
Butylated Hydroxy Anisole Chemical by Sahkar Pharmaceuticals Private Limited Ahmedabad

Disruption of the Ugt1 Locus in Mice Resembles HumanCrigler-Najjar Type I Disease*
Received for publication, November 9, 2007, and in revised form, December 21, 2007 Published, JBC Papers in Press, January 7, 2008, DOI 10.1074/jbc.M709244200
Nghia Nguyen‡, Jessica A. Bonzo‡, Shujuan Chen‡, Sarah Chouinard§, Michael J. Kelner¶, Gary Hardiman�,Alain Belanger§, and Robert H. Tukey‡1
From the ‡Laboratory of Environmental Toxicology, Departments of Chemistry & Biochemistry and Pharmacology, University ofCalifornia, San Diego, La Jolla, California 92093, the ¶Department of Pathology, University of California, San Diego,La Jolla, California 92093, the �Biomedical Genomics Microarray Facility, Department of Medicine, School of Medicine,University of California, San Diego, La Jolla, California 92093, and the §Oncology and Molecular Endocrinology Research Center,Centre Hospitalier Universitare de Quebec, Quebec G1V 4G2, Canada
The 9 UDP-glucuronosyltranferases (UGTs) encoded by theUGT1 locus in humans are key enzymes in the metabolism ofmost drugs as well as endogenous substances such as bile acids,fatty acids, steroids, hormones, neurotransmitters, and biliru-bin. Severe unconjugated hyperbilirubinemia in humans thatsuffer from Crigler-Najjar type I disease results from lesions inthe UGT1A1 gene and is often fatal. To examine the physiolog-ical importance of the Ugt1 locus in mice, this locus was ren-dered non-functional by interrupting exon 4 to create Ugt1�/�
mice. BecauseUGT1A1 inhumans is responsible for 100%of theconjugated bilirubin, it followed that newborn Ugt1�/� micedeveloped serum levels of unconjugated bilirubin that were40–60 times higher thanUgt1�/� or wild-type mice. The resultof extremeunconjugated bilirubin inUgt1�/�mice, comparableto the induced levels noted in patients withCrigler-Najjar type 1disease, is fatal in neonatalUgt1�/� mice within 2 weeks follow-ing birth. The extreme jaundice is present as a phenotype in skincolor after 8 h. Neonatal Ugt1�/� mice exhibit no detectableUGT1A-specific RNA, which corresponds to a completeabsence ofUGT1Aproteins in livermicrosomes.Conserved glu-curonidation activity attributed to theUgt1 locus can be definedin Ugt1�/� mice, because UGT2-dependent glucuronidationactivity is unaffected. Remarkably, the loss of UGT1A function-ality in liver results in significant alterations in cellular metabo-lism as investigated through changes in gene expression. Thus,the loss ofUGT1A function inUgt1�/�mice leads to ametabolicsyndrome that can serve as a model to further investigate thetoxicities associatedwithunconjugatedbilirubin and the impactof this disease in humans.
Jaundice is frequently observed and stems from a variety ofhepatic and non-hepatic conditions such as infection, biliary
obstruction, hemolysis, liver disease, and genetic diseases thatinfluence hepatic metabolism and transport. It is symptomaticclinically by the accumulation of indirect or unconjugated bili-rubin (UCB),2 which accumulates resulting in hyperbiliru-binemia. The most benign genetic disease leading to hyperbil-irubinemia is Gilbert syndrome, which is a common inheritablecondition resulting in transient levels of UCB (1). In humans,bilirubin is conjugated with glucuronic acid, which is catalyzedsolely by UDP-glucuronosyltransferase (UGT) 1A1 (2). In Gil-bert syndrome, hyperbilirubinemia is tied to a TA insertion inthe promoter region of the UGT1A1 gene (3, 4), characterizedgenetically as UGT1A1*28, and is felt to lead to reduced tran-scriptional activation of the gene and lowering of hepaticUGT1A1 levels.More detailed analysis of genetic variants asso-ciated with UGT1A1*28 indicate that Gilbert syndrome is partof a haplotype of four UGT1A variants spanning at least threeUGT1A genes (5). Themore serious of the genetic defects asso-ciated withUGT1A1 is Crigler-Najjar type 1 (CN1) disease (6),an autosomal recessive syndrome where mutations in the cod-ing region render the protein completely non-functional. Theaccumulation of UCB within focal brain regions leads to neuraldysfunction followed by cell death and permanent disability (7,8). If untreated by liver transplantation (9, 10), the completeinactivation ofUGT1A1 in humans is fatal (11), and lethality isdirectly attributed to toxic concentrations of UCB.The UGT1A1 gene is part of the UGT1 locus (12), which
encodes nine functional UGT1Aproteins (13), together playinga crucial role in the metabolism of drugs, xenobiotics, environ-mental toxicants, aswell as endogenous substances such as fattyacids, bile acids, hormones, steroids, neurotransmitters, andbilirubin (14). The organization of theUGT1 locus on chromo-some 2 is encoded by �220 kDa of DNA, with four conservedexons located at the 3�-end and flanked consecutively by anarray of nine cassette exons that encode theN-terminal portionof each UGT1A protein. Thus, the UGT1 locus encodes ninefunctional UGT1A proteins, each composed of a unique N-ter-minal region encoded by one of the 5�-flanking exons while
* This work was supported in part by United States Public Health ServiceGrant ES10337 and by Grant 15RT-0251 from the Tobacco Related DiseaseResearch Program from the State of California. The costs of publication ofthis article were defrayed in part by the payment of page charges. Thisarticle must therefore be hereby marked “advertisement” in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
1 To whom correspondence should be addressed: Laboratory of Environmen-tal Toxicology, Departments of Chemistry & Biochemistry and Pharmacol-ogy, University of California, 9500 Gilman Dr., Leichtag BiomedicalResearch Bldg., La Jolla, CA 92093-0722. Tel.: 858-822-0288; Fax: 858-822-0363; E-mail: [email protected].
2 The abbreviations used are: UCB, unconjugated bilirubin; UGT, UDP-glucu-ronosyltransferase; CN1, Crigler-Najjar type 1 disease; 4-MU, 4-methylum-billeferone; MPA, mycophenolic acid; Tg-UGT1, transgenic UGT1; Bis-Tris,2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol; T3,triiodothyroxine; T4, thyroxine; CYP7A1, cholesterol 7�-hydroxylase;WT, wild type.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 283, NO. 12, pp. 7901–7911, March 21, 2008© 2008 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
MARCH 21, 2008 • VOLUME 283 • NUMBER 12 JOURNAL OF BIOLOGICAL CHEMISTRY 7901
by guest on August 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from

sharing an identical C-terminal 245 amino acids encoded byexons 2–5. EachUGT1A gene is regulated independently fromeach other based upon tissue-specific control (15–17), which isconsidered to be the biochemical basis of organ-specific glucu-ronidation activity. Development of transgenic mice thatexpress the entire human UGT1 locus (18) confirms that thehumanUGT1A genes can be regulated in a tissue-specific fash-ion similar to that observed in human tissues. In addition, thehumanUGT1A genes are regulated by the xenobiotic receptorspregnane X receptor (18), the peroxisome proliferator-acti-vated receptor� (19), the liver X receptor (20), as well as the Ahreceptor (18). Each of these receptors, including the activationof Nrf2 by an antioxidant response (21), target the UGT1A1gene for regulation and potentially contribute to the steady-state balance of important endogenous substrates for glucu-ronidation such as bilirubin.Although jaundice and hyperbilirubinemia can lead to drug
toxicities associated with the impairment of functionalUGT1A1 (22) as well as encephalopathy or irreversible braindamage (23), animal models that resemble CN1 deficiencieshave not been used to examine the biochemical and physiolog-ical conditions associated with defects in either the murineUgt1a1 gene or the Ugt1 locus. Although the Gunn rat modelexhibits a genetic defect in theUgt1 locus (24, 25) and developshyperbilirubinemia (26, 27), the animals are healthy based upontheir ability to reproduce. To assess the functional role of theUgt1 locus inmice, we have created amousemodel inwhich thecoding region of exon 4 has been interrupted. Results are pre-sented demonstrating complete interruption of the Ugt1 locusin Ugt1�/� mice leading to levels of UCB that equate to com-parable increases observed in patients with CN1 disease. Theabsence of functional UGT1A protein predisposes neonatalmice to fatal consequences associated with non-hemolyticunconjugated hyperbilirubinemia.
