Docking small molecules with Autodocktm
45
Instructor: Elon Yariv Autodock Tutorial
Transcript of Docking small molecules with Autodocktm

Instructor: Elon YarivAutodock Tutorial
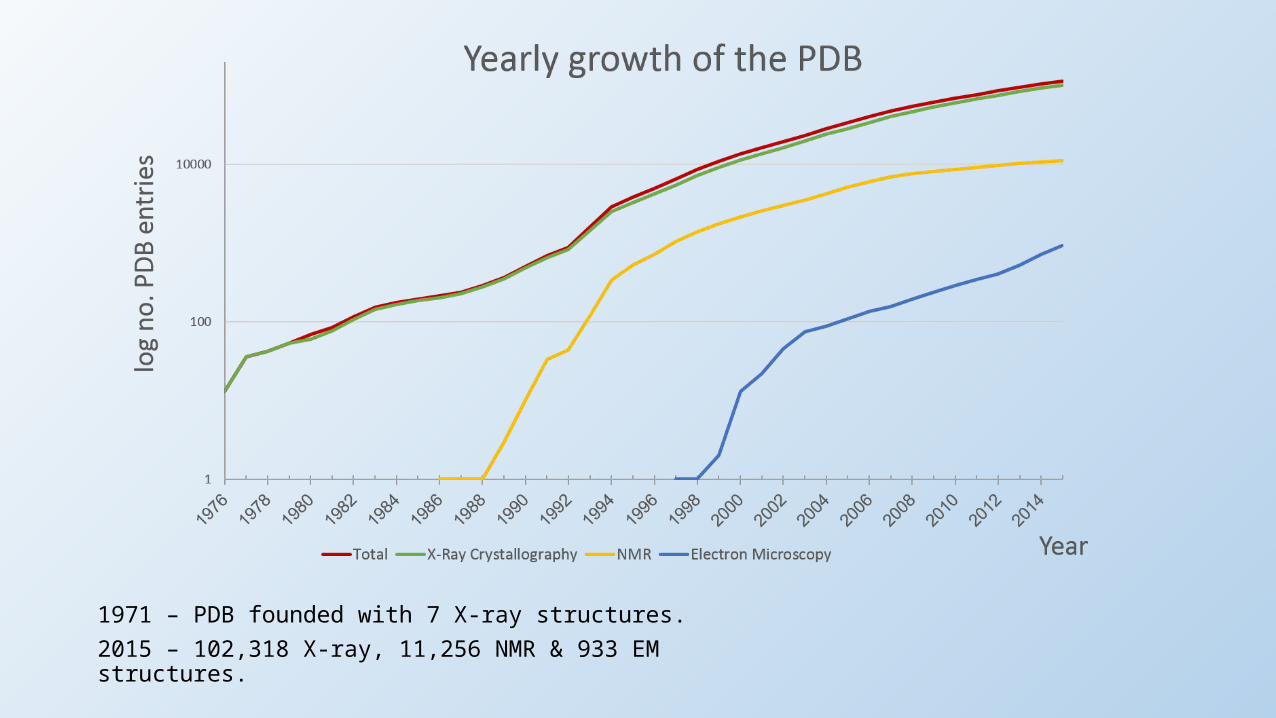
1971 – PDB founded with 7 X-ray structures.2015 – 102,318 X-ray, 11,256 NMR & 933 EM structures.
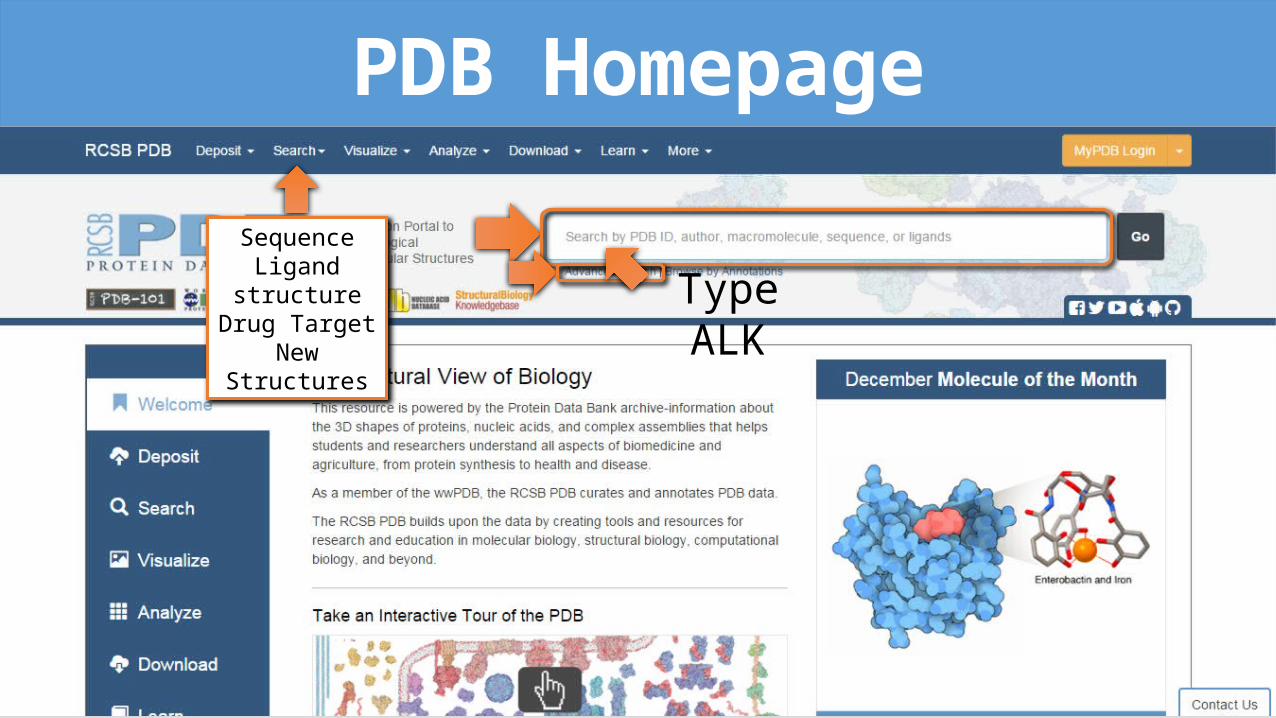
PDB HomepageSequence
Ligand structureDrug Target
New StructuresType ALK
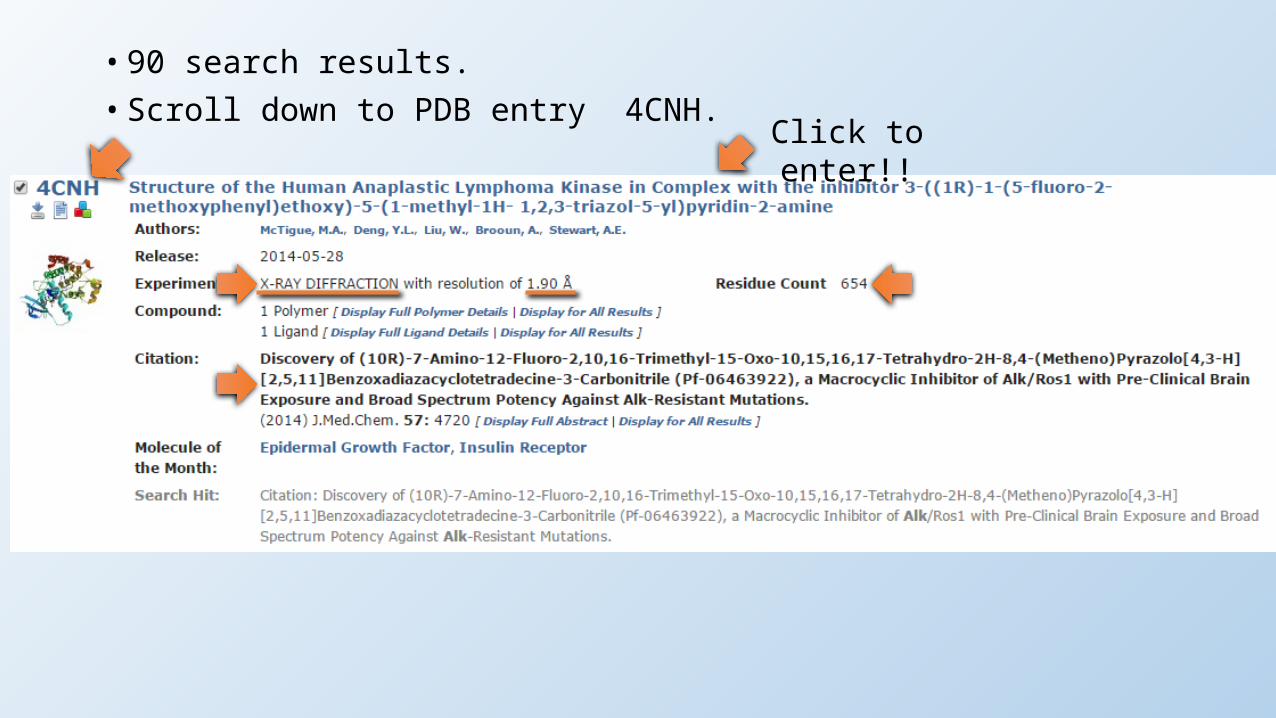
• 90 search results.• Scroll down to PDB entry 4CNH.
Click to enter!!

