
Direct electrochemical regeneration of the enzymatic cofactor 1,4-NADH employing nano-patterned...
8
Chemical Engineering Journal 188 (2012) 173–180 Contents lists available at SciVerse ScienceDirect Chemical Engineering Journal j ourna l ho mepage: www.elsevier.com/locate/cej Direct electrochemical regeneration of the enzymatic cofactor 1,4-NADH employing nano-patterned glassy carbon/Pt and glassy carbon/Ni electrodes Irshad Ali, Amardeep Gill, Sasha Omanovic ∗ Department of Chemical Engineering, McGill University, 3610 University Street, Montreal, Quebec, Canada H3A 2B2 a r t i c l e i n f o Article history: Received 4 December 2011 Received in revised form 27 January 2012 Accepted 1 February 2012 Keywords: Electrochemical NADH regeneration Glassy carbon Platinum Nickel Electrode surface modification Nano-particles a b s t r a c t A glassy carbon electrode surface was patterned with electrochemically deposited platinum and nickel nano-particles, with the aim of increasing the electrode efficiency in the process of in situ electrochemical regeneration of enzymatically active 1,4-NADH from NAD + , in a batch electrochemical reactor. The role of Pt and Ni was to provide ‘active’ adsorbed hydrogen (Pt–H ads or Ni–H ads ) for the fast protonation of elec- trochemically formed NAD-radical. The effect of nano-particle size, surface coverage, and the electrode potential on the percentage of 1,4-NADH recovered from NAD + was investigated. It was demonstrated that up to a 100% recovery of 1,4-NADH from NAD + was possible on the produced electrodes at relatively low electrode potentials, making them excellent candidates for regeneration of 1,4-NADH in industrial bioreactors. © 2012 Elsevier B.V. All rights reserved. 1. Introduction In enzymatic biocatalysis, enzymatic cofactors are usually required along with specific enzymes. One of the cofactors used in redox enzymatic reactions is nicotinamide adenine dinucleotide NAD(H). About 700 enzymes are known to use it as a coenzyme [1]. In these reactions, NAD(H) serves as a proton and electron trans- port molecule. Consequently, NAD(H) can be found in two redox forms; in an oxidized, NAD + , and in a reduced, NADH, form. In nature, NAD(H) is found in all living cells and it is used during cellular respiration involving redox enzymes [2–4]. It also participates in aerobic biotransformation of toxic Cr(VI) to non- toxic Cr(III) [5]. In industry, it is of importance in the field of chiral compounds preparation [6]. It participates in enzymatic catalysis of industrially important synthetic reactions where conventional chemical catalysts fail [7]. NAD(H) is also used for posttranslational modifications, therapy of certain medical conditions and diseases such as Alzheimer’s and Parkinson, in pharmacology, biotech- nology, biosensors, and the synthesis of new high-value-added compounds such as pharmaceuticals, food additives, perfumes, insecticides and pesticides [8–16]. In all these enzymatic reactions, it is critical to provide NAD(H) at stoichiometric quantities. How- ever, due to its very high cost, especially that of the reduced form, NADH, its industrial use is very limited. Therefore, efforts have been made to develop processes for in situ regeneration of NADH and ∗ Corresponding author. Tel.: +1 514 398 4273; fax: +1 514 398 6678. E-mail address: [email protected] (S. Omanovic). thus lower the production cost of NADH and therefore, the final product [7]. Several NADH regeneration approaches have so far been employed: biological, chemical, photochemical, electrochemical and enzymatic [17]. Electrochemical methods are of particular interest due to their potentially low cost and there is no need to add a reducing agent, thus enabling relatively simple product isolation. Many research groups have studied fundamental aspects of the mechanisms and kinetics of NAD + reduction and NADH oxidation using mostly bare (non-modified) metallic electrodes, such as mer- cury [18–27] and a variety of carbon materials [28,29]. In many of these cases the goal was to develop methods for the reduc- tion of NAD + to enzymatically active NADH isomer, 1,4-NADH, i.e. for the in situ regeneration of 1,4-NADH. However, the literature states that the major problem in the electrochemical regeneration of 1,4-NADH on bare (unmodified) electrodes is the formation of an enzymatically inactive dimer, NAD 2 as shown in Scheme 1. In Step 1, reduction of NAD + results in the formation of an NAD- free radical. In Step 2, two reaction pathways are possible; (i) two neighboring free radicals can combine in a very fast dimerization reaction to form enzymatically inactive NAD 2 , or (ii) the radical can be protonated and further reduced to form enzymatically active 1,4 NADH. Unfortunately, on bare (unmodified) electrodes, the kinetics of path (ii) is significantly slower than that of path (i), and thus the major product of NAD + reduction on these electrodes is the enzymatically inactive NAD 2 . More precisely, the protonation step is considered to be the rate determining step [19,23,24,26,30–35]. This formation of the dimer thus reduces the yield of enzymatically active 1,4-NADH that could be produced (regenerated). 1385-8947/$ – see front matter © 2012 Elsevier B.V. All rights reserved. doi:10.1016/j.cej.2012.02.005
-
Upload
irshad-ali -
Category
Documents
-
view
213 -
download
0
Transcript of Direct electrochemical regeneration of the enzymatic cofactor 1,4-NADH employing nano-patterned...

De
ID
a
ARRA
KEGPNEN
1
riNIpf
dptcocmsnciieNm
1d
Chemical Engineering Journal 188 (2012) 173– 180
Contents lists available at SciVerse ScienceDirect
Chemical Engineering Journal
j ourna l ho mepage: www.elsev ier .com/ locate /ce j
irect electrochemical regeneration of the enzymatic cofactor 1,4-NADHmploying nano-patterned glassy carbon/Pt and glassy carbon/Ni electrodes
rshad Ali, Amardeep Gill, Sasha Omanovic ∗
epartment of Chemical Engineering, McGill University, 3610 University Street, Montreal, Quebec, Canada H3A 2B2
r t i c l e i n f o
rticle history:eceived 4 December 2011eceived in revised form 27 January 2012ccepted 1 February 2012
a b s t r a c t
A glassy carbon electrode surface was patterned with electrochemically deposited platinum and nickelnano-particles, with the aim of increasing the electrode efficiency in the process of in situ electrochemicalregeneration of enzymatically active 1,4-NADH from NAD+, in a batch electrochemical reactor. The role ofPt and Ni was to provide ‘active’ adsorbed hydrogen (Pt–Hads or Ni–Hads) for the fast protonation of elec-trochemically formed NAD-radical. The effect of nano-particle size, surface coverage, and the electrode
eywords:lectrochemical NADH regenerationlassy carbonlatinumickellectrode surface modification
potential on the percentage of 1,4-NADH recovered from NAD+ was investigated. It was demonstratedthat up to a 100% recovery of 1,4-NADH from NAD+ was possible on the produced electrodes at relativelylow electrode potentials, making them excellent candidates for regeneration of 1,4-NADH in industrialbioreactors.
© 2012 Elsevier B.V. All rights reserved.
ano-particles. Introduction
In enzymatic biocatalysis, enzymatic cofactors are usuallyequired along with specific enzymes. One of the cofactors usedn redox enzymatic reactions is nicotinamide adenine dinucleotideAD(H). About 700 enzymes are known to use it as a coenzyme [1].
n these reactions, NAD(H) serves as a proton and electron trans-ort molecule. Consequently, NAD(H) can be found in two redoxorms; in an oxidized, NAD+, and in a reduced, NADH, form.
