
Durham E-Theses Ring opening metathesis polymerisation of
112
• • •
Transcript of Durham E-Theses Ring opening metathesis polymerisation of

Durham E-Theses
Ring opening metathesis polymerisation of
phenylnorbornene dicarboxyimide derivatives
Mzanywa, Neeba Lucky
How to cite:
Mzanywa, Neeba Lucky (2003) Ring opening metathesis polymerisation of phenylnorbornene dicarboxyimide
derivatives, Durham theses, Durham University. Available at Durham E-Theses Online:http://etheses.dur.ac.uk/3685/
Use policy
The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission orcharge, for personal research or study, educational, or not-for-pro�t purposes provided that:
• a full bibliographic reference is made to the original source
• a link is made to the metadata record in Durham E-Theses
• the full-text is not changed in any way
The full-text must not be sold in any format or medium without the formal permission of the copyright holders.
Please consult the full Durham E-Theses policy for further details.

Academic Support O�ce, Durham University, University O�ce, Old Elvet, Durham DH1 3HPe-mail: [email protected] Tel: +44 0191 334 6107
http://etheses.dur.ac.uk
2

Ring Opening Metathesis Polymerisation of
phenylnorbornene dicarboxyimide
derivatives
A thesis submitted for the degree of Master of Science
at the University of Durham
By
N ceba Lucky, Mzanywa
First degree in Chemistry (Fort Hare, South Africa)
A copyright of this thesis rests with the author. No quotation from it should be published without his prior written consent and information derived from it should be acknowledged.
September 2003
2 3 JUN 2004

Nceba lLuncky M.ZANYW A
JRiDll.g Opening Metathesis Polymerisation of lPhenylnorbornene Dicarboxyimide Derivatives
M§c 2003
ABSTRACT
The work described in this thesis illustrates that a series of exo-phenyl
norbomene dicarboxyimide derivatives are synthesised and characterised using several
spectroscopic methods. It also demonstrates that polymeric material can be prepared
from phenylnorbomene dicarboxyimide derivatives using ROMP. Three well-defined
initiators, ruthenium t-butoxy molybdenum
Mo[CHC(CH3)3][CIOH17N][OC(CH3)3h, Mo (1), and hexafluoro-t-butoxy molybdenum
Mo[CHC(CH3)2C6Hs][CwH!7N][OC(CF3)2CH3h, Mo (2), were used. The polymers
prepared were fully characterised using 1H NMR, 13CNMR, elemental analysis, GPC
and DSC.
The cis I trans contents of the polymers are readily determined from the 1 H
NMR spectrum from the integration of the resonance attributable to cis and trans
olefinic hydrogen. Generally the trans vinylene content for polymers prepared by
ruthenium initiator is about 83%, for the polymers prepared by t-butoxy molybdenum
initiator is about 97% and for the polymers prepared by hexafluoro-t-butoxy
molybdenum initiator is about 31%. The t-butoxy molybdenum initiator produced
polymers with narrow PDI compared to hexafluoro-t-butoxy molybdenum and
ruthenium initiators. This is an indication of a well-controlled living polymerisation
reaction. The hexafluoro-t-butoxy molybdenum initiator produced polymers with
broader PDI. This can be attributed to the secondary metathesis reactions or to the rate
of propagation being faster than the rate of initiation. Polymers obtained by ruthenium
initiator also gave polymers with broader PDI due to secondary metathesis reaction.
The Tg for the polymers prepared by t-butoxy molybdenum initiator were about
l5°C higher than the polymers produced using hexafluoro-t-butoxy molybdenum and
the ruthenium initiators, suggesting that the higher the trans content of the polymer, the
higher the Tg. It was also found that the longer the alkyl chain in the polymer, the lower
the glass transition temperatures. The Tg for the polymers prepared from of exo-phenyl
norbomene dicarboxyimides are generally higher than the polymers obtained from exo
alkyl norbomene dicarboxyimides.
11

ACKNOWLEDGEMENTS
First, I would like to thank my supervisor, Dr Ezat Khosravi for his support and
guidance throughout my work in the department. I would like to thank the NMR staff:
Dr Alan Kenwright, Mr Ian McKeag and Mrs Catherine Heffeman, GPC staff, mass
spectroscopy staff: Miss Lara Turner and Dr M. Jones, Leeds University for DSC: Peter
Hine, and elementary analysis staff: Mrs Joroslava Dostal. I would like also to thank my
colleagues in Dr Ezat's group and all the IRC colleagues. I would also like to thank
Ruth First scholarship friends, African & Caribbean society friends, Durham Amigos
friends, and Dr Luthando Adams for their support throughout my stay in Durham and,
finally my family for their prayers.
MEMORANDUM
The work of this thesis was carried out in the IRC Chemistry laboratories of the
University of Durham between October 2002 and September 2003. This work has not
been submitted for any other degree and is the original work of the author, except where
acknowledged by reference.
FINANCIAL SUPPORl!
I gratefully acknowledge the funding provided by Ruth First Scholarship and Durham
University.
lV

Contents Page
Abstract .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ... ii
Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. iv
Memorandum .. .. . . .. . .. . .. . .. . .. . .. . .. .. .. .. . .. . .. . .. .. .. . .. .. .. .. . .. .. . .. .. .. .. . .. .. .. .. . .. .. iv
Financial support .. . .. . .. . .. . .. . .. . .. . . .. .. . .. . .. . .. .. .. . .. .. . .. .. . . .. . .. .. .. .. . . . .. .. .. .. .. .. . iv
Chapter 1: General introduction and background
1.1 Aims and objectives . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2 Ring opening Metathesis polymerisation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
1.2.1 Definition and historical background of olefin
metathesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
1.2.2 ROMP mechanism . . .. .. .. . . . . . .. .. . .. . .. . .. . . .. .. .. .. . . . .. . . .. .. . .. ..... 7
1.2.3 ROMP initiators . .. . . . .. . .. . .. . . .. .. . .. . .. . . . . .. . .. . . . . . . . . .. .. .... .. .... .. .. 8
1.2.3.1 Ill-defined initiators .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. ....... 8
1.2.3.2 Well-defined initiators .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. .. ........ 9
1.2.4 Microstructure and polymer chains ................................... 15
Chapter 2: Synthesis and characterisation o~ monomers
2.1 Introduction .... 0 0 .... 0 0 0 0 ..... 0 0 00. 00 •••• 0 ... 00 •••••••••••••••••••••• 0 .. 0 .... 00 0 .... 19
2.2 Diels-Alder reaction 0 ................ 00 ............................................ 19
2.2.1 Synthesis and characterisation of
exo-norbomene-5,6 dicarboxyanhydride. 0. 00 ......................... 21
2.3 Synthesis and characterisation of exo-phenylrtorbomene
dicarboxyimide derivatives oooooooooooooo•••••••oo• 24
2.4 Experimental .. 0 ............................................. 0 ....... 0 0 0 ....... 0..... 27
2.4.1 Synthesis and characterisation of
exo-norbomene-5,6 dicarboxyanhydride .. 0 .......... 0 0 ........ 0 0 0 0 ... 27
2.402 Synthesis and characterisation of
exo-N-phenylnorbomene-5,6-dicarboxyimide . 0 ...... 0. 0 ............ 0 28
2.4.3 Synthesis and characterisation of
exo-N-benzylnorbomene-5,6-dicarboxyimide .... 0 0. 0 ... . . .......... 29
2.4.4 Synthesis and characterisation of
exo-N-phenylethylnorbomene-5,6-dicarboxyimide 0 0 0 0 0 0 0 0 0 .. 0. 0 0 30
2.405 Synthesis and characterisation of
V

exo-N-phenylpropylnorbomene-5,6-dicarboxyimide . . . . . ... . . .... 31
2.4.6 Synthesis and characterisation of
exo-N-phenylbutylnorbomene-5,6-dicarboxyimide .. . . . .... . .. .. . 31
2.4.7 Synthesis and characterisation of
exo-N-tert-butylphenylnorbomene-5,6-dicarboxyimide . . . . . . . ... 32
2.4.8 Synthesis and characterisation of
exo-N -pentylphenylnorbomene-5, 6-dicarboxyimide . . . . . . . . . . . . . . 3 3
2.4.9 Synthesis and characterisation of
exo-N-hexylphenylnorbomene-5,6-dicarboxyimide ................ 34
2.4.1 0 Synthesis and characterisation of
exo-N-hexylnorbomene-5,6-dicarboxyimide ........................ 35
Chapter 3: Polymer synthesis and characterisation
3.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. . .. . 3 7
3.2 NMR scale solution polymerisation of
exo-phenylnorbomene dicarboxyimide monomers . . . . . . . . . . . .............. 37
3 .2.1.Polymerisation using ruthenium initiator. .................... 3 7
3.2.2 Polymerisation using t-butoxy molybdenum initiator ...... 39
3.2.3 Polymerisation using hexafluoro-t-butoxy
molybdenum initiator......................................... 40
3.2.4 Copolymerisation of exo-ethylphenylnorbomene
dicarboxyimide and exo-hexylnorbomene dicarboxyimide ..... .41
3 .2.5 Block copolymerisation of exo-ethylphenylnorbomene
dicarboxyimide and exo,endo-norbomene
dimethylcarboxylate ......................................... .44
3.3 Scale up polymerisation reaction .................................................. 47
3.3.1 Polymer characterisation by 1H and 13C
spectroscopy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. . . . . . . . 49
3. 3.2 Polymer characterisation by elemental analysis . . . . . . . . . 61
3.3.3 Polymer characterisation by GPC .. .. . .. .. . .. .. .... 62
3.3.4 Polymer characterisation by DSC . . . . . . . . . . . . . . .... .. . . ... 63
Chapter 4: Overall conclusion and proposal for future work
4.1 Overall conclusion................................................................. 66
4.2 Proposal for future work. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. . . . . 67
VI

Appendix 1: Instrumentation and procedure for measl!lrements ..................... 68
Appendix 2: Analytical data for chapter 2 ................................................. 71
Appendix 3: Analytical data for chapter 3. .. .. .. .. ..... .. . .. .. .. .. ...... .. .. .. .. .. . ...... . 84
Appendix 4: Conferences and seminars attended ....................................... 100
REFERENCE§ .............................................................................. 102
VII

Chapter 1
General introduction and background
Chapter I

1.1 Aims and objectives
Dicyclopentadiene (DCPD) has been widely used as a monomer for polymer processing
methods like Ring Opening Metathesis Polymerisation (ROMP)-Reaction Injection
Moulding (RIM) and ROMP-Resin Transfer Moulding (RTM) for producing material
with good mechanical properties, figure 1.1. 1, 2 Although this has resulted in important
commercial applications, there are some drawbacks like strong monomer odours,
strongly exothermic reaction and problems surrounding the cross-linked material.3
ROMP
Dicyclopentadiene
Poly(dicyclopentadiene)
f-·0~
Cross-linked polymer
Figure 1.1: Cross linking in dicyclopentadiene
In the earlier work4, a series of mono functional n-alkyl norbornene dicarboxyimides
and difunction n-alkylene dinorbornene dicarboxyimides was successfully synthesised,
figure 1.2. The monomers were subjected to ROMP using classical initiators and also
well-detlned molybdenum initiators; the t-butoxy molybdenum initiator
Chapter I 2

Mo[CHC(CH3h][C 10H17N][OC(CH3)3]2, and hexafluoro-t-butoxy molybdenum initiator
Mo[CHC(CH3)2C6Hs][CJOH17N][OC(CF3)2CH3]2. This work served as a benchmark
towards the synthesis and characterisation of polymeric material using RIM-ROMP and
RTM-ROMP.
Figure 1.2: Structure of the (a) monofuntional monomer and (b) the difuntional monomer
The work5 that followed explored further the physical and thermal properties and
provided guidelines and basic parameters for ROMP-RTM processing of norbomene
dicarboxyimides. The initiator used for ROMP reactions was well defined ruthenium
initiator Ch[(C6H 11 )3P]2Ru=CHC6Hs. The Copolymerisation of monofunctional and
difunctional monomers produced well-defined crosslinked materials, figure 1.3.
Chapter I 3

+
RTM-ROMP
Figure 1.3: Cross linking in substituted norbomene
The physical and mechanical properties of the cross linked materials were comparable or
better than some engineering polymers such as polycarbonates and Telene 1100, table
1.1.
I
Polymer Tg Density Modulus Yield/Fracture
kg/m3 Strength
(oC) GP a (Mpa) C4M/2%C12D ~120 1136 2.7+- 0.2 61 +- 15 (F)
C5M/2%Cl2D 106 1120 2.2+- 0.2 72 +- 2 (Y)
C6M/2%C12D 87 1092 1.9 +- 0.1 59+- 3 (Y)
Poly carbonate 150 1200 2.3 62 (Y)
Poly(DCPD) 145 1030 1.9 45 Telene1100
Table 1.1: Shows the comparison of' the new polymeric material with poly(dicyclopentadiene)
However, the Tg of the crosslinked materials were lower than the commercial materials
based on Poly(DCPD).
Chapter I 4

The aim of this work was to improve the glass transition temperature of the material
produced by ROMP-RTM. According to the literature the presence of the phenyl ring
on the side chain of the polymer would raise the glass transition temperature.6
Therefore the objective of the work presented in this thesis was to synthesise and fully
characterise a series of exo-phenyl norbornene dicarboxyimide derivatives, figure 1.4,
subject them to ROMP using well-defined Grubbs ruthenium initiator, and evaluate
their Tgs of the resulting polymers.
Figure 1.4: Structures of phenyl norbornene dicarboxyimide
This work is divided into 4 chapters. The first chapter deals with the historical
background to ROMP and the latest developments on the initiators used in this process.
The second chapter deals with the synthesis and characterisation of monomer, the third
chapter deals with the polymer synthesis and characterisation and the last chapter deals
with conclusion and proposal for future work.
1.2 Ring Opening Metathesis polymerisation (ROMP)
1.2.1 Definition and! historican background of olefin metathesis
Olefin metathesis was discovered in 1920s. It was only 50 years later that it found
usefulness in polymer chemistry and only recently that a lot of useful materials are
produced through this method. Olefin metathesis can be described as the catalytically
induced bond reorganisation reaction that lead to the breaking and making of carbon
carbon double bonds without losing the total and type of chemical bond in the process.
For acyclic olefins the process lead to the exchange of alkylidene units (a) and, for
Chapter I 5

cyclic olefins the process lead to ring scission and the formation of linear polymer (b),
figure 1.5.
(a) +
Figure 1.5: Schematic presentation of olefin metathesis
(a) Schematic representation for acyclic olefins
(b) Schematic representation for cyclic olefins
The reaction for acyclic olefins was first reported by Banks and Barley in 1964 and was
termed 'olefin disproportionation' .7 Three years later, in 1967, Calderon et a/
acknowledged that acylic disproportionation of olefins and ring scission of cyclic
olefins to form linear polymers were example of the same process and hence they
commonly name these reactions 'olefin metathesis' .8 Anderson and Merkling, in 1955,
discovered evidence of olefin metathesis although it was only reported in the 1960s.9 In
the experiment they successfully polymerised bicyclo[2.2.1 ]hept-2-ene using a mixture /
of titanium tetrachloride and ethylmagnesium bromide to initiate the polymerisation,
figure 1.6.
nab TiC14 / EtMgBr
Bicyclo[2.2.1]hept-2-ene Poly(norbornene)
Figure 1.6: Romp of norbornene
Chapter l 6