Click!!

Visualizing structures in Autodock Tools

File->Preferences->Set…
Type working directory here
3D Viewer
DashBoard
Menu

File->Read molecule
Or Click
New molecule appears
Hold to rotate around Axis
Hold to move
Hold/Scroll to zoom + Shift
Hold to move in/out
Select
You can change selection level through this
menu to atoms, residues, chains
or molecules.
Select 4CNH.pdb you’ve just
downloaded
+ Shift
+ Shift
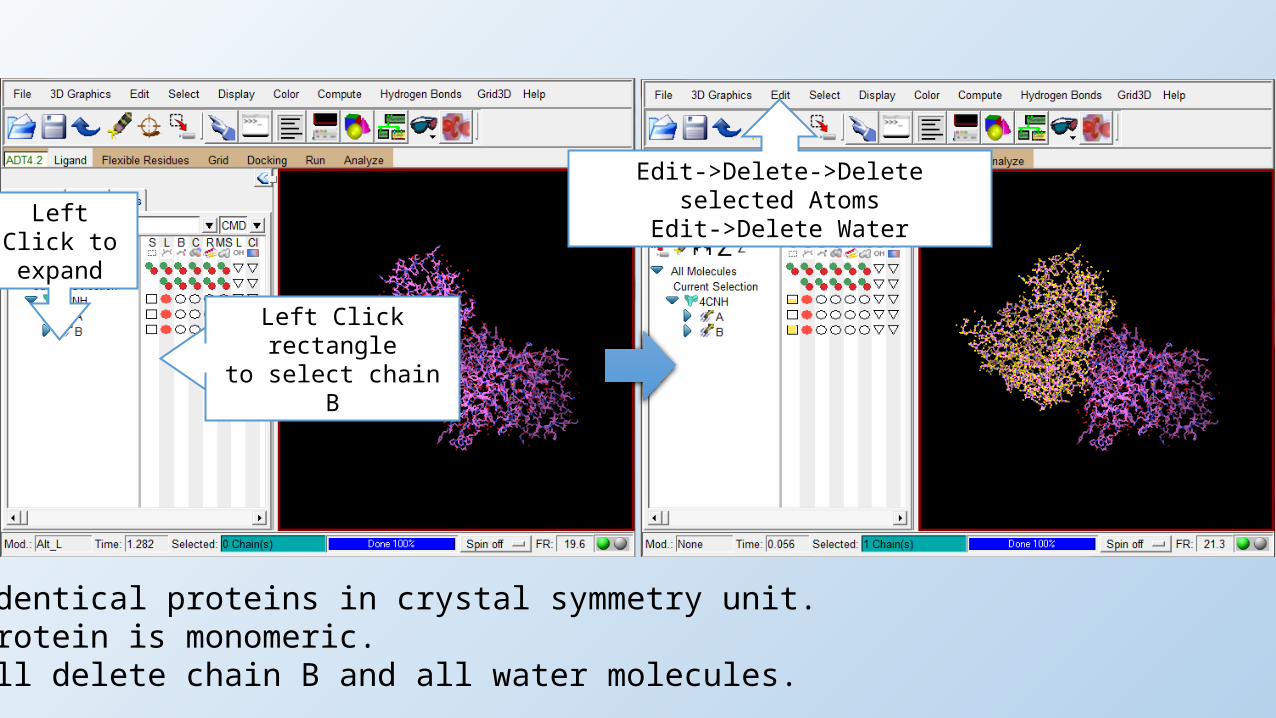
Left Click rectangleto select chain B
Edit->Delete->Delete selected AtomsEdit->Delete Water