EXPERIMENTAL PROCEDURES
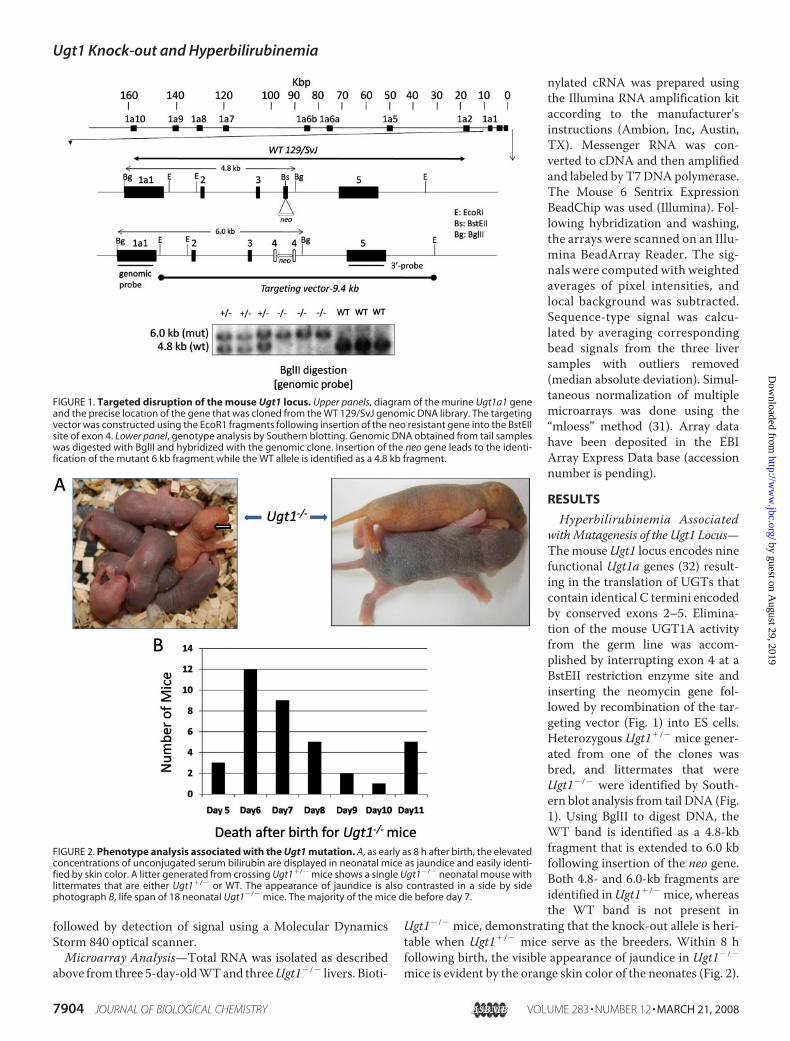
Generation of the Ugt1�/� Mice—A 14-kb Ugt1a1�-genomic DNA clone from a 129/SvJ genomic DNA library(Stratagene, 946313) was isolated by screening the library witha cDNA fragment to exons 2–5 of the mouse Ugt1a6 cDNA(28). This Ugt1a1-� DNA fragment contained exon 1 throughexon 5 with additional 3� intronic sequence. From this � DNA,we cloned two EcoRI fragments (see Fig. 1) that spanned 9.4 kbinto pBluescript II KS (Stratagene) and generated the targetingvector by interrupting amino acids Thr-413/Leu-414 of exon 4(at the BstEII site) with the G418 (neo)-resistant gene. The5�-arm was 3.2 kb, and the 3�-arm extended 6.2 kb. Followingelectroporation of the targeting construct into 129J ES cells andselection of clones with G418, clones were analyzed by South-ern blotting after BglII digestion using the 5� probe as indicatedin Fig. 1. �3% of the clones were positive for homologousrecombination. Four ES clones were injected into C57BL/6blastocysts. Male chimaeras were backcrossed to C57BL/6females to generate heterozygous founders that were bred toproduce Ugt1�/� mice. For genotype analysis, tail DNA wasdigested with BglII and determined by Southern blotting. Micewere maintained under standard 12-h light/12-h dark cyclewith access to standard rodent food and givenwater ad libitum.
All protocols for mouse experiments were approved by theInstitutional Animal Care andUseCommittee at theUniversityof California San Diego.Blood Chemistry Analysis—Blood was taken from 5-day-old
neonatal mice by decapitation and allowed to clot for 20–30min in the dark. Following centrifugation at 1000 � g for 10 min,serum was collected under minimal light to reduce bilirubindecomposition. Blood chemistries were carried out for bilirubin(total anddirect), alanineaminotransferase, aspartate aminotrans-ferase, �-glutamyltranspeptidase, albumin, total protein, lipase,lactatedehydrogenase, cholesterol, iron, alkalinephosphatase, andamylase at theUCSanDiegoMedicalDiagnosticLaboratoryusingaBeckmanLX-20analyzerwithT4,T3, andTSH levels performedwith a Bayer Centaur analyzer.Protein Preparation and Immunoblot Analysis—AllWestern
blots were prepared using 4–12% NU-PAGE Bis-Tris polyac-rylamide gels (Invitrogen). Livermicrosomal preparationsweregenerated as previously described (18) with 10 �g of proteinloaded into each well. Following electrophoresis at 200 V, theresolved proteins were transferred onto nitrocellulose mem-brane (Immobilon-Millipore), and the membrane was blockedwith 5% nonfat drymilk in Tris-buffered saline solution (10mMTris, pH 8.0, 150 mM NaCl, 0.05% Tween 20) for several hours.The membranes were then incubated with primary humanUGT1A (29) or UGT2B7 (30) antibodies overnight at 4 °C, fol-lowed by incubation with horseradish peroxidase-conjugatedsecondary antibody. Each membrane was washed again, and theconjugated horseradish peroxidase was detected using the ECLplusWestern blotting detection system (Amersham Biosciences),and the proteins were detected using a MolecularImager�ChemiDocTM XRS system (Bio-Rad).UDP-glucuronosyltransferase Activity Determination—Liver
microsomes were prepared from 5-day-old neonatal mice.Liver from three animals of each genetic stain (WT,Ugt1�/�, orUgt1�/�) were combined and homogenized at 4 °C in 0.5 ml ofphosphate-buffered saline (pH7.4) using amotorized glass Tef-lon homogenizer. The tissue homogenate was completed at 4ml with microsome buffer (2.62 mM KH2PO4, 1.38 mMK2H1PO4, 2% glycerol, and 0.5 mM dithiothreitol) and was firstcentrifuged at 12,000 � g for 20 min at 4 °C, and this resultingsupernatant was centrifuged at 105,000 � g for 60 min at 4 °C.The pellet was suspended inmicrosome buffer, and the proteinconcentrationwas determined by the Bradfordmethod. Glucu-ronidation assays were performed for 1 h in triplicate with 50mM Tris-HCl (pH 7.5), 10 mM MgCl2, 8.5 mM saccharolactone(except for serotonin, MPA, imipramine, bilirubin, and T4), 10�g/ml phosphatidylcholine (except for T4), 1 mM UDP-glucu-ronic acid (Sigma), 2.5 �g/ml pepstatin (except for T4), 0.5�g/ml leupeptin (except forMPAandT4), 10�g ofmicrosomalprotein, and 10 �M of the substrate (bilirubin, testosterone,2-OH-estrone, 4-nitrophenol, 4-methylumbilleferone, and1-naphthol) and 200 �M of the substrate (serotonin, MPA, imi-pramine and T4) in a total volume of 100 �l. Assays werestopped with 100 �l of methanol (1-naphthol, 4-nitrophenol,4-MU, testosterone, 2-OH-estrone, and bilirubin (with 0.02%butylated hydroxytoluene)) or methanol with 0.02 M HCl(MPA) or acetonitrile with 0.02% butylated hydroxytoluene(serotonin) or acetonitrile with 0.2% formic acid (imipramine)
Ugt1 Knock-out and Hyperbilirubinemia
7902 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 12 • MARCH 21, 2008
by guest on August 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from

or acetonitrile with 6% formic acid and 0.02% butylatedhydroxytoluene (T4) prior to centrifugation at 10,000� g for 10min at 4 °C, and supernatants were collected for high-perfor-mance liquid chromatography analyses.Mass Spectrometry Analysis of Enzymatic Assays—Each sam-
ple (50 �l) was diluted 1:1 with water, vortexed, and then trans-ferred into a conical vial for injection into the mass spectrom-eter. The high-performance liquid chromatography systemconsisted of a mass spectrometer (model API 3200,PerkinElmer Life Sciences/MDS Sciex, Thornhill, Ontario,Canada) and was used to detect glucuronide conjugatesformed. It was operated in the multiple reactions monitoringmode and equippedwith an electrospray ionization interface innegative or positive ion mode and a high-performance liquidchromatography pump plus autosampler model 1200 series(Agilent, Montreal, Canada). For testosterone-glucuronideanalysis, chromatographic separation was achieved with a 10mm� 4.6mm, 4-�mparticle size SynergyHydro-RP column ata flow rate of 1.0 ml/min (Phenomenex, Torrance, CA). Themobile phaseAwaswater plus 1mMammonium formate, Bwasmethanol plus 1 mM ammonium formate, and C was tetrahy-drofuran. Separation was achieved using an isocratic conditionof 48% A, 48% B, and 4% C in 7min at a flow rate of 0.9 ml/min.Afterward, the columnwas flushed 1minwith 4%A, 92%B, and4% C and re-equilibrated to initial conditions over 3 min. For2-hydroxyestrone-glucuronides, samples were analyzed with a4- to 6-mm inner diameter � 50-mm Luna Phenyl-Hexyl col-umn (3-�m particle size, Phenomenex) using the mobile phase(solvent A, water; solvent B, methanol plus 0.1% ammoniumhydroxide; and C, methanol). Separation was achieved using alinear gradient of 85% A, 5% B, and 10% B to 70% A, 5% B, and20% C in 2 min at a flow rate of 0.9 ml/min. After 2 min, thecolumnwaswashedwith 5%A, 5%B, and 90%C.Afterward, thecolumnwas re-equilibrated to initial conditions over 3min. Forimipramine-glucuronide, samples were analyzed with a3.9-mm inner diameter � 100-mm Xterra MS C18 (3.5-�mparticle size) using the mobile phase solvent A, water plus 0.1%formic acid; solvent B, acetonitrile. Separation was achievedusing a linear gradient of 30% to 50% B in 1min at a flow rate of0.9 ml/min. After 1 min, the column was washed with 90% B.Afterward, the column was re-equilibrated to initial conditionsover 3 min. For 1-naphthol-glucuronide, 4-nitrophenolglucu-ronide, and 4-MU-glucuronide, samples were analyzed with a3-mm inner diameter � 50-mm Synergie Polar column(2.5-�mparticle size, Phenomenex) using themobile phase sol-vent A, water plus 0.02% formic acid; solvent B, methanol plus0.02% formic acid. Separation was achieved using an isocraticseparation of 50% A and 50% B (3.5 min). For serotonin-glucu-ronide, 60% A and 40% B (2 min) was used with the same chro-matographic system. For MPA-glucuronide and MPA acylglu-curonide, samples were analyzed with a 4.6-mm innerdiameter � 100-mm Gemini C18 column (5-�m particle size,Phenomenex). The mobile phases were solvent A, water plus 3mM ammonium formate plus 0.5% acetic acid and B methanolplus 3mMammonium formate plus 0.5% acetic acid. Separationwas achieved using a linear gradient of 65% to 85% B in 3min ata flow rate of 0.9 ml/min. After 3 min, the column was washedwith 85%B.Afterward, the columnwas re-equilibrated to initial
