Development of Novel Small Molecule Inhibitors by A ...
258
Development of Novel Small Molecule Inhibitors of Neurotropic Alphavirus Replication by Scott Barraza A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Medicinal Chemistry) in the University of Michigan 2014 Doctoral Committee Research Professor Scott D. Larsen, Co-Chair Associate Professor David J. Miller, Co-Chair Professor John Montgomery Professor Henry I. Mosberg
Transcript of Development of Novel Small Molecule Inhibitors by A ...

Development of Novel Small Molecule Inhibitors
of Neurotropic Alphavirus Replication
by
Scott Barraza
A dissertation submitted in partial fulfillment
of the requirements for the degree of
Doctor of Philosophy
(Medicinal Chemistry)
in the University of Michigan
2014
Doctoral Committee
Research Professor Scott D. Larsen, Co-Chair
Associate Professor David J. Miller, Co-Chair
Professor John Montgomery
Professor Henry I. Mosberg

ii
Table of Contents
List of Figures .................................................................................................................... iv
List of Schemes .................................................................................................................. vi
List of Tables ..................................................................................................................... ix
List of Abbreviations ......................................................................................................... xi
Introduction ........................................................................................................ 1 Chapter I.
Arboviruses: An Emerging Public Health Threat ........................................................... 1
Neurotropic Alphavirus Virus Biology ........................................................................... 4
Project History ................................................................................................................ 6
Reduction of Polar Surface Area ...................................................................... 9 Chapter II.
Rationale ......................................................................................................................... 9
SAR ............................................................................................................................... 12
Synthesis ....................................................................................................................... 20
Conclusion .................................................................................................................... 24
Reduction of Molecular Weight .................................................................... 26 Chapter III.
Rationale ....................................................................................................................... 26

iii
SAR ............................................................................................................................... 29
Synthesis ....................................................................................................................... 49
Conclusion .................................................................................................................... 59
Conformational Restriction ........................................................................... 60 Chapter IV.
Rationale ....................................................................................................................... 60
SAR ............................................................................................................................... 63
Synthesis ....................................................................................................................... 73
Conclusion .................................................................................................................... 88
Metabolic Stability ......................................................................................... 89 Chapter V.
Rationale ....................................................................................................................... 89
SAR ............................................................................................................................... 90
Synthesis ....................................................................................................................... 98
Conclusion .................................................................................................................. 100
Future Directions ......................................................................................... 101 Chapter VI.
Experimental Data ..................................................................................... 104 Chapter VII.
Bibliography ................................................................................................................... 233

iv
List of Figures
Figure 1. Equine Encephalitis Virus Maintenance Cycle. .................................................. 3
Figure 2. Alphavirus Single-Cell Reproductive Cycle. ...................................................... 5
Figure 3. Natural WEEV Genome and WEEV Replicon. .................................................. 6
Figure 4. HTS Discovery Hit CCG-32091 and Initial Development.................................. 8
Figure 5. Polar Surface Area Contribution per Heteroatom. ............................................ 11
Figure 6. Lead Compounds. .............................................................................................. 12
Figure 7. Right-Hand Chain Length-Dependence. ........................................................... 18
Figure 8. Development of Lead CCG-205432. ................................................................. 26
Figure 9. Sites for Molecular Weight Reduction. ............................................................. 29
Figure 10. WEEV Antiviral Activity for CCG-205432 and CCG-206381. ..................... 34
Figure 11. In Vivo Pharmacokinetic Data for CCG-205432 and CCG-206381. .............. 35
Figure 12. Conformational Restriction Plan for Arene Analogs. ..................................... 42
Figure 13. Spectrum Antiviral Data for Selected Analogues. .......................................... 46

v
Figure 14. Effect of Selected Analogues on Transfected Gene Expression in BSR-T7
Cells. ................................................................................................................................. 48
Figure 15. Steric Approach to Conformational Restriction. ............................................. 61
Figure 16. Tethering Approach to Conformational Restriction. ....................................... 62
Figure 17. Ring-Locking Approach to Conformational Restriction. ................................ 62
Figure 18. Activity of Various Analogs Against Live Virus. ........................................... 69
Figure 19. Predicted Sites of Metabolism in CCG-205432, Rank Ordered...................... 90

vi
List of Schemes
Scheme 1. General Preparation of Low TPSA Analogs. .................................................. 20
Scheme 2. Preparation of CCG-204055 and CCG-205476. ............................................. 21
Scheme 3. Preparation of CCG-205421 and CCG-205475. ............................................. 22
Scheme 4. Preparation of CCG-204020 and CCG-205420. ............................................. 22
Scheme 5. Preparation of CCG-211826............................................................................ 23
Scheme 6. General Preparation of Secondary Amine Analogs. ....................................... 24
Scheme 7. Preparation of Pyrrole and Imidazole Analogs. .............................................. 49
Scheme 8. Preparation of Azetidine Analogs. .................................................................. 50
Scheme 9. Preparation of Acyclic Amide and Urea Analogs. .......................................... 51
Scheme 10. Preparation of the Right-Hand Amine Dihydrochloride 36. ......................... 52
Scheme 11. Preparation of N-Benzyl and N-Benzoyl Pyrrolidine Analogs. .................... 52
Scheme 12. Preparation of Anthranilamide and Salicylamide Analogues. ...................... 54
Scheme 13. Preparation of Benzyl Anthranilamide Analogues........................................ 55
Scheme 14. Preparation of Phenethyl Anthranilamide Analogue CCG-222983. ............. 55

vii
Scheme 15. Preparation of Benzyl and Benzoyl Benzoate Analogues. ............................ 56
Scheme 16. Preparation of Carbazole Analogues. ............................................................ 57
Scheme 17. Preparation of Quinolone Analogues. ........................................................... 58
Scheme 18. General Preparation of Rigid Benzamide Analogs. ...................................... 74
Scheme 19. Preparation of 2-(aminomethyl)indane 75. ................................................... 74
Scheme 20. Preparation of Racemic 2-(2-Aminoethyl)indane. ........................................ 75
Scheme 21. Preparation of 7-Methylindole CCG-206447. ............................................... 76
Scheme 22. Preparation of 3-Methylindole CCG-205431. ............................................... 76
Scheme 23. Preparation of pyrrolidopiperidine Analogs CCG-206485 and CCG-212394.
........................................................................................................................................... 77
Scheme 24. Preparation of Inverse Amide CCG-212052. ................................................ 78
Scheme 25. Preparation of Shifted Amide CCG-224001. ................................................ 78
Scheme 26. Preparation of (E)- and (Z)-alkene Analogs. ................................................. 80
Scheme 27. Preparation of Urea Analog CCG-212390. ................................................... 81
Scheme 28. Preparation of Tetrahydropyridine Analog CCG-211823. ............................ 82
Scheme 29. Preparation of Methylpiperidine Analog CCG-211829. ............................... 83

viii
Scheme 30. Preparation of Exo Bicyclic Analog CCG-222980. ...................................... 84
Scheme 31. Preparation of Bridged Analog CCG-224000. .............................................. 84
Scheme 32. Preparation of the Spiro Analog CCG-224220. ............................................ 85
Scheme 33. Preparation of Azetidine-Containing CCG-224002. ..................................... 86
Scheme 34. Preparation of Azapane Analog CCG-222661. ............................................. 87
Scheme 35. Preparation of 4-Fluoropyrrole Analogs. ...................................................... 98
Scheme 36. Preparation of 6-Azaindole Analog CCG-211751. ....................................... 99

ix
List of Tables
Table 1. WEEV Replicon Data for Alicyclic Low TPSA Analogs. ................................. 13
Table 2. WEEV Replicon Data for Aromatic Low TPSA Analogs. ................................. 15
Table 3. MDR1 Recognition Data for Indole and Thienopyrrole leads. .......................... 16
Table 4. WEEV Replicon Data for Amine Analogs. ........................................................ 19
Table 5. WEEV Replicon and ADME Data for Pyrrole and Imidazole Analogs. ............ 30
Table 6. WEEV Replicon Data for Acyclic Low MW Analogs. ...................................... 36
Table 7. WEEV Replicon Data for Pyrrolidine Analogs. ................................................. 37
Table 8. WEEV Replicon and In Vitro ADME Data for Arene Analogs. ........................ 39
Table 9. WEEV Replicon Date for Rigid Arene Analogs. ............................................... 43
Table 10. Antiviral Data for Select Analogues ................................................................. 45
Table 11. WEEV Replicon Data for Methylindole Analogs. ........................................... 64
Table 12. WEEV Replicon Data for Rigid Benzamide Analogs. ..................................... 65
Table 13. WEEV Replicon Data Comparisons of Analogs Based on CCG-205432 and
CCG-206382. .................................................................................................................... 67

x
Table 14. WEEV Replicon Data for Central Piperidine Analogs. .................................... 71
Table 15. MLM Metabolic Stabilities for Thienopyrrole and Indole Leads. .................... 91
Table 16. MLM Metabolic Stabilities For Indole and Pyrrole Analogs. .......................... 93
Table 17. MLM Stability and WEEV Replicon Data for Select Analogs. ....................... 94
Table 18. MLM Stability and WEEV Replicon Data for Select Analogs. ....................... 95
Table 19. WEEV Replicon Data for Fluorinated Analogs. .............................................. 97

xi
List of Abbreviations
ADME Absorption, distribution, metabolism, excretion
BBB Blood-brain barrier
BSL Bio-safety level
CC50 Cytotoxic concentration, half maximal
CEV California encephalitis virus
CNMR Carbon nuclear magnetic resonance spectroscopy
CNS Central nervous system
CPE Cytopathic effect
CYP Cytochrome P450
DMSO Dimethylsulfoxide
EEV Equine encephalitis virus
EEEV Eastern equine encephalitis virus
EMCV Encephalomyocarditis virus
fLUC Firefly luciferase

xii
FMV Fort Morgan virus
HNMR Proton nuclear magnetic resonance spectroscopy
HPI Hours after infection
HPLC High-performance liquid chromatography
HTS High-throughput screen
IC50 Inhibitory concentration, half maximal
IR Infrared spectroscopy
LC Liquid chromatography
MDCK Madin-Darby canine kidney cells
MDR1 Multidrug resistance protein 1
MLM Mouse liver microsome
MOA Mechanism of action
MOI Multiplicity of infection
MS Mass spectrometry
MS/MS Tandem mass spectrometry
MTT 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide

xiii
MW Molecular weight
NMR Nuclear magnetic resonance spectroscopy
PAMPA Parallel artificial membrane permeability assay
PFU Plaque-forming unit
Pgp P-glycoprotein
Rho123 Rhodamine 123
SAR Structure-activity relationship
SEAP Secreted embryonic alkaline phosphatase
SEM Standard error of the mean
SD Standard deviation
SINV Sindbis neuroadapted virus
TPSA Topological polar surface area
VEEV Venezuelan equine encephalitis virus
WEEV Western equine encephalitis virus

1
Introduction Chapter I.
Arboviruses: An Emerging Public Health Threat
New and emerging viruses pose an immense public health risk to the world
population,1, 2
due in large part to a lack of effective vaccines and treatments, although
inadequate public health responses may also play a significant role.3 Mosquito-borne
viruses, collectively called arboviruses (arthropod-borne viruses), constitute a large
portion of this risk because of the ubiquity of mosquitoes and the potential for mosquito-
human transmission.4-6
Increasing urbanization and mosquito range expansion further
complicates the threat of potential arboviral pandemic.4-6
Among emerging New World
arboviruses, the equine encephalitis viruses (EEVs) (Western, Eastern, and Venezuelan)
are of particular interest, and are listed as category B agents of biodefense concern due in
part to their potential use in bioterrorism.7-9
EEVs are neurotropic arboviruses that infect the central nervous system (CNS) of
vertebrates, causing debilitating inflammation and damage to the brain.10-12
They are
members of the genus Alphavirus of the Group IV family Togaviridae,13
of which more
than ten members cause significant disease in humans, and all are arboviruses.1 They are
also RNA viruses, and their emerging status may be attributed to this characteristic,2
largely due to poor genome replication fidelity and high mutation rates common to RNA
viruses.14
In nature, EEV existence is maintained by a cycle between passerine birds and
the mosquitoes that prey exclusively on them (Figure 1).15, 16
Infection in humans and

2
animals occurs when other mosquitoes, the so-called bridging vectors, pass the virus
along from birds.17
The natural infection rate in humans, while underreported, is
nonetheless low: since 1964, only 639 cases of Western equine encephalitis virus
(WEEV)18
and 220 cases of Eastern equine encephalitis virus (EEEV) infection have
been confirmed.19
Still, the clinical manifestations of infection can be severe. For victims
of WEEV, the fatality rates may be as low as 3%,20
but neural sequelae may persist well
after infection.21
Young children exhibit seizures and paralysis,22-24
for example, and
upwards of 60% continue to do so for months or years after the virus has disappeared.25
Adults, too, may experience persistent disease-associated impairment.24
Of greater
concern is EEEV, which boasts a mortality rate in excess of 30-40%, and even as high as
70% among some strains,19
and is also likely to cause permanent neurological damage in
survivors.26-28
On the other hand, Venezuelan equine encephalitis virus (VEEV), while
least likely to cause debilitating symptoms in humans,29-31
may have high agricultural
costs associated with infection as equine mortality rates have been observed to exceed
80-90% during epizootic pandemics. Furthermore, unlike with WEEV and EEEV
infections, horses and humans are not dead-end hosts and may continue to spread the
virus.16

3
Figure 1. Equine Encephalitis Virus Maintenance Cycle.
Passerine birds serve as reservoirs for many of the New World alphaviruses, but all
alphaviruses are transmitted to humans and susceptible animals by arthropods.
After World War II, many of the early developers of biological warfare found that
the equine encephalitis viruses and related alphaviruses were highly amenable to
weaponization.9, 16, 32
As stated from a report by the US Army Medical Research Institute
for Infectious Diseases, “Few [viruses] possess as many of the required characteristics for
strategic or tactical weapon development as the alphaviruses.”16
The reasons are simple:
they can be produced in large quantities, they are stable and many are infectious as
aerosols, many are fatal (or at the very least, incapacitating), they are relatively simple to
engineer (and make more lethal), and defensive vaccine development is hindered by the
existence of multiple serotypes. Many nations have long since shut down their offensive
bioweapon programs (the United States in 1969), but today there is a growing threat from
small groups of bioterrorists.9, 16, 33
There currently exist vaccines for WEEV34
and a few of the other alphaviruses,35,
36 but they require multiple shots, confer little immunoprotection, and are available only

4
to researchers.15, 16, 35
It has also been demonstrated that potential vaccines against one
alphavirus may interfere with vaccination against other alphaviruses.37-39
From a public
health perspective, this underlines the need for an effective antiviral, but there are no
therapeutics available for treating infection once the virus has colonized the CNS.15, 40
Therefore, the focus of our research is the development of new, novel compounds for the
pharmacological intervention of CNS infection by alphaviruses.
Neurotropic Alphavirus Virus Biology
The reproductive life cycle of neurotropic alphaviruses is among the most simple
of viruses, and is largely localized to the cytoplasm of the host cell.13
As shown in Figure
2, the first step (step 1) of reproduction involves binding of the virion to specific host
receptors displayed on the exterior of the cell membrane, and receptor-mediated
endocytosis draws the virion into the cell. Upon acidification of the vesicle, the virion is
uncoated and released into the cellular environment (step 2), where it may release its
simple positive-sense single-stranded RNA ((+)ssRNA) genome (step 3.) The genome
may be directly translated upon cellular entry by cytoplasmic ribosomes to produce
protein and enzyme products that facilitate amplification of the genome (step 4), or it
may be transcribed to produce many copies of the original genome (step 5). The copies
may be involved in further translation, or they may be packaged in the capsid of new
virion particles (steps 5, 9, and 10.) Some copies are involved in the translation of
membrane-bound proteins in the secretory pathway, where they will be involved in
recruitment of capsid proteins and assembly of new virion particles, and will ultimately

5
form the viral envelope (steps 6 through 8). Finally, budding occurs and the mature virion
is released (step 11.)13, 41-45
Figure 2. Alphavirus Single-Cell Reproductive Cycle.
Overview of the alphavirus life cycle. Our research focuses on the development of
inhibitors of replication (step 5).
Mosquitoes transmit EEVs to hosts upon biting, causing a localized infection that
may escalate to observable viremia after several days.46-48
The mechanism of invasion of
the CNS is a contentious subject, with some evidence suggesting that the olfactory nerves
are the major route as opposed to the direct crossing of the blood-brain-barrier (BBB).49-
54 Nevertheless, once the virus is established in the CNS, the recruitment of
immunological cells is responsible for encephalitis, but the majority of serious brain
destruction is caused by the induction of neuronal cell death.43, 55
Other cell types are

6
susceptible as well, but form of death is not uniform: glial cells chiefly undergo
apoptosis, whereas neuronal cells usually die via necrotic pathways.55-59
Project History
High-Throughput Discovery. In search of potential antiviral leads, the David
Miller lab executed a high-throughput screen (HTS) of a >50,000-compound library at
the University of Michigan Center for Chemical Genomics (CCG).60
Central to the screen
was a cell-based assay that utilized WEEV replicon plasmids encoding the firefly
luciferase reporter gene.
Figure 3. Natural WEEV Genome and WEEV Replicon. Structural genes were excised and replaced with the reporter gene fLUC, coding for
firefly luciferase. In this way, the WEEV replicon is incapable of producing infectious
virus and may be used without special precautions.
WEEV Replicon Assay. Inhibitory concentrations (IC50 values) were determined
using a WEEV replicon assay developed by our collaborators in the lab of Prof. David
Miller.60
BSR-T7/C3 cells were transfected with a complementary plasmid (pWR-LUC)
encoding a majority of the WEEV genome (Figure 3); however, most of the structural
genes are replaced with the reporter gene firefly luciferase.61
In this way, the replicon
cannot produce infectious virus and thus requires less stringent biosafety containment

