina escasia marcata con un epitopo per il quale è disponibile unbuon anticorpo (FLAG, HA, Myc, etc. ). L'esca, trasfettatanella particolare linea cellulare dove si vuole condurre lostudio, viene sovraespressa nella cellula dove formerà uncomplesso multiproteico interagendo con i suoi partnersspecifici. Una volta formatosi, il complesso viene immuno-precipitato utilizzando un anticorpo specifico per l'epi-topo con cui è stata marcata la proteina esca. Le proteinecostituenti il complesso immunoprecipitato sono quindifrazionate mediante SDS-PAGE ed identificate utilizzan-do metodologie di spettrometria di massa secondo la pro-cedura descritta.
Infine, nella metodologia denominata Tandem AffinityPurification (TAP), il gene codificante per la proteina diinteresse viene modificato mediante aggiunta di un
68
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre

69
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
costrutto contenente due differenti tags di affinità. Adesempio, nella proposta originale del metodo il costruttomodificante conteneva la Proteina A, specificamentericonosciuta dalle Immunoglobuline G (IgG) e un peptidecapace di interagire fortemente con la Calmodulina. Tra leregioni codificanti per i due tags viene inserito un sito diidrolisi specifico per la Proteasi del Virus del tabacco (TEV).La manipolazione genetica non altera la funzionalità delgene e quindi la proteina esca doppiamente marcata puòessere espressa nella cellula in condizioni fisiologiche eda livelli normali. Una volta espressa l'esca interagisce coni suoi specifici partners intracellulari ed il complesso for-mato può essere purificato mediante due successivi pas-saggi di cromatografia di affinità. Nel primo, il complessoè isolato dall'intero estratto cellulare mediante interazionecon particelle di agarosio derivatizzate con IgG; una vol-ta allontanate le proteine non interagenti, il complessosempre legato al supporto di agarosio è trattato con TEVche idrolizza il legame con la Ptorina A e causa il rilasciodel complesso. A questo punto il secondo passaggio dipurificazione è condotto utilizzando l'affinità del secon-do tag per la Calmodulina. Il complesso così purificato èfrazionato nei suoi componenti mediante SDS-PAGE e leproteine partecipanti vengono identificate come descrit-to in precedenza.
Osteopetrosi
Annalisa FrattiniSegrate, Milano
Il termine osteopetrosi definisce un eterogeneo gruppodi malattie ossee la cui base risiede in un difetto a caricodegli osteoclasti, le cellule responsabili del riassorbimen-to della matrice ossea.
Sono note varie forme di osteopetrosi classificate sia inbase alla loro gravità che al tipo di trasmissione ereditaria.
Sono conosciute due forme autosomiche dominanti(ADOI ed ADOII) benigne, caratterizzate da una progressi-va sclerosi ossea generalizzata, con insorgenza o meno difratture non traumatiche.
La forma dominante di tipo I (ADOI), spesso asintomat-ica, è caratterizzata da una sclerosi pronunciata soprat-tutto a livello della volta cranica, mentre la forma domi-nante di tipo II (ADOII, detta anche malattia di Albers-Schomberg) è caratterizzata da una sclerosi ossea pro-nunciata alla base del cranio, a livello vertebrebrale e delleossa pelviche. I pazienti affetti da quest'ultima forma pre-sentano frequenti fratture non traumatiche e osteomieli-ti.
Esistono forme di gravità intermedia con insorgenza del-la patologia in età infantile e che presentano un'ampiavariabilità sintomatologica. Infatti possono manifestare omeno anemia, difetti visivi, insorgenza di fratture sponta-nee. Queste forme sono a tutt'oggi non ben classificabilisu base genetica, data la loro eterogenicità.
La forma autosomica recessiva (ARO) colpisce gli affet-ti già prima della nascita e porta a morte entro i primi annidi vita se non viene effettuato tempestivamente untrapianto di midollo osseo, che non sempre è risolutivo.
Dal punto di vista del quadro clinico, le forme recessivesono generalmente molto severe, perché l'assoluta man-canza di riassorbimento della matrice ossea provoca unprogressivo ispessimento dell'osso, con conseguenteriduzione dello spazio midollare. Questo causa anemia,pancitopenia ed epatosplenomegalia; inoltre i pazientisono colpiti da progressiva cecità e sordità dovute alrestringimento dei forami cranici attraversati dai nervi otti-
Malattie orfane in Ematologia ed Immunologia
Tabella.
Malattia Localizzazione Gene cromosomica responsabile
Osteopetrosi con acidosi tubulare renale 8q22 Anidrasi carbonica IIADOI 11q13. 4 LRP5ADOII 16p13 ClCN7Intermedia autosomica dominante 16p13 ClCN7Intermedia autosomica recessiva 16p13 ClCN7Maligna autosomica recessiva 16p13 ClCN7Maligna autosomica recessiva 11q13. 4-q13. 5 ATP6a3Maligna autosomica recessiva 6q21 GL

70
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre
ci ed acustici. Negli ultimi anni sono stati individuati due geni coinvolti
nella ARO umana: ATP6a3 (Atp6i, TCIRG1, OC116) e ClCN7. Il gene ATP6a3 codifica per la subunità a3 di un comp-
lesso proteico che costituisce una pompa protonica local-izzata nella membrana degli osteoclasti. Tale complesso èin grado di creare un ambiente acido idoneo per il fun-zionamento degli enzimi proteolitici necessari per ladegradazione della matrice ossea.
Il gene ClCN7 codifica per un canale del cloro presentenella membrana plasmatica degli osteoclasti, la cui fun-zione principale è quella di creare un ambiente acido extra-cellulare.
Analizzando una grande casistica di pazienti con pre-sentazione clinicamente severa, abbiamo potutodimostrare come circa il 60% di essi abbia mutazioni nelgene ATP6a3, mentre solo circa il 10% presenta unamutazione nel gene CLCN7. Tuttavia, mentre pazientiATP6a3-dipendenti appaiono omogenei nel quadro clini-co e hanno entrambi gli alleli mutati, le forme ClCN7-dipendenti sono molto più eterogenee.
Mutazioni nel gene ClCN7 sono responsabili sia delleforme più severe (letali nella maggior parte dei casi) in cuientrambi gli alleli sono mutati ed il quadro clinico è ampia-mente compromesso da un punto di vista osseo, emato-logico e presenta deficit nervosi. sia di forme severe(osteosclerosi generalizzata, anemia,cecità e fratture) incui un solo allele è mutato, fino a forme intermedie (reces-sive e dominanti) e a forme benigne o addirittura asin-tomatiche rappresentative della classica ADOII, forma dicui il gene ClCN7 risulta responsabile nel 90% dei casi.
Infine la recente identificazione del gene responsabileper l'osteopetrosi murina denominata Grey Lethal ha per-messo di dissecare ulteriormente l'eterogeneità delle AROumane, anche se il coinvolgimento del gene GL nell'uomorisulta limitato all'1-3% di casi.
Le porfirie
Maria Domenica Cappellini, E. DiPierro, E. Piatti, D.Tavazzi, G. FiorelliCentro Anemie Congenite, Ospedale Maggiore PoliclinicoIRCCS, Università di Milano
Con il termine di porfirie si indica un gruppo di patolo-gie derivanti da un difetto enzimatico ereditario o acquisi-to della via biosintetica dell'eme. 1,2 Tutte le porfirie sonocaratterizzate dal difetto di almeno uno degli enzimi coin-volti in questa via con conseguente iperproduzione, accu-mulo ed escrezione dei prodotti intermedi. Difetti geneticisono stati dimostrati per ognuno degli otto enzimi dellavia biosintetica dell'eme, con il conseguente blocco della viain corrispondenza dell'enzima coinvolto e l'accumulo delprecursore a monte dell'enzima difettivo; ne conseguonodiversi pattern di aumentata escrezione urinaria dei pre-cursori dell'eme, ognuno dei quali caratterizza uno dei set-te tipi conosciuti di porfiria. Il difetto enzimatico eredita-rio è nella maggioranza dei casi solo parziale (allo stato dieterozigosi) in quanto il blocco completo della sintesi del-l'eme sarebbe letale.
Le porfirie sono classificate in acute o croniche in rap-porto alle caratteristiche cliniche della malattia e in eri-tropoietiche o epatiche a seconda del sito di espressioneprincipale del difetto enzimatico. La sola vera porfiria eri-tropoietica è una forma rara definita Porfiria Eritropoieti-ca Congenita (CEP) o Malattia di Gunther, caratterizzatadalla ipersecrezione e dall'accumulo di porfirine prevalen-temente negli eritrociti, nel midollo, nel plasma e in eleva-ta quantità nelle urine fin dalla nascita, mentre nella Pro-toporfiria Eritropoietica (EPP) si osserva un accumulo diporfirine sia nel tessuto eritropoietico che epatico. NellaPorfiria Acuta Intermittente (AIP), nella Coproporfiria Ere-ditaria (HCP) e nella Porfiria Variegata (VP), i difetti enzi-matici favoriscono l'accumulo di porfirine prevalentemen-te nel fegato. 1
Le principali manifestazioni cliniche di queste patologiesono attacchi intermittenti di alterata funzione del siste-ma nervoso e/o fotosensibilità della cute esposta alla luce.La sindrome neurologica è solitamente scatenata da far-maci quali i barbiturici, estrogeni, etc. e può manifestarsisia a carico del sistema nervoso centrale che di quello vege-tativo oltre che interessare i nervi periferici. La fotosensi-bilità è invece correlata generalmente all'aumentato accu-mulo cutaneo di porfirine, anche se il tipo di lesioni osser-vate è diverso nelle varie forme della malattia. 2
Nella Tabella 1 sono riassunte le caratteristiche biochi-miche e cliniche delle principali forme di porfirie. La mag-gior parte degli individui portatori di una porfiria eredita-ria sono in realtà asintomatici e solo l'intervento di altricofattori genetici o ambientali è in grado di provocare gliattacchi acuti.
La prevalenza delle diverse forme di porfiria varia da unazona geografica all'altra e non sempre è definitivamentestabilita. Nel complesso si tratta di malattie ritenute pocofrequenti che possono però raggiungere un'alta prevalen-za in alcune aree geografiche. In Scozia un soggetto su50.000 è affetto da una forma di porfiria, con prevalenzadella acuta intermittente e della cutanea tarda. In Sud Afri-

ca la forma prevalente è la porfiria variegata con un inci-denza di 1 su 1.000 nella popolazione bianca, mentre i Ban-tu soffrono prevalentemente di porfiria cutanea tarda. 1
Le porfirie acuteAttacchi acuti molto simili tra loro sono stati descritti per
la porfiria acuta intermittente (AIP), la coproporfiria eredi-taria (HCP), la porfiria variegata (VP) e la porfiria da deficitdi ALA-D (ADP). Essi si manifestano principalmente con sin-tomatologia gastrointestinale (vomito e costipazione) spes-so associata a disturbi psichici (ansia, depressione, diso-rientamento, confusione, delirio) e neurologici (neuropatieperiferiche, insufficienza respiratoria, epilessia). Talvolta lemanifestazioni neurologiche sono da considerarsi compli-cazioni dovute ad interventi terapeutici non appropriati.Nonostante il carattere autosomico di queste porfirie essesi manifestano soprattutto nelle donne (80% dei casi) tra
i 18 e i 45 anni di età; questo dato è supportato dal ruoloscatenante riconosciuto agli estrogeni. Nella Tabella 2 sonoelencati i sintomi ricorrenti negli attacchi acuti di porfiria.Per una più corretta diagnosi si rendono quindi necessarialtri esami di laboratorio più specifici basati sull'analisi bio-chimica dei pattern di escrezione delle porfirine e più recen-temente sull'analisi molecolare dei geni coinvolti nei difet-ti enzimatici. 5 Tra le forme acute l'AIP è la più frequente.
Le porfirie cronicheTra le porfirie croniche, la porfiria cutanea tarda (PCT) e
la protoporfiria eritropoietica (EPP) sono le uniche ad ave-re una rilevanza clinica in termini di incidenza, in quantola porfiria epatoeritropoietica (HEP) e la porfiria eritro-poietica congenita (CEP) sono estremamente rare. La sin-tomatologia cutanea è la manifestazione clinica predomi-nante in queste forme di porfiria. Le lesioni cutanee da
71
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
Tabella 1.Caratteristiche biochimiche e cliniche delle porfirie.
Principali alterazioni biochimichePorfiria Enzima Ereditarietà Manifestazioni Eritrociti Urine Feci
cliniche
AAccuutteeDeficit di ALA-D ALA-D autosomica recessiva Neuroviscerale Zn-protoporfirine ALA −
P. acuta intermittente PBGD autosomica dominante Neuroviscerale − ALA, PBG −
Coproporfiria CPOX autosomica dominante Neuroviscerale ereditaria ± fotosensibilità − ALA,PBG, coproporfirine −
P. variegata PPOX autosomica dominante Neuroviscerale ± fotosensibilità − ALA,PBG, coproporfirine coproporfirine, protoporfirine
CCrroonniicchheeP. cutanea tarda URO-D Variabile Fotosensibilità − uroporfirine, isocopro-porfirine
7-carbossiliche
P. epatoeritro-poietica URO-D autosomica recessiva Neuroviscerale Zn-protoporfirine uroporfirine, isocopro-porfirine± fotosensibilità 7-carbossiliche
P. eritropoietica congenita URO-S autosomica recessiva Fotosensibilità uroporfirine, coproporfirine uroporfirine, coproporfirine coproporfirine
Protoporfiria eritropoietica FECH autosomica dominante Fotosensibilità protoporfirine − protoporfirine
Tabella 2.Principali manifestazioni cliniche nelle porfirie acute.
Disturbi neuroviscerali Iperattività del sistema nervoso simpatico Disturbi del sistema nervoso centrale Disturbi del sistema nervoso periferico
Dolori addominali Tachicardia Depressione Deficit motorioNausea Ipertensione arteriosa Disorientamento CrampiVomito Ipotensione ortostatica (rara) Irrequietezza ParaplegiaStipsi Tremori Instabilità emotiva TetraplegiaDiarrea (rara) Ritenzione urinaria Allucinazioni Atrofia del nervo otticoFebbre Sudorazione eccessiva Ansia Disfagia
Squilibri elettrolitici Insonnia OftalmoplegiaDelirio Riflessi tendinei assenti
Convulsioni ParaestesiaCrisi epilettiche Ipoestesia
Paralisi respiratoria

72
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre
fotosensibilità sono generalmente limitate alle superficiesposte al sole (dorso delle mani, collo, gambe e caviglienelle donne). Si osservano inoltre frequentemente fragilitàcutanea (croste, bolle, cicatrici), ipertricosi (parte superio-re delle guance, orecchie, braccia) e alopecia. Le principalimanifestazioni cliniche delle porfirie croniche sono rias-sunte in Tabella 3. Anche in questo caso la diagnosi diffe-renziale risulta difficile in quanto lo stesso tipo di sinto-matologia cutanea è osservabile anche nella VP e nella HCPe sono quindi necessari esami specifici più approfonditi. 1
Bibliografia
1. Kappas A, Sassa S, Gelbraith RA, Nordmann Y. The porphyrias. In:Scriver CA, Beaudet AL, Sly WS, Valle D, editors. The metabolicbasis of inherited disease, 7th edn. McGraw-Hill, New York; 1995.p. 2103-59.
2. Bonkovsky HL. Porphyrin and heme metabolism and the porphyr-ias. In: Hepatology: a textbook for liver disease. Editori: Zakim D., Boyer T. D. , W. B. Saunders, Philadelphia; 1982.p. 351-93.
3. McDonagh AF, Bissel M. Porphyria and Porphyrinology. The past fif-teen years. Semin Liv Dis 1998;18:3-16.
4. Bonkovsky HL, Barnard G. Diagnosis of porphyric syndromes: apratical approach in the era of molecular biology. Semin Liv Dis1998;18:57-66.
5. Meyer UA, Schuurmans MM, Lindberg RLP. Acute porphyrias:pathogenesis of neurological manifestations. Semin Liv Dis 1998;18:43-52.
Le stomatocitosi ereditarie
Achille Iolascon, Loredana BoschettoGenetica Medica, Dipartimento di Biochimica e TecnologieBiomediche, Università Federico II di Napoli, CEINGE- Bio-tecnologie Avanzate, Napoli
Molte malattie emolitiche sono associate a cambia-menti di forma e di volume delle emazie. Gli stomatoc-iti sono globuli rossi che presentano, al posto dell'areacentrale di pallore, una zona allungata che fornisce allecellule l'aspetto di una bocca (dal greco stomatocita).1,2,3 Questa anomalia della forma è correlata alla con-centrazione dei cationi monovalenti intracellulari, che ècritica per il mantenimento della forma e deformabilitàdelle cellule. Un incremento di Na e K causa un richiamodi acqua all'interno della cellula, con formazione di unostomatocita o idrocita; mentre una perdita di cationi dàvita ad un globulo rosso disidratato o xerocita. 2-4 Talecondizione emolitica associata alla presenza in circolo distomatociti viene ereditata con modalità autosomicodominante. Si tratta di un gruppo di malattie che pos-sono essere divise in due raggruppamenti principali: ilprimo è formato dalla forma overidrata di stomatocitosi(OHS), in cui l'emolisi è associata ad un flusso asimmet-rico di cationi con incremento del passaggio passivo diNa, che sovrasta l'efflusso di K. Tale malattia emoliticaè associata alla scomparsa della stomatina o proteina 7.2b, una proteina transmembrana che ha una funzioneancora sconosciuta. L'emazia appare rigonfia da uneccesso di ioni e di acqua, con diminuzione della con-centrazione emoglobinica (MCHC) ed un aumento dellafragilità osmotica. Recentemente è stato dimostrato cheil deficit di stomatina compare in una fase tardiva dellaeritropoiesi e che il gene della stomatina non sembraessere implicato nella patogenesi della malattia. 5
La forma deidrata (DHSt) detta anche xerocitosi ered-itaria (HX) (MIM194380) rappresenta la condizioneopposta in termini di difetto della permeabilità. Leemazie sono come disseccate con un basso contenutointerno di ioni ed acqua ed un corrispondente aumentodell'MCHC. La fragilità osmotica è aumentata, mentre iltest dell'autoemolisi è aumentato e corretto dalla pre-senza di glucosio. Nella DHSt/HX il difetto fondamentaleè un aumento dell'efflusso del potassio dall'eritrocita.L'attività di pompa non è in grado di compensare taledifetto, e la concentrazione intracellulare di cationi e diacqua è diminuita, con conseguente disidratazione cel-lulare. Eventi conseguenti sono rappresentati anche dal-l'aumento di Ca intracellulare e dalla formazione di lega-mi incrociati tra le proteine del citoscheletro. Questaentità clinica fu descritta per primo da Glader nel 1974e confermata dalla descrizione di altri autori. Il quadroclinico è eterogeneo variando dalla asintomaticità allamalattia emolitica grave. Per quanto concerne il tratta-mento, anche se le esperienze cliniche sono limitate dal-la rarità della malattia, si può affermare che la splenec-tomia non riduce in maniera significativa la emolisi inpazienti con DHSt/HX, mentre sembrano beneficiarsiquelli con OHS.
Il difetto molecolare della DHSt/HX è ignoto. Il nostro
Tabella 3. Principali manifestazioni cliniche nelle porfirie croniche.
Sintomatologia cutanea Sintomatologia eritroepatica
Fotosensibilità Epatopatia cronicaFragilità cutanea Calcolosi biliareBolle sulle mani Anemia normociticaEritema Iperplasia eritroblastica midollareEczemaErosioniCicatriciCrosteCalcificazione dermicaIspessimento dermicoLichenificazioneAlterazioni unguealiIperpigmentazioneIpertricosiAlopeciaPrurito (EPP)Eritrodonzia (CEP)

gruppo di ricerca ha avuto l'occasione di collaborare conil gruppo francese di J. Delaunay e con quello inglese diG. Steward e di poter ottenere i campioni provenientidalla loro esperienza. I gruppi più numerosi sono quelliirlandesi, che da un punto di vista genetico sono moltoinformativi. Grazie alla creazione di un registro inter-nazionale per le Stomatocitosi ereditarie e alla annessabanca di materiale biologico abbiamo affrontato il prob-lema del clonaggio posizionale di queste patologie.
Mediante un genome wide search ed utilizzando un setdi 400 microsatelliti distribuiti ad intervalli di 10 cM suogni cromosoma abbiamo potuto mappare il locus per laDHSt/HX su braccio lungo del cromosoma 16. Abbiamopoi saturato la regione che conteva il microsatelliteD16S520 con altri marcatori in modo da poter megliodefinire la localizzazione del gene malattia (D16S402,D16S422,D16S3037, D16S498, D16S3074, D16S413,D16S3026 and D16S3121 (10). Il lod score (punteggio diassociazione) più alto è stato ottenuto con il marcatoreD16S520 (Zmax=5. 41 per q=0.00), mentre il limite cen-tromerico della regione è stato definito grazie ad alcunimembri delle famiglie che presentavano una ricombi-nazione.
Per l'identificazione del gene localizzato sul cromos-ma 16 abbiamo seguito l'approccio per geni candidati. Leemazie dei pazienti sono caratterizzate da elementimolto interessanti che possono essere di grande aiutonell'identificazione di geni candidati. In particolare leemazie presentano un aumentata concentrazione intra-cellulare di Na e corrispondenti ridotti livelli di K. Glistudi mediante traccianti mostrano un aumento del flus-so e delle capacità di pompa. In particolare il flussoinsensibile alla ouabaina più bumetanide (obi) e la pom-pa del K ouabaina sensibile (OS) sono aumentati di 2-3volte rispetto al normale, riflettendo il flusso passivoaumentato e la conseguente accelerazione della pompaNa/K caratteristiche di questa malattia. 11 Quindi i genicoinvolti nel trasporto del Na e/o degli ioni K possonoessere considerati tutti dei buoni candidati per spiegaretale patologia. Fino ad oggi abbiamo definito la sequen-za esoni-introni di 6 possibili geni candidati e ne abbi-amo effettuato la sequenza del DNA. Purtroppo nessunodi questi ha mostrato mutazioni in grado di poter spie-gare la patologia ed in linkage con la ereditarietà dom-inante.
Più recentemente grazie ad un genome wide searcheffettuato sul DNA di un nucleo familiare proveniente daLylle ed affetto da una forma di pseudoikerkalemiafamiliare (una variante di xerocitosi) abbiamo potutolocalizzare un secondo gene sul cromosoma 2.L'analisicomparata dei geni codificati in queste due aree puòportare alla identificazione di questi geni-malatti checausano le stomatocitosi.
Bibliografia
1. Delaunay J, Stewart G, Iolascon A. Hereditary dehydrated andoverhydrated stomatocytosis: recent advances. Curr OpinHematol 1999;6:110-4.
2. Iolascon A, Perrotta S, Gordon W. Steward- Red blood cellmembrane defects Rev Clin Exp Hematol 2003;7:1-35.
3. Lock SP, Sephton Smith R, Hardisty RM. Stomatocytosis: ahereditary haemolytic anomaly associated with haemolyticanaemia. Br J Haematol 1961;7:303-14.
4. Carella M, Stewart GW, Ajetunmobi JF, Perrotta S, GrootenboerS, Tchernia G, et al. Genomewide search for dehydrated hered-itary stomatocytosis (hereditary xerocytosis) mapping of locusto chromosome 16 (q23-qter). Am J Hum Genet 1998;63:810-6.
5. Fricke B, Argent AC, Chetty MC, Pizzey AR, Turner EJ, Ho MM,ET AL. The stomatin gene and protein in overhydrated heredi-tary stomatocytosis. Blood 2003;102:2268-77.
6. Iolascon A, Stewart GW, Ajetunmobi JF. Familial pseudohy-perkalemia maps to the same locus as dehydrated hereditarystomatocytosis (hereditary xerocytosis) Blood 1999;93:3120-3.
7. Grootenboer S, Schischmanoff P-O, Laurendeau I, Cynober T,Tchernia G, Dommergues J-P, et al. Pleiotropic syndrome ofdehydrated hereditary stomatocytosis, pseudohyperkalemia,and perinatal edema maps to 16q23-q24 Blood 2000;96 2599-605.
8. Carella M, Stewart GW, Ajetunmobi JF, Schettini Jr F. , Delau-nay J, Iolascon A. Genetic heterogeneity of stomatocytosis syn-dromes showing pseudohyperkalemia Haematologica1999;84:862-4.
9. Jarvis HG, Chetty MC, Nicolaou A, Fisher J, Miller A, StewartGW. A novel stomatocytosis variant showing marked abnor-malities in intracellular [Na] and [K] with minimal haemolysis.Eur J Haematol 2001;66:412-4.
10. Haines PG, Jarvis HG, King S, Noormohamed FH, Chetty MC,Fisher J, et al. Two further British families with the 'cryohy-drocytosis' form of hereditary stomatocytosis. Br J Haematol2001;113:932-7.
11. Chetty MC, Stewart GW. Pseudohyperkalaemia and pseudo-macrocytosis caused by inherited red-cell disorders of the'hereditary stomatocytosis' group. Br J Biomed Sci 2001;58:48-55.
73
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

Neutropenie congenite
Fabio TucciUO Oncoematologia, Azienda Meyer, Firenze
Le neutropenie congenite sono un gruppo di malattierare caratterizzate dalla riduzione marcata nella conta deineutrofili circolanti (Neutrofili < 0.5×109/L) presente giàalla nascita. la S. di Kostmann e la neutropenia ciclicacostituiscono le forme più frequenti di neutropenia con-genita grave isolata. In altri casi (p. e. Glicogenosi tipo I Be S. di Shwachman-Diamond, WHIM Syndrome, S. diGriscellì, Sindrome di Barth, Discheratosi congenita) laneutropenia è presente in associazione manifestazioniclinico/metaboliche più complesse.
Kostmann per primo descrisse una agranulocitosi infan-tile genetica a trasmissione autosomica recessiva in unafamiglia svedese nel 1956. 1 La sindrome di Kostmann (SK)è caratterizzata da grave e persistente neutropenia (N <0.2×109/L), presente anche fin dalla nascita, frequentemomocitosi relativa, infezioni batteriche ricorrenti, arrestomaturativo a livello di promielocito/mielocito all'esamedell'aspirato midollare.
In seguito è diventato chiaro che la neutropenia con-genita ereditaria rappresenta un gruppo eterogeneo dimalattie sia da un punto di vista genetico che, probabil-mente, fenotipico, cosìcchè attualmente si preferisce par-lare di Neutropenie Congenite Gravi (NCG) che compren-dono , oltre ai casi a trasmissione autosomica recessivi(SK) anche quelli a trasmissione autosomica dominante(rari) ed i casi sporadici (frequenti). 2 La frequenza stima-ta delle NCG è di 1 -2 casi×106. 9
I pazienti con NCG soffrono di infezioni batteriche graviche compaiono usualmente nel corso del primo anno divita. Una onfalite, subito dopo la nascita, può essere il pri-mo segno di NCG. Frequenti sono le infezioni, spesso mor-tali nonostante un'adeguata terapia antibiotica, delle altee basse vie respiratorie, le celluliti e gli ascessi epatici. Lamaggior parte dei pazienti presenta inoltre stomatitiaftose ricorrenti, iperplasia gengivale con perdita precocedei denti definitivi. I germi più frequentemente isolati sonostafilococci e streptococci, seguiti da Pseudomonas e Pep-tostreptococcus. Relativamente frequenti sono inoltre leinfezioni da Clostridium, mentre molto più raro è l'isola-mento di funghi.
In era pre-rHuG-CSF il 42% dei casi pubblicati morivaentro i 2 anni di vita generalmente per sepsi o polmonite.
I pazienti con SK presentano livelli di G-CSF normali oaumentati, con normale attività biologica; i recettori peril G-CSF sono normoespressi sulle cellule mieloidi e lacostante di legame tra recettore e G-CSF è normale. 2
Il difetto genetico alla base della SK e delle NCG è anco-ra in gran parte sconosciuto.
In gran parte dei pazienti con NCG sono state eviden-ziate mutazioni eterozigoti puntiformi costituzionali delgene ELA2 che codifica per la elastasi dei neutrofili, cos-tituente principale dei granuli azzurrofili dei promieloci-ti. Il pattern di localizzazione delle mutazioni delle NCGnon è sovrapponibile a quello descritto precedentementenella neutropenia ciclica. In questo secondo tipo di neu-tropenia congenita a trasmissione autosomica dominante,
le mutazioni, presenti nella totalità dei casi, sono con-centrate prevalentemente negli esoni 4 e 5 o alla giun-zione tra esone 4 ed introne 4. 4
Le mutazioni puntiformi del gene ELA2 descritte piùrecentemente nei casi di NCG a trasmissione autosomicadominante, nei casi sporadici ed in parte dei casi di SKsono invece più disperse. Fino ad oggi sono state descritte47 differenti mutazioni del gene ELA2.5 Non è ancorachiaro il rapporto tra presenza/tipo di mutazione e svilup-po della NCG, anche se sembra che mutazioni che portanoalla sintesi di una proteina incompleta o che interessinoil sito catalitico dell'enzima possano contribuire ad unaaccelerata apoptosi dei precursori mieloidi.
Il trapianto di cellule staminali emopoietiche da dona-tore HLA compatibile ha rappresentato in passato l'unicaopzione terapeutica nei pazienti con NGG. 3
Dal 1987 l'rHuG-CSF è stato impiegato nel trattamen-to della NCG. Dai dati del Severe Chronic NeutropeniaInternational Registry (SCNIR) istituito nel1994, cheattualmente raccoglie prospetticamente i dati di oltre 400casi di NCG, risulta che oltre il 90% dei pazienti, indipen-dentemente dal tipo di NCG, risponde al trattamento con-tinuativo con rHuG-CSF con un aumento dei neutrofili >a 1.0×109/L con conseguente riduzione drastica delnumero e della gravità degli episodi infettivi, dei giorni diricovero, del consumo di antibiotici e con aumento dellasopravvivenza e della qualità di vita. Nella maggior partedei casi è sufficiente una dose di rHuG-CSF compresa trai 3 ed i 10 µg/Kg/die2 sottocute.
Per tali motivi la terapia con G-CSF rappresenta attual-mente la terapia di prima scelta nei pazienti con NCG(2), mentre permane la indicazione al trapianto di cellulestaminali emopoietiche nei casi di NCG che non rispon-dono al trattamento con rHuG-CSF. 2,13
Con l'aumento della sopravvivenza sono diventati evi-denti nuove caratteristiche cliniche tra cui la presenza diosteoporosi/osteopenia, vasculiti, splenomegalia e possi-bile evoluzione in MDS/LMA. 2
Già in era pre-G-CSF erano stati descritti casi isolati diNCG con evoluzione in LAM. 6,7 Con l'aumento dellasopravvivenza grazie alla terapia con G-CSF, secondo idati del SCNIR Europeo, una evoluzione in MDS/AML èstata osservata nel 12% dei casi (mediana di follow up di6 anni). La trasformazione in MDS/AML è preceduta oassociata alla comparsa di una o più alterazioni geniche,come la perdita parziale o completa del cromosoma 7 (7q-o monosomia 7), mutazioni attivanti dell'oncogene ras emutazioni del gene per il recettore per il G-CSF, tutti seg-ni di una instabilità genetica delle NCG. 11 In particolareuna mutazione puntiforme del gene per il recettore del G-CSF (G-CSF-r) è stata inizialmente descritta in un caso diNCG sporadica scarsamente responsiva alla terapia conrHuG-CSF. 8 In seguito è risultato che le mutazioni delgene per il G-CSF-r coinvolgono per lo più la regione crit-ica (dal nucleotide 2300 al 2500) che codifica per laporzione intracitoplasmatica del recettore. Le mutazionidescritte determinano la formazione di un codone di stop,che porta alla perdita di una porzione del dominio car-bossilico del recettore, con conseguente aumento, almenonel topo, della risposta proliferativi al G-CSF, arresto del-la differenziazione ed effetto anti apoptotico. 11 Lemutazioni del gene per il G-CSF-r , che risultano sempre
74
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre

acquisite nel corso della malattia, sono state documentatenel 20% circa dei pazienti con NCG10 mentre sono presentinell'80% dei casi con evoluzione in MDS/AML. 11 Secon-do i dati del SCNIR non è stata evidenziata una corre-lazione statisticamente significativa tra comparsa diMDS/AML e dose di G-CSF o durata del trattamento conG-CSF. 11
In considerazione della possibile evoluzione inMDS/AML è consigliato di monitorare annualmente questipazienti con indagini citogenetiche su sangue midollare econ la ricerca delle mutazioni del gene per il G-CSF-r, pereffettuare, in caso di comparsa di alterazioni, un trapiantodi cellule staminali emopoietiche12 prima della comparsadi AML.
Dai dati del SCNIR Europeo al 2003, 28 pazienti conNCG sono stati sottoposti a trapianto di cellule staminaliemopoietiche per MDS/AML, mentre 6 pazienti con AML,dei quali nessuno è sopravvissuto, sono stati trattati solocon CT. Nei pazienti sottoposti al trapianto , prevalente-mente da donatore non correlato o familiare aploidenti-co, entro 3 mesi dalla diagnosi di MDS/AML la percentualedi sopravvivenza è risultata del 59% (Report annuale delSCNIR Europeo - 2003).
Bibliografia
1. Kostmann R. Infantile genetic agranulocytosis: a new recessivelethal disease in man. Acta Paediatr Scand 1956;105:1-78.
2. Zeidler C, Welte K. Kostmann syndrome and severe congenital neu-tropenia. Semin Hematol 2002;39:82-8.
3. Rappeport J, Parkman R, Newburger P, Camitta BM, Chusid MJ.Correction of infantile agranulocytosis (Kostmann syndrome) byallogeneic bone marrow transplantation. Am J Med 1980;68:605-9.
4. Dale DC, Bolyard AA, Aprikyan A. Cyclic neutropenia. Semin Hema-tol 2002;39:89-94
5. Bellanne-Chantelot C, Clauin S, Leblanc T, Cassinat B, Rodrigues-Lima F, Beaufils S, et al. Mutations in the ELA2 gene correlate withmore severe expression of neutropenia: a study of 81 patients fromthe French Neutropenia Register. Blood 2004;103:4119-25.
6. Gilman PA, Jackson DP, Guild HG. Congenital agranulocytosis: pro-longed survival and terminal acute leukemia. Blood 1970;36:576-85.
7. Rosen R, Kang S. Congenital agranulocytosis terminating in acutemyelomonocytic leukaemia. J Pediatr 1979;94:406-8.
8. Ward A, van Aesch YM, Schelen AM, de Koning JP, van LeeuwenD, et al. Novel point mutation in the extracellular domain of thegranulocyte colony-stimulating factor (G-CSF) receptor in a caseof severe congenital neutropenia hyporesponsive to G-CSF treat-ment. J Exp Med 1999;190:497-507.
9. Dale CD, Cottle TE. Severe chronic neutropenia: treatment and fol-low up of patients in the Severe Chronic Neutropenia Interna-tional Registry. Am J Hematol 2003;72:82-93.
10. Van de Geijn GJM, Gits J. G-csf receptor truncations found inSCN/AML releve SOCS3-controlled inhibition of STAT6 but leavesuppression of STAT3 intact. Blood 2004;103:667-74.
11. Freedman MH, Alter BP. Risk of Myelodysplastic syndrome andacute myeloid leukemia in congenital neutropenia. Semin Hema-tol 2002;39:128-38.
12. Zeidler C, Boxer L. Management of Kostmann syndrome in the G-CSF era. Br J Haematol 2000;109:490-5.
13. Zeidler C, Schwinzer B, Welte K. Congenital neutropenias. Rev ClinExp Hematol 2003;7:72-83.
La sindrome di Wiskott-Aldrich
Luigi D. NotarangeloClinica Pediatrica Università di Brescia, Ospedale dei Bam-bini, Brescia
La sindrome di Wiskott-Aldrich (WAS), descritta per laprima volta da Wiskott nel 1937,1 è rappresentata nellasua forma clinica completa dalla triade clinica: eczema,infezioni ricorrenti e manifestazioni emorragiche (legatea piastrinopenia). La natura X-recessiva della sindromevenne riconosciuta da Aldrich nel 1954. 2 Un quadro clin-ico meno severo, caratterizzato da piastrinopenia isolata,anch'esso a trasmissione X-recessiva (e definito pias-trinopenia X-recessiva, XLT) venne descritto per la primavolta nel 1956 da Wooley,3 ma la sua natura allelicarispetto alla sindrome di Wiskott-Aldrich venne riconosci-uta solo nel 1967 da Canales4 e confermata nel 1995. 5
La fisiopatologia della WAS e della XLT è stata almenoin parte chiarita a seguito del clonaggio del gene da partedi Derry e coll. nel 1994. 6 Tale gene (WASP, per Wiskott-Aldrich Syndrome Protein) codifica per una proteina di502 aminoacidi espressa selettivamente nel sistemaematopoietico e coinvolta nei processi di riarrangiamen-to del citoscheletro e di maturazione, attivazione e traf-fico degli elementi del sangue. 7
Mutazioni di WASP hanno importanti conseguenze sul-la funzione delle cellule del sistema ematopoietico. In par-ticolare, i linfociti T dei soggetti WAS hanno protrusionicellulari tozze, sono deficitari nella risposta proliferativavia CD3 e presentano importanti difetti nella formazionedi sinapsi immunologiche. 8,9 I linfociti NK dei pazientiWAS presentano una ridotta attività citotossica. 10 Impor-tanti difetti di migrazione sono stati descritti per macrofa-gi e cellule dendritiche, come conseguenza della alterataformazione di protrusioni cellulari e di una minora capac-ità di risposta a stimoli chemotattici. 11
Meno chiaro è il ruolo delle alterazioni di WASP sullaproduzione e sulla funzione delle piastrine. Apparente-mente, la capacità dei megacariociti di soggetti WAS/XLTdi produrre pro-piastrine è intatta; tuttavia, in risposta astimoli di adesione, i megacariociti dei soggetti WAS/XLTmostrano importanti difetti nella formazione di filopodi enella polimerizzazione e compartimentalizzazione di F-actina.
Genetica molecolare della WAS e della XLT - Correlazionegenotipo-fenotipo
Mutazioni a carico del gene WASP possono compro-mettere in tutto o in parte l'espressione e la funzione del-la proteina. Vi è crescente evidenza che la severità di com-promissione dell'espressione proteica sia direttamentecorrelata alla gravità del fenotipo clinico. 12 In partico-lare, mutazioni missenso negli esoni 1 e 2 del gene (chedi regola riducono, ma non abrogano, l'espressione prote-ica) sono associati ad un fenotipo lieve, la cui principalemanifestazione è rappresentata dalla piastrinopenia (XLT).Più recentemente, sono stati descritti casi nei qualimutazioni missenso del gene WASP, permissive per l'e-spressione di livelli adeguati di proteina con una singolasostituzione aminoacidica, si associano ad un fenotipo di
75
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

piastrinopenia intermittente, e rappresentano quindil'estremo più lieve dello spettro fenotipico. 13 Anche alcunemutazioni di splicing, specie al di fuori delle posizioni con-servate (-2, -1, +1, +2) ai siti donatore ed accettare displicing, possono talvolta consentire l'espressione di bassilivelli di proteina con normale sequenza aminoacidica edeterminare fenotipi clinici lievi o moderati. Al contrario,mutazioni nonsenso o mutazioni che compromettono ilsenso di lettura dell'RNA messaggero e quindi alterano lasequenza aminoacidica della proteina) abrogano oriducono fortemente l'espressione proteica e di regola siassociano ad un fenotipo clinico severo (WAS). Nel com-plesso, quindi, appare sempre più evidente che WAS e XLTnon sono due quadri clinici nettamente distinti tra di loro,ma piuttosto sembra che mutazioni del gene WASP pos-sano dar luogo ad un continuum fenotipico, nel quale sonomeglio riconoscibili forme oligosintomatiche (XLT inter-mittente, XLT) e forme cliniche complete e severe (WAS).
Epidemiologia della sindrome di Wiskott-AldrichIn base a segnalazioni storiche in vari Registri Nazion-
ali per le Immunodeficienze Primitive, è stato stimato chela WAS ha un'incidenza di circa 4 casi per 1.000.000maschi nati vivi. Tuttavia, tale dato appare largamentesottostimato, soprattutto in considerazione del fatto cheè ormai ben assodato che lo spettro fenotipico della sin-drome è molto più esteso di quanto un tempo siammettesse. Nelle descrizioni classiche, la WAS è con-traddistinta dalla triade: eczema, piastrinopenia conmicrotrombociti ed immunodeficienza. Tuttavia, questofenotipo completo si osserva solo in meno di un terzo deicasi. 14 Per cercare di ovviare alle difficoltà di inquadra-mento clinico della sindrome (e quindi anche per tentaredi predisporre linee-guida terapeutiche mirate sul fenotipoclinico), Zhu e coll. hanno proposto un sistema di classi-ficazione della malattia basato esclusivamente su rilieviclinici. 15
In base ai dati della letteratura, relativi ad un ampiostudio multicentrico americano,14 l'età media alla diagnosiera di 21 mesi (range: nascita-24. 8 anni) .
La mortalità legata alla malattia si è andata progressi-vamente riducendo nel corso degli anni. Dati retrospettivipubblicati nel 1980 riportavano infatti una sopravviven-za media di soli 8 mesi per i maschi nati prima del 1935,ma di 6. 5 anni per quelli nati dopo il 1964;16 in uno stu-dio del 1994, la sopravvivenza media risultava di 11 anni,14
mentre era di 14. 5 anni nello studio più recente, pubbli-cato nel 2003. 17 Anche tali dati, tuttavia, non sono rap-presentativi dell'intera coorte di pazienti con WAS e XLT,ma riflettono una maggiore inclusioni di pazienti confenotipo clinico più severo (WAS). Le cause prevalenti dimorte relative ai pazienti non sottoposti a trapianto dicellule staminali ematopoietiche comprendono: emorragiesevere (23% dei casi), infezioni (44%) e tumori (26%).Non esistono dati solidi in merito alla mortalità nellacoorte di soggetti con fenotipo clinico più lieve (XLT).
Fenotipo clinicoGli episodi emorragici costituiscono il segno più comune
della WAS, in tutto il suo spettro fenotipico, ma essi ten-dono ad essere più comuni e precoci nei soggetti confenotipo WAS rispetto ai pazienti XLT. 17 L'incidenza media
delle manifestazioni emorragiche prima della diagnosi è dioltre l'80%; nella maggioranza dei casi (78%), queste sonorappresentate da petecchie e porpora. Tuttavia, una quo-ta rilevante (30%) di soggetti si presenta con episodiemorragici significativi, tra i quali i più comuni sonoematemesi e melena; particolare rilievo ai fini del sospet-to diagnostico va in questo senso attribuito alla diarreaematica nel neonato-lattante. Le emorragie endocraniche,benché poco frequenti se rapportate al totale delle emor-ragie significative, tuttavia costituiscono un problemaestremamente rilevante ai fini prognostici.
La severità degli episodi emorragici è strettamente lega-ta all'entità della piastrinopenia. Il numero di episodi sig-nificativi di emorragie è di 3. 85 per paziente-anno neipazienti con valori di piastrine all'esordio inferiori a10.000/ºL, mentre scende a 1.08 episodi significativi perpaziente-anno nei pazienti con valori di piastrine all'e-sordio compresi tra 50.000 e 100.000/µL. A questa dif-ferenza si associa un rischio diverso di emorragiaendocranica ed un diverso impatto sulla mortalità totale.Nel complesso, gli incidenti emorragici costituiscono dal4 al 20% delle cause di morte nelle varie casistiche.
I soggetti con WAS presentano una aumentatasuscettibilità ad infezioni batteriche, ma anche virali (Her-pes simplex 1 e 2, papillomavirus, mollusco contagioso) efungine. Tra i batteri, un rischio particolare è legato alleinfezioni sostenute da germi capsulati, in quanto è ridot-ta la capacità di produrre anticorpi di sottoclasse IgG2 neiconfronti degli antigeni polisaccaridici. Inoltre, sono fre-quenti anche le infezioni da germi opportunisti (polmoniteda Pneumocystis jerovicii).
L'eczema interessa l'80% dei pazienti WAS nel corsodella malattia. E' più comune nei primi due anni di vita etende a regredire nel tempo. Mancano dati solidi circal'incidenza di eczema nella coorte di soggetti XLT, manumerose segnalazioni riportano che anche tali soggettipossono sviluppare eczema, in genere di grado modesto eper un periodo di tempo più breve.
L'autoimmunità costituisce una delle complicanze piùcomuni e al contempo temibili della WAS. La sua inci-denza appare tuttavia variabile nelle varie casistiche: nel-la serie multicentrica analizzata da Sullivan e coll. nel1994, il 40% dei pazienti aveva almeno una manifes-tazione autoimmune e il 25% dei pazienti aveva più man-ifestazioni. 14 In una serie di pazienti analizzati in un sin-golo centro (Hospital Necker, Parigi) e pubblicata recen-temente,18 ben il 72% dei pazienti risultava presentarealmeno una manifestazione autoimmune e il 36% avevamanifestazioni multiple. Nel recente lavoro di Imai e coll., manifestazioni autoimmuni o infiammatorie erano pre-senti nel 24% dei pazienti. 17
Tra le manifestazioni cliniche di autoimmunità preval-gono l'anemia emolitica autoimmune, le vasculiti cutanee(compresa la sindrome di Schoenlein-Henoch), lenefropatie, l'artrite, che nel loro insieme danno conto dioltre l'80% delle manifestazioni autoimmuni. 19
Sia dati storici che casistiche recenti17 segnalano comeuna complicanza frequente nel sottogruppo XLT sia rap-presentata da nefropatia ad IgA, che peraltro compare diregola oltre la seconda decade di vita.
La presenza di autoimmunità assume un significatoprognostico sfavorevole: il 25% dei soggetti WAS con
76
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre

autoimmunità sviluppa tumori (contro il 5% dei soggettiWAS senza manifestazioni autoimmuni) e, per converso,il 75% dei tumori in pazienti WAS riguarda soggetti cheavevano precedentemente sviluppato autoimmunità. 18
Inoltre, la presenza di malattie autoimmuni comporta unrischio di mortalità aumentato; ciò è particolarmente veroper l'anemia emolitica autoimmune e per la recidiva dipiastrinopenia post-splenectomia.
I tumori costituiscono, assieme alle emorragie severe eall'autoimmunità, la principale complicanza della WAS.Nello studio multicentrico pubblicato da Sullivan e coll.nel 1994, l'incidenza dei tumori nella popolazione WASera del 13%, con un'età media alla diagnosi di 9. 5 anni.14 Nel 90% dei casi si tratta di leucemie, mielodisplasia elinfomi. Dati recenti indicano che il genotipo possa rapp-resentare un ulteriore elemento di rischio. Infatti, l'inci-denza di linfomi è del 3. 2% nei soggetti WAS conmutazioni missenso, del 9. 2% tra quelli con mutazioninonsenso o errori del senso di lettura, ed è del 12.1% trai soggetti con mutazioni di splicing. In particolare, un ele-vato rischio (44% di linfomi B) è segnalato tra i pazienticon mutazioni di splicing dell'esone 6. 20
Per definizione, i soggetti XLT non dovrebbero pre-sentare tumori (vedi in seguito: correlazione genotipo-fenotipo); tuttavia, mancano studi prospettici sulla coortedi soggetti XLT ed al momento non è quindi possibiledefinire con certezza il rischio di degenerazione neoplas-tica in tali soggetti.
Alterazioni di laboratorioL'unica alterazione di lboratorio consistente nella popo-
lazione WAS/XLT è rappresentata dalla piastrinopenia conmicrotrombociti. Il numero delle piastrine in circolo è diregola inferiore a 70.000/mL. In rari casi, bassi valori dipiastrine si osservano periodicamente, intervallati a con-te normali (piastrinopenia intermittente X-recessiva). Ilvolume piastrinico risulta tipicamente ridotto ed è ingenere <5fL (v. n. : 7-10 fL).
Oltre a queste alterazioni, altre anomalie di laboratoriosi osservano in una quota di pazienti, e più spesso in quel-li con WAS rispetto a quelli con XLT. In base allo studiomulticentrico di Sullivan e coll. (1994), una linfopenia sig-nificativa (<1000/µL) è presente nel 22% dei pazientiWAS, mentre eosinofilia (>500 eosinofili/µL) si osservanel 31% dei casi. 14 L'anemia è frequente ed è conseguentea microsanguinamenti o a fenomeni autoimmuni. Ancheil volume eritrocitario medio è spesso ridotto, in analogiacon quello piastrinico. Nei pazienti WAS, sono statedescritte diverse alterazioni immunologiche. Classica-mente, i livelli di IgM sieriche sono ridotti, mentre quellidi IgA ed IgE sono aumentati. Tuttavia, queste alterazionisono incostanti e riguardano soprattutto i soggetti piùgrandi. Una quota significativa di soggetti WAS presentaIgM normali o addirittura aumentate; elevati valori di IgMrappresentano un fattore di rischio per lo sviluppo diautoimmunità. La linfopenia tende ad accentuarsi conl'età, soprattutto nella popolazione WAS, ed è più marca-ta per la sottopopolazione di linfociti T CD8+, che risultanopresenti in numero ridotto nel 61% dei pazienti. Un difet-to di risposta proliferativa a mitogeni si osserva nel 46%dei casi ed è più frequente nei confronti della stimolazionevia CD3. Inoltre, i soggetti WAS presentano spesso lin-
fonodi ipoplasici, con una deplezione linfoide che si rendesempre più evidente all'aumentare dell'età del paziente. E'assente o fortemente ipoplasica la zona marginale dellamilza, deputata alla formazione di anticorpi anti-polisac-caridi.
Trattamento: alternative disponibili e prove di efficaciaLa rarità e l'eterogeneità della sindrome ha precluso la
realizzazione di studi clinici controllati mirati a definirel'efficacia di diverse misure terapeutiche. L'esperienza rac-colta deriva quindi soprattutto da piccole serie di casiseguiti presso singoli Centri o da studi retrospettivi mul-ticentrici, ma con impiego di strategie terapeutiche diversee basate su diversi criteri di inclusione.
Oltre al trapianto di cellule staminali ematopoietiche(per il quale: vedi oltre), diverse strategie sono state uti-lizzate per far fronte alla piastrinopenia. Nello studio mul-ticentrico di Sullivan et al. ,14 gli steroidi, somministrati acicli, sono risultati efficaci in un terzo dei casi, inducen-do una elevazione della conta piastrinica di almeno20.000/µL. L'efficacia degli steroidi sembra superiore neisoggetti splenectomizzati che sviluppino PTI post-splenec-tomia.
Sempre nello stesso studio, l'utilizzo delle immunoglob-uline endovena ad alte dosi (in assenza di altri tratta-menti) è risultato inefficace in tutti i pazienti trattati,mentre ha indotto un incremento della conta piastrinicasolo in una quota di soggetti precedentemente splenec-tomizzati e che aveva in seguito sviluppato PTI. Lasplenectomia costituisce invece una terapia potenzial-mente efficace ed è certamente in grado di prolungare lasopravvivenza. Tuttavia, a fronte di una rilevante quota disoggetti che mostra una risposta iniziale soddisfacentealla splenectomia, il 13-22% sviluppa in seguito recidivedi piastrinopenia, ritenute assimilabili ad episodi di PTI.Ancora più importante è sottolineare come nelle varieserie considerate, la splenectomia si associ ad un rischiosignificativo di sepsi e di mortalità legata a sepsi, nonos-tante antibiotico-profilassi. 14,21
I mezzi più comunemente utilizzati per ridurre l'inci-denza delle infezioni nella WAS sono rappresentati dallaprofilassi antimicrobica e dalle immunoglobuline endove-na. La profilassi antimicrobica viene comunemente con-dotta con impiego di co-trimossazolo.
L'utilizzo di immunoglobuline endovena (IVIG) per con-trastare il rischio infettivo è largamente diffuso e giusti-ficato dal difetto di produzione anticorpale, benchémanchino dati solidi che consentano una valutazione diefficacia. La terapia delle manifestazioni autoimmuni nel-la WAS è stata oggetto di una recente revisione. 18 Glisteroidi sono efficaci nella PTI post-splenectomia e nell'anemia emolitica autoimmune; altre terapie utilizzate (maper le quali manca sufficiente esperienza) sono rappre-sentate da ciclofosfamide e da azatioprina. Non vi è suf-ficiente esperienza in merito all'impiego di anticorpi mon-oclonali anti-CD20 nelle forme severe di autoimmunità,stante anche il fatto che tale complicanza costituisce ingenere indicazione al trapianto di cellule staminali.
Il trattamento dei tumori nella WAS non differisce daquello della popolazione generale, ma particolare cautelava posta sia nella prevenzione delle infezioni sia nel con-trollo del rischio emorragico, anche al di fuori delle fasi di
77
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

chemio- e/o radio-terapia. . Il trapianto di cellule staminali ematopoietiche costitu-
isce al momento l'unica terapia potenzialmente risoluti-va della WAS. I risultati migliori sono stati ottenuti coltrapianto da familiare HLA genotipicamente-identico. Lasopravvivenza a lungo termine (3-5 anni) con tale tipo ditrapianto è dell'81% e dell'87%, rispettivamente, nellecasistiche europea e americana. 22,23 Risultati inferiori siottengono con il trapianto da familiare HLA non-identico(45% e 52% di sopravvivenza nei registri europei e amer-icano, rispettivamente). Inoltre, questo tipo di trapianto ègravato, nella popolazione WAS, da un elevato rischio disindromi linfoproliferative EBV-correlate. Per ovviare airisultati non molto soddisfacenti del trapianto da dona-tore familiare HLA non identico, alcuni gruppi hannointrapreso la strada del trapianto da donatore volontarioe compatibile (MUD, Matched Unrelated Donor). In parti-colare, Filipovich e coll. hanno riportato con tale tipo ditrapianto una sopravvivenza a 5 anni pari al 71%, conrisultati molto migliori se il ricevente al momento deltrapianto ha un'età <5 anni (79% di sopravvivenza) rispet-to a riceventi >5 anni di età (38% di sopravvivenza). 23 Piùrecentemente, interessanti risultati (pur se su una casis-tica ancora esigua) sono stati riportati anche con impiegodi cellule staminali cordonali, anche se parzialmente com-patibili. 24
Al momento, non esistono esperienze di terapia genicaper correggere la WAS nell'uomo. Risultati preliminari invitro dimostrano tuttavia che il trasferimento del genewasp in cellule staminali ematopoietiche di topi wasp-/ydà luogo ad una piena ricostituzione immunologica. 25,26
Nel complesso, l'esperienza accumulata sulla WASdimostra la grande eterogeneità clinica, immunologica edi difetto molecolare della sindrome. Emerge in modochiaro l'opportunità di studi clinici longitudinali che per-mettano di apprezzare meglio la storia naturale dellamalattia nelle sue diverse varianti. Al contempo, l'etero-geneità del fenotipo clinico suggerisce che vengano uti-lizzate strategie terapeutiche differenziate. Per risponderea questi obiettivi, il CSS Immunodeficienze dell'AIEOP hadi recente approvato un protocollo diagnostico e ter-apeutico della WAS/XLT. Ci attendiamo quindi che dalRegistro collegato a tale protocollo emergano prestonuove ed importanti acquisizioni per garantire una diag-nosi e un trattamento sempre più efficaci e tempestivi aipazienti affetti da questa sindrome.
Bibliografia
1 Wiskott A. Familiärer, angeborener Morbus Werlhofii? MonatsschrKinderheilkd 1937;68:212-6.
2 Aldrich RS, Steinberg AG, Campbell DC. Pedigree demonstrating asex-linked recessive condition characterized by draining ears,eczematoid dermatitis and bloody diarrhea. Pediatrics 1954;13:133-9.
3 Wooley EJS. Familial idiopathic thrombocytopenic purpura. Br MedJ 1956;1:440-7.
4 Canales ML, Mauer AM. Sex-linked hereditary thrombocytopeniaas a variant of Wiskott-Aldrich syndrome. N Engl J Med 1967;277:899-901.
5 Villa A, Notarangelo L, Macchi P. X-linked thrombocytopenia andWiskott-Aldrich sindrome are allelic diseases with mutations in theWASP gene. Nat Genet 1995, 9:414-7.
6 Derry JMJ, Ochs HD, Francke U. Isolation of a novel gene mutated
in Wiskott-Aldrich syndrome. Cell 1994;78:635-44. 7 Notarangelo LD, Ochs HD. Wiskott-Aldrich syndrome: a model for
defective actin reorganization, cell trafficking and synapse for-mation. Curr Opin Immunol 2003;15:585-91.
8 Molina IJ, Sancho J, Terhorst C, Rosen FS, Remold-O'Donnell E. Tcells of patients with the Wiskott-Aldrich syndrome have arestricted defect in proliferative responses. J Immunol 1993;151:4383-90.
9 Dupré L, Aiuti A, Trifari S, Martino S, Saracco P, Bordignon C, et al.Wiskott-Aldrich sindrome protein regulates lipid raft dynamicsduring immunological synapse formation. Immunity 2002;17:157-66.
10 Gismondi A, Cifaldi L, Mazza C, Giliani S, Parolini S, Morrone S, etal. Impaired natural and CD16-mediated NK cell cytotoxicity inpatients with WAS and XLT: ability of IL-2 to correct NK cell func-tional defect. Blood 2004;104:436-43.
11 Badolato R, Sozzani S, Malacarne F, Bresciani S, Fiorini M, Borsat-ti A, et al. Monocytes from Wiskott-Aldrich patients displayreduced chemotaxis and lack of cell polarization in response tomonocyte chemoattractant protein-1 and formyl-methionyl-leucyl-phenylalanine. J Immunol 1998;161:1026-33.
12 Jin Y, Mazza C, Christie JR, Giliani S, Fiorini M, Mella P, et al. Muta-tions of the Wiskott-Aldrich syndrome protein (WASP) : hot spots,effect on transcription and translation and phenotype/genotypecorrelation. Blood 2004;in press.
13 Notarangelo LD, Mazza C, Giliani S, D'Aria C, Gandellini F, RavelliC, et al. Missense mutations of the WASP gene cause intermittentX-linked thrombocytopenia. Blood 2002;99:2268-9.
14 Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinsti-tutional survey of the Wiskott-Aldrich sindrome. J Pediatr 1994;125:876-85.
15 Zhu Q, Watanabe C, Liu T, Hollenbaugh D, Blaese RM, Kanner SB,et al. Wiskott-Aldrich syndrome/X-linked thrombocytopenia :WASP gene mutations, protein expression, and phenotype. Blood1997;90:2680-9.
16 Lum LG, Tubergen DG, Corash L, Blaese RM. Splenectomy in themanagement of the thrombocytopenia of the Wiskott-Aldrich syn-drome. N Engl J Med 1980;302:892-5.
17 Imai K, Morio T, Zhu Y, Jin Y, Itoh S, Kajiwara M, et al. Clinicalcourse of patients with WASP gene mutations. Blood 2004;15:456-64.
18 Dupuis-Girod S, Mediani J, Haddad E. Autoimmunity in Wiskott-Aldrich sindrome: risk factors, clinical features, and outcome in asingle-center cohort of 55 patients. Pediatrics 2003, 111:622-627.
19 Schurman SH, Candotti F. Autoimmunity in Wiskott-Aldrich sin-drome. Curr Opin Rheumatol 2003;15:446-53.
20 Shcherbina A, Candotti F, Rosen FS, Remold-O'Donnell E. Highincidence of lymphomas in a subgroup of Wiskott-Aldrich sin-drome patients. Br J Haematol 2003;121:529-30.
21 Mullen CA, Anderson KD, Blaese MR. Splenectomy and/or bonemarrow transplantation in the management of the Wiskott-Aldrichsyndrome: Long-term follow-up of 6 cases. Blood 1993;82:2961-6.
22 Antoine C, Muller S, Cant A, Cavazzana-Calvo M, Veys P, VossenJ, et al. Long-term survival and transplantation of haematopoiet-ic stem cells for immunodeficiencies: report of the European expe-rience 1968-1999. Lancet 2003;361:553-60.
23 Filipovich AH, Stone JV, Tomany SC, Ireland M, Kollman C, Pelz CJ,et al. Impact of donor type on outcome of bone marrow trans-plantation for Wiskott-Aldrich syndrome: collaborative study ofthe International Bone Marrow Transplant Registry and theNational Marrow Donor Program. Blood 2001;97:1598-603.
24 Knutsen AP, Steffen M, Wassmer K, Wall DA. Umbilical cord bloodtransplantation in Wiskott Aldrich syndrome. J Pediatr 2003;142:519-23.
25 Strom TS, Cunningham JM, Nienhuis AW. Correction of the murineWiskott-Aldrich syndrome phenotype by hematopoietic stem celltransplantation. Blood 2002;99:4626-8.
26 Klein C, Nguyen D, Liu C-H, et al. Gene therapy for Wiskott-Aldrichsindrome: rescue of T-cell signaling and amelioration of colitisupon transplantation of retrovirally transduced hematopoieticstem cells in mice. Blood 2003;201:2159-66.
78
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre

Caspofungin: un nuovo farmaco antifunginoa disposizione del pediatra oncoematologo
Franco Locatelli, Stefania TelliOncoematologia Pediatrica, IRCCS Policlinico San Matteo,Pavia
Le infezioni fungine rappresentano un problema pro-gressivamente emergente nei pazienti pediatrici con difet-ti congeniti dell'immunità, così come nei soggetti affettida neoplasie che richiedono prolungati e intensivi tratta-menti citostatici, o sottoposti a trapianto allogenico dicellule staminali emopoietiche (TCSE). In particolare, infe-zioni da Aspergillus sp e da Candida, albicans o non albi-cans, sono diagnosticate con sempre maggior frequenzanei pazienti affetti da neoplasie, durante la fase di neu-tropenia o in corso di trattamento steroideo o con altri far-maci dotati di spiccata attività linfolitica. Allo stessomodo, l'aumentato riscontro di questo tipo di infezioninei pazienti trapiantati è principalmente dovuto alla rea-lizzazione di procedure trapiantologiche che comportanouno stato di profonda e prolungata immunodepressione,quali il trapianto da donatore non consanguineo e il tra-pianto con deplezione T-linfocitaria delle cellule staminaliche vengono infuse al paziente. Sempre nei soggetti sot-toposti a TCSE, l'impiego di farmaci immunosoppressori,magari per l'insorgenza di malattia del trapianto control'ospite (GVHD) acuta o cronica, non vi è dubbio favoriscalo sviluppo di infezioni fungine invasive. Secondo alcunirecenti studi, per esempio, la frequenza delle aspergillosiinvasive, nei pazienti sottoposti a TCSE e che abbiano svi-luppato GVHD, tale da richiedere un trattamento steroi-deo ad una dose di almeno 2 mg/Kg/die, raggiunge il 30%.
A dispetto dell'impiego di protocolli diagnostici artico-lati e che prevedono l'impiego di valutazioni TC o di inda-gini colturali su lavaggio bronco-alveolare, piuttosto chesu sangue o fluidi ottenuti da siti usualmente sterili, ladiagnosi di queste complicanze infettive non è sempreimmediata e, spesso, lo sviluppo di queste patologie è pre-ceduto da quadri di febbre senza apparente spiegazione eresistente al trattamento antibiotico ad ampio spettro(cosiddette febbri di origine sconosciuta o FUO). Sullascorta di questo dato, è largamente impiegata la prassi disottoporre i pazienti con immuno-deficit congeniti cheinteressano il compartimento T-cellulare, così come lafunzione granulocitaria, o che abbiano una neoplasia trat-tata con farmaci a spiccata attività mielodepressiva olinfocitolitica, o che abbiano ricevuto un TCSE, a un trat-tamento antifungino empirico se, nel contesto di un qua-dro di neutropenia (conta granulocitaria assoluta <500elementi/mm3), si abbia lo sviluppo di febbre persistiten-te per almeno 72 ore. In particolare, fino a pochi anniorsono, quasi tutti i pazienti con le caratteristiche sopra-menzionate e con FUO venivano sottoposti a trattamen-to empirico con amfotericina desossicolato, mentre, piùrecentemente, si è largamente introdotto l'uso di amfote-ricina B liposomiale, farmaco dotato della stessa efficacia
dell'amfotericina desossicolato, ma con minori effetti col-laterali, soprattutto a carico della funzione renale, pur segravato da costi significativamente molto più elevati.
Caspofungin è un nuovo farmaco antifungino apparte-nente al gruppo delle cosiddette echinocandine, le qualisvolgono la loro azione antimicotica attraverso un'inibi-zione della sintesi del beta (1,3)-D-glucano, costituentefondamentale della parete cellulare di molti funghi fila-mentosi e lieviti. Questo farmaco, dimostratosi in labora-torio ampiamente attivo su numerosi ceppi fungini (purcon qualche significativa eccezione quale, per esempio, ilFusarium), è stato ampiamente impiegato per il tratta-mento di soggetti adulti neutropenici affetti da quadri diinfezione fungina documentata o considerata come pro-babile secondo i criteri recentemente pubblicati per la dia-gnosi di infezione fungina nei pazienti neutropenici. Aquesto riguardo, sono stati resi recentemente noti anchecasi di pazienti pediatrici trattati con successo per quelche riguarda infezioni fungine documentate attraversol'impiego di Caspofungin. Inoltre, studi preliminari di far-macocinetica relativi all'uso di Caspofungin sono stati rea-lizzati in pazienti pediatrici e, grazie ad essi, è stato pos-sibile identificare la dose consigliata per il trattamento dicasi documentati di infezione fungina (solitamente 70mg/m2 il primo giorno, 50 mg/m2 i giorni a seguire, conuna dose massima giornaliera pari a 70 mg/die). In raricasi, caratterizzati da infezioni particolarmente gravi, iltrattamento con Caspofungin è stato proseguito con unadose di 70 mg/m2, senza rilevanti effetti collaterali per ilpaziente.
I dati finora disponibili indicano che Caspofungin è unamolecola dotata di eccellente tollerabilità, essendo scar-se e di non straordinaria gravità clinica le segnalazionirelative a effetti collaterali osservati in corso d'impiego delfarmaco. Anche le riserve inizialmente proposte circa l'u-so concomitante di Caspofungin e ciclosporina sembranoessere in corso di significativo ridimensionamento. A que-sto proposito, va anche menzionato che, rispetto ad altrifarmaci con elevata efficacia anti-fungina recentementeresi disponibili sul mercato, Caspofungin non mostra cosìrilevanti interferenze farmacologiche con differenti mole-cole, ciò rendendo meno problematico l'impiego clinicoin pazienti che necessariamente debbano essere sottopo-sti a molteplici trattamenti.
Negli ultimi mesi, sono stati resi noti i risultati di unostudio internazionale, multicentrico, randomizzato in cuiveniva comparata l'efficacia e la sicurezza di impiego diCaspofungin rispetto all'Amfotericina B liposomiale inpazienti neutropenici con FUO. Questo studio, effettuatoesclusivamente su pazienti adulti, ha dimostrato che: i)Caspofungin è egualmente efficace rispetto ad Amfoteri-cina B liposomiale nel trattamento della FUO del pazien-te neutropenico; ii) Caspofungin è stato più efficace neltrattamento di infezioni fungine già presenti all'inizio deltrattamento; iii) il numero e la severità di effetti collate-rali nei pazienti trattati con Caspofungin è ridotto rispet-to ai pazienti trattati con Amfotericina B liposomiale; iv)Caspofungin era superiore all'Amfotericina B liposomialein termini di numero di pazienti che hanno dovuto sospen-dere prematuramente il trattamento. Sulla scorta di que-sti dati, l'indicazione all'uso di Caspofungin della FUO inpazienti neutropenici appare ampiamente giustificata.
79
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
Nuovi Farmaci per la Terapia di Supporto

Ulteriori interessanti prospettive possono derivare dal-l'impiego combinato di Caspofungin con altri farmacidotati di attività anti-fungina. Infatti, il differente mec-canismo d'azione rende ragionevole e razionale un'inte-razione sinergica fra Caspofungin e Amfotericina B lipo-somiale, piuttosto che Voriconazolo. Al riguardo, nonmancano preliminari segnalazioni, anche in pazientipediatrici. Ovviamente, ulteriori studi nel merito potran-no meglio definire la reale efficacia e l'importanza tera-peutica delle combinazioni sopra menzionate.
Se è vero che di grosso interesse e significativa pro-spettiva è l'uso di Caspofungin nei pazienti con docu-mentata infezione fungina o a rischio di svilupparne percondizioni di alterata funzionalità immunitaria, l'impiegoin Pediatria di Caspofungin richiede, tuttavia, studi speci-ficamente condotti, non essendo certo traslabili tout courtle informazioni derivate da studi in soggetti adulti. Unavolta acquisiti questi ulteriori dati pertinenti all'età pedia-trica, sarà più precisamente definibile l'ambito di impie-go di questo nuovo farmaco anti-fungino, anche tenendoconto di valutazioni comparative che considerino valuta-zioni farmaco-economiche, non certo trascurabili in tem-pi di limitate risorse e per farmaci, ivi compreso Caspo-fungin, gravati da significativi costi.
Bibliografia
Ascioglu S, Rex J, DePauw B, et al. Defining opportunistic invasive fun-gal infections in immunocompromised patients with cancer andhematopoietic stem cell transplants: an international consensus.CID 2002:34 ;7-14.
Baddley J, Stroud T, Salzman D, Pappas P. Invasive mold infections in allo-geneic bone marrow transplant recipients. Clin Infect Dis.2001;32:1319-1324.
Boucher HW, Groll AH, Chiou CC, Walsh TJ. Newer systemic antifungalagents : pharmacokinetics, safety and efficacy. Drugs. 2004;64:1997-2020.
Castagnola E, Machetti M, Cappelli B, et al. Caspofungin associated withliposomal amphotericin B or voriconazole for treatment of refrac-tory fungal pneumonia in children with acute leukaemia or under-going allogeneic bone marrow transplant. Clin Microbiol Infect.2004; 10:255-7.
Cesaro S, Toffolutti T, Messina C, et al. Safety and efficacy of caspofun-gin and liposomal amphotericin B, followed by voriconazole in
young patients affected by refractory invasive mycosis. Eur JHaematol. 2004; 73:50-5.
Denning DW. Echinocandin antifungal drugs. Lancet. 2003; 362:1142-51.
Elanjikal Z, Sorensen J, Schmidt H, et al. Combination therapy withcaspofungin and liposomal amphotericin B for invasive aspergillo-sis. Pediatr Infect Dis J. 2003; 22:653-56.
Goodman JL, Winston DJ, Greenfield RA, et al. A controlled trial of flu-conazole to prevent fungal infections in patients undergoing bonemarrow transplantation. N Engl J Med. 1992;326:845-851.
Goodrich JM, Reed C, Mori M, et al. Clinical features and analysis of riskfactors for invasive candidal infection after marrow transplanta-tion. J Infect Dis. 1991;164:731-740.
Locatelli F, Burgio GR. Transplant of haematopoietic stem cells in child-hood.: where we are and where we are going. Haematologica1998; 83: 550-563.
Marr KA, Carter RA, Boeckh M, Martin P, Corey L. Invasive aspergillosisin allogeneic stem cell transplant recipients: changes in epidemi-ology and risk factors. Blood, 2002; 100: 4358 - 4366.
Marr KA, Seidel K, Slavin M, et al. Prolonged fluconazole prophylaxis isassociated with persistent protection against candidiasis-relateddeath in allogeneic marrow transplant recipients: long-term fol-low-up of a randomized, placebo-controlled trial. Blood.2000;96:2055-2061.
Mora-Duarte J, Betts R, Rotstein C, et al. Comparison of caspofungin andamphotericin B for invasive candidiasis. N Engl J Med. 2002;347:2020-9.
Rubin ZA, Somani J. New options for the treatment of invasive fungalinfections. Semin Oncol. 2004; 31(Suppl 4):91-8.
Sanz-Rodriguez C, Lopez-Duarte M, Jurado M,et al. Safety of the con-comitant use of caspofungin and cyclosporin A in patients withinvasive fungal infections. Bone Marrow Transplant. 2004; 34:13-20.
Schwartz M et al., Determination of a cyclic hexapeptide (L-743 872), anovel pneumocandin antifungal agent in human plasma and urineby high performance liquid chromatography with fluorescencedetection. Analitica Chimica Acta 1997; 352: 299-307.
Walsh T, Sable C, dePauw B, et al. Trial of caspofungin (CAS) vs liposo-mal Amphotericin B (LAMB) for empirical antifungal therapy ofpatients with persistent febrile neutropenia. ICAAC 2003.
Walsh TJ. Echinocandins--an advance in the primary treatment of inva-sive candidiasis. N Engl J Med 2002; 347:2070-2.
Walsh TJ, Adamson PC, Seibel NL, et al. Pharmacokinetics of Caspofun-gin in pediatric patients. ICAAC 2002.
Zecca M, Prete A, Rondelli R, et al. Chronic graft-versus-host disease inchildren: incidence, risk factors, and impact on outcome. Blood2002;100 1192-1200.
80
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre
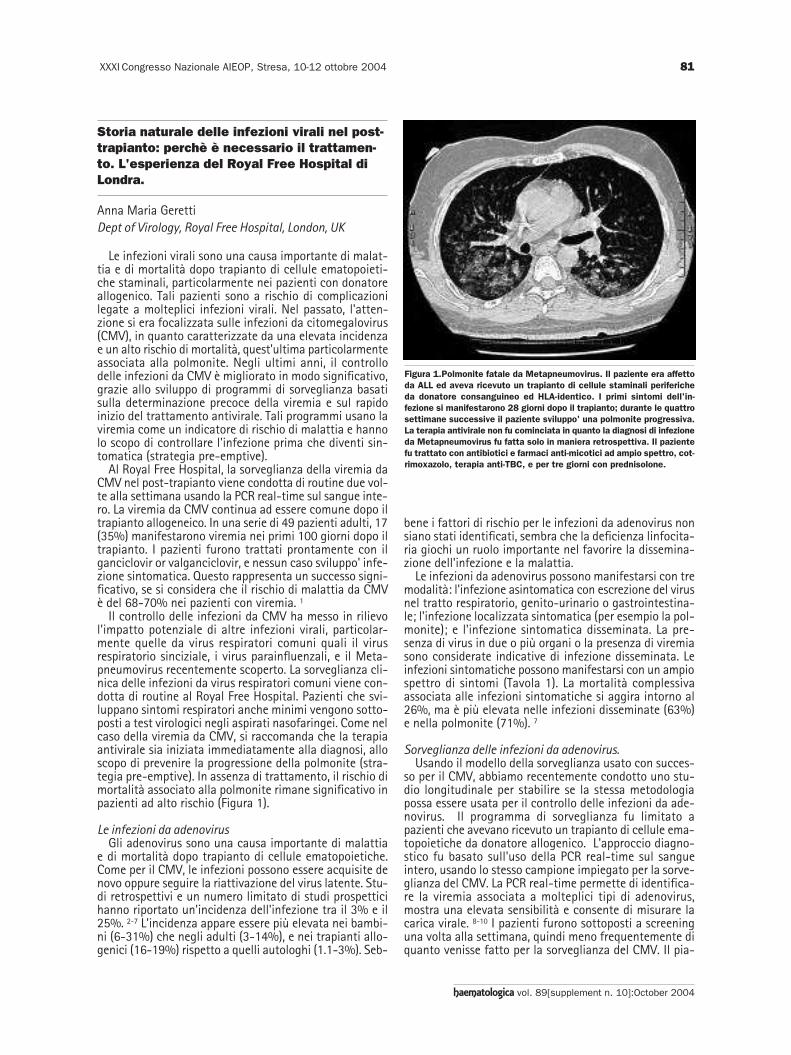
81
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
Storia naturale delle infezioni virali nel post-trapianto: perchè è necessario il trattamen-to. L'esperienza del Royal Free Hospital diLondra.
Anna Maria GerettiDept of Virology, Royal Free Hospital, London, UK
Le infezioni virali sono una causa importante di malat-tia e di mortalità dopo trapianto di cellule ematopoieti-che staminali, particolarmente nei pazienti con donatoreallogenico. Tali pazienti sono a rischio di complicazionilegate a molteplici infezioni virali. Nel passato, l'atten-zione si era focalizzata sulle infezioni da citomegalovirus(CMV), in quanto caratterizzate da una elevata incidenzae un alto rischio di mortalità, quest'ultima particolarmenteassociata alla polmonite. Negli ultimi anni, il controllodelle infezioni da CMV è migliorato in modo significativo,grazie allo sviluppo di programmi di sorveglianza basatisulla determinazione precoce della viremia e sul rapidoinizio del trattamento antivirale. Tali programmi usano laviremia come un indicatore di rischio di malattia e hannolo scopo di controllare l'infezione prima che diventi sin-tomatica (strategia pre-emptive).
Al Royal Free Hospital, la sorveglianza della viremia daCMV nel post-trapianto viene condotta di routine due vol-te alla settimana usando la PCR real-time sul sangue inte-ro. La viremia da CMV continua ad essere comune dopo iltrapianto allogeneico. In una serie di 49 pazienti adulti, 17(35%) manifestarono viremia nei primi 100 giorni dopo iltrapianto. I pazienti furono trattati prontamente con ilganciclovir or valganciclovir, e nessun caso sviluppo' infe-zione sintomatica. Questo rappresenta un successo signi-ficativo, se si considera che il rischio di malattia da CMVè del 68-70% nei pazienti con viremia. 1
Il controllo delle infezioni da CMV ha messo in rilievol'impatto potenziale di altre infezioni virali, particolar-mente quelle da virus respiratori comuni quali il virusrespiratorio sinciziale, i virus parainfluenzali, e il Meta-pneumovirus recentemente scoperto. La sorveglianza cli-nica delle infezioni da virus respiratori comuni viene con-dotta di routine al Royal Free Hospital. Pazienti che svi-luppano sintomi respiratori anche minimi vengono sotto-posti a test virologici negli aspirati nasofaringei. Come nelcaso della viremia da CMV, si raccomanda che la terapiaantivirale sia iniziata immediatamente alla diagnosi, alloscopo di prevenire la progressione della polmonite (stra-tegia pre-emptive). In assenza di trattamento, il rischio dimortalità associato alla polmonite rimane significativo inpazienti ad alto rischio (Figura 1).
Le infezioni da adenovirus Gli adenovirus sono una causa importante di malattia
e di mortalità dopo trapianto di cellule ematopoietiche.Come per il CMV, le infezioni possono essere acquisite denovo oppure seguire la riattivazione del virus latente. Stu-di retrospettivi e un numero limitato di studi prospetticihanno riportato un'incidenza dell'infezione tra il 3% e il25%. 2-7 L'incidenza appare essere più elevata nei bambi-ni (6-31%) che negli adulti (3-14%), e nei trapianti allo-genici (16-19%) rispetto a quelli autologhi (1.1-3%). Seb-
bene i fattori di rischio per le infezioni da adenovirus nonsiano stati identificati, sembra che la deficienza linfocita-ria giochi un ruolo importante nel favorire la dissemina-zione dell'infezione e la malattia.
Le infezioni da adenovirus possono manifestarsi con tremodalità: l'infezione asintomatica con escrezione del virusnel tratto respiratorio, genito-urinario o gastrointestina-le; l'infezione localizzata sintomatica (per esempio la pol-monite); e l'infezione sintomatica disseminata. La pre-senza di virus in due o più organi o la presenza di viremiasono considerate indicative di infezione disseminata. Leinfezioni sintomatiche possono manifestarsi con un ampiospettro di sintomi (Tavola 1). La mortalità complessivaassociata alle infezioni sintomatiche si aggira intorno al26%, ma è più elevata nelle infezioni disseminate (63%)e nella polmonite (71%). 7
Sorveglianza delle infezioni da adenovirus. Usando il modello della sorveglianza usato con succes-
so per il CMV, abbiamo recentemente condotto uno stu-dio longitudinale per stabilire se la stessa metodologiapossa essere usata per il controllo delle infezioni da ade-novirus. Il programma di sorveglianza fu limitato apazienti che avevano ricevuto un trapianto di cellule ema-topoietiche da donatore allogenico. L'approccio diagno-stico fu basato sull'uso della PCR real-time sul sangueintero, usando lo stesso campione impiegato per la sorve-glianza del CMV. La PCR real-time permette di identifica-re la viremia associata a molteplici tipi di adenovirus,mostra una elevata sensibilità e consente di misurare lacarica virale. 8-10 I pazienti furono sottoposti a screeninguna volta alla settimana, quindi meno frequentemente diquanto venisse fatto per la sorveglianza del CMV. Il pia-
Figura 1.Polmonite fatale da Metapneumovirus. Il paziente era affettoda ALL ed aveva ricevuto un trapianto di cellule staminali perifericheda donatore consanguineo ed HLA-identico. I primi sintomi dell'in-fezione si manifestarono 28 giorni dopo il trapianto; durante le quattrosettimane successive il paziente sviluppo' una polmonite progressiva.La terapia antivirale non fu cominciata in quanto la diagnosi di infezioneda Metapneumovirus fu fatta solo in maniera retrospettiva. Il pazientefu trattato con antibiotici e farmaci anti-micotici ad ampio spettro, cot-rimoxazolo, terapia anti-TBC, e per tre giorni con prednisolone.

82
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre
no di studio prevedeva che pazienti con viremia venisse-ro immediatamente sottoposti a test diagnostici per deter-minare l'escrezione di virus nel tratto respiratorio (aspira-ti nasofaringei e tamponi faringei per cultura virale,immunofluorescenza diretta e PCR), urinario (campionedi urine per cultura virale e PCR) e gastrointestinale (cam-pione di feci per cultura virale e PCR).
Quarantanove pazienti presero parte alla prima partedello studio. Le caratteristiche dei pazienti sono mostra-te nella Tavola 2. Tutti erano stati trattati con il Campath-
1H. I pazienti entrarono il programma di sorveglianza avari intervalli dopo il trapianto, con una media del giorno21+ (mediana giorno O). I pazienti furono seguiti setti-manalmente fino ad una media del giorno 113+ dopo il
Tavola 1.Spettro delle infezioni sintomatiche da adenovirus dopotrapianto di cellule ematopoietiche staminali.
– Febbre – Cistite emorragica (risolve spontaneamente)– Congiuntivite– Infezione del tratto respiratorio superiore– Polmonite – Enterite, colite – Epatite (anche fulminante)– Pancreatite– Nefrite (tubulo-interstiziale)– Meningo-encefalite (rara)– Emofagocitosi– Microangiopatia trombotica
Tavola 2.Caratteristiche dei 49 pazienti sottoposti a sorveglianza del-la viremia da adenovirus.
Sesso Uomini 30Donne 19
Età (anni) Media 50Mediana 54Intervallo 19-69
Malattia AML 23MDS 10NHL 7ALL 5CML 3Mieloma Multiplo 1
Trapianto PSCT 34BMT 15
Intensità Ridotta 37Standard 12
Donatore Consanguineo, HLA-identico 12Donatore alternativo, HLA-identico 26Donatore alternativo, non HLA-identico 11
Tavola 3. Caratteristiche dei quattro pazienti con viremia da adenovirus.
Paziente 1 Paziente 2 Paziente 3 Paziente 4
Età (anni) 62 57 37 23Sesso M M F MDiagnosi NHL NHL AML AMLTrapianto PSCT PSCT PSCT BMTDonatore Consanguineo Consanguineo Alternativo AlternativoHLA Identico Identico Non identico Non-identicoCondizionamento Fludarabina, Melfalan BEAM Fludarabina, Busulfan Irradiazione corporea totale, Busulfan,
Ciclofosfamide, MethotrexateIntensità Ridotta Ridotta Ridotta RidottaTrapianto precedente Autologo Allogeneico No NoViremia da CMV No No 33+ NoGVHD No No No NoData di insorgenza della viremia da adenovirus Giorno 12+ Giorno 12+ Giorno 26+ Giorno 48+Tipo di adenovirus C A12 C E4Infezione disseminata Si Si Si SiSistemi coinvolti Respiratorio Respiratorio Respiratorio Respiratorio
Gastrointestinale Gastrointestinale Gastrointestinale GastrointestinaleUrinario Urinario Urinario Urinario
Febbre Si Si Si SiDiarrea Si Si Si NoPolmonite Si Si No NoEpatite No Si (AST 5000 IU/ml) Si (AST 4000 IU/ml) Si (AST 200 IU/ml)Terapia antivirale No No Ganciclovir (da 41+ a 55+) Cidofovir (da 68+ a 131+)
Foscarnet (da 56+ a 69+)Conta linfocitaria <0.1 x109/L <0.1 x109/L <0.25 x109/L >1 x109/L dal giorno 101+Mortalità Giorno 17+ Giorno 30+ Giorno 76+ Vivo

83
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
trapianto (mediana giorno 102+), per un totale di 748person-weeks di sorveglianza. Durante tale periodo, laviremia da adenovirus fu riscontrata in quattro pazienti,dando un'incidenza dell'8. 3%. La mortalità associataall'infezione fu di tre pazienti su quattro (Tavola 3). I quat-tro pazienti viremici non sembrarono presentare fattoricomuni di rischio, sebbene tutti fossero affetti da un ele-vato grado di depressione immunitaria associato a fatto-ri quali il fallimento di un precedente trapianto, il dona-tore non consanguineo con disparità HLA, la GVHD trat-tata con farmaci steroidei, la contemporanea riattivazio-ne del CMV, la terapia di condizionamento con la fluda-rabina, o l'irradiazione corporea totale.
Nella seconda parte dello studio, 25 pazienti adulti ebambini trattati con trapianto allogenico furono sottopo-
sti alla medesima sorveglianza. Viremia fu osservata in 2casi, confermando che l'incidenza di viremia da adenovi-rus è dell'8% dopo trapianto allogenico. La mortalità asso-ciata alla viremia fu di due su due pazienti. In entrambigli studi, i pazienti viremici manifestarono febbre, caricavirale nel sangue in eccesso di 106 copie/ml ed escrezio-ne del virus nei tratti respiratorio, gastrointestinale e uri-nario. Queste osservazioni confermano che la viremia è unmarker dell'infezione disseminata. Altri sintomi comuni,sebbene non presenti in tutti i pazienti, furono la diarrea,la polmonite e l'epatite. Tutti i casi di mortalità mostra-rono una conta linfocitaria costantemente al di sotto di0.25x109/L; l'unico paziente che fu in grado di controlla-re la viremia mostro' una migliore conta linfocitaria, convalori che salirono al di sopra di 1x109/L in coincidenza con
Figura 2.Cinetiche della viremia da adenovirus dopo trapianto allo-geneico. Il primo caso dimostra un rapido insorgere della viremiaaccompagnato da rapida disseminazione (a). Il secondo caso dimostraun graduale aumento della viremia con lenta disseminazione progres-siva. Il paziente dimostrò viremia da CMV il giorno 33+ e fu trattato conil ganciclovir dal giorno 41+ al giorno 55+, seguito dal Foscarnet dalgiorno 56+ al giorno 69+, fino alla risoluzione della viremia da CMV (b).
Figura 3. Cinetiche della viremia da adenovirus in risposta al tratta-mento antivirale. La figura A mostra le cinetiche della viremia dopotrapianto allogeneico durante il trattamento con il cidofovir per viaendovenosa, quest'ultimo iniziato il giorno 67+. La scomparsa dellaviremia e il miglioramento clinico coincisero con l'aumento della con-ta linfocitaria. La figura B mostra le cinetiche della viremia da aden-ovirus in un paziente HIV-sieropositivo con polmonite ed epatite da ade-novirus, che fu trattato con il cidofovir per via endovenosa a partire dalgiorno successivo alla diagnosi della viremia. Il paziente morì con un'in-fezione disseminata. In entrambi i pazienti la dose del cidofovir fu di 5mg/kg una volta alla settimana per due settimane, seguita dalla stes-sa dose una volta ogni due settimane.
a
b
a
b

la risoluzione della viremia e dell'escrezione virale, e conil miglioramento sintomatico.
Cinetiche della viremia da adenovirus in relazione alla pro-gressione dell'infezione
Le cinetiche della viremia da adenovirus seguirono duemodelli principali: il primo modello fu caratterizzato dal-l'insorgere precoce della viremia, con elevate cariche vira-li, evidenza di simultanea disseminazione virale, e rapidaprogressione clinica (Figura 2a). Per tali pazienti ad altorischio sembra che la determinazione della viremia fattauna volta alla settimana sia insufficiente a garantire unadiagnosi precoce Il secondo modello fu quello caratteriz-zato da un basso carico virale iniziale e una lenta pro-gressione (Figura 2b). Tali pazienti offrono le miglioriopportunità per un intervento precoce, con riduzione del-la terapia soppressiva quando possibile, ed uso di farma-ci antivirali.
Cinetiche della viremia da adenovirus in relazione alla tera-pia antivirale
La mancanza di studi clinici controllati rende difficile lascelta del trattamento antivirale per pazienti con viremiada adenovirus. I farmaci proposti correntemente includo-no la ribavirina e il cidofovir, da usare per via endoveno-sa. 11,12 Entrambi i farmaci, ma particolarmente il cidofo-vir, pongono problemi di tossicità che ne rendono diffici-le l'uso e problematica la raccomandazione in mancanzadi dati controllati sull'efficacia. Nel nostro studio, l'unicopaziente che riuscì a controllare la viremia fu trattato conil cidofovir (Figura 3a). Questa osservazione sembra sug-gerire che il cidofovir possa contribuire a controllare laviremia da adenovirus. Tuttavia, la scomparsa della vire-mia coincise con l'aumento della conta linfocitaria, sug-gerendo che il miglioramento delle funzione immunolo-gica ebbe un ruolo importante. Pertanto, non è possibilegiudicare l'efficacia del trattamento antivirale in questocaso. Altri dati contribuiscono a generare dubbi sull'effi-cacia del cidofovir. In un paziente HIV-sieropositivo conbassi linfociti CD4 (44 cellule per microlitro) ed affetto dainfezione adenovirale (polmonite ed epatite), la caricaadenovirale nel sangue rimase immutata nonostante l'u-so del cidofovir. Il paziente morì con infezione dissemina-ta (Figura 3b).
Conclusioni- La storia naturale dell'infezione da adenovirus dopo
trapianto di cellule ematopoietiche staminali da dona-tore allogenico è potenzialmente aggressiva.
- La viremia è un marker di infezione disseminata e per-ciò di elevato rischio di mortalità.
- La storia naturale dell'infezione da adenovirus e il suc-cesso del modello di sorveglianza e trattamento pre-emptive del CMV suggeriscono che l'approccio da pre-ferire nel controllo dell'adenovirus sia una strategiapre-emptive, con intervento precoce allo scopo di pre-venire la progressione dell'infezione.
- La sorveglianza della viremia offre l'opportunità diidentificare pazienti ad alto rischio di mortalità.
- Tali pazienti possono essere trattati con una riduzio-ne della terapia immunosoppressiva quando possibi-le, e con farmaci antivirali.
- La ribavirina ed il cidofovir sono stati proposti comepossibilità terapeutiche. Tuttavia mancano dati con-trollati sulla loro efficacia clinica nei pazienti con infe-zione da adenovirus.
- Le cinetiche del carico virale nel sangue sono alta-mente correlate alla progressione o risoluzione del-l'infezione e offrono un marker surrogato ideale perdeterminare l'efficacia dei trattamenti antivirali in stu-di clinici controllati.
Bibliografia
1. Meyer-Koenig U, Weidmann M, Kirste G, Hufert FT.Cytomegalovirus infection in organ-transplant recipients: diag-nostic value of pp65 antigen test, qualitative polymerase chainreaction (PCR) and quantitative Taqman PCR. Transplantation.2004;77:1692-8.
2. Flomenberg P, Babbitt J, Drobyski WR, et al. Increasing incidenceof adenovirus disease in bone marrow transplant recipients. J InfectDis. 1994;169:775-81.
3. Hale GA, Heslop HE, Krance RA, et al. Adenovirus infection afterpaediatric bone marrow transplantation. Bone Marrow Transplant.1999;23:277-82.
4. Howard DS, Phillips II GL, Reece DE, et al. Adenovirus infections inhematopoietic stem cell transplant recipients. Clin Infect Dis.1999;29:1494-501
5. Baldwin A, Kingman H, Darville M, et al. Outcome and clinicalcourse of 100 patients with adenovirus infection following bonemarrow transplantation. Bone Marrow Transplant. 2000;26:1333-8.
6. Runde V, Ross S, Trenschel R, et al. Adenoviral infection after allo-geneic stem cell transplantation (SCT): report on 130 patients froma single SCT unit involved in a prospective multi center surveillancestudy. Bone Marrow Transplant. 2001;28:51-7.
7. La Rosa AM, Champlin RE, Mirza N, et al. Adenovirus infections inadult recipients of blood and marrow transplants. Clin Infect Dis.2001;32:871-6.
8. Heim A, Ebnet C, Harste G, Pring-Akerblom P. Rapid and quanti-tative detection of human adenovirus DNA by real-time PCR. JMed Virol. 2003;70:228-39
9. Lion T, Baumgartinger R, Watzinger F, et al. Molecular monitoringof adenovirus in peripheral blood after allogeneic bone marrowtransplantation permits early diagnosis of disseminated disease.Blood. 2003;102:1114-20.
10. Teramura T, Naya M, Yoshihara T, Kanoh G, Morimoto A, ImashukuS. Adenoviral infection in hematopoietic stem cell transplanta-tion: early diagnosis with quantitative detection of the viralgenome in serum and urine. Bone Marrow Transplant. 2004;33:87-92.
11. Miyamura K, Hamaguchi M, Taji H, et al. Successful ribavirin ther-apy for severe adenovirus hemorrhagic cystitis after allogeneicmarrow transplant from close HLA donors rather than distantdonors. Bone Marrow Transplant. 2000;25:545-8.
12. Legrand F, Berrebi D, Houhou N, et al. Early diagnosis of adenovirusinfection and treatment with cidofovir after bone marrow trans-plantation in children. Bone Marrow Transplant. 2001;27:621-6.
84
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre

Uso del concentrato di proteina C nel paziente settico
Armando D'Angelo, Patrizia Della Valle, Silvana Vigano' D'AngeloServizio di Coagulazione ed Unita' Ricerca Trombosi, IRCCSH S. Raffaele, Milano
L'organismo risponde all'infezione od al trauma con unareazione infiammatoria che è volta al contenimento loca-le del danno ed alla sua riparazione. Qualora questa rispo-sta sia esagerata e si amplifichi fino a coinvolgere l'inte-ro organismo, si ha uno stato d'infiammazione sistemica(SIRS) che si auto-sostiene in misura essenzialmente indi-pendente dall'insulto iniziale. La sepsi e le sue sequelecostituiscono quindi stadi successivi di una stessa entitànosologica per la quale la risposta sistemica ad un'infe-zione, sostenuta e amplificata da mediatori endogeni, puòportare ad una reazione infiammatoria generalizzata inorgani anche molto lontani da quelli colpiti in origine, finoad esitare nella disfunzione e nell'insufficienza di uno opiù organi.
La sepsi non riconosce una singola causa, né un quadroclinico univoco; non e' quindi soprendente che la stimaepidemiologica della sua prevalenza sia molta varia secon-do i punti d'osservazione e le categorie di definizione uti-lizzate. 1 Per quanto riguarda gli adulti, i dati del SepsisItalian Study raccolti nel 1994 indicavano che il 10% ditutti i pazienti ricoverati nelle 99 Unità di Terapia Inten-siva partecipanti presentavano o sviluppavano sepsi,2 edun indagine più recente condotta nel terzo millennio in 83unità di terapia intensive italiane (Studio Replay3) hadimostrato la presenza di sepsi all'ingresso nel 12.6% deipazienti, con la comparsa di sepsi durante la degenza inun ulteriore 10% dei casi. La mortalità per sepsi varia mol-to in funzione della gravità della sindrome, passando dal20% circa per i pazienti con SIRS al 45-80% - secondo lecasistiche - nei pazienti con shock settico. Dati america-ni recenti riferiscono 800-900.000 casi di sepsi ogni annocon una mortalità compresa tra il 23 ed il 26%. 4 In Ita-lia, la mortalità per shock settico è senz'altro più elevatache in altri paesi, pur se ridotta da oltre 80% (2) a 73. 5%.3
La mortalita' per sepsi in eta' pediatrica e' sicuramenteinferiore a quella dell'adulto, anche se, ad esempio nelcaso della sepsi meningococcica, gravi sequele possonocomplicare la guarigione. E' in corso un indagine epide-miologica nelle terapie intensive pediatriche italiane attaa definire la situazione attuale nel nostro paese.
Gli studi di fisiopatologia della sepsi hanno messo inrelazione la perdita di controllo delle risposte infiamma-toria e coagulativa con la progressione del quadro clinicofino al collasso d'organo e la morte. L'attivazione dellarisposta infiammatoria sistemica caratteristica della sepsiattiva di pari passo una risposta del sistema coagulativoe fibrinolitico. I due sistemi sono strettamente correlati traloro e si attivano e amplificano a vicenda in una sorta dicircolo vizioso cui consegue lo squilibrio emostatico insenso procoagulante, il collasso vascolare, l'ischemia, ildanno d'organo fino alla completa insufficienza d'organo
e la morte. La cascata coagulativa consiste di una serie di reazioni
proteoliti che concatenate, localizzate sulla membrana diglobuli bianchi, piastrine o cellule endoteliali, tramite lequali degli zimogeni circolanti sono convertiti in enzimitripsino-simili. L'innesco alla coagulazione proviene dalcontatto del sangue circolante con un recettore tran-smembranario per il fattore VII (FVII), il fattore tessutale(FT), una proteina costituzionalmente espressa da speciecellulari extravascolari (fibroblasti, cellule muscolari lisceecc), ma inducibile da vari stimoli (batteri, endotossine,citochine) anche alla superficie di monociti e cellule endo-teliali. Alla formazione del complesso FVII-FT segue l'at-tivazione efficiente del FVII, l'interazione con gli zimoge-ni fattore X (tenasi estrinseca) e IX e la loro trasformazio-ne in enzimi. A loro volta, i fattori IXa e Xa rappresenta-no la componente enzimatica di due complessi, la tenasiintrinseca e la protrombinasi, che portano alla formazio-ne rispettivamente d'ulteriore Xa e di trombina. In entram-bi questi complessi un ruolo fondamentale è giocato dadue proteine non enzimatiche, i cofattori V e VIII che favo-riscono l'assemblaggio del complesso enzima-zimogenosulla superficie cellulare. Il Fattore VIII circola normal-mente in complesso con il fattore von Willebrand; perazione di Xa e trombina esso subisce un processo di pro-teolisi con perdita d'alcuni domini (tra i quali quello dilegame al fattore von Willebrand) e trasformazione inFVIIIa. Analoga proteolisi subisce il fattore V nella tra-sformazione a FVa. Pertanto, la cascata coagulativa puòessere rappresentata come una serie di reazioni nelle qua-li un enzima, un cofattore ed uno zimogeno interagisco-no a formare un complesso multimolecolare su di unasuperficie fisiologica. I quattro componenti del comples-so debbono essere tutti presenti affinché la reazione diconversione dello zimogeno ad enzima possa avvenire conrapidità sufficiente per essere efficace in vivo.
Una serie di meccanismi anticoagulanti interagiscono aciascun livello della cascata coagulativa; in assenza diessi, i processi emostatici, continuamente operanti per ilmantenimento dell'integrità' vascolare, proseguirebberoindisturbati fino a trasformare il tappo emostatico intrombo. Per il controllo delle reazioni che portano alla for-mazione della fibrina esistono 3 principali meccanisminaturali di regolazione. Il primo meccanismo consiste nel-l'inibizione della generazione di fattore Xa e IXa da partedell'inibitore della via estrinseca (denominato Tissue Fac-tor Pathway Inhibitor, TFPI). Il secondo meccanismo con-siste nella neutralizzazione delle proteasi della coagula-zione per opera di reazioni irreversibili con una serie d'i-nibitori. Di questi, il più importante è senza dubbio l'an-titrombina (AT). Il terzo meccanismo consiste nell'inatti-vazione proteolitica di FVa e FVIIIa da parte della protei-na C attivata. L'endotelio gioca un ruolo fondamentalenel controllo della coagulazione. Tutti i principali mecca-nismi d'anticoagulazione naturale sono concentrati al suolivello tramite recettori transmembranari espressi ed atti-vi in condizioni fisiologiche. La localizzazione endotelialene permette il funzionamento efficace a livello del micro-circolo, dove il rapporto tra superficie endoteliale e san-gue circolante raggiunge il valore massimo (0.5 m2 disuperficie endoteliale per ml di sangue). L'endotelio èanche centrale ai meccanismi di fibrinolisi.
85
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