Two identical proteins in crystal symmetry unit.The protein is monomeric.We will delete chain B and all water molecules.
Left Click to expand
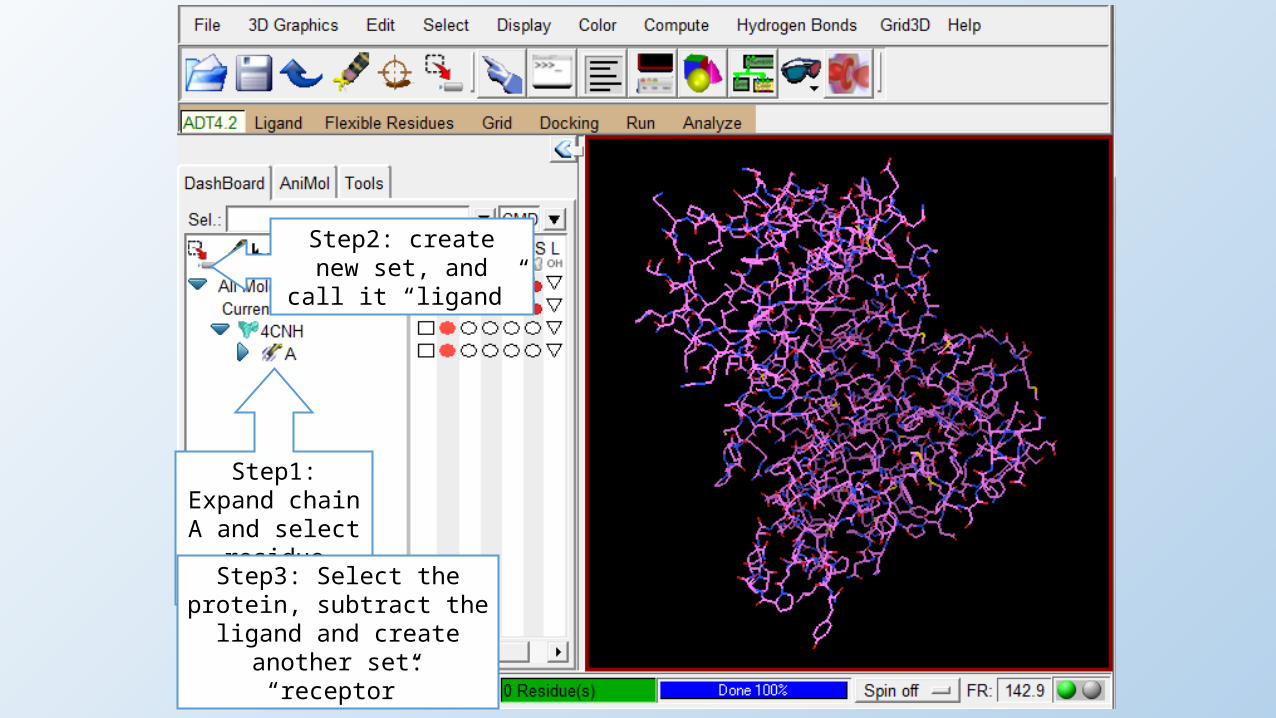
Step1: Expand chain A and select residue 3U92402.
Step2: create new set, and call it “ligand”
Step3: Select the protein, subtract the ligand and create
another set: “receptor”
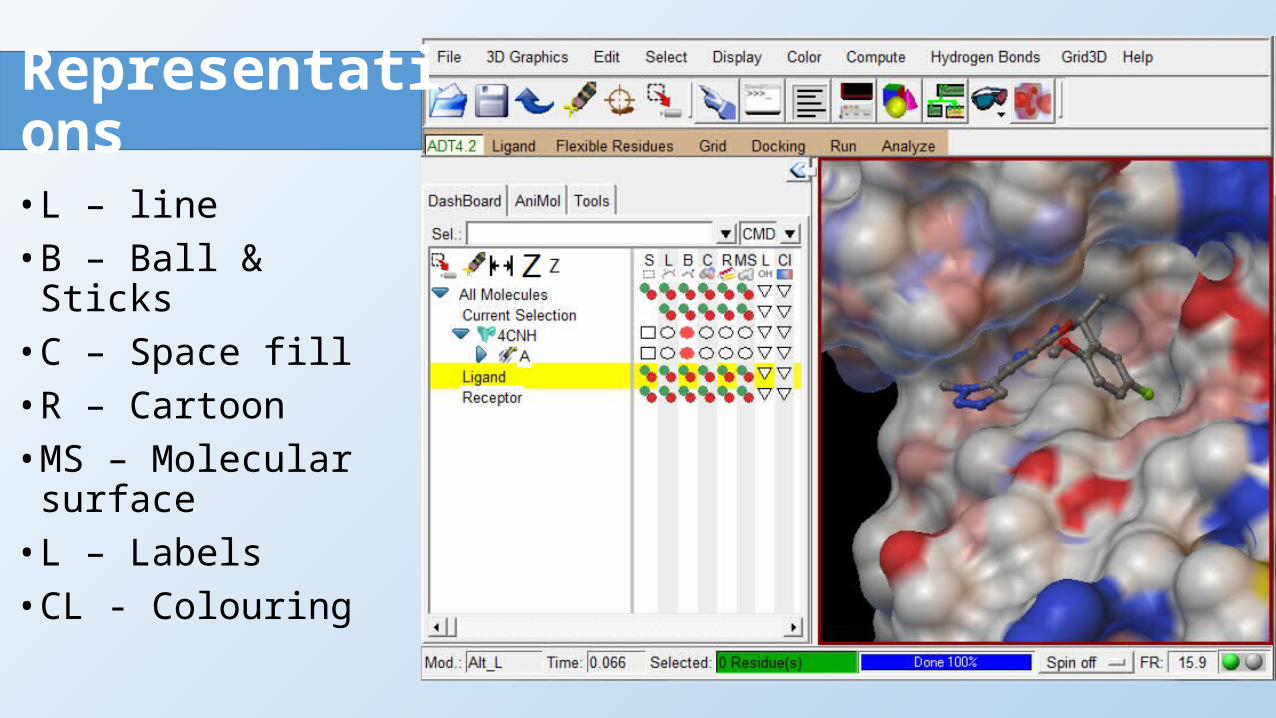
Representations• L – line• B – Ball & Sticks• C – Space fill• R – Cartoon• MS – Molecular surface• L – Labels• CL - Colouring