conditions over 3 min. For bilirubin monoglucuronide anddiglucuronide, samples were analyzed with a 4.6-mm innerdiameter � 100-mm Luna C18 column (3-�m particle size,Phenomenex). Themobile phaseAwaswater plus 1mMammo-nium formate; B was methanol plus 1 mM ammonium formate.Separationwas achieved using a linear gradient of 70%B to 95%B in 2.5 min at a flow rate of 0.9 ml/min. After 0.5 min, thecolumnwas re-equilibrated to initial conditions over 3min. Forthyroxine (T4)-glucuronide, samples were analyzed with a4.6-mm inner diameter� 100-mmSynergie RP-Hydro column(4-�m particle size, Phenomenex). The mobile phase A waswater plus 0.1% formic acid, and B was acetonitrile plus 0.1%formic acid. Separation was achieved using an isocratic separa-tion of 70% A and 30% B (3.5 min).Detection of Ugt1a RNAs by Reverse Transcription-PCR and
Northern Blot—Total RNA for Northern blot and reverse tran-scription-PCR was prepared using the RNeasy RNA purifica-tion kit from Qiagen. For reverse transcription-PCR analysis, 2�g of total RNAwas used for generation of cDNA following theOmniscript RT kit protocol (Qiagen) with oligo(dT) primers ina 20-�l reaction. Following cDNA synthesis, 2�l of the reactionwas used for PCR amplification using 2 units of ChoiceTaqDNA polymerase (Denville Scientific). For detection ofmouse UGT1A1 RNA, the forward primer was designed to ahighly specific region of exon 1a1 (5�-GGTGGAACTTG-GACGGACTG-3�). For detection of the common UGT1Atranscripts the forward primer was designed toward exon 2(5�-GCCTATGTCAACGCCTCTGG-3�). The reverse primerannealing in exon 5 (5�-GGCGCATGATGTTCTCCTTG-3�)was used for both UGT1A1 and UGT1A amplification. Fordetection of UGT1A6, the forward primer was designed toanneal to an identical region in exons 1a6a and 1a6b (5�-CAG-GATGGCTTGCCTTCTTCC-3�). The reverse primer forUGT1A6 was directed toward exon 5 (5�-TGTTCTTGGCAA-GATTCGATG-3�). Expression of actin was used as a loadingcontrol. For detection of UGT1A1, UGT1A, and actin RNAexpression, the polymerase was activated for 2 min at 95 °Cfollowed by 23 cycles of 95 °C 30 s, 58 °C 30 s, 72 °C 45 s, and afinal 72 °C extension for 7 min. The PCR for detection ofUGT1A6 RNA expression was performed under the same con-ditions except the annealing temperature was 55 °C. Eightmicroliters of the PCR product was resolved on a 1.5% agarosegel and imaged using a Bio-Rad ChemiDoc XRS.ForNorthern blot analysis, 10�g of RNA from each liver was
separated in 1% formaldehyde-agarose gels with the resolvedRNA transferred to GeneScreen membrane (PerkinElmer LifeSciences) by capillary diffusion. The membrane was pre-hy-bridized at 42 °C for several hours in a solution containing 5 �SSC, 5 � Denhardt’s solution, 1% SDS, 100 �g/ml single-stranded DNA, and 50% formamide. A 1.2-kb DNA probe thatspans a portion of exon 5 was amplified from the targetingvector by PCR (5�-AAAGGAACACCTCCAGAGACC-3� and5�-GCTACAAGGAGAACATCATGCG-3�), purified, andlabeled using [�-32P]ATP and a Rediprime II Random PrimeLabeling System (Amersham Biosciences), followed by hybrid-ization to the membrane overnight at 42 °C. The membranewas then washed with 0.5% SDS and 2� SSC several times andexposed to phosphorimaging screens (Molecular Dynamics)
Ugt1 Knock-out and Hyperbilirubinemia
MARCH 21, 2008 • VOLUME 283 • NUMBER 12 JOURNAL OF BIOLOGICAL CHEMISTRY 7903
by guest on August 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from
followed by detection of signal using a Molecular DynamicsStorm 840 optical scanner.Microarray Analysis—Total RNA was isolated as described
above from three 5-day-oldWTand threeUgt1�/� livers. Bioti-
nylated cRNA was prepared usingthe Illumina RNA amplification kitaccording to the manufacturer’sinstructions (Ambion, Inc, Austin,TX). Messenger RNA was con-verted to cDNA and then amplifiedand labeled by T7DNApolymerase.The Mouse 6 Sentrix ExpressionBeadChip was used (Illumina). Fol-lowing hybridization and washing,the arrays were scanned on an Illu-mina BeadArray Reader. The sig-nals were computed with weightedaverages of pixel intensities, andlocal background was subtracted.Sequence-type signal was calcu-lated by averaging correspondingbead signals from the three liversamples with outliers removed(median absolute deviation). Simul-taneous normalization of multiplemicroarrays was done using the“mloess” method (31). Array datahave been deposited in the EBIArray Express Data base (accessionnumber is pending).
RESULTS
Hyperbilirubinemia AssociatedwithMutagenesis of the Ugt1 Locus—ThemouseUgt1 locus encodes ninefunctional Ugt1a genes (32) result-ing in the translation of UGTs thatcontain identical C termini encodedby conserved exons 2–5. Elimina-tion of the mouse UGT1A activityfrom the germ line was accom-plished by interrupting exon 4 at aBstEII restriction enzyme site andinserting the neomycin gene fol-lowed by recombination of the tar-geting vector (Fig. 1) into ES cells.Heterozygous Ugt1�/� mice gener-ated from one of the clones wasbred, and littermates that wereUgt1�/� were identified by South-ern blot analysis from tail DNA (Fig.1). Using BglII to digest DNA, theWT band is identified as a 4.8-kbfragment that is extended to 6.0 kbfollowing insertion of the neo gene.Both 4.8- and 6.0-kb fragments areidentified inUgt1�/� mice, whereasthe WT band is not present in
Ugt1�/� mice, demonstrating that the knock-out allele is heri-table when Ugt1�/� mice serve as the breeders. Within 8 hfollowing birth, the visible appearance of jaundice in Ugt1�/�
mice is evident by the orange skin color of the neonates (Fig. 2).
FIGURE 1. Targeted disruption of the mouse Ugt1 locus. Upper panels, diagram of the murine Ugt1a1 geneand the precise location of the gene that was cloned from the WT 129/SvJ genomic DNA library. The targetingvector was constructed using the EcoR1 fragments following insertion of the neo resistant gene into the BstEIIsite of exon 4. Lower panel, genotype analysis by Southern blotting. Genomic DNA obtained from tail sampleswas digested with BglII and hybridized with the genomic clone. Insertion of the neo gene leads to the identi-fication of the mutant 6 kb fragment while the WT allele is identified as a 4.8 kb fragment.
FIGURE 2. Phenotype analysis associated with the Ugt1 mutation. A, as early as 8 h after birth, the elevatedconcentrations of unconjugated serum bilirubin are displayed in neonatal mice as jaundice and easily identi-fied by skin color. A litter generated from crossing Ugt1�/� mice shows a single Ugt1�/� neonatal mouse withlittermates that are either Ugt1�/� or WT. The appearance of jaundice is also contrasted in a side by sidephotograph B, life span of 18 neonatal Ugt1�/� mice. The majority of the mice die before day 7.