Several mechanisms were proposed for the olefin metathesis reaction. I0-!2 The widely
accepted mechanism was proposed by Chauvin. The mechanism involved cycloaddition
reaction between metal alkylidene complex and the olefin. The addition proceeds via an
intermediate metallacyclobutane that breaks up to give new alkylidene and a new olefin,
figure 1. 7. This mechanism was a break through to catalyst development as it provided
the basis for the understanding of the way the catalyst system work. This resulted in the
development of single component and well-defined homogeneous catalyst in 1970s and
1980s.14 These new catalysts will be discussed under ROMP initiators
R
[M]~
~~~ --........
Figure 1.7: Chauvin's mechanism for olefin metathesis
1.2.2 ROMP mechanism
Ring opening metathesis polymerisation (ROMP) is a variant of olefin metathesis in
which cyclic olefins are used and new olefin remains attached to the catalyst as part of
the growing chain, figure 1.8. Although ROMP has the same mechanism as the one
Chauvin proposed, the difference is with the second step of ROMP, which is in most
cases is irreversible. With advancement in catalyst development, ROMP system are
considered as living system as there is no termination step, which means that the
polymer chain ends remain active and on addition of the same monomer, the chain
continue to grow until tem1ination is induced. The sequential addition ofthe monomers
results in the synthesis of block polymers.6
Chapter 1 7

0 + -----il>
0 I I
[M]-CHR
-----il>
[M]=CHR
j n 0
Figure 1.8: Mechanistic pathway for ROMP
1.2.3 ROMP initiators
The initiator systems used for ROMP are based on group IV to group IX transition
elements such as Ti, V, Nb, Ta, Mo, W, Re, Ru, Os, and Ir.4'15 The history of initiators
started as early as in 1960's with development of catalyst that were ill-defined but
useful at the time to the present catalyst that are well-defined. The initiators are
therefore categorised into two systems, namely classical (ill-defined systems) and single
component initiators (well-defined systems).
1.2.3.1 Ill-defined initiators
Classical initiators are multiple component systems and are derived from chlorides,
oxides, and oxychlorides of Mo, W, and Re. Cocatalyst for these systems are usually
EtAlCh and ~Sn were R= Ph, Bu or Me and promoters being 0 2, EtOH and PhOH.
These systems can be homogeneous, e.g. WCl6/EtAlCh/EtOH and heterogeneous, e.g.
Mo03/Al03. These initiators suffer from a lot of drawbacks such as:
Chapter 1 8

e The precise nature of the active site at the metal centre is unknown, thus the system
is ill-defined
• The metal carbene must be generated before initiation and subsequent propagation
can commence and this process usually proceeds in a very low yield
• The activity of a given initiation system is dependent upon its chemical, thermal and
mechanical history, and upon the order and rate of mixing ofthe catalyst, eo-catalyst
and monomer
• They have limited tolerance towards functional groups in the monomer or solvent,
• There is a lack of control of molecular weight and molecular weight distribution due
to intra and intermolecular reactions with the double bond
• They display an element of irreproducibility
1.2.3.2 Well-defined initiators
Although classical initiators are still having their place in commercial applications, the
development of single component initiators has promoted a lot of research in ROMP
and resulted in synthesis of polymers with complex architectures that would not have
been possible with ill-defined initiators. Most of this success is due to the fact that the
development of single component initiators has set a benchmark for more improved
initiators with greater functional group tolerance.
The Fischer carbene, which was heteroatom stabilised complex, was the first isolated
metal cm·bene species to be reported in 1964, figure 1.9. 16 The discovery of Casey
carbene, a diphenyl complex followed this in 1973, figure 1.9. 17 Although Casey
carbene was not heteroatom stabilised complex, it was proved to be more reactive
capable of polymerising less strained olefins.
Fischer carbene Casey carbene
Figure1.9: Structural representation of Fischer and Casey car bene Chapter I 9

Table 1.2 show the advancement of functional group tolerance of the initiators from
Titanium initiator, which was only reactive to olefins but could not survive other
functional groups to ruthenium, which is tolerant to wide range of functional groups.
Due to this tolerance, ruthenium has gained a lot of attention in ROMP and this has lead
to more research done to improve the initiation capability of this initiator. Researchers
have noticed that the development of more reactive initiators has a cost on the stability
of the initiator. There is a growing interest to develop initiators that would be more
reactive but maintain the stability.
;, ftt'lhlillm , ; f ifimgst~n iii\16IY.tia~~~rtl : "l,tlltlieniUn,t, '
,>
Alcohol, water Alcohol, water Alcohol, water OLEFINS
Acids Acids Acids ALCOHOL, WATER
Aldehydes Aldehydes Aldehydes ACIDS
Ketones Ketones OLEFINS ALDEHYDES
Esters, amides OLEFINS KETONES KETONES
OLEFINS ESTERS, AMIDES ESTERS, AMIDES ESTERS, AMIDES
Figure 1.2: Outline of different initiators and their respective tolerance to a range
offunctional groups. The tolerance is increased from left to right. The
reactivity of the functional groups is increased from bottom to top.
Tebbe et al prepared a bimetallic methyledene18•19 (Tebbe reagent), which can be
classified as a well defined Ti carbene that is stabilised by coordination of Me2A1Cl and
this carbene set as the benchmark for the discovery of living polymerisation.
Titanacyclobutane complexes shown in figure 1.10 produced the first living
polymerisation of cycloolefin?0-23 These Ti complexes were prepared from Tebbe
reagent. Although these complexes were capable of forming polynorbornene with
narrow molecular weight distribution, the disadvantages were that the polymerisation
required a temperature of 50°C and the functional group tolerance was limited to
olefins.
Chapter 1 10

Tebbe reagent
Cp';,- ~ l c~ II_JJ0
Cp". )r crflj
Tit~nacyclobutane complexes
Figure 1.10: Structural representation ofTebbe reagent and Titanacyclobutane
complexes
Tungsten alkylidene complexes were the next to be isolated. Kress and Osborn prepared
the first well-defined tungsten alkylidene complex24, which required a strong Lewis acid
eo-catalyst such as GaBr3 for increased activity, figure 1.11 The functional group
tolerance was extended esters and amides. Schrock and eo-workers reported well
defined tungsten and molybdenum complexes with bulky alkoxide and arylimido
ligands, figure 1.12.25 The bulky alkoxide groups and imido ligand of both complexes
served as a shield towards intermolecular reactions. Moreover, molybdenum complexes
showed better tolerance, compared to tungsten, which extended functional group
tolerance to ketones.
Figure 1.11: Structural representation ofKress and Osborn complex
m--N M= Wor Mo
11 R = CM8:J or CMe2Ph
R'O-M =CHR R' =CM~ or CM~CF3 or CMe(CF3)2
I R'O
Figure 1.12: Structural representation ofSchrock's initiators
Chapter I 11

The most widely studied are t-butoxy substituted molybdenum initiator (complex 1) and
hexafluoro-t-butoxy substituted molybdenum initiator (complex 2), figure 1.13. 4'25
-29
Molybdenum complex 2 was found to be the most reactive of all the molybdenum
derivatives.
1 2
Figure 1.13: Structural representation of two molybdenum complexes and ruthenium
complex.
The discovery of ruthenium alkyledene complexes extended the scope to a wide range
of functional group tolerance and hence they have been widely studied in recent years.
The history of ruthenium based initiators started with the simple salts to well-defined
ruthenium alkyledene complexes. Ruthenium salts did not get much exposure in ROMP
because of low reactivity and limited functional group tolerance. Other attempts resulted
in ROMP but with longer reaction time and the complexes were only limited to highly
strained substrates14. The isolation of ruthenium alkyledene complex 3 was a
breakthrough in ruthenium catalyst development, figure 1.14. 30 Although the discovery
of this complex was a step forward, the drawback was that the complex was limited to
only highly strained monomers. The research work that followed, led to the discovery of
the second series of these ruthenium alkylidene complexes, complex 431 and 532,
figure 1.14. The work focused on modification of the ligand environment so as to extend
the activity to low strained monomers. Complex 4, was found to have a broader
metathesis activity that incorporated metathesis of acyclic olefins and an increased33
tolerance to most functionalities. However, complex 5 was found to be the most active
complex of the series.
Chapter I 12

PPh3 PCy3 PC~ Cl" I Cl" I Cl" I I: /Ru~Ph CI/R~Ph CI/Ru
Cl I - Ph I Ph I H PPh3 PCy3
PCy3
3 4 5
Figure 1.14: Series of ruthenium complexes
Further work led to the discovery of a family of mesityl-substituted N-heterocyclic
carbeme, figure 1.15. It was reported that complexes 634 and 735 were more reactive
than complex 5 and complex 735 was the most reactive. These complexes expanded the
scope ofthe monomers to sterically hindered olefins?4,3
6
6 7
Figure 1.15: Mesityl-substituted N-heterocyclic carbenes
The drawback to complex 5 or its most active derivative 7 was broad polydispersity
indices (between 1.3 and 1.5), which are attributed to unfavourable rate of initiation (k;)
as compared to propagation (kp) and secondary metathesis. This has resulted in
synthesis of a new family of Ru-based initiators by substituting one of PCy3 ligand with
different phosphines. Gibson and eo-worker established that the initiation efficiency of
5 can be enhanced by replacing one of the PCy3 ligand with Cy2PCH2Si(CH)3 to form a
new initiation complex 8, figure 1.16?7 The enhanced initiation was attributed to lower
basicity and smaller size of Cy2PCH2Si(CH3)3.
Chapter 1 13

Figure 1.16: TMS ligand exchange in ruthenium initiator
Work by Grubbs et al showed that the olefin metathesis reaction catalysed by ruthenium
complexes 5 or 7 proceed via a dissociative mechanism which can either bind to free
PCy3 to form the initial alkyledene complex or bind to the substrate and undergo
metathesis, figure 1.17.38
L
Cl" I (J Cl/ Ru-====<:::
H
k2 +olefin
-olefin
k_2
Figure 1.17: Dissociative mechanism of olefin metathesis
L
Cl" I 11) Cl/ Ru-====<::: / I H
R
Recent research by Bielawski and Grubbs has shown that addition of phosphine in the
polymerisation reaction can enhance the initiation efficiency, without having to
synthesize a new complex. The PDis in a particular experiment improved from 1.25
without the added phosphine to 1.04, depending on the type and number of equivalents
of phosphine added and also to the number of equivalents of monomer. This was due to
the fact that the rate of phosphine exchange is faster, as the result the formation of a new
complex with labile phosphine can occur before initiation. They concluded that
polymers with narrow polydispersity could be achieved this way, figure 1.18.39
Chapter 1 14

PR3 (fast)
Figure 1.18: Schematic representation of phosphine exchange mechanism
1.2.4 Microstructure of polymer chains and stereo regularity
For ROMP systems there are three main factors that define the microstructure of the
polymers:
o Cis/trans vinylene ratios and distribution
o Tacticity effect
o Head to head, head to tail and tail-to-tail arrangements
(i) Cis/trans vinylene ratios and distribution.
The repeating units in the backbone of the polymer chain are distributed in a cis and
trans configuration, figure 1.19. The distribution depends on the type of solvent,
monomer and catalyst system and concentration. For example for molybdenum catalyst
systems, the molybdenum (1) catalyst gives high trans distribution whereas
molybdenum (2) gives high cis configuration.4
Cis
Trans
Figure 1.19: Cis I trans double bonds in polynorbomene
Chapter 1 15

(ii) Tacticity effect