Il rilascio di citochine (TNF-α, IL-1, IL-6, IL-8) da partedi monociti e leucociti a seguito dell'insulto infettivo cau-sa l'aumentata espressione di fattore tessutale e l'incre-mento dei livelli circolanti d'alcuni fattori della coagula-zione che in concerto con l'attivazione dei neutrofili e del-le piastrine inducono un'aumentata formazione di trom-bina. Questi processi sono inizialmente contrastati dal-l'attività dei sistemi anticoagulanti naturali (TFPI, anti-trombina, proteina C), particolarmente attivi nella micro-circolo. Quando però il potenziale anticoagulante si esau-risce per il consumo di proteina C ed antitrombina, latrombino-formazione diviene incontrollata, con il preva-lere degli effetti di feed-back positivo della trombina sulprocesso coagulativo, e dello stimolo infiammatorio che latrombina stessa esercita su monociti, leucociti e celluleendoteliali, che porta al rilascio di citochine, fattore atti-vante le piastrine (PAF) ed all'aumentata espressione sul-la membrana endoteliale di molecole d'adesione, quali l'e-selettina, che favoriscono in un circolo vizioso il rolling el'attivazione dei granulociti neutrofili. Pertanto, la soffe-renza endoteliale causata dall'esaurimento dei sistemianticoagulanti endogeni e dal feed-back positivo d'in-fiammazione e coagulazione ostacola gli scambi sangue-tessuti e porta quindi di fatto all'ischemia d'organo primaancora che la deposizione di fibrina occluda il microcir-colo. 6 L'endotelio risulta quindi in ultima analisi il primoorgano bersaglio del processo settico.
Il coinvolgimento del sistema della proteina C nella sepsiLa trombina viene inattivata dall'organismo sostanzial-
mente tramite due meccanismi. Il primo è rappresentatodalla formazione di complessi neutralizzanti irreversibilicon l'antitrombina. Il secondo meccanismo è legato allaformazione del complesso della trombina con un recetto-re endoteliale, la trombomodulina (TM) che ne cambia laspecificità di substrato trasformandola da procoagulantein anticoagulante. Il complesso trombina-trombomoduli-na costituisce l'attivatore fisiologico della proteina C. Laproteina C è uno zimogeno plasmatico vitamina K-dipen-dente la cui concentrazione fisiologica è circa pari a 60nmol/l. Una volta attivata essa interagisce con un cofat-tore anch'esso vitamina K-dipendente, la proteina S, aneutralizzare i fattori Va e VIIIa, cofattori essenziali per laformazione di trombina. Così la stessa trombina, tramiteil sistema della proteina C, contribuisce a limitare la pro-pria generazione. 7
In alcuni segmenti del letto vascolare l'attivazione del-la proteina C risulta aumentata dalla presenza sulle cel-lule endoteliali di un recettore endoteliale per la proteinaC (EPCR). L'espressione di questo recettore è tanto piùsignificativa quanto maggiore è il lume vasale. Al contra-rio, il numero di molecole di TM nell'endotelio è relativa-mente costante in tutto il sistema vascolare, e quindi lasua concentrazione è determinata dal numero di celluleendoteliali (o superficie cellulare endoteliale) a contattocol sangue. Nei capillari, ciò corrisponde a circa 0.3-0.5 m2
d'endotelio/ml sangue (pari ad una concentrazione di cir-ca 500 nM), mentre in un vaso delle dimensioni appros-simative di un'arteria coronarica tale numero rapportoscende a circa 1,5 cm2 (pari ad una concentrazione di soli0,1-0,2 nM). L'aumentata espressione d'EPCR nei grossivasi compensa quindi la diminuzione nell'attivazione del-
la proteina C determinata dalla relativa scarsità di super-ficie endoteliale. 7 Il sistema della proteina C gioca ancheun ruolo nei processi di fibrinolisi. L'APC compete con il t-PA nel complesso con il PAI-1 e, riducendo la formazionedi trombina, previene l'attivazione di TAFI8 stimolando cosìla fibrinolisi con due diversi meccanismi. Non ultimo, l'APCesplica una azione antinfiammatoria specifica, riducendoil rilascio di citochine dai monociti probabilmente a cau-sa dell'inibizione della traslocazione nucleare di NF-B. 9 Lariduzione della produzione di TNF-α non interferisce conle attività antimicrobiche naturali dei monociti, quali ade-sione, fagocitosi, attacco dei batteri gram negativi,10 men-tre il legame dell'APC ad un recettore specifico monoci-tario inibisce il segnale intracellulare del calcio e le risul-tanti risposte proliferative. 11 Con un meccanismo proba-bilmente mediato anch'esso dall'inibizione di NF-kB, l' APCblocca l'espressione di fattore tissutale da parte di neu-trofili leucemici attivati in un processo che si ritienemediato dal recettore endoteliale della proteina C. 12 Infi-ne, la proteina C zimogeno compete con i neutrofili per lae-selettina, inibendo cosi' il loro contatto con l'endotelio.13 E' assai probabile che l'inibizione della attivazione deigranulociti neutrofili rappresenti un meccanismo fonda-mentale tramite il quale si esplica l'attivita' infiammato-ria della APC; va notato che l'attivita' antinfiammatoriadella APC e' stata osservata a concentrazioni inferiori diquelle richieste per un efficace attivita' anticoagulante eprofibrinolitica. 14
Una serie di studi sperimentali hanno condotto allacomprensione della rilevanza del sistema della proteina Cnella sepsi. Babbuini infettati con dosi letali d'EscherichiaColi sviluppavano rapidamente coagulazione intravasco-lare disseminata, danno d'organo multiplo, shock setticoe giungevano a morte. Contemporaneamente, si eviden-ziava l'aumento in circolo, con sequenze temporali diver-se, di una serie di citochine ad azione pro- ed anti-infiam-matoria. 15,16 Nello stesso modello animale, il blocco delprocesso coagulativo tramite l'infusione di un anticoagu-lante diretto (DEGR Xa), preveniva la coagulazione intra-vascolare disseminata ma non aveva effetto sulla morta-lità di babbuini trattati con dosi letali d'E. Coli ne' sui livel-li di TNF-α. 17 Peraltro, il trattamento con anticorpi diret-ti contro il FT o con VIIa attivato ed inibito al sito attivo(DEGR-VIIa) riduceva di circa il 50% la mortalità deglianimali trattati con dosi letali di E. Coli, attenuando larisposta coagulativa e la salita nei livelli plasmatici di IL-6 e IL-8, ma non mostrando effetto significativo su quel-li di TNF-α. 18 Questi dati indicano che il blocco della for-mazione di trombina, pur inibendo la deposizione di fibri-na nella microcircolazione non è sufficiente ad impedirela risposta infiammatoria ne' ad avere effetto sulla mor-talità. Al contrario, l'infusione in cani di basse dosi ditrombina (0.5 U/Kg/min) per 90 minuti non aumentava lasopravvivenza dal 7% al 64% a seguito dell'infusione didosi letali di E Coli. All'infusione di trombina si associavala comparsa di attività anticoagulante e fibrinolitica nonosservabile con l'infusione di placebo. 19 Questo natural-mente fece supporre un ruolo protettivo del sistema del-la proteina C nella sepsi. In esperimenti successivi si dimo-strò in effetti che l'infusione di proteina C purificata daplasma umano ed attivata con complesso trombina-trom-bomodulina risultava in grado di proteggere dallo shock
86
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre

settico e dalla morte babbuini trattati con dosi letali di E.Coli. 20 A questi primi esperimenti ne seguirono altri inbabbuini trattati con dosi sub-letali di E. coli, ma interfe-rendo con l'attivazione della proteina C, ossia rendendoinaccessibile con un anticorpo monoclonale il dominio diattivazione della proteina C al complesso trombina-trom-bomodulina e mimando quindi uno stato di carenza seve-ra della proteina. In queste condizioni gli animali svilup-pavano coagulazione intravascolare disseminata, shocksettico e morivano rapidamente, salvo che non venisseloro infusa APC, in grado di ripristinare il quadro coagu-lativo e di garantire la sopravvivenza dell'animale. 20 Inol-tre, gli animali trattati con dosi sub-letali di E. coli neiquali si preveniva l'attivazione della proteina C mostrava-no livelli di TFN-α marcatamente aumentati, quasi pari aquelli rilevati negli animali trattati con dosi letali del bat-terio. 20 Studi condotti più recentemente nel topo con l'in-fusione di una dose letale mediana di endotossina (LD50),hanno ulteriormente evidenziato il ruolo fondamentaledell'attivazione in vivo della proteina C nella sepsi. Utiliz-zando metodiche di ingegneria genetica, sono stati otte-nuti topi con una ridotta attività della trombomodulinaendoteliale, a causa della sostituzione di un singolo ami-no acido,21 che causa la completa assenza di attivazionedella proteina C ad opera del complesso trombina-trom-bomodulina. In questi topi, la LD50 causava morte nel100% dei casi. 22
Questi dati hanno messo in luce il ruolo essenziale delsistema della proteina C nel determinare la risposta del-l'ospite in corso di sepsi. Una carenza acquisita di protei-na C si riscontra in più dell' 85% dei pazienti con sepsi. 23
Ciò è dovuto all'attivazione in vivo della proteina ed al for-marsi di complessi tra APC ed i suoi inibitori plasmatici(α1-antitripsina, PAI-3, β2-macroglobulina) che vengonorapidamente rimossi dal circolo. Quando tale processoprevale sulla sintesi di nuova proteina C i livelli circolan-ti scendono rapidamente al 50-70% della norma. Poichél'attivazione della proteina C, alle concentrazioni fisiolo-giche, è linearmente proporzionale alla sua concentrazio-ne plasmatica, bassi livelli dello zimogeno saranno asso-ciati nel paziente settico ad una ridotta capacità dellaproteina C di attivarsi ad opera del complesso trombina-trombomodulina. La riduzione dei livelli di proteina C none' quindi soltanto un indicatore transitorio dello stato set-tico ma compare precocemente e progredisce con l'ag-gravarsi della patologia, dimostrandosi un indicatore pro-gnostico sfavorevole nel paziente settico. 23 Va inoltrericordato che il progredire della patologia potrebbe anchelimitare la disponibilità di TM ed EPCR endoteliali. 24-26
Infine, il principale inibitore dell'APC, l'α1-antitripsina,una proteina di fase acuta, aumenta in corso di sepsi, ridu-cendone così la permanenza in circolo, di norma intornoai 10-15 minuti.
L'utilizzo dei concentrati di proteina C nella sepsi severaProteina C attivata. Una preparazione ricombinante di
proteina C umana, attivata con il complesso trombina-trombomodulina (gia' Drotrecogin alfa activated, oraXigris) si e' dimostrata il primo rimedio efficace nel ridur-re significativamente la mortalita' in un ampio studio indoppio cieco condotto in 1690 pazienti con sepsi severa(Prowess Trial27). I pazienti venivano randomizzati a rice-
vere APC (0.024 mg/kg/h) o placebo per 96 ore. La mor-talita' a 28 giorni e' risultata del 24. 7% nel gruppo trat-tato con APC e del 30.8% nel gruppo trattato con place-bo, con una riduzione del rischio relativo di morte del 19.4% (p = 0.005). L'incidenza di complicanze emorragichemaggiori risultava circa raddoppiata dal trattamento atti-vo (3. 5% contro 2.0%, p = 0.06). Il trattamento induce-va una piu' rapida discesa dei livelli di D-dimero e IL-6rispetto al placebo e risultava egualmente efficace neipazienti che avevano all'ingresso livelli normali o ridottidi proteina C (zimogeno) circolante, indipendentementedai valori di score APACHE II, dal numero di organi insuf-ficienti, da eta', sesso, localizzazione e tipo di infezione.L'effetto antinfiammatorio del trattamento con APC nonsi associava inoltre ad un' aumentata incidenza di nuoveinfezioni durante la degenza dei pazienti in terapia inten-siva. Un documento della Food and Drug Administration28
riporta che circa 2/3 dei pazienti erano in trattamentoprofilattico con eparina. Il documento non evidenzia unaumento del rischio emorragico nei pazienti trattati conAPC ed eparina a dosi profililattiche, ma rileva un atte-nuarsi dell'efficacia della APC quando somministratainsieme all'eparina, con una variazione del rischio relati-vo dal 15% al 50%. Il documento inoltre chiarisce unmaggior vantaggio dall'uso della APC nei soggetti con piu'di 50 anni, con APACHE II score superiore a 25, con 2 opiu' disfunzioni d'organo e con presenza di coagulazioneintravascolare disseminata. Queste osservazioni hannoindotto la FDA a richiedere nuovi studi per la registrazio-ne di un piu' ampio pannello di indicazioni per lo Xigris.
Proteina C zimogeno. Gli studi Optimist,29 Kybersept30 eProwess27 hanno tutti riportato una maggiore incidenza dicomplicanze emorragiche nei pazienti trattati con il far-maco attivo, indipendente dal contemporaneo uso di epa-rina a dosi profilattiche, nonostante l'esclusione di pazien-ti con un aumentato rischio emorragico. Peraltro, l'effi-cacia dei trattamenti sembra essere maggiore nei pazien-ti piu' gravi, che non vengono sottoposti a profilassi epa-rinica e che avendo un disturbo maggiore della coagula-zione risultano a maggior rischio emorragico. In questicasi, ed in particolare ove sussistano controindicazioni altrattamento con Xigris, che e' da intendersi come il far-maco di scelta, e' possibile che la normalizzazione deilivelli di proteina C con l'infusione di concentrati dellozimogeno, in associazione o meno alla normalizzazionedei livelli di antitrombina31 risulti di notevole efficacia.Sebbene non esistano per i concentrati di proteina C zimo-geno studi controllati in doppio cieco, l'osservazione del-la necessita' di livelli di APC inferiori a quelli anticoagu-lanti per una efficace attivita' antinfiammatoria14 sugge-risce infatti un ruolo importante per l'attivazione endo-gena della zimogeno, che puo' verificarsi, in presenza disufficienti concentrazioni di trombina, a livello endote-liale, nonostante la potenziale downregulation della trom-bomodulina,26 od alla superficie dei monociti attivati. 9,10
I concentrati di proteina C zimogeno rappresentano la pri-ma scelta terapeutica nei casi di purpura fulminans neo-natale associati a carenza omozigote di proteina C, masono stati utilizzati con successo anche in pazienti concoagulazione intravascolare disseminata. 32,33 La maggio-re esperienza circa l'impiego di questo concentrato e' sta-ta ottenuta nel trattamento della sindrome di Waterhou-
87
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

se Friedrichsen su base meningococcica. Un razionale alsuo impiego e' fornito dal riscontro in questa sindrome diun elevatissimo consumo di proteina C a seguito della suaattivazione, con il risultato di livelli particolarmente bas-si di zimogeno circolante, simili a quelli osservabili nellecarenze omozigoti della proteina. 34 Altre comunicazionirelative a casi di shock e purpura da meningococco in eta'pediatrica hanno riportato risultati favorevoli con un trat-tamento basato sulla infusione di 100 UI/kg di zimogenoogni 6-8 ore — l'emivita della proteina C e' fisiologica-mente di circa 8-10 ore35 — per 4-6 giorni. 36,37 White etal. hanno condotto uno studio prospettico su 36 pazientidi eta' compresa tra 3 mesi e 72 anni con shock da menin-gococco, purpura fulminans e coagulazione intravascola-re disseminata, con livelli di proteina C inferiori al 30%della norma (38). Dopo la somministrazione di una dosetest, il concentrato di proteina C (Ceprotin®) veniva som-ministrato in tutti i pazienti — in aggiunta alla terapiatradizionale — tramite bolo di 100 UI/Kg seguito dall'in-fusione continua di 10-15 UI/Kg/h, per mantenere livellidello zimogeno tra 80 e 120%. La maggior parte deipazienti veniva anche sottoposto a trattamento emodia-litico e profilassi con eparina. Rispetto a dati storici (50%di mortalita', 30% di amputazioni entro le prime 24 ore)si e' ottenuto un miglioramento impressionante dell'an-damento clinico dei pazienti, di cui solo l'8% deceduti edil 6. 5% richiedenti amputazioni. 38 La coagulopatia sirisolveva generalmente entro il quinto giorno di tratta-mento, con aumento di conta piastrinica e fibrinogeno eriduzione significativa dei livelli di D-dimero. Risultatianaloghi sono stati ottenuti in casistiche piu' ridottemirando a livelli di zimogeno superiori al 60%, con moda-lita' di trattamento piu' protratte nel tempo e prevedentiboli di 50 UI/Kg fino a 6 volte al giorno. 39,40 Uno studiorecente ha dimostrato che i livelli di APC circolanti dopol'infusione del concentrato sono chiaramente correlati allaquantita' di zimogeno infusa. 41 In questo studio, 40 bam-bini con sepsi severa da meningococco e purpura fulmi-nans sono stati randomizzati al trattamento con placeboo con 3 diversi dosaggi di Ceprotin (50, 100 o 150 UI/Kg)somministrati ogni 6 ore per almeno 3 giorni (estensibilia 7). I livelli medi pretrattamento di proteina C erano com-presi tra il 15% ed il 21% della norma nei 4 gruppi, men-tre quelli di proteina C attivata variavano tra 2.3 e 8 ng/ml(livelli normali compresi tra 0.2 e 0.8 ng/ml). Con i dosag-gi di Ceprotin di 50, 100 e 150 UI/Kg si raggiungevanolivelli di picco di proteina C circolante rispettivamente del63%, 133% e 174%, mentre i corrispondenti livelli di APCaumentavano, rispetto ai valori pretrattamento, del 80%,104% e 350%. Non sorprendentemente, la discesa deilivelli di D-dimero era tanto maggiore quanto maggiore ildosaggio del concentrato. Questi dati indicano la neces-sita' di raggiungere livelli di proteina C sopranormali perottenere un significativo aumento della quota di APC cir-colante. 41
Per i pazienti con sepsi severa di origine non meningo-coccica non esistono che dati aneddotici, e l'estrapola-zione dei risultati favorevoli ottenuti negli studi succita-ti ad altri tipi di sepsi potrebbe rivelarsi non corretta, seb-bene nello studio Prowess non si siano osservate diffe-renze di efficacia della APC in funzione della natura del-la sepsi.
Bibliografia
1. Matot I, Sprung CL. Definition of sepsis. Int Care Med 2001;27:S3-S9.
2. Salvo I, de Cian W, Musico M, Langer M, Piadena R, Wolfler A, Mon-tani C, Magni E. The Italian Sepsis Study: preliminary results on theincidence and evolution of SIRS, severe sepsis and septic shock. IntCare Med 1995;21:S244-S249.
3. Fumagalli R. Personal communication. 4. Wenzel RP. Treating sepsis. N Engl J Med 2002;347:966-7. 5. Buzan J. Structural design and molecular evaluation of a cytokine
receptor family. Proc Natl Acad Sci USA 1990;87:6934-8. 6. Aird WC. The role of the endothelium in severe sepsis and multiple
organ dysfunction syndrome. Blood 2003;101:3765-77. 7. Esmon CT. The protein C pathway. Crit Care Med 2000;28:S44-S48. 8. Mosnier LO, Meijers JC, Bouma BN. Protein C inhibitor regulates the
thrombin-thrombomodulin complex in the up- and down regula-tion of TAFI activation. Thromb Haemost 2001;86:1057-64.
9. White B, Schmidt M, Murphy C, Livingstone W, O'Toole D, LawlerM, O'Neill L, Kelleher D, Schwarz HP, Smith OP. Activated proteinC inhibits lipopolysaccharide-induced nuclear translocation onnuclear factor kB (NF-kB) and tumor necrosis factor ± (TNF±) pro-duction in the THP-1 monocytic cell line. Br J Haematol2000;110:130-4.
10. Grey ST, Tsuchida A, Hau H, Orthner CL, Salem HH, Hancock WW.Selective inhibitory effects of the anticoagulant activated proteinC on the responses of human mononuclear phagocytes to LPS, IFN-gamma or phorbol esther. J Immunol 1994;153:3664-72.
11. Hancock WW, Grey ST, Hau L, Akalin E, Orthner C, Sayegh MH,Salem HH. Binding of activated protein C to a specific receptor onhuman mononuclear phagocytes inhibits intracellular calcium sig-nalling and monocyte-dependent proliferative responses. Trans-plantation 1995;60:1525-32.
12. Shu F, Kobayashi H, Fukudome K, Tsuneyoshi N, Kimoto M, Terao T.Activated protein C suppresses tissue factor expression on U937cells in the endothelial protein C receptor-dependent manner. FEBSLetters 2000;477:208-12.
13. Grinnell BW, Hermann RB, Yan SB. Human protein C inhibitsselectin-mediated cell adhesion: role of unique fucosylatedoligosaccharide. Glycobiology 1994;2:221-5.
14. Hirose K, Okajima K, Taoka Y, Uchiba M, Tagami H, Nakano K-Y etal. Activated protein C reduces the ischemia/reperfusion-inducedspinal cord injury in rats by inhibiting neutrophil activation. AnnSurg 2000;232:272-80.
15. Creasey AA, Stevens P, Kenney J, Allison AC, Warren K, Catlett R,Hinshaw L, Taylor FB Jr. Endotoxin and cytokine profile in plasmaof baboons challenged with lethal and sublethal Escherichia coli.Circ Shock 1991;33:84-91.
16. Taylor FB Jr, Chang AC, Esmon CT, Hinshaw LB. Baboon model ofEscherichia coli sepsis: description of its four stages and the role oftumor necrosis factor, tissue factors, and the protein C system inseptic shock. Curr Stud Hematol Blood Trasfus 1991;58:8-14.
17. Taylor FB Jr, Chang AC, Peer GT, Mather T, Blick KE, Catlett R, Lock-hart MS, Esmon CT. DEGR-factor Xa blocks disseminated intravas-cular coagulation initiated by Escherichia coli without preventingshock or organ damage. Blood 1991;78:364-8.
18. Taylor FB Jr, Chang AC, Peer G, Li A Ezban M, Hedner U. Active siteinhibited factor VIIa (DEGR VIIa) attenuates the coagulant and din-terelukin-6 and -8, but not tumor necrosis factor, responses of thebaboon to LD 100 Escherichia coli. Blood 1998;91:1609-15.
19. Taylor FB Jr, Chang A, Hinshaw LB, Esmon CT, Archer LT, Beller BK.A model for thrombin protection against endotoxin. Thromb Res1984;36:177-85.
20. Taylor FB Jr, Chang A, Esmon CT, D'Angelo A, Vigano'-D'Angelo S,Blick KE, Protein C prevents the coagulopathic and lethal effects ofE. Coli infusion in the baboon. J Clin Invest 1987;79:918-25.
21. Weiler H, Lindner V, Kerlin B, Isermann BH, Hendrickson SB, Coo-ley BC, Meh DA, Mosesson MW, Shworak NW, Post MJ, Conway EM,Ulfman LH, von Andrian UH, Weitz JI. Characterization of a mousemodel for thrombomodulin deficiency. Arteriosl Thromb Vasc Biol2001;21:1531-37.
22. Weiler-Guettler H, Christie PD, Beeler DL, Heally AM, Hancock WW,Rayburn H, Edelberg JM, Rosenberg RD. A targeted point mutation
88
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre

89
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
in thrombomodulin generates viable mice with a prethromboticstate. J Clin Invest 1998;101:1301-9.
23. Fisher CJ Jr, Yan SB. Protein C levels as a prognostic indicator of out-come in sepsis and related diseases. Crit Care Med 2000;28:S49-S56.
24. Takano S, Kimura S, Ohdama S, Aoki N. Plasma thrombomodulin inhealth and diseases. Blood 1990;76:2024-9.
25. Kurosawa S, Stearns-Kurosawa DJ, Carson CW, D'Angelo A, DellaValle P, Esmon CT. Plasma levels of endothelial cell protein C recep-tor are elevated in patients with sepsis and systemic lupus erythe-matosus: lack of correlation with thrombomodulin suggests involv-ment of different pathological processes. Blood 1998;91:725-7.
26. Faust SN, Levin M, Harrison OB, Goldin RD, Lockhart MS, Kondav-eeti S, Laszik Z, Esmon CT, Heyderman RS. Dysfunction of endothe-lial protein C activation in severe meningococcal sepsis. N Engl Jmed 2001;345:408-16.
27. Bernard GR, J-L Vincent, Laterre P-F, La Rosa SP, et al for theRecombinant Human Acitvated Protein C Worldwide Evaluation inSevere Sepsis (PROWESS) Study Group. Efficacy and safety ofrecombinant human activated protein C for severe sepsis. N Engl Jmed 2001;344:699-709.
28. Anti-infective Advisory Committee. FDA Briefing Document:Drotrecogin alfa (activated) (Recombinant human activated proteinC (rhAPC)) Xigris. Rockville, MD: Food and Drug Administration,September 12, 2001.
29. Abraham E, Reinhart K, Opal S, Demeyer I, et al for the Optimist Tri-al Study Group. Efficacy and safety of Tifacogin (Recombinant tis-sue factor pathway inhibitor) in severe sepsis. JAMA 2003;290:238-47.
30. Warren BL, Eid A, Singer P, Pillay SS et al, for the Kybersept TrialStudy Group. Caring for the critically ill patient. High-doseantithrombin III in severe sepsis: a randomized controlled trial.JAMA 2001;286:1869-78.
31. Giudici D, Baudo F, Palareti G, Ravizza A, Ridolfi L, D' Angelo A.Antithrombin III replacement in patients with sepsis and septicshock. Haematologica, 1999; 84:452-60.
32. Okajima K, Imamura H, Koga S, Inoue M, Takatsuki K, Aoki N. Treat-ment of patients with disseminated intravascular coagulation byprotein C. Am J Hematol 1990;33:277-8.
33. Gerson WT, Dickerman JD, Bovill EG, Golden E. Severe acquired pro-tein C deficiency in purpura fulminans associated with dissemi-nated intravascular coagulation: treatment with protein C con-centrate. Pediatrics 1993;91:418-22.
34. Fijnvandraat K, Derkx B, Peters M, et al. Coagulation activation andtissue necrosis in meningococcal septic shock : severely reducedprotein C levels predict a high mortality. Thromb Haemost1995;73:15-20.
35. Vigano' D'Angelo S, Comp PC, Esmon CT, D'Angelo A. Relationshipbetween protein C antigen and anticoagulant activity during oralanticoagulation and in selected disease states. J Clin Invest1986;77:416-25.
36. Rivard GE, David M, Farrell C, Schwarz HP. Treatment of purpura ful-minans in meningococcemia with protein C concentrate. J Pediatr1995;126:646-52.
37. Rintala E, Seppala OP, Kotilainen P, Pettila V, Rasi V. Protein C inthe treatment of coagulopathy in meningococcal disease. Crit CareMed 1998;26:965-8.
38. White B, Livingstone W, Murphy C, Hodgson A, Rafferty M, SmithOP. An open-label study of the role of adjuvant hemostatic supportwith protein C replacement therapy in purpura fulminans-associ-ated meningococcemia. Blood. 2000;96:3719-24.
39. Kreuz W, Veldman A, Escuriola-Ettinghausen C, Schneider W, BeegT. Protein C concentrate for meningococcal purpura fulminans.Lancet 1998;351:986-7.
40. Ettinghausen CE, Veldman A, Beeg T, Schneider W, Jager G, KreuzW. Replacement therapy with protein C concentrate in infants andadolescents with meningococcal sepsis and purpura fulminans.Semin Thromb Hemost 1999;25:537-41.
41. de Kleijn ED, de Groot R, Hack CE, Mulder PG, Engl W, Moritz B,Joosten KF, Hazelzet JA. Activation of protein C following infusionof protein C concentrate in children with severe meningococcalsepsis and purpura fulminans: a randomized, double-blinded, place-bo-controlled, dose-finding study. Crit Care Med. 2003;31:1839-47.
Uso della PEG asparaginasi nell'età pediatrica
C. Rizzari, R. Chiesa, M. Citterio, T. Coliva, E. Viganò,V. ConterClinica Pediatrica, Università di Milano-Bicocca, Ospeda-le S. Gerardo dei Tintori, Monza
IntroduzioneLa l-asparaginasi (ASP) rappresenta oggi un importante
farmaco nella cura della leucemia linfoblastica acuta (LLA)e dei linfomi dell'età pediatrica. L'ASP è un enzima le cuiproprietà spiegano, in parte, gli specifici effetti farmacolo-gici, tossici e terapeutici che la contraddistinguono. L'atti-vità terapeutica della ASP è attribuita all'idrolisi enzimati-ca della l-asparagina (ASN), nutriente fondamentale che iblasti leucemici non sono in grado di produrre autonoma-mente, che viene trasformata in acido aspartico ed ammo-niaca. I livelli di ASN vengono di fatto azzerati nel plasmadel paziente già alcuni minuti dopo la somministrazionedel farmaco, con conseguente inibizione della sintesi pro-teica e morte cellulare per apoptosi. 1 Già in studi effettuatinegli anni '80 passato è stato riportato che livelli plasma-tici di attività asparaginasica di 100 UI/L permetterebberouna riduzione dei livelli di ASN plasmatici e liquorali taleda assicurare un'adeguata efficacia del farmaco. 2 L'emivi-ta del prodotto nativo è peraltro relativamente breve e sonoquindi necessarie somministrazioni ravvicinate perché ven-ga esercitato un effetto terapeutico ottimale. Le ripetuteesposizioni favoriscono però l'insorgenza di reazioni aller-giche che rappresentano il fenomeno maggiormente limi-tante per il completamento del trattamento programmato.
Il processo di “pegilazione”, cioè il legame di tipo cova-lente di molecole di polietilenglicole a proteine utilizzate ascopo terapeutico, è un metodo comunemente utilizzatonella chimica farmaceutica per ridurre, attraverso un effet-to di mascheramento, il potenziale immunogenico delleproteine stesse. Altro effetto della pegilazione è quello dideterminare un'emivita plasmatica sensibilmente piu' lun-ga che, nel caso della PEG ASP, risulta di circa quattro vol-te più lunga di quella dei prodotti nativi. 3 Ciò consenteintervalli maggiori tra le singole dosi (ed in definitiva quin-di un numero minore di somministrazioni) e dosaggi ridot-ti (e quindi ridotte quantità di farmaco); in conseguenza diciò il potere antigenico del farmaco nativo diminuisce e sideterminano meno frequentemente fenomeni di ipersensi-bilità sia clinica che biologica.
Il farmaco PEG ASP è stato introdotto nella pratica cli-nica sin dall'inizio degli anni '90 soprattutto in pazienti infase di recidiva o con pregressa reazione allergica al pro-dotto nativo. Fin dalle fasi iniziali del suo uso il farmaco èstato sottoposto a numerose valutazioni di tipo farmaco-cinetico e farmacodinamico nonchè di efficacia e tollera-bilità. Scopo di questa sintesi è quello di effettuare unavalutazione del profilo farmacologico e clinico emerso daglistudi riportati negli ultimi 15 anni sull'uso della PEG ASPnell'età pediatrica.
Monitoraggio farmacocinetico
Nuovi Farmaci Antitumorali

L'introduzione sul mercato europeo della PEG ASP deno-minata Oncaspar™ (medac, Amburgo, Germania prodottodalla Kyowa Hakko, Tokyo, Giappone) è stata caratterizza-ta da una fase di monitoraggio farmacologico essenzial-mente volta alla misurazione della attività enzimatica pla-smatica per dosi comprese tra 500 e 2.500 UI/mq. Va peral-tro sottolineato che dagli studi condotti è emersa una cer-ta variabilità intra ed inter paziente sia in soggetti alla pri-ma diagnosi che in fase di recidiva. Lo studio condotto su66 pazienti trattati con la dose di 1.000 UI/mq nella fasedi reinduzione di un protocollo BFM per le LLA (nella fasedi induzione avevano ricevuto il prodotto nativo) hamostrato che solo i 2/3 dei pazienti mostravano adeguataattività enzimatica (> 100 UI/L) due settimane dopo la som-ministrazione del farmaco. Pur non essendo riportata alcu-na reazione allergica, l'attività farmacologica del prodottorisultava invece assai ridotta nel restante terzo dei pazien-ti, verosimilmente a seguito di fenomeni immunologici chedeterminavano una “silent inactivation” del farmaco. 4
Anche in pazienti recidivati il programma di monitorag-gio farmacologico ha evidenziato una certa difficoltà nel-la individuazione di una chiara correlazione tra tipo di pro-dotto usato nel trattamento front-line, insorgenza di rea-zioni allergiche ed attività farmacologica. In altre parole lapossibile perdita di attività enzimatica è risultata difficil-mente prevedibile anche in pazienti trattati con cicli ripe-tuti di PEG ASP. 5
Dai numerosi studi farmacologici effettuati si è definitoper la PEG ASP un profilo di attività farmacologica e di effi-cacia che, per somministrazioni caratterizzate da dosi edintervalli adeguati, è considerato sovrapponibile a quello delprodotto nativo. In particolare una somministrazione di PEGASP al dosaggio di circa 500-1000 UI/mq è sostanzialmenteritenuta in grado di sostituire, in termini di attività farma-cologica, 4 dosi di prodotto nativo da E. Coli somministra-to alla dose di 5. 000-10.000 UI/mq e di surrogarne quin-di gli effetti terapeutici. L'attività farmacologica della PEGASP decade dopo 3 settimane dalla somministrazione, indi-pendentemente dalla dose utilizzata; per tale motivo sem-bra giustificato, qualora si volesse ottenere un appropria-to target farmacologico (>100 UI/L) in soggetti potenzial-mente a maggior rischio di sviluppare reazioni allergiche ofenomeni di “silent inactivation”, ridurre l'intervallo tra lesomministrazioni (per esempio da 14-21 gg a 7-14 gg)piuttosto che aumentare il dosaggio della singola dose. Aseguito di queste considerazioni emerge l'importanza delmonitoraggio farmacologico quale unico strumento capa-ce di individuare precocemente una riduzione dell'attivitàfarmacologica e quindi una inadeguata efficacia terapeu-tica del farmaco.
Monitoraggio farmacodinamico ed efficacia terapeuticaSono state riportate in letteratura numerose esperienze
volte ad identificare, attraverso studi di farmacodinamicae di efficacia, quale sia l'ottimale modalità d'uso della PEGASP. A questo proposito va sottolineata l'importanza cheriveste, nella valutazione dei risultati, il tipo di prodottoutilizzato. Analogamente infatti a quanto noto per i pro-dotti nativi, anche i prodotti di PEG ASP possono differirein relazione al tipo di prodotto nativo che è stato utilizza-to per la coniugazione con il polietilenglicole. Dati di let-teratura riportati in studi condotti con il tipo di ASP nati-
va e pegilata prevalentemente utilizzata in Europa riporta-no infatti livelli di deplezione plasmatica e liquorale di ASNnettamente maggiori rispetto a quanto riportato con l'usodel prodotto nativo e pegilato utilizzato negli USA. 6-9
Per quanto riguarda l'efficacia, uno studio randomizza-to condotto negli USA ha dimostrato che, in pazienti conLLA recidivata, la somministrazione settimanale di PEG ASPalla dose di 2.500 UI/mq permette di ottenere percentualidi remissione completa significativamente superiori rispet-to a quelle ottenute con la stessa dose di farmaco ma conun intervallo di due settimane. 10 Un altro studio condottodal Dana Farber Cancer Institute in bambini affetti da LLAed in trattamento front line prevedeva una randomizza-zione tra l'uso di PEG ASP (2.500 UI/mq ogni due settima-ne per 15 dosi) e di ASP nativa da E. Coli somministrata alladose di 25. 000 UI/mq una volta la settimana per 30 setti-mane nella fase di intensificazione di un intensivo proto-collo di polichemioterapia. L'EFS a 5 anni tra i due gruppidi pazienti non ha mostrato differenze significative. 11 In unaltro studio randomizzato condotto dal Children's CancerGroup la comparazione tra pazienti trattati nella fase diinduzione con una dose di PEG ASP di 2.500 UI/mq epazienti trattati con un prodotto nativo da E. Coli sommi-nistrato ripetute volte alla dose di 6. 000 UI/mq ha dimo-strato che l'EFS a 3 anni ottenuto nei due gruppi risultavasovrapponibile. In relazione a quest'ultimo studio vacomunque sottolineato che pur essendo state riportate, neisoggetti trattati con quel tipo di PEG ASP, attività enzima-tiche >100 UI/L anche per 3 settimane, non è stata otte-nuta una adeguata deplezione di ASN sia a livello plasma-tico che liquorale. Tali dati suggeriscono che, relativamen-te al tipo di PEG ASP utilizzato, lo schema e la dose adot-tati nello studio possano essere stati inadeguati. 7
Uno studio di tipo window, condotto in Europa dal grup-po DCLSG sull'uso della PEG ASP Oncaspar™ medac som-ministrata alla dose di 1.000 UI/mq in bambini affetti daLLA in trattamento front line, ha dimostrato un'adeguataattività asparaginasica per almeno 10 gg ed una consen-suale completa deplezione plasmatica di ASN. La deplezio-ne di ASN nel liquor è risultata invece ridotta, non essen-do stato possibile rilevare in nessuno dei campioni unacompleta assenza di ASN. Il significato di questo aspetto intermini di ridotta efficacia nella profilassi della recidiva diLLA a livello neurologico resta ancora da chiarire. 12
Un altro capitolo di rilievo nel monitoraggio dell'attivitàfarmacologica del farmaco, ed in definitiva anche della suaefficacia, resta quello della misurazione degli anticorpi antiASP prodotti dall'organismo in risposta all'esposizione ripe-tuta e frequente sia ai prodotti nativi che a quelli pegilati.I risultati finora riportati sembrano indicare questo datocome un surrogato potenzialmente utilizzabile per modifi-care nella maniera più appropriata lo schema di tratta-mento. 7,13
Conclusioni e prospettive futureI dati di farmacocinetica, farmacodinamica, efficacia e
tossicità accumulati dalle esperienze riportate in letteraturasull'uso della PEG ASP permettono oggi (peraltro analoga-mente a quanto già noto per i prodotti nativi) di poterapplicare schemi e dosaggi diversificati sulla base di spe-cifici razionali farmacologici.
Da quanto riportato in questa breve sintesi emerge dun-
90
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre

que e soprattutto l'importanza di conoscere le differenticapacità dei singoli prodotti nel determinare da una parteuna adeguata attività enzimatica plasmatica e dall'altrauna soddisfacente deplezione plasmatica e liquorale diASN; ciò dovrebbe consentire la realizzazione di schemi ditrattamento modellati sulle caratteristiche del singolo pro-dotto utilizzato. L'eventuale riaggiustamento di dosi eintervalli o anche del tipo di prodotto di ASP da utilizzare,dovrebbero essere basati su un monitoraggio farmacologi-co ad hoc che potrebbe consentire inoltre di rifinire “in iti-nere” il trattamento anche per ogni singolo paziente, sfrut-tando quindi al massimo le potenzialità terapeutiche delfarmaco.
Bibliografia
1) Broome JD. Evidence that the L-asparaginase of guinea pig serumis responsible for its antilymphoma effects. J Exp Med 1963; 118:99-120.
2) Riccardi R, Holcenberg JS, Glaubiger DL, Wood JH, Poplack DG. L-Asparaginase pharmacokinetics and asparagine levels in cere-brospinal fluid of rhesus monkeys and humans. Cancer Res 1981; 41:4554-4558.
3) Asselin BL, Whitin JC, Coppola DJ, et. al. Comparative pharmacoki-netic studies of three L-Asparaginas preparations. J Clin Oncol 1993;1l: 1780-1786.
4) Müller HJ, Loning L, Horn A, et al. Pegylated asparaginase (Oncas-par) in children with ALL: drug monitoring in reinduction accordingto the ALL/NHL-BFM 95 protocols. Br J Haematol 2000; 110(2):379-84.
5) Vieira Pinheiro JP, Muller HJ, Schwabe D, et al. Drug monitoring oflow-dose PEG-Asparaginase (Oncaspar™) in children with relapsedacute lymphoblastic leukemia. Br J haematol 2001; 113: 115-119.
6) Boos J, Weber G, Ahlke E, et al. Monitoring of asparaginase activi-ty and asparagine levels in children on different asparaginase prepa-rations. Eur J Cancer 1996; 32A: 1544-1550.
7) Avramis VI, Spencer S, Periclou AP, et al. A randomized compari-son of native Escherichia Coli asparaginase and polyethylene glycolconjugated asparaginase for treatment of children with newly diag-nosed standard risk ALL: a Children's Cancer Group Study. Blood2002; 99: 1986-1994.
8) Ahlke E, Nowak-Göttl U, Schülze-Westhoff P, et al. Dose reductionof asparaginase under pharmacokinetic and pharmacodynamic con-trol during induction therapy in children with acute lymphoblasticleukemia. Br J Haematol 1997; 96: 675-681.
9) Rizzari C, Zucchetti M, Conter V, et al. L-Asparagine depletion andL-Asparaginase activity in children with acute lymphoblasticleukemia receiving i. m. or i. v. Erwinia C. or E. Coli L-Asparaginaseas first exposure. Ann Oncol 2000; 11: 189-193
10) Abshire TC, Pollock BH, Billett AL, et al. Weekly polyethylene glycolconjugated L-Asparaginase compared with biweekly dosing pro-duces superior induction remission rates in childhood relapsed acutelymphoblastic leukemia: A Pediatric Oncology Group study. Blood2000; 96(5): 1709-1715.
11) Silverman LB, Gelber RD, Dalten VK, et al. Improved outcome forchildren with acute lymphoblastic leukemia: results of Dana farberConsortium protocol 91-01.Blood 2001; 97: 1211-1218.
12) Appel IM, Pinheiro JP, den Boer ML, et al. Lack of asparagine deple-tion in the cerebrospinal fluid after one intravenous dose of PEG-asparaginase: a window study at initial diagnosis of childhood ALL.Leukemia. 2003; 17(11): 2254-2256.
13) Woo MH, Hak LJ, Storm MC, et al. Anti-asparaginase antibodiesfollowing E. Coli asparaginase therapy in pediatric acute lym-phoblastic leukemia. Leukemia 1998; 12: 1527-1533.
Attività del temozolomide nel medulloblasto-ma recidivato
G. Cefalo*, A. Ruggiero#, M. E. Abate°, M. Massimino*, P.Zucchetti,+ M. Mascarin§, M. L. Garrè^, F. Spreafico*, S.Mastrangelo#, A. Clerico, V. Ridola#, G. Mazzarella#, C.Di Rocco#, A. Donfrancescoî, G. Perilongo*, I. Laz-zareschi#, S. Sandri", F. Fossati-Bellani*, R. Riccardi#Istituto Nazionale Tumori, Milano; #Universita' Cattolica,Roma; °Casa Sollievo Della Sofferenza, S. Giovanni Roton-do; +Ospedale Silvestrini, S. Andrea Delle Fratte ; §CentroDi Riferimento Oncologico, Aviano; ^Istituto G. Gaslini,Genova; Universita' La Sapienza, Roma; îOspedale BambinGesù, Roma; *Universita' Di Padova, Padova; "OspedaleRegina Margherita, Torino
IntroduzioneIl Temozolomide è un agente alchilante orale di secon-
da generazione. Questa nuova molecola possiede unampio spettro di attività antitumorale con un profilo ditossicità relativamente limitata. 1,2 Questo derivato diseconda generazione dell'imidazotetrazina non necessitadi metabolismo epatico, poichè la metilazione nell'agen-te alchilante citotossico, il 5-(3-methyltriazen-1-yl) imi-dazole-4-carboxamide (MTIC), avviene spontaneamentea pH fisiologico, pertanto la farmacocinetica e la distri-buzione tissutale del farmaco non mostrano un'elevatavariabilità individuale. 3 Il Temozolomide è rapidamente ecompletamente assorbito per via orale con ampia distri-buzione tissutale, attraversa la barriera ematoencefalicae raggiunge adeguate concentrazioni nel sistema nervo-so centrale con un rapporto plasma-liquor fra il 30 e 40%.4 Quest'ultima caratteristica ha reso di particolare inte-resse lo studio di questa molecola nei tumori cerebrali. 5
La sua attività antitumorale nei gliomi maligni dell'adul-to è già stata ben documentata. 6 Il suo favorevole profi-lo farmacocinetico e l'attività osservata negli studi pedia-trici di fase I già condotti negli Stati Uniti e in Gran Bre-tagna,7,8 ci ha indotto a effettuare uno studio cooperati-vo multicentrico di fase II del temozolomide nei bambinie giovani adulti affetti da tumori cerebrali refrattari aitrattamenti convenzionali. Questo lavoro riporta unica-mente i dati relativi ai pazienti con diagnosi di medullo-blastoma o PNET cerebrale.
ObiettiviGli obiettivi dello studio erano valutare l'attività anti-
tumorale del Temozolomide, determinando le risposteobiettive, nei bambini e giovani adulti affetti da medullo-blastoma/PNET ricaduto e di valutarne la sua tossicità.
Pazienti e MetodiSono stati arruolati nello studio bambini e giovani adul-
ti affetti da medulloblastoma/PNET — documentato isto-logicamente al momento della diagnosi iniziale — refrat-tario ai trattamenti di prima, seconda e in alcuni casi ter-za linea, comprendenti anche terapia ad alte dosi con rein-fusione di cellule staminali. Poichè l'obiettivo principaledello studio era rappresentato dalla valutazione dellerisposte obiettive, requisito essenziale per l'arruolamentoera la presenza di malattia attiva documentabile radiolo-
91
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

gicamente. Il Temozolomide è stato somministrato adigiuno per via orale per 5 giorni consecutivi in 3 sommi-nistrazioni giornaliere ad un dosaggio compreso tra 120e 200 mg/mq/die in relazione ai precedenti trattamenti edai valori ematologici, con cicli successivi effettuati ogni 28giorni, fino a progressione o fino a un massimo di 36 ciclicomplessivi. La valutazione della risposta obiettiva è sta-ta effettuata ogni 2 mesi con risonanza megnetica, men-tre la valutazione neurologica e la tossicità sono stateeffettuate dopo ogni ciclo di trattamento.
RisultatiSono stati arruolati nello studio 42 pazienti e di questi
34 sono valutabili per la risposta e per la tossicità emato-logica. L'età mediana era di 11 anni, con un range com-preso tra 2 e 28 anni. Sono stati somministrati un totaledi 196 cicli, con una mediana di 4. 4 cicli (range 2-36). Larisposta obiettiva, determinata valutando centralmente leimmagini di risonanza magenetica, è stata del 47% con 6risposte complete (RC), 7 risposte parziali (RP), 3 risposteminori (RM), 7 malattie stabili (MS) e 11 progressioni(PRO). Fra i pazienti responsivi, la sopravvivenza libera daprogressione è stata del 67% a 6 mesi. Trombocitopeniadi grado III-IV è stata registrata in 12 su 186 cicli valuta-bili. Con una riduzione delle dosi del farmaco la tromoci-topenia si è verificata raramente e sono state comunqueosservate risposte obiettive anche con dosi ridotte del40% rispetto alle dosi raccomandate negli studi di fase IIcondotti nei gliomi ad alto grado.
ConclusioniIl Temozolomide è un farmaco attivo e ben tollerato nei
bambini i giovani adulti affetti da medulloblastoma, conuna tossicità ematologica ridotta.
Bibliografia
1. O'Reilly SM, Newlands ES, Glaser MG, Brampton M, Rice-EdwardsJM, Illingworth RD, Richards PG, Kennard C, Colquhoun IR, LewisP, et al: Temozolomide: a new oral cytotoxic chemotherapeuticagent with promising activity against primary brain tumours. EurJ Cancer 29A(7):940-2, 1993.
2. Clark AS, Deans B, Stevens MS, Tisdale MJ, Wheelhouse Rt, Den-ny BJ, and Hartley JA: Antitumor imidazotetrazine. 32.Synthesis ofnovel imidazotetrazinones and related bicyclic heterocycles toprobe the mode of action of the antitumor drug temozolomide. JMed Chem 38:1493-1504, 1995
3. Stevens MFG, Hickman JA, Langdon SP, Chubb, et al: Antitumoractivity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045:M&B39831), a novel drug with potential as an alternative todacarbazine. Cacer Res 47:5846-5852, 1987
4. Patel M, Mc Cully C, Godwin K, and Balis F: Plasma and cere-brospinal fluid pharmacokinetics of temozolomide. Proc Am SocClin Oncol 14:461, 1995
5. Friedman HS, Dolan ME, Pegg EA, Marcelli S, Keir S, Catino JJ, etal: Activity of temozolomide in the treatment of central nervoussystem tumor xenografts. Cancer Res 55:2853-2857, 1995
6. Yung WK, Prados MD, Yaya-Tur R, Rosenfeld SS, Brada M, Fried-man HS, Albright R, Olson J, Chang SM, O'Neill AM, Friedman AH,Bruner J, Yue N, Dugan M, Zaknoen S, Levin VA: Multicenter phaseII trial of temozolomide in patients with anaplastic astrocytomaor anaplastic oligoastrocytoma at first relapse. Temodal BrainTumor Group. J Clin Oncol 17(9):2762-71,1999
7. Estlin EJ, Lashford L, Ablett S, Price L, Gowing R, Gholkar A, KohlerJ, Lewis IJ, Morland B, Pinkerton CR, Stevens MC, Mott M, StevensR, Newell DR, Walker D, Dicks-Mireaux C, McDowell H, ReidenbergP, Statkevich P, Marco A, Batra V, Dugan M, Pearson AD: Phase Istudy of temozolomide in paediatric patients with advanced can-cer. United Kingdom Children's Cancer Study Group. Br J Cancer78(5):652-61, 1998
8. Nicholson HS, Krailo M, Ames MM, Seibel NL, Reid JM, Liu-MaresW, Vezina LG, Ettinger AG, Reaman GH: Phase I study of temo-zolomide in children and adolescents with recurrent solid tumors:a report from the Children's Cancer Group. J Clin Oncol16(9):3037-43, 1998
92
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre

Studio di fase 2 con Irinotecan inpazienti con sarcomi delle parti molli ricadutio resistenti
Gianni BisognoClinica di Oncoematologia, Dipartimento di Pediatria,Azienda Ospedaliera, Università di Padova
Nonostante il miglioramento dei risultati ottenuti nelleultime decadi nel trattamento dei pazienti affetti da rab-domiosarcoma (RMS) o da altri sarcomi delle parti molli(SPM), circa un terzo dei pazienti presenta una recidiva dimalattia. Nonostante ulteriori trattamenti la maggior partedi questi pazienti non ha possibilità di cura.
E' quindi necessario individuare farmaci con nuovi mec-canismi di azione che siano maggiormente efficaci controquesti tumori, in modo da poter essere utilizzati sia in cor-so della terapia di prima linea che in caso di recidiva.
L'Irinotecan (IRN), un derivato semisintetico della piantaCamptotheca, ha mostrato attività antitumorale in mod-elli preclinici di tumori pediatrici. 1
L'IRN, e in particolare l'SN38, un composto da 200 a1000 volte più attivo in cui viene convertito l'IRN dall'en-zima carbossilesterasi, svolgono un'azione inibente sullatopoisomerasi I, bloccando l'azione di rottura-ricostruzionedel filamento di DN. 2 Infatti, le camptotecine si legano inmaniera specifica al complesso topoisomerasi I-DNA impe-dendo la dissociazione dell'enzima dal DNA e laricostruzione della molecola di DNA integra. Il risultato èuna frammentazione del DNA, morte cellulare in fase S eaberrazioni cromosomiche. 3
Studi di farmacocinetica mostrano che il picco di con-centrazione plasmatica dell'IRN si verifica immediatamentedopo il completamento dell'infusione. Il picco plasmaticodel metabolita attivo SN38 è più tardivo, verificandosi da30 a 90 minuti dal termine dell'infusione. La via principaledi eliminazione sia dell'IRN sia dell'SN38 è quella biliare.
L'IRN si è dimostrato attivo in diversi modelli preclinicidi tumori, sia dell'adulto (neoplasie del colon, stomaco,mammella e polmone), che del bambino (rabdomiosarco-ma e neuroblastoma). 4 L'utilizzo dell'IRN in studi di fase Ie II in adulti ha inizialmente rivelato una tasso di rispostameno soddisfacente rispetto a quanto atteso in base airisultati degli studi preclinici. Questa differenza è stataspiegata dalla schedula-dipendenza dell'IRN, riconducibilealla sua alta specificità per la fase S del ciclo cellulare, cherichiede una prolungata esposizione a concentrazioniadeguate per ottenere la massima attività citotossica.
Differenti schedule di somministrazione sono state adop-erate: un'infusione singola ogni 3 settimane, un'infusionesettimanale per 4 settimane, un'infusione giornaliera per3 giorni ogni 3 settimane o un'infusione giornaliera per 5giorni ogni 3 settimane. 5-7
La tossicità dose-limitante (DLT) è stata la mielodepres-sione quando si è usata l'infusione singola ogni 3 setti-mane, e la diarrea nelle altre schedule. Tuttavia in accor-do con i risultati di studi preclinici, l'IRN l'attività citoto-ssica sembra maggiore in caso di somministrazioni prol-ungate. Infatti, una più recente schedule di somminis-trazione proposta da Houghton e coll. si basa sulla som-
ministrazione di basse dosi giornaliere per un periodo pro-lungato (5 giorni x 2, con 2 giorni di intervallo ogni 3 set-timane) con un aumento dell'attività antitumorale rispet-to a schedule che utilizzano lo stesso dosaggio ma con untempo di somministrazione più ristretto. 4,8 Nel loro studiodi fase I su 23 pazienti pediatrici con tumori solidi refrat-tari, l'IRN è stato somministrato a 3 differenti dosaggi: 20,24 e 29 mg/mq/die × 5 giorni × 2, ogni 3 settimane. Ladiarrea di grado 3/4 con/senza crampi addominali è statala DLT in 6 di 12 pazienti trattati a 24 mg/mq/die pertan-to una dose di 20 mg/mq/die × 5 giorni × 2, ogni 3 setti-mane è stata suggerita nei bambini. 8
Con l'obiettivo di valutare l'attività e la tossicità dell'IRNin pazienti affetti da sarcoma delle parti molli, su iniziati-va dell' Oncologia Pediatrica dell'Università Cattolica diRoma, abbiamo avviato uno studio multiistituzionale difase II.
Materiali e metodiSono stati considerati eleggibili pazienti di età compre-
sa fra 1 e 18 anni affetti da sarcoma delle parti molli inrecidiva o in progressione di malattia, con una buona fun-zione d'organo e con un'aspettativa di vita di almeno 8settimane e che avessero dato un consenso informato allostudio.
L'IRN è stato somministrato alla dose di 20 mg/m2/dieper via e. v. in infusione di 1 ora per 5 giorni + 2 giorni diintervallo + 5 giorni, ogni 4 settimane (considerando i pri-mi 5 giorni come la prima settimana di trattamento).
Il trial è stato condotto utilizzando il metodo di Gehanche prevede 2 fasi consecutive: sono stati stimati neces-sari 14 pazienti per la prima fase e di 25 per la seconda perciascun gruppo, RMS e SPM non RMS.
RisultatiDal Maggio 2002 al giugno 2004 sono stati arruolati 25
pazienti, tutti precedentemente trattati per neoplasiesolide tra cui 11 RMS, e 14 SPM non RMS. In quest'ultimogruppo sono inclusi: 9 pazienti con tumore neuroectoder-mico periferico (PNET), 3 con Tumore desmoplatico a pic-cole cellule rotonde (TDPCR), 1 con tumore rabdoideextrarenale e 1 con sarcoma a cellule chiare.
In quattro pazienti (due RMS e 2 PNET) la risposta altrattamento non è risultata valutabile (perché in tratta-mento o per tumore non misurabile). Un'analisi prelim-inare dei dati ha mostrato che nel gruppo di pazienti affet-ti da RMS si sono evidenziate 2 risposte parziali in 9 pazi-enti valutabili (tasso di risposta 22%). Sono state inoltredocumentate 2 risposte minori. Nel gruppo dei SPM non-RMS sono state dimostrate 2 remissioni complete di malat-tia e 1 risposta parziale nei 7 pazienti valutabili affetti daPNET (tasso di risposta 43%); nessuna risposta nei 3 pazi-enti affetti da TDPCR una progressione di malattia nelpaziente affetto da tumore rabdoide e 1 risposta minore nelpaziente affetto da sarcoma a cellule chiare.
La tossicità è rientrata nell'atteso e si è manifestatacome tossicità gastro-intestinale, con necessità di ricoveroin 2 pazienti, e mielotossicità, ma senza episodi settici.
ConclusioniLa nostra esperienza dimostra la possibilità di valutare
l'attività di nuovi farmaci avvalendosi della collaborazione
93
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

94
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre
dei Centri AIEOP, e conferma l'attività dell'irinotecan con-tro il RMS. Di notevole interesse è l'azione dimostrata con-tro i SPM non RMS e in particolare contro il PNET. E' quin-di è necessario arruolare un numero maggiore di pazien-ti per completare la seconda fase dello studio allo scopodi confermare e sostanziare meglio questi dati preliminari
Bibliografia
1. Houghton PJ, Cheshire PJ, Hallman JD, et al. Efficacy of topoiso-merasi I inhibitors, topotecan and irinotecan, administered at lowdose levels in protracted schedules to mice bearing xenografts ofhuman tumors. Cancer Chemother Pharmacol 1995;36:393.
2. Kawato Y, Aonuma M, Hirota Y, et al. Intracellular roles of SN-38,a metabolite of the camptothecin derivative CPT-11, in the anti-tumor effect of CPT-11.Cancer Res 1991;51:4187.
3. Liu LF. Biochemistry of camptothecin. In Potmesil M, Pinedo H,eds. Camptothecins: New Anticancer Agents. Boca Raton, CRCPress; 1995. p. 11-4.
4. Houghton PJ, Cheshire PJ, Hallman JC. Therapeutic efficacy of thetopoisomerase I inhibitor 7-ethil-10-(4-(1-piperidino)-1-piperidi-no)-carbonyloxy-camptothecin against human tumor xenografts:Lack of cross-resistance in vivo in tumors with acquired resistanceto the topoisomerase I inhibitor 9-dimethylaminomethyl-10-hydroxycamptothecin. Cancer Res 1993;53:2823.
5. Kunimoto T, Nitta AK, Tanaka T, et al. Antitumor activity of &-eth-yl-10-[4-(1-piperidino)-1-piperidino] carboxylated-camptothecin,a novel water soluble derivate of camptothecin, against murinetumors. Cancer Res 1987;47:5944.
6. Potmesil M. Camptothecins: from bench research to hospitalwards. Cancer Res 1994;54:1433.
7. Blaney S, Berg SL, Pratt C, Weitman S, Sullivan J, Luchtman-JonesL, et al. A Phase I Study of Irinotecan in Pediatric Patients: a Pedi-atric Oncology Group Study. Clinical Cancer Res 2001;7:32.
8. Furman WL, Stewart CF, Poquette CA, Pratt C, Santana V, ZamboniWC, et al. Direct translation of a protracted Irinotecan schedulefrom a xenograft model to a Phase I trial in children. J Clin Oncol1999;17:1815.
Le leucemie negli infants: perchè sono cosìdiverse
Giuseppe BassoDipartimento di Pediatria Università di Padova
La leucemia linfoblastica acuta (LLA) è probabilmente lamalattia oncologica in cui i progressi legati ai protocollidi terapia hanno dimostrato i maggiori successi negli ulti-mi 30 anni. Infatti da qualche sporadica guarigione neiprimi anni 70 si è passati attualmente ad una possibilitàdi guarire dell'80%. I grandi successi sono dipesi dall'usodi protocolli chemioterapici più intensivi, all'introduzionedel trapianto di midollo e alle maggiori conoscenzederivate dalla “conoscenza biologica” della malattia.Purtroppo questo successo non vale per tutte le forme diLLA, infatti esistono sottoclassi ora ben identificate di LLA,in cui i progressi terapeutici non sono stati così evidentinegli ultimi anni. Questo è dovuto ad una biologia diver-sa che rende le cellule leucemiche più resistenti alla ter-apia con maggior difficoltà ad ottenere una remissionecompleta ed una possibilità di ripresa di malattia elevata.Tra queste la leucemia dei bambini sotto l'anno di età(“infants”) ha aspetti assolutamente peculiari che hannostimolato la ricerca. Infatti mostra caratteristiche non pre-senti in nessuna altra forma di LLA. 1
La prima caratteristica è quella legata all'età, infatti ladefinizione di infants per tutti i bambini sotto l'annoinclude due sottoinsieme di pazienti diversi; al di sottodei sei mesi e al di sopra dei sei mesi,ed è assolutamentepeculiare che leucemie siano cosi omogenee utilizzandocome denominatore l'età.
I pazienti sotto i 6 mesi mostrano caratteristiche di pre-sentazione clinica e di risposta alla terapia e di biologiaassolutamente simili tanto da essere definiti come unicamalattia. 1-4 Queste caratteristiche tendono a sfumare conl'incremento dell'età, infatti nei bambini con età superio-re ai 6 mesi anche la risposta alla terapia è meno negati-va. 2,3
La prima caratteristica che li accomuna è l'elevatonumero di globuli bianchi alla presentazione, l'epato-splenomegalia e il frequente coinvolgimento del sistemanervoso centrale. Anche l'aspetto morfologico dei blastiappare caratteristico all'analisi microscopica; infatti cisono costantemente 2 popolazioni, una di piccoli linfoci-ti, e una di linfociti più grandi con talora l'aspetto di cel-lule “mielo-monocitoidi”, evidenziando una eterogeneitàmorfologica.
Gli studi di citogenetica e genetica molecolare hannoidentificato che esiste una lesione genetica caratteristica,anche se non esclusiva di questa forma leucemica; il riar-rangiamento della regione 11q23 (gene MLL) presentenell' 80% di questi pazienti e caratteristicamente traslo-cato nei pazienti più piccoli( < 6 mesi) nel cromosoma 4(t(4;11)(q21;q23) MLL/AF4). 1-3 Assolutamente intriganteda un punto di vista dell'etiopatogenesi di queste forme èche questa lesione genetica è identica alla lesione genet-ica (11q23) che caratterizza le leucemie acute secondarie
Le leucemie negli Infants

95
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
al trattamento con chemioterapici come le epipodofiloto-ssine, facendo ipotizzare il primo evento leucemogeniconelle forme infants ad una esposizione prenatale adinibitori della topoisomerasi-II attraverso il contatto confarmaci o sostanze naturali. 1 Che questo sia rilevante hauna indiretta conferma dal fatto che queste forme sonopresenti anche in età più avanzata, ma sono estrema-mente rare ed anche la prognosi è meno severa. 2,3
La presenza di una identica lesione genetica in un sub-set di pazienti così omogeneo per caratteristiche cliniche(età) è assolutamente peculiare nel quadro dell'oncologia.Nonostante il gene MLL possa legarsi ad un alto numerodi partners dando origine a riarrangiamenti diversi, gli stu-di effettuati mediante tecniche di espressione genica han-no evidenziato come il pattern di espressione sia suffi-cientemente omogeneo e assolutamente diverso dalle LLAche non presentano un riarrangiamento di MLL. 5 Unacaratteristica peculiare è la frequente espressione del genedella mieloperossidasi, gene caratteristicamente espressonelle cellule mieloidi,questa espressione “anomala” suf-fraga l'ipotesi che alla origine di queste leucemie sia ilcoinvolgimento di una cellula staminale non ancora com-pletamente “commited” in senso linfoide. 1 Questo è con-fermato da numerosi casi riportati in letteratura in cui siassiste ad uno shift in senso mieloide dopo alcune fasi diterapia, o alla coesistenza di fenotipi linfoidi e mieloidianche talora con un identico riarrangiamento clonale. Nonsolo l'aspetto morfologico e la genetica sono simili, maanche l'espressione di antigeni di superficie è peculiare inquesti pazienti; le LLA infants T sono praticamenteeccezionali4 mentre per oltre il 95% sono di origine B, e perla maggior parte (al si sotto dei 6 mesi) non esprimono ilCD10, antigene comunemente espresso nella maggiorparte delle leucemie B al di sopra dei 6 mesi. Poiché l'e-spressione di questo antigene è stata correlata con il ciclocellulare e la possibilità di andare in apoptosi,6 questaassenza può essere in relazione con la peggior risposta allaterapia in questi pazienti. Altra caratteristica peculiare diquesti pazienti è la frequente espressione di antigenimieloidi. 1,4
Gli studi più recenti hanno confermato queste osser-vazioni, infatti studi di espressione genica mediante l'usodi micro arrays , hanno dimostrato un pattern intermediodelle LLA-MLL+, talvolta sovrapponibile a LLA-B e talvol-ta a LMA , infatti per alcuni geni come ad esempio il CD10,CD22, TDT, l'espressione del LLA-MLL+ è fortemente ridot-ta o assente analogamente a quanto si osserva nelle LMA,mentre sono iperespressi nelle LLA-B MLL-, comporta-mento diverso si può osservare per altri geni come adesempio FLT3, iperespresso nelle leucemie degli infants enelle mieloidi confermando la definizione di mixedleukemia per queste forme. 5
Dalle osservazioni ricavate dagli studi di micro arrays ilnostro gruppo ha studiato l'espressione di alcuni antigeni,caratteristici di popolazioni cellulari B e mieloidi, utiliz-zando un approccio quantitativo; ed ha dimostrato comeanche l'espressione proteica di alcune molecole risultaestremamente diversa tra queste leucemie, tanto è veroche si possono riconoscere dei patterns assolutamentespecifici che permettono di identificare con alta speci-ficità e sensibilità alla diagnosi le LLA-MLL/AF4 dalleMLL/AF4– e dalle ALL-MLL–. 7
La iperespressione di fattori trascrizionali identificatigrazie allo studio di espressione genica o di proteine8 sem-bra essere una strada promettente per esplorare nuoveopportunità terapeutiche in questi pazienti, in cui l'ap-proccio chemioterapico e trapiantologico non ha datorisultati del tutto soddisfacenti, inoltre potrebbe spiegarealcuni quesiti non risolti come ad esempio la diversa prog-nosi di LLA di età diversa ance se genotipicamente e mol-ecolarmente identiche come la LLA MLL/AF4, facendochiarezza su quali meccanismi sono attivati negli infantsche rendono anche la t(4;11) cosi diversa.
Bibliografia
1. Biondi A, Cimino G,Pieters R, Pui CH. Biological and therapeuticsaspects of infant leukemia. Blood 2000; 96: 24-33.
2. Pui CH, Gaynon PS, Boyett JM,Chessells JM et al. Outcome of treat-ment in childhood acute lymphoblastic leukemia withrearrangemets of the 11q23 chromosomal region. Lancet 2002; 59:1909-15.
3. Pui CH,Chessells JM, Camitta B,et al. Clinical etherogeneity inchildhood acute lymphoblastic leukemia with 11q23 rearrange-ments. Leukemia 2003; 17: 700-6.
4. Basso G, Putti MC, Cantù-Rajnoldi A,et al. The immunophenotypein infant acute lymphoblastic leukaemia: correlation with clinicaloutcome. An Italian multicentre study. (AIEOP). British Journal ofHaematology 1992; 81: 184-191.
5. Amstrong AS, Stauton JE, Silverman LB et al. MLL translocationspecify a distinct gene expression profile that distinguishes aunique leukemia. Nature Genetics 2002; 30: 41-7.
6. Cutrona G,Tasso P, Dono M, , et al . CD10 is a marker for cyclingcells with propensity to apoptosis in childhood ALL. British Jour-nal of Cancer 2002; 86 :1776-85.
7. De Zen L, Bicciato S ,te Kronnie G, Basso G. Computational analy-sis of flow-cytometry antigen expression profiles in childhoodacute lymphoblastic leukemia: an MLL/AF4 identification.Leukemia 2003; 17:1557-65.
8. Te Kronnie G,Biccato S, Basso G. Acute Leukemia subclassifica-tion: a marker proteine expression perpspective. Hematology 2004:9: 65-70.

Il gene MLL nelle leucemie acute del bambino
Luca Lo NigroDivisione di Ematologia ed Oncologia Pediatrica, Catania
La banda 11q23 del cromosoma 11 è coinvolta in mec-canismi di traslocazione reciproca nel 5-10% dei bambi-ni ed adulti affetti da leucemia linfoblastica acuta (LLA) oleucemia mieloide acuta (LMA). 1 Quasi tutti i riarrangia-menti coinvolgono il gene MLL (da mixed lineageleukemia) che risiede proprio nella banda 11q23 (1). Letraslocazioni avvengono all'interno di una regione cluster(bcr) di 8. 3 kb, e sono caratterizzate dalla sostituzionedella sequenza codificante di MLL in 3' con la sequenza delgene partner. 2 In un secondo tipo di riarrangiamentomeno frequente, il gene MLL subisce una auto-dupli-cazione parziale (partial tandem duplication) con il risul-tato di produrre una proteina più grande. 3 Le più comu-ni traslocazioni che coinvolgono il gene MLL sono lat(4;11), t(9;11), t(11;19), t(6;11) e la t(10;11), le quali ven-gono riscontrate rispettivamente nel 40%, 27%, 12%, 5%e 5% dei casi definiti MLL positivi. Ad ogni modo, ilnumero totale dei loci che sono coinvolti in traslocazionicon MLL è in continua e costante crescita, raggiungendoquota 50.4 E' facilmente riscontrabile un'associazione trauna particolare traslocazione e uno specifico sottotipo dileucemia. Infatti, la quasi totalità delle t(4;11) è ascrivi-bile alla LLA, la t(9;11) è invece tipica della LMA, così comela t(11;19) (q23;p13. 1), la t(6;11) e la t(10;11). Di controla t(11;19) (q23;p13. 3) è riscontrabile sia nella LLA chenella LMA. 1
Inoltre le leucemie MLL positive mostrano particolaritànel pattern immunofenotipico; infatti nel caso delle LLAè caratteristico l'immunofenotipo pre-preB con negativ-ità del CD10 ed una elevata frequenza di espressione diantigeni mieloidi (CD15 e/o CD65). Mentre nella LMA ilgene MLL risulta esser riarrangiato nel 60% dei casi consottotipo morfologico mielomonocitico (FAB M4) e/omonoblastico (FAB M5). 1
La prognosi nei casi con anomalie della banda 11q23non è buona, e l'età svolge un ruolo importante, mostran-do differenze sostanziali tra il gruppo degli infants (< 12mesi) con una sopravvivenza globale inferiore al 30% edil gruppo di bambini con età compresa tra 2-9 anni(sopravvivenza superiore al 50%). 5
I riarrangiamenti del gene MLL si concentrano princi-palmente nelle leucemie degli infants, con incidenzadell'80% nelle LLA e del 65% nelle LMA. 6 Le stesse per-centuali si riscontrano nelle leucemie secondarie a trat-tamento chemioterapico o therapy-related leukemias, lequali insorgono nel 5-15% dei casi di pazienti con tumoreprimitivo trattati con chemioterapia intensiva e caratter-izzata dalla presenza di farmaci quali epipodofillotossine(VP16) e antracicline (doxorubicina). 7 Questi farmaci sonoinibitori della topoisomerasi tipo II ed agiscono stabiliz-zando le fratture della doppia catena del DNA che ven-gono generate da questo enzima. Il parallelismo tra le dueforme di leucemia (infants vs therapy-related) si fa intri-gante quando si confrontano i tempi di latenza ed addirit-tura le sequenze genomiche coinvolte nei riarrangiamen-ti di MLL. 8 Infatti sia nelle leucemie degli infants che nelle
leucemie secondarie il periodo di latenza è breve: nel pri-mo gruppo i riarrangiamenti del gene MLL possono esserriscontrati già in utero e l'età media d'insorgenza è di cir-ca 6 mesi, mentre nel secondo il periodo di latenza medioè compreso tra 18 e 30 mesi. 7,9 Il breve periodo compre-so tra l'evento iniziale e l'evidenza clinica della malattiainsieme all'elevata concordanza nei gemelli monocorialiinfant con la leucemia indica nella proteina di fusione diMLL il ruolo cardine nel condurre il clone iniziale verso l'in-sorgenza della malattia. In base a tali dati, è stato ipotiz-zato che l'esposizione transplacentare del feto a sostanzenaturali e non che inibiscono la topoisomerasi II possonogiocare un ruolo chiave nella insorgenza della leucemianegli infants. Soprattutto i geni che codificano per alcu-ni enzimi detossificatori, quali NQO1 che converte il tossi-co benzochinone in idrossichinone, sembrano giocare unruolo chiave nella popolazione infants dove recentementesono stati riscontrati specifici polimorfismi che neriducono l'attività. 10
Profili di espressione genica nelle leucemie con riarrangia-menti di MLL
La tecnologia del DNA microarrays è stata recentementeapplicata con lo scopo di definire il profilo di espressionegenica delle leucemie con riarrangiamenti di MLL. 11 Siale LLA che le LMA con MLL riarrangiato presentavano pre-cisi quadri di espressione genica che li distinguono dallealtre forme di LLA e LMA.
In particolare, nella valutazione dei casi con t(4;11) èstata messa in evidenza l'aumentata espressione di onco-geni che intervengono nella regolazione dell'apoptosiquali HOX A9, HOX A10, MYC, o di geni coinvolti nellaresistenza ai farmaci, e la ridotta espressione di geniproapoptotici e di oncosoppressori oltre che di inibitoridella crescita cellulare (FHIT e DAPK1). La trasposizione diquesti risultati in esperimenti con i topi ha permesso distabilire che i geni HOX A9 e HOX A7 sono fondamentaliper la immortalizzazione mieloidi in vitro mediata dallaproteina di fusione MLL-ENL, trasdotta da un vettoreretrovirale nelle cellule di midollo osseo. 12
Struttura della proteina MLL e possibile ruolo nel processodi leucemogenesi
Il gene MLL è omologo del gene Trithorax (TRX) dellaDrosophila: entrambi sono geni di conservazione evoluti-va del gruppo Trx, regolatori positivi del complesso deigeni omeotici (HOX) durante lo sviluppo e la cui attività èin contrasto con l'attività repressiva del gruppo Polycomb(PcG). 13
Il gene MLL codifica per una proteina nucleare e diver-si domini funzionali sono stati identificati; le regioniomologhe con TRX consistono in due domini zinc-fingerscentrali, con in più un dominio SET, una regione C-termi-nale di 210 aminoacidi che condivide il 61% di identitàcon i geni del Polycomb group, i quali codificano per pro-teine coinvolte nello sviluppo embrionale. 13
Il dominio SET viene perso durante il fenomeno dellaformazione delle proteine di fusione e viene rimpiazzatodalla porzione C-terminale della proteina partner. In più,la regione N-terminale di MLL contiene 3 domini A-T hooksimili a quelli del gruppo proteico definito high mobility(HMG), il quale influenzerebbe la struttura cromatinica
96
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre

mediante legami con siti minori del DNA ricchi di residuiA e T. 13 La porzione N-terminale di MLL inoltre contieneuna regione di 47 aminoacidi omologa alla regione noncatalitica della DNA metiltransferasi (MT), un enzima coin-volto nel mantenimento della metilazione del DNA nellecellule somatiche. 13 E' inoltre presente una regioneomologa ad una proteina di 70-kDa delle particelle U1small nuclear ribo-nucleo-protein (SnRNP). 13 Tutte questeindicazioni suggeriscono che MLL agisce mediante diret-ta interazione con il DNA. Studi funzionali di MLL lo han-no indicato come ruolo chiave durante lo sviluppo embri-onale in accordo con le omologie con trx e PcG.
Qual'è il possibile meccanismo molecolare che starebbealla base della leucemogenesi MLL-mediata? Un aspettopotenzialmente rilevante delle proteine di fusione di MLLè l'associazione con i complessi rimodellanti la cromati-na, i quali coinvolgono le proteine SWI-SNF, che agiscononel mantenimento della conformazione della cromatinarendendola pronta per la trascrizione. La proteinaDrosophila Brama, membro del complesso SWI-SNF, inter-agisce con i geni omeotici così come MLL omologo di trx,riducendo l'effetto repressivo sulla cromatina (13). MLLpotrebbe interagire con un complesso SWI-SNF umano inuna maniera simile; infatti il dominio SET di MLL sembr-erebbe legarsi a INI1, il gene omologo umano di SNF5 dilievito. 13 La proteina di lievito ANC1 che presenta regioniomologhe con AF9 e ENL è stata vista interagire con SNF5.13 Sebbene il dominio SET di MLL viene perso durante laformazione delle proteine di fusione, esso viene rimpiaz-zato dalla regione C-terminale di AF9, omologa a ANC1,nel generare MLL-AF9, conservando molto probabilmentela capacità di interagire con il complesso SWI-SNF. InoltreAF10 e AF17 sono associati a BR140, il quale fa parte del-la famiglia bromo-domain che include i membri SWI-SNFSWI2, brm e BRG-1.13 Inoltre, alcuni geni partner di MLL,quali CBP e p300 possiedono la proprietà di acetilazionedegli istoni,13 rendendo la cromatina più accessibile adaltri fattori di regolazione.
Pertanto, emerge un modello di azione legato alle pro-teine di fusione di MLL che nella leucemia agirebbero atti-vando o reprimendo alcuni geni intervenendo sul rimodel-lamento della cromatina.
Modelli murini per lo studio della leucemogenesi da MLLDue metodiche sono state utilizzate per generare mod-
elli murini con MLL riarrangiato per la valutazione dell'ef-fetto leucemogeno. L'inserimento (knock in) del gene part-ner AF9 nel locus di MLL mediante una ricombinazioneomologa genera animali che sviluppano la LMA in circa 6mesi. 14 Un altro metodo è rappresentato dalla transfezioneretro-virale in colture di cellule ematopoietiche progeni-trici, le quali vengono poi trapiantate nei topi: mediantequesta tecnica è stato dimostrato che i geni di fusione diMLL agiscono come alleli dominanti e la malattia sembraesser dovuta ad un guadagno di funzione (gain-of-func-tion) di MLL. Gli studi di struttura-funzione di numeroseproteine di fusione di MLL hanno dimostrato che partico-lari elementi all'interno delle proteine partner, quali domi-ni di trascrizione e motivi di interazione proteine-proteine,giocano un ruolo critico per l'attività leucemogenica. 15 Inparticolare la presenza di domini di dimerizzazione inqualche partner (AF6, AF10, AF17, AF1p, GAS7 e AFp21)
sembrano indicare nella omo-dimerizzazione uno dei mec-canismi che stanno alla base del processo di leucemogen-esi di MLL. Di contro, i più comuni geni di fusione (AF4, AF9e ENL) non possiedono domini di dimerizzazione. Alcuniricercatori hanno visto che proteine di fusione transdottein un midollo osseo in metil-cellulosa conferiscono unacapacità di crescita a lungo termine ai progenitori multi-potenti. 16 Pertanto è stato proposto che le proteine difusione di MLL potenziano la capacità di auto-rinnova-mento dei progenitori multipotenti i quali mantengonoun'abilità a differenziarsi verso gli stadi successivi. 16
ConclusioniIn conclusione si può affermare che il gene MLL sta alla
base del processo leucemogenico nei casi MLL positivi.Sebbene vi sia una elevata eterogeneità nel numero digeni partner coinvolti nelle traslocazioni, diventa man-datario continuare ad identificare i nuovi partner alloscopo di incrementare le conoscenze sui meccanismid'azione. Un grosso passo avanti è stato fatto grazie aglistudi di espressione genica e molto ci si aspetta dalla pro-teomica, alla quale è affidato il compito di identificare leproteine, insieme a quella di fusione chimerica, che inter-vengono nel processo di leucemogenesi. Quest'ultimosembrerebbe indubitabilmente esser legato alla confor-mazione della cromatina, la quale permette o meno latrascrizione di alcuni geni che regolano fenomeni chiavequali l'apoptosi.
Bibliografia
1. Canaani E, Nakamura T, Rozovskaia T, Smith ST, Mori T, Croce CMand Mazo A. ALL-1/MLL1, a homologue of Drosophila TRITHORAX,modifies chromatin and is directly involved in infant acuteleukaemia. Br J Canc 2004;90:756-60.
2. Gu Y, Nakamura T, Alder H, Prasad R, Canaani O et al. The t(4;11)chromosome translocation of human acute leukemia fuses theALL-1 gene, related to a Drosophila trithorax, to the AF4 gene.Cell 1992;71:701-8.
3. Schichman SA, Canaani E, Croce CM. Self fusion of the ALL-1 gene.A new genetic mechanism of acute leukemia. JAMA 1995;273:571-6.
4. Huret JL, Dessen P, Bernheim A. An atlas of chromosomes in hema-tological malignancies. Example: 11q23 and MLL partners.Leukemia 2001;15:987-9.
5. Pui CH, Gaynon PS, Boyett JM, Chessells JM, Baruchel A et al. Out-come of treatment in childhood acute lymphoblastic leukemiawith rearrangements of the 11q23 chromosomal region. Lancet,2002;359:1909-15.
6. Biondi A, Cimino G, Pieters R, Pui CH. Biological and therapeuticaspects of infant leukemia. Blood 2000;96:24-33.
7. Pui CH, Relling MV. Topoisomerase II inhibitor-related acutemyeloid leukemia. Br J Haematol 2000;109:13-23.
8. Lovett B D, Lo Nigro L, Rappaport E F, Blair IA, Osheroff N et al.Near-precise interchromosomal recombination and functionalDNA topoisomerase II cleavage sites at MLL and AF-4 genomicbreakpoints in treatment-related acute lymphoblastic leukemiawith t(4;11) translocation. Proc Natl Acad Sci USA 2001;98:9802-7.
9. Greaves M. Molecular genetics, natural history and the demise ofchildhood leukaemia. Eur J Canc 1999;35:1941-53.
10. Smith MT, Wang Y, Skibola CF, Slater DJ, Lo Nigro L. LowNAD(P)H:quinone oxidoreductase activity is associated withincreased risk of leukemia with MLL translocations in infants andchildren. Blood 2002;100:4590-3.
11. Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML,et al. MLL translocations specify a distinct gene expression profilethat distinguishes a unique leukemia. Nat Gen 2002;30:41-7.
97
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

98
haematologica vol. 89[supplement n. 10]:October 2004
Martedi 12 ottobre
12. Ayton PM, Cleary ML. Transformation of myeloid progenitors MLLoncoproteins is dependent of Hoxa7 and Hoxa9. Genes Dev 2003;17:2298-307.
13. Collins EC, Rabbitts TH. The promiscuous MLL gene links chromo-somal translocations to cellular differentiation and tumour tro-pism. Trend Mol Med 2002;8:436-42.
14. Dobson CL, Warren AJ, Pannel R, Forster A, Lavenir I, Corral J, etal. The MLL-AF9 gene fusion in mice controls myeloproliferationand specifies acute myeloid leukemogenesis. EMBO J1999;18:3564-74.
15. Ayton PM, Cleary ML. Molecular mechanisms of leukemogenesismediated by MLL fusion proteins. Oncogene 2001;20:5695-707.
16. So CW, Lin M, Ayton PM, Chen EH, Cleary ML. Dimerization con-tributes to oncogenic activation of MLL chimeras in acuteleukemias. Cancer Cell 2003;4:99-110.
Risultati e prospettive nel trattamento delleleucemie negli infants
Giulio De RossiOspedale Pediatrico Bambino Gesù, Roma
Le leucemie acute al di sotto dell'anno di età sono sicu-ramente estremamente rare con una incidenza inferiore a0.5/100.000 nati/anno, costituendo ~ il 3% delle leucemieacute in età pediatrica. Non esistono dati certi sulla inci-denza delle forme mieloidi e linfoidi, anche se queste ultimehanno una frequenza maggiore.
Un discorso completamente a parte spetta alle formetransitorie spesso connatali o delle prime settimane di vita,correlate, nella gran maggioranza dei casi, a sindrome diDown e ad ontogenesi mieloide, (magacarioblastica,promielocitica monoblastica).
Le peculiari caratteristiche epidemiologiche e biologiche,e la problematica risposta clinica hanno creato nelleLeucemie Infant un vasto interesse che ha spinto numerosigruppi nazionali ed internazionali a collaborare nell'otticadi chiarire la leucemogenesi e migliorarne la prognosi.
Nelle forme mieloidi (LANL) studi nazionali europei, giap-ponesi e nord americani riportano una EFS a 3-5 anni trail 35 e il 73%, utilizzando e non il trapianto di cellule sta-minali (SCT). Tali dati si riferiscono però in genere a casis-tiche retrospettive, talora dei primi anni '90, relativamentedi pochi pazienti, generalmente inclusi in protocolli nazion-ali per le LANL pediatriche. I dati (1986-1994) retrospettivi(non pubblicati) AIEOP di 39 pazienti con EFS ~ 16% a 5anni, sono poi stati raccolti senza che fosse in corso unprotocollo LANL nazionale, e quindi sono relativi a bambi-ni trattati a discrezione dei singoli Centri.
Escludendo i dati AIEOP, le prospettive di guarigione inambito Infant LANL, non sembrano essere molto lontane daquelle dei bambini tra 2 e 15 anni e quindi è necessario unosforzo cooperativo internazionale per stabilire parametribiologici progonostici ed uniformare la terapia delle LANLInfant.
Le Infant Leucemie Acute Linfoidi (LAL) sono invece unsubset ben distinto, isolato biologicamente e clinicamentedalle comuni LLA pediatriche.
La EFS a 5 yrs delle LLA Infant negli studi retrospettivi èintorno al 30%: ben al di sotto delle LLA tra 1 e 15 anni.L'infant LLA all'esordio si presenta con elevata massa neo-plastica, iperleucocitosi, elevata incidenza di localizzazioniextraemopoietiche, fenotipo immunologico mixed (linfoide+ mieloide). Più del 70% dei casi presentano una ben notaalterazione genetica nel cromosoma 11 alla banda q23, checoinvolge il gene ALL1/MLL. Tale dato ha peraltro notevoliimplicazioni epidemiologiche e nello studio dellaleuchemogenesi. Infatti è stato possibile dimostrare chenell'Infant LLA l'evento leucemogeno si è verificato un peri-odo ristretto di tempo (nove mesi della gravidanza): gli stu-di epidemiologici mirati a questo periodo stanno con-tribuendo appunto a migliorare le conoscenze sugli agen-ti leucemogeni. Studi in vitro hanno poi dimostrato che lascarsa risposta alla chemioterapia correla talora con lachemioresistenza in vitro.
Peraltro escludendo il 30% dei pazienti che non presen-tano alterazioni molecolari (MLL/ALL1 neg. ) si evidenzia

99
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
una popolazione di Infant ALL (MLL/ALL1 pos. ) a prognosidrammatica, con una EFS tendenzialmente inferiore al15%.
Perciò nel 1998 un gruppo Europeo, poi divenuto Inter-nazionale, ha varato un Protocollo Terapeutico Interfant99 con l'obiettivo di migliorare la prognosi e concentrareun cospicuo numero di ricercatori sulle peculiarità bio-logiche della Infant LLA.
Il Protocollo Interfant 99 è nato appunto nell'ottica dimigliorare l'EFS dei pazienti con Infant LLA e di stratificar-li secondo fasce di rischio. In un primo momento ha coin-volto centri Europei, successivamente si è esteso, ed attual-mente coinvolge circa 20 Gruppi Cooperativi in tutto ilmondo.
Il più recente Study Report è stato effettuato a maggio2004, in occasione del BFM Meeting 2004, da Rob Pietersche è il Coordinatore Centrale dell'Interfant 99.
Il protocollo ha utilizzato un alternarsi di farmacimieloide-linfoide specifici ed in particolare la Citosina-Ara-binoside, viste le premesse biologiche ed i dati in vitro,disponibili dalla letteratura.
Peraltro la randomizzazione ha seguito i criteri BFM eAIEOP della risposta al Prednisone (Prednisone GoodResponders - PGR; Prednisone Poor Responders - PPR).
Lo schema terapeutico ALL-like prevede Induzione, Con-solidamento, Profilassi CNS, Reinduzione, Intensificazionee Mantenimento; data l'età dei pazienti non viene utilizzatala Radioterapia craniale.
Tutti i pazienti (sia PPR che PGR) sono randomizzati aricevere o no prima del mantenimento un Ciclo simile alblocco di chemioterapia utilizzato nel Consolidamento, macon la aggiunta della Vincristina. Solo i pazienti PPRricevono VP16 durante la terapia di mantenimento. E' pre-vista una riduzione di dose-farmaci secondo l'età (<6m:2/3; 6-12m: 3/4 dose; >12m full dose). Le indicazioni altrapianto di cellule staminali sono solo per il gruppo HR--PPR.
L'analisi ad interim dei risultati, su di un totale di 321pazienti al Maggio 2004, mostra una overall EFS a 3 yrs del47. 7%, del 54. 5% per il rischio standard, e del 34. 6% perl'alto rischio.
Considerando i dati retrospettivi della letteratura intornoal 30-40% e considerando che attualmente per l'Interfant99 si tratta di una EFS a 3 yrs, i risultati sono soddisfacen-ti ma non entusiasmanti. Certo è che l'importanza di avercoinvolto più Centri in tutto il mondo (per la prima volta incampo ematologico-terapeutico) è quella di aver mobilita-to un ampio gruppo di ricercatori che coopereranno perportare un risultato terapeutico ed anche un contributobiologico nella comprensione di questa peculiare forma dileucemia e della leucemogenesi in genere. Appunto anal-izzando i risultati ad interim del Protocollo Interfant 99emergono quattro variabili significative in termini di prog-nosi, sia nella analisi statistica univariata che in quella mul-tivariata: l'età (<6m), l'MLL/ALL1 riarrangiamento, i WBC (e300.000), e la risposta al Prednisone. In particolare i pazi-enti MLL riarr. con età <6m hanno una EFS a tre anni del< 27% e se con WBC e 300.000 hanno una EFS di < 16%.
Il problema che si pone per il futuro, è quello di impostareun nuovo protocollo terapeutico che tenga conto di questidati e consideri che indubbiamente vi è una popolazione dibambini non indifferente che non ha significative possibil-
ità di guarigione con le convenzionali strategie chemioter-apiche. Quand'anche si intensifichi la terapia e non si attuila dose reduction age-related dell'Interfant 99, è improb-abile che si ottengano significativi risultati e non si paghiuna elevata tossicità farmacologica. In questo gruppo dipazienti terapie sperimentali e/o il trapianto di cellule sta-minali emopoietiche possono giocare un ruolo determi-nante.
I pazienti MLL/ALL1 non riarrangiati raggiungono EFSvicine alle classiche EFS delle LLA pediatriche, ed aggius-tamenti chemioterapici potranno migliorare ulteriormentela loro risposta terapeutica e le loro possibilità di guari-gione.
In conclusione l'Infant LLA è una patologia che ha anco-ra molte problematiche, è il fanalino di coda delle LLA pedi-atriche in quanto a successi terapeutici, ma è anche unapatologia leucemica piena di importanti risvolti biologiciche potranno avere ripercussioni significative nello studiodella leucemogenesi e della terapia delle leucemie linfoidiresistenti.
Bibliografia
1. Lampkin BC. The newborn infant with leukemia. J Pediatrics1197;131:176. 2..
2. Biondi A, Cimino G, Pieters R, Pui CH. Blood 2000;96:24. 3. Marco F, Bureo E, Ortega JJ, et al. High Survival Rate in Infant
Acute Leukemia treated with early high-dose chemotherapy andStem-Cell Support. J Clin Oncol 2000;18:3256.
4. Kawasaki H, Isoyama K, Eguchi M, et al. Superior outcome of InfantAcute Myeloid Leukemia with intensive chemotherapy: results ofJapan Infant Leukemia Study Group. Blood 2001;98:3589.
5. Creutzig V, Ritter J, Zimmermann M, et al. Improved TreatmentResults in High-Risk Pediatric Acute, Myeloid Leukemia Patientsafter intensification with high-dose Cytarabine and Mitoxantrone:Results of Study Acute Myeloid Leukemia - Berlin - Frankfurt -Munster 93. J Clin Oncol 2001;19:2705.
6. Stevens RF, Hann IM, Wheatley K, et al. Marked improvements inoutcome with chemotherapy alone in paediatric acute myeloidleukaemia: results of the United Kingdom Medical Research Coun-cil's 10th AML trial. Br J Haematol 1998;101:130.
7. Pui BH, Evans WE. Acute Lymphoblastic Leukemia in Infants. Br JClin Oncol 1999;17:438.
8. Wiemels JL, Cazzaniga G, Daniotti M, et al. Prenatal origin of AcuteLymphoblastic Leukemia in children. Lancet 1999;1:1499.
9. Luciani M, Cimino G, Angioni A, et al. A retrospective evaluationof infant patients with acute lymphoblstic leukemia treated atsingle institution. Haematologica 1999;84:464.
10. Reaman GH, Sposto R, Sensel MG, et al. Treatment outcome andPrognostic Factors for Infants with Acute Lymphoblastic Leukemiatreated on two consecutive trial of the Childrens Cancer Group. JClin Oncol 1999;17:445.
11. Pieters R, den Boer ML, Durian M, et al. Relation between age,immunophenotype and in vitro resistance in 395 children withacute lymphoblastic leukemia-implication for treatment of infants.Leukemia 1998;12:1344.

