
E2 is most favorable (lowest activation energy) when H and Lv are anti and coplanar Stereochemistry...
37
• E2 is most favorable (lowest activation energy) when H and Lv are anti and coplanar CH 3 O: - C C H Lv CH 3 O H C C Lv -H and -Lv are antiand coplanar (dihedral angle 180°) Stereochemistry of E2 A B D E E D A B
-
date post
22-Dec-2015 -
Category
Documents
-
view
215 -
download
1
Transcript of E2 is most favorable (lowest activation energy) when H and Lv are anti and coplanar Stereochemistry...
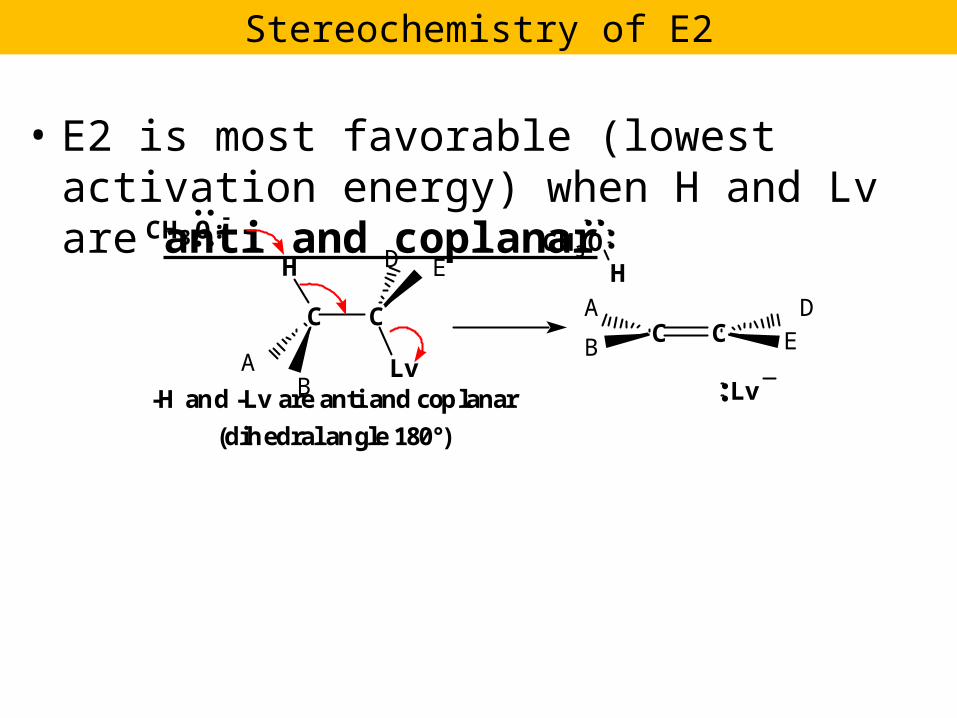
• E2 is most favorable (lowest activation energy) when H and Lv are anti and coplanar
CH3O:-
C C
H
Lv
CH3OH
C C
Lv-H and -Lv are anti and coplanar
(dihedral angle 180°)
Stereochemistry of E2
AB
D E
E
DA
B
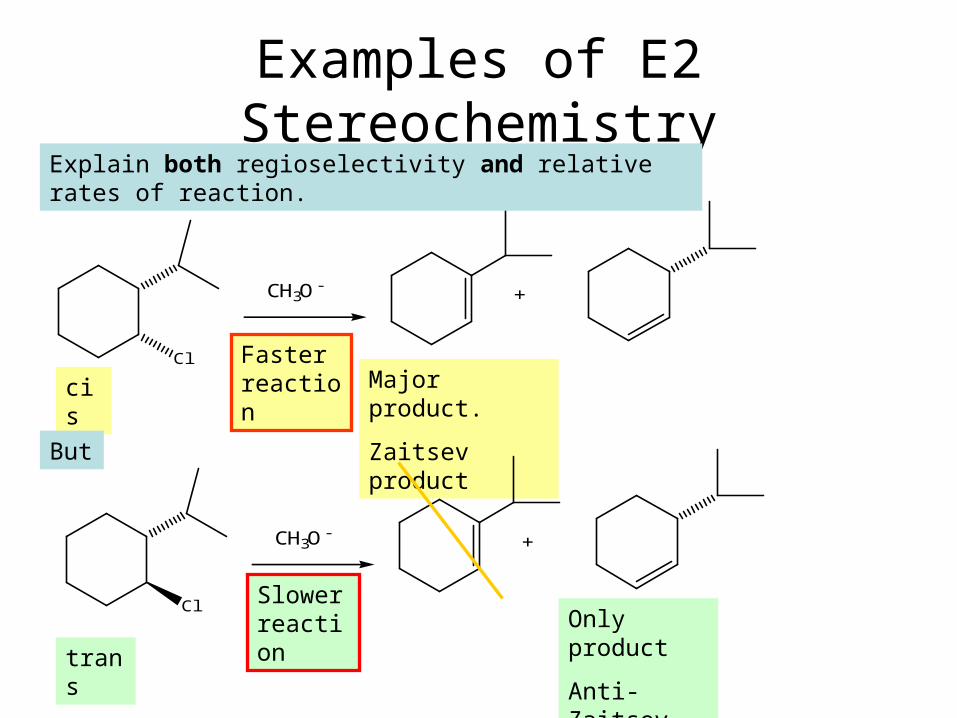
Examples of E2 Stereochemistry
Cl
CH3O -
+
cis Major product.
Zaitsev product
Cl
CH3O -
+
trans
Only product
Anti-Zaitsev
Explain both regioselectivity and relative rates of reaction.
But
Faster reaction
Slower reaction
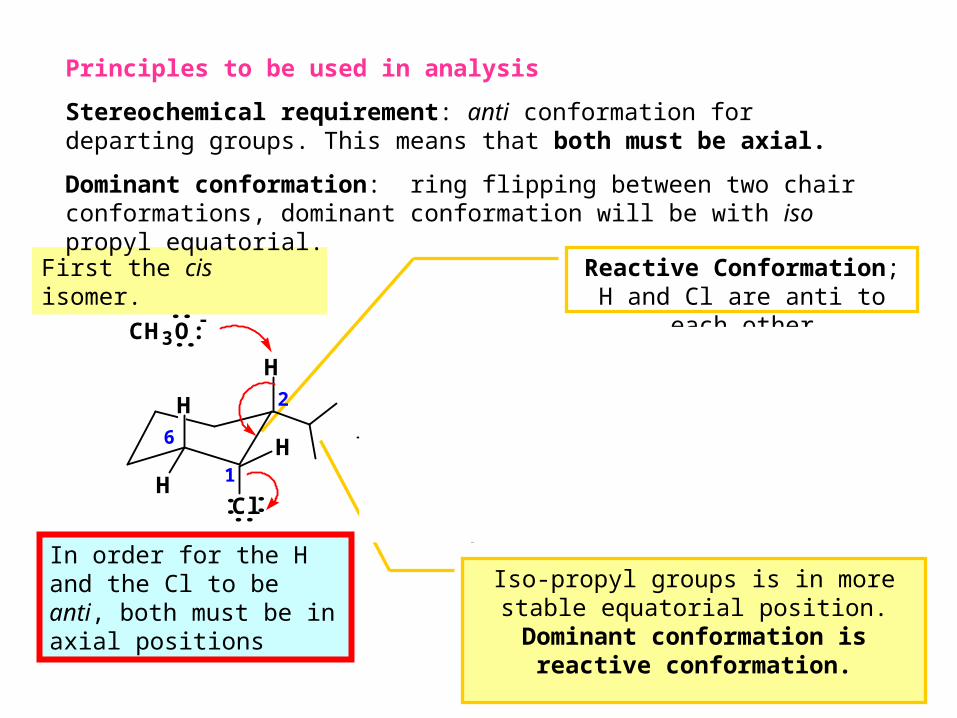
In order for the H and the Cl to be anti, both must be in axial positions
First the cis isomer. Reactive Conformation; H and Cl are anti to each other
Iso-propyl groups is in more stable equatorial position. Dominant
conformation is reactive conformation.
CH3O:-
H
H
H
H
Cl
CH3OH :Cl
1-Isopropyl-cyclohexene
2
1
6 + +E2
Principles to be used in analysis
Stereochemical requirement: anti conformation for departing groups. This means that both must be axial.
Dominant conformation: ring flipping between two chair conformations, dominant conformation will be with iso propyl equatorial.
