
Differential Time-Dependent Inactivation of P450 3A4 and P450 3A5 by Raloxifene: A Key Role for C239...
9
Differential Time-Dependent Inactivation of P450 3A4 and P450 3A5 by Raloxifene: A Key Role for C239 in Quenching Reactive Intermediates Josh T. Pearson, †,§ Jan L. Wahlstrom, †,§ Leslie J. Dickmann, † Santosh Kumar, ‡ James R. Halpert, ‡ Larry C. Wienkers, † Robert S. Foti, † and Dan A. Rock* ,† Department of Pharmacokinetics and Drug Metabolism, Amgen Inc., 1201 Amgen Court West, Seattle, Washington 98119-3105, and Department of Pharmacology and Toxicology, UniVersity of Texas Medical Branch, 301 UniVersity BouleVard, GalVeston, Texas 77555-1031 ReceiVed June 7, 2007 The role of C239 as the active-site residue responsible for forming the covalent linkage with raloxifene during P450 3A4 time-dependent inactivation (TDI) was recently identified. The corresponding residue in CYP3A5 is S239, and when the potential for TDI in P450 3A5 was investigated, only reversible inhibition was observed against midazolam and testosterone, with median inhibitory concentration (IC 50 ) values of 2.4 and 2.9 µM, respectively. In a similar fashion, when C239 was replaced with alanine in P450 3A4, TDI was successfully engineered out, and the reversible inhibition was characterized by IC 50 values of 3.7 and 3.5 µM against midazolam and testosterone, respectively. Metabolism studies confirmed that the reactive diquinone methide intermediate required for P450 3A4 inactivation formed in all of the P450 3A enzymes investigated. Furthermore, the absence of TDI in P450 3A5 led to an increase in the formation of GSH-related adducts of raloxifene compared with that for P450 3A4. Consequently, the absence of the nucleophilic cysteine leads to differential TDI and generation of reactive metabolites in the P450 3A enzyme, providing the foundation for pharmacogenetics that contributes to individual differences in susceptibility to adverse drug reactions. Introduction The pharmacogenetics associated with cytochrome P450 enzymes represents an important determinant in relating inter- individual variability of drug exposure to clinical pharmacologi- cal and toxicological end points. Initial insights into the role of genetic variability of P450 enzymes were first linked with the P450 2D6 polymorphism (1, 2). In this instance, patients receiving debrisoquine for the treatment of high blood pressure who also possessed the poor metabolizer phenotype of P450 2D6 were susceptible to an unsafe drop in blood pressure due to the reduced clearance of debrisoquine (3, 4). Additional population studies revealed that the inactive P450 2D6 enzyme was based on a homozygous null variant found in 5–10% of Caucasians and 1–2% of Southeast Asians (5). More recently, enzyme activities from single amino acid variants were linked with over-anticoagulation due in part to P450 2C9 polymor- phism in patients taking (S)-warfarin (6, 7). Despite a strong understanding of the importance of phar- macogenetics and associated toxicity based on varying exposure to parent or active metabolites, there is a gap in linking potential mechanisms of toxicity related to the effects of pharmacoge- netics on the generation of reactive intermediates. P450-mediated bioactivation generally leads to formation of reactive metabolites that inactivate the enzyme via a covalent linkage with residue(s) of the P450 (apoprotein) or through modification of the heme by either direct covalent linkage or coordination of the heme iron to form a metabolic intermediate complex (MIC). 1 Alter- natively, bioactivated intermediates may migrate from the endoplasmic reticulum to react with other cellular constituents, including DNA. Both events have the propensity to increase the risk of adverse drug reactions (8). In the liver, P450 3A4 and P450 3A5 account for ap- proximately 50% of the total P450 content (9). Moreover, P450 3A4 and P450 3A5 are the predominant P450 contributors to metabolism in the adult human liver, accounting for 40–60% of the oxidative metabolism of marketed drugs. While the importance of the role of P450 3A4 is well characterized for both liver and intestinal metabolism, the role of P450 3A5 in drug metabolism remains controversial. Initial reports suggested that P450 3A5 content was minimal, representing up to 20% of adult human livers (10). More recent data suggest that P450 3A5 may account for more than 50% of the P450 3A content in 25–30% of human livers (11). Although P450 3A4 and P450 3A5 share ∼85% sequence homology, enzyme activity and regioselectivity differences have been observed for a variety of substrates, suggesting that differing active-site architecture may lead to distinct inhibition profiles for the two P450 3A enzymes. Several studies have reported preferential inactivation of P450 3A4 over P450 3A5. Notably, mifepristone exhibited time- dependent inactivation (TDI) of P450 3A4 but not P450 3A5 (12). Furthermore, the potency of MIC formation is increased in P450 3A4 compared with P450 3A5 for diltiazem, erythro- mycin, nicardipine, and verapamil (13, 14). Reaction rates and * To whom correspondence should be addressed: 1201 Amgen Court West, Seattle, WA 98119. Tel: (206) 265-7139. Fax: (206) 265-1149. E-mail: [email protected]. † Amgen Inc. ‡ University of Texas Medical Branch. § These authors contributed equally to this work. 1 Abbreviations: MIC, metabolic intermediate complex; DLPC, L-R- dilauroylphosphatidylcholine; DLPS, L-R-dilauroyl-sn-glycero-3-phospho- serine; DOPC, L-R-dioleoyl-sn-glycero-3-phosphocholine; TDI, time- dependent inactivation; TFA, trifluoroacetic acid. Chem. Res. Toxicol. 2007, 20, 1778–1786 1778 10.1021/tx700207u CCC: $37.00 2007 American Chemical Society Published on Web 11/15/2007
Transcript of Differential Time-Dependent Inactivation of P450 3A4 and P450 3A5 by Raloxifene: A Key Role for C239...

Differential Time-Dependent Inactivation of P450 3A4 and P4503A5 by Raloxifene: A Key Role for C239 in Quenching Reactive
Intermediates
Josh T. Pearson,†,§ Jan L. Wahlstrom,†,§ Leslie J. Dickmann,† Santosh Kumar,‡
James R. Halpert,‡ Larry C. Wienkers,† Robert S. Foti,† and Dan A. Rock*,†
Department of Pharmacokinetics and Drug Metabolism, Amgen Inc., 1201 Amgen Court West,Seattle, Washington 98119-3105, and Department of Pharmacology and Toxicology, UniVersity of Texas Medical
Branch, 301 UniVersity BouleVard, GalVeston, Texas 77555-1031
ReceiVed June 7, 2007
The role of C239 as the active-site residue responsible for forming the covalent linkage with raloxifeneduring P450 3A4 time-dependent inactivation (TDI) was recently identified. The corresponding residuein CYP3A5 is S239, and when the potential for TDI in P450 3A5 was investigated, only reversibleinhibition was observed against midazolam and testosterone, with median inhibitory concentration (IC50)values of 2.4 and 2.9 µM, respectively. In a similar fashion, when C239 was replaced with alanine inP450 3A4, TDI was successfully engineered out, and the reversible inhibition was characterized by IC50values of 3.7 and 3.5 µM against midazolam and testosterone, respectively. Metabolism studies confirmedthat the reactive diquinone methide intermediate required for P450 3A4 inactivation formed in all ofthe P450 3A enzymes investigated. Furthermore, the absence of TDI in P450 3A5 led to an increase inthe formation of GSH-related adducts of raloxifene compared with that for P450 3A4. Consequently, theabsence of the nucleophilic cysteine leads to differential TDI and generation of reactive metabolites inthe P450 3A enzyme, providing the foundation for pharmacogenetics that contributes to individualdifferences in susceptibility to adverse drug reactions.
Introduction
The pharmacogenetics associated with cytochrome P450enzymes represents an important determinant in relating inter-individual variability of drug exposure to clinical pharmacologi-cal and toxicological end points. Initial insights into the role ofgenetic variability of P450 enzymes were first linked with theP450 2D6 polymorphism (1, 2). In this instance, patientsreceiving debrisoquine for the treatment of high blood pressurewho also possessed the poor metabolizer phenotype of P4502D6 were susceptible to an unsafe drop in blood pressure dueto the reduced clearance of debrisoquine (3, 4). Additionalpopulation studies revealed that the inactive P450 2D6 enzymewas based on a homozygous null variant found in 5–10% ofCaucasians and 1–2% of Southeast Asians (5). More recently,enzyme activities from single amino acid variants were linkedwith over-anticoagulation due in part to P450 2C9 polymor-phism in patients taking (S)-warfarin (6, 7).