7
measures. BSR cells constitutively express bacteriophage T7 RNA polymerase, for which
promoters and terminators exist in the plasmid to maintain high levels of transcription of
the WEEV replicon. Therefore, control cells will produce a baseline luminescence while
cells treated with active inhibitors will show a marked decrease from baseline, from
which an IC50 can be calculated.
MTT Reduction Assay. Cytotoxic concentrations (CC50 values) were established
using a standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazol-3-ium bromide
(MTT) assay in BSR cells used in the WEEV replicon assay by the lab of Prof. David
Miller.60, 62
In this classical measure of cellular viability, the tetrazolium compound MTT
is reduced to a dark purple formazan dye by oxidoreductase enzymes in living cells. Cells
stressed or killed by cytotoxic compounds exhibit diminished metabolic activity and, as a
result, are less or not at all capable of reducing the MTT to the colorful dye. Therefore,
CC50 values are calculated from the drop in absorbance relative to a healthy baseline
control.
HTS Triage. Because luciferase served as a marker for disruption of genome
replication, those compounds with the lowest IC50 values in the initial HTS were
submitted to secondary and tertiary validation assays. The secondary assay utilized
replicons containing the SEAP reporter gene to indicate transcriptional activity and
confirm that activity was not an artificial construct of luciferase inhibition,63
and the
tertiary assay screened for compounds with selectivity indices (CC50/IC50) greater than
five. One compound, CCG-32091, possessed favorable potency and selectivity, and its
activity was further verified in cultured human neurons in the presence of the live
alphaviruses SINV (Sindbis virus) and FMV (Fort Morgan Virus), which are surrogates

8
for WEEV that require lower level biosafety containment facilities. Limited structure-
activity relationships (SAR) among commercial thienopyrroles related to CCG-32091
showed promising activities and selectivity ratios, as well as a clearly defined SAR
spanning two orders of magnitude potency,60
so the compound was selected for further
optimization.
Figure 4. HTS Discovery Hit CCG-32091 and Initial Development.
The thienopyrrole was substituted with bioisosteric indole (blue), and the known
metabolism-prone furan was replaced with benzyl functionality (red).
Initial SAR. The initial approach was replacement of the thienopyrrole core of
CCG-32091 with an indole core (Figure 4).64
This was undertaken for several reasons:
indoles are generally more metabolically stable than thienopyrroles,65
are cheaper and
synthetically more robust, and phenyl and thiophene are well established bioisosteres.66, 67
CCG-102516, synthesized by Kyle Bolduc, was developed from this rationale. It
contains an indole core, a more lipophilic chloro-substituted N-benzyl group, and a
benzylamide predicted to be more metabolically stable relative to the furylmethylamide
of CCG-32091.68, 69
This initial strategy was successful, as CCG-102516 proved to be
nearly twice as potent as CCG-32091.

9
Reduction of Polar Surface Area Chapter II.
Rationale
The Blood-Brain Barrier. Neurotropic alphaviruses replicate to high titer within
the CNS,70
necessitating development of CNS-penetrant antiviral agents. This is
especially important because clinical manifestations may appear well after the systemic
virus titer has dropped to immeasurable levels, days after initial transmission.71-73
This
places an enhanced emphasis upon retaining physical properties predictive of both good
pharmacokinetics and CNS penetration while optimizing both in vitro and in vivo
activity. The most challenging barrier to CNS entry is the blood-brain barrier (BBB).74-77
The BBB is structurally distinct from other membrane obstacles; due to diminished
pinocytosis and the presence of tight-junctions, most drugs must cross the BBB via
transcellular passive diffusion alone.74, 77
However, there are a number of common
features among successful CNS-active drugs that enhance passive BBB transit, including
low molecular weight (< 400-450), low topological polar surface area (< 60–70 Å2), and
positive logD (~1-3).74-76, 78, 79
MDR1 Recognition Assays. Transporter-mediated efflux is a major hurdle to a
CNS drug’s biological activity in vivo.80, 81
In its protective role, the blood-brain barrier
(BBB) has a number of efflux transporters to facilitate removal of xenobiotics from the
CNS, of which MDR1 is generally the most important.80, 82
Neurotropic alphaviruses
replicate to high titer in the CNS, thereby necessitating the development of transporter-

10
evasive inhibitors to elicit the greatest antiviral effect. It is therefore important to generate
analogs with little to no affinity for MDR1 early in the drug development process.
Over the course of the project, two recognition assays were employed to evaluate
MDR1 recognition of WEEV inhibitors in the lab of Prof. Richard Keep.83
Initially, our
collaborators utilized a simple experiment that measured the impact of our analogs on the
efflux of tritiated (3H) vinblastine, a known MDR1 substrate, from MDCK cells that
expressed human MDR1 (MDR1-MDCKII cells).83, 84
This specific assay was employed
in early evaluations of WEEV inhibitor MDR1 interactions, such as those reported for
low topological polar surface area (TPSA) analogs in this chapter, but later the
Rhodamine 123 uptake assay was employed (see Chapter III). In the 3
H-vinblastine
uptake assay, if the test compound of interest has no interaction with MDR1, the 3H-
vinblastine concentration inside the cell will drop as the substrate is pumped out. If the
test compound does interact with MDR1, the measured 3H-vinblastine concentration will
be higher within the cell. However, due to the nature of the experiment, it cannot be said
whether or not analogs are simple blockers of MDR1 or actual substrates. Regardless, the
data are useful for identifying compounds with low recognition by MDR1 (and thus
potential for efflux) and was used for the prioritization of compounds for further
development.
Initially, we were most interested in maximizing potential for achieving CNS
penetration by reducing the TPSA of the inhibitors. TPSA is a measure of the surface
area of all polar atoms in a molecule, predominately heteroatoms like oxygen and
nitrogen.79, 85, 86
Additionally, polar substituents possess high desolvation penalties and
reduce passive permeability due to repulsive interactions with highly nonpolar lipid

11
membranes.87, 88
High TPSA is also associated with greater recognition by efflux
transporters.89, 90
The total heteroatom contribution to the TPSA of CCG-102516 is
presented in Figure 5; it is evident that heteroatoms with electrons engaged in resonance
have little impact, whereas heteroatoms with a complement of free electrons contribute
more to the TPSA.
Figure 5. Polar Surface Area Contribution per Heteroatom.
Total TPSA and individual contributions were calculated using ChemDraw’s chemical
properties calculator.
BBB-PAMPA Assay. Due to the importance of BBB penetration to our
compounds, and the role of TPSA in enhancing this property, we estimated the passive
membrane permeability for select compounds using BBB-PAMPA (blood-brain barrier
parallel artificial membrane permeability assay) from pION, Inc.,91, 92
and utilized a
cosolvent system due to the poor aqueous solubility of early compounds.93
The traditional
PAMPA assay was developed as a useful low-cost tool for assessing potential passive
membrane permeability for drug-like compounds in the gut, and was found to correlate
well with higher-cost cellular assays.94, 95
However, the BBB is structurally and
compositionally distinct from other membranes, causing PAMPA to often report false
positives; for this reason, BBB-PAMPA was developed and was found to possess

12
superior correlation with BBB passive permeability.96
It is also an important complement
to the MDR1 recognition assay because the MDR1 assay correlates poorly with BBB
passive permeability – due to the different lipid composition of the cells – but contains
efflux transporters not present in BBB-PAMPA.97
While the TPSA of CCG-102516 is within the range of successful CNS drugs,
which have TPSAs significantly lower than other drug classes,74
it is on the higher end of
that range, and we felt it was important to identify low TPSA features that could serve as
new templates throughout other aspects of the SAR campaign in order to maintain
optimal properties predictive of BBB-penetration. Furthermore, it was important to
determine whether TPSA could be reduced with maintenance of anti-viral activity.
SAR
Initial analogs were based on CCG-203880, the equipotent N-4-fluorobenzyl
analog of CCG-102516 (Figure 6) because of its lower molecular weight (see Chapter
III). A general feature of these analogs was the replacement of the right-hand secondary
amide with alternative linkers.
Figure 6. Lead Compounds.
CCG-102516 and its equipotent fluoro analog CCG-203880.

13
Table 1. WEEV Replicon Data for Alicyclic Low TPSA Analogs.
CCG No. X R IC
50
(µM)a
CC50
(µM)b
MDR1
(% vinblastine
uptake)c
TPSAd
203880† F 15 ± 2 >100 1,495 53
203941 F >100 >100 --- 27
205421 F 14 ± 1 24 ± 1 --- 44
205475 F >100 >100 --- 41
203945 F 23 ± 2 >100 --- 44
204055 F 12 ± 1 96 821 27
205476 Cl 10 ± 2 38 --- 27
aInhibition of luciferase expression in WEEV replicon assay. bCell viability determined
by the MTT reduction assay. Values are mean of at least n=3 independent experiments.
dTPSA was predicted using ChemDraw’s chemical properties calculator. †Synthesized by
Janice Sindac.
Alicyclic Low TPSA SAR. As seen in Table 1, we were able to demonstrate that
TPSA could be reduced substantially in some cases without impacting activity or toxicity
(e.g. alicyclic analogs CCG-203945 and 204055). Aromatic rings are capable of
engaging in numerous types of bonding interactions – like ring-stacking or weak CH

14
hydrogen bonding – not available to alicycles. That the active low TPSA analogs bear
non-aromatic heterocycles and carbocycles suggests that the binding in this region may
be largely dependent on hydrophobic interactions between the ring and lipophilic
residues in the unknown binding site. Also of interest is the difference in activities
between CCG-205421 and CCG-205475. Both compounds are hydrogen bond acceptors,
but only CCG-205421 is also a hydrogen bond donor, perhaps forming a key contact
with an acceptor in the binding site. CCG-205421 is also tetrahedral about the sp3
alcohol carbon, unlike the sp2 carbonyl carbon in CCG-205475. This could indicate that
CCG-205421 has geometry that enhances interactions with unknown binding site
contacts.
Aromatic Low TPSA SAR. As seen in Table 2, like the alicyclic analogs, we
found TPSA could be halved in the aromatic analogs while maintaining favorable
potency and toxicity profiles (e.g. aromatic analogs CCG-203942 and CCG-203945),
placing WEEV inhibitor properties well within the range of successful CNS drugs.
Intriguingly, the SAR of ketone CCG-203943 and alcohol CCG-204056 matched that of
their alicyclic counterparts CCG-205475 and CCG-205421, respectively (Table 1). We
also noted that benzylamine CCG-205420 was substantially more potent than its
benzylether analog CCG-203944, but this result may be an artificial product of its high
toxicity. Curiously, benzyl piperazine analogs (e.g. CCG-203942) were eqipotent with
the lead, in contrast to the inactive cyclohexylmethyl piperazine CCG-203941 (Table 1).
This challenged the assumption that lipophilicity was the most important factor for
binding in this region of molecule.

15
Table 2. WEEV Replicon Data for Aromatic Low TPSA Analogs.
CCG No. X R IC
50
(µM)a
CC50
(µM)b
MDR1
(% vinblastine
uptake)c
TPSAd
203880† F 15 ± 2 >100 1,495 53
203943 F >100 >100 --- 41
204056 F 15 ± 0.4 40 --- 44
204020 F >100 >100 --- 27
205420 F 22 ± 3 39 --- 36
203944 F >100 >100 --- 33
203942 F
12 ± 1 >100 764 27
206372 Cl
12 ± 1 >100 --- 27
211827 Cl
7.2 >100 --- 27
211826 Cl
12.4 >100 --- 39
aInhibition of luciferase expression in WEEV replicon assay.
bCell viability determined
by the MTT reduction assay. Values are mean of at least n=3 independent experiments.
†Synthesized by Janice Sindac. dTPSA was predicted using ChemDraw’s chemical
properties calculator.

16
Table 3. MDR1 Recognition Data for Indole and Thienopyrrole leads.
CCG No. R MDR1 Recognition (%
vinblastine uptake)a
102516†
534.0
203880†
1495.6
203881†
433.3
32088*
694.1
aMDR1 recognition was assessed by measuring uptake
3H-vinblastine at 30 µM of test
anti-viral or vehicle. †Synthesized by Janice Sindac. *Synthesized by Bryan
Yestrepsky.
MDR1 Recognition. It was hoped that the best low TPSA candidates from this
assay may serve as new templates for further development. After our initial survey of the
activity of these analogs was complete, CCG-204055 and CCG-203942 were graduated
to the MDR1/vinblastine assay because they showed promising inhibition and low
cytotoxicity. Both of these compounds were found to possess improved MDR1-efflux
results over CCG-203880 (which may be attributed to their lower TPSA compared to
their amide counterparts), and were therefore considered potential new leads. However,

17
initial trends among other scaffolds suggested that chloro-substituted compounds may be
more MDR1-evasive than their fluoro-substituted counterparts (Table 3), prompting us to
prepare and evaluate chloro-substituted analogs of some of the better fluoro-substituted
compounds (e.g. CCG-205476 and CCG-206372, Table 1 and Table 2, respectively).
These compounds were found to be equipotent to their fluoro-substituted cousins.
Unfortunately, these compounds were never submitted to the MDR1 recognition assay
due to the discovery of the significantly more potent 4-pyridylethylamide inhibitor class,
described briefly below and in detail in Chapter III.
TPSA Analog Length-Activity Dependence. While many of the TPSA analogs
feature low polar surface area with retention of activity, it was apparent that there was
still room for potency optimization. Janice Sindac, working on other aspects of the
WEEV project, had established that the right-hand portion of the template must be of a
specific length, where the intervening alkyl chain between the amide and the aromatic is
optimally 2-carbons long (Figure 7).83
Interestingly, propyl linkers abolished activity
entirely. The low TPSA compounds in Table 1 and Table 2 were significantly truncated,
so longer analogs were investigated. Interestingly, analogs with ethyl and propyl linkers
did not perform significantly better than their shorter counterparts (e.g. CCG-206372
compared to CCG-211827, Table 2), nor were they worse. Even CCG-211826, which
bears a 4-pyridylethyl substituent crucial to the activity of later analogs (see Chapter
III), merely maintained activity. This suggested that either important structural features
were not present among the low TPSA analogs or that they were not binding in the same
way as the amide leads. Indeed, later work established the importance of the right-hand

18
secondary amide to activity (see Chapter IV) which is lacking in the low TPSA inhibitor
class.
Figure 7. Right-Hand Chain Length-Dependence.
Chain-length dependence of the right-hand region “R” was found to decrease in activity
such that n=2>1>>3, where n is an intervening number of methylene units.
Secondary Amine Analog SAR. The low TPSA analogs are highly lipophilic.
While this aids in BBB permeation, it decreases water solubility such that aqueous
delivery of compound is impeded. Interestingly, analogs containing ionizable
functionalities, such as amines, exhibited the greatest activity (e.g. piperazine-containing
analogs CCG-203942 and CCG-204055), perhaps because of improved aqueous
solubility. Alternatively, the analogs may simply possess improved permeability, as very
lipophilic compounds may be retained by the membrane, inhibiting their ability to
penetrate the cell. In an effort to exploit this trend, a new generation of right-hand amine-
bearing analogs was prepared (Table 4), but these proved to be quite toxic, especially
those containing secondary amines (e.g. CCG-206486 and CCG-206500), perhaps via
detergent activity (disruption of cellular membranes). On the other hand, tertiary amines
(e.g. CCG-206502 and CCG-206503) were better tolerated but displayed markedly
decreased activity. All of the analogs possessed lower CC50 values than those seen in

19
their amide counterparts, and there was no significant activity improvement. For these
reasons, the amine-containing series was discontinued early. This did not undermine
development efforts of the low TPSA WEEV inhibitor class; overall, we demonstrated
that activity could be maintained with reduced TPSA and – potentially – increased
hydrophilicity.
Table 4. WEEV Replicon Data for Amine Analogs.
CCG No. R IC50 (µM)a CC50 (µM)
b
203880†
14.8 ± 2.1 >100
206486
6.1 ± 0.7 9
206499
>100 >100
206500
6.5 ± 0.3 12
206501
8.9 ± 0.4 16
206502
40.6 ± 0.5 96
206503
25.1 ± 1.9 85
aInhibition of luciferase expression in WEEV replicon assay.
bCell
viability determined by the MTT reduction assay. Values are mean
of at least n=3 independent experiments. †Synthesized by Janice
Sindac.

