
Division of Project Management - gphaonline.org · Division of Project Management ... considered...
72
1 Division of Project Management Office of Regulatory Operations Office of Generic Drugs, FDA GPhA Fall Tech Conference, 2016
Transcript of Division of Project Management - gphaonline.org · Division of Project Management ... considered...

1
Division of Project Management Office of Regulatory Operations
Office of Generic Drugs, FDA GPhA Fall Tech Conference, 2016

2

3
Office of Generic Drugs Division of Project Management (DPM): Things
That Make You Go Hmmmm
1. Form FDA 356h: Joe Shin, PharmD 2. Secure Email: Mandy Kwong, PharmD 3. Transfer of Ownership (TOO): Megan Tychinski, PharmD, CAPM 4. ANDA Consolidation: Kathleen Melendez, RPh 5. Endorsement/Clearance Phase: CDR Lakeeta Carr, MSN/MHA,
BSN, RN, NE-BC 6. Withdrawal Process: Tre Ferguson, PharmD

DPM: Things That Make You Go Hmmmm
----Form FDA 356h---
Joe Shin, Pharm.D. Regulatory Project Manager Division of Project Management Office of Regulatory Operations Office of Generic Drugs, FDA

5
Form FDA 356h

6
Purpose of Form FDA 356h • Accompanies regulatory submissions to new drug
applications (NDAs), biologic license applications (BLAs), abbreviated new drug applications (ANDAs), and supplements
• Describes the reason for, and content of, the submission
• Captures information used to populate FDA systems

7
Regulation 21 CFR 314.94 (a)(1): The applicant shall submit a completed and signed application form that contains the information described under CFR 314.50(a)(1), (a)(3), (a)(4), and (a)(5). The applicant shall state whether the submission is an abbreviated application under this section or a supplement to an abbreviated application under CFR 314.97.

8
Sections of Form FDA 356h
• There are 4 sections to the FDA 356h form – Applicant Information – Product Description – Application Information – Certification

9
Applicant Information
• Applicant Information section: – Fields 2-6

10
Applicant Information
– Field 6: Authorized U.S. Agent • Required for non-U.S.
applicants. See 21 CFR 314.50 (a)(5)
• Include the name of the individual along with their company name.
• Provide a secure email address • Phone number (should include
extension if applicable)
• Common Issues: – Field 2: Name of Applicant
• Inconsistency of Applicant name (e.g. Acme Drugs and Acme Drugs, Inc. are considered separate companies by our Document Room)

11
Product Description
• Product Description section: – Fields 7-15

12
Product Description • Common Issues:
– Field 9: Established Name • Ensure proper use of the USP
name
– Field 11: Chemical/Biochemical/Blood Product Name (If any)
• Provide chemical name (e.g. 2-[(2,6-dichlorophenyl)amino]-benzeneacetic, monosodium salt)
– Field 13: Strengths • “Injections” or “for Injection”
drug products, include total strength and fill volume in addition to mg/ml

13
Application Information
• Application Information section: – Fields 16-31

14
Application Information • Common Issues:
– Field 20: Identify the listed drug product that is the basis for the submission
• Fill in the name of the RLD and its application number
• Ensure that the patent certification checkboxes are complete and accurate. This should match information found in Module 1.3.5.

15
Application Information • Common Issues cont.:
– Field 29: Establishment Information • List all facilities associated with
the manufacturing, packaging, and control sites for both drug substance and drug product
• Provide accurate address information for all establishments.
• Provide accurate FEI and DUNS numbers

16
Application Information • Common Issues cont.:
– Field 29: Establishment Information • Select appropriate checkbox for
establishment status – Pending: New
establishments at time of submission
– If not a new establishment, indicate current status (e.g. active, inactive, or withdrawn).

17
Application Information • Common Issues cont.:
– Field 29: Establishment Information • Establishment contact person
should be an individual at the site, not a corporate office or the U.S. Agent.
• Provide accurate telephone number for the contact person.
• Provide a brief description of the manufacturing steps and/or types of testing conducted at the site.
– Field 30: Cross References • Should include all cross
references, including all DMFs

18
Certification
• Certification section: – Fields 32-39

19
Certification • Common Issues:
– Field 32: Name & Title of Applicant’s Responsible Official
• This individual is responsible for certifying compliance with applicable laws and regulations.
• U.S. Agent from Field 6 may also act as the applicant’s Responsible Official.
– Field 38 & 39: Signature & Countersignature
• If the applicant’s Responsible Official does not reside or have a place of business within the U.S. then Field 39 must be countersigned by an attorney, agent, or authorized official who does reside or have business within the U.S.

