
DFT Calculations Shaun Swanson. The Game Plan DFT Basics Job File Structure Sample Structure: Solid...
29
DFT Calculations Shaun Swanson
-
Upload
camron-fletcher -
Category
Documents
-
view
226 -
download
3
Transcript of DFT Calculations Shaun Swanson. The Game Plan DFT Basics Job File Structure Sample Structure: Solid...

DFT CalculationsShaun Swanson

The Game Plan
DFT Basics
Job File Structure
Sample Structure: Solid Mg
Performing ‘scf’ Calculations on Solid Mg
Converging ‘scf’ Energies
Performing ‘relax’ calculations on Solid Mg
The Limits of DFT
Current work: GST structure and CNT properties

What is Density Functional Theory (DFT)?
• In Summary: “Density Functional Theory is a phenomenally successful approach to finding solutions to the fundamental equation for the quantum behavior of atoms and molecules, the Schrodinger equation, in settings of practical value.”
• Problem: Solving the Schrodinger equation is a many-body problem (3N bodies to be exact).
• Solution: Electrons are indistinguishable. So the quantity of physical interest is instead the probability that N electrons in any order have specific coordinates. So the electron density, which is a function of only 3 coordinates, contains a great deal of information that is physically observable from the full solution to the Schrodinger equation.

The T in DFT
• The entire field of Density Functional Theory rests on two fundamental mathematical theorems proven by Kohn and Hohenberg, and the derivation of a set of equations by Kohn and Sham.
• Theorem #1: There exists a 1-to-1 mapping between the ground state wave function and the ground state electron density.
• Theorem #2: The electron density that minimizes the energy of the above functional is the true electron density corresponding to the full solution to the Schrodinger equation.
Guess the functional form and vary the electron density iteratively until the energy is minimized!

The Math
• A useful way to write down the functional described by Theorem #1 is in terms of the single electron wave functions (according to the Hartree Product approximation). The Schrodinger equation then becomes the Kohn-Sham equations:

Pseudocode
1. Define an initial, trial electron density.
2. Solve the Kohn-Sham equations using the trial electron density to find the single particle wave functions.
3. Calculate the electron density defined by the Kohn-Sham single particle wave functions from step 2.
4. Compare the calculated electron density with the electron density used to solve the Kohn-Sham equations. If they agree with some specified error, then compute the total energy from this density, otherwise update the trial electron density in some way.
5. Repeat.
This iterative method can lead to a solution of the Kohn-Sham equations that is self-consistent!

Methods for Efficient Computation
• K-points (k)– Discrete points specified in Brillouin Zone used to perform
numerical integration during calculation.
• Energy Cut-off Value (ecut)– Energy value for maximum energy state included in a
summation over electron states.
• Pseudopotentials (pp)– Offers specific exchange-correlation functional form which
represents frozen “core electrons.” They are based largely on empirical data.

The Game Plan
DFT Basics
Job File Structure
Sample Structure: Solid Mg
Performing ‘scf’ Calculations on Solid Mg
Converging ‘scf’ Energies
Performing ‘relax’ calculations on Solid Mg
The Limits of DFT
Current work: GST structure and CNT properties

Job File Structure
Input variables thatcontrol the amount ofI/O on the hard driveand on the screen
Input variables thatspecify the system
under study.
Input variables thatcontrol the algorithms
used to reach a self-consistentsolution of the Kohn-Sham
equations.
Name, mass, and pseudopotentialused for each atomic species present
in the system
Type and coordinates of each atom in the unit cell
Coordinates and weights of theK-points used for BZ integration

The Game Plan
DFT Basics
Job File Structure
Sample Structure: Solid Mg
Performing ‘scf’ Calculations on Solid Mg
Converging ‘scf’ Energies
Performing ‘relax’ calculations on Solid Mg
The Limits of DFT
Current work: GST structure and CNT properties

Solid Mg (hcp)
Space group: P63/mmc
a = 3.21 Angstromc = 5.21 Angstrom

Inputting Structures into Quantum ESPRESSO
• There are multiple options in QE, for inputting structures.• In the &SYSTEM namelist, there is a parameter called ‘ibrav’
– ibrav=4 refers to the index for an internally-specified hcp Bravais lattice.
– Ibrav=0 allows one to self-define their unit cell.
Ibrav = 0Ibrav = 4
•Internally specifies lattice vectors•Requires the celldm() parameters to be specified [alat = celldm(1)]•Better recognizes symmetry operations
•Requires CELL_PARAMETERS card for lattice vector input.•alat = the length of the first lattice vector