Step1: Select ligand (green dot)
Step2: Select->Spherical region
Step3: left click
Step4: Click
Step5: select from protein
Step6: click select, then closeBefore closing make sure the selection level is at residues.
Add the selection to a new set: “protein near ligand”
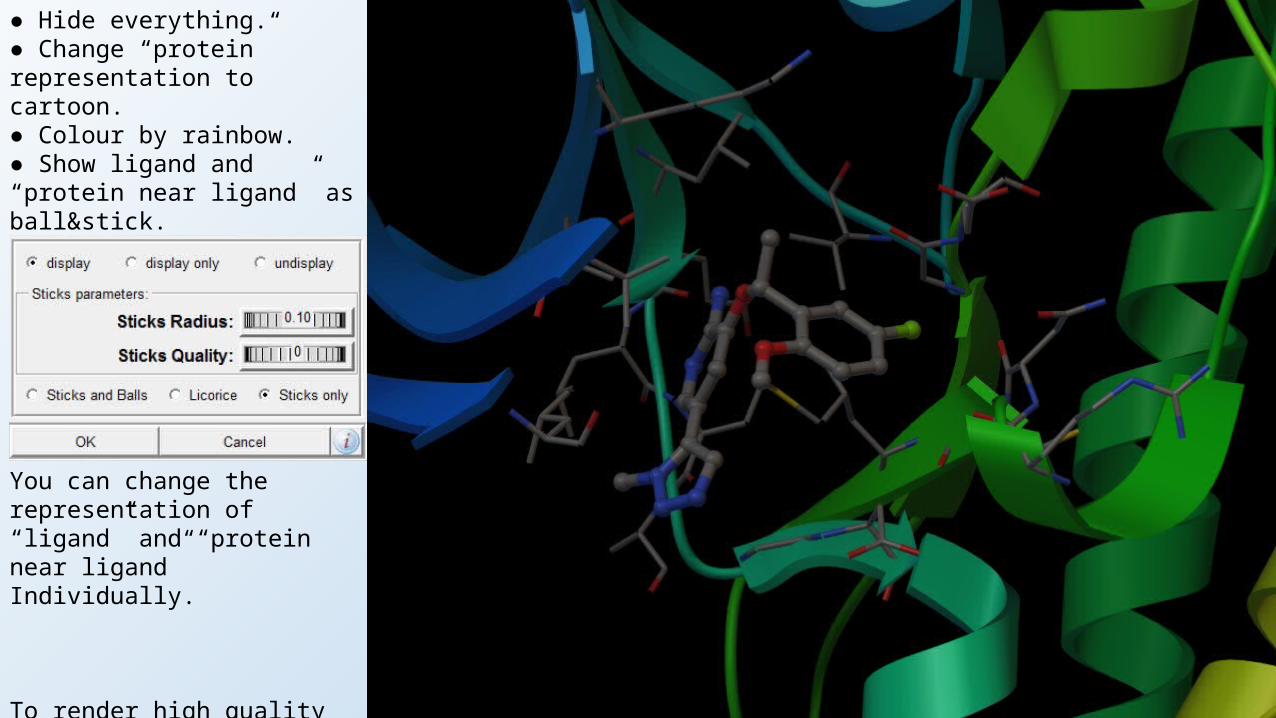
● Hide everything.● Change “protein” representation to cartoon.● Colour by rainbow.● Show ligand and “protein near ligand” as ball&stick.● Right click the show ball&stick button next to “ligand”.
You can change the representation of “ligand” and “protein near ligand”Individually.
To render high quality images go to:3D graphics->Render large image

File->Save->Current session
Save to a folder of your liking.
Session file ends with .psf
To load an old session:
File-> Read session
Saving Your Progress:

File->Save->Write PDB
Call the new file – 4CNH_monomer.pdb
Confirm
Saving structure files:

Break

PDB structures aren’t perfect• Missing residues.• Missing sidechains.
Hydrogens cannot be resolved in most X-ray structures
• Hydrogens aren’t present in PDB structures.• Histidine protonation.• Asp/Gln orientation.