20
Possible Consequences
1. For Initial ANDA submissions, failure to include a complete and signed Form FDA 356h may result in a refuse-to-receive notification.

21
Possible Consequences – Cont. 2
2. If FDA discovers during technical review that the applicant failed to identify all facilities on the Form FDA 356h, the applicant will be asked to update the Form FDA 356h in an IR or CRL. – If the facility requires an inspection, a 10-month
review goal may be added to the review of the IR or CR amendment.

22
Possible Consequences – Cont. 3
3. For amendments and resubmissions, an incomplete Form FDA 356h may require further communication with the applicant to provide updated and complete Form FDA 356h forms leading to delays in review.

23
Takeaways • Clearly complete and update the Form FDA 356h
with every submission • Follow the Instructional Supplement for proper
completion of the Form FDA 356h • Ensure that the most up-to-date version of the
Form FDA 356h is being utilized.

24
Takeaways – Cont. • Provide accurate contact information for the
applicant’s Responsible Official and/or U.S. Agent • Include complete information on the locations of
all manufacturing, packaging, and control sites for both drug substance and drug product

25
References • Regulations:
– 21 CFR 314.94(a)(1) – 21 CFR 314.50 (a)(1), (a)(2), (a)(3), (a)(4), & (a)(5) – 21 CFR 314.101 (d)(1)
• Forms: – Form FDA 356h
(http://inside.fda.gov:9003/downloads/administrative/forms/fda/ucm008209.pdf)
– Form FDA 356h Supplement (http://inside.fda.gov:9003/downloads/administrative/forms/fda/ucm321898.pdf)
• Guidances: – ANDA Submission-Refuse-to-Receive Standards: Guidance for Industry (http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm370352.pdf)

DPM: Things That Make You Go Hmmm
----Secure Email---
Lt. Mandy C. Kwong, PharmD Regulatory Project Manager- Team Leader Division of Project Management

27
What is Secure E-mail? • FDA uses Secure E-mail Technology to safeguard
confidential information, and help protect our machines against incoming e-mail threats or otherwise harmful, unsolicited e-mail.
www.fda.gov

28
FDA Secure Email Partners • You are strongly encouraged to become a
Secure Email Partner • Why?
– Ensure that our messages will not be readable by a third-party
– E-mails are only secure if they are sent to a Secure E-mail partner
www.fda.gov

29
FDA Secure Email Partners • Allows for TIMELY informal communications
that may include commercial confidential information (CCI) – If we are unable to confirm that your email is
secure, we will send the communication containing any CCI via mail or fax

30
FDA Secure Email Partners Must be a business email address
We cannot secure internet service provider accounts such as:

31
Options for Setting Up Secure Email 1) S/MIME (Secure/Multipurpose Internet Mail
Extension) Encryption 2) Secure SMTP (Simple Mail Text Protocol) over
TLS (Transport Layer Security) Encryption

32
Options for Setting Up Secure Email 1) S/MIME Encryption
- More secure but more complex to set up - Done at workstation-level (without e-mail administrators) - Certificate will need to be repurchased/renewed once-a-year
- One year Digital ID costs $20.00 per person - For each FDA user or mailbox you wish to securely
communicate with, a one-time setup process is required to create an FDA Outlook contact and corresponding FDA proxy certificate
- Set up time is as little as a few hours - To set up, email [email protected]
www.fda.gov

33
Options for Setting Up Secure Email 2) Secure SMTP over TLS encryption
- A one year Digicert SSL certificate is $144. A three year certificate is $345
- Configuration involves your email administrator - Everyone at your organization will be able to send email
securely to the FDA - Set up time can be a few days or longer, especially if your
email system is outsourced. - To set up, email [email protected]
www.fda.gov

34
S/MIME vs TLS User
Computer User
Mailbox
Company Email Server
Cloud /3rd Party Email Server
Internet FDA
S/MIME protection
TLS protection
FDA Internet Company
Email Server
User Mailbox
User Computer
S/MIME protection
TLS protection For additional questions, email [email protected]

Things That Make You Go Hmmm… Transfer of Ownership
Megan Tychinski, PharmD, CAPM Regulatory Project Manager
Division of Project Management Office of Generic Drugs, FDA

36
What is a Transfer of Ownership (TOO)? • The Code of Federal Regulations section 314.99(a)
states that: An applicant shall comply with the requirements of 314.65 regarding withdrawal by the applicant of an unapproved abbreviated application and 314.72 regarding a change in ownership of an abbreviated application.
• TOO: Change in ownership of an application – Applicant A Applicant B – Subsidiaries
• Parent company Subsidiary • Subsidiary 1 Subsidiary 2