101
haematologica vol. 89[supplement n. 10]:October 2004
EMATOLOGIA E COAGULAZIONE
C001ASSENZA DI ELASTASI GRANULOCITARIA IN PAZIENTI CON NEUTROPENIASEVERA CONGENITABadolato R, Notarangelo L, Fontana S, Donini M, Dusi S,Fausti R, Caldiani C, Savoldi G, Vermi W, Ferrari D,Facchetti F, D’ippolito C, Notarangelo LDClinica Pediatrica, Istituto di Medicina Molecolare“Angelo Nocivelli”, e Cattedra di Anatomia Patologicadell’Università di Brescia; Istituto di Patologia Generale,Università di Verona
La neutropenia severa congenita è una malattia auto-somica dominante causata in circa 80% dei casi da muta-zioni in eterozigosi del gene ELA2 codificante per elasta-si granulocitaria. I pazienti affetti presentano infezionibatteriche ricorrenti e neutropenia persistente con unnumero di granulociti neutrofili < a 500 cell/mcL. L’ana-lisi del midollo osseo dimostra generalmente un arrestomaturativi allo stadio promielocita-metamielocita. Seb-bene la causa genetica sia stata chiarita, il meccanismofisiopatologico della neutropenia rimane largamente nondefinito. In questo studio, abbiamo valutato l’espressionedi elastasi granulocitaria e la sua localizzazione cellularein quattro pazienti affetti da neutropenia cronica severaportatori di mutazioni del gene ELA2.Tutti i pazienti han-no presentato un esordio di malattia entro tre mesi di vitacon infezioni cutanee e/o respiratorie caratterizzate dafebbre e da una rapida evoluzione di tipo settico. Due diquesti pazienti presentavano la stessa mutazione di tipomissense (sostituzione aminoacidica a carico del residuo185, G185R) che era stata già associata con un decorsosevero della malattia ed una risposta clinica insoddisfa-cente al trattamento con G-CSF. Al contrario, gli altri duepazienti presentavano una risposta terapeutica normaledopo il trattamento con G-CSF. Lo studio di espressione dielastasi granulocitaria tramite immunofluorescenza deigranulociti isolati dal sangue dei pazienti con mutazioneG185R ha evidenziato completa assenza della proteina inpiù del 90% delle cellule isolate dal sangue periferico tra-mite centrifugazione su gradiente di densità. Inoltre, l’a-nalisi quantitativa delle concentrazioni plasmatiche di ela-stasi dimostrava una riduzione significativa dei livelli cir-colanti dell’enzima nei soggetti con mutazione di ELA2.Alfine di valutare lo stadio differenziativo dei granulociticircolanti abbiamo analizzato l’espressione di mieloperos-sidasi e lattoferrina tramite immunofluorescenza e/owestern blot. Attraverso queste due tecniche abbiamoosservato la mancata espressione nei pazienti affetti da
neutropenia severa congenita. Nel complesso, i nostri datisuggeriscono che le mutazioni di elastasi comportano unamancata formazione dei granuli primari e quindi determi-nano il blocco del programma differenziativo dei granu-lociti neutrofili dei pazienti con neutropenia severa con-genita.
C002LA POLICITEMIA DI TIPO CHUVASH IN ITALIA: ANALISI MOLECOLARE DELGENE DI VON HIPPEL-LINDAUPerrotta S, Ferraro M, Migliaccio C, Ladogana S,*Cirillo P, Punzo F, Maresca P, Nobili B, Della Ragione F°Dipartimento di Pediatria e °Dipartimento di Biochimicae Biofisica, Seconda Università di Napoli, Napoli *Oncoe-matologia pediatrica, S. Giovanni Rotondo, Foggia
Il termine policitemia indica un aumento nel numero dieritrociti circolanti. Esistono diversi tipi di policitemie,distinguibili in primarie e secondarie. Quelle primarieincludono condizioni acquisite o ereditarie che determi-nano un’eccessiva risposta dei progenitori eritroidi allecitochine circolanti. Le policitemie secondarie, invece, sonocausate da fattori ormonali (prevalentemente l’eritro-poietina, EPO) e l’aumento nella massa degli eritrociti èconseguenza dell’ipossia tissutale o della produzione ano-mala di EPO. Una forma particolare di policitemia secon-daria è rappresentata da quella definita di Chuvash, malat-tia autosomica recessiva osservata con una alta inciden-za in una particolare popolazione russa (Chuvash). Essapresenta caratteristiche comuni sia alle forme primarieche secondarie: un elevato livello di emoglobina (di soli-to più di 200g/L), livelli plasmatici di eritropoietina normalio aumentati, e ipersensibilità delle cellule eritroidi all’EPO.Clinicamente si presenta con emangiomi vertebrali, venevaricose, bassa pressione sanguigna, elevata concentra-zione serica di VEGF (vascular endothelial growth factor)e mortalità prematura correlata ad eventi vascolari cere-brali e trombosi periferiche. La mancata regolazione del-l’omeostasi dell’ossigeno appare responsabile di questaforma di policitemia. Il complesso HIF1 (hypoxia-induci-ble factor 1), costituito da due subunità alfa e beta, è ilprincipale fattore trascrizionale coinvolto nella rispostadelle cellule allo stato ipossico. In particolare, il livello del-la subunità α viene finemente controllato, in maniera ossi-geno-dipendente, attraverso la modulazione della velo-cità di degradazione, mentre l’espressione della subunitàbeta è costitutiva. Infatti, la proteina HIF1alfa appare sta-bile in condizioni di ipossia, al contrario, essa è soggettaad ubiquitinazione (attivata dalla proteina VHL o di vonHippel-Lindau) e successiva degradazione in condizioni di
ABSTRACTSselezionati per la presentazione come:
Comunicazioni (C), Poster (P)

102
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Ematologia e Coagulazione
normossia. HIF1α regola l’espressione di una serie di genitra cui EPO (eritropoietina) e VEGF (vascular endotelialgrowth factor) Pertanto, l’inattivazione funzionale delgene VHL ed in particolare la mutazione Arg200Trp iden-tificata nella policitemie di Chuvash, impedisce la degra-dazione della proteina HIF1α, aumentando l’espressionedei geni target (EPO e VEFG, in particolare) e causando diconseguenza un’eccessiva eritropoiesi, Abbiamo effettua-to una analisi molecolare del gene VHL in alcune famigliecon policitemia tipo- Chuvash del sud Italia, per verifica-re la presenza di mutazioni in tale gene. In una famigliacon due casi (madre e figlia) affetti dalla policitemia tipo-Chuvash abbiamo dimostrato la presenza della mutazio-ne Arg200Trp in omozigosi. In altre due famiglie analiz-zate, i soggetti malati (due casi in una prima famiglia edun singolo soggetto affetto nella seconda) sono risultatieterozigoti composti per la mutazione Arg200Trp prece-dentemente descritta e per le nuove mutazioni Leu89Phe(famiglia uno) e Pro146Arg (famiglia due), rispettivamen-te localizzate nell’esone uno e due del gene VHL. Succes-sivamente, abbiamo testato la frequenza delle tre muta-zioni in una popolazione di controlli sani. La mutazioneArg200Trp elimina un sito di taglio per l’enzima di restri-zione Fnu4HI permettendo in questo modo lo screeningper digestione enzimatica. Per le altre due mutazioni loscreening è stato effettuato mediante sequenziamentodiretto delle regioni esoniche. Le mutazioni non sono pre-senti nella popolazione di controllo. Sono in corso un sag-gio di attività di HIF1alfa su linee cellulari umane deriva-te da linfociti B degli individui affetti e l’analisi di espres-sione di alcuni dei geni target di HIF1 tra cui VEGF ed EPO.In conclusione, lo studio riportato rappresenta la primadescrizione di casi di policitemia di tipo Chuvash in Italia,Nel contempo, i risultati ottenuti descrivono nuove muta-zioni del gene VHL e forniscono nuove indicazioni sullaregolazione dell’eritropoiesi e sulla fisiologia dell’omeo-stasi dell’ossigeno.
P001ANEMIA DI BLACKFAN-DIAMOND (DBA) : STUDIO DELL’ESPRESSIONEGENICAQuarello P, Campagnoli MF, Garelli E, Carando A,Crescenzio N, Doria A, Martino S, Renga D, Ramenghi U,Dianzani I*Divisione di Ematologia, Dipartimento di ScienzePediatriche, Università di Torino, *Dipartimento diScienze Mediche, Università del Piemonte Orientale
Introduzione e obiettivi. La DBA è la più frequente aplasiacongenita pura della serie rossa. La maggior parte dei casiè sporadica, nel 25% è presente mutazione in eterozigosidel gene codificante per la proteina ribosomiale (RP)S19.L’eziopatogenesi della DBA presenta ancora questioni irri-solte: le funzioni di RPS19 e il suo ruolo nell’eritropoiesinon sono chiari. Inoltre è necessario ricercare altri genicausali. Scopo del lavoro è stato analizzare l’espressionedi geni coinvolti nella proliferazione in colture cellulari dipazienti DBA, mutati e non in RPS19, per valutare se esi-stesse un’alterata attività proliferativa nella DBA. Metodi.L’analisi dell’espressione genica sui fibroblasti di 5 pazien-
ti è stata effettuata mediante la tecnica dei macroarrays,utilizzando il Panorama Human Cytokine Gene Array, con-tenente 847 geni coinvolti nella proliferazione cellulare.L’intensità dei singoli segnali ottenuti, corrispondenti aigeni esaminati, convertita in valore numerico medianteanalisi densitometrica, è stata confrontata col controllosano. Risultati. Alcuni geni sono risultati costantementeiperespressi, altri ipoespressi. MMP14 (metalloproteinasi14) e TIMP2 (inibitore tissutale delle metalloproteinasi 2),iperespressi nei pazienti DBA, sono coespressi e coregola-ti durante lo sviluppo muscoloscheletrico, cardiovascola-re e urogenitale. MMP14 ha un ruolo fondamentale nelladismissione in circolo delle cellule CD34+; TIMP2 stimolala crescita dei progenitori della serie rossa, MMP14 è ipe-respressa durante la differenziazione eritroide. IGFBP3(proteina di trasporto delle somatomedine), ipoespressanei pazienti con DBA, modula la crescita cellulare siamediante un meccanismo IGF-dipendente sia in mododiretto. E’ ipoespressa anche nella Cartilage-Hair Hypo-plasia, condrodisplasia metafisaria autosomica recessiva.E’ possibile che il difetto eritropoietico di questa sindro-me sia dovuto ad un meccanismo simile a quello dellaDBA. Conclusioni. I dati dimostrano che il difetto di RPS19presenta manifestazioni anche a livello di cellule non emo-poietiche. Abbiamo osservato un’alterata espressione geni-ca in colture di fibroblasti cutanei e stromali di 5 pazien-ti DBA; è in corso l’analisi per confermare che all’incre-mento o riduzione dei trascritti, corrisponda una effettivavariazione della quota proteica.
P002CARATTERIZZAZIONE MOLECOLARE DI PAZIENTI AFFETTI DAAGAMMAGLOBULINEMIA AUTOSOMICA RECESSIVAFerrari S,1 Sourkova V,1 Soresina A,2, Lougaris V,2Meini A,2 Bottelli C,2 Cattaneo G,2 Naselli A,3Notarangelo LD,2 Plebani A2
1Laboratorio di Genetica Medica, Policlinico S. Orsola-Malpighi, Bologna; 2Clinica Pediatrica, Università degliStudi di Brescia 3Clinica Pediatrica, Istituto GianninaGaslini, Genova
Introduzione e Obiettivi. Difetti nei primi stadi di svilup-po dei linfociti B danno origine ad una condizione patolo-gica nota come agammaglobulinemia, caratterizzata da unprofondo deficit nella produzione di anticorpi e una graveriduzione nel numero dei linfociti B maturi periferici. Laforma legata all'X (XLA) rappresenta il tipo più comune diagammaglobulinemia ed è causata da mutazioni nel geneBTK. Mutazioni nei geni che codificano per la catenapesante mu (IGHM), la catena leggera surrogata Lambda 5(IGLL1) e le proteine MB1 (CD79A), BLNK, LRRC8, causanouna patologia pressochè identica a XLA detta Agamma-globulinemia Autosomica Recessiva (Ag-AR). Mutazioni nelgene IGHM rappresentano la causa più frequente di Ag-AR.Lo scopo del nostro lavoro è quello di identificare le muta-zioni causative in pazienti affetti da Ag-AR, tramite l'ap-proccio classico cosidetto del gene candidato. L'obiettivo alungo termine è quello di identificare nuovi geni coinvoltiin questa patologia. Metodi. Dieci pazienti affetti da Ag-AR e seguiti presso la Clinica Pediatrica dell'Università di

103
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
Brescia sono stati analizzati dapprima per la presenza dimutazioni nei geni IGHM, IGLL1, CD79A. Lo screening èstato poi esteso ai geni CD79B e VpreB. Per alcuni di que-sti pazienti è stato analizzato il fenotipo immunologico deiprecursori midollari dei linfociti B. Risultati e Conclusioni.L'analisi dei geni candidati ha permesso di identificare ine-quivocabilmente la mutazione responsabile della patologiain due pazienti di sesso femminile. In entrambe i casi sitratta di mutazioni nel gene IGHM, una delle quali maidescritta prima. Inoltre abbiamo osservato in un pazientela presenza contemporanea di numerosi polimorfismi acarico del gene IGHM, che potrebbero avere un ruolo pato-genetico. Non abbiamo identificato nessuna mutazione deigeni IGLL1, CD79A, CD79B e VpreB. L'analisi del fenotipoimmunologico dei precursori midollari dei linfociti B dialcuni pazienti nei quali non abbiamo trovato alcunamutazione dei geni analizzati, ha mostrato un quadro feno-tipico peculiare e differente da quelli finora riportati. Que-sto suggerisce che il blocco differenziativo possa esseredovuto a mutazioni presenti in geni differenti rispetto aquelli finora analizzati.
P003NEUTROPENIA E MIELOCATESSI COME SINTOMI DI PRESENTAZIONE DELLASINDROME DI ZUELZER-KRILLNotarangelo L, Tassone L, Bonomi V, Moratto D,Tettoni K, Domenghini S, Gualeni C, Imberti L, Bajia M,Sciotto A, Sensi A, Plebani A, Notarangelo LD,Badolato RClinica Pediatrica dell’Università di Brescia, Istituto diMedicina Molecolare "Angelo Nocivelli"; 3° LaboratorioSpedali Civili, Brescia; Clinica Pediatrica dell'Universitàdi Catania; Cattedra di Genetica Medica dell'Universitàdi Ferrara
La sindrome di Zuelzer-Krill è una malattia genetica atrasmissione autosomica dominante che si caratterizzaper l’associazione di leucopenia a mielocatessi, ipogam-maglobulinemia e comparsa diverruche, ma anche notacon l’acronimo WHIM (Warts, Hypogammaglobulinemia,Infections, Myelokathexis). Recentemente, la causa gene-tica è stata identificata nella presenza di mutazioni ete-rozigoti del gene codificante per il recettore per chemo-chine CXCR4. Sulla base di questi elementi abbiamo ricer-cato mutazioni del gene CXCR4 in pazienti con neutrope-nia e/o ipogammaglobulinemia per i quali erano stateescluse altre cause di immunodeficienza primitiva. L’ana-lisi genetica ha consentito di identificare 7 pazienti di etàcompresa tra i 4 e i 36 anni che avevano presentato sin-tomi riferibili ad immunodeficit quali infezione respirato-ria, o gastroenterite in un’epoca compresa tra i 4 mesi e i9 anni di vita. La presenza di verruche, che viene general-mente considerata un elemento caratterizzante dellamalattia, veniva riscontrata nei 4 pazienti di età superio-re a 15 anni, mentre non era riscontrabile nei 3 pazientipiù giovani, suggerendo che l’assenza di verruche non èsufficiente per escludere la malattia. In tutti i pazienti lemutazioni erano di tipo nonsense o delezione di singolonucleotide ed avvenivano a carico della regione codifi-cante per il dominio carbossi-terminale determinando laperdita del frammento intracellulare della proteina. Tutti
i pazienti studiati presentavano neutropenia ( granuloci-ti: 213±127) e leucopenia (1361±302) nel corso dell’epi-sodio infettivo di esordio, con l’eccezione di uno di essi cheaveva un numero normale di granulociti. Inoltre, il tratta-mento con G-CSF effettuato in 4 pazienti determinava uneffettivo aumento a valori normali in 3 di questi (dopo G-CSF: 2966±1003). La presenza di ipogammaglobulinemiaa carico di tutte le classi di Ig veniva riscontrata in 6 deisette pazienti, ma, in due di questi la vaccinazione con tos-soide tetanico era sufficiente per indurre la produzione diimmunoglobuline specifiche. L’analisi del compartimentoB evidenziava la presenza di una percentuale ridotta dilinfociti B rispetto ai soggetti sani di pari età con una ridu-zione più marcata a carico dei linfociti B di memoria. Alfine di valutare l’effetto delle mutazioni sull’espressionedella proteina, abbiamo analizzato l’espressione di CXCR4su linfociti e granulociti neutrofili. L’analisi citofluorime-trica dimostrava che le mutazioni in eterozigosi non alte-rano l’espressione di membrana del recettore per chemo-chine. Tuttavia, lo studio della risposta chemiotatticadimostra che la perdita della porzione intracellulare delrecettore CXCR4 nei pazienti comporta una aumento del-la risposta chemiotattica dei linfociti T e dei granulocitineutrofili verso la chemochina CXCL12 (SDF-1). La che-mochina CXCL12 viene espressa dalle cellule dello stromadegli organi linfoidi primari come midollo osseo e timo enei linfonodi ed ha la funzione di mantenere l’omeostasidella circolazione dei leucociti tra questi organi. Pertan-to, sulla base di questi dati ipotizziamo che l’aumento del-la risposta cellulare a questa chemochina delle cellule deipazienti è alla base della formazione di un midollo iper-cellulato e delle alterazioni della distribuzione dei linfoci-ti tra il sangue e i tessuti, spiegando cosi le manifestazio-ni cliniche ed immunologiche della malattia.
P004ESPRESSIONE DI TRAIL E SUOI RECETTORI IN LINEE FANCONI E LOROMODULAZIONE DOPO TRATTAMENTO CON TNF-ααCorcione A,* Ferretti E,* Pigullo S, Lanciotti M, Swahn J,Cavillo M, Pistoia V,* Dufour C*Laboratorio di Oncologia; U. O. S. Ematologia,Dipartimento di Emato-Oncologia Pediatrica,Istituto G. Gaslini, Genova
Introduzione e obiettivi. TRAIL (TNF-related apoptosis-inducing ligand) è una molecola appartenente alla super-famiglia del TNF (tumor necrosis factor)-α che induceapoptosi nelle cellule bersaglio legandosi a recettori spe-cifici. Sono noti differenti recettori per TRAIL (TRAILRs):due di questi, TRAILR1/DR4 e TRAILR2/DR5 contengonoun dominio citoplasmatico di morte, mentre altri due(TRAILR3/DcR1 e TRAILR4/DcR2) sono privi di tale domi-nio e quindi non in grado di indurre apoptosi (decoy). Stu-di precedenti hanno dimostrato un’espressione elevata diTNF-α in cellule midollari di pazienti affetti da anemia diFanconi (FA), suggerendo un ruolo patogenetico del TNF-α nella genesi dell’insufficienza midollare propria di que-sta malattia. In questo studio è stata analizzata l’espres-sione di TRAIL e TRAILRs, in linee Fanconi in condizionibasali e dopo trattamento con TNF-α. Metodi. Lo studio èstato effettuato mediante citofluorimetria su 3 linee Fan-coni appartenenti al gruppo di complementazione A

104
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Ematologia e Coagulazione
(HSC72, GM13022A e GM16749), su una linea C (HSC536)e sulle relative controparti corrette. Risultati: (vedi Tabel-la 1). Conclusioni: Questi risultati suggeriscono che: A) Incondizioni basali, TRAIL è espresso a bassi livelli da tuttele linee Fanconi e up-regolato nelle linee FAA dopo trat-tamento con TNF-α; B) I recettori di morte (TRAILR1 eTRAILR2) risultano più espressi e up-regolati dal TNF-αnelle linee FAA rispetto alla controparte corretta; C) Irecettori decoy (TRAILR3 e TRAILR4) sono espressi a livel-li inferiori nelle linee FAA rispetto alla controparte corret-ta. Questi dati suggeriscono che le linee FAA potrebberoessere più sensibili agli effetti proapoptotici di TRAIL siaper l’elevata espressione di TRAILR1 e TRAILR2 che per laminore espressione dei recettori decoy (TRAILR3 eTRAILR4) e che questa sensibilità possa essere modulatadal TNF-α.
Tabella 1.
P005LA PRIMA MUTAZIONE (E417X) DEL GENE EPO-R IN UNA FAMIGLIAITALIANA CON POLICITEMIA CONGENITA FAMILIARE PRIMITIVAPerrotta S, Migliaccio C, Rossi F, Amendola G,*Ferraro M, Di Pinto D, Cennamo L, D’urso L, Nobili BDipartimento di Pediatria, Seconda Università di Napoli,Napoli; *Oncoematologia pediatrica, Ospedale Umberto I,Nocera Inferiore, Salerno
Il termine policitemia raggruppa una serie di patologieaventi come denominatore comune l’aumento del nume-ro di globuli rossi nel sangue periferico. Le policitemie pos-sono essere primarie (policitemia familiare) o secondariea patologie congenite (emoglobine a bassa affinità perl’ossigeno, disordini del metabolismo del 2,3-difosfoglice-rato, policitemia di Chuvash) o acquisite (disordini epati-ci, polmonari, cardiaci). La policitemia primaria familiarecongenita (PFCP) è una patologia proliferativa a carico deiprogenitori della serie eritroide tipicamente a trasmissio-ne autosomica dominante, caratterizzata clinicamente daelevata massa eritrocitaria e concentrazione dell’emoglo-bina, ipersensibilità dei progenitori eritroidi all’eritropoie-tina (EPO), bassi livelli di EPO circolanti, normale disso-ciazione dell’emoglobina dall’ossigeno e assenza di pro-
gressione verso la leucemia. L’eritropoietina (EPO) è unormone glicoproteico che stimola la proliferazione e ladifferenziazione dei globuli rossi. La sua produzione daparte del rene è stimolata dalla riduzione della tensione diossigeno nel sangue periferico. L’EPO agisce andandosi alegare alla parte N-terminale del dominio extracellularedel suo recettore (EPO-R) presente sui precursori eritroidi(cellule BFU-E e cellule CFU-E) a livello midollare. In rispo-sta all’EPO si ha l’omodimerizzazione del recettore e l’at-tivazione della chinasi JAK2.Tale attivazione porta allafosforilazione di alcune delle otto tirosine presenti neldominio citoplasmatico di EPOR con conseguente attiva-zione dei pathway di trasmissione del segnale. Tra i tra-sduttori del segnale riveste un ruolo particolare STAT5(signal transducer and activator of transcription 5) che,quando fosforilato, dimerizza e trasloca nel nucleo dove vaa legarsi ai suoi specifici elementi responsivi e induce latrascrizione dei geni bersaglio. Alcune fosfatasi specifi-che, in particolare SHP1 (src homology-2 containing pro-tein tyrosine phosphatase-1) funzionano come regolatorinegativi di tali processo di signalling a valle dei recettori.La fosfatasi SHP1, legandosi alla tirosina in posizione 429della proteina EPOR inibisce la proliferazione cellulare,defosforilando JAK2 e spegnendo conseguentemente l’at-tivazione della trasduzione del segnale. Il gene codifican-te per il recettore dell’eritropoietina (EPOR) è costituitoda otto esoni e 508 amminoacidi che vanno a formare undominio extracelllulare, un dominio transmembrana uni-co ed il dominio citoplasmatico con le funzioni preceden-temente descritte. Il gene EPOR è risultato perciò un otti-mo candidato per studiare la base molecolare della PFCP.Sono state descritte dieci mutazioni del gene EPOR, ottodelle quali danno come risultato funzionale il troncamen-to della proteina. Le mutazioni troncanti la proteina inat-tivano il complesso meccanismo di regolazione mediato daEPOR producendo quindi un segnale continuo per la pro-liferazione e differenziazione cellulare. Abbiamo analiz-zato il caso di un ragazzo di otto anni la cui anamnesifamiliare evidenziava la presenza di policitemia nella sorel-la, nel padre ed in alcuni zii e cugini paterni. Il dosaggiodell’Eritropoietina sierica risultava ai limiti bassi in tutti ipazienti ed i progenitori eritroidi del probando presenta-vano una ipersensibilità all’eritropoietina (EPO). Il geneEPO-R è stato analizzato mediante PCR e sequenziamen-to diretto dei prodotti di amplificazione utilizzando oligo-nucleotidi fiancheggianti i singoli esoni. E’ stata identifi-cata in corrispondenza dell’esone 8, codificante per ildominio citoplasmatico regolatorio negativo, una nuovamutazione nonsense in posizione 1249 (GAG−>TAG;E417X) che determina una proteina tronca di 92 aminoa-cidi. Tale mutazione, la prima descritta in Italia, segregacon la malattia in tutti i componenti affetti della famigliae rappresenta un tipico esempio di mutazione nonsenseassociata a policitemia congenita familiare primitiva. Lalocalizzazione della mutazione, determinando un notevo-le troncamento del dominio citoplasmatico, impedirebbeil legame di SHP-1 nella posizione necessaria per iniziareil processo di regolazione negativa del segnale di EPORattraverso la regolazione del pathway Jak/Stat. Sono incorso gli studi funzionali per dimostrarne l’effetto funzio-nale.

105
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
P006UTILITÀ DI ALCUNI PARAMETRI DELL’ESAME EMOCROMOCITOMETRICONELLA IDENTIFICAZIONE DELLA PATOGENESI DI UN’ANEMIAGarbarini L, Parodi E, Quarello P, Alba L, Martino S,Renga D, Ramenghi UDivisione di Ematologia, Dipartimento di ScienzePediatriche, Università di Torino
L’esame emocromocitometrico eseguito con i modernicontaglobuli fornisce nuovi parametri utilizzati per la clas-sificazione delle anemie. L’indice di distribuzione volume-trica dei globuli rossi (RDW), valutando il grado di etero-geneità volumetrica, è considerato parametro importanteper differenziare l’ eterozigosi talassemica dalla siderope-nia. Il basso contenuto emoglobinico reticolocitario (CHr)è riferito quale indice di sideropenia. Abbiamo valutatol’utilità di tali parametri nella diagnosi patogenetica dianemia in età pediatrica. Abbiamo esaminato le cartellecliniche di tutti i pazienti ricoverati per anemia presso lanostra Divisione negli ultimi 4 anni (2000-2003). Consi-derato i pazienti con Hb all’ingresso inferiore a 2DS rispet-to al valore medio per l’età. Analizzati RDW e CHr pervalutarne l’attendibilità nella diagnosi di anemia sidero-penica e nel discriminare tra carenza marziale e trattotalassemico. Sono risultati valutabili 140 casi (55 F e 85M). La diagnosi è stata: anemia sideropenica (S) in 62/140casi, di cui 33 dovuti a ridotta assunzione, 11 a malassor-bimento, 8 a perdita gastrointestinale, 7 a epistassi ripe-tute, in 4 non identificata la causa; 15/140 erano etero-zigoti per talassemia (T). I restanti 63 erano anemie daaltra causa (A): 22 anemie emolitiche non immuni, 19 ipo-rigenerative, 12 emolitiche immuni, 5 post emorragiche, 1deficit di folati, 1 CDA II e 3 di n. n. d. I valori dell’ RDW eCHr sono stati confrontati tra queste 3 popolazioni. I valo-ri medi di RDW non sono risultati significativamente dif-ferenti nei tre gruppi (S 17. 9±4. 4, A 16. 8±4. 7, T 17,8±3,6%). Un CHr >26 è stato riscontrato nel 9 % dei siderope-nici e nel 75 % delle altre anemie; un CHr<20 è statoriscontrato solamente in pazienti sideropenici. Nella nostracasistica l’RDW, contrariamente a quanto riportato in let-teratura, non risulta un parametro attendibile per la dia-gnosi differenziale tra anemia sideropenica e tratto talas-semico. Confermiamo invece come il CHr basso sia pre-dittivo di carenza marziale in alternativa alle indagini bio-chimiche; in particolare CHr < 20 è risultato avere un valo-re predittivo positivo e una specificità del 100% per side-ropenia.
P007IMPIEGO DI RITUXIMAB NELLA PIASTRINOPENIA CRONICA REFRATTARIAParodi E,^ Farinasso L,^ Quarello P,^ Licciardello M,°Bombace V,° Russo G,° Zecca M,* Locatelli F,*Ramenghi U^^Divisione di Ematologia, Dipartimento di ScienzePediatriche, Università di Torino, °Centro di RiferimentoRegionale di Ematologia ed Oncologia Pediatrica,Università di Catania; *Divisione di Onco-ematologiapediatrica, Policlinico San Matteo, Pavia
L'azione di interferenza o abolizione della produzioneanticorpale da parte del rituximab, anticorpo monoclona-le specifico verso l'antigene CD20 dei linfociti B, è utiliz-zata anche nel trattamento di patologie autoimmuni. Datirecenti ne confermano l'efficacia nell'anemia emoliticaautoimmune in età pediatrica. Riferiamo dati preliminarirelativi alla nostra esperienza nel trattamento della pia-strinopenia immune cronica, sintomatica e refrattaria.Casistica e metodiche. sono stati trattati 6 pazienti con PTIcronica, tutti di sesso femminile, con uno o più cicli dirituximab (375mg/m2/dose/settimana per 1-4 infusioni).Trattamento precedente: uno o più cicli di IVIg (6/6),mPDN (5/6), desametasone (3/6), CSA (2/6) e Ig antiD (1/6).In 3/6 la somministrazione è stata successiva al fallimen-to della splenectomia (splenectomia a +6,+11,+41 mesidalla diagnosi; rituximab a +11, +12, +73 mesi dalla dia-gnosi); in 3/6 precedente alla splenectomia (rituximab a+21, +30, +26 mesi dalla diagnosi; splenectomia a +33,+34, +30 mesi dalla diagnosi). La risposta positiva al trat-tamento è stata definita come incremento delle piastrinesuperiore a 50.000/mm3 per almeno 7 giorni. Risultati. Iltrattamento è sempre stato ben tollerato; un solo caso hapresentato fugace reazione pomfoide. Il follow-up media-no dall'ultima infusione di rituximab è di 9,5 mesi. Hannorisposto al trattamento 2/3 pazienti trattati dopo sple-nectomia e 0/3 pazienti non splenectomizzati (rispettiva-mente al giorno +19; +26 dall'infusione). Entrambi i sog-getti hanno presentato un incremento superiore a150.000/mm3. Dei 2 pazienti responsivi solo uno mantie-ne valori piastrinici >150.000/mm3; il secondo è ricadutoa 120 giorni, sottoposto a nuovo ciclo è nuovamente reci-divato dopo 180 giorni. Conclusioni: I nostri dati prelimi-nari evidenziano un'efficacia del rituximab nelle piastri-nopenie inferiore a quella osservata nelle anemie emoli-tiche autoimmuni (Zecca,Blood 2003). Risposta è statainoltre osservata solo in pazienti splenectomizzati. E'necessario un maggior numero di casi per avere informa-zioni più conclusive; tuttavia, in considerazione della buo-na tolleranza, e della gravità di alcuni casi, riteniamo ilrituximab possa essere considerato un farmaco da testa-re in caso di fallimento della terapie convenzionali.
P008PROTOCOLLO DIAGNOSTICO DI SORVEGLIANZA PER SINDROMIMIELODISPLASTICHE SECONDARIE IN ETÀ PEDIATRICAFarinasso L, Timeus F, Garbarini L, Basso ME, Aschero S,Alba L, Iavarone A,* Linari A,* Gottardi E,° Crescenzio N,Saracco PDipartimento di Scienze Pediatriche e dell' Adolescenza,Università di Torino; *Dipartimento di Patologia Clinica,Ospedale Infantile Regina Margherita; °Dipartimento diScienze Biomediche, Università di Torino
Introduzione e Obiettivi: le sindromi mielodisplastiche(MDS) sono disordini clonali rari dell’ematopoiesi suscet-tibili di evoluzione in leucemia mieloide acuta (LAM); laloro frequenza in età pediatrica è stimata intorno al 10%delle emopatie maligne. Le MDS possono presentarsi denovo o essere secondarie a pre-esistenti malattie delmidollo osseo (aplasia midollare congenita o acquisita-AA),pregresso trattamento con chemioterapici (soprattutto

106
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Ematologia e Coagulazione
agenti alchilanti o inibitori della topoisomerasi II). Abbia-mo elaborato un protocollo di sorveglianza per l’ identifi-cazione delle MDS secondarie di cui vengono descritti irisultati relativi ai pazienti con aplasia midollare congeni-ta o acquisita. Metodi Attualmente il nostro protocolloprevede: valutazione morfologia dello striscio periferico,studio delle proteine GPI-linked, conta delle cellule CD34+
circolanti e del loro indice apoptotico, morfologia mieloa-spirato e osteomielobiopsia, test clonogenici, immunofe-notipo, cariotipo con citogenetica standard e FISH, anali-si molecolare (identificazione di traslocazioni suggestive diLMA, valutazione delle copie del gene WT1). Il protocolloè stato applicato in maniera completa dal 2000, con con-trolli semestrali-annuali a 14 Anemia di Fanconi-FA, 1 S.di Kostmann, 2 Discheratosi Congenite, 17 AA, 3 anemiedi Blackfan Diamond, 2 aplasie pure della serie rossa-PRCA. Risultati: nell’ambito dei pz affetti da aplasia midol-lare abbiamo osservato 5 AREB, 4 in pz. in trattamentocon G-CSF (1 AA e 3 FA) e 1 in un pz con PRCA. Le altera-zioni citogenetiche osservate sono state: -7 in 3 casi. Intutti i casi di MDS abbiamo riscontrato un’elevata contadi cellule CD34+ circolanti con basso indice apoptotico.Conclusioni: nella nostra esperienza l’associazione dell’analisi citogenetica, del quadro morfologico della biopsiaossea e la valutazione delle CD34+ circolanti è altamentepredittiva per evoluzione in mielodisplasia.
P009LA MALATTIA DA EMOGLOBINA H IN ETÀ PEDIATRICACampus S, Mandas C, Barella S, Giagu N, Sollaino C,Origa R, Galanello RStruttura Complessa Microcitemie ed altre MalattieEmatologiche Ospedale Microcitemico ASL8, Universitàdegli Studi, Cagliari
Introduzione. La malattia da emoglobina H (HbH) è unaforma intermedia di α-talassemia caratterizzata da ane-mia emolitica microcitica ed ipocromica di grado variabi-le. E’ causata dalla perdita di funzione di 3 dei 4 geni alfaglobinici, per cui si ha un grave deficit di catene alfa, coneccesso relativo di gamma globine nel neonato e di betaglobine nelle età successive, che formano rispettivamen-te tetrametri γ 4 (Hb di Bart) e β 4 (HbH). I tetrameri di βcatene sono relativamente instabili e precipitano neglieritrociti causando emolisi. Esistono due tipi di malattia daHbH: da delezione (--/-α) e da non delezione (--/α nd α).Le forme da delezione sono in genere clinicamente piùlievi. Obiettivi. Scopo dello studio è di valutare la malat-tia da emoglobina H in età pediatrica. Pazienti. Presso l’O-spedale Microcitemico vengono seguiti regolarmente 239pazienti (107 maschi e 132 femmine) con malattia daHbH, di età compresa tra 1 e 82 anni. 215 presentano laforma da delezione e 34 quella da non delezione. Il sinto-mo di esordio più comune è risultato essere l’anemia, piùraramente la splenomegalia; a volte la diagnosi è stataoccasionale, specie in età adulta. Risultati. 56 pazientisono stati seguiti longitudinalmente in età pediatrica. Diquesti il 18% presenta epatomegalia, il 66% splenome-galia ed in 3 casi è stata riscontrata una calcolosi dellacolecisti. Nel 25% è presente una dilatazione delle cavitàcardiache, dovuta verosimilmente all’anemia cronica. Il
14. 3% ha un ritardo dell’accrescimento (Z score per l’al-tezza inferiore a –1,8%). Tutti i pazienti hanno un’anemiamicrocitica ed ipocromica (Hb 9. 1±0.8 g/dl, MCV=56±3fl),con reticolocitosi (3. 2±1.1%) e nel 24% dei pazienti viè una modesta iperbilirubinemia indiretta. In 12 casi èstata necessaria terapia trasfusionale per crisi emolitichein corso di infezione (7), crisi aplastica da Parvovirus B19(1), anemia neonatale (3), intervento chirurgico (1). In nes-suno dei bambini è stato riscontrato un accumulo di fer-ro, che invece è presente spesso nelle età successive. Con-clusioni La malattia da HbH è una forma intermedia dialfa-talassemia relativamente frequente nelle regionimediterranee, che presenta una notevole eterogeneitàfenotipica e che per le possibili complicanze necessita diun accurato follow-up soprattutto in età pediatrica.
P010AUMENTATA PROTEOLISI DEL FATTORE VON WILLEBRAND IN CORSO DITERAPIA DELLA LEUCEMIA LINFOBLASTICA ACUTA DEL BAMBINOPerutelli P, Amato S, De Mattia D,* Del Vecchio GC,*Giordano P,* Russo G,§ Saracco P,° Molinari ACDipartimento di Emato-Oncologia, IRCCS G. Gaslini, Genova;*Dipartimento di Biomedicina dell’Età Evolutiva, Universitàdi Bari; §Clinica Pediatrica, Università di Catania; °Diparti-mento di Pediatria, Ospedale Regina Margherita, Torino
Introduzione e obiettivi. Gli eventi tromboembolici rap-presentano una grave complicanza della leucemia linfo-blastica acuta (LLA) del bambino. Nei pazienti arruolati nelprotocollo terapeutico AIEOP LLA 2000 eventi sono statiregistrati soprattutto nella fase Ia. Il fattore von Willebrand(VWF) ha un ruolo chiave nel promuovere l’aggregazione el’adesione piastrinica. VWF è una glicoproteina multimeri-ca di grandi dimensioni. In condizioni di elevato shear stressla proteolisi del VWF aumenta, determinando una modifi-cazione della composizione multimerica. Abbiamo ricerca-to modificazioni del VWF, quali marcatori di trombofilia,durante la fase di induzione della terapia della LLA. Meto-di. 58 bambini con LLA (17 mesi -15,2 anni, mediana 48mesi) arruolati presso i Centri partecipanti all’AIEOP di Bari,Catania, Genova e Torino. Sono stati raccolti campioni ema-tici all’esordio della malattia (T1) e ai giorni +24 (T2), +36(T3), +52 (T4). Abbiamo valutato frammenti di proteolisi ecomposizione multimerica del VWF. Risultati. I frammentidi proteolisi del VWF erano significativamente aumentati aT3 (p<0,01): T1 19,5%; T2 20,2%; T3 22,3%; T4 20,0%. Perquanto concerne la composizione multimerica, si osserva-va una variazione della componente a più elevato pesomolecolare (multimeri >15): T1 16,6%; T2 15,5%; T3 14,5%(p<0,025); T4 15,8%. I prodotti della degradazione deglialti pesi molecolari confluivano principalmente nelle com-ponente a peso molecolare intermedio (multimeri 6-10):T1 27,6%; T2 29,0% (p<0,05); T3 29,4% (p<0,02); T427,8%. Conclusioni. In bambini leucemici, abbiamo osser-vato il progressivo incremento di proteolisi del VWF nelleprime fasi della terapia di induzione, con riduzione dellacomposizione multimerica della molecola. Questo risulta-to ci porta ad ipotizzare un danno endoteliale nei pazien-ti. Nella casistica oggetto dello studio non sono stati regi-strati eventi tromboembolici, ma il reperto di aumentata

107
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
proteolisi del VWF potrebbe offrire una ulteriore chiave dilettura degli eventi registrati in altri pazienti arruolati nel-lo stesso protocollo. L’ampliamento della casistica e l’e-stensione delle indagini laboratoristiche potrebbero miglio-rare la comprensione dell’eziopatogenesi del tromboembo-lismo in corso di LLA ponendo le basi per la prevenzione.
P011DUBBIA EFFICACIA DELLA TERAPIA STEROIDEA SHORT COURSE AD ALTEDOSI NELLA PORPORA TROMBOCITOPENICA IDIOPATICAGualeni C, Arrighetti A, Caldiani C, Tettoni K,Notarangelo Lucia D, Notarangelo Luigi D, Porta FUnità dipartimentale di terapia cellulare e genica delleoncoemopatie infantili “Angelo Nocivelli”, Clinicapediatrica, Spedali Civili, Brescia
Introduzione. La PTI è una malattia immunomediatacaratterizzata da ridotta conta piastrinica dovuta alla pre-senza di autoanticorpi diretti contro antigeni piastriniciper la quale esistono tuttora incertezze sulle strategieterapeutiche. Le controversie riguardano principalmente lemodalità di trattamento, in particolare la necessità di trat-tare o meno paziente gravemente piastrinopenici ma nonsintomatici, nonché il tipo di farmaco da utilizzare. I diver-si studi riportati in letteratura hanno dimostrato inoltreche i vari tipi di trattamenti utilizzati non influenzano inalcun modo né la possibilità di risoluzione né l’evoluzioneverso la forma cronica. Obiettivi. L’utilizzo di steroidi adalte dosi (4 mg/Kg) per un breve periodo di tempo (4 gior-ni) è in grado di aumentare la conta piastrinica entro valo-ri > 20.000 plt/mm3 in una settimana; abbiamo volutoverificare tale dato su alcuni pazienti giunti alla nostraosservazione. Metodi e pazienti. Sono stati trattati 4pazienti affetti da PTI (2 maschi e 2 femmine) di età com-presa tra 2 aa 10/12 e 11 aa e 9/12 (età mediana 4 aa e5/12); i valori delle piastrine all’esordio erano tutti < a20.000/mm3 (mediana 3. 000/mm3); tutti i pazienti pre-sentavano ecchimosi e petecchie diffuse, in un caso erapresente sanguinamento mucoso (epistassi). I pazientisono stati sottoposti a trattamento con con metilpredni-solone ad alte dosi (4 mg/Kg) per 4 giorni, previa esecu-zione di aspirato midollare risultato compatibile con PTI. Ipazienti sono stati successivamente valutati sia per quan-to riguarda la conta piastrinica, sia per l’aspetto clinico.Risultati. 3 pazienti hanno presentato una transitoriarisposta piastrinica tra il 4° e il 6° giorno (piastrine com-prese tra 26. 000 e 36. 000/mm3) con ritorno a valori infe-riori alle 20.000 plt tra l’8° e il 15° giorno mentre il 4°paziente non ha mostrato alcun incremento piastrinico; uncaso ha necessitato di trattamento con immunoglobulinea distanza di 30 gg per epistassi marcata parzialmenteresponsiva ai comuni trattamenti; un paziente ha effet-tuato trattamento con immunoglobuline e. v. per traumacranico. Conclusioni. Sia pure su una casistica molto pic-cola, la nostra esperienza dimostra la transitorietà dellarisalita della conta piastrinica dopo terapia steroidea adalte dosi per 4 giorni nella PTI. Tale risalita temporaneapone dei dubbi sul reale ruolo di un trattamento steroideoper un periodo così breve.
P012LIVELLI DI LIPOPROTEINA (A) IN PAZIENTI PEDIATRICI CON LEUCEMIALINFOBALSTICA ACUTA ALLA DIAGNOSI E DURANTE IL TRATTAMENTOSaracco P,° Del Vecchio GC,* Bertorello N,° Farinasso L,°Russo G,** Uberti B,^ Bo M,° Giordano P*°Dipartimento di Scienze Pediatriche Università diTorino; * Dipartimento di Biomedicina dell’età evolutiva,Università di Bari, **Centro di Riferimento Regionale diEmatologia ed Oncologia Pediatrica Università diCatania, ^Lab. Diabetologia e Malattie del Ricambio,ASO S. Giovanni Battista, Torino
Introduzione ed Obiettivi. I livelli ematici di lipoprotei-na a, Lp(a), in parte geneticamente determinati, se > 30mg/dL correlano con un aumentato rischio trombotico,anche in età pediatrica. Le sue concentrazioni ematiche,però, possono essere modificate da fattori esogeni, siaaumentando in corso di infiammazione, sia diminuendoin corso di trattamento con alcuni farmaci, quali la L-Asparaginasi (L-Asp). Secondo alcuni autori non può, per-tanto, essere considerato un marker protrombotico stabi-le, nei bambini con leucemia linfoblastica acuta (LLA),durante terapia con L-Asp. (Korte et al. , Blood 2001).Abbiamo dosato i livelli di Lp(a) durante la terapia con L-Asp e a distanza in pazienti pediatrici con LLA, al fine diconfrontare l’andamento dei valori di Lp(a), stratificandoi pazienti sulla base dei livelli riscontrati all’esordio. Meto-di. In 54 pazienti consecuitivi, affetti da LLA, trattatisecondo il protocollo, AIEOP-BFM 2000 (età media 6anni,24 M e 15 F) e in 27 pazienti di controllo, di pari etàsono stati dosati all’esordio, nei giorni +24, +36, +52 diinduzione (comprensiva di 8 dosi bisettimanali di L-Asp. )e in mantenimento i livelli della Lp(a), secondo metodonefelometrico (Dade Behring nefelometric assay, Ham-burg, Germany). I dati sono stati analizzati con test stati-stici non parametrici, Friedman test e Mann-Whitney Utest, (STAT view program, Abacus Concepts, CA). I pazien-ti sono stati stratificati in quartili in base ai livelli Lp(a)all’esordio. Risultati. I livelli di Lp(a) sono risultati elevatialla diagnosi rispetto ai controlli (p<0,05), con una ridu-zione significativa in tutti i tempi dello studio e nadir nelgiorno +24, (p<0,01). Il 57% dei pazienti con livelli Lp(a)all’esordio superiori al 3° quartile, ha mantenuto, nono-stante una riduzione significativa al giorno +24, livelli ele-vati durante il mantenimento. Conclusioni. Considerandola casistica complessiva si conferma la riduzione dei livel-li di Lp(a) durante la terapia con L-Asp. in bambini con LLA.Nei pazienti con livelli persistentemente elevati, pertanto,la presenza di alte concentrazioni ematiche di Lp(a) duran-te le diverse fasi dello studio può costituire un fattore dirischio pro-trombotico.
P013RISCONTRO DI MALATTIA DI VON WILLEBRAND ACQUISITA IN UN PAZIENTECON TUMORE DI WILMSNotarangelo Lucia D, Tettoni K, Caldani C, Gualeni C,Porto C,1 Martini G,2 Tonegatti L,3 Ekema G,3Federici A,4 Notarangelo Luigi D,5 Porta FUnità Dipartimentale di Terapia Cellulare e Genica delle

108
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Ematologia e Coagulazione
Oncoemopatie Infantili “Angelo Nocivelli”, ClinicaPediatrica Spedali Civili, Brescia; 1Servizio di Anestesiae Rianimazione, Ospedale dei Bambini, Brescia;22°Laboratorio, Spedali Civili, Brescia;3 Chirurgia Pediatrica, Ospedale dei Bambini, Brescia;4Centro Emofilia e Trombosi “Angelo Bianchi Bonomi,Ospedale Maggiore Policlinico, Milano;5Clinica Pediatrica, Spedali Civili, Brescia
La malattia di Von Willebrand è una condizione emorra-gica congenita dovuta ad una riduzione o alterata funzio-ne del fattore di Von Willebrand (VWF) su base genetica.Una condizione acquisita, con meccanismi fisiopatologicidiversi, è stata descritta in associazione a malattie linfo emieloproliferative, gammopatie monoclonali e, meno comu-nemente, a tumori solidi. Descriviamo il caso di una pazien-te affetta da tumore di Wilms che ha presentato test dilaboratorio compatibili con malattia di Von Willebrand ditipo I normalizzatisi in seguito ad intervento chirurgico echemioterapia. F. M. , anni 3 e 7/12, giunge alla nostraosservazione per riscontro di massa addominale compati-bile con tumore di Wilms. Lo screening preoperatorio dimo-stra valori di PTT ripetutamente alterati (51 sec, 55 sec, 53sec) in assenza di segni clinici e con anamnesi familiare epersonale negativa per coagulopatie. La valutazione dei fat-tori della coagulazione dimostra valori di fattore VIII coa-gulabile (FVIII:C) pari al 39% (v. n. 50-150), fattore di VonWillebrand antigenico (VWF Ag) pari al 37% (v. n. 50-150),VWF Ristocetin co-factor (VWF:RCo) pari al 38% (v. n. 50-150) con normalità dei restanti fattori della via intrinseca.Il gruppo sanguigno è O positivo. Viene effettuato un testalla desmopressina che normalizza i dati di laboratorio. Lapiccola viene sottoposta ad intervento chirurgico con aspor-tazione in blocco della neoplasia e del surrene. La diagno-si istologica conferma il sospetto di tumore di Wilms; lastadiazione depone per uno stadio II e viene pertanto ini-ziato trattamento con vincristina settimanale e actinomi-cina D ogni due settimane. La valutazione dei test emo-coagulativi, effettuata VWF:RCo ancora alterati (rispetti-vamente 36% e 51%) mentre, a distanza di due settimanedall’inizio del trattamento chemioterapico, tali valori risul-tano normalizzati (FVIII:C 115%, VWF Ag 58%, vWF Ri:Co82%) con normalità dell’agglutinazione alla ristocetina. Aconferma della condizione acquisita è stata effettuata adentrambi i genitori la valutazione del PTT, FVIII:C, VWF:Ag,VWF:RCo e agglutinazione alla ristocetina risultati nellanorma. Il caso da noi riportato conferma la possibile asso-ciazione, sebbene rara, tra sindrome di von Willebrandacquisita e tumore di Wilms, e sottolinea la necessità dieffettuare un accurato screening della coagulazione nelleoncoemopatie, in particolare nel tumori di Wilms, onderidurre il rischio di sanguinamenti intraoperatori e per effet-tuare una appropriata terapia evitando il ricorso ad emo-derivati qualora non indicato.
P014ANEMIA DISERITROPOIETICA E TROMBOCITOPENIA: NUOVA MUTAZIONEDEL GENE GATA-1Del Vecchio GC, Giordani L, de Santis A, De Mattia DDipartimento di Biomedicina dell’Età Evolutiva,Università di Bari
Introduzione: l’emopoiesi è un processo complesso rego-lato da proteine nucleari che coordinano l’espressionegenica di specifici profili di linee cellulari. La mutagenesimirata ha evidenziato il ruolo critico che possiede il fat-tore trascrizionale GATA-1 nella differenziazione eritroci-taria e megacariocitaria. Il GATA-1 possiede due dominiche sono essenziali per il suo funzionamento. Il dominioC-terminale è necessario per il legame al DNA. Il dominioN-terminale interagisce con il FOG-1, un cofattore per ilGATA-1.Le mutazioni del dominio N-terminale sonoresponsabili di anomalie nell’emopoiesi. Obiettivo: ripor-tiamo una famiglia con una nuova mutazione nel domi-nio N-terminale del GATA-1 associata ad anemia diseri-tropoietica e macrotrombocitopenia. Metodo: il probando,di 17 anni, terzogenito di genitori non consanguinei, pre-sentava anemia sin dalla nascita trattata con occasionaliemotrasfusioni sino all’età di 5 anni e trombocitopeniaresistente alle terapie, con emorragie cutanee e mucose lacui frequenza e gravità sono migliorate con lo sviluppo. E’giunto alla nostra osservazione con presenza di lieve ane-mia macrocitica (Hb 9,6 g/dL, MCV 103 fl), macrotrombo-citopenia (PST 12x109/L) e criptorchidismo. La madre pre-sentava lieve trombocitopenia, entrambi i fratelli criptor-chidismo. E’ stata studiata la sequenza codificante delgene GATA-1 nel probando, in un fratello e nei genitorimediante sequenziamento diretto. Conclusioni: Un’altramutazione descritta nello stesso codone 208 è stata cor-relata alla presenza di sola trombocitopenia. I nostri datisupportano ed estendono il ruolo della sostituzione ami-noacidica per la funzione del GATA-1 non solo nella dif-ferenziazione megacariocitaria, ma anche in quella eri-trocitaria.
P015VALUTAZIONE DEL FERRO NON LEGATO ALLA TRANSFERRINA (NTBI) INSOGGETTI AFFETTI DA ββ-TALASSEMIA TRATTATI CON TERAPIAFERROCHELANTE COMBINATA (DESFERRIOXAMINA + DEFERIPRONE)O CON MONOTERAPIA (DESFERRIOXAMINA O DEFERIPRONE) Meo A, La Rosa MA, Zanghì L, Ruggeri A, Duca L,*Nava I,* Cappellini MD*Dipartimento di Pediatria, Università di Messina; *CentroAnemie Congenite, Dipartimento di Medicina Interna,Ospedale Maggiore Policlinico IRCCS di Milano
Introduzione e obiettivi: la β-talassemia major (TM) èun’ anemia emolitica ereditaria dovuta alla riduzione oall’ assenza totale della sintesi delle catene β-globiniche.Il sovraccarico di ferro conseguente alle trasfusioni di san-gue, nonostante l’ intensiva terapia chelante, rimane lacausa principale di morte nei pazienti affetti da TM. Inalcuni studi è stata documentata la presenza di una fra-zione di ferro tossico a basso peso molecolare (Non-tran-sferrin-bound iron o NTBI) in pazienti con sovraccarico diferro. Il trattamento convenzionale con infusioni sottocu-tanee di desferrioxamina (DFO) ha migliorato in modosignificativo la sopravvivenza dei soggetti TM, per quan-to recentemente sia stato ipotizzato che la terapia ferro-chelante combinata con DFO e deferiprone (L1) possaessere ancor più efficace della monoterapia. Metodi: lostudio ha indagato lo stato del ferro e l’NTBI in 12 pazien-

109
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
ti affetti da TM trattati con DFO 50 mg/kg/die 5 giorni allasettimana per almeno 10 anni, in 12 pazienti con TM sot-toposti a terapia con L1 75 mg/kg/die per un periodo di 12mesi e in 10 pazienti TM sottoposti a trattamento combi-nato DFO per via sottocutanea 40 mg/kg/die, 3/7 giorni, +L1 75 mg/kg/die, per un anno. Tutti i parametri sono sta-ti controllati a 2 anni di distanza dalla prima valutazione.Lo stato del ferro è stato determinato con procedure stan-dard di laboratorio, mentre l’ NTBI è stato valutato su sie-ro mediante HPLC dopo chelazione con acido nitrilotria-cetico (NTA); i valori dell’ NTBI nei soggetti normali di con-trollo risultano sempre negativi (-0,72±0,70 µM) a causadella sottrazione del bianco formato da NTA ed acqua checontiene piccole quantità di ferro che non vengono lega-te dalla transferrina; nel campione del soggetto sano inve-ce, la transferrina non completamente saturata capta ilferro dal complesso ferro-NTA. Risultati: alla prima valu-tazione non si sono evidenziate differenze significative perquanto riguarda lo stato del ferro (ferritina e % di satura-zione della transferrina); viceversa nel caso dell’ NTBI sisono rilevati valori significativamente inferiori nei pazien-ti sottoposti a terapia combinata (1,59±1,03 µM) rispet-to ai soggetti trattati con il solo L1 (2,61±1,31 µM)(p=0,038) o DFO (2,31±1,05 microM) (p=0,035). A distan-za di due anni la ferritina sierica non ha mostrato varia-zioni significative; i livelli di NTBI invece sono diminuiti del31% nei pazienti trattati con DFO (1,59±1,03 µM) e del79% nei pazienti sottoposti a terapia combinata(0,24±1,23 µM), mentre sono rimasti pressoché invariatinei soggetti trattati con L1 (2,73±1,59 µM). Conclusioni:DFO ed L1 potrebbero agire con meccanismi di tipo siner-gistico o additivo migliorando la chelazione del ferro emantenendo la sua frazione tossica entro livelli limitati,come dimostrato dai risultati ottenuti.
P016EMOCROMATOSI EREDITARIA E MALATTIA DREPANOCITICARusso G, Samperi P, Bertuna G, Barbagallo MlCentro di Riferimento di Ematologia ed OncologiaPediatrica, Università di Catania
Introduzione e obiettivi. L'emocromatosi è una malattiaereditaria caratterizzata da aumento dell’assorbimento delferro alimentare. La sua manifestazione dipende dal pro-gressivo accumulo di ferro con conseguente danno a variorgani (cuore, fegato, ghiandole endocrine etc). Sono sta-ti identificati diversi geni responsabili, tra cui HFE, TFR2,FPN1.Esistono anche condizioni (anemie emolitiche, epa-topatie etc. ) che comportano un’emocromatosi seconda-ria. Scopo del lavoro è valutare la presenza e l’effetto del-le più frequenti mutazioni dei geni su menzionati neipazienti con malattia drepanocitica, che sono a rischio disiderosi sia per l’emolisi cronica che per le eventuali tra-sfusioni. Metodi. Abbiamo studiato 42 pazienti (22M, 11SS e 31Sth) con malattia drepanocitica, di età tra 4 e 46anni. Nessuno dei soggetti aveva avuto crisi dolorose, infe-zioni e/o trasfusioni nelle 6 settimane precedenti. Di que-sti, 16 erano stati politrasfusi in passato; 26 mai o solooccasionalmente avevano ricevuto trasfusioni. Sono statidosati ferritina e saturazione della transferrina. Sono sta-te ricercate le mutazioni V53M, V59M, H63D, H63H, S65C,
Q127H, P160del C, E168Q, E168X, W169X, C282Y, Q283Prelativamente al gene HFE; le mutazioni E60X, Y250X,M172K, AVAQ594-597 del relativamente al gene TFR2; lemutazioni N144H, V162del relativamente al geneFPN1.Risultati. Ventisei pazienti risultano negativi per lemutazioni testate (gruppo A) e 16 sono positivi (gruppo B);in particolare 1 soggetto è omozigote e 13 eterozigoti perla mutazione H63D del gene HFE, 2 eterozigoti per lamutazione S65C dello stesso gene. I soggetti politrasfusiin passato sono 8 nel gruppo A (31%) e 8 nel gruppo B(50%). La saturazione della transferrina è 31.2 ± 8 % nelgruppo A e 39. 5±11% nel gruppo B. La ferritina è 378.3±428 ng/dL nel gruppo A e 472±378. 3 ng/dL nel grup-po B. Nessuna differenza raggiunge la significatività sta-tistica. Conclusioni. Emerge l’assenza della mutazioneC282Y, più diffusa nel Nord Italia e nel Nord Europa ingenerale; spicca invece l’alta prevalenza della mutazioneH63D (33% dei soggetti), notoriamente diffusa nel sudItalia. La presenza di tale mutazione non sembra compor-tare un maggiore rischio di sovraccarico marziale nellanostra popolazione.
LEUCEMIE E LINFOMI
C003LE REINDUZIONI CICLICHE CON VINCRISTINA (VCR) E DESAMETAZONE(DXM) DURANTE LA TERAPIA DI MANTENIMENTO NON MIGLIORANO IRISULTATI NELLA LEUCEMIA LINFOBLASTICA ACUTA (LLA) A RISCHIOINTERMEDIO (RI) TRATTATA CON PROTOCOLLI BFM: RISULTATI DELLOSTUDIO I-BFM-SG ALL IR 95Conter V,1 Valsecchi MG,1 Silvestri D,1 Campbell M,2Dibar E,3 Magyarosy E,4 Gadner H,5 Stary J,6 Riehm H,7Masera G,1 Schrappe M7
International BFM Study Group
Obiettivo: scopo di questo studio è di valutare l’effica-cia della somministrazione di reinduzioni con VCR-DXMdurante il mantenimento in bambini con LLA trattati conprotocolli BFM orientati. Mtodi e pazienti: questo studiointergruppo, prospettico e randomizzato è stato condottocon una metodologia basata sui criteri dalla meta-analisinei seguenti Paesi: Italia, Cile, Argentina, Ungheria,Austria, Repubblica Ceca, Svizzera e Germania. Tutti iGruppi partecipanti appartengono all’International BFMStudy Group (I-BFM-SG) e utilizzano protocolli terapeu-tici basati sullo schema BFM, che include il protocollo I perla fase di Induzione, il protocollo M (methotrexate ad altedosi, 2 o 5 g/m2) come consolidamento, e il protocollo IIper la fase di reinduzione. Il RI è definito da: età < 1 annoo a 6 anni o globuli bianchi alla diagnosi 20,000/mm3 edalla assenza di caratteristiche ad alto rischio (scarsarisposta alla prefate steroidea, presenza delle traslocazio-ni t(9;22) o t(4;11), no remissione completa dopo la faseIA). I pazienti a RI sono stati randomizzati all’inizio delmantenimento per ricevere oltre al purinethol e al metho-trexate VCR (1,5 mg alla settimana x 2) e DXM (6 mg/m2
al giorno per 7 giorni) ogni 10 settimane per 6 volte duran-

110
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Leucemie e Linfomi
te il mantenimento. Risultati: nel periodo Maggio 1995 -Dicembre 2000 sono entrati nello studio 3080; di essi 163hanno presentato un evento prima della randomizzazio-ne, 295 non sono stati randomizzati, 1327 sono stati ran-domizzati nel Braccio A e 1295 nel Braccio B. Con follow-up mediano di 4,6 anni si sono osservati 254 eventi, di cui240 ricadute in entrambi i bracci. La DFS (SE) a 7 anni è77,4 (1,5) nel braccio A e del 78,3 (1,3) nel braccio B (log-rank p=0,67) con un hazard-ratio di 0,97 (Braccio A vs B;95% CI 0,82-1,16). Conclusioni: In questo studio l’ag-giunta di reinduzioni con VCR-DXM durante il manteni-mento non è stata associata a differenze a lungo terminerispetto a DFS o tasso di ricadute o mortalità in remissio-ne. Questi dati pertanto non evidenziano alcun beneficioassociato all’intensificazione della terapia di manteni-mento in pazienti con LLA a RI trattati con una terapia ini-ziale intensiva basata sulla strategia BFM.
C004RUOLO DELL’IMMUNOSORVEGLIANZA ANTI-TUMORALE NEL CONTROLLODELLA MALATTIA RESIDUA MINIMA: VALUTAZIONE DELLA COMPARSA DIPRECURSORI LINFOCITARI CITOTOSSICI (CTLP) SPECIFICI PER I BLASTILEUCEMICI IN PAZIENTI AFFETTI DA LEUCEMIA LINFATICA ACUTADaudt L, Locatelli F, Turin I, Montini E, Zecca M,Bonetti F, Telli S, Lisini D, Moretta A, Maccario R,Montagna DLaboratorio di Immunologia e Trapianti, Dipartimento diScienze Pediatriche, Università di Pavia, e Unità diOncoematologia Pediatrica, IRCCS Policlinico SanMatteo, Pavia
Introduzione e obiettivi. Negli ultimi anni si sono fattemolte ipotesi sul ruolo giocato dal sistema immunitario nelcontrollo della crescita delle cellule neoplastiche, anche se,al momento, vi sono poche evidenze che dimostrano lacomparsa di una risposta immunitaria specifica in pazien-ti affetti da leucemia. Il nostro gruppo ha precedente-mente dimostrato che precursori linfocitari citotossici(CTLp), di origine del donatore e diretti verso i blasti leu-cemici (BL) del paziente, compaiono nel sangue perifericodi pazienti affetti da leucemia acuta, sottoposti a tra-pianto allogenico di cellule staminali emopoietiche e pos-sono contribuire al mantenimento di uno stato di remis-sione di malattia. Sulla base di questi presupposti, nel pre-sente studio, abbiamo valutato se un attività anti-leuce-mica autologa può essere evidenziata in un gruppo dipazienti affetti da leucemia linfatica acuta (LLA) trattaticon la sola chemioterapia. Abbiamo, inoltre, valutato ilsuo eventuale ruolo nell’immunosorveglianza anti-tumo-rale. Metodi. A questo scopo abbiamo allestito degli espe-rimenti in diluizione limite per misurare la frequenza diCTLp diretti verso i BL autologhi in un gruppo di pazientipediatrici affetti da LLA durante le varie fasi della che-mioterapia, fino oltre allo stop terapia. Sono stati finoraarruolati 40 pazienti esorditi dal giugno 2001 presso l’u-nità di Oncoematologia Pediatrica dell’IRCCS PoliclinicoSan Matteo. Al momento sono state analizzate le fre-quenze di CTLp in 15/40 pazienti il cui follow up varia dai18 ai 30 mesi. Risultati. I risultati di questo studio dimo-strano come nel sangue periferico di questi pazienti, già
durante la fase di induzione e consolidamento sono docu-mentabili frequenze apprezzabili di CTLp (range: 1/5,000-1/41,000). L’analisi completa dei 15 pazienti ha, inoltre,dimostrato come 11/15 hanno mantenuto alte frequenzedi CTLp per tutto il periodo di osservazione e rimangonoin remissione di malattia. Dei 4 pazienti in cui sono statedocumentate frequenze inferiori a 1/100,000 durante ilperiodo di mantenimento 2 sono ricaduti prima dello stopterapia, mentre i rimanenti 2 sono al momento in remis-sione di malattia. Conclusioni. I risultati preliminari di que-sto studio dimostrano (i) che un’attività anti-LLA media-ta da linfociti T e diretta verso le cellule neoplastiche auto-loghe può svilupparsi già durante le prime settimana ditrattamento (ii) rimane da dimostrare il ruolo di questecellule nel controllo della malattia residua minima.
P017PROTOCOLLO AIEOP-BFM LLA2000.RISULTATI PRELIMINARI DELLA STRAT-IFICAZIONE, MEDIANTE VALUTAZIONE DELLA MALATTIA RESIDUA MINIMA(MRM), DEI PAZIENTI ARRUOLATI NELLO STUDIO AIEOP-BFM 2000Cazzaniga G,° Corral L,° Spinelli M,* Silvestri D,°D’ aniello E,° Del Giudice L,* Songia S,° Germano G,*Manenti M,° Giarin E,* Villa T,° André V,° Musarò A,°Valsecchi MG,° Basso G,* Masera G,^ Biondi A°°Centro Ricerca M. Tettamanti, Clinica PediatricaUniversità Milano-Bicocca, Ospedale S. Gerardo, Monza,Italia*Clin. di Oncoemat. Ped. , Dip. di Pediatria, Univ. diPadova, Italia;^ Clinica Pediatrica Università Milano-Bicocca, Ospedale S. Gerardo, Monza, Italia
Introduzione e obiettivi: L’ analisi molecolare dellaMalattia Residua Minima (MRM) mediante monitoraggiodel riarrangiamento somatico dei geni per i T-cell recep-tor (TCR) ed immunoglobuline (Ig) è il principale fattoreprognostico utilizzato per la stratificazione dei pazienticon leucemia linfoblastica acuta (LLA) nel protocolloAIEOP-BFM LLA2000, il primo protocollo clinico della LLAin età pediatrica comune a Italia, Germania, Austria e Sviz-zera ed attivato in Italia dal Settembre 2000.Risultati: Incirca 3 anni sono stati arruolati nei quattro paesi 2564bambini (1053 italiani); Nel 97% dei casi è stato possibi-le eseguire l’ analisi di MRM. Complessivamente la strati-ficazione secondo MRM è stata possibile nel 73. 7% deipazienti; L’ analisi di MRM ha consentito di identificare laseguente distribuzione di pazienti: 41% SR, 50% IR, 9%HR. Considerando altre caratteristiche cliniche che defini-scono l’ alto rischio, la stratificazione finale complessivaè risultata la seguente: 29% SR, 55% IR, 16% HR; Le prin-cipali cause di insuccesso nella stratificazione secondoMRM sono state: mancanza di materiale di follow up,assenza di marcatori, assenza di marcatori sensibili, modi-fiche di trattamento. Tali aspetti verranno presentati ediscussi. Conclusioni: Lo studio ha dimostrato la fattibilitàdella valutazione della MRM per la stratificazione deipazienti affetti da LLA, in uno studio prospettico, multi-centrico ed internazionale. La distribuzione ottenuta èsimile a quella identificata nello studio retrospettivo euro-peo (Lancet, 1998) che ha costituito il razionale dell’ appli-cazione clinica in corso. L’ analisi della sopravvivenza alungo termine indicherà se questa stratificazione avrà con-

111
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
sentito di migliorare la prognosi dei bambini con LLA. P018DISORDINI LINFOPROLIFERATIVI DOPO TRAPIANTO EPATICO IN ETA'PEDIATRICA: ESPERIENZA DI UN SINGOLO CENTROGiraldi E, Provenzi M, Cornelli P, Stroppa P, Cavalleri L,Altobelli M, Riva S, Colledan M,** Spada M,* Gridelli B,*Rambaldi A,° Torre GU. O. Pediatria, ** U. O. Chirurgia III, °U. O. EmatologiaOspedali Riuniti di Bergamo, *ISMETT UPMC Palermo
I disordini linfoproliferativi post-trapianto (PTLD) costi-tuiscono un gruppo eterogeneo di malattie, che va dalleiperplasie linfocitarie benigne a quadri di franco linfoma,insorgenti in corso di terapia immunosoppressiva (IS) pertrapianto d’organo. L’incidenza delle PTLD nell’ambito deltrapianto epatico (OLTx) pediatrico è compresa tra 4 e 20%ed è tuttora gravata da un’elevata mortalità (10-20%). Lamaggior parte delle PTLD, soprattutto se precoci, è correla-ta con l’infezione da EBV. Nel Centro Trapianti di Fegato diBergamo dal 1987 a giugno 2004 su 267 OLTx sono statediagnosticate 38 PTLD (14%) suddivise, secondo la classif-ficazione WHO, in early lesions (N°31), PTLD polimorfichepoliclonali (1) e monomorfiche monoclonali (LNH) (6). L’etàmediana dei pz era di 47 mesi (range 9-134) ed il tempomediano intercorso tra il trapianto e la PTLD pari a 27 e 24mesi rispettivamente per le forme poli- e monoclonali. 12pz assumevano Ciclosporina-A e 26 Tacrolimus. La correla-zione con EBV è stata dimostrata nel 76% dei casi. Nelle for-me policlonali il trattamento di scelta è consistito nella ridu-zione o sospensione temporanea della IS, eventualmenteassociata ad exeresi chirurgica adeno-tonsillare e/o linfo-nodale, con completo controllo della malattia ma successi-vo rigetto in 8 pz. Per la PTLD polimorfica policlonale, reci-divata dopo 1 anno dalla remissione (RC),si è reso necessa-rio, oltre alla sospensione della IS, l’impiego dell'Ab mono-clonale anti-CD20 (Rituximab). A seguito di rigetto incon-trollabile è stato eseguito ritrapianto epatico con successi-vo exitus. Nei 6 LNH-B la IS è stata sospesa;in 2 casi è sta-ta associata exeresi chirurgica addominale + Rituximab edin 3 pz chemioterapia + Rituximab. 5 pz hanno raggiuntola RC ma sviluppato rigetto. Sono vivi ed in RC tutti i pz conearly lesions e 5 su 6 LNH con follow up mediano di 35 (2-71) e di 46 mesi (14-79), rispettivamente. Nel 37% si è veri-ficato, ad una distanza mediana di 10 mesi (0.5-26), riget-to epatico, incontrollabile solo in 2 casi. Dalla nostra espe-rienza viene confermato che la riduzione della IS, eventual-mente associata a chirurgia locale,rappresenta il gold stan-dard del trattamento delle PTLD policlonali. Nei LNH la RCdi malattia è stata ottenuta e mantenuta grazie alla sospen-sione della IS associata achemioterapia + Rituximab.
P019STUDIO CITOFLUORIMETRICO DELLA MALATTIA RESIDUA MINIMA (MRM)NELLE LEUCEMIE LINFOBLASTICHE ACUTE (LLA): STANDARDIZZAZIONEMULTICENTRICAMaglia O,^ Leoni V,^ Veltroni M,° Spinelli M,° Bugarin C,^Michelotto B,^ Buldini B,° Basso G,° Biondi A,^ Gaipa G^^Centro Ricerca “Tettamanti”, Clinica Pediatrica,Università di Milano/Bicocca, Ospedale S. Gerardo,Monza (MI); °Laboratorio di Oncoematologia Pediatrica,Dipartimento di Pediatria, Università Di Padova

112
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Leucemie e Linfomi
Introduzione: La valutazione della MRM nelle leucemielinfoblastiche acute (LLA) pediatriche costituisce un fat-tore prognostico indipendente. Nell’ambito del protocol-lo AIEOP-BFM LLA 2000 il dato di MRM ottenuto in cito-fluorimetria (CFM) è oggetto di uno studio collaborativo(4 centri europei) iniziato nell’anno 2000.Obiettivi: Nel-l’ambito di una possibile applicazione clinica della CFM sisono valutati: (1) la concordanza nel reclutamento e nel-l’incidenza di pazienti MRM positivi e (2) la riproducibi-lità interlaboratorio del dato CFM. Metodi 1-Dal Giu-gno‘03 al Dicembre ‘03 sono stati arruolati 413 pazienti(88, 61, 110, 154 rispettivamente a Monza, Padova, Ber-lino, Vienna) e 617 dai centri AIEOP e centralizzati a Pado-va. I time-points MRM sono stati: aspirato midollare (BM)giorno+15, +33, +78. 2- L’analisi di 93 files relativi a cam-pioni di BM (+15, +33, +78) e PB (diagnosi, +8, +15, +33)di 15 pazienti (13 B-LLA, 2 T-LLA) è stata confrontata trai 4 laboratori in cieco. Risultati 1-Il reclutamento medioè stato 92,5%, 91,25%,88% al giorno +15, 33,78 rispet-tivamente. Il reclutamento dei singoli centri è risultatoaltamente paragonabile. Il valore medio di MRM (% bla-sti/cellule nucleate) è stato 0,58 (range 0.28-1.1) alBM+15; 0,044 (range 0.025-0.063) al +33; 0.17 (range0.02-0.2) al +78. La percentuale di pazienti MRM positi-vi è risultata del 86,75% al BM +15, 33,75% al +33 e7,25% al +78. 2- 66/93 files (71%) hanno mostrato unaconcordanza qualitativa completa tra i 4 centri, mentre laconcordanza di almeno 3 centri è stata del 95,7%. La con-cordanza quantitativa è stata del 84,8%. La minor con-cordanza è stata riscontrata ai livelli più bassi di MRM (tra0,01 e 0,1%) e nei time-points BM+33, PB+15. Conclu-sioni: L’alto grado di concordanza ottenuto ha dimostra-to la riproducibiltà del protocollo di acquisizione e anali-si dei campioni. L’elevato indice di reclutamento neipazienti ha evidenziato la fattibilità dell’indagine di MRMmediante CFM almeno quando eseguita a livello di singo-le istituzioni.
P020RADIOTERAPIA CRANIALE (RTC) IN PAZIENTI AFFETTI DA LEUCEMIALINFOBLASTICA ACUTA (LLA) T CON IPERLEUCOCITOSI E PREDNISONEGOOD RESPONSE (PGR) ARRUOLATI NELLA FASCIA A RISCHIOINTERMEDIO (RI)Conter V, Arico’ M, Barisone E, Basso G, Biondi A,Casale F, D’incalci M, De Rossi G, Dini G, Favre C,Ladogana S, Lo Nigro L, Locatelli F, Messina C,Micalizzi C, Pession A, Pinta MF, Rizzari C, Rondelli R,Santoro N, Testi AM, Valsecchi MG, Zecca M, Masera Gper l’AIEOP
Obiettivo: Il confronto tra i risultati ottenuti nei duecontemporanei protocolli AIEOP-ALL 91 ed ALL-BFM 90aveva dimostrato che nei pazienti trattati nella fascia a RIla somministrazione protratta di una triplice terapia intra-tecale (TIT, AIEOP) aveva determinato risultati sovrappo-nibili a quelli ottenuti con l’uso di RTC e 3 dosi di MTX IT(BFM) nei pazienti con PGR T-LLA e <100.000 WBC/mm3
ma peggiori in quelli con PGR T-LLA e 100.000 WBC/mm3
(Conter V et al. , J Clin Oncol 15:2786-2791;1997). Sullabase di questa esperienza nel protocollo AIEOP LLA 95 la
RTC è stata somministrata nei pazienti con PGR T-LLA ed100.000 WBC/mm3 alla diagnosi. Vengono qui riportati irisultati ottenuti in questi pazienti. Pazienti e metodi: Nelprotocollo AIEOP LLA 95 sono stati reclutati complessiva-mente, nel periodo Aprile 1995-Agosto 2000, 1746pazienti dei quali 1338 classificati a rischio intermedio; traquesti ultimi 121 erano T-LLA di cui 77 con < 100.000WBC/mm3 e 44 con 100.000 WBC/mm3. Il trattamentochemioterapico era caratterizzato da un back-bone BFM:Induzione: Protocollo IA e IB; Consolidamento HD-MTX a5g/m2x4; Reinduzione: Protocollo II; Mantenimento: 6-MP e MTX con o senza pulses di VCR e Desametazonesomministrate nell’ambito di uno studio randomizzato. Iltrattamento profilattico sul SNC consisteva di una doseall’esordio di IT-MTX e da 16 successive dosi di TIT inpazienti con < 100.000 WBC/mm3 alla diagnosi e di unadose iniziale di IT-MTX, 10 successive dosi di TIT + RTC(18Gys) in pazienti con 100.000 WBC/mm3. I risultati sonostati aggiornati a Dicembre 2003. Risultati: Dei 77 pazien-ti con PGR T-LLA e < 100.000 WBC/mm3 alla diagnosi 4sono morti in remissione completa (CR), 13 sono recidi-vati e 60 rimangono in RC continua; il loro EFS (SE) a 5anni è del 79. 0% (4. 9). Dei 44 pazienti con PGR T-LLA e100,000 WBC/mm3 alla diagnosi 1 è morto in CR, 14 sonorecidivati e 29 rimangono in RC continua; il loro EFS (SE)a 5 aa è del 65. 9% (7. 2). Conclusioni: Questi dati con-fermano che, come riportato nello studio comparativo trai protocolli AIEOP-LLA 91 ed ALL-BFM 90, la RTC non ènecessaria per i pazienti PGR T-LLA con < 100.000WBC/mm3 alla diagnosi. Viene confermato inoltre chel’aggiunta della RTC ad uno schema convenzionale di tipoBFM contribuisce a migliorare i risultati nei pazienti conPGR T-LLA e 100.000 WBC/mm3 alla diagnosi.
P021VALUTAZIONE FARMACOLOGICA DELLA PEG L-ASPARAGINASI (PEG ASP)IN BAMBINI AFFETTI DA LEUCEMIA LINFOBLASTICA ACUTA (LLA) TRATTATICON IL PROTOCOLLO AIEOP LLA 2000Rizzari C,1 Citterio M,1 Zucchetti M,2 Conter V,1Chiesa R,1 Colombini A,1 Malguzzi S,1 Masera G,1D'Incalci M2
1Clinica Pediatrica, Università di Milano-Bicocca,Ospedale S. Gerardo, Monza e 2Istituto M. Negri perle Ricerche Farmacologiche, Milano
Introduzione: La PEG ASP è stata prevalentemente usa-ta ed estensivamente studiata in soggetti affetti da pato-logie emato-oncologiche con reazione allergica al pro-dotto nativo. Dati assai limitati sono invece disponibili inletteratura sulla farmacocinetica e farmacodinamica del-l’uso front-line della PEG ASP in bambini con prima dia-gnosi di LLA. Obiettivi dello studio: Valutare l’attività enzi-matica plasmatica della PEG ASP ed i livelli di asparagina(ASN) nel plasma e nel liquor di bambini con LLA trattaticon la PEG ASP nell’ambito del protocollo AIEOP LLA2000.Pazienti e metodi: Venti bambini con LLA consecu-tivamente diagnosticati presso il centro di Monza hannoricevuto durante la fase di induzione (prima esposizione)due somministrazioni (gg +12 e gg +27) di PEG L-ASP(1.000 UI/m2 e. v. ) al posto di 8 dosi del prodotto nativo

113
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
(5. 000 UI/m2 i. m. ogni 3 gg) e, durante le fasi di reindu-zione (seconda esposizione), una sola somministrazione diPEG ASP (1.000 UI/mq e. v. ) al posto di quattro dosi delprodotto nativo (10.000 UI/m2 i. m. ogni 3 gg). L’attivitàplasmatica della PEG ASP è stata misurata con metodicaspettrofotometrica mentre i livelli plasmatici e liquorali diASN sono stati dosati con HPLC. Risultati: In induzionetutti i pazienti hanno presentato un’attività plasmatica100IU/L per almeno 12 gg dopo ciascuna delle due dosi delfarmaco; durante la reinduzione 11/15 pazienti hanno pre-sentato livelli di attività plasmatica 100 IU/L per almeno15 gg. Una completa deplezione plasmatica (<0.2 M/L) diASN è stata osservata in tutti i pazienti che durante laprima e la seconda esposizione esposizione avevano man-tenuto adeguati livelli di attività plasmatica di ASP. I livel-li liquorali di ASN sono risultati in tutti i casi, tranne uno,al di sopra del limite di misurazione della metodica usata.Nessuna reazione allergica è stata osservata nei soggettistudiati. Conclusioni: L’uso front-line della PEG ASP inbambini con LLA al posto di ripetute dosi convenzionali delprodotto nativo permette un’adeguata attività plasmati-ca enzimatica ed una completa deplezione plasmatica diASN sia durante la prima che la seconda esposizione al far-maco. L’insoddisfacente dato di mancata deplezione liquo-rale di ASN conferma quanto già riportato in letteratura.
P022ALTERAZIONI A LUNGO TERMINE DEL METABOLISMO DEI CARBOIDRATI INSOGGETTI TRATTATI PER LEUCEMIA LINFOBLASTICA ACUTADi Marzio A,° Capanna R,* Di Marzio D,*De Berardinis A,* Mohn A,* Fioritoni G,° Chiarelli F*°Divisione di Ematologia, Università Chieti-Pescara*Clinica Pediatrica, Università Chieti-Pescara
Introduzione: il nostro gruppo ha dimostrato che la tera-pia chemioterapica nei soggetti con leucemia linfoblasti-ca acuta (LLA) può indurre un’alterazione della funziona-lità delle beta-cellule e del metabolismo dei carboidratidurante il trattamento; questa alterazione può persistereanche a distanza di 2 anni dalla interruzione della terapia(Lancet 2004;10:127-8). Obiettivi: scopo di questo studioè stato quello di valutare se le alterazioni descritte peg-giorano in questi bambini a distanza di 4 anni dalla inter-ruzione della terapia. Materiali e metodi: dopo due anni dalprecedente studio tutti i pazienti arruolati sono stati nuo-vamente contattati per effettuare una rivalutazione delmetabolismo dei carboidrati mediante un carico orale diglucosio (OGTT), condotto dopo un digiuno di almeno 8ore. Sono stati dosati glicemia ed insulinemia ai tempi 0,+30’, + 60’, +90’ e + 120’; è stata inoltre valutata l’emo-globina glicosilata (HbA1c). Dei 32 pazienti precedente-mente arruolati, 2 (1M, 1F) non hanno partecipato allostudio perché sottoposti a trapianto di midollo osseo (BMT)per recidiva di malattia e 2 (1M, 1F) hanno rifiutano diripetere la valutazione. Sono stati rivalutati pertanto 28ragazzi (16 M, 12 F). Risultati: dei 28 soggetti esaminati,13 hanno presentato una ridotta tolleranza ai carboidra-ti (glicemia al tempo +120’ > 140 mg/dL), 1 dei quali hapresentato un diabete secondo i criteri dell’ADA (glicemiaal tempo + 120’ > 200 mg/dL). I dati evidenziano che,rispetto al controllo precedente, la percentuale di sogget-
ti con ridotta tolleranza ai carboidrati è aumentata dal34,3% (11 soggetti su 32) al 46,4% (13 soggetti su 28);non sono stati evidenziati invece nuovi casi di diabete cli-nicamente manifesto. Nessuna differenza significativa èemersa inoltre in termini di HbA1c. Conclusioni: i risulta-ti del nostro studio evidenziano un peggioramento del-l’intolleranza ai carboidrati nel tempo non associato allosviluppo di un diabete manifesto. Sebbene i dati del nostrostudio necessitino di conferme mediante ulteriori studi difollow-up, la persistenza dell’alterazione del metabolismodei carboidrati sembra essere una importante sequela alungo termine nei bambini trattati per LLA, per la quale èindispensabile una valutazione periodica della funziona-lità pancreatica e del metabolismo dei carboidrati.
P023ETEROGENEITÀ DELLA STRATIFICAZIONE SECONDO MALATTIA RESIDUAMINIMA (MRM) NEI PAZIENTI CON LEUCEMIA LINFOBLASTICA ACUTA(LLA) E TRASLOCAZIONE T(12;21)Cazzaniga G,° Palmi C,° Frascella E,* Manenti M,° Spinel-li M,* Villa T, ° Musarò A,° Fazio G,° Masera G,^ Basso G,*Biondi A°°Centro Ricerca M. Tettamanti, Clinica PediatricaUniversità Milano-Bicocca, Ospedale S. Gerardo, Monza,Italia ^ Clinica Pediatrica Università Milano-Bicocca,Ospedale S. Gerardo, Monza, Italia *Clin. di Oncoemat.Ped. , Dip. di Pediatria, Univ. di Padova, Italia
Introduzione e obiettivi: La traslocazione t(12;21) è lapiù comune anomalia cromosomica strutturale dei pazien-ti pediatrici con LLA (circa 25% dei casi). Nel precedenteprotocollo AIEOP LAL95, l’ analisi di EFS a 5 e 8 anni con-ferma la t(12;21) come fattore prognostico favorevole.Tuttavia, sono stati osservati casi positivi e ricaduti pre-coci e tardivi. Più precisamente, in una serie prospettica di184 pazienti ricaduti, la t(12;21) è stata riscontrata nel15% dei casi (23. 5% dei casi con ricaduta tardiva). Abbia-mo analizzato l’ eterogeneità dei pazienti con t(12;21)valutandone l’ analisi molecolare di malattia minima resi-dua (MRM) mediante monitoraggio del riarrangiamentodei geni dei T-cell receptor; (TcR) e delle immunoglobuli-ne (Ig) nel protocollo AIEOP-BFM ALL2000.Metodi: la RT-PCR della fusione TEL/AML1 e il riarrangiamento di TcR eIg sono stati eseguiti in 847 pazienti eleggibili e valutabi-li al protocollo. Risultati: Nel protocollo corrente, 52.2%dei positivi TEL/AML1 sono stati stratificati nel gruppostandard (SR) per MRM e 45. 2% nel gruppo a rischiointermedio (IR) per MRM. Tre casi (2.6%) sono stati arruo-lati al rischio alto (HR) per MRM. 31% degli SR, 21% degliIR e 9% degli HR per MRM erano rispettivamente t(12;21)positivi. Considerando la stratificazione finale (basata suMRM e caratteristiche cliniche), 32% degli SR, 24% degliIR e 8% degli HR erano t(12;21) positivi. Tra i casi IR perMRM, senza caratteristiche cliniche da HR, 51 casi eranoTEL/AML1 positivi, con livelli variabili di MRM ai punti +33e +78 del protocollo. Conclusione: E’ stata riscontrata unanotevole eterogeneità di risposta alla terapia iniziale deipazienti con t(12;21) arruolati al protocollo AIEOP-BFMALL2000.In particolare, un sottogruppo di casi t(12;21)possono essere HR. Di conseguenza, è necessaria cautela

114
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Leucemie e Linfomi
nell’ interpretare la t(12;21) come fattore prognostico indi-pendente, sul quale basare eventuali proposte di riduzio-ne della terapia, almeno in protocolli BFM.
P024ESPRESSIONE DI C-MET NELLE B-LLA: POSSIBILE COINVOLGIMENTONELL'APOPTOSI DEI BLASTI LEUCEMICIAccordi B,* Campo Dell'orto M,* Franceschini L,*Pistollato F,* Pillozzi S,° Cazzaniga G,^ Arcangeli A,°Te Kronnie G,* Basso G**Dipartimento di Pediatria, Clinica di OncoematologiaPediatrica, Università di Padova; °Dipartimento diPatologia e Oncologia Sperimentali, Università di Firenze;^Clinica Pediatrica, Università di Milano Bicocca, CentroRicerca Tettamanti
Introduzione: L’oncogene c-Met è un recettore tirosin-chinasico che lega il fattore di crescita HGF (hepatocytegrowth factor). c-Met attivato aumenta la motilità e laproliferazione cellulare, protegge dall’apoptosi e stimolal’angiogenesi. Alterazioni di questo recettore si osservanoin numerosi tumori di origine epiteliale. E’ stata però avan-zata l’ipotesi che c-Met possa anche stimolare l’apoptosisecondo meccanismi ancora sconosciuti. Recentemente èstata infatti descritta un’associazione tra c-Met e la pro-teina pro-apoptotica Fas in alcuni tipi cellulari, ma non èstato dimostrato il ruolo di c-Met in questo complesso.Materiali: In questo studio è stata valutata mediante SYBRGreen Real Time Quantitative PCR (RQ-PCR) l’espressionedi c-Met e Fas in 64 bambini affetti da B-LLA, di cui 22portanti la traslocazione t(12;21) (TEL-AML1). Come con-fronto sono state scelte cellule B normali isolate da buffycoat. Il modello in vitro consiste in due linee cellulari di B-LLA, la REH t(12;21)+ e l’MHH-CALL2 non traslocata, sul-le quali sono stati condotti esperimenti di RQ-PCR, immu-noprecipitazione e Western blot per c-Met e Fas. Risulta-ti: Dagli esperimenti di RQ-PCR c-Met è risultato esseresottoespresso nei blasti rispetto alle cellule B normali. Inparticolare la sua espressione è molto diversa nei due sot-togruppi t(12;21)+ e t(12;21)- (χ2 test, p<0,001): neipazienti t(12;21)- c-Met è espresso solo nel 35,7% deicasi, mentre nei pazienti t(12;21)+ è espresso nel 100% deicasi. Il livello medio di espressione è più alto e simile ailivelli normali nel gruppo t(12;21)+. Fas è risultato sem-pre leggermente sottoespresso nei blasti in modo non dif-ferente nei due gruppi. E’ stata osservata nelle REH tramiteRQ-PCR l’espressione sia di c-Met che di Fas, mentre nel-le MHH-CALL2 solo di Fas: le due linee risultano così simi-li ai corrispondenti gruppi di pazienti. Questo risultato èstato confermato anche dagli esperimenti di immunopre-cipitazione e Western blot condotti sulle linee. Conclusio-ni: L’osservazione che sorprendentemente l’oncogene c-Met venga espresso maggiormente nel gruppo di leucemiet(12;21)+ a prognosi migliore, favorisce l’ipotesi che c-Met possa cooperare nel processo apoptotico mediato daFas indotto dalla terapia farmacologica.
P025IDENTIFICAZIONE DELLE CLASSI DI RISCHIO CON CITOMETRIA A FLUSSO
IN PAZIENTI PEDIATRICI AFFETTI DA LEUCEMIA LINFOBLASTICA ACUTA ECONFRONTO CON LA STRATIFICAZIONE OTTENUTA MEDIANTE BIOLOGIAMOLECOLAREVeltroni M,* Gaipa M,° Leoni V,° Maglia O,° Buldini B,*Spinelli M,* Cazzaniga G,° Michielotto B,* Benetello A,*Biondi A,° Basso G** Laboratorio di Oncoematologia Pediatrica, Dip. Pediatria, Università di Padova; °Centro Ricerca"Tettamanti", Clinica Ped, Università di Milano/Bicocca,Ospedale S. Gerardo, Monza (MI)
Introduzione. Nell'attuale protocollo AIEOP LLA 2000 ladeterminazione della Malattia Residua Minima (MRM),ottenuta tramite indagine molecolare (PCR), rappresentauno dei criteri di stratificazione dei pazienti pediatriciaffetti da leucemia linfoblastica acuta (LLA). L'importan-za clinica della MRM nelle LLA è stata dimostrata anchein studi condotti mediante citofluorimetria (CFM). Obiet-tivi. Identificazione delle diverse classi di rischio median-te citometria a flusso e confronto con i risultati ottenutidalla stratificazione dei pazienti effettuata attraverso PCR.Metodi. Nell'ambito di un progetto cooperativo per lo stu-dio della MRM nelle LLA pediatriche sono stati seleziona-ti, da gennaio 2001 a ottobre 2003, 376 pazienti pedia-trici con LLA, di cui 112 provenienti da Monza e Padova,e 264 da altri centri e centralizzati nel laboratorio di Pado-va. I criteri di selezione includevano un follow-up MRMcompleto con CFM [aspirato midollare (BM) +15, +33,+78], presenza di almeno due marcatori immunofenotipi-ci specifici, e contemporanea analisi mediante PCR. Ipazienti sono stati stratificati mediante CFM (Dworzak etal. , Leuk Lymphoma 2003): standard risk (SR): MRM < 10blasti/mmc al BM+l5 e negativa al +33 e +78; Alto Rischio(HR): MRM 1 blasto/mm3 al BM+78; Rischio Intermedio(MR): tutti i casi non inquadrabili nelle precedenti classidi rischio. Risultati. La stratificazione dei pazienti ottenu-ta mediante CFM è stata la seguente: SR 37,76%, MR53,98%, HR 8,24%. Il confronto della stratificazione inCFM vs quanto ottenuto in PCR ha permesso di evidenziareche gli SR-CFM si distribuiscono per il 40,84% in SR,28,16% MR, 0% HR; i MR-CFM 22,66% SR, 47,29% MR,4,43% HR; infine, gli HR-CFM 16,12% SR, 19,35% MR,45,16% HR. Inoltre, globalmente, il 27,12% dei pazientiesaminati non risulta valutabile mediante PCR e si strati-fica con CFM per il 43% in SR, 51% MR, 6% HR. Conclu-sioni. L'osservazione prospettica permetterà di valutarel'impatto clinico di una stratificazione con CFM. Inoltre, laCFM consente di classificare una parte considerevole dipazienti in cui non è attuabile una stratificazione median-te PCR.