Ugt1 Knock-out and Hyperbilirubinemia
7904 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 12 • MARCH 21, 2008
by guest on August 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from
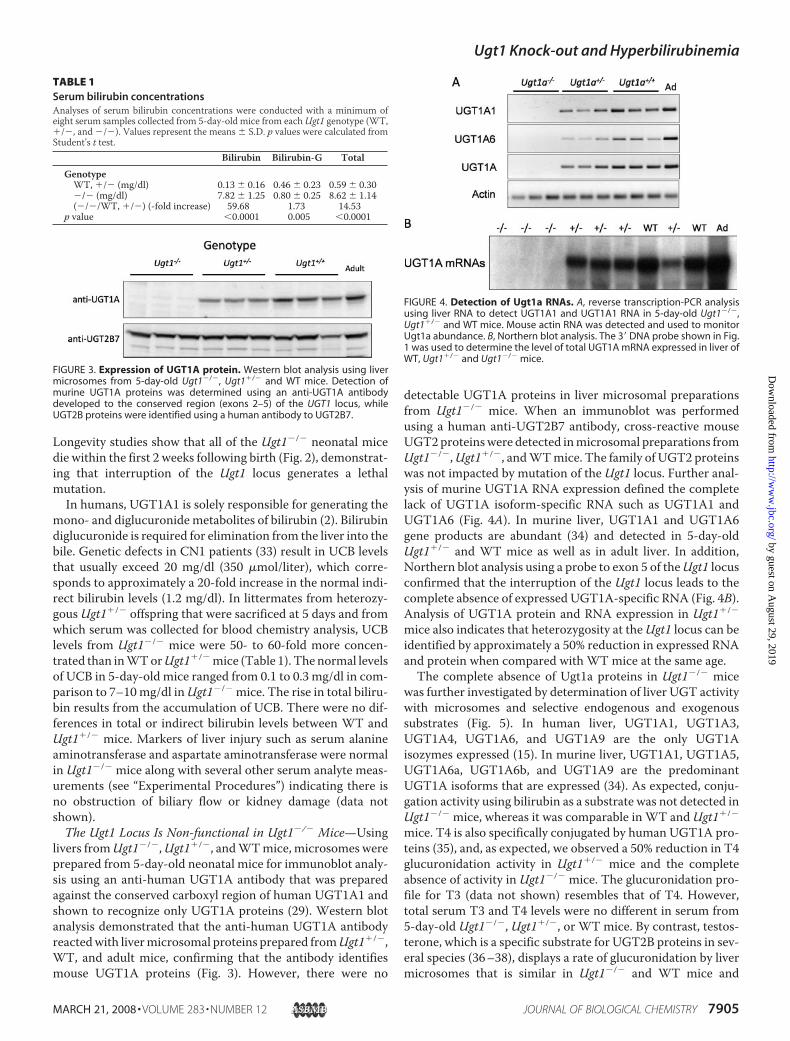
Longevity studies show that all of the Ugt1�/� neonatal micedie within the first 2 weeks following birth (Fig. 2), demonstrat-ing that interruption of the Ugt1 locus generates a lethalmutation.In humans, UGT1A1 is solely responsible for generating the
mono- and diglucuronidemetabolites of bilirubin (2). Bilirubindiglucuronide is required for elimination from the liver into thebile. Genetic defects in CN1 patients (33) result in UCB levelsthat usually exceed 20 mg/dl (350 �mol/liter), which corre-sponds to approximately a 20-fold increase in the normal indi-rect bilirubin levels (1.2 mg/dl). In littermates from heterozy-gous Ugt1�/� offspring that were sacrificed at 5 days and fromwhich serum was collected for blood chemistry analysis, UCBlevels from Ugt1�/� mice were 50- to 60-fold more concen-trated than inWTorUgt1�/�mice (Table 1). The normal levelsof UCB in 5-day-old mice ranged from 0.1 to 0.3 mg/dl in com-parison to 7–10mg/dl inUgt1�/� mice. The rise in total biliru-bin results from the accumulation of UCB. There were no dif-ferences in total or indirect bilirubin levels between WT andUgt1�/� mice. Markers of liver injury such as serum alanineaminotransferase and aspartate aminotransferase were normalin Ugt1�/� mice along with several other serum analyte meas-urements (see “Experimental Procedures”) indicating there isno obstruction of biliary flow or kidney damage (data notshown).The Ugt1 Locus Is Non-functional in Ugt1�/� Mice—Using
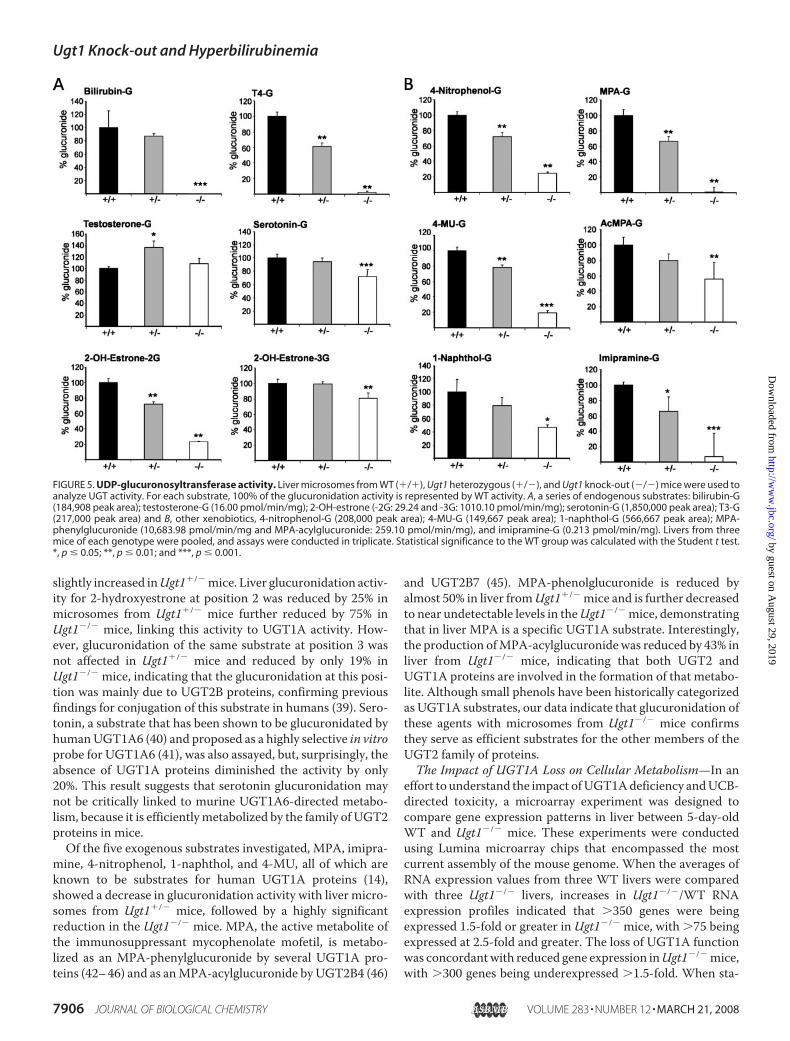
livers fromUgt1�/�,Ugt1�/�, andWTmice, microsomes wereprepared from 5-day-old neonatal mice for immunoblot analy-sis using an anti-human UGT1A antibody that was preparedagainst the conserved carboxyl region of human UGT1A1 andshown to recognize only UGT1A proteins (29). Western blotanalysis demonstrated that the anti-human UGT1A antibodyreactedwith livermicrosomal proteins prepared fromUgt1�/�,WT, and adult mice, confirming that the antibody identifiesmouse UGT1A proteins (Fig. 3). However, there were no
detectable UGT1A proteins in liver microsomal preparationsfrom Ugt1�/� mice. When an immunoblot was performedusing a human anti-UGT2B7 antibody, cross-reactive mouseUGT2proteinswere detected inmicrosomal preparations fromUgt1�/�,Ugt1�/�, andWTmice. The family of UGT2 proteinswas not impacted by mutation of the Ugt1 locus. Further anal-ysis of murine UGT1A RNA expression defined the completelack of UGT1A isoform-specific RNA such as UGT1A1 andUGT1A6 (Fig. 4A). In murine liver, UGT1A1 and UGT1A6gene products are abundant (34) and detected in 5-day-oldUgt1�/� and WT mice as well as in adult liver. In addition,Northern blot analysis using a probe to exon 5 of theUgt1 locusconfirmed that the interruption of the Ugt1 locus leads to thecomplete absence of expressed UGT1A-specific RNA (Fig. 4B).Analysis of UGT1A protein and RNA expression in Ugt1�/�
mice also indicates that heterozygosity at theUgt1 locus can beidentified by approximately a 50% reduction in expressed RNAand protein when compared with WT mice at the same age.The complete absence of Ugt1a proteins in Ugt1�/� mice
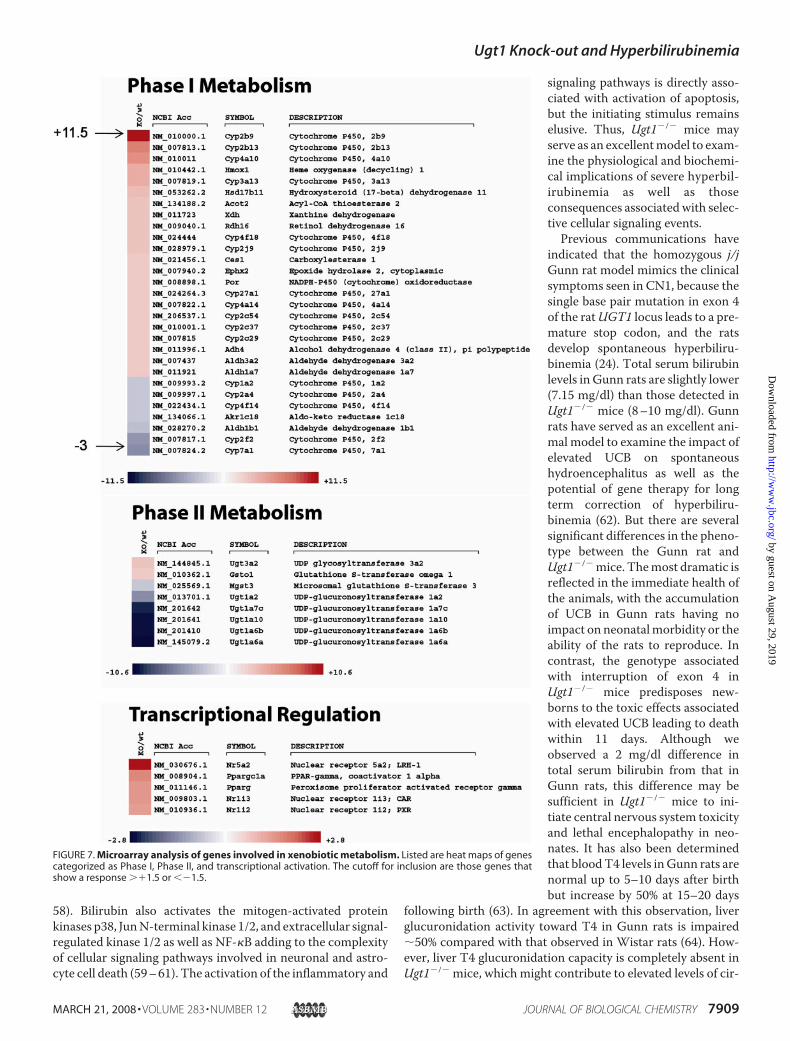
was further investigated by determination of liver UGT activitywith microsomes and selective endogenous and exogenoussubstrates (Fig. 5). In human liver, UGT1A1, UGT1A3,UGT1A4, UGT1A6, and UGT1A9 are the only UGT1Aisozymes expressed (15). In murine liver, UGT1A1, UGT1A5,UGT1A6a, UGT1A6b, and UGT1A9 are the predominantUGT1A isoforms that are expressed (34). As expected, conju-gation activity using bilirubin as a substrate was not detected inUgt1�/� mice, whereas it was comparable in WT andUgt1�/�
mice. T4 is also specifically conjugated by human UGT1A pro-teins (35), and, as expected, we observed a 50% reduction in T4glucuronidation activity in Ugt1�/� mice and the completeabsence of activity in Ugt1�/� mice. The glucuronidation pro-file for T3 (data not shown) resembles that of T4. However,total serum T3 and T4 levels were no different in serum from5-day-old Ugt1�/�, Ugt1�/�, or WT mice. By contrast, testos-terone, which is a specific substrate for UGT2B proteins in sev-eral species (36–38), displays a rate of glucuronidation by livermicrosomes that is similar in Ugt1�/� and WT mice and
FIGURE 3. Expression of UGT1A protein. Western blot analysis using livermicrosomes from 5-day-old Ugt1�/�, Ugt1�/� and WT mice. Detection ofmurine UGT1A proteins was determined using an anti-UGT1A antibodydeveloped to the conserved region (exons 2–5) of the UGT1 locus, whileUGT2B proteins were identified using a human antibody to UGT2B7.