37
I have submitted my TOO… What happens next?
• FDA Process
(1) Ensure all required information has been submitted to the application (2) Issue TOO Acknowledgement Letter to applicant

38
FDA Obstacle
• Ensuring all required information has been submitted • 356h form(s) • Transfer of ownership from former owner • Acceptance of ownership from new owner

39
21 CFR 314.72 Change in ownership of an application
• (a) An applicant may transfer ownership of its
application. At the time of transfer the new and former owners are required to submit information to the Food and Drug Administration as follows: – (1) The former owner shall submit a letter or other
document that states that all rights to the application have been transferred to the new owner.

40
21 CFR 314.72 Change in ownership of an application
• (2) The new owner shall submit an application form signed by the new owner and a letter or other document containing the following: – (i) The new owner's commitment to agreements, promises, and
conditions made by the former owner and contained in the application;
– (ii) The date that the change in ownership is effective; and – (iii) Either a statement that the new owner has a complete copy of
the approved application, including supplements and records that are required to be kept under 314.81, or a request for a copy of the application from FDA's files. FDA will provide a copy of the application to the new owner under the fee schedule in 20.45 of FDA's public information regulations.
www.fda.gov

41
21 CFR 314.72 Change in ownership of an application
• (b) The new owner shall advise FDA about any change in the conditions in the approved application under 314.70, except the new owner may advise FDA in the next annual report about a change in the drug product's label or labeling to change the product's brand or the name of its manufacturer, packer, or distributor.

42
To be noted… • If the new owner requests a copy of the application, they
must make a Freedom of Information Act request. – Directions found at:
http://www.fda.gov/RegulatoryInformation/FOI/HowtoMakeaFOIARequest/ucm2007229.htm
• If former and new owners provide separate submissions for the transfer, a 356h form should be included with each submission.
• If one submission is sent for the transfer that includes all requirements, only one 356h form with the new owner’s information is needed.

43
Withdrawn ANDAs • An applicant’s decision to withdraw an ANDA
pursuant to 21 CFR 314.150(c) is without prejudice to refiling.
• Once notice of withdrawal is published in the Federal Register, withdrawal is final.

44
OGD Recommendations
• Submit TOO Amendment in a separate submission – i.e. Do not combine with CR responses, other administrative
changes, etc. • When submitting multiple TOOs:
– Ensure accuracy of information submitted to reduce processing delays
– Please include a list of all ANDAs being transferred on each of the cover letters
• List the ANDA # and the established name – Ex. Atenolol Tablets USP, 25 mg, 50 mg, and 100 mg
• Contact RPM for all ANDA-specific questions

45
ANDA Consolidation
Kathleen Melendez, Pharmacist Primary Filing Reviewer, ORO/DFR Center for Drug Evaluation and Research Office of Generic Drugs U.S. Food and Drug Administration

46
Information needed to process the request
• Submit a separate copy of the request for consolidation to each ANDA applicable.
• Request submitted to the parent ANDA should be identified as a “Correspondence.”
• Request submitted to the child ANDAs should each be identified as “Request for Consolidation.”

47
Identify all formulations and ANDAs • Indicate that all inactive ingredients are
proportional for all formulations identified in the request by including a chart of all ingredients, quantity of the ingredient per dosage form, and the weight to weight percentage.

48
Submit a chart like this in the Cover Letter for each ANDA submitted.

49
Provide BE documentation • Provide documentation that BE studies were
performed. • Provide documentation of any BE waivers
received.

50
Denial of the Request The most common reasons for holds or denials: • Pending supplements such as an open issue or
pending review. • Consolidation requests should not be submitted
as a CBE-30. They should be submitted as a Correspondence/Request for Consolidation.

51
Approval • If all the information has been provided, and
there are no open issues or pending reviews associated with the ANDAs, then the request for consolidation will be approved.

52
• FDA MAPP 5241.2 describes FDA’s process for reviewing and approving or denying requests to consolidate previously approved ANDAs: – http://www.fda.gov/aboutfda/centersoffices/officeofme
dicalproductsandtobacco/cder/manualofpoliciesprocedures/default.htm
• MAPP stands for Manual of Policies and Procedures.
• MAPPs describe internal policies and procedures and are made available to the public to increase transparency. www.fda.gov
For more information

Endorsement/Clearance Phase
Lakeeta Carr, MSN/MHA, BSN, RN, NE-BC Commander, United States Public Health Service
Regulatory Project Manager