In nature, NAD(H) is found in all living cells and it is useduring cellular respiration involving redox enzymes [2–4]. It alsoarticipates in aerobic biotransformation of toxic Cr(VI) to non-oxic Cr(III) [5]. In industry, it is of importance in the field of chiralompounds preparation [6]. It participates in enzymatic catalysisf industrially important synthetic reactions where conventionalhemical catalysts fail [7]. NAD(H) is also used for posttranslationalodifications, therapy of certain medical conditions and diseases
uch as Alzheimer’s and Parkinson, in pharmacology, biotech-ology, biosensors, and the synthesis of new high-value-addedompounds such as pharmaceuticals, food additives, perfumes,nsecticides and pesticides [8–16]. In all these enzymatic reactions,t is critical to provide NAD(H) at stoichiometric quantities. How-
ver, due to its very high cost, especially that of the reduced form,ADH, its industrial use is very limited. Therefore, efforts have beenade to develop processes for in situ regeneration of NADH and∗ Corresponding author. Tel.: +1 514 398 4273; fax: +1 514 398 6678.E-mail address: [email protected] (S. Omanovic).
385-8947/$ – see front matter © 2012 Elsevier B.V. All rights reserved.oi:10.1016/j.cej.2012.02.005
thus lower the production cost of NADH and therefore, the finalproduct [7].
Several NADH regeneration approaches have so far beenemployed: biological, chemical, photochemical, electrochemicaland enzymatic [17]. Electrochemical methods are of particularinterest due to their potentially low cost and there is no need to adda reducing agent, thus enabling relatively simple product isolation.
Many research groups have studied fundamental aspects of themechanisms and kinetics of NAD+ reduction and NADH oxidationusing mostly bare (non-modified) metallic electrodes, such as mer-cury [18–27] and a variety of carbon materials [28,29]. In manyof these cases the goal was to develop methods for the reduc-tion of NAD+ to enzymatically active NADH isomer, 1,4-NADH, i.e.for the in situ regeneration of 1,4-NADH. However, the literaturestates that the major problem in the electrochemical regenerationof 1,4-NADH on bare (unmodified) electrodes is the formation ofan enzymatically inactive dimer, NAD2 as shown in Scheme 1.
In Step 1, reduction of NAD+ results in the formation of an NAD-free radical. In Step 2, two reaction pathways are possible; (i) twoneighboring free radicals can combine in a very fast dimerizationreaction to form enzymatically inactive NAD2, or (ii) the radical canbe protonated and further reduced to form enzymatically active 1,4NADH. Unfortunately, on bare (unmodified) electrodes, the kineticsof path (ii) is significantly slower than that of path (i), and thusthe major product of NAD+ reduction on these electrodes is the
enzymatically inactive NAD2. More precisely, the protonation stepis considered to be the rate determining step [19,23,24,26,30–35].This formation of the dimer thus reduces the yield of enzymaticallyactive 1,4-NADH that could be produced (regenerated).
174 I. Ali et al. / Chemical Engineering Journal 188 (2012) 173– 180
N
H H
NH2
C
O
N N
C O
H2N
CO
N
H
NH2C
O
N
H
NH2C
O
e
ADPR ADPR
AD
PR
AD
PR
ADPR
dim
erizat
ion e -
+H +
(NAD free radica l)(NAD+)
NH2
fast
slow
ically
ipao8eNta
ferdmne1
g‘o1bgfsNcsRofi
brtn1Sre
and all the potentials in this paper are referred to MSE. As a workingelectrode, a GC plate embedded in epoxy resin, to give a two-dimensional surface exposed to the electrolyte (2.5 cm × 2.5 cm),
Scheme 2. Representation of the bifunctional character of GC–Pt and GC–Ni elec-
(inac tive NAD2 di mer)
Scheme 1. Reduction of NAD+ to NAD2 and enzymat
To obtain higher yields of enzymatically active 1,4-NADH, chem-cally modified electrodes have been used in the NAD+ reductionrocess. For example, Long and Chen [36] attached l-histidine to
silver electrode by a covalent bond. The electrode was capablef producing enzymatically active 1,4-NADH at a yield of about2%. Similarly, Baik et al. [37] used an unmodified gold-amalgamlectrode and obtained a very low yield of enzymatically active 1,4-ADH (ca. 10%). With a bare platinum electrode, the yield increased
o 50%, while the cholesterol modified gold amalgam electrode gave yield of almost 75%.
Electrochemical systems that utilize enzyme-based electrodesor 1,4-NADH regeneration from NAD+ have also attracted consid-rable scientific attention [12,38–44]. However, these systems areather complex and expensive and lack long-term stability, mostlyue to the denaturation of the enzyme and loss of the electronediator [2,31]. Therefore, for industrial purposes, there is still a
eed to develop a stable and cheap electrode surface that wouldnable direct electrochemical regeneration of enzymatically active,4-NADH at high yields [45,46].
To address the above-mentioned problems, many researchroups [2,36,37,47–49], including ours [30–32], have developed
surface-modified’ electrodes. Our group has pioneered the devel-pment of bi-functional metal nano-patterned electrode surface for,4-NADH regeneration [30–32]. The design of these surfaces haseen based on the hypothesis that if were able to provide hydro-en (H+) at the electrode site adjacent to the site of NAD-radicalormation, the kinetics of pathway (ii) in Step 2 (Scheme 1) wouldignificantly increase, and the ratio of the production active 1,4-ADH over inactive NAD2 would increase. To provide high surfaceoverage of hydrogen at the NAD-radical reduction/hydrogenationurface reaction site, good hydrogen evolution catalysts (e.g. Pt, Ru,h, Ir, Re, Ni) can be used to form nano-islands (i.e. nano-particles)n a high-overpotential (i.e. poor hydrogen catalyst) electrode sur-ace, such as glassy carbon (GC) or gold (Au) [30–32]. The hypothesiss graphically presented in Scheme 2.
In this paper we report our results on the development ofi-functional GC–Pt and GC–Ni electrodes for electrocatalyticegeneration of enzymatically active 1,4-NADH. It will be shownhat when the GC electrode was patterned with Pt and Niano-islands, a very high yield of 1,4-NADH was achieved (a
00% recovery form NAD+), proving the hypothesis presented incheme 2. It will also be shown that the percentage of 1,4-NADHecovery is dependent on the nano-island surface coverage and thelectrode potential.(active 1.4 NADH)
active 1,4-NADH. ADPR, adenosine diphosphoribose.
2. Materials and methods
2.1. Chemicals and reagents
Regeneration of enzymatically active 1,4-NADH from 1 mMNAD+ using GC–Pt and GC–Ni electrodes was studied in 0.1 Mphosphate buffer at pH of 5.8 and temperature of 295 K. NAD+
solutions were prepared by dissolving a proper amount of �-NAD+
(sodium salt, purity 98%) in phosphate buffer. High purity hydrogenhexachloroplatinate (H2PtCl6 × 6H2O) and nickel(II)nitrate hex-ahydrate (Ni(NO3)2 × 6H2O) were used for the formation of metalnano-islands on the GC surface. Aqueous solutions were preparedusing deionized water of resistivity 18.2 M� cm.
2.2. Electrode surface patterning
Formation of Pt and Ni nano-islands (nano-particles, NPs) ona GC electrode surface (i.e. electrode surface patterning) wasperformed by electrochemical deposition of Pt and Ni. A conven-tional two-compartment, three-electrode batch electrochemicalcell, connected to a potentiostat/galvanostat, was used for this pur-pose. The counter electrode was a graphite rod, which was, priorto each use, sonicated for 30 min in ethanol, followed by thoroughrinsing with water. During measurements, the counter electrodewas separated from the working and reference electrode elec-trolyte compartment by a glass frit. A mercury/mercurous sulfateelectrode (MSE; +0.642 V vs. SHE) was used as a reference electrode,
trodes used for 1,4-NADH regeneration, in this manuscript. The purpose of Ptnano-particles is to provide ‘active’ adsorbed hydrogen (Pt–Hads) at the site ofNAD-radical formation. This increases the radical protonation kinetics, and henceminimizes the probability of dimerization of two neighboring radicals, leading tothe preferential formation of enzymatically active 1,4-NADH.