The repeating units of polymers like polynorbornene have chiral centres and each
configurational unit of a polymer chain is the dyad, figure 1.20. A meso dyad is
observed when the chiralities of the two chiral carbons adjacent to the double bond are
opposite and a series of meso dyads gives isotactic polymers. A racemic dyad on the
other hand is observed when the chiralities of the two chiral carbons adjacent to double
bond are the same and a series of racemic dyads gives syndiotatic polymers.
Cis - isotactic
Cis - syndiotactic
Trans - isotactic
Trans - syndiotactic
Figure 1.20: Tacticity effect in polynorbornene
(iii) Head to head, head to tail and tail-to-tail arrangements.
Unsymmetrically substituted monomers give rise to head to head, head to tail or tail-to
tail form. Figure 1.21 shows the different fonns. The polymers prepared from this thesis
are synthesized from symmetrically substituted monomer and therefore, the polymers
will not experience the tail I head effects.
Chapter 1 16

R TTTT HH HH TH HT
Figure 1.21: Head I Tail effects in the polymer ofunsymmetrical
substituted monomer
Chapter 1 17

Chapter 2
Synthesis and characterisation of the monomers
18

2.1 Introduction
The focus of this chapter is on the synthesis of monofunctional phenylnorbornene
dicarboxyimide derivatives. These are synthesised in two steps. The first step is the
Diels-Alder cycloaddition reaction of maleic anhydride and dicyclopentadiene to give a
mixture of exo I endo -norbornene-5,6-dicarboxyanhydride. This is followed by
rec1ystallisation in acetone for several times (at least five times) to give pure exo
norbornene dicarboxyanhydride (exo-AN). The second step is the reaction of the pure
exo-AN with phenylamines to produce phenylnorbornene dicarboxyimide derivatives.
Only exo-monomers are synthesized in this work, as they have proved in previous work
to be very reactive for ROMP reactions.5•40
2.2 Diels-Alder reaction
The reaction involves conjugate addition of a conjugated diene to an alkene, dienophile,
to produce a cyclohexene. It is one of the few methods available for forming cyclic
molecules and hence it is also called cycloaddition as the reaction produce cyclic
products.41 The simplest example is the reaction of 1,3-butadiene with alkene to form
cyclohexene. The reaction proceeds through a cyclic transition state in a single step
process.42
+ 11
heat 0 1 ,3-butadiene ethene cyclohexene
Figure 2.1: The Diels-Alder reaction between a conjugated diene and dienophile.
It is a widely used and studied method in organic synthesis ever since it was first
reported in 1928.43 It has gained synthetic applicability in the synthesis of compounds
such as drugs, dyes and polymers chemistry. The experimental procedures for this
reaction are usually simple and it produces good yields. For Diels-Alder cycloaddition
19

to occur under normal conditions the dienophile must contain electron withdrawing
group and the diene must contain electron-donating group.
Diels-Alder reaction is stereospecific with respect to both the diene and dienophile as
the result the stereochemistry of the dienophile determines the stereochemistry of the
resulting product.41 Most importantly the diene must be in the cis conformation in order
to react.44 This relationship is illustrated in figure 2.2.
( +
R'
( R
- CCR R
cis-dienophile cis-substituted product
R'
f R --trans-dienophile trans-substituted product
Figure 2.2: A schematic representation showing the dependence of the stereochemistry of the
product on the stereochemistry of the dienophile in a Diels-Alder reaction.
The stereoisomeric product of cyclic dienes depends on the orientation of dienophile to
the diene in the transition state and that results in two possible additions that is endo and
exo.45 If the dienophile lies under the diene an endo product is formed and if the
dienophile lies away form the diene the exo is formed. Endo product is usually the
major product due to kinetic control and due to the best overlap when the two reactant
lies on top of each other.46'47
20

~0 0
exo addition
Orientation of dienophile in the transition state
Hesunder I
0
~saway
~ 0~ 0
--- ~0 0
Endo addition
Figure 2.3: The schematic representation showing the dependence of type of addition on the orientation of the
dienophile from the diene in the transition state of a Diels-Alder reaction.
2.2.1 Synthesis and characterisation
dicarboxyanhydride
~: 0
Maleic anhydride
+ Dichlorobenzene
180°C
Dicyclopentadiene
of exo-norbornerne-5,6-
~0 0
Mixture of exo and endo-AN
Recrystallisation (in acetone)
~0 0
Pure exo-AN
Figure 2.4: Outline ofthe synthetic route for exo-AN
The synthesis of exo-norbomene-5,6-dicarboxyanhydride (exo-AN) was the result of a
Diels-Alder reaction between maleic anhydride and dicyclopentadiene.4·5•48 The
21

reaction was carried out at 180°e in dichlorobenzene as the solvent for 16 hours. At this
temperature, which is the boiling point of dichlorobenzene, dicyclopentadiene was
cracked into two cyclopentadiene, which then react with the maleic anhydride .. The
product formed by the reaction was the mixture of exo and endo-norbornene-5,6-
dicarboxyanhydride. In order to get a pure exo-adduct, the mixture was recrystallised
several times from acetone (at least five times). The product was clear, crystalline solid.
Analytical methods like 1 H and 13e NMR, elemental analysis, and mass spectroscopy
were used to confinn the product. The mass spectrum showed a parent peak (M) at
164, which corresponded with the molecular weight of exo-AN and the peak at 66
correspond to cyclopentadiene. The mass spectrum is shown in figure 2.5. The peaks in
the 1H and 13e NMR are similar to the peaks for exo-AN in the previous work5 and the
spectrum is shown in figure 2.6. The results are found in the experimental section ofthis
chapter.
1001 66 4.0E6
95 M+-C,H,01 3.8E6
90 91 3.6E6
85 3.4E6
80 '3 .2E6
75 3.0E6
70 2.8E6
65 2 .6E6
60 2.4E6
55 2.2E6
50 2.0E6
45 1.8E6
40 1.6E6
35 _1.4E6
30 1.2E6
25 l.OE6
20 8.0E5 51 120
6.0E5 15
10
,J 11
M·-co, 164 4.0ES
5 J~ ~~~~ y -f. M•, C,H801 2.0ES
0 I O.OEO
40 50_ 60 70 80 90 160 iio 1 0 l:lo 140 lSO 160 i?o 1ao i9o 260 2 0 m/z
Figure 2.5: Mass spectrum of exo-norbornene-5,6-dicarboxyanhydride
22

(a)
2,3
7 6 5 4
2,3
8,9
5,6
1,4
3 2
1,4 5,6
7
*
I I
pp m
I) 11 iljfil ijlli I ji ill Jllii jlliijii I 1]1 illjlllljillljlllfjll I I) ill 1 jlliijll iljllllji 11 ijlllljililj
180 160 140 120 100 80 60 40
Figure 2.6: (a) 1HNMR spectrum of exo-AN in acetone
(b) 13CNMR spectrum ofexo-AN in acetone
* Residual hydrogen in acetone
o Water in acetone
pp m
23

2.3 Synthesis and characterisation of exo-phenylnorbornene dicarboxyimides
~0 118-120°C AcOH
~N-R 0 0
Pure Exo-AN Exo-monomer
Figure 2.7: Outline of the synthetic route for exo-monomers where R-NH2 is phenyl amines
Synthesis of exo-monofuntional and exo-difuntional monomers using aliphatic amines
has been done successfully in the previous work using Diels-Alder reaction giving
product in high yields. In this work the focus was on the use of a series of phenyl
amines. The synthesis of the monomers was the result of a reaction between the pure
exo-AN and a series of phenyl amines. The reaction was carried out at l20°C using the
solvent glacial acetic acid. At this temperature, amines were added drop wise for a
certain period of time to react with exo-AN. After the addition was complete, the
reaction mixture was left for a further two. hours to reflux. All monomers were
recovered as solids except exo-C6M, which was recovered as the clear liquid. A single
recrystallisation was necessary to purify the. solid monomers, while exo-C6M was
purified by distillation. All monomers were soluble in chlorinated solvents. Analytical
methods like 1H and 13C NMR, elemental analysis, and mass spectroscopy were used to
confirm the product. Figure 2.8 shows an example of one of the monomers.
~0 AcOH
~N-o 0 0
Pure Exo-AN Exo-PhM
Figure 2.8: Outline ofthe synthetic route for exo-N-phenylnorbornene-5,6-dicarboxyimide
24

The NMR analysis of these monomers was similar to that of the previous work, with the
phenyl protons found in the region from 7.26 to 7.47ppm and the phenyl carbons in the
region from 126.03-151.89ppm. The result for mass spectroscopy showed that the
parent peak (M+) of each spectrum corresponded with the molecular weight of the
corresponding monomer and in addition there were two peaks, (66) and ~-66), which
are characteristic peak of reverse Diels-Alder reaction. The result found for elemental
analysis was close and in some cases the same as the calculated values. The results of
these monomers are found in the experimental section of this chapter. To illustrate the
characterisation of the monomers, figure 2. 9 shows the mass spectrum of the exo-PhM
monomer and figure 2.10 shows the 1H NMR and 13C NMR. The expanded part in
figure 2.10 (a) and (b) shows the proton and carbon signal in the phenyl ring
respectively.
'10 eo ~0 T?O J 0 T10 J~O r~o :SQO :s~o 3 0 3~o - :s~o. JQO 3~0 3f0 3~0 W\~
0 11'" 11'111' 11 :sfo· O'OEO
2 .l'JE<J
TO J"2E2
12 3':SE2 103
:so ~ f'-C QH,No,na 3'31!2
:i2 2'1 .l 3".lE2
30 J:Sil M·-c,H,; <f"<fE2
32 2"JE2
'10 2"8E2
'12 e·eE2
20 M·-c.H.o, .l"3E2
22 M', C15H13NO, 8"0E2
eo 8"81!2
e2 333 il"2E2
.lO T"OEe
.l2 ilJ r·rEe
eo r:sEe
82 r:SEe
ilO J"JEe
32 T.l3 r<rEe
JOOi ee r·2Ee
Figure 2.9: Mass spectrum of exo-N-phenlynorbomene-5,6-dicarboxyimide
25

0
(a)
(b)
11 12
N~13 15 14
12,14 11,15
13
I I I I' I I I I I I I I I I I I I I' I I I I' I I I I' I I I I' I I I I' I I •
7
7.:/J
I I
I
10
I
I I
I I
I I
I 2,3
7.40 7.30 ppm
1,4
6 5 4
12,14 11,15
13
5,6
3
I I I I I I I I I 11 J I I I I I I 1 I I I I I I 1 I I I I I I I ) I I I I I I I I I I I I I I I I
• 134 131 128 pp m
2,3
* 8,9
1 I
7 7'
' I '
2 pp m
1,4 5,6
7
_l_ -'----4---l-'l: I _.e____;.-.---__}L_ _ _A.J._J___
jtlllj I I 11j I iltji it I j I 11 f[llltjll I I (1 I !1 j If I lj I 11 I if I I ljlltljtlltjl ri ij I 1 I tjt I I! j 11 ttjlf I I
180 160 140 120 100 80 60
Figure 2.10: (a) 1HNMR spectrum of exo-PhM in CDCh
(b) 13CNMR spectrum ofexo-PhM in CDCh
* Residual hydrogen in CDCh
40 pp m
26

2.4 Experimental
Reagents
Maleic anhydride, dicyclopentadiene, 1.2-diclorobenzene, n-alkylamine and n
phenylamines were purchased from A ldrich Company Ltd, acetic acid, acetone were
purchased from BDH Chemical Company Ltd. All reagents and solvents were used
without further purification.
2.4.1 Synthesis and characterisation of exo-norbornerne-5,6-dicarboxyanhydride
Maleic anhydride (490.2g, 5.0mol) and 1,2-dichlorobenzene (500ml) were placed in a,
3-necked round bottom flask (lOOOml). The flask was fitted with a stopper, a condenser
and a dropping funnel. The mixture was heated to 180°C and dicyclopentadiene (335ml,
2.5mol) was added via a dropping funnel. The mixture was left overnight to reflux and
brown solution was formed. The product was recovered from the solution by filtration
and dried in the vacuum oven. The product was yellow crystals, which was a mixture of
exo and endo-norbomene dicarboxyanhydride. The product was recrystallised five times
in hot acetone to give 100% pure exo-norbomene-5,6-dicarboxyanhydride.
(a) Elemental analysis of C9Hs03: calculated (found)
C = 65.85% (65.75%), H = 4.91% (4.89%), 0 = 29.24% (29.36%)
(b) 1H NMR (see appendix x), CDCh, 500 MHz, o (ppm)
6.37 (s, 2H, Hz,3 ), 3.34 (s, 2H, H1,4), 3.13 (s, 2H, Hs,6), 1.59 (d, 1H, H7),
1.39 (d, 1H, HT)
(c) 13C NMR (see appendix y), CDCh, 500 l.VIHz, o (ppm)
177.37 (Cs,9), 138.05 (Cz,3), 49.13 (Cs,6), 46.73 (CI,4), 43.95 (C7).
(d) Mass spectrum (see appendix z), (Ell:
164 (M+, C9Hs03), 120 (M+- COz), 66 (M+- C4H203)
27

Assignment of C and H atoms of exo-AN
2.4.2 Synthesis and characterisation of exo-N-phenylnorbornene-5,6-
dicarboxyimide: (exo-PhM)
Exo-norbornene-5,6-dicarboxyanhydride (5.003g, 0.03mol) and glacial acetic acid
(35ml) were placed in a 3-necked round-bottomed flask (lOOml). The flask was fitted
with a suba-seal, condenser and a thermometer. The top of the condenser was fitted with