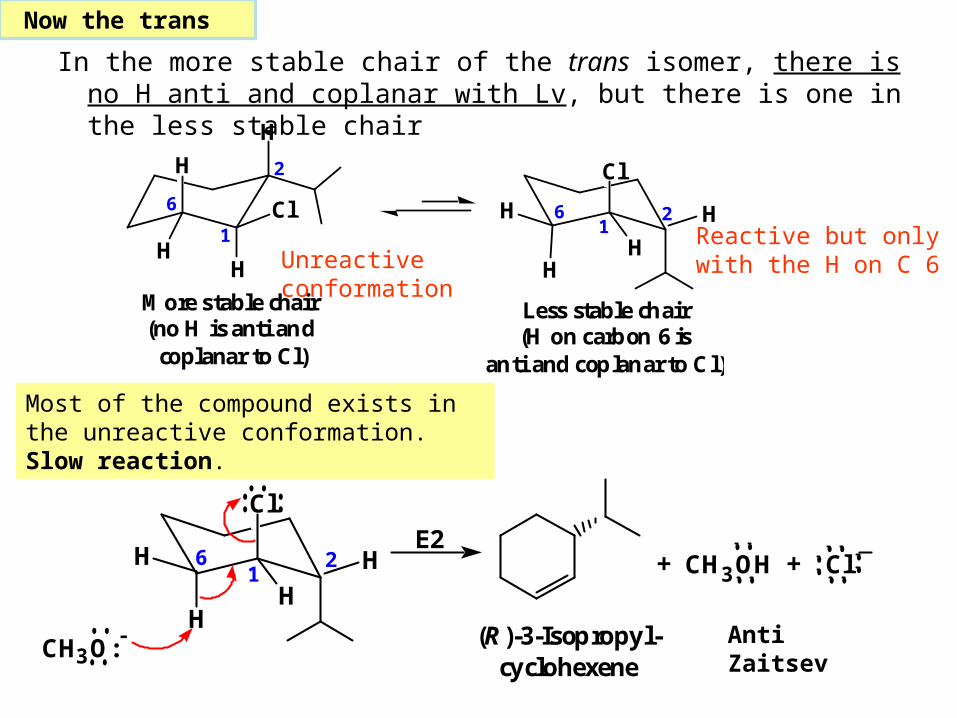
In the more stable chair of the trans isomer, there is no H anti and coplanar with Lv, but there is one in the less stable chair
More stable chair (no H is anti and coplanar to Cl)
Less stable chair(H on carbon 6 is
anti and coplanar to Cl)
2
2
11
6 6Cl
H
H
H
H
Cl
HH
HH
Now the trans
Unreactive conformation
Reactive but only with the H on C 6
Most of the compound exists in the unreactive conformation. Slow reaction.
H
Cl
HH
HCH3O:
-
CH3OH Cl
(R)-3-Isopropyl-cyclohexene
21
6E2
+ +
Anti Zaitsev
More stable chair (no H is anti and coplanar to Cl)
Less stable chair(H on carbon 6 is
anti and coplanar to Cl)
2
2
11
6 6Cl
H
H
H
H
Cl
HH
HH

Example, Predict Product
Problem!: Fischer projection diagram represents an eclipsed structure.
Task: convert to a staggered structure wherein H and Br are anti and predict product. We will convert to a Newman and see what we get…
Ph
H CH3
Ph
H3C Br
base
Ph
H CH3
Ph
H3C Br
Ph
H CH3
Ph
H3C Br
=
Ph
H3C H
Ph
H3C Br
rotate upper chiral C by 180
H3C
Ph
H
Br
Ph
H3C
CH3
H Ph
Ph
H3C Br
H
CH3
Ph
Br
Ph
H3C
rotate 120further
H3C Ph
H3C Phbase
H3C Ph
H3C Ph
H & Br not anti yet!
Now anti and we can see where the pi bond will be.
Ph
H CH3
Ph
H3C Br
base
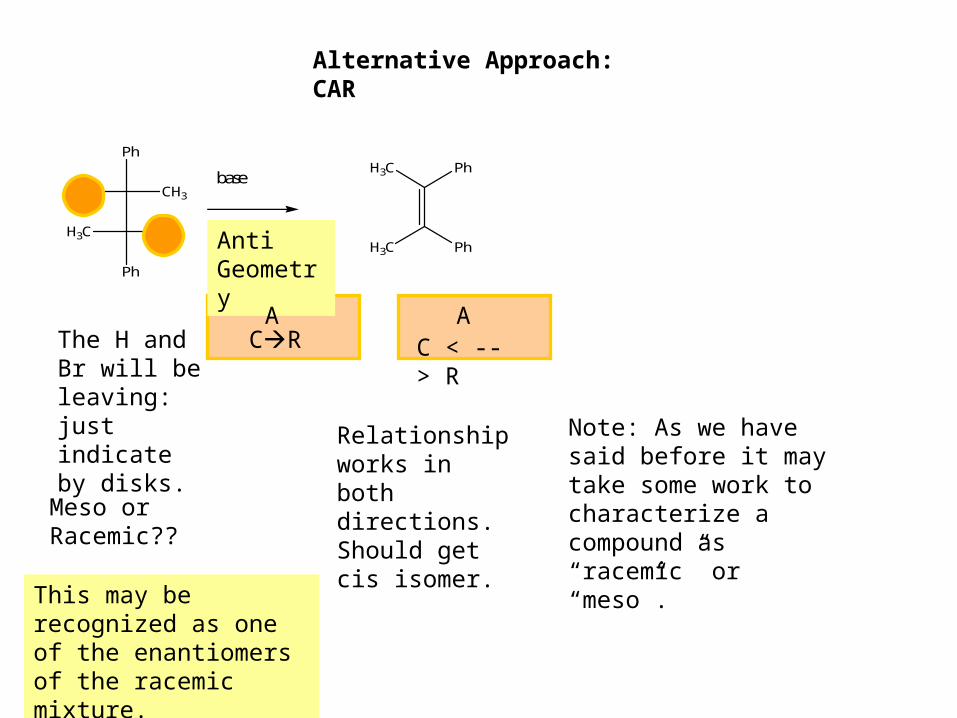
Alternative Approach: CAR
The H and Br will be leaving: just indicate by disks.
Meso or Racemic??
Anti Geometry
CRA
Relationship works in both directions. Should get cis isomer.
H3C Ph
H3C Ph
Note: As we have said before it may take some work to characterize a compound as “racemic” or “meso”.
This may be recognized as one of the enantiomers of the racemic mixture.
AC < -- > R
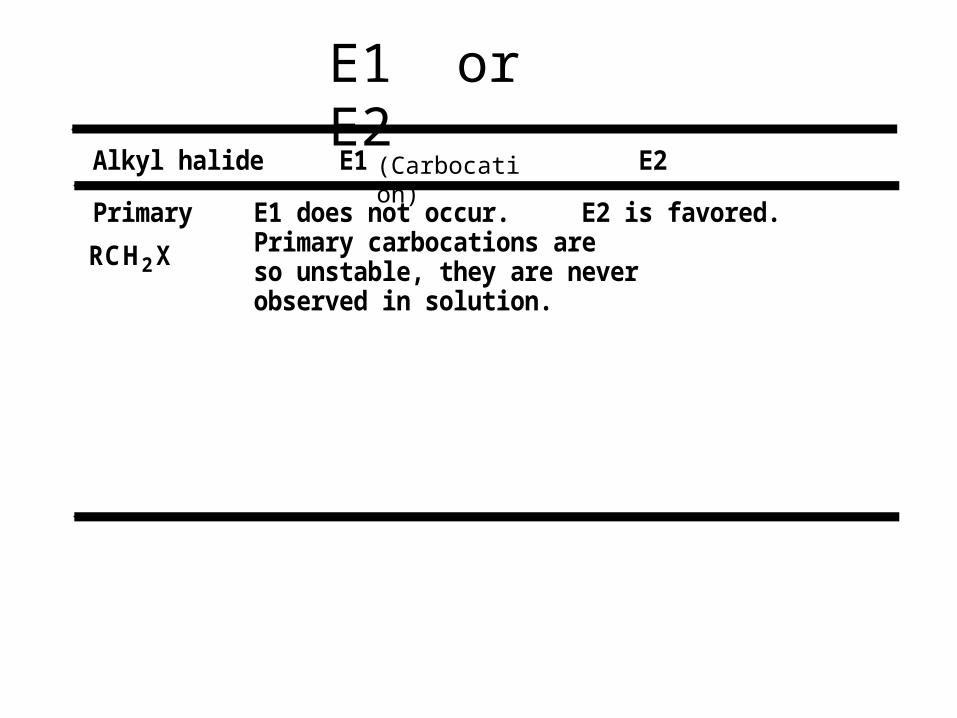
RCH2X
R2CHX
R3CX
Alkyl halide E1 E2
Primary
Secondary
Tertiary
E1 does not occur.Primary carbocations areso unstable, they are neverobserved in solution.
E2 is favored.
Main reaction with strong bases such as OH- and OR-.
Main reaction with weak bases such as H2O, ROH.
Main reaction with strong bases such as OH- and OR-.
Main reaction with weak bases such as H2O, ROH.