ering Journal 188 (2012) 173– 180 175
wcpboei−ib1−onmp1r
2
rctwtGeipfpaowie
cow(sw
aC(t
3
3
gt(niwTnd
Fig. 1. (a) A SEM micrograph of Pt NPs deposited on a freshly prepared and electro-chemically activated GC electrode surface. Pt NPs were electrodeposited on the GCsurface from 0.5 M H2SO4 containing 1 mM H2PtCl6 × 6H2O by cycling the electrodefrom −0.6 to 0.1 V at a scan rate of 50 mV s−1 for 5 scans. (b) Pt NP size distribution.
I. Ali et al. / Chemical Engine
as used. Before each surface nano-patterning (electrode modifi-ation), the GC surface was carefully wet-polished with polishingaper (grid 1200/4000) until a mirror finish was obtained, followedy degreasing with ethanol and sonication for 5 min in ethanol inrder to remove polishing residues. To ensure a clean GC surface,lectrochemical pretreatment of the GC surface was carried outn 0.5 M H2SO4 by cyclic potentiodynamic polarization between1.5 V and 1.1 V at a scan rate of 100 mV s−1, for 50 cycles. Nano-
slands of Pt were formed on such freshly prepared GC surfacey performing electrochemical cyclic voltammetry in a solution of
mM H2PtCl6 × 6H2O in 0.5 M H2SO4, in the potential range from0.6 V to 0.1 V, at a scan rate of 50 mV s−1, for a specific numberf cycles (specified for each electrode, later in the text). Similarly,ano-islands of Ni on GC were formed by performing cyclic voltam-etry in 2 mM Ni(NO3)2 × 6H2O in acetate buffer pH 4, in the
otential range from −0.8 V to 0 V, at a scan rate of 50 mV s−1, for0 cycles [50]. The two electrodes are termed as GC–Pt and GC–Ni,espectively.
.3. Electrochemical regeneration of 1,4-NADH
Electrochemical reduction of NAD+, i.e. the electrochemicalegeneration of 1,4-NADH, was performed in a three-electrode, twoompartment batch electrochemical reactor of the same configura-ion used for the electrochemical nano-patterning of the GC surfaceith Pt and Ni. The electrolyte volume was 80 mL and the ini-
ial NAD+ concentration was 1 mM. As a working electrode, eitherC–Pt or GC–Ni was used. The total geometric area of the workinglectrode was 12.5 cm2 (two GC–Pt or GC–Ni electrodes connectedn a series were used). Two carbon rod counter electrodes werelaced opposite of the two working electrodes, to ensure the uni-orm electric field. All 1,4-NADH regeneration experiments wereerformed in 1 mM NAD+ in 0.1 M phosphate buffer, at pH of 5.8nd temperature of 295 K, and under potentiostatic conditions. Inrder to maintain an oxygen-free electrolyte, argon (99.998% pure)as purged through the electrolyte prior and during electrochem-
cal measurements. This also ensured convective mass transport oflectroactive species to/from the electrode surface.
Cyclic voltammetry and potentiostatic measurements werearried out using a potentiostat/galvanostat. The surface morphol-gy of the prepared bi-component GC–Pt and GC–Ni electrodesas analyzed by a field-emission scanning electron microscope
FE-SEM). For the surface image processing, open-source ImageJoftware was used. The progress of the NAD+ reduction reactionas monitored by UV/vis spectrophotometry.
To determine the enzymatic activity of the regenerated NADH,ctivity tests were made according to the regular Sigma Qualityontrol Test Procedure (EC 1.8.1.4) using lipoamide dehydrogenase5.3 U/mg, Calzyme Laboratories, Inc.) as an enzyme and dl-6,8-hioctic acid amide (Sigma) as a substrate [30,32,33,35].
. Results and discussion
.1. Characterization of a GC–Pt electrode
A SEM micrograph of Pt nanoparticles (NPs) deposited on alassy carbon surface is presented in Fig. 1a. The micrograph showshat the surface distribution of Pt NPs on the GC surface is uniformEDX analysis confirmed that the NPs are indeed platinum). Onlyegligible agglomeration of NPs is present. The mean Pt NP size
s 79 nm, and the size distribution is relatively narrow (Fig. 1b). It
as determined that 4.6% of the GC surface is covered by Pt NPs.his electrode is termed “Electrode A”, further in the text. Small (i.e.ano) Pt particle size, good physical separation and uniform surfaceistribution of the NPs, could lead to a highly electrocatalytically
This electrode is termed “Electrode A”, in the text.
active electrode surface capable of efficiently reducing NAD+ to1,4-NADH, which will indeed be evidenced later in the text.
In order to characterize the electrochemical behavior of GC–PtElectrode A, a cyclic voltammogram (CV) was recorded in 0.5 MH2SO4 solution in a wide potential region, between hydrogen andoxygen evolution, and presented together with the CVs of bare GCand bare Pt, in Fig. 2. The CV of the bare GC electrode (dashed lines)does not show any redox peaks in the potential region investigated,demonstrating its electrochemical inertness, as expected. On theother hand, the CV of the bare Pt electrode (dotted line) showscharacteristic peaks related to hydrogen desorption and adsorp-tion, Pt–Hads (potential regions a and b, respectively) and Pt oxidefilm formation and reduction (potential regions c and d, respec-tively) [51–54]. The CV of GC–Pt Electrode A (solid line) is almost thesuperposition of the bare GC and Pt response. Namely, it shows thecharacteristic hydrogen adsorption and desorption peaks (regionsa and b) and Pt oxide formation and reduction peaks (regions c andd), and also an anodic ‘hump’ in the potential region between ca.−0.35 V and 0.1 V, which is the response of the GC surface. Thus,
Fig. 2 further confirms the presence and electrochemical activity ofPt on a glassy carbon electrode surface.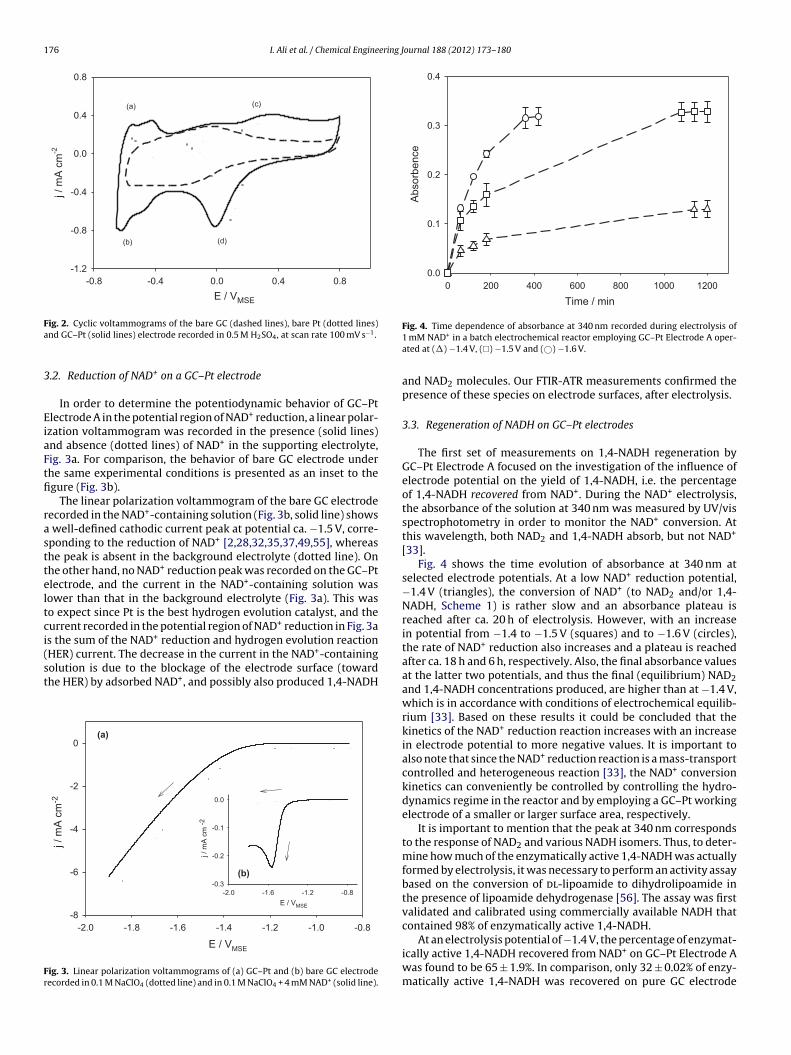
176 I. Ali et al. / Chemical Engineering Journal 188 (2012) 173– 180
E / VMSE
-0.8 -0.4 0.0 0.4 0.8
j /
mA
cm
-2
-1.2
-0.8
-0.4
0.0
0.4
0.8
(a)
(b) (d)
(c)
Fa
3
EiaFtfi
rastteltci(st
Fr
Time / min
0 20 0 40 0 60 0 80 0 100 0 120 0
Ab
so
rbe
nce
0.0
0.1
0.2
0.3
0.4
ig. 2. Cyclic voltammograms of the bare GC (dashed lines), bare Pt (dotted lines)nd GC–Pt (solid lines) electrode recorded in 0.5 M H2SO4, at scan rate 100 mV s−1.