drying tube. The mixture was heated to 120°C and aniline (2.84g, 0.03mol) was added
drop wise for 25min using a disposable syringe via a suba seal. The resulting yellow
solution was left to reflux for 2 hours. The solution was then poured into cold distilled
water (50ml) and a white precipitate was formed. Dichloromethane (50ml) was added
twice to extract the monomer. The monomer was then washed twice with distilled water
(50ml). The monomer was then dried over anhydrous magnesium sulphate.
Dichloromethane was removed under reduce pressure to give pale yellow crystals. The
monomer was recrystallised from toluene and was dried using a vacuum oven (5.6g,
0.023mol, 77%, mpt: 201 °C).
(a) Elemental Analysis of C15H13N02: calculated (found)
C = 75.30% (75.40%), H = 5.48% (5.47%), N = 5.85% (5.89%), 0 = 13.37% (13.24%)
(b) 1H NMR (see appendix x), CDCh, 500 MHz, o (ppm)
7.47 (t, 2H, H12,14), 7.39 (t, 1H, H 13), 7.26 (d, 2H, Hll,Is), 6.35 (s, 2H, H2,3 ),
3.41 (s, 2H, HI,4), 2.86 (s, 2H, Hs,6), 1.62 (d, lH, H7), 1.49 (d, 1H, HT)
(c) 13C NMR (see appendix y), CDCh, 500 MHz, o (ppm)
177.32 (Cs,9), 138.23 (C2,3), 132.05 (C 10), 129.42 (Cl2,I4), 128.91 (C13), 126.60 (CII,I5 ),
48.10 (Cs,6), 46.06 (CI,4), 43.20 (C7).
28

(d) Mass spectrum (see appendix z), (E:r).
239 ~' CisH13N02), 173 (~- CsH6), 91 ~- C9Hs02), 66 (~- CwH1N02)
7
11 12 ~ 11 12
3~---'013
Hh.!7 Hr 3 N~13
2 0 15 14 2 0 15 14
Assignments for C and H atoms of exo-PhM
2.4.3 Synthesis and characterisation of exo-N-benzylnorbornene-5,6-
dicarboxyimide: ( exo-PhCM)
The same procedure as in section 2.4.2 was followed using exo-AN (5.004g, 0.03mol)
and benzylamine (3.26g, 0.030mol). The monomer was recovered as white crystals,
which was recrystallised in hexane to give white solid (4.93g, 0.020mol, 64% yield,
mpt: 103°C).
(a) Elemental Analysis of C16H1sN02: calculated (found)
C = 75.87% (75.92%), H =5.97% (5.96%), N = 5.53% (5.53%), 0 =12.63% (12.59%)
(b) 1H NMR (see appendix x), CDCh, 500 MHz, o (ppm)
7.38 (d, 2H, H 12,t6), 7.30 (t, 2H, Hl3,I5), 7.28 (t, 1H, H14), 6.27 (s, 2H, H2,3), 4.61 (s, 2H,
H 10), 3.24 (s, 2H, H 1,4), 2.67 (s, 2H, H5,6), 1.40 (d, 1H, H1), 1.06 (d, 1H, HT)
(c) 13C NMR (see appendix y), CDCb, 500 MHz, o (ppm)
177.93 (C8,9), 138.16 (C2,3), 136.13 (C 11), 129.11 (CI2,I6), 128.88 (CI3,I5), 128.15 (C14),
48.02 (Cs,6), 45.52 (Ct,4), 42.87 (C1), 42.58 (Cw)
(d) Mass spectrum (see appendix z), (El+).
253 (~, C16HtsN02), 187 (~- CsH6), 91 (~- C9HsN02), 66 (M+- C1IH9N02)
29

H
12 13
8 10 -u11j \\ N-cH \ 14
9 2 ---
16 15 0
Assignment of C and H atoms for exo-PhCM
2.4.4 Synthesis and characterisation of exo-N-phenyletlllylnorbornene-5,6-
dicarboxyimide: ( exo-PhC2M).
The same procedure as in section 2.4.2 was followed using exo-AN (5.002g, 0.03mol)
and phenylethylamine (3.69g, 0.030mol). The monomer was recovered as yellow liquid,
which then solidified at room temperature. The monomer was recrystallised in ether to
give white solid (5.29g, 0.020mol, 65% yield, mpt: 94°C).
(a) Elementary Analysis of C17H17N02: calculated (found)
C = 76.38% (76.41 %), H = 6.41% (6.41 %), N = 5.24% (5.03%), 0 = 11.97% (12.15%)
(b) 1H NMR (see appendix x), CDCh, 500 MHz, c) (ppm)
7.28 (t, 2H, Ht4,t6), 7.23 (d, 2H, H13,t7), 7.22 (t, 1H, Hts), 6.26 (s, 2H, H2,J ),
3.72 (t, 2H, Hw), 3.22 (s, 2H, Ht,4), 2.88 (t, 2H, H 11 ), 2.63 (s, 2H, H5,6), 1.41 (d, IH,
H1), 1.02 (d, 1H, HT)
(c) 13C NMR (see appendix y), CDCh, 500 MHz, c) (ppm)
178.10 (Cs,9), 138.04 (C2,3), 137.93 (C12), 129.06 (Cn,!7), 128.76 (Ct4,t6), 126.92 (Cts),
48.00 (Cs,6), 45.33 (Ct,4), 42.86 (C7), 39.91 (Cw), 33.70 (Ctt).
(d) Mass spectrum (see appendix z), (Ell.
267 (~, Ct7H17N02), 104* (~- C9H9N02), 91 (M+- CwHwN02), 66 (~- C12HttN02)
13 14
8 N~Hb~~15 9 2 2~---
17 16 0
Assignment of C and H atoms of exo-PhC2M
30

2.4.5 Synthesis and characterisation of exo-N-phenylpropylnorbornene-5,6-
dicarboxyimide: ( exo-PhC3M)
The same procedure as in section 2.4.2 was followed using exo-AN (5.00lg, 0.03mol)
and phenylpropylamine ( 4.12g, 0.030mol). The monomer was recovered as white
crystals, which was recrystallised in ether to give pale yellow solid (6.49g, 0.023mol,
76% yield, mpt: 64°C)
(a) Elementary Analysis of C18H 19N02: calculated (found)
C = 76.84% (76.86%), H = 6.81% (6.82%), N = 4.98% (4.88%), 0 = 11.37% (11.44%)
(b) 1H NMR (see appendix x), CDCh, 500 Mllz, o (ppm)
7.26 (t, 2H, His,J7), 7.23 (d, 2H, H14,Is), 7.22 (t, 1H, HI6), 6.26 (s, 2H, H2,3 ), 3.52 (t, 2H,
H10), 3.25 (s, 2H, H1,4), 2.63 (t, 2H, H12), 2.61 (s, 2H, Hs,6), 1.89 (p; 2H, HII), 1.48 (d,
1H, H1), 1.20 (d, 1H, H7')
(c) 13C NMR (see appendix y), CDCh, 500 Mllz, o (ppm)
178.28 (Cs,9), 141.17 (Cn), 138.04 (C2,3), 128.66 (Cis,!7), 128.52 (CI4,I8), 126.30 (C16 ),
48.02 (Cs,6), 45.37 (CI,4), 42.98 (C1), 38.75 (Cw), 33.55 (C12), 29.33 (C11).
(d) Mass spectrum (see appendix z), (El+).
281 (M\ C1sHI9N02), 177 (MH+-CsH9), 117* ~- C9H10N02), 91 (M+- C11H12N02),
66 (M+- C13H13N02)
H 7 7 Hr
0
Assignment of C and H atoms of exo-PhC3M
2.4.6 Synthesis and characterisation of exo-N-phenylbutylnorbornene-5,6-
dicarboxyimide: (exo-PhC4M)
The same procedure as in 2.4.2 was followed using exo-AN (5.002g, 0.03mol) and
phenylbutylamine (4.553g, 0.030mol). The monomer was recovered as white crystals,
which was recrystallised in toluene to give white solid (4.93g, 0.02lmol, 68% yield,
mpt: 115°C).
31

(a) Elementary Analysis of e 19H21N02: calculated (found)
C = 77.26% (77.32%), H = 7.17% (7.19%), N = 4.74% (4.73%), 0 = 10.83% (10.76%)
(b) 1H NMR (see appendix x), eDCh, 500 MHz, o (ppm)
7.26 (t, 2H, HI6,Is), 7.16 (t, 1H, H17), 7.15 (d, 2H, His,I9),6.27 (s, 2H, H2,3), 3.48 (t, 2H,
Hw), 3.25 (s, 2H, H1,4), 2.65 (s, 2H, Hs,6), 2.62 (t, 2H, H13), 1.60 (t, 4H, H 11 ,12), 1.49 (d,
1H, H1), 1.20 (d, 1H, HT)
(c) ne NMR (see appendix y), eDCh, 500 MHz, o (ppm)
178.33 (Cs,9), 142.17 (CI4), 138.06 (C2,3), 128.64 (CI5,I9), 128.59 (CI6,I8), 126.08 (C!7 ),
48.04 (Cs,6), 45.39 (CI,4), 42.98 (C7), 38.67 (Cw), 35.58 (CII), 29.05 (C12), 27.64 (C13).
(d) Mass spectrum (see appendix z), (El+).
295 (M\ C19H21N02), 230 (M.l-t-CsH6), 91 (M+- C12H14N02), 66 (M+- C14HisN02)
Assignment of C and H atoms of exo-PhC4M
2.4. 7 Synthesis and characterisation of exo-N-tert-butylphenylnorbornene-5,6-
dicarboxyimide: ( exo-e4PhM)
The same procedure as in figure 2.4.2 was followed using exo-AN (5.008g, 0.03mol)
and 4-tert-butylaniline (3.26g, 0.030mol). The monomer was recovered as white
crystals, which was recrystallised in toluene to give white solid (4.556g, 0.020mol, 68%
yield, mpt: 165°C).
(a) Elemental Analysis of C19H21N02: calculated (found)
C = 77.26% (77.39%), H = 7.17% (7.19%), N = 4.74% (4.73%), 0 = 10.83% (10.69%)
(b) 1H NMR (see appendix x), CDC13, 500 MHz, o (ppm)
7.47 (d, 2H, H 11 ,1 5), 7.18 (d, 2H, H12,14), 6.33 (s, 2H, H2,J ), 3.39 (s, 2H, H1,4), 2.84 (s,
2H, H5,6), 1.60 (d, 1H, H1), 1.48 (d, 1H, HT), 1.32 (s,9H, H17).
(c) ne NMR (see appendix y), eDCh, 500 MHz, o (ppm)
32

177.51 (Cs,9), 151.89 (C JO), 138.23 (Cz,J), 129.33 (C13), 126.46 (CII,Is), 126.03 (CI2,I4),
48.08 (Cs,6), 46.05 (CI,4), 43.19 (C7), 34.98 (CI6), 31.51 (C17).
(d) Mass spectrum (see appendix z), (E:r).
295 (~, C19HziNOz), 280 ~-CH3), 229 ~- CI4HisNOz), 214 ~-CH3-CsH6), 66
(M+- C14H1sNOz)
Analysis data for exo-C4PhM
2.4.8 Synthesis and characterisation of exo-N-pentylphenylnorbornene-5,6-
dicarboxyimide: ( exo-C5PhM).
The same procedure as in figure 2.4.2 was followed using exo-AN (5.006g, 0.03mol)
and benzylamine (4.978g, 0.03mol). The monomer was recovered as pink crystals,
which was recrystallised in hexane to give pinkish solid (8.lg, 0.026mol, 86% yield,
mpt: 108°C)
(a) Elemental Analysis of C20H23NOz: calculated (found)
C = 77.64% (77.90%), H = 7.49% (7.53%), N = 4.53% (4.55%), 0 = 10.34% (10.02%)
(b) 1H NMR (see appendix x), CDCb, 500 MHz, () (ppm)
7.27 (d, 2H, Hll,Is), 7.15 (d, 2H, H12,14), 6.34 (s, 2H, H2,3 ), 3.39 (s, 2H, H1,4), 2.84 (s,
2H, H5,6), 2.62 (HI6), 1.61 (m, 3H, H17 and H7), 1.48 (d, 1H, Hr), 1.32 (m, 4H, His,I9),
0.89 (t, 3H, Hzo).
(c) 13C NMR (see appendix y), CDCb, 500 MHz, () (ppm)
177.50 (Cs,9), 143,91 (C 10), 138.22 (Cz,J), 129.52 (C13), 129.42 (CII,Is), 126.32 (Cn,I4),
48.07 (Cs,6), 46.03 (CI,4), 43.18 (C7), 35.86 (CI6), 31.67 (C17), 31.21 (Cis), 22.75 (C19),
14.26 (Czo).
(d) Mass spectrum (see appendix z)
309 (~, CzoH23NOz), 243 (~-CsH6), 186 (M+-C6Hs-C4H9), 66 (~- C1sH17NOz)
33

0 11 12
HM7 H7' 16 17 18 19 20
3 ~of ~ CHCHCHCHCH 2 2 2 2 3 -
2 15 14 0
Assignment of C and H atoms for exo-C5PhM
2.4.9 Synthesis and characterisation of exo-N-hexylphenylnorbornene-5,6-
dicarboxyimide: ( exo-C6PhM).
The same procedure as in 2.4.2 was followed using exo-AN (4.631g, 0.028mol) and
benzylamine (5.000g, 0.028mol). The monomer was recovered as white crystals, which
was recrystallised in hexane to give white solid (7.7g, 0.024mol, 84% yield, mpt: 96°C)
(a) Elemental Analysis of C21H25N02: calculated (found)
C = 77.99% (77.93%), H = 7.79% (7.73%), N = 4.33% (4.23%), 0 = 9.89% (10.11%)
(b) 1H NMR (see appendix x), CDCh, 500 MHz, o (ppm)
7.26 (d, 2H, Hll,Js), 7.15 (d, 2H, H12,I4), 6.34 (s, 2H, H2.3 ), 3.39 (s, 2H, H1,4); 2.84 (s,
2H, Hs,6), 2.62 (H16), 1.61 (m, 3H, H17 and H7), 1.48 (d, lH, HT), 1.30 (m, 6H, H18_20),
0.88 (t, 3H, H21).
(c) 13C NMR (see appendix y), CDCb, 500 MHz, o (ppm)
177.49 (Cs,9), 143.92 (C JO), 138.22 (C2,3), 129.51 (C13), 129.41 (CII,Is), 126.31 (CI2,I4),
48.07 (Cs,6), 46.03 (CI,4), 43.18 (C7), 35.90 (CI6), 31.93(CI7), 31.50 (Cis), 29.20 (C19),
22.83 (C20), 14.35 (C21).
(d) Mass spectrum (see appendix z)
323 (~, c21H2sN02), 257 (M+-CsH6), 186 (M+- C6Hs- CsHJI), 66 (~- CI6HI9N02)
Assignment of C and H atoms for exo-C6PhM
34

2.4.10 Synthesis and characterisation of exo-N-hexylnorbornene-5,6-
dicarboxyimide: ( exo-C6M).
The same procedure as in 2.4.2 was followed using exo-AN (1 0.09g, 0.060mol) and
hexylamine (6.16g, 0.060mol). The monomer was recovered as pale yellow liquid,
which was distilled to give colourless liquid (87% yield, bpt: 130@ 4mmbar).
(a) Elemental Analysis of C1sH21N02: calculated (found)
C = 72.84% (72.49%), H = 8.56% (8.52%), N = 5.66% (6.80%), 0 = 12.94% (12.19%)
(b) 1H NMR (see appendix x), CDCh, 500 MHz, () (ppm)
6.28 (s, 2H, H2,3 ), 3.45 (t, 2H, H10), 3.27 (s, 2H, H1,4), 2.67 (s, 2H, Hs,6), 1.52 (m, 3H,
H11 and H7), 1.28 (m, 6H, H 12_14), 1.23 (d, 1H, HT), 0.87 (t, 3H, His).
(c) 13C NMR (see appendix y), CDCh, 500 MHz, <> (ppm)
178.33 (Cs,9), 138.04 (C2,3), 48.00 (Cs,6), 45.36 (CI,4), 42.91 (C7), 38.96 (CIO), 31.52
(CII), 27.93 (C12), 26.82 (C13), 22.68 (CI4), 14.20 (Cis).
(d) Mass spectrum (see appendix z)
247 (M+, CisH21N02), 182 (M+-CsH6), 186 ~- CsH6- CsHII), 66 (~- C10HisN02)
7
10 11 12 13 14 15 3 N-CH2CH2CH2CH 2CH2CH3
2 0
Assignment of C and H atoms for exo-C6M
35

Chapter 3
Polymer synthesis and characterisation
Chapter 3 36

3.1 Introduction
The focus of this chapter is on the synthesis of linear polymers via ROMP of exo
phenyl norbomene dicarboxyimide monomers using well-defined initiators. The
initiators used for polymerisation reaction were well defined ruthenium carbene initiator
{Ch[(C6HII)3PhRu=CHC6Hs}, the t-butoxy substituted molybdenum initiator
{Mo[CHC(CH3)3][CIOH17N][OC(CH3)3]2}, Mo (1), and hexafluoro-t-butoxy substituted
molybdenum initiator {Mo[CHC(CH3)2C6Hs][CwHl7N][OC(CF3)2CH3]2}, Mo (2).
NMR scale ROMP reactions were carried out to provide information on the reactivity of
a series of exo- phenyl norbomene dicarboxyimides. The ROMP reactions were scaled
up to provide polymer samples for 1 H NMR, 13C NMR, and elemental analysis, the
molecular weight measurement by GPC and thermal properties evaluation by DSC
(differential Scanning Calorimetry) and DMTA (dynamic mechanical thermal analysis).
The cis and trans content of the polymers were estimated from 1HNMR and 13CNMR
spectra.
3.2 NMR scale solution polymerisation of exo-phenylnorbornene dicarboxyimide
monomers
3.2.1 Polymerisation using ruthenium initiator
Polyn1erisation reactions were performed in the glove box (Braun) at room temperature.
The initiator (10 mg) and monomer (10 equivalents) were dissolved in CDCh; in two