Adding explicit hydrogensOpen ‘Edit’ menu
Press to add hydrogens
Quick solution but not the best.
We will not do this in this workshop.

Molprobity - molprobity.biochem.duke.edu/• Adds explicit hydrogens.• Asp/Gln flips.• Changes Histidine protonation.

type ‘4CNH’
Or upload 4CNH_monomer.pdb

Select this option
Fix Asn/Gln/His residues
Hydrogen placement for X-ray structures
Click to add hydrogens

Review Results
Confirm results and download .pdb file
Choose which residues you want to flip.

Steric Clashing in PDB structures
Structures must be minimized to relax protein strain.
Chimera built-in minimization: www.cgl.ucsf.edu/chimera/3Drefine (webserver): sysbio.rnet.missouri.edu/3Drefine/Chiron (webserver): troll.med.unc.edu/chiron/login.phpPDB-Redo (webserver): www.cmbi.ru.nl/pdb_redo/

Select the ligand, and then remove it
Don’t forget to set selection level to residues.
• Read the molprobity output file.• Delete the ligand
residue within the structure.
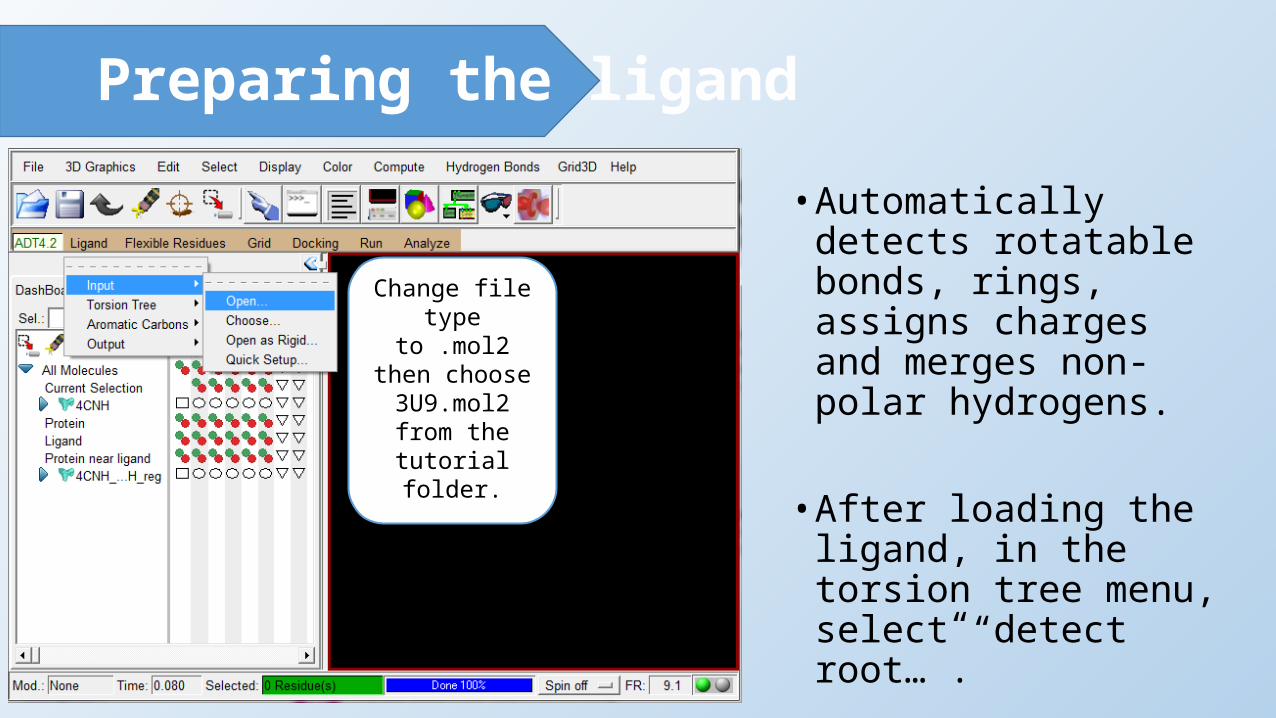
Preparing the ligand• Automatically detects
rotatable bonds, rings, assigns charges and merges non-polar hydrogens.
• After loading the ligand, in the torsion tree menu, select “detect root…”.
Change file type to .mol2 then
choose 3U9.mol2 from the tutorial
folder.
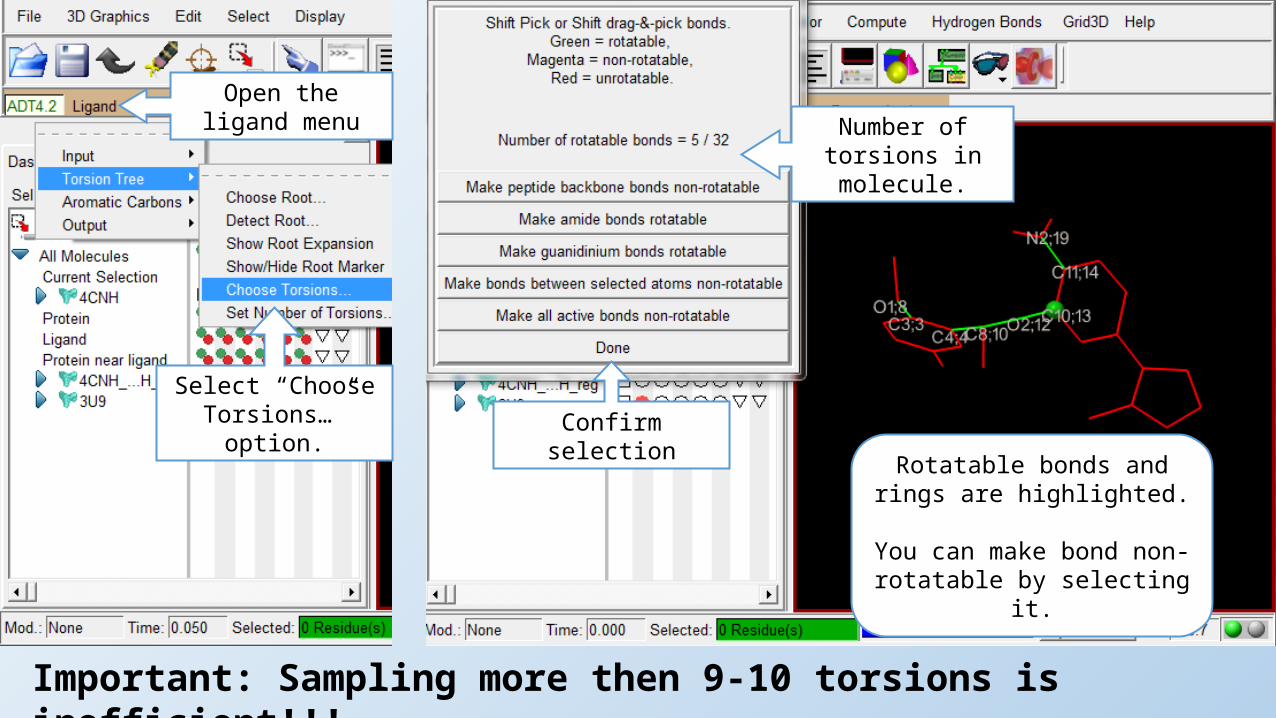
Open the ligand menu
Select “Choose Torsions…” option.
Rotatable bonds and rings are highlighted.
You can make bond non-rotatable by selecting it.
Number of torsions in molecule.
Confirm selection
Important: Sampling more then 9-10 torsions is inefficient!!!