Inputting Vectors into Quantum ESPRESSO
• For the required ATOMIC_POSITIONS card and the optional CELL_PARAMETERS card, the input of basis and lattice vectors follow a general format:
• Note: While the three lattice vectors in the CELL_PARAMETERScard can only be specified in alat-normalized Bohr units, there arefour options for inputting basis vectors in ATOMIC_POSITIONS:
“alat” – alat-normalized Bohr units (default)“bohr” – Bohr units
“angstrom” – Angstrom units“crystal” – fractional coordinates

Performing Self-consistent Energy Calculations on Solid Mg

LDA vs. GGA Pseudopotential Approximations
• “Local-density approximations (LDA) are a class of approximations to the exchange-correlation (XC) energy functional in DFT that depend solely upon the value of the electronic density at each point in space (and not, for example, derivatives of the density or the Kohn-Sham orbitals).” This is more of a first-order approximation.
• “Generalized gradient approximations (GGA) are still local but also take into account the gradient of the density at the same coordinate.”
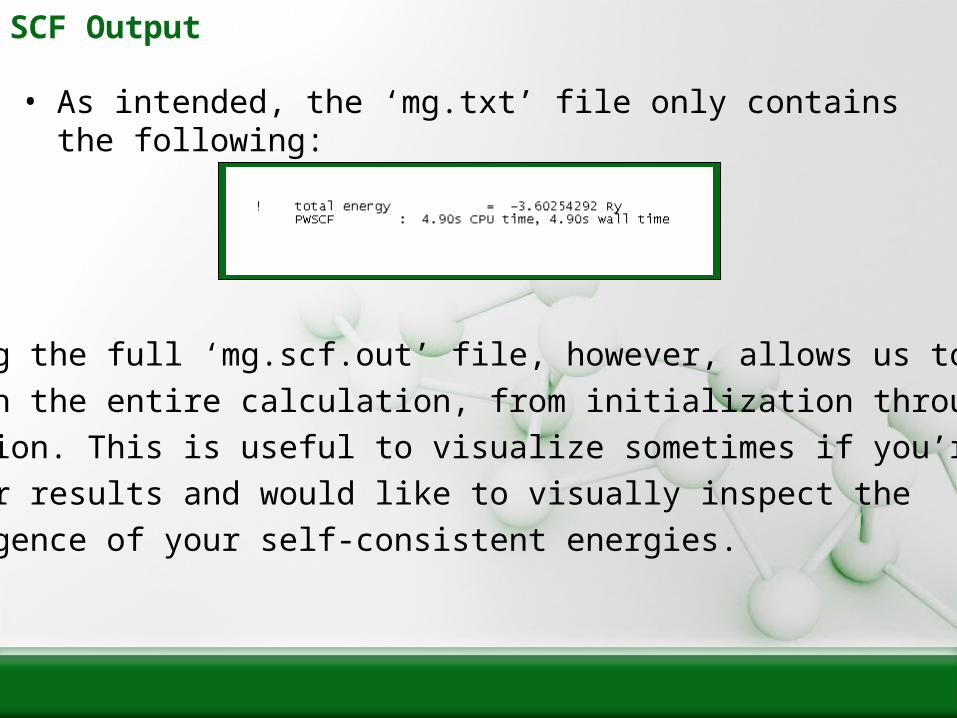
SCF Output
• As intended, the ‘mg.txt’ file only contains the following:
• Reading the full ‘mg.scf.out’ file, however, allows us to step
through the entire calculation, from initialization through each
iteration. This is useful to visualize sometimes if you’re unsure
of your results and would like to visually inspect the
convergence of your self-consistent energies.