separate vials and were stirred for few minutes to allow them to dissolve. The initiator
solution was transferred into the monomer vial and the mixture was stirred for few
minutes. The reaction mixture was then transferred to the screw capped NMR tube. The
NRM tube was removed from the glove box and the spectrum was recorded. The
polymerisation reaction is illustrated in figure3 .1. · In all cases, the monomer was
consumed within ten minutes of the reaction. This was shown by the disappearance of
the monomer signal at 6.30ppm in the 1H NMR spectra of the reaction mixture.
The alkylidene region in the 1H NMR spectra showed signals due to initiator alkylidene
hydrogens at 19.98 pp m and signals due to the propagating alkylidene hydrogens was at
19.52 ppm and 18.67 ppm. A typical example is shown in figure 3.2. On addition of20
equivalents of the monomer, the polymerisation was continued; the initiator signal was
reduced with subsequent growth in the propagation signal. This suggested that the
polymerisation was living. The initiator signal was still observable after the addition of
Chapter 3 37

70 equivalents of the monomer. This indicates that the rate of propagation is faster than
the rate of initiation i.e. kp >> ki.
(a) Initiation
PC::p PCy3 I""
cr......__ I I.& ~N-o Cl" I .& Ru + - Ru
Cl/ I H Cl/ I H
PCy3 0 Cy3P 0
N Ru-initiator Exo-monomer 0
Exo-propagating species
(b) Propagation
PCy3 "" ~N-o
PCy3
Cl" I h Cl" I Ru + - Ru
cr .. ..-- I H Cl/ I Gyp 0 Gyp
N Exo-monomer N
0 0 Exo-propagating species Poly(exo)
Figure 3.1: (a) Outline of the initiation reaction by well defined Ru-initiator.
(b) Outline of the propagation step.
Alkylidene hydrogen of Ru initiator
Exo-propagating alkylidene hydrogen
r
19.4 ppm
: :
'TiTfiTiTfiTi"'l'[illljllll[lllllllll[lllllllll[lllllllllflllllllll[lllllllll[lllllllll[lllllllll[lllllll
20 18 16 14 12 8 6 4 pp m
Figure 3.2: 1H NMR spectra from polymerisation of exo-monomer by well-defined ruthenium initiator.
Chapter 3 38

3.2.2 Polymerisation using t-butoxy substituted molybdenum initiator
The same procedure as in section 3.2.1 was followed using t-butoxy substituted
molybdenum initiator. The polymerisation reaction is illustrated in figure 3.3. The
alkylidene region in the 1H NMR spectra showed signals due to initiator alkylidene
hydrogens at 11.21 ppm and signals due to the propagating alkylidene hydrogens was at
11.59 ppm. A typical example is shown in figure 3.4.
(a) Initiation
+ ~N-o 0
Me-initiator Exo-monomer
Exo-propagating species
(b) Propagation
Exo-monomer
Exo-propagating species Poly(exo)
Figure 3.3: (a) Outline of the initiation reaction by well defined Mo (1) initiator.
(b) Outline of the propagation step.
Chapter 3 39

Alkylidene hydrogen ofMo (I) initiator ?
(b) Exo-propagating alkylidene hydrogen ~
11f1111f1111f1111f1111f1111f1111f1111f1111f1111f1111f111
+ 11.8 11.4 ppm
11 10 9 8 6 5 4 pp m
Figure 3.4: 1H NMR spectra from polymerisation of exo-monomer by well-defined Mo (1) initiator.
3.2.3 Polymerisation using hexafluoro-t-butoxy substituted molybdenum initiator
The same procedure as in section 3.2.1 was followed using t-butoxy substituted
molybdenum initiator. The polymerisation reaction is illustrated in figure 3.5. The
alkylidene region in the 1H NMR spectra showed signals due to initiator alkylidene
hydrogens at 12.40 ppm and signals due to the propagating alkylidene hydrogens was at
12.77 ppm. A typical example is shown in figure 3.6.
(a) Initiation
~N-o 0
Me-initiator Exo-monomer
(b) Propagation Exo-propagating species
Exo-monomer
Exo-propagating species Poly(exo)
Figure 3.5: (a) Outline of the initiation reaction by well defined Mo (2)-initiator.
(b) Outline of the propagation step.
Chapter 3 40

Alkylidene hydrogen ofMo (2) initiator
Exo-propagating alkylidene hydrogen
Q
iijiilljillijitltjlllljili:Jilitjtttijtlltjlllijli
12.5 t
: :
r I l:·rr--r .. ,. .. , .. ,..TT ,. .. 1: I I I I I I l. I I \ I I I I I I r I 1 I ) I I I I I I I I I I I 1 I I I I I I I I I 1 r I I I I I I I I I
13 12 11 10 9 8 7 6 5 4 3 2 ppm
Figure 3.6: 1H NMR spectra from polymerisation of exo-monomer by well-defined Mo (1) initiator.
3.2.4 Copolymerisation of exo-ethylphenyl norbornene dicarboxyimide and exo
hexyl norbornene dicarboxyimide
The alkyl norbomene dicarboxyimides prepared in the previous work were liquid and
therefore it possible to carry out ROMP using well-defined ruthenium initiator in both
solution and bulk. However, all the phenyl norbomene dicarboxyimide monomers in i
this work were solids. The exo-ethylphenyl norbomene dicarboxyimide (exo-PhC2M)
monomer was found to be soluble in exo-hexyl norbornene dicarboxyimide (exo-C6M)
monomer. The aim of this section was to test the possibility of carrying out ROMP on a
mixture of monomers and obtain some information on the reactivity ratios of the
monomers in the mixture. Three experiments were performed using the ruthenium
initiator (lOmg): the first polymerisation reaction contain 20 equivalents of exo-C6M,
the second polymerisation reaction contain 20 equivalents of exo-PhC2Mand the third
polymerisation reaction contained a mixture of 20 equivalents of exo-C6M and exo
PhC2M. All the experiments were analysed by 1H NMR within the first ten minutes. All
the monomers were consumed within the first ten minutes and no resonances due to the
monomer could be found. It appears that in the case of the experiment using a mixture
of two monomers both monomers are polymerising at a same rate. The alkylidene
region in the NMR spectra of the homopolymerisation of the two monomers showed
expected signals for initiator alkylidene and propagating alkylidene hydrogens. The
polymerisation of the mixture is a random copolymerisation and we expect to see two
Chapter 3 41

propagating alkylidene signals, see figure 3.7. The alkylidene region in the NMR
spectrum showed a multiplet signal for the propagating alkylidene, which appears to be
the results of overlapping of the two propagating signals characteristic of the two
monomers. The 1H NMR spectra for the three ROMP reactions are shown in figure 3.8.
PC::p3
"" Cl......._ I I .0
Ru Cl/ I H +
PCy3
Ru-initiator
exo-C1;M c
0
~~CH2CH2--o 0
mix
exo-Phc;M propagating species
or
exo-C6M propagating species
Figure 3.7: Expected propagation reaction in a mixture of exo-C6M and exo-PhC2M
Chapter 3 42

Alkylidene hydrogen of Ru initiator
.......
(a) .····
!..20 ............... 18 .....•
Alkylidene hydrogen of Ru initiator
~-.
(b)
"' ~
~ 20 18
Alkylidene hydrogen of Ru initiator
(c)
20 18
Propagating alkylidene hydrogen
.··"
16 I
14 12
Propagating alkylidene hydrogen
:tf
1S.O
16
16
""'
14 12
Propagating alkylidene hydrogen
14 12
10 B 6 4 pp m
I'
10 B 6 4 2 ppm
10 8 6 4 2 ppm
Figure 3.8: 1H NMR spectra of the polymerisation of a mixture of exo-C6M and exoPhC2M. (a) Exo-C6M spectra. (b) Exo-PhC2M (c) Mixture of exo-C6M and exo-PhC,M
Chapter 3 43

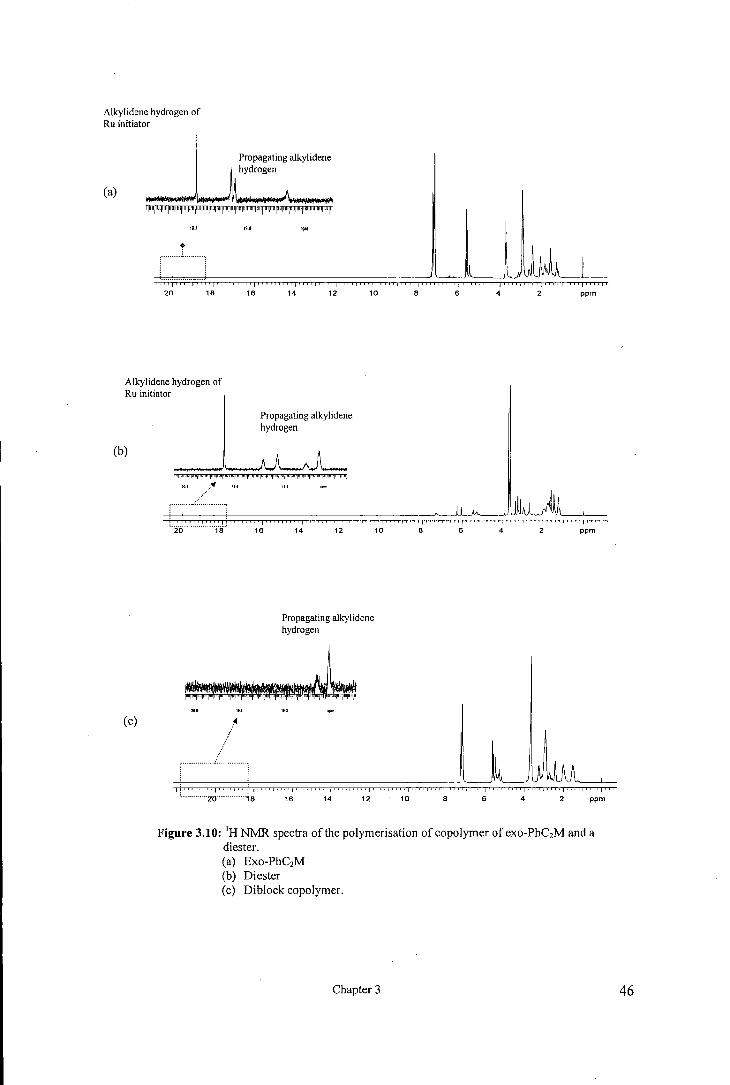
3.2.5 Block copolymerisation of exo-phenyl norbornene dicarboxyimides and exo,
endo-norbornene dimethylcarboxylate
To establish the living nature of the ROMP of exo-phenyl norbomene dicarboxyimides
block copolymers were synthesised via sequential addition of monomers using exo,
endo-norbomene dimethylcarboxylate as the co-monomer, see figure 3.9.
In a typical block copolymerisation reaction, ruthenium initiator (lOmg) was dissolved
in CDCh and was added to exo-ethyl phenyl norbomene dicarboxyimide (1 00
equivalents) dissolved in CDCh. The reaction was stirred for 1 hr to allow all the
monomer to be consumed m the reaction. Then exo, endo-norbomene
dimethylcarboxylate ( 100 equivalents) dissolved in C DCh was added to the reaction
mixture. The reaction was allowed to stir for 1 hr and the reaction mixture was
analysed by 1H NMR. Figure 3.10 shows the 1HNMR spectra ofthe living ROMP
reaction for exo-ethyl phenyl norbomene dicarboxyimide (a), exo, endo-norbomene
dimethylcarboxylate (b) and block copolymer (c). The alkylidene region in figure
3 .1 0( c) shows that the resonances due the propagating alkylidene for exo-ethyl phenyl
norbomene dicarboxyimide have disappeared and new set of resonances are appeared.
Two sets of signals are observed in the alkylidene region for the homopolymerisation of
exo, endo-norbomene dimethylcarboxylate figure 3.1 O(b ), one at 19.3 5ppm and
19.12ppm and the other at 18.65ppm and 18.45ppm probably due to cis and trans
vinylene contents although not absolute certain. The alkylidene region for the block
copolymer shows only one set of signals at 18.64ppm and 18.43ppm, figure 3.10(c).
This suggests that a block copolymer have been formed. It appears that the propagating
ends in poly( exo-ethyl phenyl norbomene dicarboxyimide) polymerise exo, endo
norbomene dimethylcarboxylate in a different fashion to that of the initiator.
Chapter 3 44

PC::p3
""" Cl'-.. I I ~
Ru Cl/ I H
PCy3
Ru-initiator
0
+ n ~N-cH,cH,--Q 0
0
n H
Exo-propagating species
Exo-propagating species
+ m ~COOMe ~COO Me
Exo,endo-diester
block copolymer
Figure 3.9: Outline of the polymerisation reaction between exo, endo-diester and exo-PhC2M
Chapter 3 45
Alkylidene hydrogen of Ru initiator
(a)
19.!
20 18
Alkylidene hydrogen of Ru initiator
(b)
1"'111 "1"' .ff .... ··
··2o ·· ·······Ti(
(c)
Propagating alkylidene hydrogen
\9.0
16 14 12 10
Propagating alkylidene hydrogen
16 14 12
Propagating alkylidene hydrogen
10
'·············zo ..... · ·······18 I
16 14 12
8 6 4 2 pp m
8 6 4 pp m
10 8 6 4 2 pp m
Figure 3.10: 1H NMR spectra of the polymerisation of copolymer of exo-PhC2M and a diester. (a) Exo-PhC2M (b) Diester (c) Diblock copolymer.
Chapter 3 46
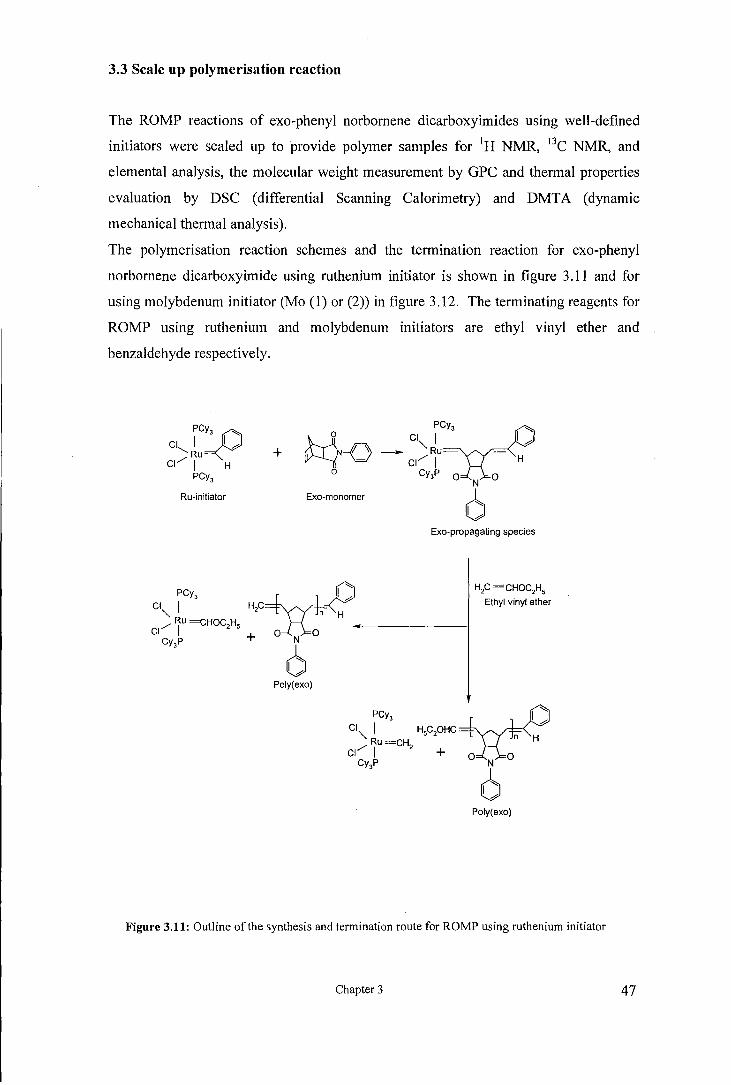
3.3 Scale up polymerisation reaction
The ROMP reactions of exo-phenyl norbornene dicarboxyimides using well-defined
initiators were scaled up to provide polymer samples for 1H NMR, 13C NMR, and
elemental analysis, the molecular weight measurement by GPC and thermal properties
evaluation by DSC (differential Scanning Calorimetry) and DMTA (dynamic
mechanical thermal analysis).
The polymerisation reaction schemes and the termination reaction for exo-phenyl
norbornene dicarboxyimide using ruthenium initiator is shown in figure 3.11 and for
using molybdenum initiator (Mo (1) or (2)) in figure 3.12. The terminating reagents for
ROMP using ruthenium and molybdenum initiators are ethyl vinyl ether and
benzaldehyde respectively.
+ ~N-o 0
Ru-initiator Exo-monomer
Poly(exo)
PCX<?3
..., Cl" I I A
---- Ru- -Cl/ I H Cy3P 0 0
N
0 Exo-propagating species
H2C =cHOC2H5
Ethyl vinyl ether
Poly(exo)
Figure 3.11: Outline of the synthesis and termination route for ROMP using ruthenium initiator
Chapter 3 47

+ ~N-o 0
Me-initiator Exo-monomer
R
Exo-propagating species
Poly(exo)
0 11
e-H
6 R
+ [Mo}=o
Figure 3.12: Outline of the polymerisation and termination route for ROMP using Mo (I) or Mo (2)
initiator
For ruthenium initiator, 0.75 g and for both molybdenum initiators, 0.5g of each
monomer was used. The ratio of monomer to initiator was 150:1. The polymerisation
reactions were carried out in the glove box (Braun) at room temperature. Both initiator
and monomer were dissolved in separate vials in dichloromethane and stirred for few
minutes. The monomer solution was added to the initiator solution and the reaction
mixture was stirred overnight. The polymerisation reaction was terminated by the
addition of the tenninating reagent. The polymers were recovered by precipitation
twice in methanol and dried under vacuum resulting in white solids. All the polymers
were film forming. The polymers formed from ruthenium initiator were soluble in THF,
chloroform, dichloromethane and DMF.
Chapter 3 48

3.3.1 Polymer characterisation using 1H and 13C NMR spectroscopy
The cis I trans contents of the polymers are readily determined from the 1H NMR
spectrum from the integration of the resonance attributable to cis and trans olefinic
hydrogen (H2,3). The resonances due to trans and cis vinylenes for the polymers
obtained by the well defined initiators are similar and for the trans vinylenes appears at