115
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
P026IMPLICAZIONI DIAGNOSTICHE E PROGNOSTICHE DEL GENE NPM-ALK NELLINFOMA ANAPLASTICO A GRANDI CELLULEMussolin L,° Pillon M,° Garaventa A,^ Lombardi A,"Conter V,# Tettoni K,°° D'amore E,^^ Zanesco L,°Rosolen A°°Clinica di Onco-ematologia Pediatrica AziendaOspedaliera, Università di Padova; ^ Istituto G. GasliniGenova; " Ospedale Bambino Gesù Roma;# Ospedale S.Gerardo xUniversità Bicocca Monza Milano; °°Clinicadi Onco-ematologia pediatrica Ospedale di Brescia; ^^Divisione di Anatomia Patologica Ospedale S. BortoloVicenza
I linfomi anaplastici a grandi cellule (ALCL) rappresen-tano circa il 15% dei linfomi non Hodgkin pediatrici. L’a-berrazione cromosomica più frequente è la t(2;5)(p23;q35)presente in oltre il 70% degli ALCL. Questa traslocazionecoinvolge il gene NPM sul cromosoma 5, che codifica perla proteina ubiquitaria nucleofosmina e il gene ALK sulcromosoma 2, che codifica per un recettore tirosin-china-sico. La traslocazione porta alla formazione sul cromoso-ma derivativo 5 di un gene di fusione NPM-ALK il cui tra-scritto è identificabile mediante RT-PCR. Gli obiettivi del-la nostra ricerca sono: 1)ricerca del trascritto NPM-ALKnegli ALCL diagnosticati e trattati all’interno del proto-collo nazionale AIEOP-LNH97 ed europeo ALCL-99;2)valutazione prospettica dell’interessamento midollarealla diagnosi nell’ALCL. Abbiamo analizzato 40 biopsie diALCL pediatrici e l’87,5% (35/40) sono risultate positiveper NPM-ALK in RT-PCR. Lo studio di malattia minima dis-seminata (MMD) è stato condotto sull’aspirato midollareall’esordio di 30 pazienti, 18 (60%) sono risultati positivi.Solo 2/30 erano infiltrati morfologicamente. I risultati del-l’analisi univariata sulla sopravvivenza libera da eventimostrano, in una proiezione a 5 anni, una percentuale disopravvivenza del 42% per i pazienti con BM positivo inRT-PCR e del 100% per i pazienti negativi. Analogamen-te, la sopravvivenza libera da malattia è del 46% per ipazienti con BM positivo in RT-PCR all’esordio e del 100%per i pazienti negativi. Dal nostro studio emerge che l’in-filtrazione minima midollare è sensibilmente più frequen-te di quanto riportato in letteratura. Inoltre i risultati otte-nuti dall’analisi univariata evidenziano un probabile ruo-lo prognostico negativo della MMD nell’ALCL.
P027TRAPIANTO AUTOLOGO DI CELLULE STAMINALI IN PAZIENTI AFFETTIDA LINFOMA DI HODGKIN IN ETÀ PEDIATRICAAmendola A, Moleti ML, Capria S, Ribersani M,Barberi W, Cardarelli L, Palumbo G, Malandruccolo L,Russo C, Giona F, Testi AM, Foà R, Meloni GUniversità La Sapienza Roma; Dipartimento diBiotecnologie Cellulari ed EmatologiaSezione Ematologia
Il linfoma di Hodgkin (LH) è una patologia rara in etàpediatrica. La chemioterapia convenzionale da sola o inassociazione a basse dosi di radioterapia ha permesso di
ottenere lunghe sopravvivenze libere da malattia in oltrel’85% dei casi. Ciononostante, per i pazienti che nonottengono una prima e persistente remissione completa,sono necessarie strategie terapeutiche alternative. Le altedosi di chemioterapia seguite da autotrapianto di cellulestaminali emopoietiche, come terapia di salvataggio, sisono dimostrate efficaci nel trattamento dei LH refratta-ri o in recidiva in ampie casistiche su pazienti adulti. Leesperienze in pazienti con diagnosi di LH in età pediatri-ca sono limitate. Riportiamo la nostra esperienza relativaall’impiego di una terapia mieloablativa e trapianto auto-logo in 34 pazienti di età <18 anni alla diagnosi, con LHrefrattario o in recidiva seguiti presso la cattedra di Ema-tologia dell’Università La Sapienza di Roma dall’Ottobre1979 al Maggio 2002.Caratteristiche alla diagnosi: maschi21 e femmine 13; età mediana 12,5 anni (range 8,1-17,9);stadio II in 12, III 13, IV 9; sintomi B 30; diagnosi istolo-gica LH scleronodulare 20, LH a cellularità mista 6, LH apredominanza linfocitaria 1, non specificata 7; coinvolgi-mento mediastinico 31, extranodale 11; malattia bulky11.La terapia di prima linea consisteva negli schemi ABVDin 15, MOPP/ABVD in 12, MOPP in 3, COPP/ABVD in 2,OPPA/COP/COPP in 1, altro in 1.La radioterapia è stataeffettuata in 27 pazienti. Venti pazienti avevano ottenu-to una remissione completa (RC), 9 pazienti una remissio-ne parziale (RP), 5 una progressione di malattia. Almomento del trapianto l’ età mediana era 17,3 anni (ran-ge 11,5-33,5). Sedici pazienti erano in II RC, 11 pazientiin RP, 1 NR, 2 in progressione di malattia e 4 in recidiva L’intervallo tra diagnosi e trapianto è stato di 32 mesi (ran-ge 9-277). Il regime di condizionamento è stato lo sche-ma BEAM in 16 pazienti, CVB in 17, Vancouver in 1.La sor-gente di cellule staminali è stata in 17 pazienti sanguevenoso periferico e in 17 sangue midollare. Allo statoattuale 22/34 pazienti (65%) sono vivi (20 RC, 2 RP) conun follow-up mediano di 47,5 mesi (18-216),1 paziente èin progressione di malattia, 3 pazienti sono persi al follow-up, 8 pazienti sono deceduti. I nostri risultati confermanoil ruolo del trapianto di cellule staminali autologhe nellaterapia di salvataggio del LH in età pediatrica.
P028ANALISI COMPARATIVA DELLE REAZIONI ALLERGICHE ALLAL-ASPARAGINASI (ASP) NATIVA OSSERVATE IN BAMBINI AFFETTI DALEUCEMIA LINFOBLASTICA ACUTA TRATTATI NEI PROTOCOLLI AIEOPE BFM ALL 2000Rizzari C, Moericke A,* Silvestri D, Conter V,Zimmermann M,* Citterio M, Colombini A, Chiesa R,Coliva T, Viganò E, Valsecchi MG, Masera G,Schrappe M*Per i gruppi BFM-ALL* ed AIEOP-LLA
Background: La ASP è un un polipeptide di origine bat-terica molto efficace nel trattamento della LLA. Uno deimaggiori limiti nel suo uso è rappresentato dalle reazioniallergiche. Tali fenomeni, che sono osservati fino al 50%dei pazienti trattati, rappresentano, in alcuni casi, un pro-blema rilevante potendo determinare uno scadimento del-l’attività del farmaco ed in qualche modo una compro-

116
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Leucemie e Linfomi
missione della sua efficacia terapeutica. Obiettivo: Valu-tare 1.l’incidenza delle reazioni allergiche dopo la som-ministrazione intramuscolare (i. m. , AIEOP) o endoveno-sa (e. v. , BFM) di ASP da E. Coli in uno schema terapeuti-co identico per la restante parte chemioterapica e 2.l’im-patto di tale fenomeno sul completamento del tratta-mento con ASP previsto dal protocollo. Pazienti e meto-di: Sono stati valutati per questo studio tutti i bambiniarruolati nei protocollo AIEOP nel periodo 1.9. 2000-31.10.2003 mentre per il protocollo BFM sono stati con-siderati i bambini per i quali era disponibile sufficientedocumentazione. Si è trattato di un’analisi retrospettivabasata sull’uso di schede ad hoc per l’AIEOP e sui dati ditossicità disponibili nella banca dati del protocollo per ilBFM. Risultati: Dei 997 vs 675 bambini valutati rispetti-vamente nei protocolli AIEOP e BFM, 68(7%, 11 sistemi-che gravi) vs 308 (46%, tutte sistemiche a gravità varia-bile) (p<0,001) hanno sviluppato almeno una rezioneallergica. Attraverso un’analisi ad hoc non sono state iden-tificate variabili clinico-biologiche capaci di predire ilrischio di sviluppare una reazione allergica ad eccezionedell’appartenenza alla fascia di rischio HR (il che è presu-mibilmente dovuto al più elevato numero di esposizioni alfarmaco previste in questa fascia di rischio). Oltre il 90%delle reazioni allergiche sono avvenute alla prima sommi-nistrazione del farmaco prevista nella seconda esposizio-ne (protocollo II o III). Complessivamente, grazie alla sosti-tuzione del prodotto nativo con altri prodotti di ASP il92% (97% in SR/MR, 65% in HR) dei pazienti ha potutocompletare il previsto schema di trattamento con ASP.Conclusioni: In questa valutazione comparativa la via disomministrazione endovenosa è risultata associata conun’incidenza di reazioni allergiche significativamentesuperiore a quella osservata con la via intramuscolare.
P029STUDIO DI CLONALITÀ IN PAZIENTI CON LEUCEMIA LINFOBLASTICA ACUTA(LLA) E TRASLOCAZIONE T(12;21)Songia S,° Palmi C,° Fazio G,° Panzer-Grumayer R,*Biondi A,° Cazzaniga G°°Centro Ricerca M. Tettamanti, Clinica Pediatrica Univer-sità Milano-Bicocca, Ospedale S. Gerardo, Monza, Italia;*Children’s Cancer Research Institute St. AnnaKinderspital, Vienna, Austria
Introduzione e obiettivi: La traslocazione t(12;21) è lapiù comune anomalia cromosomica strutturale dei pazien-ti pediatrici con LLA. La sua origine prenatale, la latenzaa volte elevata e la delezione secondaria dell’ allele nonriarrangiato del gene TEL suggeriscono che la fusioneTEL/AML1 rappresenti un evento iniziatore del processotumorigenico. Nonostante la t(12;21) sia associata ad unaprognosi favorevole, si riscontrano casi positivi che rica-dono, anche se tali casi rispondono di nuovo rapidamen-te al trattamento. Scopo del lavoro è determinare l’ effet-tiva rilevanza clinica dell’ ipotesi che un clone minorita-rio positivo per la traslocazione t(12;21), già presente alladiagnosi, possa accumulare ulteriori mutazioni che lo ren-dono leucemico, prevalente e responsabile della ricaduta.
Metodi: RT-PCR e RQ-PCR del trascritto di fusioneTEL/AML1; analisi molecolare del riarrangiamento soma-tico dei geni dei T-cell receptor (TcR) e delle Immunoglo-buline (Ig) nei casi arruolati al protocollo AIEOP-BFMALL2000 in Germania, Austria ed Italia. Risultati: Sonostati analizzati complessivamente 30 pazienti t(12;21)positivi ricaduti. In 7 dei casi italiani analizzati è stataosservata una differenza nel tipo di riarrangiamenti tradiagnosi e ricaduta. In particolare in 6 dei 7 casi è statoindividuato almeno un clone presente solo nella fase diricaduta e non evidente al’ esordio. L’ analisi di PCR quan-titativa incrociata di tutti i cloni di esordio e ricaduta ètuttora in corso per valutare l’ evoluzione della malattiaa livello clonale. Conclusioni: Tale studio consentirebbe diindagare con ulteriore dettaglio la naturale evoluzione delclone t(12;21) e di avere una ricaduta applicativa nellastrategia di monitoraggio della malattia residua minima(MRM) nei casi LLA t(12;21) positivi.
P030GLI EFFETTI DEL DESAMETAZONE SUL COMPORTAMENTO DEI BAMBINIDURANTE LA FASE DI INDUZIONE DELLA TERAPIA PER LEUCEMIALINFOBLASTICA ACUTAPozzi L, Nichelli F, Barisone E, Baronci C, Caprino D,Consarino C, Favara C, Giuliano M, Lautizi L, Lippi A,Mura R, Varotto S, Jankovic MCSD Psicosociale dell'AIEOP
Introduzione: Gli steroidi trovano un ampio ed efficaceimpiego nel trattamento della LLA dell’infanzia. Il farma-co steroide più utilizzato nell’Induzione è sempre stato ilPrednisone (PDN), anche se diversi studi indicano unamaggiore efficacia del Desametazone (DXM) soprattuttoper quanto riguarda il passaggio della barriera ematoen-cefalica. L’uso del DXM sembra essere causa però di mag-giori effetti collaterali a livello comportamentale e neu-ropsicologico. L’obiettivo del presente studio è la valuta-zione della tossicità comportamentale in uno studio ran-domizzato (PDN vs DXM) nella terapia di Induzione delleLLA, allo scopo di 1) quantificare il fenomeno 2) adottareeventuali terapie di supporto (es. terapia ansiolitica, anti-depressiva), nel rispetto dell’efficacia della terapia in atto.Metodi: E’ stato condotto uno studio prospettico a misu-re ripetute, nel quale a 230 bambini affetti da LLA, di etàcompresa tra 1,5 e 18 anni, arruolati nel protocollo AIEOPLLA 2000 in 12 diverse Cliniche Pediatriche italiane, è sta-to somministrato il questionario CBCL (Child BehaviorCheckList) in 3 tempi: T1 (precedente al 1° random), T2(dopo 15 giorni del 1° random) e T3 (dopo 1 mese dal 1°random e, di conseguenza, 7-10 giorni prima della fase diconsolidamento). La somministrazione è stata curata daglipsicologi. Risultati: In nessun caso si sono ottenuti pun-teggi patologici nei soggetti trattati con PDN, mentre neisoggetti trattati con DXM si sono riscontrati punteggi for-temente al di sopra dei valori di norma in alcune scale, maesclusivamente al tempo T2.Per tutti i soggetti trattaticon DXM si sono registrati punteggi patologici nella sca-la della Internalizzazione e, in particolare, nell’area del-l’Introversione; inoltre si è potuto rilevare che i bambini

117
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
piccoli (1,5 – 5 anni) sono quelli che risentono maggior-mente del trattamento, evidenziando punteggi elevatianche nell’area della reattività emotiva e nel punteggiototale del questionario. Conclusioni: Possiamo conclude-re che, in uno studio quantitativamente numeroso, glieffetti di tossicità comportamentale si sono riscontratisolo nei bambini trattati con DXM, in modo più evidentein quelli più piccoli e comunque solo al tempo T2.
P031ANALISI COMPARATIVA DEI MARCATORI DI CLONALITÀ TRA ESORDIO ERICADUTA IN PAZIENTI PEDIATRICI LAL-TGermano G, Del Giudice L, Polato K, Giarin E, Basso GLaboratorio di Emato Oncologia, Dipartimento diPediatria, Università di Padova
Introduzione. L’utilizzo dei marcatori di clonalità nellemalattie linfoproliferative rappresenta un importante tooldiagnostico per il monitoraggio della malattia residuaminima (MRM). Attualmente il protocollo AIEOP-BFMLAL2000 per i pazienti con leucemia acuta linfoblastica(LAL) utilizza i riarrangiamenti somatici dei geni delleImmunoglobuline (Ig) e del recettore T per l’antigene (TCR)per la stratificazione dei pazienti. Un limite di questametodica è rappresentato dall’ ongoing dei riarrangia-menti Ig/TCR dovuto all’attività delle ricombinasi ancoraattive nelle cellule leucemiche. Questo potrebbe genera-re dei falsi negativi durante il monitoraggio della MRM.Obiettivi Abbiamo analizzato la stabilità dei principalimarcatori di clonalità utilizzati per lo studio della MRMnelle LAL-T. Abbiamo poi valutato la stabilità dei riarran-giamenti di TCR beta che potrebbe essere utilizzato comenuovo marcatore per il monitoraggio della MRM nelleLAL-T. Metodi. Tramite analisi di sequenza delle regionigiunzionali paziente-specifiche (N region) abbiamo con-frontato la stabilità tra esordio e ricaduta dei riarrangia-menti genici di TCR β, TCR γ, TCR δ, delezione di TAL1 e iriarrangiamenti incompleti delle Ig (DH family) in 33pazienti LAL-T. Risultati Un totale di 122 riarrangiamen-ti genici Ig/TCR sono stati identificati alla diagnosi in32/33 (96%) pazienti. L’82% (101/122) dei riarrangia-menti genici identificati alla diagnosi si sono conservatialla ricaduta. In 20/32 pazienti (62%) tutti i riarrangia-menti genici Ig/TCR trovati all’esordio si conservano allaricaduta. In 29/32 (90%) pazienti almeno un riarrangia-mento genico Ig/TCR rimane stabile alla ricaduta. TCR β èriarrangiato nell’87% (29/33) dei pazienti. In 25/29 (86%)dei pazienti almeno un riarrangiamento di TCR β rimanestabile alla ricaduta (Tabella 1).
L’analisi di TCR v risulta utile per 5 pazienti negativi alladiagnosi per tutti gli altri riarrangiamenti Ig/TCR. Infatti4/5 (80%) pazienti presentano almeno un riarrangiamen-to di TCR β stabile alla ricaduta. Conclusioni. Le LAL-Tpresentano una maggiore stabilità complessiva dei riar-rangiamenti Ig/TCR rispetto alle LAL-B (90% vs 71%). IlTCR β è stabile e frequentemente riarrangiato nelle LAL-T.
Questa ricerca è stata finanziata dalla FondazioneCittà della Speranza
Tabella 1.Stabilità dei riarrangiamenti Ig e TCR in 33 pazienti LAL-Tricaduti.
Stabilità dei riarrangiamenti Stabilità nei pazienti
Tutti riarrangiamenti Almeno unconservati riarrangiamento
conservato
TCRBcompleto 25/30 83% 17/21 81% 19/21 90%incompleto 13/19 68% 9/15 60% 11/15 73%totale 38/49 77% 20/29 69% 25/29 86%
TCRG 38/44 86% 21/26 80% 23/26 88%TCRD
completo 12/12 100% 3/4 75% 3/4 75%incompleto 3/4 75% 11/11 100% 11/11 100%totale 15/16 93% 12/13 92% 12/13 92%
Tal1 3/3 100% 3/3 100% 3/3 100%IGH
incompleto 7/9 77% 6/8 75% 7/8 87%
P032TEL/ARG INDUCE ANOMALIE CITOSCHELETRICHE NELLA LINEA CELLULARE293TPalmi C,° Fazio G,° Bonamino M,° Cassetti A,* Villa A,*Biondi A,° Cazzaniga G°°Centro Ricerca M. Tettamanti, Clinica Pediatrica,Università Milano-Bicocca, Ospedale San Gerardo,Monza (MI), Italia; *Consorzio MIA Dipartimento diNeuroscienze, Università Milano-Bicocca, Monza (MI)
Introduzione e obiettivi: Il gene Ets variant gene 6(ETV6/TEL), che codifica per un repressore della trascrizio-ne, è frequentemente coinvolto in anomalie cromosomi-che associate a leucemie, dove risulta riarrangiato congeni partners localizzati su diversi cromosomi. Abbiamoprecedentemente identificato il gene Abelson-relatedgene (ARG) come nuovo partner di TEL in un pazienteaffetto da leucemia mieloide acuta con traslocazionet(1;12)(q25;p13). Il trascritto TEL-ARG, che deriva dallatraslocazione, codifica per una chinasi costitutivamenteattiva che possiede il dominio di oligomerizzazione di ETV6e il dominio tirosin chinasico e di legame all’actina di ARG.Scopo del progetto é lo studio dell’attività trasformante diquesto gene chimerico e in particolare delle sue interazionicon gli elementi del citoscheletro. Metodi: Il cDNA diTEL/ARG derivato dal paziente è stato clonato nel vettorelentivirale bicistronico pRRLPPT. CMV. iresGFPpre. Talecostrutto è stato quindi utilizzato per trasdurre la lineacellulare 293T (fibroblasti renali umani). Risultati: Trami-te analisi con microscopio confocale è stato dimostratoche la proteina TEL/ARG è localizzata nel citoplasma del-le cellule 293T trasdotte e, in particolare, co-localizza conl’actina. Inoltre l’espressione della proteina chimerica neifibroblasti induce un cambiamento morfologico: le cellu-

118
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Leucemie e Linfomi
le diventano tondeggianti, perdono la capacità di aderireal supporto di coltura e muoiono in pochi giorni. Tale feno-tipo viene revertito in modo transiente tramite crescita sufibronectina. Conclusioni: Il trascritto TEL/ARG sembraindurre anomalie citischeletriche, un meccanismo poten-zialmente importante per la sua attività oncogenica. Stu-di futuri saranno rivolti ad indagare gli effetti dell’espres-sione di TEL/ARG in precursori ematopoietici, dove altriautori hanno descritto un ruolo dell’omologo BCR/ABLnell’indurre anomalie citoscheletriche e un’alterata ade-sione alla matrice extracellulare del midollo osseo.
P033ANALISI DELLA MALATTIA RESIDUA MINIMA (MRM) IN PAZIENTI AFFETTIDA RICADUTA S2 DI LEUCEMIA LINFOBLASTICA ACUTA (LLA) SOTTOPOSTIA TERAPIA DI INDUZIONE IASongia S,° Chiesa R,^ Spinelli M,* Coliva T,°Cazzaniga G,° Basso G,* Masera G,^ Locatelli F,§Conter V,^ Biondi A°°Centro Ricerca M. Tettamanti, Clinica PediatricaUniversità Milano-Bicocca, Ospedale S. Gerardo, Monza,Italia, *Dipartimento di Pediatria, Università di Padova,Italia, ^ Clinica Pediatrica Università Milano-Bicocca,Ospedale S. Gerardo, Monza, Italia; §IRCCS PoliclinicoSan Matteo, Pavia, Italia
Introduzione e obiettivi. La malattia residua minima(MRM) nelle prime fasi del trattamento dopo ricaduta diLLA rappresenta un parametro prognostico rilevante.Abbiamo valutato la MRM come marcatore surrogato dirisposta alla terapia di induzione di tipo continuativo (FaseIA) in pazienti con LLA stratificati nel gruppo di rischio S2(ricadute extramidollari precoci e molto precoci, ricadutemidollari o combinate tardive e ricatute combinate pre-coci non-T). Metodi. Sono stati studiati 19 pazienti. 15pazienti presentavano una ricaduta midollare tardiva, 2pazienti una ricaduta combinata e 2 una ricaduta extra-midollare isolata (una SNC e una testicolare). Due pazien-ti risultavano essere positivi alla diagnosi per la trasloca-zione t(12;21) mentre nessuno dei pazienti presentavatraslocazione t(9;22) e t(4;11). La valutazione della MRMè stata effettuata mediante PCR quantitativa del riarran-giamento clonale dei geni codificanti le catene dei T-cellreceptor (TcR) e Immunoglobuline (Ig). 14 pazienti hannoseguito lo schema di Induzione IA prevista dal protocolloAIEOP LLA REC 03 mentre 5 quello del protocollo AIEOPLLA REC 98. Questi 2 schemi sono molto simili; entrambiusano infatti una terapia steroidea continuativa, 4 dosi divincristina e idarubicina e 8 dosi di L-asparaginasi. Risul-tati. Nei pazienti con ricaduta midollare o combinata, laMRM al termine della fase di Induzione è risultata infe-riore a 10-3 in 11/17 (64. 7%). I due pazienti con t(12;21)risultano entrambi negativi al termine dell'Induzione euno presentava MRM negativa anche ad un punto inter-medio prima della fine di Induzione. I 2 pazienti con reci-diva extramidollare presentano entrambi valori di MRMnel BM inferiori a 10-3 al termine dell'induzione. Conclu-sioni. Questi dati suggeriscono che la risposta alla terapiadi Induzione di tipo continuativo (Fase IA) utilizzata neiprotocolli per la ricaduta di LLA S2 risulta paragonabile a
quella ottenuta per lo stesso tipo di ricaduta negli studiBFM con 2 blocchi di chemioterapia. L’ osservazione è rile-vante se si tiene conto del fatto che la terapia secondo loschema IA può essere somministrata a regime ambulato-riale, mentre la terapia a blocchi necessita di ospedaliz-zazione.
P034ANALISI DEI RIARRANGIAMENTI CLONALI E MONITORAGGIO DELLAMALATTIA RESIDUA MINIMA (MRM) NEI PAZIENTI ITALIANI ARRUOLATIAL PROTOCOLLO INTERNAZIONALE INTERFANT-99Corral L,° Luciani M,§ de Lorenzo P,° Spinelli M,*Leszl A,* Rossi V,° Fazio G,° Lo Nigro L, Cazzaniga G,°Basso G,* De Rossi G,§ Biondi A,° Task Force Infant, Aieop°Centro Ricerca M. Tettamanti, Clinica PediatricaUniversità Milano-Bicocca, Ospedale S. Gerardo, Monza,Italia *Clin. di Oncoemat. Ped. , Dip. di Pediatria, Univ. diPadova, Italia Centro di Ematooncol. Ped. , Univ. diCatania, Italia; §Divisione Ematologica, OspedalePediatrico Bambin Gesù, Roma, Italia
Introduzione e obiettivi: Le LLA dei bambini di età infe-riore ad 1 anno (infanti) costituiscono un gruppo peculia-re per caratteristiche biologiche e di risposta alla terapia.L’ oligoclonalità per i geni dei T-cell receptor (TcR) eImmunoglobuline (Igs) costituisce una di tali caratteristi-che. Non è nota la sua implicazione nello studio dellamalattia residua minima (MRM). Metodi: La frequenza deiriarrangiamenti dei TcR e Igs è stata valutata in 37 casi diLLA infant, arruolati in Italia nel protocollo internaziona-le Interfant-1; il pattern di tali riarrangiamenti è statoconfrontato con 649 casi di LLA arruolati nel protocolloAIEOP BFM LLA 2000 e con le stese caratteristiche immu-nofenotipiche. Risultati: I casi con fenotipo pre-B hannopattern e frequenze di riarrangiamenti simili nei due grup-pi di età. Al contrario i casi pro-B LLA mostrano una mino-re incidenza di riarrangiamento di Igκ (8% vs 41% perVk-Kde e 0 vs 19% per intron-Kde) e minore incidenza diTcR γ (16% vs 36%). TcR γ risulta essere meno riarrangia-to anche in infant common (12% vs 57%), mentre IgKintron-Kde è più riarrangiato rispetto ai non-infant (43%vs 15%). L’ analisi di MRM è stata eseguita nei pazientiindicati mediante monitoraggio molecolare di TcR/Ig e/odei geni di fusione di MLL. In 35/37 (95%) dei casi è sta-to individuato almeno un marcatore molecolare sensibi-le, con un sufficiente follow up analizzabile. Conclusioni:I pazienti infant con immunofenotipo più immaturo han-no la tendenza ad avere una minore attività di riarran-giamento somatico. La presenza di oligoclonalità riduce lapossibilità di utilizzo di Ig/TcR e rende necessario l’ utiliz-zo di primers genomici della giunzione di MLL.
P035IL POLIMORFISMO DEL GENE MIF NON È PIÙ FREQUENTE NEI BAMBINICON LEUCEMIA LINFOBLASTICA ACUTA (LLA) PREDNISONE POORRESPONDERS (PPR)Ziino O, D’urbano LE, De Benedetti F, Giarin E,D'angelo P, Caselli D, Farruggia P, Russo D, Trizzino A,

119
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
Basso G, Aricò MOnco Ematologia Pediatrica, Ospedale dei Bambini “G. Di Cristina”, Palermo; Laboratorio di Reumatologia,Direzione Scientifica, IRCCS Ospedale PediatricoBambino Gesù; Onco Ematologia Pediatrica, Universitàdi Padova
Macrophage migration inhibitory factor (MIF) possiedenumerose attività proinfiammatorie ed immunostimolan-ti. Induce la produzione delle citochine proinfiammatorieIL-1, IL-6, TNF e IL-8 dai monociti-macrofagi e stimola laproliferazione e la produzione di IL-2 di linfociti T. E’ l’u-nica citochine in grado di inibire in vitro ed in vivo glieffetti anti-infiammatori ed immunosoppressivi dei glu-cocorticoidi. E’ stato recentemente dimostrato che un SNPdel promotore di MIF (MIF-173G/C), recentemente iden-tificato, è funzionalmente rilevante in vitro ed in vivo conl’allele MIF-173*C associato ad una maggiore produzionedi MIF. In pazienti con artrite idiopatica giovanile siste-mica, la presenza dell’allele MIF-173*C è associata ad unapeggior risposta terapeutica ai glucocorticoidi. Oggettodello studio è l’analisi della frequenza del polimorfismo–173G/C di MIF nei pazienti affetti da leucemia linfaticaacuta (LLA) prednison poor responders (PPR). L’analisi delSNP-173G/C è stata effettuata su DNA genomico di 12pazienti PPR trattati con protocollo AIEOP-LLA-2000;amplificazione mediante primers specifici e successivaanalisi in DHPLC. Come controlli sono stati utilizzati 259soggetti sani di razza caucasica studiati in precedenza.Dei 12 pazienti studiati, le cui caratteristiche sono rias-sunte nella Tabella 1, 3 (25%) sono portatori dell’alleleMIF-173*C. Tale allele è presente peraltro nel 20.8% deicontrolli. Non sono state individuate caratteristiche spe-cifiche dei pazienti con polimorfismo relativamente a ses-so, età, GB alla diagnosi, presenza di traslocazioni (t(4;11)o t(9;22), immunofenotipo, risposta alla terapia di indu-zione. L’allele MIF-173*C non è significativamente piùfrequente nei pazienti con LLA PPR rispetto a quantoosservato nella popolazione italiana di controllo.
Tabella 1.Caratteristiche dei pazienti.
Caso MIF -173 Età/Sesso Fenotipo Trasl. GB diagnosi Blasti SNC Blasti (%) Blasti (%)g. +8 BM +15 BM +33
1 C/G 1.4/F CALL NO 72.520 2.244 NO 2 12 C/G 5. 6/M EARLY-T NO 264. 800 11.435 NO 10 113 G/G 2.9/M T-ALL NO 219. 500 21.250 NO 5 34 G/G 4. 0/M CALL NO 11.650 1.504 NO 3 15 G/G 14. 0/M PRE-PREB 4;11 583. 400 24. 327 NO 2 26 G/G 11.1/M CALL NO 28. 730 1.024 NO 92 607 G/G 3. 7/M EARLY-T NO 75. 310 13. 265 NO 80 28 G/G 11.9/F THYM-T NO 465. 700 29. 520 NO 67 29 G/G 11.9/M EARLY-T NO 30.500 3. 220 SI 90 110 G/G 3. 11/F THYM-T NO 146. 700 1.081 SI 0 011 C/G 9. 0/M T NO 16. 290 2.160 NO 0 012 G/G 1.1/F PRE-PREB 4;11 156. 600 2.516 NO 51 3
P036IL POLIMORFISMO C609T DEL GENE NAD(P)H:CHINONE OSSIDOREDUTTASI(NQO1) È SIGNIFICATIVAMENTE ASSOCIATO AD UN AUMENTATO RISCHIODI INFANT ALL INDIPENDENTE DAL RIARRANGIAMENTO DEL GENE MLLLanciotti M,* Haupt R,** Corral L,*** Di Michele P,*Pigullo S,* De Rossi G,° Basso G,°° Luciani M,°Lo Nigro L,°°° Micalizzi C,* Biondi A,*** Dufour C**Dipartimento di Emato-Oncologia Pediatrica, IstitutoG. Gaslini, Genova **Sezione di Epidemiologia e Biostati-stica, Direzione Scientifica Istituto G. Gaslini, Genova;°Divisione di Ematologia, Ospedale Pediatrico BambinGesù, Roma, °°Emato-Oncologia Pediatrica, Universitàdi Padova, Padova; °°°Centro di Emato-oncologiaPediatrica, Università di Catania, Catania; ***CentroRicerca Tettamanti, Clinica Pediatrica Università diMilano Bicocca, Monza
Introduzione e obbiettivi. L'enzima NAD(P)H:chinoneossidoreduttasi (NQO1) protegge le cellule dagli stressossidativi e da molecole tossiche derivanti dal metaboli-smo di sostanze dannose come il benzene. Nel gene NQO1esiste un polimorfismo (C609T) che provoca una riduzio-ne (CT) o abolizione (TT) dell'attività proteica. Questo stu-dio si propone di verificare se questo polimorfismo puòessere un fattore associato al rischio di leucemie linfo-blastiche acute infantili (età fino a 12 mesi) (iALL). Meto-di. Abbiamo studiato mediante PCR e taglio con enzimadi restrizione, la distribuzione dei genotipi NQO1 in unapopolazione Italiana di 50 pazienti con iALL, 32 dei qualierano positivi per il riarrangiamento del gene MLL (MLL+)e 18 erano negativi (MLL-). Questa distribuzione è stataconfrontata con i genotipi di 106 bambini (età 1-18 anni)con ALL e di 147 controlli normali. Risultati. La frequen-za del genotipo inattivante (CT+TT) è risultata significati-vamente più alta nei pazienti iALL/MLL- rispetto ai con-trolli normali (72% vs 38% p=0.006; OR 4. 22; 95% LC1.43-12.49), mentre nelle iALL/MLL+ la frequenza deigenotipi NQO1 è simile a quella dei controlli normali (44%vs. 38% p=0.553; OR 1.26; 95% LC 0.58-2.74). I risultatinon sono differenti se i genotipi dei pazienti pediatricisono usati come controlli. Conclusioni. I nostri risultatimostrano che il polimorfismo inattivante (CT+TT) di NQO1è significativamente associato con iALL senza riarrangia-mento del gene MLL. Ciò suggerisce un possibile ruolo delgene NQO1 come fattore di rischio, nel processo di leu-chemogenesi delle iALL che è indipendente dal riarran-giamento del gene MLL.
P037LO STUDIO DEL RIARRANGIAMENTO DEL GENE DEL TCRβ CONSENTE DIIMPLEMENTARE LA STRATIFICAZIONE SECONDO MRM DI PAZIENTI CONLLA-TAndré V,° Songia S,° Mollica E,° Spinelli M,* Musarò A,°Basso G,* Biondi A,° Cazzaniga G°°Centro Ricerca M. Tettamanti, Clinica PediatricaUniversità Milano-Bicocca, Ospedale S. Gerardo, Monza,Italia *Clin. di Oncoemat. Ped. , Dip. di Pediatria, Univ. di

120
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Leucemie e Linfomi
Padova, Italia
Introduzione e obiettivi: Lo studio AIEOP-BFM ALL2000si propone di stratificare i pazienti in tre diversi gruppi dirischio in base al risultato di Malattia Residua Minima(MRM) ottenuto mediante l’ analisi di 2 marcatori clona-li con sensibilità 10-4, in due diversi punti della terapia neiprimi 3 mesi. La necessità di due marcatori sensibili richie-de un’ analisi molto dettagliata di tutti i possibili riarran-giamenti dei geni codificanti le catene dei T-cell receptor(TCR) e Immunoglobuline (Ig) all’ esordio di malattia limi-tando l’ attribuzione ad uno specifico gruppo di rischiosulla base del risultato di MRM. Tale limitazione è soprat-tutto rilevante nei casi LLA della linea T, in cui è possibi-le stratificare in base alla MRM solo nell 50% dei casi. LeLLA-T presentano un pannello ridotto di riarrangiamenti.Tra questi, il riarrangiamento di TcR gamma, il più fre-quente nelle LLA-T, risulta un marcatori generalmentepoco sensibile. Metodi: Abbiamo valutato la presenza diriarrangiamenti di TcR β in 137 pazienti con LLA-T delprotocollo AIEOP-BFM LLA 2000, mediante multiplex PCRe utilizzato tale marcatore per monitorare la MRM trami-te RQ-PCR. Risultati: TcR β è risultato riarrangiato nei casicon T-LLA analizzati (70%). La frequenza del riarrangia-mento di TcR β non correla a caratteristiche cliniche ebiologiche particolari. TcR β si è inoltre dimostrato unmarcatore sensibile nei casi analizzati (>=10-4). Conclu-sioni: L’ introduzione dell’ analisi del riarrangiamento diTcR β ha permesso di implementare l’ attribuzione correttaad un gruppo di rischio terapeutico dei pazienti con LLA-T arruolati al protocollo AIEOP-BFM ALL2000.
P038ANALISI FUNZIONALE DELLA PROTEINA CHIMERICA PAX5/TELFazio G,° Palmi C,° Bonamino M,° Cassetti A,* Villa A,*Biondi A,° Cazzaniga G°°Centro Ricerca M. Tettamanti, Clinica PediatricaUniversità Milano-Bicocca, Ospedale S. Gerardo, Monza,Italia *Consorzio MIA Dipartimento di Neuroscienze, Uni-versità Milano-Bicocca, Milano, Italia
Introduzione e obiettivi: Il gene chimerico PAX5/TEL, ori-ginato dalla traslocazione t(9;12)(q11;p13) è stato iden-tificato in un paziente adulto affetto da leucemia linfo-blastica acuta. Dati recenti indicano che la fusionePAX5/TEL definisce l’entità citogenetica dic(9;12)(p13;p13). È ipotizzabile che PAX5/TEL sia un fattore ditrascrizione aberrante, derivato dalla regione 5’ del genePAX5 (un fattore di trascrizione essenziale per il differen-ziamento delle cellule B) fusa con la regione 3’ del geneTEL/ETV6 (Ets-family DNA binding domain). Scopo dellavoro è determinare se la proteina chimerica PAX5/TELpresenta attività trasformante. Metodi: Inizialmenteabbiamo clonato il cDNA di FLAG-PAX5/TEL nel vettoreretrovirale pMSCV-IRES-GFP (MigR1). Abbiamo trasdottola linea cellulare di fibroblasti murini NIH3T3 e la linea cel-lulare murina proB IL-3 dipendente, Ba/F3, ai fini di ana-lizzare la localizzazione subcellulare e l’attività trasfor-mante della proteina chimerica. Risultati: Medianteimmunofluorescenza è stato determinato che la proteina
chimerica PAX5/TEL si localizza nel nucleo delle celluleNIH3T3, trasdotte con MigR1-PAX5/TEL; la presenza del-la proteina chimerica PAX5/TEL non ha mostrato unasignificativa attività trasformante, determinata mediantesaggio di trasformazione in terreno semisolido (colonie insoft-agar). La linea cellulare Ba/F3 trasdotta con MigR1-PAX5/TEL ha mostrato una modulazione della curva di cre-scita; infatti singoli subcloni che esprimono la proteinachimerica mostrano una diminuizione nella crescitarispetto a cellule controllo tradotte con MigR1-IRES-GFP.Inoltre in tale linea cellulare l’espressione della proteinachimerica PAX5/TEL non induce crescita indipendente daIL-3. Conclusioni: Tali risultati preliminari suggerisconoche la proteina PAX5/TEL non svolga direttamente un’at-tività trasformante. Ulteriori studi funzionali permette-ranno di comprendere la funzione della proteina chimeri-ca PAX5/TEL in modelli ematopoietici
P039STUDIO DELLE MUTAZIONI DI BCR/ABL IN PAZIENTI CON LEUCEMIE PH+
PER IDENTIFICAZIONE DI FENOTIPI POTENZIALMENTE RESISTENTI ALTRATTAMENTO CON GLEEVEC MEDIANTE TECNOLOGIA NANOGENCorradi B,1 Monn K,2 Piazza R,3 Gambacorti-Passerini C,3Masera G,1 Biondi A,1 Cazzaniga G1
1Centro Ricerca M. Tettamanti, Clinica PediatricaUniversità Milano-Bicocca, Ospedale San Gerardo,Monza (MI), Italia; 2Nanogen Europe, Stuttgard,Germania; 3Dip. Oncologia Sperimentale, IstitutoNazionale Tumori, Milano, Italia
Introduzione e obiettivi: Recentemente è stato dimo-strato che la resistenza al trattamento con il Gleevec neicasi di leucemia Ph+ è associata a sostituzioni aminoaci-diche in differenti posizioni all’interno del sito chinasicodi Abelson. Il breve periodo entro cui è possibile rilevarela resistenza al trattamento fa supporre che esista unasub-popolazione di cellule leucemiche in cui sia già preesistente tale mutazione e che divenga selezionabile solodurante il trattamento farmacologico. L’identificazionedelle mutazioni pre- e post- trattamento farmacologiconei pazienti con leucemia Ph+ avviene spesso con tecni-che che non sono sufficientemente sensibili o richiedonomolto tempo. Scopo del progetto è testare la possibilità diutilizzare il BioChip della Nanogen per la rapida indivi-duazione delle mutazioni puntiformi di BCR/ABL. Metodi:Nanogen (San Diego, CA, USA) ha sviluppato un metodoper lo studio delle mutazioni genetiche non note, inclusii polimorfismi di un singolo nucleotide (SNPs) mediantel’utilizzo di un microchip elettronico. Il metodo permetteil controllo dell’ibridazione del DNA mediante l’applica-zione di un campo elettromagnetico sul BioChip e ladetection della mutazione attraverso l’utilizzo di una pro-teina legata ad un fluoroforo che riconosce e si lega sta-bilmente ai mismatch del DNA. L’applicazione di tale stru-mento permetterebbe di rivelare, in modo rapido e sensi-bile, la presenza delle mutazioni nel sito chinasico di ABLdei pazienti con leucemie Ph+; analizzando simultanea-mente pazienti diversi o diverse mutazioni nello stessoindividuo. Risultati: In una serie di esperimenti prelimina-ri, si è dimostrato che le mutazioni puntiformi di ABL pos-sono essere identificate in una diluizione contenente 10

121
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
copie totali di un plasmide in cui è presente la mutazio-ne; e in diluizioni seriali fino a 10-4 di cDNA di pazientiresistenti al Gleevec. Il confronto tra le due metodiche èstato esteso ad ulteriori casi di leucemia mieloide croni-ca (LMC) che hanno presentato resistenza al Gleevec edin cui la ricerca di mutazioni è stata eseguita mediantesequenziamento diretto. Conclusioni: Per la sensibilitàdimostrata dal metodo e per il breve tempo richiesto perl’analisi di diversi campioni, Nanogen rappresenta un’al-ternativa al DHPLC o al clonaggio, per l’identificazionedelle mutazioni di BCR/ABL prima e durante il trattamen-to con il Gleevec.
P040ANALISI DELLE MUTAZIONI A CARICO DEI GENI RAS E FLT3 NELLE LLA DELBAMBINO DI ETÀ INFERIORE ALL’ ANNO (INFANT)Dell'oro MG,° Martinelli S,^ Scicchitano B,° Corral L,°Leszl A,* Beretta C,° Spinelli M,* Fazio G,° Basso G,*De Rossi G,§ Tartaglia M,^ Cazzaniga G,° Biondi A°°Centro Ricerca M. Tettamanti, Clinica PediatricaUniversità Milano-Bicocca, Ospedale S. Gerardo, Monza,Italia* Clin. di Oncoemat. Ped. , Dip. di Pediatria, Univ. diPadova, Italia ^Biologia cellulare e Neuroscienze, IstitutoSuperiore di Sanita, Roma, Italia §Divisione Ematologica,Ospedale Pediatrico Bambin Gesù, Roma, Italia
Introduzione e obiettivi: Diversi geni accomunati dallavia di segnale che attraverso RAS porta all’ attivazionedella proliferazione cellulare sono stati trovati alterati insottogruppi di leucemie pediatriche e non. Abbiamo direcente dimostrato un’ elevata incidenza di mutazionimutualmente esclusive dei geni PTPN11, KRAS e NRAS inpazienti pediatrici con LLA. Mutazioni di PTPN11 sonopreferenzialmente associate a fenotipo common e sonoalternative alle più frequenti traslocazioni cromosomiche.Inoltre, mutazioni di PTPN11 si osservano in LAM pedia-triche con FAB M5. Mutazioni somatiche e overespressio-ne del gene per FLT3 sono stati altresì osservati in sotto-gruppi di LLA infant e non. Abbiamo analizzato 37 casi diLLA nfant per verificare l’ ipotesi che mutazioni di diversigeni accomunati dalla via di segnale di RAS possano iden-tificare sottogruppi specifici in questa popolazione etero-genea di pazienti. Metodi: DHPLC e sequenziamento perle mutazioni puntiformi di PTPN11 e RAS, amplificazionee restrizione enzimatica per FLT3, RQ-PCR per l’ overe-spressione di FLT3, citogenetica e FISH (Splitted signal)per riarrangiamenti 11q23. Risultati: MLL (11q23) è sta-to trovato riarrangiato in 23/28 (82%) casi; nessun casoha mostrato mutazioni di PTPN11, 1/25 (4%) e 5/28 (18%)casi hanno evidenziato mutazioni di KRAS2 e NRAS,rispettivamente. Mutazioni puntiformi di FLT3 sono statetrovate in 2/35 (6 %) casi alla diagnosi, mentre nessuncaso era positivo per FLT3/ITD. 11/14 (79%) casi con riar-rangiamento del gene MLL, presentavano overespressio-ne di FLT3. Conclusioni: mutazioni di PTPN11 sono assen-ti nelle LLA infant; l’ incidenza di mutazioni di RAS èsovrapponibile nei casi di LLA infant e nei bambini di etàsuperiore all’ anno. Studi più approfonditi su casisticheprospettiche più estese potranno chiarire se le alterazio-ni genetiche dei segnali di RAS e FLT3 possano influenza-
re il comportamento clinico eterogeneo dei casi LLA infant.
P041CLASSIFICAZIONE DI PAZIENTI PEDIATRICI CON LEUCEMIA ACUTAMIELOIDE APPLICANDO L’ANALISI COMPUTAZIONALE A DATI QUANTITATIVIACQUISITI MEDIANTE CITOMETRIA A FLUSSOZangrando A,1 Luchini A,2 Michielotto B,1 Buldini B,1Bicciato S,2 Te Kronnie G,1 Basso G1
1Dipartimento di Pediatria; 2Dipartimento di Processi Chi-mici dell'Ingegneria, Università di Padova
Con leucemia acuta mieloide (LAM) si identifica ungruppo eterogeneo di malattie ematologiche classificatemorfologicamente in otto sottogruppi da M0 ad M7(nomenclatura F. A. B. , 1976). Di recente, l’Organizzazio-ne Mondiale per la Salute (WHO) ha proposto una nuovaclassificazione che risalta il ruolo delle alterazioni gene-tiche nella definizione dei sottogruppi F. A. B. Abbiamoapplicato l’analisi computazionale sui dati raccolti da 89casi con LAM, utilizzando un nuovo approccio multipara-metrico per l’analisi di dati citofluorimetrici di pazientileucemici. Dal regolare pannello di indicatori per la dia-gnosi di leucemie acute non linfoblastiche mediante cito-fluorimetria, abbiamo analizzato i valori di intensità mediadi fluorescenza (MFI) ed il coefficiente di variazione a pic-co intero (FPCV) di 35 antigeni. Una prima Analisi delleCorrispondenze è stata applicata ai 7 antigeni più discri-minanti con valori di p<0.0005: gli antigeni CD64, CD33e CD61 hanno mostrato forte correlazione rispettivamen-te con i sottotipi M5, M3-M3v ed M7. Successivamente,abbiamo utilizzato un clustering Gerarchico Non Super-visionato per correlare i pazienti con i 18 antigeni speci-fici per i riarrangiamenti cromosomici (con valori dip<0.07). Il dendrogramma ha separato con successo ipazienti in 4 gruppi principali: t(8;21)(q22;q22) AML1-ETO, t(15;17)(q22;q11) PML-RARa, inv(16)(p13;q22)CBFB-MYH11 ed i negativi per queste traslocazioni. L’a-nalisi multiparametrica non supervisionata permette l’i-dentificazione oggettiva di nuove categorie in ogni ambi-to di ricerca. L’applicazione di questa metodica in ambitoclinico-diagnostico potrebbe avere un impatto significa-tivo nell’identificazione di nuovi sottogruppi LAM.
P042L’ACIDO VALPROICO INDUCE IPERACETILAZIONE DEGLI ISTONI E,IN COMBINAZIONE CON ACIDO ALL-TRANS RETINOICO, CAUSA AUMENTODI P21CIP1 E CASPASI 8, ARRESTO IN G1, E APOPTOSI NELLA LEUCEMIAMIELOIDE ACUTA CON RIARRANGIAMENTI DI MLLFronza R,*Tonelli R,* Freccero F,* Ballarini M,°Franzoni M,* Ferrari S,^ Sartini R,* Locatelli F,#Minucci S,°§ Pession A**Dipartimento di Pediatria, Università di Bologna,Ospedale S. Orsola, Bologna, Italia; °Istituto Europeo diOncologia, Dipartimento di Oncologia Sperimentale,Milano, Italia; ^ Divisione di Genetica Medica, Universitàdi Bologna, Ospedale S. Orsola, Bologna, Italia;#Ematologia/Oncologia Pediatrica, IRCCS PoliclinicoSanMatteo, Università di Pavia, Pavia, Italia;

122
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Leucemie e Linfomi
§Dipartimento di Scienze Biomolecolari e Biotecnologie,Università di Milano
Introduzione e obiettivi. I pazienti pediatrici affetti daleucemia mieloide acuta (LAM) associata a riarrangia-menti del gene MLL, localizzato alla banda cromosomica11q23, hanno ancora prognosi sfavorevole e pertantonecessitano di nuovi approcci terapeutici. Le iston-dea-cetilasi (HDAC) possono rappresentare un target comuneper il trattamento delle LAM. L’effetto dell’acido valproi-co (VPA), un inibitore delle HDAC ben tollerato e già uti-lizzato in clinica come antiepilettico, è stato valutato sin-golarmente o in combinazione con acido all-trans-reti-noico (ATRA) nelle LAM con i riarrangiamentiMLL/HRX/ALL-1.Metodi. Per lo studio sono stati usati iblasti di un paziente infant affetto da LAM con riarran-giamento MLL e le linee cellulari THP-1, MM6 e MOLM-13 (positive per MLL-AF9). La vitalità cellulare è statavalutata con Trypan Blu. Apoptosi, ciclo cellulare e diffe-renziamento sono stati determinati tramite analisi cito-fluorimetrica. Con tecniche di Western Blot e PCR quan-titativa sono stati valutati l'acetilazione degli istoni, l'e-spressione di p21Cip1 e la quantità di messaggero dip21Cip1, p27Kip1 e caspasi 8. Risultati In tutte le tre lineecellulari e' stato riscontrato che il VPA induce iperaceti-lazione degli istoni. Inoltre, il trattamento con VPA deter-mina anche apoptosi e arresto del ciclo cellulare senzainduzione del differenziamento. L'aggiunta dell'ATRA, cheda solo ha un’azione meno evidente nei campioni valuta-ti, potenzia questi effetti. Il VPA (ma non l'ATRA) aumen-ta i livelli di mRNA di p21Cip1 e di p27Kip1, e il tratta-mento combinato porta all'aumento sinergico dei livelli dip21Cip1, ma non di p27Kip1.Il VPA induce anche l'e-spressione del gene caspasi 8. Nelle tre linee cellulari, iltrattamento combinato con l'ATRA rafforza l'effetto delVPA. Conclusioni. Nelle LAM con riarrangiamenti di MLL,l'inibizione di HDAC indotta da VPA aumenta la rispostaall'ATRA, causando citotossicità, induzione di p21Cip1 ecaspasi 8, inibizione del ciclo cellulare ed apoptosi, manon differenziamento.
P043IL GENE WT1 COME MARCATORE DI MALATTIA RESIDUA MINIMA (MRM)NELLA LEUCEMIA MIELOIDE ACUTA (LAM): ESPERIENZA DISTANDARDIZZAZIONE DELLA METODICA NELL'AMBITO DELL'I-BFM SGCazzaniga G,* Coliva T,* Rossi V,* Viganò E,* Spinelli M,°Frascella E,° Bader P,^ Trka J,^^ Gruhn B,# Basso G,°Rizzari C,* Biondi A*Clinica Pediatrica di Monza*, Padova°, Tubingen^, Praga^^, Jena#
Introduzione e obiettivi: La valutazione della MRM harilevanza in ematoncologia per valutare la risposta precocealla terapia, per identificare la ripresa della malattia stes-sa e indirizzare precocemente l'intervento terapeutico.Nella maggior parte dei casi di LAM non sono disponibilimarcatori genetici per il monitoraggio della MRM. Il geneWT1 è overespresso in diversi tipi di leucemie, incluse leLAM. Per standardizzare la valutazione dell'espressionedel gene WT1 mediante PCR quantitativa e verificare il
suo utilizzo come marcatore di MRM nelle LAM si è costi-tuito un gruppo di studio internazionale (Italia, Germania,Repubblica Ceca). Metodi: Le condizioni ottimali di ana-lisi quantitativa del gene WT1 sono state definite in segui-to ad un controllo di qualità eseguito da 4 diversi labora-tori. Sono stati valutati due diversi plasmidi e tre diversiset di primer e probe. La stabilità dei trascritti di WT1 (tar-get) e ABl (reference) è stata valutata mediante analisiquantitativa di campioni di midollo all'esordio estratti a 0-24-48 ore dal momento del prelievo. Per valutare il signi-ficato prognostico della MRM e confrontare diversi pro-tocolli terapeutici adottati nei singoli Paesi ci si proponedi studiare il livello di espressione di WT1 nel BM e PB neipazienti affetti da LAM durante le diverse fasi del tratta-mento, sull'aspirato midollare che precede il trapianto dimidollo allogenico o sull'espianto del midollo autologo ea 1,3,6,12 mesi dal trapianto di midollo autologo e allo-genico o dallo stop terapia. Risultati: In 37/38 casi è sta-to possibile quantificare l'espressione di WT1 nel BM all'e-sordio e in 22/38 si è osservata overespressione del gene.In 3 casi finora analizzati i valori di MRM si sono dimo-strati coerenti con l'andamento della malattia. Conclu-sioni: La standardizzazione delle metodiche di studio del-l'espressione del gene WT1 a livello internazionale renderàpossibile l'analisi del suo valore prognostico anche in pro-tocolli terapeutici differenti.
P044DENSITÀ MINERALE OSSEA (DMO) IN BAMBINI E ADOLESCENTIGUARITI DA LINFOMA MALIGNO (LM)Sala A,*°# Coliva T,# Citterio M,# Vigano E#
Conter V,# Webber C,° Posgate S,° Barr RD°#
*Clinica Pediatrica, Università di Milano-Bicocca,Ospedale S. Gerardo, Monza; °McMaster University;#McMaster Children’s Hospital, Hamilton, Ontario, Canada
Introduzione: I pazienti trattati per LM prima dellamaturazione scheletrica potrebbero presentare una ridu-zione della massa ossea. E’ stata dunque analizzata la cor-relazione tra la diminuzione della massa ossea e la che-mioterapia somministrata in una popolazione di bambinied adolescenti fuori terapia. Pazienti e metodi: Sono sta-ti studiati 42 pazienti (27 maschi e 15 femmine), trattatiper malattia di Hodgkin (HD = 22 pazienti) o linfoma non-Hodgkin (NHL = 20 pazienti), fuori terapia (da almeno 1anno) e senza malattia residua o ricorrente nota. La DMOè stata misurata a livello del rachide lombare medianteDXA (dual X-ray absorptiometry) ed espressa come z–sco-re specifico per età e sesso. Una particolare TAC (periphe-ral quantitated computer tomography, strumento StratecXCT2000) è stata utilizzata per lo studio dell’osso corticalee trabecolare del radio. Sono stati inoltre valutati para-metri biochimici di turnover osseo, come calcio serico,fosfato organico, TSH, osteocalcina, telopeptide C-termi-nale e vitamina D, ma non sono stati riportati qui. Risul-tati: Lo z –score mediano di DMO lombare per HD e NHLè risultato – 0,55 e -0,98 (range da -2,49 a +1,37 e da -4,5 a +1,47 rispettivamente). Osteopenia (definita come z-scores della DMO < minus 1,00) è stata riscontrata in 9/22pazienti con HD e in 10/20 pazienti con NHL. La DMO tra-becolare del radio è risultata compresa nei limiti di nor-

123
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
ma per i pazienti di sesso maschile, mentre nelle femmi-ne con età > 20 anni i risultati sono stati con valori infe-riori rispetto a quelli della popolazione di controllo. Nonsono state invece trovate differenze tra maschi e femmi-ne riguardo alla DMO corticale del radio. Conclusioni:Importanti differenze nell’esposizione ai farmaci, in par-ticolare alle dosi cumulative di farmaci steroidei, potreb-bero giustificare le differenze di massa ossea e di meta-bolismo osseo osservate tra i guariti da HD o NHL.
P045PERSISTENZA DELLA RISPOSTA ANTICORPO-SPECIFICA (ANTI EPATITE B)IN UNA POPOLAZIONE PEDIATRICA DOPO CHEMIOTERAPIA PER LEUCEMIALINFOBLASTICA ACUTAArrighetti A, Gualeni C, Caldiani C, Tettoni K, Sinelli MT,D’Ippolito C, Porta F, Notarangelo LDUnità dipartimentale di terapia cellulare e genica delleoncoemopatie infantili “Angelo Nocivelli”, ClinicaPediatrica, Brescia
Introduzione e obiettivi: la chemioterapia comporta unatemporanea riduzione della funzione immunitaria. Il pre-sente lavoro ha lo scopo di valutare la possibile persisten-za della risposta anticorpo-specifica dopo chemioterapiaper leucemia linfoblastica acuta. Metodi e pazienti: sonostati esaminati retrospettivamenti tutti i pazienti trattatisecondo i protocolli LLA 95 e 2000 per un totale di 64pazienti (27 femmine e 37 maschi, ). E’ stata scelta la rispo-sta alla vaccinazione anti epatite B perché routinariamen-te ricercata in tutti i pazienti all’esordio e dopo 1 mese dal-lo stop terapia. Criteri di inclusione: vaccinazione control’epatite B, disponibilità dei titoli anti epatite B pre e postchemioterapia, completamento dei 2 anni di trattamentoprevisti. Criteri di esclusione: immunodeficienza primitiva,trapianto di midollo osseo allogenico. Sono risultati per-tanto eleggibili allo studio 42 pazienti (23 maschi e 19femmine, di età compresa tra i 4 anni + 5/12 e 14 anni +6/12); i motivi di esclusione erano da ricondursi a: trapiantodi midollo osseo (6 pz. ), decesso per complicanze duranteil trattamento (3 pz. ), ricaduta in 1° remissione completadurante il mantenimento (4 pz), immunodeficienza primi-tiva (1 pz), mancata vaccinazione anti epatite B (6 pz. ), nondisponibilità del titolo post chemioterapia (2 pz. ), trasfe-rimento presso altro centro (2 pz. ). Degli eleggibili, 12pazienti avevano seguito il protocollo di terapia LLA 2000,28 il protocollo LLA 9502 e 2 il protocollo LLA 9501.Risul-tati: 32 pazienti (76%) hanno mostrato una persistenza deltitolo anticorpale dopo la chemioterapia; 5 pz. (12%) pre-sentavano una negatività del titolo anticorpale nonostan-te l’avvenuta vaccinazione; 5 pz. (12%), hanno dimostratouna negativizzazione del titolo post chemioterapia; uno diquesti pazienti, sottoposto a richiamo vaccinale, ha dimo-strato una sieroconversione. Conclusioni: questo studiodimostra la persistenza della risposta anticorpo specificaanti epatite B in un’alta percentuale di soggetti vaccinati.La quota di non responder alla vaccinazione è risultatasostanzialmente sovrapponibile a quanto riportato in let-teratura in riferimento alla popolazione generale. Sia purein un solo caso documentato, il richiamo vaccinale effet-tuato come da calendario per l’età, si è dimostrato in gra-
do di produrre una risposta efficace, confermando la per-sistenza della memoria immunologica dopo chemioterapiaantiblastica per LLA. P046RELAZIONE TRA CREATININA SERICA (CS) E MASSA MAGRA CORPOREA(MMC) IN BAMBINI AFFETTI DA LEUCEMIA LINFOBLASTICA ACUTA (LLA)Sala A,1,2,3 Citterio M,1 Coliva T,1, Vigano’ E,1,
Conter A,1 Tarnopolsky M,2 Webber C,2 Barr RD2,3
1Clinica Pediatrica Università di Milano-Bicocca,Ospedale S. Gerardo, Monza; 2McMaster University e3McMaster Children’s Hospital, Hamilton, Ontario,Canada
Introduzione: In pazienti con tumore sono state dimo-strate modificazioni nel metabolismo, in particolare vi èun’alterazione del turnover proteico, risultante soprattut-to in una perdita della MMC. Un parametro ematico chepotrebbe monitorare la MMC è la CS, dal momento che lacreatinina è prodotta quasi esclusivamente nel muscoloscheletrico. Mentre è nota la correlazione tra MMC edescrezione urinaria di creatinina nelle 24 ore, esistonopochi dati riguardanti la correlazione tra MMC e CS. Inconsiderazione della difficoltà nell’ottenere una correttaraccolta dell’urina delle 24 ore nei bambini, è stata ese-guita una valutazione della relazione tra CS e MMC, deter-minata mediante valutazione con dual X-ray absorptio-metry (DXA). Pazienti e metodi: Trentasette bambini affet-ti da LLA sono stati studiati con analisi seriata e 20 bam-bini con patologia muscolare primitiva (PMP – 15 condistrofia muscolare di Duchenne e 5 con altre miopatie)sono stati utilizzati come gruppo di controllo, essendodimostrato che questi pazienti presentano un’alterazionenella MMC. Sono stati analizzati tre gruppi di dati neipazienti con LLA (alla diagnosi, durante la terapia e allostop-terapia) ed un singolo gruppo di dati nei pazienti conPMP. Risultati: Sono state dimostrate correlazioni signi-ficative tra CS e MMC nei pazienti con PMP (r = 0,77, p <0,001) e in quelli con LLA (r = 0,77, p < 0,001 alla diagnosi;r = 0,75, p < 0,001 durante la terapia; r = 0,59, p = 0,02allo stop-terapia). La correlazione tra CS e indice di mas-sa corporea è risultato più debole (r = 0,38; r = -0,91; r =0,29 alle successive osservazioni nella coorte dei pazien-ti con LLA). Conclusioni: Questi risultati permettono diconsiderare la CS come una misura indiretta della MMC,e conseguentemente dello stato nutrizionale, in quellesituazioni dove la valutazione della MMC mediante DXAnon è disponibile, come nei paesi con risorse limitate in cuivive la maggior parte dei bambini con cancro.
P047CONTROLLO DI QUALITÀ EUROPEO PER LO STUDIO DELLA MALATTIARESIDUA MINIMA (MRM) IN PAZIENTI AFFETTI DA LEUCEMIALINFOBLASTICA ACUTA (LLA)Manenti M,° Corral L,° Villa T,° Musarò A,°D’ aniello E,° Songia S,° Van Dongen J, * Biondi A,°Cazzaniga G°°Centro Ricerca M. Tettamanti, Clinica PediatricaUniversità Milano-Bicocca, Ospedale S. Gerardo, Monza,Italia *Dept. of Immunology, Erasmus MC UniversityMedical Center, Rotterdam, Netherlands

124
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Tumori Solidi
Introduzione e obiettivi: l’ applicazione della MRM aprotocolli clinici e l’ apertura di studi multicentrici per lavalutazione della MRM hanno reso necessaria l’ introdu-zione di una procedura di standardizzazione delle meto-diche e di controllo di qualità (QC) al fine di rendere irisultati di gruppi diversi più uniformi. A tale scopo è sta-to costituito il gruppo europeo European Study Group onMRD detection in childhood ALL; (ESG-MRD-ALL). Meto-di: al gruppo ESG-MRD-ALL partecipano 25 laboratorieuropei, tra i quali i laboratori di Monza, Heidelberg eVienna, responsabili per la valutazione di MRM nel proto-collo AIEOP-BFM ALL2000.E’ stato introdotto un control-lo di qualità semestrale, seguito da incontri dedicati alladiscussione delle conclusioni a cui sono giunti i differen-ti gruppi. Per ogni caso di QC proposto viene inviato ilDNA dell’ esordio di malattia di 2 pazienti per lvdentifi-cazione dei target Ig e TCR, e altri 2 per l’ analisi dellaMRM dei relativi follow-up e per l’ interpretazione deirisultati di PCR quantitativa. Risultati: Il protocollo meto-dologico (identificazione dei target paziente specifici, scel-ta dei 2 target con sensibilità 10-4 per la PCR quantita-tiva e interpretazione finale dei risultati) è stato standar-dizzato e uniformato nel corso degli anni e semestral-mente vengono discusse ed aggiornate le linee guida chepermettono una comune interpretazione dei risultati otte-nuti con PCR quantitativa.
Figura 1.Risultati dell’analisi di MRM nei follow up di due pazienti,ottenuti nei diversi laboratori.
Conclusioni: I risultati dei successivi QC si sono rivela-ti sempre più omogenei, dimostrando che la collaborazio-ne europea dei laboratori che eseguono l’ analisi di MRMè necessaria per uniformare l’ analisi e l’ interpretazionedei dati. Ciò consente di poter confrontare in modo piùcontrollato risultati ottenuti in studi diversi.
TUMORI SOLIDI
C005L'ESPRESSIONE DI PAX3-FKHR E' INDICE PROGNOSTICO NEGATIVO INPAZIENTI AFFETTI DA RABDOMIOSARCOMASartori F, Carli M, Tridello G, Alaggio R,* Ninfo V,*Rosolen AClinica di oncoematologia pediatrica, AziendaOspedaliera; *Dipartimento di Scienze Oncologiche e Chi-rurgiche, Università di Padova
Il rabdomiosarcoma (RMS) è un tumore pediatrico a pic-cole cellule rotonde blu caratteristico delle parti molli. Trai RMS vengono riconosciuti distinti istotipi: il rabdomio-sarcoma embrionale (ERMS) è l'istotipo più frequente men-tre il rabdomiosarcoma alveolare (ARMS) presenta unamaggiore aggressività locale ed una più spiccata tendenzaalla disseminazione. Mediante analisi citogenetica classi-ca sono state descritte anomalie cromosomiche in tutti isottotipi di RMS, tuttavia alcune regioni cromosomichesono risultate coinvolte in modo non casuale. In particola-re, il sottotipo alveolare presenta la traslocazione cromo-somica reciproca t(2;13)(q35;q14) o, meno frequentemen-te, la t(1;13)(q36;q14) danno origine ai geni di fusionePAX3-FKHR o PAX7-FKHR, rispettivamente. Scopo delnostro lavoro è quello di valutare se vi sia una correlazio-ne tra la presenza del gene di fusione e la prognosi. Abbia-mo perciò studiato in modo prospettico, mediante RT-PCR,una coorte di 154 pazienti affetti da rabdomiosarcomaarruolati in due protocolli AIEOP consecutivi, 57 dei qualicon istologia alveolare. Il trascritto di fusione PAX3-FKHRè stato trovato in 29/57 pazienti, PAX7-FKHR 10/57, men-tre 18/57 erano negativi. L'analisi univariata evidenza unEFS a 5 anni del 30% (CI 9. 86-48. 89) per i pazienti chepresentano il trascritto chimerico PAX3-FKHR e del 60% (C.I. 33. 31-85. 95) per i pazienti che non presentano il tra-scritto di fusione (p=0.03). La OS è del 27% (C. I. 8. 35-45.98) per i pazienti positivi per il trascritto PAX3-FKHR e del63. 5% (C. I. 37. 65-89. 47) per i pazienti negativi (p=0.03).I pazienti positivi per il trascritto PAX7-FKHR hanno unandamento intermedio. Questi dati suggeriscono che,pazienti affetti da RMSA, positivi per il trascritto chimeri-co hanno un andamento più sfavorevole rispetto al grup-po di pazienti negativi.
C006MECCANISMO DI AZIONE DEGLI ACIDI PEPTIDO-NUCLEICI (PNA)CON AZIONE ANTI-GENE N-MYC IN CELLULE DI NEUROBLASTOMAPurgato S,* Tonelli R,* Fronza R,* Camerin C,*Franzoni M,* Corradini R,° Sforza S,° Faccini A,°Marchelli R,° Pession A**Oncoematologia Pediatrica e Terapia Cellulare, ClinicaPediatrica S. Orsola-Malpighi, Bologna °Dipartimentodi Chimica Organica, Università di Parma
Introduzione e Obiettivi. Il 30% dei neuroblastomi pri-mari non trattati presenta un'amplificazione del gene N-

125
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
myc, associata a stadi avanzati della malattia, rapida pro-gressione del tumore e prognosi sfavorevole. Inibitoriselettivi di N-myc possono essere utilizzati come agentiterapeutici specifici per i neuroblastomi con over-espres-sione di N-myc. I PNA sono analoghi sintetici degli acidinucleici, in cui il normale scheletro fosfodiesterico è sosti-tuito da una catena pseudo-peptidica formata da mono-meri di N-(2-amminoetil)glicina legati covalentementealle basi del DNA. Metodi. Sono state utilizzate 4 lineecellulari di neuroblastoma: GI-LI-N e IMR-32 (N-mycamplificato e overespresso), GI-CA-N e GI-ME-N (N-mycnon amplificato). E' stato sintetizzato un PNA anti-genediretto contro una sequenza unica dell'esone 2 di N-mycsul filamento antisenso del DNA e coniugato, all'estre-mità N-terminale, ad un segnale di localizzazione nuclea-re (NLS). La veicolazione del PNA rhodaminato nel nucleoè stata verificata mediante microscopia a fluorescenza.Gli effetti del PNA sono stati valutati mediante analisidella vitalità cellulare, western blotting e analisi citofluo-rimetriche del ciclo cellulare e dell'apoptosi. Risultati.Mediante la microscopia a fluorescenza è stato dimostra-to il trasporto nucleare del PNA nelle quattro linee cellu-lari. Nelle IMR-32 e GI-LI-N un rilevante effetto anti-pro-liferativo è già osservabile dopo 24 ore, aumenta a 48 oree raggiunge un picco a 72 ore (80% in GI-LI-N e 90% inIMR-32); nessuna riduzione é stata registrata nelle GI-CA-N e GI-ME-N. Dopo 3 ore dal trattamento il PNA cau-sa una riduzione di NMYC del 50%, come dimostrato conil western blotting. L'analisi citofluorimetrica sulle IMR-32ha mostrato che il PNA induce accumulo nella fase G1 delciclo cellulare ed apoptosi. L'attività selettiva del PNA èstata dimostrata osservando che un PNA anti-gene con tremutazioni puntiformi e un PNA contro il filamento sensodel DNA non hanno effetto. Conclusioni. Il PNA anti-genemostra un effetto inibitorio specifico sull'espressione di N-myc che riduce la proliferazione cellulare e induce accu-mulo delle cellule nella fase G1 del ciclo ed apoptosi. Con-frontando con un PNA antisenso, il PNA anti-gene mostraun effetto inibitorio più forte e persistente a concentra-zioni più basse.
P048MUTAZIONI GERMINALI DEL GENE POU6F2 IN TUMORI DI WILMSCON PERDITA DI ETEROZIGOSI SUL CROMOSOMA 7p14Perotti D, De Vecchi G, Testi MA, Lualdi E, Modena P,Mondini P, Ravagnani F, Collini P, Di Renzo F,*Spreafico F, Terenziani M, Sozzi G, Fossati-Bellani F,Radice PIstituto Nazionale Tumori, Milano;*Dipartimento di Biologia, Università di Milano
Introduzione ed Obiettivi. Il tumore di Wilms (TW) è unaneoplasia pediatrica caratterizzata da una elevata etero-geneità genetica. In precedenza avevamo riportato lacaratterizzazione citogenetica e molecolare di un TW (no.30) che mostrava una delezione interstiziale sul cromo-soma 7p14 tra i marcatori D7S555 e D7S668. Studi di per-dita di eterozigosi (PE) avevano dimostrato che questastessa regione veniva persa in 8/38 casi di TW esaminati,suggerendo che nell’intervallo identificato fosse localiz-
zato un putativo gene oncosoppressore. Metodi e risulta-ti: Per confermare questa ipotesi, in questo lavoro abbia-mo analizzato 35 nuovi casi di TW, in quattro dei quali èstato possibile identificare PE per la regione di interesse.Inoltre, siamo stati in grado di definire in maniera piùaccurata l’estensione della delezione del caso no 30, map-pandola in un intervallo non superiore alle 390 kb a valledi D7S555. Ad oggi, un solo gene, POU domain, class 6,transcription factor 2 (POU6F2), è stato mappato in que-sto intervallo. Il sequenziamento di questo gene nei 12casi con PE nella regione ed in un numero corrisponden-te di TW senza delezione ha portato alla identificazione didue mutazioni germinali. La prima era presente nellaregione 5’ non tradotta del gene, mentre la seconda pro-vocava una sostituzione aminoacidica in un dominio diripetizioni di glutamine. Queste mutazioni, che non sonstate riscontrate in più di 100 individui di controllo, sonostate identificate in due pazienti in cui l’allele normaleveniva perso a livello del tumore. Inoltre, abbiamo dimo-strato l’espressione dell’analogo murino del gene POU6F2sia nel rene embrionale che nel rene adulto. Conclusioni:Le nostre osservazioni suggeriscono che il gene POU6F2sia un oncosoppressore e che sia coinvolto nella predi-sposizione ereditaria al TW.
Con parziale finanziamento di: Associazione BiancaGaravaglia, AIRC, CNR.
P049BLASTOMI PLEURO-POLMONARI: STUDIO CLINICO-PATOLOGICOSU 21 CASIIndolfi P, Casale F, Carli M,* Bisogno G,* Cecchetto G,*Ferrari A,** Martone A, D’onofrio V, Santoro N,§Donfrancesco A,° Inserra A,° Conte M,°° Riccardi R,^di Cataldo A,^^ D’Angelo P,§§ Cellini M,+ Izzi G,++
Rondelli R,*** Pota E, di Martino M, Di Tullio MTGruppo Cooperativo AIEOP-TREP, Napoli; *Padova;**Milano; §Bari; °OPBG Roma; °°Genova; ^ CattolicaRoma; ^^Catania; §§Palermo; +Modena; ++Parma;***Bologna
Introduzione. Il blastoma pleuro-polmonare (PPB) è unarara ed aggressiva neoplasia intratoracica dell’età pedia-trica che usualmente si osserva in bambini con meno di 5anni. Tale neoplasia è caratterizzata istologicamente daquadri misti con isole blastematose ad alta attività mito-tica che si associano ad aree con cellule fusate mesen-chimali indifferenziate o in vari gradi di differenziazionemaligna. Nonostante l’introduzione di un approccio tera-peutico multimodale che prevede la chirurgia, la chemio-terapia e, talvolta, la radioterapia, il PPB conserva, anco-ra oggi, una prognosi sfavorevole. Materiali e Metodi: Incollaborazione con il Gruppo cooperativo italiano per iSarcomi delle parti molli e con il Gruppo AIEOP-TREPabbiamo analizzato le caratteristiche cliniche dei casi diPPB diagnosticati e trattati presso Centri AIEOP distribui-ti sul territorio nazionale. Risultati: Lo studio ha riguardato21 pazienti (14 maschi e 7 femmine) la cui età medianaè risultata essere di 29 mesi (range 2-109 mesi). In 5pazienti il tumore si era sviluppato su malformazioni cisti-che polmonari. Era presente coinvolgimento di strutture

126
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Tumori Solidi
extrapolmonari in 11 casi e lesioni metastatiche (ossee) inun paziente. In un caso la localizzazione primitiva erasolamente pleurica. Una resezione completa del tumore èstata possibile, alla diagnosi, in 7 casi. Di essi, 5 pazientisono vivi in continua completa remissione tra 9 e 154mesi dalla diagnosi. Otto pazienti (38%) sono recidivati (5localmente e 3 a distanza). In due pazienti (9. 5%) si èavuta una progressione locale di malattia. Di essi, 2pazienti sono liberi da malattia, in seconda remissione,rispettivamente a 70 e 115 mesi ed uno è vivo con malat-tia a 12 mesi dalla diagnosi. La sopravvivenza globale a 3anni è risultata essere del 61.2% mentre la sopravviven-za libera da eventi a 3 anni del 47. 3%. Conclusioni:Laprognosi del PPB resta globalmente severa. I pazienti consottotipo istologico di tipo 2 o con coinvolgimento extra-polmonare alla diagnosi hanno una prognosi peggiore.L’intervento chirurgico radicale alla diagnosi costituisce ilmigliore indicatore prognostico. La chemioterapia post-operatoria può costituire un’arma aggiuntiva nella pro-gnosi di questi pazienti, mentre la radioterapia sembrasvolgere un ruolo marginale nel trattamento del PPB.
P050VALUTAZIONE LONGITUDINALE DELL’INFEZIONE CRONICA DA EPATITE C INUN GRUPPO DI LUNGO SOPRAVVIVENTI DOPO TRATTAMENTO PER TUMOREFioredda F,* Gigliotti A, Hanau G, Faraci M, Grisolia F,°Haupt R, °Calevo MG,* Giacchino RUnità Operativa Emato-Oncologia e Trapianto di MidolloOsseo°, Servizio di Epidemiologia e Biostatistica, UnitàOperativa Malattie Infettive, Istituto Giannina Gaslini,Genova
Introduzione. La storia naturale dell’infezione da HCVnell’infanzia è poco conosciuta soprattutto nella popola-zione dei guariti da malattia oncologica. Il tempo di pro-gressione di alcune forme croniche a cirrosi ed epatocar-cinoma ed i fattori di rischio che li favoriscono sono tut-tora oggetto di studio. Obiettivo dello studio. Scopo dellavoro è descrivere l’esperienza dell’infezione cronica daHCV in un gruppo di lungo sopravviventi (oltre i 10 anni)dopo chemioterapia o trapianto di midollo osseo permalattia oncologica e correlare il tipo di trattamenti rice-vuti con l’andamento di malattia. Pazienti e Metodi. Dal-le cartelle dei pazienti infettati da HCV durante il tratta-mento per malattia neoplastica, sono stati raccolti, dovedisponibili, dati biochimici, virologici diretti ed indirettied istologici. Sono state valutate le possibili relazioni trail tipo di trattamento chemioterapico, la radioterapia, glialtri farmaci epatotossici, il sovraccarico di ferro, la dura-ta (lunghezza) dell’immunosoppressione e l’andamentopiù o meno aggressivo dell’epatite tramite i consueti teststatistici (test di Fisher e test di Mann-Withney). Risulta-ti. Sono stati inclusi nell’osservazione 17 pazienti (4 fem-mine e 13 maschi),età mediana 70 mesi (range 6-152mesi) con diagnosi di neuroblastoma 5/17, leucemia mie-loblastica acuta 5/17, leucemia linfoblastica acuta 4/17,tumore di Wilm’s 2/17 e sarcoma di Ewing 1/17. 12pazienti sono stati sottoposti a trapianto di midollo (10autologhi e 2 allogenici). Il tempo mediano di follow-upè stato di 13,7 anni (range 10-18,5 anni). L’analisi longi-
tudinale dell’intero gruppo ha consentito di osservare chei valori medi delle ALT durante i primi 4 anni dopo l’ off-therapy erano oltre 3 volte i valori normali. Il monitorag-gio successivo delle stesse ha evidenziato una tendenzaalla normalizzazione con alcuni picchi stanti ad indicarereplicazione ad andamento caratteristicamente ondulan-te. L’analisi dei parametri di replicazione (ALT medie/annoe HCV-RNA virale) ha consentito di definire un gruppo adandamento più aggressivo ed uno ad andamento piùmoderato in base al numero percentuale diALT/medie/anno più elevate di 1,5 volte la norma. 7/17pazienti (41%) hanno mostrato valori medi annui oltre1,5 la norma nel 63-100 % delle determinazioni, mentrei rimanenti 10/17 avevano valori medi annui elevati inmeno del 50% delle determinazioni (valori medi ALT dei7/17, 83. 4 [range 106-62]; valori medi 10/17, 32.7 [ran-ge 47-17]). Inoltre lo studio dell’RNA virale risultava posi-tivo nel 100% dei casi nel primo gruppo, rispetto al 70%nel secondo; 3/7 pazienti appartenenti al secondo grup-po avevano RNA costantemente negativo. Nel primo grup-po più studiato dal punto di vista istologico, le biopsiehanno identificato un moderato grado di danno tissutale(EC G1 S1–EC G2S2-3). oppure (max EC G2 S2-3) Non èstata trovata correlazione significativa nel gruppo che hamostrato andamento più aggressivo con i diversi tratta-menti chemioterapici, radioterapici, con assunzione postterapia di farmaci epatotossici, valori di ferritina all’ off-therapy. La durata dell’immunosoppressione cui sono sta-ti esposti i due gruppi è risultata tendenzialmente più lun-ga anche se non statisticamente significativa nel gruppocon epatite più severa (24 mesi/16 mesi) ed inoltre talegruppo è stato esposto mediamente ad un numero mag-giore di fattori di rischio. Conclusioni. L’andamento del-l’epatite nel lungo follow-up non ha mostrato sostanzia-li differenze rispetto alla popolazione non trattata permalattia oncologica. Non si sono osservati casi di cirrosio epatocarcinoma. Il movimento più significativo delletransaminasi nei primi 4 anni dopo l’ off-therapy è vero-similmente relativo alla ricostituzione immunologica e/oal residuo effetto epatotossico dei farmaci. Negli anni suc-cessivi il corso dell’epatite C è stato ondulante come disolito descritto. Non è stato rilevato un fattore di rischiospecifico, ma la lunghezza dell’immunosoppressione e l’e-sposizione a più fattori di rischio concomitanti sembranoessere correlati ad una maggiore attività citolitica. Talidati andranno confermati su popolazioni più numerose dipazienti arruolati in studi prospettici.
P051PROTOCOLLO COOPERATIVO AIEOP-SNC99 RS PER IL TRATTAMENTO DELMEDULLOBLASTOMA A RISCHIO STANDARD IN ETA' PEDIATRICASandri A,* Sardi N,* Basso ME,* Bertin D,* Borgogno M,*Todisco L,* Mastrodicasa L,* Ricardi U,° Genitori L,§Cordero di Montezemolo L,# Madon Eper conto dell'AIEOP* Divisione di Pediatria Oncologica,Università di Torino° Dipartimento di Radiologia eRadioterapia, Università di Torino; §Divisione diNeurochirurgia, ASO OIRM-S. Anna, Torino; #Divisionedi Ematologia Pediatrica, Università di Torino

127
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
Il presente studio riporta i risultati relativi all'applica-zione del protocollo cooperativo AIEOP-SNC 99 RS di trat-tamento del medulloblastoma rischio standard (resezionetotale/subtotale e M0/1) in età pediatrica. Il trattamentoconsiste di 3 fasi: (1) chemioterapia pre-radioterapica (CTpre-RT: Methotrexate ad alte dosi (8 gr/mq)e Vincristina(1,5 gr/m2) il giorno 1 (ciclo 1), seguiti da Carboplatino(600 mg/mq)e Vincristina (1,5 mg/m2) il giorno 8 ed Eto-poside (150 mg/m2)i giorni 8 e 9 (ciclo 2); questo bloccochemioterapico è ripetuto dopo 3 settimane (cicli 3 e 4);ad ogni ciclo viene somministrato Methotrexate intrate-cale alla dose di 12 mg; 2) radioterapia craniospinale iniperfrazionamento, 1 Gy 2 volte al giorno per una dosetotale di 36 Gy sull'encefalo e sul midollo spinale e di 66Gy sulla fossa cranica posteriore; 3)chemioterapia post-radioterapica (CT post-RT): Vincristina (1,5 mg/m2) i gior-ni 1, 8, e 15, CCNU (80 mg/m2) e Cisplatino (70 mg/m2) ilgiorno 1; questo ciclo è ripetuto ogni 6 settimane per untotale di 4 cicli. Sono stati arruolati 54 pazienti, 33 maschie 21 femmine, di età mediana di 8 anni ed 1 mese; 41pazienti sono stati sottoposti ad asportazione totale del-la neoplasia, 13 ad asportazione subtotale; 4 pazienti pre-sentavano il liquor positivo per cellule neoplastiche. Perquanto riguarda la tossicità di grado III-IV OMS, nella fasedi CT pre-RT si è registrata una tossicità prevalentemen-te epatica dopo i cicli 1 (15 pazienti su 51 valutati) e 3 (16su 45) ed ematologica dopo i cicli 2 (30 su 51) e 4 (33 su47); durante la radioterapia 14 pazienti su 48 hanno pre-sentato piastrinopenia, 7 anemia e 6 neutropenia; più del50% dei pazienti è andato incontro a tossicità ematolo-gica durante la CT post-RT. Trentasette pazienti hannoterminato il trattamento al momento dell'ultimo control-lo, con un tempo mediano di follow-up dopo l'off-the-rapy di 29 mesi (range: 0-59 mesi). Ventisette di questi 37pazienti sono in remissione completa continua di malat-tia, mentre 10 sono recidivati ad un tempo mediano di 7mesi (range 2-24 mesi) dall' off-therapy. Un paziente èrecidivato in corso di trattamento. Le sopravvivenze glo-bale e libera da eventi a 5 anni dalla diagnosi (Figura 1)sono rispettivamente dell'85,1% (SE: 5,7%) e del 73,0%(SE: 7,2%).
Parzialmente finanziato da CNR, MIUR ex-40%,grant E. M.
P052VALUTAZIONE NEUROCOGNITIVA IN BAMBINI CON TUMORI CEREBRALIIuvone L, Peruzzi L,* Ruggiero A, Attinà G, Lazzareschi I,Maurizi P, Caldarelli M,° Tamburrini G,° di Rocco C,°Guzzetta F,* Riccardi Riv. Oncologia Pediatrica, °Div. Neurochirurgia Infantile, *Div. Neuropsichiatria Infantile, Università Cattolica delSacro Cuore, Roma
Introduzione e Obiettivi: Numerosi fattori, oltre l’età del-la diagnosi ed il trattamento radioterapico, quali sede edestensione del tumore, trattamento medico e chirurgico,possono causare deficit neurocognitivi a lungo termine neibambini affetti da tumori cerebrali. Una valutazione pro-spettiva neuropsicologica può pertanto permettere di iden-tificare l’influenza dei singoli fattori sullo sviluppo neuro-cognitivo. Metodi: Sono stati valutati in maniera prospet-tiva 85 bambini (54 maschi e 31 femmine), con un’etàmedia alla diagnosi di 8. 3 anni. 44 pazienti sono stati valu-tati alla diagnosi prima di procedure mediche o chirurgiche;12 nei primi 6 mesi dopo la chirurgia; 29 pazienti sono sta-ti valutati dopo più di 6 mesi dall’inizio del trattamento. In45 pazienti il tumore era a livello della fossa cranica poste-riore, in 15 casi la localizzazione era sovratentoriale a livel-lo della linea mediana, in 25 bambini il tumore interessa-va la corteccia emisferica. I principali istotipi erano rap-presentati da medulloblastoma ed astrocitoma di bassogrado. In tutti i bambini sono state valutate, oltre al quo-ziente intellettivo (verbale e di performance), specificheabilità cognitive quali memoria (test verbale e spaziale),linguaggio (Token test e batteria per l’analisi di deficit afa-sici), funzioni visuo-spaziali (Bell test revisionato, Beerytest, Rey Figure copy, Progressive Colored Matrices byRaven), funzioni frontali (Tower of London e Wisconsin CardSorting test) e lateralizzazione (Piaget-head test, questio-nario di Oldfield). Risultati: La sede del tumore appare stret-tamente correlata con il profilo cognitivo. I tumori dellafossa cranica posteriore non determinano deficit linguisti-ci, influenzando però le funzioni frontali e l’integrazionevisuo-motoria. I tumori sovratentoriali profondi produco-no riduzioni significative dei test di memoria. La durata deisintomi influenza in maniera negativa il quoziente intel-lettivo. Conclusioni: I deficit cognitivi sono già presenti almomento della diagnosi e sono principalmente associatialla sede del tumore. Una completa valutazione neuroco-gnitiva al momento della diagnosi è importante per poteridentificare il ruolo specifico del tumore, della chirurgia edella chemioterapia nello sviluppo cognitivo dei bambinicon tumori cerebrali.
P053TOPOTECAN-VINCRISTINA-DOXORUBICINA (TVD) PER I PAZIENTI NONRESPONSIVI AL PROTOCOLLO EUROPEO NEUROBLASTOMA ALTO RISCHIOBiasotti S,# Luksch R,• Podda M,• Viscardi E,@;Bianchi M,* Aru B,° Nantron M,# Garaventa A#
#U. O. Ematologia ed Oncologia Pediatrica Istituto G.Gaslini Genova; •U. O. di Pediatria Istituto NazionaleTumori Milano; @; Dipartimento di Pediatria Universitàdi Padova; *Dipartimento di Pediatria Oncologica

128
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Tumori Solidi
Ospedale Regina Margherita Torino, °Dipartimentodi Pediatria Università di Cagliari
Introduzione e Obiettivi: Studi di fase I e II nell'adulto enel bambino hanno dimostrato l’attivita' antitumorale delTopotecan, analogo semisintetico delle Camptotecine ini-bitore specifico della Topoisomerasi I. Recentementeabbiamo condotto uno studio di fase 2 dell' associazionedi Topotecan/Vincristina e Doxorubicina, in pazienti conNeuroblastoma resistente alla terapia di prima linea orecidivato concludendo che questa associazione e' attivae ben tollerata. Scopo del presente studio e' stato valuta-re l’attivita' dell’associazione TVD in pazienti con neuro-blastoma scarsamente responsivi alla fase di induzione(COJEC) del protocollo europeo E-SIOP NB AR01 e perciònon eleggibili alla randomizzazione sul tipo di chemiote-rapia mieloablativa(R1). Metodi: pazienti con neurobla-stoma alto rischio arruolati nel protocollo E-SIOP NB AR01che, al termine degli 8 cicli di chemioterapia d'induzione(COJEC), abbiano ottenuto una risposta inadeguata perprocedere alla terapia di consolidamento ad alte dosi.Terapia:Topotecan 1,5 mg/mq/die in 30' ev giorni 1-5,Vin-cristina 2 mg/mq in 48 h ic giorni 5-6, Doxorubicina 45mg/mq in 48 h ic giorni 5-6. I cicli sono stati erogati ogni21 giorni con le medesime modalità. Risultati: Sono statitrattati 19 pazienti affetti da Neuroblastoma stadio 4 cheal termine del COJEC presentavano il seguente stato dimalattia:6 RP (remissione parziale con piu' di 3 lesioniMIBG positive),10 in MR (risposta minore), 2 in MS(malat-tia stabile),1 non responsivo. Complessivamente sono sta-ti erogati 70 cicli di TVD. Risposta: un paziente ha otte-nuto RC, 2 VGPR, 12 RP, 2 MR e 2 hanno presentato PM.In 11 pazienti al termine della terapia con TVD è statadocumentata una risposta di malattia che consentiva laripresa del Protocollo E-SIOP NB AR01 con il programmadi consolidamento ad alte dosi. Tossicita': tutti i pazientihanno presentato tossicità ematologica di grado 4. Unpaziente ha sviluppato sepsi CVC correlata, uno mucositedi grado 3. Nessun paziente e' deceduto per tossicita'. In4 pazienti è stata eseguita raccolta di PBSC. Conclusioni:La combinazione TVD si conferma attiva e ben tollerata inpazienti affetti da Neuroblastoma alto rischio ed in gra-do di migliorare la risposta ottenuta con la fase di indu-zione del protocollo ESIOP AR 01.
P054RECIDIVA LOCALE TARDIVA A 20 ANNI DALLA DIAGNOSI DIOSTEOSARCOMA TIBIALEBrach del Prever A,* Comandone A,** Berta M,* Ferrari S,# Boglione A,** Brach Del Prever E,##
Gino G,## Pagano M,* Mercuri M#
*Dipartimento di Onco - Ematologia ed Immuno-Infetti-vologia, Università degli Studi di Torino, A. S. O. O. I. R. M.S. Anna; **Ospedale Gradenigo, Divisione di OncologiaMedica, Torino; #Dipartimento di OncologiaMuscoloscheletrica, Istituti Ortopedici Rizzoli, Bologna;##Ospedale CTO - M. Adelaide, Dipartimento di Ortopedia,Università degli Studi di Torino
A seguito del riscontro di lesione litica metafisaria dista-le della tibia destra, nel giugno 1984 giunse alla nostra
attenzione un ragazzo di 13 anni e 8 mesi di età. L’esamebioptico incisionale pose diagnosi di osteosarcoma. LaTomografia Computerizzata (TC) del torace risultò nellanorma, la scintigrafia ossea non rilevò la presenza di lesio-ni secondarie. Il paziente fu quindi trattato secondo il pro-tocollo IOR/NEO I comprendente chemioterapia (CT) preo-peratoria con Vincristina, Methotrexate (MTX) 7. 5 g/m2, eCisplatino (CDP) i. a. , seguito da resezione tibiale ampia,sostituzione con innesti ossei autoplastici e rinforzomediante chiodo di Kuntscher tibio-calcaneare. L’esameistologico rivelò margini chirurgici di resezione ampi enecrosi tumorale del 95%; il paziente fu pertanto tratta-to con CT postoperatoria comprendente MTX, CDP e. v. edAdriamicina 90 mg/m2.Gli esami strumentali al termine deltrattamento e nel corso del follow-up dimostrarono lo sta-to di remissione completa (RC) di malattia. Nel maggio2003 comparve tumefazione dolorosa a livello del terzodistale della gamba destra. Gli esami radiologici rilevaro-no la presenza di ampia lesione litico-addensante tibiale.La scintigrafia ossea confermò la presenza di area di iper-captazione tibiale destra. L’esame istologico su biopsiaincisionale dimostrò trattarsi di osteosarcoma teleangec-tasico. Fu pertanto somministrata CT neoadiuvante conMTX, CDP ed Ifosfamide (IFO), seguita da intervento diamputazione, e CT post-operatoria con CDP ed IFO. Gliesami strumentali al termine del trattamento hanno dimo-strato la RC di malattia. Attualmente il paziente è in buo-ne condizioni generali. Conclusioni. La prognosi dei pazien-ti con osteosarcoma localizzato è significativamentemigliorata nel corso degli ultimi decenni. Questo caso, conrecidiva locale molto tardiva (229 mesi dalla diagnosi) sot-tolinea le difficoltà tuttora presenti nello stabilire quandoconsiderare un paziente definitivamente guarito.
P055NEOPLASIA CEREBRALE EMBRIONARIA RICCA IN NEUROPILO E ROSETTEVERE: UNA VARIANTE O UNA NUOVA ENTITÀ NEOPLASTICA?La Spina M,° Perilongo G,* Skrap M, Russo GCentro di Riferimento Regionale di Emato-OncologiaPediatrica. Azienda Policlinico – Università di Catania,Via S. Sofia 78, 95124 Catania: °Divisione diEmato-Oncologia Pediatrica, Università di Padova -Ospedale di Padova, Via Giustiniani 3, 35128 Padova.*Divisione di Neurochirurgia, Ospedale S. Maria dellaMisericordia, Udine, Italy
Introduzione: la neoplasia cerebrale embrionaria riccain neuropilo e rosette vere costituisce una rara entità conpeculiari caratteristiche istologiche, immunoistochimichee ultrastrutturali, esordio tra 1-3 anni, localizzazione soprae sottotentoriale, prognosi infausta. Sono descritti solo 7casi: 5 deceduti a 5-14 mesi dalla diagnosi, 1 in evidenzadi malattia a 7 mesi da resezione e chemioterapia, 1 inassenza di malattia a 30 mesi dalla diagnosi. Nessuno diquesti ha eseguito radioterapia, per età inferiore a 3 anni.Caso clinico: RA, anni 5, maschio, da circa venti giornimodificazioni del comportamento e disturbi dell’equilibrioai quali, si sono aggiunti iperemia congiuntivale, tumefa-zione palpebrale, strabismo convergente occhio sinistro eprogressiva incapacità a deambulare. Esame obiettivo:

129
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
emiplegia emisoma destro, mano ad artiglio, strabismoconvergente occhio sn, ptosi palpebrale e deviazione delcapo a dx. Incapacità di deambulare, mantenere la stazio-ne eretta e seduta. ROT ipoelicitabili a sn. RM encefalo:lesione espansiva ponto-mesencefalica sn, a contenutonecrotico-colliquativo di circa 25 mm di diametro max concomponente nodulare a carico della base della compo-nente cistica della stessa. Intervento neurochirurgico diasportazione radicale. Obiettività neurologica post-inter-vento invariata. TC e RM post-intervento confermavano laradicalità dell’intervento. L’esame istologico e le indaginidi immunoistochimica sono risultati compatibili con unaneoplasia neuroepiteliale embrionaria ad alto grado, conaspetti suggestivi per tumore embrionario ricco in neuro-pilo e rosette vere. In accordo con il gruppo neurooncolo-gico AIEOP è stata eseguita chemioterapia secondo Proto-collo Tumori Maligni SNC Alto Rischio con rescue di sta-minali (HD-MTX + VCR, VP16, EDX + VCR, CBDCA) e radio-terapia craniospinale iperfrazionata; attualmente ha com-pletato il settimo degli otto cicli di chemioterapia di man-tenimento secondo protocollo SIOP/PNET (CDDP/CCNU/VCR). Ultimo controllo RM: remissione completa a 17 mesidalla diagnosi. xConclusioni: l’interpretazione del caso sin-golo e il follow-up ancora breve non permettono di trarrerisultati definitivi, ma la remissione completa dopo 17 mesisuggerisce che, dopo asportazione radicale, tale schemachemioterapico associato alla radioterapia, rappresentauna valida strategia per il controllo di questa rara e aggres-siva neoplasia.
P056SARCOMI DELLE PARTI MOLLI DI TIPO ADULTO IN ETA’ PEDIATRICA:ANALISI RETROSPETTIVA DI 182 PAZIENTI TRATTATI ALL’ISTITUTONAZIONALE TUMORI DI MILANOFerrari A, Casanova M, Meazza C, Luksch R,Massimino M, Cefalo G, Terenziani M, Spreafico F,Polastri D, Podda M, Catania S, Cereda S, Marcon I,Gandola L, Collini P, Gronchi AIstituto Nazionale per lo Studio e la Cura dei Tumori
Introduzione. I sarcomi delle parti molli non-rabdomio-sarcoma rappresentano circa il 3% dei tumori pediatrici ecostituiscono un eterogeneo gruppo di neoplasie la cuistrategia terapeutica ottimale non è ancora definita. Inparticolare, non vi è un consenso generale sul ruolo dellachemioterapia: questi tumori sono in genere consideratinon-chemiosensibili, ma dati recenti derivati da esperien-ze nei sarcomi dell’adulto sembrano suggerire che i pazien-ti ad alto rischio (alto grado, dimensioni elevate) possanoavere un vantaggio significativo in termini di sopravviven-za quando trattati con chemioterapia intensiva (con ifo-sfamide e adriamicina). Pazienti e Metodi. Al fine di iden-tificare un gruppo di tumori il più omogeneo possibile,sono stati definiti come sarcomi delle parti molli di tipoadulto quei sarcomi a) tipici dell’età adulta (escludendocosì ad esempio i fibrosarcomi infantili), b) sicuramentemaligni (escludendo istotipi borderline come l'emangioen-dotelioma), c) con morfologia riferibile a tessuti maturidifferenziati (escludendo i sarcomi a piccole cellule roton-de come rabdomiosarcoma, sarcoma di Ewing/pPNET
extraosseo e tumore desmoplastico a piccole cellule). Sonostati analizzati 182 pazienti di età inferiore a 18 anni, trat-tati presso il nostro Istituto in un periodo di 25 anni. La chi-rurgia ha rappresentato il cardine del trattamento di que-sti pazienti; la radioterapia è stata utilizzata in 73 casi,mentre 114 hanno ricevuto chemioterapia (70 come trat-tamento adiuvante). Risultati. La sopravvivenza a 5 anni èrisultata essere del 89% nei pazienti sottoposti a chirur-gia completa alla diagnosi (IRS gruppo I), 79% in quelli sot-toposti a chirurgia marginale (gruppo II), 52% nei pazien-ti non-resecabili alla diagnosi (gruppo III), 17% nei pazien-ti metastatici (gruppo IV). I pazienti con tumore ad altogrado (G3) e di dimensioni > 5 cm hanno evidenziato unalto rischio di diffusione metastatica anche dopo chirur-gia iniziale macroscopicamente adeguata (sopravvivenzalibera da metastasi 36%); in questo sottogruppo, la che-mioterapia adiuvante è sembrata essere utile (tutti ipazienti che non hanno ricevuto chemioterapia hanno svi-luppato metastasi). Tra i pazienti inizialmente non-rese-cabili (sopravvivenza libera da eventi 45%), il tasso dirisposta alla chemioterapia è stato del 58%; i pazienti chehanno risposto alla chemioterapia e che hanno beneficia-to di una chirurgia differita completa hanno evidenziatouna sopravvivenza libera da eventi intorno al 70%. Con-clusioni. Nei sarcomi delle parti molli l’identificazione difattori prognostici è fondamentale per permettere di dise-gnare terapie adattate al gruppo di rischio. I pazienti contumore ad alto grado di malignità e di elevate dimensioni(anche se asportato chirurgicamente alla diagnosi), oltreche i pazienti con tumore non-resecabile, presentano unalto rischio di diffusione metastatica di malattia. Purrestando la chirurgia il cardine del trattamento di questitumori, i nostri dati – assieme ad alcune recenti esperien-ze nei sarcoma dell’adulto – sembrano poter cautamentesuggerire che una chemioterapia intensiva possa avere unruolo forse più significativo di quanto generalmente rico-nosciuto
P057TOPOTECAN E CICLOFOSFAMIDE SEGUITI DA ICE IN PRIMA LINEA NEIPAZIENTI AFFETTI DA NEUROBLASTOMA AD ALTO RISCHIODonfrancesco A, Jenkner A, Castellano A, Ilari I,Milano GM, de Ioris MA, Serra A, Dominici C, de Sio L,Fidani P, Cozza R, De Laurentis CU. O. di Oncologia, Ospedale Pediatrico Bambino Gesù,IRCCS - Roma
Introduzione ed obiettivi: il trattamento del neurobla-stoma avanzato continua ad essere insoddisfacente, manuovi agenti e nuove associazioni sono risultate attive instudi preclinici e clinici. Uno studio pilota con Topotecanimpiegato in prima linea è stato iniziato nell'Aprile2001.Metodi: due cicli di Topotecan 6 mg/m2 con Ciclofo-sfamide 4,2 g/m2 (140 mg/kg) erano seguiti da 2 cicli ICE(Ifosfamide 9 g/m2, Carboplatino 800 mg/m2 ed Etoposi-de 500 mg/m2), raccolta di CSSP (immunoselezione), chi-rurgia, un ciclo CAV (Ciclofosfamide 3 g/m2, Doxorubici-na 75 mg/m2 e Vincristina 1,5 mg/m2), terapia mieloa-blativa ETC (Etoposide 600 mg/m2, Tiotepa 750 mg/m2 eCiclofosfamide 120 mg/kg), reinfusione di CSSP, radiote-

130
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Tumori Solidi
rapia iperfrazionata locale (21 Gy in 7 giorni) ed infineCRA (160 mg/m2 per 12 mesi). Sono stati arruolati 14pazienti consecutivi affetti da neuroblastoma ad altorischio oltre 1 anno di età: maschi/femmine 6/8, etàmediana 32,5 mesi (range 13 - 66). 1 INSS stadio 2 (MYC-NA e 1pdel), 1 stadio 3 (MYCNA e 1pdel), 12 stadio 4 (1MYCNA e 1pdel, 1 MYCNA). 4 ulteriori pazienti non han-no ancora completato l'induzione. Risultati: la tossicitàera prevedibile e quasi esclusivamente ematologica, gra-do WHO 3-4 in tutti i cicli. Mediana dell'intervallo tra ciclitopo-ciclo era di 29 giorni (range 23 - 35), le risposteINRC osservate erano 1 CR, 1 VGPR, 6 PR e 6 MR (RR57%). Mediana dell'intervallo tra cicli ICE era di 32 gior-ni (range 23 - 40), le risposte osservate al termine dell'in-duzione erano 1 CR, 1 VGPR, 9 PR e 3 MR (RR 79%). 13/14paz. hanno raccolto CSSP. Dopo chirurgia abbiamo osser-vato 7 CR, 6 PR, 1 paz. è TETE (RR 100%). Mediana del-l'intervallo tra reinfusione di CSSP e radioterapia era di 45giorni (range 31 - 62). Dopo un follow-up mediano di 17mesi (range 7 - 29), nei 10 pazienti che hanno completa-to il trattamento abbiamo osservato 1 DOD (stadio 3 MYC-NA e 1pdel) a 12 mesi dalla diagnosi, 2 CR (stadio 4) a 27e 24 mesi; 2 SD (stadio 4, 1 MYCNA) sono PFS a 15 e 12mesi, 2 PD (stadio 4) a 26 e 14 mesi sono AWD a 29 e 19mesi, 2 RD (stadio 4 e stadio 2, MYCNA e 1pdel) a 28 e 12mesi sono AWD a 29 e 13 mesi; 1 DOC (stadio 4 MYCNAe 1pdel) dopo chirugia a 7 mesi dalla diagnosi. Conclu-sioni: dopo induzione e chirurgia abbiamo osservato neiprimi 14 pazienti una RR del 100%; non abbiamo osser-vato morti tossiche ne PD durante l'induzione ed è statopossibile raccogliere CSSP in oltre il 90% dei pazienti. Lacombinazione topo-ciclo seguita da ICE risultava attiva eben tollerata in questa popolazione ad alto rischio, conuna qualità della vita paragonabile ad altri protocolli diinduzione convenzionali. Il semplice schema, accompa-gnato da una tossicità prevedibile, si presta all'associa-zione con modificatori della risposta biologica ed altreterapie innovative, anche in forma randomizzata, nellafase post-induzione.
P058SCOMPENSO CARDIACO ACUTO COME PRIMO SINTOMO DIPARAGANGLIOMA PELVICO IN ETA’ PEDIATRICAD’angelo P, Caselli D, Russo D, Tropia S, Ziino O,Cipriani A,° Alaggio R,* lo Cascio M,** Aricò MOncoematologia Pediatrica, °Cardiochirurgia Pediatrica,**Chirurgia Pediatrica, Ospedale dei Bambini, Palermo;*Anatomia Patologica, Università di Padova
Il paraganglioma, varietà extra-surrenale del feocro-mocitoma, è rarissimo nei bambini e di solito localizzatoin cavità addominale. Descriviamo un caso di paragan-glioma pelvico esordito con scompenso cardiaco acuto. R.R., femmina, 12 anni; dopo circa 1 mese di episodi di cefa-lea e distermia presenta prostrazione, pallore, tachicardia;l’ospedale periferico, posta diagnosi di scompenso cardia-co acuto, la trasferisce presso la nostra UTIC ove giungecon intenso pallore cutaneo e delle mucose, sudorazioneprofusa algida, lieve polipnea, tachicardia 190/m. Cineti-ca ventricolare depressa, frazione di eiezione 30%. Dopo
terapia con isotropi, vasodilatatori, beta-bloccanti, miglio-ra rapidamente (FE>65%), dimessa in 10° giornata edinviata in Mal. Infettive per sospetta miocardite. L’eco-grafia mostra una neoformazione retrovescicale, per cui civiene inviata. All’E. O. l’unico dato patologico è l’iperten-sione arteriosa (155/110 mmHg); AVM urinario di 37mg/24 h. (0.8 mg/Kg/24 h. ) con rapporto AVM/creat. 45.29 (v. n. < 12); NSE 48 ng/ml (v. n. <15). TC addome:neoformazione solida di 5 cm nello scavo pelvico a mar-gini lobulati, con disomogenea captazione del mdc.Durante l’intervento chirurgico di exeresi radicale dellaneoplasia, già nella fase iniziale presenta severe crisi iper-tensive (sistolica 160-240, diastolica 85-140 mmHg),nonostante il trattamento farmacologico. La gravità del-le crisi ipertensive aumenta con la manipolazione dellaneoplasia. Al contrario, nelle fasi finali dell’intervento,dopo l’asportazione del tumore, sviluppa 2 gravissime cri-si ipotensive con bradicardia fino all’arresto cardiaco,superate solo grazie a procedure rianimatorie estreme delcardiologo, tra cui la somministrazione di 19 + 16 fiale diadrenalina nelle fasi di arresto. Al termine dell’interventola bambina è stata ricoverata per 4 giorni in U. O. di Tera-pia Intensiva e successivamente dimessa in ottime condi-zioni in 9° giornata. Tutti gli accertamenti eseguiti inseguito hanno permesso di escludere la presenza di esiticardiocircolatori e/o neurologici delle fasi critiche diiper/ipotensione. L’esame istologico della neoplasia docu-menta un paraganglioma. A 11 mesi dall’intervento labambina è in ottime condizioni, con livelli pressori, indicidi contrattilità cardiaca, amine urinarie ed NSE stabil-mente normali ed imaging negativo. Questo caso di para-ganglioma pediatrico è eccezionale per la sede pelvica ati-pica e l’esordio con grave scompenso cardiaco acuto. Iltrattamento chirurgico pur indispensabile va affrontatoin modo multidisciplinare per l’elevato rischio di morta-lità secondaria alla capacità secernente di queste neopla-sie neuroendocrine.
P059TUMORI MALIGNI A CELLULE GERMINALI DELLA VAGINA: OMISSIONEDEL TRATTAMENTO LOCALE DAL PROGRAMMA TERAPEUTICOTerenziani M, Spreafico F, Piva L, Massimino M,Cereda S, Casanova M, Cefalo G, Ferrari A, Luksch R,Polastri D, Podda M, Fossati-Bellani FUO Pediatria, Istituto Nazionale per lo Studio e la Curadei Tumori, Milano
Introduzione e obiettivi. I tumori maligni a cellule ger-minali (TMCG) ottengono un’ alta percentuale di guari-gione anche negli stadi avanzati, grazie a definiti approc-ci multidisciplinari basati principalmente sulla chemiote-rapia e la chirurgia. I TMCG della vagina sono rari e rap-presentano una sede critica per la decisione riguardanteil trattamento locale, per il rischio di mutilazioni o di gra-vi sequele tardive. Metodi. Descriviamo 2 case reports diTMCG della vagina trattati con sola chemioterapia. Risul-tati. Case 1.Bimba di 6 mesi giunta alla diagnosi di TMGCin seguito alla comparsa di perdite ematiche dalla vagi-na. La Tac dell’addome e pelvi dimostra adenopatie retro-peritoneali e una massa a partenza vaginale e il valore

131
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
della FP, corretto per l’età, risulta francamente patologi-co (3000 UI/ml). Dopo l’accertamento bioptico eseguitosulla massa, la pz inizia la chemioterapia secondo lo sche-ma PEB (cisplatino, VP16 e bleomicina), di cui esegue 6cicli ogni 4 settimane ottenendo la remissione completadella malattia alla TAC e la normalizzazione della FP. Labimba è viva e libera da malattia 14 anni dopo il terminedelle cure. Case 2.Bimba di 2 anni giunta alla diagnosi perperdite ematiche dalla vagina e disturbi sfinterici. La Tacdell’addome e pelvi dimostra una voluminosa lesione ute-rina aggettante in vagina e il valore della FP risulta fran-camente patologico (100.000 UI/ml). Dopo l’accertamen-to bioptico eseguito sulla massa, la pz inizia la chemiote-rapia secondo lo schema PEB (cisplatino, VP16 e bleomi-cina), di cui esegue 4 cicli ogni 3 settimane ottenendo lanormalizzazione della FP. Un nodulo residuo in sede cer-vico-vaginale, biopsiato al termine e dopo 6 mesi dallafine della chemioterapia, non evidenzia tumore vitale resi-duo. La bimba è viva e libera da malattia a 2 anni dal ter-mine delle cure. Conclusioni. L’omissione del trattamentolocale può essere considerato una valida opzione tera-peutica nei TMCG in remissione completa e in cui la sederisulti critica per mutilazioni e/o sequele invalidanti.
P060VALUTAZIONE DELLA FUNZIONE GONADICA IN PAZIENTI TRATTATI PERNEOPLASIA IN ETA’ PEDIATRICA: IFOSFAMIDE VS CICLOFOSFAMIDECarli OM,* Pizzato C,* Bisogno G,* Bettella A,°Ferrari A,°° Merico M,° Zanetti I,* Viscardi E,* Foresta C°
*Clinica di Oncoematologia - Dipartimento diPediatria - Azienda Ospedaliera Università di Padova;°Centro di Crioconservazione dei Gameti Maschili -Azienda Ospedaliera Università di Padova; °°Divisione diOncologia Pediatrica, Istituto Nazionale per la Cura deiTumori, Milano
Introduzione e Obiettivi: La tossicità gonadica è unodegli effetti collaterali a lungo termine secondario all’u-so dei farmaci alchilanti. Poco si sa sulla gonadotossicitàindotta dall’Ifosfamide (IFO). L’ obiettivo dello studio èstato quello di valutare la gonadotossicità dell’IFO, con-frontandola con quella della Ciclofosfamide (CPM) inpazienti trattati per tumore durante l’infanzia. Metodi:Sono stati valutati 30 soggetti maschi, postpuberi, fuoriterapia, trattati durante l’infanzia per sarcoma delle par-ti molli e dell’osso. L' età mediana era: alla diagnosi 8 anni(range 1-17), al momento della valutazione andrologica20 anni (range 15-31); follow-up mediano 12,5 anni (ran-ge 1,5-23). 22 pazienti hanno ricevuto IFO (Gruppo A), 8CPM (Gruppo B). La valutazione della funzione gonadicaha comportato: anamnesi e visita andrologica; valutazio-ne ecografica testicolare; dosaggio ormonale (FSH, Inibi-na B, LH, Testosterone, Prolattina ed Estradiolo); spermio-gramma. Risultati: Gruppo A: Tutti i pazienti hanno rag-giunto uno sviluppo puberale adeguato per l’età. Volumetesticolare medio: 12,4 ± 3,6 ml. Valori ormonali: FSH8,4±10,4 mUI/ml, Inibina B 155±144,7 ng/l, LH 3,7±2,5mUI/ml, Testosterone 645±280, 2 ng/100 ml. 6/22 pazien-ti sono risultati oligozoospermici, 2/22 criptozoospermi-ci, 1 azoospermico. 2/12 trattati in età pre-pubere hannoavuto una compromissione della fertilità (oligozoosper-
mici) rispetto a 7/10 trattati dopo la pubertà (4/7 oligo-zoospermici, 2/7 criptozoospermici, 1/7 azoospermico).Gruppo B: Tutti i pazienti, tranne uno, hanno raggiuntouno stadio di sviluppo puberale adeguato per l’età. Volu-me testicolare medio: 5,9±2,5 ml. Valori ormonali: FSH25±14,8 mUI/ml, Inibina B 64,8±78 ng/l, LH 7,2±4 mUI/ml,Testosterone 397,8±163,3 ng/100 ml. 6/8 pazienti hannoeseguito la raccolta del liquido seminale: 5 sono risultaticripto-azoospermici e 1 gravemente oligospermico. Con-clusioni: L’IFO può essere causa di gonadotossicità, tutta-via il danno testicolare sembra essere inferiore rispetto aquello provocato dalla CPM, sebbene la numerosità deipazienti sia bassa. Il danno testicolare appare maggiorequando l’IFO viene somministrata dopo la pubertà, men-tre non abbiamo evidenziato una correlazione con la dose.
P061FEOCROMOCITOMA SPORADICO COME SPIA DI SINDROMI GENETICHE:UN REPORT PRELIMINARE DAL PROGETTO TREPBonutti A, Murgia A, Cecchetto G, Opocher G,Martella M, Indolfi P, Conte M, Tamburini A, Ferrari A,Bisogno GClinica di Oncoematologia, Dipartimento di Pediatria -Azienda Ospedaliera Università di Padova
Introduzione e Obiettivi. Il feocromocitoma è una neo-plasia rara in età pediatrica, più frequentemente sporadi-ca e solo nel 10% dei casi rappresenta una manifestazio-ne di una sindrome ereditaria (MEN IIa, MEN IIb, Sindromedi von Hippel Lindau, Neurofibromatosi 1, sindrome di Stur-ge Weber). In letteratura sono descritte mutazioni geneti-che a carico dei geni ret, VHL, SDHD e SDHB nel 24% deifeocromocitomi sporadici. Abbiamo analizzato l’incidenzadi alterazioni genetiche nei pazienti con feocromocitomaregistrati nell’ambito del progetto TREP (Tumori Rari in EtàPediatrica) in database. Metodi. Dal gennaio 2000 al mag-gio 2004 sono stati registrati 10 pazienti con feocromoci-toma e paraganglioma. Un paziente è stato perso al follow-up, mentre 2 pazienti con diagnosi risalente al 1997 e 1999sono stati recuperati per l’analisi genetica. L’analisi gene-tica per mutazioni dei geni RET, VHL, SDHD e SDHB è sta-ta eseguita in 7 pazienti. Risultati. L’età media alla dia-gnosi degli 11 pazienti valutabili (6 xy, 5 xx) era 12.5 anniL’anamnesi era negativa per sindromi genetiche. Tutti ipazienti presentavano malattia localizzata (9 feocromoci-toma, di cui uno bilaterale, e 2 paraganglioma). La terapiaè stata chirurgica in tutti i casi e nessun paziente ha pre-sentato recidiva durante il follow-up. L’analisi genetica èrisultata positiva per VHL su due (1 xx, 1 xy), dei 7 pazien-ti esaminati. L’età all’esordio era 13 e 16 anni ed entram-bi presentavano localizzazione unilaterale. Conclusioni. Ilmanifestarsi di un tumore raro in età pediatrica può esse-re in alcuni casi il primo segno di una sindrome genetica.La nostra esperienza dimostra la validità dell’ indicazionealla analisi genetica routinaria per i pazienti con feocro-mocitoma sporadico. La presa in carico di questi pazientinon si deve pertanto limitare al solo aspetto terapeutico,ma deve comprendere anche l’analisi genetica e l’eventua-le counseling familiare. Lo sviluppo di un progetto TREP,nell’ambito AIEOP, permette di sviluppare un approcciocomplessivo ai pazienti con tumore raro in età pediatrica.