54
Introduction
Endorsement/Clearance Phase
Divisions/Personnel Involved
Communication from FDA to You

55
Endorsement/Clearance Phase
What does endorsement/clearance phase mean?
www.fda.gov
56
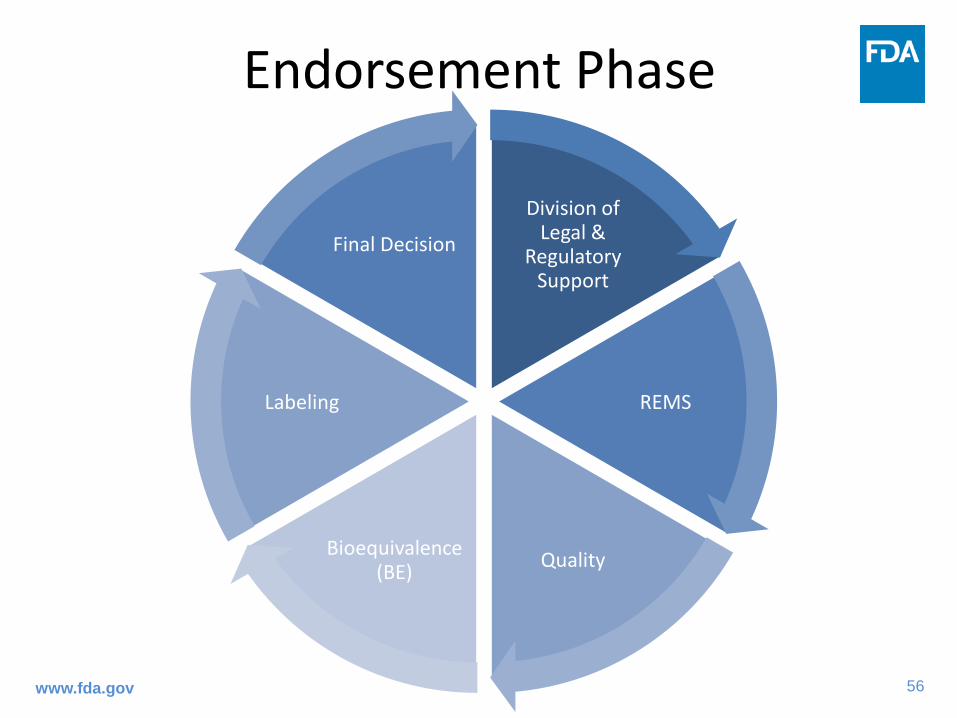
Endorsement Phase
Division of Legal &
Regulatory Support
REMS
Quality Bioequivalence (BE)
Labeling
Final Decision
www.fda.gov

57
Division of Legal & Regulatory Support
Addressed all listed patents & exclusivities
Required documentation related to patents &
exclusivities have been provided
Consistent application of Hatch Waxman policy &
precedents across applicants
All grants of 180 day exclusivity consistently applied/compliant with
“Forfeiture Provisions” of 505(j)(5)(D)(I)-(VI) of FD&C
Act
Certifications/statements to newly listed patents
All information required by 314.95(e)-notice
requirements and 314.107(e)-court actions
Changes to certifications and impact on labeling/timing of
approval

58
OGD Discipline Final Reviews
DMF & Facilities Statuses
RLD Labeling Updates
Updated BE
Guidance
REMS Quality
BE Labeling
* Applicant expected to keep all information updated
59
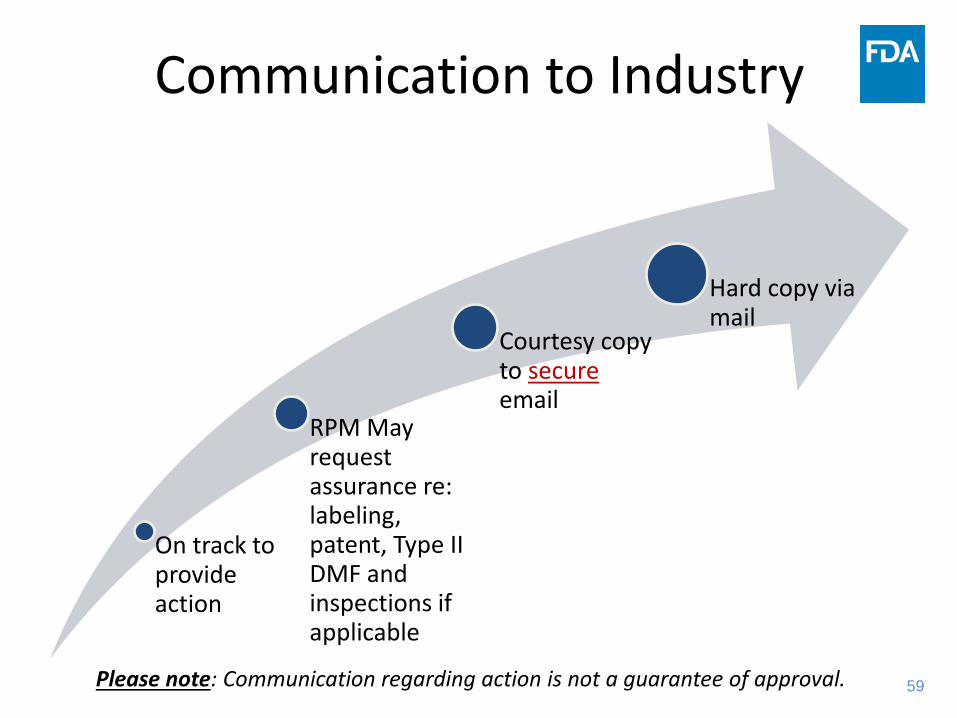
Communication to Industry
On track to provide action
RPM May request assurance re: labeling, patent, Type II DMF and inspections if applicable
Courtesy copy to secure email
Hard copy via mail
Please note: Communication regarding action is not a guarantee of approval.