• Save the ligand.• Hide root marker.• Hide it and display the
receptor – 4CNH_monomerFH_reg
PDPQT format:Contains atom coordinates of structure. This file also
contains partial charges and autodock atom types.
Both ligand and receptor must be converted to PDBQT.
Recognized atom types:H, C, N, O, F, Mg, P, S, Cl, Ca,
Mn, Fe, Zn, Br and I.
Open the ligand menu

Preparing the Grid input files
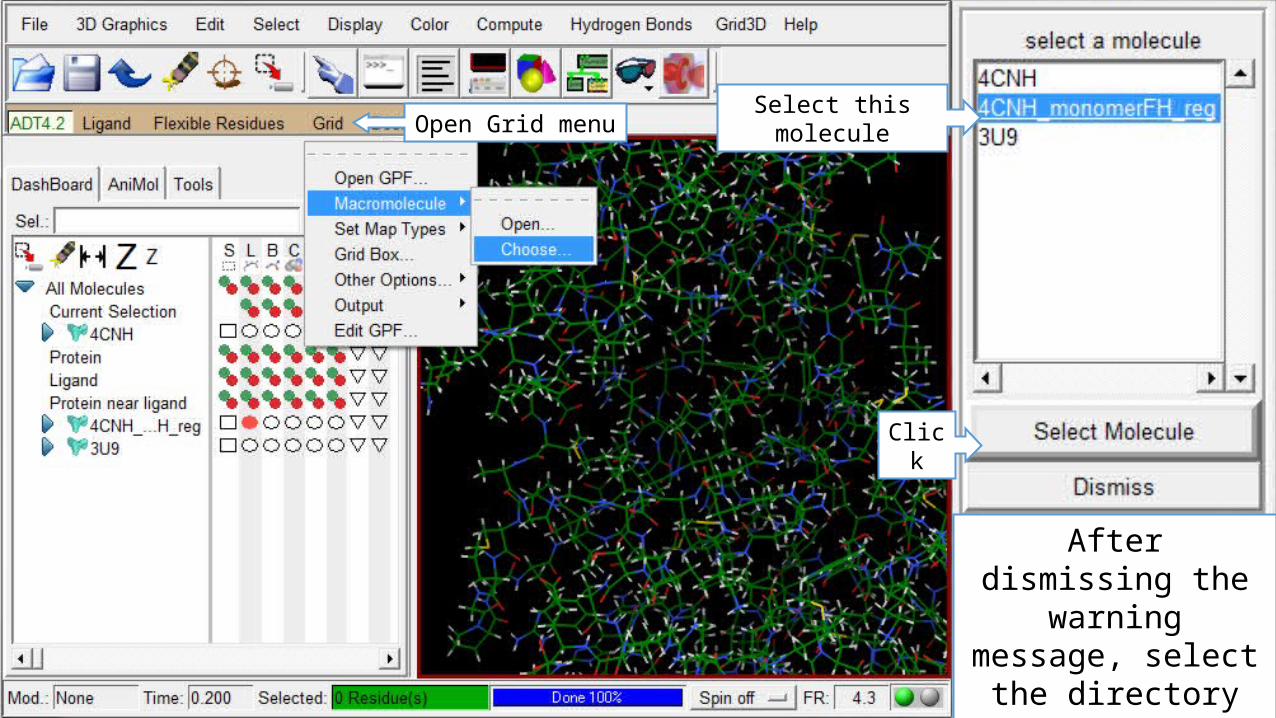
Open Grid menuSelect this molecule
Click
After dismissing the warning message,
select the directory and the name of the protein .PDBQT file.