Despite a strong understanding of the importance of phar-macogenetics and associated toxicity based on varying exposureto parent or active metabolites, there is a gap in linking potentialmechanisms of toxicity related to the effects of pharmacoge-netics on the generation of reactive intermediates. P450-mediatedbioactivation generally leads to formation of reactive metabolitesthat inactivate the enzyme via a covalent linkage with residue(s)of the P450 (apoprotein) or through modification of the heme
by either direct covalent linkage or coordination of the hemeiron to form a metabolic intermediate complex (MIC).1 Alter-natively, bioactivated intermediates may migrate from theendoplasmic reticulum to react with other cellular constituents,including DNA. Both events have the propensity to increasethe risk of adverse drug reactions (8).
In the liver, P450 3A4 and P450 3A5 account for ap-proximately 50% of the total P450 content (9). Moreover, P4503A4 and P450 3A5 are the predominant P450 contributors tometabolism in the adult human liver, accounting for 40–60%of the oxidative metabolism of marketed drugs. While theimportance of the role of P450 3A4 is well characterized forboth liver and intestinal metabolism, the role of P450 3A5 indrug metabolism remains controversial. Initial reports suggestedthat P450 3A5 content was minimal, representing up to 20% ofadult human livers (10). More recent data suggest that P4503A5 may account for more than 50% of the P450 3A contentin 25–30% of human livers (11). Although P450 3A4 and P4503A5 share ∼85% sequence homology, enzyme activity andregioselectivity differences have been observed for a variety ofsubstrates, suggesting that differing active-site architecture maylead to distinct inhibition profiles for the two P450 3A enzymes.
Several studies have reported preferential inactivation of P4503A4 over P450 3A5. Notably, mifepristone exhibited time-dependent inactivation (TDI) of P450 3A4 but not P450 3A5(12). Furthermore, the potency of MIC formation is increasedin P450 3A4 compared with P450 3A5 for diltiazem, erythro-mycin, nicardipine, and verapamil (13, 14). Reaction rates and
* To whom correspondence should be addressed: 1201 Amgen CourtWest, Seattle, WA 98119. Tel: (206) 265-7139. Fax: (206) 265-1149.E-mail: [email protected].
† Amgen Inc.‡ University of Texas Medical Branch.§ These authors contributed equally to this work.
1 Abbreviations: MIC, metabolic intermediate complex; DLPC, L-R-dilauroylphosphatidylcholine; DLPS, L-R-dilauroyl-sn-glycero-3-phospho-serine; DOPC, L-R-dioleoyl-sn-glycero-3-phosphocholine; TDI, time-dependent inactivation; TFA, trifluoroacetic acid.
Chem. Res. Toxicol. 2007, 20, 1778–17861778
10.1021/tx700207u CCC: $37.00 2007 American Chemical SocietyPublished on Web 11/15/2007

specific differences in regioselectivity between the two P4503A enzymes may offer a partial explanation of the aforemen-tioned examples. Therefore, exposure to reactive metabolitesfrom P450 3A depends not only on the enzyme’s capacity tometabolize the molecular site necessary to produce bioactivationbut also on the appropriate enzyme architecture to dictatewhether the electrophilic species acts locally (at the P450) orescapes the active site to react with other cellular constituents.
Recent studies from this laboratory established a role for C239in the mechanism-based inactivation of P450 3A4 by the second-generation selective estrogen receptor modulator raloxifene (15).From sequence alignments, S239 in P450 3A5 corresponds toC239 in P450 3A4. In light of the potential for differences inmetabolic profiles due to differences in P450 3A active-sitearchitecture, it was postulated that this variability could alsoextend to structure-based mechanisms of TDI. In the currentstudy, we hypothesized that P450 3A5 may exhibit reduced orminimal TDI inactivation at comparable rates of raloxifenemetabolism, leading to an enhanced potential for systemicexposure to bioactivated intermediates. In view of this, the P4503A genotype could play an important role in raloxifene oxidationand reactive metabolite generation.
Experimental Procedures
Materials. Raloxifene, midazolam, testosterone, reduced glu-tathione, CHAPS, potassium HEPES, MgCl2, guanidine hydro-chloride, CaCl2, ZnSO4, and NADPH were purchased from Sigma-Aldrich (St. Louis, MO). Supersomes containing P450 3A4 or P4503A5 were purchased from BD Gentest (Woburn, MA). A ZorbaxRX-C8 column (2.1 × 250 mm) was obtained from Agilent (PaloAlto, CA). The QuikChange XL site-directed mutagenesis kit wasobtained from Stratagene (La Jolla, CA). For P450 3A5, oligo-nucleotide primers for the polymerase chain reaction (PCR) wereobtained from Sigma-Genosys (The Woodlands, TX).
Instrumentation. For metabolite identification studies, an LTQmass spectrometer (Thermo Scientific, San Jose, CA) was connectedon-line with an Agilent 1100 series HPLC system equipped with adegasser, pumps, an autoinjector, a column oven, and a diode-arraydetector. For the analysis of midazolam and testosterone TDI studiesas well as raloxifene substrate depletion studies, a 4000Q-Trapsystem (Applied Biosystems, San Jose, CA) was connected to aShimadzu HPLC system equipped with a degasser.
Mutagenesis of P450 3A4 and P450 3A5. A human P450 3A4NF14 construct containing a C-terminal polyhistidine tag clonedinto the pCWori vector was a gift from Professor William Atkins(University of Washington). The C239A mutation was introducedinto the P450 3A4 clone with the sense 5′-ctc atc cca att ctt gaagta tta aat atc gct gtg ttt cca aga gaa gtt ac-3′ and antisense 5′-gtaact tct ctt gga aac aca gcg ata ttt aat act tca aga att ggg atg ag-3′oligonucleotides according to the manufacturer’s suggested protocol.The mutation was verified by whole-protein mass spectrometryusing purified protein. The S239C mutation was introduced intothe P450 3A5 clone with the sense 5′-gca tta aat gtc tgt ctg tttcca-3′ and antisense 5′-tgg aaa cag aca gac att taa tgc-3′ oligo-nucleotides according to the manufacturer’s suggested protocol. Toconfirm the formation of the desired mutation and verify the absenceof any unintended mutations, the construct was sequenced at theUniversity of Texas Medical Branch Protein Chemistry Laboratory(Galveston, TX).
Protein Expression and Purification. P450 3A4 enzymes wereexpressed in the Escherichia coli DH5R strain. Complete expressionconditions were the same as those described by Gillam et al. (16).Briefly, E. coli was freshly transformed with either the P450 3A4wild-type or the C239A mutant plasmid, plated under ampicillin(50 µg/mL) selection, and grown overnight at 37 °C. Cells wereshaken at 180 rpm for 48 h at 27 °C in a 2.8 L Fernbach flask.Pelleted cells were resuspended in buffer containing 50 mMpotassium phosphate (pH 7.4), 500 mM NaCl, 20% glycerol, 50
µM testosterone (a stabilizing ligand), 20 mM �-mercaptoethanol,1% Emulgen 911, and Sigma protease inhibitor cocktail (0.5 mL/Lof initial culture volume). Cells were lysed using a French pressoperated at 10000 psi and then spun at 150000g. The supernatantwas loaded directly onto NTA-Ni resin equilibrated with 50 mMpotassium phosphate (pH 7.4), 500 mM NaCl, 20% glycerol, 50µM testosterone, and 0.2% Emulgen 911. The column was washedwith 200 mL of wash buffer containing 50 mM potassium phosphate(pH 7.4), 20% glycerol, 20 mM imidazole, 0.2% cholate, and 50µM testosterone. P450 3A4 was eluted from the column with buffercontaining 50 mM potassium phosphate (pH 7.4), 20% glycerol,500 mM imidazole, and 0.2% cholate. The eluted protein wasdialyzed against 100 mM potassium phosphate (pH 7.4) in 20%glycerol and stored at -80 °C. P450 3A5 and the S239C mutantwere expressed as His-tagged proteins in E. coli TOPP3 and purifiedusing a Ni-NTA affinity column as described previously (17).Protein concentrations were determined using the Bradford proteinassay kit (BioRad, Hercules, CA). The specific contents of P4503A5 and S239C were 17.4 and 11.6 nmol of P450/mg of protein,respectively. Rat cytochrome b5 and P450 reductase were expressedand purified as described previously (18, 19).