20
Synthesis
Low TPSA analogs were generally prepared through the alkylation of ethyl 2-
indolecarboxylate 1 with either 4-chloro- or fluorobenzyl chloride 2a or 2b. The N-
alkylated ethyl ester intermediate 3a,b was then saponified to the corresponding
carboxylic acid 4a,b and coupled with the desired amine to give the general structure
5a,b (Scheme 1).
Scheme 1. General Preparation of Low TPSA Analogs.a
aReagents and conditions: (a) K2CO3, DMF, 60 °C, 20 h, 70%; (b) 7 M aq. NaOH, EtOH,
50 °C, 3 h, 97%; (c) HNR1R2, EDC, HOBt, DIPEA, DMF, rt, ~ 24 h.
Many amines R1R2NH had to be prepared due to commercial unavailability. The
1-(piperidin-4-ylmethyl)piperidine portion of the analogs CCG-204055 and CCG-
205476 was first accessed through the Boc-protection and Swern oxidation of
piperidinyl-4-methanol 6. This provided the aldehyde 8 that underwent reductive
amination with piperidine to give the boc-protected amine 9, and this was subsequently

21
deprotected to reveal the amine as a trifluoroacetate salt and coupled with the
appropriately halogenated indole carboxylic acid to yield the desired analogs (Scheme 2).
Scheme 2. Preparation of CCG-204055 and CCG-205476.a
aReagents and conditions: (a) Boc2O, Na2CO3, H2O, THF, reflux, 2 h, 87%; (b) DMSO,
(COCl)2, -78 °C, 30 min; then TEA, -78 °C→0 °C, 3 h, 89%; (c) piperidine, NaCNBH3,
TFE, 3 Å MS, RT, 20 h, 72%; (d) TFA, DCM, RT; (e) 4a or 4b, EDCHCl, HOBT,
DIPEA, DMF, RT, 15-24 h, 54-63%.
The alicyclic analogs CCG-205421 and CCG-205475 were accessed through a
single linear route in which the latter was a direct synthetic derivative of the former
(Scheme 3). In this fashion, the aldehyde 8 was treated with cyclohexylmagnesium
bromide and the resulting Grignard reaction adduct 10 was deprotected to reveal the
secondary amine, which was then coupled with the indole carboxylic acid to afford the
first analog, hydroxyl-containing CCG-205421, which was contaminated with ester from
competing O-acylation. The desired amide was separated from the ester side-product and
confirmed by IR spectroscopy. CCG-205475 was then prepared by simply oxidizing the
alcohol to the ketone under Swern conditions.

22
Scheme 3. Preparation of CCG-205421 and CCG-205475.a
aReagents and conditions: (a) cyclohexyl-MgBr, Et2O, RT, 2 h, 85%; (b) TFA, DCM,
RT, 15 h; (c) 1-(4-fluorobenzyl)-1H-indole-2-carboxylic acid, EDCHCl, HOBT, DIPEA,
DMF, rt, 24 h, 60%; (d) (COCl)2, DMSO, DCM, -78 °C, 30 min; then TEA, -78 °→RT,
30 min, 58%.
Scheme 4. Preparation of CCG-204020 and CCG-205420.a
aReagents and conditions: (a) BnBr, Na2CO3, H2O, DCM, reflux, 3 h, 87%; (b) TFA,
DCM, RT, 18 h; ; (c) 1-(4-fluorobenzyl)-1H-indole-2-carboxylic acid, EDCHCl, HOBT,
DIPEA, DMF, rt, 30 h, 52%; (d) Pd(OH)2, H2, HCl, EtOH, THF, RT, 1 h, 77%.

23
The benzylated amine analogs CCG-204020 and CCG-205420 were prepared in
a synthetically distinct, albeit similarly linear fashion, to the previous two compounds
(Scheme 4). The protected 4-aminopiperidine 12 was doubly benzylated with benzyl
bromide and deprotected with TFA to afford the amine salt 14. Coupling with the indole
carboxlic acid 4b gave CCG-204020, and time-sensitive selective mono-debenzylation
provided the singly substituted benzyl amine CCG-205420.
The synthesis of piperazine analog CCG-211826 required prior preparation of a
suitable alkylation partner (Scheme 5) by displacement of alcohol 15 under forcing
conditions with refluxing hydrobromic acid. 1-Boc-piperazine was then alkylated with
the resulting bromo compound 16, deprotected under acidic conditions, and coupled with
the indole carboxylic acid 4a to provide the desired final analog.
Scheme 5. Preparation of CCG-211826.a
aReagents and conditions: (a) conc. HBr, 130 °C, 2 h, 40%; (b) t-butyl piperazine-1-
carboxylate, NaH, DMF, RT, 19 h, 66%; (c) HCl, 1,4-dioxane, RT, 15 min, 100%; (d) 1-
(4-Chlorobenzyl)-1H-indole-2-carboxylic acid, TEA, EDCHCl, HOBT, DCM, RT, 14 h,
63%.

24
Secondary amine analogs were generally prepared through the coupling of the
indole carboxylic acid 4b and piperidine-4-methanol 19 (Scheme 6). The resulting
alcohol adduct was then oxidized under Swern conditions to the aldehyde 20, which
could undergo reductive amination with various amines to provide the desired diversity
of structural type 21.
Scheme 6. General Preparation of Secondary Amine Analogs.
aReagents and conditions: (a) EDCHCl, HOBT, DIPEA, DMF, 94%; (b) DMSO,
(COCl)2, DCM, -78 °C; then TEA, -78 °C→0 °C, 66%; (c) NHR1R2, Na(CN)BH3,
AcOH, THF, EtOH.
Conclusion
The primary goal of this chapter was reduction of topological polar surface area. To
this end, we succeeded in halving the TPSA while maintaining potency, bringing our
compounds well within the optimal range of successful CNS drugs. This work also
identified useful structural modifications (e.g. piperidine-to-piperazine substitutions) and

25
underlined the need to avoid secondary amine functionality due to cytotoxicity issues.
Notably, reduction of TPSA appeared to enhance MDR1 evasion, an important feature
for CNS-active drugs.

26
Reduction of Molecular Weight Chapter III.
Rationale
Janice Sindac, working on other aspects of the WEEV project, had identified
CCG-205432 as a viable new lead due to its substantially improved potency and
solubility over the earlier lead CCG-102516 (Figure 8).83
However, it was clear that
some properties of CCG-205432 required optimization due to its increased molecular
weight (MW) and TPSA, features associated with diminished passive permeability and
thus reduced activity. Due to the rather flat SAR associated with the low TPSA analogs
(see Chapter II), we elected to focus on molecular weight reduction as an alternative
course of action for correcting the physicochemical properties of CCG-205432.
Figure 8. Development of Lead CCG-205432.
Molecular weight (MW) is a property of extreme importance for CNS-acting
drugs, which tend to be of low molecular weight (<450).74, 98
This is crucial, as reduction

27
of molecular weight promotes passive permeability into membranes non-linearly;99
even
a small change in molecular weight can have a large impact on membrane penetration.
Furthermore, reduced molecular weight is associated with lower recognition by efflux
transporters like MDR1,79, 80, 100, 101
an attribute we considered essential for our WEEV
inhibitors.
MDR1 Rho123 Uptake Assay. Our collaborators in the lab of Prof. Richard
Keep discovered that the 3H-vinblastine uptake assay suffered from poor reproducibility
due to complications in acquiring the specific MDR1-MDCKII cell line used in previous
work (see Chapter II). Fortunately, they found that the Rhodamine 123 (Rho123) uptake
assay102
was a superior substitute to the 3H-vinblastine uptake assay, and this was utilized
for the reduced molecular weight analog class. Not only did the Rho123 assay feature
improved reproducibility, but Rho123 is a simple dye whose concentration may be
measured by absorbance and avoids those complications associated with radioactivity.
Both of these assays operate in the same manner; if the test compound of interest has no
interaction with MDR1, the 3H-vinblastine or Rho123 concentration inside the cell will
drop as the substrate is pumped out. If the test compound does interact with MDR1, the
measured 3H-vinblastine or Rho123 concentration will be higher within the cell. Results
for selected analogs are reported as percent effect on Rho123 intracellular concentration
relative to tariquidar, where lower values indicate lesser interaction with MDR1 and
therefore less potential for efflux. Moreover, the Rho123 assay was controlled by
comparing WEEV inhibitors to the efflux of the known MDR1 inhibitor tariquidar.
We were also interested in undertaking MDR1 SAR studies in order to define an
MDR1-recognition pharmacophore, but such work is not always trivial.89
MDR1 binds

28
many different substrates, and traditional SAR, which focuses on biomolecules that bind
specific substrates, is not always up to the task. Further complications arise when MDR1
binds only a few compounds from a class of analogs. There are very general SAR trends
associated with the transporter, but most individual chemotypes must typically be
optimized on a case-by-case basis.
Mouse Liver Microsome Metabolic Stability Assay. Metabolic stability is one
of the most crucial aspects of early drug development, and instability is a potential killer
of lead series.81
It is therefore necessary to improve the stability of a compound to
maximize its potential for in vivo efficacy, especially as half-life is related to a
compound’s biological availability. (See Chapter V for more information regarding
metabolic stability in drug development.) To this end, the half-lives of compounds were
determined by incubating in the presence of Balb/C mouse liver microsomes containing
cytochrome p450 (CYP) enzymes and monitoring the decrease in parent compound
concentration over time. Compounds more susceptible to CYP-mediated metabolism will
decrease more rapidly than those that are more stable. Concentration was measured using
an LC/MS/MS method developed specifically to maximize throughput, and all values
were controlled against a known concentration of a structurally-related internal standard.
The assay and analytical protocols were developed in our lab specifically to fit the needs
of the WEEV project, and details appear in the experimental section.
Kinetic Solubility Assay. The kinetic solubility is a measure of the metastable
solution equilibrium of a compound (pre-dissolved in a solvent like DMSO and added to
aqueous media) that begins to precipitate as its concentration exceeds its ability to
dissolve.79, 103
This can be observed by measuring the absorbance of the sample. The

29
concentration at which the compound precipitates is reported as the kinetic solubility, and
is a useful measure of aqueous solubility that allows for prioritization of compounds of
interest.
SAR
The average CNS drug has a molecular weight of about 319, but the lead
compounds CCG-102516 and CCG-205432 (Figure 8) were much higher than that. We
were therefore interested in exploring the indole and piperidine cores as potential sites for
molecular weight reduction (Figure 9), largely due to the lack of attention these regions
had received until this point. A number of analogs featuring monocyclic heterocycles
lacking the fused phenyl ring of indole, such as pyrroles and imidazoles, were prepared
(Table 5). Azetidine-containing analogs of the piperidine core were also synthesized and
evaluated.
Figure 9. Sites for Molecular Weight Reduction.

30
Tab
le 5
. W
EE
V R
epli
con
an
d A
DM
E D
ata
for
Pyrr
ole
an
d I
mid
azo
le A
nalo
gs.
MW
486
501
437
451
437
ML
M
T1
/2
(min
)e
7.3
± 3
.9
11.5
---
2.9
---
MD
R1
Rec
ognit
i
on (
%
Rho123
upta
ke)
d
---
30.9
± 1
.0
---
-1.2
± 0
.6
---
Sol
(µM
)c
---
31
-63
---
125
-250
---
BB
B-
PA
MP
A
(logP
eff)
-3.6
±
0.3
-4.1
8 ±
0.0
6
---
-5.0
1 ±
0.0
3
---
CC
50
(µM
)b
>100
65.3
>100
>100
>100
IC5
0
(µM
)a
15.6
± 1
.8
0.5
3 ±
0.0
1
77 ±
23
0.6
8 ±
0.0
4
>100
R
---
---
X
---
---
C
C
N
CC
G N
o.
102516†
205432†
204054
206381
206586

31
Tab
le 5
. C
on
tin
ued
.
MW
437
451
437
a Inhib
itio
n o
f lu
cife
rase
expre
ssio
n i
n W
EE
V r
epli
con a
ssay
. bC
ell
via
bil
ity d
eter
min
ed b
y t
he
MT
T r
educt
ion a
ssay
. V
alues
are
mea
n o
f at
lea
st n
=3 i
ndep
enden
t ex
per
imen
ts. c
Log o
f ef
fect
ive
per
mea
bil
ity (
cm/s
) det
erm
ined
usi
ng P
AM
PA
Explo
rer
(pIO
N)
wit
h B
BB
lip
id m
ixtu
re m
easu
red a
t pH
= 7
.4.
dK
inet
ic s
olu
bil
ity m
easu
red u
sing t
he
sam
e as
say m
edia
as
WE
EV
repli
con a
ssay
, ex
cept
wit
h 1
0%
fet
al b
ovin
e se
rum
. e R
hodam
ine
123 u
pta
ke
was
mea
sure
d i
n M
DR
1-M
DC
KII
cel
ls u
tili
zing
Glo
max
Mult
i D
etec
tion S
yst
em (
Pro
meg
a). ‘M
DR
1 r
ecognit
ion’
was
ass
esse
d b
y m
easu
ring u
pta
ke
of
Rhodam
ine
123 i
n t
he
pre
sence
of
MD
R i
nhib
itor,
tar
iquid
ar (
5 µ
M),
30 µ
M o
f te
st a
nti
-vir
al o
r v
ehic
le, an
d c
alcu
lati
ng:
(Cav
– C
veh
)*100/(
Cta
r-C
veh
),
wher
e C
av =
conce
ntr
atio
n o
f rh
odam
ine
123 i
n t
he
pre
sen
ce o
f an
ti-v
iral
, C
veh
= c
once
ntr
atio
n i
n t
he
pre
sen
ce o
f v
ehic
le, C
tar =
conce
ntr
atio
n o
f rh
odam
ine
123 i
n t
he
pre
sen
ce o
f ta
riquid
ar.
In t
he
pre
sen
ce o
f ta
riquid
ar, rh
od
amin
e 123 u
pta
ke
was
1123 ±
54%
of
veh
icle
contr
ols
(n=
44).
f Hal
f-li
fe i
n B
AL
B/c
mouse
liv
er m
icro
som
e in
cubat
ion. V
alues
are
hal
f-li
ves
cal
cula
ted f
rom
the
equat
ion T
1/2
=ln
(2)/
slope,
wher
e th
e sl
ope
was
ln(%
det
ecte
d)
agai
nst
tim
e, p
lott
ed f
rom
the
mea
n o
f at
lea
st n
=3
indep
enden
t ex
per
imen
ts. †
Synth
esiz
ed b
y J
anic
e S
indac
.
ML
M
T1
/2
(min
)f
---
---
---
MD
R1
Rec
ognit
ion
(% R
ho123
upta
ke)
e
0.1
3 ±
0.3
7
---
---
Sol
(µM
)d
>500
---
---
BB
B-
PA
MP
A
(logP
eff)
c
-5.0
6 ±
0.0
4
---
---
CC
50
(µM
)b
>100
>100
>100
IC5
0
(µM
)a
>100
>100
22 ±
8
R
X
N
C
C
CC
G N
o.
208916
208829
208915

32
Heterocyclic Monocycle SAR. The imidazole analogs (CCG-206586 and CCG-
208916) were completely inactive, presumably due to the enhanced ionizability and
diminished lipophilicity arising from the presence of two azine nitrogens (Table 5).
These properties would be expected to impede passive membrane permeability.
Gratifyingly, the pyrrole analog CCG-206381 was nearly equipotent with indole CCG-
205432, and boasted improved aqueous solubility and a 50 Da improvement in molecular
weight (Table 5). Attempts to reduce molecular weight of CCG-206381 further, by
replacing the central piperidine with azetidine, returned significantly less active
compounds (Table 5). This may be attributed to the length-activity dependence discussed
in Chapter II, as the small 4-membered ring is much smaller and shorter than the larger
6-membered piperidine. Curiously, the pyrrole analog of CCG-102516 (CCG-204054)
was substantially less active than its lead.
Potential for BBB Penetration. Passive permeability is an excellent predictor of
successful BBB penetration, and high permeability may also overcome the effect of
efflux transporters.79
Therefore, compounds with good BBB permeability are of central
importance to our development of CNS active inhibitors, and the pyrrole and imidazole
analogs were evaluated for their potential to permeate the BBB. The notion that the
significantly diminished activity of the imidazole analogs was related to increased
ionizability and lipophobicity was supported by the high aqueous solubility and low
BBB-PAMPA permeability of CCG-208916 seen in Table 5 (compounds with log Peff >
4.7 are considered to be highly permeable, whereas those with log Peff 6 are
considered poorly permeable). CCG-206381, as expected, possessed improved aqueous
solubility (Table 5), but appeared to have reduced permeability as measured by the BBB-