.2. Reduction of NAD+ on a GC–Pt electrode
In order to determine the potentiodynamic behavior of GC–Ptlectrode A in the potential region of NAD+ reduction, a linear polar-zation voltammogram was recorded in the presence (solid lines)nd absence (dotted lines) of NAD+ in the supporting electrolyte,ig. 3a. For comparison, the behavior of bare GC electrode underhe same experimental conditions is presented as an inset to thegure (Fig. 3b).
The linear polarization voltammogram of the bare GC electrodeecorded in the NAD+-containing solution (Fig. 3b, solid line) shows
well-defined cathodic current peak at potential ca. −1.5 V, corre-ponding to the reduction of NAD+ [2,28,32,35,37,49,55], whereashe peak is absent in the background electrolyte (dotted line). Onhe other hand, no NAD+ reduction peak was recorded on the GC–Ptlectrode, and the current in the NAD+-containing solution wasower than that in the background electrolyte (Fig. 3a). This waso expect since Pt is the best hydrogen evolution catalyst, and theurrent recorded in the potential region of NAD+ reduction in Fig. 3as the sum of the NAD+ reduction and hydrogen evolution reactionHER) current. The decrease in the current in the NAD+-containing
olution is due to the blockage of the electrode surface (towardhe HER) by adsorbed NAD+, and possibly also produced 1,4-NADHE / VMSE
-2.0 -1.8 -1.6 -1.4 -1.2 -1.0 -0.8
j /
mA
cm
-2
-8
-6
-4
-2
0
E / VMSE
-2.0 -1.6 -1.2 -0.8
j / m
A c
m -
2
-0.3
-0.2
-0.1
0.0
(b)
(a)
ig. 3. Linear polarization voltammograms of (a) GC–Pt and (b) bare GC electrodeecorded in 0.1 M NaClO4 (dotted line) and in 0.1 M NaClO4 + 4 mM NAD+ (solid line).
Fig. 4. Time dependence of absorbance at 340 nm recorded during electrolysis of1 mM NAD+ in a batch electrochemical reactor employing GC–Pt Electrode A oper-ated at (�) −1.4 V, (�) −1.5 V and (©) −1.6 V.
and NAD2 molecules. Our FTIR-ATR measurements confirmed thepresence of these species on electrode surfaces, after electrolysis.
3.3. Regeneration of NADH on GC–Pt electrodes
The first set of measurements on 1,4-NADH regeneration byGC–Pt Electrode A focused on the investigation of the influence ofelectrode potential on the yield of 1,4-NADH, i.e. the percentageof 1,4-NADH recovered from NAD+. During the NAD+ electrolysis,the absorbance of the solution at 340 nm was measured by UV/visspectrophotometry in order to monitor the NAD+ conversion. Atthis wavelength, both NAD2 and 1,4-NADH absorb, but not NAD+
[33].Fig. 4 shows the time evolution of absorbance at 340 nm at
selected electrode potentials. At a low NAD+ reduction potential,−1.4 V (triangles), the conversion of NAD+ (to NAD2 and/or 1,4-NADH, Scheme 1) is rather slow and an absorbance plateau isreached after ca. 20 h of electrolysis. However, with an increasein potential from −1.4 to −1.5 V (squares) and to −1.6 V (circles),the rate of NAD+ reduction also increases and a plateau is reachedafter ca. 18 h and 6 h, respectively. Also, the final absorbance valuesat the latter two potentials, and thus the final (equilibrium) NAD2and 1,4-NADH concentrations produced, are higher than at −1.4 V,which is in accordance with conditions of electrochemical equilib-rium [33]. Based on these results it could be concluded that thekinetics of the NAD+ reduction reaction increases with an increasein electrode potential to more negative values. It is important toalso note that since the NAD+ reduction reaction is a mass-transportcontrolled and heterogeneous reaction [33], the NAD+ conversionkinetics can conveniently be controlled by controlling the hydro-dynamics regime in the reactor and by employing a GC–Pt workingelectrode of a smaller or larger surface area, respectively.
It is important to mention that the peak at 340 nm correspondsto the response of NAD2 and various NADH isomers. Thus, to deter-mine how much of the enzymatically active 1,4-NADH was actuallyformed by electrolysis, it was necessary to perform an activity assaybased on the conversion of dl-lipoamide to dihydrolipoamide inthe presence of lipoamide dehydrogenase [56]. The assay was firstvalidated and calibrated using commercially available NADH thatcontained 98% of enzymatically active 1,4-NADH.
At an electrolysis potential of −1.4 V, the percentage of enzymat-ically active 1,4-NADH recovered from NAD+ on GC–Pt Electrode Awas found to be 65 ± 1.9%. In comparison, only 32 ± 0.02% of enzy-matically active 1,4-NADH was recovered on pure GC electrode

I. Ali et al. / Chemical Engineering J
0
20
40
60
80
100
-1.4 -1.5 -1.6 -1.7 -1.8
E / VMSE
1,4
-NA
DH
re
co
ve
ry / %
Fig. 5. The percentage recovery of enzymatically active 1,4-NADH from NAD+ onGc
uoit
aotvaup9etnFpc9teotm
tcr1btvkiintsN1weIva
C–Pt Electrode A (Fig. 1), obtained by reduction of 1 mM NAD+ in a batch electro-hemical reactor operating at different electrode potentials.
nder the same experimental conditions [33]. Thus, the patterningf a GC surface by Pt nano-particles (Fig. 1a) resulted in a ca. 100%ncrease in the amount of 1,4-NADH regenerated in comparison tohe bare GC electrode surface.