In Silico Modeling. Maestro (Schrodinger, Portland, OR) wasused to generate a homology model of P450 3A5 based on thecrystal structure of P450 3A4 (PDB entry 1TQN) (20). Sequencealignment was performed using BIOEDIT version 7.0.5.2 (IbisTherapeutics, Carlsbad, CA). The optimal alignment was obtainedby allowing the ends of both sequences to slide. The Induced FitDocking module of Maestro was applied to raloxifene with eachof P450 3A4 and P450 3A5 in order to provide a relative idea ofraloxifene binding orientations in each enzyme.
Incubations with Purified P450. Enzyme reconstitution has beendescribed previously (15, 16). Briefly, P450 (3A4, 3A5, 3A4C239A,or 3A5S239C) (100 pmol) was combined with NADPH-P450reductase (200 pmol), cytochrome b5 (100 pmol), 0.1 mg/mL Chaps,20 µg/mL liposomes [1:1:1 (w/w/w) DLPC/DOPC/DLPS], 3 mMreduced glutathione, 50 mM potassium HEPES (pH 7.4), and 30mM MgCl2 in a total volume of 0.5 mL. Raloxifene was added toa final concentration of 50 µM. Each reconstituted P450 systemwas incubated for 10 min at 37 °C prior to the addition of 1 mMNADPH. Each reaction was allowed to proceed for 20 min unlessotherwise noted.
Time-Dependent Inactivation of P450 3A Enzymes. Primaryreactions (200 µL) were carried out by incubating a P450 3Aenzyme or the corresponding mutant (10 pmol) with differentconcentrations (0–20 µM) of raloxifene, and for each reaction, 5µL aliquots were removed at 0, 2, 4, 6, 8, and 10 min and placedinto secondary incubations containing 1 mM NADPH and 25 µMmidazolam. The secondary reactions were quenched with 100µL of 1 µM tolbutamide in acetonitrile after 5 min. The finalvolume of each secondary reaction was 100 µL, providing a 1:20dilution of the primary incubation. Product formation frommidazolam was determined to be linear over the 5 min incubationperiod. The reported measurements represent the average ofduplicate incubations. Quantitation of 1′-hydroxymidazolam forma-tion by mass spectrometry is described below.
Measurement of Raloxifene IC50 Values. Median inhibitoryconcentration (IC50) values of raloxifene were determined againstboth testosterone and midazolam. Prior to the IC50 measurements,Km and kcat values for both testosterone and midazolam weredetermined for each enzyme studied (data not shown), and theresults were consistent with published data (16, 21). Varyingconcentrations of raloxifene (0–100 µM) were preincubated withenzyme (10 nM) and substrate (at the predetermined Km value) for5 min before initiation of the reaction with NADPH. After 5 minfor midazolam and 20 min for testosterone, the reactions werequenched with an equal volume (100 µL) of 1 µM tolbutamide inacetonitrile. Concentrations of 1′-hydroxymidazolam and 6�-hydroxytestosterone were measured using the multiple reactionmonitoring (MRM) scan function of the 4000Q-Trap. 1′-Hy-droxymidazolam was monitored with Q1 and Q3 set at m/z 342.1and 203.1, respectively, with a declustering potential of 66 and a
Differential Time-Dependent InactiVation of P450 3A Chem. Res. Toxicol., Vol. 20, No. 12, 2007 1779

collision energy of 39. Concentrations of 6�-hydroxytestosteronewere measured with Q1 and Q3 set at m/z 305.0 and 269.0,respectively, with a declustering potential of 55 and a collisionenergy of 25. Tolbutamide was monitored with Q1 and Q3 set atm/z 271.2 and 91.1, respectively, with a declustering potential of66 and a collision energy of 39. The following instrument settingswere the same for all three compounds: dwell time, 500 ms; curtaingas, 10; ion-spray voltage, 4500 V; source temperature, 400 °C;and ion-spray gas 1 and 2, 40. Incubations were run in triplicate,and IC50 values were determined using Prism 5.0 (GraphPadSoftware, San Diego, CA, www.graphpad.com).
Substrate Depletion of Raloxifene. Enzyme (10 pmol) waspreincubated for 5 min at 37 °C prior to the addition of 10 mMNADPH to a final concentration of 1 mM (1 mL total volume).Aliquots (50 µL) were removed from the incubation at 0, 3, 6, 9,12, and 15 min and placed into 100 µL of cold acetonitrile spikedwith 1 µM tolbutamide as an internal standard. All incubations wererun in triplicate. Concentrations of unreacted raloxifene weremeasured using the 4000Q-Trap in MRM mode with Q1 and Q3set at m/z 474.28 and 111.9, respectively, with a declusteringpotential of 66 and a collision energy of 47. Tolbutamide wasmonitored with Q1 and Q3 set at m/z 271.2 and 91.1, respectively.The following instrument settings were the same for both com-pounds: dwell time, 500 ms; curtain gas, 10; ion-spray voltage,4500 V; source temperature, 400 °C; and ion-spray gas 1 and 2,40.
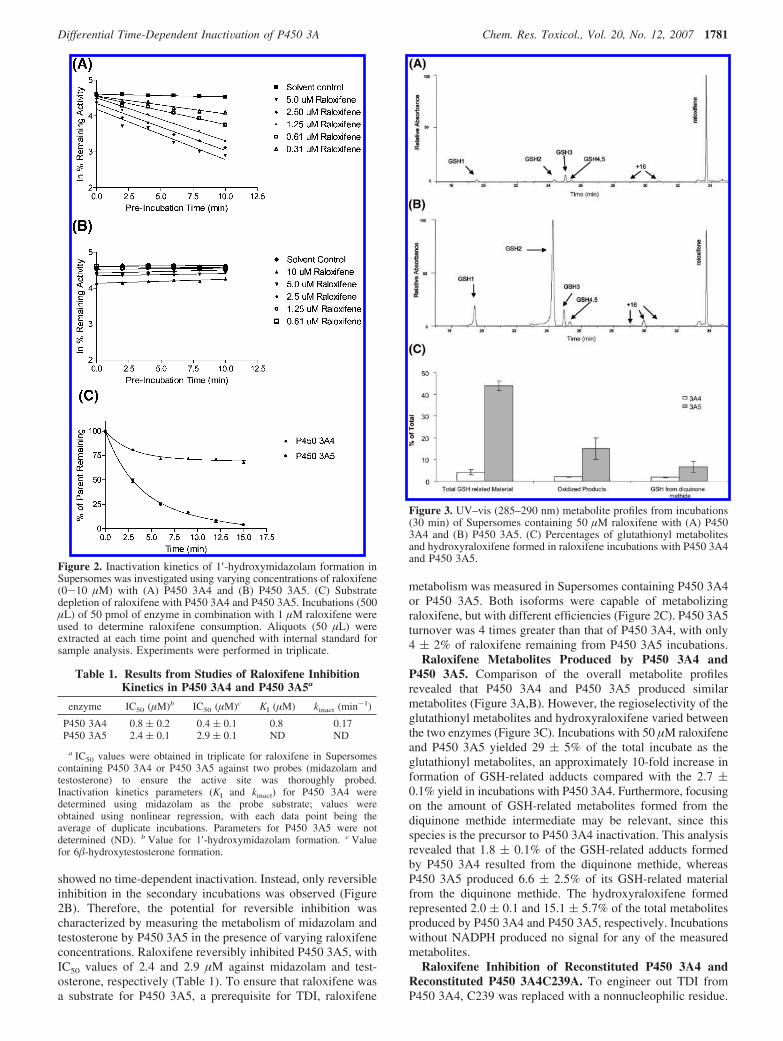
Measurement of Raloxifene Metabolites Produced by P4503A Enzymes. Following a 30 min incubation of P450 3A4, P4503A5, or P450 3A4C239A (10 pmol) with 50 µM raloxifene, 5 mMGSH, and 1 mM NADPH as described previously (15), the reactionwas quenched with 20 µL of trifluoroacetic acid (TFA). Controlreactions without NADPH were conducted simultaneously. Thesamples were centrifuged at 13000 rpm for 30 min at 4 °C.Supernatant (100 µL) was injected onto an Agilent Zorbax RX-C8column (2.1 × 250 mm) at a flow rate of 1 mL/min, with 25% ofthe flow diverted into the mass spectrometer. Initial HPLCconditions were 90% solvent A (0.05% TFA in H2O) and 10%solvent B (0.05% TFA in acetonitrile). The following elutiongradient was used: B increased from 10 to 50% over 30 min, thenincreased from 50 to 95% over 2 min, and then was held at 95%for an additional 3 min. In the absence of metabolite standards,the metabolite levels were determined on the basis of a comparisonof their UV–vis responses with the signature absorbance ofraloxifene at 285–290 nm. Even though the molar absorptivities ofthe metabolites may differ subtly from that of raloxifene as a resultof oxidation and glutathione conjugation, this method provides atleast a semiquantitative measure of metabolite levels. Metaboliteformation was reported as the percentage of the total raloxifene fromeach of the incubations. Reactions were performed in triplicate in orderto obtain standard deviations. The diglutathione metabolite (GSH1,m/z 550) eluted at 19.7 min, and the hydroxyglutathione adduct(GSH2, m/z 779) eluted at 24.4 min. The three monoglutathionemetabolites (GSH3-GSH5, m/z 779) eluted from 25.1 to 25.5 min.Hydroxyraloxifenes (m/z 490) eluted between 29 and 31 min, andraloxifene (m/z 474) eluted at 33.7 min.
