
DUEXIS® 800 mg/26.6 mg Film-coated Tablets (PL 36663/0002)
64
UKPAR DUEXIS ® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002 1 DUEXIS ® 800 MG/26.6 MG FILM-COATED TABLETS PL 36663/0002 UKPAR TABLE OF CONTENTS Lay Summary Page 2 Scientific discussion Page 3 Steps taken for assessment Page 61 Summary of Product Characteristics Page 62 Patient Information Leaflet Page 63 Labelling Page 64
Transcript of DUEXIS® 800 mg/26.6 mg Film-coated Tablets (PL 36663/0002)

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
1
DUEXIS® 800 MG/26.6 MG FILM-COATED TABLETS
PL 36663/0002
UKPAR
TABLE OF CONTENTS Lay Summary
Page 2
Scientific discussion
Page 3
Steps taken for assessment
Page 61
Summary of Product Characteristics
Page 62
Patient Information Leaflet
Page 63
Labelling
Page 64

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
2
DUEXIS® 800 mg/26.6 mg Film-coated Tablets
PL 36663/0002
LAY SUMMARY
On 6th March 2013, the MHRA granted a Marketing Authorisation (licence) for the medicinal product DUEXIS® 800 mg/26.6 mg Film-coated Tablets (PL 36663/0002). This medicine is only available on prescription from your doctor. What DUEXIS is DUEXIS contains two different medicines called ibuprofen and famotidine in a single tablet. Each of these medicines works in a different way.
- Ibuprofen belongs to a group of medicines called non-steroidal anti-inflammatory drugs (NSAIDs). It reduces pain and inflammation.
- Famotidine belongs to a group of medicines called H2-blockers. It decreases the amount of acid in your stomach therefore reducing the risk for pelptic ulcers (ulcers in your stomach or duodenum) which may develop in patients who need to take NSAIDs.
What DUEXIS is used for: DUEXIS is used for the relief of pain, swelling, redness and heat (inflammation) in people suffering from osteoarthritis, rheumatoid arthritis and ankylosing spondylitis. You will be given this medicine if
- a lower dose of ibuprofen, or another NSAID, is considered unlikely to improve your pain
- you require regular treatment, - you are at risk of getting ulcers in your stomach (gastric ulcer) and/or in the
first part of the intestine (duodenal ulcer) when taking NSAIDs, and - if treatment with a proton pump inhibitor is not considered appropriate.
You must take this medicine three times daily to provide a high enough level of famotidine, which protects against gastric side effects. This protection may be less if a dose is missed. No new or unexpected safety concerns arose from this application and it was, therefore, judged that the benefits of taking DUEXIS® 800 mg/26.6 mg Film-coated Tablets outweigh the risks and a Marketing Authorisation was granted.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
3
DUEXIS® 800 mg/26.6 mg Film-coated Tablets
PL 36663/0002
SCIENTIFIC DISCUSSION
TABLE OF CONTENTS
Introduction
Page 4
Pharmaceutical assessment
Page 6
Non-clinical assessment
Page 10
Clinical assessment
Page 11
Overall conclusions and risk benefit assessment Page 59

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
4
INTRODUCTION The Medicines and Healthcare products Regulatory Agency (MHRA) granted a Marketing Authorisation (licence) for the medicinal product DUEXIS® 800 mg/26.6 mg Film-coated Tablets (PL 36663/0002) to Horizon Pharma (UK) Limited, on the 6th March 2013. This application was submitted as an Article 10b application for a new oral fixed combination product (FCP) containing the active substances ibuprofen (800 mg) and famotidine (26.6 mg) in tablet form. The proposed name is DUEXIS® 800 mg/26.6 mg Film-coated Tablets and is referred as HZT-501 in some parts of this report. DUEXIS is indicated for the symptomatic treatment of osteoarthritis, rheumatoid arthritis and ankylosing spondylitis in patients who require regular treatment with high dose ibuprofen administered three times a day and who are at risk of developing non-steroidal anti-inflammatory drug (NSAID) associated gastric and/or duodenal ulcers. DUEXIS should only be used where treatment with lower doses of ibuprofen or of other NSAIDs is not considered sufficient and where treatment with a proton pump inhibitor (PPI) is not considered appropriate. An application was previously submitted in Europe as a Decentralised Procedure on 6th January 2011 with the United Kingdom as the reference member state and France, Germany, Italy, Luxemburg, Norway and The Netherlands as concerned member states (CMS). Following Quality and Clinical potential serious risks for public health being raised, supported by the CMSs, this procedure was withdrawn on 6th January 2012. The applicant re-submitted the application under a National Procedure. The application was referred to the Commission on Human Medicines (CHM) and was considered by the Commission at their meeting on 17th May 2012. Scientific advice was sought on a range of quality, non-clinical and clinical aspects of development. DUEXIS is an immediate release, fixed dose combination tablet of ibuprofen and famotidine. Ibuprofen possesses analgesic, anti-inflammatory and antipyretic activities. Its mode of action, like that of other NSAIDs, is not completely understood, but may be related to prostaglandin synthetase inhibition. Famotidine is a competitive inhibitor of histamine H2-receptors. The primary clinically important pharmacologic activity of famotidine is inhibition of gastric secretion. Both the acid concentration and volume of gastric secretion are suppressed by famotidine, while changes in pepsin secretion are proportional to volume output. The applicant has submitted pharmacokinetic, efficacy and safety data, including 2 pivotal phase III studies comparing the proportion of patients developing endoscopically diagnosed gastric and duodenal ulceration after 24 weeks of the FCP, compared to 800 mg ibuprofen alone. The efficacy and safety of the ibuprofen

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
5
component is supported by pharmacokinetic data claiming to demonstrate bioequivalence with an EU reference product. All the clinical studies were conducted in the United States and are in compliance with Good Clinical Practice (GCP). Details of a pharmacovigilance system have been provided with this application and are satisfactory. A satisfactory Risk Management Plan (RMP) has also been provided.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
6
PHARMACEUTICAL ASSESSMENT
DRUG SUBSTANCES
Nomenclature rINN: Famotidine Chemical Names: 3- [[[2-[(Diaminomethylene)amino]thiazol-4-yl] methyl] sulphanyl]- N'-sulphamoyl propanimidamide. or [l-Amino-3- [[[2-[(diaminomethylene) amino]-4-thiazolyl]-methyl] thio] propylidene] sulfamide or Propanimidamide, N ′-(aminosulfonyl)-3-[[[2-[(diaminomethylene) amino]-4-thiazolyl] methyl]thio Structure:
Molecular Formula: C8H15N7O2S3
Molecular Weight: 337.45 g/mol Appearance: A white to pale yellowish white, crystalline powder Solubility: Freely soluble in dimethylformamide and in glacial acetic acid; slightly soluble in methanol; very slightly soluble in water; practically insoluble in acetone, in alcohol, in chloroform, in ether and in ethyl acetate. All aspects of the manufacture and control of the active substance famotidine are covered by a European Directorate for the Quality of Medicines (EDQM) Certificate of Suitability. rINN: Ibuprofen Chemical Names: (±)-2-(p-isobutylphenyl) propionic acid (2RS)-2-[4-(2
methylpropyl)phenyl]propanoic acid Structure:

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
7
Molecular Formula: C13H18O2
Molecular Weight: 206.28 Appearance White or almost white crystalline powder or colourless crystaline Solubility: Soluble in ethanol, chloroform, ether, acetone, aqueous solutions of alkali hydroxides and carbonates, dichloromethane, methanol, and ethyl acetate. Insoluble in water All aspects of the manufacture and control of the active substance ibuprofen are covered by a European Directorate for the Quality of Medicines (EDQM) Certificate of Suitability. DRUG PRODUCT Other ingredients Other ingredients consist of the pharmaceutical excipients microcrystalline cellulose, lactose anhydrous, croscarmellose sodium, silica, colloidal anhydrous, magnesium stearate, Opadry white (titanium dioxide (E171), hypromellose, macrogol 400 and polysorbate 80) making up the tablet core. The outer tablet consists of microcrystalline cellulose, povidone, magnesium stearate, silica, colloidal anhydrous, croscarmellose sodium and Opadry II blue (poly (Vinyl Alcohol), titanium Dioxide (E171), macrogol 3350, talc, indigo carmine aluminium Lake (E132) and Brilliant Blue FCF Aluminium Lake (E133)). All excipients used comply with their respective European Pharmacopoeia monographs with the exception of Opadry white and Opadry II blue which are covered by in-house specifications. Satisfactory Certificates of Analysis have been provided for all excipients. The only excipient used that contains material of animal or human origin is lactose anhydrous. The applicant has provided a declaration that the milk used in the production of lactose anhydrous excipient is sourced from healthy animals under the same conditions as that for human consumption. Confirmation has also been given that the magnesium stearate used in the tablets is of vegetable origin. Pharmaceutical development This fixed-dose, immediate release, combination of 800 mg ibuprofen and 26.6 mg famotidine is designed to facilitate the administration of an anti-ulcer agent, famotidine, with the non-steroidal anti-inflammatory drug (NSAID), ibuprofen for the symptomatic treatment of osteoarthritis, rheumatoid arthritis and ankylosing spondylitis in patients who are at risk of developing, NSAID-associated gastric and/or duodenal ulcers and where treatment with lower doses of ibuprofen or of other NSAIDs is not considered sufficient. Comparable dissolution and impurity profiles are provided for this product versus the originator product.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
8
Manufacture Satisfactory batch formulae have been provided for the manufacture of the product, along with an appropriate account of the manufacturing process. The manufacturing process has been validated at pilot-scale and has shown satisfactory results. A process validation study was performed for the minimum commercial batch. The applicant has committed to performing process validation for the first three consecutive full- scale commercial batches. Finished product specification The finished product specifications are satisfactory. Test methods have been described and are validated adequately. Batch data have been provided and comply with the release specification. Certificates of Analysis have been provided for any working standards used. Container Closure System The tablets are packed in white bottles made of high-density polyethylene (HDPE) with child-resistant closures (polypropylene) and tamper-evident seals. The pack size is 90 film-coated tablets. Specifications and Certificates of Analysis for all packaging materials have been provided. These are satisfactory. All primary packaging complies with EU legislation regarding contact with food. Stability Finished product stability studies have been conducted in accordance with current guidelines and in the packaging proposed for marketing. Based on the results, a shelf-life of 2 years with storage condition “Do not store above +30C” is set. This is satisfactory. Summary of Product Characteristics (SmPC), Patient Information Leaflet (PIL) and Labelling The SmPC, PIL and labelling are satisfactory from a pharmaceutical perspective. A package leaflet has been submitted to the MHRA together with results of consultations with target patient groups ("user testing"), in accordance with Article 59 of Council Directive 2001/83/EC. The results indicate that the package leaflet is well-structured and organised, easy to understand and written in a comprehensive manner. The test shows that the patients/users are able to act upon the information that the package leaflet contains. The Marketing Authorisation Holder has committed to submitting mock-ups for unmarketed pack sizes to the relevant regulatory authorities for approval before those packs are marketed commercially. Marketing Authorisation Application (MAA) Form The MAA form is satisfactory.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
9
Expert Report The quality overall summary is written by an appropriately qualified person and is a suitable summary of the pharmaceutical aspects of the dossier. Conclusion There are no objections to the approval of this product from a pharmaceutical point of view.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
10
NON-CLINICAL ASSESSMENT
The pharmacodynamic, pharmacokinetic and toxicological properties of ibuprofen and famotidine are well-known. Thus, the applicant has not provided additional studies and further studies are not required. A non-clinical overview has been provided, written by an appropriately qualified person. This is satisfactory. A suitable justification has been provided for the non-submission of an environmental risk assessment (ERA) for ibuprofen. An ERA was submitted for the active substance famotidine and was concluded at phase I. The conclusion is that the proposed therapeutic use of famotidine is unlikely to pose a risk to the environment. There are no objections to the approval of this product from a non-clinical point of view.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
11
CLINICAL ASSESSMENT Clinical Pharmacology The proposed fixed combination product (FCP) comprises an immediate-release, film-coated tablet formulation containing a 26.6 mg famotidine core, compression-coated with 800 mg ibuprofen. The applicant has submitted pharmacokinetic, efficacy and safety data, including 2 pivotal phase III studies comparing the proportion of patients developing endoscopically diagnosed gastric and duodenal ulceration after 24 weeks of the FCP, compared to 800 mg ibuprofen alone. The efficacy and safety of the ibuprofen component is supported by pharmacokinetic data claiming to demonstrate bioequivalence with an EU reference product. Pharmacokinetics (PK) The Applicant has submitted data from seven Phase 1 pharmacokinetic studies involving 157 healthy volunteers as part of the development plan for HZT-501. All studies were conducted in the United States with healthy volunteer and subjects with renal insufficiency. Bioequivalence Study HZ-CA-005 Bioavailability of prototype HZT-501 Phase 1 formulation This study used an earlier formulation of HZT-501, in a single dose, fasting, cross-over study in healthy volunteers (n=20). Bioavailability was compared to a free combination of ibuprofen 800 mg (U.S. formulation) and famotidine (Pepcid syrup 26.6 mg). The PK parameters of the HZT-501 formulation used in this are less relevant to this application. However, the Pepcid single dose PK parameters (mean + SD) are of interest, as Pepcid is not usually dosed at 26.6 mg. The Applicant has submitted new efficacy and safety data to support the famotidine component of the FCP, and is not relying on the demonstration of bioequivalence to Pepcid. Study HZ-CA-010 Ibuprofen 800 mg tablet (HZT-405) use as Phase 3 comparator In the Phase 3 clinical trials (Studies HZ-CA-301 and HZ-CA-303) and the follow-on long-term safety study (Study HZ-CA-304), HZT-405 (ibuprofen 800 mg) tablet was used as the reference treatment in order to maintain the study blind. At the request of the FDA the bioequivalence of HZT-405 and a commercially available ibuprofen 800 mg tablet US reference medicinal product (800 mg IBU Tablets) was evaluated (Study HZ-CA-010). This was a randomised, single dose, open-label, two period, cross-over, bioequivalence study in healthy volunteers under fasting conditions. All subjects completed the study. The results are shown in Table below.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
12
Relative bioavailability of 2 ibuprofen formulations in healthy subjects – HZ-CA-010
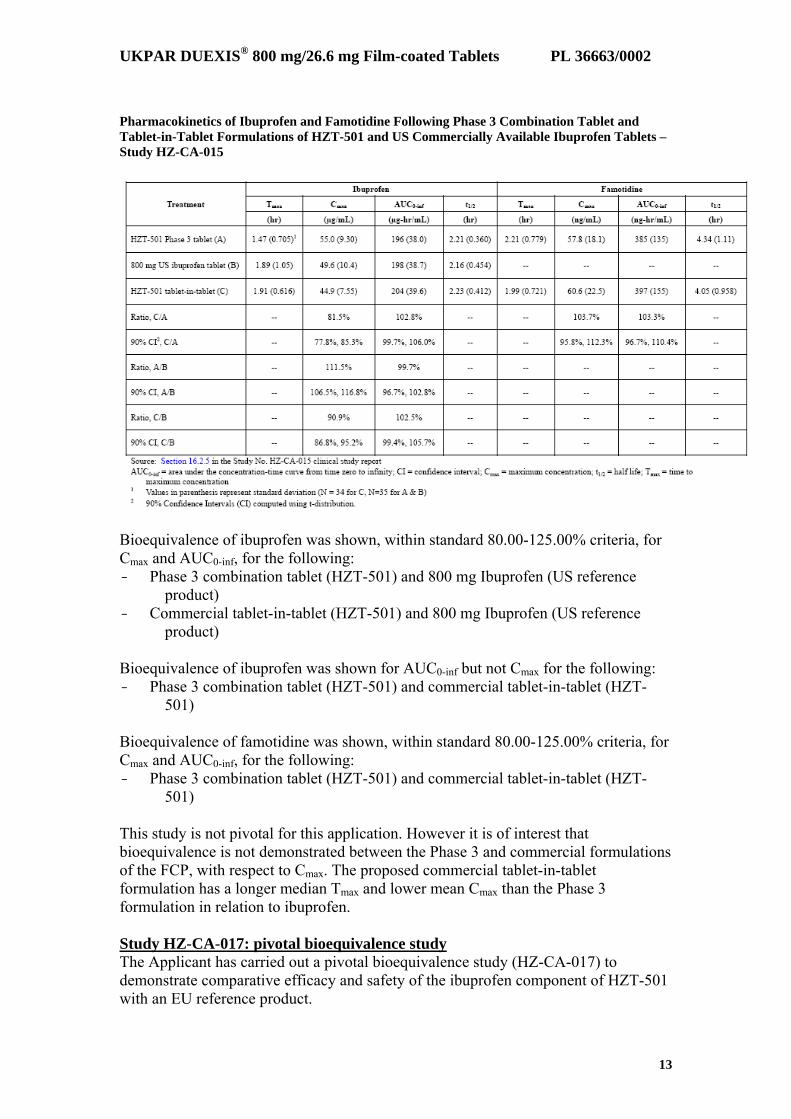
The results show that the 800 mg Ibuprofen formulation (HZT-405) used in the Phase 3 studies is bioequivalent to a commercially available 800 mg Ibuprofen from the U.S.market. The use of the ibuprofen comparator, which is bioequivalent to a U.S. marketed 800 mg ibuprofen formulation is considered acceptable. Bridging from Phase 3 FCP formulation to commercial FCP formulation A HZT-501 Phase 3 combination tablet was used in the Phase 3 studies (Studies HZ-CA-301 and HZ-CA-303), in the follow-on long-term safety study (Study HZ-CA-304), and in a Phase 1 study in subjects with renal insufficiency (Study HZ-CA-006). During the Phase 3 clinical trials, the Applicant determined that the Phase 3 combination tablet had suboptimal stability for commercial distribution. Therefore, the applicant developed a new tablet-in-tablet formulation of HZT-501 for commercialisation and conducted two Phase 1 bioequivalence studies (Study HZ-CA-015 and HZ-CA-017) to show the bioequivalence of each of these formulations (the Phase 3 combination tablet and the tablet-in-tablet commercial formulation) to each other and also to an 800 mg formulation of ibuprofen available within the US (Study HZ-CA-015) or Europe (HZ-CA-017). Study HZ-CA-015 A single-centre, randomised, open-label, three-period, six-sequence crossover bioequivalence study was conducted in 36 subjects in the fasting state. Thirty three subjects completed the study. The results are shown in the table.
UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
13
Pharmacokinetics of Ibuprofen and Famotidine Following Phase 3 Combination Tablet and Tablet-in-Tablet Formulations of HZT-501 and US Commercially Available Ibuprofen Tablets – Study HZ-CA-015
Bioequivalence of ibuprofen was shown, within standard 80.00-125.00% criteria, for Cmax and AUC0-inf, for the following: - Phase 3 combination tablet (HZT-501) and 800 mg Ibuprofen (US reference
product) - Commercial tablet-in-tablet (HZT-501) and 800 mg Ibuprofen (US reference
product) Bioequivalence of ibuprofen was shown for AUC0-inf but not Cmax for the following: - Phase 3 combination tablet (HZT-501) and commercial tablet-in-tablet (HZT-
501) Bioequivalence of famotidine was shown, within standard 80.00-125.00% criteria, for Cmax and AUC0-inf, for the following: - Phase 3 combination tablet (HZT-501) and commercial tablet-in-tablet (HZT-
501) This study is not pivotal for this application. However it is of interest that bioequivalence is not demonstrated between the Phase 3 and commercial formulations of the FCP, with respect to Cmax. The proposed commercial tablet-in-tablet formulation has a longer median Tmax and lower mean Cmax than the Phase 3 formulation in relation to ibuprofen. Study HZ-CA-017: pivotal bioequivalence study The Applicant has carried out a pivotal bioequivalence study (HZ-CA-017) to demonstrate comparative efficacy and safety of the ibuprofen component of HZT-501 with an EU reference product.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
14
This was a randomised, 3 period, 6 sequence, cross-over, single dose and open-label fasting study in healthy volunteers, to determine the bioequivalence of: - Famotidine and ibuprofen in the Phase 3 and Tablet-in-Tablet formulations of
HZT-501 - The bioequivalence of ibuprofen in the Phase 3 and Tablet-in-Tablet
formulations of HZT-501 to an EU reference ibuprofen formulation. Following a screening phase, subjects were assigned randomly in equal numbers to one of six, three-period treatment sequences. After providing informed consent, subjects received a single dose in each treatment period. Subjects fasted overnight for at least 10 hours prior to administration of each dose of study medication, and for 4 hours afterwards. Treatments were administered orally with approximately 240 mL of non-carbonated water, followed by a mouth check. Blood samples for determination of plasma ibuprofen and famotidine concentrations were collected prior to administration of each dose of study medication, and at 0.33, 0.67, 1, 1.33, 1.67, 2, 2.5, 3, 4, 6, 8, 10, 12, 15, 18, and 24 hours after administration of each dose. The washout period was at least 7 days. The study design is considered satisfactory in order to assess the bioequivalence in relation to ibuprofen of both the Phase 3 and commercial formulations of HZT-501 to an EU reference product. The famotidine component, dosed at 26.6mg three times daily, is not claimed to be bioequivalent to any EU reference product; efficacy and safety are to be established with data from 2 Phase III trials. Analytical methods The analytical method is satisfactory. Pharmacokinetic variables All subjects who completed at least 2 of the 3 treatment periods were included in the pharmacokinetic analysis. The following PK parameters of ibuprofen and famotidine were calculated: Tmax, Cmax, T1/2, AUC0-t, and AUC0-inf
. Actual time-points were used. Statistical methods After log transformation, computed pharmacokinetic parameters (Cmax, AUC0-t, and AUC0-inf) were subjected to analysis of variance (ANOVA) models appropriate for a randomised crossover design. The model included terms for sequence, subject within sequence, period, and treatment. Ninety percent (90%) confidence intervals (CIs) were computed using two one-sided tests for the difference between the treatment means for each parameter using an error term from the ANOVA. The 90% CIs of the ratios of the respective mean log-transformed values were pre-specified in the range 80.00-125.00% for ibuprofen and famotidine Cmax and AUC0-t. All randomised subjects who received study drug were analysed for safety. The PK and statistical methods are satisfactory
UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
15
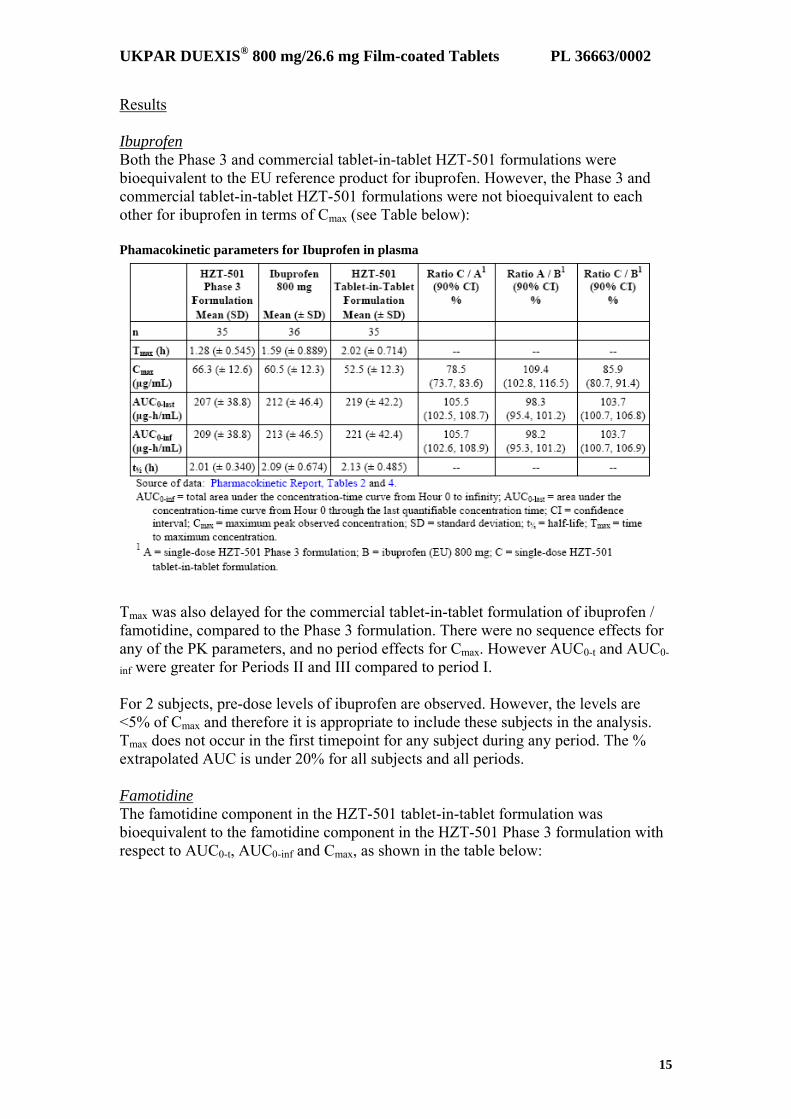
Results Ibuprofen Both the Phase 3 and commercial tablet-in-tablet HZT-501 formulations were bioequivalent to the EU reference product for ibuprofen. However, the Phase 3 and commercial tablet-in-tablet HZT-501 formulations were not bioequivalent to each other for ibuprofen in terms of Cmax (see Table below): Phamacokinetic parameters for Ibuprofen in plasma
Tmax was also delayed for the commercial tablet-in-tablet formulation of ibuprofen / famotidine, compared to the Phase 3 formulation. There were no sequence effects for any of the PK parameters, and no period effects for Cmax. However AUC0-t and AUC0-
inf were greater for Periods II and III compared to period I. For 2 subjects, pre-dose levels of ibuprofen are observed. However, the levels are <5% of Cmax and therefore it is appropriate to include these subjects in the analysis. Tmax does not occur in the first timepoint for any subject during any period. The % extrapolated AUC is under 20% for all subjects and all periods. Famotidine The famotidine component in the HZT-501 tablet-in-tablet formulation was bioequivalent to the famotidine component in the HZT-501 Phase 3 formulation with respect to AUC0-t, AUC0-inf and Cmax, as shown in the table below:

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
16
Phamacokinetic parameters for Famotidine in plasma
There were no period or sequence effects between the two HZT-501 formulations, for AUC0-t, AUC0-inf or Cmax. Safety results Two subjects had adverse events: one subject following ibuprofen 800 mg dosing (influenza), and one subject following HZT-501 tablet-in-tablet formulation dosing (abdominal pain). Both adverse events were considered mild. The abdominal pain was considered possibly related to the study medication. The influenza was considered to be probably not related to the study medication; the subject withdrew prematurely from the study due to the adverse event. No other subjects withdrew due to adverse events, and there were no deaths or SAEs. There were no safety concerns regarding laboratory values or vital signs. The commercial tablet-in-tablet FCP and the Phase 3 FCP are bioequivalent to the EU reference product with respect to ibuprofen. However the commercial tablet-in-tablet FCP formulation appears bioequivalent to the Phase 3 FCP formulation for famotidine but not ibuprofen. Cmax is lower and Tmax longer for the commercial formulation. However AUC0-t and AUC0-inf are within 80.00-125.00%. It is therefore possible to bridge from the commercial FCP to an EU reference product for efficacy and safety of the ibuprofen component. The lack of bioequivalence of the Phase 3 and commercial tablet-in-tablet formulations in relation to ibuprofen is not considered to affect the validity of the Phase 3 results. The objective of the Phase 3 program is to investigate the efficacy and safety of the famotidine component, which is not bioequivalent to an EU reference product. Had the commercial tablet-in-tablet formulation be used in Phase 3, it is likely that the results would have favoured HZT-501, due to a lower Cmax, which if anything may have reduced the incidence of upper gastrointestinal ulceration.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
17
Study HZ-CA-016 This was a Phase 1 food effect study with the commercial tablet-in-tablet formulation of HZT-501 in 28 subjects. The effect of a standardised high-fat breakfast on the absorption of ibuprofen and famotidine was examined in 28 healthy volunteers in a randomised 2 period cross-over study. Twenty five subjects completed the study. The results are shown in table below: Effect of Food on the Pharmacokinetics of Ibuprofen and Famotidine Following HZT-501 Tablet-in-Tablet in Healthy Subjects – Study HZ-CA-016
When taken in the fed state, both Cmax and AUC0-inf are reduced, for both ibuprofen and famotidine components. This is most marked for famotidine Cmax. Exposure to famotidine and ibuprofen, as measured by AUC0-inf, is not affected to a significant degree by food. Summaries of Product Characteristics (SmPCs) for the individual components, as licensed in the EU, indicate that they do not require administration with regard to food. In the Phase 3 studies, the FCP was self-administered without regard to meals. The proposed SmPC for DUEXIS includes a statement that the bioavailability of the ibuprofen and famotidine components is not affected by food. This is acceptable. Special populations Impaired renal function It is known that the clearance of famotidine may be prolonged in subjects with marked renal insufficiency (creatinine clearance <45 mL/min). As a result, these subjects were excluded from the Phase 3 studies. Study HZ-CA-006 was conducted to determine the relative bioavailability of ibuprofen and famotidine from orally administered HZT-501 (the HZT-501 Phase 3 combination tablet) versus concurrent oral administration of equivalent doses of commercially available ibuprofen and famotidine in renal insufficiency. Study HZ-CA-006 Five subjects were enrolled and completed the study, a randomised, 2 sequence 2 period cross-over single dose design. Two subjects had moderate renal insufficiency

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
18
(CLCr 30-60 mL/min) and 3 had severe renal insufficiency (CLCr <30 mL/min). The results are shown in the table below: Pharmacokinetics of Ibuprofen and Famotidine after HZT-501 Phase 3 Combination Tablet vs. Concurrent Administration of Commercially Available Ibuprofen and Famotidine in Renally Impaired Subjects – Study HZCA-006
When compared to the PK parameters derived from healthy volunteers in Study HZ-CA-017 for the Phase 3 combination tablet, the ibuprofen parameters are comparable. However with the regard to famotidine, the mean AUC0-inf is increased 2.7 fold and the half-life by 2.5 fold in renally-impaired patients. This is expected, as famotidine is primarily excreted in the urine. The SmPC of Pepcid (MSD), an EU marketed formulation of famotidine, advises that in moderate and severe renal impairment the dose should be reduced, to avoid excess accumulation. Dose reduction is not possible for DUEXIS. The following advice is proposed in the DUEXIS SmPC. Section 4.2 Ibuprofen and famotidine are known to be substantially excreted by the kidney therefore the risk of toxic reactions to DUEXIS may be greater in patients with impaired renal function. In patients with mild renal impairment DUEXIS should be used cautiously and renal function should be monitored closely. DUEXIS is contraindicated in patients with moderate to severe renal impairment (see section 4.3 and 4.4). Section 4.3 Severe and moderate renal impairment (creatinine clearance of less than 50 ml/minute). Section 4.4 Ibuprofen should be used with greater care in patients with impaired renal function and the monitoring of serum creatinine and/or creatinine clearance is advised in these patients. DUEXIS is not recommended for use in patients having a baseline creatinine clearance of less than 50 ml/minute (see section 4.3). Famotidine is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
19
Certain patients, specifically those whose renal blood flow is compromised, because of extracellular volume depletion, cirrhosis of the liver, sodium restriction, congestive heart failure, and pre-existing renal disease, should have renal function assessed before and during DUEXIS therapy. Because elderly patients are more likely to have decreased renal function, as well as patients using diuretics, care should be taken in dose selection and adjusting dose interval, and it may be useful to monitor renal function. The SmPC advice is considered adequate. Study HZ-CA-001 The potential for a drug-drug interaction when ibuprofen and famotidine are administered concurrently was evaluated in six healthy male subjects ranging in age from 21 to 34 years in an open-label, randomised, single-dose, two-period crossover study. Subjects were randomly assigned to two treatment sequences both involving either a single dose of ibuprofen 800 mg tablet (non-EU formulation) followed 24 hours later by famotidine 40 mg tablet (Pepcid®, non-EU formulation) or concurrent administration of the same tablet formulations of ibuprofen 800 mg and famotidine 40 mg. All subjects received study drug in the morning following an overnight fast. All subjects completed the study as planned. Pharmacokinetics parameters of ibuprofen and famotidine when administered alone or in combination are shown in Table below. Pharmacokinetics of Ibuprofen and Famotidine Alone vs. Combination in Healthy Subjects – Study HZ-CA-001
The Cmax for both ibuprofen and famotidine, when administered concurrently, are slightly greater than for each component when administered individually. AUC0-t for famotidine, when administered concurrently, is also higher compared to when administered individually, although not in the case of ibuprofen. However, it is difficult to draw conclusions from such a small study. The results from the pivotal bioequivalence study HZ-CA-017 do not indicate that the famotidine component affects the bioavailability of ibuprofen in the FCP, compared to an ibuprofen EU reference product. SmPCs of licensed ibuprofen and famotidine products do not contain any interaction warning of relevance. Exposure relevant for safety evaluation The Applicant has submitted data from the literaure, in the form of a conference poster, that compares steady state pharmacokinetics of famotidine twice a day (BID)

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
20
and three times a day (TID) dosing (Tidmarsh and Rodriguez 2009). The formulation study was Pepcid® Oral Suspension (40 mg/ 5mL). Mean steady state trough plasma concentrations of famotidine are summarised in the following table: Trough Plasma Concentration of famotidine following repeated oral administration of famotidine three times a day (26.6 mg/dose) or twice a day (40 mg/dose)
Plasma Famotidine Concentration (ng/mL) (Mean ± SD)
Day
Famotidine 40 mg BID Famotidine 26.6 mg TID Day 1 10.5 ± 2.8 9.7 ± 4.9 Day 5 15.7 ± 4.6 15.7 ± 8.9
Source: (Tidmarsh & Rodriguez, 2009) BID = twicew a day; SD = standard deviation; TID = three times a day Trough plasma famotidine concentrations on Day 5 were similar for TID and BID dosing, demonstrating that TID dosing did not lead to significant accumulation of famotidine over a 5-day dosing period. Data from the literature suggests that trough plasma famotidine concentrations on Day 5 were similar for 80 mg daily of Pepcid® Oral Suspension (40 mg/5 mL), whether dosed TID or BID, demonstrating that TID dosing did not lead to significant accumulation of famotidine over a 5-day dosing period. No steady state data for the proposed FCP has been submitted. Therefore it is not known whether steady state exposure to famotidine is equivalent to that from a 40 mg BID dose, for which there is clinical experience. Assessor’s overall conclusions on pharmacokinetics The applicant has submitted an extensive pharmacokinetic package in support of the proposed fixed combination product. The following conclusions can be drawn:
The proposed HZT-501 commercial formulation is bioequivalent to an 800 mg ibuprofen EU reference product. The Phase 3 HZT-501 formulation is also bioequivalent to the ibuprofen EU reference product.
The proposed commercial formulation has a lower ibuprofen Cmax and longer Tmax than the Phase 3 formulation; Cmax is outside the standard 80.00-125.00% criteria. However this is considered unlikely to affect the results of the Phase 3 study to investigate the efficacy and safety of the famotidine component.
The ibuprofen 800 mg formulation used as a comparator in the Phase 3 studies is bioequivalent to a U.S. marketed formulation of ibuprofen. It is not considered necessary for the comparator to be bioequivalent to an EU reference product.
Exposure to the ibuprofen and famotidine components of HZT-501 is not affected by food to a clinically significant degree.
An interaction study showed a slight increase in bioavailability when famotidine and ibuprofen were administered concurrently, compared to subsequent days. These findings were not reproduced in the pivotal bioequivalence study in relation to ibuprofen. SmPCs of EU approved famotidine and ibuprofen products do not contain interaction warnings of

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
21
relevance to the combination. A clinically significant interaction is considered unlikely.
No steady state data for the proposed FCP has been submitted. It is accepted, based on the evidence provided, that steady state famotidine exposure from DUEXIS is unlikely to be significantly different from that of a marketed formulation dosed BID, when total daily dose is the same.
Data from the literature suggests that trough plasma famotidine concentrations on Day 5 were similar for 80 mg daily of Pepcid® Oral Suspension (40 mg/5 mL), whether dosed TID or BID, demonstrating that TID dosing did not lead to significant accumulation of famotidine over a 5-day dosing period.
In renally-impaired volunteers, famotidine exposure from a single dose of HZT-501 is increased, compared to that observed in healthy volunteers. This is expected from the known pharmacokinetic profile of famotidine. DUEXIS is contra-indicated in moderate and severe renal impairment (creatinine clearance <50mL/min). The information provided in the SmPC regarding renal impairment is acceptable.
Pharmacodynamics Introduction The applicant considers that the clinical pharmacology profiles of ibuprofen and famotidine are well-established, and there is significant clinical pharmacology information available in the published literature. This is acceptable. Relationship between plasma concentration and effect The proposed posology is TID. This is in line with approved EU ibuprofen products. However, in relation to famotidine, there is no approved product with TID dosing. The applicant has not carried out any PK/PD studies with the proposed FCP to investigate the relationship between plasma concentration and effect. However, data from the literature (Tidmarsh & Rodriguez, 2009) is presented to support TID dosing of famotidine. This is presented in the form of a conference poster. The publication describes PK/PD modelling of intragastric pH following BID or TID famotidine dosing, using known pharmacokinetic parameters, as shown in figure 1 below:

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
22
The modelling suggests that TID dosing may lead to better maintenance of gastric suppression above an acidic threshold. The poster also describes the testing of this hypothesis with a randomised, open-label, two-period, crossover, 5-day study in 13 healthy subjects. The study used commercially available Pepcid® Oral Suspension (40 mg/5 ml). Gastric pH was maintained above pH 2.5 on Day 1 for all subjects with TID dosing, whereas three subjects had gastric pH below 2.5 with BID dosing. Inter-subject variability was reduced with TID dosing as measured by median 24-hour pH, and in all other pH measures, on Day 1 (Table 1). Median 24-hour pH while in an upright position was higher with TID dosing compared to BID dosing.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
23
The amount of time during which the pH was below the critical pH values of 3.5 and 4.0 is summarised in Table 2. In addition to an increased median 24-hour pH, the time in the acidic pH range was reduced with TID dosing on Day 1 (mean time with pH <3.5: 694 vs.713 minutes, and mean time with pH <4: 785 vs. 807 minutes, for TID vs. BID dosing, respectively). TID dosing reduced the total time during which pH was in the acidic range on Day 1.
The Authors state that an analysis of Day 5 data revealed that the values were not consistent with previously published results and therefore Day 5 data were deemed to be unreliable. A possible explanation may be a technical fault with the insertion and placement of the nasogastric probe. Both BID and TID dosing with total daily doses of 80 mg of famotidine for 5 days were well tolerated by all subjects. The source of the pH data for 80 mg daily of Pepcid® Oral Suspension (40 mg/ 5mL), dosed TID or BID is from a conference poster presentation. The Day 1 data suggests that pH values are consistently higher with TID, compared to BID dosing. However the Day 5 data, at steady state, would have been more informative. Assessor’s overall conclusions on pharmacodynamics The applicant has concluded, based on data from the literature, that gastric pH is better controlled on famotidine 26.6 mg TID compared to 40 mg BID. EFFICACY The applicant has submitted data from 2 pivotal Phase 3 clinical studies (HZ-CA-301 and HZ-CA-303) in support of the efficacy and safety of the famotidine component of the proposed FCP of HZT-501 (ibuprofen 800 mg / famotidine 26.6 mg, dose TID). Dose-response studies and main clinical studies The applicant has not carried out dose finding studies. The dose of the famotidine component of the FCP is justified based on data from the literature demonstrating