We have already shown that the percentage of enzymaticallyctive 1,4-NADH recovered by reduction of NAD+ strongly dependsn electrolysis potential [32,33]. For example, on a bare GC elec-rode, with an increase in electrode potential to more negativealues, the recovery of 1,4-NADH also increases, and reaches 100%t a highly negative potential, −2.3 V [33] (in this reference wesed a term ‘yield’ instead of ‘recovery’, and considering that theurity of initial NAD+ was 98%, the obtained 1,4-NADH yield of8% actually meant a 100% recovery). To investigate the effect oflectrode potential on the recovery of 1,4-NADH using GC–Pt Elec-rode A, electrolysis experiments were also performed at potentialsegative of −1.4 V, and the corresponding results are presented inig. 5. The results demonstrate that by increasing the electrolysisotential from −1.4 V to −1.5 V, a significant increase in the per-entage of recovered 1,4-NADH was obtained, from 65 ± 1.9% to5 ± 0.7%, respectively. A further, increase in electrolysis potentialo −1.6 V resulted in even further increase in the 1,4-NADH recov-ry, to 100%. These experiments prove our hypothesis on the rolef Pt NPs on the GC surface as the hydrogen protonation (adsorp-ion, Pt–Hads) sites, as outlined in the introduction section of the
anuscript (Scheme 2).However, a further increase in electrolysis potential from −1.6 V
o −1.7 V and then to −1.8 V resulted in a decrease in the per-entage of active 1,4-NADH recovered, to 89 ± 1.5% and 76 ± 2.1%,espectively, Fig. 5. The trend in this figure, i.e. the presence of a,4-NADH recovery maximum, could be explained by consideringoth the mechanism of NAD+ reduction reaction (Scheme 1) andhe activity of Pt in the hydrogen evolution reaction (HER). As pre-iously explained, protonation of a NAD-radical is considered to beinetically slower than the dimerization of two neighboring rad-cals (Scheme 1), resulting in the production of an enzymaticallynactive NAD2 dimer. Hence, to increase the kinetics of the proto-ation step, it is preferable to provide (adsorb) hydrogen very closeo the site of NAD-radical formation. The higher the hydrogen (Hads)urface coverage, the higher the probability the surface-adsorbedAD-radical will be protonated, thus yielding enzymatically active,4-NADH. As already explained in our previous manuscript [33],ith an increase in electrode potential to more negative values, the
lectrode surface coverage by adsorbed hydrogen also increases.n addition, the increase in electrode potential to more negativealues also enhances the rate of electron transfer to both NAD+
nd NAD-radical (Scheme 1). Consequently, one could expect to
ournal 188 (2012) 173– 180 177
also see an increase in the yield of 1,4-NADH formed, which weindeed observed on a bare GC electrode [33]. However, Fig. 5 doesnot follow this trend, but shows a presence of a maximum at −1.6 V,obviously caused by the presence of Pt NPs on the GC surface. It iswell known that Pt is the best hydrogen evolution catalyst, whichform a Pt–Hads bond of an intermediate strength [57]. Therefore,with an increase in electrode potential from −1.4 V to −1.6 V (Fig. 5)the coverage of Pt NPs by adsorbed hydrogen (Pt–Hads) increases,while the second HER step, the production of molecular hydrogen,is still a slow step on Pt NPs at these low potentials (relative to thekinetics of the first step). Thus, in accordance with Scheme 1, thekinetics of the NAD-radical protonation step also improves with anincrease in the surface coverage of Hads, producing an increase inthe percentage of enzymatically active 1,4-NADH recovered fromNAD+, as opposed to the percentage of enzymatically inactive dimerNAD2 (Fig. 5). However, the further increase in electrode poten-tial from −1.6 V to −1.8 V favors the kinetics of the second HERstep, corresponding to the production of molecular hydrogen fromPt–Hads [57], thus diminishing the amount of Hads available for theNAD-radical protonation reaction. Consequently, the yield of active1,4-NADH decreases (Fig. 5).
Therefore, −1.6 V appears to be an optimum electrode poten-tial for the regeneration of enzymatically active 1,4-NADH fromNAD+ under the experimental conditions employed, giving a 100%recovery. For comparison, literature shows that reduction of NAD+
on a gold-amalgam unmodified electrode gave only ca. 10% enzy-matically active 1,4-NADH, a bare gold electrode gave 30% of active1,4-NADH [35], while the yield in active 1,4-NADH increased to 50%and 75% when an unmodified platinum and a cholesterol-modifiedgold-amalgam electrode were used [37]. When a GC electrode wasmodified by Au, the yield in active 1,4-NADH was 67% [49]. Fur-thermore, our previous results show that when a GC electrode wasmodified with Ru NPs, the yield of 1,4-NADH achieved was 96% (ora 98% recovery) [32]. Thus, it appears that the Pt-nano-patternedGC electrode offers so far the highest electrocatalytic activity inthe conversion of NAD+ to 1,4-NADH, effectively enabling a 100%conversion.
To investigate the influence of Pt NPs surface coverage and sizeon the percentage recovery of 1,4-NADH, three different GC–Ptelectrodes were produced, Fig. 1 (Electrode A) and Fig. 6 (ElectrodesB and C).
The micrograph and Pt NPs size distribution for Electrode A(Fig. 1), which was prepared by employing 5 potential scans, wasalready discussed previously in the text. However, doubling thenumber of potential polarization cycles, from 5 to 10, resulted inthe formation of bigger Pt NPs, characterized by a mean particlesize of 97 nm and higher Pt surface coverage, 6.5%, as shown inFig. 6a and b, respectively (Electrode B). Here, NPs are not sphericalin shape, but rather represent agglomerates of a few smaller NPs.In addition, the NP size distribution is wider than on the surfaceof Electrode A (Fig. 1b). A further increase in number of potentialpolarization cycles from 10 to 20 resulted in a further increase in thePt NPs size and surface coverage, yielding a mean NP size of 155 nmand a 13.2% surface coverage, Fig. 6c and d, respectively (ElectrodeC). Also, the NP size distribution has further widened in compari-son to Electrode A (Fig. 1b) and Electrode B (Fig. 6b), especially inthe lower NP diameter range, suggesting a significant degree of NPagglomeration.
As we documented in the previous section of the manuscript,the highest recovery (100%) of enzymatically active 1,4-NADH withElectrode A was obtained at an electrode potential of −1.6 V, Fig. 5.Therefore, the electrocatalytic activities of Electrodes B and C in 1,4-
NADH regeneration, were tested at the same electrode potentialand the results are presented in Fig. 7. It is clear from the fig-ure that Electrode A shows the highest catalytic activity towardregeneration of enzymatically active 1,4-NADH, yielding a 100%
178 I. Ali et al. / Chemical Engineering Journal 188 (2012) 173– 180
Fig. 6. (a and c) SEM micrographs of GC electrodes patterned with Pt NPs of differentsize and surface coverage. Pt NPs were electrodeposited on the GC surface from 0.5 MH2SO4 containing 1 mM PtHCl6 by cycling the electrode in a potential range from−0.6 to 0.1 V at a scan rate of 50 mV s−1 for 10 scans (Electrode B) and 20 scans(Electrode C). Plots (b) and (d) show the corresponding Pt NPs size distribution,respectively.
0
20
40
60
80
100
A B C
GC-Pt electrodes with different surface coverages
1,4
-NA
DH
re
co
ve
ry / %
Fig. 7. The percentage recovery of enzymatically active 1,4-NADH from NAD+ on
GC–Pt electrodes with different Pt NP size and surface coverage (Figs. 1 and 6), byreduction of 1 mM NAD+ in a batch electrochemical reactor at a potential of −1.6 V.recovery. On the other hand, Electrodes B and C enabled a 90% and80% recovery of 1,4-NADH, respectively. A possible reason for thehigher recovery of enzymatically active 1,4-NADH on Electrode Ais a smaller size and narrower size distribution of Pt NPs on the GCsurface (Fig. 1). Namely, with an increase in Pt NP size, the proba-bility that hydrogen adsorbed on Pt far from the Pt/GC interface isused for protonation of the NAD-radical decreases, while the prob-ability for production of molecular hydrogen increases. Thus, withthe increase in distance from the Pt/GC interface to the Pt NP center,the ratio of hydrogen used for NAD-radical protonation decreases,and thus the percentage of 1,4-NADH recovery decreases.