Results
Structural Comparison of P450 3A4 and P450 3A5. BothP450 3A4 and P450 3A5 contain 503 residues; of these, 84%are identical and 92% are similar, yielding a strong alignmentscore of 2244 for these enzymes. Furthermore, from thesequence alignment, C239 in P450 3A4 corresponds to S239in P450 3A5. Residue 239 is located in substrate recognitionsite 3 (SRS-3) in both P450 3A enzymes (22). To betterelucidate the potential role of C239 in sequestering reactivemetabolites, a homology model of P450 3A5 was constructed.Overall, the three-dimensional structure of P450 3A5 accuratelyoverlaid the structural folds in P450 3A4. The distance betweenthe heme iron and the sulfur of C239 in P450 3A4 was measured
to be 21 Å, while the corresponding hydroxyl group of S239 inP450 3A5 was 19 Å from the heme iron. In the structuralrepresentations, both side chains point toward the distal face ofthe heme and closely overlap one another when the enzymesare superimposed (Figure 1A). Docking studies with raloxifeneproduced an enzyme–substrate conformation in which thebenzothiophene alcohol of raloxifene was oriented toward theheme iron at a distance of 3.9 Å, which subsequently positionedthe phenol 6.1 Å from the sulfur of C239 (Figure 1B). A similarbinding motif was observed in P450 3A5 (data not shown).
Raloxifene Inhibition of P450 3A4 and P450 3A5. Ralox-ifene inhibited P450 3A4 in Supersomes in a time-dependentmanner, with KI and kinact values of 0.78 µM and 0.18 min-1,respectively (Figure 2A). These results are similar to thosepreviously reported by Chen et al. (24). In P450 3A5, raloxifene
Figure 1. Homology model of P450 3A5 based on the crystal structureof P450 3A4 (PDB entry 1TQN) (36). (A) Residue 239 is displayed inyellow in both P450 3A4 (cyan) and P450 3A5 (green). The sulfuratom of C239 (P450 3A4) is 19.5 Å from the heme iron, and thehydroxyl oxygen atom of S239 (P450 3A5) is 22.1 Å from the hemeiron. (B) Induced-fit docking of raloxifene in P450 3A4 illustrates asingle binding mode prone to oxidation at the 6 position of thebenzothiophene, leading to formation of a diquinone methide followedby nucleophilic attack at the 3′ or 5′ position at the other end ofraloxifene.
1780 Chem. Res. Toxicol., Vol. 20, No. 12, 2007 Pearson et al.
showed no time-dependent inactivation. Instead, only reversibleinhibition in the secondary incubations was observed (Figure2B). Therefore, the potential for reversible inhibition wascharacterized by measuring the metabolism of midazolam andtestosterone by P450 3A5 in the presence of varying raloxifeneconcentrations. Raloxifene reversibly inhibited P450 3A5, withIC50 values of 2.4 and 2.9 µM against midazolam and test-osterone, respectively (Table 1). To ensure that raloxifene wasa substrate for P450 3A5, a prerequisite for TDI, raloxifene
metabolism was measured in Supersomes containing P450 3A4or P450 3A5. Both isoforms were capable of metabolizingraloxifene, but with different efficiencies (Figure 2C). P450 3A5turnover was 4 times greater than that of P450 3A4, with only4 ( 2% of raloxifene remaining from P450 3A5 incubations.
Raloxifene Metabolites Produced by P450 3A4 andP450 3A5. Comparison of the overall metabolite profilesrevealed that P450 3A4 and P450 3A5 produced similarmetabolites (Figure 3A,B). However, the regioselectivity of theglutathionyl metabolites and hydroxyraloxifene varied betweenthe two enzymes (Figure 3C). Incubations with 50 µM raloxifeneand P450 3A5 yielded 29 ( 5% of the total incubate as theglutathionyl metabolites, an approximately 10-fold increase information of GSH-related adducts compared with the 2.7 (0.1% yield in incubations with P450 3A4. Furthermore, focusingon the amount of GSH-related metabolites formed from thediquinone methide intermediate may be relevant, since thisspecies is the precursor to P450 3A4 inactivation. This analysisrevealed that 1.8 ( 0.1% of the GSH-related adducts formedby P450 3A4 resulted from the diquinone methide, whereasP450 3A5 produced 6.6 ( 2.5% of its GSH-related materialfrom the diquinone methide. The hydroxyraloxifene formedrepresented 2.0 ( 0.1 and 15.1 ( 5.7% of the total metabolitesproduced by P450 3A4 and P450 3A5, respectively. Incubationswithout NADPH produced no signal for any of the measuredmetabolites.
Raloxifene Inhibition of Reconstituted P450 3A4 andReconstituted P450 3A4C239A. To engineer out TDI fromP450 3A4, C239 was replaced with a nonnucleophilic residue.
Figure 2. Inactivation kinetics of 1′-hydroxymidazolam formation inSupersomes was investigated using varying concentrations of raloxifene(0-10 µM) with (A) P450 3A4 and (B) P450 3A5. (C) Substratedepletion of raloxifene with P450 3A4 and P450 3A5. Incubations (500µL) of 50 pmol of enzyme in combination with 1 µM raloxifene wereused to determine raloxifene consumption. Aliquots (50 µL) wereextracted at each time point and quenched with internal standard forsample analysis. Experiments were performed in triplicate.
Table 1. Results from Studies of Raloxifene InhibitionKinetics in P450 3A4 and P450 3A5a
enzyme IC50 (µM)b IC50 (µM)c KI (µM) kinact (min-1)
P450 3A4 0.8 ( 0.2 0.4 ( 0.1 0.8 0.17P450 3A5 2.4 ( 0.1 2.9 ( 0.1 ND ND
a IC50 values were obtained in triplicate for raloxifene in Supersomescontaining P450 3A4 or P450 3A5 against two probes (midazolam andtestosterone) to ensure the active site was thoroughly probed.Inactivation kinetics parameters (KI and kinact) for P450 3A4 weredetermined using midazolam as the probe substrate; values wereobtained using nonlinear regression, with each data point being theaverage of duplicate incubations. Parameters for P450 3A5 were notdetermined (ND). b Value for 1′-hydroxymidazolam formation. c Valuefor 6�-hydroxytestosterone formation.
Figure 3. UV–vis (285–290 nm) metabolite profiles from incubations(30 min) of Supersomes containing 50 µM raloxifene with (A) P4503A4 and (B) P450 3A5. (C) Percentages of glutathionyl metabolitesand hydroxyraloxifene formed in raloxifene incubations with P450 3A4and P450 3A5.