P062CHEMIOTERAPIA AD ALTE DOSI SEQUENZIALI CON CICLOFOSFAMIDE,
ETOPOSIDE, MITOXANTRONE+MELPHALAN E AUTOTRAPIANTO DI CELLULE
STAMINALI EMOPOPOIETICHE IN BAMBINI AFFETTI DA EWING’S SARCOMA
IN RICADUTA
Podda M,* Meazza C,* Cereda S,* Catania S,* Sironi O,*Bozzi F,* Barzanò E,* Ravagnani F,° Coluccia P,°Gandola L,# Luksch R*
*UO Pediatria, °UO Medicina di Laboratorio 2, #UORadioterapia B - Istituto Nazionale per lo Studio e laCura dei Tumori - Milano
Introduzione e obiettivi. Nel periodo 1994-2000, ipazienti con di sarcoma di Ewing non metastatico in pri-ma ricaduta venivano trattati con un piano di cura inten-sivo, con l’obiettivo di ottenere remissioni durature epazienti lungosopravviventi. Metodi. Lo studio prevedevaVCR+CTX+ADR+DACT x 4 cicli mensili, trattamento loca-le su base individuale (chirurgia e/o radioterapia), e suc-cessivamente una chemioterapia ad alte dosi sequenzialicon CTX 7g/m2+G-CSF e successiva leucaferesi per la rac-colta di precursori emopoietici circolanti (PEC), VP162g/m2 + G-CSF, e infine mitoxantrone 60mg/m2+L-PAM180mg/mq e autotrapianto. Sono stati arruolati 13pazienti consecutivi (età mediana: 15 anni; tempo media-no alla ricaduta: 26 mesi, range 12-126). Le sedi dellaricaduta erano: scheletro-6, scheletro+polmoni-3, pol-moni-3, ripresa locale-1pz. Risultati. Durante il tratta-mento non si sono verificate né progressioni né eventiseveri avversi, ed è stato possibile raccogliere in tutti unnumero adeguato di PEC. Il tempo medio di attechimen-to dopo autotrapianto è stato 11 giorni per PMN e 19giorni per le piastrine. Il follow-up mediano dal momen-to della ricaduta è di 61 mesi, con probabilità di soprav-vivenza a 3 anni dall’autotrapianto = 0.4. Suddividendo ipazienti per tempo alla ricaduta, la probabilità è 0.7 per ipazienti con ricaduta tardiva (>24 mesi dalla diagnosi) e0 quelli con ricaduta precoce (??24 mesi dalla diagnosi).In questa casistica nessuna delle altre variabili analizza-te (età, sesso, sede/i di ricaduta) ha un impatto progno-stico. Conclusioni. Pur con i limiti della numerosità delcampione, il piano di cura si è dimostrato fattibile. I risul-tati suggeriscono che pazienti con sarcoma di Ewing nonmetastatico all’esordio che ricadono a oltre due anni dal-la diagnosi possono beneficiare di un trattamento inten-sivo con chemioterapie ad alte dosi, mentre questa stra-tegia non offre vantaggi significativi per i pazienti conricaduta precoce.
P063STUDIO DELLA TIROSINA IDROSSILASI (TH) NELLA VALUTAZIONE DIINFILTRAZIONE MIDOLLARE IN PAZIENTI AFFETTI DA NEUROBLASTOMA(NBL) MEDIANTE REAL-TIME RT-PCRLibri V, Conforti R, Fronza R, Mignani E, Prete A,Melchionda F, Pession AOncoematologia Pediatrica e Terapia Cellulare, ClinicaPediatrica S. Orsola-Malpighi, Bologna
Introduzione e obiettivi: Il NBL è il più comune tumoresolido extracranico dell'età pediatrica, costituendo circa il6% delle neoplasie maligne dell'infanzia. Le cellule di NBLhanno la caratteristica di sintetizzare catecolamine, cherisultano alterate nel 95% dei pazienti. La TH, primo enzi-ma limitante il pathway di biosintesi delle catecolamine, èuno dei marcatori tumorali tessuto-specifici di tale neo-plasia maligna. Oltre all'impiego di tecniche qualitative, ilricorso a metodi quantitativi di real-time RT-PCR dell'e-spressione del mRNA della TH può essere utilizzato peridentificare infiltrazione midollare di NBL alla diagnosi e permonitorare la malattia minima residua (MRD). Materiali eMetodi: E’ stata utilizzata Real-Time RT-PCR per quantifi-care il grado di espressione della TH su sangue midollare(BM) in: 7 campioni di 5 pazienti con NBL stadio I; 16 cam-pioni di 10 pazienti in stadio II; 17 campioni derivati da 9pazienti in stadio III, ed in 37 campioni di 24 pazienti in sta-dio IV. Il grado di espressione del gene della TH è stato defi-nito mediante il rapporto tra mRNA di tale gene e quellodella β-actina. Risultati: L’espressione positiva di mRNAdella TH è stata individuata nei seguenti pazienti suddivisiper stadio: 1/5 nel gruppo in stadio I; 0/10 nei pazienti instadio II; 2/9 nei pazienti in stadio III e 20/24 nei pazientiin stadio IV. La ROC analisi utilizzata ha dimostrato unasensibilità del 70.8% e una specificità del 75%. E’ statoosservato inoltre che l’espressione positiva del mRNA del-la TH non è correlata con l’amplificazione di N-myc e ladelezione dell' 1p in tutti i pazienti portatori di tali altera-zioni. Conclusioni: La concordanza con le analisi diagno-stiche standard evidenziata nello stadio IV consente di con-siderare la TH quale utile parametro predittivo alla dia-gnosi. Negli stadi localizzati, dove in 3/24 (12.5%) è stataosservata la positività isolata dell’espressione della TH sug-gestiva di infiltrazione midollare minima, questa indaginepotrebbe rappresentare un valido strumento di approfon-dimento diagnostico. La valutazione della TH inoltre potreb-be entrare tra le analisi diagnostiche indici di contamina-zione neoplastica di cellule staminali emopoietiche perife-riche o midollari raccolte in pazienti positivi alla diagnosi.
P064REGISTRO ITALIANO DEL RETINOBLASTOMA (RIR): UPDATING AL 31.5.2004Acquaviva A, Caini M, Marconcini S, Cozza R, Carli ML,*Aricò MGruppo Italiano Del Retinoblastoma, Dip. di Pediatria,Ostetricia e Medicina della Riproduzione - Università diSiena; *U. O. Oncologia Pediatrica, IRCCS Bambino Gesù,Città del Vaticano;* Dip. di Pediatria - Università di **Padova, ^Brescia; U. O. Onco-Ematologia Pediatrica -Ospedale dei Bambini "G. Di Cristina", Palermo
132
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Tumori Solidi

Introduzione obiettivi: Il retinoblastoma (RB) è la neo-plasia maligna oculare più frequente in età pediatrica (3%di tutti i tumori infantili ed 8° per frequenza). Nel maggio1985 è stato avviato su tutto il territorio nazionale un’in-dagine epidemiologica retrospettiva e prospettica sul RB.Pazienti e Metodi. Una scheda di raccolta dati è stata invia-ta a tutte le cliniche pediatriche e oculistiche universita-rie e ospedaliere, a centri di anatomia e istologia patolo-gica, a servizi di radioterapia e di genetica, a registri tumo-ri italiani, a centri oculistici europei, a venditori di protesioculari. E’ stato di fatto costituito il RIR. Al 31.05. 2004sono stati reclutati 1107 pazienti (pz), segnalati da 118centri e diagnosticati da 1.1.1942.Si tratta della 2a piùnumerosa casistica nazionale finora nota in letteratura. E’stato eseguito controllo anagrafico per tutti i pz. Risulta-ti: Sono attualmente valutabili 1088 pz. SEGNI E SINTO-MI: La leucocoria da sola o associata allo strabismo é pre-sente nel 68. 9% dei casi. Il secondo sintomo per frequen-za é lo strabismo. DISTRIBUZIONE PER SESSO: 586 (54%)maschi e 502 (46%) femmine. DISTRIBUZIONE PER LATE-RALITA’: 710 unilaterali (65,25%), Bilaterali 341 (31,34%)e lateralità non nota 393,50%). DISTRIBUZIONE PER SES-SO E LATERALITA’: maschi 586 di cui 383 unilaterali, 189bilaterali e 14 con lateralità non nota; Femmine 502 di cui324 unilaterali, 155 bilaterali e 23 con lateralità non nota.SECONDE NEOPLASIE MALIGNE (SNM): sono state accer-tate 32 SNM (rappresentanti il 2,9% di tutta la casisticavalutata): sarcomi 20, tumori cerebrali 5, leucosi acute nonlinfoblastiche 3, fibroistiocitoma maligno 1, carcinomamammario 1, carcinoma intestinale 1, seminoma 1.Risul-tano deceuti 28/32.STATO DEI PAZIENTI: 875 (80.4%) vivi,178 (16. 2%) deceduti, 36 (3. 4%) persi al follow-up.SOPRAVVIVENZA: La percentuale di sopravvivenza globa-le a 5 anni è del 75,8%. La ricerca eseguita e tuttora in cor-so non può considerarsi esaustiva e non ha permesso dicalcolare l’incidenza del RB in Italia. Conclusioni: I registridi patologia tumorale sono di enorme importanza, oltreche per gli epidemiologi, anche per i clinici. Infatti tali regi-stri raccolgono la memoria clinica di pz indispensabile perdisegnare clinical trias. E’ auspicabile una maggiore colla-borazione dei vari specialisti che osservano i casi di RB conil RIR allo scopo anche di aumentare le informazioni su unapatologia neoplastica relativamente rara ma così impor-tante dell’età pediatrica.
P065INCIDENZA DI ENDOCRINOPATIE IN PAZIENTI PEDIATRICI PORTATORIDI NEOPLASIE CEREBRALIPallotti F,1 Massimino M,2 Seregni E,1 Spreafico F,2Martinetti M,2 Sironi O,1 Ballabio G,1 Bombardieri E,1Fossati-Bellani F2
1U. O. Medicina Nucleare; 2U. O. Oncologia Pediatrica,Istituto Nazionale Tumori Milano
Introduzione ed obbiettivi. Circa il 60% dei pazienti pedia-trici portatori di neoplasie cerebrali guarisce e raggiungel'età adulta. La maggior parte dei pazienti manifesta seque-le (deficit neuropsicologici, alterazioni dello sviluppo, endo-crinopatie) imputabili al tumore e ai trattamenti adottati.Scopo del presente studio è definire l'incidenza di endocri-
nopatie in un'ampia coorte di pazienti pediatrici affetti daneoplasie cerebrali. Pazienti e metodi. Dal gennaio 2002 almaggio 2004 sono stati considerati 85 pazienti (paz. ): 46maschi, 39 femmine; età 3-45 anni (mediana 12); tempointercorso dalla diagnosi: 6- 240 mesi (mediana 36). Istolo-gia: 29 medulloblastomi, 21 neoplasie gliali, 11 tumori ger-minali, 5 ependimomi, 4 altri istotipi. Sede: 37 fossa poste-riore, 21 sovra-tentoriale, 12 linea mediana. Terapie: chirur-gia 37 paz., radioterapia 60 (23 craniale, 37 cranio-spinale),chemioterapia 66. Funzioni endocrine esplorate (metodi):tiroide (fT3, fT4, TSH, ecografia collo), corticosurrene (ACTH,cortisolemia, cortisoluria), gonadi (gonadotropine basali edopo stimolo con GnRH, prolattina, testosterone, estradiolo,progesterone, sviluppo puberale, ecografia mammaria e pel-vica), ormone della crescita (IGF-I, GH seriato dopo stimolo,auxologia), ADH, metabolismo fosfo-calcico ed osseo (PTH,vit. D, marcatori di riassorbimento e formazione, DEXA).Risultati. Presenza di almeno un'alterazione endocrina in 60degli 80 pazienti esaminati (75%). In particolare: alterazio-ni tiroidee in 30 pazienti (42%), deficit di GH in 29 (41%),ipogonadismo in 22 (30%), deficit di mineralizzazione in 15(21%), insufficienza cortico-surrenalica in 14 (20%), diabe-te insipido in 12 (17%) e pubertà precoce in 6 (9%). I tumo-ri della linea mediana comportano la più elevata incidenzadi endocrinopatie (50 in 12 pazienti) seguiti dai tumori del-la fossa posteriore (51 in 37 pazienti) e dai tumori sovraten-toriali (26 in 21 pazienti). Conclusioni. Questo studio dimo-stra l'elevata incidenza di endocrinopatie nei pazienti porta-tori di neoplasie cerebrali.
P066RUOLO DEL PROFILO DI ESPRESSIONE GENICA NEI SARCOMI DI EWINGAD ALTO RISCHIO MEDIANTE ANALISI DEI MICROARRAYSardi I, Capanna R,* Faulkner LB, Lippi A, Tamburini A,Tintori V, Tondo A, Tucci F, Iannalfi A, Sanvito C,Marchi C, Bernini GU. A. U, Oncoematologia Pediatrica Az. Ospedaliero,Universitaria A. Meyer, Dipartimento di Pediatria,Università degli Studi di Firenze. *Divisione di ChirurgiaOrtopedica Oncologica Ricostruttiva, CTO, Az. O. U. Careggi, Firenze
Introduzione e Obiettivi. Il sarcoma di Ewing è un tumo-re a piccole cellule rotonde che colpisce prevalentemente ilsistema scheletrico di giovani pazienti. L’ uso di terapie mul-timodali ha migliorato la prognosi nelle forme localizzatenon metastatiche, anche se essa rimane ancora molto gra-ve per i pazienti metastatici alla diagnosi o che vanno in pro-gressione durante il trattamento. Il nostro studio si propo-ne di valutare, mediante microarray, l’ espressione genica deisarcomi di Ewing ad alto rischio che non rispondono alleterapie tradizionali e verificare alla luce di questi risultati lapossibilità di nuove strategie terapeutiche per i sarcomi no-responders approntando trattamenti specifici. MetodiSonostati selezionati, per l’ analisi del profilo di espressione geni-ca, sette pazienti, età media 15,4. Al momento dello studioquattro pazienti erano liberi da malattia, due erano dece-duti e uno era recidivato durante la terapia. L'RNA totale deicampioni di tessuto è stato amplificato e ibridato su chipcontenente 650 geni ben caratterizzati. La valutazione dei
133
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

livelli espressione genica era eseguita mediante scannerAXON GenPix 4000B confrontando ciascun campione conun RNA totale standard. I risultati ottenuti dall’ esame deimicroarray erano confermati da un’ analisi quantitativamediante Real Time-PCR. Risultati. L’ analisi multigenicadei sette sarcomi di Ewing che presentavano il trascritto difusione t(11;22)(q24;q12) ha rivelato che in tutti i tumoriera presente un aumento dell’ espressione di alcuni geni trai quali: TGFβ, TNFα, INFγ, Erb b2 ed enzimi quali adenina-fosforibosil ttransferasi e glicogeno sintasi kinasi β3. Nume-rosi altri geni, possibili bersagli terapeutici, erano accesi inquei sarcomi che poi hanno mostrato una prognosi sfavo-revole. Nei pazienti tuttora liberi da malattia si sono evi-denziati possibili target genici per il monitoraggio dellaripresa di malattia. ConclusioniIl nostro studio indica che ilmonitoraggio dell’ espressione genica può apportare nuovirisultati su molti aspetti dei sarcomi di Ewing, quali la com-prensione di alcuni meccanismi molecolari e la caratteriz-zare di nuovi bersagli terapeutici. L’ informazione così otte-nuta apre nuove e suggestive prospettive circa l’ impiego distrategie innovative per combattere forme altrimenti noncontrollabili.
P067PROTOCOLLO EUROPEAN INFANT NEUROBLASTOMA STUDY:RECLUTAMENTO IN ITALIADi Cataldo A, Bombace V, Angelini P, Arcamone G,Aru B, Bertuna G, Bianchi M, Bonetti F, Castellano A,Cellini M, Conte M, Cosmi C, D' Angelo P, de Leonardis F,Fagnani A, Giuliano M, Luksch R, Marradi P,Mastrangelo S, Miglionico L, Migliorati R, Miraglia V,Nardi M, Paone G, Pericoli R, Parigi GB, Pierani P, ProvenziM, Serino E, Tamburini A, Tettoni K,Tonegatti L, Torrisi V, Viscardi E, Zanazzo GA,De Bernardi B per il GcinbCentri AIEOP di: Ancona, Bari I, Bari II, Bergamo, Bologna,Brescia, Cagliari, Catania, Ferrara, Firenze, Genova,Milano Alfieri, Milano INT, Modena, Napoli I, Napoli II,Padova, Palermo, Pavia, Pisa, Rimini, Roma BG, RomaGemelli, Sassari, S. G. Rotondo, Torino, Trieste, Verona
Nel dicembre 1999 è stato aperto al reclutamento il pro-tocollo European Infant Neuroblastoma Study con l’obietti-vo di migliorare la sopravvivenza degli infant con neurobla-stoma a)trattando quelli con malattia localizzata inopera-bile, con chemioterapia poco intensa ed approccio conser-vativo (trial 1); b)utilizzando, nei pazienti con metastasi insedi diverse da polmone, ossa e SNC, la chemioterapia soloin casi di estrema gravità clinica (trial 2); c)non ricorrendoad alte dosi nei pazienti con metastasi polmonari, ossee,SNC (trial 3); d)ricorrendo ad alte dosi nei casi con MYCNamplificato (trial 4). Il reclutamento nei trial 1 e 4 è già sta-to chiuso, perché si è raggiunto il numero di pazienti neces-sario per la valutazione, mentre gli altri trial si chiuderan-no entro il 2004. In Italia, in 28 Centri AIEOP, sono statiregistrati 182 pazienti, rispettivamente 36 nel trial 1, 35 neltrial 2, 9 nel trial 3, e 6 nel trial 4. Altri 96 casi, non eleggi-bili, sono stati studiati separatamente. La causa di non eleg-gibilità è stata: malattia localizzata operabile in 74 casi,mancanza del dato di MYCN in 12 casi, ritardo di registra-
zione in 2 casi, incompleta stadiazione in 3 casi, chiusura alreclutamento del trial 1 in 5 casi. In 8 bambini la diagnosiè seguita ad un sospetto ecografico prenatale. In altri 30 casila diagnosi è stata posta a 0-1 mesi di età. Lieve è stata laprevalenza del sesso femminile, in oltre l'80% dei casi iltumore primitivo aveva sede addominale Il tempo di osser-vazione mediano è di 24 mesi, con range 0-48. Nei Centriitaliani è apparsa chiara l’importanza di una diagnosi isto-logica centralizzata e della determinazione di MYCN. Infat-ti, la centralizzazione del materiale tumorale è avvenuta intutti i casi che hanno potuto eseguire una biopsia o rese-zione all'esordio, e la valutazione di MYCN è stata eseguitain 168 casi.
P068IMPIEGO DELLA 18F-FDG PET NELLA DIAGNOSTICA E NEL FOLLOW-UPDEL NEUROBLASTOMA (NB)Castellini C, Prete A, Nanni C,* Fanti S,* Franchi R,*Pession AMO Oncoematologia Pediatrica e Terapia Cellulare,Università degli Studi Bologna *UO Area Radiologica, Ospe-dale S. Orsola-Malpighi
Introduzione. La 18F-FDG PET, è una tecnica mirata allostudio del metabolismo del glucosio a livello di organi edapparati, già routinariamente impiegata nello studio dinumerose neoplasie, quali i tumori SNC dell’ età pediatrica.Nella PET-CT l’ indagine TC integra lo studio metabolico conimmagini anatomiche, aumentando la sensibilità dell’ esa-me. Obiettivo: con l’ intento di individuare l’ indagine piùspecifica, sensibile ed eventualmente caratterizzata daminor tossicità per i pazienti (pz), è stato messo a punto unprotocollo diagnostico con lo scopo di confrontare sensibi-lità e specificità di 18F-FDG PET e 131I-MIBG. Casi e meto-di tra Febbraio e Maggio 2004, tre pz affetti da NB IV sta-dio recidivato dopo trapianto di cellule staminali emopoie-tiche autologo (2) e allogenico non mieloablativo (1), sonostati sottoposti a scintigrafia con 131I-MIBG e 18F-FDG PETal momento della ricaduta e, successivamente, dopo ogni 2cicli di chemioterapia. . Risultati. In 2/3 pz al momento del-la recidiva le indagini concordavano per sede ed estensionedi malattia; in 1/3 la 18F-FDG PET documentava l’ esisten-za di una sede ulteriore, misconosciuta alla 131I-MIBG enon più rilevata dopo 2 cicli di trattamento. Alla rivaluta-zione la 18F-FDG PET, eseguita troppo a ridosso della tera-pia, è risultata falsamente negativa in 2 pz, mentre la 131I-MIBG documentava la presenza di malattia. Conclusioni: la18F-FDG PET non ha ancora spazio routinario nella diagno-stica del NB, essendo il suo limite maggiore rappresentatoda una minore disponibilità rispetto alla 131I-MIBG, tutta-via mostra promettenti risultati. Le caratteristiche di sensi-bilità e specificità associate al fatto che gran parte dei NBassume il FDG in maniera indipendente rispetto alla capta-zione delle catecolamine, ne fanno intravedere la potenzia-lità applicativa, soprattutto nell’ individuazione delle reci-dive precoci. Inoltre, potrebbe risultare particolarmente uti-le nei casi di NB che non captino la MIBG. Una più accura-ta tempistica nell’ esecuzione ed un maggior numero di pzpotranno sancirne il potenziale vantaggio nella diagnosi,staging e follow-up di pz affetti da NB.
134
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Trapianto

135
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
TRAPIANTO
C007VARIAZIONI TEMPORALI DEI FLUSSI MIGRATORI PER TRAPIANTO DICELLULE STAMINALI EMOPOIETICHE (TCSE) TRA CENTRI ADERENTEALL’ ASSOCIAZIONE ITALIANA DI EMATOLOGIA E ONCOLOGIA PEDIATRICA Rondelli R, Gualandi L, Locatelli F, Ronchetti F, Dini G,Zecca M, Fagioli F, Cesaro S, Prete A, Uderzo C,Luksch R, Porta F, Donfrancesco A, Meloni G, Favre C,Faulkner L, Andolina M, Ripaldi M, Zucchetti P,Di Cataldo A, Pierani P, Ladogana S, Argiolu F,De Rossi G, Di Bartolomeo P, Marradi P, Paolucci P,La Nasa G, Messina C, Pession ARegistro AIEOP-TMO
Introduzione e obiettivi: Sono ormai noti i flussi migra-tori extraregionali dei pazienti pediatrici affetti da neo-plasia verso centri AIEOP, per la diagnosi e/o la terapia(DT). Il TCSE, poiché richiede centri ad alta specializzazio-ne, rappresenta la ragione di un’ ulteriore migrazione (M)verso un centro diverso da quello di DT. L’ obiettivo delpresente studio è verificare il contributo del TCSE al feno-meno migratorio extraregionale che interessa i pazientiaffetti da tumori e patologie non tumorali, residenti inItalia, con DT in centri AIEOP e sottoposti a TCSE in età <18 anni in un centro AIEOP TMO tra il 1989 e il 2003 e daquesti registrati nella banca dati AIEOP TMO. Metodi: LaM extraregionale è stata definita sulla base di: a) nonappartenenza del centro di DT e/o del centro di TCSE allaregione di residenza del paziente, b) diversità tra le regio-ni di appartenenza dei due centri. La migrazione così defi-nita è stata suddivisa in 5 categorie: A) no M; B) M perTCSE; C) M per DT+TCSE; D) M per DT e M per TCSE; E) Mper DT e no M per TCSE. Il fenomeno migratorio è statovalutato sia globalmente, sia per singola patologia e pertipo di TCSE, confrontandolo per periodo di TCSE(P1:1989-1993, P2:1994-1998, P3:1999-2003). Risulta-ti: La popolazione valutabile è composta da 2535 pazien-ti, di cui il 53. 3% è risultato avere avuto almeno una Mdalla regione di residenza. I casi che hanno eseguito ilTCSE nello stesso centro di DT dove erano migrati (C) sonopari al 28. 9%, mentre la M di tipo B è pari al 20.6%, ridu-cendosi dal 27. 9% nel P1 al 16. 6% nel P3 (p=. 000).Migrazioni multiple (D) si sono osservate solo nel 2.9% deicasi e solo lo 0.8% è risultato appartenere alla categoriaE. La M extraregionale dal centro di DT al centro di TCSE(B+D+E) pari al 24. 3% è andata diminuendo significati-vamente nel tempo, passando dal 32.7% nel P1 al 19. 4%nel P3 (p=. 000). Conclusioni: La riduzione del fenomenomigratorio extraregionale per TCSE che si è osservata nelcorso degli ultimi 15 anni consente di affermare che l’incremento dei centri AIEOP con competenze di TCSEabbia in parte assorbito il costante aumento dei casi dasottoporre a TCSE dovuto all’ aumento delle indicazioni alTCSE in età pediatrica. Solo l’ analisi dei risultati ottenu-ti per tipologia di M fornirà la garanzia in merito al man-tenimento degli standard qualitativi di riferimento nazio-nali e internazionali.
C008ISOLAMENTO ED ESPANSIONE IN VITRO DELLE CELLULE STAMINALIMESENCHIMALI ISOLATE DAL MIDOLLO OSSEO DI DONATORI PEDIATRICIE ADULTIMareschi K, Ferrero I, Rustichelli D, Venturi C, Montin D,Barat V, Asaftei S, Fagioli F, Madon EDipartimento di Scienze Pediatriche e dell'Adolescenza,Università degli Studi di Torino
Introduzione e obiettivi. L’enorme plasticità delle cellu-le staminali mesenchimali (CSM) suggerisce il loro utiliz-zo in terapia cellulare. CSM ottenute dal midollo osseo didonatori sani sono state isolate ed analizzate durante laloro espansione cellulare per verificare se esistano diffe-renze tra le CSM isolate da donatori pediatrici e quelleottenute da donatori adulti. Metodi. Le CSM sono stateisolate dal midollo osseo di donatori sani, pediatrici (età2-13) e adulti (età 20-50), mediante stratificazione sugradiente Percoll ed espanse in vitro. Ad ogni passaggiosono stati valutati: sterilità, vitalità, immunofenotipo,cariotipo e lunghezza del telomero. Risultati. Sono statiraccolti ed analizzati 6 campioni di midollo osseo da dona-tori pediatrici e 12 campioni da donatori adulti. Le CSMsono state espanse per una mediana di 20 passaggi (dona-tori pediatrici) e di 18 passaggi (donatori adulti). Durantetutti i passaggi non sono stati evidenziati segni di conta-minazione batterica, fungina o da micoplasma e la vita-lità cellulare variava dal 98% al 100%. Dopo 10 passag-gi di coltura le CSM ottenute da donatori pediatrici rag-giungevano un valore mediano di espansione di 2.2 LOG,mentre quelle ottenute da donatori adulti un valore infe-riore a 2 LOG. Al primo passaggio le CSM ottenute dadonatori pediatrici erano negative per CD45, CD14 emostravano un’alta espressione per CD90, CD29, CD44,CD105, che rimaneva costante durante l’espansione invitro, e bassa espressione per CD106, CD166. Le CSM otte-nute da donatori adulti, invece, mostravano al primo pas-saggio una lieve contaminazione di cellule ematopoieti-che che diminuiva nei passaggi successivi, ed alti livelli diCD90, CD44, CD105 e CD29 al primo passaggio e nei pas-saggi successivi, ad eccezione del CD29, la cui espressio-ne diminuiva ad ogni passaggio. In entrambi i gruppi nonsono state evidenziate alterazioni cromosomiche, né signi-ficative diminuzioni della lunghezza dei telomeri. Conclu-sioni. Le CSM sono facilmente isolabili dal midollo osseoe sono in grado di proliferare ed espandersi in vitro man-tenendo inalterate le loro caratteristiche immunofenoti-piche senza la comparsa di alterazioni cromosomiche oevidenze di senescenza cellulare. L’età del donatore è cor-relata con una minor crescita (2 LOG) ma non con la capa-cità funzionale delle CSM. Parzialmente supporto da AIRC,MIUR ex-40% e 60%.

P069LA CERTIFICAZIONE DI QUALITÀ IN SANITÀ: L’ESPERIENZA DEL CENTROTRAPIANTI DI MIDOLLO OSSEO – UNITÀ CLINICA E LABORATORIO – DELDIPARTIMENTO DI SCIENZE PEDIATRICHE E DELL’ADOLESCENZADELL’UNIVERSITÀ DI TORINO, AZ. OSPEDALIERA O. I. R. M. S. ANNAVassallo E, Ferrero I, Vagliano L, Rustichelli D,Mareschi K, Niceforo C, Gastaldo L, Venturi C, Biasin E,Nesi F, Berger M, Fagioli F, Madon EDipartimento di Scienze Pediatriche e dell'Adolescenza.Università degli studi di Torino
Introduzione e obiettivi. Come altre organizzazioni pro-duttrici di beni e servizi, anche la strutture sanitarie sonochiamate a realizzare e assicurare la qualità del servizio,in misura proporzionata ai bisogni che sono chiamate asoddisfare. Le esigenze che la qualità in ambito sanitarioè chiamata a soddisfare rientrano nella categoria dei biso-gni primari, tutelati da apposite leggi e norme cogentidello Stato, e sono strettamente connesse con problema-tiche di natura accessoria. La qualità del servizio sanita-rio può essere intesa come l’insieme di elementi scienti-fici, tecnici e tecnologici, organizzativi, procedurali, rela-zionali e di comunicazione, che dovrebbero contribuire alraggiungimento della soddisfazione dell’utente. La crea-zione di un sistema di assicurazione della qualità confor-me alle norme ISO 9001:2000 è nata dall’esigenza di otti-mizzare la qualità del servizio offerto dal Centro Trapian-ti di Midollo Osseo (CTMO) pediatrico relativamente alleproblematiche cliniche e di laboratorio legate alla raccol-ta, manipolazione, conservazione ed infusione delle cellulestaminali emopoietiche. Metodi. Sono stati definiti gliobiettivi di qualità e sono stati predisposti i documentidel sistema (procedure, istruzioni operative, modulistica dibase). Dopo una fase di implementazione e verifica, si ègiunti alla redazione del Manuale Qualità, il documentoconclusivo del sistema qualità in cui sono descritti l’ap-plicazione del sistema, la struttura, le responsabilità, leregole base. Per ogni processo sono stati inoltre definitiindicatori di qualità in grado di valutare l’efficacia delsistema nell’ottica del miglioramento continuo. Risultati.L’iter certificativo intrapreso con BVQI ITALIA S. r. l. , enteaccreditato SINCERT leader nel settore sanitario, si è con-cluso a dicembre 2003. Il certificato di conformità ha vali-dità 3 anni e prevede verifiche ispettive di mantenimen-to ogni 12 mesi. Conclusioni. La scelta della certificazio-ne rappresenta il punto di partenza per il raggiungimen-to dell'accreditamento di eccellenza JACIE (Joint Accredi-tation Committee of ISCT-Europe and EBMT), un pro-gramma di accreditamento dei due maggiori gruppi coo-perativi di trapianto in Europa, in allineamento scientifi-co e normativo con quelli americani.
Parzialmente supportato da AIRC, ADISCO Sez. Pie-monte.
P070RUOLO DEL MISMATCH PER SESSO E DELLE VARIAZIONI DEL CROMOSOMAY NELLA GVHD IN SARDEGNAOrofino MG, Contu D, Argiolu F, Sanna MA, Gaziev D,Rizzo F, Cucca F, Cao ACentro TMO 2°Clinica Pediatrica Ospedale per le Microci-temie Cagliari
La graft versus host disease (GVHD) è una severa com-plicanza del TMO allogenico, presentandosi con una inci-denza tra il 20-35% nel trapianto da donatore HLA iden-tico. Sono stati descritti vari fattori predittivi correlati allaGVHD quali: malattia di base, regime di condizionamen-to, età del donatore (D) e ricevente (R), diversità di sessotra D e R. In modo particolare il mismatch per sesso (Dfemmina versus R maschio) è stato riportato come uno deifattori di rischio maggiori, potendo sottintendere unadiversità per gli antigeni minori di istocompatibilità(mHag). I risultati in differenti studi sono però contraddi-tori. Noi abbiamo valutato l’impatto del mismatch per ses-so D/R in una serie di 176 pazienti sardi trapiantati dafratello HLA identico. Fra questi pazienti, diciotto maschiche hanno presentato GVHD tra il II°-III° e ricevuto unTMO da una sorella HLA identica erano inseriti in un pro-getto sperimentale di caso-controllo, per la valutazionedel ruolo della variazione della porzione non ricombinan-te del cromosoma Y (NRY. L’analisi dei nostri dati non sup-porta il mismatch per sesso come fattore di rischio indi-viduale per la GVHD. Inoltre nel gruppo dei pazienti disesso maschile con donatrice femmina non abbiamo nep-pure osservato alcuna significativa associazione tra allelie aplotipi NRY e GVHD acuta. Questi nostri risultati sonoin accordo con alcuni studi riportati in letteratura, ma incontrasto con altri. Tali risultati contradditori in differen-ti popolazioni potrebbero essere giustificati dalla presen-za in alcune popolazioni di una specifica mutazione fon-datrice in siti funzionalmente rilevanti per i loci di mHagdel cromosoma Y, non presente in altre come quella sar-da. Anche se un maggior numero di casi appare necessa-rio per aumentare il potere statistico, noi concludiamoche in Sardegna il mismatch per sesso e le variazioni delcromosoma Y non hanno un ruolo significativo nella GVHDnel TMO da fratello HLA identico.
P071TERAPIA CELLULARE PRENATALE NELE IMMUNODEFICIENZE PRIMITIVEE NELL'OSTEOGENESI IMPERFETTA: REPORT DI 7 CASIPorta F,* Lanfranchi A,° Verardi R,° Berta S,° Baggio E,°Bolda F,° Baffelli R,° Easton J,* Tettoni K,* Zucca S,^Guandalini F,^ Mazzolari E,§ Notarangelo LD§
*Centro di Terapia Cellulare e Genica "A. Nocivelli";°Lab TMO, III servizio analisi; ^Clinica Ostetricoginecologica; §Clinica Pediatrica, Spedali Civili, Brescia
Dal 1984 sono stati realizzati più di 40 trapianti di cel-lule staminali in utero. Ad oggi gli unici successi clinici concura della malattia di base sono stati ottenuti in feti affet-ti da immunodeficienze primitive. Tuttavia nel corso deglianni le tecnologie di manipolazione delle cellule staminali,
136
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Trapianto

137
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
le modalità di infusione ed il tipo e numero delle celluleinfuse sono cambiate rendendo difficile una valutazionedell'effettiva efficacia clinica di questa strategia. Dal 1996abbiamo effettuato 7 trapianti in utero: in 5 casi il fetoera affetto da immunodeficienza combinata grave (2 SCIDT-B+NK-; SCID T-NK+, 1 sindrome di Omenn), in 2 casi daosteogenesi imperfetta (OI). Tutti i feti hanno ricevuto lecellule staminali del donatore mediante iniezione intra-peritoneale ripetuta due volte, ad una distanza di una set-timana, tra la 19a e la 24a settimana di gestazione. I duefeti affetti da SCID T-B+NK- hanno ricevuto cellule CD34+
paterne ed alla nascita hanno mostrato ricostituzione siadel comparto T che B cellulare. Nel caso dei due feti affet-ti da SCID T-NK++, dal momento che nel primo caso la solainfusione di cellule CD34+ del padre aveva portato ad unattecchimento modesto (10%), nel secondo è stato effet-tuata una coinfusione di cellule CD34 e di cellule stami-nali mesenchimali midollari paterne che ha portato a pie-na ricostituzione immunnologica. Nel caso del feto affet-to dalla sindrome di Omenn il trapianto in utero è statorealizzato con successo mediante cellule da sangue peri-ferico di origine materna. Sulla base dei risultati ottenutiche dimostrano un ruolo delle cellule mesenchimali e del-la tolleranza materno-fetale, abbiamo realizzato due tra-pianti in utero in due feti affetti da OI coinfondendo cel-lule CD34+ e cellule mesenchimali resting prelevate dallamadre. Il trapianto è stato effettuato in entrambi i casi al5 mese di gravidanza. Entrambi i bambini sono nati sen-za segni di reazione da trapianto, nè la procedura a cau-sato sequelae. Mentre nel primo caso è stato possibilemettere in evidenza un chimerismo del donatore nei fibro-blasti pari al 4%, nel secondo le analisi su biopsia ossea ecutanea sono in corso.
P072LA FOTOAFERESI EXTRACORPOREA PER IL TRATTAMENTO DELLA GVHDREFRATTARIA. ESPERIENZA MONOCENTRICABerger M, Albiani R,° Giubellino C,° Incarbone E,°Vassallo E, Nesi F, Fagioli F, Madon EDipartimento di Scienze Pediatriche e dell'Adolescenza,Università di Torino. ° Servizio di Immunoematologia eCentro Trafusionale, A. S. O. O. I. R. M. -S. Anna, Torino
La fotoaferesi extracorporea (ECP) costituisce una nuo-va strategia terapeutica per il trattamento della GvHDresistente al trattamento convenzionale (Messina et al. BrJ Haematol 2003). In questo studio si è voluto studiarel’effetto della ECP per il trattamento della GvHD resisten-te al trattamento di prima linea dopo trapianto di cellulestaminali emopoietiche allogeniche (TCSE). Pazienti eMetodi: da Giugno 2001 a Giugno 2004 sono stati trat-tati 21 pazienti con GvHD acuta e cronica trattati in pri-ma linea con ciclosporina e cortisone. Lo schema del trat-tamento ha previsto 2 sedute/settimana per le prime 4settimane, 2 sedute/bisettimanali per il 2°-3° mese, indi acadenza mensile per i successivi 3 mesi. La ECP è stataeseguita utilizzando lo strumento Fresenius Hemo-CareCom. Tec con flusso continuo. Sono state processate 2volemie per ogni paziente ed il volume finale della frazio-ne mononucleata aggiustato a 300 mL. Sono stati quindi
aggiunti 3 ml di 8-MOP per una concentrazione finale di8-MOP di 200 mg/ml. Le cellule sono state quindi trasfe-rite in una sacca permeabile ai raggi UVA (Maco Pharma,Tourcoing, Francia) e quindi sottoposte a fotoattivazionecon raggi UVA per 4 cicli di 5 minuti attraverso un irra-diatore PUVA Combi light, e successivamente reinfuse alpaziente entro 30 minuti. Due pazienti adulti sono statitrattati con lo strumento Terakos, in cui 240 mL di buffycoat e 300 mL di plasma sono stati incubati con 100 µgdi 8-MOP e successivamente esposti a raggi UVA a 2 J/cm2
per 90 minuti. I criteri di risposta sono stati valutati inaccordo a Greinix et al. , 1998, 2000)(Tabella 1). Risultati:I pazienti trattati per GvHD acuta hanno ottenuto laremissione completa nel 66% dei casi. In questi pazientiè stato possibile ridurre progressivamente la terapia ste-roidea sino alla sospensione. Per i restanti pazienti si sonoresi necessari ulteriori trattamenti immunosoppressivi. Ipazienti trattati per GvHD cronica hanno ottenuto laremissione completa nel 44% dei casi, mentre un ulterio-re 33% ha ottenuto una stabilizzazione clinica delle mani-festazioni della GvHD cronica. Dei rimanenti pazienti unonon ha risposto ed un altro ha rifiutato il proseguimentodell’ECP. La sopravvivenza libera da eventi a 36 mesi peri pazienti che hanno risposto al trattamento con ECP èstata del 87% rispetto al 50% per coloro che non hannorisposto (p=0.02). Nessun incremento di infezioni è statodocumentato ed in generale le procedure con gli accorgi-menti indicati nei metodi sono state ben tollerate da tut-ti i pazienti. Conclusioni. Il trattamento con ECP costitui-sce una valida scelta terapeutica per i pazienti affetti daGvHD acuta che non rispondono alla terapia di primalinea. I risultati ottenuti per i pazienti affetti da GvHDcronica refrattaria a varie linee di terapia, meritano altret-tanta considerazione.
Tabella 1.
GvHD acuta GvHD cronica
N° Pazienti 12 9Eta' 10 (5,9-15,9) 8,2 (4,5-21)Sesso M/F 4/8 6/3Diagnosi
LLA 4 4LMA 0 2LMC 4 1THAL 1 1Immunodeficienza 1 -LNH 1 -MDS 1 -SAA - 1
DonatoreFamiliare Identico 3 4Aploidentico 1 1Non Consanguineo 5 7TBI Si/No 9/3 6/3Intervallo GvHD-ECP (gg) 15 (6-82) 172 (2-1187)Grado GvHD pre-ECP 0-I=25% Lim=33%
II-IV=75% Est=66%

Parzialmente supportato da AIRC, MIUR ex-40% e60% grant EM.
P073TRAPIANTO DI MIDOLLO OSSEO DA DONATORE VOLONTARIO NEL LINFOMAA GRANDI CELLULE ANAPLASTICHE RICADUTO/REFRATTARIO IN PAZIENTIPEDIATRICI: ESPERIENZA MONOCENTRICACesaro S,* Pillon M,* Calore E,* Gazzola MV,* Destro R,*Mussolin L,* Visintin G,* Surico G,° Zanesco L,*Rosolen A,* Messina C**Clinica di Oncoematologia Pediatrica, Università ofPadova; °II Clinica Pediatrica, Università di Bari
Introduzione: il linfoma anaplastico a grandi cellule(ALCL) in età pediatrica rappresenta una rara malattiamaligna generalmente chemio-sensibile, con una soprav-vivenza del 70-90%. I dati riguardanti l’intensificazionecon trapianto di midollo osseo (TMO) per le ricadute diALCL sono scarsi, principalmente limitati al TMO autolo-go con risultati non ottimali. Metodi: noi riferiamo di 4pazienti (2 F, 2 M; di 4, 7, 12, 16 anni, rispettivamente),affetti da ALCL (2 st. III, 1 st. II, 1 st. IV alla diagnosi) sot-toposti a TMO allogenico da donatore volontario HLAidentico. Le indicazioni al TMO erano: ricaduta precoce, 2;ricadute multiple, 1; progressione di malattia, 1.Lo statodei pazienti pre-TMO era: 2^RC, 2; 5^RC, 1; 1^RC, 1, dopoprogressione di malattia. Lo studio della malattia residuaminima aveva riscontrato in 1 paziente la positività deltrascritto NPM-ALK mediante RT-PCR sul midollo e sulsangue periferico. Il regime di condizionamento è stato TBI(1200 cGy) e VP16 (60 mg/kg) in 3 pazienti (2 con boostcraniale di 1200cGy); e TBI, thiotepa (10 mg/kg) e cyclofo-sfamide (120 mg/kg) in 1 paziente. Ciclosporina, bassedosi di MTX e pre-TMO ATG 2,5-3 mg/kg sono stati usaticome profilassi della GVHD. Risultati: tutti i pazienti han-no avuto un rapido attecchimento per PMN e PLT. La tos-sicità precoce è stata limitata a mucosite (grado II secon-do Bearman), GVHD acuta di grado I; II in tutti, riattiva-zione virale (CMV 2; EBV, 2). Dopo un follow-up di 0.5, 1.5,2 e 3. 5 anni, rispettivamente, tutti i pazienti sono vivi. Ilpaziente in 5^RC presenta GVHD cronica con bronchioli-te obliterante e fibrosi polmonare ed è in terapia contacrolimus, micofenolato e dosi intermittenti di predniso-ne. Questo paziente ricevette bleomicina nel trattamentopre-TMO. Due pazienti sono in RC, senza immunosop-pressione e in assenza di GVHD. L’ultimo paziente nonpresenta segni di GVHD e sta riducendo la terapia immu-nosoppressiva. Conclusioni: questa limitata esperienza,suggerisce che il TMO allogenico può essere una opzionevalutabile nel trattamento del ALCL ad alto rischio.
P074QUALITÀ DI VITA (QDV) DEI PAZIENTI LUNGO SOPRAVVIVENTI DOPOTRAPIANTO DI CELLULE STAMINALI EMOPOIETICHE (TCSE): ESPERIENZAMONOCENTRICAPillon M, Cesaro S, Calore E, Tridello G, Visintin G,Pelosin A, Brugiolo A, Varotto S, Greggio N, Cereda C,Errigo G, Zanesco LE, Messina CUnità TMO - Clinica di Oncoematologia Pediatrica,Dipartimento di Pediatria, Università - AziendaOspedaliera di Padova
Introduzione: il termine QdV rappresenta la percezionedell’ individuo per quanto riguarda la sua posizione nellavita e nel contesto culturale, i suoi sistemi di valori, leaspettative, gli standard e le proprie preoccupazioni.Metodi: è stato eseguito uno studio pilota mediante inter-vista telefonica su un campione di 49 su 88 pazienti fuo-ri terapia, vivi e in RC dopo TCSE (allogenico, 24%; MUD,19%; autologo, 44%; PBSC, 13%) dal 1983 al 2000, perleucemia (72%), linfoma (11%) o tumore solido non cere-brale (17%). Il questionario riguardava: scolarità, capacitàcognitive, lavoro, attività sportiva, relazioni sociali, per-cezione corporea-sociale. Veniva richiesta inoltre unaauto-valutazione della propria QdV espressa in termini disoddisfazione e punteggio. Risultati: dei 49 ragazzi inter-vistati (18 F e 31 M, età mediana 18 anni, range 10-32),il 7% frequenta la scuola media, il 33% la scuola supe-riore soprattutto tecnico-professionale, l’11% l’università,mentre il 49% lavora come impiegato o operaio. E’ statoriferito buon interesse e profitto scolastico in oltre la metàdei casi, mentre il 43% ha presentato difficoltà e 10pazienti sono stati bocciati: si trattava quasi esclusiva-mente di pazienti sottoposti a TBI RT craniale. Difficoltàdi tipo cognitivo sono state riferite più dagli studenti chedai lavoratori (48% vs 25%). Il 48% dei pazienti praticasport, in uguale misura di squadra o individuale; solo nel9% dei casi non viene praticata alcuna attività fisica percause legate alla malattia. L’ analisi delle relazioni socia-li evidenzia un piccola percentuale di ragazzi (22%) condifficoltà a fare amicizia o ad avere relazioni sociali a cau-sa di timidezza e solo il 29% ha un scarso gradimentodella propria immagine corporea per eccessiva magrezzaod obesità. La maggior parte degli intervistati mostra otti-mismo e buon umore e riferisce una soddisfazione gene-rale per la vita. Il 67% dà un punteggio da 8 a 10 nell’autovalutazione della propria QdV. Conclusioni: lo studiopilota dimostra che la percezione della QdV del pazientelungo sopravvivente è complessivamente positiva. Que-sto costituisce un prezioso stimolo per migliorare ulte-riormente l’ assistenza globale, ricordando che la sorve-glianza del paziente lungo sopravvivente dopo TCSErichiede un approccio multidisciplinare.
138
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Trapianto

P075CARDIOTOSSICITÀ A DISTANZA DA TRAPIANTO DI MIDOLLO OSSEO. RUOLO DELL’ECODOBUTAMINA NELLA VALUTAZIONE DI PAZIENTIPEDIATRICIUderzo C,* Cavatorta E,* Trocino G,° Rovelli A,*Balduzzi A,* Longoni D,* Viganò E,* Bonanomi S,*Tagliabue A,* Corti P* *Clinica Pediatrica, Ospedale San Gerardo, Monza,Italy°Divisione di Cardiologia,Ospedale San Gerardo,Monza, Italy
Introduzione e Obiettivi. Lo scopo di questo studio èquello di valutare la riserva contrattile cardiaca a distan-za di più di 10 anni da TMO eseguito in età pediatrica. Siè poi voluto individuare il ruolo dell’Ecodobutamina nel-la diagnosi di disfunzioni cardiache subcliniche. Metodi:Ventinove pazienti, 24 maschi e 5 femmine,15 affetti daLLA, 5 da LMA, 4 da LMC, 2 da AREB, 1 da LNH, 1 daLLA+LMA, 1 da AAS, sono stati seguiti prospetticamentetramite ecocardiogramma a partire dal TMO (età media-na al TMO 9,2 anni, range 1,1-17,9). 20/29 sono stati sot-toposti a TMO allogenico da fratello HLA compatibile, 1/29a TMO singenico, 1/29 a TMO aploidentico, 6/29 a TMOautologo e 1/29 a TMO unrelated. 22/29 pazienti sonostati trattati con antracicline preTMO (dose medianacumulativa 300 mg/m2, range 120-610); 23/29 hannoricevuto regime di condizionamento con TBI frazionata e29/29 con Ciclofosfamide (dose mediana 135 mg/kg, ran-ge 40-330). 20/29 hanno manifestato aGVHD e 7/29cGVHD. Tutti i pazienti sono stati rivalutati ad una distan-za di più di 10 anni da TMO (follow-up mediano 14,8 anni,range 9,9-23,3) con ecocardiogramma a riposo e doposomministrazione di dobutamina a basse dosi (10mcg/kg/min), quale stimolo inotropo. Risultati: All’esecu-zione dell’Ecostress l’età mediana dei nostri pazienti era20,9 anni (range 14,2-32). 28/29 pazienti sono risultatiasintomatici e con ecocardiogramma a riposo nella nor-ma, tuttavia 7/29 pazienti testati con Ecodobutaminahanno dimostrato una ridotta riserva contrattile cardiaca(incremento di Frazione di Eiezione -FE- dopo stimolo<10%). Questi 7 pazienti mostravano, confrontati con ilgruppo che presentava una normale riserva contrattile(22/29):1.minor incremento di FE dopo stimolo (5,8% ver-sus 18,9%)2.minor incremento di diametro e volume tele-diastolico del ventricolo sinistro 3. minor ispessimentosistolico di parete posteriore e setto interventricolare.Conclusioni. Ecodobutamina è una importante metodicaper la valutazione di effetti cardiaci tardivi in pazientiguariti in età pediatrica mediante TMO. L’evidenza che il24,2% della nostra popolazione, seppurasintomatica,mostra una ridotta risposta allo stress, concompromissione sistolica e diastolica. Questo impone unasorveglianza continua nel tempo al fine di conoscere emigliorare la qualità di vita a lungo termine.
P076TRAPIANTO ALLOGENICO DI MIDOLLO OSSEO E DLI PER IL TRATTAMENTODELLA CITOPENIA AUTOIMMUNE REFRATTARIACaselli D, D’Angelo P, Farruggia P, Russo D, Trizzino A,Ziino O, Locatelli F,* Aricò MOnco Ematologia Pediatrica, Ospedale dei Bambini “G. DiCristina”, Palermo *Onco Ematologia Pediatrica, IRCCSPoliclinico San Matteo, Pavia
Il trapianto di cellule staminali emopoietiche (TCSE) èstato progressivamente introdotto nel trattamento di unampio spettro di malattie autoimmuni refrattarie alle tera-pie convenzionali. Si preferisce in genere il trapianto auto-logo per la minore tossicità, ma in letteratura sono ripor-tati alcuni casi trattati con trapianto allogenico seguito daremissione completa e persistente della malattia di base.La sindrome di Evans è una combinazione di anemia emo-litica e trombocitopenia autoimmuni che è generalmenteè più difficile da trattare delle singole entità separate. R.A. , 20 mesi, sviluppa citopenia trilineare autoimmune percui viene trattato con cortisone + IG ad alte dosi; la rispo-sta è pronta ma transitoria, con recidiva 1 mese dopo lasospensione. Trattato con altri 3 cicli di IgG ad alte dosiha una risposta modesta e transitoria. Viene trattato quin-di in successione con: Rituximab; ciclosporina + predni-sone; ciclofosfamide; micofenolato. Mentre la piastrino-penia si è risolta dopo 6 mesi, il bimbo rimane trasfusio-ne-dipendente. Essendo il fratellino HLA-identico, si deci-de di avviare il piccolo al TCSE dopo condizionamento conMelphan (140 mg/m2) + Thiotepa (10 mg/kg in 2 dosi) +Fludarabina (40 mg/m2 x4); la profilassi della GVHD è sta-ta condotta con MTX (4 dosi) e ciclosporina. Vengono infu-se 3. 5x108/kg cellule mononucleate con attecchimentoin +18; il chimerismo in +23 mostra 85% donatore. Per-siste tuttavia l’anemia emolitica ed una riattivazione del-l’infezione da CMV, che viene trattata con successo, mache si accompagna ad una progressiva riduzione del chi-merismo fino al 40% donatore in +34. In +39 prima infu-sione di linfociti del donatore (DLI; 1.6x107/kg del rice-vente) ripetuta in +59 (5x107/kg); il chimerismo in +53 è60% donatore. Attualmente, in +90, piastrine e neutrofi-li sono stabilmente normali e l’Hb è in lento recupero, adoltre 45 giorni dall’ultima trasfusione. Lo svezzamentodalla necessità di emotrasfusioni di GRCF in presenza diun chimerismo misto, sostenuto anche con l’uso di DLIsembra suggerire che il TCSE allogenico ha avuto nei mesisuccessivi al trapianto un effetto favorevole sui meccani-smi della emolisi immunomediata. Un follow-up più pro-lungato è necessario prima di stabilire che la remissionepossa persistere in effetti a lungo termine, confermandola validità di questa opzione terapeutica inusuale.
139
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

P077RUOLO DELLA MALATTIA RESIDUA MINIMA NELLA LEUCEMIALINFOBLASTICA ACUTA DOPO TRAPIANTO ALLOGENICOBonanomi S,* Manenti M,° Coliva T,* Songia S,° Dassi M,'Riva MR,' Corti P,* Cazzaniga G,° Perseghin P,'Mascaretti L,' Uderzo C,* Biondi A,° Balduzzi A**Clinica Pediatrica dell'Università di Milano Bicocca,Ospedale San Gerardo, Monza; °Centro Ricerca M. Tettamanti, Ospedale San Gerardo, Monza; 'CentroTrasfusionale, Ospedale San Gerardo, Monza
Introduzione: La recidiva di LLA dopo trapianto alloge-nico ha prognosi infausta; l’identificazione precocemediante analisi della malattia residua minima (MRM)potrebbe offrire possibilità di trattamento. Pazienti eMetodi: Sono stati monitorati 11 pazienti (9 maschi, etàmediana 11 anni, range 2-16) trapiantati da donatorefamiliare (5 HLA identici, 1 incompatibile ad un locus, 1aploidentico) o da banca (4) per LLA, 5 in 1°, 4 in 2°, 2 in3° RC, dopo regime di condizionamento contenente TBI. Ipazienti sono stati monitorati per MRM a 1, 3, 6, 9 e 12mesi dopo trapianto, oppure in base all’andamento clini-co. Cinque pazienti hanno sviluppato GVHD acuta di II-IVgrado, trattata in 4 casi con ATG. Sei pazienti sono vivi inRC (mediana 14 mesi, range 6-19), uno è deceduto in RC(7 mesi), 4 sono ricaduti (mediana 7 mesi, range 3-23), edi questi 3 sono deceduti. Risultati: All’analisi della MRMal momento del trapianto 8 pazienti erano positivi, men-tre 3 erano negativi. Dei 3 pazienti negativi per MRM, 1 èrimasto negativo ed è in RC a 19 mesi, 1 si è positivizza-to 6 mesi dopo trapianto da banca ed è ricaduto due mesidopo, mentre 1 è ricaduto 30 mesi dopo trapianto aploi-dentico. Degli 8 pazienti positivi per MRM, 2 sono rima-sti sempre positivi e sono ricaduti a 3 e 7 mesi dopo tra-pianto, mentre 6 hanno presentato negativizzazione. Dei6 pazienti, 4 si sono negativizzati al primo o al terzo mese,3 di loro sono vivi in RC a 9, 12, e 13 mesi dal trapiantomentre 1 è deceduto in RC; uno dei 6 ha presentato MRMpositiva a 6 mesi da trapianto dopo una transitoria nega-tivizzazione ed è in RC, mentre uno ha alternato positivitàe negatività ed è in RC a 6 mesi. L’analisi quantitativa del-la MRM ha permesso in 1 paziente l’infusione dei linfoci-ti del donatore (DLI) e in 5 un precoce scalo/interruzionedella terapia immunosoppressiva, comportando GVHDsevera in 1 paziente. Di questi 5 pazienti 2 sono in RC,mentre 3 sono ricaduti, nonostante uno abbia presentatouna transitoria riduzione quantitativa della MRM ed unonegativizzazione della MRM. Conclusioni: Il monitoraggiodella MRM nei pazienti sottoposti a trapianto per LLA adalto rischio potrebbe fornire l’indicazione per un tratta-mento immunologico, quale una precoce interruzione del-la terapia immunosoppressiva o DLI come prevenzionedella recidiva di malattia.
P078ANALISI DELL'ESPRESSIONE GENICA DELLE IMMUNOGLOBULINE NELLECELLULE B CD27+ DI MEMORIA DOPO TRAPIANTO DI CELLULE STAMINALIEMATOPOIETICHE NEI BAMBINIScuderi F, Terranova MP, Di Martino D, Valetto A,*Miano M, Lanino E, Faraci M, Dini G, Dallorso S,Morreale GDipartimento di Ematologia e Oncologia Pediatrica,Istituto G. Gaslini, Genova; Dipartimento di GeneticaMolecolare U. O. S. Chiara, Pisa
Introduzione e obiettivi: La ricostituzione immunologiadopo trapianto di cellule staminali ematopoietiche (TCSE)è un processo lento e complesso che coinvolge tutte lecomponenti del sistema immunitario. Nei primi mesi dopoil trapianto si verifica pertanto una fase di immunodefi-cienza con conseguente elevato rischio di complicanzeinfettive. Scopo del nostro lavoro è la valutazione del coin-volgimento dei linfociti B di memoria mediante l’analisidell’espressione della terza regione ipervariabile (CDR3)della catena pesante delle immunoglobuline IgM nellasottopopolazione di cellule B CD27+/CD19+. Metodi: Sonostati inseriti nello studio 7 pazienti sottoposti a TCSE allo-genico e 9 pazienti sottoposti a TCSE autologo presso l’I-stituto G. Gaslini di Genova. A +180gg dopo il trapianto,sono stati selezionati linfociti CD27+ mediante separazio-ne in campo magnetico (MACS). L’espressione del CDR3della catena pesante delle IgM è stata analizzata con latecnica dell’IgH CDR3 fingerprinting sui linfociti B dimemoria e sulla popolazione totale di cellule B, isolata dasangue periferico prima del trapianto e a vari tempi dopotrapianto. Risultati: Nei pazienti studiati, l’analisi del CDR3fingerprinting, nei primi 30-90 giorni dopo il TCSE, ha evi-denziato una situazione di mono e/o oligoclonalità (pre-senza di una o poche bande) rispetto alla normale situa-zione di policlonalità (16-20 bande) che si osserva neldonatore sano. Nella maggior parte dei pazienti il reper-torio delle cellule B ha raggiunto una situazione di nor-male policlonalità al tempo +90 nel trapianto autologo eal tempo +180 nell’allogenico. A +180 giorni post TCSE,il repertorio IgH CDR3 delle IgM per le famiglie VH3/VH4risulta policlonale sia per le cellule CD27+ che per la popo-lazione totale di linfociti B, mentre per la famiglia VH6nelle cellule CD27+ si evidenzia una diversa oligoclonalitàrispetto alla popolazione totale di linfociti B. Conclusio-ni: Il nostro studio evidenzia che dopo 6 mesi dal TCSE siaallogenico che autologo, il compartimento di linfociti B dimemoria ricostituisce come la totalità della popolazionedi cellule B, per quanto riguarda le famiglie VH3 e VH4,mentre si ha una ricostituzione immunologia più lenta perquanto riguarda la famiglia VH6. Sulla base di tale risul-tato è possibile ipotizzare una implicazione della famigliaVH6 nella ricostituzione dei linfociti B di memoria dopoTCSE.
140
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Trapianto

141
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
P079TRAPIANTO DI CELLULE EMATOPOIETICHE NELLE MALATTIE METABOLICHECONGENITE: I PRIMI 5 ANNI DI ESPERIENZA DEL MOMINRovelli A, Corti P, Furlan F, Bonanomi S, Gaipa G,Venturi N, Perseghin P, Dassi M, Sersale G, Parini R,Bertagnolio B, Uziel G, Longoni D, Balduzzi A, Uderzo C,Biondi A, Masera GMonza-Milano Network for Lysosomal and PeroxisomalDiseases (MOMIN)
Le malattie metaboliche congenite si trovano alla con-vergenza di molte scoperte di biologia di base con ladisponibilità di trattamenti sperimentali e sono conside-rate un modello per affrontare molti dei problemi dellemalattie orfane. I trattamenti oggi disponibili includono iltrapianto di cellule ematopoietiche (HCT) e la terapia enzi-matica sostitutiva, mentre trattamenti combinati o altrialternativi costituiscono in modo realistico la frontiera deiprossimi anni. Al nostro network, attivo da oltre 5 anni,sono stati riferiti sinora 50 casi con la richiesta di valu-tarne l’eleggibilità all’HCT. Dodici pz. con diverse malat-tie sono stati sottoposti a 14 HCT, nella maggioranza dadonatore non correlato e in genere non “fully matched”.Escludendo un caso con f. u. troppo breve, 10/11 pz. sonovivi con un f. u. mediano di 3. 3 (1.2-5. 7) anni. In 7/11l’HCT è risultato di successo nel ricostituire l’enzima difet-tivo e modificare sensibilmente la storia naturale dellamalattia. L’obbiettivo dell’HCT oggi non può essere sem-plicemente considerato la sopravvivenza con correzionedei livelli enzimatici, ma la sopravvivenza con un’adegua-ta qualità di vita e la maggior autonomia possibile. Infat-ti i 4/11 insuccessi sono così considerati: 2 per ragionitrapiantologiche, ma 2 (chimera completa con stabilizza-zione neuroradiologica) per sviluppo di sequele (mioclo-no negativo) di tale entità da compromettere il recuperoneuropsichico e la qualità di vita. Riteniamo comunquenotevoli i risultati conseguiti (positivi in 7/11 pz. con bas-sa TRM, 1/11), peraltro in patologie altrimenti inesorabil-mente fatali. L’HCT non è il trattamento ottimale, per lomeno nelle sue modalità applicative attuali, ma per mol-ti casi è l’unica terapia possibile. Il successo dipende dal-l'efficienza della cross-correction e dalla plasticità tran-sdifferenziativa della staminale ematopoietica di cui que-sti pz. rappresentano il modello in vivo. La scarsa effi-cienza di questi fenomeni e la particolare evolutività dialcune malattie rende ragione dei fallimenti osservabiliquando non si sia provveduto ad un’accurata selezionedei casi. Quest’ultima e una gestione multidisciplinarecoordinata e complessiva di tutti gli aspetti clinici corre-lati alla malattia di base costituiscono i fattori principaliper il successo trapiantologico.
P080EFFETTI SULLA FUNZIONALITÀ ENDOCRINA POST-TRAPIANTO:DIMENSIONE DEL PROBLEMA E PREVENZIONEBarbieri E, Prete A, Pasini A,* Castellini C, Marino F,Pession AOncoematologia Pediatrica e Terapia Cellulare,Clinica Pediatrica S. Orsola-Malpighi. Bologna,*Endocrinologia Pediatrica. Clinica PediatricaS. Orsola-Malpighi, Bologna
Obiettivi: Monitorare, prevenire e curare gli effetti avver-si endocrini della chemio-radioterapia precedente e di pre-parazione al trapianto di cellule staminali emopoietiche(tCSE) nel bambino oncologico lungo-sopravvivente permigliorarne la qualità di vita. Metodi: 74 pz, 48M (25 pre-puberi, p0, 12 all’inizio, p1, e 11 in pubertà avanzata oadulti, p2), e 26F (15 p0, 11 p1), sottoposti ad allo (36) eauto (38) tCSE ad una età media 7. 4 aa (0.8-16. 5), doporadio-chemioterapia di I/II linea, perché affetti da patolo-gia oncoematologica (67) e non (7). La terapia di prepara-zione al tCSE comprendeva alchilanti, tra cui Busulfano(Bu; 38) e/o TBI (20). E’ stata valutata la funzionalità tiroi-dea, gonadica e, in 35 pz, l’osteosonografia con determi-nazione di AD-SoS, amplitude-dependent speed of sound,espresso in Z-score per età. Risultati: FUP medio dal tCSE:5. 2 aa (1-18). Funzionalità tiroidea: dopo un tempo medio(Tm) post-tCSE di 4. 3 aa (1-10), 31 pz, 41.8%, (11 TBI, 7Bu al tCSE, 8 Rx-terapia craniospinale, mediastinica o pol-monare; 5 MIBG) mostravano segni laboratoristici (23 conipertireotropinemia, 19 sottoposti a terapia sostitutiva) e/oecografici (27) di alterata funzionalità tiroidea. Funziona-lità gonadica: 22 pz M (5 p0, 6 p1, tutti gli 11 p2), 45. 8%,presentavano elevati valori di FSH, 8 (2 p1, 6 p2), 16. 6%,elevati livelli di LH. Solo per i 2 pz sottoposti a irradiazio-ne testicolare si è resa necessaria una terapia sostitutiva.19 pz F (8 p0, tutte le 11 p1), 73%, mostravano elevatilivelli di gonadotropine. 8 pz puberi con insufficienza ova-rica hanno richiesto terapia sostitutiva. Osteosonografia:15 pz (42.8%) presentavano gradi variabili di osteoporo-si/osteopenia (AD-SoS Z-score <-2DS in 8 pz. , tra <-2DSe <-1DS in 7). Conclusioni: Si conferma il ruolo della tera-pia radiante nello sviluppo del danno tiroideo, precoce-mente individuato, nei pazienti eutiroidei, mediante eco-grafia. Il danno gonadico si evidenzia in pubertà e mag-giore è la predisposizione delle femmine a sviluppare unsevero danno ovarico. L’Eco-doppler, con valutazione del-la pulsatilità ovarica e uterina, potrebbe essere un mezzoper individuare precocemente le pz che richiedono inter-venti terapeutici. Tanto più accurata e precoce è la rileva-zione della tossicità endocrina, tanto più efficace è la curadel danno tiroideo o gonadico.

ALTRO
C009L’ASSETTO PSICOSOCIALE DEI CENTRI AIEOPJankovic MClinica Pediatrica Università di Milano-Bicocca,Ospedale S. Gerardo, Monza (con il contributo di AMGEN– ISIS Research) a nome del CSD Psicosociale AIEOP
Introduzione: La finalità di questa indagine è stata quel-la di avere una fotografia reale dell’assetto psicosociale deinostri Centri per poter a) sapere come e dove migliorareo integrare certe realtà, b) organizzare studi ad hoc in taleambito. Metodi: I Centri contattati sono stati 56: da tut-ti questi Centri sono state ricevute notizie sul flusso dibambini, sulla tipologia dei tumori trattati, sull’assettomedico ed infermieristico comprendendo anche operato-ri non strutturati. Risultati: Assistenti sociali presenti nel73. 2% dei Centri; servizio scolastico presente nel 76. 8%(62.7% scuola materna, 97. 6% scuola elementare, 76.8% scuola media, 41.9% scuola superiore); supporto psi-cologico presente nel 87. 5%; riunioni periodiche tra com-ponenti del gruppo oncologico (87. 5%); disponibilità disala giochi per i bambini degenti (85. 7%), disponibilità disala giochi per i bambini in Day-Hospital (75. 0%), dispo-nibilità di personale dedicato al gioco (82.1%), altre atti-vità ricreazionali (64. 2%). Tipo di sostegno alla famiglia:logistico (82.1%), economico (69. 6%), lavorativo (33.9%); presenza di una struttura esterna per accogliere lefamiglie (SI 64. 3%); carenze di personale per tale assi-stenza: psicologi (66. 6%), insegnanti (21.4%), assistentisociali (33. 9%). Conclusioni: Questa fotografia moltopuntuale della situazione consentirà riflessioni specifiche.Ulteriori dettagli dell’indagine verranno presentati in sededi Congresso.
C010ANALISI DEI POLIMORFISMI NEI GENI PER LE CITOCHINE IN PAZIENTIAFFETTI DA ISTIOCITOSI A CELLULE DI LANGERHANS (ICL)De Filippi P,1 Danesino C,1 Ferrarese D,2 De Silvestri A,3Badulli C,3 Trizzino A,4 Fidani P,5 Garaventa A,6Lombardi A,7 Cuccia M,2 Martinetti M,3 Aricò M4
1Genetica Medica, Università di Pavia ed I. R. C. C. S. SanMatteo, Pavia; 2Laboratorio di Immunogenetica,Dipartimento di Genetica e Microbiologia, Università diPavia; 3Laboratorio HLA, Servizio di Immunoematologiae trasfusione, I. R. C. C. S. Policlinico San Matteo, Pavia;4Onco Ematologia Pediatrica, Ospedale dei Bambini“G. Di Cristina”, Palermo; 5Oncologia Pediatrica ed7Ematologia Pediatrica, Ospedale Pediatrico BambinoGesù, Roma; 6I. G. Gaslini, Genova
Introduzione ed Obiettivi. L’istiocitosi a cellule di Lan-gerhans (ICL) è caratterizzata dalla proliferazione ed accu-mulo locale di cellule infiammatorie citologicamente beni-gne, tra cui hanno un ruolo primario le cellule di Lan-
gerhans (CL), cellule dendritiche che risiedono nell’epi-dermide. Alterazioni di citochine e chemochine, che nedeterminano la mobilizzazione, sembrano implicate nellapatogenesi dell’ICL. Nostri studi precedenti suggerisconol’importanza di una componente genetica nello sviluppodella ICL. Abbiamo quindi studiato una serie di polimorfi-smi in differenti geni per le citochine in pazienti con ICLed in una popolazione italiana di controllo. Pazienti emetodi. Quarantuno pazienti, età media alla diagnosi 6. 1(±5. 0) anni, 15 con malattia single-system (SS) diagno-sticata a 7. 5 (±3. 9) anni, 26 multi-system (MS), età media5. 3 (±5. 3) anni. Polimorfismi genici analizzati (CytokineGenotyping Tray; One Lambda Inc. ): Il-1a-889 C/T, Il-1b(–511 C/T; +3962 T/C), IL-1R pst1 1970 C/T, IL 1RA mspa111100 T/C, IL 4Ra +1902 G/A, IL 12 -1188 C/A, γ IFN UTR5644 A/T, TNFα (-308 G/A; -238 G/A), IL 2 (-330 T/G; +160G/T), IL 4 (-1098T/G; -590 T/C; -33 T/C), IL 6 (-174 G/C;nt565 G/A), IL 10 (-1082 G/A; -819 C/T; -592 C/A). Risul-tati. I seguenti polimorfismi hanno una distribuzione ano-mala nei pazienti rispetto ai controlli: IL-4-33 T/C; IL-4-590 T/C; Il-10 –819C/T; IL-1β+3962T/C; Il-1a-889 C/T;TNFα-238 G/A. L’analisi multivariata di queste variabiliha dimostrato che i polimorfismi IL-4 –33TT, IL-4 –590 TCed IL-1β+3962TT sembrano conferire un rischio aumen-tato di sviluppare la forma SS di LCH, mentre i polimorfi-smi TNF-α-238GA, IL-1α-889CC ed IL-10-819CC corre-lano con la probabilità di sviluppare la forma MS. Con-clusioni. Questi dati confermano che le manifestazioni cli-niche della ICL sono condizionate anche da fattori gene-tici che interferiscono con la rete delle citochine.
P081L'ART THERAPY COME SUPPORTO AI BAMBINI SOTTOPOSTI AL TRAPIANTODI MIDOLLO OSSEOFavara Scacco C, Italia S, Pace P, Bonanno A,Di Cataldo ACentro di Riferimento Regionale di Emato-OncologiaPediatrica, Azienda Policlinico di Catania
Introduzione. I bambini candidati al trapianto di midol-lo osseo (TMO) vengono ricoverati in un apposito repartoper almeno 21 giorni incontrando esclusivamente l’ equi-pe sanitaria ed un genitore per volta. Al disagio fisico, siassocia quello emotivo per l’ isolamento e la riduzione distimoli. Pertanto, presso il nostro Centro, durante il TMOoffriamo a bambini e genitori un supporto con Art Therapy(AT), per limitare ansie, paure e mantenere attivi i processidi pensiero e le capacità relazionali. La variabilità dell’ AT,la rende adatta alle diverse età e bisogni. Attraversomodalità creative l’ AT facilita l’ espressione di vissuti inmodo non traumatico così da prevenire accumuli ansio-geni promuovendo continuità nella crescita psico-affet-tiva. Pazienti e Metodi. Dal 3/2000 al 5/2004, sono statitrattati 34 bambini di età 1-14 anni. Il supporto com-prendeva un pre-ricovero che forniva informazioni neces-sarie a rendere il TMO prevedibile e meno ansiogeno, eduna seconda fase di supporto psico-affettivo quotidiano,con tecniche di AT: PAESAGGIO. Quotidianamente si rea-lizza il particolare di un paesaggio, offrendo al bambinouno scopo da raggiungere per mantenere costante la
142
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Altro

motivazione ad agire. BURATTINI. Personaggi ottenutidagli indumenti sterili del reparto favoriscono la confi-denza con essi e l’ elaborazione di nodi emotivi e aggres-sività. MEDICAL PLAY. Una bambola di pezza per il bam-bino ed interviene con materiali medici per sentire il con-trollo sulla malattia. COLLAGE. Tagliare per rilasciare ansiae poi incollare i pezzi per esperire l’ integrazione. COMU-NICARE. Si invitano i bambini a comunicare tra loro com-pletando disegni o frasi per mantenere attive le capacitàrelazionali. RISULTATI. La maggior parte dei bambini harichiesto la presenza dello psicologo durante il TMO e rife-risce soddisfazione per quello che realizza allontanandosolitudine e noia, ansia e rabbia. I genitori rivelano l’ uti-lità del’ AT perché li aiuta a sentirsi attivi e partecipi albenessere del proprio bambino.
P082PROTOCOLLO CGD AIEOP: CARATTERISTICHE CLINICHE E STORIANATURALE DI 59 PAZIENTI CON MALATTIA GRANULOMATOSA CRONICADe Mattia D, Martire B, Rondelli R, Plebani A,Soresina A, Gattorno M, Finocchi R, Rabusin M,Pietrogrande MC, Pignata C, Martino S, Amato GM,Azzari C, Candusso M, Cossu F, Pierani P, Trizzino A,Varotto S, Pession A, Ugazio AGPer il Comitato Strategico e di Studio ImmunodeficienzePrimitive CSS-AIEOP
Introduzione. A maggio 2001 è iniziato l'arruolamentodei pazienti con Malattia Granulomatosa Cronica nel Pro-tocollo CGD AIEOP, attraverso la centralizzazione dei datipresso il Centro Operativo AIEOP-FONOP, secondo un dise-gno operativo teso ad individuare un apprroccio ottimalealla diagnosi e alla cura della malattia. Risultati. A mag-gio 2004, 16 centri afferenti al CSS ImmunodeficienzePrimitive hanno arruolato 59 pazienti (57 M, 2 F), con etàmediana attuale di 13. 5 anni (2 a-38 a). Il periodo mediodi osservazione è stato di 9 anni. Una storia familiare posi-tiva per CGD era presente in 19 pazienti (31%) e in 9 grup-pi familiari c'era più di un probando affetto nella stessafratria. Un pattern di ereditarietà X recessivo è stato dimo-strato nel 66% dei pazienti, AR nel 10%, sconosciuto nelrestante 24% dei casi. L'età mediana alla diagnosi era di2 anni (1 m-38 a), l'età mediana di esordio dei sintomi di7. 5 mesi (1 m-10 a). L'età mediana alla diagnosi e all'e-sordio dei sintomi è significativamente più bassa neipazienti con CGD XR rispetto a quelli con malattia AR (2a vs 5. 5 a e 0.1 a vs 0.5 a). Le infezioni più frequenti alladiagnosi sono state: infezioni della cute (dermatiti e asces-si sottocutanei) 62%, dell'apparato respiratorio e linfo-ghiandolare,47% rispettivamente. Durante il follow-up ilnumero di infezioni totali registrate è stato di 147, di cui70 (47%) infezioni gravi (che hanno richiesto ospedaliz-zazione, terapia parenterale e/o intervento chirurgico). Leinfezioni più frequenti sono state: infezioni dell'apparatorespiratorio (45%), infezioni cutanee (36%) e linfoadeni-ti (25%). Il 13% dei pazienti ha sviluppato complicanzeinfiammatorie croniche intestinali (enterocoliti). L'inci-denza di infezioni per paziente per anno di follow up è sta-ta di 0.5. Il numero di infezioni totali e di infezioni graviè significativamente maggiore nei pazienti con CGD XR
rispetto a quelli con malattia AR (rispettivamentep=0.0001 e p=0.016). Non c'è differenza significativa nei2 gruppi di pazienti per quanto riguarda le infezioni mino-ri; fra queste le piu' frequenti sono state: infezioni dellebasse vie urinarie 35%, stomatogengiviti 27. 5% e con-giuntiviti 23%. Lo S. Aureo è la causa maggiore di infe-zione (35% degli isolamenti microbiologici), mentre l'Aspergillus spp. è la causa più frequente di polmonite(50% degli isolamenti microbiologici). Il numero di pazien-ti che interrompe la profilassi antinfettiva con cotrimos-sazolo, itraconazolo e gamma-interferone, usati singolar-mente o in associazione, aumenta nel corso del follow up(11% vs 2% alla diagnosi). Durante il periodo di osserva-zione 5 pazienti sono morti(4 per complicanze infettive, 1per GVHD cronica post TMO); l'età mediana all'exitus è di14 anni (0-18 a). Il rate di sopravvivenza a 5 e 20 anni èrispettivamente del 90% e 80%. Sei pazienti con CGD XRhanno eseguito TMO da fratello HLA identico; l'età media-na al trapianto era di 9. 5 anni (0.7 a-20 a). Due pazientisono deceduti per complicanze infettive; i restanti 4 sonovivi e senza malattia ad un periodo medio di osservazio-ne di 9. 7 anni (4 a-18 a). Conclusioni. I dati acquisiti daquesta ampia coorte di pazienti potranno consentire unamigliore definizione della storia naturale della malattia edi individuare approcci terapeutici più efficaci al fine dimigliorarne la qualità di vita.
P083LE RACCOMANDAZIONI AIEOP PER LA DIAGNOSI E LA TERAPIADELL'AGAMMAGLOBULINEMIA X-RECESSIVA: CARATTERISTICHECLINICHE E STORIA NATURALE DI 129 PAZIENTIPlebani A, Soresina A, Rondelli R, Notarangelo LD,Duse M, Quinti I, Pietrogrande MC, Martino S,De Mattia D, Martire B, Rossi P, Moschese V,Cardinale F, Masi M, Azzari C, Pignata C, Amato GM,Cazzola Ga, Consolini R, Locatelli F, Nigro G, Stabile A,Marseglia Gl, Sciotto A, Di Nardo R, Strisciuglio P,Aricò M, Ambrosioni G, Nespoli L, Felici P, Spadaro G,Orlandi P, Fiorini M, Pession A, Ugazio AG*Per il Comitato Strategico e di Studio Immunodeficienze(CSS ID)AIEOP. *Coordinatore CSS ID AIEOP
L'arruolamento dei pazienti con AgammaglobulinemiaX-recessiva (XLA) ha avuto inizio nel gennaio 2000 daparte dei Centri del Comitato Strategico e di Studio Immu-nodeficienze AIEOP, con sistematica registrazione dei datinella banca dati web based AIEOP XLA. A maggio 2004, 25/ 54 Centri hanno arruolato 129 pazienti, che corrispon-dono al 95% dell'atteso nazionale per questa patologiarara. L'analisi di mutazione del gene BTK è stata eseguitain 122/128 casi valutabili e la mutazione risulta presentein 103 (85%). L'età mediana alla diagnosi risulta pari a 3.5 anni (range 3 mesi-58 anni) con esordio dei sintomi a16. 5 mesi (range 3 mesi-11 anni). Tra le patologie prin-cipali alla diagnosi le più frequenti risultano essere: otiti(32%) e broncopolmoniti (42%), seguite dalle infezionicutanee (21%), infezioni gastrointestinali (20%), artriti(20%), sepsi (6%) e meningiti (4%). L'età mediana attua-le è di 15 anni (range 1-74 anni); la durata mediana del-la terapia sostitutiva con Immunoglobuline per via endo-
143
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

venosa (IVIG) ed il tempo di osservazione risultanoentrambi pari a 9,5 anni (range 2 mesi - 32 anni) con untempo cumulativo di osservazione di 1287 anni. La curvadi sopravvivenza a 30 anni dalla diagnosi è del 78% consegnalati 7 casi di morte, metà dei quali dovuti a compli-canze broncopolmonari. Durante il periodo di follow-up,in corso di terapia con IVIG, risulta molto ridotta l'inci-denza di infezioni sistemiche gravi, quali sepsi e menin-giti (tasso di incidenza annuale: 0.002), mentre, perman-gono problemi di carattere infettivo sia broncopolmonariche a livello dei seni paranasali (tasso di incidenza annua-le rispettivamente di 0.08 e 0.15). Inoltre, all'ultimo fol-low-up un quadro di broncopneumopatia cronica è risul-tata presente in 46/128 casi (35%) e di sinusite cronicain 56/128 casi (44%). A 4 anni di operatività, il principa-le obiettivo terapeutico previsto, cioè garantire a tutti untrattamento terapeutico adeguato, è stato raggiunto:all'ultimo follow-up la percentuale di pazienti trattati cor-rettamente è salita dal 86% al 99%. Ora il nostro impe-gno è concentrato nel cercare gli strumenti più adeguatiper prevenire e controllare le complicanze a lungo termi-ne più invalidanti per i nostri pazienti, come le compli-canze a carico dell'apparato respiratorio. Si ringrazia peril supporto la Associazione AIP.
P084L’IMPATTO DELL’INTRODUZIONE DI RACCOMANDAZIONI PER LA DIAGNOSIE IL TRATTAMENTO DELL’AGAMMAGLOBULINEMIA X-RECESSIVA (XLA)SULLE MIGRAZIONI EXTRAREGIONALI: ESPERIENZA DEI CENTRIAIEOP-IMMUNODEFICIENZESoresina A, Rondelli R, Pession A, Plebani A,° Ugazio A*Data Review Committee Comitato Strategico e di StudioImmunodeficienze (CSS ID)AIEOP; °Responsabile XLAAIEOP; *Coordinatore CSS ID AIEOP
L’XLA è una malattia congenita del sistema immune(incidenza 1/100000). Perché rara, la diagnosi può esseredifficile e tardiva e richiede raccomandazioni comuni pergarantire a tutti i pazienti su tutto il territorio nazionaleun approccio diagnostico-terapeutico ottimale, il più pos-sibile vicino a casa. Le raccomandazioni per XLA, operati-ve da Gennaio 2000, e la crescita di una rete di CentriAIEOP ID (CAID) in gran parte d’Italia, dovrebbero contri-buire a ridurre il fenomeno migratorio extraregionale(FME) di pazienti dal 2000 in poi. Ci proponiamo di misu-rare il FME di casi XLA e verificare se l’introduzione di rac-comandazioni nazionali influenzi nel tempo il FME. Si èanalizzato il FME per diagnosi e terapia dei casi XLA, regi-strati nella banca dati AIEOP XLA a Dicembre 2003, resi-denti in Italia, in regioni dove sono presenti CAID. Si è cal-colato il rapporto % tra casi residenti diagnosticati in CAIDdella stessa regione e casi residenti per ciascuna regioneconsiderata, confrontandolo per periodo di diagnosi(<2000 vs >2000). Stesso rapporto si è calcolato consi-derando i CAID dove i pazienti sono stati trattati dopo ladiagnosi. Il FME è analizzato anche per aree (Nord, Cen-tro, Sud, Isole) di residenza e di appartenenza dei CAID didiagnosi e di terapia. I casi sono 117, registrati da 24 CAID:99 (84. 6%) sono diagnosticati dal 1971 al 2000 e 18 dal2000 al 2003. Il FME per diagnosi di XLA interessa il 28.
2% di casi, ma risulta essersi ridotto (da 29. 3% a 22.2%).Il FME risulta elevato per i pazienti del Sud e Isole, che sirivolgono a CAID del Centro-Nord nel 43. 9% dei casi, masi è notevolmente ridotto negli anni (da 50% all’attuale14. 3%). Il FME dovuto al trattamento successivo alla dia-gnosi interessa il 26. 5% dei casi, riducendosi notevol-mente nel tempo da 28. 3% a 16. 7% del periodo 2000-2003. Anche in questo caso, pur restando elevata per icasi del Sud e Isole (41.5%), vi è una riduzione progressi-va del FME negli anni fino ai valori attuali di 14. 3%. Lamigrazione sia per diagnosi che terapia si è ridotta neltempo, soprattutto per i casi residenti al Sud e Isole. Nonvi sono differenze significative di età alla diagnosi tra i dueperiodi (anche se in quello >2000 non ci sono casi con età>15 anni). La migrazione per diagnosi è superiore a quel-la per trattamento. Più Centri, grazie alle raccomandazio-ni, si sentono in grado di curare i pazienti.
P085INFEZIONI DEL TUNNEL DEL CATETERE VENOSO CENTRALE (CVC) ETERAPIA ANTIBIOTICA IN INFUSIONE CONTINUA IN BAMBINI CONPATOLOGIA NEOPLASTICAGiacchino M,* Bezzio S,* Valera M,* Barisone E,*Marengo M,** Kanga Leunga GG,* Tovo PA**Dipartimento di Onco-Ematologia ed Immuno-Infetti-vologia Ospedale Infantile Regina Margherita Torino**Servizio Farmacia Ospedale Infantile Regina MargheritaTorino
Le infezioni del tunnel rappresentano una complicanzadifficile da trattare. Le linee guida americane consiglianola rimozione immediata del catetere associata a terapiaantibiotica sistemica in quanto i tentativi conservativisono gravati da un alto tasso di insuccesso (>80%). Obiet-tivo dello studio è valutare nella nostra casistica l'effica-cia dell'antibioticoterapia in infusione continua (i. c. ) chepermette di: evitare frequenti manipolazioni del CVC eboli possibili generatori di emboli settici, somministrareantibiotici la cui attività battericida è tempo dipendente,in modo da avere costante concentrazione battericidasuperiore alla MIC. Nel periodo in studio (01/01/2000-31/12/2003) sono stati inseriti 519 CVC in 427 pazientiafferenti al Dipartimento di Onco-Immuno-Ematologia-dell'Ospedale Infantile Regina Margherita di Torino. Leinfezioni del tunnel hanno costituito il 3% delle compli-canze infettive con 15 eventi; 7 (47%) con diagnosi micro-biologica 8 (53%) con diagnosi clinica. Si sono manife-state in 12 bambini (3%), uno ha presentato 3 episodi euno ne ha presentati 2; in media ogni bambino ha pre-sentato 0.02 infezioni del tunnel nei 4 anni di studio. L'in-cidenza per catetere introdotto è stata di 0.071/1000 gior-ni CVC. Il 100% delle infezioni è stato causato da Grampositivi, in prevalenza S. coagulasi-negativi (6) e in parti-colare S. hominis (4). La terapia antibiotica è stata effet-tuata mediante:- i. c. di Vancomicina 40 mg/kg/die in 24halla concentrazione di 4 mg/ml in media per 10 giorni nei7 CVC con diagnosi microbiologica di infezione da Gram+. - Cefalosporina di terza generazione e. v. per via peri-ferica in boli di 15-30 minuti in media per 10 giorni asso-ciata a Vancomicina i. c. per via centrale negli 8 con infe-
144
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Altro

145
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
zione clinicamente documentata- terapia locale conAureomicina Il successo di tale terapia è stato 80% aduna prima analisi, ridotto a 60% comprendendo anche lereinfezioni (3, di cui due nello stesso paziente). In lette-ratura è riportato un successo terapeutico del 5-20%. Lanostra esperienza ha dimostrato che è possibile tentareuna terapia antibiotica sistemica. Attualmente riteniamoche sia consigliabile il ricorso alla rimozione solo in casodi insuccesso o di infezione fungina pur essendo oppor-tuno un ampliamento della casistica per giungere a delleconclusioni definitive.
P086VARIAZIONI TEMPORALI DELLE MIGRAZIONI PER AREA GEOGRAFICA INBAMBINI CON TUMORE MALIGNO REGISTRATI DAI CENTRI AIEOP CONMOD. 1.01Dama E,* Rondelli R,“ Pastore G,*° Magnani C,*°Pession A“*Registro dei Tumori Infantili del Piemonte. Sezione diEpidemiologia dei Tumori, CPO Piemonte, A. O. S. Giovanni e Università di Torino°Struttura SempliceOncoematologia Pediatrica - Ospedale S. AndreaVercelli°°Dipartimento di Scienze Mediche, Università delPiemonte Orientale“Dipartimento di Pediatria, Universitàdi Bologna
Introduzione e Obiettivi. Obiettivo dello studio è la valu-tazione della concordanza tra area di residenza ed areadove è collocato il centro di cura, al fine di indagare ilfenomeno delle migrazioni sanitarie tra i casi di tumorepediatrico curati presso i Centri afferenti all’AssociazioneItaliana di Ematologia Oncologia Pediatrica (AIEOP).Metodi. Nello studio sono stati inclusi i pazienti con dia-gnosi di tumore in età pediatrica (0-14 anni) registratinella banca dati AIEOP Mod. 1.01 tra il 1989 e il 1998.Sono stati considerati nell’analisi 10.791 soggetti, esclu-dendo i casi con diagnosi di Istiocitosi benigna, LeucemiaAcuta Mieloblastica Secondaria e Nefroma Mesoblastico.E’ stata analizzata la distribuzione dei soggetti per areageografica di residenza (Nord, Centro, Sud e Isole) almomento della diagnosi e area geografica di appartenen-za del Centro AIEOP (Nord, Centro, Sud e Isole) che hapreso in carico il bambino, stratificando per periodo didiagnosi (1989-1993, 1994-1998) e per tipo di tumore(Leucemie e Linfomi: LL, Tumori Solidi: TS). Risultati. Con-siderando il periodo di diagnosi, la migrazione è risultataridotta del 2.3% nel 1994-1998 rispetto al 1989-1993(p=. 005). Tale riduzione è stata registrata sia per LL (1.9%,p=. 039) sia per TS (3. 1%, p=. 017). Tra i bambini residentinel Nord la migrazione è risultata pressoché assente perentrambe le categorie di tumore e in entrambi i periodi didiagnosi considerati. Tra i residenti nel Centro le migra-zioni sono diminuite in modo non significativo nel perio-do di diagnosi 1994-1998, con un incremento di sogget-ti che sono rimasti per le cure nella propria area di resi-denza del 1.4% per le Leucemie e i Linfomi, e del 0.9% peri Tumori Solidi. Nel Sud e nelle Isole infine il fenomeno èdiminuito considerevolmente nel periodo di diagnosi1994-1998, con un incremento di soggetti che sono rima-sti per le cure nella propria area di residenza pari al 3. 8%
per le Leucemie e i Linfomi, e al 5. 6% per i Tumori Solidi(p=. 013). Conclusioni. Le migrazioni, per diagnosi e/o trat-tamento, in area geografica diversa da quella di residen-za dei bambini affetti da tumore maligno registrati daiCentri AIEOP interessano circa il 20% dei casi e risultanopiù evidenti nelle aree meridionali e insulari della nostrapenisola. Tuttavia, nel corso degli anni, si è registrata unacostante riduzione di questo fenomeno, più significativoal Sud e nelle Isole (p=. 009) dove la spinta migratoria èstoricamente maggiore.
P087TRASDUZIONE DI CELLULE KILLER INDOTTE DA CITOCHINE (CIK)MEDIANTE VETTORE RETROVIRALEMarin V, Biondi A, D’Amico GCentro Ricerca M. Tettamanti, Dipartimento di Pediatria,Università Milano Bicocca, Italy
Introduzione e obiettivi: le CIK rappresentano una popo-lazione di cellule effettrici che co-esprimono i marcatoriCD3 e CD56 e mostrano un’elevata attività citotossica nonMHC-ristretta contro cellule tumorali derivanti sia datumori solidi sia da alcuni tumori ematopoietici, come laleucemia mieloide acuta (LMA) e la leucemia melodie cro-nica (LMC). E’ stato dimostrato come le CIK, tuttavia, nonsiano in grado di lisare i blasti di leucemia linfoblasticaacuta (LLA). Scopo del presente studio è quello di valuta-re la possibilità di trasdurre le CIK mediante un vettoreretrovirale, al fine di introdurre geni potenzialmente utilia promuovere la loro attività citotossica contro i blasti diLLA. Metodi: Cellule Mononucleate (MNC) di donatori sanisono state incubate per 21 giorni in presenza di IFNγ, dianticorpo monoclonale anti CD-3 (OKT3) e di IL-2.Al ter-mine della coltura il 30% delle cellule co-esprimeva CD3e CD56. Per ottimizzare le condizioni di infezione sonostati confrontati tre diversi metodi di trasduzione con ilvettore retrovirale pMSCV-IRES-GFP. In un pozzetto pre-cedentemente rivestito con retronectina è stato aggiun-to 1 mL di surnatante virale per 18h che è stato succes-sivamente rimosso e sostituito con 25x105 cellule/ml diterreno addizionato con IL-2 e polybrene. Successiva-mente le cellule sono state sottoposte a tre diversi meto-di di centrifugazione: 2 cicli di centrifugazione a 1800rpm per 45‘ a 25°C; 2 cicli a 3700 rpm per 90‘ a 30°C; 4cicli a 1800 rpm per 45‘ a 25°C. Risultati: L’infezione è sta-ta effettuata al giorno +3 e +8 della coltura e la percen-tuale di cellule fluorescenti è stata valutata mediante ana-lisi citofluorimetrica. Indipendentemente dagli rpm uti-lizzati e dal giorno di infezione, il 15% delle cellule sot-toposte a 2 cicli di centrifugazione appariva fluorescente.La migliore efficienza di trasduzione è stata ottenuta uti-lizzando 4 cicli di centrifugazione al giorno +3 di coltura(>25% delle cellule trasdotte appariva fluorescente). Taleefficienza è risultata sostanzialmente ridotta quando l’in-fezione è stata effettuata al giorno +8 di coltura. In tuttii metodi utilizzati non sono state osservate alterazionifenotipiche delle CIK. Conclusioni: Tali risultati prelimina-ri mostrano come le CIK siano suscettibili alla traduzionecon vettore retrovirale. La possibilità di manipolare gene-ticamente tali cellule potrebbe rappresentare un bersaglioinnovativo nella terapia biologica della LLA.