3.4. Regeneration of NADH on a GC–Ni electrode
The previous section demonstrated that GC–Pt Electrode A wascapable of achieving a 100% recovery of enzymatically active 1,4-NADH, from NAD+. Although the very high cost of 1,4-NADH canjustify the use of expensive Pt at the surface coverage relevant tothat on Electrode A (Fig. 1), it would be beneficial if Pt was replacedby a less expensive metal, while still achieving the high (preferably100%) 1,4-NADH recovery. Considering the hypothesis we postu-lated in the introduction of the manuscript (Scheme 2), i.e. the ideaof providing “active” (i.e. electrode-adsorbed) hydrogen, M-Hads,for the NAD-radical protonation by patterning a GC surface with agood hydrogen evolution catalyst, we made an attempt of replac-ing Pt by Ni. The rationale for choosing Ni is that, this metal has thehighest hydrogen evolution electrocatalytic activity among non-noble metals [57], it is considerably cheaper than Pt, it is muchmore abundant, and it is stable in a neutral media in the potentialregion of NADH regeneration.
Patterning of a GC surface by Ni NPs was done by cyclic voltam-metry, as described in the experimental part of the manuscript,and an example of the GC–Ni electrode surface is presented inFig. 8, together with the Ni NP size distribution. The micrographdemonstrates that a good Ni NPs surface coverage and uniform NPdistribution was achieved. The mean particle size is 83 nm and thesize distribution is relatively narrow, while the achieved surfacecoverage is 40.5%. Despite the large Ni NP surface coverage, it willbe demonstrated further in the text that the GC–Ni electrode shownin Fig. 8 is an extremely efficient 1,4-NADH regeneration electrode.Furthermore, to the best of the authors’ knowledge, the results inFig. 8 represent the smallest Ni NP size, the narrowest NP size dis-
tribution, and the most uniform Ni NP electrode surface coveragereported in the literature, on the formation of Ni NP on an electrodesurface by electrodeposition [50,58–61].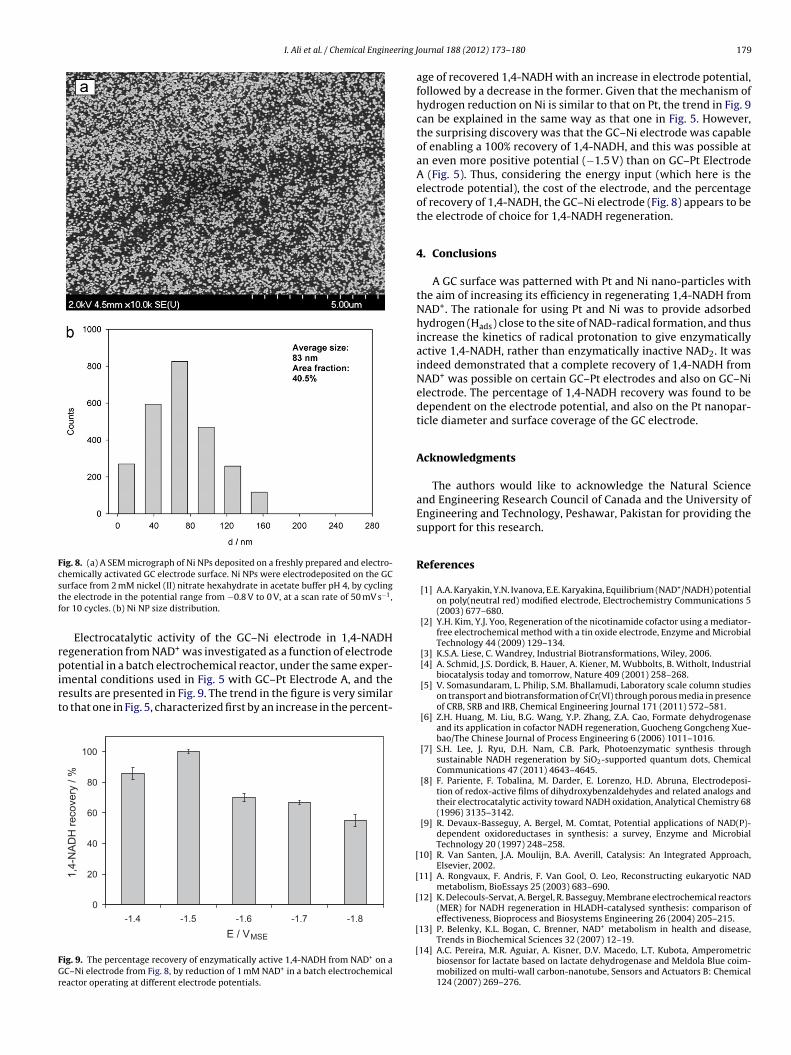
I. Ali et al. / Chemical Engineering J
Fig. 8. (a) A SEM micrograph of Ni NPs deposited on a freshly prepared and electro-chemically activated GC electrode surface. Ni NPs were electrodeposited on the GCstf
rpirt
FGr
urface from 2 mM nickel (II) nitrate hexahydrate in acetate buffer pH 4, by cyclinghe electrode in the potential range from −0.8 V to 0 V, at a scan rate of 50 mV s−1,or 10 cycles. (b) Ni NP size distribution.
Electrocatalytic activity of the GC–Ni electrode in 1,4-NADHegeneration from NAD+ was investigated as a function of electrode
otential in a batch electrochemical reactor, under the same exper-mental conditions used in Fig. 5 with GC–Pt Electrode A, and theesults are presented in Fig. 9. The trend in the figure is very similaro that one in Fig. 5, characterized first by an increase in the percent-
0
20
40
60
80
100
-1.4 -1.5 -1.6 -1.7 -1.8
E / VMSE
1,4
-NA
DH
re
co
ve
ry / %
ig. 9. The percentage recovery of enzymatically active 1,4-NADH from NAD+ on aC–Ni electrode from Fig. 8, by reduction of 1 mM NAD+ in a batch electrochemical
eactor operating at different electrode potentials.
[
[
[
[
[
ournal 188 (2012) 173– 180 179
age of recovered 1,4-NADH with an increase in electrode potential,followed by a decrease in the former. Given that the mechanism ofhydrogen reduction on Ni is similar to that on Pt, the trend in Fig. 9can be explained in the same way as that one in Fig. 5. However,the surprising discovery was that the GC–Ni electrode was capableof enabling a 100% recovery of 1,4-NADH, and this was possible atan even more positive potential (−1.5 V) than on GC–Pt ElectrodeA (Fig. 5). Thus, considering the energy input (which here is theelectrode potential), the cost of the electrode, and the percentageof recovery of 1,4-NADH, the GC–Ni electrode (Fig. 8) appears to bethe electrode of choice for 1,4-NADH regeneration.
4. Conclusions
A GC surface was patterned with Pt and Ni nano-particles withthe aim of increasing its efficiency in regenerating 1,4-NADH fromNAD+. The rationale for using Pt and Ni was to provide adsorbedhydrogen (Hads) close to the site of NAD-radical formation, and thusincrease the kinetics of radical protonation to give enzymaticallyactive 1,4-NADH, rather than enzymatically inactive NAD2. It wasindeed demonstrated that a complete recovery of 1,4-NADH fromNAD+ was possible on certain GC–Pt electrodes and also on GC–Nielectrode. The percentage of 1,4-NADH recovery was found to bedependent on the electrode potential, and also on the Pt nanopar-ticle diameter and surface coverage of the GC electrode.