Differential Time-Dependent InactiVation of P450 3A Chem. Res. Toxicol., Vol. 20, No. 12, 2007 1781
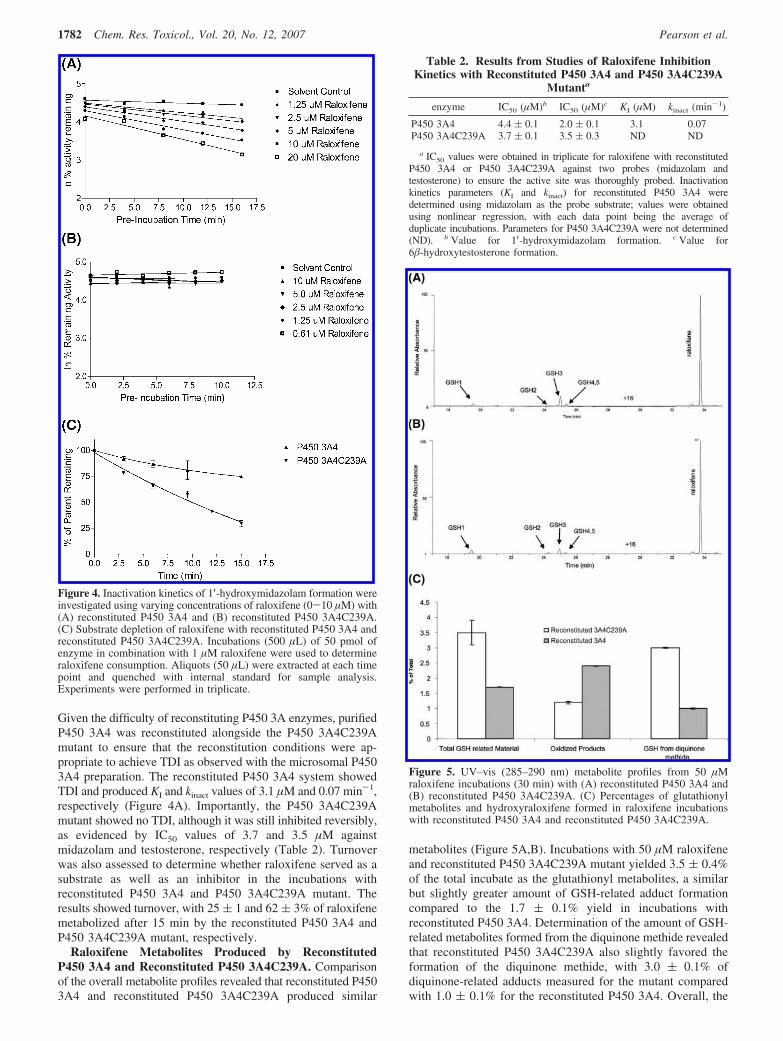
Given the difficulty of reconstituting P450 3A enzymes, purifiedP450 3A4 was reconstituted alongside the P450 3A4C239Amutant to ensure that the reconstitution conditions were ap-propriate to achieve TDI as observed with the microsomal P4503A4 preparation. The reconstituted P450 3A4 system showedTDI and produced KI and kinact values of 3.1 µM and 0.07 min-1,respectively (Figure 4A). Importantly, the P450 3A4C239Amutant showed no TDI, although it was still inhibited reversibly,as evidenced by IC50 values of 3.7 and 3.5 µM againstmidazolam and testosterone, respectively (Table 2). Turnoverwas also assessed to determine whether raloxifene served as asubstrate as well as an inhibitor in the incubations withreconstituted P450 3A4 and P450 3A4C239A mutant. Theresults showed turnover, with 25 ( 1 and 62 ( 3% of raloxifenemetabolized after 15 min by the reconstituted P450 3A4 andP450 3A4C239A mutant, respectively.
Raloxifene Metabolites Produced by ReconstitutedP450 3A4 and Reconstituted P450 3A4C239A. Comparisonof the overall metabolite profiles revealed that reconstituted P4503A4 and reconstituted P450 3A4C239A produced similar
metabolites (Figure 5A,B). Incubations with 50 µM raloxifeneand reconstituted P450 3A4C239A mutant yielded 3.5 ( 0.4%of the total incubate as the glutathionyl metabolites, a similarbut slightly greater amount of GSH-related adduct formationcompared to the 1.7 ( 0.1% yield in incubations withreconstituted P450 3A4. Determination of the amount of GSH-related metabolites formed from the diquinone methide revealedthat reconstituted P450 3A4C239A also slightly favored theformation of the diquinone methide, with 3.0 ( 0.1% ofdiquinone-related adducts measured for the mutant comparedwith 1.0 ( 0.1% for the reconstituted P450 3A4. Overall, the
Figure 4. Inactivation kinetics of 1′-hydroxymidazolam formation wereinvestigated using varying concentrations of raloxifene (0-10 µM) with(A) reconstituted P450 3A4 and (B) reconstituted P450 3A4C239A.(C) Substrate depletion of raloxifene with reconstituted P450 3A4 andreconstituted P450 3A4C239A. Incubations (500 µL) of 50 pmol ofenzyme in combination with 1 µM raloxifene were used to determineraloxifene consumption. Aliquots (50 µL) were extracted at each timepoint and quenched with internal standard for sample analysis.Experiments were performed in triplicate.
Table 2. Results from Studies of Raloxifene InhibitionKinetics with Reconstituted P450 3A4 and P450 3A4C239A
Mutanta
enzyme IC50 (µM)b IC50 (µM)c KI (µM) kinact (min-1)
P450 3A4 4.4 ( 0.1 2.0 ( 0.1 3.1 0.07P450 3A4C239A 3.7 ( 0.1 3.5 ( 0.3 ND ND
a IC50 values were obtained in triplicate for raloxifene with reconstitutedP450 3A4 or P450 3A4C239A against two probes (midazolam andtestosterone) to ensure the active site was thoroughly probed. Inactivationkinetics parameters (KI and kinact) for reconstituted P450 3A4 weredetermined using midazolam as the probe substrate; values were obtainedusing nonlinear regression, with each data point being the average ofduplicate incubations. Parameters for P450 3A4C239A were not determined(ND). b Value for 1′-hydroxymidazolam formation. c Value for6�-hydroxytestosterone formation.
Figure 5. UV–vis (285–290 nm) metabolite profiles from 50 µMraloxifene incubations (30 min) with (A) reconstituted P450 3A4 and(B) reconstituted P450 3A4C239A. (C) Percentages of glutathionylmetabolites and hydroxyraloxifene formed in raloxifene incubationswith reconstituted P450 3A4 and reconstituted P450 3A4C239A.
1782 Chem. Res. Toxicol., Vol. 20, No. 12, 2007 Pearson et al.
hydroxyraloxifene metabolites were favored in the reconstitutedP450 3A4 compared to the P450 3A4C239A mutant, whichpredominantly formed the glutathionyl metabolites (Figure 5C).Incubations without NADPH produced no signal for any of themeasured metabolites.
P450 3A5 and Reconstituted P450 3A5S239C. In an effortto confer susceptibility to TDI onto P450 3A5, the S239C mutantwas created. Reconstituted P450 3A5 wild type showed an IC50
value for inhibition of testosterone hydroxylation comparableto that for the enzyme in Supersomes. However, the IC50 valuefor the reconstituted mutant was 10-fold higher, and theefficiency of midazolam and testosterone oxidation was greatlyimpaired, precluding accurate assessment of TDI.