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
24
efficacy of famotidine 40 mg BID in the prevention of NSAID-associated gastroduodenal ulceration, although no H2RAs are licensed for this indication in Europe. In addition, famotidine 26.6 mg TID is not approved in any indications in Europe. Steady state PK data is not available for HZT-501, to allow comparison with marketed famotidine formulations. Some data from the literature has been submitted in support of TID dosing of famotidine. Study HZ-CA- 301 This was a multi-centred, randomised, double-blind, parallel group, phase 3 study of the efficacy and safety of HZT-501 compared to ibuprofen alone in subjects requiring NSAID treatment. Subjects were randomised 2:1 to HZT-501 or ibuprofen for 24 weeks or until they developed endoscopically diagnosed upper gastrointestinal ulcer. Study Participants Planned enrolment was n=600. Subjects provided informed consent. Subjects who completed the 24-week treatment period without developing an endoscopically-diagnosed upper gastrointestinal ulcer were eligible to participate in a follow-on study with HZT-501 (HZ-CA-304). Subjects who did not enter HZ-CA-304 were monitored for safety for an additional four weeks via a telephone visit. The inclusion and exclusion criteria were acceptable. The intended patient population, according to the proposed indication, is patients with a previous history, or who are at risk of developing, non-steroidal anti-inflammatory drug (NSAID) associated upper gastrointestinal ulcers. Risk-factors to develop NSAID related gastro-intestinal complications include high age, concomitant use of anticoagulants, corticosteroids, low-dose aspirin, debilitating cardiovascular disease, and a history of gastric and/or duodenal ulcers. The study population, as defined by the inclusion and exclusion criteria, would be expected to be of lower risk of NSAID-associated ulceration than the target population. However, the applicant did stratify at randomisation according to ulcerrisk. Treatments HZT-501 (ibuprofen 800 mg/famotidine 26.6 mg) or HZT-405 (ibuprofen 800 mg) was self-administered orally, on a double-blind basis, TID for up to 24 consecutive weeks. There were no restrictions on dosing with regard to food. Subjects were prohibited from taking any NSAIDs other than study drug. Subjects were prohibited from taking any drugs or interventions that neutralize gastric acid for more than three days during any two-week period and from taking any H2-receptor antagonists, proton pump inhibitors (PPIs) or misoprostol other than study drug. Subjects taking low dose aspirin and/or other anticoagulant medication could continue to use these medications, on their usual regimen, during the treatment period. The rationale for the proposed FCP is considered to be a substitution indication. It is established clinical practice to prescribe gastroprotection, such as PPIs, to patients at risk of NSAID-associated upper GI ulceration requiring long-term high dose NSAID

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
25
therapy. The aim of the Phase 3 program is to establish the efficacy and safety of the famotidine component of the FCP. Therefore a non-inferiority comparison with a licensed regimen such as ibuprofen 2400 mg daily in conjunction with a PPI would have been more informative. Objectives Primary objective To evaluate the efficacy of HZT-501 in reducing the proportion of subjects who develop at least one endoscopically-diagnosed upper gastrointestinal (i.e. gastric and/or duodenal) ulcer during the 24-week treatment period, as compared to ibuprofen, in subjects at risk for NSAID-induced ulcers. All randomised subjects who received at least one dose of study treatment and underwent a baseline and at least Week 8 endoscopic examination were included in the primary population for all primary and secondary efficacy analyses All patients who did not undergo the Week 8 endoscopic examination were excluded from the efficacy analysis. This would be a concern if it was felt these very early withdrawals could be for unreported GI related reasons. It makes sense to exclude them the primary life-table based analysis as in that analysis they would simply be censored at time 0- and therefore contribute nothing to the analysis anyway. However they should be accounted for in a sensitivity analysis to test the robustness of the results. Secondary objectives To evaluate the efficacy of HZT-501 in reducing endoscopically diagnosed gastric and duodenal ulceration, and the incidence of serious gastrointestinal (GI) complications. The safety objective of this study was to compare the adverse event (AE) incidence rates between HZT-501 and ibuprofen in subjects at risk for NSAID-induced ulcers. Outcomes/endpoints Endoscopic examinations were performed during screening (baseline) and at Weeks 8, 16 and 24. Subjects were terminated early from the study in the event they developed an endoscopically-diagnosed upper gastrointestinal ulcer of unequivocal depth and at least 3 mm in diameter. Subjects who terminated early for reasons other than development of an endoscopically-diagnosed upper gastrointestinal ulcer were to undergo an endoscopic examination as soon as possible after administration of their final dose of study drug. Primary efficacy endpoint − the proportion of subjects who developed at least one endoscopically-
diagnosed upper gastrointestinal ulcer of unequivocal depth and at least 3 mm in diameter during the 24-week treatment period (defined as up to and including week 26.7).
Secondary efficacy endpoints − the proportion of subjects who develop at least one endoscopically diagnosed
gastric ulcer during the 24-week treatment period − the proportion of subjects who develop at least one endoscopically diagnosed
duodenal ulcer during the 24-week treatment period

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
26
− the incidence rate of NSAID-associated serious gastrointestinal complications (perforation, obstruction, bleeding) during the 24 week treatment period
The primary endpoint is well-validated and considered appropriate. The secondary endpoints are adequate. Sample size A sample size of 600 subjects was selected in order to provide approximately 90% power to detect a difference of 6% versus 16% in the proportion of subjects in the two treatment arms who developed at least one upper gastrointestinal ulcer during the 24-week Treatment Period, with a two-sided α = 0.05. This sample size accounted for the 2:1 randomisation and a 15% dropout rate for reasons other than ulcer development. Subjects were randomised 2:1 to receive HZT-501 or ibuprofen. Randomisation was stratified for high risk of development of ulceration based on (1) concomitant use of low dose aspirin or other anticoagulant medication, (2) history of upper GI ulcer. Randomisation was not stratified at the site level given the large number of sites used. The HZT-501 and the ibuprofen tablets were comparable to each other with respect to size, shape, colour, and weight, to enable administration of both drugs on a double-blind basis. All subjects, Investigators, study centre personnel, sponsor personnel and CRO personnel remained blinded to all subjects’ treatment assignments until after the study database had been locked. The primary efficacy analysis was the comparison between HZT-501 and ibuprofen of the proportion of subjects who developed at least one endoscopically-diagnosed upper gastrointestinal (i.e., gastric and/or duodenal) ulcer of unequivocal depth and at least 3 mm in diameter by the completion of the 24-week treatment Period. Subjects who terminated early from the study after the Week 8 (visit window allowance ≥ 6.7 weeks) endoscopic examination without having developed an endoscopically-diagnosed upper gastrointestinal ulcer of unequivocal depth and at least 3 mm in diameter were considered for the analyses to have their time to ulcer censored at the last time of observation in estimating the Week 24 proportion. Subjects for whom data for their final on-study endoscopic examination were missing and who had not previously developed an endoscopically-diagnosed upper gastrointestinal ulcer were considered for the analyses similarly. The proportions at Week 24 and their standard errors were estimated within treatment groups using life table estimation to provide estimates in the context of incomplete data e.g. due to dropouts, loss to follow-up, end of study, etc. The following sensitivity analyses were conducted. 1) The crude rates for the ulcer endpoints were explored as sensitivity analyses for the primary population. The number of subjects who experienced an ulcer through the Week 24 estimate, up to and including Week 26.7, in each treatment arm was tested for statistical significance using a Fisher’s Exact test, a Chi-Square test adjusted for continuity, and a Cochran-Mantel-Haenszel (CMH) test.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
27
2) The crude rates for all ulcer endpoints for the primary population were explored for sensitivity to ulcer definition by examining ulcer incidence crude rates when subjects with an ulcer plus subjects who withdrew from the study early were both considered as having experienced an ulcer. The number of subjects who experienced an ulcer, plus those who terminated early through the Week 24 estimate, up to and including Week 26.7, in each treatment arm was tested for statistical significance using a Fisher’s Exact test, a Chi-Square test adjusted for continuity, and a Cochran-Mantel-Haenszel test. Life table estimation (similar to Kaplan-Meier estimation) is an appropriate choice for the primary analysis to cope with the situation where many patients will leave the study early without experiencing an event. This method uses a censoring approach to help account for the differing time on study for different patients. However the method does operate under certain assumptions – namely that leaving the study early gives no information about whether the patient would subsequently have had an event. This is possibly a questionable assumption, as it seems possible that some withdrawals could be because of undocumented GI problems. The two sensitivity analyses will help this to be assessed – the first assumes all early withdrawals without an event are successes, the second that they are all failures (perhaps the most realistic of the assumptions). If both these results are positive this will provide some confidence in the robustness of the results. We should also take account of the patients who withdraw before week 8 when looking at the results. Results Participant flow All randomised subjects received at least one dose of study medication and are therefore included in the safety population (n=627). 34.5% of those in the HZT-501 arm terminated early, 5.8% due to adverse event, 8.0% due to endoscopically-diagnosed UGI ulcer, 10.4% withdrew consent. 42.5% of those in ibuprofen arm terminated early, 7.1% due to adverse event, 16.0% due to endoscopically-diagnosed UGI ulcer and 12.3% withdrew consent. In the sub-group taking low dose aspirin or oral anticoagulant (n=90), 55.2% on ibuprofen only terminated early, compared to 32.8% on HZT-501 (p=0.0428). The difference was due mainly to increased adverse events and endoscopically diagnosed UGI ulcer. A greater proportion of patients terminated early in the Ibuprofen group, which provides some suggestion of efficacy in itself if it is felt that many of the withdrawals were either directly or indirectly because of GI problems. From the Safety Population database, comprising pivotal studies 301 and 303, the use of concurrent medications that might have an effect on the GI tract was reported as follows: Medication HZT-501 group(n=1022)
Number (%) Ibuprofen group (n=511) Number (%)
Oral bisphosphonates 48 (4.7) 27 (5.3) Corticosteroids 75 (7.3) 31 (6.1) Anticoagulants 16 (1.6) 8 (1.6)

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
28
The baseline characteristics of the primary population were similar for the two treatment groups. Although the use of concomitant medications has not been reported for the individual pivotal studies, it appears from pooled data that use is low and balanced between the two treatment groups. Outcomes and estimation Primary efficacy analysis Proportion of Subjects Who Developed at Least One Upper Gastrointestinal (i.e., Gastric and/or Duodenal) Ulcer Overall (Primary Population)
There was a statistically significant reduction in the proportion of subjects who developed at least one upper gastrointestinal ulcer, the primary study objective, in the HZT-501 group (13.8%) compared to the ibuprofen group (22.6%),p-value = 0.0304. Similar results were seen for the per protocol population, in which the proportion developing upper gastrointestinal ulceration was 13.8% in the HZT-501 group compared to 23.2% in the ibuprofen group (p= 0.0248). Sensitivity analyses Crude incidence rates Crude Incidence Rate of Subjects Who Developed at Least One Upper Gastrointestinal, Gastric, or Duodenal Ulcer (Primary Population)

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
29
The applicant has also calculated crude incidence rates for subjects who developed at least one upper GI ulcer or terminated the study early. These sensitivity analyses help assess the robustness of the assumptions made for the primary analysis. Focusing on upper gastrointestinal ulcers, the first analysis shows us that if withdrawals without an event are assumed to be successes (rather than censored) the difference between the treatment groups is increased. If early withdrawals without an event are assumed to be failures the difference decreased and statistical significance was lost, though the trend was strong and still favoured HZT-501. The sensitivity analyses create no major concerns, as all the trends are strongly favourable, but it would have been more reassuring if the analysis with withdrawal = failure had also reached statistical significance – especially as this may be the most realistic assumption. Fortunately there is a second study (303), and in that study both sensitivity analyses were positive. The positive trend here, combined with the positive result in the second study combine to give strong reassurance on the robustness of the results to the handling of early withdrawals. We should also consider here the patients who withdrew before week 8 and so were completely excluded from these analyses. There were 35 such patients randomised to HZT-501 and 22 to ibuprofen. If we repeat the first sensitivity analysis this gives 40/415 (9.6%) vs. 38/212 (17.9%), p=0.0046 (Fisher’s exact test). Repeating the second sensitivity analysis gives 150/415 (36.1%) vs. 93/212 (43.9%), p=0.0688. These findings are very similar to those seen without these patients suggestion their inclusion/exclusion is unimportant for the overall conclusion. The following table demonstrates the primary outcome measure according to randomisation strata: Proportion of Subjects Who Developed at Least One Upper Gastrointestinal Ulcer By Randomisation Strata (Primary Population)