FIGURE 4. Detection of Ugt1a RNAs. A, reverse transcription-PCR analysisusing liver RNA to detect UGT1A1 and UGT1A1 RNA in 5-day-old Ugt1�/�,Ugt1�/� and WT mice. Mouse actin RNA was detected and used to monitorUgt1a abundance. B, Northern blot analysis. The 3� DNA probe shown in Fig.1 was used to determine the level of total UGT1A mRNA expressed in liver ofWT, Ugt1�/� and Ugt1�/� mice.
TABLE 1Serum bilirubin concentrationsAnalyses of serum bilirubin concentrations were conducted with a minimum ofeight serum samples collected from 5-day-old mice from eachUgt1 genotype (WT,�/�, and �/�). Values represent the means � S.D. p values were calculated fromStudent’s t test.
Bilirubin Bilirubin-G TotalGenotypeWT, �/� (mg/dl) 0.13 � 0.16 0.46 � 0.23 0.59 � 0.30�/� (mg/dl) 7.82 � 1.25 0.80 � 0.25 8.62 � 1.14(�/�/WT, �/�) (-fold increase) 59.68 1.73 14.53
p value �0.0001 0.005 �0.0001
Ugt1 Knock-out and Hyperbilirubinemia
MARCH 21, 2008 • VOLUME 283 • NUMBER 12 JOURNAL OF BIOLOGICAL CHEMISTRY 7905
by guest on August 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from
slightly increased inUgt1�/�mice. Liver glucuronidation activ-ity for 2-hydroxyestrone at position 2 was reduced by 25% inmicrosomes from Ugt1�/� mice further reduced by 75% inUgt1�/� mice, linking this activity to UGT1A activity. How-ever, glucuronidation of the same substrate at position 3 wasnot affected in Ugt1�/� mice and reduced by only 19% inUgt1�/� mice, indicating that the glucuronidation at this posi-tion was mainly due to UGT2B proteins, confirming previousfindings for conjugation of this substrate in humans (39). Sero-tonin, a substrate that has been shown to be glucuronidated byhumanUGT1A6 (40) and proposed as a highly selective in vitroprobe for UGT1A6 (41), was also assayed, but, surprisingly, theabsence of UGT1A proteins diminished the activity by only20%. This result suggests that serotonin glucuronidation maynot be critically linked to murine UGT1A6-directed metabo-lism, because it is efficientlymetabolized by the family of UGT2proteins in mice.Of the five exogenous substrates investigated, MPA, imipra-
mine, 4-nitrophenol, 1-naphthol, and 4-MU, all of which areknown to be substrates for human UGT1A proteins (14),showed a decrease in glucuronidation activity with liver micro-somes from Ugt1�/� mice, followed by a highly significantreduction in the Ugt1�/� mice. MPA, the active metabolite ofthe immunosuppressant mycophenolate mofetil, is metabo-lized as an MPA-phenylglucuronide by several UGT1A pro-teins (42–46) and as anMPA-acylglucuronide by UGT2B4 (46)
and UGT2B7 (45). MPA-phenolglucuronide is reduced byalmost 50% in liver fromUgt1�/� mice and is further decreasedto near undetectable levels in theUgt1�/�mice, demonstratingthat in liver MPA is a specific UGT1A substrate. Interestingly,the production ofMPA-acylglucuronidewas reduced by 43% inliver from Ugt1�/� mice, indicating that both UGT2 andUGT1A proteins are involved in the formation of that metabo-lite. Although small phenols have been historically categorizedas UGT1A substrates, our data indicate that glucuronidation ofthese agents with microsomes from Ugt1�/� mice confirmsthey serve as efficient substrates for the other members of theUGT2 family of proteins.The Impact of UGT1A Loss on Cellular Metabolism—In an
effort to understand the impact ofUGT1Adeficiency andUCB-directed toxicity, a microarray experiment was designed tocompare gene expression patterns in liver between 5-day-oldWT and Ugt1�/� mice. These experiments were conductedusing Lumina microarray chips that encompassed the mostcurrent assembly of the mouse genome. When the averages ofRNA expression values from three WT livers were comparedwith three Ugt1�/� livers, increases in Ugt1�/�/WT RNAexpression profiles indicated that �350 genes were beingexpressed 1.5-fold or greater in Ugt1�/� mice, with �75 beingexpressed at 2.5-fold and greater. The loss of UGT1A functionwas concordantwith reduced gene expression inUgt1�/�mice,with �300 genes being underexpressed �1.5-fold. When sta-
FIGURE 5. UDP-glucuronosyltransferase activity. Liver microsomes from WT (�/�), Ugt1 heterozygous (�/�), and Ugt1 knock-out (�/�) mice were used toanalyze UGT activity. For each substrate, 100% of the glucuronidation activity is represented by WT activity. A, a series of endogenous substrates: bilirubin-G(184,908 peak area); testosterone-G (16.00 pmol/min/mg); 2-OH-estrone (-2G: 29.24 and -3G: 1010.10 pmol/min/mg); serotonin-G (1,850,000 peak area); T3-G(217,000 peak area) and B, other xenobiotics, 4-nitrophenol-G (208,000 peak area); 4-MU-G (149,667 peak area); 1-naphthol-G (566,667 peak area); MPA-phenylglucuronide (10,683.98 pmol/min/mg and MPA-acylglucuronide: 259.10 pmol/min/mg), and imipramine-G (0.213 pmol/min/mg). Livers from threemice of each genotype were pooled, and assays were conducted in triplicate. Statistical significance to the WT group was calculated with the Student t test.*, p � 0.05; **, p � 0.01; and ***, p � 0.001.