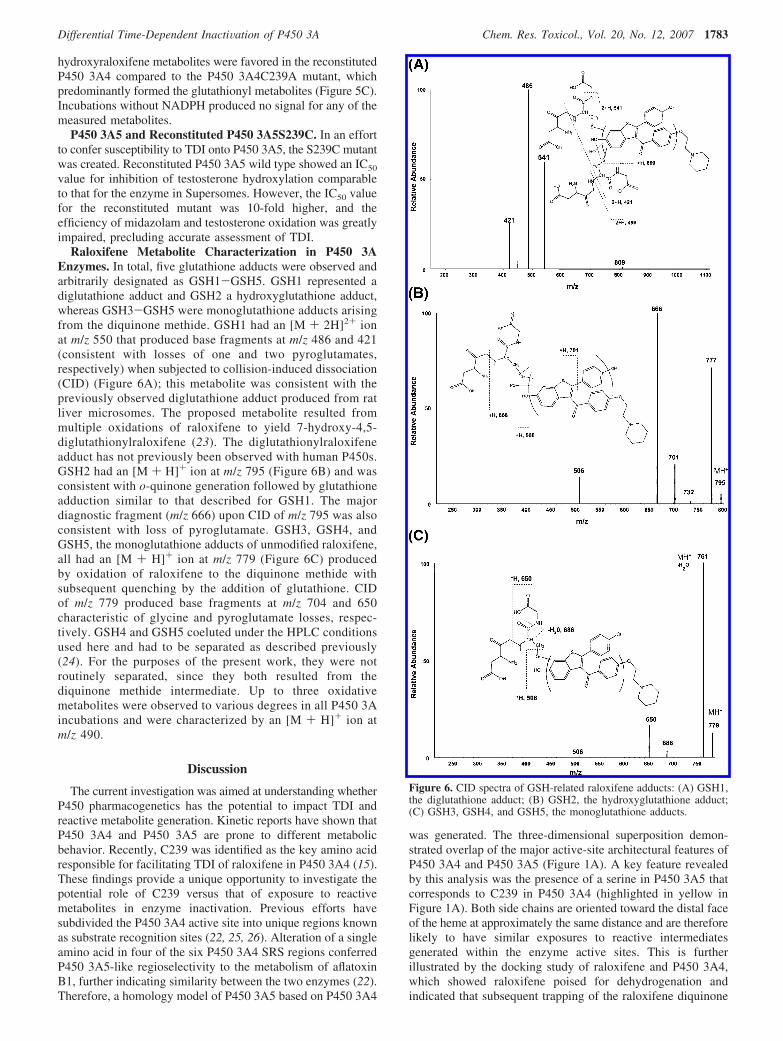
Raloxifene Metabolite Characterization in P450 3AEnzymes. In total, five glutathione adducts were observed andarbitrarily designated as GSH1-GSH5. GSH1 represented adiglutathione adduct and GSH2 a hydroxyglutathione adduct,whereas GSH3-GSH5 were monoglutathione adducts arisingfrom the diquinone methide. GSH1 had an [M + 2H]2+ ionat m/z 550 that produced base fragments at m/z 486 and 421(consistent with losses of one and two pyroglutamates,respectively) when subjected to collision-induced dissociation(CID) (Figure 6A); this metabolite was consistent with thepreviously observed diglutathione adduct produced from ratliver microsomes. The proposed metabolite resulted frommultiple oxidations of raloxifene to yield 7-hydroxy-4,5-diglutathionylraloxifene (23). The diglutathionylraloxifeneadduct has not previously been observed with human P450s.GSH2 had an [M + H]+ ion at m/z 795 (Figure 6B) and wasconsistent with o-quinone generation followed by glutathioneadduction similar to that described for GSH1. The majordiagnostic fragment (m/z 666) upon CID of m/z 795 was alsoconsistent with loss of pyroglutamate. GSH3, GSH4, andGSH5, the monoglutathione adducts of unmodified raloxifene,all had an [M + H]+ ion at m/z 779 (Figure 6C) producedby oxidation of raloxifene to the diquinone methide withsubsequent quenching by the addition of glutathione. CIDof m/z 779 produced base fragments at m/z 704 and 650characteristic of glycine and pyroglutamate losses, respec-tively. GSH4 and GSH5 coeluted under the HPLC conditionsused here and had to be separated as described previously(24). For the purposes of the present work, they were notroutinely separated, since they both resulted from thediquinone methide intermediate. Up to three oxidativemetabolites were observed to various degrees in all P450 3Aincubations and were characterized by an [M + H]+ ion atm/z 490.
Discussion
The current investigation was aimed at understanding whetherP450 pharmacogenetics has the potential to impact TDI andreactive metabolite generation. Kinetic reports have shown thatP450 3A4 and P450 3A5 are prone to different metabolicbehavior. Recently, C239 was identified as the key amino acidresponsible for facilitating TDI of raloxifene in P450 3A4 (15).These findings provide a unique opportunity to investigate thepotential role of C239 versus that of exposure to reactivemetabolites in enzyme inactivation. Previous efforts havesubdivided the P450 3A4 active site into unique regions knownas substrate recognition sites (22, 25, 26). Alteration of a singleamino acid in four of the six P450 3A4 SRS regions conferredP450 3A5-like regioselectivity to the metabolism of aflatoxinB1, further indicating similarity between the two enzymes (22).Therefore, a homology model of P450 3A5 based on P450 3A4
was generated. The three-dimensional superposition demon-strated overlap of the major active-site architectural features ofP450 3A4 and P450 3A5 (Figure 1A). A key feature revealedby this analysis was the presence of a serine in P450 3A5 thatcorresponds to C239 in P450 3A4 (highlighted in yellow inFigure 1A). Both side chains are oriented toward the distal faceof the heme at approximately the same distance and are thereforelikely to have similar exposures to reactive intermediatesgenerated within the enzyme active sites. This is furtherillustrated by the docking study of raloxifene and P450 3A4,which showed raloxifene poised for dehydrogenation andindicated that subsequent trapping of the raloxifene diquinone
Figure 6. CID spectra of GSH-related raloxifene adducts: (A) GSH1,the diglutathione adduct; (B) GSH2, the hydroxyglutathione adduct;(C) GSH3, GSH4, and GSH5, the monoglutathione adducts.
Differential Time-Dependent InactiVation of P450 3A Chem. Res. Toxicol., Vol. 20, No. 12, 2007 1783

methide is feasible on the basis of a single binding conformation(Figure 1B). Furthermore, solvent accessibility of C239 in P4503A4 was determined on the basis of selective alkylation ofthe protein at C239 with the addition of iodoacetamide (15).The differing nucleophilicities of the cysteine thiol group andthe serine hydroxyl group may contribute to the differences intheir abilities to trap reactive metabolites (22).
Precedence has been established for preferential reactivitiesof electrophilic reactive intermediates with nucleophiles thatretain a similar degree of “hardness” or “softness” (27–29).Iminium ions formed from the metabolism of cyclic amines,such as nicotine, react preferentially with nucleophiles such ascyanide (30). Reactive intermediates such as aldehydes are oftentrapped with amines (31). Glutathione has a weaker propensityto trap hard electrophiles but reacts well with soft electrophilessuch as arene oxides, quinones, and thiophene sulfoxides (29).Correspondingly, it may be expected that protein residues suchas cysteine will trap soft electrophiles such as the diquinonemethide intermediate of raloxifene, while lysine and serineresidues are more likely to trap harder electrophiles. In orderto test this hypothesis, TDI experiments using raloxifene werecarried out using P450 3A4, P450 3A5, and P450 3A4C239A.
Studies of raloxifene inactivation kinetics to compare TDIin P450 3A4 and P450 3A5 were conducted in order to examinethe potential for differential inhibition of P450 3A4 and P4503A5. Raloxifene exhibited TDI of midazolam 1′-hydroxylationin P450 3A4 but did not show TDI in P450 3A5 at concentra-tions up to 10 µM, which is well above the measured IC50 valuesagainst midazolam and testosterone (Table 1). The metaboliccapacities of P450 3A4 and P450 3A5 toward raloxifene werealso investigated by measuring raloxifene depletion, and theresults showed that raloxifene was a substrate for both P4503A4 and P450 3A5. The activity from P450 3A4 decreasedrapidly, consistent with enzyme inactivation and the moderatecapacity for P450 3A4 to metabolize raloxifene (Figure 2C).Under these circumstances, it may have been expected thatminimal amounts of bioactivated raloxifene would be generated,since the activity of P450 3A4 was simultaneously being reducedwithin the first few minutes of the incubation as a result ofirreversible inhibition of P450 3A4. The rapid enzyme inactiva-tion resulted from efficient trapping of a large percentage ofthe reactive intermediate and was consistent with a low partitionratio of 1.8 (32).
The exponential decay of raloxifene resulting from P450 3A5-mediated metabolism (Figure 2C) was consistent with the resultsshown in Figure 2B, which indicate that raloxifene did notdisplay TDI in P450 3A5. Consequently, P450 3A5 was capableof continually producing reactive species from raloxifenethroughout the course of the experiment. Extending thesefindings to a potential in vivo scenario suggests that the cysteineof P450 3A4 could minimize exposure to P450 3A4-generatedreactive metabolites by inactivating the enzyme, the source ofthe reactive metabolites, via a pathway akin to a biologicalfeedback inhibition mechanism (Scheme 1). Alternatively, inthe case of P450 3A5 where enzyme inactivation does not occur,the reactive intermediates could be continually produced. Thishypothesis may be extended to explain the correlation betweenaflatoxin-albumin adducts and P450 3A5 enzyme levels (33).Moreover, this notion is also consistent with the lack of TDIobserved for aflatoxin in P450 3A5 (data not shown).