Acknowledgments
The authors would like to acknowledge the Natural Scienceand Engineering Research Council of Canada and the University ofEngineering and Technology, Peshawar, Pakistan for providing thesupport for this research.
References
[1] A.A. Karyakin, Y.N. Ivanova, E.E. Karyakina, Equilibrium (NAD+/NADH) potentialon poly(neutral red) modified electrode, Electrochemistry Communications 5(2003) 677–680.
[2] Y.H. Kim, Y.J. Yoo, Regeneration of the nicotinamide cofactor using a mediator-free electrochemical method with a tin oxide electrode, Enzyme and MicrobialTechnology 44 (2009) 129–134.
[3] K.S.A. Liese, C. Wandrey, Industrial Biotransformations, Wiley, 2006.[4] A. Schmid, J.S. Dordick, B. Hauer, A. Kiener, M. Wubbolts, B. Witholt, Industrial
biocatalysis today and tomorrow, Nature 409 (2001) 258–268.[5] V. Somasundaram, L. Philip, S.M. Bhallamudi, Laboratory scale column studies
on transport and biotransformation of Cr(VI) through porous media in presenceof CRB, SRB and IRB, Chemical Engineering Journal 171 (2011) 572–581.
[6] Z.H. Huang, M. Liu, B.G. Wang, Y.P. Zhang, Z.A. Cao, Formate dehydrogenaseand its application in cofactor NADH regeneration, Guocheng Gongcheng Xue-bao/The Chinese Journal of Process Engineering 6 (2006) 1011–1016.
[7] S.H. Lee, J. Ryu, D.H. Nam, C.B. Park, Photoenzymatic synthesis throughsustainable NADH regeneration by SiO2-supported quantum dots, ChemicalCommunications 47 (2011) 4643–4645.
[8] F. Pariente, F. Tobalina, M. Darder, E. Lorenzo, H.D. Abruna, Electrodeposi-tion of redox-active films of dihydroxybenzaldehydes and related analogs andtheir electrocatalytic activity toward NADH oxidation, Analytical Chemistry 68(1996) 3135–3142.
[9] R. Devaux-Basseguy, A. Bergel, M. Comtat, Potential applications of NAD(P)-dependent oxidoreductases in synthesis: a survey, Enzyme and MicrobialTechnology 20 (1997) 248–258.
10] R. Van Santen, J.A. Moulijn, B.A. Averill, Catalysis: An Integrated Approach,Elsevier, 2002.
11] A. Rongvaux, F. Andris, F. Van Gool, O. Leo, Reconstructing eukaryotic NADmetabolism, BioEssays 25 (2003) 683–690.
12] K. Delecouls-Servat, A. Bergel, R. Basseguy, Membrane electrochemical reactors(MER) for NADH regeneration in HLADH-catalysed synthesis: comparison ofeffectiveness, Bioprocess and Biosystems Engineering 26 (2004) 205–215.
13] P. Belenky, K.L. Bogan, C. Brenner, NAD+ metabolism in health and disease,
Trends in Biochemical Sciences 32 (2007) 12–19.14] A.C. Pereira, M.R. Aguiar, A. Kisner, D.V. Macedo, L.T. Kubota, Amperometricbiosensor for lactate based on lactate dehydrogenase and Meldola Blue coim-mobilized on multi-wall carbon-nanotube, Sensors and Actuators B: Chemical124 (2007) 269–276.

1 ering J
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
80 I. Ali et al. / Chemical Engine
15] N. Pollak, C. Dolle, M. Ziegler, The power to reduce: pyridine nucleotides –small molecules with a multitude of functions, Biochemical Journal 402 (2007)205–218.
16] A.A. Sauve, NAD+ and vitamin B3. From metabolism to therapies, Journal ofPharmacology and Experimental Therapeutics 324 (2008) 883–893.
17] H.K. Chenault, G.M. Whitesides, Regeneration of nicotinamide cofactors foruse in organic synthesis, Applied Biochemistry and Biotechnology 14 (1987)147–197.
18] J.N. Burnett, A.L. Underwood, Electrochemical reduction of diphosphopyridinenucleotide, Biochemistry 4 (1965) 2060–2064.
19] C.O. Schmakel, K.S.V. Santhanam, P.J. Elving, Nicotinamide adenine dinucle-otide (NAD+) and related compounds. Electrochemical redox pattern andallied chemical behavior, Journal of the American Chemical Society 97 (1975)5083–5092.
20] C.O. Schmakel, M.A. Jensen, P.J. Elving, Effect of the adenine moiety onthe electrochemical behavior of nicotinamide adenine dinucleotide; possi-ble reduction of the adenine, Bioelectrochemistry and Bioenergetics 5 (1978)625–634.
21] W.T. Bresnahan, J. Moiroux, Z. Samec, P.J. Elving, Nucleotides and relatedsubstances: conformation in solution and at solution|electrode interfaces, Bio-electrochemistry and Bioenergetics 7 (1980) 125–152.
22] W.T. Bresnahan, P.J. Elving, The role of adsorption in the initial one-electronelectrochemical reduction of nicotinamide adenine dinucleotide (NAD+), Jour-nal of the American Chemical Society 103 (1981) 2379–2386.
23] H. Jaegfeldt, 420 – a study of the products formed in the electrochemi-cal reduction of nicotinamide-adenine-dinucleotide, Bioelectrochemistry andBioenergetics 8 (1981) 355–370.
24] P.J. Elving, W.T. Bresnahan, J. Moiroux, Z. Samec, NAD/NADH as a model redoxsystem: mechanism, mediation, modification by the environment, Bioelectro-chemistry and Bioenergetics 9 (1982) 365–378.
25] M.A. Jensen, W.T. Bresnahan, P.J. Elving, Comparative adsorption ofadenine and nicotinamide adenine dinucleotide (NAD+) at an aqueoussolution|mercury interface, Bioelectrochemistry and Bioenergetics 11 (1983)299–306.
26] J. Moiroux, S. Deycard, T. Malinski, Electrochemical reduction of NAD+ andpyridinium cations adsorbed at the mercury/water interface. Electrochemicalbehavior of adsorbed pyridinyl radicals, Journal of Electroanalytical Chemistry194 (1985) 99–108.
27] M. Studnickova, H. Paulova-Klukanov, J. Turanek, J. Kovar, Reduction of NAD+
and NADP+ and reductive cleavage of NADH and NADPH yielding NAD andNADP, Journal of Electroanalytical Chemistry 252 (1988) 383–394.
28] J. Moiroux, P.J. Elving, Adsorption phenomena in the NAD+/NADH system atglassy carbon electrodes, Journal of Electroanalytical Chemistry 102 (1979)93–108.
29] B. Qi, L. He, X. Bo, H. Yang, L. Guo, Electrochemical preparation of free-standingfew-layer graphene through oxidation–reduction cycling, Chemical Engineer-ing Journal 171 (2011) 334–340.
30] A. Damian, K. Maloo, S. Omanovic, Direct electrochemical regeneration of NADHon Au, Cu and Pt–Au electrodes, Chemical and Biochemical Engineering Quar-terly 21 (2007) 21–32.
31] F. Man, S. Omanovic, A kinetic study of NAD+ reduction on a ruthenium modi-fied glassy carbon electrode, Journal of Electroanalytical Chemistry 568 (2004)301–313.
32] A. Azem, F. Man, S. Omanovic, Direct regeneration of NADH on a rutheniummodified glassy carbon electrode, Journal of Molecular Catalysis A: Chemical219 (2004) 283–299.