E1 or E2
(Carbocation)
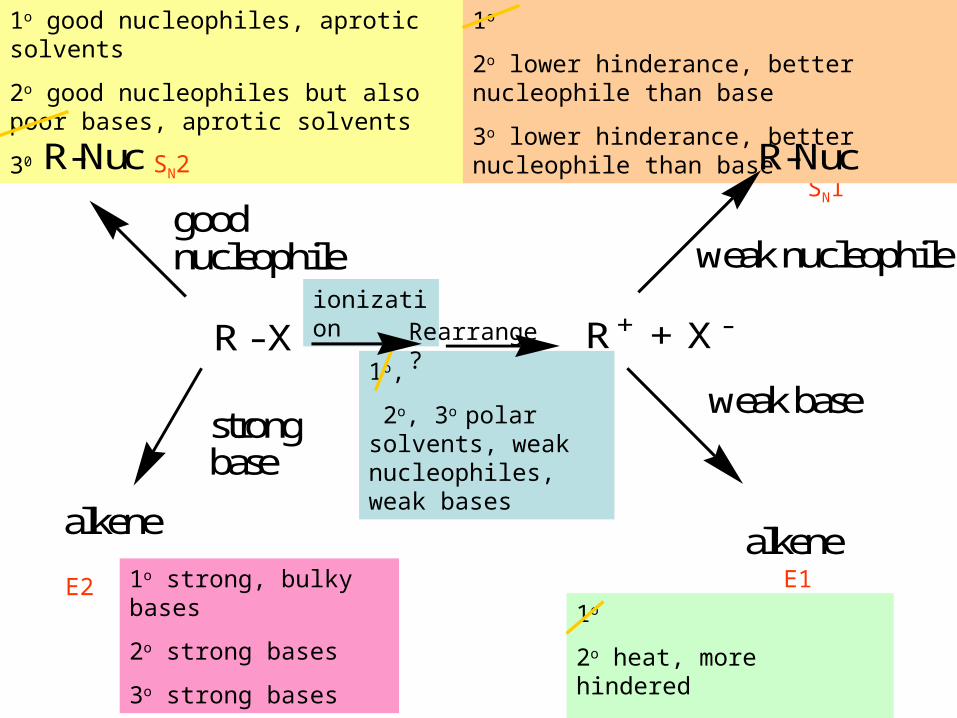
ionization
1o,
2o, 3o polar solvents, weak nucleophiles, weak bases
1o strong, bulky bases
2o strong bases
3o strong bases
1o good nucleophiles, aprotic solvents
2o good nucleophiles but also poor bases, aprotic solvents
30
SN2SN1
E2 E1
Rearrange ?
1o
2o heat, more hindered
3o heat, more hindered
1o
2o lower hinderance, better nucleophile than base
3o lower hinderance, better nucleophile than base
R - X R + + X -
good nucleophile
strong base
R-Nuc
alkene
weak nucleophile
weak base
alkene
R-Nuc
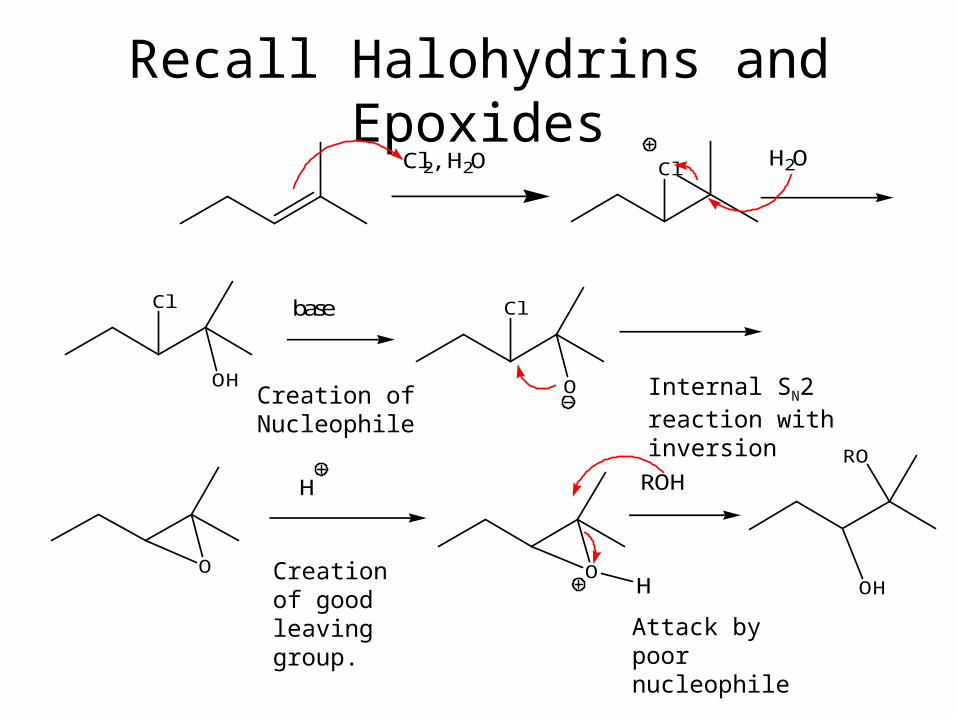
Recall Halohydrins and EpoxidesCl2, H2O Cl H2O
Cl
OH
base Cl
O
O
H
OH
ROH
OH
RO
Creation of Nucleophile Internal SN2 reaction with inversion
Creation of good leaving group.
Attack by poor nucleophile
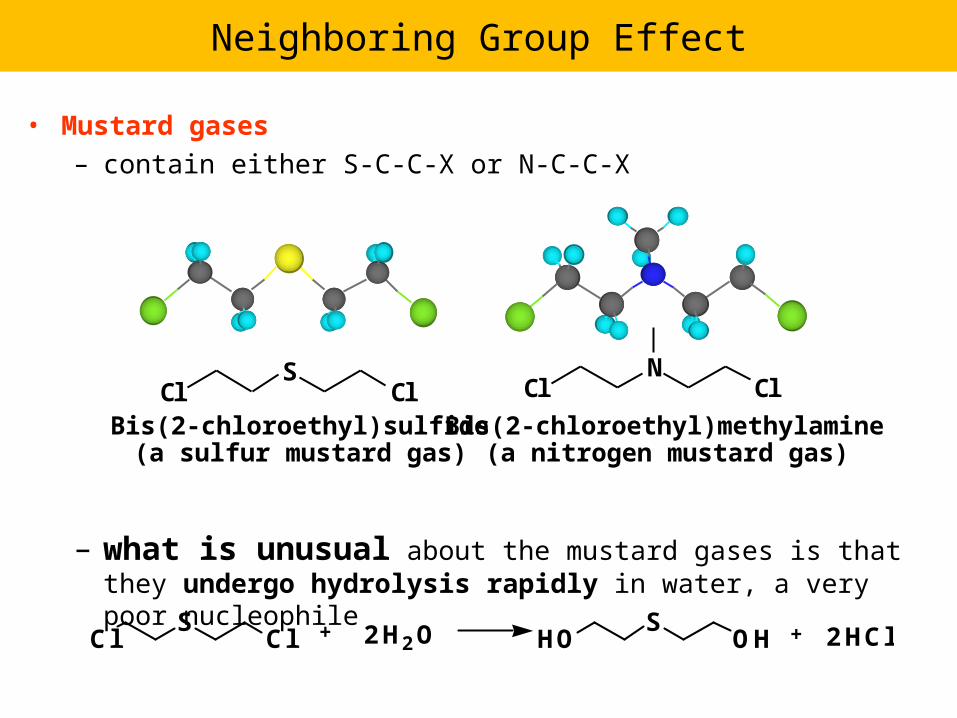
Neighboring Group Effect
• Mustard gases – contain either S-C-C-X or N-C-C-X
– what is unusual about the mustard gases is that they undergo hydrolysis rapidly in water, a very poor nucleophile
ClS
Cl 2H2O HOS
OH 2HCl+ +
Bis(2-chloroethyl)sulfide(a sulfur mustard gas)
Bis(2-chloroethyl)methylamine(a nitrogen mustard gas)
ClS
Cl ClN
Cl
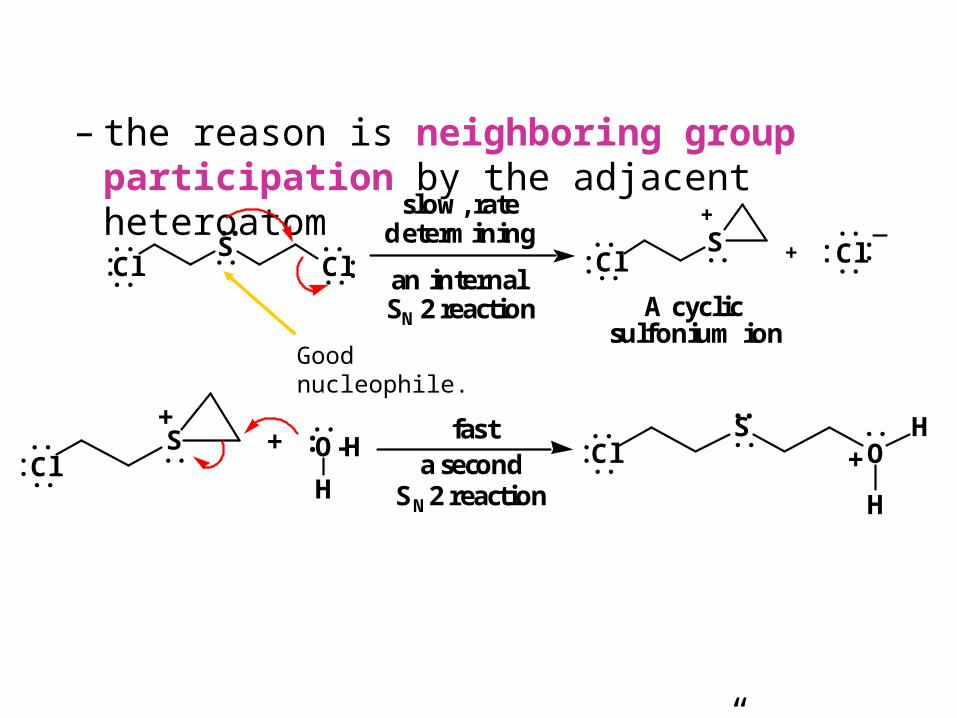
– the reason is neighboring group participation by the adjacent heteroatom
– proton transfer to “solvent” completes the reaction
ClS
Cl
ClS O-H
H
ClS
ClS
O
H
H
Cl+
+
A cyclic sulfonium ion
an internal SN2 reaction
slow, ratedetermining
++
a secondSN2 reaction
fast+
:
:
:
Good nucleophile.