5.74ppm and for the cis vinylenes at 5.51ppm., see figures 3.13-3. Table 3.1 shows the
cis and trans vinylene content of polymers prepared by each initiator. Generally the
trans vinylene content for polymers prepared by ruthenium initiator is about 83%, for
the polymers prepared by t-butoxy molybdenum initiator is about 97% and for the
polymers prepared by hexafluoro-t-butoxy molybdenum initiator is about 31%. These
results are consistent with the previous result49 that ruthenium initiator give polymers
with 80% trans content and that t-butoxy molybdenum initiator give polymers with high
trans content while hexafluoro-t-butoxy molybdenum initiator give polymers with high
cis content.
Chapter 3 49
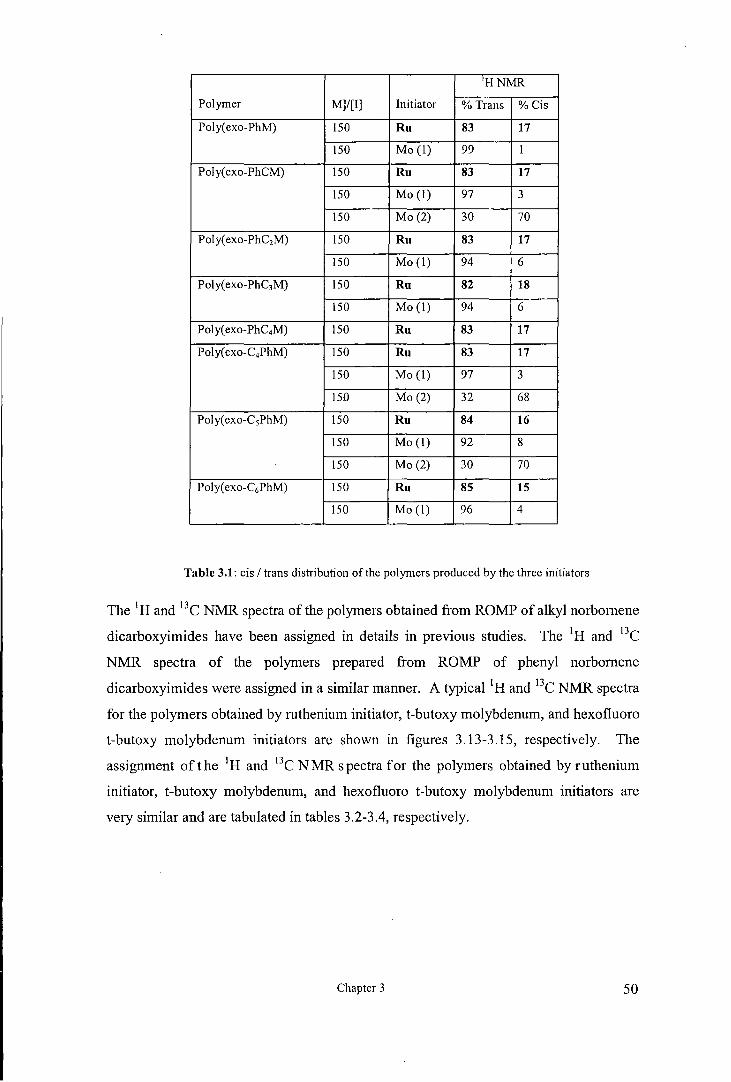
HNMR
Polymer M]/[I] Initiator % Trans %Cis
Poly(exo-PhM) 150 Ru 83 17
150 Mo (1) 99 1
Poly(exo-PhCM) 150 Ru 83 17
150 Mo (1) 97 3
150 Mo (2) 30 70
Poly(exo-PhC2M) 150 Ru 83 17
150 Mo (1) 94 6
Poly(exo-PhC3M) 150 Ru 82 18
150 Mo (1) 94 6
Poly(exo-PhC4M) 150 Ru 83 17
Poly( exo-C4PhM) 150 Ru 83 17
150 Mo (1) 97 3
150 Mo (2) 32 68
Poly( exo-C5PhM) 150 Ru 84 16
150 Mo (I) 92 8
150 Mo (2) 30 70
Poly( exo-C6PhM) 150 Ru 85 15
150 Mo (I) 96 4
Table 3.1: cis I trans distribution of the polymers produced by the three initiators
The 1H and 13C NMR spectra ofthe polymers obtained from ROMP of alkyl norbomene
dicarboxyimides have been assigned in details in previous studies. The 1H and 13C
NMR spectra of the polymers prepared from ROMP of .phenyl norbomene
dicarboxyimides were assigned in a similar manner. A typical 1H and 13C NMR spectra
for the polymers obtained by ruthenium initiator, t-butoxy molybdenum, and hexofluoro
t-butoxy molybdenum initiators are shown in figures 3.13-3.15, respectively. The
assignment of the 1H and 13C NMR spectra for the polymers obtained by ruthenium
initiator, t-butoxy molybdenum, and hexofluoro t-butoxy molybdenum initiators are
very similar and are tabulated in tables 3.2-3.4, respectively.
Chapter 3 50

N I
10CH I '
11yH2
()
12CH2
18 14
17 15 16
(a)
5
r---------: a=Hl,4cis : b=Hs,6cis 1---------
H,,,...,
e 14,18
0 B 9 0 e,,,,
18
17
IN 10CH
I 11 CH
I
2
2
2
14
15 en 612CH
16
(b) e,. en
8,9 r---------., I I I a=C1,4cis I
e I ----------' e"
e,,.,,., Cl,41ru1S e,,,~
e,.",J je, je,
Cs,6<Js 1r
I' If I I' I If I' I If I' If I I'' I I I'' ''I'' If I''' 'I' I I I I' I If I',, I I''' 'I' '''I'''' I' I If I'' 160 140 120 100 90 80 70 60 50
Figure 3.13: (a) 1H NMR spectrum of poly (exo-PhC3M) using Ru initiator in CDCI3
(b) 13C NMR spectrum of poly (exo-PhC3M) using Ru initiator in CDCI3
* Residual hydrogen in CDCh
• Solvent CH2Ciz
Chapter 3
40 ppm
51
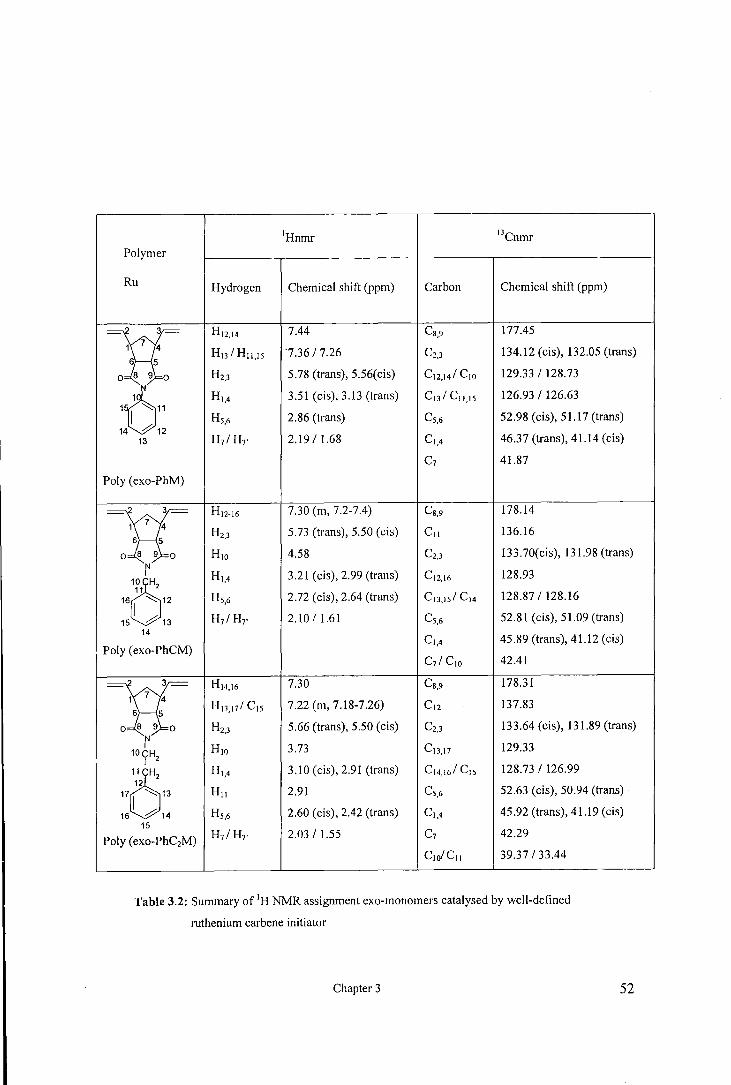
1Hnmr 13 Cnmr Polymer
Ru Hydrogen Chemical shift (ppm) Carbon Chemical shift (ppm)
sr Hl2,t4 7.44 Cs,9 177.45
Hn I H11.ts 7.36 I 7.26 c2.3 134.12 (cis), 132.05 (trans)
H2,J 5.78 (trans), 5.56(cis) c,2,141 c10 129.33 I 128.73
H,,4 3.51 (cis), 3.13 (trans) c,JI c,,,,s 126.93 I 126.63
'6" Hs,6 2.86 (trans) Cs,6 52.98 (cis), 51.17 (trans) 14 12
13 H1IH7' 2.19 I 1.68 c,,4 46.37 (trans), 41.14 (cis)
c1 41.87
Poly (exo-PhM)
};[ H12-t6 7.30 (m, 7.2-7.4) Cs,9 178.14
H2,J 5.73 (trans), 5.50 (cis) c,, 136.16
Hw 4.58 C2,J 133.70(cis), 131.98 (trans) N I Ht,4 3.21 (cis), 2.99 (trans) C12,t6 128.93
1~1 H2
161 '"-': 12 Hs,6 2.72 (cis), 2.64 (trans) CIJ,tsl Ct4 128.87 I 128.16
15 ~13 H1IH7' 2.1011.61 Cs,6 52.81 (cis), 51.09 (trans) 14
Poly (exo-PhCM) Ct,4 45.89 (trans), 41.12 (cis)
C71 CIO 42.41
};[ Ht4,t6 7.30 Cs,9 178.31
H 13,171 Cts 7.22 (m, 7.18-7.26) cl2 137.83
Hz,J 5.66 (trans), 5.50 (cis) C2.J 133.64 (cis), 131.89 (trans) N I Hw 3.73 CIJ,t7 129.33 10CH I 2
"6" Ht,4 3.10 (cis), 2.91 (trans) Ct4,t61 Cts 128.73 I 126.99
H11 2.91 Cs,6 52.63 (cis), 50.94 (trans)
16 14 Hs,6 2.60 (cis), 2.42 (trans) Ct,4 45.92 (trans), 41.19 (cis) 15
Poly (exo-PhC2M) H1IH1' 2.03 I 1.55 c1 42.29
CIOICII 39.37 I 33.44
Table 3.2: Summary of 1H NMR assignment exo-monomers catalysed by well-defined
ruthenium cat·bene initiator
Chapter 3 52

1Hnmr 13Cnmr
Polymer
Ru Hydrogen Chemical shift (ppm) Carbon Chemical shift (ppm)
=rr H1s,11 7.27 Cs,9 178.50
H14,1s I C16 7.18 I 7.17 Cn 141.21
Hz,3 5.74 (trans), 5.51 (cis) c2,3 133.79 (cis), 132.00 (trans) N I Hto 3.51 C1s,n 128.68
10CH I 2
Hl,4 3.22 (cis), 2.92 (trans) cl4,ls/ C16 128.52 I 126.30 11 CH
I 2
12CH Hs,6 2.71 (cis), 2.62 (trans) Cs,6 52.78 (cis), 51.03 (trans)
"6" H12 2.62 cl,4 45.94 (trans), 41.04 (cis)
17 15 H7/ HT 2.11 I 1.61 c7 42.37
16 H11 1.91 C1o/C12 38.68 I 33.43
Poly (exo-PhC3M) C11 29.13
_2 - H16,1s 7.26 Cs,9 178.54
1 7 4 Hn/ C1s,19 7.16 C14 142.21
6 5
0 6 9 0 H2,3 5.75 (trans), 5.52 (cis) Cz,3 133.82 (cis), 132.01 (trans) N I Hto 3.46 C1s.19 128.68
10CH I 2
Hl,4 3.24 (cis), 2.97 (trans) C161s I Cn 128.62 I 126.11 11 CH
2 I 12CH Hs,6 2.75 (cis), 2.62 (trans) Cs,6 52.83 (cis), 51.08 (trans)
I 2
0 Hn 2.62 cl,4 46.10 (trans), 41.13 (cis)
19 15 H 7 I H1· 2.13 I 1.65 c7 42.44
16 16 H11,12 1.59 Ctol Cn 38.51 I 35.57
17 c11/ c12 28.87 I 27.53 Poly (exo-PhC4M)
13: H11,1s 7.45 Cs,9 177.62
Hl2,14 7.18 C1o 151.69
Hz,3 5.81 (trans), 5.56 (cis) c2,3 134.27 (cis), 132.45 (trans)
'i)" Hl,4 3.55 (cis), 3.15 (trans) Cn 129.33
14 12 Hs,6 2.86 (trans) C11,1S I C1z.14 126.35 I 126.05
C~CH3 ) 3 H1/HT 2.19 I 1.70 Cs,6 53.04 (cis), 51.16 (trans) 16 7
Poly (exo-C4PhM) Hn 1.31 cl,4 46.42 (trans), 41.12 (cis)
c7 42.23
c16/C17 34.97 I 31.53
Table 3.2: Summary continued ....
Chapter 3 53

1Hnmr 13 Cnmr
Polymer
Ru Hydrogen Chemical shift (ppm) Carbon Chemical shift (ppm)
:;[ H11,15 7.24 Cs,9 177.65
H12.16 7.15 Cw 143.64
Hz,J 5.81 (trans), 5.56 (cis) c2.1 134.26 (cis), 132.32 (trans)
'Q" HI,4 3.53 (cis), 3.12 (trans) Cn 129.52
14 12 Hs,6 2.86 (trans) cll,Is 1 cl2,14 129.30 I 126.34
169H2 H16 2.60 Cs,6 53.02 (cis), 51.13 (trans)
179H2 H1/HT 2.1911.68 cl,4 46.40 (trans), 41.12 (cis) 1BCH
I 2 Hl1 1.60 c1 41.91 19CH2 20tH 3
HlS-19' 1.31 c161c11 35.84 I 31.67
Hzo 0.88 C1s I C19 31.221 22.76
Poly (exo-C5PhM) Czo 14.28
}[[ HILlS 7.24 Cs,9 177.57
H12.14 7.15 Cw 143.64
H2,J 5.81 (trans), 5.56 (cis) c2,3 134,22 (cis), 132.31 (trans)
HI,4 3.53 (cis), 3.12 (trans) Cn 129.51
'Q" Hs,6 2.85 (trans) cll,Isl cl2,14 129.29 I 126.34 14 12
H16 2.61 Cs,6 53.02 (cis), 51.13 (trans) 16CH
I 2 H1IHT 2.19 I 1.69 cl,4 46.40 (trans), 41.11 (cis)
17CH I 2
Hl1 1.59 c1 41.91 1BCH I 2
19CH HlS-20 1.29 c16/ c11 35.88 I 31.94 I 2
20CH2 H21 0.88 C1s I C19 31.51129.20 21 CH
3 Czo I C21 22.85 I 14.37 Poly (exo-C~hM)
Table 3.2: Summary continued ....
Chapter 3 54
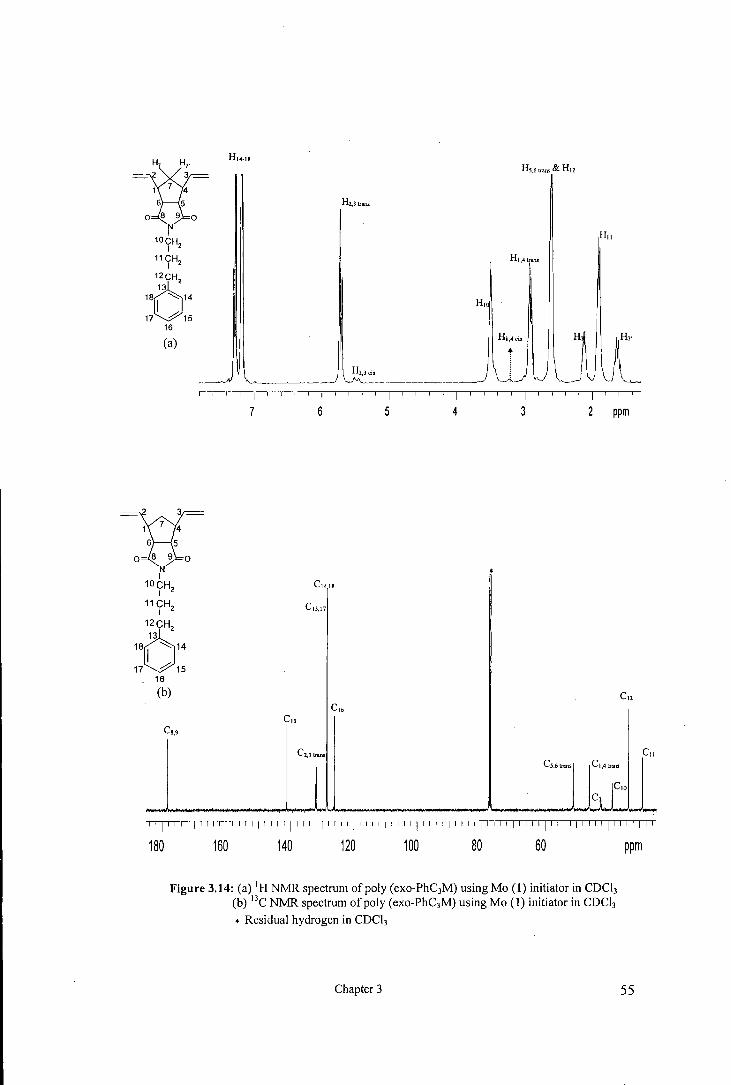
18
17
I 10CH
I 2 c"'' 11 CH
2 I c,,,,,
B 14
15 16
(b) c,, c,o
ell Cs,9
Cz,Jtraru: c,, Cs,o u.ru Cl,4trans
cl jc!O
''I' I I I I'' ''I' I I I I' I I I I'' I 'I' I I 'I',, I )I 11 'I' I I I I' 11 I I' 11 I I' I I 'I''' 'I' '''I' I 11 I' I
180 160 140 120 100 80 60
Figure 3.14: (a) 1H NMR spectrum of poly (exo-PhC3M) using Mo (I) initiator in CDC13
(b) 13C NMR spectrum of poly (exo-PhC3M) using Mo (I) initiator in CDCh
• Residual hydrogen in CDCh
Chapter 3
pp m
55

Polymer 1Hnmr 13Cnmr
Mo (1) Hydrogen Chemical shift (ppm) Carbon Chemical shift (ppm)
13: H,z,I4 7.44 Cs,9 177.53
Hn I Hll,IS 7.36 I 7.26 Cz,3 132.37 (trans)
H2.3 5.80 (trans), 5.57(cis) c101 c,2,14 132.11 I 129.30
H,,4 3.51 (cis), 3.16 (tra11s) Cn I cii,IS 128.71 I 126.63 1611 Hs,6 2.87 (trans) Cs,6 51.19 (trans)
14 12 H1IH7' 2.1911.70 c,,4 46.42 (trans), 41.21(cis) 13
Poly (exo-PhM) c1 42.08
~s:i-H12-16 7.30 (m, 7.24-7.34) Cs,9 178.13
H2.3 5.74 (trans), 5.50 (cis) c" 136.19
.ko HIO 4.58 Cz.3 132.03 (trans) N I H,,4 3.20 (cis), 2.99 (trans) c,2,16 128.92
10 H2 11 Hs,6 2.65 (trans) Cn,IS I c,4 128.84 I 128.14
161 ~12
15 ~13 H1IH7' 2.11 I 1.62 Cs,6 51.13 (trans) 14
c,,4 45.90 (trans) Poly (exo-PhCM)
C71 CIO 42.30
1f{: H,4,16 7.30 Cs,9 178.34
Hn,I7 I C,s 7.23 (m, 7.18-7.26) cl2 137.83
H2.3 5.65 (trans), 5.50 (cis) Cz,3 131.92 (trans)
I HIO 3.73 Cn,I7 129.33 10CH I 2
17~5, H,,4 3.11 (cis), 2.92 (trans) C,4,161 C,s 128.73 I 126.98
H11 2.92 Cs,6 52.62 (cis), 50.94 (trans)
16 14 Hs,6 2.60 (cis), 2.42 (trans) c,,4 45.93 (trans), 41.15 (cis) 15
Poly (exo-PhCzM) H71H7' 2.04 I 1.57 c7 42.16
c101 c" 39.38 I 33.44
Table 3.3: Summary of 1H NMR assignment exo-monomers catalysed by t-butoxy
substituted molybdenum initiator
Chapter 3 56
Table 3.3: Summary continued ....
Chapter 3 57
1Hnrnr 13Cnmr
Polymer
Mo (1) Hydrogen Chemical shift (ppm) Carbon Chemical shift (ppm)

0 8 9 0
15 11
14 12
16CH I 2
17CH I 2
18CH I 2
19CH I 2
20CH3
0 8 9 0
15~11 14~12
16yH2
11yH2
18yH2
19CH2 20CH
3
180
*
160
H,,,,s & 12,14
7 6
r-----------, : a=C2.Jcis : I a' = c2 3 tnns I
b=Cil. I
c=CII,I5 d=CI2,14
140
,---t--, I I I I C I d
b
120
5 4
100
Hl,4 & Hs,6
l ___ t __ , I I
*
80
c 8
60 40
Figure 3.14: (a) 'HNMRspectrumofpoly (exo-CsPhM) using Mo (2) initiator in CDCb (b) 13CNMRspectrum of poly (exo-CsPhM) using Mo (2) initiator in CDCb
* Residual hydrogen in CDCb
Chapter 3
pp m
pp m
59

1Hnmr 13Cnmr
Polymer
Mo (2) Hydrogen Chemical shift (ppm) Carbon Chemical shift (ppm)
}([ H12-16 7.23 (m, 7.12-7.33) Cs,9 178.13
Hz,J 5.71 (trans), 5.48 (cis) ell 136.41
Hw 4.51 c2,3 133.80 (cis), 131.99 (trans)
I Ht,41 Hs,6 3.06 (m) C12,16 128.92 10 H2 11
H?l HT 161 ~12
2.04 I 1.48 Cn,ts 1 C14 128.75 I 127.93
15 ~13 Cs,6 53.22 (cis), 51.22 (trans) 14
Poly (exo-PhCM) Ct,4 45.89 (trans), 41.28 (cis)
C1l Cw 43.15 I 42.64
- 3- Hn,tsl Ct2,14 7.28 (m, 7.11-7.47) Cs,9 177.56
1 7 4 H2,J 5.76 (trans), 5.52 (cis) CIO 151.31 6 5
0 8 9 0 Ht,41 Hs,6 3.13 (m) c2,3 134.14 (cis), 132.39 (trans)
H?l HT 2.17 I 1.69 Cn 129.68
'Q" H!7 1.29 Ctt,ts1Cl2t4 126.12 (m) 14 12
C ~CH 3 ) 3
Cs,6 53.31 (cis), 51.33 (trans)
16 7 Ct,4 46.48(trans), 42.09 (cis) Poly (exo-C4PhM)
c1 43.59
Ct61C17 34.93 I 31.54
)[ Hn,ts I C12,t6 7.09 (m, 7.04-7.16) Cs,9 177.55
H2,J 5.78 (trans), 5.52 (cis) CIO 143.06
Ht,4/Hs,6 3.14 (m) c2,3 134.06 (cis), 132.24 (trans)
'i~t Ht6 2.59 Cn 129.86
14 12 H?l HT 2.16 I 1.60 Ctt,ts I Ct2,t4 128.89 I 126.38
16CH H!7 1.60 Cs,6 53.28 (cis), 51.31 (trans) I 2
11yH2 Hts-19 1.33 Ct,4 46.41 (trans), 41.97 (cis) 1BCH
I 2 Hzo 0.89 C1 43.12 19CH2 20tH
3 Ct6IC11 35.83 I 31.75
Poly (exo-C5PhM) Cts I Ct9 31.24 I 22.77
Czo 14.26
Table 3.4: Summary of 1H NMR assignment exo-monomers catalysed by hexafluoro-t-butoxy substituted molybdenum initiator
Chapter 3 60

3.3.2 Polymer characterisation by elemental analysis
The polymers obtained from ROMP . of exo-phenyl norbornene dicarboxyimide derivatives using the well-defined initiators were characterised by elemental analysis. The results are tabulated in table 3.5. Generally a good agreement between the calculated and found values has been obtained for all the polymer samples.
Polymer Initiator Calculated(found) in%
Poly(exo-PhM) Ru c = 75.30(73.84) H = 5.48(5.54) N = 5.85(5.82)
Mo (1) c = (72.86) H = (5.30) N = (5.58)
Poly( exo-PhCM) Ru c = 75.87(74.30) H =5.97 (5.86) N =5.53 (6.15)
Mo (1) c = (74.76) H = (5.84) N = (5.52)
Mo (2) c = (74.46) H = (5.79) N = (5.20)
Poly( exo-PhC2M) Ru c =76.38(75.33) H =6.41(6.32) N = 5.24(5.22)
Mo (1) c = (75.91) H = (6.54) N = (6.25)
Poly( exo-PhC3M) Ru c = 76.84(75.59) H = 6.81(6.71) N = 4.98( 4.94)
Mo (1) c = (76.33) H = (6.87) N = (6.13)
Poly( exo-PhC4M) Ru c = 77.26(76.56) H =_7.17(7.14) N = 4. 74(5.86)
Poly( exo-C4PhM) Ru c = 77.26(77.07) H = 7.17(7.25) N = 4.74(4.57)
Mo (1) c = (75.89) H = (6.99) N = (4.60)
Mo (2) c = (75.89) H = (7.05) N = (4.50)
Poly( exo-C5PhM) Ru c = 77.64(76.45) H = 7.49(7.43) N = 4.53(5.41)
Mo (1) c = (76.45) H = (7.38) N = (4.36)
Mo (2) c = (76.52) H = (7.34) N = (4.13)
Poly( exo-C6PhM) Ru c = 77.99(78.81) H = 7.79(7.96) N = 4.33( 4.96)
Mo (1) c = (77.06) H = (7.74) N = (5.49)
Table 3.5: Elemental analysis results of the polymers
Chapter 3 61

3.3.3 Polymer characterisation by GPC
The polymers obtained from ROMP of phenyl norbomene dicarboxyimide derivatives
using the well-defined initiators were subjected to Gel Permeation Chromatography
(GPC) to determine their molecular weights and Polydispersity Indices (PDI). The
results are tabulated in table 3.6. It can be seen that the t-butoxy molybdenum initiator
produced polymers with narrow PDI compared to hexafluoro-t-butoxy molybdenum and