P088UN CASO DI IMMUNODEFICIENZA AD ESORDIO TARDIVOSinelli MT,* D'ippolito C,* Schumacher F,* Soresina R,*Ravelli C,* Ferrè R,^ Facchetti F," Benetti A,"Lanfranchi A,# Bolda F,§ Hershfield MS,§ Rizzari C,°Chiesa R,° Dell'orto MG,° Masera G,° Porta F,*Notarangelo LD**Clinica pediatrica Spedali Civili BS° Clinica pediatricaS. Gerardo Monza^ U. O. Esine (BS)" Dipartimento di 1^Anatomia Patologica Spedali Civili BS# Laboratorio TMOSpedali Civili BS § Laboratory of genetics Duke UniversityNC USA
Descriviamo il caso di un ragazzo giunto per sospet-taimmunodeficienza primitiva. I genitori non sono con-sanguinei ma provengonodallo stesso comune. Il bambi-no ha presentato due episodi di anemia emolitica autoim-mune (AEA) da anticorpi freddi e un discreto numero diepisodi infettivi alle prime vie aeree. A 11 anni nuovo rico-vero per polmonite interstiziale trattato presso la clinicapediatrica di Monza; gli accertamentiimmunologici ese-guiti evidenziavano linfocitopenia con deficit di tuttelesottopopolazioni, particolarmente marcato per CD4+ eCD19+, ed aumentatonumero di CD3+ γ/δ+; il dosaggio del-le immunoglobuline era adeguato mavi era assenza dititolo anticorpale specifico. La risposta proliferativaaimitogeni risultava assente, il dosaggio dell'Adenosin Dea-minasi era nella norma, mentre quello della PurinaNucleoside Fosforilasi (PNP) eraridotto (= 8 U/Hb, con v.n. >330), dato confermato anche dal laboratoriodella DukeUniversity (NC, USA) dove veniva inoltre individuata omo-zigosiper la mutazione 257 Hys>Arg. A distanza di 6 mesiil ragazzo sviluppavaiperpiressia non rispondente a tera-pia antiinfettiva e lesioni polmonaried epatiche dal cuiesame istologico si evidenziava un linfoma B, EBV corre-lato;veniva pertanto iniziata terapia con rituximab da cuiil paziente traevasolo un temporaneo beneficio. L'esameistologico aveva inoltre mostratoquattro elementi ZiehlNielsen positivi per cui si intraprendevaanche terapia anti-micobatterica. Dopo circa un mesesi iniziava un tratta-mento polichemioterapico a dosi ridotte con buonarispo-sta. La terapia veniva discretamente tolleratama contem-poraneamente si assisteva a riattivazione dell'infezioneda CMV(263. 000 copie/mL). Il paziente risulta attual-mente in terapiacon foscavir e ganciclovir ed in attesadella remissione per poter effettuaretrapianto di midolloosseo da donatore volontario. Il deficit di PNP, enzimaubiquitario maggiormente concentrato nei tessutilinfoidiè un raro disordine ereditario a carattere autosomicorecessivoche di regola si manifesta nei primi anni di vitacon disturbi neurologici, autoimmunità (AEA) e infezioni.La prognosi è estremamente severa. Unicaterapia risolu-tiva è rappresentata dal trapianto allogenico di cellule-staminali ematopoietiche (CSE). A tutt'oggi, non sonoriportati sopravviventioltre i 12 anni di vita, non sottopo-sti a trapianto di CSE. Questo casorappresenta quindi unevento eccezionale e suggerisce (in presenza di sintomie-vocativi) l'utilità di un'attenta valutazione immunologia ebiochimicaper SCID anche al di là dei primi anni di vita.
P089ANSIA ANTICIPATORIA E MANOVRE DOLOROSE IN ONCOEMATOLOGIAPEDIATRICA: EFFICACIA DELLA GIOCOTERAPIA COME SUPPORTOPSICOLOGICOScarponi D, Fini MC, Berni B, Agresta S, Mazza S, Pession ASezione di Oncoematologia Pediatrica, Università degliStudi di Bologna
Premessa: Il supporto emotivo al paziente oncologico ealla sua famiglia, che affrontano il dolore, ha un ruoloimportante nelle linee guida in oncologia (Clinical Practi-ce Guidelines in Oncology- v. 1.2004). In pediatria il dolo-re legato alle tecniche procedurali risente degli interven-ti psicologici di sostegno. L'ansia per le manovre ha, nel-la giocoterapia, una collocazione ideale che permette alpaziente di comunicare sentimenti: paura, rabbia e ango-scia, che la sostengono. Dopo tale intervento la manovraviene affrontata dal bambino con maggiore competenzaemozionale e minore sofferenza. Scopo: Valutare in modoprospettico e controllato (giocoterapia si vs no) l'espe-rienza del paziente, dei genitori e degli operatori, attra-verso la Scala delle Facce di Wong/Baker, durante lemanovre dolorose (mediamente: puntura al dito, endove-nosa, intramuscolo; moderatamente: puntura lombare;severamente: aspirato midollare) effettuate senza anal-gesia o anestesia, per mancata indicazione (poco o media-mente dolorose), per rifiuto o non eleggibilità all'inter-vento farmacologico (severamente dolorose). Campione:Da gennaio 2001 a gennaio 2004, 74 pazienti oncologicisono stati seguiti dallo psicologo: prima della manovra,con una seduta di giocoterapia; dopo la manovra, attra-verso il test. I punteggi ottenuti dal gruppo sperimentalesono stati confrontati con quelli del gruppo di controllo,75 pazienti oncologici sottoposti alle stesse manovre, nonseguiti con giocoterapia. Sono stati valutati inoltre: ilgenitore, gli operatori che hanno compiuto la manovra elo psicologo. Risultati: La giocoterapia ha reso la mano-vra subita dai pazienti meno drammatica e la valutazio-ne ha mostrato differenze, nella percezione del dolore,statisticamente significative tra i due campioni di pazien-ti. La stessa differenza è stata espressa dagli adulti (fami-liari ed operatori). Conclusioni: La giocoterapia riduce, coneffetto a cascata, l'ansia di tutti gli attori coinvolti, faci-litandone la partecipazione al percorso di cura: si riducela passività nei pazienti, i sensi di colpa negli adulti. Lo stu-dio ha poi incoraggiato l'équipe a dare spazio ad interventidi supporto, così come indicato dal National Comprehen-sive Cancer Network, per i pazienti che con dolore inoncoematologia.
146
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Altro

147
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
P090FARMACI SU MISURA PER IL PAZIENTE ONCOLOGICO PEDIATRICOAndresciani A, Belardinelli P, Ciuccarelli F, Moretti V,Pierani P, Pompilio A, lucchesi B, Sarteschi A,Formicola F, Rossi R, Miglietta M, Andreuzzi F,Morosini E, Di Lenardo E, loardi GOspedale G. Salesi Ancona, Ospedale Pediatrico MeyerFirenze, Ospedale Santobono di Napoli, Istituto Gaslini diGenova, OIRM S. Anna Torino, O. P. Bambin Gesù diRoma, Azienda Ospedaliera di Padova, Spedali Civili diBrescia
Introduzione. Gli Ospedali Pediatrici e Materno-Infan-tili italiani hanno affrontato la necessità di un coordina-mento delle problematiche e dei progetti nelle diverse areein cui emerge maggiormente la propria peculiarità. L’ areadel farmaco è stata evidenziata come un’ area di vitaleimportanza: ridotto numero si sperimentazioni cliniche,carenza di formulazioni adatte per dosaggio o per formafarmaceutica al paziente pediatrico, uso off-label, farma-ci con eccipienti controindicati in pediatria, esigenza dipersonalizzazione dei dosaggi. Obiettivi. Partendo dallapositiva esperienza maturata in alcuni Centri, la Confe-renza Permanente ha attivato un gruppo di lavoro pervalutare le esigenze e le risposte fornite dalle UU. OO. diFarmacia Ospedaliera negli Ospedali Pediatrici Italiani.Scopo del presente lavoro è quello di 1.analizzare le pre-parazioni galeniche allestite dai diversi Ospedali Pediatri-ci. 2.verificarne la rispondenza alle finalità ed ottimizza-re le scelte metodologiche e pratiche adottate, 3. studia-re le modalità idonee per assicurare la continuità assi-stenziale del farmaco personalizzato anche nella fase didimissione e a domicilio del paziente,4. rendere omogeneasul territorio nazionale la disponibilità di farmaci su misu-ra. Materiali e Metodi. Sono state individuate all’ internodelle formulazioni galeniche pediatriche allestite dagliOspedali Pediatrici quelle di interesse in campo oncologi-co, riguardanti sia la chemioterapia antitumorale, che leterapie di supporto. Si è proceduto a verificare ed otti-mizzare la tecnica di allestimento e l’ adozione delle Nor-me di Buona Preparazione (NBP). E’ stato individuato ilpercorso per la stesura di un Prontuario Galenico Pedia-trico che potesse agevolare l’ applicazione dei protocollie di conseguenza la disponibilità dei preparati di interes-se in tutte le sedi. Risultati. Di seguito sono presentate lepreparazioni pediatriche di maggior interesse in campooncologico realizzate dalle UU. OO di Farmacia Ospeda-liera negli Ospedali Pediatrici Italiani: Principio Attivo(dosaggi personalizzati) Formulazioni già disponibilic/oBusulfano (cartine, capsule, sospensione bloccata):Gaslini Meyer - SalesiCalcio levofolinato (cartine, capsu-le, soluzione): Bambin Gesù ; S. Anna ; Meyer - SalesiCo-deina fosfato 2% (sciroppo, gocce): SalesiEtoposide (solu-zione orale): GasliniIdrossiurea (cartine, capsule): Gaslini;Salesi ; S. Anna - SantobonoIndometacina (cartine, capsu-le): Gaslini; Meyer AO. Padova Isotretinoina 5 mg/mlsospensione bloccata: SalesiLidocaina 2% soluzione sprayper mucosa orale, gel: Salesi; AO. PadovaLomustina (car-tine, capsule): S. Anna - SalesiMercaptopurina (cartine):MeyerMetotressato soluzione/sospensione orale: Salesi-Micofenolato (sciroppo): Gaslini - SantobonoMorfina (sci-
roppo): AO. Padova - SalesiNaprossene (capsule): AO.Padova Orzel cps personalizzate (ac folinico+ uracile-tegafur): SalesiOxibuprocaina (soluzione): O. C. Brescia-Procarbazina (cartine): S. Anna - GasliniTacrolimus (car-tine, capsule, sospensione): Gaslini - SalesiThioguanina(cartine): Gaslini. Conclusioni. Al paziente oncologicopediatrico occorre assicurare farmaci specifici e di sup-porto adatti che facilitino l’ assunzione e la precisione deidosaggi, assicurando la migliore efficacia-sicurezza-com-pliance La collaborazione tra l’ U. O. di Oncoematologia el’ U. O. di Farmacia permette di affrontare e risolvere i sin-goli aspetti della terapia. La Conferenza Permanente sipone come strumento di approfondimento, validazione ediffusione di una migliore assistenza farmaceutica su tut-to il territorio nazionale.
P091IL BAMBINO ONCOEMATOLOGICO E SUO FRATELLOBallarini V, Sciarra P, Di Marzio ADip. Ematologia, O. C. di Pescara
Da oltre due anni presso il reparto di OncoematologiaPediatrica dell'O. C. di Pescara è stato intrapreso un moni-toraggio rivolto a tutti i bambini con lo scopo di indagareessenzialmente su alcuni indici di benessere psicologico deipiccoli pazienti (vedi Atti del Cong. AIEOP, 2002). Si è scel-to di indagare nell'ambito dello sviluppo del concetto delsé, dell'autostima, della capacità del controllo dell'ansia edinoltre della Da oltre due anni presso il Reparto di Oncoe-matologia Pediatrica dell'O. C. di Pescara è stato intrapre-so un monitoraggio rivolto a tutti i bambini con lo scopodi indagare essenzialmente su alcuni indici di benesserepsicologico dei piccoli pazienti (vedi Atti del Congr. AIEOP,2002). Si è scelto di indagare nell'ambito dello sviluppo delconcetto del sé, dell'autostima, della capacità del control-lo dell'ansia ed inoltre della organizzazione delle relazionifamiliari. Per quello che riguarda quest'ultimo punto in par-ticolare, abbiamo utilizzato il Family Relations Test (E.Bene, J. Antonj). Ad oggi il campione complessivo a cui èstato somministrato questo test è di 42 bambini di etàcompresa tra i 6 ed i 17 anni. Tutti quanti i soggetti sonopazienti in carico al nostro servizio che si trovano in diver-se fasi del programma terapeutico o in follow up, fattaeccezione per 5 soggetti che nel frattempo sono deceduti.L'obiettivo del Family Relations Test è di individuare lanatura e la qualità dei rapporti del soggetto con tutti glialtri componenti della sua famiglia, attraverso l'utilizza-zione di alcune figure di cartoncino rappresentanti indivi-dui adulti e bambini di ambedue i sessi, per ciascuno deiquali viene chiesto al bambino di assegnare una colloca-zione fisica ed una serie di commenti, anch'essi rappresen-tati su cartoncino. Come è facile intuire, il bambino nor-malmente colloca ai primi posti della sua composizionefamiliare le due figure genitoriali e, a seguire, fratelli, non-ni etc. Diversamente da quello che ci si poteva aspettare, ilnostro campione ha prodotto un piccolo ma significativosconvolgimento di questa teorica graduatoria, riservando,si, il primo posto alla figura materna, ma dando la succes-siva preferenza, là dove ce ne siano, al proprio fratello osorella piuttosto che al padre come ci si sarebbe potutoaspettare (Figura 1). Il fratello assume quindi il delicato

148
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Altro
ruolo di colui che diventa, all'interno del gruppo dei pari,quello con cui identificarsi e vivere attraverso di lui ciò cheal bimbo malato normalmente non viene consentito. E'importante sottolineare che la rilevanza del fratello vale siaper i sentimenti positivi che per quelli negativi; in assolu-to egli rappresenta una sorta di alter ego. Nella stragran-de maggioranza dei casi il fratello è stato definito il miomigliore amico.
P092NODULI DI IPERPLASIA RIGENERATIVA EPATICA: EFFETTO TARDIVODOPO TRATTAMENTO PER TUMOREGrisolia F,# Hanau G,# Fioredda F,# Faraci M,#Tomà P,* Magnano GM,* Caruso S,° Haupt R°#Divisione di Emato-Oncologia e Trapianto di MidolloOsseo ° Servizio di Epidemiologia e Biostatistica;*Servizio di Radiologia, Istituto Giannina Gaslini, Genova
INTRODUZIONE e OBIETTIVO: E’ nota la possibilità dirilievo di noduli di iperplasia rigenerativa epatica (IRE) inpazienti oncologici trattati con chemio-radioterapia. Ladiagnosi è sostanzialmente radiologica mentre l’ eziologiaè poco chiara così come la patogenesi. Si sono quindi riva-lutate retrospettivamente le caratteristiche di tali lesionie dei pazienti ai quali sono state diagnosticate. METODI:Sono state riviste le cartelle dei pazienti trattati presso ilnostro centro per neoplasia in età pediatrica ai quali,durante il follow-up, nel periodo 1990-2004, era statadiagnosticata la presenza di almeno 1 nodulo di IRE, docu-mentato all’ ecografia come immagine ipo-iso o ipereco-gena frequentemente lobulata e con zona centrale a for-ma di stella. Per ogni paziente con IRE sono stati raccoltidati sui trattamenti ricevuti e su possibili fattori di rischio.RISULTATI: Sono stati individuati 21 pazienti con noduliepatici, 3 dei quali sono stati esclusi dallo studio perchéaffetti da epatite C. Dei 18 pazienti inclusi, 11 erano sta-ti affetti da neuroblastoma, 2 da t. a cellule germinali, 2da t. renali, 1 da sarcoma di Ewing, 1 da LMA e 1 da LLA.L’ età mediana alla diagnosi di tumore era di 4 anni (ran-ge 1-14 anni), e l’ intervallo mediano dalla fine delle curealla diagnosi di IRE e’ stato di 9,7 anni (range 3,2-15,7);
la maggior parte delle IRE è stata diagnosticata nel perio-do 2001-2004. L’ intervallo mediano rispetto all’ ultimocontrollo ecografico normale è stato di 15 mesi (range 4-60 mesi). I noduli erano singoli in 5 casi, multipli in 13, condimensioni variabili tra 4 e 60 mm. 11 pazienti (61%) era-no stati sottoposti a trapianto di cellule staminali TCS- (10auto, 1 allo) ed in 1 si era avuta una VOD. 8 pazienti(44,4%) avevano ricevuto radioterapia sul fegato (6 TBI e2 radioterapia addominale), 3 (14%) avevano effettuatoradioterapia metabolica, 16 (88,8%) antaraciclinici, 16(88,8%) alchilanti, e 1 (5,5%) antimetaboliti. Quattropazienti erano in trattamento ormonale sostitutivo peripogonadismo. La biopsia epatica che ha confermato l’ipotesi diagnostica è stata eseguita in 5 pazienti. CON-CLUSIONI: L’ IRE è un riscontro abbastanza raro in pazien-ti trattati per neoplasia in età pediatrica che sembra esse-re diagnosticata sempre più frequentemente per l’ ausilodi tecniche di imaging più raffinate. Sembra presentarsicon maggior frequenza in soggetti sottoposti a TCS e inpazienti che hanno ricevuto radioterapia sul fegato. Nel-la nostra esperienza tale complicanza è stata diagnosti-cata con maggiore frequenza nei pazienti che hanno rice-vuto una trapianto di cellule staminali, e nei pazienti chehanno ricevuto radioterapia sul fegato.
P093ALINFOCITOSI B, DISFUNZIONE MULTI-ORGANO ED ERITEMA MULTIFORMEIN UN CASO DI INFEZIONE DA MYCOPLASMA PNEUMONIAEMartire B, Foti C,* Cassano N,* de Mattia DDipartimento di Biomedicina dell'Età Evolutiva,Università di BariDipartimento M. I. D. M, Sezione diDermatologia II, Università di Bari
Descriviamo il caso di una bambina di sei anni giuntaalla nostra osservazione per iperpiressia, tosse e algieaddominali. All'esame obiettivo presentava pallore inten-so, epatosplenomegalia, fini rantoli inspiratori e ipofone-si bilaterale delle basi polmonari. Le indagini di laborato-rio mostravano pancitopenia e alterazione della funzio-nalità epatica con indici di colestasi elevati: Hb 7 g/dl,RBC 2700000/mm3, WBC 2300/mm3 (N40%, L 58%, M2%, PLT 103000/mm3), GOT 237 UI/l, GPT 290 UI/l, γGT153 UI/l, Trigliceridi 268 mg/dL, Bilirubina totale 1.7 mg/dl,Bilirubina diretta 1.2 mg/dL. L'esame morfologico dell'a-spirato midollare escludeva la presenza di blasti e di figu-re di eritrofagocitosi. L'immunofenotipo linfocitario evi-denziava assenza di linfociti B CD19+ (0.7%) e aumentodei linfociti T citotossici CD3+CD8+CD57+ (40%). Le cellu-le NK e i livelli sierici di immunoglobuline erano nella nor-ma. All'Rx del torace erano presenti opacità parenchima-li a livello dei lobi polmonari inferiori e versamento pleu-rico destro. L'ecografia addominale mostrava una marca-ta epatosplenomegalia con dilatazione delle vie biliariintraepatiche ed ispessimento della parete colecistica,compatibile con un quadro di colecistite acuta. Le inda-gini infettivologiche documentavano la presenza di IgM adalto titolo anti-Mycoplasma pneumoniae (4 UI/ml, testimmunoenzimatico). Il trattamento con Claritromicinadeterminò un rapido miglioramento delle condizioni cli-niche della paziente e normalizzazione dei parametri di

laboratorio, ad eccezione della conta dei B linfociti. Incorso di terapia antibiotica (12° giorno) la piccola pre-sentò rash cutaneo polimorfo maculopapulare distribuitosimmetricamente su tronco ed arti, con diffuso eritema ededema degli avambracci. Fu fatta diagnosi di eritema mul-tiforme e iniziata terapia orale con prednisone e oxato-mide, con completa regressione delle lesioni cutanee dopo10 giorni. Nei successivi mesi di follow up non non c'èstata recidiva dell'eritema multiforme, anche in seguito asomministrazione di claritromicina per comuni affezionirespiratorie. Il deficit dei B linfociti è persistito fino a 5mesi dall'esordio della infezione. Discussione. Il Mycopla-sma pneumoniae è uno dei patogeni più freqentementeresponsabili di polmonite atipica in bambini e adolescen-ti. L'insorgenza di complicanze extrapolmonari durantel'infezione da M. pneumoniae è un evento ben noto, chepuò verificarsi anche in soggetti immunocompetenti e cheè probabilmente legato a fenomeni di mimicria antigeni-ca. Nel nostro caso, la presentazione atipica dell'infezio-ne con epatosplenomegalia, pancitopenia, e disfunzioneepatica lasciava immaginare la possibilità di una leucemiao di una sindrome da attivazione macrofagica. Resta dachiarire il meccanismo immunopatogenetico causa delladeplezione B linfocitaria. Recenti studi in vitro hannodimostrato una interazione tra M. pneumoniae e linfoci-ta B mediata da molecole HLA di classe II; è quindi ipo-tizzabile un meccanismo di citotossicità anticorpo dipen-dente. Peraltro, anche l'eritema multiforme riconosce unmeccanismo di citotossicità nei confronti di cheratinoci-ti esprimenti antigeni non self. L'espansione dei linfociti Tcitotossici può inoltre spiegare l'interessamento sistemi-co dell'infezione, simile a quello che si verifica in corso disindrome da attivazione macrofagica. Conclusioni. Ripor-tiamo una nuova entità clinica a nostra conoscenza maidescritta finora,caratterizzata da disfunzione multi-orga-no,eritema multiforme e alinfocitosi B legata ad infezio-ne da M. pneumoniae; lo studio dell'attività linfocitotos-sica e NK potrebbe essere indicato nei casi di infezionesistemica da M. pneumoniae.
P094L’ IPOGAMMAGLOBULINEMIA TRANSITORIA: CARATTERISTICHE CLINICHEED IMMUNOLOGICHEMoschese V, Rossi P, Graziani S, Chini L, Pignata C,Plebani A, Soresina AR, Consolini R, Livadiotti S,Martino S, Aricò M, De Mattia D, Martire B, Bossi G,Rondelli R, Pession A, Ugazio A*Per Il comitato strategico e di studioImmunodeficienze/aieop. * Coordinatore Del ComitatoStrategico e di studio Immunodeficienze/aieop
Introduzione e obiettivi: L’ipogammaglobulinemia tran-sitoria (THI) è un difetto di sintesi di uno o più isotipi del-le immunoglobuline, dei primi anni di vita del bambino,generalmente autolimitantesi, in quanto le ridotte con-centrazioni anticorpali tendono a normalizzarsi con l’età.La base patogenetica è tuttora oscura così come sonoestremamente eterogenee le informazioni sulla prevalen-za e sulle caratteristiche cliniche ed immunologiche diquesta condizione. Nell’ambito dello Studio Nazionale
Multicentrico sulla THI del Comitato Strategico e di Stu-dio Immunodeficienze/AIEOP è stato proposto un proto-collo di diagnosi e terapia della THI al fine di meglio defi-nire la condizione clinica e la storia naturale della malat-tia. Metodi: Sono stati arruolati 44 pazienti di sessomaschile e femminile (31 M; 13 F) di età compresa tra i12 ed i 36 mesi con: 1) marcata riduzione (al di sotto del-le 2DS dei valori normali per l’età) delle IgG associato omeno ad un difetto (al di sotto delle 2DS dei valori nor-mali per l’età) di IgA o di IgM ; e 2) valori di linfociti Bsuperiori al 2%. Risultati: Una familiarità positiva perimmunodeficienze è stata rilevata in 3/44 bambini. 40/44(89%) pazienti hanno presentato un decorso sintomaticodi cui 22/40 (55%) < 6 mesi di vita. 36/44 (82%) bambi-ni con THI presentavano patologie infettive prima delladiagnosi con un interessamento dell’apparato respiratorionel 68% dei casi e nel 46% dopo la diagnosi. Le patolo-gie allergiche ed il reflusso gastro-esofageo erano asso-ciati rispettivamente in 21/44 (48%) e in 7/44 (16%) bam-bini. 17/44 pazienti avevano un difetto isolato di IgG,12/44 un difetto coinvolgente tutti e 3 gli isotipi delle Ig,10/44 un difetto combinato IgG/IgM e 5/44 un difettocombinato IgG/IgA. Lo studio delle sottopopolazioni linfo-citarie ha rilevato una riduzione sia in termini assoluti che% dei linfociti CD8 in 1 paziente e dei linfociti B CD19+ inun altro paziente. Al momento della diagnosi è statoriscontrato un difetto di IgG2 (29%), di IgG3 (13%), dirisposta alla vaccinazione antitetanica (17%) e alla vac-cinazione antiepatite B (20%). Conclusioni: I dati clinicied immunologici ottenuti ci danno un quadro estrema-mente eterogeneo della THI. Tale disordine si presentainoltre frequentemente associato a patologie allergiche, asupporto di una potenziale disregolazione del sistemaimmunitario. La valutazione prospettica di questi pazien-ti ci consentirà di definire meglio questa condizione e diacquisire ulteriori informazioni sulla evoluzione della THIverso forme più complesse di immunodeficienza.
Si ringrazia per il supporto l’Associazione Immunodefi-cienze Primitive (AIP).
P095QUALITÀ DELLA VITA E PROFILO PSICOPATOLOGICO IN SOGGETTI CONIMMUNODEFICIENZA CONGENITANeri F,° Micheli S,° Soresina A,* Meini A,* Cantoni C,*Ravelli C,* Cattalini M,* Bottelli C,* Cattaneo G,*Plebani A*°Scuola di Specialità in Neuropsichiatria InfantileUniversità degli Studi Brescia; *Unità di ImmunologiaPediatrica, Presidio Ospedale dei Bambini, Universitàdegli Studi di Brescia
Introduzione: La letteratura sottolinea come la patolo-gia cronica, ad es. diabete, influenzi la qualità di vita (QDV)e abbia un'impatto psicologico importante sui pazienti.Per le Immunodeficienze congenite (ID), malattie croniche,non esiste ad oggi nessun dato in proposito e neppurestrumenti validati specifici sulla QDV. Obiettivi: valutarel'impatto delle ID, partendo dai difetti umorali (Agamma-globulinemia X-recessiva, XLA e Immunodeficienza Comu-ne variabile, CVID) che richiedono terapia sostitutiva conImmunoglobuline per via endovenosa continuativamente
149
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

150
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Altro
in ospedale, sulla QDV dei soggetti affetti, confrontando-la con il profilo psicologico emerso da colloquio clinico etest di personalità. Metodi: il questionario WHOQOL-100(World Health Organisation Quality of Life Assesment) perla QVD e l'MMPI-2 (Minnesota Multiphasic PersonalityInventory-2), test costruito per valutare le caratteristichestrutturali della personalità e disturbi emozionali, fornen-do una misura obiettiva del funzionamento psicologico.Sono stati valutati 12 soggetti, 3 femmine ; 9 maschi; 6con XLA e 6 con CVID. Età media :27,8 +/- 8,8 anni. Risul-tati : degli 11/12 pazienti studiati con valutazione com-pleta, i 4 pazienti con QDV discreta sono sintomaticiall'MMPI-2; dei 7 pazienti con QDV buona solo 3 sonosintomatici all'MMPI-2.Conclusioni: premessa l'esiguitàdel campione, dall'analisi descrittiva emerge che tra ipazienti sintomatici all'MMPI-2 (7/11) con un disturbopsichico, 4/11 hanno punteggi di QDV discreti. Sembraesistere una correlazione tra presenza di sintomi depres-sivi, disturbi somatici all'MMPI-2 e punteggi più bassi allaQDV, soprattutto nell'area psichica del WHOQOL-100.Chinon ha sintomi psichici (4/11) presenta punteggi di QDVbuoni: un migliore giudizio di realtà sembra determinarepiù equilibrio nella situazione di malattia. I soggetti sin-tomatici all'MMPI-2 e comunque con punteggi elevati diQDV presentano sintomi ipomaniacali e disturbi compor-tamentali, associati a negazione del vissuto di malattia.L'applicazione di questi due strumenti appare efficace nelmeglio definire l'impatto della ID sulla QDV e quindi lestrategie terapeutiche di supporto. Come già concordatonel CSS ID AIEOP, questo studio sarà applicato a tutti isoggetti arruolati nei diversi protocolli ID specifici.
P096ASPETTI METODOLOGICI DELLA VALUTAZIONE DELLA QUALITÀ DI VITAE DELLO STATO DI SALUTE DELLE PERSONE ADULTE GUARITE DOPO UNTUMORE MALIGNO IN ETÀ PEDIATRICA: UNO STUDIO DEL REGISTROTUMORI INFANTILI DEL PIEMONTEAlessi D,* Mosso ML,* Pastore G,*° Magnani C*°°*Registro dei Tumori Infantili del Piemonte. Sezione diEpidemiologia dei Tumori, CPO Piemonte, A. O. SanGiovanni Battista, Università di Torino°StrutturaSemplice di Oncoematologia Pediatrica, OspedaleS. Andrea Vercelli°°Dipartimento di Scienze Mediche,Università del Piemonte Orientale
Introduzione ed Obiettivi. Il miglioramento della pro-gnosi dei bambini affetti da tumore maligno ha determi-nato un notevole incremento nella proporzione di perso-ne guarite o con lunghe sopravvivenze e di conseguenzaun crescente interesse verso la qualità di vita (QoL), inte-sa come valutazione non solo degli aspetti fisici, ma anchepsicologici e sociali. Obiettivo di questo studio è la valu-tazione della QoL dei lungo-sopravvissuti mediante la rac-colta di dati presso le persone interessate e i loro medicidi medicina generale; questo permetterà il confronto didati soggettivi (auto-valutazione della QoL) e oggettivi(stato di salute). Obiettivo secondario è la descrizione deisoggetti che hanno rifiutato di partecipare, che possonopresentare caratteristiche significativamente diverse dacoloro che hanno aderito allo studio. METODI. Dal Regi-
stro dei Tumori Infantili del Piemonte (RTIP) si sono estrat-ti 1123 nominativi di soggetti con età alla diagnosi < 15anni posta nel 1967-1998, residenza in Piemonte alla dia-gnosi, età >= 15 anni nel 2000 e sopravvivenza >= 5 anni.Presso gli Uffici Anagrafe si è reperita la loro residenza eattraverso le Aziende Ospedaliere i dati anagrafici dei loromedici. Ai pazienti è stato inviato un questionario otte-nuto integrando l'HEALTH UTILITIES INDEX MARK2 &MARK3 - formulato dalla Health Utilities Inc. OntarioCanada - con domande riguardanti l'inserimento sociale;ai medici un questionario riguardante stato di salute eaccesso ai servizi sanitari dei loro assistiti. Pazienti e medi-ci erano informati del reciproco coinvolgimento nello stu-dio. I pazienti e i medici che non hanno risposto dopo unmese dall'invio del questionario sono stati sollecitati duevolte. Per i medici si è inoltre effettuato un sollecitotelefonico. RISULTATI. Dei 1123 pazienti estratti dal RTIP,13 sono risultati deceduti, 3 trasferiti all'estero, 5 irrepe-ribili e 17 senza medico: sono entrati nello studio 1085pazienti e i rispettivi medici. I risultati relativi all'invio deiquestionari sono presentati in tabella 1.Saranno presen-tate le prime elaborazioni dei questionari e la distribuzio-ne delle caratteristiche delle persone che hanno rifiutatodi partecipare allo studio. CONCLUSIONI. I risultatimostrano una buona rispondenza da parte di medici epazienti, che potrà essere ulteriormente incrementata daisolleciti ancora da effettuarsi.
P097STUDIO PROSPETTICO OSSERVAZIONALE SULLE COMPLICANZE CORRELATECON LA PRESENZA DI 418 CATETERI VENOSI CENTRALI A PERMANENZA INBAMBINI CON MALATTIA EMATO-ONCOLOGICAHaupt R,* Molinari AC,* Parodi S,* Longo S,° Saracco P,°Castagnola E,* Fratino G**Istituto "G. Gaslini", Genova° Ospedale "ReginaMargherita S. Anna", Torino
Lo studio ha valutato prospetticamente l'incidenza e latipologia delle complicanze correlate con la presenza di418 cateteri (180 Hickman-Broviac monolume, HB-M,162 Hickman-Broviac biluma, HB-B, e 76 catetri con val-vola pressione-attivata, PASV) inseriti nel periodo gen-naio 2000 - maggio 2002.I catetri sono stati posizionatiin 368 bambini, 190 con tumore solido, 162 con leucemiaa cuta e 16 con malattia ematologica non maligna sotto-posta a trapianto di midollo, di età media 7. 3 anni, con203 cateteri (49%) posizionati in soggetti < 6 anni. Il tota-le delle giornate di follow up è stato di 107012, con unrange compreso tra 3 e 868 giorni. Sono state osservate234 complicanze con una incidenza globale di 2.2 episo-di/1000 giorni-catetere. I pazienti con malattia ematolo-gia (maligna o non maligna) hanno presentato un ainci-denza di complicanze significativamente maggiore rispet-to a quelli con tumore solido (p<0.0001). Gli episodi osser-vati sono stati: 93 infezioni (0.87/1000 giorni catetere), 84malfunzionamenti (0.78/1000 giorni-catetere), 48 com-plicanze meccaniche (0.45/1000 giorni catetere) e 9 trom-bosi clinicamente sintomatiche (0.08/1000 giorni catete-re). Almeno 1 complicanza è stata osservata in 169 cate-tri (40%): 46% dei HB-B, 46% dei PASV e 33% dei HB-M

151
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
(p=0.02). In una analisi multivariata i fattori più impor-tanti per lo sviluppo di una qualsiasi complicanza catete-re-correlata sono risultati essere la diagnosi di malattiaematologia (RR 3. 0, 95% CI 1.8-4. 8) e l’età < 6 anni (RR2.5, 95%CI 1.5-4. 1). I cateteri HB-B hanno presentato unrischio di complicanza assai più elevato dei cateteri HB-M (RR 2.1, 95%CI 1.2-3. 6). In conclusione il cateterevenoso può causare complicanze nel 40% dei pazientiportatori; il tipo di catetere, la malattia di base e l’etàall’inserzione sono 3 fattori importanti per determinarel’incidenza di queste complicanze. I cateteri HB-M sonoquelli gravati dal minor numero di problemi.
P098COMPLICANZE LEGATE AL CATETERE VENOSO CENTRALE IN BAMBINIAFFETTI DA PATOLOGIA ONCOLOGICA: STUDIO EPIDEMIOLOGICO DI 4 ANNIGiacchino M,* Bezzio S,* Kanga Leunga GG,* Gay F,*Longo S,** Saracco P,* Alice A,* Madon E**Dipartimento di Onco-Ematologia ed Immuno Infettivo-logia Ospedale Infantile Regina Margherita Torino;**Cardiochirurgia Pediatrica Ospedale Infantile ReginaMargherita Torino
Nel periodo dal 01/01/2000 al 31/12/2003 è stato con-dotto uno studio prospettico che ha coinvolto i pazientiafferenti al Dipartimento di Onco-ematologia ed Immu-no-infettivologia dell'O. I. R. M. , nei quali è stato inseri-to un CVC. Risultati: sono stati inseriti 519 CVC in 427pazienti, 488 (94%) parzialmente impiantabili, 31 (6%)totalmente impiantabili tipo Port. Il 79% dei bambini(338) avevano patologia oncologica, il restante 21% (89)patologia immuno-ematologica. I CVC sono rimasti in situda un minimo di 0 ad un massimo di 1451 giorni (media369 giorni; mediana 320 giorni). In totale si sono mani-festati 407 eventi complicanze con un'incidenza di1.95/1000 giorni CVC, di cui 54% infettive, 46% mecca-niche. Si sono verificati 219 eventi infettivi con inciden-za pari a 1.05/ 1000 giorni CVC. Il 42% dei cateteri intro-dotti è stato interessato da tale complicanza. Le sepsi, con135 eventi, hanno interessato il 22% dei cateteri rappre-sentando il 62% delle complicanze infettive; 114 (85%)hanno avuto diagnosi microbiologica, 21 (15%) esclusi-vamente clinica. Un solo catetere totalmente impiantabi-le è stato responsabile di sepsi. L'incidenza è risultata paria 0.65/1000 giorni CVC. 75 (55%) sepsi sono state daGram+, 27 (20%) da Gram -, 4 (3%) polimicrobiche e 8(6%) da miceti. Gli S. coagulasi negativi rappresentavanoil 50% dei Gram +, tra i Gram - erano prevalenti i germiappartenenti al gruppo delle Enterobatteriacee. Le 15 infe-zioni del tunnel sottocutaneo hanno costituito il 3% del-le complicanze infettive. L'incidenza per catetere intro-dotto era di 0.071/1000 giorni catetere. Il 100% di que-ste infezioni era causato da Gram positivi. Le infezioni delforo di uscita hanno rappresentato il 13% (con incidenzadi 0.33/1000 giorni CVC) delle complicanze se si conside-rano anche i semplici arrossamenti, il 2% valutando sologli eventi con isolamento microbiologico o con necessitàdi terapia sistemica. Sono state registrate 13 complican-ze trombotiche pari al 3% delle complicanze totali, conincidenza di 0.06/1000 giorni CVC. Hanno presentato
trombosi lo 0.02% dei CVC inseriti. Le complicanze occlu-sive rappresentano il 42% delle totale Hanno coinvolto il34% dei cateteri introdotti con una incidenza di 0.8/1000giorni CVC. Nel nostro centro si è avuta una incidenza dieventi complicanza minore rispetto alla letteratura.
P099UTILIZZO DI UNA MEDICAZIONE CON NANOCRISTALLI DI ARGENTO NELTRATTAMENTO DI UNA CELLULITE DA STAPHILOCOCCUS AUREUS IN UNPAZIENTE PEDIATRICO ONCOLOGICODel Carlo TIP, Antonioli A, Santarlasci I, Di Donato CI,Gambacciani S, Gallo E, Funghi C, Bonacchi LCentro di Ematologia ed Oncologia Pediatrica conTrapianto di Midollo Osseo-Azienda OspedalieraUniversitaria Pisana/Pisa
INTRODUZIONE e OBIETTIVI. Le complicanze infettive,locali e sistemiche, rappresentano una delle più impor-tanti cause di mortalità e morbosità nei pazienti oncolo-gici. La comparsa di un’infezione in soggetti così grave-mente immunocompromessi, impone ovviamente un trat-tamento antibiotico parenterale ad ampio spettro che pre-vede l’associazione di più agenti antimicrobici. Nel caso diimportanti infezioni superficiali, tuttavia, una concentra-zione adeguata dei farmaci a livello dei tessuti interessa-ti è difficile da raggiungere ed è quindi spesso necessarioricorrere ad un approccio combinato medico-chirurgico.L’esperienza relativa all’impiego di antimicrobici topici inquesto settore è ancora limitata. Presentiamo, di seguitoun caso di cellulite batterica in un paziente pediatricoaffetto da Leucemia Linfoblastica Acuta (LLA), trattatacon successo mediante l’impiego di una medicazione arilascio continuo di argento nanocristallino. METODI. C. C., 16 anni, rumeno, giunge alla nostra osservazione perrecidiva midollare di LLA diagnosticata, nel paese di ori-gine, 18 mesi prima. Le condizioni generali del ragazzoall’ingresso si presentano compromesse e gli esami ema-tochimici mostrano una spiccata iperleucocitosi (GB121.000/mm3) con severa neutropenia (N 1%). Vieneintrapresa chemioterapia intensiva secondo protocolloAIEOP LLA REC 2003 (prednisone, fludarabina, daunoxo-me, HD-AraC) che determina una sensibile riduzione del-la conta leucocitaria con il rapido instaurarsi di un qua-dro di aplasia midollare. In 4° giornata di trattamento vie-ne evidenziata, a livello della superficie volare dell’avam-braccio destro, in corrispondenza della sede di una pre-gressa venipuntura, una lesione cutanea infiltrata, dolen-te e dolorabile, ad evoluzione rapidamente peggiorativache si associa, nelle ore successive, ad iperpiressia (TC >39. 5 °C). Nonostante il tempestivo inizio di una antibio-ticoterapia parenterale empirica ad ampio spettro (amika-cina, teicoplanina, ceftazidime sostituito dopo 48 ore conmeropenem), si assiste ad un ulteriore scadimento dellecondizioni generali del ragazzo con persistenza delle pun-tate febbrili, estensione della lesione cutanea alla quasitotalità dell’arto superiore e comparsa di parestesie e defi-cit di forza alla mano. Gli esami strumentali eseguiti (eco-grafia e RMN), mostrano un quadro compatibile con cel-lulite-fascite ed evidenziano la presenza di una estesa rac-colta fluida compresa tra il tessuto sottocutaneo ed il pia-

152
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Altro
no fasciale. A 72 ore dall’esordio, in attesa di attuare uneventuale approccio chirurgico, viene applicata local-mente una medicazione a rilascio continuo di argentonanocristallino previa scarificazione della cute in cinquedistinti punti. Già dopo poche ore si assite ad un arrestodell’estensione del processo flogistico con sensibile ridu-zione del turgore cutaneo e della sintomatologia sogget-tiva. La raccolta fluida prefasciale risulta sottile e filifor-me al controllo ecografico effettuato a 12 ore di distan-za, e non è più apprezzabile dopo 6 giorni. Parallelamen-te si assiste ad una progressiva riduzione, in frequenza edelevazione, delle puntate febbrili. Un tampone cutaneoeffettuato sulla lesione in fase di acuzie è risultato posi-tivo per Staphilococcus aureus. Negative sono invece risul-tate tutte le emocolture. RISULTATI E CONCLUSIONI. Leproprietà antimicrobiche dell’argento, note da secoli, siesplicano contro un largo spettro di microrganismi com-prendente funghi e batteri. I mediatori diretti dell’effettobattericida sono gli ioni di argento libero o i suoi radicaliche agiscono attraverso l’istantaneo blocco del sistemarespiratorio enzimatico, l’alterazione del DNA e della pare-te cellulare batterica. Le cellule dell’organismo umanovengono, invece, caratteristicamente risparmiate e nonsono noti fenomeni di resistenza. All’azione antibattericasi associano inoltre una efficace proprietà antinfiamma-toria ed un effetto favorente sul processo di riparazionetissutale. Nel caso clinico presentato è stata utilizzata unamedicazione a rilascio continuo di argento nanocristalli-no. La particolare forma dei nanocristalli permette diaumentare la superficie ricoperta di argento messa adisposizione della lesione mentre il rilascio continuato diuna dose bassa ma costante di argento garantisce un’a-zione prolungata e diminuisce le possibilità di danno cel-lulare e tissutale. Tale tipo di medicazione si è dimostra-ta efficace nell’arrestare l’ inesorabile progressione delprocesso flogistico favorendo, una immediata riduzionedella sintomatologia soggettiva ed, al tempo stesso, unarapida risoluzione del quadro. E’ stato in tal modo possi-bile, evitare il ricorso ad un approccio di tipo chirurgicosicuramente non scevro di rischi in un paziente così gra-vemente compromesso dal punto di vista immunitario.
P100PROGRAMMA DI SCREENING SENOLOGICO PER GIOVANI DONNE IRRADIATESULLA PARETE TORACICA PER UN TUMORE MALIGNO DELL’INFANZIA OADOLESCENZATerenziani M,* Cereda S,* Lepera P,° Gennaro M,°Bergonzi S,§ Collini P,# Gandola L,+ Clerici C,*Spreafico F,* Massimino M,* Cefalo G,* Luksch R,*Casanova M,* Ferrari A,* Polastri D,* Fossati-Bellani F*UO Pediatria*, ° UO Chirurgia Generale 4: §UO Radiologia,#UO Anatomia Patologica, +UO Radioterapia B, IstitutoNazionale per lo Studio e la Cura dei Tumori, Milano
Il carcinoma della mammella rappresenta la prima cau-sa di morte per tumore nella donna con una età medianadi insorgenza tra i 45 e i 50 anni , mentre meno del 5% simanifesta al di sotto dei 30 anni. Le radiazioni ionizzantirappresentano un noto fattore di rischio per l'insorgenzadi un carcinoma mammario. Sulla scorta dei dati epide-
miologici, della letteratura e della nostra personale casi-stica, che documentano un aumento del rischio di svilup-pare un tumore mammario nelle pazienti irradiate in gio-vane età di circa 40-50 volte e una età media di insor-genza notevolmente inferiore rispetto alla forma sporadi-ca, abbiamo iniziato un programma di screening senolo-gico per tutte le nostre pazienti pediatriche lungosoprav-viventi, irradiate sulla parete toracica. Lo studio si propo-ne di valutare sia l'utilità dello screening in termini di dia-gnosi precoce in questa casistica selezionata, sia l'effica-cia diagnostica delle singole procedure proposte (auto-palpazione, visita senologica, ecografia mammaria, mam-mografia e RMN) sulla mammella di giovani donne. Gliesami strumentali sono pianificati secondo l'età attualedel soggetto e l'età in cui è stata effettuata la radiotera-pia (infanzia o adolescenza). Questionari per la valutazio-ne dell'impatto psicologico in relazione alla possibilità disviluppare un tumore iatrogeno, per la raccolta dell'a-namnesi famigliare e di altri fattori di rischio collegati allosviluppo di un carcinoma mammario sono rilasciati almomento del primo incontro del programma di screening.Vengono altresì valutati gli esiti estetici connessi ai pre-cedenti trattamenti e la necessità di una eventuale chi-rurgia ricostruttiva. Grazie alla possibilità di utilizzare lecompetenze cliniche e di ricerca nell'ambito del carcino-ma della mammella disponibili presso il nostro Istituto,questo programma di screening potrebbe divenire riferi-mento per tutte le giovani donne irradiate sulla paretetoracica durante l'infanzia, in relazione alla problematicasempre più emergente delle sequele iatrogene tardive.
P101FATTIBILITÀ DI UN TRATTAMENTO FISIOTERAPICO DOMICILIARE PRESSOIL DIP. DI EMATOLOGIA ED ONCOLOGIA PEDIATRICA DELL’ ISTITUTOG. GASLINISavio C,° Garaventa A,° Manfredini L,° Fieramosca S,°Tanasini R,* Leimer M,° Dini G,° Gremmo M,*Camoriano R,* Miano M°IRCCS G. Gaslini, Genova
I pazienti affetti da patologie emato-oncologiche richie-dono frequentemente un trattamento fisioterapico chespesso prolunga la durata dei ricoveri in reparto o in dayhospital e per il quale è inoltre difficile mantenere unacontinuità assistenziale. Al fine di ridurre la durata deiricoveri e migliorare la qualità di vita dei pazienti assisti-ti presso il Dip. di Ematologia ed Oncologia dell’ IstitutoG. Gaslini di Genova, nell'Aprile 2000 è stato attivato unprogramma di Assistenza Domiciliare (AD) cui sono staticonsiderati eleggibili tutti i bambini in condizioni generalistabili con necessità di essere sottoposti a terapie e. v. ,nutrizione parenterale, supporto trasfusionale, controlliemato-chimici e gestione del catetere venoso centrale(CVC) oltre a tutti i bambini con malattia in fase termi-nale in trattamento con terapie palliative e di supporto. Ilservizio è attivo dal Lunedì al Venerdì dalle 8. 00 alle 16.00.Nell’ equipe dell'AD costituita da 2 medici, 3 infermie-re, una psicologa dal Giugno 03 è stato inserito un fisio-terapista. Nei primi 4 anni di attività sono stati trattati adomicilio 190 pazienti di età compresa tra 1 mese e 22

153
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
anni (mediana 6 anni), dal Giugno 03, 33 pazienti di etàmediana di 8. 5 anni (estremi 6 mesi -22), affetti da tumo-ri SNC (10), NB (6), LLA (5), tumori ossei (4), LMA (2) o altrepatologie (6) hanno ricevuto 607 sedute di trattamentofisioterapico per rieducazione neuromotoria con eventua-le prescrizione di ausili e/o ortesi (248), ricondizionamen-to e riallenamento allo sforzo (189), rieducazione moto-ria (139) o miglioramento del comfort e qualità di vita inpazienti in fase terminale (31). Tali trattamenti sono sta-ti somministrati a domicilio (358), in reparto (139) o pres-so il Servizio di Fisioterapia (110). Questa esperienza dimo-stra che la somministrazione di fisioterapia a domicilio èfattibile, può garantire un'importante continuità del trat-tamento e ridurre considerevolmente i disagi dei pazien-ti e delle loro famiglie.
P102LA PSICO-ONCOLOGIA QUALE NUOVA SPECIFICITÀ DELL’AREAD’INTERVENTO CLINICOPinto A, Camera F, Poggi VDipartimento di Oncologia, Azienda Santobono,Pausilipon, Napoli
Introduzione. Anche in Italia, la Psicologia Ospedalieraha subito un notevole sviluppo, la domanda di assistenzapsicologica si è notevolmente ampliata, gli interventi cli-nici hanno assunto una loro specificità a seconda dei cam-pi di applicazione medica e della complessa evoluzione diprotocolli clinico-assistenziali. Il crescente insorgere dimalattie tumorali e la grave incidenza sui sistemi socialinel quale l’individuo è coinvolto ha sostenuto gli psicolo-gi nella ricerca di peculiari modalità d’intervento, favo-rendo lo sviluppo di una specifica area: la psiconcologia.Metodologia. Da circa due anni è stata implemenatata, trale attività del Dipartimento di Oncoematologia Pediatri-ca, quella psicologico/clinica, quale area funzionale dicompetenza, incardinando nel proprio organico una equi-pe di psicologi – tra cui due dirigenti – impegnati a tem-po pieno in attività cliniche, di ricerca e formazione. Ilmodello teorico di riferimento è stato attinto dai piùmoderni studi di psiconcologia nei quali si sottolinea l’al-to rischio di frantumazione dell’unità mente- corpo ed ilricorso a strategie di copyng rigide primitive e quindi ina-deguate. Il modello operativo d’intervento si è articolatosu due livelli: il primo di tipo clinico-assistenziale direttoal bambino ed alla sua famiglia e teso a sostenere il recu-pero di migliori qualità di vita; il secondo di formazione ericerca diretto agli operatori dell’equipè e finalizzato amigliorare la qualità della relazione con il paziente e apermettere l’attività integrata. Risultati. Dopo due anni dilavoro sono stati avviate significative procedure, tra que-ste: - banca dati al fine di consentire la creazione di unosservatorio psicologico relativo a particolari condizioniemotive del bambino oncologico e della sua famiglia; -percorsi integrati, diagnostico- terapeutici secondo lineeguida ministeriali; - formazione di operatori sanitari e sco-lastici - supervisione didattica post-laurea per le Univer-sità di Roma e Napoli - collaborazione a progetti di uma-nizzazione- indagini di ricerca e studio. Conclusioni. L’e-sperienza condotta sino ad oggi, rappresenta un signifi-
cativo cambiamento di prospettiva nell’odierno panoramaregionale, garantendo ad un crescente numero di pazien-ti a rischio un’assistenza medico/psicologica, omogenea eintegrata. L’organizzazione lavorativa tutt’ora in via di svi-luppo, ha come obiettivo il raggiungimento di standardsempre più adeguati, così come realizzato negli Ospedalipiù avanzati e accreditati (HPH) a livello Europeo.
P103ESPERIENZA PRELIMINARE NELL’USO DELLA PROTEINA C ATTIVATARICOMBINANTE UMANA NELLE SEPSI IN ETÀ PEDIATRICARusso G, Branciforte F,° Disma N, Tumino M, La Spina MCentro di Riferimento Regionale di Emato-OncologiaPediatrica. Azienda Policlinico, Università di Catania,Via S. Sofia 78, 95124 Catania; °Divisione di anestesia erianimazione. Azienda Policlinico, Università di Catania
La sepsi grave è, ancora oggi, condizione gravata daaltissima mortalità in adulti e bambini, a causa di shocke/o disfunzione multiorgano, la cui patogenesi viene ricon-dotta, tra gli altri, alla carenza di proteina C. La sommini-strazione di Proteina C Attivata ricombinante umana (PCA)ha permesso un miglioramento dell’evoluzione clinica disoggetti adulti con sepsi grave. Caso 1: SL, anni 13, rab-domiosarcoma alveolare atipico vescicale; 6 settimanedopo il primo ciclo chemioterapico IVAdo ha presentatosepsi da Stafilococco Aureo, leucocitosi neutrofila, feb-bre, sensorio obnubilato, soffusioni emorragiche cutanee,cianosi periferica. Nonostante la terapia antibiotica, pro-gressiva disfunzione multiorgano (dispnea, ipossia, oligu-ria, ipotensione, alterazioni emocoagulative). E’ stata ini-ziata somministrazione di PCA (24 mcg•kg-1•h-1) per 96ore consecutive. Dopo 48 ore, lento e progressivo miglio-ramento delle condizioni cliniche e dei parametri di labo-ratorio. Caso 2: DP, anni 10, LMA M1, dieci giorni dopoHDAra-C/Mitoxantrone, in neutropenia, quadro di sepsigrave con disfunzione d’organo (febbre, dispnea, polmo-nite interstiziale), in assenza di colture positive. Intrapre-sa terapia con PCA; dopo 48 ore evidente miglioramentodelle condizioni cliniche e dei parametri di laboratorio.Caso 3: NG, anni 4, LLA, recidiva testicolare a due mesidallo stop-therapy; due giorni dopo la fine del blocco R1(REC2003), in neutropenia, febbre, gastroenterite acuta,shock ipovolemico, acidosi metabolica e oliguria, in assen-za di colture positive. Iniziata terapia con PCA, dopo 72 orelento e progressivo miglioramento dei parametri vitali.Caso 4: RA, 16 mesi, LMA M7, primo ciclo ICE interrottodopo i primi 4 giorni per polmonite lobare e febbre. In set-tima giornata, febbre continuo-remittente, evidente tos-sicità epatica (iperbilirubinemia, ascite), alterazioni emo-coagulative ed soffusioni cutanee emorragiche, in assen-za di colture positive. In nona giornata inizia terapia conPCA. Dopo 24 ore condizioni stabili; dopo 36 ore grave aci-dosi respiratoria, scompenso cardiocircolatorio ed exitus.Conclusioni: l’uso della PCA in 4 bambini si è dimostratosicuro e nessun effetto collaterale, lieve o grave si è veri-ficato. La nostra esperienza suggerisce che l’uso della PCAnel contesto clinico di una sepsi grave è una strategiaterapeutica attuabile anche in età pediatrica.

154
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Altro
P104TERAPIE INNOVATIVE NELLA CURA DELL'EPATITE AUTOIMMUNEGIGANTOCELLULARE ASSOCIATA AD ANEMIA EMOLITICA AUTOIMMUNEBeretta C,* Bovo G,* Rovelli A,* Leoni V,* Ronzoni S,*Viganò E,* Coliva T,* Conter V,* Mieli G°*Clinica Pediatrica Università Milano Bicocca, Monza °Institute of Liver Studies, King’s College Hospital, London,UK
Introduzione: L’epatite autoimmune gigantocellulare(EAG) è una rara patologia ad eziopatogenesi sconosciuta.L’ evoluzione della malattia è fatale nel 100% dei casi senon trattata; la terapia convenzionale si avvale dell’ usocombinato di steroidi e azatioprina. La terapia dell’EAGassociata ad anemia emolitica autoimmune non è ancoraben definita, soprattutto per i casi poco tolleranti o resi-stenti alla terapia convenzionale. In questo contesto rive-stono notevole interesse terapie immunosoppressive inno-vative con anticorpi monoclinali. Caso: Maschio. A 10 mesi
diagnosi di morbo celiaco. A 14 mesi diagnosi di anemiaemolitica autoimmune e inizio terapia con Metilprednoso-lone. A 17 mesi (Gennaio 2003) ricovero per vomito e itte-ro, SGOT = 4025 U/L, SGPT = 5972 U/L, bilirubina tot = 6mg/dL: diagnosi tramite biopsia epatica di EAG e inizio tera-pia con deltacortene (DTC) a 2 mg/kg/die somministrato invia continuativa e associato inizialmente ad azatioprina(AZT) a 0,5 mg/kg/die, dopo 2 settimane a Metil Micofeno-lato ed infine a Rituximab. Il decorso clinico ha evidenzia-to una dipendenza dagli steroidi che dopo 10 mesi di som-ministrazione non erano ulteriormente utilizzabili a causadei gravi effetti collaterali. Per tale ragione nell' Ottobre2003 si è deciso di trattare il bambino con Alemtuzumab15 mg/die per 6 giorni. Risultati: In seguito ad Alemtuzu-mab il bambino ha presentato aplasia profonda della dura-ta di circa 5 mesi. Il 15/3/04 la biopsia epatica evidenzia-va la scomparsa dei focolai a cellule giganti. Le sottopopo-lazioni linfocitarie, a distanza di 7 mesi dalla terapia (Mag-gio 2004), appaiono in iniziale lenta ripresa. Al momento ilbambino è in buone condizioni generali e da 5 mesi nonassume alcuna terapia immunosoppressiva. Conclusioni:

haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
L’Alemtuzumab può essere considerato una valida alterna-tiva nell’approccio terapeutico dell’ EAG rispetto alle tera-pie immunosoppressive piu’ convenzionali (DTC, AZT, Ciclo-sporina, Rituximab, Metil Micofenolato), qualora tali tera-pie risultassero inefficaci o non tollerabili. L’ apparente effi-cacia della terapia con Alemtuzumab contribuisce, inoltre,a chiarire la patogenesi dell'EAG, suggerendo un meccani-smo di tipo cellulo-mediato alla base della disfunzioneautoimmunitaria.
P105GIOCO ED EMOZIONI IN UN REPARTO DI ONCOLOGIA PEDIATRICAMarotta RS, Camera F, Pinto A, Poggi VDipartimento di Oncologia, Azienda Santobono,Pausilipon, Napoli
Introduzione. All’interno del Dipartimento di Oncologiadell’Ospedale Pausilipon di Napoli, è stata condotta unaricerca, oggetto di tesi di Laurea, in collaborazione con lacattedra di Tecniche di Osservazione del ComportamentoInfantile dell’Università “La Sapienza” di Roma, facoltà diPsicologia. La ricerca è divisa in due studi: il primo ha comeobiettivo la descrizione delle reazioni di bambini alle pro-poste di gioco e di animazione in ospedale; il secondo esa-mina le emozioni che i piccoli pazienti associano allamalattia. Il campione è composto da 15 bambini di etàcompresa tra i 3 e i 6 anni affetti da LLA nel primo anno diterapia e un gruppo di controllo, per il secondo studio, dibambini non malati reclutati all’interno di una scuolamaterna. Il primo studio è stato condotto mediante osser-vazione etologica (tre per bambino), il secondo con l’ausi-lio di un breve colloquio che ha utilizzato come stimoloquattro vignette, due raffiguranti un bambino e una bam-bina nel letto e due le loro faccine che esprimono diverseemozioni. Risultati. Lo studio osservativo ha evidenziatoche le proposte di gioco e di animazione sono di aiuto albambino durante il ricovero, mentre nei controlli ambula-toriali il ritorno in ospedale a distanza di tempo slatentiz-za comportamenti regressivi e aggressivi che riducono l’in-teresse verso il gioco. Questo sottolinea la necessità, anchenelle attività ludiche, di mirare l’intervento per ottenereuna reale risposta positiva da parte del bambino, sorreg-gendo quindi il gioco con un pensiero specifico e indivi-dualizzato. Il nostro studio ha ancora evidenziato la diffi-coltà di questi bambini nel rapporto con i coetanei, il cuicontatto è risultato limitato ed essenzialmente di tipodistale, senza reale condivisione del gioco e delle attività.Il colloquio ha evidenziato che rispetto ai coetanei sani, ibambini malati associano la rabbia alla malattia, sono
meno fiduciosi sulla possibilità che gli adulti possano leg-gere correttamente le loro emozioni e sulla loro capacità dicomunicarle, adottano come strategia per affrontare leemozioni negative il gioco, pur valutando la possibilità chenulla riesca ad aiutarli. Nell’imminente futuro si estenderàil secondo studio ad un campione più vasto con l’obiettivodi approfondire le strategie che i bambini mettono in attoper modificare le emozioni negative.
ABSTRACTSselezionati per la presentazione come
dati per letti (L)

156
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Letti
L001PORPORA TROMBOCITOPENICA IDIOPATICA (PTI): TERAPIA CON ALTDOSI DI DESAMETASONE IN ETÀ PEDIATRICAAmendola A, Palumbo G, Giona F, Moleti ML, Testi AM,Bernasconi S, Demasi B, Foà RUniversità La Sapienza Roma, Dipartimento BiotecnologieCellulari ed Ematologia, Sezione Ematologia
L’efficacia delle alte dosi di desametasone nel tratta-mento della PTI refrattaria o ricorrente dell’adulto e delbambino è stata descritta in letteratura ma i risultati sonoancora controversi. Al fine di valutarne l’efficacia e la tol-lerabilità in età pediatrica, abbiamo utilizzato il Desame-tasone, al dosaggio di 30 mg/m2/die per 4 giorni consecu-tivi ogni 28 giorni, per un massimo di 6 cicli, in uno studiopilota in bambini affetti da PTI all’esordio. I pazienti cheottenevano una risposta completa dopo il I ciclo, esegui-vano solo 4 cicli di terapia. Abbiamo trattato 13 pazienti (8maschi, 5 femmine) con età mediana di 12 anni (range 6-18 anni). La conta piastrinica mediana era di 5x109/L (ran-ge 1-20x109/L). Dodici pazienti presentavano sintomatolo-gia emorragica (WHO 2). La risposta è stata valutata alg+28 dell’ultimo ciclo effettuato; è stata definita rispostacompleta (RC) una conta piastrinica >150x109/L, rispostaparziale (RP) una conta piastrinica compresa tra 50109/L e150109/L, non risposta (NR) una conta piastrinica <50109/L. Dei 13 bambini trattati, 10 (77%) hanno ottenu-to una RC e 3 (23%) una RP. In tutti i bambini si è verifi-cato un incremento del numero delle piastrine. La RC si èottenuta dopo il I ciclo in 5, dopo il II in 3, dopo il III in 1 edopo il V in 1.Dieci bambini (71%) sono persistentementerispondenti con un follow-up mediano di 76 mesi (range29-83); tre (2 RC e 1 RP) sono recidivati dopo un tempomediano di 2 mesi (range 1-3 mesi). Una tossicità gastroin-testinale (WHO 2) è stata osservata in 2 pazienti. La nostraesperienza su un numero limitato di bambini ha dimostra-to la tollerabilità delle alte dosi di Desametasone in etàpediatrica. Nei pazienti con risposta precoce dopo il I ciclo,potrebbe essere indicata una riduzione del numero di ciclidi terapia, al fine di evitare eventuali tossicità.
L002LA RISONANZA MAGNETICA NUCLEARE(RMN)CON SEQUENZA T2STAR:UNA NUOVA METODICA UTILE NELLA DETERMINAZIONE DELL'ACCUMULOMIOCARDICO DI FERROBonato S,1 Tregnaghi T,2 Putti MC,1 Zanesco L1
1Dipartimento di Onco-Ematologia Pediatrica di Padova;2Istituto di Radiologia dell'Ospedale di Padova
Introduzione. Nonostante in pazienti talassemici l’uso dichelanti sia costante, l’accumulo parenchimale di ferro é laprincipale causa di morbidità e mortalità. Mentre l’accu-mulo epatico é stimato accuratamente tramite biopsia emetodiche magnetiche (SQUID), per quello miocardico nonesiste ancora una tecnica adeguataAbbiamo studiato 17pazienti con RMN (risonanza magnetica nucleare) T2starper valutarne le possibilità Metodi10 maschi e 7 femmine(età 7-39 anni), affetti da talassemia major in regime tra-sfusionale regolare e terapia con desferrossamina sottocu-
tanea in infusione continua, sono stati valutati con ferriti-nemia, funzionalità epatica,cardiaca, endocrina e accumu-lo intraepatico di ferro tramite SQUID. Sono state inoltrecondotte scansioni T2star assiali oblique passanti per lacupola epatica ed intersecanti le camere ventricolari car-diache. Il segnale epatico e miocardico,in assenza di valo-re misurabile assoluto,vengono confrontati con il segnaledi un tessuto di riferimento quale il muscolo e/o con quel-lo di una fiala di lipiodol (contrasto oleoso) Risultati Il rap-porto fra segnale muscolare e miocardico é risultato <1 in11 pazienti. Tale dato,che indica un basso accumulo di fer-ro,concorda col fatto che 10 pazienti presentano una nor-male funzionalità cardiaca. Solo uno è affetto da insuffi-cienza cardiaca moderata6 pazienti presentano un rappor-to >1.Di questi 2 hanno insufficienza cardiaca lieve,unomedia,i restanti 3 funzione cardiaca ai limiti inferiori dinormaIl rapporto segnale muscolare/epatico è <4 in 7pazienti,i quali hanno funzionalità epatica normale e con-centrazione intraepatica di ferro tra 1186 e 1904 ug/g(SQUID). Il rapporto negli altri 10 pazienti è >4. Essi han-no funzionalità epatica da normale a moderatamente alte-rata e concentrazione epatica di ferro tra 1139 e 4123Ilrapporto segnale lipiodol/fegato è concordante con quellomuscolo/fegato. Conclusioni. La RMN T2star appare unametodica utile nella determinazione dell’accumulo mio-cardico di ferro
L003PIASTRINOPENIA IDIOPATICA E NEUTROPENIA AUTOIMMUNE IN DUE GEMEL-LI ETEROZIGOTIBuldini B, Martini G, Bernuzzi G,* Flora G,* Veltroni M,Zanesco L, Sainati LClinica di Oncoematologia Pediatrica, Dipartimento diPediatria di Padova*Divisione di Pediatria, S. Donà di Piave(VE)
La patogenesi autoimmune delle citopenie ematopoieti-che è ben nota; l’incidenza, tuttavia è sporadica, a diffe-renza delle patologie autoimmuni che non interessano ilsistema ematopoietico, per le quali un’aggregazione fami-liare è descritta. Riportiamo il caso di due gemelli eterozi-goti affetti da citopenia periferica di natura autoimmune.All’anamnesi familiare si segnala cugina materna affetta daDiabete mellito di tipo I. E. B. ha presentato piastrinopeniaisolata (9000/mm3) a due anni. All’esame obiettivo: milzaa 3 cm, micropoliadenia inguinale ed ascellare, non malfor-mazioni somatiche. Le indagini eseguite deponevano peruna PTI (Porpora Trombocitopenica Idiopatica) con Ac anti-piastrine IgG associate (PAIG) positivi, i restanti anticorpiorgano e non organo specifici erano negativi con l’ecce-zione di ICA IgG debolmente positivi. E. B. ha presentato neiquattro anni successivi altri episodi di piastrinopenia sem-pre trattati con IgGvena con buona risposta. G. B. gemelloeterozigote ha presentato piastrinopenia isolata(15000/mm3) all’età di 5 anni. All’esame obiettivo emato-mi diffusi e petecchie; milza a 4 cm, non evidenti malfor-mazioni somatiche. G. B. veniva quindi trattato con IgGev(1g/kg) con discreta risposta. 4 mesi dopo comparsa di neu-tropenia e moderata anemia emolitica associate alla PTI: GB3900/mm3 (PMN 300), Hb 10.6 g/dL, PLT 53000/mmc. Reti-colociti>120000/mmc, aptoglobina 0.08 g/l. In questa

157
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
occasione si riscontrava la presenza di anticorpi anti-neu-trofili (IgG adese e sieriche), anti emazie nel siero e PAIGnegativi al precedente controllo. In entrambi veniva esclu-so deficit della funzionalità immunitaria (immunoglobuli-ne, sottopopolazioni linfocitarie, complemento); la tipizza-zione sierologica HLA di classe I e II non evidenziava aplo-tipi comunemente associati a patologie autoimmuni; il DEBtest era negativo; l’aspirato midollare e la biopsia osteo-midollare dimostravano morfologia compatibile con cito-penia rigenerativa. Riportiamo il caso di due gemelli concitopenia periferica, in precedenza solo un caso analogoera stato descritto ( Eur J Hematol 2002:69). La storia deidue pazienti indica la necessità di ripetere i controlli perdocumentare la patogenesi autoimmune non sempre subi-to evidente e che l’ espressione clinica può essere variabi-le nella stessa famiglia.
L004LINEE GUIDA AIEOP SULLA PORPORA TROMBOCITOPENICA IMMUNE ACUTA:GRADO DI IMPLEMENTAZIONE 1999-2002de Mattia D,* Del Vecchio GC,* de Santis A,* Giordano P,*Amendola G,** Arrighini A,° Baronci C,°° Del Principe D,°°°De Santis R,# Molinari AC,## Jankovic M,### Magro S,§Nespoli L,§§ Nobili B,§§§ Nardi M, Paolucci P, Paone G,Ramenghi U, Russo G, Sabato V, Tucci F, Zecca M*Dipartimento di Biomedicina dell’Età Evolutiva,Università di Bari; **Ematologia e Oncologia Pediatrica,Ospedale Umberto I, Nocera inferiore ° Clinica Pediatrica,Università di Brescia; °°Dipartimento di OncoEmatologiaPediatrica e Medicina Trasfusionale, Ospedale Pediatrico“Bambin Gesù”, Roma: °°°Dipartimento di Sanità Pubblica,Clinica Pediatrica, Università Tor Vergata, Roma: #IRCCS“Casa Sollievo della Sofferenza”, San Giovanni Rotondo;##Dipartimento di Emato-Oncologia, Ospedale Pediatrico“G. Gaslini”, Genova; ###Clinica Pediatrica dell'Universitàdi Milano-Bicocca, Ospedale San Gerardo, Monza;§Divisione di Ematologia, Azienda Ospedaliera“Pugliese-Ciaccio”, Catanzaro; §§Dipartimento di Pediatria,Università degli Studi Dell'Insubria, Varese; §§§Dipartimen-to di Pediatria, II Università di Napoli; Dipartimento diPediatria, Azienda Ospedaliera-Universitaria, Pisa;Clinica Pediatrica II, Università di Modena; ClinicaPediatrica, Università di Bologna; Dipartimento di ScienzePediatriche, Università di Torino; Centro di Riferimento diEmatologia e Oncologia Pediatrica, Università di Catania;Medicina Pediatrica III, Ospedale Giovanni XXIII, Bari;Unità Operativa di Oncoematologia Pediatrica, Ospedale“A. Meyer”, Firenze; Dipartimento di Scienze Pediatriche,Università di Pavia
Introduzione: L’Associazione Italiana di Ematologia eOncologia Pediatrica (AIEOP) ha pubblicato le linee guidaper la diagnosi e il trattamento della Porpora Trombocito-penica Immune (PTI) in età pediatrica nel 1999. Obiettivo:verificare l’implementazione delle linee guida nella praticaclinica dei centri AIEOP. Metodi: è stato inviato un que-stionario a tutti i centri AIEOP partecipanti alla stesura del-le linee guida e a tutti gli altri centri che hanno aderitoall’utilizzo delle stesse. Nel questionario erano richiesti datisul sesso, l’età anagrafica e l’età di diagnosi, il numero di
piastrine, il tipo di ricovero e di terapia effettuata all’esor-dio per i casi di PTI acuta osservati da Gennaio 1999 aDicembre 2002.Risultati: hanno risposto al questionario 17dei 22 (77%) centri partecipanti alla stesura delle linee gui-da e altri 9 aderenti alle stesse. Dei 609 casi valutabili (309maschi, 300 femmine), 346 (56,8%) erano forme A asinto-matiche-paucisintomatiche, 262 (43%) forme B a sinto-matologia intermedia e solo una (0,2%) forma C grave. Laconta delle piastrine alla diagnosi è risultata maggiore(p<0,001) nella forma A rispetto alla forma B. 498 sogget-ti (81,7%) sono stati ospedalizzati alla diagnosi, con unnumero di giorni di ospedalizzazione maggiore (p<0,01)nella forma B rispetto alla forma A. 151 soggetti (24,8%)non hanno effettuato alcuna terapia farmacologica, 135(38,6%) hanno praticato immunoglobuline, 146 (23,9%)steroidi e 77 (12,6%) altri tipi di terapie. Il ricorso ai varitipi di terapia farmacologica è risultato diverso tra le for-me A e B (p<0,001). Il trattamento è risultato appropriatoin 428 (70,4%) soggetti, non unanimemente consigliato in71 (11,6%), non appropriato in 95 (15,6%), non valutabilein 14 (2,4%). La qualità del trattamento è risultata diversatra le forme A e B (p<0,001). Conclusioni: il grado di imple-mentazione è stato soddisfacente nel periodo esaminato.
L005UN RARO CASO DI SINDROME IPEREOSINOFILA IDIOPATICASTEROIDE-RESPONSIVOMarconcini S, D'Ambrosio A, Acquaviva ADipartimento di Pediatria, Ostetricia e medicina dellariproduzione, Università degli Studi di Siena
Introduzione e obiettivi. La sindrome ipereosinofila idio-patica (HES) è caratterizzata da persistente conta assolu-ta di eosinofili (AEC) > a 1500/mm3 e/o dati clinici-labora-toristici d’infiltrazione e danno d’organo, in assenza di cau-sa nota d’ipereosinofilia. Gli Autori (AA) riportano un rarocaso di HES in età pediatrica. Caso clinico: R. M. , maschio,di 11 anni e 5 mesi ricoverato per febbre serotina (da 10gg. TC max 38. 5°C), intensa sudorazione notturna, Obiet-tività all’ingresso: Peso Kg 48. 9, Altezza cm 156, cuore etorace nella norma, linfoadenopatie laterocervicali bilate-rali (diametro max 2 cm) e ascellare sn (diametro 3 cm),modesta epatosplenomegalia, scialorrea, lesioni orticarioi-di agli arti. Esame neurologico normale. PA 110/75 mmHg.Esami di laboratorio: emocromo GB 38. 000/mm3, E 80 %,AEC (30.400/mm3), PLT 477. 000/mmc, Hb 14. 7 g/dL; VES10 mm 1a ora; sierologia negativa per CMV, EBV, Toxopla-sma G, Toxocara C, Leishmania, Borrelia, Strongiloides S.Esame coproparassitologico negativo. Mieloaspirato: cel-lularità aumentata con iperplasia mieloide orientata in sen-so eosinofilo (2/3 del totale); analisi molecolare dei riar-rangiamenti genici negativa per BCR-ABL, BCR-FGR1, TEL-PDGFRbeta, FIP1L1-PDGFRα. Osteomielobiopsia: midolloipercellulare (cellularità 80-90%) cospicua iperplasia del-la linea eosinofila con numerose forme mature; Biopsialinfonodale: morfologia conservata con cospicui infiltratieosinofili focali; ecoaddome: splenomegalia. In base allaclinica e agli esami è stata avvalorata la diagnosi di HES.Dopo un’attesa di 45 gg, persistendo l’ipereosinofilia (AEC= 3706/mmc), pur ridottasi la sintomatologia iniziale, per

158
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Letti
il possibile sviluppo di lesioni miocardiche e/o neurologiche,è stata iniziata terapia con Prednisone (PDN) 1,5 mg/kg/die.Dopo 7 gg di terapia AEC = 401/mmc, ridottasi dopo 3 set-timane (AEC = 292/mm3), è stata iniziata riduzione del PDN(in 3 settimane) fino a sospensione. Dopo un inizialerebound (AEC = 689/mm3), l’AEC si è stabilizzata sponta-neamente dopo 3 mesi a 450/mmc con obiettività clinicanormale. Conclusioni: La rarità della HES e la severa pro-gnosi nei casi non responsivi alla terapia, hanno indotto gliAA. a segnalare il caso. La buona risposta al PDN deve tut-tavia imporre un regolare e frequente follow-up per la pos-sibilità di recidive.
L006FIBROSI EPATICA CONGENITA (FEC): UNA RARA CAUSA DI PANCITOPENIANEL BAMBINOTrizzino A, Craxì A,* D’angelo P, Caselli D, Farruggia P,Russo D, Ziino O, Aricò MOncoematologia Pediatrica, Ospedale dei Bambini “G. DiCristina”, *Clinica Medica I Università degli Studi, Palermo
La malattia renale policistica autosomica recessiva(ARPKD) ha una frequenza di 1:20.000.Nonostante un’am-pia variabilità clinica, recentemente è stato identificato ununico gene responsabile della malattia (PK1D), codificanteuna proteina, la fibrocistina, indispensabile per la correttadifferenziazione terminale dell’albero biliare e dei dottirenali collettori. Caratteristiche della malattia sono dilata-zione e allungamento dei dotti collettori con formazione dicisti renali; dilatazione e malformazione dei dotti biliari;fibrosi epatica e renale. Se 90% di dotti è malformato, lamalattia si manifesta nella primissima infanzia, con unagrave insufficienza renale ed un alto tasso di mortalità. Seil numero è <10%, la funzionalità renale si deteriora neltempo, con esordio nell’adolescenza o oltre, e la FEC simanifesta con sintomi dell’ipertensione portale: epato-splenomegalia, ipersplenismo (anemia, piastrinopenia e leu-copenia), sanguinamento da varici esofagee, con test difunzionalità epatica normali. L’ecografia mostra ipereco-genicità del fegato e cisti renali, al doppler anche iperten-sione portale e inversione del flusso della vena porta. MS,16 anni, genitori sani non consanguinei; dopo banale trau-ma faciale modesta emorragia retinica ed ampio ematomaperiorbitario. Riscontrata pancitopenia ci viene inviato persospetta leucemia. All’ingresso: Hb 10 g/dL, piastrine40.000/mm3, GB 2.000/mm3, con epatomegalia (3 cm) esplenomegalia (10 cm). L’aspirato midollare esclude la leu-cemia, con cariotipo normale anche al DEB. Esami di rou-tine con funzionalità epatica e renale, e sierologia per virus(CMV, EBV, HBV, HCV, HIV, parvo), toxoplasma e leishma-nia: normali. Eco: iperecogenicità del fegato e cisti renalibilaterali, con ipertensione portale; varici esofago–gastri-che. Agobiopsia: fibrosi epatica. Diagnosi: ARPKD. In unbambino con epato-splenomegalia e pancitopenia, esclu-sa la leucemia, la diagnosi di FEC va considerata tra le altrecause rare.
L007MUTAZIONE MTHFR ED ESPRESSIONE FENOTIPICA: TRE CASI PEDIATRICI
Luciani M,* Coletti V,* Soldati M,° Pansini V,* Caruso R,*Baronci C,* Madafferi S,^ De Rossi G**Divisione Ematologia, Ospedale Pediatrico Bambino Gesù;°Laboratorio di Ematologia- Ospedale Pediatrico BambinoGesù; ^Divisione di Chirurgia Generale, Ospedale Pediatri-co Bambino Gesù
Introduzione e obiettivi: Il tromboembolismo venoso (TEV)è una patologia seria e complessa soprattutto per le suepotenziali complicanze ed è favorito da fattori sia acquisi-ti che congeniti. Tra i fattori congeniti sono stati identifi-cati una serie di mutazioni che determinano una riduzioneanche superiore al 50% dell'attività di alcuni enzimi coin-volti nella via metabolica omocisteina-metionina, quali laMetiltetraidrofolato Reduttasi (MTHFR). Ancora discusso èil legame tra TEV, iperomocisteinemia e mutazione del-l'MTHFR. Descriviamo tre casi clinici di pazienti pediatricicon un'importante e complicata storia trombotica, nei qua-li l'unico fattore di rischio evidenziato è stato quello dellamutazione del MTHFR. Metodi e Risultati: Tra Dicembre2002 e Dicembre 2003 sono stati ricoverati presso il nostroOspedale, tre bambini di età 3, 5 e 7 anni con trombosivenosa profonda. In tutti e tre i bambini e nei loro genito-ri è stata rilevata la presenza di polimorfismo del MTHFRcon sostituzione 677C->T. In particolare due pazienti risul-tavano omozigoti per il gene mutato, un paziente (l'unicoperaltro che presentava anche aumento dell'omocisteinaplasmatica e urinaria) era eterozigote. L'evento tromboti-co è stato rilevato mediante Angio TC, Ecocolordoppler eCavografia digitale. Un paziente presentava trombosi del-la vena cava fino alla biforcazione delle vene renali appa-rentemente sine causa, gli altri due pazienti, probabilmen-te in corso di episodio settico, hanno presentato trombosidella vena mesenterica con infarto intestinale, che ha resonecessaria una resezione intestinale. Tutti e tre i pazientiinizialmente sono stati trattati con eparina a basso pesomolecolare. Successivamente un paziente ha proseguitocon terapia anticoagulante orale, mentre gli altri due, valu-tato l'assorbimento intestinale, sono stati trattati con aci-do folico e vitamine del gruppo B. Conclusioni: I risultatiriguardo all'associazione tra la mutazione omozigote delMTHFR e la trombosi non sono ancora concordi, anche per-chè non sempre questo polimorfismo comporta elevatilivelli di omocisteina nel sangue. D'altronde ciò non esclu-de, che in concomitanza di particolari condizioni si possa-no verificare dei picchi di iperomocisteinemia, non sempredimostrabili, ma con una loro rilevanza clinica. E' quindiancora difficile predire l'impatto di questi polimorfismi sulfenotipo clinico, però è anche vero che l'iperomocisteine-mia è uno dei pochi fattori, se non l'unico, facilmente cor-regibile con una terapia semplice, non dannosa e pococostosa, quale quella vitaminica.
L008CATETERE VENOSO CENTRALE E QUALITÀ DELLA VITA NEI BAMBINILEUCEMICI AL PRIMO RICOVERO E NEI LORO GENITORIAxia G,* Tremolada M,** Scrimin S,* Zanesco L***Dipartimento di Psicologia dello Sviluppo, Università diPadova; **Dipartimento di Pediatria, Clinica diOncoematologia Pediatrica, Università di Padova

159
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
Introduzione e obiettivi. Nei molti studi medici sull’inse-rimento del catetere venoso centrale nei bambini leucemi-ci, non viene considerato l’effetto psicologico della drasti-ca riduzione delle ripetute esperienze di dolore acuto. L’i-potesi principale di questo studio è che esista una correla-zione tra il tempo trascorso dall’inserimento del CVC(numero di giorni) e una migliore capacità di bambini egenitori nell’affrontare le complicazioni del primo ricoveroper leucemia. Il metodo è un’intervista in profondità con 30madri di bambini leucemici (età media, 6. 0, ds 4. 5)) rico-verati da 9 giorni, ds 3. 7. Le variabili dipendenti si ricava-no dall’analisi quantitativa delle narrazioni (accordo tra igiudici. 71%) e sono composte da 4 o 5 item. Per esempio,QdV (contatti con i coetanei, tolleranza fisica delle terapie,mantenimento di routine precedenti, gioco in ospedale);Coping del bambino (coping al dolore, monitoraggio delleprocedure, scarso ritiro, bisogno espresso ai medici di spie-gazioni, accettazione spiegazioni sulle procedure). Risulta-ti. Sia il Coping del bambino (r=. 32*), che la sua Qualità divita (r=. 47**) sono positivamente correlati con il tempotrascorso dall’inserimento del CVC e con l’età (rispettiva-mente r=. 77***, r=. 42**), ma non con il tempo trascorsodal ricovero. Il tempo trascorso dall’inserimento del CVCcorrela con il Coping dei genitori (r=. 50**) e con il lorolivello di Fiducia nell’ospedale (r=. 44**). , Né l’età del bam-bino, né i giorni di ricovero influenzano il coping dei geni-tori o la loro fiducia nelle cure mediche. Una regressionelineare step-wise sulla QdV del bambino ricoverato ha con-siderato come variabili indipendenti. Età del bambino, gior-ni di ricovero, giorni di CVC, coping del bambino, coping delgenitore e fiducia nelle cure mediche. Il miglior predittoredella QdV del bambino è risultato la Fiducia dei genitori nel-le cure mediche (Beta =. 59, p<. 001) e il secondo migliorpredittore è, sorprendentemente, il tempo trascorso dal-l’inserimento del CVC (Beta=. 47, p<. 01). In breve, l’inse-rimento del CVC nei primissimi giorni di ricovero sembradare un sostegno notevole alle capacità e alla QdV del bam-bini leucemici.
L009LA RAPAMICINA INDUCE APOPTOSI IN CELLULE DI LEUCEMIA ACUTALINFOBLASTICA PEDIATRICARomano MF, Parasole R,* Avellino R, Romano S,Bisogni R, Venuta S, Poggi V*Dipartimento di Biochimica e Biotecnologie Mediche, Uni-versità di Napoli, Federico II; Dipartimento di Oncologia,AORN Santobono-Pausilipon*
In ambito ematologico, la leucemia acuta linfoblastica(LLA) in età pediatrica è considerata a buona prognosi inquanto risponde ai tradizionali schemi di polichemiotera-pia. Tuttavia il trattamento delle recidive e delle formerefrattarie costituisce ancora un problema irrisolto. I recen-ti studi sulla caratterizzazione di vie di trasduzione delsegnale intracellulare, spesso alterate nelle patologie neo-plastiche, rappresentano una base di partenza fondamen-tale per identificare bersagli specifici e per una più mirataed efficace terapia. Un difetto nella produzione di citochi-ne o dei segnali di risposta alle stesse sono stati riscontra-
ti in molti tipi di neoplasie ematologiche; ad esempio unaattivazione costitutiva del fattore trascrizionale STAT5 èstata descritta da vari ricercatori come un evento frequen-te nelle LLA pediatriche. La rapamicina, antibiotico appar-tenente alla famiglia dei macrolidi, è classicamente usatocome immunosoppressivo in quanto inibisce la risposta allecitochine. L'attività del farmaco è mediata dal legame conuna immunofillina, FKBP12, formando un complesso cheinibisce mTOR, una serin-treonin chinasi a valle di PI3k edAkt cioè della via dei fosfoinositoli, che come è noto rego-la la proliferazione e la sopravvivenza delle cellule in rispo-sta a citochine, mitogeni, aminoacidi. La via dei fosfoino-sitoli è ampiamente descritta essere coinvolta nella tumo-rigenesi e la rapamicina ha un riconosciuto effetto antitu-morale nelle neoplasie che presentano una attivazionecostitutiva di PI3k/Akt. Viste le proprietà di inibire sia l'at-tivazione linfocitaria che la sopravvivenza di cellule mali-gne, si è investigato se la rapamicina fosse in grado in vitrodi esercitare una attività anti-tumorale nei confronti delleALL pediatriche. A questo scopo sono state coltivate, inpresenza di rapamicina a dosi scalari, cellule mononuclea-te separate su gradiente di Ficoll-Hypaque da 22 campio-ni di midollo osseo di pazienti con LLA (19 LLA all'esordio,2 recidive e 1 refrattaria) e si è analizzata l'apoptosi convarie tecnologie. La percentuale di blasti nei campioni dicellule purificate era superiore al 90% eccetto che per laforma refrattaria in cui era pari al 59. 1%. L'apoptosi è sta-ta dimostrata in 13 su 22 campioni (59. 0%) incubati conrapamicina. L'analisi del potenziale di membrana mitocon-driale rivelava una spiccata depolarizzazione già dopo 6ore di coltura. Dopo 24 ore più del 70% delle cellule espo-nevano fosfatidilserina sulla membrana cellulare ed allostesso tempo l'attività catalitica di caspasi 3 apparivaaumentata del 170%. La rapamicina si dimostrava essereattiva in un range di concentrazione compreso tra 20 e 100ng/ml. La risposta pro-apoptotica della rapamicina eradimostrata anche nei 2 campioni di LLA recidivata e in quel-la refrattaria ed in particolare in quest'ultima forma è sta-to possibile dimostrare la scarsa sensibilità all'azione apop-totica della rapamicina della quota midollare residua nor-male. Per verificare se l'azione antileucemica del macroli-de potesse essere connessa all'inibizione della via dei fosfoi-nositoli si è analizzato se la wortmannina, uno specifico ini-bitore di PI3k, avesse lo stesso effetto apoptotico dellarapamicina. L'analisi è stata effettuata su nove campionisensibili alla rapamicina. In 6 casi la wortmannina induce-va apoptosi della stessa entità della rapamicina, in 3 casil'effetto della wortmannina era poco apprezzabile o assen-te. In conclusione il nostro studio dimostra che la rapami-cina induce apoptosi dei blasti di LLA pediatrica, preser-vando la controparte midollare normale, con un meccani-smo che coinvolge, ma non solo, l'inibizione di PI3k/Akt eche richiede ulteriori approfondimenti.
L010ALTERAZIONE DELL’ ESPRESSIONE DEL GENE CREB (CYCLIC ADENOSINEMONOPHOSPHATE (CAMP) RESPONSE-ELEMENT BINDING PROTEIN) NELLALEUCEMIA ACUTA PEDIATRICA: NUOVO GENE TARGET?Pigazzi M,1 Germano G,1 Ricotti E,2 Chiesa M,3 Disarò S,1Polato K,1 Basso G1

1Università di Padova, Dipartimento di Pediatria, Padova;2Università di Torino, Dipartimento di Pediatria, Torino;3Ospedale "Regina Margherita", Dipartimento diNefrologia, Torino
Introduzione: L'approfondimento biologico e geneticodella leucemia acuta (LA) pediatrica, grazie anche a nuovetecnologie di gene expression, ha consentito di individua-re dei possibili geni target che sembrano caratterizzare for-me diverse di leucemia e migliorare l'inquadramento dia-gnostico. CREB è un fattore di trascrizione (TF) attivato dal-la fosforilazione nel sito Serina133. Come TF riconosce ilpromotore, detto CRE, e regola la trascrizione di molti genicoinvolti nei principali processi, tra i quali differenziazionee sopravvivenza cellulare. Studi hanno dimostrato che CREBè fosforilato in risposta a fattori di crescita attivando latrascrizione in cellule di leucemie mieloidi, indicandolocome gene target per LA. Metodi/Risultati: Per definire ilpossibile ruolo del gene CREB nelle LA, è stata studiata lasua espressione in linee cellulari leucemiche di tipo B, T emieloide (JURKAT, KASUMI2, CEM, 1301, 697, HL-60) e inesordi di leucemia acuta linfoide (LLA) e mieloide (LMA)verso l'espressione di CREB in prelievi di controllo di dona-tori sani, di altre patologie ematologiche non neoplastichee di midolli in remissione morfologica allo stop terapia. Irisultati dell'espressione del trascritto di CREB mediantePCR quantitativa mostrano che è aumentato negli esordi diLA in media da 2 a 5 volte. La proteina risulta overespres-sa in tutte le linee cellulari leucemiche, in 26/28 (92%) diLLA e in 20/23 (86%) LMA rispetto ai controlli, sia inWestern Blot che in ELISA (significatività p<0.001). L'ove-respressione è tale anche per la forma CREB[pSer133], aconferma che CREB negli esordi di LA è fosforilato cioè atti-vo. Infine, per dimostrare il ruolo biologico e funzionale diCREB è stato analizzato il suo legame con il promotore CREin Gel Shit Assay; l'eccesso di legame CREB/CRE dimostra-to nell'83% degli esordi di LA rispetto ai controlli, ci fa ipo-tizzare una maggior attivazione della trascrizione dei genia valle di CREB nei pazienti. Conclusioni: I nostri risultatisuggeriscono che CREB possa contribuire all'eziopatoge-nesi della LA. La sua overspressione all'esordio ammette l'i-potesi che possa essere studiato come possibile nuovo genetarget alla diagnosi. Inoltre, l'espressione normale nei pre-lievi in remissione vede un possibile ruolo per CREB nellaleucemia attiva come marcatore molecolare.
L011LINFOMA ANAPLASTICO A GRANDI CELLULE (ALCL): ESISTE UN RUOLOPER L’ANALISI CITOGENETICA ?Sainati L, Leszl A, Pillon M, Bonato P, Tridello G,Mussolin M, Zanesco L, Rosolne A, Basso GClinica di Oncoematologia Pediatrica, Dipartimento diPediatria, Università di Padova
I linfomi anaplastici a grandi cellule (ALCL) presentano lat(2;5)(p21;q35), identificata come anomalia specifica inuna percentuale variabile dal 16 al 83%. L’ analisi moleco-lare permette la più rapida identificazione del gene ibridoNPM-ALK, prodotto della traslocazione e attualmenteancora più semplici tecniche immunoistochimiche sono in
grado di identificare la presenza della proteina chimerica el’ espressione ectopica di ALK nel tessuto linfoide, specifi-ca dei linfomi ALCL. L’ introduzione delle tecniche moleco-lari ed immunoistochimiche ha reso più semplice e fattibi-le la identificazione di questa anomalia. Queste metodi-che, tuttavia, non sono informative nei confronti delle ano-malie eventualmente associate. In età pediatrica gli ALCLpresentano alcune caratteristiche cliniche e biologichepeculiari rispetto all’ adulto: sottotipo istologico, estensio-ne della malattia all’ esordio e prognosi. La ricorrenza del-la traslocazione t(2;5) è molto più elevata nei pazienti inetà pediatrica raggiungendo il 17;83% in alcune casistiche(Dressler 2000). Allo scopo di individuare eventuali ano-malie citogenetiche, associate o meno alla traslocazioneprincipale, che ricorrano negli ALCL pediatrici e che even-tualmente li caratterizzino, abbiamo studiato il cariotipo di15 casi di ALCL e li abbiamo confrontati insieme a quelliriportati in letteratura in età pediatrica (25 casi), con icariotipi di pazienti adulti. I risultati hanno confermato unapiù elevata ricorrenza della t(2;5) nelle casistiche pediatri-che riportate e nella nostra serie rispetto all’ adulto; infat-ti in età pediatrica la traslocazione principale risulta asso-ciata ad altre anomalie semplici. Nell’ adulto invece la mag-gior parte dei cariotipi è complessa in assenza di t(2;5);mentre i casi dell’ adulto con t(2;5) presentano una minorcomplessità e più frequentemente che in età pediatricamostrano l’ anomalia principale isolata. La distribuzionedelle anomalie associate è inoltre diversa in età pediatricae nell’ adulto. Verrà discusso il possibile ruolo clinico diqueste anomalie.
L012GRAVE NEUROTOSSICITA’ DURANTE IL TRATTAMENTO DELLA LEUCEMIALINFOBLASTICA ACUTA: L’ESPERIENZA DELL’AIEOPJankovic M, Barisone E, Lippi A, Lo Nigro L, Varotto S,Reciputo A, Poggi V a nome dell’AIEOP
Introduzione: Con riferimento ai protocolli AIEOP LLA 95e 2000 si è osservato lo sviluppo, da parte di alcuni bam-bini, di un grave danno al sistema nervoso centrale (SNC)quantificato secondo i gradi 3 e 4 della scala dell’OMS (es.plegie, paresi, convulsioni complesse). Tali eventi hannocreato particolari difficoltà sia per la loro comprensioneeziopatogenetica sia per la loro implicazione nel prosegui-mento della terapia in atto. Metodi: Abbiamo raccolto que-sti gravi eventi con uno specifico form. Lo studio si è svol-to dal 1995 al 2002.Risultati: 30 bambini su circa 3000(incidenza 1%) hanno presentato problemi neurologici cosìsuddivisi per fase di terapia: Induzione 8 bambini; Conso-lidamento 6; Reinduzione 6; Mantenimento 10.24/30 lesio-ni sono state considerate lesioni ischemiche transitoriebenigne da danno vascolare mediato da un aumento pla-smatici e liquorale di omocisteina e neurotramettitori. 6/10sono state invece lesioni trombotico-emorragiche da L-Asparaginasi. Evoluzione: In 27/30 casi si è osservata unanormalizzazione sia clinica che neurologica (tempo mediodi regressione 3 settimane, range 1-10). Comparsa del dan-no: Da 24 ore a 3 settimane dalla somministrazione di MTXi. t. Quadro radiologico alla TAC e alla RMN: ipodensità del-la sostanza bianca, lesioni quasi sempre bilaterali e sim-metriche, a volte multifocali; all’EEG: presenza di foci uni
160
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Letti