Ugt1 Knock-out and Hyperbilirubinemia
7906 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 12 • MARCH 21, 2008
by guest on August 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from

tistically significant alterations in gene profiling were catego-rized into functional groups, it could be seen that aspects ofnormal cellular metabolism and function were potentiallyaffected in Ugt1�/� mice (Table 2). For example, genes linkedto cell cycle dependence are significantly repressed, whereasgenes linked to the inhibition of cellular kinases are induced(Fig. 6). Other important pathways such as those linked to fattyacid and pyrimidine metabolism as well as steroid metabolismshow a significant degree of change in Ugt1�/� liver. Thesecombined changes in gene expression in developing hepato-cytes would be anticipated to have a significant impact on theearly phases of liver development, and potentially contribute tothe overall toxicity associated with the mutation.Examination of gene expression profiles linked to xenobiotic
metabolism shows a number of impressive regulated geneexpression patterns (Fig. 7). We have confirmed similar pat-terns of expression for CYP2B9, heme oxygenase (HO-1), cho-lesterol 7�-hydroxylase (CYP7A1), andCYP4A10 RNAby real-time PCR (data not shown). Induction of HO-1, whichcatabolizes heme to bilivirdin, occurs under situations of oxi-dative stress and is felt to be an adaptive response to injury anddisease (47, 48). However, with increasing concentrations ofserumbilirubin resulting fromUgt1a1deficiency, the inductionof HO-1 would add to the already heightened bilirubin accu-mulation. Cholesterol is metabolized to either steroids or bileacids in mammals. CYP7A1 is liver-specific and converts cho-lesterol into 7�-hydroxycholesterol producing primarily cholicacid (49). In the intestine, bile acids play an important role infacilitating the uptake of fats, sterols, and fat-soluble vitamins(49). The only other genes that are repressed to the same degreeas Cyp7a1 (�3.0) are the deficient Ugt1a genes. In Cyp7a1�/�
mice, the mutation leads eventually to liver failure and postna-tal lethality (50). We might predict from this result that theUgt1�/� mice are also deficient in bile acid synthesis, whichmight contribute to a lack of nutrient and vitamin uptakethrough their diet (49). It is also interesting to note that severalof the genes that encode some of themajor regulatory factors ofxenobioticmetabolism, such as PXR (51) andCAR (52), are alsoinduced inUgt1�/� mice, potentially linking their regulation tothe induced levels of several of the Phase I genes.
DISCUSSION
An animal model has been developed by introducing agenetic lesion into the common exon 4 of the Ugt1 locus thatdisplays physiological conditions similar to what is observed inhuman CN1 patients. Neonatal Ugt1�/� mice accumulatesevere levels of UCB that results in deathwithin 2weeks follow-ing birth. The interruption of the reading frame in exon 4 leadsto a complete absence of UGT1A RNA and protein, similar towhat is observed in a CN1 patients. The accumulation of UCBresulting from non-hemolytic hyperbilirubinemia is attributedto the presence of the UGT1A1*28 polymorphism, which islinked toGilbert syndrome, or sensemutations in any of the fiveUGT1A1 exons that results in either CN1 or CN2 disease (1).Crigler-Najjar type 2 is less severe resulting from less dramaticmissense mutations or heterozygous expression of mutant andnormal alleles and is treatable by phototherapy or inducing theUGT1A1 gene with phenobarbital therapy (53). More severelesions are associated with CN1, an inborn error of metabolismdisease that results in stop mutations or frameshifts and ren-ders either the UGT1A1 gene or the entire UGT1 locus non-functional. Interruption of exon 4 in Ugt1�/� mice inactivatesthe entire murine Ugt1 locus resulting in neonatal death, thusresembling the toxicity patterns seen in CN1 patients.The most pronounced phenotype in the Ugt1�/� mice is
observable neonatal jaundice and serum UCB levels that rangefrom 40- to 60-fold normal neonatal indirect bilirubin values.Indirect bilirubin values in humans that are 20- to 40-fold thosemeasured in normal individuals (14) have been linked to braindamage following the accumulation of bilirubin in the basalganglia and brainstem nuclei. Experiments have not been per-formed at this time to examine the impact of the hyperbiliru-binemia onCNS toxicity inUgt1�/�mice. The role of UGT1A1as the sole enzyme involved in bilirubin glucuronidation inhumans has been definitively established in patients with CN1through linkage studies ofmutations only in the unique exon oftheUGT1A1 gene (2). Although it is unclear if mouse UGT1A1serves as the only enzyme tied to bilirubin glucuronidation,interruption of the Ugt1 locus through lesions in one of theconserved exons disrupts all of theUgt1a genes followed by theearly development of hyperbilirubinemia. There is no detecta-ble bilirubin glucuronidation activity in liver microsomes fromUgt1�/� mice. Over 77 genetic lesions in exons 1 through 5 ofthe humanUGT1A1 gene have been reported and tied to eitherCN1 or CN2 (54). Although there exists a wide spectrum ofUGT1A1mutations that are associatedwith the onset of hyper-bilirubinemia, little information exists on the biochemical out-come of the UGT1A1 mutations that lead to bilirubin inducedtoxicities.In post-mortem evaluation of patients with hyperbiliru-
binemia, necrosis as well as neuronal degeneration is evident inregions of bilirubin deposition (55). Studies using cultured neu-rons and astrocytes have provided some evidence for the induc-tion of apoptosis after elevated bilirubin exposure. Low concen-trations of unconjugated bilirubin (�5 �M) that moreaccurately reflect in vivo exposures demonstrate poly(ADP-ri-bose) polymerase cleavage, DNA fragmentation, cytochrome crelease, and mitochondrial membrane permeabilization (56–
TABLE 2List of functional pathways that are altered significantly in Ugt1�/�
liver compared to WT liverIn ranking genes of interest we utilized amethod similar to that described by Cole etal. (71) for their software package Focus, and we calculated p values for groups ofgenes. For pathway analysis, the groups of genes are defined by the individual path-ways. The pathways included are all those listed in the Kyoto Encyclopedia of Genesand Genomes and present in the commercial BioCarta Database (BioCarta, SanDiego, CA). As the p values increase, the pathways become less and less significant.Based upon the ordering, the cell cycle pathways are themost significant. Shown areonly those pathways with a p value of �10�7, because p values greater than thissubstantially increase the possibility of false positives.
Pathway description p valueCell cycle 0.504 E-18Pentose and glucuronate interconversions 0.340 E-14Porphyrin metabolism 0.472 E-13Starch and sucrose metabolism 0.134 E-12Androgen and estrogen metabolism 0.337 E-12Metabolism of xenobiotics 0.327 E-11Fatty acid metabolism 0.810 E-09Pyrimidine metabolism 0.336 E-07
Ugt1 Knock-out and Hyperbilirubinemia
MARCH 21, 2008 • VOLUME 283 • NUMBER 12 JOURNAL OF BIOLOGICAL CHEMISTRY 7907
by guest on August 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from

FIGURE 6. Microarray analysis of cell cycle genes. Total RNA from 5-day-old neonatal WT and Ugt1�/� mice were used in microarray analysis. Using theIllumina Mouse 6 Sentrix Expression BeadChip, the expression of each gene in either WT of Ugt1�/� mice was analyzed in triplicate using three independentliver RNA samples. Shown is a heat map for gene clusters that are linked to the cell cycle.
Ugt1 Knock-out and Hyperbilirubinemia
7908 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 12 • MARCH 21, 2008
by guest on August 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from
58). Bilirubin also activates the mitogen-activated proteinkinases p38, JunN-terminal kinase 1/2, and extracellular signal-regulated kinase 1/2 as well as NF-�B adding to the complexityof cellular signaling pathways involved in neuronal and astro-cyte cell death (59–61). The activation of the inflammatory and
signaling pathways is directly asso-ciated with activation of apoptosis,but the initiating stimulus remainselusive. Thus, Ugt1�/� mice mayserve as an excellentmodel to exam-ine the physiological and biochemi-cal implications of severe hyperbil-irubinemia as well as thoseconsequences associatedwith selec-tive cellular signaling events.Previous communications have
indicated that the homozygous j/jGunn rat model mimics the clinicalsymptoms seen in CN1, because thesingle base pair mutation in exon 4of the ratUGT1 locus leads to a pre-mature stop codon, and the ratsdevelop spontaneous hyperbiliru-binemia (24). Total serum bilirubinlevels in Gunn rats are slightly lower(7.15 mg/dl) than those detected inUgt1�/� mice (8–10 mg/dl). Gunnrats have served as an excellent ani-mal model to examine the impact ofelevated UCB on spontaneoushydroencephalitus as well as thepotential of gene therapy for longterm correction of hyperbiliru-binemia (62). But there are severalsignificant differences in the pheno-type between the Gunn rat andUgt1�/�mice. Themost dramatic isreflected in the immediate health ofthe animals, with the accumulationof UCB in Gunn rats having noimpact on neonatalmorbidity or theability of the rats to reproduce. Incontrast, the genotype associatedwith interruption of exon 4 inUgt1�/� mice predisposes new-borns to the toxic effects associatedwith elevated UCB leading to deathwithin 11 days. Although weobserved a 2 mg/dl difference intotal serum bilirubin from that inGunn rats, this difference may besufficient in Ugt1�/� mice to ini-tiate central nervous system toxicityand lethal encephalopathy in neo-nates. It has also been determinedthat bloodT4 levels inGunn rats arenormal up to 5–10 days after birthbut increase by 50% at 15–20 days
following birth (63). In agreement with this observation, liverglucuronidation activity toward T4 in Gunn rats is impaired�50% compared with that observed in Wistar rats (64). How-ever, liver T4 glucuronidation capacity is completely absent inUgt1�/� mice, which might contribute to elevated levels of cir-
FIGURE 7. Microarray analysis of genes involved in xenobiotic metabolism. Listed are heat maps of genescategorized as Phase I, Phase II, and transcriptional activation. The cutoff for inclusion are those genes thatshow a response ��1.5 or ��1.5.