ruthenium initiators. This is an indication of a well-controlled living polymerisation
reaction. The hexafluoro-t-butoxy molybdenum initiator produced polymers with
broader PDI. This can be attributed to due to the secondary metathesis reactions50 or to
the rate of propagation being faster than the rate of initiation. Polymers obtained by
ruthenium initiator also gave polymers with broader PDI due to secondary metathesis
reaction. Similar trend have bee previous been observed.4
GPC
Polymer Initiator [M]/[I] Mn Mw PDI
Poly(exo-PhM) Ru 150 62,400 83,200 1.3'
Mo (1) 150 11,400 11,700 1.03
Poly( exo-PhCM) Ru 150 46,500 61,600 1.3
Mo (1) 150 39,400 45,500 1.1
Mo (2) 150 18,600 31,000 1.7
Poly(exo-PhC2M) Ru 150 44,300 56,900 1.3
Mo (1) 150 35,800 47,200 1.1
Poly( exo-PhC3M) Ru 150 48,800 59,000 1.2
Mo (1) 150 42,400 53,000 1.2
Poly( exo-PhC4M) Ru 150 53,000 54,600 1.03
Poly( exo-C4PhM) Ru 150 51,000 64,600 1.3
Mo (1) 150 59,700 66,500 1.1
Mo (2) 150 45,000 94,500 2.1
Poly( exo-C5PhM) Ru 150 47,800 59,000 1.2
Mo (1) 150 35,800 41,000 1.1
Mo (2) 150 19,400 47,600 1.7
Poly( exo-C6PhM) Ru 150 45,700 55,300 1.2
Mo (1) 150 45,100 54,000 1.2
Table 3.6: GPC results of the polymers
Chapter 3 62

3.3.4 Polymer characterisation using DSC
The glass transition temperatures, Tgs, for all the polymers have been obtained by DSC
analysis and the results are shown in table 3.7. It can be seen that the Tgs for the
polymers prepared by t-butoxy molybdenum initiator were about l5°C higher than the
polymers produced using hexafluoro-t-butoxy molybdenum and the ruthenium
initiators. The results suggest that for the systems studied the higher the trans content of
the polymer, the higher the glass transition temperature. It was also found that the
longer the alkyl chain in the polymer, entry l-5 and 6-8, the lower the glass transition
temperatures. This is consistent with the previous result by Feast52 et a/ and earlier
work by Hoffthat followed the same trend. 53
DSC
Entry Polymer Initiator [M]/[I] Mn Tg (°C)
1 Poly( exo-PhM) Ru 150 62,400 223
Mo (1) 150 11,400 241
2 Poly(exo-PhCM) Ru 150 46,500 143
Mo (1) 150 39,400 161
Mo (2) 150 18,600 148
3 Poly( exo-PhC2M) Ru 150 44,300 103
Mo (1) 150 35,800 121
4 Poly( exo-PhC3M) Ru 150 48,800 88
Mo (1) 150 42,400 98
5 Poly( exo-PhC4M) Ru 150 53,000 69
6 Poly( exo-C4PhM) Ru 150 51,000 245
Mo (1) 150 59,700 260
Mo (2) 150 45,000 242
7 Poly( exo-C5PhM) Ru 150 47,800 162
Mo (1) 150 35,800 175
Mo (2) 150 19,400 173
8 Poly( exo-C6PhM) Ru 150 45,700 142
Mo (1) 150 45,100 148
Table 3. 7: DSC results showing Tg of the polymers
Chapter 3 63

The results also show that the Tg for the polymers prepared from ROMP of exo-phenyl
norbomene dicarboxyimides are generally a lot higher than the polymers obtained from
exo-alkyl norbomene dicarboxyimides studied previously.
Chapter 3 64

Chapter 4
Overall conclusion and proposal for future work
Chapter 4 65

4.1 Overall conclusion
The work described in this thesis illustrates that a series of exo-phenyl norbornene
dicarboxyimide derivatives are synthesised and characterised usmg several
spectroscopic methods. It also demonstrates that polymeric material can be prepared
from phenylnorbornene d icarboxyimide derivatives using ROMP. Three well-defined
initiators, ruthenium Ch[(C6H11 ) 3P]zRu=CHC6H5, t-butoxy molybdenum
Mo[CHC(CHJ)J](CtoH17N][OC(CH3hh, Mo (1), and hexafluoro-t-butoxy molybdenum
Mo[CHC(CH3)zC6Hs][CwH17N][OC(CF3)2CH3h, Mo (2), were used.
The polymers prepared were fully characterised using lH NMR, 13CNMR, elemental
analysis, GPC and DSC.
The cis I trans contents of the polymers are readily determined from the 1H NMR
spectrum from the integration of the resonance attributable to cis and trans olefinic
hydrogen. Generally the trans vinylene content for polymers prepared by ruthenium
initiator is about 83%, for the polymers prepared by t-butoxy molybdenum initiator is
about 97% and for the polymers prepared by hexafluoro-t-butoxy molybdenum initiator
is about 31%.
The t-butoxy molybdenum initiator produced polymers with narrow PDI compared to
hexafluoro-t-butoxy molybdenum and ruthenium initiators. This is an indication of a
well-controlled living polymerisation reaction. The hexafluoro-t-butoxy molybdenum
initiator produced polymers with broader PDI. This can be attributed to due to the
secondary metathesis reactions or to the rate of propagation being faster than the rate of
initiation. Polymers obtained by ruthenium initiator also gave polymers with broader
PDI due to secondary metathesis reaction. Similar trend have bee previous been
observed.
The elemental analysis of polymers showed a good agreement between the calculated
and found values has been obtained for all the polymer samples.
The Tgs for the polymers prepared by t-butoxy molybdenum initiator were about l5°C
higher than the polymers produced using hexafluoro-t-butoxy molybdenum and the
ruthenium initiators, suggesting that the higher the trans content of the polymer, the
higher the glass transition temperature. It was also found that the longer the alkyl chain
in the polymer, the lower the glass transition temperatures. The results also showed that
the Tg for the polymers prepared from ROMP of exo-phenyl norbornene
Chapter 4 66

dicarboxyimides are generally a lot higher than the polymers obtained from exo-alkyl
norbomene dicarboxyimides studied previously.
4.2 Proposal for future work
The prevwus work showed that exo-alkyl norbomene dicarbcixyimides could
successfully be subjected to ROMP using well-defined ruthenium initiator resulting in
well-defined linear polymers in both solution and bulk. It was also demonstrated that
the copolymerisation of exo-alkyl norbomene dicarboxyimides with exo-alkyl
dinorbomene dicarboxyimides difunctional monomers results in a well-defined
crosslinked materials, using resin transfer moulding process (RTM). However, the Tg
of the crosslinked materials were lower than the commercial materials based on
Poly(DCPD). The linear polymeric materials produced here from the ROMP of exo
phenyl norbomene dicarboxyimides exhibited higher Tgs than Poly(DCPD). The
monomers were all solid but soluble in exo-alkyl norbomene dicarboxyimides. It will
therefore be possible to carry out ROMP-RTM processing of the monomers to obtain
linear materials and also well-defined crosslinked materials using exo-alkyl
dinorbomene dicarboxyimides difunctional monomers with higher Tgs.
Chapter 4 67

Appendix 1
Instrumentation and procedures for measurements
Appendix 1 68

i. NMR spectroscopy: 1H, 13C, COSY, DEPT
The samples were analysed either on a V ARIAN INOV A 500 NMR spectrometer at
499.78 eH) I 125.67 (13C) MHz, or V ARIAN MERCURY 400 NMR spectrometer at
399.96 eH) I 100.57 (13C) MHz, or BRUKER AV ANCE 400 NMR spectrometer at . I .
400.13 (H). The solvents used were deuterated chloroform and deuterated acetone.
ii. GEL PERMEATION CHROMATOGRAPHY (GPC)
o The samples were dissolved in one of the solvents; chloroform (CHCh),
dimethylformamide (DMF), and tetrahydrofuran (THF). The choice of solvent
depends on the solubility of the polymer. Each solvent was run at a different
instrument.
• THF: Viscotek 200 with refractive index, viscosity and light scattering detectors,
and 2 x 300mm PLgel 5flm mixed C columns; THF was used as the eluent with a
flow rate of 1.0mllmin and a constant temperature of 30°C. Molecular weights were
obtained using a conventional calibration curve generated from narrow molecular
weight distribution PEG/PEO standards.
• DMF: Viscotek TDA 302 with refractive index detector, and 2 x 300mm PLgel
5flm mixed C colunms; DMF was used as the eluent with a flow rate of l.Omllmin
and a constant temperature of 80°C. Molecular weight were obtained using triple
detection, the detectors were calibrated with a single narrow molecular weight
distribution polystyrene standard.
• CHCI3 : Viscotek TDA 301 with refractive index, viscosity and light scattering
detectors, and 1 x 300mm PLgel 5!lm mixed C columns; CDCh was used as the
eluent with a flow rate of 1.0mllmin and a constant temperature of 30°C. Molecular
weight were obtained using triple detection, the detectors were calibrated with a
single narrow molecular weight distribution polystyrene standard.
iii. MASS SPECROSCOPY
The samples were analysed on an EXETER ANALYTICAL, Inc CE-440 elemental
analyser.
Appendix 1 69

iv. DIFFERENTIAL SCANNING CALORIMETER (DSC)
The samples were analysed on a Perkin Elmer DSC 7 or a Pyris 1 differential
scanning calorimeter at a ramp rate of 1 0°C/min.
Appendix 1 70

Appendix 2
Analytical data for chapter 2
Appendix 2 71

_j I ~ ~
I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I
7 6 5 4 3 2 ~~m
Appendix 2.1: 1H NMR spectrum of exo-PhCM
I lljllllllllljlllllll I ljlll lllllljllllllllljllllllllljlllllll 11 jllll llllljlllllll
180 160 140 120 100 80 60 ppm
Appendix 2.2: 13C NMR spectrum of exo-PhCM
Appendix 2 72

1001 66 8.285
95 7.885
90 7.4E5
as· 7.0ES
80 6.6E5
7s 187 6.185
70 5.7ES
65 91 253 S.JBS
60 4.9ES
55 4.SES
50 4.1ES
45 J.?ES
40 3.3ES
35 2.985
30- 2.585
2s 2.085
20 106 1.6ES
15 1.285
10
r, ~ ~I TT 8.2E4
159 5 sh .. -1 1f9 4.1E4
0 . [, O.OEO 40 6o 80 160 1Jo i!o 160 iao 260 220 240 2/;o 2so 300 3:\o 3!o J6o m/z
Appendix 2.3: Mass spectrum of exo-PhCM
I I I I
8 7 6 5 4 3 2 pp m
Appendix 2.4: 1H NMR spectrum of exo-PhC2M
Appendix 2 73

I111111111Ji 111111 IIJIIII 1 illlJIIiljll r 11''' r 11 ltijlillj 1 t 111 t t 1 t 1''''1'1 1 ijlllll 1 t 11
180 160 140 120 100 80 60 40 pp m
Appendix 2.5: 13 C NMR spectrum of exo-PhC2M
1001 1 4 7.1'1!6
95 7.4E6
90 7.0E6
85 6.6E6
80 6,2E6
75 5.8E6
70 5.4E6
65 5.0E6
60 4.6E6
55 4.3E6
50 3.9E6
45 3.5E6
40 91 267 3.1E6
35 :i.7E6
30 66 2.3E6
J5 1.9E6
20 110 l.SE6
15 1.2E6
10 7.7E5
:bl J,
77 5 Jil, 2r 3.9ES
0 1r 176 O.OEO 40 ·6o 80 160 120 140 160 u\o 260 220 240 260 280 360 3:20 340 360 m/z
Appendix 2.6: Mass spectrum of exo-PhC2M
Appendix 2 74

__ __) L__ __ __l\._ _________ _JL_JL. .--' \._ _ __!lJL__ll__ __ I I I
8 7 6 5 4 3 2 pp m
Appendix 2.7: 1H NMR spectrum of exo-PhC3M
I lllljllllllllljllllllllljllllllllljllllllllljllllllllljllll llllljllllllllljllllllllljll
180 160 140 120 100 80 60 40 pp m
Appendix 2.8: 13 C NMR spectrum of exo-PhC3M
Appendix 2 75

1001
95
90
85
80
75
70
65
60
55
50
45
40
35
30
25
20
15
10
5
0 40
l 1 .9.3E5
8.BE5
8.4E5
7.9E5
1.4E5
7.0ES
91 6.5E5
6.0E5
5.685
5.1E5
111 4.6E5
4.2E5 66 3. 7E5
3.3ES 281
177 2.8E5
2.3E5
216 1.9E5
l.4ES 82
.·~ 164
9.3E4
~~( .1, 9~8J~ I I~ :i4
1t8 ·r 4.6E4 1~0 O.OEO
60 80 160 1 0 14o 1/;0 180 260 22o 240 26o 2 0 360 32o · J4o Jlo m/z
Appendix 2.9: Mass spectrum of exo-PhC3M
'--------'L_ _____ _J~ L__)'---,_1...____, ll1L__)L__ I
7 I
6
I I I I I
5 4
I I I
3
I I
Appendix 2.10: 1H NMR spectrum of exo-PhC4M
Appendix 2
2 ppm
76

I I lllllllljl 11111111111111111111111111111111111111111 I I I' I Ill I 1111111 I 111111111111111
180 160 140 120 100 80 60 40 pp m
Appendix 2.11: 13 C NMR spectrum of exo-PhC4M
100l 91 5.2E6
95 4.9E6
90 4.7E6 66 4.4E6 85
80 295 4.2E6
75 3.9E6
70 3.6E6
65 3.4E6
60 3.1E6
55 2.9E6
50 2.6E6
45 230 2.3E6
40 I 2.1E6 110 131
35 204 1.8Efi
30 194 138 1.6E6
25 1.3E6 164
20 1.0E6
15 82 7.8E5
10
lilt I~ 98 177 5.2E5
~5 II I 5 d 1fl6 2.6E5
0 '~. . I ll 2f7 O.OEO
40 60 80 100 120 1 0 1~0 1~0 2bo 2Io 2!o 2~o 280 360 32o 3!o 36o m/•
Appendix 2.12: Mass spectrum of exo-PhC4M
Appendix 2 77

----~L~L----~~--------------------~'--~·~-~----~Illu_ __ __
8 I
7 6 5 4 3
Appendix 2.13: 1H NMR spectrum of exo-C4PhM
2 pp m
lllllllllllllllllllllllllllllllllllllllllll!tlllllllllllllllllijilllllllllillllllllltillllillll
180 160 140 120 100 80 60 40 pp m
Appendix 2.14: 13C NMR spectrum of exo-C4PhM
Appendix 2 78

10 2 4 3.8E6
95 3.6E6
90 3.4E6
85 3.2E6
80 3.0E6
75 2.8E6
70 2.7E6
65 2.5E6
60 2.3E6
55 2.1E6
50 1.9E6
45 280 i.7E6 295
40 1.5E6
35 1.3E6
30 91 66
1.1E6
25
20 132
229 9.5E5
7.6E5 186
15 5.7E5 115
10 3.8E5
5 l.9E5
m/z
Appendix 2.15: Mass spectrum of exo-C4PhM
•u \.___j
7 6 5 4 3 2 pp m
Appendix 2.16: 1H NMR spectrum of exo-C5PhM
Appendix 2 79

I I I I I It '1''''1'' I 'I I llljlll I 1''''1''''1''''1''' I l I I 11 I I I ''I'' I 'I''') jll I 'I'' I I I I llljl I I 'I I