161
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
o bilaterali. Previsto solo trattamento sintomatico (antico-miziali), la risoluzione è spontanea. Cosa abbiamo fatto: 1)interruzione momentanea della somministrazione i. t. o e.v. di MTX; 2) ripresa di tale terapia solo in presenza di nor-malizzazione del quadro clinico e strumentale; 3) se l’e-vento è avvenuto nelle fasi iniziali ella malattia si è ripre-sa la terapia preventiva al SNC, altrimenti la si è interrot-ta definitivamente. Conclusioni: 1) Sono lesioni che spa-ventano ma che sono benigne; 2) Occorre avere una valu-tazione radiologica dell’encefalo all’esordio, prima dellaterapia; 3) Fare attenzione a: quando avviene l’evento, met-terlo in correlazione con la terapia ricevuta nelle settima-ne precedenti, senza necessariamente sospendere la tera-pia preventiva al SNC, ma, eventualmente, solo posticipar-la.
L013VALUTAZIONE DELL'ESPRESSIONE CITOPLASMATICA DEL CD94 NELLALEUCEMIA MIELOIDE ACUTA DEL BAMBINO: CORRELAZIONE CON IL FABE L'ESORDIO DI MALATTIALo Nigro L, Mirabile E, Poli A, Munda S, Sciotto A,Bottino D, Schilirò GCentro di Riferimento Regionale di Ematologia edOncologia Pediatrica, Azienda Policlinico, Università diCatania
Background. Recentemente abbiamo riportato che l'e-spressione citoplasmatica dell'NCAM nei bambini con LMA(AIEOP 2002) risulta esser correlata con specifici sotto-gruppi FAB e con un esordio della malattia più aggressivo.Per confermare tali dati, abbiamo studiato l'espressione diun altro gene Natural-Killer-related, il CD94. Materiali.Sono stati analizzati 28 casi di LMA: 8 con FAB M1; 4 conM2; 3 con M3; 3 con M3v; 3 con M4; 5 con M5 e 2 M7,rispettivamente. Tutti questi bambini sono stati diagnosti-cati e trattati presso il nostro Centro dal 1999 al 2004, uti-lizzando i vigenti protocolli terapeutici. Metodi. Abbiamoapplicato una metodologia semiquantitativa nel tentativodi quantificare l'espressione del CD94 nelle cellule di LMAal momento della diagnosi. Come controllo positivo è sta-to utilizzato il cDNA di un caso di leucemia NK e di un casodi linfoma NK, già segnalati (MPO, 2000; Leuk Lymphoma2004). Abbiamo amplificato una diluizione seriale del cDNANK da 100ng a 0,01ng, assegnando uno score che riflette-va un elevata (high: ++++ o +++), bassa (low: ++ o +)oppure assente espressione del CD94. Come controllo inter-no di qualità abbiamo usato il gene della Beta-actina. Risul-tati E' stata riscontrata una forte correlazione tra elevataespressione di CD94 ed il FAB M3v. Abbiamo inoltre osser-vato che i casi che hanno presentato elevati livelli di espres-sione di CD94 sono deceduti all'esordio od in prossimità diesso per eventi legati alla malattia. Di contro non è statariscontrata alcuna correlazione tra l'espressione del CD94ed una prognosi infausta. Conclusioni I nostri dati confer-mano che alcuni sottogruppi di leucemia mieloide acutapossono derivare da un precursore comune sia al lineagemieloide che a quello NK. Pertanto, il nostro metodo d'a-nalisi può esser applicato per studiare una grande coorte dibambini con LMA, allo scopo di confermare i nostri datipreliminari e di valutare attentamente la correlazione traCD94 ed esordio fatale della malattia.
L014ASPETTI METODOLOGICI DELL'IDENTIFICAZIONE E CARATTERIZZAZIONEDELLE INTERNAL TANDEM DUPLICATION DI FLT3Zampieron C, del Giudice L, Pigazzi M, Disaro' S,Frascella EClinica Oncoematologica, Dipartimento di Pediatria,Università di Padova
Flt3 è il gene più frequentemente mutato nelle leucemiemieloidi acute (LMA). Internal Tandem Duplication (ITD) siriscontrano in circa il 25% dei casi e sembrano definire unsottogruppo a prognosi sfavorevole. Abbiamo studiato Flt3retrospettivamente su 261 casi di LMA, mediante RT-PCR,comparando l’ efficienza di diverse tecniche (elettroforesisu gel di poliacrilamide e agarosio, sequenziamento e valu-tazione mediante Genescan) nel definire presenza, lun-ghezza e numero degli ITD trovati. 54 (20%) casi sono risul-tati positivi, tutti individuati con gel agarosio e conferma-ti sia con gel di acrilamide che mediante Genescan. I cam-pioni positivi nel gel di poliacrilamide mostravano uno spe-cifico pattern di migrazione delle bande con due o più pro-dotti di peso molecolare apparentemente elevato, costitui-ti da eterodimeri. Il sequenziamento degli ITD è stato pos-sibile dopo eluizione delle bande da gel di poliacrilamide,mentre le bande eluite da agarosio sono sempre risultatecontaminate dalla presenza del trascritto wild-type. 50 casisono stati sequenziati, 3 mancavano del trascritto wild-type e 6 presentavano due trascritti mutanti. La lunghez-za delle ITD variava tra 16 e 116 bp e la regione duplicataandava dal nuleotide 1704 al 1857, in 12 casi vi eranoinserzioni nucleotidiche ed in 2 anche una sequenza intro-nica. In 2 casi erano presenti due ITD della stessa lunghez-za ma con sequenze differente. L’ analisi con Genescan hasempre confermato la lunghezza dell’ ITD dedotta dallasequenza ed ha permesso valutare il rapporto tra il tra-scritto ITD e wild-type che nella nostra serie è risultatocompreso tra 0.02 e 8. 884 In conclusione nella nostraesperienza l’ elettroforesi su gel di agarosio è sempre risul-tata adeguata per identificare Flt3-ITD. Abbiamo osserva-to che la forte propensione di Flt3-ITD a costituire etero-dimeri determina un particolare pattern di migrazione deicampioni positivi in gel di poliacrilamide e limita la purez-za delle bande eluite dall’ agarosio. Pertanto per il sequen-ziamento è raccomandato L’ utilizzo di bande eluite da geldi poliacrilamide. L’ analisi mediante Genescan ha permes-so l’ identificazione di tutti gli ITD, tranne che nei casi incui i 2 ITD presentavano la stessa lunghezza ma diversasequenza, ed inoltre consente di valutare il rapporto tra iltrascritto mutato ed il wild-type.
L015RIPRODUCIBILITÀ DELL’ ANALISI DI MALATTIA RESIDUA MINIMA (MRM)NEL PROTOCOLLO AIEOP-BFM LLA2000Songia S,° Germano G,* Corral L,° Spinelli M,*Manenti M,° Del Giudice L,* Basso G,* Van Dongen J,§Biondi A,° Cazzaniga G°°Centro Ricerca M. Tettamanti, Clinica Pediatrica

162
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Letti
Università Milano-Bicocca, Ospedale S. Gerardo, Monza,Italia; *Clin. di Oncoemat. Ped. , Dip. di Pediatria, Univ. diPadova, Italia; §Dept. of Immunology, Erasmus MCUniversity MedicCenter, Rotterdam, Netherlands
Introduzione e obiettivi: l’ applicazione della MRM a pro-tocolli clinici e l’ apertura di studi multicentrici per la valu-tazione della MRM hanno reso necessaria la verifica dellariproducibilità dell’ analisi di MRM, sia per quanto riguar-da la ripetizione di singoli esperimenti, che per la accura-tezza dell’ interpretazione dei risultati di RQ-PCR, al fine direndere i risultati di gruppi diversi più uniformi. Metodi:più di 200 test di RQ-PCR nei follow up di pazienti arruo-lati al protocollo AIEOP-BFM LLA2000 sono stati ripetutinei 4 laboratori incaricati per le analisi in Monza, Padova,Vienna e Heidelberg. La ripetizione è stata eseguita in con-dizioni che riproducessero il più possibile l’ esperimentooriginario, sul quale è stata decisa la stratificazione delpaziente. La ripetizione dell’ esperimento è stata reinter-pretata, oltre che nel laboratorio di origine, anche da unesperto di un secondo laboratorio del gruppo AIEOP-BFM,secondo le linee guida del gruppo europeo ESG-MRD-ALL.Risultati: Ad una prima analisi degli esperimenti ripetuti,27/201 test (13%) sono risultati discordanti, comprenden-do casi negativi/positivi e casi positivi quantificabili versopositivi-non quantificabili. La revisione delle linee guida ela rianalisi dei casi ha ulteriormente diminuito i casi discor-danti (positivi quantificabili verso positivi-non quantifica-bili), portando la riproducibilità circa al 95%. L’ adesionealle linee guida europee consente un’ ulteriore incrementonella riproducibilità dell’ interpretazione dei dati prodottiin laboratori diversi. Conclusioni: L’ analisi molecolare del-la MRM mediante RQ-PCR si dimostra essere una metodo-logia altamente riproducibile in un contesto molto con-trollato di laboratori esperti, soggetti a controlli di qualitàperiodici, alla discussione frequente dei casi di difficileinterpretazione, all’ adesione a linee guida europee di ese-cuzione ed interpretazione dei risultati.
L016IL MONITORAGGIO COMPARATO DI BCR/ABL MEDIANTE PCR QUALITATIVE QUANTITATIVA INDICA ETEROGENEITÀ DI RISPOSTA ALLA TERAPIA NELLALLA PH+
Rossi V, ° Fazio G,° Vallinoto C,^ Spinelli M,* Masera G,^Basso G,* Cazzaniga G,° Biondi A°°Centro Ricerca M. Tettamanti, Clinica PediatricaUniversità Milano-Bicocca, Ospedale S. Gerardo, Monza,Italia^ Clinica Pediatrica Università Milano-Bicocca,Ospedale S. Gerardo, Monza, Italia; *Clin. di Oncoemat.Ped. , Dip. di Pediatria, Univ. di Padova, Italia
Introduzione e obiettivi: La traslocazione t(9;22) è asso-ciata a rischio elevato di fallimento terapeutico con i pro-tocolli terapeutici convenzionali. Il monitoraggio moleco-lare del trascritto di fusione BCR/ABL può indicare unadiversa propensione di pazienti con LLA-Ph+ a raggiunge-re la remissione completa. Un precedente studio prospet-tico compiuto mediante PCR qualitativa sulla casisticaAIEOP ha dimostrato la presenza di pazienti con diversa
cinetica di riduzione della malattia Ph+ e diversa propen-sione alla ricaduta. La recente introduzione del Gleeveccome ulteriore farmaco specifico rinforza la necessità diidentificare pazienti con diversa cinetica di risposta, permeglio indirizzare le scelte cliniche. Scopo dello studio è diconfrontare i risultati ottenuti mediante PCR qualitativa equantitativa, come continuazione del precedente studio,mediante un più preciso monitoraggio. Metodi: RT-PCR diBCR/ABL secondo metodo Biomed; RQ-PCR secondo pro-tocollo EAC. Gene di controllo ABL. Al momento sono sta-ti monitorati 13 pazienti LLA Ph+. Risultati: Abbiamo con-fermato con PCR quantitativa i risultati ottenuti nel pre-cedente studio con PCR qualitativa. Inoltre, l’incremento insensibilità ha consentito di individuare casi debolmentepositivi per RQ-PCR e negativi per RT-PCR. In altri casi lavalutazione quantitativa ha consentito di evidenziare unacinetica di incremento o decremento del segnale nel followup, non visibile per RT-PCR. Conclusioni: La RQ-PCR puòsostituire la RT-PCR nel monitoraggio della LLA Ph+. I prin-cipali vantaggi sono: i) la possibilità di monitorare in paral-lelo un gene di controllo per valutare la qualità e quantitàdel campione in analisi e correggere la stima della caricatumorale; ii) la possibilità di monitoraggio quantitativoaccurato, con potenziali ricadute applicative nelle scelteterapeutiche. L’ampliamento della casistica prospettica e lavalutazione di un lungo follow-up permetteranno di con-fermare queste osservazioni preliminari.
L017INV(11)(P15Q22) CON FUSIONE NUP98-DDX10 IN LEUCEMIA MIELOIDEACUTA PEDIATRICAMorerio C, Rapella A, Acquila M, Tassano E, Rosanda C,Panarello CDipartimento di Ematologia ed Oncologia Pediatrica, Istituto G. Gaslini, Genova
Introduzione e obiettivi. L'inversione del cromosoma 11(p15q22) è una rara anomalia cromosomica associata amielodisplasia (MDS) e leucemia mieloide acuta (LMA) denovo o secondaria a terapia. L'inversione risulta nella fusio-ne del gene NUP98 (11p15) che codifica per una proteinadel complesso del poro nucleare con il gene DDX10 (11q22)che codifica per una RNA-elicasi putativa probabilmentecoinvolta nell'assemblaggio ribosomiale. Presentiamo uncaso di LMA pediatrica con inv(11)(p15q22) come unicaanomalia cromosomica, con identificazione dei geni difusione mediante ibridazione in situ fluorescente (FISH).MetodiUn ragazzo di 12 anni con diagnosi di LMA FAB-M5b, trattato secondo il protocollo BFM87, ottenne unaprima remissione completa dopo due mesi. A 16 mesi dal-l’esordio si verificò una ricaduta midollare ed oculare.Nonostante la terapia con mitoxantrone e AraC dopo quat-tro mesi nel midollo erano ancora presenti blasti leucemi-ci con lo stesso immunofenotipo dell’esordio. Fu eseguitoil protocollo AIEOP AML 2001/01 relapse, che determinòremissione completa, seguito da trapianto allogenico. Ilpaziente attualmente è in buone condizioni cliniche ed inremissione ematologica. L'analisi citogenetica con ban-deggio Q fu eseguita su sangue midollare alla ricaduta esangue periferico stimolato. Le sonde utilizzate per la FISH

163
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
erano: per il gene NUP98, RP5-1173K1 specifica per gliesoni 10-20, p9R1 e p6G2 specifiche rispettivamente per gliesoni 10-12 e 13-14; per il gene DDX10, RP11-976P22 eRP11-25I9. Risultati. Il cariotipo risultò 46,XY,inv(11)(p15q22) nel 50% delle metafasi esaminate, mentre ilcariotipo costituzionale su sangue periferico risultava46,XY. La FISH mostrava segnali splittati per entrambi igeni NUP98 e DDX10 sul cromosoma derivativo 11.Con-clusioniAd oggi sono stati descritti sei pazienti adulti epediatrici, tutti di origine giapponese, con inv(11)(p15q22)e fusione NUP98-DDX10, di questi, due pediatrici con MDS-LMA de novo, gli altri quattro secondari a terapia. In baseall'analisi dei trascritti chimerici sono stati descritti duediversi tipi di fusione: nel nostro caso l'analisi dei segnaliFISH collocherebbe il punto di rottura nel gene NUP98 tragli esoni 15 e 20.L'analisi con RT-PCR permetterà di defi-nire l'esatto nucleotide coinvolto.
L018ALTERAZIONI DI FLT3 E OVERESPRESSIONE DEL GENE WT1 IN PAZIENTIAFFETTI DA LEUCEMIA MIELOIDE ACUTA (LAM) ARRUOLATI NELPROTOCOLLO AIEOP LAM 2002/01: RISULTATI PRELIMINARI ECONSIDERAZIONE METODOLOGICHEColiva T,° Dell’ oro MG,° Beretta C, ° Spinelli M,* Rossi V,°Frascella E, * Scicchitano B,° Basso G,* Cazzaniga G,°Rizzari C, ^ Biondi A,° Pession A*°Centro Ricerca M. Tettamanti, Clinica PediatricaUniversità Milano-Bicocca, Ospedale S. Gerardo, Monza,Italia*Dipartimento di Pediatria, Università di Padova,Italia^ Clinica Pediatrica Università Milano-Bicocca,Ospedale S. Gerardo, Monza, Italia
Introduzione e obiettivi: La presenza, attesa in un 20-25% dei casi di LAM in soggetti adulti, della duplicazioneinterna in tandem (FLT3/ITD) del recettore di FLT3 e muta-zione puntiforme (aa 835-836) così come l’ overespressio-ne del gene WT1 sembrano rappresentare importanti fat-tori prognostici e/o marcatori universali di malattia residuaminima (MRM) in pazienti pediatrici con LAM. Per talemotivo, queste anomalie sono in corso di valutazione pro-spettica nei pazienti arruolati nel protocollo AIEOP LAM2002/01.Metodi: Il materiale biologico (BM, PB) è statosottoposto a PCR su DNA per FLT3/ITD, amplificazione erestrizione enzimatica per mutazione FLT3, e a RQ-PCR perWT1.I pazienti con FLT3/ITD e/o overespressione di WT1sono stati monitorati durante la terapia mediante RQ-PCRsu DNA (FLT3/ITD) o RNA (WT1). Risultati: All’ esordio dimalattia: FLT3/ITD: 11/87 casi (12%); mutazioni di FLT3:2/83 casi (2%); overespressione di WT1: 37/38 pazienti stu-diati (97%). I valori di WT1 normalizzati rispetto a K562sono i seguenti: 8,441E+01 (4,886E-01 - 1,608E+03), cor-rispondenti a circa due logaritmi più elevati rispetto adonatori sani. Per quanto concerne i metodi di analisi: a)entrambi i metodi hanno dimostrato valori di MRM coerenticon l’ andamento della malattia, b) il monitoraggio per ana-lisi quantitativa dell’ espressione del gene WT1 è di utiliz-zo più frequente ma meno sensibile di FLT3/ITD, c) entram-bi i metodi possono potenzialmente essere utilizzati inpazienti senza ulteriori anomalie clonali, d) il monitorag-gio di FLT3/ITD eseguito su DNA ha il vantaggio di mante-
nere la relazione univoca tra numero di molecole e nume-ro di cellule alterate, e) il riarrangiamento FLT3/ITD si èdimostrato essere relativamente poco stabile. Il monito-raggio mediante RQ-PCR può anche essere utilizzato perindagare evoluzioni clonali di FLT3/ITD nel corso della tera-pia. Conclusioni: La frequenza di alterazione di FLT3 in que-sta popolazione pediatrica è del 14%, al di sotto di quellaattesa nella LAM dell’ adulto. La overespressione di WT1 èosservabile nella maggior parte dei pazienti all’ esordio. L’analisi prospettica uni- e multi-variata del rischio di reci-diva, quando si raggiungerà un sufficiente periodo di osser-vazione, permetterà di valutare il potenziale utilizzo delmonitoraggio molecolare di FLT3/ITD e/o WT1 come fatto-ri prognostici e marcatore di MRM.
L019INSUFFICIENZA CARDIACA CONGESTIZIA (CHF) DOPO TRATTAMENTO PERLINFOMA DI HODGKIN IN ETA’ PEDIATRICA: LA RADIOTERAPIAMEDIASTINICA COSTITUISCE UN FATTORE DI RISCHIO AGGIUNTIVOALLA DOXORUBICINA?
Indolfi P, Casale F, Cesaro S,* Santoro N,** De Rosa E,Pota E, Giuliano M, Pelosin A,* Greco N,° Di Tullio MTServizio Oncologia Pediatrica Dipartimento di Pediatria,Seconda Università di Napoli *Clinica di OncoematologiaPediatrica Dipartimento di Pediatria, Università di Padova;*Dipartimento di Biomedicina dell’Età Evolutiva, Univer-sità di Bari: °Unità Operativa di Radioterapia, Dipartimen-to medico-chirurgico di Internistica Clinica e Sperimenta-le “F. Magrassi-A. Lanzara” Seconda Università di Napoli
Introduzione: Le antracicline rappresentano una catego-ria di farmaci efficaci e molto utilizzati in Oncologia Pedia-trica, ma gravati da una tossicità cardiaca subclinica che,talvolta, può sfociare in CHF. Alcuni reports hanno sugge-rito che la radioterapia sul mediastino può predisporre aduna prematura malattia coronarica. L’evenienza di malat-tia cardiaca può costituire un problema nei pazienti affet-ti da Linfoma di Hodgkin trattati con l’associazione antra-cicline-radioterapia sul mediastino. Scopo del nostro stu-dio è stato quello di verificare, in un’ampia popolazione dipazienti lungo-sopravviventi dopo trattamento per Linfo-ma di Hodgkin, l’eventuale incidenza di CHF. Pazienti eMetodi: Sono stati inclusi nello studio pazienti trattati pri-ma dei 18 anni di età con i protocolli AIEOP MH-83; MH-89; MH-96. L’analisi retrospettiva, condotta nei 3 centriAIEOP di Napoli, Padova e Bari, ha identificato 193 pazien-ti, diagnosticati tra il 1980 ed il 2001.Di essi, 69 pazientisono stati esclusi per diverso trattamento o persi al fol-low-up o deceduti (nessuno per problemi cardiaci). Per-tanto lo studio ha riguardato 124 pazienti, 83 maschi e 41femmine, la cui età media alla diagnosi era di 11.4 anni (1-17. 8). I pazienti erano in continua completa remissione efuori terapia dopo una mediana di osservazione di 7. 4 anni(1.10-19. 11) ed avevano ricevuto un dosaggio medio di150 mg/mq di doxorubicina cui era stata associata (92 casi)radioterapia sul mediastino al dosaggio medio di 20 Gy(20-40.5). Quindici pazienti erano in stadio IA, 42 IIA, 16IIB, 21 IIIA, 14 IIIB, 6 IVA e 10 IVB. Per lo stile di vita abbia-mo analizzato: il fumo, l’assunzione di alcool, l’eventualeattività sportiva ed il tipo di occupazione. Risultati: Nessu-

164
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Letti
na differenza statisticamente significativa è emersa tra età,sesso, stadio e dosaggio medio di antracicline tra i pazien-ti trattati o meno con radioterapia. Nessun paziente soffri-va di malattie cardiache all’inizio del trattamento chemio-radioterapico. Per quanto riguarda lo stile di vita purtrop-po i dati analizzati sono incompleti. Possiamo però segna-lare che 48/55 pazienti sono risultati non fumatori e 53/55non bevitori. La pratica di un’attività sportiva è stata segna-lata in 35/72 pazienti. Di 71 pazienti, 48 erano studenti e23 esplicavano un’attività lavorativa. Durante il periodo diosservazione nessun paziente inserito nello studio ha svi-luppato una Insufficienza Cardiaca Congestizia conclama-ta. Conclusioni: Il nostro studio conferma che la radiotera-pia sul mediastino, associata all’impiego di doxorubicina, aidosaggi erogati nei tre protocolli in studio, non sembracostituire un fattore aggiuntivo di tossicità cardiaca. E’necessario però confermare tale affermazione su di unacasistica più ampia e con un più lungo follow-up. E’ inte-ressante inoltre segnalare, pur tenendo conto dell’esiguitàdei dati raccolti, lo stile di vita dei nostri pazienti teso adevitare fattori di rischio aggiuntivi di tossicità cardiaca qua-li il fumo e/o l’assunzione di alcool.
L020I GLUCOCORTICOIDI INDUCONO IN VIVO L’ARRESTO IN G1 DEI LINFOBLASTILEUCEMICI ATTRAVERSO LA DEFOSFORILAZIONE DELLA PROTEINA RB-1NELLA LEUCEMIA LINFOBLASTICA ACUTA (LLA) DELL’ETÀ PEDIATRICACasale F, Crisci S, D’Angelo V, Addeo R, Pota E,Romano MT, Siano M, Martini A, Indolfi P, di Tullio MTServizio di Oncologia Pediatrica Dipartimento diPediatrica Seconda Università degli Studi di Napoli
Introduzione: I meccanismi molecolari coinvolti nellapatogenesi e nella chemioresistenza della LLA sono in lar-ga parte oscuri. La proliferazione dei blasti leucemici potreb-be essere legata alla deregolazione del ciclo cellulare ed iglucocorticoidi (GC), farmaci attivi nelle LLA, potrebberoagire come regolatori in alcune fasi del ciclo. Scopo delnostro studio è stato quello di analizzare, in bambini conLLA, alla diagnosi e dopo monoterapia steroidea, le fasi delciclo cellulare e l’espressione di alcune molecole regolatri-ci del ciclo. Materiali e Metodi: Sono stati studiati 32 bam-bini non consecutivi affetti da LLA, diagnosticati e seguitipresso il Servizio di Oncologia Pediatrica Dipartimento diPediatria II Università, tra il 1995 e il 2000.I pazienti sonostati suddivisi in: good responders (GR) e poor responders(PR), in rapporto al numero di blasti circolanti se < 1000/mLo > 1000/mL rispettivamente, dopo una settimana di mono-terapia con GC. Il ciclo cellulare e l’espressione proteica dipRb1 e delle CDKI (p15, p16, p21 e p27) sono stati valuta-ti alla diagnosi e dopo due giorni (48 h) di monoterapia consteroidi, sui linfoblasti del sangue periferico. Le fasi del ciclocellulare sono state analizzate in citofluorimetria (FACScan).I blasti leucemici, isolati, sono stati lisati in Buffer conte-nente NP40 0.1%. Le proteine sono state separate su gel diAcrilamide al 12%, trasferite su filtro di nitrocellulosa erivelate utilizzando un substrato chemioluminescente (ECL).L’analisi quantitativa dei campioni è stata normalizzata conun anticorpo anti a-actina. L’analisi statistica è stata con-dotta utilizzando il t-test di Student per le variabili conti-nue, mentre le categorie variabili sono state comparate uti-
lizzando il χ2.Risultati: I risultati confermano la complessitàe la molteplicità d’azione dei GC sulle cellule leucemiche;infatti dopo trattamento con GC, è stato registrato unaumento significativo della percentuale dei blasti leucemi-ci in fase G0/G1 in 22/32 pazienti, di cui 18 erano anchegood responders. Inoltre, in 29/32 pazienti è stato eviden-ziato un aumento dell’espressione delle quattro CDKI ana-lizzate, senza variazioni dell’espressione dei substrati (CDK2e CDK4). Tutti i pazienti alla diagnosi presentavano livellisvelabili di Rb-fosforilato. Venti/trentadue pazienti, dopoterapia con GC, mostravano una diminuzione della fosfori-lazione di p110/Rb-1, due pazienti presentavano un aumen-to e 10 nessuna variazione. All’analisi univariata la riduzio-ne della fosforilazione di pRb-1 era significativamente piùalta nei pazienti con fenotipo immunologico B e nei GR.Conclusioni: I nostri dati suggeriscono che la ipofosforila-zione di Rb-1 e, conseguentemente, la sua ridotta attivitàsi correla in maniera statisticamente significativa alla rispo-sta alla monoterapia con GC. La ipofosforilazione di Rb arre-sterebbe la progressione del ciclo cellulare bloccando le cel-lule nella fase G1 mediante un’inibizione della trascrizionedel DNA.
L021IPERESPRESSIONE DI FLT3 CARATTERIZZA LE LEUCEMIE ACUTEPROMIELOCITICHE DELL’ INFANZIA INDIPENDENTEMENTE DALLA PRESENZADI INTERNAL TANDEM DUPLICATIOFrascella E, Zampieron C, Germano G, Basso GClinica di Oncoematologia Pediatrica, Dipartimentodi Pediatria, Università di Padova
Le leucemie acute promielocitiche (APL) sono il gruppodi leucemie acute mieloidi che presenta più frequente-mente aberrazioni di Flt3, tuttavia proprio in questo grup-po non è mai stato dimostrato un significato prognosticosfavorevole legato alla presenza di queste aberrazioni.Abbiamo analizzato retrospettivamente 52 APL pediatri-che, mediante RT-PCR, individuando internal tandem dupli-cation (ITD) in 25 casi (48%) e mutazioni in posizione 835in 3 casi (5,8%). La presenza di Flt3-ITD è risultata signifi-cativamente correlata con iperleucocitosi (p=0,024),variante microgranulare (p=0,003) e trascritto bcr3(p<0,001), ma non con sesso, età alla diagnosi e presenzadi malattia extramidollare. Le ITD presentavano una lun-ghezza variabile da 18 a 116 nucleotidi. In nessun caso eraassente il trascritto wild-type e 4 casi presentavano 2 ITD.L’ analisi mediante Genscan ha dimostrato in tutti i casi unamaggiore espressione del trascritto normale rispetto almutato, con un rapporto tra Flt3ITD/Flt3wild-type com-preso tra 0,02 e 0,66. L’ analisi quantitativa dell’ espressionedi Flt3, è stata eseguita in 23 pazienti considerando glo-balmente l’ espressione sia del trascritto mutato che diquello normale, ed ha mostrato un’ iperespressione in tut-ti i casi studiati. Flt3 è risultato espresso mediamente 5,07volte in più rispetto al midollo normale, senza significati-ve differenze nella mediana di espressione in relazione atipo di trascritto PML-RARa (4,92 nei casi con bcr1-2 e4,58 in quelli con bcr3) e conta di WBC alla diagnosi (4,92e 4,81 rispettivamente nei casi con WBC<=20000 e>20000). Il gruppo positivo per Flt3-ITD è risultato avere

165
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
una mediana di espressione inferiore rispetto al grupposenza mutazioni (3,7 versus 5,2), tuttavia la differenza nonè statisticamente significativa. In conclusione nella seriestudiata abbiamo notato un’ incidenza di ITD particolar-mente elevata che potrebbe tuttavia dipendere da un biasdi selezione dovuto alla maggiore disponibilità di campio-ni con iperleucocitosi. Sono state rilevate significative cor-relazioni tra la presenza di ITD e elevata conta di WBC, sot-totipo M3 variante e trascritto bcr3. L’ iperespressione diFlt3 caratterizza tutte le APL, dovrà essere confermato suuna più ampia serie se l’ entità di espressione sia diversa trale APL Flt3 -ITD positive e le negative.
L022IPERPLASIA A CENTRI GERMINATIVI PROGRESSIVAMENTETRASFORMATI:CONDIZIONE A RISCHIO DI TRASFORMAZIONE NEOPLASTICAMaranella E, Filippini B, Facchini E, Sabattini E,*Semeraro M, Paone G, Pession A, Burnelli ROncoematologia Pediatrica e Terapia Cellulare - Dip. diPediatria, Policlinico S. Orsola-Malpighi; *Sez. di IstologiaEmolinfopatologica, Ist. Ematologia e Oncologia Medica"L&A Seragnoli", Policlinico S. Orsola-Malpighi, Bologna
Introduzione. Il centro germinativo progressivamente tra-sformato (CGPT) è un follicolo iperplastico presente nel 3.5% delle linfadeniti croniche aspecifiche diagnosticate biop-ticamente. L’iperplasia a CGPT si manifesta come linfoade-nomegalia latercervicale spesso singola, frequente nei gio-vani maschi. Nel 50% circa dei pazienti pediatrici recidivaanche dopo anni dalla diagnosi. Riscontrata un’associazio-ne non casuale tra CGPT e linfoma di Hodgkin (LH) a preva-lenza linfocitaria nodulare (PLN), il 10-30% dei pazienti conLHPLN hanno una pregressa diagnosi di CGPT. L’iperplasiafollicolare con CGPT può riscontrarsi prima, in concomitan-za della neoplasia o dopo chemioterapia. Obiettivi Valutarela correlazione tra CGPT e linfomi, evidenziando le caratte-ristiche che predicono la possibile trasformazione maligna.Risultati. Dal Dic. ’84 al Giu. ’04 sono stati diagnosticati 3casi (2 M 11 aa, 1 F 4 aa) di iperplasia follicolare con CGPT.L’esordio era rappresentato da un’adenomegalia isolata,inguinale in 2 casi e laterocervicale in uno. 2 pazienti han-no sviluppato rispettivamente un linfoma non Hodgkin(LNH) a larghe cellule anaplastiche ed un LH sclerosi nodu-lare, a distanza di 3 mesi ed 1 anno dalla diagnosi di CGPT.Quest’ultimo paziente era stato sottoposto a due biopsie equella eseguita 5 mesi prima della diagnosi di linfoma pone-va forti dubbi interpretativi nel differenziare un processolinfoproliferativo iperimmune da un LH. La terza paziente,già sottoposta ad una 2° biopsia in sede inguinale per reci-diva dopo 23 mesi, presenta un’adenomegalia inguinale per-sistente a 55 mesi dalla diagnosi. Discussione e conclusio-ni. La nostra esperienza conferma come i CGPT rappresen-tino una condizione di rischio di neoplasia, con evoluzionein linfoma in 2/3 casi: 1/66 LH e 1/147 LNH registrati nellostesso periodo, identificando una correlazione con istotipidiversi da quanto segnalato in letteratura. Non sono stateidentificate caratteristiche predittive di evoluzione neopla-stica, ma recidive in 2 su 3 pazienti. Esistono però casi in cuila reazione iperimmune può persistere per anni senza segnidi trasformazione. Una stretta sorveglianza, anche con bio-psie ripetute, è dunque necessaria per una corretta diagno-
si differenziale e per poter identificare tempestivamente l’e-voluzione neoplastica.
L023CISPLATINO ED ETOPOSIDE AD ALTE DOSI E TRAPIANTO DI FEGATO DACADAVERE NEL TRATTAMENTO DI EPATOBLASTOMA (EB) RESISTENTE ALPROTOCOLLO SIOPEL 3Farruggia P, D’Angelo P, Spada M,* Caselli D, Trizzino A,Gridelli B,* Aricò MOncoematologia Pediatrica, Ospedale dei Bambini “G. Di Cristina”; *Sezione Trapianti di Fegato, Is. Me. T. T. , A. R.N. A. S. Palerm
I tumori maligni del fegato costituiscono circa l’1% deitumori infantili; l’ EB i 2/3 di questi. Per lo più si presentanei primi 2 anni di vita come massa palpabile, il riscontropuò essere occasionale; sintomi di accompagnamento comeanoressia, perdita di peso, nausea, vomito e addominalgiasono presenti nelle fasi avanzate. In circa il 90% dei casi sirileva un cospicuo aumento dell’a-FP. La chirurgia ha sem-pre un ruolo fondamentale nel trattamento, ma la che-mioterapia (CT), soprattutto con i derivati del platino, haprofondamente cambiato l’approccio terapeutico e la pro-gnosi dell’EB non resecabile o metastatico. Descriviamo ilcaso di un bambino con EB non resecabile, non responsivoal prot. Siopel 3, che è stato curato con trapianto di fega-to dopo terapia di salvataggio con alte dosi di cisplatino edetoposide. Il destro viene sottoposto a TC addome: volumi-nosa massa epatica del dm massimo di cm 18. 4 x 11.8 chedisloca in basso il rene di dx e le anse intestinali; altre gros-se modularità in corrispondenza della cupola con interes-samento di tutti i segmenti epatici tranne il III; a-FP 18. 400ng/mL. La biopsia ecoguidata della lesione, ha permesso diporre diagnosi di EB. Iniziata chemioterapia sec. SIOPEL 3HR (pretext IV), regime superPLADO. Dopo i primi 3 cicliprogressione delle dimensioni del tumore alla TC, con mag-giore coinvolgimento di tutte e tre le vene sovraepatiche,in concordanza con un progressivo innalzamento dell’a-FP(18. 000 à 20.700 à 22.900 à 26. 700 ng/mL). Scartata lapossibilità di trapianto di fegato (TF) per la progressione dimalattia, è stata intrapresa CT secondo schema POG 9345,omettendo il 1° ciclo con il solo carboplatino già praticatocon il Siopel 3: VCR 1.5 mg/mq/die gg 1, 8, 15 + Carbopla-tino 700 mg/mq/die gg 1 + 5-FU 3 g/m2 i. c. in 72 ore. Dopoquesto primo ciclo il bambino ha presentato una sindromeda encefalopatia posteriore reversibile, per cui il tratta-mento è stato proseguito con l’altra combinazione: CDDP40 mg/mq/die gg. 1-5 + VP16 100 mg/m2/die gg. 2-4, som-ministrato ogni 21 giorni. Dopo 3 cicli, seguiti da una tos-sicità accettabile (ematologica e gastrointestinale) unarivalutazione TC ha permesso di rilevare una discreta ridu-zione della neoplasia: voluminosa neoformazione epaticacon satellitosi bilaterale che interessa i segmenti VII, VIII, V;cranialmente comprime senza invaderla la vena epaticamedia; minima dilatazione delle vie biliari intraepatiche adestra; asse spleno-porto-mesenterico pervio; non versa-mento libero in addome. Il livello dell’a-FP è progressiva-mente sceso fino a 970 ng/mL. A 23 giorni dall’ultimo ciclodi CDDP+VP16 il ragazzo è stato sottoposto a TF da dona-tore cadavere secondo tecnica standard. L’esame istologi-

166
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Letti
co sul fegato asportato ha confermato la diagnosi di EBvarietà transizionale. Il decorso post-TF è stato privo dicomplicanze. Il pz. dopo 34 giorni dal TF, a-FP 6 ng/mL, haricevuto un ultimo ciclo di chemioterapia con Carboplati-no 500 mg/m2 gg 1 + VP16 70 mg/m2/die gg. 2-4. Attual-mente il ragazzo sta bene, in RC e l’ultimo valore di a-FP èrisultato 2.4 ng/mL. L’EB, come la maggior parte dei tumo-ri germinali, è solitamente un tumore molto chemiosensi-bile. Nel nostro paziente la progressione di malattia in cor-so di trattamento con il Prot. Siopel 3 è stata documenta-ta sia con l’imaging che con il costante aumento dell’a-FP.Il ricorso a uno schema con CDDP e VP16 ad alte dosi haconsentito di ottenere una remissione parziale, con unatossicità tutto sommato accettabile, e avviare il ragazzo alTF, unica possibilità di guarigione definitiva.
L024IPERCALCEMIA SINTOMATICA IN CORSO DI TERAPIA CON ACIDO13-CIS-RETINOICOSinelli MT,° Tettoni K,* D'Ippolito C,° Notarangelo LD,*Spinoni V,° Fogazzi B,° Gualeni C, Arrighetti A,* Dughi S,#Porta F**Day Hospital di Onco-ematologia Pediatrica, SpedaliCivili Brescia°Clinica Pediatrica, Spedali Civili Brescia;#Servizio di Radiologia Pediatrica, Spedali Civili Brescia
Descriviamo due casi di ipercalcemia durante tratta-mento con acido 13-cis-retinoico. Il primo caso riguardauna bimba trattata presso questo reparto da marzo 2003con protocollo NB AR01 per neuroblastoma surrenalico edesteso coinvolgimento osseo (c-Myc amplificato). Durantela fase di mantenimento a undici giorni dall’inizio dell’as-sunzione di acido 13-cis-retinoico la bambina ha presen-tato ipercalcemia (>15 mg/dL) con intensi dolori ossei,desquamazione furfuracea periorale, alle estremità eprofonda astenia. Su indicazione del Centro Coordinatoreè stato deciso di sospendere la terapia per due settimane;alla successiva ripresa del farmaco a dosaggio dimezzato èstata nuovamente rilevata ipercalcemia importante. Sonostati eseguiti vari tentativi di prosecuzione delle cure condosaggi ulteriormente ridotti, ma la calcemia risaliva giàdopo pochi giorni di assunzione del farmaco. E’ stata quin-di valutata la possibilità che fosse l’interazione con altrifarmaci a scatenare tale processo: alla sospensione del flu-conazolo per la profilassi antifungina si è assistito alla sta-bilizzazione della calcemia, con la prosecuzione e comple-tamento del ciclo. Il secondo bambino ha presentato iper-calcemia e desquamazione alle estremità già dopo i primigiorni di terapia, se pur con sintomatologia meno impor-tante. I valori di calcemia si sono normalizzati dopo sospen-sione del fluconazolo e il primo ciclo è stato terminato adose di acido retinoico dimezzata. Successivamente la tera-pia è continuata senza modifiche. L’ipercalcemia in corsodi terapia con derivati dell’acido retinoico è stata più vol-te descritta in letteratura con un meccanismo di tossicitàa livello dei recettori del paratormone non ancora chiaritocompletamente. Esiste la segnalazione di effetti collatera-li dovuti all’interazione fra i derivati dell’acido retinoico eil fluconazolo dovuti al meccanismo competitivo a livellodel citocromo P-450.E’ stato evidenziato inoltre che l’as-
sunzione di triazolici prima di acido retinoico determini unaumento dei valori plasmatici di quest’ultimo di quattrovolte. Suggeriamo, dunque, un’attenta valutazione deglieffetti collaterali descritti in caso di somministrazione diquesti farmaci associati.
L025NON PUBBLICATO
L026COMPARSA DI METASTASI OSSEE MULTIPLE A 14 ANNI DALLA DIAGNOSIDI SARCOMA ALVEOLARE DEI TESSUTI MOLLIBrach del Prever A,* Comandone A,** Linari A,## Berta M,*Boglione A,** Gino G,# Brach Del Prever E,# Pagano M,*Forni M,## Madon E**Dipartimento di Onco - Ematologia ed Immuno - Infettivo-logia, Università degli Studi di Torino, A. S. O. O. I. R. M. S.Anna; **Ospedale Gradenigo, Divisione di Oncologia Medica,Torino; #Ospedale CTO - M. Adelaide, Dipartimento di Orto-pedia, Università degli Studi di Torino; ##Servizio di AnatomiaPatologica, Ospedale Infantile Regina Margherita, Torino
Nel novembre 1988 giunse alla nostra attenzione unaragazza di 13 anni ed 11 mesi di età, cui era stato diagno-sticato sarcoma alveolare dei tessuti molli della regionescapolare sinistra. La biopsia escissionale della lesione erastata eseguita presso altro Centro; la revisione della feritachirurgica confermò margini di resezione ampi. Gli agoa-spirati midollari e la Tomografia Computerizzata (TC) deltorace risultarono nella norma, la scintigrafia ossea nonrilevò la presenza di lesioni. La paziente fu pertanto trat-tata secondo il protocollo AIEOP RMS 88 (Ifosfamide Vin-cristina ed Act-D). Considerati i margini ampi di resezione,non fu eseguita radioterapia (RT). Gli esami strumentali altermine del trattamento e nel follow-up dimostrarono laremissione completa (RC). Nel marzo 2003, dopo sforzofisico, la ragazza lamentò gonalgia destra (dx); la radio-grafia dimostrò la presenza di una lesione litica nella regio-ne meta-diafisaria distale d dimostrò ipercaptazione a livel-lo del terzo distale del femore dx; l’esame istologico su bio-psia incisionale dimostrò trattarsi di metastasi ossea di sar-coma alveolare con ampie aree di necrosi. Fu pertanto ese-guita resezione ampia del femore distale dx, con impiantodi protesi di Kotz-Campanacci; l’esame istologico confermòla presenza di ampie aree di necrosi. La PET-TC, eseguita nelfollow-up a settembre 2003, rilevò la presenza di area diipercaptazione a livello della quarta vertebra dorsale, con-fermata alla RMN. La paziente fu trattata con RT (30 Gy) alivello di T4 e chemioterapia comprendente Epirubicina 120mg/m2 ed Ifosfamide 9 g/m2.La paziente è attualmente inbuone condizioni generali, in RC a 15 anni e 7 mesi dalladiagnosi e 15 mesi dalla recidiva. La PET-TC al termine deltrattamento è risultata nella norma. Il sarcoma alveolare èpatologia rara in età pediatrica; ci è parsa opportuna lasegnalazione di questo caso per la metastatizzazione osseamultipla a più di 14 anni dalla diagnos.
L027

167
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
VALUTAZIONE DEL MIDOLLO OSSEO NEI PAZIENTI AFFETTI DANEUROBLASTOMA: STUDIO MORFOLOGICO, ISTOLOGICO EIMMUNOCITOCHIMICO DELLA MALATTIA MIDOLLARE ALLA DIAGNOSI EDURANTE IL TRATTAMENTORosanda C,1 Fawlkner L,2 Sementa A,1 Gambini C,1Garaventa A,1 Manzitti C,1 Dau D,1 Luksch R,3 Viscardi E,4Di Cataldo A,5 Prete A,6 Giuliano M,7 Aricò M,8Bianchi M,9 Tettoni K,10 Provenzi M,11 Zanazzo GA,12
De Bernardi B1
1Oncologia Pediatrica Istituto G. Gaslini Genova e 3IstitutoNazionale Tumori Milano, 2Clinica Pediatrica di Firenze,4Padova, 5Catania, 6Bologna, 7Napoli, 8Palermo, 9Torino,10Brescia, 11Bergamo, 12Trieste
Introduzione e obiettivi. La malattia midollare condizionala prognosi dei pazienti affetti da Neuroblastoma. La valu-tazione del midollo osseo si basa sull'esame morfologico del-l'aspirato e su quello istologico della biopsia, recentementesono state rese disponibili metodiche in immunocitochimi-ca e di biologia molecolare che hanno dimostrato un eleva-to grado di sensibilita'. Scopo del presente lavoro e' statovalutare il ruolo clinico dello studio immunocitochimico del-la malattia midollare alla diagnosi e nella valutazione dellarisposta alla terapia. Materiali e metodiSono stati inclusinello studio 218 pazienti diagnosticati e trattati presso cen-tri AIEOP. Secondo l'International Neuroblastoma StagingSystem sono stati valutati l'aspirato midollare e la biopsiaosteomidollare in due sedi confrontati con lo studio immu-nocitochimico con anticorpo monoclonale anti GD2 all'e-sordio, e per i pazienti stadio 4 dopo induzione, prima e dopoterapia ad alte dosi. RisultatiSono risultati valutabili per le 3metodiche 169 pazienti alla diagnosi: 58 con malattia loca-lizzata, 77 stadio 4 e 34 stadio 4s (ritenuti valutabili qualo-ra fossero valutabili almeno due aspirati midollari), e 55pazienti stadio 4 in 113 valutazioni durante il trattamento.Alla diagnosi in 148 casi vi è stata concordanza tra morfo-logia-istologia e immunocitochimica e discordanza in 21casi: immunocitochimica positiva, morfologia istologicanegativa in 17 casi ed in 4 casi morfologia-istologia positi-ve con immunocitochimica negativa. Durante il trattamen-to vi è stata concordanza in 70/113 valutazioni, in 20 casil'immunocitochimica è risultata positiva a fronte di morfo-logia-istologia negative e viceversa in 23 casi l'immunoci-tochimica era negativa con morfologia-istologia positive.Conclusioni I nostri dati sembrerebbero indicare che all'e-sordio considerata la maggiore invasivita' della biopsiaosteomidollare, l'immunocitochimica possa essere conside-rata l'esame da affiancare all'esame morfologico dell'aspiratomidollare. Qualora i risultati contrastino con le altre indagi-ni di stadiazione e' opportuna l'esecuzione della biopsia osseain due sedi. Durante il trattamento vi e' una maggiore inci-denza di discordanza per cui sarebbero indicate le 3 meto-diche.
L028TUMORE DI WILMS (TW) BILATERALE: ESPERIENZA DELL’ISTITUTONAZIONALE TUMORI DI MILANOSpreafico F,* Terenziani M,* Piva L,§ Gandola L,°Collini P,+ Marchianò A,# Massimino M,* Luksch R,*
Casanova M,* Cefalo G,* Ferrari A,* Polastri D,* Podda M,* Meazza C,* Perotti D,& Radice P,&Fossati-Bellani F*Unità Operative di Oncologia Pediatrica,* Urologia,§Radioterapia,° Anatomia Patologica,+ RadiologiaDiagnostica,# Oncologia Sperimentale,& Istituto NazionaleTumori, Milano, Italy
Introduzione e obiettivi. I 14 casi di TW bilaterale segui-ti nel periodo 1987-2003 sono oggetto della presente ana-lisi. Metodi Pazienti: età mediana 28 mesi (range, 6-77); 11femmine, 3 maschi; metastasi alla diagnosi in 4 casi. In 10casi è stato eseguito accertamento istologico (agobiopsia,nefrectomia d’urgenza per emoperitoneo 1 caso). 12/14hanno effettuato CT primaria, con 3 farmaci (VCR, ACTD,ADM) in 9 casi, con 2 farmaci (VCR, ACTD) in 3 casi, per unadurata mediana di 12 sett. Risultati Risposta alla CT preo-peratoria: RP in 9 casi, malattia stabile in 2, progressionein 2.Chirurgia: nefrectomia + enucleazioni (5 casi), enu-cleazioni bilaterali multiple (3), nefrectomia + nefrectomiaparziale (2), enucleazioni + nefrectomia parziale (2), nefrec-tomia + solo biopsia del rene controlaterale (2) (entrambideceduti per progressione). 12/14 hanno eseguita CT com-plementare, con 3-farmaci in 8 pz, 2-farmaci in 2 pz, e altrifarmaci (IFO, CBDCA, VP16) in 2 casi; 2 pz hanno rifiutatoulteriori terapie. 8 pz hanno ricevuto RT sull’addome (5 log-gia renale, 3 addome, 12 Gy). L’esame istologico sul pezzooperatorio, nei 12 casi di cui abbiamo informazioni com-plete, ha evidenziato: TW a predominanza blastematosa(6), con differenziazione rabdomioblastica (3), epiteliale (2),aspetti necrotici (1); in 4 pz è stata documentata la pre-senza di residui nefrogenici, in 2 casi di anaplasia. In 5 pz(4/4 metastatici alla diagnosi) si è verificata recidiva/pro-gressione di malattia a 11 mesi (mediana) dalla diagnosi (4sono deceduti); le sedi di ripresa sono: fegato, progressio-ne locale (in malattia solo biopsiata), polmone, sede nonnota perché pz perso al f-up. Un paziente ha sviluppatoun’enterite fatale post ADM. Entrambi i casi con anaplasiasono deceduti per malattia. OS a 5 anni (f-up mediano 25mesi) 51%; 15%, EFS 53%; 14%. Tutti i pz. hanno una fun-zione renale conservata. Conclusioni La presenza di meta-stasi e/o di anaplasia costituiscono il principale fattore pro-gnostico sfavorevole. L’approccio con CT primaria devesempre essere perseguito. La chirurgia conservativa (anchel’enucleo-resezione) non ha determinato un aumentatorischio di recidiva locale, anche se l’impiego della RT in 8casi non permette di formulare un giudizio univoco sullepossibilità di controllo locale della malattia in caso di chi-rurgia conservativa
L029TUMORI ADRENOCORTICALI IN ETA PEDIATRICA. L'ESPERIENZAMULTICENTRICA ITALIANADall' Igna P,* Gasparella M, Bisogno G, Ferrari A,Indolfi P, Inserra A, De Salvo Gl, Alaggio R, di Cataldo A,Bernini G, Cecchetto G Per Il Gruppo Di Studio TREP(Tumori Rari in Età Pediatrica)*Divisione di Chirurgia Pediatrica, Dipartimento diPediatria, Università di Padova

168
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Letti
Scopo dello studio. Analisi dei casi italiani di tumoreadrenocorticale osservati negli ultimi 18 anni, nell’ inten-to di verificare il significato prognostico di età dei pazien-ti, dimensioni ed istologia. Materiali e metodi. 29 pazienti(22F, 9M, età media 54 mesi) sono stati osservati in 11centri di Chirurgia ed Oncologia Pediatrica (1984-2003).Una sindrome virilizzante era evidente in 19/29; in 1 casoil tumore è stato scoperto nel periodo prenatale; in 2 era-no presenti alla diagnosi metastasi epatiche/polmonari. Ilvalore medio del massimo diametro tumorale all’ imagingera di 6. 5 cm (range 2.5-25). In 26/29 pazienti è stata ese-guita una resezione completa (3 nefrectomie), in 1/29 unabiopsia (metastasi alla diagnosi) e in 2 una resezione conresidui microscopici (rottura all’intervento). In 24 pazientinon è stato eseguito alcun altro trattamento; negli altri èstato adottato l’ oDDD associato a chemioterapia secondodiversi regimi. La radioterapia è stata utilizzata in unpaziente con metastasi. Resultati. I pazienti valutabili sono28: 21/28 (età 1-180) sono in remissione completa (fu 5-196 mesi) (in 20 l’ exeresi era stata radicale; 1 paziente conresidui microscopici è stato trattato con CDDP-o, pDDD);6/28 (età 3-26) sono deceduti a 4-18 mesi dalla diagnosi:2 per progressione di malattia (metastasi alla diagnosi), e4 per recidiva locale dopo asportazione completa di unamassa di 6-8 cm; 1 (94 mesi) è vivo con malattia a 32 mesidalla diagnosi, dopo trattamento per metastasi comparse2 mesi dopo la chirurgia. Conclusioni. L’ età non ha influen-zato i risultati. Il volume tumorale è strettamente in rela-zione con la possibilità di un’ asportazione completa, tut-tavia abbiamo osservato risultati buoni con tumori >200cm3 e risultati scarsi con tumori più piccoli. Anche se l’ isto-logia non è in generale dirimente, alcune caratteristiche(invasione della vena cava, necrosi, attività mitoticaaumentata) suggeriscono un comportamento maligno: talicartteristiche, non note per i pazienti trattati prima del2000, sono state riscontrate in 3/11 pazienti con cattivooutcome trattati negli ultimi anni nell’ ambito dello studiocooperativo TREP. L’asportazione completa rimane il trat-tamento di scelta. L’ efficacia della chemioterapia e deltrattamento litico adottati finora rimane incerta.
L030TUMORI GERMINALI MALIGNI IN ETÀ PEDIATRICA. RISULTATI DEL SECONDOSTUDIO COOPERATIVO ITALIANO TCG-98Dall’ Igna P,* Lo Curto M, Fiori GM, Alaggio R, Cesca E,Basso E, Siracusa F, De Bernardi B, Tamaro P, Boglino C,Cecchetto G Per Il Gruppo Cooperativo Nazionale*Divisione di Chirurgia Pediatrica, Dipartimento diPediatria, Università di Padova
Scopo. Analisi del trattamento e dei risultati riguardantii pazienti registrati nello Studio Italiano TCG-98 (tuttoraaperto) e confronto dei dati con quelli ottenuti dallo Stu-dio precedente TCG-91.Materiali e metodi. Sono stati ana-lizzati 25 pazienti valutabili (11M, 14F, età media 20.5 mesi)arruolati nello studio dal gennaio 1998 al marzo 2003. Itumori erano gonadici in 16 (10 testicolo, 6 ovaio), extra-gonadici in 9 (7 sacrococcigei, 2 retroperitoneali); semino-matosi in 3 (ovaio) e non-seminomatosi in 22.Le linee gui-da terapeutiche raccomandavano: ST-I (tumore completa-
mente asportato, no estensione locale) CT solo in caso dimarkers ancora positivi; ST-II (tumore completamenteasportato, estensione locale) e IIIa (residui microscopici,linfonodi negativi) CT con carboplatino+VP16; ST- IIIb-c(residui macroscopici o biopsia iniziale) CT con carboplati-no+ VP16+dactinomicina+ifosfamide e chirurgia differita;ST-IV CT come per ST IIIb-c e chirurgia del tumore primiti-vo/metastasi. Risultati. ST-I 13: 11 (10 gonadici, 1 retrope-ritoneale) remissione completa (CR) (FU 12-72 mesi), 1(testicolo) vivo con malattia (AWD) dopo trattamento peruna recidiva locale (LR) insorta 7 mesi dopo la diagnosi, 1(retroperitoneale) deceduto (DOD) 35 mesi dopo la diagno-si per LR/metastasi. ST-II 1(sacrococcige) CR (FU 17 mesi).ST-IIIa 1 (sacrococcige) CR dopo LR trattata con secondachirurgia (FU 23). ST-IIIb 2 (ovaio) CR (FU 25,29). ST-IIIc 3:2 (ovaio) CR (FU 9,22), 1 (sacrococcige) DOD 22 mesi doporeintervento per LR. ST-IV 5: 2 (sacrococcige) CR (FU 27,39),2 (testicolo, sacrococcige) AWD (FU 18,43), 1 (sacrococci-ge) DOD per progressione di malattia 13 mesi dopo la dia-gnosi. Conclusioni. No discordanze maggiori tra linee-gui-da e approccio terapeutico adottato. Risultati favorevoliper: escissione completa o con residui microscopici alla dia-gnosi, tumori seminomatosi, sede gonadica. La chirurgiadifferita ha ottenuto il controllo locale. AFP alla diagnosi haun valore prognostico: 8000 mg/L nei pazienti in CR, 80000nei pazienti DOD. Confronto tra gli Studi TCG-91 (dati giàpubblicati) e TCG-98: registrazione inferiore, sopravviven-za totale simile, prognosi migliore per ST-IV.
L031FECONDAZIONE IN VITRO E SVILUPPO DI RETINOBLASTOMAAcquaviva A, Marconcini S, Hadijistilianou T,*De Francesco S,* Toti PDipartimento di Pediatria, Ostetricia e Medicina dellaRiproduzione, *Dipartimento di Scienze Oftalmologiche eNeurochirurgiche, **Dipartimento di Patologia Umana eOncologia - Università degli Studi di Siena
Introduzione e Obiettivi: Le tecniche di fecondazione invitro (IVF) sono estesamente applicate per la cura della ste-rilità nei paesi industrializzati. Oltre ai rischi già noti (gra-vidanze multiple, prematurità, basso peso alla nascita,malformazioni, difetti neurologici, aumento di aneuploidiee altre anomalie genetiche trasferite ai figli maschi) recen-temente è stata segnalata l’occorrenza di tumori in etàpediatrica. In particolare AC Moll e coll. 1 e D. Ben Ezra. 2
nel gennaio 2003 hanno segnalato su Lancet una maggio-re incidenza di retinoblastoma (RB) nei bambini nati da IVFrispettivamente in Olanda e in Israele. Gli Autori (AA) han-no voluto verificare l’occorrenza di retinoblastoma nei natida IVF e diagnosticati nel loro Centro. Pazienti, Metodi eRisultati: Da 1.1.2001 al 31.12.2003 nel ns. Centro sonostati osservati 75 casi di RB: di questi due erano nati dopoIVF ed entrambi erano maschi con forme non familiari, dicui uno unilaterale (diagnosticato all’età di 3 mesi) e l’al-tro bilaterale (diagnosticato all’età di 10 mesi). Conclusio-ni: E’ possibile che il numero di casi di RB nati dopo IVF pos-sa essere superiore: infatti, come riferito anche da BenEz-ra,2 le donne tendono a non dichiarare che il bambino sianato da IVF, specie se non richiesto in modo esplicito. IlRegistro Italiano del Retinoblastoma, che ha reclutato dal

169
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
1.1.1942 al 30.05. 2004 1107 casi di RB, ha attivato unaricerca finalizzata ai seguenti obiettivi: a) determinare l’in-cidenza di RB associata a IVF; b) possibili variazioni d’inci-denza nel tempo di RB IVF-correlati; c) identificare i centridi fecondazione assistita per stabilire se specifiche tecni-che di IVF sono correlate a aumentata incidenza di RB.
References
1. Moll AC, Imhorf SM, Cruysberg JRM, Schouten- van Meeteren AYN,Boers M, van Leeuwen FE: Incidence of retinoblastoma in childrenborn after in-vitro fertilisation. Lancet 2003;361:309-10.
2. BenEzra D. In-vitro fertilisation and retinoblastoma. Lancet 2003;361:273-4.
L032TRAPIANTO DI CELLULE EMATOPOIETICHE (HCT) NELLA LEUCODISTROFIAMETACROMATICA (MLD) DI TIPO ADULTORovelli A, Corti P, Furlan F, Bonanomi S, Gaipa G, Venturi N, Perseghin P, Dassi M, Sersale G, Parini R,Bertagnolio B, Uziel G, Longoni D, Balduzzi A, Uderzo C,Biondi A, Masera GMonza-Milano Network for Lysosomal and PeroxisomalDiseases (MOMIN)
Il decorso della MLD ha caratteristiche e velocità diver-se a seconda dell’età d’insorgenza, ma è comunque severoe inesorabile. L’HCT non è in grado di modificare l’evolu-zione dei casi ad esordio precoce; i migliori risultati si sonoottenuti nei casi asintomatici. Nel fenotipo giovanile eadulto, la minor velocità evolutiva può fornire una finestradi opportunità per l’HCT. Una donna, dopo 2 anni di distur-bi comportamentali progressivi mal definiti, veniva ricono-sciuta affetta da MLD all’età di 32 anni. Si tratta dell’uni-co caso di MLD adulta sinora sottoposto ad HCT in Italia.All’HCT la pz. presentava alcuni disturbi di tipo piramidalee frontale che la rendevano ormai incapace di attività pro-fessionale, ma ancora autonoma per molti pattern dellavita familiare e, se supportata, sociale. La curva della malat-tia faceva prevedere l’evoluzione verso la demenza grave elo stato vegetativo nell’arco di pochi anni. Pur non trat-tandosi del candidato migliore, con la famiglia si è condi-visa l’ipotesi, in assenza di altre terapie ragionevoli, chel’HCT potesse progressivamente stabilizzare la malattiaconsentendo alla donna non solo la sopravvivenza, ma dimantenere una sufficiente autonomia nel contesto fami-liare e sociale da loro ritenuta accettabile. Trapiantata(thiotepa, ciclofosfamide, ATG) con CD34+ positivamenteselezionate da donatore non correlato, la pz. è attualmen-te a +14 mesi con normali livelli di arilsulfatasi, RMN ence-falo, segni deficitari motori e autonomia stazionari con unmodesto grado di progressione del malfunzionamento fron-tale rispetto al pre-HCT. Dai pochi casi con MLD di tipoadulto sottoposti ad HCT è noto che un evidente rallenta-mento/arresto della progressione non si osserva se non ver-so almeno i 2 anni dall’HCT. Nel nostro caso, la curva evo-lutiva post-HCT sembra far ritenere possibile che una sta-bilizzazione possa essere raggiunta nel prossimo periodo.Nell’MLD forse non sono lontani possibili diversi approcciterapeutici, ma sinchè questi non saranno testati al di là delmodello animale, l’HCT rimane l’unica terapia praticabile e
per migliorarne l’efficienza nella MLD (inferiore a quellaosservata in altre malattie metaboliche) rimane fonda-mentale l’estrema precocità della diagnosi (necessità disensibilizzazione del mondo medico) o meglio la diagnosiprenatale (studi in corso).
L033VALUTAZIONE DELLA FUNZIONALITÀ CARDIACA E RESPIRATORIA IN BAMBINISOTTOPOSTI A TRAPIANTO DI MIDOLLO OSSEO : STUDIO PROSPETTICO DELGRUPPO EUROPEO EBMTUderzo C,* Fedeli S,* Fagioli F,° Faraci M,^ Varotto S,'Galambrun C,°° Zintl F,^^ Nysom K,** Verdeguer A,°°°Urban C,*** Corti P,* Rovelli A**Clinica Pediatrica, Università di Milano Bicocca, OspedaleS. Gerardo,Monza, Italy °Clinica Pediatrica,Università diTorino, Ospedale Regina Margherita,Torino, Italy; ^IstitutoG. Gaslini,Genova, Italy; 'Clinica di OncoematologiaPediatrica, Dipartimento di Pediatria,Padova, Italy;°°Hopital Debrousse, Immuno-Hematologie Pediatrique etTransplantation de Moelle Osseuse, Lyon, France;^^Dept. of Pediatrics, University of Jena, Jena, Germany;**BMT Unit Dept. of Hematology L4042, Copenhagen,Denmark °°°Hospital Infantil La Fe, Oncologia Pediatrica,Valencia, Spain ***Division of Pediat. Hematol. /Oncology,University Children's Hospital, Graz, Austria
Introduzione ed Obiettivi. Lo studio, prospettico e poli-centrico, si riferisce a pazienti pediatrici sottoposti a tra-pianto di midollo osseo (TMO) e ha come scopi principali divalutare l’incidenza, la severità, l’evoluzione ed i fattori dirischio delle complicanze respiratorie e cardiache tardive.METODI : Sono stati reclutati 220 soggetti (130 maschi e90 femmine), sottoposti a TMO (191 allogenico,29 autolo-go) per patologie maligne (188) e non maligne (32), in 9centri EBMT dal marzo 1994 al dicembre 1997. I pazientitrattati con antraciclinici pre-TMO (dose mediana cumula-tiva 259,7 mg/m2) sono stati 161.Hanno ricevuto TBI duran-te il regime di condizionamento pre-TMO 120 soggetti.Hanno presentato GVHD acuta 106 pazienti e GVHD cro-nica 43 pazienti. La valutazione della funzione cardiore-spiratoria è stata eseguita prima del TMO, a 6 mesi didistanza dal TMO ed in seguito annualmente per i primi 5anni dal TMO con osservazioni cliniche, spirometria ed eco-cardiografia M-mode. E’ stata compiuta una analisi dellavarianza per evidenziare eventuali alterazioni degli indiciconsiderati (VC,FEV1,TLC e SF) rispetto ai valori attesi. Risul-tati. Il follow up mediano è di 5 anni, 53 su 220 soggettisono deceduti,nessuno di essi per cause cardiache o pol-monari. Dei 134 pazienti valutabili per la funzione cardia-ca 11(8,2%) hanno mostrato alterazioni tardive della fra-zione di accorciamento ( SF < 30%). Dei 114 soggetti con-siderati per la funzione respiratoria,13(11,4%) hannomostrato alterazioni tardive (valori < 80 %), 9 soggetti han-no sviluppato una forma restrittiva e 4 soggetti una formaostruttiva. Si osserva un peggioramento di tutti gli indiciconsiderati entro il primo anno dal trapianto, quindi unprogressivo miglioramento nelle seguenti osservazioni. L’u-nica correlazione statisticamente significativa riscontratatra fattori di rischio e complicanze tardive riguarda il TMOallogenico (p value = 0.05 per CV e 0.03 per FEV1). Con-clusioni. Benché le alterazioni riscontrate siano solo sub-

cliniche e mostrino incidenza ridotta, questo studio evi-denzia la necessità di un monitoraggio continuo nel tem-po per prevenire una maggiore severità delle stesse.
L034INCIDENZA DI SCLERODERMIA IN CORSO DI GVH CRONICA NEI PAZIENTISOTTOPOSTI A TRAPIANTO ALLOGENICO DI CELLULE STAMINALIEMATOPOIETICHELenhardt A, Rabusin M,* Patarino F, Maximova N,Andolina MU. O Clinica Pediatrica Centro Trapianti; *U. O Emato-oncologica, IRCCS Burlo Garofolo, Trieste
Obiettivi: valutare l’incidenza, le caratteristiche cliniche ela risposta a vari regimi terapeutici della sclerodermia cuta-nea o sistemica in corso di GvH cronica nei pazienti sotto-posti a TCSE allogenico. Metodi e Pazienti: Abbiamo analiz-zato retrospettivamente tutti i trapianti di midollo alloge-nico eseguiti presso il Centro Trapianti del nostro Istituto dal1984 al 2003, valutando l’incidenza di GVH cronica, l’even-tuale insorgenza di sclerodermia cutanea o sistemica, e larisposta al trattamento farmacologico. La GvH cronica èstata divisa in limitata od estesa secondo le normali classi-ficazioni. Risultati: Dei 146 trapianti eseguiti, sono risulta-ti valutabili per GvH cronica (sopravvivenza almeno a 100giorni post-trapianto) 75 trapianti. In 19/75 pazienti (25%)è comparsa GvH cronica, di cui 7 in forma limitata e 12 informa estesa. I 19 soggetti (8 femmine, 11 maschi) con GvHcronica avevano un’età media al trapianto di 11 anni (ran-ge 8 mesi-36 anni) e di questi 19 soggetti 6 sono stati sot-toposti a un trapianto da familiare HLA identico, 9 da fami-liare parzialmente compatibile e 4 da donatore compatibi-le non familiare. La sclerodermia è comparsa in 6 dei 19soggetti (8% del totale, 31% dei casi con GvH). Le manife-stazioni cutanee erano presenti generalmente in forma este-sa, compresa l’alopecia e l’onicodistrofia, mentre in due casierano limitate alle mani o alle caviglie. In 2 casi vi è statoinoltre un coinvolgimento viscerale: nel primo coinvolgi-mento del pericardio che si è manifestato con pericarditiricorrenti e un episodio di tamponamento cardiaco acuto;nel secondo le manifestazioni viscerali hanno coinvolto ilpolmone, causando un’insufficienza respiratoria di tiporestrittivo che necessita di c-PAP notturna, e l’esofago, perla cui importante discinesia con disfagia è stata avviata sup-plementazione enterale notturna con SNG. Il trattamentofarmacologico della sclerodermia è risultato estremamentedifferenziato. In un caso vi è stata risposta al trattamentocon ciclosporina A e steroide, mentre negli altri casi è sta-to necessario associare un terzo immunosoppressore (mico-fenolato-mofetil, azatioprina, talidomide) o ricorrere a stra-tegie immunosoppressive più aggressive, quali il trapiantoautologo di CSE e la fotoaferesi. Commento: La percentua-le di GvH cronica e poi di sclerodermia nella nostra casisti-ca concorda con i dati della letteratura, nonostante l’eleva-ta quota (67%) di TCSE eseguiti da donatore parzialmentecompatibile o da donatore non familiare. Le manifestazio-ni cliniche di sclerodermia in corso di GvH cronica sono sta-te estremamente eterogenee. I vari trattamenti immuno-soppressivi messi in atto hanno avuto un ottimo risultatosulla componente cutanea della malattia, mentre scarso èstato il miglioramento sulla componente viscerale. Segna-
liamo quindi come la sclerodermia in corso di GvH sia unamalattia trattabile con successo sulla componente cutaneamentre rimangono importanti gli esiti a lungo termine del-la componente viscerale.
L035ALLOTRAPIANTO DI CELLULE STAMINALI EMOPOIETICHE NONMIELOABLATIVO (NON-MYELOABLATIVE STEM CELL TRANSPLANTATION-NST) NEI TUMORI SOLIDI DELL’ INFANZIA: EFFICACIA E TOLLERABILITÀCastellini C, Prete A, Franzoni M, Libri V, Assirelli E,Pession AMO Oncoematologia Pediatrica e Terapia Cellulare,Università degli Studi di Bologna
Introduzione: al fine di indurre un effetto Graft versusTumor (GvT) è stato attivato un programma di NST in tumo-ri solidi nelle forme pluri-recidivate. Pazienti e metodi: duepz affetti da NB refrattario ed 1 da RMS metastatico, sonostati sottoposti a NST da fratello HLA-identico, dopo con-dizionamento con thiotepa (300 mg/m2) e melphalan (140mg/m2). La profilassi della GvHD è stata eseguita con CsA(2 mg/kg dal g-1), associata a short term MTX in 1 pz. E’stata effettuata una valutazione delle sottopopolazionilinfocitarie (SL) e dei livelli sierici di TNF-α, IL-6, IL-8 e IL-2R prima del NST e dopo 1-3-6-9-12 mesi. Risultati: il nadirdei GB si è realizzato in g +6 (1) e +8 (2), con valori di 20,60 e 70 GB/mm3 rispettivamente. L’ attecchimento dei PMNè stato ottenuto ai g +12 (2) e +13 (1), quello delle PLT aigiorni +15, +20, +24 rispettivamente. Non è stata docu-mentata tossicità trapianto correlata; in 2 casi è insortaGvHD acuta grado I, trattata con steroidi e risoltasi in g +18e +19; 1 pz ha presentato GvHD cronica a 8 mesi dal NSTtrattata con Fk506+MMF. In 3/3 pz l’ analisi degli STR hadocumentato chimerismo completo ai giorni +15, +30 e+60.Riguardo ai livelli di citochine circolanti è stato evi-denziato un picco al giorno +15: TNF-α ha raggiunto finoa 20 volte i valori normali (v. n. ), IL-2R fino a 3 volte, IL-6fino a 100 volte, IL-8 fino a 700 volte, per poi tornare ai v.n. 6-12 mesi dopo il NST. I linfociti T CD4+ e CD8+ hannoraggiunto v. n. per età entro 3 mesi dal NST; le cellule NK,che riconoscono gli antigeni MHC di classe I, presenti sututte le cellule nucleate del’ organismo, hanno presentatoun incremento fino a 20 volte i livelli basali durante i pri-mi 2 mesi, per poi tornare ai v. n. dopo 6-12 mesi. I 2 pz inRC sono andati incontro a recidiva rispettivamente a 5 e 15mesi dal NST, e sono attualmente in terapia di reinduzio-ne; il pz in RP, dopo un’ iniziale risposta, a 5 mesi dal tra-pianto ha presentato progressione di malattia ed è dece-duto a 6 mesi dal NST. Conclusioni: il NST è una procedu-ra attuabile ed efficace in pz resistenti, pluritrattati ed incondizioni cliniche scadute, essendo gravato da scarsa tos-sicità, inoltre in grado di indurre un’’ azione immunotera-pica e un effetto GvT, documentato nel nostro studio dal-la riduzione della massa in un pz e dal concomitante incre-mento delle cellule NK e dei linfociti T.
L036STUDIO EPIDEMIOLOGICO DELLA PREVALENZA DELLE MALATTIE TIROIDEE,TUMORALI ED AUTOIMMUNITARIE NELLE FAMIGLIE DI PAZIENTI CON
170
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Letti

171
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
ISTIOCITOSI A CELLULE DI LANGERHANS (ICL) Trizzino A,* D’angelo P,* Trizzino A,* Varotto S,°°Lombardi A,° Fidani P,° Tettoni K,°°° Brach Del Prever A,**Aricò M**OncoEmatologia Pediatrica, Ospedale dei Bambini “G. DiCristina”, Palermo; °Oncologia Pediatrica, OspedaleBambino Gesù, Roma; °°OncoEmatologia Pediatrica,Padova;°°°Clinica PEdiatrica Brescia; **OncoEmatologiaPediatrica, Torino; CSS AIEOP Istiocitosi
L’ICL è caratterizzata da un’ampia variabilità clinica e daetio-patogenesi non ancora ben definite. Nostri studi pre-cedenti suggeriscono una componente gentica nella malat-tia. Studi epidemiologici precedenti hanno preso in consi-derazione il possibile effetto di vari fattori di rischio pre epost natali e l’associazione con malattie presenti nella fami-glia. In particolare Hamre et al. hanno osservato un’asso-ciazione con una storia familiare di tumori benigni, mentreBathia et al. con una storia familiare di patologie tiroidee.In ultimo è stata osservata un’associazione con varie pato-logie maligne (linfomi, leucemie e tumori solidi). Abbiamocondotto uno studio caso – controllo policentrico per con-fermare l’associazione della ICL con la presenza nella storiafamiliare di malattie tiroidee, autoimmuni e/o atopia, tumo-ri benigni o maligni. Sono stati raccolti, dal Gennaio 2003,41 alberi genealogici di pazienti con ICL. Il gruppo di con-trollo è consistito in 35 bambini trattati per neoplasia nel-lo stesso periodo nella nostra U. O. La raccolta della storiafamiliare è stata estesa ad almeno tre generazioni, inclu-dendo nella prima i fratelli dei casi, nella seconda, genitorie fratelli dei genitori, nella terza i nonni ed i fratelli dei non-ni. La prevalenza di patologia tiroidea è del 4,9 % nellefamiglie di soggetti affetti da ICL (n=850) e del 3,3% nellefamiglie dei soggetti affetti da neoplasia (n=842). La pre-valenza della patologia tumorale è stata del 10% nelle fami-glie dei soggetti con ICL ed 8% nelle famiglie dei controlli.Le differenze osservate non sono statisticamente significa-tive. Per quanto riguarda le patologie autoimmuni e/o aller-giche è stata osservata una prevalenza del 4. 2 % nel grup-po dei pazienti con ICL e del 2% nei controlli (RR 2,10; IC1,10-3,71). Non è stata osservata, a differenza di ciò che erastato precedentemente segnalato, una differenza statisti-camente significativa, della prevalenza di patologie tiroidee,e di disordini proliferativi, nelle famiglie dei soggetti conLCH presi in considerazione, rispetto al gruppo di controllo.I soggetti con atopia e/o disordini autoimmunitari sonoinvece significativamente più frequenti nelle famiglie deisoggetti con LCH. È in corso la raccolta della storia familia-re in un gruppo di controlli sani per paragonare l’incidenzadi patologie tiroidee o tumorali.
L037INDAGINE CONOSCITIVA SULLE PROBLEMATICHE PSICO-SOCIALI DIPAZIENTI AFFETTI DA LLA DURANTE LA FASE DI MANTENIMENTO. Guardascione M, Pinto A, Camera F, D’avino E,De Leonibus F, Menna GDipartimento di Onco-Ematologia, AziendaSantobono-Pausilipon, Napoli
Introduzione e obiettivi. Nel Dipartimento di Oncologia
dell’AORN Santobono-Pausilipon è stato condotto un’in-dagine conoscitiva per valutare i vissuti dei genitori di bam-bini affetti da LLA rispetto al reinserimento nell’ambientescolastico ludico e sociale, a distanza di 12-18 mesi dal-l’insorgenza della malattia. Metodi. A tal fine è stato som-ministrato ad un campione di 66 famiglie un questionariodi 14 domande chiuse rivolte ai genitori e relative a pro-blematiche psicosociali dei figli. Risultati e conclusioni. Adistanza di 12/18 mesi lo stato emotivo dei pazienti secon-do la percezione dai genitori è risultato: - per il 46% diripresa - per il 37% di lento recupero - per il 15% di gran-de difficoltà Tra i motivi di forte stress vengono attribuitiai bambini le seguenti condizioni emotive: 1.ricordi diffici-li da tollerare 2.difficoltà di accettazione dell’evento trau-matico 3. sentimenti di diversità Lo stato emotivo dei geni-tori risulta essere di preoccupata attesa nonostante fossepassato un anno dalla comunicazione della diagnosi; que-sto dato indica come la fase di mantenimento determininelle famiglie uno stato costante di allarme allorché il con-tatto con la struttura ospedaliera diventa sempre meno fre-quente. La metà dei pazienti non riprende la normale atti-vità scolastica per il persistere di condizioni immuno-depressive ma anche per scelta dei genitori a causa del lorostato d’ansia. Quelli che riprendono presentano problemilegati soprattutto all’apprendimento e al recupero del pro-gramma, anche nel caso in cui abbiano ricevuto assisten-za scolastica ospedaliera e/o domiciliare; mentre i bambi-ni più piccoli che non accedono alla scuola materna pre-sentano poi problemi di socializzazione. Rispetto al reinse-rimento sociale dei pazienti si evidenzia un sentimentogenerale di abbandono e in taluni casi anche di discrimi-nazione sociale. Le famiglie si rinchiudono nel loro conte-sto familiare limitando i contatti sociali e le attività ludi-che del bambino.
L038NON PUBBLICATO
L039EFFICACIA DI LYNEZOLID NEL TRATTAMENTO DELLA BATTERIEMIA DACOCCHI GRAM-POSITIVI RESISTENTI AI GLICOPEPTIDI IN PAZIENTIPEDIATRICI SOTTOPOSTI A CHEMIOTERAPIAMelchionda F, Fernicola P, Filippini B, Castellini C,Rondelli R, Pession AOncoematologia Pediatrica e Terapia Cellulare,Clinica Pediatrica S. Orsola-Malpighi, Bologna
Introduzione e obiettivi: Il rischio di complicazioni infet-tive in pazienti neutropenici in terapia immunosoppressi-va è in continuo aumento. Studi epidemiologici delle infe-zioni nosocomiali hanno visto emergere l’eziologia media-ta da patogeni Cocchi Gram+, che appaiono tra le cause piùinsidiose di batteriemia nosocomiale. Scopo dello studio èvalutare la tollerabilità e l’efficacia della terapia antibioti-ca di terza linea con Lynezolid in pazienti neutropenici condocumentata infezione da cocchi Gram+ multiresistenti nonresponsivi a terapia con glicopepdide. Metodi: Sono statitrattati con Lynezolid 10 mg/kg x 3 volte/die pazienti di età

172
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Letti
pediatrica affetti da leucemia acuta in fase di neutropeniasevera (<200 PMN) conseguente a chemioterapia citotos-sica con infezione da patogeni cocchi Gram+ multiresistentinon responsivi a terapia antibiotica standard. Tutti i pazien-ti trattati sono portatori di catetere venoso centrale (CVC).La batteriemia è stata documentata mediante multipleemocolture eseguite sia da CVC che da via periferica. Risul-tati: Tutti i 4 casi trattati hanno ben tollerato la terapia ese-guita con Lynezolid; nessun effetto collaterale è stato rile-vato durante la somministrazione endovenosa ed orale. Lasintomatologia di esordio è stata iperpiressia in corso dineutropenia in 4/4 pazienti, in assenza di altra documen-tata sede di infezione. Il trattamento di seconda linea conglicopeptide + aminoglicoside eseguito è stato guidato dal-l’antibiogramma che documentava sensibilità a teicoplani-na/vancomicina in tutti i pazienti. Il dosaggio della teico-planinemia è stato eseguito in 5° e 6° giornata in 2 pazien-ti e ha confermato un livello ematico adeguato >10 ng/ml.Dopo un trattamento con Lynezolid e. v. per 11 giorni (ran-ge 10-14) è stata ottenuta risoluzione clinica e microbio-logica dell’infezione in tutti i pazienti. Conclusioni: La tera-pia con Lynezolid è stata ben tollerata e capace di risolve-re in tutti i pazienti trattati il quadro clinico e microbiolo-gico di una batteriemia da cocchi Gram+ multiresistenti enon responsivi alla terapia standard di seconda linea conglicopeptide.
L040LE SINDROMI MIELODISPLASTICHE IN ETÀ PEDIATRICACaruso R, Coletti V, Lombardi A, Luciani M, Pansini V,Rana I, De Rossi GDivisione di Ematologia - Ospedale Pediatrico BambinoGesù
Introduzione: Le Sindromi mielodisplastiche (MDS) sonoun gruppo eterogeneo di disordini emopoietici rappresentatida un disordine clonale della cellula staminale emopoieti-ca. Recentemente sono state classificate secondo critericitogenetici, immunologici, e citomorfologici (classificazio-ne WHO); è da valutare se tale classificazione abbia o menouna riproducibilità in età pediatrica. Obiettivi dello studio:- Valutare l'incidenza delle MDS (WHO) in età pediatrica. -Valutare l'associazione con altre patologie. - Valutare l'an-damento nei 6 mesi successivi alla diagnosi. - Definire iparametri che indicano la necessità di una terapia specifi-ca. - Definire le alterazioni biologiche all'esordio in grado dicaratterizzare l'evento mielodisplastico come transitorio oneoplastico/clonale. Metodi e Risultati: Tra il 1997 e il 2004abbiamo studiato 16 pazienti, di età inferiore a 15 anni, conMDS, che presentavano anemia arigenerativa o citopeniaassociate ad un quadro midollare di displasia. Quattro sonostati classificati come Anemia Refrattaria (AR) e dodici comebicitopenia o pancitopenia refrattaria con midollo ipercel-lulare. Sei erano sindromici: uno con monosomia 7 e l'altrocon delezione 22q11.2; i rimanenti quattro con cariotiponormale. Nessun paziente presentava riarrangiamenti tipi-ci delle LAM. Quattro pazienti sono stati sottoposti a tra-pianto di midollo osseo (TMO) o sono in attesa. Un pazien-te è evoluto in LAM dopo 9 mesi dalla diagnosi. Tre pazien-
ti hanno mostrato una regressione spontanea. Gli altri tre-dici presentano una situazione stabile e non necessitano dinessun trattamento specifico. Conclusioni: Le MDS pedia-triche sono spesso associate ad anomalie cromosomichecostituzionali inquadrabili in situazioni sindromiche; taloranella prima infanzia si evidenziano disordini midollari chemimano classiche MDS e rendono problematico l'inquadra-mento nosografico nella classificazione WHO. Dal punto divista morfologico nella nostra casistica pediatrica sembraevidente un'eterogeneità in cui la displasia bi o trilinearesenza eccesso di blasti è preminente pur rimanendo l'ARuna delle forme più comuni. In una parte dei nostri pazien-ti la MDS coesiste con altri disordini costituzionali e non èquindi correlabile ad una patologia della cellula staminaleemopoietica. L'approccio terapeutico deve essere precedeu-to da un periodo di attenta osservazione in assenza di defi-niti markers di clonalità. Il trapianto di cellule staminali èl'opzione preferibile per ottenere una guarigione in etàpediatrica.
L041IL FUMETTO COME STRUMENTO DI RILEVAZIONE ED ELABORAZIONE DICONDIZIONI EMOTIVE CRITICHE: RIFLESSIONI SULL’UTILIZZO DISTRATEGIE DI COMUNICAZIONE Pinto A, Camera F, Misuraca A, Guardascione M, Poggi VDipartimento di Oncologia, Azienda Santobono-Pausili-pon, Napoli
Introduzione. Lavorare in un Centro di Alta Specializza-zione, nel quale afferiscono paz. con gravi patologie, pro-pone allo psicologo clinico innumerevoli quesiti legati all’u-tilizzo del setting clinico. In particolare è stata presa inconsiderazione la condizione di crisi intendendo per essa lostato di disequilibrio emotivo e cognitivo, nel quale si tro-vano alcuni pazienti, nell’avvertire la minaccia per la pro-pria vita. L’estrema difficoltà a comunicare in questimomenti ha spinto l’èquipe psicologica a ricercare nuovemodalità di comunicazione, utilizzando sin dalla diagnosilinguaggi diversi, diretti e creativi, come può essere quellodei fumetti. I personaggi dei Globemon nati per informarei bambini e a creare con loro un clima di fiducia e consa-pevolezza, è diventato col tempo uno strumento di ulterioreconoscenza del paziente occasione di approfondimento ericerca. Metodologia. Ad ogni paziente e’ stata offerta lapossibilità di elaborare il proprio fumetto in epoca succes-siva alla somministrazione di test proiettivi e successiva-mente alla presentazione del fumetto informativo, liberi diaderire o meno alla proposta creativa, secondo una grigliadi lavoro – stimolo. Risultati. Gli AA nel ricercare strumen-ti di comunicazione più adeguati hanno incentrato l’atten-zione su quei momenti di crisi durante i quali i pazienti siscontrano con realtà dure e dolorose e nei quali l’approc-cio diretto al tema non lascia spazi di facile accessibilità edelaborazione. Gli AA riflettono sul caso di dieci bambini incui il fumetto si è rilevato uno strumento utile ad eviden-ziare emozioni altrimenti inconfessabili come nel caso di M.7 anni arrabbiato per la ricaduta, di A. 8 anni in conflittocon i medici per la terapia anticonvulsivante, di V. 15 anniin contrasto con l’ingestibilità degli effetti cortisonici, di D.