Determining grid dimensionsOpen menu through
Grid->Gridbox…
Set box size to
50x50x50
Set centroid to:(45,-4.5,-49)
The Grid box appears at origin
of axis (0,0,0).
The position and size of the box must be altered
When finished press the file tab in the Grid Box window.
Choose: “Close saving current”

Select the Ligand
Autogrid will use the atom types found in
the ligand.It will place them at
each grid point
Open Grid menu
Confirm

• Save the grid file as “4CNH.gpf”.
This file includes the grid box dimensions and the atom types found in the ligand molecule.
Open Grid menu
Save the grid parameters file
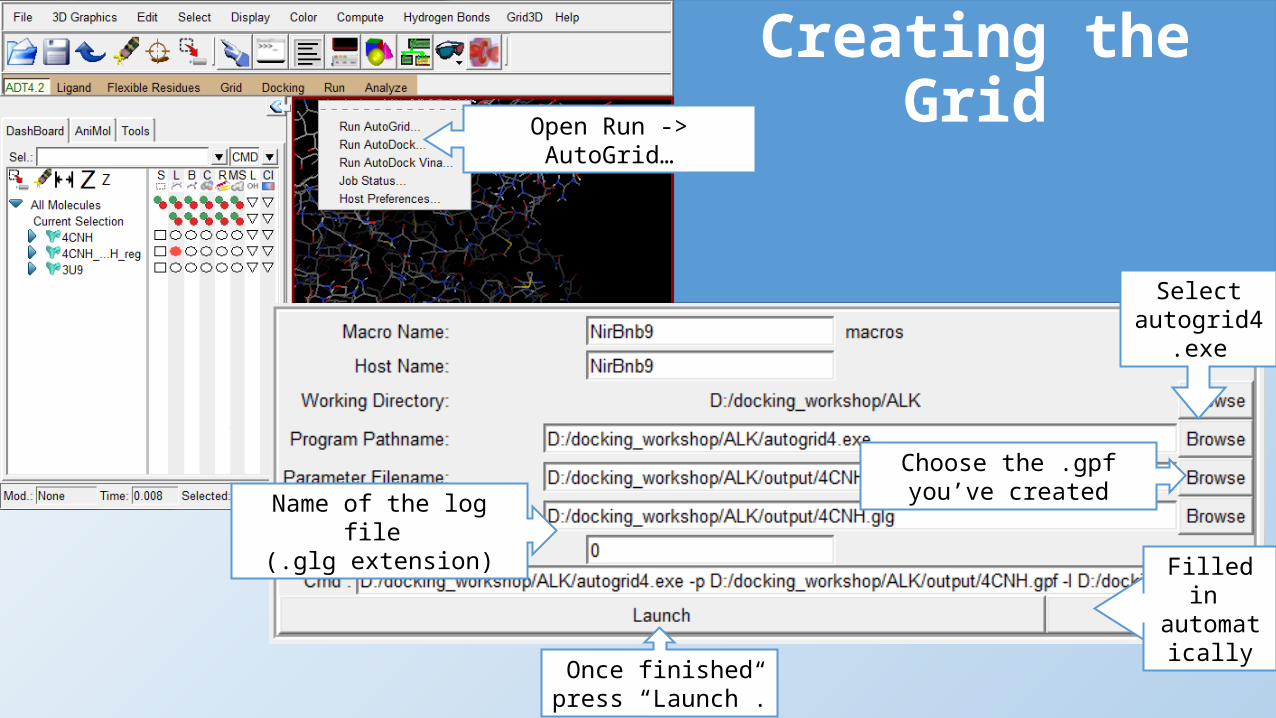
Choose the .gpf you’ve created
Creating the Grid
Name of the log file (.glg extension)
Filled in automaticall
y
Once finished press “Launch”.
Open Run -> AutoGrid…
Select autogrid4.exe

Monitoring results• Job last about 15 seconds.• Can be monitored by opening 4CNH.glg with Notepad++.• Or with the ‘tail’ command in Unix.• Here you will find the error messages.
• Output: Grid map file for each atom type in the ligand..map file: List of the scores of every point in the grid, most values are zero or large positive numbers.

Initializing the docking simulations
(But first we must prepare the parameter files)

Open Docking menu and select Open Docking menu
and select
Choose the receptor structure file:4CNH_monomerFH_reg.pdpqt
Choose ligand “3U9”.
A new window will open that allows you to review ligand properties, set the initial
location of the ligand and the initial number of torsions.
Press “Accept” without changing anything.

Docking -> Search Parameters -> Genetic Algorithm
New window opensNumber of output results
Change duration to short
Confirm to exit
Short simulation: about 1 minMedium simulation: about 10-20 min
Long simulation: Can only guess…
Always generate more then 1 ligand pose.