33
PAMPA assay. It is important to note that PAMPA assays are not perfect models of
membranes because of reduced surface area and the presence of multiple (>100) lipid
layers. Therefore, it could not be confidently concluded that CCG-206381 was in fact
less permeable than CCG-205432.
Recognition by efflux transporters is highly correlated with diminished BBB
permeation, so selected analogs were tested for recognition by MDR1 in the Rho123
assay, using the known MDR1 substrate tariquidar as a control (Table 5). Significantly,
CCG-205432 interacted with MDR1 to an extent equal to 30% that of tariquidar, whereas
CCG-206381 had virtually no detectable interaction with the transporter. This indicated
that CCG-206381 could potentially evade MDR1 and thus achieve a higher in vivo brain
concentration than CCG-205432.
Antiviral Activity. Due to its improved aqueous solubility and reduced molecular
weight, CCG-206381 was advanced into live virus studies and compared to CCG-
205432. In these assays, cells were infected with live WEEV and the number of plaques –
often visually distinct zones caused by infection – were counted and used to extrapolate
viral concentration after treatment with varying concentrations of test compound.
Viability was determined by measuring the number of living cells with the MTT assay.
CCG-205432 reduced viral titer by over two log units, and CCG-206381 was able to
successfully reduce viral titer by about one log unit (Figure 10A). Both compounds also
comparably conferred protection to infected cells (Figure 10B). Together, these results
indicated that, while only modestly less active, pyrrole could serve as a viable reduced
molecular weight alternative to indole.
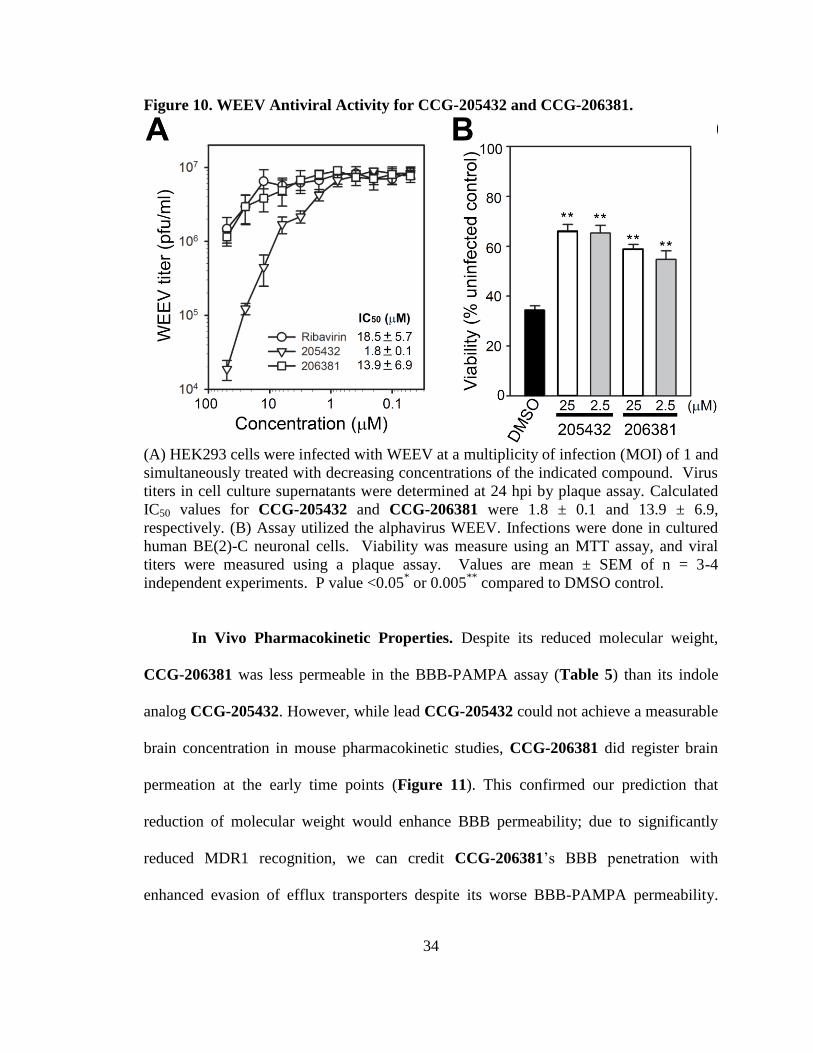
34
Figure 10. WEEV Antiviral Activity for CCG-205432 and CCG-206381.
(A) HEK293 cells were infected with WEEV at a multiplicity of infection (MOI) of 1 and
simultaneously treated with decreasing concentrations of the indicated compound. Virus
titers in cell culture supernatants were determined at 24 hpi by plaque assay. Calculated
IC50 values for CCG-205432 and CCG-206381 were 1.8 ± 0.1 and 13.9 ± 6.9,
respectively. (B) Assay utilized the alphavirus WEEV. Infections were done in cultured
human BE(2)-C neuronal cells. Viability was measure using an MTT assay, and viral
titers were measured using a plaque assay. Values are mean ± SEM of n = 3-4
independent experiments. P value <0.05* or 0.005
** compared to DMSO control.
In Vivo Pharmacokinetic Properties. Despite its reduced molecular weight,
CCG-206381 was less permeable in the BBB-PAMPA assay (Table 5) than its indole
analog CCG-205432. However, while lead CCG-205432 could not achieve a measurable
brain concentration in mouse pharmacokinetic studies, CCG-206381 did register brain
permeation at the early time points (Figure 11). This confirmed our prediction that
reduction of molecular weight would enhance BBB permeability; due to significantly
reduced MDR1 recognition, we can credit CCG-206381’s BBB penetration with
enhanced evasion of efflux transporters despite its worse BBB-PAMPA permeability.

35
However, the instability of CCG-206381 towards CYP-mediated metabolism compared
to CCG-205432 in the mouse liver microsomal (MLM) assay (see Chapter V for more
information) was reflected in its pharmacokinetic profile (Figure 11). CCG-205432 had
an MLM half-life of 11.5 minutes and was measurable in rat plasma for over 20 hours,
whereas CCG-206381 had an MLM half-life of 2.9 minutes and was measurable in rat
plasma or brain samples only up to one hour. Overall, this data indicated that reduction of
molecular weight was a viable tactic in achieving brain penetration and, therefore, in vivo
efficacy.
Figure 11. In Vivo Pharmacokinetic Data for CCG-205432 and CCG-206381.
CCG-205432 and CCG-206381 possess different pharmacokinetic profiles. Balb/C mice
were dosed and sacked at various time points (David Irani lab). Compound
concentrations in plasma and brain samples were evaluated via LC/MS/MS by the lab of
Prof. Duxin Sun.
0
20
40
60
80
100
120
0 10 20
Con
cen
trati
on
(n
g/m
L)
Time (hours)
CCG-205432 Plasma/Brain
Concentration-Time
Plasma
Brain
0
20
40
60
80
100
120
0 10 20
Con
cen
trati
on
(n
g/m
L)
Time (hours)
CCG-206381 Plasma/Brain
Concentration-Time
Plasma
Brain

36
Table 6. WEEV Replicon Data for Acyclic Low MW Analogs.
No. R IC50
(µM)a
CC50
(µM)b MW
206381 --- 0.68 ± 0.04 >100 451
208825
60.2 ± 4.6 >100 401
208827
40.9 ± 5 >100 415
208848
>100 >100 386
208846
>100 >100 400
aInhibition of luciferase expression in WEEV replicon assay.
bCell
viability determined by the MTT reduction assay. Values are mean of
at least n=3 independent experiments.
Acyclic Analog SAR. Having established that a smaller scaffold than indole
could retain antiviral activity and improve BBB permeability, a variety of additional
analogs was synthesized (Table 6) that featured complete excision of the left-hand
heterocycle. Many of these analogs possess molecular weights in the low 400 to high 300
g/mol range, bringing these values substantially closer to successful CNS drugs than any
WEEV inhibitor synthesized to this point. However, while the pyrrole analogs indicated
that the indole left-hand core was not a necessary component of the SAR, it was
immediately evident that complete removal of the heterocycle eliminated activity. Only
ureas CCG-208825 and CCG-208827 retained any activity in the WEEV replicon assay,
and those were greatly diminished relative to pyrrole CCG-206381. Amides 208848 and

37
208846 were completely inactive. These results suggested that some degree of rigidity is
likely necessary to retain good antiviral activity.
Table 7. WEEV Replicon Data for Pyrrolidine Analogs.
No. X IC50
(µM)a
CC50
(µM)b
MW
206381 --- 0.68 ± 0.04 >100 451
(S)-211758 CH2 13.5 ± 2 91.6 455
(R)-211757 CH2 53.5 ± 12 >100 455
(S)-211754 CO >100 >100 467
(R)-211756 CO >100 >100 467 aInhibition of luciferase expression in WEEV replicon assay.
bCell
viability determined by the MTT reduction assay. Values are mean
of at least n=3 independent experiments.
Pyrrolidine Analog SAR. We therefore turned our attention to cyclic, but non-
aromatic, pyrrolidine analogs (Table 7). Our rationale was that the saturated heterocycles
would possess greater stability towards oxidation than pyrrole, yet provide the rigidity
that was apparently required. While significantly less active than pyrrole CCG-206381,
N-benzyl pyrrolidines CCG-211758 and CCG-211757 still retain some activity. Worth
noting is the small but significant chiral preference for the S-enantiomer, which is four
times more potent than the R-enantiomer. This was consistent with our previous

38
observation that the unknown molecular target for this series of antiviral compounds
possesses some degree of enantiospecificity.64, 83
N-benzoyl pyrrolidines CCG-211754
and CCG-211756 by contrast were completely inactive. This could suggest a need for a
basic nitrogen in the linker, but more likely reflects an unfavorable conformational bias
induced by the planar nature of the amide.
Arene Monocycle SAR. We then synthesized and evaluated a series of arene
derivatives (Table 8) to more closely mimic the pyrrole scaffold but with greater
predicted stability to oxidative metabolism. Several of these compounds possessed low
micromolar activity in the replicon assay, including salicylamide CCG-212392 and
anthranilamide CCG-211824. We anticipated that the aniline moiety might be the most
suitable bioisosteric replacement for pyrrole based on its electronic similarity, and indeed
CCG-211824 was only two-fold less potent than CCG-206381. The benzyl aniline
homologue CCG-222660 proved to be equipotent with the lead (compound CCG-
206381), while further homologation of the anthranilamide (analog CCG-222983)
resulted in a diminishment of activity. This observation of an optimal length is consistent
with what we observed while developing CCG-20543283
and suggests either a well-
defined limit to the size of the unknown binding site, or simply reflects an excessive loss
of entropy needed for binding a long acyclic substituent. Removal of the chloride
(intended to improve metabolic stability by reducing lipophilicity) resulted in compounds
(CCG-222981 and CCG-222982) with greatly reduced potency. Replacement of the
aniline nitrogen of CCG-211824 with oxygen (ether analog CCG-212392) resulted in a
modest loss of activity, while replacement with methylene (benzyl analog CCG-222821)
provided an equipotent analogue.

39
Tab
le 8
. W
EE
V R
epli
con
an
d I
n V
itro
AD
ME
Data
for
Are
ne
An
alo
gs.
MW
501
451
464
463
429
Sol
(µg/m
L)f
31-6
3
125-2
50
>250
ML
M
T1
/2
(min
)e
9
3
18
19
---
MD
R1
Rec
ognit
ion
(% R
ho
123
upta
ke)
d
30.9
± 1
.0
-1.2
± 0
.6
9.3
± 3
.2
47.7
± 7
.2
10.9
± 1
.7
BB
B-
PA
MP
A (
log
Pef
f)c
-4.1
8 ±
0.0
6
-5.0
1 ±
0.0
3
-4.6
8 ±
0.0
9
-4.4
5 ±
0.1
1
-4.7
5 ±
0.0
2
CC
50
(µM
)b
49.9
>100
56.0
>100
>100
IC5
0
(µM
)a
0.5
3 ±
0.0
8
0.6
8 ±
0.0
4
6.1
± 0
.8
1.6
± 0
.2
28.8
± 5
.2
G
---
---
No.
205432
206381
212392
211824
222981

40
Tab
le 8
. C
on
tin
ued
.
MW
477
477
443
491
476
462
Sol
(µg/m
L
)f
>250
>250
>250
ML
M
T1
/2
(min
)e
---
15
---
---
--- 3
MD
R1
Rec
ognit
ion
(% R
ho123
upta
ke)
d
3.9
± 1
.5
59.6
± 7
.5
2.5
± 0
.3
30.7
± 4
.7
1.8
± 1
.3
22.6
± 2
.7
BB
B-
PA
MP
A (
log
Pef
f)c
-4.7
1 ±
0.0
9
-4.3
9 ±
0.0
5
-4.9
3 ±
0.0
3
-4.4
9 ±
0.3
5
-5.0
8 ±
0.0
2
-4.4
0 ±
0.0
6
CC
50
(µM
)b
>100
75.2
92.2
86.0
>50
70.7
IC5
0
(µM
)a
15.4
± 2
.8
0.5
6 ±
0.0
8
19.0
± 1
.7
3.4
9 ±
0.1
>50
1.4
6 ±
0.0
9
G
No.
212393
222660
222982
222983
212391
222821

41
Tab
le 8
. C
on
tin
ued
.
a Inhib
itio
n o
f lu
cife
rase
expre
ssio
n i
n W
EE
V r
epli
con a
ssay
. bC
ell
via
bil
ity d
eter
min
ed b
y t
he
MT
T r
educt
ion a
ssay
. V
alues
are
mea
n o
f at
lea
st n
=3 i
ndep
enden
t ex
per
imen
ts. c
Log o
f ef
fect
ive
per
mea
bil
ity (
cm/s
) det
erm
ined
usi
ng P
AM
PA
Explo
rer
(pIO
N)
wit
h B
BB
lip
id m
ixtu
re m
easu
red a
t pH
= 7
.4.
dR
hodam
ine
123 u
pta
ke
was
mea
sure
d i
n M
DR
1-M
DC
KII
cel
ls u
tili
zing
Glo
max
Mult
i D
etec
tion S
yst
em (
Pro
meg
a). ‘M
DR
1 r
ecognit
ion’
was
ass
esse
d b
y m
easu
ring u
pta
ke
of
Rhodam
ine
123 i
n t
he
pre
sence
of
MD
R i
nhib
itor,
tar
iquid
ar (
5 µ
M),
30 µ
M o
f te
st a
nti
-vir
al o
r v
ehic
le, an
d c
alcu
lati
ng:
(Cav
– C
veh
)*100/(
Cta
r-C
veh
),
wher
e C
av =
conce
ntr
atio
n o
f rh
odam
ine
123 i
n t
he
pre
sen
ce o
f an
ti-v
iral
, C
veh
= c
once
ntr
atio
n i
n t
he
pre
sen
ce o
f v
ehic
le, C
tar =
conce
ntr
atio
n o
f rh
odam
ine
123 i
n t
he
pre
sen
ce o
f ta
riquid
ar. In
the
pre
sen
ce o
f ta
riquid
ar, rh
od
amin
e 123 u
pta
ke
was
1123 ±
54%
of
veh
icle
contr
ols
(n=
44)
. e H
alf-
life
in B
AL
B/c
mouse
liv
er m
icro
som
e in
cubat
ion. V
alues
are
hal
f-li
ves
cal
cula
ted f
rom
the
equat
ion T
1/2
=ln
(2)/
slope,
wher
e th
e sl
ope
was
ln(%
det
ecte
d)
agai
nst
tim
e, p
lott
ed f
rom
the
mea
n o
f at
lea
st n
=3
indep
enden
t ex
per
imen
ts. f K
inet
ic s
olu
bil
ity m
easu
red u
sing t
he
sam
e as
say m
edia
as
WE
EV
rep
lico
n a
ssay
, ex
cept
wit
h 1
0%
feta
l bovin
e se
rum
. S
ee E
xper
imen
tal
Sec
tion f
or
det
aile
d m
ethods
and s
ynth
etic
pro
cedu
res.

42
Interestingly, the N-methylated analogue CCG-212393 was ten times less active
than its parent anthranilamide CCG-211824, suggesting that the aniline nitrogen may be
engaged in internal hydrogen bonding with the ortho carbonyl oxygen, resulting in a
specific conformation favorable for activity. Further evidence for this conformational
hypothesis is provided by comparing benzylbenzamide analogue CCG-222821, with
inactive benzoylbenzamide CCG-212391. The latter would not be expected to be able to
achieve a conformation similar to the internally hydrogen bonded conformation of CCG-
211824.
Figure 12. Conformational Restriction Plan for Arene Analogs.
Analogs were designed to tether the arene rings (red, carbazole derivatives) or tie the
amide to the aniline nitrogen (blue, quinolone derivatives).
Conformational Restriction. Several rigid analogues were designed to evaluate
our conformational hypothesis (Figure 12 and Table 9). Carbazole derivatives (CCG-
222824 and CCG-222825) were intended to tether the two arenes in the molecule and
prevent rotation around the two C-N bonds. Surprisingly, these compounds were
remarkably less active than their more flexible parent, which may indicate that the
putative internal NH hydrogen bond is not sufficient for achieving the biologically active

43
conformation and that in fact the two tethered aromatic rings need to be orthogonal rather
than planar. It is also possible that tight binding to the unknown molecular target entails
induced fit, which the rigid carbazole cannot accommodate. The quinolone derivatives
(CCG-222823 and CCG-222822), on the other hand, were designed to freeze rotation
about the aniline-amide C-C bond. Their poor activity, however, indicates either a poor
match for the biologically active conformation, or a key role for the benzamide carbonyl
of CCG-211824 that has been disrupted.
Table 9. WEEV Replicon Date for Rigid Arene Analogs.
No. G IC50 (µM)a CC50 (µM)
b
222824
59.2 ± 6.5 >100
222825
18.7 ± 5.4 66.1
222823
88.3 ± 11.7 >100
222822
10.9 ± 2.0 34.1
aInhibition of luciferase expression in WEEV replicon assay.
bCell viability determined by the MTT reduction assay.
Values are mean of at least n=3 independent experiments.