33] I. Ali, B. Soomro, S. Omanovic, Electrochemical regeneration of NADH on a glassycarbon electrode surface: the influence of electrolysis potential, Electrochem-istry Communications 13 (2011) 562–565.
34] K. Takamura, A. Mori, F. Kusu, 411 – role of adsorption in the electrochemicalbehaviour of nicotinamide adenine dinucleotide at a gold electrode, Bioelec-trochemistry and Bioenergetics 8 (1981) 229–238.
35] A. Damian, S. Omanovic, Electrochemical reduction of NAD+ on a polycrys-talline gold electrode, Journal of Molecular Catalysis A: Chemical 253 (2006)222–233.
36] Y.T. Long, H.Y. Chen, Electrochemical regeneration of coenzyme NADH on ahistidine modified silver electrode, Journal of Electroanalytical Chemistry 440(1997) 239–242.
37] S.H. Baik, C. Kang, I.C. Jeon, S.E. Yun, Direct electrochemical regeneration ofNADH from NAD+ using cholesterol-modified gold amalgam electrode, Biotech-
nology Techniques 13 (1999) 1–5.38] K. Cheikhou, T. Tzedakis, Electrochemical microreactor for chiral synthesesusing the cofactor NADH, AIChE Journal 54 (2008) 1365–1376.
39] X. Chen, J.M. Fenton, R.J. Fisher, R.A. Peattie, Evaluation of in situ electroen-zymatic regeneration of coenzyme NADH in packed bed membrane reactors:
[
ournal 188 (2012) 173– 180
biosynthesis of lactate, Journal of the Electrochemical Society 151 (2004)E56–E60.
40] S. Kim, S.-E. Yun, C. Kang, The formation of a diaphorase enzyme multilayerbound to a self-assembled monolayer for the mediated enzyme-catalyzedreduction of NAD+, Electrochemistry Communications 1 (1999) 151–154.
41] S.B. Sobolov, M.D. Leonida, A. Bartoszko-Malik, K.I. Voivodov, F. McKinney, J.Kim, A.J. Fry, Cross-linked LDH crystals for lactate synthesis coupled to elec-troenzymatic regeneration of NADH, Journal of Organic Chemistry 61 (1996)2125–2128.
42] K.I. Voivodov, S.B. Sobolov, M. Draganoiu Leonida, A.J. Fry, Enzymatic trans-formation in an electrochemical reactor utilizing a redox mediator-modifiedenzyme electrode for NAD(H) regeneration, Bioorganic and Medicinal Chem-istry Letters 5 (1995) 681–686.
43] A.A. Karyakin, O.A. Bobrova, E.E. Karyakina, Electroreduction of NAD+ to enzy-matically active NADH at poly(neutral red) modified electrodes, Journal ofElectroanalytical Chemistry 399 (1995) 179–184.
44] A.J. Fry, S.B. Sobolov, M.D. Leonida, K.I. Voivodov, Electroenzymatic synthe-sis (regeneration of NADH coenzyme): use of nafion ion exchange films forimmobilization of enzyme and redox mediator, Tetrahedron Letters 35 (1994)5607–5610.
45] C. Kohlmann, W. Maerkle, S. Luetz, Electroenzymatic synthesis, Journal ofMolecular Catalysis B: Enzymatic 51 (2008) 57–72.
46] F. Hollmann, A. Schmid, Electrochemical regeneration of oxidoreductases forcell-free biocatalytic redox reactions, Biocatalysis and Biotransformation 22(2004) 63–88.
47] A. Salimi, M. Izadi, R. Hallaj, S. Soltanian, H. Hadadzadeh, Electrocatalytic reduc-tion of NAD+ at glassy carbon electrode modified with single-walled carbonnanotubes and Ru(III) complexes, Journal of Solid State Electrochemistry 13(2009) 485–496.
48] E. Siu, K. Won, B.P. Chan, Electrochemical regeneration of NADH using conduc-tive vanadia–silica xerogels, Biotechnology Progress 23 (2007) 293–296.
49] G. Rahman, J.Y. Lim, K.D. Jung, O.S. Joo, NAD+ hydrogenation on Au electrodedeposited on modified glassy carbon, Electrochemistry Communications 12(2010) 1371–1374.
50] A. Salimi, E. Sharifi, A. Noorbakhsh, S. Soltanian, Direct voltammetry and elec-trocatalytic properties of hemoglobin immobilized on a glassy carbon electrodemodified with nickel oxide nanoparticles, Electrochemistry Communications8 (2006) 1499–1508.
51] S. Szabo, I. Bakos, Investigation of ruthenium deposition onto a platinized plat-inum electrode in sulfuric acid media, Journal of Electroanalytical Chemistry230 (1987) 233–240.
52] J.B. Raoof, R. Ojani, S.A. Esfeden, S.R. Nadimi, Fabrication of bimetallic Cu/Ptnanoparticles modified glassy carbon electrode and its catalytic activity towardhydrogen evolution reaction, International Journal of Hydrogen Energy 35(2010) 3937–3944.
53] M. Farcas, N.P. Cosman, D.K. Ting, S.G. Roscoe, S. Omanovic, A comparativestudy of electrochemical techniques in investigating the adsorption behaviourof fibrinogen on platinum, Journal of Electroanalytical Chemistry 649 (2010)206–218.
54] T.S. Mkwizu, M.K. Mathe, I. Cukrowski, Automated electrodeposition ofbimetallic noble-metal nanoclusters via redox-replacement reactions for elec-trocatalysts, ECS Transactions 19 (2009) 97–113.
55] Y. Nakamura, S.I. Suye, J.I. Kira, H. Tera, I. Tabata, M. Senda, Electron-transferfunction of NAD+ immobilized alginic acid, Biochimica et Biophysica Acta –General Subjects 1289 (1996) 221–225.
56] V. Massey, Q.H. Gibson, C. Veeger, Intermediates in the catalytic action of lipoyldehydrogenase (diaphorase), Biochemical Journal 77 (1960) 340–341.
57] S. Trasatti, Work function, electronegativity, and electrochemical behaviour ofmetals. III. Electrolytic hydrogen evolution in acid solutions, Journal of Electro-analytical Chemistry 39 (1972) 163–184.
58] N.F. Heinig, N. Kharbanda, M.R. Pynenburg, X.J. Zhou, G.A. Schultz, K.T. Leung,The growth of nickel nanoparticles on conductive polymer composite elec-trodes, Materials Letters 62 (2008) 2285–2288.
59] A. Mohammadi, A. Bayandori Moghaddam, M. Kazemzad, R. Dinarvand, J.Badraghi, Synthesis of nickel oxides nanoparticles on glassy carbon as anelectron transfer facilitator for horseradish peroxidase: direct electron trans-fer and H2O2 determination, Materials Science and Engineering C 29 (2009)1752–1758.
60] W. Zhu, G. Wang, X. Hong, X. Shen, D. Li, X. Xie, Metal nanoparticle chainsembedded in TiO2 nanotubes prepared by one-step electrodeposition, Elec-
trochimica Acta 55 (2009) 480–484.61] A. Bayandori Moghaddam, M.R. Ganjali, A.A. Saboury, A.A. Moosavi-Movahedi,P. Norouzi, Electrodeposition of nickel oxide nanoparticles on glassy carbonsurfaces: application to the direct electron transfer of tyrosinase, Journal ofApplied Electrochemistry 38 (2008) 1233–1239.













![REVIEW Development in Voltammetric Analysis with Chemically … cyclodextrin/Nafion modified glassy carbon electrode was investigated [67]. The tetrachlorophthalate(III) was bound](https://static.fdocuments.in/doc/165x107/606b582dd7f4be62e9012efa/review-development-in-voltammetric-analysis-with-chemically-cyclodextrinnafion.jpg)