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
30
In subjects with a positive UGI ulcer history, 23.5% of subjects in the HZT-501 developed upper gastrointestinal ulceration compared to 20.0% in the ibuprofen arm. This adverse subgroup finding was not repeated in the second pivotal trial (303), so it seems likely to be a chance result. When looking at a large number of sub-groups it is to be expected that some comparisons will favour Ibuprofen by chance. The following table demonstrates the primary outcome measure according to demography: Proportion of Subjects Who Developed at Least One Upper Gastrointestinal Ulcer by Demographic Strata (Primary Population)
In men (n=184), the proportion developing upper gastrointestinal ulceration was 18.4% in the HZT-501 group compared to 11.7% in the ibuprofen group. This adverse subgroup result was not repeated in the second pivotal trial (303), so we can be confident that it is a chance finding. Primary outcome data is presented for subgroups of relevance to the target population at risk of NSAID-associated upper GI ulceration. There are trends strongly in favour of HZT-501 for the > 65 years (n=100) and subjects using LDA and/or OAC (n=84). A reverse trend is seen for patients with an upper GI ulcer history (n=29) but this is likely to be a chance finding. Secondary efficacy analyses Gastric ulcers Proportion of Subjects Who Developed at Least One Gastric Ulcer Overall (Primary Population)

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
31
In the primary population there was a trend toward reduction in the proportion of subjects who developed at least one gastric ulcer in the HZT-501 group (13.0%) compared to the ibuprofen group (19.7%); p=0795. The study was not powered to show a statistical difference for the secondary efficacy endpoints. Duodenal ulcers Proportion of Subjects Who Developed at Least One Duodenal Ulcer Overall (Primary Population)
There was a reduction in the proportion of subjects who developed at least one duodenal ulcer in the HZT-501 group (0.9%) compared to the ibuprofen group (6.6%). Significance testing was not carried out due to hierarchical testing rules. NSAID-associated serious gastrointestinal complication There were no occurrences during this study. Ulcer incidence by time on study Ulcer Incidence by Time on Study (Primary Population)

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
32
The data presented at Weeks 8, 16 and 24 are generally consistent with the primary outcome. Conclusion The study met the pre-specified primary objective and demonstrated a statistically significant reduction in the proportion of subjects who developed at least one upper gastrointestinal (i.e., gastric and/or duodenal) ulcer in the HZT-501 group (13.8%) compared to the ibuprofen group (22.6%) at Week 24 (p-value = 0.0304). This result is supported by the crude incidence rate analysis and is supported to some extent by the analysis where early withdrawal or developing an ulcer is counted as failure, where there was a positive trend though statistical significance was just missed. Study HZ-CA-303 This was a multicentred, randomised, double-blind, parallel group, phase 3 study of the efficacy and safety of HZT-501 compared to ibuprofen alone in subjects requiring NSAID treatment. This study was very similar in design to Study 301, the main difference being that the primary endpoint was endoscopically-diagnosed gastric ulcer, not upper gastrointestinal (gastric or duodenal) ulcer. Subjects were randomised 2:1 to HZT-501 or ibuprofen for 24 weeks or until they developed endoscopically diagnosed upper gastrointestinal ulcer. Planned enrolment was n=875. Subjects provided informed consent. Subjects who completed the 24-week treatment period without developing an endoscopically-diagnosed upper gastrointestinal ulcer were eligible to participate in a follow-on study with HZT-501 (HZ-CA-304). Subjects who did not enter HZ-CA-304 were monitored for safety for an additional four weeks via a telephone visit. Inclusion and exclusion criteria were acceptable. Treatments HZT-501 (ibuprofen 800 mg/famotidine 26.6 mg) or HZT-405 (ibuprofen 800 mg) was self-administered orally, on a double-blind basis, TID for up to 24 consecutive weeks. There were no restrictions on dosing with regard to food. Subjects were prohibited from taking any NSAIDs other than study drug. Subjects were prohibited from taking any drugs or interventions that neutralize gastric acid for more than three days during any two-week period and from taking any H2-receptor antagonists, PPIs or misoprostol other than study drug. Subjects taking low dose aspirin and/or other anticoagulant medication could continue to use these medications, on their usual regimen, during the treatment period. Objectives Primary objective To evaluate the efficacy of HZT-501 in reducing the proportion of subjects who develop at least one endoscopically-diagnosed gastric ulcer (of unequivocal depth and at least 3 mm in diameter) during the 24-week treatment period, as compared to ibuprofen, in subjects at risk for NSAID-induced ulcers.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
33
All randomised subjects who received at least one dose of study treatment and underwent a baseline and at least Week 8 endoscopic examination were included in the primary population for all primary and secondary efficacy analyses The population definitions were the same as for HZ-CA-301. Please see comment in that study on exclusion of patients withdrawing before week 8. Secondary objectives To evaluate the efficacy of HZT-501 in reducing endoscopically diagnosed upper gastrointestinal (i.e. gastric and/or duodenal) ulcer, duodenal ulceration and the incidence of serious GI complications. The safety objective of this study was to compare the adverse event (AE) incidence rates between HZT-501 and ibuprofen in subjects at risk for NSAID-induced ulcers. Outcomes/endpoints Endoscopic examinations were performed during screening (baseline) and at Weeks 8, 16, and 24. Subjects were terminated early from the study in the event they developed an endoscopically-diagnosed upper gastrointestinal ulcer of unequivocal depth and at least 3 mm in diameter. Subjects who terminated early for reasons other than development of an endoscopically-diagnosed upper gastrointestinal ulcer were to undergo an endoscopic examination as soon as possible after administration of their final dose of study drug. Primary efficacy endpoint − the proportion of subjects who developed at least one endoscopically-
diagnosed gastric ulcer of unequivocal depth and at least 3 mm in diameter during the 24-week treatment period (defined as up to and including week 26.7).
Secondary efficacy endpoints
the proportion of subjects who develop at least one endoscopically diagnosed upper gastrointestinal ulcer during the 24-week treatment period
the proportion of subjects who develop at least one endoscopically diagnosed duodenal ulcer during the 24-week treatment period
the incidence rate of NSAID-associated serious gastrointestinal complications (perforation, obstruction, bleeding) during the 24 week treatment period
The primary endpoint is well-validated and considered appropriate. The secondary endpoints are adequate. A sample size of 875 subjects was selected in order to provide approximately 90% power to detect a difference of 6% versus 14% in the proportion of subjects in the two treatment arms who developed at least one gastric ulcer during the 24-week Treatment Period, This sample size accounted for the 2:1 randomisation, a 15% dropout rate for reasons other than ulcer development, and the estimated 5% occurrence of non-gastric (i.e., duodenal) ulcers that were not included in the primary efficacy endpoint definition.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
34
The sample size was changed from 600 to 875 subjects. The expected proportion for ulcer incidence in the ibuprofen treatment group was corrected from 16% to 14% based on a re-review of the data from the pilot study in the published literature with regard to the anticipated rate of occurrence of gastric (i.e., non-duodenal) ulcers. The estimated power remained at 90% as originally planned. All study data remained fully blinded at the time this change was made. Subjects were randomised 2:1 to receive HZT-501 or ibuprofen. Randomisation was stratified for high risk of development of ulceration based on (1) concomitant use of low dose aspirin or other anticoagulant medication, (2) history of upper GI ulcer. Randomisation was not stratified at the site level given the large number of sites used. The HZT-501 and the ibuprofen tablets were comparable to each other with respect to size, shape, colour, and weight, to enable administration of both drugs on a double-blind basis. All subjects, Investigators, study centre personnel, Sponsor personnel and CRO personnel remained blinded to all subjects’ treatment assignments until after the study database had been locked. The statistical methods were the same as for trial HZ-CA-301. Results All randomised subjects received at least one dose of study medication and are therefore included in the safety population (n=906). 28.7% of those in the HZT-501 arm terminated early, 6.6% due to adverse event, 8.4% due to endoscopically-diagnosed UGI ulcer, 7.9% withdrew consent. 43.1% of those in the ibuprofen arm terminated early, 8.0% due to adverse event, 17.1% due to endoscopically-diagnosed UGI ulcer, 8.7% withdrew consent. In the sub-group taking low dose aspirin or oral anticoagulant (n=140), 55.0% on ibuprofen only terminated early, compared to 28.0% on HZT-501 (p=0.0026). The difference was due mainly to endoscopically diagnosed UGI ulcer. A greater proportion of patients terminated early in the Ibuprofen group, which provides some suggestion of efficacy in itself if it is felt that many of the withdrawals were either directly or indirectly because of GI problems. From the Safety Population database, comprising pivotal studies 301 and 303, the use of concurrent medications that might have an effect on the GI tract was reported as follows: Medication HZT-501 group (n=1022)
Number (%) Ibuprofen group (n=511) Number (%)
Oral bisphosphonates 48 (4.7) 27 (5.3) Corticosteroids 75 (7.3) 31 (6.1) Anticoagulants 16 (1.6) 8 (1.6)
The baseline characteristics of the primary population were similar for the two treatment groups. Although the use of concomitant medications has not been reported

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
35
for the individual pivotal studies, it appears from pooled data that use is low and balanced between the two treatment groups. Outcomes and estimation Primary efficacy analysis Proportion of Subjects Who Developed at Least One Gastric Ulcer Overall (Primary Population)
There was a statistically significant reduction in the proportion of subjects who developed at least one gastric ulcer, the primary study objective, in the HZT-501 group (12.9%) compared to the ibuprofen group (25.3%). Similar results were seen for the per protocol population, in which the proportion developing gastric ulceration was 12.5% in the HZT-501 group compared to 24.8% in the ibuprofen group (p= 0.0014). Sensitivity analyses Crude incidence rates Crude Incidence Rate of Subjects Who Developed at Least One Gastric, Upper Gastrointestinal or Duodenal Ulcer (Primary Population)

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
36
The applicant has also calculated crude incidence rates for subjects who developed at least one upper GI ulcer or terminated the study early. These sensitivity analyses help assess the robustness of the assumptions made in for the primary analysis. The clear conclusion here is that regardless of whether the early withdrawals are considered as successes or failures, the difference between the treatment groups is still clear. We should also consider here the patients who withdrew before week 8 and so were completely excluded from these analyses. There were 57 such patients randomised to HZT-501 and 37 to ibuprofen. Focussing on gastric ulcers if we repeat the first sensitivity analysis this gives 55/607 (9.1%) vs. 52/299 (17.4%), p<0.0001 (Fisher’s exact test). Repeating the second sensitivity analysis gives 186/607 (30.6%) vs. 136/299 (45.5%), p<0.0001. These findings are just as extreme as those seen without these patients suggesting their inclusion/exclusion is unimportant for the overall conclusions. The following table demonstrates the primary outcome measure according to randomisation strata: Proportion of Subjects Who Developed at Least One Gastric Ulcer By Randomisation Strata (Primary Population)
In the sub-groups likely to be at higher risk of gastric ulceration, such as those using LDA and or OAC (n=129), and those with a positive UGI ulcer history (n=57), the proportion of subjects developing gastric ulcers was lower in the HZT-501 arm compared to the Ibuprofen arm. The potentially concerning finding in subjects with a positive upper GI ulcer history from study 301 was not repeated here, so we can confidently dismiss that as a chance finding.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
37
The following table demonstrates the primary outcome measure according to demography: Proportion of Subjects Who Developed at Least One Gastric Ulcer by Demographic Strata (Primary Population)
In patients aged > 65 years (n=149), 21.7% developed gastric ulceration on HZT-501, compared to 20.3% on Ibuprofen. This subgroup is considered at higher risk of NSAID-associated upper GI ulceration, the target population for the proposed indication. A similar trend was not seen in study 301 so this is likely to be a chance result. Similarly the potentially concerning finding in men that was seen in study 301 was not repeated here. The study was not powered to show a statistical difference for sub-group analyses. Secondary efficacy analyses Upper gastrointestinal ulcers Proportion of Subjects Who Developed at Least One Upper Gastrointestinal (i.e. Gastric and/or Duodenal) Ulcer Overall (Primary Population)

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
38
There was a statistically significant reduction in the proportion of subjects who developed at least one upper gastrointestinal ulcer in the HZT-501 group (14.7%) compared to the ibuprofen group (29.1%) with a p-value = 0.0002 in the primary population. Similar results were seen for the per protocol population. Duodenal ulcers Proportion of Subjects Who Developed at Least One Duodenal Ulcer Overall (Primary Population)
There was a reduction in the proportion of subjects who developed at least one duodenal ulcer in the HZT-501 group (2.1%) compared to the ibuprofen group (7.1%), p=0.0226. Similar results were seen for the per protocol population. NSAID-associated serious gastrointestinal complications There were 3 reports of NSAID-associated serious gastrointestinal complications (0.6%), all gastrointestinal bleeding in the HZT-501 arm, compared to none in the Ibuprofen arm. All 3 cases were diagnosed following endoscopy, one at Week 8, two at Week 16.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
39
Ulcer incidence by time on study Ulcer Incidence by Time on Study (Primary Population)
The data presented at Weeks 8, 16 and 24 is consistent with the primary outcome. There no clear time to event pattern. Conclusion The study met the pre-specified primary objective and demonstrated a statistically significant reduction in the proportion of subjects who developed at least one gastric ulcer in the HZT-501 group (12.9%) compared to the ibuprofen group at Week 24 (25.3%; p-value = 0.0009). This result is supported by the crude incidence rate analysis and the analysis where both early withdrawal and developing an ulcer is considered a failure. Clinical studies in special populations No clinical studies have been carried out in special populations. Analysis performed across trials (pooled analyses AND meta-analysis) The applicant has pooled the primary and secondary efficacy outcomes across studies 301 and 303, as summarised in the Table below:

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
40
Proportion of Subjects Who Developed at Least One Upper Gastrointestinal, Gastric, or Duodenal Ulcer Overall (Primary Population) - Pooled Data from Studies HZ-CA-301 and HZ-CA-303
The results of the pooled analysis are in line with the outcomes of the pivotal studies 301 and 303. The applicant has also carried out a multivariate analysis of risk factors for development of upper GI ulcers. For the risk factors Age > 65 years, positive ulcer history and use of low dose aspirin, there are trends in favour of HZT-501. The benefit in LDA users reaches statistical significance. Supportive efficacy data from the literature The applicant claims that the ability of H2RAs to decrease the risk for NSAID-associated ulcers has been demonstrated in multiple clinical trials, with reference to the literature: Hudson et al 1997 Patients with osteoarthritis or rheumatoid arthritis who used NSAIDs were endoscoped. Those found to have ulceration (excavated mucosal break >3mm diameter) were treated with famotidine 40 mg bid. 78 patients with healing by 12 weeks, who wished to continue NSAID were randomised to famotidine 40 mg bid or placebo (double-blind). The risk of gastric ulceration after a further 24 weeks was 19.1% (95% CI 6.3% to 31.9%) for famotidine compared to 41.4% (95% CI 24.0% to 58.7%) for placebo; p=0.026. This study of secondary prevention was in a population who had already responded to famotidine. The results are considered less relevant for the proposed target population of DUEXIS, but do provide some evidence of the efficacy of famotidine dose at 40 mg twice daily.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
41
Kobata et al 2009 This paper reports a beneficial effect on Lanza scores after 4 weeks of treatment with famotidine, compared to rebamipide. The results are of less relevance because of the lack of validated endpoint and short duration of the study. Laine et al 2009[b] This is a published report of the applicant’s own clinical data. Leandro et al 2001 This is a meta-analysis to investigate the prevention of acute NSAID-related gastroduodenal damage (< 30 days of NSAID use). The authors conclude that misoprostol and PPIs are more effective than H2RAs. Taha et al 2006 In this double-blind, parallel group randomised controlled trial, patients with osteoarthritis or rheumatoid arthritis using NSAIDs for more than 1 month, with normal baseline endoscopy, were eligible. 285 patients were randomised to famotidine 40 mg bid, famotidine 20 mg bid or placebo. The primary endpoint was cumulative incidence of gastric or duodenal ulceration at 24 weeks (excavated mucosal break > 3mm diameter). The mean age in the famotidine 40 mg bid arm was 55 years. 13% had a previous ulcer history. The outcomes are summarised in the following table:
The study population included 50% H. pylori positive patients, compared to none for the pivotal DUEXIS studies (HZ-CA-301/303). Twice as many had a positive ulcer history. Mean age was similar.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
42
When comparing the results of Taha et al 1996 and the applicant’s pooled pivotal studies, it is more relevant to consider gastric ulcer prevention, since duodenal ulcers are generally considered easier to prevent (in fact standard dose ranitidine is licensed in the UK for the prevention of NSAID-associated duodenal ulceration). The relative risk reduction is 44% for the pivotal studies, compared to 56% for Taha et al. It should be noted that the placebo gastric ulcer rates are higher in the pivotal studies, despite a lower risk population. This may be due to the administration of high dose ibuprofen. However, the data from Taha et al (1996) provides evidence of the efficacy of famotidine dosed at 40 mg bid for the prevention of NSAID associated gastric and duodenal ulceration. Rostom et al 2009 This Cochrane Collaboration meta-analysis concludes that double dose H2RAs are effective in reducing the risk of NSAID-associated gastric and duodenal ulceration, with an expected relative risk reduction of 56%. The meta-analysis considers data from Taha et al 1996, Hudson et al 1997, (both described above) and Ten Wolde 1996, a small study (n=30) of double dose ranitidine in which an advantage over placebo was not demonstrated. The results of this meta-analysis appear to be driven largely by the data from Taha et al 1996. The meta-analysis is the reference for the recommendation in some guidelines, e.g. EULAR, that double dose H2RAs should be considered for the prevention of NSAID-associated upper gastrointestinal ulceration. The applicant has not submitted steady state pharmacokinetic data for the famotidine component of DUEXIS, dosed at 26.6 mg TID, to allow a comparison with that expected from marketed formulations of famotidine dosed at 40 mg bid. It is accepted, based on the evidence provided, that steady state famotidine exposure from DUEXIS is unlikely to be significantly different from that of a marketed formulation dosed BID, when total daily dose is the same. Overall conclusions on clinical efficacy The efficacy of the ibuprofen component of DUEXIS is accepted, based on the demonstration of bioequivalence with an EU marketed formulation of ibuprofen. The efficacy of the famotidine component of DUEXIS is supported by 2 pivotal clinical efficacy and safety studies. DUEXIS is associated with less NSAID-associated upper gastrointestinal, gastric and duodenal ulceration, measured by endoscopy at 24 weeks, compared to ibuprofen 800mg TID. The risk reductions are both statistically and clinically significant. Subgroup analyses provide supportive evidence of efficacy in higher risk patients. When considering whether the demonstrated risk reduction for the famotidine component of DUEXIS is adequate, it is relevant to compare with that expected for PPIs. However, the applicant has not compared DUEXIS directly with a combination of ibuprofen and PPI. Based on pooled data from the pivotal studies, a risk reduction of 44% for endoscopic gastric ulceration at 24 weeks is expected. This is less than an estimate of 60% for proton pump inhibitors, based on the literature. The CHM have proposed an amendment to the indication to restrict use to patients for whom treatment with a PPI is not considered appropriate:

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
43
DUEXIS is indicated for the symptomatic treatment of osteoarthritis, rheumatoid arthritis and ankylosing spondylitis in patients who require regular treatment with high dose ibuprofen administered three times a day and who are at risk of developing non-steroidal anti-inflammatory drug (NSAID) associated gastric and/or duodenal ulcers. DUEXIS should only be used where treatment with lower doses of ibuprofen or of other NSAIDs is not considered sufficient and where treatment with a proton pump inhibitor (PPI) is not considered appropriate.
CLINICAL SAFETY The applicant has submitted Study HZ-CA-304 and Study HZ-CA-401 as supportive safety studies. Study HZ-CA-304 This Phase 3, multicenter, double-blind, parallel group, follow-on study was designed to evaluate the safety of long-term treatment with HZT-501. Patients completing Studies 301 and 303 with no endoscopic evidence of upper gastrointestinal ulceration by 24 weeks were enrolled. The study duration was 24 weeks and treatment allocation remained blinded. Crossover between the two study treatments was not allowed. A total of 179 subjects were enrolled into the study. 132 subjects received HZT-501 (84.8% of these subjects completed the study) and 47 subjects received ibuprofen (80.9% of these subjects completed the study). Efficacy was not assessed. Endoscopy was not performed routinely, but only if clinically indicated. Study HZ-CA-401 Study 401 is an open-label 54 week safety study of the proposed commercial tablet-in-tablet formulation of HZT-501 in patients aged 40-80 years requiring long-term NSAIDs. The study completed in July 2011. 86 subjects were enrolled, of which 56 (65.1%) completed one year. This is termed the ‘Open Label Safety Population’. The safety database comprises 1,022 subjects treated for 6 months and 163 treated for 1 year. The applicant claims that the addition of famotidine improves the safety profile of high-dose ibuprofen by reducing upper GI ulcers, improving the GI adverse effect profile and increasing tolerability. Safety profiles of the individual components are well known in the EU. According to the SmPC of Pepcid, the following adverse effects are common or uncommon, and therefore may be reported more frequently in patients receiving HZT-501, compared to ibuprofen 800 mg alone: Common: headache, dizziness, constipation, diarrhoea Uncommon: taste disorder, dry mouth, nausea and/or vomiting, abdominal discomfort/distention, flatulence, anorexia, rash, pruritis, urticaria, fatigue

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
44
Patient exposure As well as the patients treated in the pivotal studies, 128 healthy volunteers, and 5 volunteers with renal impairment were also exposed to HZT-501. These data have not been pooled and are presented individually. The primary safety dataset presented, termed the ‘Safety Population’ are pooled data from the 2 pivotal studies 301 and 303. 1022 subjects received at least one dose of HZT-501 and 511 subjects received at least one dose of 800 mg ibuprofen. The mean (±SD) exposure to HZT-501 was 139.1 (± 51.6) days. The ‘Safety Follow-on Population’ comprises the 179 subjects recruited for Study 304, the 24 week extension to Studies 301 and 303. 132 subjects received at least one dose of HZT-501 and 47 subjects received at least one dose 800 mg ibuprofen. Of these, 107 in the HZT-501 arm and 36 subjects in the ibuprofen arm completed Study 304, and therefore 1 year of dosing. The mean (±SD) exposure to HZT-501 was 348.5 (± 48.0) days. Study 401 is termed the ‘Open Label Safety Population’. The following table shows the cumulative drug exposure for the Phase 3 program: Cumulative drug exposure in the Safety Population, Safety Follow-on Population, and Open Label Safety Population
Exposure is in line with the minimum requirements of ICH E1 and is considered adequate. The Safety Population and Safety Follow-in Populations were exposed to the Phase 3 formulation of HZT-501. This is not bioequivalent to the proposed commercial tablet-in-tablet formulation as Cmax is lower, and Tmax is longer with regard to ibuprofen in the commercial formulation. It is not expected that this formulation would result in

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
45
more ibuprofen-related adverse events, therefore the safety results can be extrapolated to the proposed commercial tablet-in-tablet formulation. Study 401 (open label safety population) was conducted using the commercial formulation. Adverse events Safety Population At least one adverse event (AE) was reported by 55.0% and 58.7% of subjects in the HZT-501 and Ibuprofen groups respectively. The following table summarises AEs occurring in 1% or more by SOC and preferred term. Treatment Emergent Adverse events in 1% or more of the Safety Population by Preferred Term