44
Potential for CNS Penetration. To measure the potential for MDR1 recognition
of the arene analogs, we used the Rho123 assay.83
In Table 8, replacement of the pyrrole
of CCG-206381 with phenyl (CCG-211824) markedly increased recognition by MDR1.
Surprisingly, simple N-methylation of CCG-211824 (CCG-212393) almost completely
abated MDR1 recognition. Reductions in MDR1 recognition were also observed upon
replacement of the NH of CCG-211824 with O, C=O, or CH2 (CCG-212393, CCG-
212391 and CCG-222821, respectively) suggesting that the NH may be a key factor for
recognition. This is supported by the strong MDR1 recognition of NH homologs CCG-
222660 and CCG-222983. However, it is notable that simple removal of the aromatic
chloro group strongly attenuated MDR1 recognition (CCG-222981 and CCG-222982),
indicating that overall lipophilicity may also be a factor.
Permeability is an excellent predictor of potential BBB penetration, so selected
compounds were evaluated using pION’s BBB-PAMPA assay and PAMPA Explorer
program as previously described.83
Compounds with log Peff > 4.7 are considered to be
highly permeable, whereas those with log Peff 6 are considered poorly permeable. In
general, our compounds exhibit moderate to high BBB permeability, and all but one
(CCG-212391) possess modestly better permeability than pyrrole lead CCG-206381
(Table 8).
In vitro Metabolic Stability. Based on their WEEV replicon activities, selected
compounds were advanced into the mouse liver microsome (MLM) assay to assess their
stabilities towards Phase I oxidative metabolism (Table 8). (See Chapter V for more
information regarding metabolism.) The stability of aniline CCG-211824 was improved
with a half-life of 19 minutes, a six-fold improvement over the slightly more electron-rich

45
pyrrole CCG-206381 (3 minutes) and a two-fold improvement over the indole CCG-
205432 (9 minutes). The chlorobenzyl homologue CCG-222660 was slightly less stable
towards metabolism than CCG-211824, perhaps because of the introduction of a benzylic
carbon and increased lipophilicity. Phenolic ether CCG-212392 was as stable towards
microsomal metabolism as aniline CCG-211824. Interestingly, replacement of the NH of
CCG-211824 with CH2 (CCG-222821) markedly attenuated metabolic stability, due
either to increased lipophilicity or the creation of an additional site for oxidation.
Table 10. Antiviral Data for Select Analoguesa
FMV WEEV
CCG No. Conc. (µM) Titer (x 10
6
pfu/ml)
Viability (%
uninfected control)
Titer (x 104
pfu/ml)
DMSO NA 30.7 ± 9.0 35.6 ± 4.7 37.3 ± 7.3
205432 25 ND 69.5 ± 10.0* 1.1 ± 0.1
**
211824 25 3.7 ± 1.4* 58.1 ± 9.0 3.2 ± 2.2
*
5 10.6 ± 4.1 54.6 ± 9.2 49.2 ± 19.4
222660 25 3.2 ± 1.0**
66.6 ± 9.5* 1.6 ± 0.5
**
5 9.4 ± 5.6* 73.3 ± 9.1
* 11.7 ± 3.6
*
aAssay utilized the alphaviruses Fort Morgan Virus (FMV) and western equine
encephalitis virus (WEEV). Infections were done in cultured human BE(2)-C neuronal
cells. Viability was measure using an MTT assay, and viral titers were measured using a
plaque assay. Values are mean ± SEM of n = 3-4 independent experiments. P value
<0.05* or 0.005
** compared to DMSO control. NA, not applicable. ND, not determined.
Data from the David Miller lab.
Antiviral Activity. Based on their WEEV replicon potencies, CCG-211824 and
CCG-222660 were advanced into assays to evaluate their ability to inhibit cellular
replication of infectious live virus. Our collaborators used two parallel assays to measure
activity against infectious virus: reduction in cytopathic effect (CPE) and extracellular

46
virus titers, and examined activity against both WEEV and Fort Morgan Virus (FMV), a
WEEV-serogroup alphavirus that can be used safely under reduced biosafety level
conditions compared to WEEV. Against FMV, both CCG-211824 and CCG-222660
reduced virus titers by ten-fold when used at 25 µM, and also reduced FMV-induced CPE
similar to previous lead CCG-205432 (Table 10). Furthermore, both CCG-211824 and
CCG-222660 also reduced WEEV titers by at least 10-fold when used at 25 µM.
Although we did not do full titration curves with these experiments, CCG-222660
maintained antiviral activity against both FMV and WEEV when used at 5 µM, whereas
CCG-211824 had reduced activity at this lower concentration (Table 10).
Figure 13. Spectrum Antiviral Data for Selected Analogues.
CCG-211824 and CCG-222660 have potent and broad spectrum antiviral activity. (A)
HEK293 cells were infected with FMV at an MOI of 1 and simultaneously treated with
decreasing concentrations of the indicated compound. Virus titers in cell culture
supernatants were determined at 24 hpi by plaque assay. The horizontal dashed line
indicates FMV titers in cells treated with control DMSO. Calculated IC50 values for
CCG-205432, CCG-211824, and CCG-222660 were 1.0 ± 0.5, 5.2 ± 1.0, and 2.3 ± 1.2
µM, respectively. (B) HEK293 cells were infected with the indicated viruses at an MOI
of 0.1, simultaneously treated with the indicated compound at 25 µM, and virus titers in
tissue culture supernatants were determined by plaque assay at 24 hpi. P <0.05* or
0.005** compared to DMSO-treated controls. Data from the David Miller Lab.

47
To further evaluate the antiviral potency and breadth of activity for CCG-211824
and CCG-222660, we completed both full titration studies with FMV in HEK293 cells
(Figure 13A) and examined their inhibitory activity against viruses derived from three
different families: Venezuelan equine encephalitis virus (VEEV, TC-83 vaccine strain;
Togaviridae), California encephalitis virus (CEV; Bunyaviridae), and
encephalomyocarditis virus (EMCV; Picornaviridae) (Figure 13B). Both CCG-211824
and CCG-222660 had dose-dependent antiviral activity against FMV, where CCG-
222660 had an IC50 similar to previous lead CCG-205432 (Figure 13A). In addition,
both CCG-211824 and CCG-222660 had demonstrable antiviral activity against VEEV,
CEV, and EMCV in cultured cells, with an approximate 10-fold reduction in infectious
virus titers for all three pathogens, similar to previous lead CCG-205432 (Figure 13B).
Mechanism of Action Studies. The use of the WEEV replicon assay, a
phenotypic cell-based assay, increases the probability of selecting compounds having a
cellular rather than a viral target for antiviral activity. There are several advantages of
targeting host factors over viral proteins. The most significant advantage is a decreased
probability of the targeted virus developing resistance to therapeutics.105
Our
collaborator’s studies with the lead CCG-205432 and related carboxamide analogs
indicated that the mechanism of action for their antiviral activity is through the
modulation of cellular cap-dependent translation.104
To examine whether our monocyclic
analogs used a similar mechanism, we evaluated the ability of CCG-211824 and CCG-
222660 to modulate cellular cap-dependent translation (Figure 14A). For these
experiments, we used BSR-T7 cells and two reporter plasmids: pSV40-LUC, which
produces a capped mRNA through nuclear transcription and export, and pCITE-LUC,

48
which produces an uncapped mRNA containing a cap-independent translational element
(CITE) through cytoplasmic transcription that is driven by a T7 promoter (Figure
14B).106
Similar to lead CCG-205432 and previous generation compounds, both CCG-
211824 and CCG-222660 had no effect upon cap-independent translation, but potently
inhibited cellular cap-dependent translation of transcripts produced by the pSV40-LUC
reporter plasmid (Figure 14B). These results suggest that the anthranilamide analogs
CCG-211824 and CCG-222660 also function as antiviral compounds, in part, through
suppression of host cell cap-dependent translation.
Figure 14. Effect of Selected Analogs on Transfected Gene Expression in BSR-T7
Cells.a
9a and 11a modulate cellular gene expression. BSR-T7 cells were transfected with the
expression plasmids pSV40-LUC (circles) or pT7/CITE-LUC (triangles), treated with
decreasing concentrations of the indicated compound, and fLUC activity was measured
20 h later. Calculated IC50 values for suppression of pSV40-LUC activity for 9a and 11a
were 2.2 ± 0.2 and 1.0 ± 0.1 µM, respectively. 1 has an IC50 value of 0.1 µM in this
assay.104
N=3 or 4 for all data, and results are the mean ± SEM. Data from the David
Miller lab.

49
Synthesis
The piperidine-containing pyrrole analogs CCG-204054, CCG-206381, and
CCG-208914 and the imidazole analogs CCG-206586, CCG-208913, and CCG-208916
were first prepared by alkylating the appropriate azole with 4-chlorobenzyl chloride to
provide the methyl ester. Saponification of this ester and amide coupling with ethyl
isonipecotate gave the intermediate. This ethyl ester was then hydrolyzed and coupled
with the appropriate amine (Scheme 7).
Scheme 7. Preparation of Pyrrole and Imidazole Analogs.
Reagents and conditions: (a) p-chlorobenzyl chloride, K2CO3, DMF, 60 °C, 36 h; (b) 7M
NaOH, EtOH, 70 °C, 4 h; (c) 26, EDCHCl, HOBt, DIPEA, DMF, rt, 24 h; (d) 7M
NaOH, EtOH, 60 °C, 8 h; (e) 27, EDCHCl, HOBt, DIPEA, DMF, rt, 24 h.

50
Scheme 8. Preparation of Azetidine Analogs.a
aReagents and conditions: (a) methyl 4-azetidinecarboxylate hydrochloride, EDC, HOBT,
DIPEA, DCM, rt; (b) 10% aq. NaOH, EtOH; (c) R-NH2, EDC, HOBT, TEA, DCM, rt.
The azetidine-containing pyrroles CCG-208793, CCG-208829, and CCG-
208915 were prepared through the union of N-benzyl pyrrole 28 and methyl azetidine-3-
carboxylate, followed by base hydrolysis and amidation with the appropriate amine
(Scheme 8). Urea analogs CCG-208825, CCG-208826, CCG-208827, and CCG-
208828 were synthesized by the addition of ethyl isonipecotate 26 to isocyanates 30a,b,
followed by hydrolysis and amidation with the appropriate amine (Scheme 9). The
acyclic amides CCG-208846, CCG-208847, CCG-208848, and CCG-208849 were
synthesized from the amide coupling of the appropriate carboxylic acid 32a,b and ethyl
isonipecotate 26, followed by hydrolysis/amidation with the appropriate amine similar to
the previous syntheses (Scheme 9).

51
Scheme 9. Preparation of Acyclic Amide and Urea Analogs.a
aReagents and conditions: (a) 26, DCM, rt; (b) 10% aq. NaOH, EtOH, rt; (c) R-NH2,
EDC, HOBT, TEA, DCM, rt; (d) 26, EDCHCl, HOBT, TEA, DCM, RT.
The right-hand portion of the pyridine-bearing analogs, N-(2-(pyridin-4-
yl)ethyl)piperidine-4-carboxamide dihydrochloride 36 (Scheme 10), was assembled by
first boc-protecting ethyl isonipecotate 26. The protected ethyl ester was then hydrolyzed
to the carboxylic acid and coupled with 4-(2-aminoethyl)pyridine 27. The protecting

52
group was then removed under dry acidic conditions to yield the dihydrochloride salt 36
of the desired amine.
Scheme 10. Preparation of the Right-Hand Amine Dihydrochloride 36.a
aReagents and conditions: (a) Boc2O, Na2CO3, H2O, THF, reflux; (b) NaOH, H2O, EtOH,
RT; (c) 4-(2-aminoethyl)pyridine, EDCHCl, HOBT, DCM, RT; (d) HCl, dioxane, RT.
N-benzylpyrrolidine enantiomers CCG-211757 and CCG-211758 were prepared
through base-catalyzed alkylation of proline with 4-chlorobenzylchloride to protect
stereochemical integrity via the carboxylate anion,107
followed by amide coupling with
amine dihydrochloride 3683
to yield the appropriate (R)- and (S)- analogs (Scheme 11).
Pyridine-bearing enantiomers CCG-211754 and CCG-211756, and indane-bearing
enantiomers CCG-211753 and CCG-211755, were synthesized in similar fashion, first
by acylating proline under classical Schotten-Baumann conditions108, 109
followed by
amide coupling with ethyl isonipecotate 26 (Scheme 11). Hydrolysis of the ethyl ester
(R)- or (S)-40 through a non-racemizing protocol110
provided the carboxylic acid, and
amidation with amine 27 gave the desired (R)- and (S)- benzoyl analogs.

53
Scheme 11. Preparation of N-Benzyl and N-Benzoyl Pyrrolidine Analogs.a
aReagents and conditions: (a) 2a, KOH, IPA, 40 °C; (b) 36, HATU, TEA, DCM, RT; (c)
4-chlorobenzoylchloride, IPA, KOH, H2O, 0 °C; (d) 26, HATU, TEA, DCM, RT; (e) 9:1
DCM:MeOH, NaOH, rt; (f) 27, HATU, TEA, DCM, RT.
Salicylamide analogue CCG-212392 was prepared from the Chan-Lam
coupling111
of methyl salicylate 41 and 4-chlorophenyl boronic acid, providing the ether
intermediate 42 in low yield, followed by ester hydrolysis and amidation (Scheme 12).
Secondary phenyl anthranilamides CCG-211824 and CCG-222981 were assembled from
Buchwald amination112, 113
of triflated methyl salicylate and the appropriate aniline,
followed by saponification/amidation. The N-methyl anthranilamide derivative CCG-

54
212393 was accessed through methylation of coupling intermediate 43a, followed by the
usual saponification and amide coupling.
Scheme 12. Preparation of Anthranilamide and Salicylamide Analogues.a
aReagents and conditions: (a) Cu(OAc)2, 4-chlorophenylboronic acid, TEA, pyridine,
DCM, rt; (b) 10% aq. NaOH, EtOH, RT; (c) 36, EDC, HOBT, TEA, DCM, RT; (d)
PhN(Tf)2, TEA, DMF, RT; (e) 4-chloroaniline or aniline, Pd(OAc)2, DPPP, Cs2CO3,
toluene, reflux; (f) ethyl isonipecotate, EDC, HOBT, TEA, DCM, RT; (g) 4-(2-
aminoethyl)pyridine, EDC, HOBT, TEA, DCM, rt; (h) MeI, Cs2CO3, DMF, RT.

55
Benzyl anthranilamides CCG-222660 and CCG-222982 were obtained from the
alkylation of methyl 2-aminobenzoate 46 and the corresponding benzyl halide, followed
by the base hydrolysis and amidation (Scheme 13).
Phenethyl anthranilamide CCG-222983 could not be prepared through direct N-
alkylation of methyl 2-aminobenzoate because the electrophile was highly prone to
elimination. Conveniently, the transformation could be achieved through a Jourdan-
Ulmann coupling114
of 2-bromobenzoic acid 48 and 1-(2-aminoethyl)-4-chlorophenyl
amine in ethylene glycol dimethyl ether (Scheme 14). Base hydrolysis and amidation
then provided the desired analog.
Scheme 13. Preparation of Benzyl Anthranilamide Analogues.a
aReagents and conditions: (a) 4-chlorobenzyl chloride or benzyl bromide, tBuOK or
Cs2CO3, DMF; (b) 10% aq. NaOH, EtOH, rt; (c) 36, EDC, HOBT, TEA, DCM, rt.
Scheme 14. Preparation of Phenethyl Anthranilamide Analogue CCG-222983.a
aReagents and conditions: (a) 1-(2-aminoethyl)-4-chlorophenyl amine, Cu2O, Cu
0,
K2CO3, EDME, 130 °C; (b) 36, EDC, HOBT, TEA, DCM, rt.

56
Scheme 15. Preparation of Benzyl and Benzoyl Benzoate Analogues.a
aReagents and conditions: (a) AlCl3, PhCl, reflux; (b) 36, EDC, HOBT, TEA, DCM, rt;
(c) Zn0, CuSO45H2O, 28% aq. NH3, reflux.
The preparation of benzyl- and benzoyl benzamide derivatives (CCG-222821 and
CCG-212391 respectively) entailed the known Friedel-Crafts acylation of chlorobenzene
with phthalic anhydride, 115
which delivered ketone intermediate 51 from which a simple
amidation with 36 provided the benzoylbenzamide analogue CCG-212391 (Scheme 15).
The related benzylbenzamide analogue CCG-222821 was accessed through a dissolving
metal reduction of the ketone intermediate 51116, 117
to afford benzyl benzoic acid 52,
which was followed by amidation with amine 36. Interestingly, the ketone could not be
reduced without concomitant reduction of the chloride under several heterogenous
hydrogenation conditions.115, 118
Also, attempts to reduce the ketone to the respective
alcohol with NaBH4 failed,119
even with excess borohydride and vigorous conditions.
Thionation with Lawesson’s Reagent and subsequent Raney-Ni reduction also could be