13 anni spaventata per il trapianto. Naturalmente risulterànecessario validare i risultati attraverso la comparazionecon altri test standardizzati.
L042ESPERIENZE DI GRUPPO CON GLI ADOLESCENTI IN UN CENTRO DIONCO-EMATOLOGIACamera F, Pinto A, Misuraca A, Cataldi A, Marotta RS,Maltese ADipartimento di Onco-Ematologia, Azienda Santobono-Pausilipon, NapolI
Introduzione. Gli AA riportano l’esperienza di gruppo con-dotta con adolescenti seguiti presso il Dipartimento diOncologia dell’Ospedale Pausilipon di Napoli. Il gruppo èstato lo strumento attraverso il quale poter accogliere econdividere le loro vicissitudini emotive e visualizzare gliinfiniti livelli e articolazioni dei processi mentali, talvoltaappiattiti da un dolore fisico e psicologico, quale quelloprodotto dalla malattia, la cui comunicazione è talvoltaresa complessa dalla difficoltà a trovare parole veramentesignificanti per descriverlo. La creazione di un setting all’in-terno di un reparto di oncologia ha dovuto tener conto del-le resistenze emotive caratteristiche dell’età, della fre-quenza discontinua e delle diverse fasi della terapia. Meto-di. Il nostro percorso è stato mediato dal fare. Il coinvolgi-mento all’interno di attività pratiche quali la redazione diun giornale, il laboratorio teatrale e pittorico ha fatto sì chei ragazzi accogliessero l’idea di un gruppo e portassero alsuo interno tutti i possibili linguaggi. Abbiamo, quindi, ten-tato attraverso l’esperienza gruppale di ricomporre le variesoggettività e di dare un senso alle loro esperienze. I grup-pi di creatività, sono stati il modello che ci ha permesso diarticolare un intervento psicoterapeutico dotato dellanecessaria incisività e capacità trasformativo - elaborativapartendo dal creare, dal fare, dal giocare. I gruppi, di un’etàcompresa tra i 10 e i 16 anni, si incontrano a cadenza set-timanale, aperti ad accogliere nuove partecipazioni ma for-temente caratterizzati intorno ad alcuni componenti, sonocondotti da due psicologi; osservatori partecipanti (tiroci-nanti psicologi) e un insegnante si avvicendano per crearee sostenere le attività. Risultati e Conclusioni. L’esperienza,nonostante la possibilità che nel gruppo del fare si ripro-ponesse la fuga dal pensare, è stata un’esperienza interio-re; seppure con l’insofferenza tipica degli adolescenti. Leparole che non potevano essere dette sono state dipinteraccontate, drammatizzate in uno scenario creativo cheperò ha trovato la possibilità di rispecchiamento, come diceWinnicott, ed ha permesso ai ragazzi di ritrovare se stessiin un territorio che è intermedio tra la realtà interiore del-l’individuo e la realtà condivisa del mondo che è esterna agliindividui.
L043TELENGIECTASIA EMORRAGICA EREDITARIA: ESPERIENZA PEDIATRICAGiordano P, Nigro A, Russo FG, Sabbà C,* Saracco P,°De Mattia DDip. di Biomedicina dell’Età Evolutiva, Università di Bari.
*Dip. di Medicina Interna e di Medicina Pubblica,Università di Bari; °Div. Universitaria EmatologiaPediatrica, Az. Osp. O. I. R. M. , S. Anna, Torino
La teleangiectasia emorragica ereditaria (HHT) o malat-tia di Rendu-Osler-Weber si configura quale fibrodisplasiavascolare trasmessa come carattere autosomico dominan-te, a penetranza incompleta ed espressività variabile. Essasi caratterizza per la presenza di angiodisplasie localizzatea livello muco-cutaneo e di organi interni e viene ricono-sciuta in presenza di tre dei quattro criteri clinici di Shov-lin: epistassi ricorrenti, teleangiectasie cutanee, lesioniviscerali, storia familiare positiva. I geni coinvolti nella pato-genesi sono l’endoglina (ENG), che mappa in 9q33 ed ALK1, che mappa in 12q13. I prodotti di entrambi i geni sonocomponenti dei complessi recettoriali del trasforminggrowth factor. Abbiamo arruolato soggetti pediatrici sinto-matici o asintomatici di nuclei familiari con mutazione notae nuovi casi pediatrici sintomatici. Le teleangiectasie muco-cutanee sono state ricercate attraverso capillaroscopia alivello cutaneo e fibrorinoscopia a livello nasale. Le malfor-mazioni artero-venose polmonari (PAVMs) sono state valu-tate mediante ECOCARDIO-bubble (BE) e/o TAC torace mul-tislice; quelle epatiche (HAVMs) mediante ECO-color-dop-pler e/o TAC addome. Le malformazioni cerebrali (CAVMs)sono state screenate attraverso angio-RMN. La nostra casi-stica comprende 17 casi pediatrici familiari (F:10 M:7; 1-18anni di età, età media: 10,6 +/- 5,12) di cui 13 (76,5%) conmutazione causale identificata e 4 (23,5%) in corso di iden-tificazione. In 9/13 (69,2%) casi la mutazione era in ALK1 ein 4/13 (30,8%) in ENG. I risultati dello screening clinico-diagnostico sono ripotati in Tabella 1.In un singolo caso conpresentazione di ematemesi e melena è stata eseguita EGDSche ha rivelato teleangiectasie multiple in tutto il trattogastro-intestinale. La nostra casistica pediatrica sia pur limi-tata ha evidenziato un’alta percentuale di pazienti con epi-stassi e malformazioni artero-venose che ci impone lanecessità di adottare appropriati screening clinico-diagno-stici e approcci terapeutici età-correlati.
Tabella 1.
EPISTASSI 12/17 (70,6%)
TELANGIECTASIE MUCO-CUTANEE 14/17 (82,3%)
CAVMs 1/16 (6,25%)
PAVMs 10/16 (BE+) (62%)7/11 (TAC+) (63,6%)
HAVMs 10/17 (ECO+) (58,8%)6/11 (TAC+) (54,5%)
L044RICONOSCIMENTO PRECOCE DEL DISAGIO EMOTIVO ED INVIO ALLACONSULTAZIONE PSICOLOGICA CLINICA; ANALISI DI 5 ANNI DI ATTIVITÀ INUN REPARTO DI ONCOLOGIA PEDIATRICAClerici CA,°* Massimino M,° Luksch R, ° Cefalo G, °
173
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

Terenziani M,° Ferrari A,° Casanova M, ° Spreafico F,°Polastri D,° Meazza C,° Podda M,° Casiraghi C,°Armiraglio M,° Fossati-Bellani F°°Istituto Nazionale per lo Studio e la Cura dei Tumori,Milano*Dipartimento di Psicologia, Università degli Studi,Milano-Bicocca
Introduzione e obiettivi: Obiettivo di questo lavoro è dieffettuare un’analisi delle procedure d’invio alla consulta-zione psicologica clinica svolte in un reparto di OncologiaPediatrica negli ultimi 5 anni, alla luce di una revisione del-le teorie sull’invio all’intervento psicologico nelle malattiegravi dell'età evolutiva. Metodi: E’ stata esaminata la casi-stica degli invii alla consultazione e ad interventi di psico-terapia avvenuti presso l’Unità Operativa di Pediatria dell’I-stituto Nazionale per lo Studio e la Cura dei Tumori di Mila-no dal 1 gennaio 1999 al 31 dicembre 2003, consistente in252 pazienti (1031 colloqui svolti) e 430 famiglie di pazien-ti (1284 colloqui svolti). La letteratura sulla ricerca empiri-ca sul tema dell’invio alla consultazione psicologica inPediatria è stata raccolta anche attraverso le banche datiMedline, PsychINFO e Cochrane Library. Risultati: E’ illu-strata la distribuzione delle motivazioni dell’invio, differen-ti a seconda delle categorie di invianti. In generale, fra lemotivazioni più frequenti sono state indicate: difficoltà diadattamento alla malattia e alle cure (34%), problemi psi-cosociali (18%), situazioni particolari come chemioterapiead alte dosi e interventi mutilanti (16%), difficoltà relazio-nali nell’ambito familiare (9%), disagio emotivo e difficoltàdi adattamento dei pazienti lungo-sopravviventi (8%), scar-sa compliance alle terapie (5%), malattia in fase terminale(4%), difficoltà relazionali con l’equipe curante (3%), con-dizioni psicopatologiche soggettivamente disturbanti o checreano difficoltà nello svolgimento dell’attività di cura (3%).Conclusioni: L’invio alla consultazione psicologica è un atto,con valenze emotive, di natura tecnica (diagnostico), rela-zionale (ha un successo diverso a seconda degli invianti edell’accordo inviante – paziente), organizzativo (sono diver-se le procedure e l’organizzazione degli invii a seconda del-le diverse istituzioni). E’ necessario sviluppare ulteriori ricer-che, con metodologie adeguate, che consentano di perfe-zionare le possibilità d’identificazione precoce delle condi-zioni di disagio emotivo, e di affrontare il problema dellavalutazione di efficacia degli interventi.
L045LA MORTE IN REPARTO: IL TEMPO DELLA RIFLESSIONECirillo F, Gurreri Ra, De Benedetta G, Indolfi P,di Martino M, Di Tullio MTServizio di Oncologia Pediatrica Dipartimento di PediatriaII Ateneo di Napoli
I legami affettivi, i legami di conoscenza con chi è giàmorto ci accompagnano per tutto il ciclo vitale e questilegami tengono in vita il mondo dei morti, che si popolasempre di più di persone conosciute, facendolo temere dimeno. Tuttavia gli uomini temono la morte e si difendonotenendone lontano il pensiero. Curare pazienti affetti dapatologie neoplastiche significa per l’équipe curante con-vivere ed accettare l’altra dimensione del vivere che è il
morire. La morte, e soprattutto la morte di bambini pone ilgruppo oncologico di fronte ad un carico di dolore tale daattivare la consapevolezza della perdita e meccanismi didifesa dall’angoscia. Come viene gestito dagli operatori delreparto di oncologia pediatrica il vissuto della morte e del-la perdita? L’équipe psicologica ha osservato che di fronteall’evento morte di un bambino il dolore avvertito dal grup-po oncologico viene, a volte, agito con reazioni di fuga (intesa come il fare per non sentire il dolore) ed, altre vol-te, con la esasperata condivisione della pena dei genitori.Da qui è nata l’esigenza di attivare un tempo della rifles-sione, della condivisione e dell’elaborazione del lutto. Iltempo della riflessione presuppone il superamento di moda-lità reattive di tipo evacuativo e/o riparatorio all’eventocatastrofico costruendo, così, il presupposto per condividereemozioni e vissuti di sofferenza senza restarne invischiati.Scrollarsi di dosso il dolore, allontanandolo da sé per la dif-ficoltà di tollerarlo o attivarsi con atteggiamenti freneticidi tipo consolatorio e riparativo non permette la creazionedi quello spazio di pensiero necessario per poter condividerela sofferenza e restituire il dolore elaborato. È stato osser-vato un tempo, successivo all’agire, in cui la sofferenza vie-ne quasi nascosta dagli operatori, sia a se stessi sia aglialtri, e delimitata con la costruzione di confini per evitaredi esserne inondati. Questo tempo sembra essere funzionalealla possibilità di entrare in contatto con il proprio dolorepsichico. Infatti, la funzione di contenimento esercitatadalla struttura ospedaliera ed, allo stesso tempo, il vivereall’esterno esperienze che stimolano energia vitale dà aglioperatori la forza di contattare il proprio dolore, accettarei propri limiti, rendendo pensabile la condivisione dell’an-goscia. Soltanto la consapevolezza dei propri limiti con-sente all’équipe curante di non sentire i vissuti di impotenzaed accettarsi nel ruolo di testimoni della sofferenza psichi-ca propria e dell’altro. Riconoscere l’importanza di questoruolo permette di accogliere la storia del bambino e dellafamiglia conservando per sempre nella memoria il legame.
L046QUANTI FARMACI PER UN DOLORE?Pierinelli S,* Bachiocco V,° Placci A,* Melchionda F,*Filippini B,* Facchini E,* Burnelli R,* Castellini C,*Vivarelli F,* Bolognini S,* Pession A**MO Oncoematologia Pediatrica e Terapia CellulareDipartimento Scienze Pediatriche Mediche e ChirurgichePoliclinico S. Orsola Malpigli Bologna°servizio Anestesiae Rianimazione Pediatrica Policlinico S. Orsola MalpighiBologna
Introduzione. Il dolore neoplastico in età pediatrica rico-nosce molteplici cause e differenti meccanismi, la cui com-binazione dà spesso luogo a dolori severi per intensità equalita’ e soprattutto resistenti alla terapia. La distinzionenei meccanismi fisiopatologici in base al tipo di dolore per-mette di aderire con un razionale al loro trattamento. Nesegue la necessità di una terapia con diverse classi di far-maci, ciascuna delle quali ha un target neurobiologico.Obiettivi. Riportiamo la storia clinica di tre piccoli pazien-ti, affetti da dolore nocicettivo e neuropatico. Metodi e
174
haematologica vol. 89[supplement n. 10]:October 2004
Abstracts | Letti

Risultati. SF. Maschio, 10 anni, affetto da Linfoma non
Hodking con lesioni ossee plurifocali (comprese L3-L4-L5),
infiltrazione testicolare e liquorale. Il paziente presentava
alla Visual Analogue Scale un punteggio di 10 (VAS; 0-10)
e alla Faces Scale di 5 (FS; 1-5). Erano presenti allodinia ed
iperalgesia. La somministrazione di farmaci adiuvanti
(gabapentin, amitriptilina, clonazepam) associati ad un mu-
agonista e ad un FANS ha permesso un rapido controllo del-
l’intensità del dolore (Punteggio minimo VAS=0). Terapia
antineoplastica con ciclofosfamide. SM. Maschio, anni 3.
Neuroblastoma addominale IV stadio (metastasi midollo
osseo, rachide dorso-lombare, infiltrazione meningea,
nevrite post-erpetica). VAS 10, FS 5, iperalgesia diffusa. La
combinazione di gabapentin, amitriptilina, clonazepam e
morfina ha rapidamente attenuato la severità del dolore
(Punteggio Minimo VAS=0). Somministrazione di melpha-
lan, come chemioterapico. MD. Maschio, 10 anni. Sarcoma
di Ewing IV stadio con lesioni ossee diffuse (cranio-faccia-
li, rachide dorso-lombare, arti inferiori e superiori). VAS 10,
FS 5. Il dolore è stato efficacemente controllato dalla som-
ministrazione di farmaci adiuvanti (gabapentin, amitripti-
lina, clonazepam) e morfina (Punteggio Minimo VAS=0).
Terapia antineoplastica con melphalan. Conclusioni. Agen-
ti farmacologici appropriati al meccanismo neurobiologico
permettono di controllare dolori difficilmente trattabili. Un
attento monitoraggio condiviso dall’equipe medico-infer-
mieristica della risposta alla terapia farmacologica è fon-
damentale per una ottimale modulazione della terapia
antalgica.
L047TRATTAMENTO DI ASPERGILLOSI INVASIVA CON L’IMPIEGO DI UNAASSOCIAZIONE DI ANTIMICOTICI: UTILITA’ DEL MONITORAGGIODELL’ATTIVITA’ FUNGICIDA E FUNGOSTATICA SIERICANardi M, Tascini C,* Casazza G, Menconi M, Najajreh M,Favre C, Menichetti F* Centro Trapianti Midollo Osseo, U. O. OncoematologiaPediatrica Azienda Ospedaliera Universitaria Pisana*UO Malattie Infettive, Osp. Cisanello, Pisa
Introduzione. L’impiego combinato di antimicotici nelleinfezioni fungine invasive dell’immunodepresso non è stato, adoggi, estesamente valutato. In particolare, l’introduzione nel-la pratica clinica del Caspofungin, appartenente ad una nuo-va classe di antimicotici (echinocandine), con meccanismo diazione peculiare, apre la prospettiva di un sinergismo con altrifarmaci. E’ stata utilizzata, a tale scopo, la valutazione del-l’attività fungostatica sierica (SFA) e fungicida (SFCA) prima edopo l’aggiunta di un trattamento sequenziale ad un regimeterapeutico dato. E’ stato inoltre utilizzato il metodo E-test pervalutare il sinergismo anti-fungino. Metodi. Una paziente di18 aa affetta da sindrome di Shwachman-Diamond, presentòfebbre e tosse in corso di aplasia post-chemioterapia di Indu-zione per LMA. Fu intrapresa terapia antibiotica a largo spet-tro. Una TAC torace mostrava una lesione triangolare a con-tenuto liquido, suggestiva di aspergillosi, a livello del lobosuperiore del polmone destro. La ricerca di antigene sierico diAspergillus (Platelia) risultò positiva, e al trattamento anti-biotico fu aggiunta la somministrazione di caspofungin edamphotericina B liposomiale. Si ottenne rapidamente la scom-parsa della febbre e, dopo la risoluzione della neutropenia, fusospesa la terapia antibiotica, mentre la terapia antimicoticafu proseguita per 40 gg. Al termine di tale periodo, la TACdimostrava la persistenza della lesione, la cui biopsia transto-racica dette esito a coltura positiva per A. fumigatus e A. niger.La sensibilità dei ceppi è stata testata mediante micrometo-do (linee guida NCCLS) e risultava una sensibilità ad amfote-ricina B, itraconazolo, voriconazolo e caspofungin. Fu esegui-ta terapia con amfotericina B liposomiale, caspofungin e vori-conazolo per 2 settimane. Durante tale trattamento il picco diattività fungostatica sierica (SFA) e il picco di attività fungi-cida sierica (SFCA) erano, rispettivamente, 1:32 e 1:8 per A.fumigatus e 1:16 e 1:2 per A. niger. Trattandosi di una unicalesione, in prospettiva di sottoporre la paziente a TMO, si è pro-ceduto a resezione chirurgica del lobo polmonare interessato.La coltura del materiale prelevato risultò positiva per le stes-se 2 specie di Aspergillo. La terapia antimicotica combinata fuulteriormente protratta per 14 gg, poi fu proseguito tratta-mento con il solo Voriconazolo. Durante tale trattamento ilpicco di attività fungostatica sierica (SFA) e il picco di attivitàfungicida sierica (SFCA) erano, rispettivamente, 1:32 e 1:2 perA. fumigatus e 1:16 e > 1:2 per A. niger. E-test dimostravaassenza di sinergismo tra Amfotericina B e Voriconazolo, sug-gerendo il ruolo determinante del caspofungin nella terapiacombinata. La paziente ha successivamente ricevuto un TMOda donatore volontario, senza che vi fossero ulteriori segni diAspergillosi nel decorso post-TMO. Conclusioni. Nel nostrocaso, l’impiego dell’associazione di più antimicotici e della chi-rurgia ha consentito l’eradicazione dell’infezione in un sog-getto particolarmente a rischio, oltre che per la leucemia,anche a causa della patologia di base.
175
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

176
haematologica vol. 89[supplement
kind and sessionXXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
Umanizzazione in ospedale
Dorella ScarponiMedico Dirigente di Primo Livello Psichiatra, presso laSezione di Oncoematologia Pediatrica (Prof. G. Paolucci)dell'Azienda Policlinico S. Orsola, di Bologna
L'umanizzazione degli ambienti di cura corrisponde alladisposizione collettiva della equipe a tutelare se stessa ei pazienti da una scissione oltremodo dannosa tra mentee corpo.
Per i curanti tale assetto, estremizzato e difensivo, difunzionamento rischia di allontanare il proprio sè dal ruo-lo che si riveste, con conseguenti sofferenze psicosoma-tiche, tipiche del fenomeno del “Burn out”.
Nei pazienti il rischio è quello di sentire prevalere l'a-spetto concreto della esperienza di malattia senza possi-bilità di comprensione e metabolizzazione di essa.
L'umanizzazione degli ospedali, fenomeno più frequen-te tra quelli pediatrici, assicura l'equilibrio tra gli aspettitecnici e quelli emotivi della cura elargita, fornendo già,a chi soffre, un modello di “integrazione possibile” dell'e-sperienza, dentro la propria storia evolutiva. Soprattuttoin pediatria, infatti, la continuità del percorso di crescitadel piccolo paziente, insieme alla convinzione che in luisono presenti risorse, oltre che bisogni, rappresentano glielementi di base sui quali pensare all'umanizzazione comeuno dei percorsi insostituibili della cura.
Scuola in reparto... Integrazione di esperienze
C. Ferri, M. SverberiSpazio Scuola Primo Circolo Didattico di Modena
Uno dei punti di forza della nostra scuola è sicuramen-te il lavoro di tessitura di relazioni costruite sulla rete disolidarietà, sia all'interno che all'esterno della strutturaospedaliera. Parallelamente si sono sviluppate le attivitàdi coordinamento e collaborazione con tutti gli operato-ri, gli enti e le associazioni di volontariato che operano nele per il reparto di oncoematologia.
SCUOLA IN REPARTO COME…scuola aperta al confron-to, al dialogo, all'interazione fra le diverse figure profes-
sionali e di volontariato. SCUOLA IN REPARTO COME…gruppo di lavoro che coo-
pera con la preoccupazione comune del benessere psico-fisico del bambino malato e della sua famiglia.
Principali attività…SCAMBI CON:
- il personale medico e infermieristico: RIUNIONI PERIO-DICHE per la programmazione del percorso educativo, inbase ai tempi della malattia, CORSI DI AGGIORNAMEN-TO sulle diverse patologie, RIUNIONI DELLA COMMIS-SIONE MAEV (medici, agenzie educative, volontari)
- gli operatori degli altri servizi educativi (insegnantiscuola infanzia e operatori Biblioteca Delfini)
COLLABORAZIONE CON:- le scuole di appartenenza del bambino (attivazione
istruzione domiciliare)- le scuole del Circolo Didattico e del territorio (incon-
tri-lezioni, iniziative di solidarietà, )- altre scuole ospedaliere- l'Azienda Ospedaliera- l'Associazione Aseop- altre associazioni, enti, istituzioni operanti sul territo-
rio
COORDINAMENTO DI:- volontari e tirocinanti- interventi di esperti o rappresentanti di gruppi o asso-
ciazioni- spettacoli teatrali e/o feste legate alla tradizione
SCUOLA IN REPARTO COME…scuola che rivede, modi-fica il proprio operato e il ruolo stesso degli insegnanti
Scuola che si pone obiettivi trasversali ad ogni età (l'au-tonomia, la comunicazione con gli altri, la costruzionedelle identità e delle abilità in ambiti specifici)
Insegnanti che imparano ad operare all'interno di duecornici: flessibilità e discontinuità.
Insegnanti che diventano:- mediatori tra scuola dentro e fuori l'ospedale- facilitatori di relazioni all'interno e all'esterno della
clinica pediatrica- supporto emotivo alle famiglie
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Congresso Nazionale Associazione Italiana Ematologia Oncologia Pediatrica
Programma Infermieristico
L'AGIRE INFERMIERISTICO QUOTIDIANO:
ATTRAVERSO LA FORMAZIONE, LA COMUNICAZIONE
L'UMANIZZAZIONE DELLE STRUTTURE DI CURA

SCUOLA IN REPARTO COME…scuola che cerca di pro-porre attività coinvolgenti, stimolanti ed attiva in que-st'ottica vari progetti:
1) Informatica Camere sterili: non più soli con la rete2) Animazione con pupazzi di gommapiuma e burattini2) Animazione musicale3) Manualità ed educazione ambientale Creo e ricreo:
materiali in gioco 4) Comico-terapia
Curriculum insegnantiFerri Carla scuola elementareSverberi Marisa scuola elementare
Le insegnanti operano nel reparto di Pediatria del Poli-clinico di Modena dal 1999. Entrambe hanno scelto dioperare nella scuola ospedaliera dopo aver insegnato nel-le scuole elementari comuni per vent'anni. L'intesa e lacollaborazione tra le due insegnanti è iniziata circavent'anni fa (hanno insegnato gli ultimi quindici anni nel-la stessa scuola).
Numerosi sono i corsi d'aggiornamento sia di tipo disci-plinare sia inerenti alla specificità della scuola ospedalie-ra ai quali le insegnanti hanno partecipato. Svariate lepartecipazioni a convegni, sia a carattere internazionaleche nazionale. Negli ultimi due anni sono state impegna-te nelle diverse iniziative di formazione messe in atto alivello regionale per tutti gli insegnanti ospedalieri stata-li. In occasione della mostra Documentaria che si tiene aModena ogni due anni hanno presentato, con l'ausilio difilmati e cd-rom, alcune delle loro esperienze più signifi-cative. Collaborano al giornalino dell'Aseop e alla rivista“Il bambino in ospedale”. Promuovono incontri e cicli dilezioni con personale medico sia nelle scuole del CircoloDidattico di appartenenza, sia in quelle del territorio. Par-tecipano a progetti di carattere più ampio promossi daComune, Circoscrizione. . .
Diagnosi infermieristiche in OncoematologiaPediatrica
C. FerautInfermiera Professionale Centro Trapianti Ospedale Regi-na Margherita Torino
I problemi di salute effettivi o potenziali, conducibili asoluzioni mediante intervento infermieristico, vengonoidentificati come “diagnosi infermieristiche”.
La diagnosi infermieristica, secondo la NANDA (1990),costituisce la base sulla quale scegliere gli interventi infer-mieristici volti a conseguire degli esiti di cui l'infermiereè responsabile.
Il processo di nursing, definito come l'essenza dell'assi-stenza infermieristica, prevede una serie di fasi pianifica-te finalizzate all'identificazione ed alla soluzione dei pro-blemi per soddisfare i bisogni di assistenza sanitaria edinfermieristica dei pazienti.
Anche il bambino oggi viene considerato come sogget-
to portatore di bisogni specifici, tipicamente pediatrici,legati alle dinamiche familiari ed all'età evolutiva. E'importante nell'ambito della formulazione di un piano diassistenza infermieristica, tenere conto di aspetti fonda-mentali quali:- età e grado di sviluppo cognitivo ed emotivo del bam-
bino- l'esigenza di gioco, occupazione, svago e crescita- la “triade” genitori e bambino
Nell'ambito della pediatria oncoematologica le princi-pali diagnosi infermieristiche codificate dalla NANDA eda noi identificate sono:- rischio di infezione- rischio di lesioni- compromissione dell'integrità tissutale- alterazione della mucosa del cavo orale- rischio di alterazione della temperatura corporea (iper-
termia)- paura- alterazione del confort- disturbo del concetto di se- nutrizione alterata inferiore al fabbisogno- rischio elevato di gestione inefficace del regime
terapeutico
Nella nostra realtà le diagnosi infermieristiche cosìcome proposte dalla NANDA non sono ancora utilizzate:questa relazione può essere un incentivo per poterle inse-rire nella quotidianità dell'assistenza infermieristica.
Metodologia per la rilevazione della comples-sità assistenziale infermieristica. Calcolodell'indice di complessità assistenziale (ICA)
I. I. D. Cavaliere Bruno Prof. a. c in infermieristica clinica presso il corso di Laureaper Infermieri e Prof. a. c. di organizzazione - managementinfermieristico presso la scuola D. D. S. I. - Facoltà di Medi-cina e Chirurgia dell'Università degli studi di Genova
Introduzione Da alcuni anni a questa parte, le innovazioni introdot-
te nei meccanismi di gestione delle aziende sanitarie pub-bliche e private, hanno determinato la necessità di realiz-zare nuovi strumenti organizzativi finalizzati alla razionalerilevazione di indicatori dell'assistenza infermieristicabasati su criteri oggettivi, riproducibile e verificabili. Taleapproccio si rende indispensabile in un'organizzazione chesi basa sempre di più su logiche di “governo clinico” fina-lizzate alla determinazione e alla misurazione delle lineedi produzione delle prestazioni sanitarie.
Questa esigenza, tuttavia, può essere soddisfatta solo acondizione che si proceda alla ridefinizione dei modelligestionali attualmente in uso e alla formulazione di solu-zioni più aderenti al nuovo assetto conferito al sistemasanitario e alle nuove competenze che, in particolare, nel-la professione infermieristica prevedono profondi cam-biamenti in termini di metodologie, strumenti da applica-
haematologica vol. 89[supplement
XXXI Con-gressoNazionale
Programma Infermieristico
haematologica vol. 89[supplement n. 10]:October 2004

re per garantire nuovi standard di risultato. In quest'ottica il metodo del calcolo dell'indice di com-
plessità si pone come “sistema organizzativo integrato “capace di facilitare alcuni meccanismi di cambiamentoattraverso la creazione di basi-dati efficienti ed efficaci dalpunto di vista metodologico.
Il sistema I. C. A. si configura quindi come parte inte-grante del “sistema informativo” del professionista fina-lizzato a garantire un adeguato processo decisionale del-l'infermiere.
I parametri presi in considerazione per lo sviluppo del-lo studio in argomento, si identificano con le undici “pre-stazioni infermieristiche” indicate dal “modello delle pre-stazioni” e ne misurano la complessità in relazione all'e-spressione del continuum salute/malattia; attraverso le“finalità assistenziali” che l'infermiere realizza rispetto alsoddisfacimento dei bisogni d'assistenza infermieristica(Figura 1).
Tabella 1.Finalità assistenziali del modello delle prestazioni.
Indirizzare Guidare Sostenere Compensare Sostituire| | | | | |
Come si può vedere dalla tabella 1, che rappresenta ilcontinuum autonomia/dipendenza, nello studio in discus-sione sono ipotizzati cinque livelli di finalità assistenzialii quali possono essere facilmente interpretati come livel-li di “complessità assistenziale” in quanto si correlano allacondizione della persona assistita configurando, perciò,un criterio di misurazione il “Numero Indice di Comples-sità Assistenziale”. Tale criterio consente di definire alcu-ni standard che determinano la possibilità di classificaredifferenti tipologie di prestazioni che possono essere misu-rate attraverso la rilevazione degli indicatori specifici perogni malato.
Tali livelli definiscono sia le finalità dell'intervento infer-mieristico che le azioni che ne derivano e che si raccor-dano alla complessità intrinseca dell'intervento infermie-ristico da parte del professionista infermiere, ovvero:1. INDIRIZZARE: orientare in funzione di un esplicito e
conveniente criterio di scelta. L'azione di indirizzo sifonda sul presupposto che la persona, acquisite deter-minate conoscenze, sia in grado di soddisfare i propribisogni;
2. GUIDARE: sorreggere la scelta con un intervento teo-rico e/o pratico. L'azione di guida si fonda sul presup-posto che la persona, compiute le scelte ed acquisitespecifiche abilità, sia in grado di agire efficacementeper soddisfare i propri bisogni;
3. SOSTENERE: contribuire al mantenimento di una con-dizione di relativa stabilità e sicurezza. L'azione disostegno si fonda sul presupposto che la persona, mes-sa nelle condizioni di poterlo fare,, mantenga o met-ta in atto le conoscenze e le abilità acquisite per sod-disfare il bisogno;
4. COMPENSARE: intervenire per ristabilire un equilibrioprecedente tramite una parziale sostituzione. L'azio-ne di compensazione si fonda sul presupposto che lapersona necessiti costantemente di interventi infer-mieristici di parziale sostituzione nello svolgere le atti-
vità collegate al soddisfacimento del bisogno; 5. SOSTITUIRE: espletare completamente una o più fun-
zioni di una persona in sua vece. L'azione di sostitu-zione si fonda sul presupposto che la persona neces-siti costantemente di interventi infermieristici di tota-le sostituzione, che può avvenire anche mediante l'im-piego di ausili, presidi, attrezzature da parte dell'in-fermiere.
Questa classificazione ci consente di affermare che lacomplessità assistenziale infermieristica può essere misu-rata attraverso specifici criteri in grado di determinarestandard che possono essere rilevati attraverso la regi-strazione degli indicatori specifici per ogni singolo mala-to e intrinsecamente collegati alle prestazioni erogate,come rappresentato in Tabella 3.
L'intento della metodologia dell'I. C. A , è quello di for-nire con poche operazioni importanti informazioni di sin-goli o raggruppamenti di malati e della struttura interes-sata attraverso l'impiego di strumenti semplici ed intuiti-vi che consentono di:— definire e pianificare la propria attività — misurare la complessità assistenziale per malato
(intervalli di classi per gravità)— determinare le competenze necessarie (mix delle com-
petenze)— definire strumenti di integrazione organizzativa (pro-
cedure e istruzioni operative) — comparare strutture organizzative basate sull'indice
di complessità.
Definire e Pianificare la propria attività La definizione della propria attività si basa sulla conte-
stualizzazione delle prestazioni infermieristiche. ovverosull'analisi delle proprie attività in relazione al modelloassistenziale impiegato.
In questa fase i professionisti, attraverso gruppi di lavo-ro, individuano e specificano le proprie funzioni e il pro-prio operare scegliendo un modello assistenziale (sono inatto sperimentazioni dell'I. C. A. per l'impiego di altrimodelli e/o di altri sistemi di determinazione delle attivitàcome ad esempio l'utilizzo del tariffario nomenclatore)idoneo al loro campo specifico di attività e all'impiego dispecifici strumenti di integrazione organizzativa finalizzatialla pianificazione delle attività .
Si tratta di un momento molto importante e qualifi-cante perché permette al gruppo professionale di discu-tere e condividere in modo concreto le proprie conoscen-ze, le strategie e le soluzioni più appropriate per renderelo strumento di rilevazione il più attendibile possibile.
Questa fase, molto delicata, consente di delineare gliobiettivi assistenziali infermieristici (le linee di attivitàproprie- nel nostro caso le undici prestazioni ) rispettoall'erogazione delle prestazioni complessive all'interno diuna determinata struttura (reparto, dipartimento, ospe-dale, domiciliare, distretto ecc…) .
La sua realizzazione viene facilitata attraverso l'impie-go di strumenti di definizione delle attività come ad esem-pio le “procedure” che ci aiutano a definire meglio il cosafare, il come farlo, quando farlo, chi lo deve fare e con chi.
In particolare nella definizione delle attività, almeno in
178
haematologica vol. 89[supplement
kind and sessionXXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
haematologica vol. 89[supplement n. 10]:October 2004

una prima fase, si sono largamente utilizzate le tabelledelle prestazioni pubblicate sul testo di Cantarelli . Que-sta documentazione è stata utilizzata come punto di par-tenza per mappare le attività (azioni) rispetto alle finalità(complessità) realizzando una operazione di modifica/con-testualizzazione del loro contenuto a tal fine si allega unatabella n. 2
La struttura della tabella presenta due fondamentaliunità: la prestazione, che a sua volta definisce delle azio-ni e degli atti infermieristici e le finalità assistenziali chea loro volta definiscono la complessità della prestazioneerogata come rappresentato a titolo d'esempio nellatabella 2 relativa alla prestazione Assicurare l'alimenta-zione
In definitiva il peso dell'attività espressa in un valorevariabile da 1 a 5 indica il livello di complessità ovveroindica come attività più lieve l'indirizzo valore uno (1)(informazione al malato, famiglia …) rispetto alla sostitu-zione valore cinque (5) (sostituirsi integralmente al mala-to ) la quale è più complessa in quanto ricomprende tut-ti le altre finalità.
Il gruppo professionale come già indicato precedente-mente, in questa fase, attraverso lavori di gruppo, condi-vide il contenuto ed il significato delle attività variando,integrando, contestualizzando le attività in esso contenute
al fine di produrre un documento condiviso dal gruppodisciplinare (meglio se anche gli altri gruppi disciplinaricon livelli di conoscenza differenti) e completa il lavororealizzando le procedure legate alle attività .
Queste fasi consentono quindi di definire le attività pro-prie dell'infermiere il quale a questo punto può attraver-so la rilevazione puntuale dell'indice di complessità assi-stenziale per singolo malato tracciare anche una pianifi-cazione dell'assistenza infermieristica.
Con una adeguata casistica ricavata dalla rilevazionedella complessità assistenziale per singolo malato saràpossibile trarre in breve tempo dei profili di cura di assi-stenza infermieristica che faciliteranno la pianificazionedegli interventi assistenziali.
Potremmo finalmente iniziare anche degli studi piùesaustivi rispetto all'efficacia (qualità dei risultati ) e all'ef-ficienza ( costo) dell'assistenza infermieristica.
Misurare la complessità assistenziale per malatoAttraverso il costante rilevamento dei dati (vedi la tabel-
la 3), gli infermieri ottengono quotidianamente la misu-razione della complessità per singolo malato, attraverso unsistema di misurazione obiettivo, verificabile e riproduci-bile.
L'assegnazione del livello di gravità per singola presta-zione è realizzata attraverso la compilazione quotidiana diuna scheda (vedi tabella 3) che registra la rilevazione del-lo stato del malato in rapporto alle attività e alla gravitàdelle stesse definite dai professionisti, come precedente-
haematologica vol. 89[supplement
XXXI Con-gressoNazionale
Programma Infermieristico
haematologica vol. 89[supplement n. 10]:October 2004
Tabella n. 3 Rilevazione dell'I. C. A. per singolo malato
Malato Bianchi Letto 9999 Matricola 99999 Reparto: 9999999 descrizione : chirurgia
PRESTAZIONI 10 . 10 . 2004 11.10.20041.Assicurare la respirazione I. C. A Livello
1 22 3 4 52.Assicurare alimentazione e idratazione I. C. A Livello
1 2 3 4 553. Assicurare l'eliminazione urinaria e intestinale I. C. A. Livello
1 22 3 4 54. Assicurare l'igiene I. C. A. Livello
1 22 3 4 55. Assicurare il movimento I. C. A. Livello
1 22 3 4 6. Assicurare il riposo e il sonno I. C. A. Livello
1 22 3 7. Assicurare la funzione Cardiocircolatoria I. C. A. Livello
1 2 33 4 58. Assicurare un ambiente sicuro I. C. A. Livello
2 559. Assicurare l'interazione nella comunicazione I. C. A. Livello
1 22 3 4 510 Applicare le procedure terapeutiche I. C. A. Livello
2 3 5511. Eseguire le procedure diagnostiche I. C. A. Livello
22 532
Tabella 2. Definizione delle attività secondo il modello delle Prestazioni- Modificato
Prestazione infermieristica: ASSICURARE L'ALIMENTAZIONE E L'IDRATAZIONE.
Finalità Azioni
1.Indirizzare Illustrare alla persona i fattori che possono influenzare l'alimentazione e l'idratazioneOmissis
2.Guidare Istruire la persona sulle modalità per mantenere l'alimentazione e l'idratazioneOmissis
3. Sostenere Favorire l'assunzione di alimenti e/o bevande (dieta libera)OmissisFavorire l'assunzione degli alimenti e delle bevande consentiteOmissis
4. Compensare Alimentare e idratare per via enterale naturaleImboccareOffrire da bereAlimentare il neonato con biberon
5. Sostituire Alimentare e idratare per via enterale artificialeIntrodurre il sondino nasogastricoSomministrare alimenti e/o liquidi tramite sondino
Monitorare il bilancio idrico e l'apporto alimentareSomministrare alimenti e/o liquidi tramite stomiaRilevare lo spessore delle pliche cutaneeRilevare peso e/o altezzaMisurare le entrate per via enteraleMisurare le entrate per via parenterale BILANCIOMisurare le urine COMPLETOPesare le feciQuantificare il vomito

180
haematologica vol. 89[supplement
kind and sessionXXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
haematologica vol. 89[supplement n. 10]:October 2004
mente descritto e mostrato a titolo d'esempio nella tabel-la 2 , nelle tabelle di definizione delle undici prestazionecodificate per azioni, atti e finalità assistenziali.
Una volta assegnati tutti i livelli per tutte e undici leprestazioni si passa al calcolo della classe di gravità cheviene calcolato attraverso la sommatoria di tutte le rile-vazioni quotidiane come illustrato nella tabella 3.
Come si può notare la determinazione della classe digravità si consegue attraverso la sommatoria di tutti eundici i livelli rilevati ( contrassegnati in grassetto e sot-tolineati nella colonna del giorno 10/10/2004 ) che nel-l'esempio della tabella n. 3 è appunto pari a 32.
Oltre a questa rilevazione è possibile rilevare anche laspecifica attività assicurata apponendola nella casella afianco al livello . Ad esempio sempre nella tabella n. 3 nel-la prestazione n. 2 Assicurare l'alimentazione e l'idrata-zione oltre al livello pari a 5 è stato anche assegnata l'a-zione ovvero l'attività da garantire come descritto nellatabella 2 : 5. 2 Monitorare il bilancio idrico e l'apporto ali-mentare
Questa operazione può sembrare complessa ma è piùcomplesso scriverla che farla perché proprio grazie ai lavo-ri di gruppo e alla condivisione della constestualizzazio-ne delle attività erogate questo aspetto diviene subitochiaro e immediato in quanto i professionisti compilanola scheda facendo riferimento all'interiorizzazione deicontenuti piuttosto che alla continua consultazione del-la tabella delle attività definite (solo in casi poco frequenti
si ricorre alla consultazione diretta) ecco perché la fase dicondivisione e di contestualizzazione è fondamentale aifini dell'applicabilità e dell'attendibilità del metodo.
Come abbiamo già visto il giorno 10/10/2004 il signorBianchi ha un I. C. A. di 32 questo valore ci consente diclassificare il malato per classi di gravità. Attraverso latabella 4 nel nostro esempio L'I. C. A. di 32 equivale ad unaclasse di gravità tre ovvero a una classe di gravità mode-rata.
Con tale sistema si possono raggruppare tutti i malatiper classi di gravità anche utilizzando terminologie diffe-renti da quella proposta l'obiettivo è rendere questo siste-ma significativo al gruppo professionale.
Questi dati possono essere rappresentati anche grafi-camente attraverso istogrammi e/o aerogrammi comerappresentato nelle tabelle n. 5 e n. 6.
Queste rappresentazioni grafiche dimostrano in modoevidente la possibilità di creare sistemi sintetici di verifi-ca sull'efficacia delle cure infermieristiche .
Il grafico n. 6 consente di determinare i mix di compe-tenza maggiormente ricorrenti all'interno della strutturaorganizzativa osservata o all'interno di un specifico cam-pione di malati osservati e quindi molto utile per la deter-minazione della classe modale delle attività, informazio-ne molto importante per la determinazione delle compe-tenze specifiche della struttura e di conseguenza anche unutile elemento per la determinazione delle competenzedegli stessi professionisti. .
Determinare le competenze necessarieIl metodo adottato assicura la registrazione puntuale
delle attività pianificate e di quelle realizzate, assegnan-do ad ognuna un peso di complessità, ed ovviamenteanche un livello di competenza. Da queste informazioni èpossibile dedurre le competenze necessarie (che cosaoccorre saper fare), le conoscenze da possedere, le neces-sità strumentali, i tempi e i costi.
Ovviamente la base -dati che si viene a comporre conuna rilevazione di un numero sufficiente di frequenze(significativo per il campione osservato) consentirà dideterminare le attività prevalenti erogate in una determi-nata struttura e quindi di conseguenza si potranno intra-prendere sistemi di rilevazione permanenti orientati allecompetenze e alla valutazione del potenziale dei profes-
Tabella n. 4 Classe di gravità.
Classi DESCRIZIONE Intervallo Numero Malati
1 Complessità relativa assente 14 - 212 Complessità lieve 22 - 293 Complessità moderata 30 - 374 Complessità grave 38 - 455 Complessità critica 46 - 53
Tabella n. 5 Grafico Indice di complessità assistenziale per malato
Tabella n. 6 grafico livelli assegnati per singola prestazione

sionisti. Con questo sistema si può intraprendere un progetto di
assegnazione del personale basato non più solo sul ruoloma bensì un sistema che renda prioritaria la competenzarispetto al ruolo.
Il mix di competenze rilevato sui malati sarà il riferi-mento (il criterio/standard) sul quale si potrà realizzareuna ulteriore base dati rappresentata dalle competenze inpossesso dei professionisti (indicatori)
Strumenti di integrazione organizzativaLa metodologia del calcolo dell'indice di complessità fa
largo uso di strumenti di integrazione organizzativa perfavorire la definizione delle funzioni, le modalità operati-ve di esecuzione, le competenze, le responsabilità, ladeterminazione degli strumenti di verifica.
Con queste metodologie e strumenti il gruppo interdi-sciplinare, in piena autonomia, determina preventiva-mente la “standardizzazione delle attività”, che come èben noto è il più efficace sistema di coordinamento tra iprofessionisti sia per quanto riguarda le scelte operative,sia per quanto riguarda l'applicazione delle tecniche.
Sono da ritenersi strumenti indispensabili per lo stessofine anche la “matrice di processo” e la riunione. La primaè utilissima per facilitare l'integrazione con le altre pro-fessioni la seconda , se impiegata in modo corretto, è unfantastico strumento di comunicazione e condivisione del-le informazioni e soprattutto delle decisioni assunte.
Comparazione tra struttureQuesta dimensione del sistema consente di realizzare
una metodologia di calcolo comparativo tra strutture oaree di attività aggregabili.
Partendo dalla base dati raccolta per il calcolo dell'in-dice di complessità assistenziale per singolo malato comerappresentato nel paragrafo tre e rappresentato nell'e-sempio della tabella n. 3.
Si procede alla distribuzione dei dati dei singoli malatiraccolti attraverso la scheda rappresentata in tabella n. 7.
La compilazione di questa scheda viene realizzatamediante il raggruppamento di tutti i dati dei malati.
La tabella così strutturata è in grado di dirci come imalati di quella struttura (reparto, dipartimento, ospeda-le, gruppo di assistiti domiciliare ecc…) si distribuisconoall'interno delle variabili di prestazione e livello di gravità.
Il risultato che si ottiene è del tutto simile a quellomostrato in tabella n. 7 che d'ora in poi utilizzeremo comeesempio per esemplificare lo sviluppo della metodologia.
Riprendendo il nostro esempio analizziamo ora l'ultimariga della tabella. Si noti l'ultimo dato in basso a destra:il numero contenuto (1244) è l'I. C. A. di struttura che vie-ne calcolato attraverso le operazioni: di sommatoria e diprodotto. Prendiamo ad esempio la colonna del livello cin-que, sommiamo i valori contenuti 4+12+12+12+12+22+10+28+15 la loro somma è pari a 127 che a questopunto viene moltiplicata per il livello stesso di gravità cheè cinque conseguendo il risultato di 635 come rappresen-tato nell'ultimo rigo della colonna del livello cinque. Tut-ti gli altri livelli vengono calcolati allo stesso modo Lasomma di questi valori così calcolati ci consente di deter-minare l'I. C. A. di struttura che è pari a 1244 come indi-cato nell'ultimo rigo in basso a destra.
Ora procederemo al calcolo per la comparazione tra piùstrutture: si osservi la tabella n. 8 la prima colonna con-tiene il numero di strutture analizzate e quindi i relativi I.C. A. (colonna due ) che sono stati calcolati attraverso la
haematologica vol. 89[supplement
XXXI Con-gressoNazionale
Programma Infermieristico
haematologica vol. 89[supplement n. 10]:October 2004
Tabella n. 7 griglia di rilevazione dell'indice di complessità assistenziale per struttura
PRESTAZIONE 1 2 3 4 5
ASSICURARE LA RESPIRAZIONE 6 5 7 10 4ASSICURARE L'ALIMETAZIONE EIDRATAZIONE - 10 - 10 12ASSICURARE L'ELIMINAZIONE URINARIA E INTESTINALE - - 10 10 12ASSICURARE L'IGIENE - 5 10 5 12ASSICURARE IL MOVIMENTO - - 10 10 12ASSICURARE IL RIPOSO E IL SONNO 17 10 5ASSICURARE LA FUNZIONE CARDIOCIRCOLATORIA 5 5 10 12 -ASSICURARE UN AMBIENTE SICURO 10 22ASSICURARE L'INTERAZIONE NELLA COMUNICAZIONE 5 5 2 10 10APPLICARE LE PROCEDURE TERAPEUTICHE - 4 28ESEGUIRE LE PROCEDURE DIAGNOSTICHE 17 15
SOMMATORIA ( N. MAL * IND. DI COMPL. ) 33 134 174 268 635 1244
Tabella 8. Calcolo della media ponderata.
Strutture A) B) C) D)INDICE DI COMPLESSITA' N. MALATI IND. COMPL. PONDERATO MEDIA PONDERATA di C)
Struttura 1 1230 30 41 38Struttura 2 1244 32 38,875Struttura 3 1195 35 34,142

modalità appena descritta. La struttura due è quella delnostro esempio che abbiamo appena finito di calcolare. Lealtre due strutture sono state calcolate con la stessamodalità.
Il calcolo dell'indice di complessità ponderato ci con-sente di trasformare l'ICA in un valore comparabile attra-verso una semplice operazione che consiste nel dividerel'ica calcolato per il numero dei malati sottoposti a rile-vazione. Questa ponderazione è necessaria in quanto èovviamente comune trovare un numero variabile di mala-ti in strutture differenti (per numero di malati non si inten-de il posto letto ma bensì i ricoverati ) per un periodo rela-tivo (si tenga conto che questo calcolo che state osser-vando può essere fatto per qualsiasi intervallo di tempo ungiorno un mese un anno )
Successivamente, nella colonna cinque viene calcolataanche la media ponderata, elemento che ci consente dideterminare lo standard di complessità/gravità del cam-pione osservato.
Proseguiamo nel nostro esempio e calcoliamo lo scartocome rappresentato dalla tabella n. 9 che è appunto larealizzazione di quanto già affermato in precedenza ovve-ro il calcolo del delta delle strutture dallo standard rap-presentato dalla media ponderata dell'ICA che nel nostrocaso corrisponde a 38 e al loro posizionamento sotto lozero o sopra lo zero.
Le riflessioni su questi risultati possono essere molteplici
la più importante è sicuramente rappresentata dalla pos-sibilità di misurare l'assistenza infermieristica con siste-mi obiettivi e riproducibile in una qualsiasi struttura.
Questa metodologia aiuta ad orientare il sistema digestione della risorsa verso la competenza e non solo ver-so il ruolo. Spostare la gestione verso la competenza, ver-so il “come fare” completa il riduttivo attuale approcciolimitato solo all'aspetto quantitativo del “quanto fare”.
Il potere di misurare è sinonimo di qualità è sinonimodi miglioramento continuo una base dati di questo tipopuò consentire di intraprendere studi di analisi delle atti-vità erogate sia dal punto di vista qualitativo e che dalpunto vista quantitativo .
ConclusioniAd oggi il metodo dell'indice di complessità assistenzia-
le (I. C. A. ) si pone non solo come un metodo di raccoltadati ma piuttosto come un sistema integrato di supportoorganizzativo in grado di favorire l'applicazione dei model-li professionali assistenziali e soprattutto la raccolta di datilegati alla qualità della prestazione infermieristica pergarantire la definizione e pianificazione delle attività, misu-rare la complessità, determinare le competenze e compa-rare le strutture. .
Chi scrive è convinto che oggi più che mai la professio-ne infermieristica ha bisogno di applicare metodi e stru-menti capaci di essere efficaci (orientati ai risultati) e allostesso tempo sufficientemente sintetici (orientati all'inte-grazione e all'efficienza), per essere facilmente realizzatianche in contesti dove la complessità organizzativa nonconsente di assegnare specifiche risorse ai nuovi progetti.
Alcune sperimentazioni del metodo fanno ben sperareper il futuro in particolare, si può dire che questa metodo-logia ha trovato dei notevoli punti di forza attraverso l'im-piego dell'informatica in quanto il suo impiego velocizzatutti i passaggi e quindi garantisce automatismi su tutto ilprocesso di calcolo attraverso un unico inserimento di dati.La base dati che si crea ci da la possibilità di realizzare mol-ta ricerca.
Un altro punto di forza verificato è la maggiore coesio-ne del gruppo, una maggiore presa di coscienza del proprioruolo e l'impiego di strumenti di integrazione organizzati-va e standardizzazione delle attività più consone ai pro-fessionisti.
Altra tendenza ormai chiara è la possibilità di staccarsicompletamente da un unico modello professionale (neltesto presentato si adotta unicamente il “Modello dellePrestazioni”) e di potere applicare questa metodologiaanche con altre strutture concettuali. In merito a questopunto è in atto un'ipotesi di lavoro rispetto all'impiego deltariffario nomenclatore come “core” delle attività. Si diceche ogni ricerca porta sempre a nuove strade e anche que-sta volta è accaduto, l'ICA fa intravedere, attraverso la suaapplicazione, la possibilità di realizzare successivi step disviluppo del metodo che ci consentiranno di realizzare deilavori applicabili all'attuale sistema di classificazione e tarif-fazione delle prestazioni sanitarie. Ad oggi i sistemi impie-gati infatti non prevedono il riconoscimento di attività auto-noma dell'infermiere ma considerano solo l'attività infer-mieristica come attività delegata dal medico e calcolatasolo in termini di ore lavoro (logica quantitativa).
I nuovi sviluppi di questa metodologia si stanno orien-
182
haematologica vol. 89[supplement
kind and sessionXXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
haematologica vol. 89[supplement n. 10]:October 2004
Tabella 9. Calcolo dello scarto della media ponderata dell'Indice dicomplessità assistenziale.
REPARTI A) B) C)INDICE DI COMPLESSITA' PONDERATO MEDIA PONDERATA SCARTO
REPARTO 1 44 38 6REPARTO 2 38,875 38 0,875REPARTO 3 34,142 38 -3,858
Tabella n 10.Grafico del calcolo dello scarto dell'ICA di struttura

tando verso questa strada con la convinzione che soloattraverso la sperimentazione sistematica si potranno con-fermare le ipotesi appena descritte.
Per una più completa analisi di tale metodologia sirimanda alla bibliografia e alle pubblicazioni di più artico-li sull'argomento.
Bibliografia
Marisa Cantarelli, “Il Modello delle Prestazioni Infermieristiche”, Masson,Milano, 1998
Monica Casati, “La documentazione infermieristica”, Mc Graw Hill,Milano, 1999
Snaidero, Cavaliere “Il lavoro per turni dell'infermiere “ , Carocci Faber ,Roma, 2003
Focarile F. ”indicatori di qualità nell'assistenza sanitaria”Centro Scien-tifico Editore,Torino-1998
Cavaliere Bruno, Snaidero Diego, “Metodologia per la rilevazione dellacomplessità assistenziale infermieristica: calcolo dell'indice dicomplessità assistenziale”, Management Infermieristico, n°1, 1999,pp 32-36
Bruno Cavaliere, Maurizio Susmel, “La qualità nell'assistenza infer-mieristica: uno strumento di rilevazione e di elaborazione del-l'Indice di Complessità Assistenziale (I. C. A. )”, Nursing Oggi, n°2,2001
Antonella guerra, Bruno Cavaliere, Applicazione della metodologia del-l'indice di complessità assistenziale (ICA) presso l'ospedale“Pasquinucci”, Massa, A. O. IFC CNR - CREAS, Pisa” ManagementInfermieristico, Milano , 2003 numero 1 pag. 4 -11
Cavaliere Bruno, “Il sistema informativo dell'infermiere dirigente”, L'in-fermiere dirigente, n° 2, 1995
Cavaliere Bruno, “Il Patrimonio informativo come elemento determi-nante del processo decisionale: le base dati”, L'infermiere diri-gente, 1° trimestre, 1997, pp. 54-56
Gli emoderivati, riflessioni sulla normativavigente
Antonio Errico Servizio Trapianti Midollo Osseo U. O. Ematologia, Azien-da Ospedaliera Careggi, Firenze
La trasfusione è un atto terapeutico che consiste nellasomministrazione di sangue intero o di suoi costituenti pervia endovenosa.
Le diverse persone e strutture coinvolte nella raccolta,conservazione, trasporto e utilizzo del prodotto trasfusio-nale sono regolamentate e tutelate attraverso la legisla-zione inerente alle trasfusioni.
Il panorama legislativo italiano, riguardante il problematrasfusionale, si presenta costellato da un notevole nume-ro di norme: dal 1990 ad oggi se ne possono contare alme-no 24 tra leggi (2), decreti (20), ordinanze (1), e circolari (1)emanate per garantire e tutelare non solo la persona rice-vente la trasfusione ma anche il donatore.
La disciplina delle trasfusioni di sangue è tuttoggi rego-lamentata dalla Legge quadro n° 107 del 4 Maggio 1990(G. U. n° 108 dell'11 Maggio 1990) ”Disciplina per le atti-vità trasfusionali relative al sangue umano ed ai suoi com-ponenti e per la produzione di plasmaderivati. ”
Tale legge, malgrado tentativi di aggiornamento e revi-sione attraverso la Legge 28 gennaio 1994 n. º 63 e unaserie di decreti ministeriali, rimane il testo principale anche
se non completamente esaustivo ( soprattutto per la disci-plina delle attività di somministrazione della trasfusione) edè probabilmente oggi inadeguato rispetto ai cambiamentiavvenuti negli ultimi anni in particolar modo in relazionealle responsabilità del personale sanitario (abrogazione delmansionario, disposizioni in materia di professioni sanita-rie…).
Sarebbe auspicabile, tenendo conto delle ultime leggiemanate, una nuova legge che raccogliesse e aggiornassetutte le precedenti fino ad ora pubblicate ( progetti di leg-ge n°640, 660,1983 del 2001) ma i tentativi fino ad ora fat-ti non hanno però portato nessuna variazione.
Dovendo presentare la legislazione attuale possiamo ini-ziare evidenziando che la normativa (d. m. 26 gennaio2001)distingue il sangue e suoi prodotti in:
Sangue: unità di sangue intero omologo ed autologo. Emocomponenti: prodotti ricavati dal frazionamento del
sangue con mezzi fisici semplici o con aferesi. Farmaci plasmaderivati: farmaci estratti dal plasma
mediante processo di lavorazione industriale. Le strutture che svolgono attività trasfusionali, nello stes-
so decreto, sono distinte in:1.servizi di immunoematologia e trasfusione;2.centri trasfusionali;3. unità di raccolta. 4. A livello regionale ed interregionale sono altresì pre-
visti:a. centri di coordinamento e compensazioneb. centri ed aziende convenzionate per la produzione di
emoderivati.
A livello nazionale è inoltre prevista la Commissionenazionale per il servizio trasfusionale.
Per donazione di sangue e di emocomponenti, la legge107/90, intende l'offerta gratuita di sangue intero o plasma,o piastrine, o leucociti, previo il consenso informato e laverifica della idoneità fisica del donatore. Il donatore puòconsentire ad essere sottoposto indifferentemente ai diver-si tipi di donazione, sulla base delle esigenze trasfusionalied organizzative (art3).
Le competenze mediche e quelle infermieristiche relati-ve alla procedura della donazione sono descritte nel D. M.26 gennaio 2001: “Protocolli per l'accertamento della ido-neità del donatore di sangue e di emocomponenti. ” doveall'articolo n° 3 dice che il personale sanitario delle strut-ture trasfusionali e di raccolta e' tenuto:
a) a garantire che il colloquio con il candidato donatoresia effettuato nel rispetto della riservatezza;
b) a adottare tutte le misure volte a garantire la riserva-tezza delle informazioni riguardanti la salute fornite dalcandidato donatore e dei risultati dei test eseguiti sulledonazioni, nonché nelle procedure relative ad indaginiretrospettive, qualora si rendessero necessarie;
c) a garantire al donatore la possibilità di richiedere alpersonale medico della struttura trasfusionale o di raccol-ta di non utilizzare la propria donazione, tramite una pro-cedura riservata di autoesclusione;
d) a comunicare formalmente al donatore qualsiasi signi-ficativa alterazione clinica riscontrata durante la valuta-zione predonazione e/o negli esami di controllo.
Le altre uniche competenze infermieristiche sono citatenell'articolo 6 : “Il medico responsabile della selezione, o
haematologica vol. 89[supplement
XXXI Con-gressoNazionale
Programma Infermieristico
haematologica vol. 89[supplement n. 10]:October 2004

personale sanitario appositamente formato operante sottola responsabilità del predetto, effettua la compilazione delquestionario di cui all'allegato n. 2, parte A” (dati anagra-fici e domande di tipo anamnestico del questionario mini-steriale).
La legge 107/94 definisce che: “Il prelievo di sangue inte-ro è eseguito da un medico, o sotto la sua responsabilità edin sua presenza, da un infermiere professionale”. Tale com-ma potrebbe essere ampliamente discusso in quanto l'abo-lizione del mansionario e la capacità tecnica dell'infermie-re di eseguire un prelievo e di reperire un accesso venosoadeguato, non possono essere messe in dubbio dato chesono manovre quotidiane per la nostra professione. Rima-ne certamente di competenza medica la prescrizione di unsalasso, così come il prelievo di aferesi dove la raccolta diemocomponenti mediante separatori cellulari rende la pro-cedura non priva di rischi, tanto da dichiarare nell'art. 2 delD. M. 25/1/2001: “ Durante l'intera procedura il donatoredeve essere attentamente osservato e deve essere assicu-rata la disponibilità di un medico esperto in tutte le pro-blematiche dell'aferesi onde fornire assistenza adeguata einterventi d'urgenza in caso di complicazioni o di reazioniindesiderate. ”
Il Decreto del Presidente del Consiglio dei Ministri 1º set-tembre 2000 nell'acclusa “tabella A” riporta esempi di defi-nizione delle competenze del personale (vedi allegato).
Il decreto del 25 gennaio 2001 regola le modalità di rac-colta, di lavorazione e di conservazione del sangue interodegli emocomponenti nonché delle caratteristiche dei fri-goriferi e dei congelatori adibiti alla loro conservazione.
Non meno importanti sono le modalità per il trasportodelle sacche dal centro trasfusionale al reparto: il sangueintero e gli emocomponenti debbono essere trasportati incontenitori termoisolanti dotati di appositi sistemi di con-trollo della temperatura interna: quelli allo stato liquido aduna temperatura compresa tra + 1oC e + 10oC, quelli con-servati a 22oC (+ o - 2oC) a temperatura ambiente quan-do la temperatura esterna risulta compatibile con quella diriferimento…
I contenitori per il trasporto di unità di sangue debbonoessere preraffreddati a + 4oC; i contenitori utilizzati per iltrasporto di piastrine debbono essere mantenuti a tempe-ratura ambiente per almeno 30 minuti prima del loro impie-go. L'esame ispettivo delle sacche deve essere ripetuto dachi riceve i preparati inviati, unitamente alla verifica deidispositivi di controllo della temperatura interna dei con-tenitori.
Il decreto del 26 gennaio 2001 impone che presso ognistruttura trasfusionale deve essere predisposto un sistemadi registrazione e di archiviazione dati che consenta di rico-struire il percorso di ogni unità di sangue o emocompo-nenti, dal momento del prelievo fino alla sua destinazionefinale. (art. n°15) ; e che la documentazione relativa al per-corso di ogni unità di sangue o la determinazione del grup-po o altro ancora, abbiano una conservazione che varia, inbase al documento, da dodici mesi a tempo illimitato.
La richiesta di sangue e/o emocomponenti, secondo ildecreto del 25 gennaio 2001, contenente le generalità delpaziente e l'indicazione alla trasfusione, deve essere firma-ta dal medico su apposito modulo fornito dalla strutturatrasfusionale o su propria carta intestata o su quella dellastruttura di degenza del ricevente. La richiesta deve essere
accompagnata da un campione di sangue del ricevente diquantità non inferiore a 5 ml; per pazienti pediatrici pos-sono essere accettati volumi inferiori. Il campione deveessere raccolto in provetta sterile entro 72 ore precedentila trasfusione, contrassegnato in modo da consentire l'i-dentità del soggetto cui appartiene e firmato dal responsa-bile del prelievo. Il D. M. in questione annulla il decreto del27 dicembre 1990 che obbligava la firma del campione disangue “ dal medico che ha la responsabilità del prelievo” .
L'informazione e l'acquisizione del consenso informatodel paziente sono regolamentati dal decreto ministeriale del1 settembre 1995 “Costituzione dei comitati per il buonuso del sangue”.
In questo decreto si stabilisce che ai pazienti deve esse-re comunicata la possibilità di effettuare, quando indicata,l'autotrasfusione e deve essere richiesto il consenso infor-mato alla trasfusione di sangue ed emocomponenti ed allasomministrazione di emoderivati”
Nell'art. 4 viene inoltre detto: “Quando vi sia un perico-lo imminente di vita, il medico può procedere a trasfusio-ne di sangue anche senza consenso del paziente. Devonoessere indicate nella cartella clinica, in modo particolareg-giato, le condizioni che determinano tale stato di necessità.
Nei casi che comportano trattamenti trasfusionali ripe-tuti, il consenso si presume formulato per tutta la duratadella terapia, salvo esplicita revoca da parte del paziente. ”
Per le unità non utilizzate il decreto del 25 gennaio 2001si esprime chiaramente: “Qualora l'unita' di sangue o diemocomponente richiesta non venga utilizzata, il richie-dente deve provvedere alla restituzione della stessa allastruttura trasfusionale fornitrice nel più breve tempo pos-sibile. L'unita' restituita deve essere accompagnata da unadocumentazione attestante la sua integrità e l'osservanzadei protocolli stabiliti dal responsabile della struttura tra-sfusionale relativamente alla sua conservazione e traspor-to.”
Dell'esecuzione della trasfusione, considerata da sempredi pertinenza medica, nessuna legge dal 1990 in poi neparla in maniera esplicita. Motivo di questo si deve al D. P.R. 24 agosto 1971, n°1256 “Regolamento per l'esecuzionedella legge 14 luglio1967, n°592, concernente la raccolta,conservazione e distribuzione del sangue umano” dove l'art.91 dice che: “la trasfusione del sangue e degli emoderiva-ti deve essere eseguita sotto il costante controllo del medi-co. ”
Nelle Linee-Guida per la Preparazione, l'Uso e la Garan-zia di Qualità degli Emocomponenti (Capitolo 28 Terapiatrasfusionale ) edita dal Consiglio d'Europa nel 1995, sidichiara inoltre che: “Il medico che effettua una trasfusio-ne a un paziente è responsabile del controllo di identità edi tutte le altre misure di sicurezza. ”
Solamente le linee guida redatte dalla CommissioneNazionale per le trasfusioni ed emanate dal Ministero del-la Sanità nel 1993, in applicazione dell'art. 12 della legge107/1990 affermano che:
“La trasfusione é un atto medico e va pertanto prescrit-ta ed effettuata dal medico, che é responsabile dei seguen-ti atti: - accertamento dell'indicazione; - valutazione per l'autotrasfusione; - ottenimento del consenso informato; - richiesta di sangue;
184
haematologica vol. 89[supplement n. 10]:October 2004
kind and sessionXXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004

- verifica e sottoscrizione della corretta compilazione deidati anagrafici del paziente sulla richiesta e sui campio-ni di sangue;
- accertamento della compatibilità teorica di gruppo AB0e tipo Rh, tra l'unità da trasfondere e il ricevente;
- ispezione dell'unità prima della trasfusione; - registrazione dell'ora di inizio della trasfusione e del
numero di carico dell'unità; - trasfusione di sangue (sorveglianza del paziente e valu-
tazione di efficacia); - segnalazione di eventuali complicanze della trasfusione.
Inoltre il medico é corresponsabile con il personale infer-mieristico dei seguenti atti: - identificazione del paziente al momento dei prelievi di
sangue e della trasfusione; - verifica dell'identità tra il paziente che deve ricevere la
trasfusione ed il nominativo del ricevente riportato sul-l'unità;
- registrazione dei dati.
Il personale infermieristico di reparto é responsabile deiseguenti atti: - compilazione della parte anagrafica della richiesta di
sangue o di gruppo sanguigno; - esecuzione dei prelievi di sangue e compilazione delle
relative etichette; - invio della richiesta e dei campioni di sangue al servi-
zio trasfusionale; - gestione in reparto delle unità consegnate sino al
momento della trasfusione; - registrazione dell'ora in cui termina la trasfusione ed
eliminazione del contenitore; - invio al servizio trasfusionale di una copia del modulo
di assegnazione e trasfusione; - invio al servizio trasfusionale delle segnalazioni di rea-
zione trasfusionale e dei materiali necessari alle inda-gini conseguenti.
Pertanto, al fine di garantire la sicurezza trasfusionale ela tutela del proprio operato, nell'attesa di normative mag-giormente precise, diventa indispensabile fare riferimentoalle procedure operative obbligatorie previste dalla strut-tura trasfusionale dell'Azienda Ospedaliera o Sanitaria d'ap-partenenza.
Allegato:Tabella A: Esempi di definizione delle compe-tenze del personale (D. P. C. M. 1/9/2000)
Personale medico Ha le responsabilità dellaqualifica e relative competenze professionali con partico-lare riferimento alle seguenti attività: attività di raccolta,validazione e qualificazione biologica delle donazioni, pro-duzione di emocomponenti e assegnazione degli stessi,laboratorio di immunoematologia (e altre attività di labo-ratorio se previste), medicina trasfusionale, direzione ecoordinamento.
Attività di diagnosi e cura dei pazienti ambulatoriali e inregime di day-hospital
Personale laureato non medico Ha la responsabilitàdella qualifica e relative competenze professionali con par-ticolare riferimento alle seguenti attività: validazione e
qualificazione biologica delle donazioni, produzione diemocomponenti, laboratorio di immunoematologia, altreattività di laboratorio se previste.
Particolare rilievo assume la figura del biologo per l'at-tuazione dei programmi di controllo di qualità delle proce-dure di laboratorio.
Personale Tecnico di Laboratorio Ha le responsabilitàdella qualifica e relative competenze professionali con par-ticolare riferimento alle seguenti attività: supporto alle atti-vità di raccolta, validazione e qualificazione biologica delsangue raccolto, produzione di emocomponenti, assegna-zione e distribuzione, laboratorio di immunoematologia,altre attività di laboratorio se previste.
Attività di supporto ,amministrativo, per quanto di com-petenza, con particolare riferimento alla tenuta e compi-lazione dei registri di legge ed alla informatizzazione.
Esecuzione tecnica delle procedure, gestione/manuten-zione ordinaria delle apparecchiature
Personale Infermieristico (Caposala, Infermiere profes-sionale, Assistente sanitaria)Ha le responsabilità dellaqualifica e relative competenze professionali.
Svolge le funzioni infermieristiche inerenti la raccolta disangue ed emocomponenti, l'aferesi terapeutica, le vacci-nazioni necessarie ai donatori e politrasfusi (es. anti-epa-tite B), l'attività assistenziale in day-hospital, se previsto.
Nell'ambito del day hospital si rende inoltre garante del-l'igiene ambientale, dell'attivazione e gestione delle proce-dure di ammissione e dimissione degli utenti, della verifi-ca periodica della qualità dell'assistenza e del grado di sod-disfazione dei bisogni dell'utenza.
Collabora con le Associazioni e Federazioni di volonta-riato per l'organizzazione di campagne di propaganda, pre-venzione ed educazione alla salute nei confronti dei dona-tori e pazienti.
Effettua le rilevazioni statistiche necessarie, ivi compre-se quelle inerenti il registro nazionale sangue.
Svolge altresì funzioni di carattere organizzativo e le atti-vità amministrative legate all'informatizzazione di specifi-ca competenza infermieristica.
Personale ausiliario Ausiliario socio sanitario specializ-zato, Operatore tecnico addetto all'assistenza Ef fe t tuaquanto previsto dal profilo dell'ausiliario socio-sanitariospecializzato, con particolare attenzione all'igiene ambien-tale e alla gestione delle scorte di materiali.
Personale amministrativo L'assistente amministrativoeffettua tutte le attività legate alla propria figura profes-sionale comprese: le attività amministrative conseguentialla corretta valorizzazione delle prestazioni della struttu-ra trasfusionale nonchè ai corretti rapporti con le industrieconvenzionate addette alla lavorazione degli emoderivati equelle relative alla informatizzazione dati e alla gestionemagazzino scorte materiali e reagenti.
AutistaHa le responsabilità della qualifica e relative com-petenze professionali con particolare riferimento alleseguenti attività: raccolta mobile.
Trasporto del sangue, degli emocomponenti a scopo tra-sfusionale e dei campioni biologici, sia in condizioni ordi-narie che in situazioni di urgenza.
Bibliografia
Anceschi S. “Il parere dell'avvocato”QUESITO DEL 27/01/03 “Trasfusioni disangue ed emoderivati a domicilio” risposta del Legale del Collegio
haematologica vol. 89[supplement n. 10]:October 2004
XXXI Con-gressoNazionale
Programma Infermieristico

186
haematologica vol. 89[supplement
kind and session
IPASVI di R. E. Sito web IPASVI Reggio Emilia. Benci L. “Competenze e responsabilità nel servizio trasfusionale”in: Obbi-
ettivo n°3 e 4 del 2002, rivista IPASVI Firenze. Bottega A. “Emotrasfusioni: novità legislative” in: Infermieri Informati,
Anno II n. 2, maggio-agosto rivista IPASVI Vicenza. Legge n°107 del 4 Maggio 1990 “Disciplina per le attività trasfusionali rel-
ative al sangue umano ed ai suoi componenti e per la produzionedi plasmaderivati “
Decreto 26 gennaio 2001 “Protocolli per l'accertamento della idoneità deldonatore di sangue e di emocomponenti. ”
Decreto 25 gennaio 2001 “Caratteristiche e modalità per la donazione disangue e di emocomponenti. ”
Decreto 1 settembre 2000 “Atto di indirizzo e coordinamento in materiadi requisiti strutturali, tecnologici ed organizzativi minimi per l'e-sercizio delle attività sanitarie relative alla medicina trasfusionale.”
Decreto 5 novembre 1996 “Integrazione al decreto ministeriale 1° set-tembre 1995 concernente la costituzione e compiti dei comitati peril buon uso del sangue presso i presidi ospedalieri. ”
Decreto 5 novembre 1996 “Indicazioni per l'istituzione del registro delsangue e del plasma in ciascuna regione e provincia autonoma. ”
Decreto 1 settembre 1995 “Linee guida per lo svolgimento di attività miratedi informazione e promozione della donazione di sangue nelleregioni che non hanno conseguito l'autosufficienza. ”
Decreto 1 settembre 1995 “Disciplina dei rapporti tra strutture pubblicheprovviste di servizi trasfusionali e quelle pubbliche e private, accred-itate e non accreditate, dotate di frigoemoteche. ”
Decreto 1 settembre 1995 “Schema-tipo di convenzione tra le regioni e leimprese produttrici di dispositivi emodiagnostici per la cessione disangue umano od emocomponenti. ”
Legge 28 gennaio 1994 “Conversione in legge, con modificazioni, deldecreto legge 29 novembre 1993, n° 480, recante la modifica del-l'articolo 10, comma 3, della legge 4 maggio 1990, n° 107, concer-nente disciplina per le attività trafusionali relative al sangue umanoed ai suoi componenti e per la produzione di plasmaderivati. ”
Decreto 29 novembre 1993 “Modifica dell'articolo 10, comma 3, dellalegge 4 maggio 1990, n° 107, concernente disciplina per le attivitàtrasfusionali relative al sangue umano ed ai suoi componenti e perla produzione di plasmaderivati. ”
Decreto 22 novembre 1993 “Aggiornamento del prezzo unitario di cessionedelle unità di sangue tra servizi sanitari, uniforme per tutto il ter-ritorio nazionale. ”
Decreto 27 settembre 1993 “Modifica dell' articolo 10, comma 3, dellalegge 4 maggio 1990, n° 107, concernente la disciplina per le attiv-ità trasfusionali relative al sangue umano ed ai suoi componenti eper la produzione di plasmaderivati. ”
Decreto 12 febbraio 1993 “Individuazione dei centri di produzione diemoderivati autorizzati alla stipulazione di convenzioni con i cen-tri regionali di coordinamento e compensazione per la lavorazionedi plasma nazionale raccolto in Italia. ”
Legge 25 febbraio 1992 “Indennizzo a favore dei soggetti danneggiati dacomplicanze di tipo irreversibile a causa di vaccinazioni obbligato-rie, trasfusioni e somministrazione di emoderivati. ”
Decreto 27 gennaio 1992 “Trasferimento dei centri trasfusionali dellaCroce Rossa italiana, ivi compreso il Centro Nazionale trasfusionesangue, alle strutture sanitarie indicate dalla regione competente. ”
Decreto 18 settembre 1991 “Determinazione dello schema tipo di con-venzione fra regioni e associazioni e federazioni di donatori volon-tari di sangue. ”
Decreto 18 settembre 1991 “Determinazione del prezzo unitario di cessionedelle unità di sangue tra servizi sanitari, uniforme per tutto il ter-ritorio nazionale. ”
Decreto 18 giugno 1991 “Indicazioni per l'istituzione del registro delsangue in ciascuna regione e provincia autonoma. ”
Decreto 15 gennaio 1991 “Protocolli per l'accertamento della idoneità deldonatore di sangue ed emoderivati. ”
Decreto 27 dicembre 1990 “Caratteristiche e modalità per la donazione delsangue ed emoderivati. ”
Presentazione dati ricerca sulla mucositeorale
A. F. D. L. VaglianoCentro Trapianti Ospedale Regina Margherita Torino
La mucosite orale rappresenta uno degli effetti collate-rali maggiormente ricorrenti, temporaneamente moltoinvalidanti e in grado di peggiorare in modo significativola qualità di vita del bambino sottoposto a chemioterapiaantiblastica , a radioterapia e/o a trapianto di cellule sta-minali emopoietiche.
Il termine mucosite orale è comunemente utilizzato perindicare l'insieme di sintomi (dolore, intolleranza a cibicaldi e freddi, acidi o piccanti ) e di segni , quali eritema,xerostomia ed ulcerazioni della mucosa.
L'insorgenza della mucosite orale è causata da diversifattori di rischio, individuali o conseguenti il trattamentoantiblastico tra cui è possibile riconoscere alcune patolo-gie orali e dei denti ( quali carie, granulomi, ecc. ) , unainadeguata igiene della cavità orale, l'età e la rispostaindividuale del paziente al trattamento, lo stato nutrizio-nale nonché la durata del trattamento antineoplastico,l'agente chemioterapico utilizzato o il campo di irradia-zione , le dosi di esposizione ed il tipo di ionizzante uti-lizzato.
L'implicazione infermieristica nella prevenzione e neltrattamento della mucosite orale è fondamentale , soprat-tutto per ciò che riguarda l'educazione all'igiene e il con-trollo dello stato delle mucose del cavo orale.
All'interno del G. I. T. M. O. (Gruppo Italiano Trapiantodi Midollo Osseo - Sez. Infermieristica ) è in corso , da set-tembre 2002, una raccolta dati sull'incidenza e la gravitàdella mucosite orale nel paziente sottoposto a trapiantodi cellule staminali emopoietiche : Lo strumento utilizza-to per la raccolta dati è la scala WHO . Oggetto della rela-zione è la presentazione dei dati, tratti dal database G. I.T. M. O. , relativi ai Centri Trapianto Pediatrici che hannoaderito allo studio.
Utilizzo della linfofotoaferesi in un caso diGvH-D
A. M. Lo Sapio, R. Pinto, A. Balboni,G. Sorrentino,L.Corvino, A. Orza,A Ruggiero, E. Scuotto. Azienda Ospedaliera Pediatrica Santobono-Pausillipon -Napoli
La GvH-D (Graft versus host disease) è una delle mag-giori complicanze che può insorgere nei pazienti sottopo-sti a Trapianto midollo osseo allogenico (T M O ),ed èresponsabile di un alto indice di morbilità e mortalità .
La GvH-D si manifesta quando le cellule immunocom-petenti del donatore riconoscono le differenze antigeni-che nei tessuti del ricevente. Le cellule coinvolte in que-sto processo sono soprattutto i T linfociti . Bisogna fareuna distinzione fra GvH-D acuta e cronica ,la differenza
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
haematologica vol. 89[supplement n. 10]:October 2004

viene fatta dal tempo della sua comparsa e dagli interes-samenti degli organi colpiti e cioè cute, intestino,fegato.
La GvH-D insorta nei primi 100 giorni, è definita acuta,quella che insorge in maniera primaria dopo i 100 giornio in continuità con una acuta, è definita cronica.
Generalmente non vengono interessati tutti e tre gliorgani, ed a seconda del livello di gravità delle manife-stazione viene classificata in quattro stadi.
Spesso è accompagnata dal sopraggiungere di graviinfezioni, blocco del midollo e una seria compromissionedel benessere generale.
Il trattamento terapeutico prevede sostanze citotossi-che, immunosoppressori, siero antilinfocitario, anticorpimonoclonali e da ultimo la linfofotoaferesi.
Caso clinico. Un bambino di anni otto sottoposto a TMOallogenico da sorella HLA compatibile per una LMA (M3)in 2° RC , dopo un rapido attecchimento dei WBC (G+9)ha sviluppato una GVH di quarto grado con interessa-mento della cute (> 75% sc) e dell'intestino (diarrea >2,5l)trattata prima con metilprednisolone (da 2 a 5 mg/Kg) esuccessivamente con Infliximab (10mg/Kg una dose perquattro settimane)
La GvH non solo non ha mostrato regressione , ma si èassistito ad un coinvolgimento epatico con bilirubina inrapida risalita che ha raggiunto i valori di 10 mg/dl.
Alla luce di questo nuovo evento si è iniziato un pro-gramma di:– linfofotoaferesi (2 sedute a settimana per quattro set-
timane) – l’infusione di Anti CD25(1 fl. 20 mg. ) ai giorni
+1,+2,+4,+7,+14,+21.
Il risultato è stato una riduzione del 50% della Gvh-dper cui si è deciso di programmare un secondo ciclo cheha conservato le stesse caratteristiche temporali e dosag-gi terapeutici ;ma in particolar modo è stato dato unaimportanza fondamentale alla linfofotoferesi terapeuti-ca. Gli effetti della fotoaferesi rilevanti per il trattamen-to della GvH-Dsono fondamentalmente tre:1. induzione dell'apoptosi di linfociti T2. stimolazione di una risposta immune anti-linfociti T3. stimolazione della secrezione di determinate citochi-
ne.
Il protocollo attualmente in uso in Italia prevede cicli dilinfofotoaferesi terapeutica ben definiti. Lo schema è ilseguente:
MESE FREQUENZA SEDUTE1°(ciclo di 8 sedute) 2 ogni 7 gg, in giorni
consecutivi2°, 3 (ciclo di 4 sedute) 2 ogni 14 gg, in giorni
consecutivi 4°,5°,6° (ciclo di 2 sedute) 2 ogni 28 gg, in giorni
consecutivi
ConclusioniIl risultato ottenuto con questo programma a 100 gior-
ni dal TMO è stato— scomparsa della GvH a livello cutaneo e dell'intestino
anche se le feci sono ancora semiliquide — un valore della bilirubina intorno a 5mg/dL
Noi riteniamo sicuramente utili l'utilizzo di schemi pro-tocollari ma, in taluni casi,l'esperienze sviluppata dai varicentri AIEOP può determinare variazioni utili al singolopaziente. Riteniamo inoltre di fondamentale importanzauna stretta collaborazione tra medici ed infermieri nelrilevare tutti i sintomi e i segnali utili ad identificare unacomplicanza e la strategia terapeutica necessaria a con-trastare l'avanzare della malattia.
Infermiere di ricerca clinica [CRN] - Funzionied interazioni con l'Infermiere di area clinica(due anni di esperienze e sviluppo)
Callegaro L. Inf-CRNHSR-TIGET - Unità di Ricerca Clinica Pediatrica, DirezioneScientifica Ospedale San Raffaele, Milano
IntroduzioneLa nascita di nuove figure infermieristiche operanti in
tutti gli ambiti della medicina, ricerca compresa, rappre-senta un evoluzione della professione in ambiti ancoranon esplorati in Italia. Tra le nuove figure, quella dell'in-fermiere di ricerca clinica sarà nel prossimo futuro unosbocco di carriera, per tale motivo lo sviluppo di un per-corso, completato da funzioni e dall'analisi delle intera-zioni tra le varie figure (Infermieristiche, mediche e non)rappresenta un primo passo verso questo futuro. L'ob-biettivo di questa presentazione è fornire una prima ana-lisi del percorso compiuto nel nostro centro e degli obbiet-tivi ancora da raggiungere in modo da dare una base didiscussione tra tutti gli infermieri che operano nell'ambi-to della ricerca clinica.
PercorsoDurante i principali momenti di uno studio (Apertura,
Screening, Baseline, Trattamento, Follow-up, Chiusura),l'infermiere di ricerca clinica (CRN) sviluppa funzioni di
haematologica vol. 89[supplement
XXXI Con-gressoNazionale
Programma Infermieristico
haematologica vol. 89[supplement n. 10]:October 2004

188
haematologica vol. 89[supplement
kind and session
formazione/Informazione nei confronti dei propri colleghidi area clinica, in rapporto al protocollo preso in carico;funzioni organizzative, gestisce le visite e gli esami previ-sti, predispone (in collaborazione con il data manager)agende personalizzate dei pazienti inseriti, recupera egestisce i dati richiesti dallo studio, verificandone la com-pletezza e l'accuratezza (se possibile), se necessario creaschede personalizzate e remainder; funzioni operative inrapporto ai campioni da raccogliere ed inviare ai labora-tori esterni (se richiesti) ed in relazione alla gestione(opzionale) e somministrazione del farmaco in sperimen-tazione, rilevazione dei parametri previsti dallo studio neivari momenti. Lo schema sottostante mostra il percorso intermini generali.
ConclusioniSe la formulazione del percorso è quasi totalmente sta-
ta creata sulla realtà del nostro centro e sui protocolliseguiti, le basi teoriche per la ricerca di funzioni ed inte-razioni sono secondarie ad un lavoro pubblicato nel 1998dal Royal College of Nursing* di Londra, da tale lavoroveniva estratta una tabella/percorso nella quale i CRNvengono suddivisi in base a titoli di studio ed esperienzelavorative ed ad ogni livello corrispondeva una retribu-zione consigliata.
*The Clinical Research Nurse in NHS Trusts and GP Prac-tices: Guidance for nurses and their employers ;The RoyalCollege of Nursing December 1998 - RCN re-order no001023
La formazione degli infermieri in Oncologia
Ivana CarpanelliOperatore Professionale Dirigente - Direzione Sanitaria I.S. T. , Genova
I requisiti professionali dell'infermiere specializzato inoncologia, messi a punto dal Comitato consultivo sullaformazione infermieristica della commissione europea,hanno guidato l'eons nella definizione di un curriculumessenziale per un corso post base di formazione in nursingoncologico.
Il curriculum è stato successivamente utilizzato per svi-luppare i programmi di formazione nei diversi stati mem-bri, tra cui l'Italia e aggiornato sulla base delle esperien-ze di formazione svolte e sulla base dei nuovi bisogni diassistenza infermieristica in oncologia sia nell'area geria-tria che in quella di assistenza all'adulto che in quellapediatrica.
Nel 1999 la Federazione dei Collegi ha costituito ungruppo di lavoro, composto da professionisti esperti nelsettore, ai quali ha affidato il compito di mettere a pun-to le linee guida per la definizione di un master in onco-logia e cure palliative.
Questo master rientra nella formazione specialistica inarea critica.
Il percorso proposto alle università ha lo scopo di for-nire al professionista competenze specifiche nel settoredisciplinare.
La metodologia utilizzata è orientata ad una formazio-ne che sia adattata ai reali bisogni della comunità.
Sono individuati tre campi di apprendimento: cogniti-vo, comunicativo-relazionale e gestuale.
L'infermiere con competenza certificata specifica è ingrado di fornire un'assistenza mirata alla soluzione deiproblemi di salute, alla necessità di aiuto alla persona assi-stita, alla collaborazione con l'equipe multiprofessionale,all'attività di ricerca in ambito specifico e multidiscipli-nare.
Il Gruppo come strumento di lavoro riflessivo
Dott. ssa Dorella ScarponiMedico Dirigente di Primo Livello Psichiatra, presso laSezione di Oncoematologia Pediatrica (Prof. G. Paolucci)dell'Azienda Policlinico S. Orsola, di Bologna
Lo spazio del gruppo è a buona ragione considerato ele-mento fondamentale per riflettere su come ognuno di noipartecipa alla professione che svolge.
Quando poi tale occupazione lavorativa prevede unaqualche relazione di aiuto, la partecipazione individuale ecollettiva al compito diventano aspetti complessi, amomenti dominanti da elementi emozionali, definiti, inambito istituzionale, “turbolenze emotive”.
L'assetto istituzionale cioè, in cui l'operatore è a con-tatto non solo con molti pazienti ma anche con molti altrioperatori, diventa l'ambiente che accoglie- o rifiuta- que-gli aspetti della professione non strettamente tecnici,semplicemente umani, che possono sfuggire alla com-prensione o addirittura alla consapevolezza del gruppoche lavora.
Il gruppo che riflette recupera la dimensione mentaledel proprio essere individuo dentro ad un team e facilitala comunicazione emotiva.
Gli elementi tipici della esperienza di gruppo: il respec-chiamento, la condivisione, la risonanza…possono diven-tare fattori con la dimensione di aiuto per chi aiuta, aven-do proprio, tra gli obiettivi principali, l'educazione alla “comunicazione”.
In questa ottica la comunicazione, indispensabile pas-saggio professionale all'ascolto del paziente non può tra-scendere ma solo comprendere l'ascolto di chi cura.
XXXI Congresso Nazionale AIEOP, Stresa, 10-12 ottobre 2004
haematologica vol. 89[supplement n. 10]:October 2004

haematologica vol. 89[supplement
XXXI Con-gressoNazionale
Programma Infermieristico
haematologica vol. 89[supplement n. 10]:October 2004

Accordi B 113Acquaviva A 132, 156, 167Acquila M 161Addeo R 163Agresta S 146Alaggio R 124, 129, 167Alba L 105Albiani R 136Alessi D 149Alice A 150Altobelli M 111Amato GM 142, 143Amato S 106Ambrosioni G 143Amendola A 114, 155Amendola G 104, 156Andolina M 134, 169André V 110, 119Andresciani A 146Andreuzzi F 146Angelini P 133Antonioli A 151Arcamone G 133Arcangeli A 113Argiolu F 134, 136Aricò M 111, 118, 129, 132, 139, 141, 143, 148, 157, 164, 166, 170Armiraglio M 173Arrighetti A 107, 122, 165Arrighini A 156Aru B 127, 133Asaftei S 135Aschero S 105Assirelli E 169Attinà G 127Avellino R 158Axia G 158Azzari C 142, 143
Bachiocco V 174Bader P 121Badolato R 101, 103Badulli C 141Baffelli R 136Baggio E 136Bajia M 103Balduzzi A 138, 139, 140, 168Ballabio G 132Ballarini M 121Ballarini V 147Barat V 135Barbagallo Ml 109Barberi W 114Barella S 106Barisone E 111, 116, 144, 159Baronci C 116, 156, 157Barr RD 122, 123Barzanò E 131Basso E 167Basso G 110, 111, 113, 114, 116, 117, 118, 119, 120, 121, 159, 161,
162, 163Basso ME 105, 126Belardinelli P 146Benetello A 114Benetti A 145Beretta C 120, 153, 162Berger M 135, 136Bergonzi S 151Bernasconi S 155Berni B 146Bernini G 133, 167Bernuzzi G 155Berta M 127, 165
Berta S 136Bertagnolio B 140, 168Bertin D 126Bertorello N 107Bertuna G 133Bettella A 130Bezzio S 144, 150Bianchi M 127, 133, 166Biasin E 135Biasotti S 127Bicciato S 121Biondi A 110, 111, 113, 114, 115, 117, 118, 119, 120, 121, 123, 139,
140, 145, 161, 162, 168Bisogni R 158Bisogno G 125, 130, 131, 167Bo M 107Boglino C 167Boglione A 127, 165Bolda F 145Bolognini S 174Bombace V 105, 133Bombardieri E 132Bonacchi L 151Bonamino M 117, 119Bonanno A 142Bonanomi S 138, 139, 140, 168Bonato P 159Bonato S 155Bonetti F 110, 133Bonomi V 103Bonutti A 131Borgogno M 126Bossi G 148Bottelli C 102, 149Bottino D 160Bovo G 153Bozzi F 131Brach Del Prever A 127, 165, 170Brach Del Prever E 127, 165Branciforte F 153Brugiolo A 138Bugarin C 111Buldini B 111, 114, 121, 155Burnelli R 164, 174
Caini M 132Caldani C 107Caldarelli M 127Caldiani C 101, 107, 122Calevo MG 125Calore E 137, 138Camera F 152, 153, 170, 171, 172Camerin C 124Camoriano R 152Campagnoli MF 102Campbell M 109Campo Dell'orto M 113Candusso M 142Cantoni C 149Cao A 136Capanna R 112, 133Cappellini MD 108Capria S 114Caprino D 116Carando A 102Cardarelli L 114Cardinale F 143Carli M 124, 125Carli ML 132Carli OM 130Caruso R 157, 171Casale F 111, 125, 162, 163
i
haematologica vol. 89[supplement n. 10]:October 2004
Indice degli Autori

Casanova M 128, 130, 151, 166, 173Casazza G 174Caselli D 118, 129, 139, 157, 164Casiraghi C 173Cassano N 148Cassetti A 117, 119Castagnola E 150Castellano A 129, 133Castellini C 134, 141, 169, 171, 174Cataldi A 172Catania S 128, 131Cattalini M 149Cattaneo G 102, 149Cavalleri L 111Cavatorta E 138Cavillo M 103Cazzaniga G 110, 113, 114, 115, 117, 118, 119, 120, 121, 123, 139,
161, 162Cazzola Ga 143Cecchetto G 125, 131, 167Cefalo G 128, 130, 151, 173Cellini M 125, 133Cennamo L 104Cereda C 138Cereda S 128, 130, 131, 151Cesaro S 134, 137, 138, 162Cesca E 167Chiarelli F 112Chiesa M 159Chiesa R 112, 115, 145Chini L 148Cipriani A 129Cirillo F 173Cirillo P 101Citterio M 112, 115, 122, 123Ciuccarelli F 146Clerici C 151Clerici CA 173Coletti V 157, 171Coliva T 115, 117, 121, 122, 123, 153, 162Colledan M 111Collini P 124, 128, 151, 166Colombini A 112, 115Coluccia P 131Comandone A 127, 165Conforti R 132Consarino C 116Consolini R 143, 148Conte M 125, 131, 133Conter A 123Conter V 109, 111, 112, 114, 115, 117, 122, 153Contu D 136Corcione A 103Cordero di Montezemolo L 126Cornelli P 111Corradi B 120Corradini R 124Corral L 110, 118, 119, 120, 123, 161Corti P 138, 139, 140, 168Cosmi C 133Cossu F 142Cozza R 129, 132Craxì A 157Crescenzio N 102, 105Crisci S 163Cucca F 136Cuccia M 141
D'Aniello E 110, 123D'Amico G 145D'Angelo P 118, 125, 129, 133, 139, 157, 164, 170D'Angelo V 163D'Avino E 170D'Incalci M 111D'Ippolito C 101, 122D'Onofrio V 125
D'Urbano LE 118D'Urso L 104Dall'Igna P 167Dallorso S 140Dama E 144D'Ambrosio A 156D'Amore E 114Danesino C 141Dassi M 139, 140, 168Dau D 166Daudt L 110De Benedetta G 173De Benedetti F 118De Berardinis A 112De Bernardi B 133, 166, 167De Filippi P 141De Francesco S 167de Ioris MA 129De Laurentis C 129de Leonardis F 133De Leonibus F 170de Lorenzo P 118De Mattia D 106, 108, 142, 143, 148, 156, 172De Rosa E 162De Rossi G 118, 119, 120, 134, 157, 171De Salvo Gl 167de Santis A 108, 156De Santis R 156De Silvestri A 141de Sio L 129De Vecchi G 124Del Carlo TIP 151Del Giudice L 110, 116, 160, 161Del Principe D 156Del Vecchio GC 106, 107, 108, 156Della Ragione F 101Dell'Oro MG 120, 145, 162Demasi B 155Destro R 137Di Bartolomeo P 134di Cataldo A 125, 133, 134, 142, 166, 167Di Donato CI 151Di Lenardo E 146Di Martino D 140di Martino M 125, 173Di Marzio A 112, 147Di Michele P 119Di Nardo R 143Di Pinto D 104Di Renzo F 124di Rocco C 127Di Tullio MT 125, 162, 163, 173Dianzani I 102Dibar E 109D'Incalci M 112Dini G 111, 134, 140, 152D'Ippolito C 145, 165Disarò S 159, 160Disma N 153Domenghini S 103Dominici C 129Donfrancesco A 125, 129, 134Donini M 101Doria A 102Duca L 108Dufour C 103, 119Dughi S 165Duse M 143Dusi S 101
Easton J 136Ekema G 107Errigo G 138Facchetti F 101, 145Facchini E 164, 174Faccini A 124
ii
haematologica vol. 89[supplement n. 10]:October 2004

Fagioli F 134, 135, 136, 168Fagnani A 133Fanti S 134Faraci M 125, 140, 147, 168Farinasso L 105, 107Farruggia P 118, 139, 157, 164Faulkner L 133, 134Fausti R 101Favara C 116Favara Scacco C 142Favre C 111, 134, 174Fawlkner L 166Fazio G 115, 117, 118, 119, 120, 161Fedeli S 168Federici A 107Felici P 143Fernicola P 171Ferrarese D 141Ferrari A 125, 128, 130, 131, 151, 167, 173Ferrari D 101Ferrari S 102, 121, 127Ferraro M 101, 104Ferrero I 135Ferretti E 103Fidani P 129, 141, 170Fieramosca S 152Filippini B 164, 171, 174Finocchi R 142Fioredda F 125, 147Fiori GM 167Fiorini M 143Fioritoni G 112Flora G 155Foà R 114, 155Fogazzi B 165Fontana S 101Foresta C 130Formicola F 146Forni M 165Fossati-Bellani F 124, 130, 132, 151, 166, 173Foti C 148Franceschini L 113Franchi R 134Franzoni M 121, 124, 169Frascella E 113, 121, 160, 162, 163, 164Fratino G 150Freccero F 121Fronza R 124, 132Funghi C 151Furlan F 140, 168
Gadner H 109Gaipa G 111, 140, 168Gaipa M 114Galambrun C 168Galanello R 106Gallo E 151Gambacciani S 151Gambacorti-Passerini C 120Gambini C 166Gandola L 128, 131, 151, 166Garaventa A 114, 127, 141, 152, 166Garbarini L 105Garelli E 102Gasparella M 167Gastaldo L 135Gattorno M 142Gay F 150Gaziev D 136Gazzola MV 137Genitori L 126Gennaro M 151Germano G 110, 116, 159, 161, 163Giacchino M 144, 150Giacchino R 125Giagu N 106
Giarin E 110, 116, 118Gigliotti A 125Gino G 127, 165Giona F 114, 155Giordani L 108Giordano P 106, 107, 156, 172Giraldi E 111Giubellino C 136Giuliano M 116, 133, 162, 166Gottardi E 105Graziani S 148Greco N 162Greggio N 138Gremmo M 152Gridelli B 111, 164Grisolia F 125, 147Gronchi A 128Gruhn B 121Gualandi L 134Gualeni C 103, 107, 122, 165Guandalini F 136Guardascione M 170, 171Gurreri Ra 173Guzzetta F 127
Hadijistilianou T 167Hanau G 125, 147Haupt R 119, 125, 147, 150Hershfield MS 145
Iannalfi A 133Iavarone A 105Ilari I 129Imberti L 103Incarbone E 136Indolfi P 125, 131, 162, 163, 167, 173Inserra A 125, 167Italia S 142Iuvone L 127Izzi G 125
Jankovic M 116, 141, 156, 159Jenkner A 129
Kanga Leunga GG 144, 150
La Nasa G 134La Rosa MA 108La Spina M 128, 153Ladogana S 101, 111, 134Lanciotti M 103, 119Lanfranchi A 136, 145Lanino E 140Lautizi L 116Lazzareschi I 127Lenhardt A 169Leoni V 111, 114, 153Lepera P 151Leszl A 118, 120, 159Libri V 132, 169Licciardello M 105Linari A 105, 165Lippi A 116, 133, 159Lisini D 110Livadiotti S 148Lo Cascio M 129Lo Curto M 167Lo Nigro L 111, 118, 119, 159, 160Loardi G 146Locatelli F 105, 110, 111, 117, 121, 134, 139, 143Lombardi A 114, 141, 170, 171Longo S 150Longoni D 138, 140, 168Lougaris V 102Lualdi E 124Lucchesi B 146
iii
haematologica vol. 89[supplement n. 10]:October 2004

Luchini A 121Luciani M 118, 119, 157, 171Luksch R 127, 128, 130, 131, 133, 134, 151, 166, 173
Maccario R 110Madafferi S 157Madon E 126, 135, 136, 150, 165Maglia O 111, 114Magnani C 144, 149Magnano GM 147Magro S 156Magyarosy E 109Malandruccolo L 114Malguzzi S 112Maltese A 172Mandas C 106Manenti M 110, 113, 123, 139, 161Manfredini L 152Manzitti C 166Maranella E 164Marchelli R 124Marchi C 133Marchianò A 166Marcon I 128Marconcini S 132, 156, 167Marengo M 144Maresca P 101Mareschi K 135Marin V 145Marino F 141Marotta RS 153, 172Marradi P 133, 134Marseglia Gl 143Martella M 131Martinelli S 120Martinetti M 132, 141Martini A 163Martini G 107, 155Martino S 102, 105, 142, 143, 148Martire B 142, 143, 148Martone A 125Marzio D 112Mascaretti L 139Masera G 109, 110, 111, 112, 113, 115, 117, 120, 140, 145, 161, 168Masi M 143Massimino M 128, 130, 132, 151, 166, 173Mastrangelo S 133Mastrodicasa L 126Maurizi P 127Maximova N 169Mazza S 146Mazzolari E 136Meazza C 128, 131, 166, 173Meini A 102, 149Melchionda F 132, 171, 174Meloni G 114, 134Menconi M 174Menichetti F 174Menna G 170Meo A 108Mercuri M 127Merico M 130Messina C 111, 134, 137, 138Miano M 140, 152Micalizzi C 111, 119Micheli S 149Michielotto B 111, 114, 121Mieli G 153Migliaccio C 101, 104Miglietta M 146Miglionico L 133Migliorati R 133Mignani E 132Milano GM 129Minucci S 121Mirabile E 160
Miraglia V 133Misuraca A 171, 172Modena P 124Moericke A 115Mohn A 112Moleti ML 114, 155Molinari AC 106, 150, 156Mollica E 119Mondini P 124Monn K 120Montagna D 110Montin D 135Montini E 110Moratto D 103Moretta A 110Moretti V 146Morosini E 146Morreale G 140Moschese V 143, 148Mosso ML 149Munda S 160Mura R 116Murgia A 131Musarò A 110, 113, 119, 123Mussolin L 114, 137Mussolin M 159
Najajreh M 174Nanni C 134Nantron M 127Nardi M 133, 156, 174Naselli A 102Nava I 108Neri F 149Nesi F 135, 136Nespoli L 143, 156Niceforo C 135Nichelli F 116Nigro A 172Nigro G 143Ninfo V 124Nobili B 101, 104, 156Notarangelo L 101, 103Notarangelo LD 101, 102, 103, 107, 122, 136, 143, 145, 165Nysom K 168
Opocher G 131Origa R 106Orlandi P 143Orofino MG 136
Pace P 142Pagano M 127, 165Pallotti F 132Palmi C 113, 115, 117, 119Palumbo G 114Panarello C 161Pansini V 157, 171Panzer-Grumayer R 115Paolucci P 134, 156Paone G 156, 164Parasole R 158Parigi GB 133Parini R 140, 168Parodi E 105Parodi S 150Pasini A 141Pastore G 144, 149Patarino F 169Pelosin A 138, 162Pericoli R 133Perilongo G 128Perotti D 124, 166Perrotta S 101, 104Perseghin P 139, 140, 168Perutelli P 106
iv
haematologica vol. 89[supplement n. 10]:October 2004

Peruzzi L 127Pession A 111, 121, 124, 132, 134, 141, 142, 143, 144, 146, 148, 162,
164, 169, 171, 174Piazza R 120Pierani P 133, 134, 142, 146Pierinelli S 174Pietrogrande MC 142, 143Pigazzi M 159, 160Pignata C 142, 143, 148Pigullo S 103, 119Pillon M 114, 137, 159Pinta MF 111Pinto A 152, 153, 170, 171, 172Pistoia V 103Pistollato F 113Piva L 130, 166Pizzato C 130Placci A 174Plebani A 102, 103, 142, 143, 148, 149Podda M 127, 128, 130, 131, 166, 173Poggi V 152, 153, 158, 159, 171Polastri D 128, 130, 151, 166, 173Polato K 116, 159Poli A 160Pompilio A 146Porta F 107, 122, 134, 136, 145, 165Porto C 107Posgate S 122Pota E 125, 162, 163Pozzi L 116Prete A 132, 134, 141, 166, 169Provenzi M 111, 133, 166Punzo F 101Purgato S 124Putti MC 155
Quarello P 102, 105Quinti I 143
Rabusin M 142, 169Radice P 124, 166Rambaldi A 111Ramenghi U 102, 105, 156Rana I 171Rapella A 161Ravagnani F 124, 131Ravelli C 145, 149Reciputo A 159Renga D 102, 105Ribersani M 114Ricardi U 126Riccardi R 125, 127Ricotti E 159Riehm H 109Ripaldi M 134Riva MR 139Riva S 111Rizzari C 112, 115, 121, 145, 162Rizzo F 136Romano MF 158Romano MT 163Romano S 158Ronchetti F 134Rondelli R 111, 125, 134, 142, 143, 144, 148, 171Ronzoni S 153Rosanda C 161, 166Rosolen A 114, 124, 137Rosolne A 159Rossi F 104Rossi P 143, 148Rossi R 146Rossi V 118, 121, 161, 162Rovelli A 138, 140, 153, 168Ruggeri A 108Ruggiero A 127Russo C 114
Russo D 118, 129, 139, 157Russo FG 172Russo G 105, 106, 107, 109, 128, 153, 156Rustichelli D 135
Sabato V 156Sabattini E 164Sabbà C 172Sainati L 155, 159Sala A 122, 123Samperi P 109Sandri A 126Sanna MA 136Santarlasci I 151Santoro N 111, 125, 162Sanvito C 133Saracco P 105, 106, 107, 150, 172Sardi I 133Sardi N 126Sarteschi A 146Sartini R 121Sartori F 124Savio C 152Scarponi D 146Schilirò G 160Schrappe M 109, 115Schumacher F 145Sciarra P 147Scicchitano B 120, 162Sciotto A 103, 143, 160Scrimin S 158Scuderi F 140Sementa A 166Sensi A 103Seregni E 132Serino E 133Serra A 129Sersale G 140, 168Sforza S 124Siano M 163Silvestri D 109, 110Sinelli MT 122, 145, 165Siracusa F 167Sironi O 131, 132Skrap M 128Soldati M 157Sollaino C 106Songia S 110, 115, 117, 119, 123, 139, 161Soresina A 102, 142, 143, 149Soresina AR 148Soresina R 145Sourkova V 102Sozzi G 124Spada M 111, 164Spadaro G 143Spinelli M 110, 111, 117, 118, 119, 120, 121, 161, 162Spinoni V 165Spreafico F 124, 128, 130, 132, 151, 166, 173Stabile A 143Stary J 109Strisciuglio P 143Stroppa P 111Surico G 137Swahn J 103
Tagliabue A 138Tamaro P 167Tamburini A 131, 133Tamburrini G 127Tanasini R 152Tarnopolsky M 123Tartaglia M 120Tascini C 174Tassano E 161Tassone L 103Te Kronnie G 113, 121
v
haematologica vol. 89[supplement n. 10]:October 2004

Telli S 110Terenziani M 124, 128, 130, 151, 166, 173Terranova MP 140Testi AM 111, 114, 155Testi MA 124Tettoni K 103, 107, 122, 133, 136, 165, 166, 170Timeus F 105Tintori V 133Todisco L 126Tomà P 147Tondo A 133Tonegatti L 107, 133Tonelli R 121, 124Torre G 111Torrisi V 133Toti P 167Tovo PA 144Tregnaghi T 155Tremolada M 158Tridello G 124, 138, 159Trizzino A 118, 139, 141, 142, 157, 164, 170Trka J 121Trocino G 138Tropia S 129Tucci F 133, 156Tumino M 153Turin I 110
Uberti B 107Uderzo C 134, 138, 139, 140, 168Ugazio A 143, 148Ugazio AG 142, 143Urban C 168Uziel G 140, 168
Vagliano L 135Valera M 144Valetto A 140Vallinoto C 161Valsecchi MG 109, 110, 111, 115Van Dongen J 123, 161Varotto S 116, 138, 142, 159, 168, 170Vassallo E 135, 136Veltroni M 111, 114, 155Venturi C 135Venturi N 140, 168Venuta S 158Verardi R 136Verdeguer A 168Vermi W 101Viganò E 115, 121, 122, 123, 138, 153Villa A 117, 119Villa T 110, 113, 123Viscardi E 127, 130, 133, 166Visintin G 137, 138Vivarelli F 174
Webber C 122, 123
Zampieron C 160, 163Zanazzo GA 133, 166Zanesco L 114, 137, 155, 158, 159Zanesco LE 138Zanetti I 130Zanghì L 108Zangrando A 121Zecca M 105, 110, 111, 156Ziino O 118, 129, 139, 157Zimmermann M 115Zintl F 168Zucca S 136Zucchetti P 134
vi
haematologica vol. 89[supplement n. 10]:October 2004









![Risk-adapted therapy of AML in younger adultsSolalettura].pdf · Risk-adapted therapy of AML in younger adults Sergio Amadori Tor Vergata University Hospital Rome Pescara 11/2010.](https://static.fdocuments.in/doc/165x107/5e2962bb73f8a5419b612bcd/risk-adapted-therapy-of-aml-in-younger-solaletturapdf-risk-adapted-therapy-of.jpg)