Ugt1 Knock-out and Hyperbilirubinemia
MARCH 21, 2008 • VOLUME 283 • NUMBER 12 JOURNAL OF BIOLOGICAL CHEMISTRY 7909
by guest on August 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from

culating T4. However, at 5 days old, the circulating levels of T3and T4 are similar in Ugt1�/� and Ugt1�/� mice, indicatingthat the influence of glucuronidation on thyroid hormones isnot impacted by deletion of the Ugt1 locus in early neonatalmice.Bilirubin has been implicated as a ligand for the xenobiotic
receptor CAR (65) as well as the Ah receptor (66). In tissueculture cells, bilirubin activates the Ah receptor as demon-strated by the ability of the receptor to bind to dioxin responseelements. In addition, basal levels of bothCYP1A1 andCYP1A2protein and mRNA are elevated in liver of Gunn rats duringneonatal development (67). Inmice, we have demonstrated thatneonatal exposure to TCDD through lactation leads to activa-tion of the Ah receptor and induction of human CYP1A1-driven luciferase activity in 2-day-old transgenicmice, confirm-ing that activation of the Ah receptor can occur in very youngmice. Thus, if bilirubin serves as an endogenous Ah receptorligand inUgt1�/�mice, wewould expect to see induction of theCYP1A proteins. However, at 5 days following birth and at ele-vated levels of UCB in serum, there is no detectable CYP1A1 orincreases in CYP1A2 protein in Ugt1�/� mice compared withsame age littermates that were WT or heterozygous for theUgt1 allele (data not shown). In neonatal liver, expression ofCYP2A10 as determined by microarray analysis is only slightlyelevated in Ugt1�/� mice, indicating that the low level of CARexpression in neonatalmice ismost likely not a factor in expres-sion of CAR target genes by bilirubin.Several lines of evidence suggest that certain drugs or com-
pounds can be used as tools to identify the expression of indi-vidual isoforms of the human UGT1A family of proteins. Forexample, recombinant expression of UGT1A6 in comparisonto the other human UGTs is highly selective for the glucu-ronidation of serotonin (41). However, when UGT1A6 is coex-pressed with the other UGT isoforms, a significant increase inserotonin glucuronidation is achieved, particularly whenUGT1A6 is coexpressed with UGT2B7 (68). Recent evidencesupports the notion that the family of UGTs oligomerize in themembrane and formdimer or heterodimer complexes (69)witha structure that can alter substrate specificity (68). However, itis unclear if this specificmechanismwithUGT1A6 is in place inmice. There is only a 20% difference in serotonin glucuronida-tion activity between WT and Ugt1�/� mice. This result leadsus to conclude that murine UGT1A6 is unable to efficientlyconjugate serotonin, even though UGT1A6 is a highly con-served protein among different species. More importantly, thisresult indicates that additional murine UGT2 proteins play amajor role in the glucuronidation of serotonin and indicate thatserotonin is not a selective substrate or probe to detect thepresence of murine UGT1A6 expression.Although the toxicity associated with hyperbilirubinemia
has been speculated to result primarily in neuronal damage,microarray analysis of gene expression patterns in livers fromUgt1�/�mice allows us to conclude that the underlying toxicitymay result from cellular and molecular alterations that areoccurring in other tissues. One of the most significant changesoccurs with those genes that are clustered for cell cycle depend-ence. Neonatal liver tissue is rapidly proliferating during devel-opment, and a host of genes tied to the control of cell cycling are
repressed. Other significant changes were noted with fatty acidand steroid metabolism. Impressively, these later changes ingene expression profiles were attributed in part to genes linkedto xenobiotic metabolism, such as the Ugt1a genes, becausethey have been tied to estrogen metabolism and the Cyp4agenes, which play a role in arachidonic acid metabolism (70).Although several of the cytochrome P450 genes such asCyp2b9are impressively induced, the repression of Cyp7a1 expressionmay contribute to poor uptake of ingested fats and fat-solublevitamins. It is interesting to note that neonatal Ugt1�/� miceappear to be malnourished about 1-day before they die as evi-dent by weight differences when compared with their WT andUgt1�/� littermates.
The entire humanUGT1 locus has recently been shown to beregulated in transgenic Tg-UGT1mice in a tissue-specific pat-tern that closely resembles expression profiles that have beendocumented in human tissues (18). For example, a significantabundance of the UGT1A proteins accumulates in the smalland large intestines of Tg-UGT1mice (18, 19), similar to whathas been observed in human tissue. However, there is littleinformation on how these genes are regulated in other impor-tant tissues associated with glucuronidation such as thoselinked to hormonal and steroid control. Having available ahumanized mouse model would help to define the role ofhuman glucuronidation as it pertains to inducibility, tissuespecificity, and its role in homeostatic control of hormones andsteroids. With the ability to cross-breed Ugt1�/� mice withTg-UGT1 mice, future experiments can be designed withTg-UGT1/Ugt1�/� mice to examine the role of human glucu-ronidation as it pertains to regulation by tissue, inducibility byxenobiotics, and its role in drug metabolism and toxicity.
REFERENCES1. Bosma, P. J. (2003) J. Hepatol. 38, 107–1172. Bosma, P. J., Seppen, J., Goldhoorn, B., Bakker, C., Oude, E. R.,
Chowdhury, J. R., Chowdhury, N. R., and Jansen, P. L. (1994) J. Biol. Chem.269, 17960–17964
3. Monaghan, G., Ryan, M., Seddon, R., Hume, R., and Burchell, B. (1996)Lancet 347, 578–581
4. Bosma, P. J., Chowdhury, J. R., Bakker, C., Gantla, S., De Boer, A., Oostra,B. A., Lindhout, D., Tytgat, G.N. J., Jansen, P. L.M., Elferink, R. P. J. O., andChowdhury, N. R. (1995) N. Engl. J. Med. 333, 1171–1175
5. Lankisch, T. O., Moebius, U., Wehmeier, M., Behrens, G., Manns, M. P.,Schmidt, R. E., and Strassburg, C. P. (2006) Hepatology 44, 1324–1332
6. Ritter, J. K., Yeatman, M. T., Ferreira, P., and Owens, I. S. (1992) J. Clin.Invest. 90, 150–155
7. Wennberg, R. P. (2000) Cell Mol. Neurobiol. 20, 97–1098. Roger, C., Koziel, V., Vert, P., and Nehlig, A. (1995) Early Hum. Dev. 43,
133–1449. Sokal, E. M., Silva, E. S., Hermans, D., Reding, R., de Ville, d. G., Buts, J. P.,
and Otte, J. B. (1995) Transplantation 60, 1095–109810. Schauer, R., Stangl, M., Lang, T., Zimmermann, A., Chouker, A., Gerbes,
A. L., Schildberg, F. W., and Rau, H. G. (2003) J. Pediatr. Surg. 38,1227–1231
11. Crigler, J. F., Jr., and Najjar, V. A. (1952) Pediatrics 10, 169–18012. Ritter, J. K., Chen, F., Sheen, Y. Y., Tran,H.M., Kimura, S., Yeatman,M.T.,
and Owens, I. S. (1992) J. Biol. Chem. 267, 3257–326113. Gong, Q. H., Cho, J. W., Huang, T., Potter, C., Gholami, N., Basu, N. K.,
Kubota, S., Carvalho, S., Pennington, M. W., Owens, I. S., and Popescu,N. C. (2001) Pharmacogenetics 11, 357–368
14. Tukey, R. H., and Strassburg, C. P. (2000) Annu. Rev. Pharmacol. Toxicol.40, 581–616
Ugt1 Knock-out and Hyperbilirubinemia
7910 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 12 • MARCH 21, 2008
by guest on August 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from

15. Strassburg, C. P., Oldhafer, K., Manns,M. P., and Tukey, R. H. (1997)Mol.Pharmacol. 52, 212–220
16. Strassburg, C. P., Nguyen, N., Manns, M. P., and Tukey, R. H. (1998)Mol.Pharmacol. 54, 647–654
17. Strassburg, C. P., Nguyen, N., Manns, M. P., and Tukey, R. H. (1999)Gastroenterology 116, 149–160
18. Chen, S., Beaton, D., Nguyen, N., Senekeo-Effenberger, K., Brace-Sinno-krak, E., Argikar, U., Remmel, R. P., Trottier, J., Barbier, O., Ritter, J. K., andTukey, R. H. (2005) J. Biol. Chem. 280, 37547–37557
19. Senekeo-Effenberger, K., Chen, S., Brace-Sinnokrak, E., Bonzo, J. A., Yueh,M. F., Argikar, U., Kaeding, J., Trottier, J., Remmel, R. P., Ritter, J. K.,Barbier, O., and Tukey, R. H. (2007) Drug Metab. Dispos. 35, 419–427
20. Verreault, M., Senekeo-Effenberger, K., Trottier, J., Bonzo, J. A., Belanger,J., Kaeding, J., Staels, B., Caron, P., Tukey, R. H., and Barbier, O. (2006)Hepatology 44, 368–378
21. Yueh, M. F., and Tukey, R. H. (2007) J. Biol. Chem. 282, 8749–875822. Tukey, R. H., Strassburg, C. P., and Mackenzie, P. I. (2002)Mol. Pharma-
col. 62, 446–45023. Strauss, K. A., Robinson, D. L., Vreman, H. J., Puffenberger, E. G., Hart, G.,
and Morton, D. H. (2006) Eur. J. Pediatr. 165, 306–31924. Iyanagi, T. (1991) J. Biol. Chem. 266, 24048–2405225. El-Awady, M., Chowdhury, J. R., Kesari, K., van Es, H., Jansen, P. L. M.,
Lederstein, M., Arias, I. M., and Chowdhury, N. R. (1990) J. Biol. Chem.265, 10752–10758
26. Yamamura, H., and Takagishi, Y. (1993) Nagoya J. Med. Sci. 55, 11–2127. Chowdhury, J. R., Kondapalli, R., and Chowdhury, N. R. (1993) Adv. Vet.