57
used to generate the desired benzyl intermediate 52, but the dissolving metal reduction in
Scheme 15 was found to be more direct and efficient.
Scheme 16. Preparation of Carbazole Analogues.a
aReagents and conditions: (a) pyrrolidine, 37% aq. HCHO, EtOH, reflux; (b) s-BuLi,
THF, CO2(g), then 1 M aq. HCl, reflux; (c) sat. methanolic HCl, reflux; (d) SO2Cl2, DCM,
0 °C; (e) 10% aq. NaOH, EtOH, THF, rt; (f) 36, EDC, HOBT, TEA, DCM, rt.
Carbazole analogues were synthesized as shown in Scheme 16. Ortho-directed
lithiation of carbazole 53 followed by carboxylation with carbon dioxide in a manner
similar to that reported by Katritzky provided key intermediate 54.120
Direct coupling
with amine 36 provided analogue CCG-222824. The carbazole carboxylic acid 54 could
not be chlorinated with NCS under a variety of conditions,121, 122
while the methyl ester
yielded a complex mixture of variously chlorinated carbazoles. Ultimately, sulfuryl
chloride123
gave clean regioselective conversion to chloro-substituted carbazole ester 55
as confirmed by 1HNMR. This ester was then hydrolyzed to the corresponding carboxylic
acid and coupled with amine 36 to give the desired analogue CCG-222825.
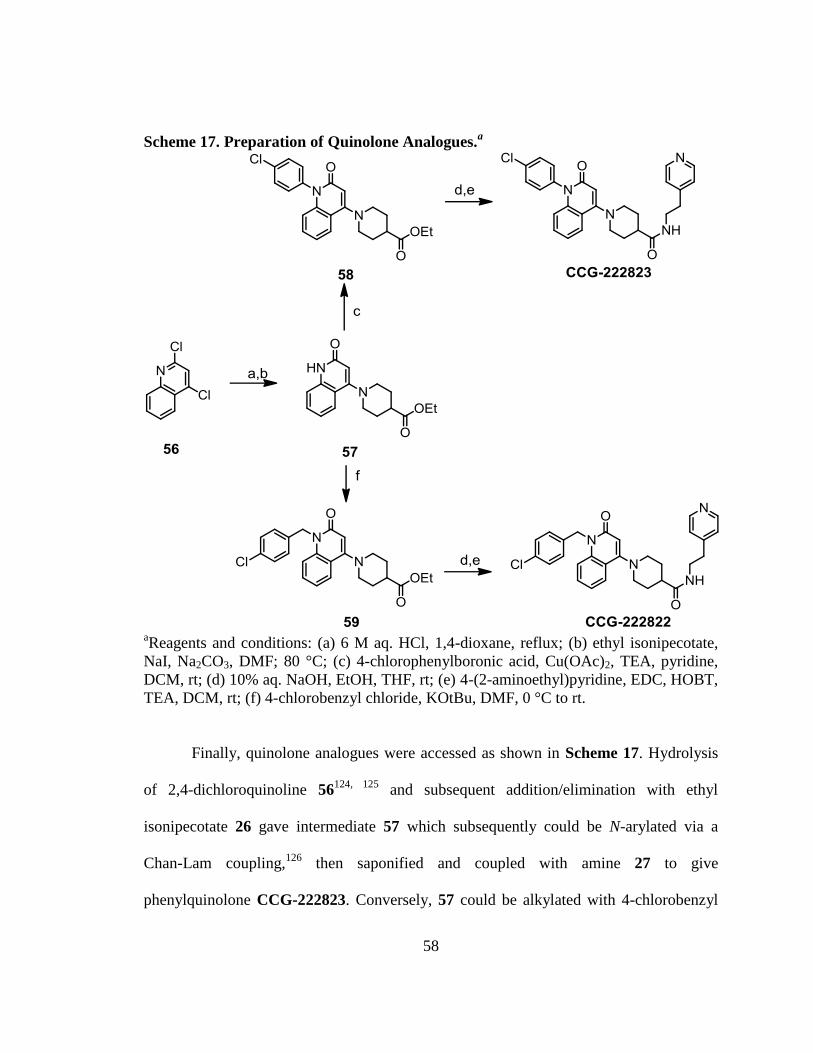
58
Scheme 17. Preparation of Quinolone Analogues.a
aReagents and conditions: (a) 6 M aq. HCl, 1,4-dioxane, reflux; (b) ethyl isonipecotate,
NaI, Na2CO3, DMF; 80 °C; (c) 4-chlorophenylboronic acid, Cu(OAc)2, TEA, pyridine,
DCM, rt; (d) 10% aq. NaOH, EtOH, THF, rt; (e) 4-(2-aminoethyl)pyridine, EDC, HOBT,
TEA, DCM, rt; (f) 4-chlorobenzyl chloride, KOtBu, DMF, 0 °C to rt.
Finally, quinolone analogues were accessed as shown in Scheme 17. Hydrolysis
of 2,4-dichloroquinoline 56124, 125
and subsequent addition/elimination with ethyl
isonipecotate 26 gave intermediate 57 which subsequently could be N-arylated via a
Chan-Lam coupling,126
then saponified and coupled with amine 27 to give
phenylquinolone CCG-222823. Conversely, 57 could be alkylated with 4-chlorobenzyl

59
chloride 2a, hydrolyzed and coupled under similar conditions to the phenylquinolone to
provide the benzylquinolone CCG-222822.
Conclusion
The primary goal of this chapter was the reduction of molecular weight, as low MW
is associated with improved BBB-permeability and is a feature of successful CNS drugs.
To this end, we successfully developed a novel MDR1-evasive pyrrole compound (CCG-
206381) with sub-micromolar potency and demonstrable in vivo CNS penetration. This
marks the first successful alphavirus antiviral with measurable CNS exposure, and
confirmed our predictions that reduction of MW was a viable tactic in achieving
favorable CNS permeability. However, this compound was remarkably susceptible to
CYP-mediated metabolism; therefore, an additional goal was improvement of metabolic
stability, and we identified the aniline moiety as a suitable substitute for pyrrole. We
unveiled phenylaniline CCG-211824 and benzylaniline CCG-222660 as low-to-sub-
micromolar inhibitors with MLM half-lives twice that of the original indole lead CCG-
205432 and five-to-six times that of the pyrrole lead CCG-206381. Importantly, these
aniline compounds demonstrated excellent activity (almost two-fold titer reduction)
against live viruses from three different families. This is an important step towards the
development of antivirals that are both potent and bioavailable.

60
Conformational Restriction Chapter IV.
Rationale
Potency is important for many aspects of drug delivery; potent compounds are
generally more amenable to oral administration because they require less material to
achieve the desired therapeutic effect, and they are less prone to off-target effects
(toxicity) because of the low doses required.79
Therefore, as with any drug, a high
therapeutic index, representing high potency and low toxicity, is a major goal for WEEV
inhibitors. Potency is also especially important for the successful action of many CNS-
active drugs; due to the brain-blood barrier (BBB), very little of the available drug will
actually penetrate the brain, as described later.
An important task in potency optimization is the identification of the bioactive
conformation of the inhibitors.127-130
In the absence of structural information, this can
only be accomplished by synthesizing and evaluating rigid analogs.131, 132
Of the multiple
conformations that a drug can adopt, only a small subset will have maximal activity if the
binding site is well defined and not highly flexible.128, 133
It is therefore informative to
lock out or freeze certain conformations and observe their effect on activity in an effort to
pinpoint the active one, especially when the binding site is unknown.134
It is also a very
useful strategy for improving selectivity versus off-target effects.135-137
When designing
such analogs, it is important to minimize the number of additional atoms used in locking
a conformation because adding molecular weight or introducing new interactions with the

61
binding site may have their own effects on activity, thereby confounding interpretation of
the actual impact of conformational restriction. Rigid analogs may be prepared utilizing
several approaches to restricting rotatable bonds: a) substituting small functionality with
sterically demanding functionality; b) bridging nonadjacent atoms; and c) restricting ring
conformations.
Figure 15. Steric Approach to Conformational Restriction.
Installation of sterically restricting functionality is most often the simplest
approach of the three tactics and retains some degree of flexibility. In the case of lead
CCG-102516, the N-benzyl encounters minimal resistance to rotation from the small
indole hydrogens and would be expected to adopt a configuration that minimizes steric
clashes with the amide carbonyl (Figure 15 A). On the other hand, methylation of the
indole 7-position hinders its ability to rotate over this region and would be expected to

62
greatly limit the available conformers, inducing a conformation (63) that is distinct from
61 (Figure 15 B).
Figure 16. Tethering Approach to Conformational Restriction.
The second tactic, bridging of nonadjacent atoms, is a much more restrictive
approach. For example, the distal amide of CCG-102516 is free to rotate about
piperidine-amide C-C bind (Figure 16), but inclusion of a bridging methylene between
the piperidine 3-position and the amide nitrogen provides a compound with an
immobilized carboxamide conformer.
Figure 17. Ring-Locking Approach to Conformational Restriction.

63
The third approach, the locking or steric restriction of ring conformers, is similar
to that of the previous tactic in that it most frequently involves bridging of nonadjacent
and (often) transannular atoms (Figure 17). The lead compound CCG-102516 features a
1,4-disubstituted piperidine, and due to the meso C2 symmetry about the ring, there
predominantly exist two major conformers: equatorial-equatorial (66) and axial-
equatorial (67). Either of these can be conveniently locked. An ethylene bridge across the
piperidine 2- and 6-positions can only axial-axial (68), resulting in a locked equatorial-
equatorial relationship between the piperidine substituents. Similarly, preparation of the
fused cyclopropane/pyrrolidine (69) allows access to the axial-equatorial analog, with
minimal impact on molecular weight.
SAR
Initial SAR focused on the restricting the indole and benzamide portions of the
lead CCG-102516. Biological results for indole core analogs appear in Table 11.
Utilizing the steric approach to conformational restriction, two compounds were designed
on the notion that strategically placed methyl groups would impede bond rotations of
interest while only contributing a small increase in molecular weight. Both the 7-
methylindole CCG-206447 and the 3-methylindole CCG-205431 possessed substantially
reduced potency, indicating that the methyl groups may in fact be blocking access to the
active conformations.
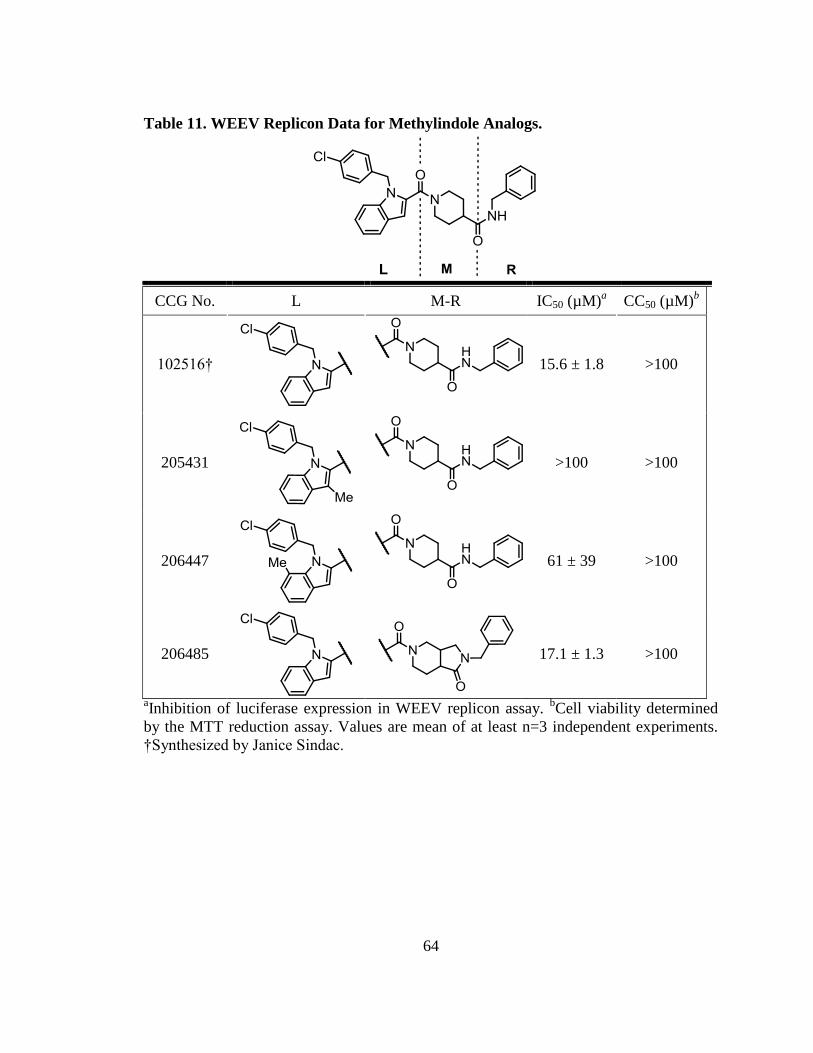
64
Table 11. WEEV Replicon Data for Methylindole Analogs.
CCG No. L M-R IC50 (µM)a CC50 (µM)
b
102516†
15.6 ± 1.8 >100
205431
>100 >100
206447
61 ± 39 >100
206485
17.1 ± 1.3 >100
aInhibition of luciferase expression in WEEV replicon assay.
bCell viability determined
by the MTT reduction assay. Values are mean of at least n=3 independent experiments.
†Synthesized by Janice Sindac.

65
Table 12. WEEV Replicon Data for Rigid Benzamide Analogs.
CCG No. R IC50 (µM)a CC50 (µM)
b
102516†
15.6 ±1.8 >100
205470
>100 >100
205471
>100 >100
205472
>100 >100
205473
>100 >100
205474
>100 >100
206328
>100 >100
206382
0.53 ± 0.04 >100
206549
1.7 ± 0.1 >100
206550
7.1 ± 0.9 >100
206397†
15.2 ± 4.7 62 ± 13
206587
>100 >50
206382
0.53 ±0.04 >100
aInhibition of luciferase expression in WEEV replicon assay.
bCell viability determined
by the MTT reduction assay. Values are mean of at least n=3 independent experiments.
†Synthesized by Janice Sindac.

66
Restricted of the Terminal Carboxamide. We then synthesized and evaluated
analogs of the benzamide featuring phenylpyrrolidines, isoindolines, indolines, and
indane substructures (Table 12) and a bicyclic compound CCG-206485 (Table 11), but
the majority of analogs exhibited no activity, which was not entirely undesirable.
Uniformly active compounds would indicate a non-selective binding site – one to which
binding optimization would be difficult. Excitingly, one compound (CCG-206382)
possessed significantly better activity than others in this series, and constituted the most
active compound in the analog arsenal at that time. This result was most likely not the
product of increased lipophilicity or chain extension because analogs of similar logP
(CCG-206550) and length (CCG-206587) have higher IC50 values (Table 12).
Interestingly, the racemic methyl analog of CCG-206382 (CCG-206549) was also
active, though just over two-fold less so than its more potent relative. Curiously, CCG-
206550 was similarly active to the lead CCG-102516, but CCG-205472 was completely
inactive despite being shorter by only a single methylene unit. Overall, the results in
Table 12 suggest that the amide binding pocket of the unknown molecular target is very
well defined and discriminating.
Indane SAR. Due to the superb in vitro potency of CCG-206382 in the WEEV
replicon assay, the indane moiety was incorporated into other WEEV inhibitor analogs,
including those of the low molecular weight class (Chapter III). As previously discussed
in that chapter, Janice Sindac had discovered the equally potent compound CCG-205432
bearing a pyridine in place of the indane. Curiously, related analogs differing only by the
indane or pyridine functionality did not possess similar activities (Table 13); in some
cases, the IC50 values differed in excess of one hundred-fold (e.g. CCG-206381

67
compared to CCG-208914, and CCG-209021 compared to CCG-209020.) The indane
compounds also behaved differently than their pyridine counterparts in the replicon
assay. For example, an IC50 value for CCG-209793 could not be deduced because the
concentration-inhibition plot was uncharacteristically linear, and more than one
compound’s plot oddly plateaued in a manner inconsistent with every other analog and
class synthesized to-date. These results questioned the validity of CCG-206382’s
activity, but our collaborators found no evidence of direct luciferase inhibition. The
compound was consequently promoted for further studies to evaluate its true efficacy
against live virus.
Table 13. WEEV Replicon Data Comparisons of Analogs Based on CCG-205432
and CCG-206382.
CCG # R IC50 (µM)a CC50 (µM)
b
205432†
0.53 ± 0.08 65.0
206382
0.53 ± 0.04 >100
206381
0.68 ± 0.04 >100
208914
61.8c 70.3
208916
>100 >100
208913
>100 >100
208915
22.0 ± 8 >100
208793
CNBDd >100

68
Table 13. Continued.
CCG # R IC50 (µM)a CC50 (µM)
b
209021
0.93 ± 0.2 8.0
209020
>100 >100
211756
>100 >100
211753
>100 >100
211754
>100 >100
211755
>100 >100
208825
60 ± 5 >100
208826
>100 >100
208827
41 ± 5 >100
208828
>100 >100
208846
>100 >100
208847
>100 >100
208848
>100 >100
208849
33 ± 15 >100
aInhibition of luciferase expression in WEEV replicon assay.
bCell viability determined
by the MTT reduction assay. Values are mean of at least n=3 independent experiments. cValue is n=1 experiment.
dCould not be determined.
†Synthesized by Janice Sindac.

69
Antiviral Activity. When cells infected with WEEV or NSV (neuro-adapted
sinbis virus) were treated with the pyridine analog CCG-205432, viral titer was reduced
by nearly two log units, and cell viability was significantly improved (Figure 18).
However, the indane analog CCG-206382 (despite being equipotent in the WEEV
replicon assay) neither decreased viral titer nor improved cell viability relative to the
negative control, suggesting that the compound was truly not active. Analogs featuring
the indane moiety were subsequently dropped.
Figure 18. Activity of Various Analogs Against Live Virus.
Data from the David Miller Lab.

70
Piperidine Amide SAR. Having determined the terminal carboxamide portion of
the analogs to be a challenging site for conformational restriction, our attention was
turned towards the central piperidine ring. As in Chapter III (low molecular weight
series), CCG-205432 was employed as the lead due to its potency and demonstrable
activity against live virus, and thus this series features the right-hand pyridylethyl amide.
Biological results appear in Table 14. In Chapter II (reduced TPSA series), it was noted
that there appeared to be a limited tolerance for TPSA reduction, perhaps because certain
structure-activity requirements were not met. It was noted that compounds like CCG-
211826 (Table 2), despite being structurally similar to potent leads, lacked the right-hand
secondary amide. To evaluate the postulated importance of this functionality, four
compounds were prepared (Table 14). Activities of the inverse amide CCG-2120532 and
the shifted amide CCG-224001 are over 30-fold less than CCG-205432, confirming that
the amide is important to the unknown pharmacophore. In particular, both of these
analogs retain the amide carbonyl in the same relative location, but differ in the location
of the amide nitrogen, which indicates that the position of the carbonyl is apparently
more important than the NH. The amide carbonyl may be engaged as a hydrogen-bond
acceptor, and, due to the dependence on the acceptor-donor angle, translocation of the
carbonyl by one methylene unit may be sufficient to deprive the molecule of a productive
hydrogen bond in the unknown target, accounting for the diminished activity.
Furthermore, complete excision of the amide (e.g. E- and Z-alkenes CCG-211823 and
CCG-211825) returned analogs with no activity, complementing this hypothesis.