Metabolite profiles (indicating regioselectivity) for P450 3A4and P450 3A5 enzymes were also examined in order to ensurethat the lack of TDI in P450 3A5 did not result simply fromthe inability of the enzyme to generate glutathione adducts, in
particular those arising from the diquinone methide, which haspreviously been determined as the precursor to inactivation ofP450 3A4 (15). Incubations with P450 3A4 confirmed diquinonemethide formation as indicated by the presence of the mono-glutathionyl adducts, which were consistent with previouslyobtained results based on mass spectrometry (23, 24). Inaddition, the monohydroxylated metabolites and trace amountsof a diglutathione adduct were also present. The diglutathionemetabolite has previously been reported in rat liver microsomesand is postulated to be formed via sequential enzyme oxidationreactions (23). P450 3A5 favored the formation of oxidativeglutathione adducts as well as significantly greater amounts ofall the other metabolites, including the monoglutathione anddiglutathione adducts, observed from P450 3A4 incubations(Figure 3B and 3C). The resultant CID fragmentations wereconsistent with previous mass spectral data generated with themono- and diglutathione raloxifene metabolites from the rat aswell as the monoglutathione adducts from human liver mi-crosomes (23, 24). Most importantly, P450 3A5 generated thediquinone methide intermediate required for P450 3A4 inactiva-tion, yet no inactivation of P450 3A5 occurred; this is consistentwith the hypothesis that P450 3A5 is capable of yieldingincreased amounts of reactive metabolites relative to P450 3A4when the resulting bioactivated compound (in this case ralox-ifene) preferentially reacts with a soft nucleophile such ascysteine (Figure 3C).
The two mutants P450 3A4C239A and P450 3A5S239C wereengineered on the basis of the unique difference between P4503A4 and P450 3A5 at residue 239 in SRS-3. The P4503A5S239C mutation significantly impacted the kinetics profileof midazolam and testosterone turnover, to the extent that furtherstudies could not be carried out because of solubility limitations.For the P450 3A4C239A mutant, alanine instead of serine waschosen to replace the cysteine in P450 3A4, in order to eliminatethe potential for any electrophilic quenching of raloxifenebioactivation at residue 239. This precaution was taken becausedifferent micro environments are known to activate the nucleo-philic behavior of serine residues (34). Furthermore, serine haspreviously been implicated in covalent binding of other P450enzyme systems (35). Raloxifene failed to produce TDI of theP450 3A4C239A mutant with respect to midazolam 1′-hydroxy-lation, whereas reversible inhibition of P450 3A4C239A wassimilar to that observed for reconstituted P450 3A4, suggestingthat raloxifene still binds to the mutant enzyme (Table 2). WhileTDI was not observed for the P450 3A4C239A mutant in theseexperiments, it was critical to determine that raloxifene is asubstrate for the enzyme prior to drawing any conclusionsregarding the significance of the cysteine/serine difference asthe determining factor for differential TDI of P450 3A4compared with P450 3A5.
The turnover of raloxifene for the P450 3A4C239A mutantwas compared to that for reconstituted P450 3A4 in order toensure that raloxifene behaved as a substrate in both systems.
Scheme 1
1784 Chem. Res. Toxicol., Vol. 20, No. 12, 2007 Pearson et al.

Both enzymes showed activity toward raloxifene (Figure 4C).A difference in activity was observed by the end of theincubation, which showed that the P450 3A4C239A mutant wasmore efficient than the reconstituted P450 3A4, consistent withthe lack of TDI observed (Figure 4B). This study confirmedthat raloxifene was a substrate for both P450 3A4C239A andthe reconstituted P450 3A4. Examination of the metaboliteprofiles further revealed that the putative precursor to P450 3A4inactivation forms in both reconstituted systems and thus shouldfacilitate TDI (Figure 5A,B). Furthermore, the P450 3A4C239Amutant produced a 2-fold increase in the amount of GSH adductsformed, with the majority of this GSH-related material origi-nating from the diquinone methide precursor. These resultsconfirm that the P450 3A4C239A is capable of forming thenecessary metabolites for inactivation, while the lack ofinactivation indicates that the C239A mutation protects theenzyme from inactivation.
Several mechanisms can be postulated for the differential TDIby raloxifene of P450 3A4 compared with P450 3A5: (1)different rates of substrate turnover, (2) alteration in regiose-lectivity due to differences in active site architecture, or (3) thepresence of varying nucleophilic active-site residues. To date,literature reports attempting to explain the differences betweenP450 3A4 and P450 3A5 have focused on differential regiose-lectivity produced by the enzymes. For instance, the antibacterialagent erythromycin forms a metabolite-inhibitor complex inP450 3A4 but is unable to inhibit P450 3A5 in this manner.Reports suggest that a sequential oxidation pathway leading tothe reactive intermediate is available in P450 3A4 but does notoccur in P450 3A5, possibly as a result of different substrateorientations or mechanisms of oxidation (13). In addition,mifepristone also selectively inactivates P450 3A4. Interestingly,examining the metabolic profile of mifepristone revealed thatboth C-hydroxylated and N-dealkylated metabolites were formedby P450 3A4, while only N-dealkylated products were formedby P450 3A5. Although the exact structural basis for theobserved difference was not elucidated, the authors proposedthat differential substrate binding did not allow the putativealkynyl reactive metabolite precursor to access the reactive ox-ygen species, thereby preventing generation of the reactiveintermediate (12). Moreover, others have reported that vera-pamil, diltiazem, and nicardipine selectively form MICs in P4503A4 compared with P450 3A5, although the mechanism of thisinactivation selectivity is not well understood (13).
We believe that the current study is the first report describingselect P450 genotype-mediated inactivation based on alkylationof active-site residues. This proposal is based on concordanceof results generated from kinetic analysis, computational model-ing, and site-directed mutagenesis. These results firmly establisha unique role for C239 in P450 3A4 (in contrast with S239 inP450 3A5) as a nucleophilic “hook” for capturing enzyme-generated reactive intermediates before they are able to leavethe active site. Furthermore, the current study also provides apossible mechanistic rationale for observed differences betweenP450 3A4 and P450 3A5 pharmacogenetic drug-related toxicitiesobserved across human populations.
Acknowledgment. This research was supported in part byNIH Grant GM54995 (to J.R.H.).
References
(1) Mahgoub, A., Idle, J. R., Dring, L. G., Lancaster, R., and Smith, R. L.(1977) Polymorphic hydroxylation of Debrisoquine in man. Lancet2, 584–586.
(2) Dayer, P., Gasser, R., Gut, J., Kronbach, T., Robertz, G. M.,Eichelbaum, M., and Meyer, U. A. (1984) Characterization of acommon genetic defect of cytochrome P-450 function (debrisoquine-sparteine type polymorphism)sincreased Michaelis constant (Km) andloss of stereoselectivity of bufuralol 1′-hydroxylation in poor metabo-lizers. Biochem. Biophys. Res. Commun. 125, 374–380.
(3) Tucker, G. T., Silas, J. H., Iyun, A. O., Lennard, M. S., and Smith,A. J. (1977) Polymorphic hydroxylation of debrisoquine. Lancet 2,718.
(4) Idle, J. R., Mahgoub, A., Lancaster, R., and Smith, R. L. (1978)Hypotensive response to debrisoquine and hydroxylation phenotype.Life Sci. 22, 979–983.
(5) Bradford, L. D. (2002) CYP2D6 allele frequency in EuropeanCaucasians, Asians, Africans and their descendants. Pharmacogenom-ics 3, 229–243.
(6) Heimark, L. D., Wienkers, L., Kunze, K., Gibaldi, M., Eddy, A. C.,Trager, W. F., O’Reilly, R. A., and Goulart, D. A. (1992) Themechanism of the interaction between amiodarone and warfarin inhumans. Clin. Pharmacol. Ther. 51, 398–407.
(7) Steward, D. J., Haining, R. L., Henne, K. R., Davis, G., Rushmore,T. H., Trager, W. F., and Rettie, A. E. (1997) Genetic associationbetween sensitivity to warfarin and expression of CYP2C9*3. Phar-macogenetics 7, 361–367.
(8) Becking, G. C. (1995) Use of mechanistic information in riskassessment for toxic chemicals. Toxicol. Lett. 77, 15–24.
(9) Shimada, T., Yamazaki, H., Mimura, M., Inui, Y., and Guengerich,F. P. (1994) Interindividual variations in human liver cytochromeP-450 enzymes involved in the oxidation of drugs, carcinogens andtoxic chemicals: studies with liver microsomes of 30 Japanese and 30Caucasians. J. Pharmacol. Exp. Ther. 270, 414–423.
(10) Wrighton, S. A., Brian, W. R., Sari, M. A., Iwasaki, M., Guengerich,F. P., Raucy, J. L., Molowa, D. T., and Vandenbranden, M. (1990)Studies on the expression and metabolic capabilities of human livercytochrome P450IIIA5 (HLp3). Mol. Pharmacol. 38, 207–213.