Sci. Comp. Med. 37, 149–17328. Lamb, J. G., Straub, P., and Tukey, R. H. (1994) Biochemistry 33,
10513–1052029. Albert, C., Vallee, M., Beaudry, G., Belanger, A., and Hum, D. W. (1999)
Endocrinology 140, 3292–330230. Guillemette, C., Levesque, E., Beaulieu, M., Turgeon, D., Hum, D.W., and
Belanger, A. (1997) Endocrinology 138, 2998–300531. Sasik, R., Woelk, C. H., and Corbeil, J. (2004) J. Mol. Endocrinol. 33, 1–932. Mackenzie, P. I., Walter, B. K., Burchell, B., Guillemette, C., Ikushiro, S.,
Iyanagi, T., Miners, J. O., Owens, I. S., and Nebert, D. W. (2005) Pharma-cogenet. Genomics 15, 677–685
33. Clarke, D. J., Moghrabi, N., Monaghan, G., Cassidy, A., Boxer, M., Hume,R., and Burchell, B. (1997) Clin. Chim. Acta 266, 63–74
34. Buckley, D. B., and Klaassen, C. D. (2007) Drug Metab. Dispos. 35,121–127
35. Yoder Graber, A. L., Ramirez, J., Innocenti, F., and Ratain, M. J. (2007)Pharmacogenet. Genomics 17, 619–627
36. Turgeon, D., Carrier, J. S., Levesque, E., Hum, D. W., and Belanger, A.(2001) Endocrinology 142, 778–787
37. Barbier, O., Belanger, A., andHum,D.W. (1999)Biochem. J. 337, 567–57438. Belanger, G., Barbier, O., Hum, D. W., and Belanger, A. (1999) Eur. J. Bio-
chem. 260, 701–70839. Lepine, J., Bernard, O., Plante, M., Tetu, B., Pelletier, G., Labrie, F., Be-
langer, A., and Guillemette, C. (2004) J. Clin. Endocrinol. Metab. 89,5222–5232
40. Krishnaswamy, S., Hao, Q., Al Rohaimi, A., Hesse, L.M., vonMoltke, L. L.,Greenblatt, D. J., and Court, M. H. (2005) J. Pharmacol. Exp. Ther. 313,1340–1346
41. Krishnaswamy, S., Duan, S. X., von Moltke, L. L., Greenblatt, D. J., and
Court, M. H. (2003) Drug Metab. Dispos. 31, 133–13942. Bernard, O., and Guillemette, C. (2004)Drug Metab. Dispos. 32, 775–77843. Mackenzie, P. I. (2000) Ther. Drug Monit. 22, 10–1344. Mojarrabi, B., and Mackenzie, P. I. (1997) Biochem. Biophys. Res. Com-
mun. 238, 775–77845. Picard, N., Ratanasavanh, D., Premaud, A., Le Meur, Y., and Marquet, P.
(2005) Drug Metab. Dispos. 33, 139–14646. Shipkova, M., Strassburg, C. P., Braun, F., Streit, F., Grone, H. J., Arm-
strong, V. W., Tukey, R. H., Oellerich, M., and Wieland, E. (2001) Br. J.Pharmacol. 132, 1027–1034
47. Nath, K. A., Balla, G., Vercellotti, G.M., Balla, J., Jacob, H. S., Levitt, M. D.,and Rosenberg, M. E. (1992) J. Clin. Invest. 90, 267–270
48. Rizzardini, M., Terao, M., Falciani, F., and Cantoni, L. (1993) Biochem. J.290, 343–347
49. Russell, D. W. (2003) Annu. Rev. Biochem. 72, 137–17450. Ishibashi, S., Schwarz, M., Frykman, P. K., Herz, J., and Russell, D. W.
(1996) J. Biol. Chem. 271, 18017–1802351. Blumberg, B., Sabbagh, W., Jr., Juguilon, H., Bolado, J., Jr., van Meter,
C. M., Ong, E. S., and Evans, R. M. (1998) Genes Dev. 12, 3195–320552. Wei, P., Zhang, J., Egan-Hafley, M., Liang, S., and Moore, D. D. (2000)
Nature 407, 920–92353. Crigler, J. F., Jr., and Gold, N. I. (1969) J. Clin. Invest. 48, 42–5554. Servedio, V., d’Apolito,M.,Maiorano,N.,Minuti, B., Torricelli, F., Ronchi,
F., Zancan, L., Perrotta, S., Vajro, P., Boschetto, L., and Iolascon, A. (2005)Hum. Mutat. 25, 325
55. Shapiro, S. M. (2003) Pediatr. Neurol. 29, 410–42156. Rodrigues, C.M., Sola, S., and Brites, D. (2002)Hepatology 35, 1186–119557. Rodrigues, C. M., Sola, S., Silva, R., and Brites, D. (2000) Mol. Med. 6,
936–94658. Ostrow, J. D., Pascolo, L., and Tiribelli, C. (2003) Pediatr. Res. 54, 92659. Fernandes, A., Falcao, A. S., Silva, R. F., Brito, M. A., and Brites, D. (2007)
Eur. J. Neurosci. 25, 1058–106860. Fernandes, A., Falcao, A. S., Silva, R. F., Gordo, A. C., Gama, M. J., Brito,
M. A., and Brites, D. (2006) J. Neurochem. 96, 1667–167961. Lin, S., Yan, C., Wei, X., Paul, S. M., and Du, Y. (2003)Neurosci. Lett. 353,
209–21262. Takahashi, M., Ilan, Y., Chowdhury, N. R., Guida, J., Horwitz, M., and
Chowdhury, J. R. (1996) J. Biol. Chem. 271, 26536–2654263. Komaki, T., Sakata, S., Kamikubo, K., Matsuda, M., Nakamura, S., Ogawa,
T., Sato,H., Keino,H., Kashiwamata, S., andMiura, K. (1991) J. Endocrinol.Invest. 14, 409–415
64. Kato, Y., Ikushiro, S., Haraguchi, K., Yamazaki, T., Ito, Y., Suzuki, H.,Kimura, R., Yamada, S., Inoue, T., and Degawa, M. (2004) 81, 309–315
65. Huang, W., Zhang, J., Chua, S. S., Qatanani, M., Han, Y., Granata, R., andMoore, D. D. (2003) Proc. Natl. Acad. Sci. U. S. A. 100, 4156–4161
66. Phelan, D., Winter, G. M., Rogers, W. J., Lam, J. C., and Denison, M. S.(1998) Arch. Biochem. Biophys. 357, 155–163
67. Kapitulnik, J., and Gonzalez, F. J. (1993)Mol. Pharmacol. 43, 722–72568. Kurkela, M., Patana, A. S., Mackenzie, P. I., Court, M. H., Tate, C. G.,
Hirvonen, J., Goldman, A., and Finel,M. (2007) Pharmacogenet. Genomics17, 115–126
69. Operana, T. N., and Tukey, R. H. (2007) J. Biol. Chem. 282, 4821–482970. Capdevila, J. H., Falck, J. R., and Estabrook, R. W. (1992) FASEB J. 6,
731–73671. Cole, S.W., Galic, Z., and Zack, J. A. (2003) Bioinformatics 19, 1808–1816
Ugt1 Knock-out and Hyperbilirubinemia
MARCH 21, 2008 • VOLUME 283 • NUMBER 12 JOURNAL OF BIOLOGICAL CHEMISTRY 7911
by guest on August 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Gary Hardiman, Alain Bélanger and Robert H. TukeyNghia Nguyen, Jessica A. Bonzo, Shujuan Chen, Sarah Chouinard, Michael J. Kelner,
Disease Locus in Mice Resembles Human Crigler-Najjar Type IUgt1Disruption of the
doi: 10.1074/jbc.M709244200 originally published online January 7, 20082008, 283:7901-7911.J. Biol. Chem.
10.1074/jbc.M709244200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/283/12/7901.full.html#ref-list-1
This article cites 70 references, 25 of which can be accessed free at
by guest on August 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from










![Antioxidant Properties of Thymol and Butylated Hydroxytoluene in … · 2010. 8. 15. · Thymol (p-methyl-isopropyl-phenol) is the main constituent of the oils of Thymus vulgaris[7].](https://static.fdocuments.in/doc/165x107/5fe1517555edc50f792e182b/antioxidant-properties-of-thymol-and-butylated-hydroxytoluene-in-2010-8-15.jpg)