180 160 140 120 100 80 60 40 pp m
Appendix 2.17: 13C NMR spectrum of exo-C5PhM
liMll 1 6 );SE6
95 3.4E6
90 3.2E6
85 3.0E6
80 2.8E6
75 2.7E6
70 243 2.5E6·
65 2.3E6
60 2.1E6
55 2.0E6
309 1.BE6 so 45
1.6E6
40 1.4E6
66 91 ·J5 1. 2E~
30 132 ! 1.1E6
·25 158 8.9E5
20 7.1ES
15 77 5.3E5
10
]f~ t T256
3.5E5 51 . T 5 l! Lu, T
l.8E5
145 214 O.'OEO 0 40 '6o so i6o i:lo 1!o ao iso :ioo 2~o :i4o 2/;o 28o .:i6o :iio :i{o 3/;o :ieo 46o 4~o 44o .. m/z
Appendix 2.18: Mass spectrum of exo-C5PhM
Appendix 2 80

7 6 5 4 3 1 ppm
Appendix 2.19: 1H NMR spectrum of exo-C6PhM
llli[IIIIJIIII[IIIIJIIII[IIIIJIIII[IIIIJIIII[IIIIJIIII[IIIIJIIII[IIIIJIIII[IIIIJIIII[IIIIJII
180 160 140 120 100 80 60 40 ppm
Appendix 2.20: 13 C NMR spectrum of exo-C6PhM
Appendix 2 81

1001 1 6 6.886
95 6.5E6
90 6.2E6
85 5.8E6
80 5.5E6
75 257
5.1E6
70 323 4.8E6
65 4.4E6
60 4.1E6
55 3.BE6
50 3.4E6
45 3.1E6
40 2.7E6
35 66 91 2.4E6
30 132 2.1E6
25 158 1. 7E6
20 1.4E6
15 l.OE6
10
l l 6.8E5
5 l 1
irlk ~ ~r T 3',4E5
0 214 O.OEO
40 '6o 80 ioo Ho iAo 1 o iao .2oo :i2o 24o 2 a :iao :ioo 3 o :i4o _:i~o :iao 4oo 42o 44o m/z
Appendix 2.21: Mass spectrum of exo-C6PhM
I I u 'L- I I I I I I I I I I I I I I I I I
8 7 6 5 4 3 2 1 .Q pp m
Appendix 2.22: 1H NMR spectrum of exo-C6M
Appendix 2 82

IIIIIIIII[IIIIIIIII[IIIIIIIII[IIIIIIIII[IIIIIIIII[IIIIIIIII[IIIIIIIII[IIIIIIIII[IIIIJIIII[II
180 160 140 120 100 80 60 40 pp m
Appendix 2.23: 13C NMR spectrum of exo-C6M
100! 66 3. SE6
95 3.6E6
90 3.4E6
85 3. 2E6
u 3.0~
7S 2. 8E6
70 2.7E6
65 182 2.5E6
60 2.3E6
ss· 2.1E6
50 1.9E6
45 1.7E6
40 1.5E6
3S 91 1.3E6
30 110 247 1.1E6
25. 98 9.5ES
20 ss 164 7. 6E5
15 82 120 5.7ES
1
: l .~: 1!1~ ,;. lk ~13
f8
l]s2
::::: 19o 2 ?4 2~8 01~~~! ~~~~~~~~~~l~l~~~~~~~~~~~~~~~O.OEO 40 6o 80 160 1 o Ho i~o 1 o :ibo 2:2o :i~o ito i~o 36o 3:lo 3~o :lto )~o 46o m/z
Appendix 2.24: Mass spectrum of exo-C6M
Appendix 2 83

Appendix 3
Analytical data for chapter 3
Appendix 3 84

L_ 7 6 5 4 3 2 pp m
Appendix 3.1: 1H NMR spectrum poly(exo-PhM)- Ru
lllllll(lllijlilijlilljllli[iilllllii[illljlrll]llil[iili[iilfjllll]litiflifi[iiiljilli[liiiliiii[iilijilii[iiiljllll[lilllll
220 200 180 160 140 120 100 80 60 40 20 pp m
Appendix 3.2: 13C NMR spectrum poly(exo-PhM)- Ru
Appendix 3 85

I
7 6 5 4 3 2
Appendix 3.3: 1H NMR spectrum poly(exo-PhCM)- Ru
ppm
'''I''' I 1''''1''''1''''1''''1''''1''''1''''1''''1''''1''''1'' 11 1''''1''''1''''1''''1 11
180 160 140 120 100 80 60 pp m
Appendix 3.4: 13C NMR spectrum poly(exo-PhCM)- Ru
Appendix 3 86

7 6 5 4 3 2 pp m
Appendix 3.5: 1H NMR spectrum poly(exo-PhC2M)- Ru
J JJJ.J I 11, 'I,, ,, 1, ,, , I',, IJI r, 1 1,,,, 1,, 11l''t, 1,, ''I, 11, 1,, ''l'l, , 11 '''!'l''l 1,, 1 I', ''l''''l' 1,
180 160 140 120 100 80 60 40 pp m
Appendix 3.6: 13C NMR spectrum poly(exo-PhC2M)- Ru
Appendix 3 87
7 6 5 4 3 2 ppm
Appendix 3.7: 1H NMR spectrum poly(exo-PhC4M)- Ru
I I I I [ I ( I I I 11 11 I' 11 I I I I I I I' I I I I I I I I I I I I I I I I I I I' I I I I I I I I I ( I I l I l ( [ I I' I I I I I I I I I' I I ( I I I I I I I I I I I
180 160 140 120 100 80 60 40 pp m
Appendix 3.8: IJC NMR spectrum poly(exo-PhC4M)- Ru
Appendix 3 88

8 7 6 5 4 3 2 ppm
Appendix 3.9: 1H NMR spectrum poly(exo-C4PhM)- Ru
llllliij I I If JIIIIJIIIIJIIIIJIIIIJII I IJII I 'I I lll[lllijili f Jllll I IIIIJ t I IIJI I I ljlllljiliijl I
180 160 140 120 100 80 60 40 pp m
Appendix 3.10: llC NMR spectrum poly(exo-C4PhM)- Ru
Appendix 3 89

7 6 5 4 3 2 pp m
Appendix 3.11: 1H NMR spectrum poly(exo-C5PhM)- Ru
I J IIJ!Illjillljlliijlllljl!iijllllllllljlllfJIIIIjllii)lilljll!l]iilljllliJIIIIjllllllllijliiiJIIIIjllif)lllljliiiJIIIIjllll)il
220 200 180 160 140 120 100 80 60 40 20 pp m
Appendix 3.12: 13C NMR spectrum poly(exo-C5PhM)- Ru
Appendix 3 90

Appendix 3.13: 1H NMR spectrum poly(exo-CJ'hM)- Ru
J I J tJ!ll 1 1]11 t tlliltJlillji 111 ]I I 1 ljllii]li 1 ljll!l]lllijllll]iitl] 111 IJII 1 'I' 1 11]111 t 1 t 1111 r 1
160 140 120 100 80 60 40 pp m
Appendix 3.14: 13C NMR spectrum poly(exo-C6PhM)- Ru
Appendix 3 91

8 I
7 6 5 4 3 2
Appendix 3.15: 1H NMR spectrum poly(exo-PhM)- Mo (I)
pp m
I I I I I I I i I I I I I I I I I' I 1 I I I I I I 1 I I I I I I I 11 I I I I I I 1 1 If I I I I I l I I I I ,. I I I I I I I I I' I I I I I I I I I I J I I I I I l I jl
180 160 140 120 100 80 60 40 pp m
Appendix 3.16: 13C NMR spectrum poly(exo-PhM)- Mo (1)
Appendix 3 92

8 7 6 5 4 3 2 ppm
Appendix 3.17: 1H NMR spectrum poly(exo-PhCM)- Mo (1)
I I I I I l I l I I I I 1 I 1 I I I I I I I I f I I I I I I I ~ I I I I I ,·I I I I I I I I I I I I I I I 1 I 1 I 1 I I 1 I I I I I I I I I I I 1 I I 1 l I I I I I I I I
160 140 120 100 80 60 40 pp m
Appendix 3.18: 13C NMR spectrum poly(exo-PhM)- Mo (1)
Appendix 3 93

8 7 6 5 4 3 2 pp m
Appendix 3.19: 1H NMR spectrum poly(exo-PhC2M)- Mo (I)
I I I I I 11 I jlllijlllljl I I ljll I I) I I I 1!1_1 I 1) I I .I I Ill I I 11 I I 1 I 11 I I) I I I I Ill I ill I I I I I llljllillll
180 160 140 120 100 80 60 40 pp m
Appendix 3.20: 13C NMR spectrum poly(exo-PhC2M)- Mo (1)
Appendix 3 94

I I
8 7 6 5 pp m
Appendix 3.21: 1H NMR spectrum poly(exo-C4PhM)- Mo (I)
I 11 I
lljlllljlllljtflljllllllllljlllljllfijlllllfllljiilf(lllljlllljlilijlll!fll!ljlillllltljlilljlllijliiiJiiiljllll!lllljllll!ll
220 200 180 160 140 120 100 80 60 40 20 pp m
Appendix 3 95

Appendix 3.22: ne NMR spectrum poly(exo-C4PhM)- Mo (1)
7 6 5 4 3 2 pp m
Appendix 3.23: 1H NMR spectrum poly(exo-C5PhM)- Mo (1)
I I L 1 1 t 1 1 I' t 1 1 1 1 1 1 1 I' 1 1 1 1 t 1 1 1 I' t 1 1 11 1 1 1 1 t 1 1 1 1 1 1 1 1 I' 1 1 1 1 1 t 1 1 1 1 1 1 1 ! 1 1 1 1 11 1 1 1 1 1 1 1 1 I' r 1 1 1 1 1 1t 1 t 1 1 1 1 1 t 1 1 jll 1
180 160 140 120 100 80 60 40 20 pp m
Appendix 3.24: ne NMR spectrum poly(exo-CsPhM)- Mo (1)
Appendix 3 96

7 6 5 4 3 2 pp m
Appendix 3.25: 1H NMR spectrum poly(exo-C6PhM)- Mo (1)
I I 1 1 I t 1 1 1 1 1 1 t 1 1 1 t 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 11 1 1 r 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 , 1 1 t 1 1 t 1 1 1 1 1 1 1 1 1 t 1 1 ' 1 1 I' t 1 t 1 1 1 1 1 I' t t 1 1 11
160 140 120 100 80 60 40 pp m
Appendix 3.26: 13C NMR spectrum poly(exo-C6PhM)- Mo (1)
Appendix 3 97

I I
7 6 5 4 3 2 pp m
Appendix 3.27: 1H NMR spectrum poly(exo-PhCM)- Mo (2)
lljllli]llii]iliijllli]lliljllfi]illljlill]iifijiilljflfijllfljtlil]iiiljiili]lli\]lilljilll]lllijlllljtiiljilii]!ill]llll]li
220 200 180 160 140 120 100 80 60 40 20 pp m
Appendix 3.28: 13C NMR spectrum poly(exo-PhCM)- Mo (2)
Appendix 3 98

' I
7 6 5 pp m
Appendix 3.29: 1H NMR spectrum poly(exo-C4PhM)- Mo (2)
220 200 180 160 140 120 100 80 60 40 20 pp m
Appendix 3.30: 1H NMR spectrum poly(exo-C4PhM)- Mo (2)
Appendix 3 99

Appendix 4
Conferences and seminar attended
Appendix4 100

CONFERENCESANDSEMINARSATTENDED
• ProfMarcetta Darensbourg: Dept of Chemistry, Texas A&M University
Functioning Catalyst Inspired by Active Sites in Bio-organometallic Chemistry:
Thew Hydrogenases (23rd October2002).
@ Prof Geoffrey Lawrance: Newcastle University, Australia
Designer L igands: M acrocyclic and alicyclic molecules for metal c omplexation
and biocatalysis (13th November 2002).
8 Dr David Procter: Dept of Chemistry, University of Glasgow
New Strategies and Methods for Organic Synthesis (22"d January 2003).
• Prof Paul Raithby: Dept of Chemistry, University of Bath
Adventures in Organometallic Polymer chemistry (1 ih February 2003).
• Prof Kingsley Cavell: University of Cardiff
(26th February 2003).
8 ProfKevin Shakesheff: The Pharmacy school, University of Nottingham
Polymer materials in tissue engineering (19th March 2003).
• Prof Joe Keddie: Dept of Physics, University of Surrey.
The sticky Business of Waterborne Pressure-Sensitive Adhesives (26th March
2003).
• Royal Society of Chemistry: Dalton division, One day symposium
Metal Mediated Polymerisation (11 1h November 2002).
Appendix 4 0 . ' . ,,'
101

REFERENCES
1. J.D. Rule and J.S. Moore, macromolecules, 35, 7878, 2002.
2. L. Matejka, C. Houtrnan and C.W. Macosko, J Appl. Polym. Sci., 30,2787, 1985.
3. P.J. Hine, T. Leejarkpai, E. Khosravi, R.A. Duckett and W.J. Feast, Polymer, 42, 9413,
2001.
4. A.M.A. Al-Hajaji, PhD Thesis, University ofDurharn, UK, 1995.
5. T. Leejarkpai, PhD Thesis, University of Durham, UK, 2000.
6. R.J. Young and P.A. Lovell, Introduction to Polymers, Nelson Thames LTD, UK,
1991.
7. R.L. Banks and G.C. Bailey, Ind. Eng. Chem. Prod. Res. Dev., 3, 170, 1964.
8. N. Calderon, Chem, Eng, News, 45, 51, 1967.
9. A.W. Anderson and N. G. Merckling., US. Patent 2, 721, 189, ChemAbstr., 50, 3008i,
1955.
10. C.P.C. Bradshaw, E.J. Howrnann, and L. Turner, J Catal., 7, 269, 1967.
11. N. Calderon, E.A. Ofstead, J.P. Ward, W.A. Judy and K.W. Scott, JAm. Chem. Soc.,
90, 4133, 1968.
12. J. C. Mol, F.R Visser and J. Boelhouwer, J Catal., 17, 114, 1970.
13. J. Herrison and Y. Chauvin, Makromol. Chem., 141, 161, 1970.
14. T.M. TRNKA and R.H Grubbs, Ace. Chem. Res., 34, 19,2001.
15. K.J. Ivin and J.C. Mol, Olefin Metathesis and Metathesis Polymerisation, Academic
Press, San Diego, USA, 1997.
16. E.O. Fischer and A. Maasbol, Angew. Chem. Inter. Ed., 3, 580, 1964.
17. C.P. Casey and T.S. Burkhardt, JAm. Chem. Soc., 95, 5833, 1973.
18. F. N. Tebbe, G.W. Parshall, G.S.J. Reddy, JAm. Chem. Soc., 100, 3611, 1978.
19. F. N. Tebbe, G.W. Parshall, D.W. Ovenall, JAm. Chem. Soc., 101, 5074, 1979.
20. L.R. Gilliom, and R.H. Grubbs, JAm. Chem. Soc., 108, 733, 1986.
21. R. H. Grubbs, Comprehensive Organometallic Chemistry, Vol6, p.499, Wilkinson, G.
(Ed), Pergamon, Oxford, 1982.
22. T.R. Howard, J.B. Lee. R.H Grubbs, JAm. Chem. Soc., 102, 6878, 1980.
23. J.B. Lee, K.C. Ott, R.H. Grubbs, JAm. Chem. Soc., 104, 7491, 1982.
24. J. Kress and J.A. Osbom., JAm. Chem. Soc., 105, 6346, 1983. _
102

25. RR Schrock, J.S Murdzek, G.C Bazan, J. Robbins, M. Dimare and M. O'Regan, J
Am. Chem., Soc., 112, 3875, 1990.
26. G.C. Fu and RH. Grubbs, JAm. Chem. Soc., 114, 7324, 1992.
27. G. C. Fu and RH. Grubbs, JAm. Chem. Soc., 114, 5426, 1992.
28. J.S. Clark and J.G. Kettle, Tetrahedron Lett., 38, 123, 1997.
29. T.A. Kerkland, RH. Grubbs, JAm. Chem. Soc., 62, 7310, 1997.
30. S.T. Nguyen, L.K. Johnson and RH. Grubbs, JAm. Chem. Soc., 114, 3974, 1992.
31. S.T. Nguyen, RH. Grubbs, J.W. Ziller, JAm. Chem. Soc., 115, 9858, 1993.
32. P. Scwab, R.H. Grubbs and J.W. Ziller, JAm. Chem. Soc., 118, 100, 1996.
33. G.C. Fu, S.T. Nguyen, RH. Grubbs, JAm. Chem. Soc., 115, 9856, 1993.
34. M. Scholl, T.M. Trnka, J.P. Morgan and RH. Grubbs, Tetrahedron. Lett., 40,2247,
1999.
35. M. Scholl, S. Ding, C.W. Ding, RH. Grubbs, Org. Lett., 1, 953, 1999.
36. C.W. Bielawski, R.W. Grubbs, Angew. Chem. In. Ed., 39,2903,2000
37. D.A. Robson, V.C. Gibson, RG. Davies, M. North, Macromolecules, 32,6371, 1999.
38. M.S. Sanford, M. Ulman and RH. Grubbs, JAm. Chem. Soc., 123, 749, 2001.
39. C.W. Bielawski and RH. Grubbs, Macromolecules, 34, 8838, 2001.
40. J. Asrar, Macromolecules, 25, 5150, 1992.
41. P.Y. Bruce, Organic Chemistry, 4th Ed., Person Educational Int., USA, 2001.
42. G. Solomon, C. Fryhle, Organic Chemistry, ih Ed., John Wiley and Sons, Inc., USA,
2000.
43. G.M. Loudon, Organic Chemistry, 2nd Ed., The Benjamin/ Cummings Publishing
Company, Inc., California, 1988.
44. C.K. Ingold, Structure and Mechanism in Organic Chemistry, G. Bell and Sons Ltd.,
London, 1953.
45. J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic Chemistry, Oxford
University Press, New Yolk, 2001.
46. J. McMURRY, Organic chemistry. znct Ed., Brooks/Cole publishing Company,
California, 1992.
47. T.H. Lowry and K.S. Richardson, Mechanism and Theory in Organic Chemistry, 3rd
Ed., Harper Collins Publishers, New Yolk, 1987.
48. D. Craig, JAm. Chem. Soc., 73 (4), 4889, 1951.
103

49. S.C.G. Biagini, M.P. Coles, V.C. Gibson, M.R. Giles, E.L. Marshall and M. North,
Polymer, 39 (5), 1007, 1998.
50. E. Khosravi and A.A. Al-Hajaji, Polymer, 39 (23), 5619, 1998.
51. J.H. Oskam and R.R. Schock, JAm. Chem. Soc., 115, 11831, 1993.
52. E. Khosravi, W.J. Feast, A.A. Al-Hajaji and T. Leejarkpai, J Mol. Cat., 160, 1, 2000.
53. E.A. Hoff, D.W. Robinson, A.H. Willboum, J Polym. Sci., 18, 161, 1955.
104


