71
Table 14. WEEV Replicon Data for Central Piperidine Analogs.
CCG No. R IC50 (µM)a CC50 (µM)
b
205432†
0.53 ± 0.08 65.0
212052
17.6c 66.7
c
224001
24.7 ± 3.5 77.3
211823
>50 >50
211825
>50 >50
212390
24.4 ± 4.0 78.0
211822
3.9 ± 0.4 74.2
211829
>50 >50
211761†
1.3 ± 0.4 63.2

72
Table 14. Continued.
CCG No. R IC50 (µM)a CC50 (µM)
b
212394
24.4 ± 6.3 61.0
222980
24.6 ± 1.6 >100
224000
0.96 ± 0.06 50.9
224220
10.9 ± 0.6 56.5
224002
73.1 ± 29.1 88.1
222661
0.49 ± 0.09 41.1
aInhibition of luciferase expression in WEEV replicon assay.
bCell viability determined
by the MTT reduction assay. Values are mean of at least n=3 independent experiments. cValue is n=1 experiment.
†Synthesized by Janice Sindac.
Core Piperidine SAR. The significantly reduced activity of the urea analog
CCG-212390 and eneamide CCG-211822 are also consistent with the postulated
importance of the secondary amide (Table 14). Both analogs were built to rigidify and
flatten the core ring, and such modifications may have changed the projection of the
amide carbonyl. The bicyclic analog CCG-212394 may display the amide in a similarly
nonproductive manner, especially in light of the better activity of the very similar (but

73
less restricted) analog CCG-211761. The α-methyl analog CCG-211829 was much less
active, presumably because the methyl was either impeding favorable chair
conformations or occluding the amide carbonyl. The exo-bicycle CCG-222980 was also
less potent than the lead. Interestingly, the ethylene-bridged piperidine CCG-224000 was
a mere two-fold less potent than CCG-205432, suggesting that the locked equatorial-
equatorial relationship between the 1- and 4-substituents may be indicative of the
unknown bioactive conformation.
The importance of length to the activity of WEEV inhibitors was discussed in
Chapter II and Chapter III, and, unsurprisingly, the azetidine-containing CCG-224002
was less active than its piperidine-containing counterpart CCG-205432 (Table 14). To
determine the effect of larger rings on activity, the azapane analog CGG-222661 was
synthesized and tested. Interestingly, this compound was equally potent with the lead
despite being less rigid and possessing an offset right-hand amide. Furthermore, the
active enantiomer could potentially be two-fold more active than the racemate, marking
this compound as the most active to-date in the WEEV replicon assay.
Synthesis
Restricted analogs based upon the benzylamide lead CCG-102516 were generally
prepared through amide coupling of various amines with the core carboxylic acid 71,
which was itself accessed via coupling of the indole-2-carboxylic acid 4a with ethyl
isonipecotate 26 followed by saponification (Scheme 18).

74
Scheme 18. General Preparation of Rigid Benzamide Analogs.a
aReagents and conditions: (a) 26, EDC∙HCl, HOBT, TEA, DCM, RT; (b) 10% aq.
NaOH, EtOH, RT; (c) HNR1R2, EDC∙HCl, HOBT, TEA, DCM, RT.
Scheme 19. Preparation of 2-(aminomethyl)indane 75.a
aReagents and conditions: (a) HMDS, HATU, DIPEA, DMF, rt, 20 h; (b) LiAlH4, THF,
RT, 8 h.
2-(Aminomethyl)indane 75 (Scheme 19, for the synthesis of CCG-206382) was
accessed first through amidation with the 2-indane carboxylic acid 73 and HMDS, after
which aqueous workup conditions resulted in hydrolytic degradation of the silyl
functionality to reveal the primary carboxamide 74. This procedure was found to be more
efficient than direct aminolysis of the carboxylic acid. Reduction of the carboxamide with
LAH provided the desired amine 75, which was coupled with indole carboxylic acid 71
as described in Scheme 18. Racemic 2-(2-aminoethyl)indane 78 was prepared first

75
through double displacement of dibromoxylene 76 with acetylacetate followed by
reductive amination of the ketone 77 (Scheme 20).
Scheme 20. Preparation of Racemic 2-(2-Aminoethyl)indane.a
aReagents and conditions: (a) ACAC, NaH, DMF; (b) 7 M NH3/MeOH, NaBH4, RT, 20
h.
The 7- and 3-methyl indole analogs were generally prepared through classical
Fischer indolization (Scheme 21 and Scheme 22). The appropriate anilines (o-toluidine
79 and aniline 84) were transformed into their respective diazonium salts and reduced to
hydrazines through the action of tin(II) chloride.138
The hydrazines were then allowed to
undergo Fischer indolization with the necessary β-ketoester 80 or 85, but only the 7-
methyl indole required a transition metal Lewis acid catalyst to facilitate cyclization to
indole 81. Overall, this route was found to be more expedient than the reported two-step
Japp-Klingemann/Fischer indolization approach139
to the indole scaffold. Alkylation of
the indole N1-position with 4-chlorobenzyl chloride 2a was found to be slow for the 7-
methyl analog 81, presumably due to steric hindrance of the nucleophile. Subsequent
saponification and amide coupling with benzylamine yielded the desired methyl indole
analogs CCG-205431 and CCG-206447.

76
Scheme 21. Preparation of 7-Methylindole CCG-206447.a
aReagents and conditions: (a) 79, NaNO2, conc. HCl, 0 °C, 30 min; then SnCl2, 4 °C, 11
h, 56%; (b) 80, ZnCl2, TsOH, EtOH, reflux, 8 h; (c) 2a, K2CO3, DMF, 60 °C, 3 d, 46%;
(d) 10% aq. KOH, EtOH, 70 °C, 4 h, 94%; (e) N-benzylpiperidine-4-carboxamide,
EDCHCl, HOBT, DIPEA, DMF, RT, 18 h, 47%.
Scheme 22. Preparation of 3-Methylindole CCG-205431.a
aReagents and conditions: (a) PhNH2, NaNO2, conc. HCl, 0 °C, 15 min; then SnCl2, 0 °C,
24 h; then 85, EtOH, TsOH, reflux, 12 h, 65%; (b) 2a, K2CO3, DMF, 60 °C, 12 h, 72%;
(c) THF, 5 M aq. NaOH, 55 °C, 18 h, 96%; (d) N-benzylpiperidine-4-carboxamide
hydrochloride, HATU, DIPEA, DMF, RT, 20 h, 51%.
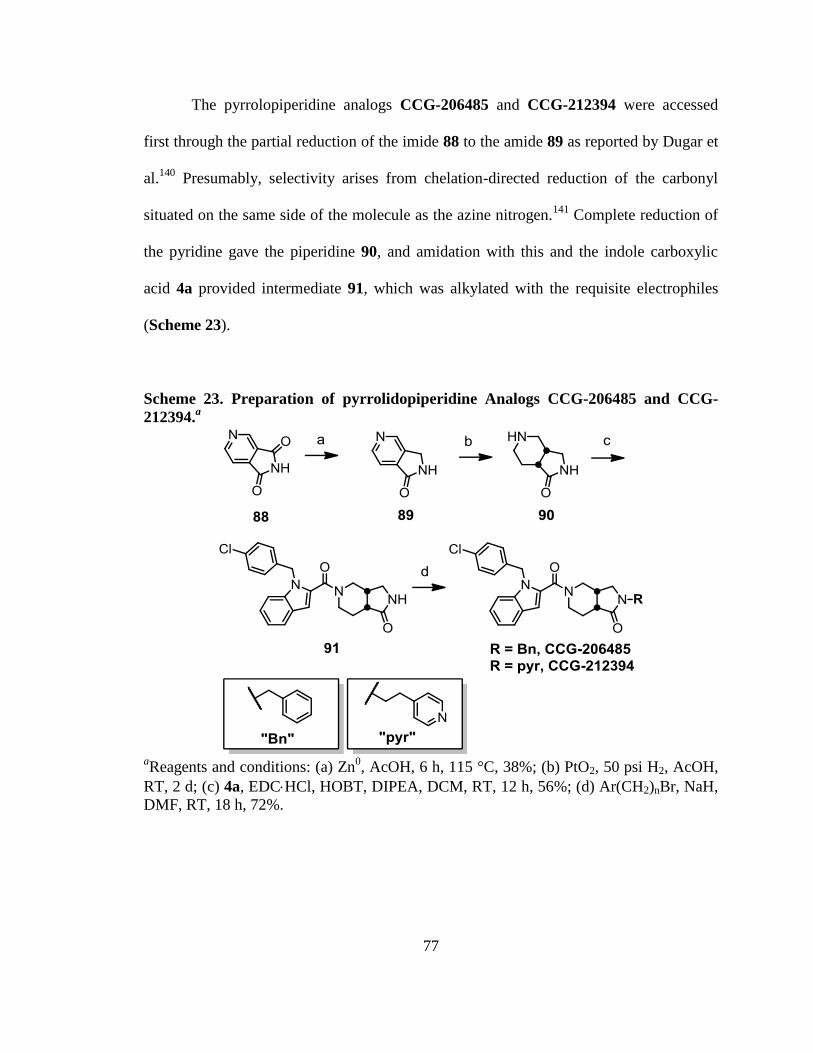
77
The pyrrolopiperidine analogs CCG-206485 and CCG-212394 were accessed
first through the partial reduction of the imide 88 to the amide 89 as reported by Dugar et
al.140
Presumably, selectivity arises from chelation-directed reduction of the carbonyl
situated on the same side of the molecule as the azine nitrogen.141
Complete reduction of
the pyridine gave the piperidine 90, and amidation with this and the indole carboxylic
acid 4a provided intermediate 91, which was alkylated with the requisite electrophiles
(Scheme 23).
Scheme 23. Preparation of pyrrolidopiperidine Analogs CCG-206485 and CCG-
212394.a
aReagents and conditions: (a) Zn
0, AcOH, 6 h, 115 °C, 38%; (b) PtO2, 50 psi H2, AcOH,
RT, 2 d; (c) 4a, EDCHCl, HOBT, DIPEA, DCM, RT, 12 h, 56%; (d) Ar(CH2)nBr, NaH,
DMF, RT, 18 h, 72%.

78
Scheme 24. Preparation of Inverse Amide CCG-212052.a
aReagents and conditions: (a) EDCHCl, HOBT, TEA, DCM, RT, 30 h, 71%; (b) HCl,
dioxane, RT, 1 h, 100%; (c) 4a, EDCHCl, HOBT, TEA, DCM, RT, 22 h, 61%.
Scheme 25. Preparation of Shifted Amide CCG-224001.a
aReagents and conditions: (a) EDCHCl, HOBT, TEA, DCM, RT; (b) KOH, H2O, EtOH,
RT; (c) 4-(aminomethyl)pyridine, EDCHCl, HOBT, TEA, DCM, RT.
The inverse amide CCG-212052 was prepared through the amide coupling of N1-
protected 4-aminopiperidine 92 and 3-(4-pyridyl)propanoic acid 93. Subsequent

79
deprotection and coupling with indole carboxylic acid 4a furnished the desired inverse
amide CCG-212052 (Scheme 24).
To prepare the shifted amide analog CCG-224001, the commercially available
amino ester 95 was first coupled with the indole carboxylic acid 4a. The ester of the
resulting intermediate was then hydrolyzed and coupled with 4-(aminomethyl)pyridine to
provide the final compound (Scheme 25).
It was anticipated that a Wittig reaction would ultimately provide the key (Z)-
alkene feature of analog CCG-211829, so the requisite phosphonium bromide 98 was
prepared from 4-(3-hydroxypropyl)pyridine 97.142
This was then deprotonated by nBuLi
to provide the ylide, which was subsequently permitted to react with the piperdinyl
aldehyde 8 to provide the desired alkene 99a with >50 Z:E selectivity. Deprotection and
subsequent coupling with the indole carboxylic acid 4a gave the (Z)-alkene analog CCG-
211829 (Scheme 26).
A large number of highly E-selective olefinations exist,143
but because the
phosphonium bromide salt was already in hand, the Schlosser modification of the Wittig
reaction was employed in order to access (E)-alkene analog CCG-211822 (Scheme 26).
In this procedure, the intermediate oxaphosphetane is deprotonated by PhLi – uniquely
suited to deprotontion of sterically shielded protons144
– and allowed to equilibrate to the
more thermodynamically favorable trans-betaine carbanion before being quenched with
tBuOH. In this way, the desired E stereochemistry was quickly accessed to provide the
(E)-alkene 99b. Deprotection and amide coupling with indole carboxylic acid 4a yielded
the desired (E)-alkene analog CCG-211822.

80
Scheme 26. Preparation of (E)- and (Z)-alkene Analogs.a
aReagents and conditions: (a) Conc. HBr, PPh3, tol, reflux, 11 h, 34%; (b) n-BuLi, THF,
RT; then 8, RT, 12 h, 52%; (c) 4 M HCl/1,4-dioxane, RT, 1 h; (d) 1-(4-chlorobenzyl)-
1H-indole-2-carboxylic acid, EDCHCl, HOBT, TEA, DCM, RT, 14 h, 56% (2 steps); (e)
PhLi, LiBr, THF, RT, 20 min; then 8, THF, -78 °C, 30 min; then PhLi, -78 °C, 30 min;
then t-BuOH, THF, -78 °CRT, 36 h, 41%; (f) 4 M HCl/1,4-dioxane, RT, 1 h; (g) 1-(4-
chlorobenzyl)-1H-indole-2-carboxylic acid, EDCHCl, HOBT, TEA, DCM, RT, 14 h,
60% (2 steps).
The piperazinyl urea analog CCG-212390 was prepared by treating boc-
piperazine 100 with phosgene and 4-(2-aminoethyl)pyridine 27 (Scheme 27). Order of
addition was critical; if 27 was treated with phosgene, the intermediate N-acyl chloride
would rapidly react with the pyridine nitrogen and no productive chemistry would arise.
Thus, the boc-piperazine 100 was treated first with phosgene, and the pyridyl amine 27
was added last. Once the urea was in hand, deprotection and coupling with indole
carboxylic acid 4a provided the desired urea analog CCG-212390.

81
Scheme 27. Preparation of Urea Analog CCG-212390.a
aReagents and conditions: (a) COCl2, TEA, tol, DCM, 0 °C, 1 h, 81%; (b) HCl, dioxane,
RT, 1 h, 100%; (c) 4a, EDCHCl, HOBT, TEA, DCM, RT,
Initial attempts to access the tetrahydropyridine core of CCG-211823 proceeded
through the partial reduction of the formyl pyridinium salt of ethyl isonicotinate with
NaBH4,145, 146
but this method was found to be poor yielding and complicated by side
products. The selenoxide elimination was then identified as a viable alternative, shown in
Scheme 28.147, 148
Treatment of protected ethyl isonipecotate 102 with LDA and
subsequent addition of phenylselenyl chloride provided the phenylselenide 103. The
compound could undergo the oxidation-elimination at this point, but it was found that
carrying the selenyl functionality through the first amidation aided purification efforts. In
this way, phenylselenide intermediate 105 was oxidized with mCPBA under cold
conditions and basified to prevent Bronsted-acid catalyzed hydrolytic decomposition via
a seleno-Pummerer pathway,149
and the resulting tetrahydropyridine ethyl ester 106 was

82
saponified then amidated with 4-(2-aminoethyl)pyridine to give the desired analog CCG-
211823.
Scheme 28. Preparation of Tetrahydropyridine Analog CCG-211823.a
aReagents and conditions: (a) Boc2O, Na2CO3, THF, H2O, reflux, 86%; (b) n-BuLi,
DIPA, -78 °C, 15 min; then PhSeCl, THF, -78 °CRT, 3 h, 58%; (c) 4 M HCl/1,4-
dioxane, RT, 2 d, 100%; (d) 4a, EDCHCl, HOBT, TEA, DCM, RT, overnight, 63%; (e)
mCPBA, DCM, -78 °C, 30 min; then TEA, DCM, -78 °C; then -78 °CRT, 1 h, 82%; (f)
10% aq. NaOH, EtOH, RT, 95%; (g) 27, EDCHCl, HOBT, TEA, DCM, RT, 91%.
In a manner similar to the preparation of the tetrahydropyridine analog CCG-
211823, Boc-protected ethyl isonipecotate 102 was alkylated with methyl iodide after
treatment with LDA to provide the methylpiperidine 107. This piperidine was
deprotected and coupled with indole carboxylic acid 4a, and the ethyl ester 109 was

83
saponified and coupled with 27 to give the desired methyl analog CCG-211829 (Scheme
29).
Scheme 29. Preparation of Methylpiperidine Analog CCG-211829.a
aReagents and conditions: (a) nBuLi, DIPA, THF, -78 °C; then MeI, THF, -78 °C; (b) 4
M HCl, dioxane, RT, 1 h; (c) 4a, EDC∙HCl, HOBT, TEA, DCM, RT; (d) 27, EDC∙HCl,
HOBT, TEA, DCM, RT.
The key cyclopropane of the exo-bicyclic analog CCG-222980 was installed by a
method previously reported.150, 151
An electron-deficient carbenoid was generated from
ethyl diazoacetate and treated with Cbz-protected 2,5-dihydropyrrole 111 over several
days. This yielded a mixture of unreacted starting material and two diastereomers of the
product 112, enriched in the exo-isomer, which was taken forward after tedious
separation and deprotected via hydrogenolysis and coupled with the indole carboxylic

84
acid 4a. Treatment with KOTMS hydrolyzed the ester to the potassium salt, which was
subsequently coupled with amine 27 to yield the final analog (Scheme 30).
Scheme 30. Preparation of Exo Bicyclic Analog CCG-222980.a
aReagents and conditions: (a) N2CHCO2Et, Rh2(OAc)4, DCM, reflux, 72 h, 3:1 dr; (b)
Pd/C, H2, EtOH, HCl, RT; (c) 4a, EDC∙HCl, HOBT, TEA, DCM, RT; (d) KOTMS, THF,
RT; (e) 27, EDC∙HCl, HOBT, TEA, DCM, RT.
Scheme 31. Preparation of Bridged Analog CCG-224000.a
aReagents and conditions: (a) KOTMS, THF, RT, 12 h; (b) 27, EDC∙HCl, HOBT, TEA,
DCM, RT; (c) HCl, dioxane, RT; (d) 4a, EDC∙HCl, HOBT, TEA, DCM, RT.