60
Conclusion
Endorsement/Clearance Phase
Divisions/Personnel Involved
Communication from FDA to You

61
DPM: Things that Make You Go Hmmm
Voluntary Withdrawal of Approved and
Unapproved ANDAs by Applicants
Presented by: Wilbert (Tre’) Ferguson III, PharmD
Regulatory Project Manager Food & Drug Administration
Office Of Generic Drugs

62
So you’ve decided to withdraw your unapproved ANDA..what now?
www.fda.gov

63
What does the Code of Federal Regulations have to say?
21 CFR 314.65 to be precise
• An applicant may at any time withdraw an unapproved application by notifying the FDA in writing.
• You may refile without prejudice. • The agency will retain the application and will
provide a copy on request under the fee schedule in 21 CFR 20.45.

64
Withdrawal of an unapproved ANDA • The application is considered withdrawn on the
date of the request. • Upon receipt of the request to withdraw an
unapproved ANDA from an applicant a withdrawal acknowledgment letter will be issued to the applicant.
• All pending reviews will be discontinued.
www.fda.gov

65
But what about approved ANDAs?

66
Back to the Code of Federal Regulations this time its 21 CFR 314.150(c)
1. FDA will withdraw approval of an application if the
application is being withdrawn because the product is no longer marketed, provided none of the conditions in 314.150 (a) and (b) apply to the drug.
2. If there are safety or efficacy issues please contact the regulatory project manager for details. If there are safety or efficacy issues, 21 CFR 314.150(c) does not apply!
www.fda.gov

67
Back to the Code of Federal Regulations this time its 21 CFR 314.150(c) – Cont.
3. FDA will consider a written request for a withdrawal to be a waiver of an opportunity for hearing otherwise provided for in this section.
4. The applicant may refile without prejudice.

68
What else do we need to know about withdrawing approval under 21 CFR
314.150(c)?
1. The withdrawal request should specify the products are no longer marketed and request that FDA withdraw approval of the application, including any supplements.
2. If the request does not include the withdrawal of any pending supplements, the RPM will call the applicant to confirm. If the applicant wishes to withdraw pending supplements, the applicant will need to submit a revised request to include them.
www.fda.gov

69
Withdrawal of an Approved ANDA Under 21 CFR 314.150(c) - cont. 2
3. Once a complete request is received, a withdrawal acknowledgment letter will be issued to the applicant. Based on the applicant’s complete request, the withdrawal acknowledgement may include all pending submissions.
4. FDA will publish a notice in the Federal Register (FR) announcing the withdrawal of the approval (usually published with several other approvals of applications being withdrawn).

70
Withdrawal of an Approved ANDA Under 21 CFR 314.150(c) - cont. 3
5. The FR notice will note the effective date of withdrawal, usually 30 days after the date of publication of the notice.
6. Until the effective date of the FR notice the application is not officially withdrawn and the applicant must still comply with all regulatory requirements including those set forth for an approved ANDA in 21 CFR 314.80 and 314.81.

71
Withdrawal of an Approved ANDA Things to consider
• If the applicant wants to rescind the request to withdraw the approved ANDA it must be done prior to the effective date of the withdrawal published in the Federal Register.
• It would be helpful if the applicant lists all pending supplements that they wish to withdraw in the cover letter including the supplement number and submission date.

72
That’s all Folks!!


