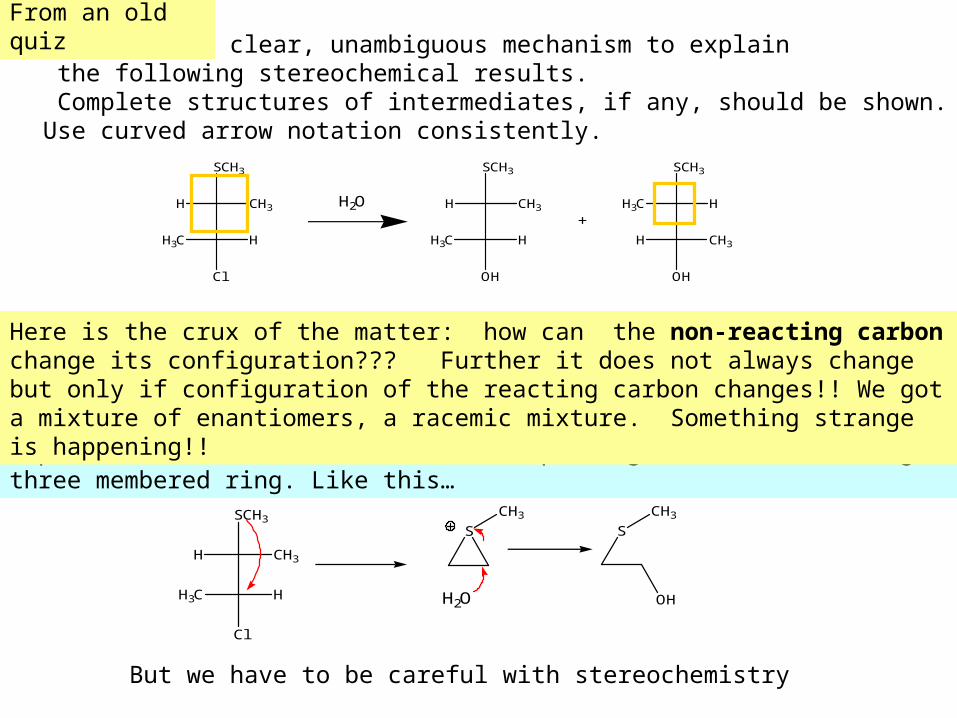
5. Provide a clear, unambiguous mechanism to explain the following stereochemical results. Complete structures of intermediates, if any, should be shown. Use curved arrow notation consistently.
H
SCH3
CH3
Cl
H3C H
H2O H
SCH3
CH3
OH
H3C H
H3C
SCH3
H
OH
H CH3
+
Expect sulfur to attack the C-Cl, displacing the Cl and forming a three membered ring. Like this…
But we have to be careful with stereochemistry
H
SCH3
CH3
Cl
H3C H
S
CH3
H2O
S
CH3
OH
Here is the crux of the matter: how can the non-reacting carbon change its configuration??? Further it does not always change but only if configuration of the reacting carbon changes!! We got a mixture of enantiomers, a racemic mixture. Something strange is happening!!
From an old quiz
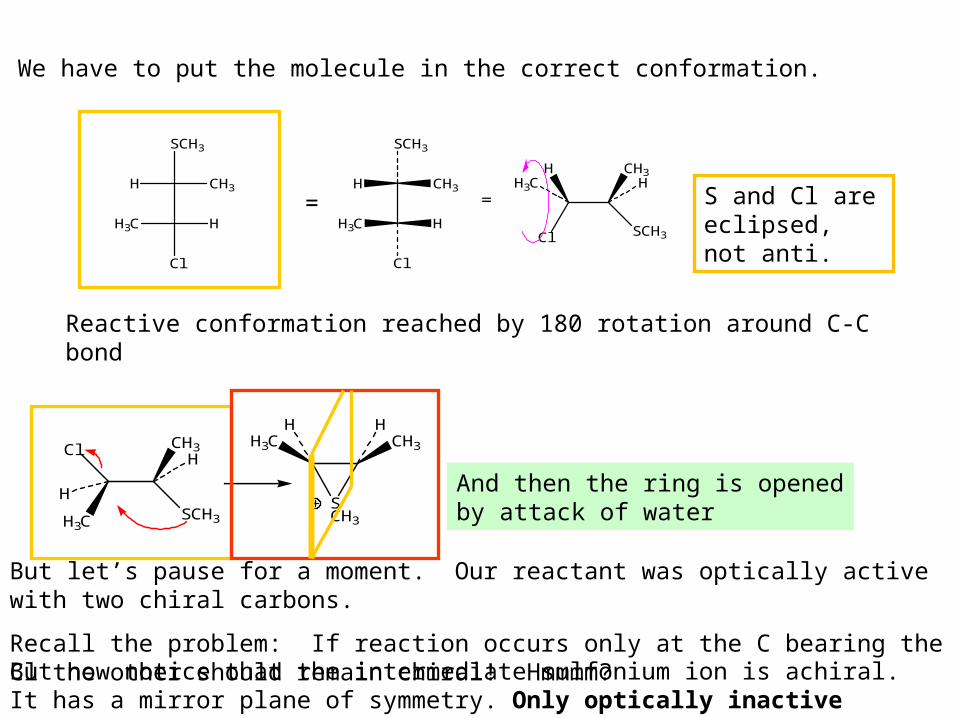
H
SCH3
CH3
Cl
H3C H
We have to put the molecule in the correct conformation.
=H
SCH3
CH3
Cl
H3C HCl SCH3
H CH3H3C H
=
Reactive conformation reached by 180 rotation around C-C bond
Cl
SCH3H3C
CH3
H
H
SCH3
H3C CH3
HH
And then the ring is opened by attack of water
S and Cl are eclipsed, not anti.
But let’s pause for a moment. Our reactant was optically active with two chiral carbons.
Recall the problem: If reaction occurs only at the C bearing the Cl the other should remain chiral! Hmmmm?But now notice that the intermediate sulfonium ion is achiral. It has a mirror plane of symmetry. Only optically inactive products will result.
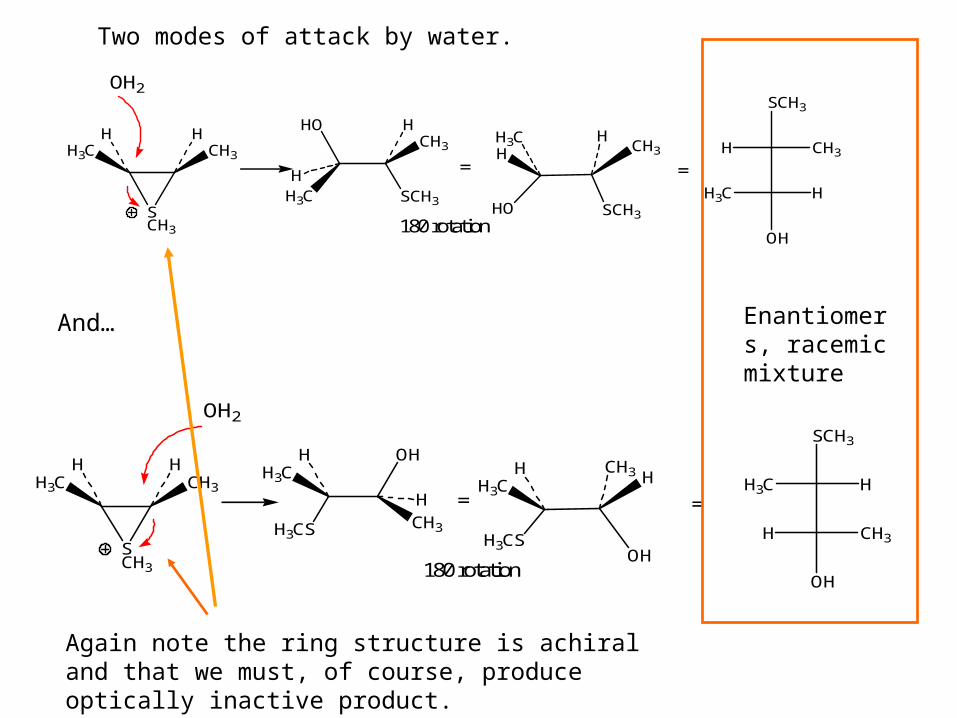
H
SCH3
CH3
OH
H3C HSCH3
H3C CH3
HH
OH2
SCH3H3C
CH3
H
H =
SCH3
H CH3HH3C
=
180 rotation
HO
HO
Two modes of attack by water.
And…
SCH3
H3C CH3
HH
OH2
H3CS
H3C
CH3
H
H OH
H3C
SCH3
H
OH
H CH3
=
H3CS
H3CH
CH3H
OH
=
180 rotation
Again note the ring structure is achiral and that we must, of course, produce optically inactive product.
Enantiomers, racemic mixture

Alcohols

Hydrogen Bonding
Three ethanol molecules.

Hydrogen Bonding & boiling pointIncreases boiling point, higher temperature needed to separate the molecules.
Hexane 69 deg.