Ensuring k and ecut Lead to a Converged Energy
• The most important skill in performing DFT calculations is the ability to get converged energies. Since the appropriate choice of k-points and ecut vary wildly among different geometries (and even different required accuracies), it is important to be able to form the following graphs every time you perform ‘scf’ calculations on new geometries.
• Converged values of ecut and k should be reported any time you publish DFT results, so that someone else may reproduce your calculation and agree on the same numerical error.
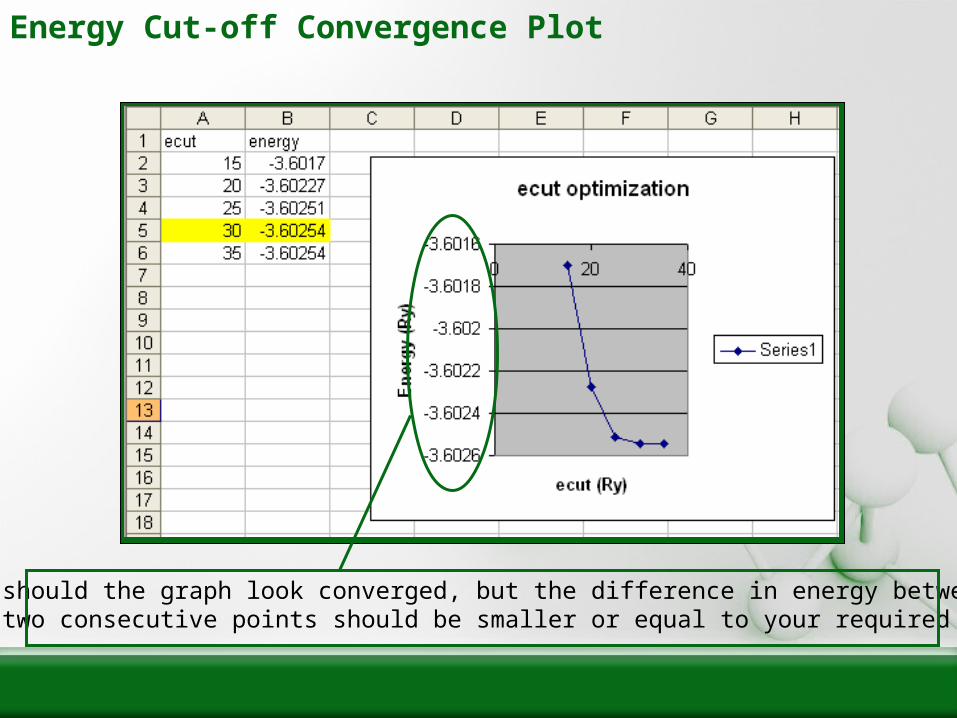
Energy Cut-off Convergence Plot
Not only should the graph look converged, but the difference in energy betweenthe last two consecutive points should be smaller or equal to your required accuracy!
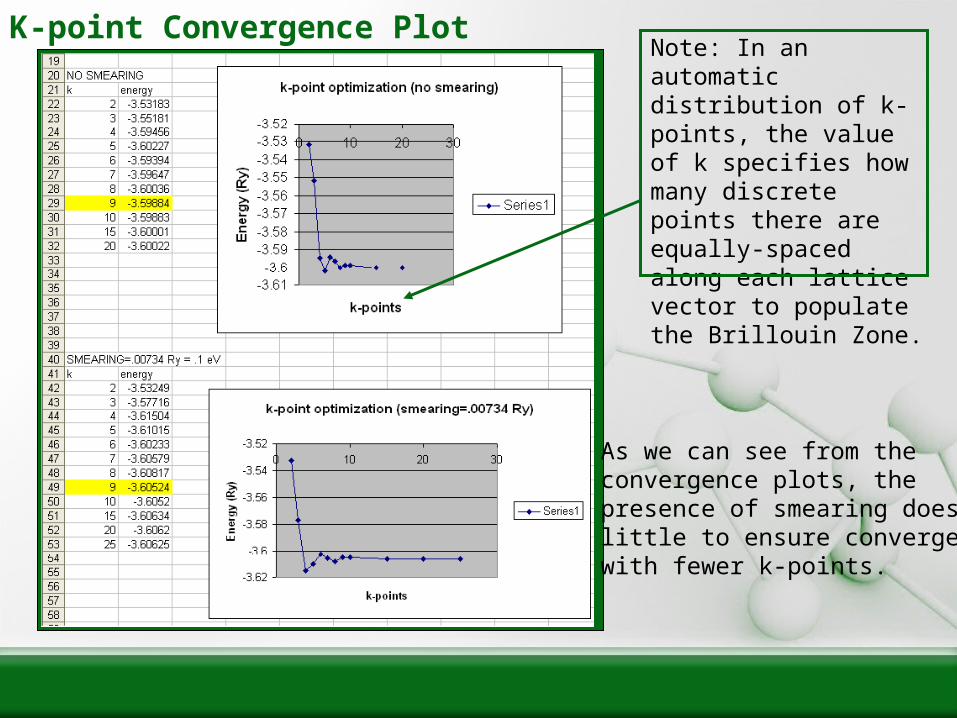
K-point Convergence Plot
As we can see from theconvergence plots, the presence of smearing doeslittle to ensure convergencewith fewer k-points.
Note: In an automaticdistribution of k-points, the value of k specifies how many discrete points there are equally-spaced along each lattice vector to populate the Brillouin Zone.