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
46

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
47
Gastrointestinal disorders were the commonest system organ class (SOC) reported. Dyspepsia, gastro-oesophageal reflux disease, gastritis, abdominal distension and abdominal tenderness were more common in the Ibuprofen group. Nausea, diarrhoea, abdominal pain upper, vomiting, stomach discomfort and flatulence were more common in the HZT-501 group. However the only notable difference was for dyspepsia. Infections and infestations also occurred commonly, as expected for a 24 week study. Renal and urinary disorders occurred more commonly in the HZT-501 group. Acute renal failure was reported in 5 subjects in the safety population: 4 (0.4%) in the HZT-501 group and 1 (0.2%) in the ibuprofen group. Other SOCs were reported with relatively low frequencies in both treatment groups. However, there were 4 cases of hepatobiliary disorder (0.4%) in the HZT-501 group, compared to 0.0% in the Ibuprofen group. These were: cholecystitis, cholelisthiasis, biliary dilatation and hepatic stenosis. Only 6.2% and 0.3% of subjects reported AEs that were severe or life-threatening, respectively. The severities of AE were balanced across the treatment groups.
Safety Follow-on Population At least one AE was reported by 68.2% and 68.1% of the HZT-501 and Ibuprofen groups respectively. The commonest reported SOC was Infections and infestations, as expected in a population exposed for up to 1 year. Gastrointestinal disorders occurred slightly more frequently in the HZT-501 group compared to the Ibuprofen group (26.5% vs 23.4%). Hypertension occurred in 8.3% of the HZT-501 population compared to 2.1% of the Ibuprofen population. There was one case of pneumonia (0.8%) and no lower respiratory tract infections reported in the HZT-501 group (compared to 2.1% and 4.3% respectively in the Ibuprofen group). Only 7.8% of subjects reported AEs that were severe. The moderate AEs were balanced across the treatment groups, but the proportion reporting mild and severe AEs was higher in the Ibuprofen group. Open-label Safety Population Overall, treatment emergent AEs were reported by 88.4% of subjects. The commonest SOC was Infections and infestations (50%). Bronchitis occurred in 1.2% (1). No cases of pneumonia were reported. 48.8% (42) reported gastrointestinal disorders, of which 2.3% (2) developed gastric ulcer, 1.2% (1) erosive oesophagitis, 1.2% (1) oesophageal stenosis and 1.2% (1) small intestinal obstruction. Under Metabolism and nutrition disorders, there was one case of hypomagnesaemia (1.2%) and one case of vitamin B12 deficiency (1.2%). There were 5 cases of renal disorder (5.8%), of which 2 (2.3%) were acute renal failure. There were 8 cases (9.8%) of treatment emergent hypertension.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
48
Safety experience in volunteers There were no deaths or SAEs reported. The AEs reported were mild and consistent with the reported AE profiles for ibuprofen or famotidine. Relationship to study drug In the Safety Population, 20.6% of subjects reported AEs considered possibly related to study drug in the HZT-501 group compared to 25.0% in the Ibuprofen group. Gastrointestinal disorders were judged possibly related for 14.8% and 20.0% of HZT-501 and Ibuprofen groups respectively. Dyspepsia was more common in the Ibuprofen group (6.5% vs 2.8%). Regarding the Investigations SOC, 2.7% were judged possibly related in the HZT-501 group compared to 1.4% in the Ibuprofen group. In the Safety Follow-on Population, 19.7% of those in HZT-501 group reported AEs judged possibly related to study treatment, compared to 23.4% in the Ibuprofen group. Gastrointestinal disorders were judged possibly related for 12.1% and 17.0% of HZT-501 and Ibuprofen groups respectively. In the Open Label Safety Population, 36.0% (31 of 86) of subjects reported a TEAE considered possibly related to HZT-501. In the GI Disorders SOC, 48.8% (42 of 86) of subjects reported an event, and 30.2% (26 of 86) of subjects reporting an event considered by the investigator to be related to study drug. Adverse events were well-balanced between the groups. Gastrointestinal disorders were commonly reported in both treatment groups, as expected from the known adverse effect profiles of ibuprofen and famotidine. Dyspepsia was more common in the Ibuprofen group, likely due to a beneficial effect of famotidine. The increase in hepatobiliary effects, largely cholestatic, is most likely a chance finding related to the demographics of the study population. An increase in renal and hypertension-related AEs was observed in the HZT-501 group, compared to the Ibuprofen group. This is most likely due to the ibuprofen component of the FCP as renal and hypertensive adverse events are a well-recognised effect of ibuprofen, particularly at higher doses. In contrast, in the SmPC of Pepcid (famotidine), there are no adverse effects reported under Renal and urinary disorders, and no reports of hypertension. It is unlikely that the combination of ibuprofen and famotidine would increase the risk of renal failure and hypertension compared to ibuprofen alone. The applicant has proposed a contraindication for patients with moderate and severe renal impairment, in whom famotidine is known to accumulate. There are also adequate warnings in Section 4.4 regarding the monitoring of renal function. Serious adverse events and deaths There was a single death in the Safety Population. A subject randomised to Ibuprofen only in Study 303 died as a result of cardio-respiratory arrest and multi-organ failure attributed to paracetamol toxicity. This event was considered by the Investigator as probably not related to study drug.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
49
Serious adverse events (SAE) Safety Population SAEs reported for the Safety Population System organ class Preferred Term
HZT-501 (N=1022) % (n)
Ibuprofen (N=511) % (n)
Total number of subjects with at least one SAE
3.2 (33) 3.3 (17)
Eosinophilia 0 0.3 (1) Coronary artery disease 0 0.4 (2) Cardio-respiratory arrest 0 0.3 (1) Oesophageal ulcer 0.1 (1) 0 Haemorrhoidal haemorrhage
0.1 (1) 0
Haemorrhoids 0.1 (1) 0 Abdominal hernia 0.1 (1) 0 Inguinal hernia 0.1 (1) 0 Chest pain 0.3 (3) 0.2 (1) Oedema peripheral 0 0.2 (1) Non-cardiac chest pain 0.1 (1) 0.2 (1) Multi-organ failure 0 0.2 (1) Abscess 0.1 (1) 0 Diverticulitis 0.1 (1) 0 Infection 0.1 (1) 0 Staphylococcal abscess 0.1 (1) 0 Pneumonia 0.1 (1) 0.4 (2) Upper respiratory tract infection
0 0.2 (1)
Cellulitis 0.2 (2) 0 Bronchitis 0 0.2 (1) Traumatic brain injury 0.2 (2) 0.2 (1) Alcohol poisoning 0.1 (1) 0 Drug toxicity 0 0.2 (1) Road traffic accident 0 0.2 (1) Wrist fracture 0.1 (1) 0 Blood creatinine increased 0.1 (1) 0 Dehydration 0.2 (2) 0 Hypoglycaemia 0 0.2 (1) Osteoarthritis 0.1 (1) 0.2 (1) Myositis 0 0.2 (1) Osteonecrosis 0 0.2 (1) Rotator cuff syndrome 0.1 (1) 0 Benign neoplasm 0 0.2 (1) Migraine 0.1 (1) 0 Syncope 0 0.2 (1) Transient ischaemic attack 0.1 (1) 0

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
50
Schizoaffective disorder 0.2 (2) 0 Bipolar disorder 0 0.2 (1) Major depression 0.1 (1) Suicide attempt 0.1 (1) 0 Renal failure acute 0.3 (3) 0 Calculus ureteric 0.1 (1) 0 Menorrhagia 0.1 (1) 0 Asthma 0.1 (1) 0 Bronchospasm 0.1 (1) 0 Chronic obstructive pulmonary disease
0.1 (1) 0
Dyspnoea 0.1 (1) 0 Dyspnoea exertional 0 0.2 (1) Hypertension 0.1 (1) 0 SAEs reported for the Safety Follow-on Population System organ class Preferred Term
HZT-501 (N=132) % (n)
Ibuprofen (N=47) % (n)
Total number of subjects with at least one SAE
6.1 (8) 6.4 (3)
Abdominal hernia 0.8 (1) 0 Diabetic gastroparesis 0.8 (1) 0 Oesophageal stenosis 0 2.1 (1) Non-cardiac chest pain 1.5 (2) 0 Cholecystitis 0.8 (1) 0 Celluitis 0.8 (1) 0 Gastroenteritis viral 0 2.1 (1) Traumatic brain injury 0.8 (1) 2.1 (1) Road traffic accident 0 2.1 (1) Diabetes 0.8 (1) 0 Hyperglycaemia 0 2.1 (1) Hyperosmolar state 0 2.1 (1) Migraine 0.8 (1) 0 Dyspnoea 0.8 (1) 0 Hypertension 0.8 (1) 0 In the Safety Population, 4 SAEs were reported in the Renal and Urinary Disorders SOC, acute renal failure (3 reports) and calculus ureteric (1 report) all in the HZT-501 group. There were no SAEs in this SOC in the Safety Follow-on Population. All recovered upon discontinuation of the study drug. Open label safety population Of the 76 treatment emergent AEs reported, 23 were considered mild, 46 moderate, 6 severe and 1 life-threatening (angina, unstable: possibly related to HZT-501). SAEs are shown in the following table:

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
51
SAEs reported for the Open-label Safety Population
Under Gastrointestinal disorders, there were 2 SAEs, one gastric ulcer (possibly related) and one small intestinal obstruction (not related). The proportion of subjects in the Safety Population and the Safety Follow-on Population reporting at least one SAE was low and balanced between the treatment arms. The proportion was higher in the Follow-on Population due to the longer exposure time. Laboratory findings In the Safety Population, blood for haematology and clinical chemistry assessment was collected at Baseline and at Weeks 8, 16 and 24. In the Safety Follow-on Population, blood was collected at 38 and 52 weeks. In the Open Label Safety Population, blood was collected at baseline, and weeks 6, 18, 30, 42 and 54. Haematology Overall mean changes from baseline were low and similar across treatment groups.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
52
Shifts from normal to low haemoglobin were observed with a slightly higher incidence in the HZT-501 group at 16 and 24 weeks. This trend was also observed in the Safety Follow-on Population. Shifts in white cell counts and lymphocytes were observed at low rates and were balanced across treatment groups. Anaemia was reported as an AE in 1.6% of the HZT-501 group and 1.4% of the Ibuprofen group. There was a single AE of neutropenia in the HZT-501 group. A decreased haemtocrit and red cell count caused the discontinuation of one subject each in the HZT-501 group. In the safety Follow-on Population, shifts from normal to low white cell counts were observed in the HZT-501 group at Weeks 8, 24 and 52, at a maximum incidence of 2.8%. This compares to a single shift to a higher white cell count in the Ibuprofen arm. Shifts from normal to low haemoglobin (and heamatocrit) levels were observed slightly more frequently in the HZT-501 group, compared to the Ibuprofen group. In the Open Label Safety Population, anaemia was reported by 2.3% (2 of 86) subjects. The most commonly observed shifts from normal at baseline to abnormal post-baseline were shifts from normal to high at the last post-baseline visit for hematocrit (6/86; 7.0%), monocytes/leukocytes (4/86, 4.7%), and eosinophils/leukocytes (3/86, 3.5%). The SmPC of Pepcid reports as very rare the adverse effects of pancytopenia, leucopenia, thrombocytopenia, agranulocytosis and neutropenia. There was an observed trend of a shift towards lower white cell counts in the HZT-501 group. However, haematological AEs were not observed to a greater degree in the HZT-501 group. It is likely that the haemoglobin changes and anaemia AEs are due to ibuprofen, with a slightly greater incidence occurring in the HZT-501 group by chance. Clinical chemistry Overall the mean changes from baseline were low and similar across treatment groups. In the Safety Follow-on Population, increased blood creatinine was reported as an AE in 2 subjects, both from the HZT-501 group. In the Open Label Safety Population blood creatinine increased, blood urea increased, glycosylated haemoglobin increased, urine uric acid increased, hypercholesterolemia, hyperkalaemia, hypoglycaemia, hypokalemia, and hypomagnesaemia were reported for 1/86 (1.2% each) subjects. Shifts in clinical chemistry parameters were unremarkable. Elevations in tranaminases of >3x upper limit of normal (ULN) are expected in clinical trials of NSAIDs with an incidence of about 1%. For the Safety Population, alanine transaminase (ALT) elevation AEs were reported in 0.3% of the HZT-501 and 0.4% of the Ibuprofen groups respectively. Aspartame (AST) elevation AEs were reported in 0.1% of the HZT-501 and 0.2% of the Ibuprofen groups respectively. In only 2 cases were transaminases >3x ULN, both in HZT-501 group. No AEs related to increased transaminases were reported in the Follow-on Population or Open Label Safety Population.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
53
Urinalysis Minor changes in urinalysis results were seen over the duration of the studies. There were no clinically significant abnormalities and no AEs reported. Vital signs Mean values were very similar across both treatment groups and the changes from baseline were small for all timepoints for systolic blood pressure, diastolic blood pressure and heart rate. Safety in special populations The Elderly Subjects > 65 years of age made up 18% of the Safety Population. The incidence of SAEs in the >65 years population was 2.2%, and 4.1% in the HZT-501 group and Ibuprofen group respectively. This compares to 3.4% and 3.1% for the <65 years population. Discontinuations due to AEs occurred slightly more frequently in the >65 years group (8.3% vs 11.2% in the HZT-501 and Ibuprofen groups respectively) compared to the <65 years group (6.3% vs 6.8% in the HZT-501 and Ibuprofen groups respectively). Similar to the overall population, the most common AEs that led to discontinuation were gastrointestinal. Of the 3 gastrointestinal complications reported, one occurred in the >65 years group. Renal impairment Subjects enrolled in the Phase 3 program had a creatinine clearance of > 45 mL/min. However, Study HZ-CA-006 enrolled subjects with creatinine clearance of <45 mL/min into a randomised, 2 period, crossover, oral single dose open label study to evaluate the relative bioavailability of ibuprofen and famotidine from HZT-501 Phase 3 combination tablet, and commercially available individual components. There were no deaths, SAEs, or AEs judged related to study treatment. No overall differences in safety were observed according to gender, race or weight. No studies have been conducted regarding the use of HZT-501 in pregnant or lactating women. The SmPC of Pepcid (MSD), an EU marketed formulation of famotidine, advises that in moderate and severe renal impairment the dose should be reduced, to avoid excess accumulation. For DUEXIS, a fixed combination product, dose reduction is not possible, as this would risk a consequential reduction in gastroprotection. However, DUEXIS is contraindicated in moderate and severe renal failure (creatinine clearance <50ml/min). This is acceptable. Immunological events In the Safety Population, 0.6% of the HZT-501 and 0.4% of the ibuprofen group reported an Immune System Disorders SOC adverse event. Drug hypersensitivity was reported by one subject in each arm. No SAEs in this SOC were reported in the Safety Population or Safety Follow-on Population, or Open-label Population. No SAEs were reported for the SOC Skin and Subcutaneous Tissue Disorders.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
54
Safety related to drug-drug interactions and other interactions Data from study HZ-CA-016 is supportive of SmPC advice that HZT-501 can be taken without regard to food. Data from Study HZ-CA-001 demonstrated no evidence of pharmacokinetic drug-drug interactions between famotidine and ibuprofen. The proposed SmPC warnings are based on EU approved SmPCs of ibuprofen and famotidine. This approach is acceptable. Discontinuation due to AES In the Safety Population, 6.7% of the HZT-501 arm and 7.6% of the Ibuprofen arm discontinued study treatment due to an AE. This is in addition to the early discontinuations due to the efficacy endpoint of endoscopically diagnosed upper GI ulceration, not counted as AEs (8.2% of the HZT-501 arm, 16.6% of the Ibuprofen arm). The highest rate of discontinuation was due to GI disorders, particularly nausea and dyspepsia. The incidence of discontinuation due to nausea was balanced across the treatment groups, but the incidence of discontinuation due to dyspepsia was higher in the Ibuprofen group. In the Safety Follow-on Population 1.5% of the HZT-501 arm and 2.1% of the Ibuprofen arm discontinued early due to an AE. In the HZT-501 arm, 3 AEs leading to discontinuation were reported: Abdominal Pain, Diabetic Gastroparesis and Blood Creatinine Increased. In the Ibuprofen, there was a single case: Throat Irritation. In the Open Label Safety Population 20.9% (18 of 86) of subjects discontinued due to a treatment emergent AE, with most subjects discontinuing due to a GI-related AE (12.8%, 11 of 86 Subjects). Gastric ulcer was reported as the reason for discontinuation in 2.3% (2 of 86) subjects. Gastrooesophageal reflux disease was also reported as the reason for study discontinuation in 2.3% (2 of 86) of subjects. Abdominal discomfort, abdominal distension, diarrhoea, dyspepsia, erosive oesophagitis, flatulence, gastritis and stomach discomfort were each reported as the reason for study discontinuation in 1.2% (1 of 86) of subjects. These AEs were predominately of mild or moderate severity. The only severe AE leading to discontinuation was a gastric ulcer. No additional safety concerns are raised following assessment of discontinuation data. Post marketing experience/Risk management HZT-501 is approved in the US since April 2011. However the applicant states that HZT-501 is not currently available for commercial sale in any country. The applicant has submitted post-marketing safety data from the U.S. Supportive safety data from the literature The proposed dose of the famotidine component is higher than that commonly used for currently licensed indications, for which a dose of 20-40 mg daily is approved. However it is stated in the SmPC of Pepcid that doses of 800 mg daily have been used for up to one year in the treatment of Zollinger-Ellison syndrome without the development of significant adverse effects or tachyphylaxis.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
55
The applicant refers to a study from the literature in support of the safety of long-term high dose famotidine. Howard et al 1987 is a non-comparative study of high dose famotidine in 32 patients with Zollinger- Ellison syndrome. Daily doses ranged from 50 mg to 800 mg daily (mean 330 mg). Mean follow-up was 10.5 months. 9 patients were treated for longer than 12 months and 6 for longer than 24 months. No haematological, biochemical or clinical evidence of toxicity was reported. There is a detailed discussion in the Clinical Overview of the safety of H2RAs, compared to PPIs, relating to interactions, fracture risk, pneumonia, infective enterocolitis, B12 deficiency and hypomagnesaemia: Interactions The applicant has provided a discussion, with reference to the literature, that all PPIs are hepatically metabolised to some degree by CYP isozymes including CYP2C19, and that inhibition of CYP2C19 occurs, decreasing the conversion of clopidrogel to its active metabolite. There is controversy regarding the clinical significance of this interaction. However, there is evidence that patients receiving clopidrogel with a PPI are at increased cardiovascular risk compared to those receiving clopidrogel alone. Omeprazole, esomeprazole and lansoprazole are licensed for the prevention of NSAID-associated gastric and duodenal ulceration. The SmPCs of esomeprazole and omeprazole contain clear warnings regarding the risk of interaction with clopidrogel, although the SmPC of lansoprazole does not. Fracture risk The applicant has provided a discussion of the evidence for an increased risk of fractures in patients taking PPIs, with reference to the literature. The referenced epidemiological studies, many case-controlled, reported an association between PPI use and fractures. Some studies also report an association for H2RAs, although this association is weaker and less consistent, with one study reporting a protective effect for H2RAs. In February 2012, the PhVWP have reported on proton pump inhibitors and risk of fractures of the hip, wrist and spine. The majority of the pharmacoepidemiology studies considered have reported a modest increase in risk. The size of estimated risk increased with increased dose and duration of use. There is currently no unequivocal pharmacological explanation for the increased risk of fractures. The following wording is agreed for inclusion in the SmPCs of prescription-only PPIs. Section 4.4 Proton pump inhibitors, especially if used in high doses and over long durations (>1 year), may modestly increase the risk of hip, wrist and spine fracture, predominantly in the elderly or in presence of other recognised risk factors. Observational studies suggest that PPIs may increase the overall risk of fracture by 10–40%. Some of this increase may be due to other risk factors. Patients at risk of osteoporosis should receive care according to current clinical guidelines and they should have an adequate intake of vitamin D and calcium. Section 4.8