1-pentanol 138
1,4-butanediol 230
Ethanol 78 deg
Dimethyl ether 24

Earlier Discussion of Acidity
Methanol Ethanol 2-Propanol 2-Methyl-2-propanol
Increasing Hinderance of Solvation
RO-H RO – (solvated) + H + (solvated)
Increasing Acidity of the alcohol
Recall: H2O + Na Na+ + OH- + ½ H2(g)
Alcohols behave similarly
ROH + Na Na+ + OR- + ½ H2(g)
Alkoxide, strong base, strong nucleophile (unless sterically
hindered)
Also: ROH + NaH Na+ + OR- + ½ H2(g)
Increasing Basicity of Alkoxide Anion, the conjugate base
Alkoxide ion, base
Alkoxides can be produced in several ways…

-OH as a Leaving Group
R-OH + H + R-OH2+
Protonation of the alcohol sets-up a good leaving group, water.
Poor leaving group, hydroxide ion.
Another way to turn the –OH into a leaving group…

Conversion to Alkyl Halide,HX + ROH RX + H2O
When a carbocation can be formed (Tertiary, Secondary alcohols) beware of rearangement. SN1
Expect both configurations.
When a carbocation cannot be formed. Methanol, primary. SN2
RCH2OH RCH2OH2
X -H +
RCH2X
R3COH R3COH2 R3C + + H2O
X -H +
R3CX

But sometimes experiment does not agree with our ideas…
Observed reaction CH2OH
HX
CH2X
X
The problem:
•Rearrangement of carbon skeleton which usually indicates carbocations.
•Reacting alcohol is primary; do not expect carbocation.
•Time to adjust our thinking a bit….
H3C
CH2OH
H + H3C
CH2OH2+
CH2.......OH2
H3C CH3
H2O
X-X
Not a primary carbocation
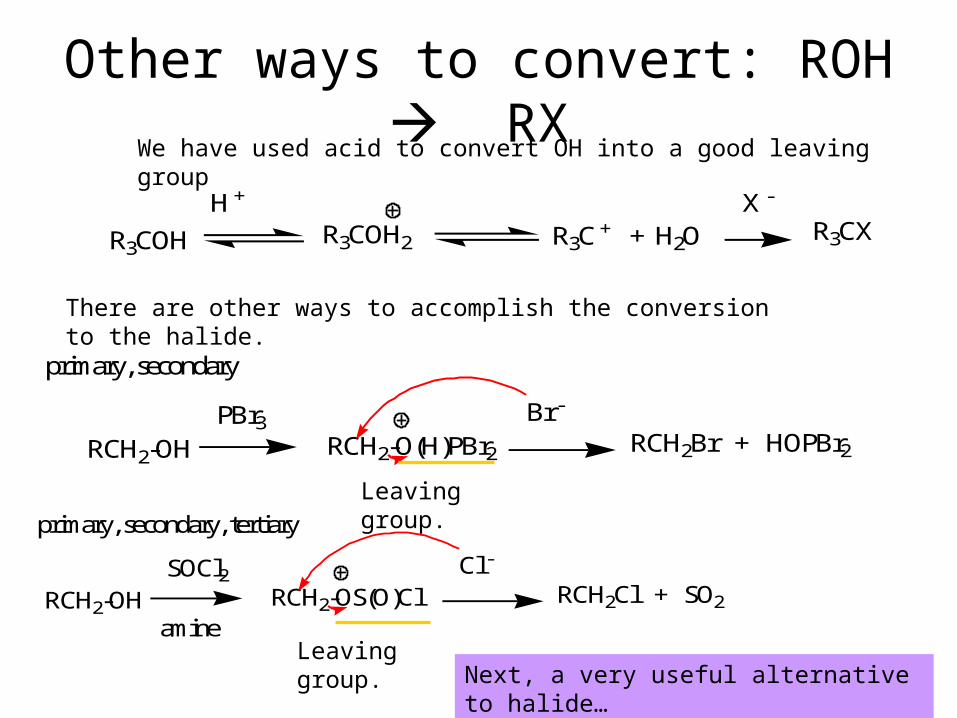
Other ways to convert: ROH RXWe have used acid to convert OH into a good leaving group
There are other ways to accomplish the conversion to the halide.
RCH2-OHPBr3
RCH2-O(H)PBr2
Br -
RCH2Br + HOPBr2
primary, secondary
RCH2-OHSOCl2
RCH2-OS(O)ClCl -
RCH2Cl + SO2
primary, secondary, tertiary
amine
R3COH R3COH2 R3C + + H2O
X -H +
R3CX
Leaving group.
Leaving group.Next, a very useful alternative to halide…
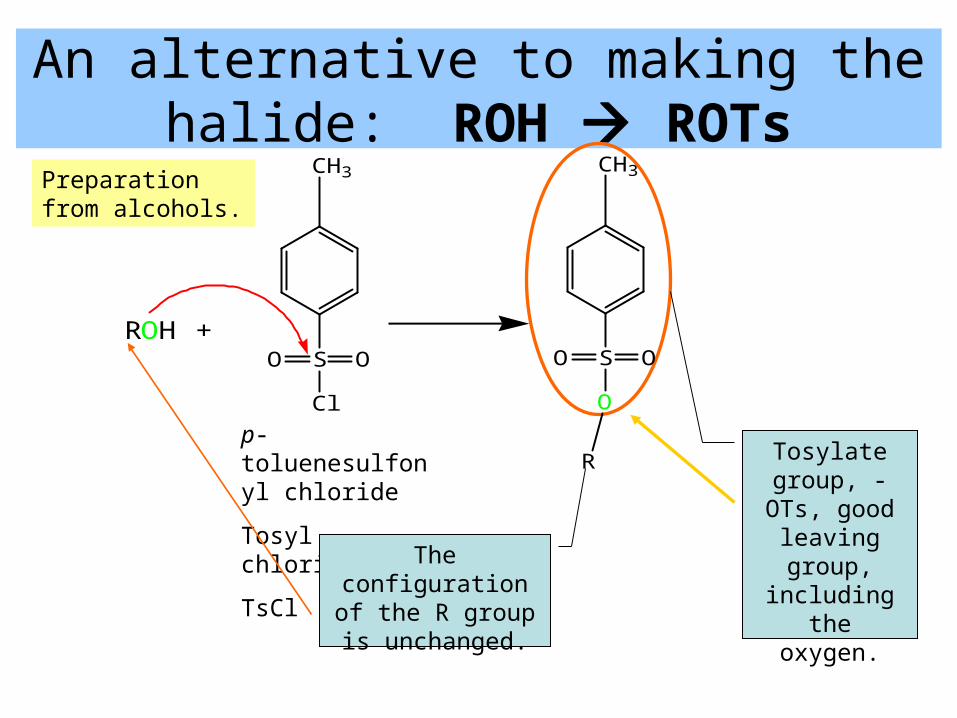
An alternative to making the halide: ROH ROTs
p-toluenesulfonyl chloride
Tosyl chloride
TsCl
ROH +S OO
Cl
S OO
O
CH3 CH3
R Tosylate group, -OTs, good leaving
group, including the
oxygen.
The configuration of the R group is unchanged.
Preparation from alcohols.
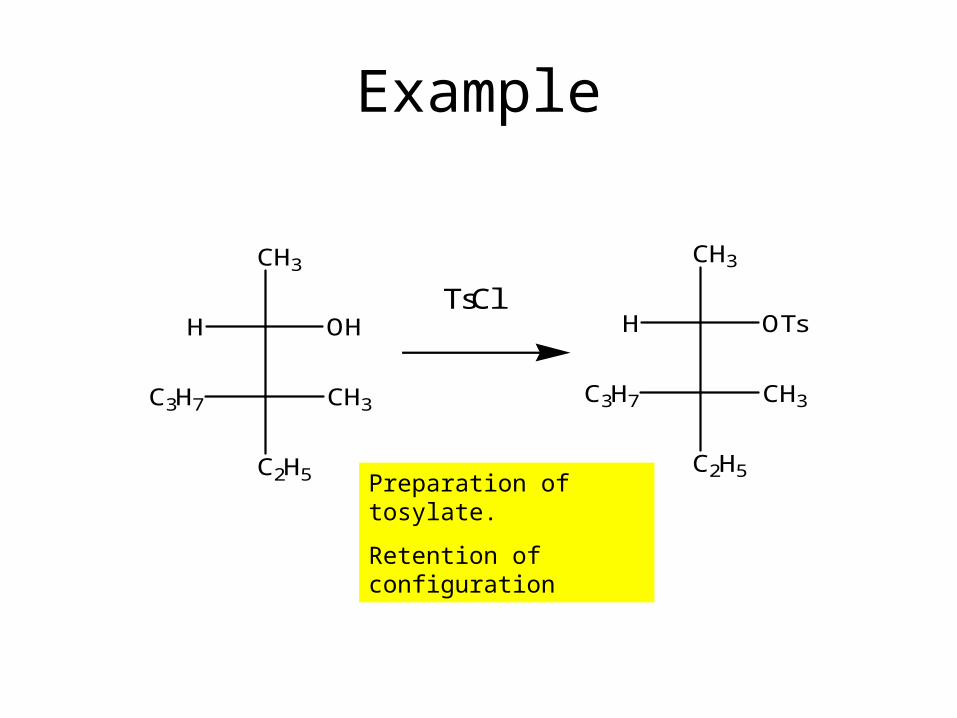
Example
CH3
H OH
C2H5
C3H7 CH3
TsCl
CH3
H OTs
C2H5
C3H7 CH3
Preparation of tosylate.