K-point Convergence Plot (cont.)
• When unit cells do not have equal-length lattice vectors, it is sometimes computationally rewarding to “geometrically-optimize” your automatic k-point distribution.
• For example, if one had a unit cell that was four times taller in one direction than its other two directions, one should specify only a quarter as many k-points along the taller direction.– This makes sense, because in reciprocal space, the taller
distance will only be a quarter as long as the other two distances.

Performing Geometry Relaxation Calculations on Solid Mg
‘vc-relax’ differs from ‘relax’ onlyin that the cell parameters can varyduring the relaxation.

VC-RELAX Output
• Reading the full ‘mg.rx.out’ file allows us to step through the entire calculation, from initialization through each iteration.
• It becomes apparent that a relaxation calculation is simply a series of scf calculations with forces specified at each step and updated positions at each iteration that correspond to the strength of the forces between the atoms in the system.

Comparing the Relaxed Structure to Literature

Comparing the Relaxed Structure to Literature (cont.)
• On top of comparing the ‘a’ and ‘c/a’ lattice parameters to literature and to experiment, it is also useful to compare a quantity called “cohesive energy”– The difference between the average energy of the atoms of
a crystal and that of the free atoms.
• It is important to compare cohesive energies and not specifically the self-consistent energies, because only differences in energies are physically meaningful.– The energy datum for an ‘scf’ calculation is specified by the
choice of pseudopotential.

The Game Plan
DFT Basics
Job File Structure
Sample Structure: Solid Mg
Performing ‘scf’ Calculations on Solid Mg
Converging ‘scf’ Energies
Performing ‘relax’ calculations on Solid Mg
The Limits of DFT
Current work: GST structure and CNT properties

What Density Functional Theory Can Do
• Applies to atoms, molecules, surfaces and periodic structures...
• Self-consistent total energies, forces, stresses (ground state)• Structural optimization• Elastic modulus• Electron band structure of many-body systems• Phonon frequencies and eigenvectors at any wave vector• Full phonon dispersions• Electron-phonon interactions• ab-initio MD• etc..

What Density Functional Theory CanNOT Do
• DFT Calculations are not exact solutions of the full Schrodinger equation (exact functional unknown).
• The Hohenberg-Kohn theorems apply only to ground state energies. Excited states can be predicted, but these predictions are not on the same footing as predictions for ground-state energies.
• There is a well-known inaccuracy in DFT involving the underestimation of calculated band gaps in semiconducting and insulating materials.
• DFT gives inaccurate results involving weak van der Waals attractions, which are a direct result of long range electron correlation.
• It is computationally expensive to perform DFT calculations. Simulations with hundreds of atoms are about the largest you can get.

Current NTPL Use of DFT Calculations
• Determining the Structure of Ge2Sb2Te5 Phase-change Material
• Electron and Phonon Properties of Carbon Nanotubes
Stable Structure
(hcp)
Meta-stable Structure(rocksalt)

Any Questions?
Please visit the related pages on the NTPL Wiki for more information!
![Large-scale first principles and tight-binding density ... · the tight-binding DFT calculations, we used the DFTB+ code [25]. ONETEP [1] is a linear-scaling approach for DFT calculations,](https://static.fdocuments.in/doc/165x107/5f58478819c084702320ff1b/large-scale-first-principles-and-tight-binding-density-the-tight-binding-dft.jpg)

