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
56
Musculoskeletal disorders Frequency (uncommon): Fracture of the hip, wrist or spin The daily dose of famotidine is higher than that commonly used in the licensed indications. In the absence of a clear mechanism of action for this effect, it cannot be ruled out that long term high dose H2RAs could be associated with a risk of fracture. Pneumonia The applicant has provided a discussion of the evidence for an increased risk of community and hospital acquired pneumonia in patients taking PPIs, with reference to the literature. The papers discussed provide consistent evidence of an association with PPI, but also some evidence of association with H2RAs, although less consistently, and with a lower relative risk. A warning regarding the risk of community-acquired pneumonia is included in the SmPC of ranitidine, based on the results of a large epidemiological study with an observed relative risk of 1.82. Similar warnings are not currently included in the SmPCs of PPIs. For the epidemiological studies discussed, it is likely that H2RAs were used at lower doses than that proposed for DUEXIS. Infective enterocolitis The applicant has provided a discussion of the evidence for an increased risk of enteric infections, in particular C. difficile, in patients using PPIs, with reference to the literature. There is less evidence for an association with H2RAs. Warnings regarding the risk of enteric infections such as Salmonella, Campylobacter and C.difficile are given in the SmPCs of lansoprazole, omeprazole and rabeprazole. The proposed SmPC of DUEXIS contains the following warning: ‘Decreased gastric acidity due to any means including H2-receptor antagonists, increases gastric counts of bacteria normally present in the GI tract.’ Again, it is likely that the epidemiological studies discussed included lower doses of H2RAs than that proposed for DUEXIS. Vitamin B12 deficiency and hypomagnesaemia The applicant has provided a discussion of the evidence for an increased risk of Vitamin B12 deficiency and hypomagnesaemia, in patients taking PPIs, with reference to the literature. There is less evidence for an association with H2RAs. Hypomagnesaemia is listed as a very rare adverse effect in the SmPCs of omeprazole and esomeprazole. There is a warning regarding the risk of vitamin B12 malabsorption in the SmPCs of omeprazole and pantoprazole. Again, it is likely that the epidemiological studies discussed included lower doses of H2RAs than that proposed for DUEXIS. Assessor’s overall conclusions on clinical safety No specific safety concerns are raised relating to the proposed fixed combination product DUEXIS, in addition to the known safety profiles of ibuprofen and famotidine. An excess of renal and hypertensive AEs and SAEs were observed in

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
57
patients exposed to DUEXIS, compared to Ibuprofen. However it is considered most likely that these events were related to the ibuprofen component of DUEXIS, and occurred more commonly in the DUEXIS arm by chance. Dyspepsia was reported less commonly by patients on DUEXIS, compared to Ibuprofen only. The daily dose of ibuprofen is the maximum licensed in the UK. Due to the dose-dependent safety profile of NSAIDs, CHMP have advised that NSAIDs should be used at the minimum effective dose for the shortest duration necessary to control symptoms (Key elements for the SmPCs of non-selective NSAIDs, EMEA/CHMP/343456/2005). In order that patients are not prescribed unnecessarily high doses of ibuprofen, the indication is restricted to patients in whom lower doses of ibuprofen or of other NSAIDs is not considered sufficient. Even within this second line population, the lack of dose flexibility means that some patients may receive higher doses than would be effective, since the conditions to be treated will fluctuate in severity. The SmPC advises switching to alternative therapeutic regimens if a daily ibuprofen dose of 2400 mg is not appropriate. The advice is in line with that provided for Vimovo, a recently licensed fixed combination of enteric coated naproxen / eosmeprazole, in which the naproxen component is dosed at 500 mg bid, the maximum licensed naproxen dose in the UK. The SmPC advice is considered acceptable. There is a lack of clinical experience with famotidine dosed at 80 mg daily for more than one year. A post-authorisation safety study is requested. Based on data from the literature, the applicant claims that there are significant safety advantages for famotidine, compared to PPIs, in particular for interactions, fracture risk, pneumonia, infective enterocolitis, B12 deficiency and hypomagnesaemia. Evidence from the literature is presented to support this conclusion. The lack of interaction of famotidine with clopidrogel may be an advantage for some patients. As regards the other safety issues discussed, there may be more evidence for association with PPIs because PPIs are more widely used long-term. In addition, H2RAs tend to be used at lower doses than that proposed for DUEXIS. A safety disadvantage of famotidine in this fixed combination, compared to PPIs, is the risk of accumulation in renal impairment. CONCLUSION The Commission considered that the gastroprotection provided by PPIs was higher than that provided by H2 receptor antagonists. However the Commission was reassured by the data provided by the Company that famotidine 80 mg would achieve sufficient gastroprotection provided adequate measures are in place to ensure that the product is taken regularly three times daily. The Commission considered that with provision of adequate measures to ensure correct dosage, DUEXIS would be useful in a patient population that require regular treatment with a high dose of ibuprofen, and in whom treatment with a proton pump inhibitor would be inappropriate. There are no specific clinical safety issues to raise following assessment of the submitted safety data.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
58
EXPERT REPORT The clinical overview is written by an appropriately qualified physician and is a suitable summary of the clinical aspects of the dossier. SUMMARY OF PRODUCT CHARACTERISTICS (SmPC) This is satisfactory. PATIENT INFORMATION LEAFLET (PIL) This is satisfactory. LABELLING This is satisfactory MAA FORM(S) This is satisfactory. CONCLUSION There are no objections to the approval of this product from a clinical point of view.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
59
OVERALL CONCLUSION AND RISK BENEFIT ASSESSMENT QUALITY The important quality characteristics of DUEXIS® 800 mg/26.6 mg Film-coated Tablets are well defined and controlled. The specifications and batch analytical results indicate consistency from batch to batch. There are no outstanding quality issues that would have a negative impact on the benefit/risk balance. NON-CLINICAL No non-clinical data on the combination of the active substances are available. There are no known interactions between ibuprofen and famotidine that would indicate any novel or synergistic adverse pharmacology, pharmaco/toxicokinetics, toxicity, physical/chemical interaction or tolerability issues as a result of their combination. CLINICAL Pharmacokinetics The ibuprofen component of the proposed DUEXIS commercial formulation is bioequivalent to an 800mg ibuprofen EU reference product. The applicant has not provided bioequivalence data for the famotidine component of DUEXIS, as there is no suitable reference product of equivalent strength. Instead, clinical efficacy and safety data has been submitted. Pharmacodynamics No pharmacodynamic studies have been conducted with DUEXIS. Based on the assessment of the efficacy and safety data, no pharmacodynamic data is required. Efficacy The efficacy of the ibuprofen component of DUEXIS is accepted, based on the demonstration of bioequivalence with an EU marketed formulation of ibuprofen. The efficacy of the famotidine component of DUEXIS is supported by 2 pivotal clinical efficacy and safety studies. DUEXIS is associated with less NSAID-associated upper gastrointestinal, gastric and duodenal ulceration, measured by endoscopy at 24 weeks, compared to ibuprofen 800mg TID. The risk reductions are both statistically and clinically significant. Subgroup analyses provide supportive evidence of efficacy in higher risk patients. When considering whether the demonstrated risk reduction for the famotidine component of DUEXIS is adequate, it is relevant to compare with that expected for PPIs. However, the applicant has not compared DUEXIS directly with a combination of ibuprofen and PPI. Based on pooled data from the pivotal studies, a risk reduction of 44% for endoscopic gastric ulceration at 24 weeks is expected. This is less than an estimate of 60% for proton pump inhibitors, based on the literature. The CHM have proposed an amendment to the indication to restrict use to patients for whom treatment with a PPI is not considered appropriate. As a result the indication in the SmPC has been updated.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
60
Safety The safety profiles of ibuprofen and famotidine are well-established. No specific safety concerns remain following assessment of the submitted clinical safety data, and the applicant’s responses to the questions raised during the assessment. The product information contains adequate safety warnings in relation to the high dose of ibuprofen in DUEXIS. Conclusion On the basis of the revised indication, the product is approvable. PRODUCT LITURATURE The SmPC, PIL and labelling are satisfactory. RISK BENEFIT ASSESSMENT The quality of the product is acceptable and no new non-clinical data were submitted. All the clinical efficacy and safety concerns raised during the assessment have been resolved. The risk benefit is, therefore, considered to be positive.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
61
DUEXIS® 800 mg/26.6 mg Film-coated Tablets
PL 36663/0002
STEPS TAKEN FOR ASSESSMENT
1 The MHRA received the Marketing Authorisation application on 31st January
2012
2 Following standard checks and communication with the applicant the MHRA considered the application valid on 20th February 2012
3 Following assessment the application was referred to CHM on 17th May 2012
4 Pre-Hearing took place on 8th November 2012
5 Oral Representation took place on 6th December 2012
6 The application was determined on 6th March 2013

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
62
Module 2 Summary of Product Characteristics
In accordance with Directive 2010/84/EU the Summaries of Product Characteristics (SmPC) and Patient Information Leaflets (PIL) for products granted Marketing Authorisations at a national level are available on the MHRA website.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
63
Module 3 Patient Information Leaflet
In accordance with Directive 2010/84/EU the Summaries of Product Characteristics (SmPC) and Patient Information Leaflets (PIL) for products granted Marketing Authorisations at a national level are available on the MHRA website.

UKPAR DUEXIS® 800 mg/26.6 mg Film-coated Tablets PL 36663/0002
64
LABELLING


