Retention of configuration
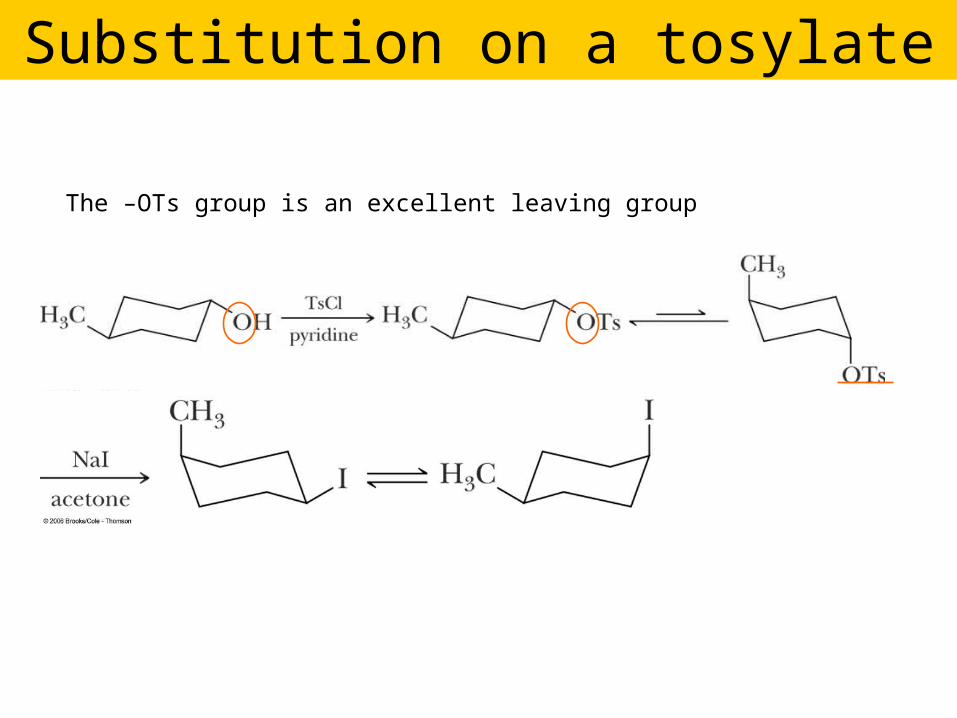
Substitution on a tosylate
The –OTs group is an excellent leaving group
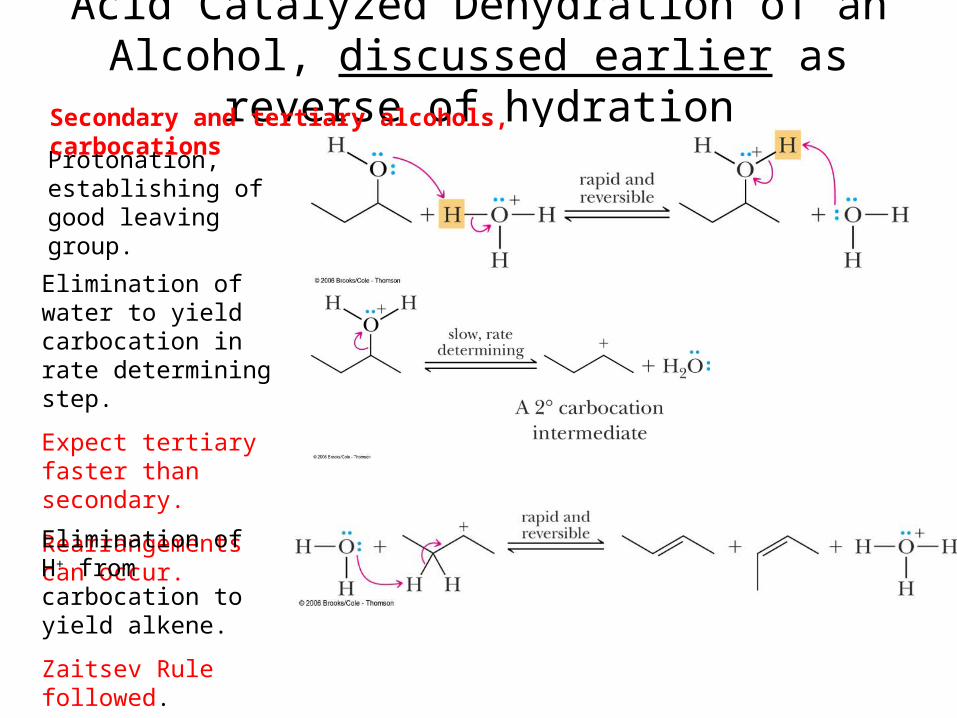
Acid Catalyzed Dehydration of an Alcohol, discussed earlier as reverse of hydration
Protonation, establishing of good leaving group.
Elimination of water to yield carbocation in rate determining step.
Expect tertiary faster than secondary.
Rearrangements can occur.
Elimination of H+ from carbocation to yield alkene.
Zaitsev Rule followed.
Secondary and tertiary alcohols, carbocations

Primary alcohols
Problem: primary carbocations are not observed. Need a modified, non-carbocation mechanism.
Recall these concepts:
1. Nucleophilic substitution on tertiary halides invokes the carbocation but nucleophilic substitution on primary RX avoids the carbocation by requiring the nucleophile to become involved immediately.
2. The E2 reaction requires the strong base to become involved immediately.
Note that secondary and tertiary protonated alcohols eliminate the water to yield a carbocation because the carbocation is relatively stable. The carbocation then undergoes a second step: removal of the H+.
The primary carbocation is too unstable for our liking so we combine the departure of the water with the removal of the H+.
What would the mechanism be???
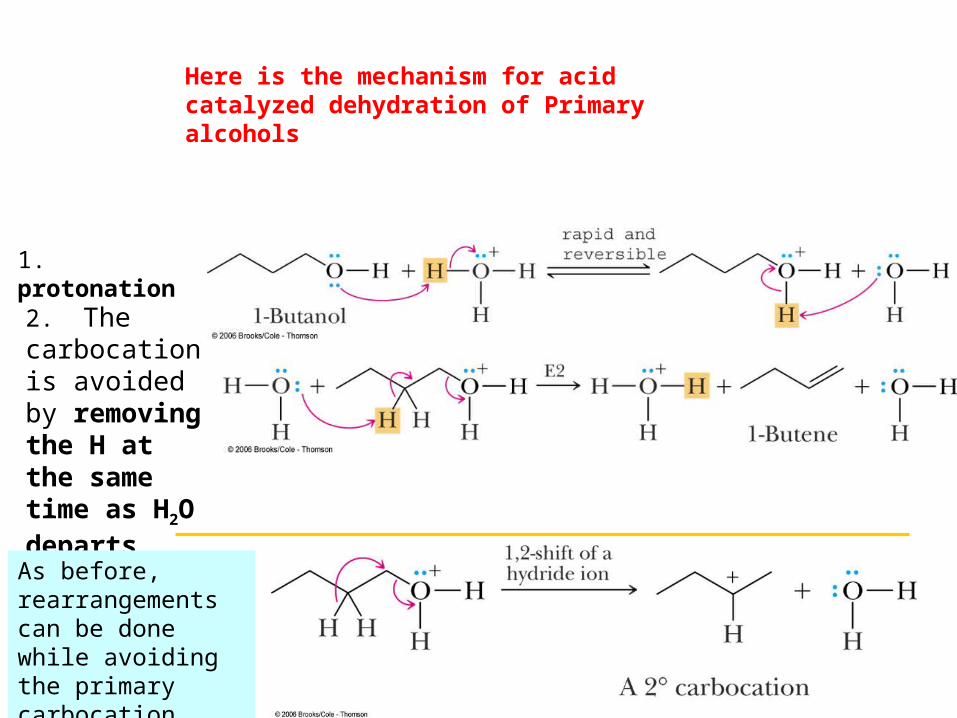
Here is the mechanism for acid catalyzed dehydration of Primary alcohols
1. protonation
2. The carbocation is avoided by removing the H at the same time as H2O departs (like E2).
As before, rearrangements can be done while avoiding the primary carbocation.
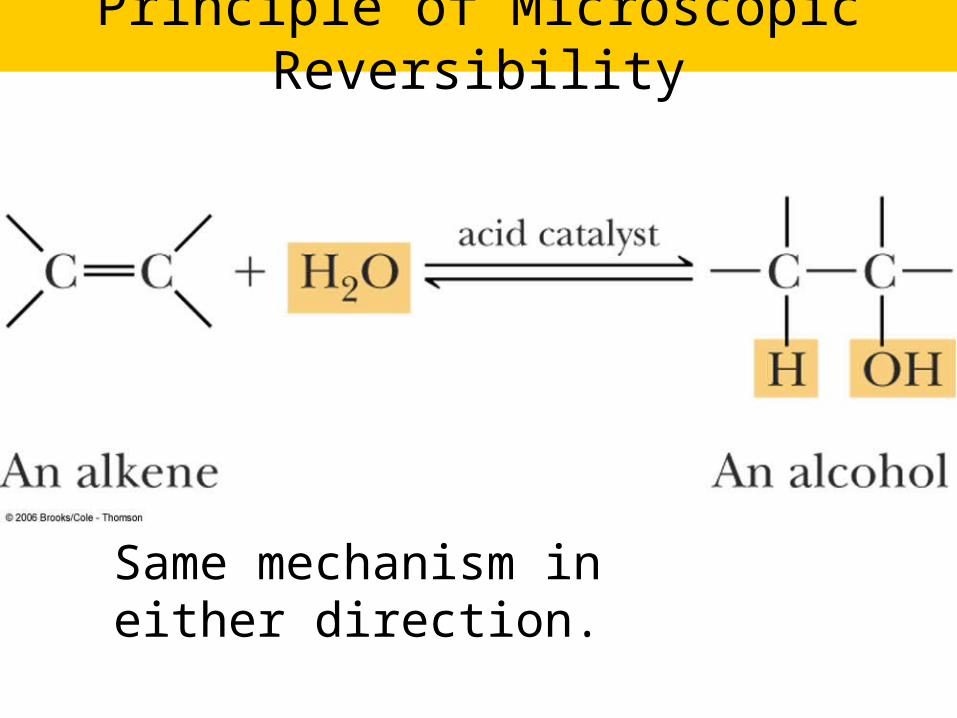
Principle of Microscopic Reversibility
Same mechanism in either direction.