“Docking Parameters…” window• For advanced users.• You shouldn’t mess with these!
• Add the crystal ligand conformation, i.e. 3U9.pdb, as a reference for RMS calculations.• This will give you an indication
how well the results were.Write the directory of
3U9.pdb
Select ‘no output’
“Accept” to Exit
Click to expand
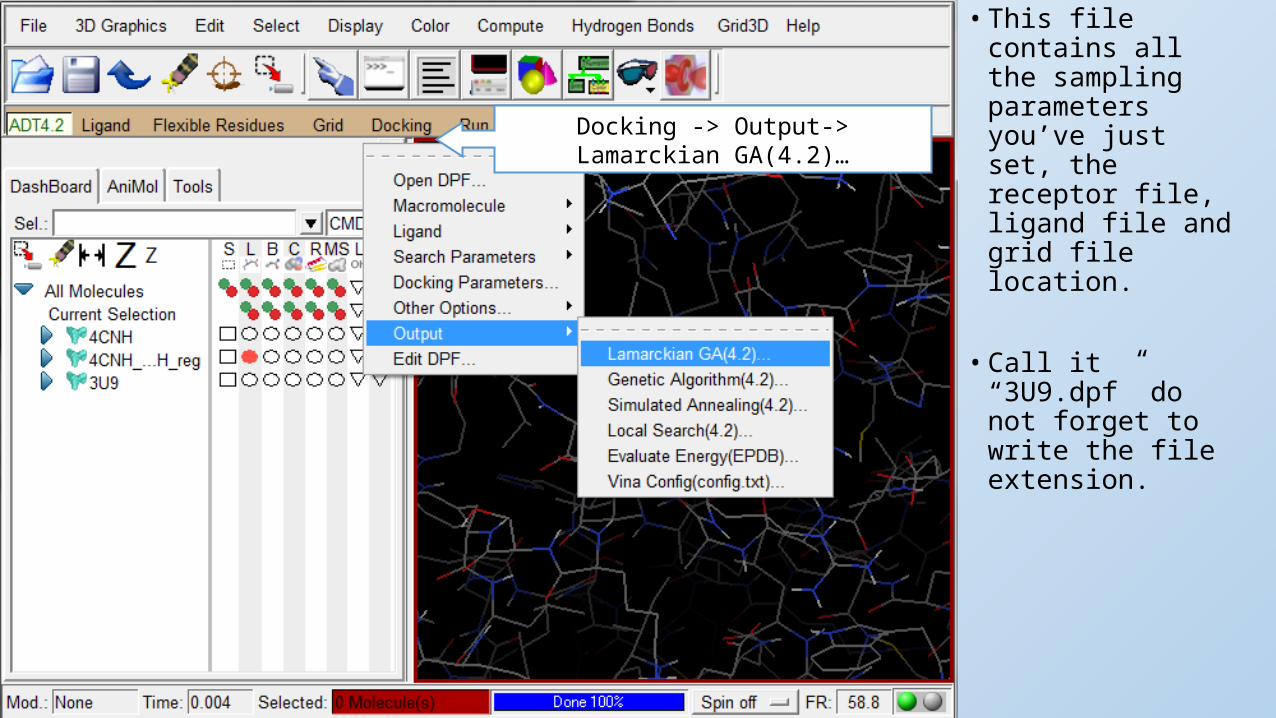
• This file contains all the sampling parameters you’ve just set, the receptor file, ligand file and grid file location.
• Call it “3U9.dpf” do not forget to write the file extension.
Docking -> Output-> Lamarckian GA(4.2)…

Running Autodock (finally!)
Choose the .dpf you’ve created
Select Autodock4.exe
Press to start simulation
Select autodock4.exe

Loading results• To load results into open the Analyze menu->Dockings->Open…• Select 3U9.dlg – the output file.• To view the different poses:Analyze->Conformations->Play, Ranked by Energy
Pose Rank
Next pose
Play/PausePose Player
optionsMove to first pose
Exit

Displaying Hydrogen bonds• Open the analyze menu->Macromolecule->Choose…• Select 4CNH_monomerFH_reg and confirm• Open the pose player options menu
Select to build Hydrogen bonds
Show energy components for
docked pose
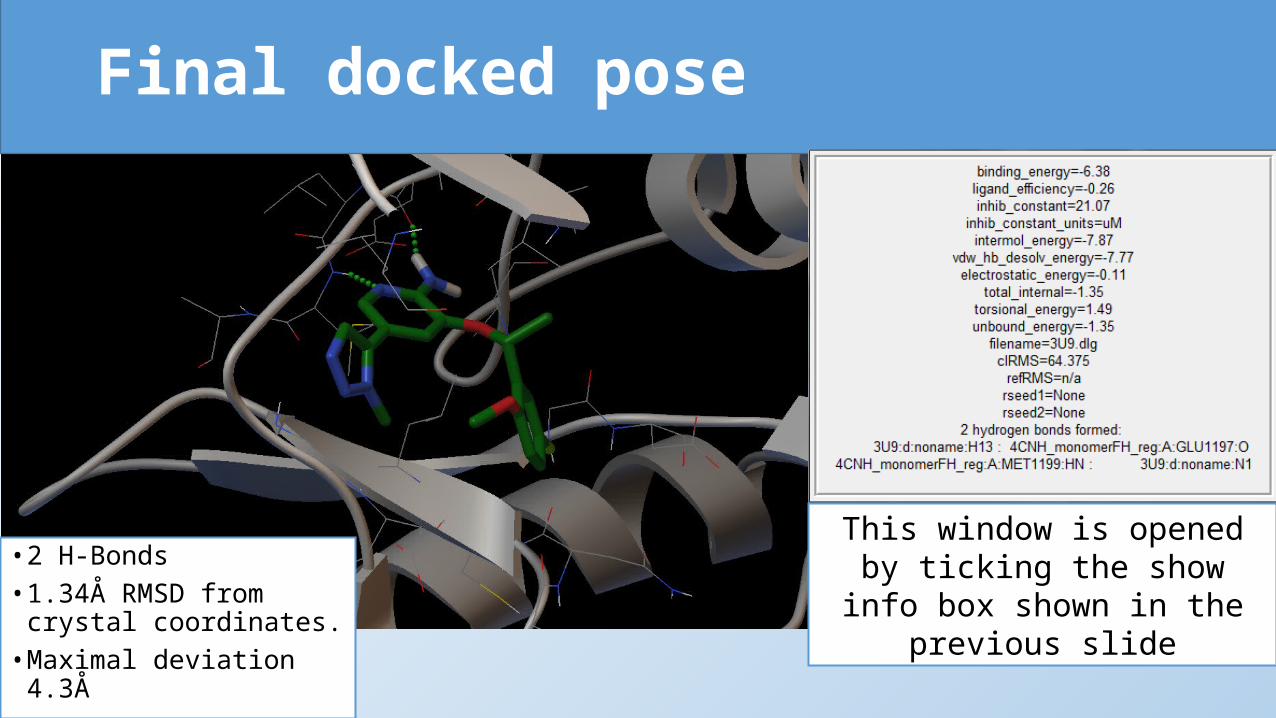
Final docked pose
• 2 H-Bonds• 1.34Å RMSD from
crystal coordinates.• Maximal deviation 4.3Å
This window is opened by ticking the show info box shown in the
previous slide

Break