(11) Lin, Y. S., Dowling, A. L., Quigley, S. D., Farin, F. M., Zhang, J.,Lamba, J., Schuetz, E. G., and Thummel, K. E. (2002) Co-regulationof CYP3A4 and CYP3A5 and contribution to hepatic and intestinalmidazolam metabolism. Mol. Pharmacol. 62, 162–172.
(12) Khan, K. K., He, Y. Q., Correia, M. A., and Halpert, J. R. (2002)Differential oxidation of mifepristone by cytochromes P450 3A4 and3A5: selective inactivation of P450 3A4. Drug Metab. Dispos. 30,985–990.
(13) McConn, D. J., 2nd, Lin, Y. S., Allen, K., Kunze, K. L., and Thummel,K. E. (2004) Differences in the inhibition of cytochromes P450 3A4and 3A5 by metabolite-inhibitor complex-forming drugs. Drug Metab.Dispos. 32, 1083–1091.
(14) Wang, Y. H., Jones, D. R., and Hall, S. D. (2005) Differentialmechanism-based inhibition of CYP3A4 and CYP3A5 by verapamil.Drug Metab. Dispos. 33, 664–671.
(15) Baer, B. R., Wienkers, L. C., and Rock, D. A. (2007) Time-dependentinactivation of P450 3A4 by raloxifene: identification of Cys239 asthe site of apoprotein alkylation. Chem. Res. Toxicol. 20, 954–264.
(16) Gillam, E. M., Baba, T., Kim, B. R., Ohmori, S., and Guengerich,F. P. (1993) Expression of modified human cytochrome P450 3A4 inEscherichia coli and purification and reconstitution of the enzyme.Arch. Biochem. Biophys. 305, 123–131.
(17) Domanski, T. L., He, Y. A., Khan, K. K., Roussel, F., Wang, Q., andHalpert, J. R. (2001) Phenylalanine and tryptophan scanning mutagen-esis of CYP3A4 substrate recognition site residues and effect onsubstrate oxidation and cooperativity. Biochemistry 40, 10150–10160.
(18) Holmans, P. L., Shet, M. S., Martin-Wixtrom, C. A., Fisher, C. W.,and Estabrook, R. W. (1994) The high-level expression in Escherichiacoli of the membrane-bound form of human and rat cytochrome b5and studies on their mechanism of function. Arch. Biochem. Biophys.312, 554–565.
(19) Rock, D., Rock, D., and Jones, J. P. (2001) Inexpensive purificationof P450 reductase and other proteins using 2′,5′-adenosine diphosphateagarose affinity columns. Protein Expression Purif. 22, 82–83.
(20) Yano, J. K., Wester, M. R., Schoch, G. A., Griffin, K. J., Stout, C. D.,and Johnson, E. F. (2004) The structure of human microsomalcytochrome P450 3A4 determined by X-ray crystallography to 2.05-Åresolution. J. Biol. Chem. 279, 38091–38094.
(21) Shaw, P. M., Hosea, N. A., Thompson, D. V., Lenius, J. M., andGuengerich, F. P. (1997) Reconstitution premixes for assays usingpurified recombinant human cytochrome P450, NADPH-cytochromeP450 reductase, and cytochrome b5. Arch. Biochem. Biophys. 348,107–115.
(22) Wang, H., Dick, R., Yin, H., Licad-Coles, E., Kroetz, D. L., Szklarz,G.,Harlow,G.,Halpert,J.R.,andCorreia,M.A.(1998)Structure-functionrelationships of human liver cytochromes P450 3A: aflatoxin B1metabolism as a probe. Biochemistry 37, 12536–12545.
(23) Yu, L., Liu, H., Li, W., Zhang, F., Luckie, C., van Breemen, R. B.,Thatcher, G. R., and Bolton, J. L. (2004) Oxidation of raloxifene to
Differential Time-Dependent InactiVation of P450 3A Chem. Res. Toxicol., Vol. 20, No. 12, 2007 1785

quinoids: potential toxic pathways via a diquinone methide ando-quinones. Chem. Res. Toxicol. 17, 879–888.
(24) Chen, Q., Ngui, J. S., Doss, G. A., Wang, R. W., Cai, X., DiNinno,F. P., Blizzard, T. A., Hammond, M. L., Stearns, R. A., Evans, D. C.,Baillie, T. A., and Tang, W. (2002) Cytochrome P450 3A4-mediatedbioactivation of raloxifene: irreversible enzyme inhibition and thioladduct formation. Chem. Res. Toxicol. 15, 907–914.
(25) Hasemann, C. A., Kurumbail, R. G., Boddupalli, S. S., Peterson, J. A.,and Deisenhofer, J. (1995) Structure and function of cytochromesP450: a comparative analysis of three crystal structures. Structure 3,41–62.
(26) Xue, L., Wang, H. F., Wang, Q., Szklarz, G. D., Domanski, T. L.,Halpert, J. R., and Correia, M. A. (2001) Influence of P450 3A4 SRS-2residues on cooperativity and/or regioselectivity of aflatoxin B1oxidation. Chem. Res. Toxicol. 14, 483–491.
(27) Coles, B. (1984) Effects of modifying structure on electrophilicreactions with biological nucleophiles. Drug Metab. ReV. 15, 1307–1334.
(28) Mutlib, A. E., Goosen, T. C., Bauman, J. N., Williams, J. A., Kulkarni,S., and Kostrubsky, S. (2006) Kinetics of acetaminophen glucuronida-tion by UDP-glucuronosyltransferases 1A1, 1A6, 1A9, and 2B15.Potential implications in acetaminophen-induced hepatotoxicity. Chem.Res. Toxicol. 19, 701–709.
(29) Stark, K. L., Harris, C., and Juchau, M. R. (1989) Influence ofelectrophilic character and glutathione depletion on chemical dysmor-phogenesis in cultured rat embryos. Biochem. Pharmacol. 38, 2685–2692.
(30) Argoti, D., Liang, L., Conteh, A., Chen, L., Bershas, D., Yu, C. P.,Vouros, P., and Yang, E. (2005) Cyanide trapping of iminium ionreactive intermediates followed by detection and structure identification
using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Chem. Res. Toxicol. 18, 1537–1544.
(31) Schnetz-Boutaud, N., Daniels, J. S., Hashim, M. F., Scholl, P., Burrus,T., and Marnett, L. J. (2000) Pyrimido[1,2-R]purin-10(3H)-one: areactive electrophile in the genome. Chem. Res. Toxicol. 13, 967–970.
(32) Zhou, S., Chan, E., Lim, L. Y., Boelsterli, U. A., Li, S. C., Wang, J.,Zhang, Q., Huang, M., and Xu, A. (2004) Therapeutic drugs thatbehave as mechanism-based inhibitors of cytochrome P450 3A4. Curr.Drug Metab. 5, 415–442.
(33) Wojnowski, L., Turner, P. C., Pedersen, B., Hustert, E., Brockmoller,J., Mendy, M., Whittle, H. C., Kirk, G., and Wild, C. P. (2004)Increased levels of aflatoxin-albumin adducts are associated withCYP3A5 polymorphisms in The Gambia, West Africa. Pharmacoge-netics 14, 691–700.
(34) Anderson, B. M., Cordes, E. H., and Jencks, W. P. (1961) Reactivityand catalysis in reactions of the serine hydroxyl group and of O-acylserines. J. Biol. Chem. 236, 455–463.
(35) Melet, A., Assrir, N., Jean, P., Pilar Lopez-Garcia, M., Marques-Soares,C., Jaouen, M., Dansette, P. M., Sari, M. A., and Mansuy, D. (2003)Substrate selectivity of human cytochrome P450 2C9: importance ofresidues 476, 365, and 114 in recognition of diclofenac and sul-faphenazole and in mechanism-based inactivation by tienilic acid. Arch.Biochem. Biophys. 409, 80–91.
(36) Yano, J. K., Denton, T. T., Cerny, M. A., Zhang, X., Johnson, E. F.,and Cashman, J. R. (2006) Synthetic inhibitors of cytochrome P-4502A6: inhibitory activity, difference spectra, mechanism of inhibition,and protein cocrystallization. J. Med. Chem. 49, 6987–7001.
TX700207U
1786 Chem. Res. Toxicol., Vol. 20, No. 12, 2007 Pearson et al.


