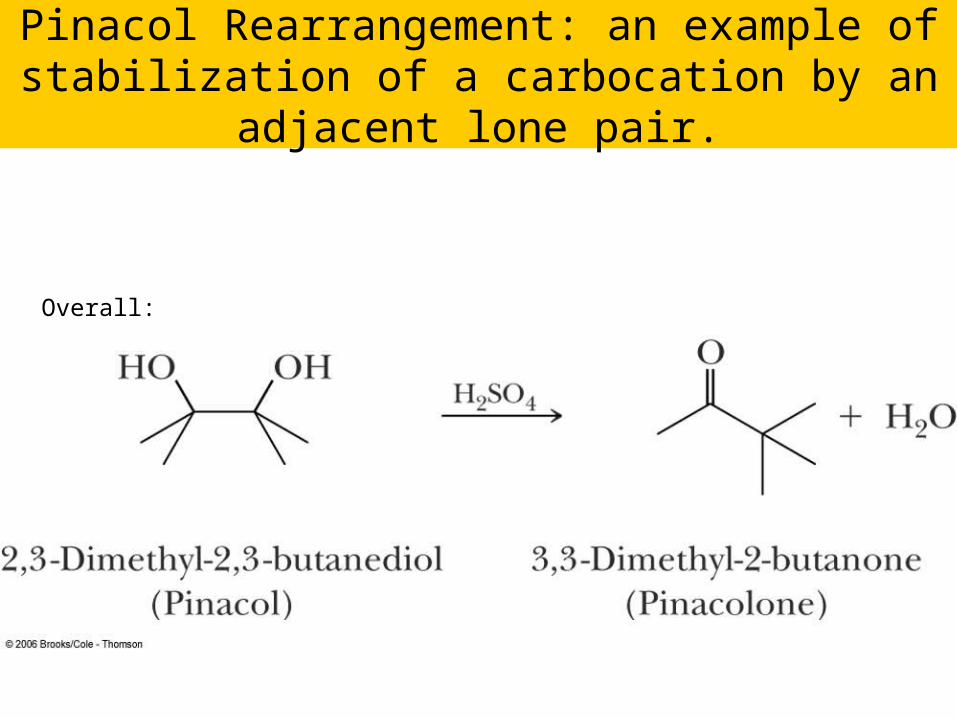
Pinacol Rearrangement: an example of stabilization of a carbocation by an adjacent lone
pair.
Overall:
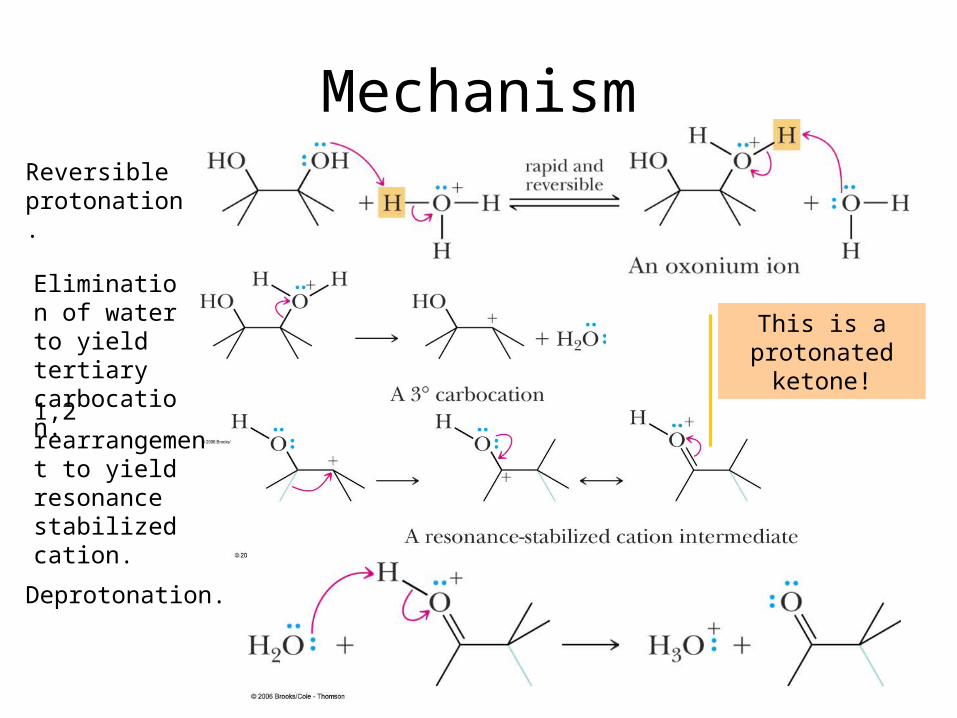
MechanismReversible protonation.
Elimination of water to yield tertiary carbocation.
1,2 rearrangement to yield resonance stabilized cation.
Deprotonation.
This is a protonated
ketone!
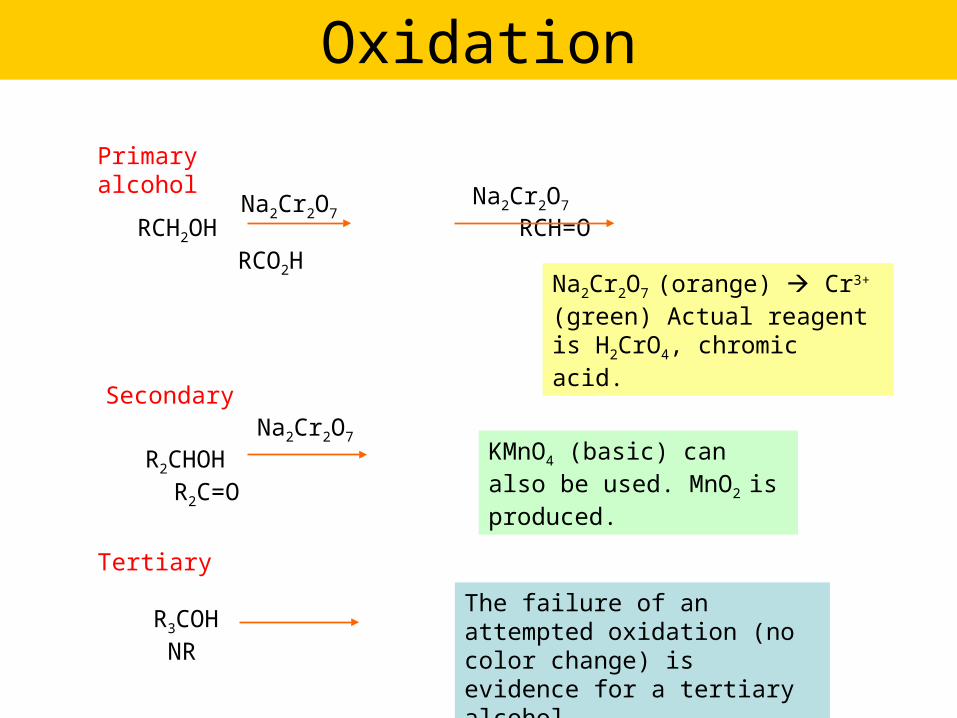
Oxidation
Primary alcohol
RCH2OH RCH=O RCO2H
Na2Cr2O7
Na2Cr2O7
Na2Cr2O7 (orange) Cr3+ (green) Actual reagent is H2CrO4, chromic acid.
Secondary
R2CHOH R2C=O
Tertiary
R3COH NR
KMnO4 (basic) can also be used. MnO2 is produced.
The failure of an attempted oxidation (no color change) is evidence for a tertiary alcohol.
Na2Cr2O7

OH
CH2OH
HO
Na2Cr2O7
acid
OH
CO2H
O
Example…
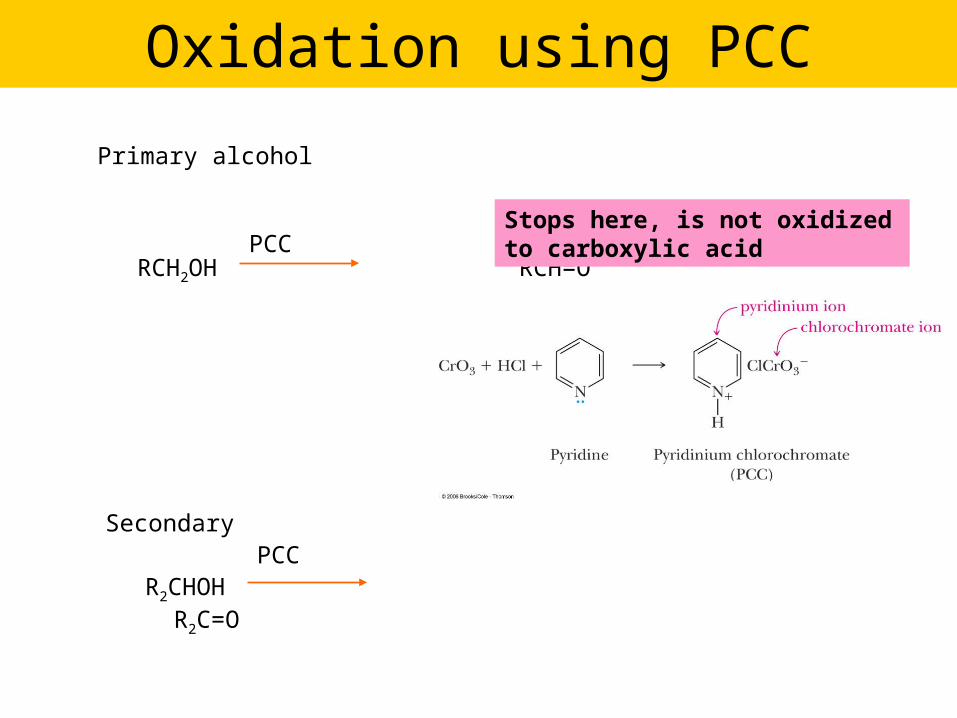
Oxidation using PCC
Primary alcohol
RCH2OH RCH=OPCC
PCCSecondary
R2CHOH R2C=O
Stops here, is not oxidized to carboxylic acid
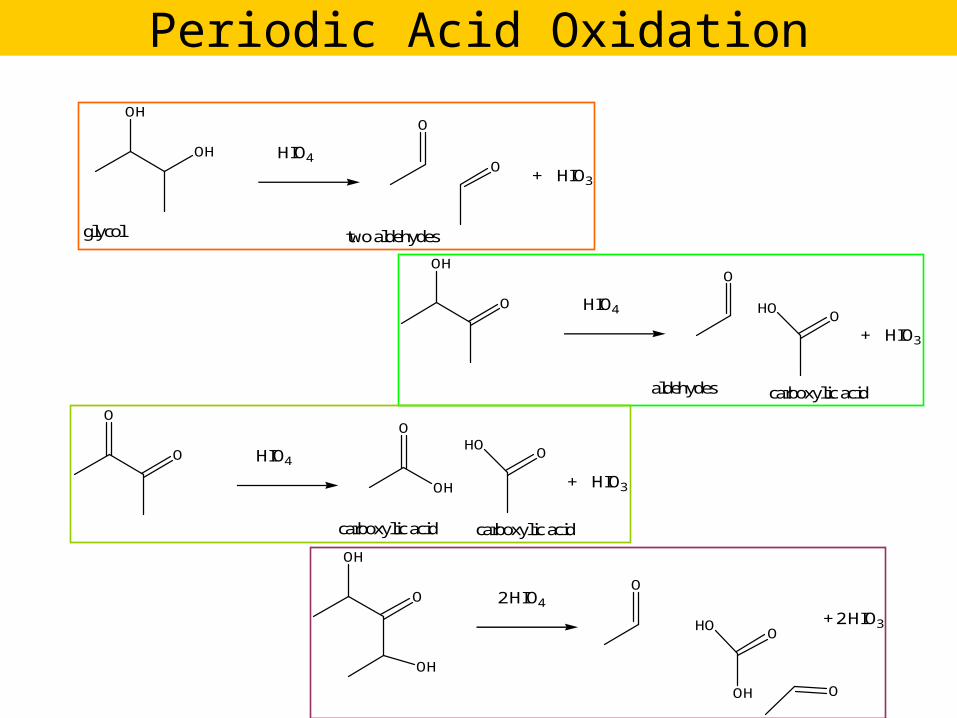
Periodic Acid Oxidation
OH
OH
glycol
HIO4
O
O
two aldehydes
+ HIO3
OH
O HIO4
O
O
aldehydes
HO
carboxylic acid
+ HIO3
O
O HIO4
O
OHO
carboxylic acidcarboxylic acid
OH + HIO3
OH
O 2 HIO4
OH
O
OH
OHO
O
+ 2 HIO3
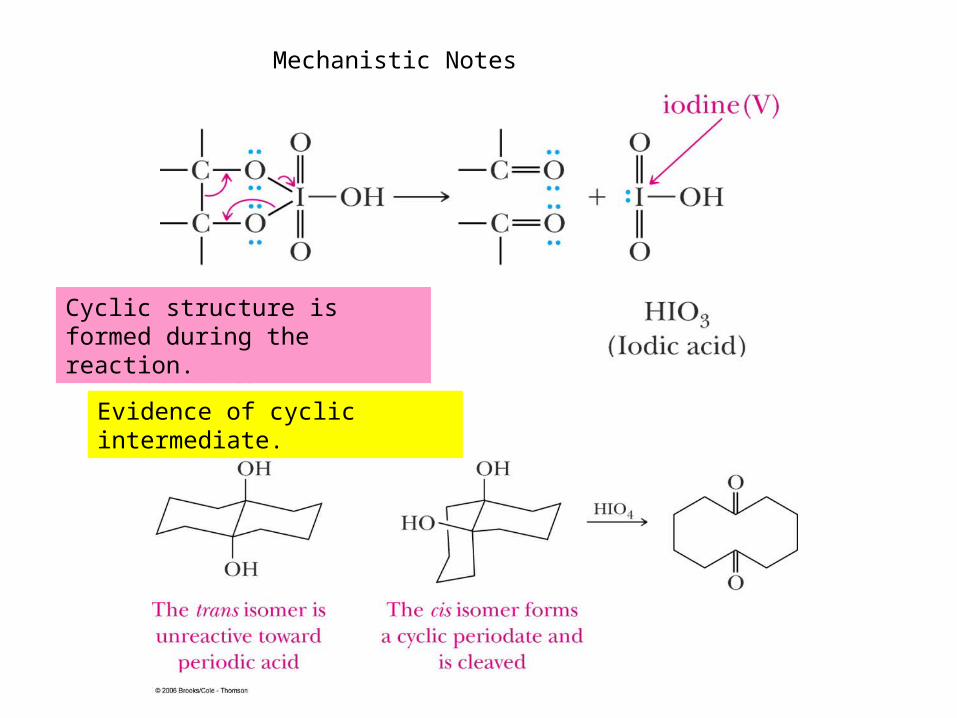
Mechanistic Notes
Cyclic structure is formed during the reaction.
Evidence of cyclic intermediate.
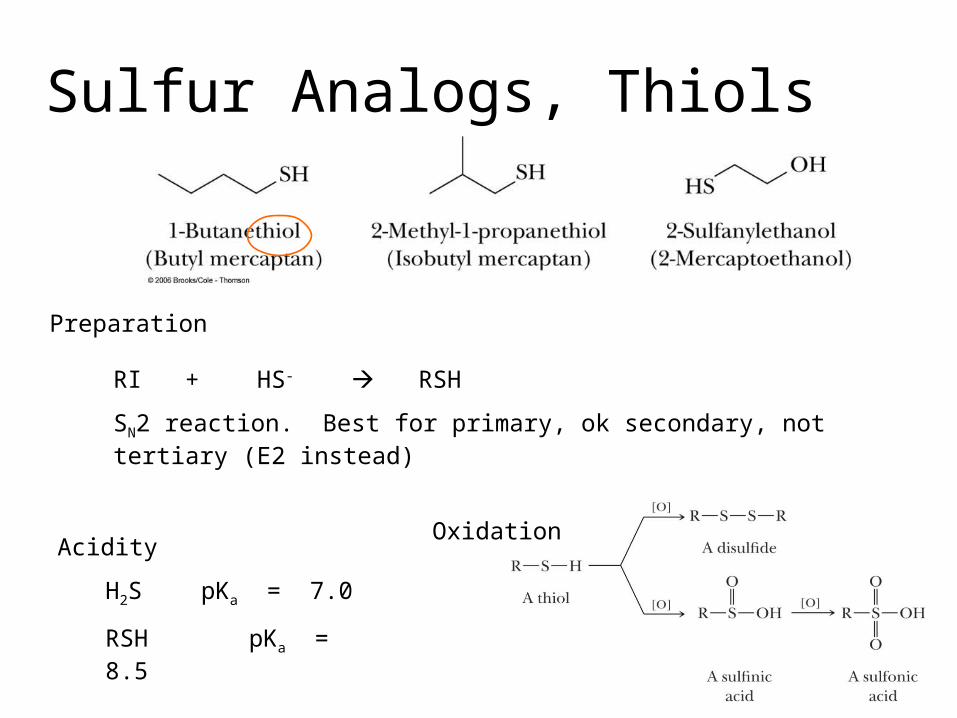
Sulfur Analogs, Thiols
Preparation
RI + HS- RSH
SN2 reaction. Best for primary, ok secondary, not tertiary (E2 instead)
Acidity
H2S pKa = 7.0
RSH pKa = 8.5
Oxidation








![Enhanced dual-beam excitation photoelectric detection of ... · E2, [NV-] ~ 20 ppm) and annealed. Coplanar electrodes with a distance of 100 µm (samples R, E2 and A) or 50 µm (sample](https://static.fdocuments.in/doc/165x107/5e14056fe10469153672bf0a/enhanced-dual-beam-excitation-photoelectric-detection-of-e2-nv-20-ppm.jpg)