85
The ethylene-bridged piperidine analog CCG-224000 was first constructed by
hydrolyzing the commercially available protected amino ester 114 and coupling the
resulting acid with amine 27 to provide the latent right-hand half 115 of the analog.
Deprotection under acidic conditions followed by amide coupling with the indole
carboxylic acid 4a provided the desired analog (Scheme 31).
Scheme 32. Preparation of the Spiro Analog CCG-224220.a
aReagents and conditions: (a) PhCHO, tol, reflux, 2 h, 85%; (b) diethyl malonate, NaH,
DMF, reflux, 12 h, 84%; (c) Pd/C, H2, MeOH, RT, 6 d, 98%; (d) MsCl, TEA, DCM, RT,
3 h; (e) TsNH2, K2CO3, DMSO, 90 °C, 5 h, 48% (2 steps); (f) NaOH, H2O, EtOH, reflux,
2 h, 91%; (g) pyridine, reflux, 12 h, 92%; (h) 4-pyridylethylamine, EDCHCl, HOBT,
TEA, DCM, RT, 12 h, 80%; (i) Na0, naphthalene, THF, RT, 5 min; (j) 4a, EDCHCl,
HOBT, TEA, DCM, RT, 14 h, 61% (2 steps).
The spiro core of CCG-224220 was assembled in manner similar to that reported
(Scheme 32).152, 153
The diol 117 was protected as an acetal,154
and the bromides were

86
doubly displaced by diethylmalonate under basic conditions to give the diester 119. The
acetal was removed to reveal the alcohols of 120, which were subsequently mesylated
and displaced by tosylamide to give the cyclized spiro product 121. Ester hydrolysis and
thermal decarboxylation furnished the mono-carboxylic acid 122 in good yield, and this
was coupled with amine 27 to give the latent right-hand half 123 of the desired final
compound. Cleavage of the tosyl proved to be a substantial challenge, however. Various
conditions with samarium (II) iodide either failed to remove the tosyl, or reduced and
cleaved the amide preferentially.155, 156
Other reported methods of tosyl deprotection were
employed with no success157, 158
until sodium napthalenide was used,159
which quickly
and cleanly cleaved the S-N bond to reveal the amine 124, which was then coupled with
the indole carboxylic acid 4a to afford the desired analog CCG-224220.
Scheme 33. Preparation of Azetidine-Containing CCG-224002.a
aReagents and conditions: (a) methyl 4-azetidinecarboxylate hydrochloride, EDC, HOBT,
DIPEA, DCM, rt; (b) 10% aq. NaOH, EtOH; (c) 27, EDC, HOBT, TEA, DCM, rt.

87
The azetidine-containing analog CCG-224002 (Scheme 33)
Scheme 33was assembled in a manner similar to that used for construction of the
azetidine-containing pyrrole analogs. The indole carboxylic acid 4a was coupled with the
azetidine methyl ester, and the intermediate 125 was saponified and coupled with 4-(2-
aminoethyl)pyridine to give the final analog.
Scheme 34. Preparation of Azapane Analog CCG-222661.a
aReagents and conditions: (a) N2CHCO2Et, BF3OEt2, Et2O, -78 °CRT, 1h; (b) NaBH4,
EtOH, 0 °CRT, 2h; (c) MsCl, TEA, DCM, 0 °CRT, 6 h; (d) DBU, THF, 80 °C, 1 h; (e)
Pd/C, 50 psi H2, EtOH, HCl, RT, 24 h; (f) ) 4a, EDCHCl, HOBT, TEA, DCM, RT, 16 h;
(g) 10% aq. NaOH, EtOH; (h) 27, EDCHCl, HOBT, TEA, DCM, RT.
The core azapane of CCG-222661 was first accessed by way of ring expansion of
N-protected 4-piperidone 126 with ethyl diazoacetate and the strong Lewis acid BF3
(Scheme 34).160
The resulting racemic γ-ketoester 127 was reduced with NaBH4 to the γ-

88
hydroxy ester and mesylated. The mesylate 129 was then easily eliminated under mild
heating with DBU, and the ene ester 130 was exposed to heterogenous reduction
conditions to both reduce the alkene and the Cbz protecting group. The product amine
131 was then coupled with indole carboxylic acid 4a, saponified, and then coupled with
amine 27 to give the azapane analog CCG-222661.
Conclusion
The primary goal of this chapter was the identification of the unknown inhibitor
pharmacophore. To achieve this, conformationally-restricted analogs were prepared and
evaluated in the WEEV replicon assay. While the majority of compounds possessed
substantially reduced potency, the data pointed towards the importance of the distal
amide carbonyl to activity and a potential equatorial-equatorial relationship between the
1,4-substituents of the core piperidine ring. Intriguingly, the azapane CCG-222661 was
identified as our most potent compound to-date, with an IC50 = 0.49 ± 0.09 for the
racemate, and with an expected IC50 up to two-fold better for the active enantiomer. It is
hoped this data may be useful for the generation of a computational 3-D pharmacophore
model that may be applied to the identification of new alphavirus inhibitor templates via
in silico screening methods.

89
Metabolic Stability Chapter V.
Rationale
Metabolism is the enzymatic modification of compounds so that an organism may
enhance clearance, and is a crucial detoxification mechanism for removing reactive or
toxic xenobiotics.161
Unfortunately, even drugs considered safe may undergo this process.
Metabolism dictates the overall biological availability of a compound, and those
compounds that undergo rapid metabolism may be depleted before reaching their
intended biological target. Metabolic stability is one of the most crucial aspects of early
drug development, and instability is a potential killer of lead series.81
The majority of metabolism occurs in the liver, where a battery of metabolic
enzymes resides, although metabolism may occur elsewhere as well (blood, epithelial
intestinal cells, etc.)162, 163
These enzymes largely consist of a class called the cytochrome
p450s, or CYPs, that are responsible for Phase I metabolism, whose purpose is the
installation of polar functional groups to improve aqueous solubility to enhance clearance
and to provide handles for further Phase II metabolic pathways.79, 164
Potential sites of metabolism can be easily predicted using computational models
like that shown in Figure 19.165-169
However, while likely, empirical methods are
required to confirm such predictions because properties like atom accessibility and
overall lipophilicity are important factors. To this end, it was necessary for us to develop

90
an in-house metabolic stability assay to facilitate our ability to rapidly identify labile
functionalities and design them out of future analogs.
Figure 19. Predicted Sites of Metabolism in CCG-205432, Rank Ordered.
Predicted sites of metabolism are rank ordered where 1 = most likely, 2 = next most
likely, etc. Predictions calculated using smartCYP v.2.4.2.165, 166
The assay utilized commercially-available mouse liver microsomes harvested
from Balb/C mice, though the line of mice used by our collaborators in the lab of Prof.
David Irani for in vivo testing were C57 BL/6, and we found that the results were
translatable. Compounds were incubated in the presence of microsomes over 15 minutes,
and aliquots were removed and quenched over this time at specific time points. We also
developed an LC/MS/MS method to maximize throughput and detection sensitivity so
that parent compound concentration could be quickly assessed at concentrations as low as
single-digit nanomolar, or less than one percent of starting incubation concentration.
SAR
As discussed in Chapter I, avoidance of predicted metabolic instability led to the
replacement of the early thienopyrrole scaffold with indole. However, it remained

91
necessary to demonstrate empirically that we had gained stability through this
substitution, so we established a mouse liver microsome-based metabolic stability assay
and a corresponding LC/MS/MS analytical method for our lab.
Table 15. MLM Metabolic Stabilities for Thienopyrrole and Indole Leads.
CCG
No. Structure
IC50
(µM)a
CC50
(µM)b
MLM
T1/2
(min)c
ClogP
203881
11.1 ± 1.3 >100 1.7 ± 1.7 4.4
102516†
15.6 ± 1.8 >100 7.3 ± 3.9 4.5
203926†
6.8 ± 1.0 >100 31 ± 7.1 4.9
aInhibition of luciferase expression in WEEV replicon assay.
bCell viability determined
by the MTT reduction assay. Values are mean of at least n=3 independent experiments. cHalf-life in BALB/c mouse liver microsome incubation. Values are half-lives calculated
from the equation T1/2=ln(2)/slope, where the slope was ln(% detected) against time,
plotted from the mean of at least n=3 independent experiments. Values are mean ± SEM
of n = 3 independent experiments. †Synthesized by Janice Sindac.
Our newly developed protocol allowed for convenient in-house metabolic
stability analysis and was able to differentiate the short half-lives of our leads (Table 15).
Key to this was optimizing the concentration of microsomes, as the compounds were so

92
unstable as to be not differentiable under the conditions originally employed. Ultimately,
we found that quartering the amount relative to established protocols worked well. In the
assay, we discovered that the indole CCG-102516 had a half-life of 7.3 ± 3.9 minutes,
whereas the thienopyrrole CCG-203881 had a half-life of only 1.7 ± 1.7 minutes,
supporting our original stability hypothesis and validating our thienopyrrole/indole
switch. Despite this, CCG-102516 was still quite metabolically unstable. Interestingly,
CCG-203926, which differs by only a methyl group from CCG-102516, had
dramatically improved stability, with a half-life of 31 ± 7.1 minutes. This was
presumably due to steric hindrance of the potentially labile benzylic site.
Table 16 shows the activities of the lead indole compound CCG-205432, its
pyrrole-substituted analog CCG-206381, and the best compound from Table 15,
CCG-203926. Note that the stability of CCG-203926 (2.3 ± 0.1 minutes) does not match
its stability in Table 15 (31 ± 7.1 minutes); this was mostly likely due to the fresher,
more active microsomes used in the former study. However, the relative stabilities are not
affected, and we can confidently conclude that CCG-206381 (3.7 ± 1.3 minutes) and
CCG-205432 (16.7 ± 1.7 minutes) are substantially more stable than CCG-203926 in the
assay. This information suggests that the right-hand benzyl functionality may be a major
site of metabolism, since stability is significantly improved by replacement of the benzyl
amide with pyridylethyl substitution, although overall reduction in ClogP might also be a
factor. The data also indicate that pyrroles are far more metabolically labile than indoles
in this template. Detail regarding the application of these data appears in Chapter III.

93
Table 16. MLM Metabolic Stabilities For Indole and Pyrrole Analogs.
CCG
No. Structure
IC50
(µM)a
CC50
(µM)b
MLM T1/2
(min)c
ClogP
205432†
0.53 ± 0.1 65 16.7 ± 1.7 3.1
206381
0.68 ± 0.04 >100 3.7 ± 1.3 1.7
203926†
6.8 ± 1.0 >100 2.3 ± 0.1 4.9
aInhibition of luciferase expression in WEEV replicon assay.
bCell viability determined
by the MTT reduction assay. Values are mean of at least n=3 independent experiments. cHalf-life in BALB/c mouse liver microsome incubation. Values are half-lives calculated
from the equation T1/2=ln(2)/slope, where the slope was ln(% detected) against time,
plotted from the mean of at least n=3 independent experiments. Values are mean ± SEM
of n = 3 independent experiments. †Synthesized by Janice Sindac.
With the metabolic stability knowledge gained from Table 15 and Table 16, a
variety of proposed metabolically stable compounds were prepared and evaluated.
Pyridine CCG-212090 was over ten-fold more stable towards metabolism than CCG-
203926 (Table 17). ClogP is highly correlated with CYP metabolism,170, 171
thus CCG-
212090’s stability may be attributed to its ten-fold lower ClogP relative to CCG-203926.
Alternatively, this could suggest that the benzyl ring is a potential site of metabolism, as
pyridines are known to be less susceptible due to their electron-deficiency and

94
polarity.172, 173
While imidazoles are expected to be more stable than pyrroles by virtue of
the electron-withdrawing property of the azine nitrogen, the improved half-life of CCG-
206582 is more likely indicative of CYP inhibition, as the imidazole moiety is known to
bind to heme iron, a crucial component of CYP enzyme function.174-176
Table 17. MLM Stability and WEEV Replicon Data for Select Analogs.
CCG No. R IC50 (µM)a CC50 (µM)
b
MLM T1/2
(min)c
ClogP
205432†
0.53 ± 0.1 65 11.5 ± 0.7 3.1
203926†
6.8 ± 1.0 >100 1.8 ± 0.5 4.9
206582†
4.9 ± 2.6 >100 17.0 ± 2.7 2.7
212090†
0.6 ± 0.3 92 23.4 ± 4.0 3.4
aInhibition of luciferase expression in WEEV replicon assay.
bCell viability determined
by the MTT reduction assay. Values are mean of at least n=3 independent experiments. cHalf-life in BALB/c mouse liver microsome incubation. Values are half-lives
calculated from the equation T1/2=ln(2)/slope, where the slope was ln(% detected)
against time, plotted from the mean of at least n=2 independent experiments. Values are
mean ± SEM of n = 2 independent experiments. †Synthesized by Janice Sindac.
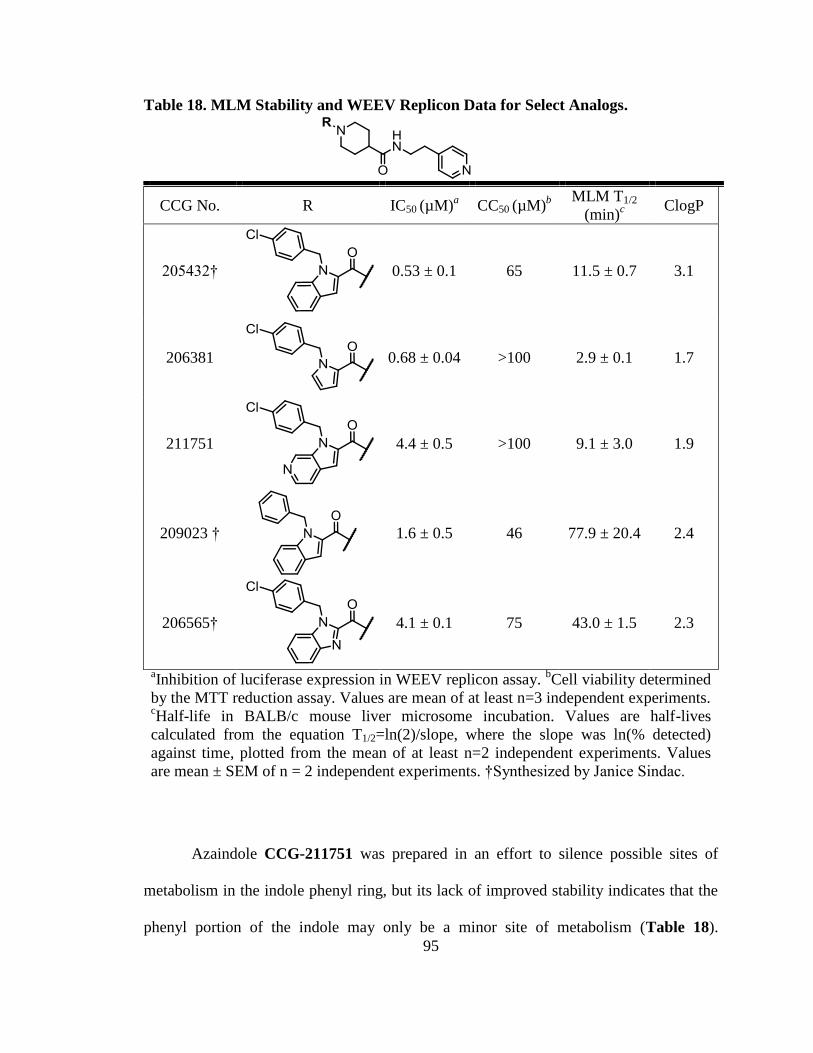
95
Table 18. MLM Stability and WEEV Replicon Data for Select Analogs.
CCG No. R IC50 (µM)a CC50 (µM)
b
MLM T1/2
(min)c
ClogP
205432†
0.53 ± 0.1 65 11.5 ± 0.7 3.1
206381
0.68 ± 0.04 >100 2.9 ± 0.1 1.7
211751
4.4 ± 0.5 >100 9.1 ± 3.0 1.9
209023 †
1.6 ± 0.5 46 77.9 ± 20.4 2.4
206565†
4.1 ± 0.1 75 43.0 ± 1.5 2.3
aInhibition of luciferase expression in WEEV replicon assay.
bCell viability determined
by the MTT reduction assay. Values are mean of at least n=3 independent experiments. cHalf-life in BALB/c mouse liver microsome incubation. Values are half-lives
calculated from the equation T1/2=ln(2)/slope, where the slope was ln(% detected)
against time, plotted from the mean of at least n=2 independent experiments. Values
are mean ± SEM of n = 2 independent experiments. †Synthesized by Janice Sindac.
Azaindole CCG-211751 was prepared in an effort to silence possible sites of
metabolism in the indole phenyl ring, but its lack of improved stability indicates that the
phenyl portion of the indole may only be a minor site of metabolism (Table 18).

96
Conversely, benzimidazole CCG-206565 was markedly more stable than the
corresponding indole CCG-205432, due either to stabilization of the pyrrole ring, or
simply reduced ClogP. Intriguingly, replacement of chlorine with hydrogen upon the N-
benzyl substituent of the template (CCG-209023) resulted in a dramatic half-life
improvement. This may simply be the result of lower ClogP, as compounds with high
logP are known to be susceptible to CYP450 enzymes. Furthermore, it suggests that
metabolism of the indole N-benzyl is not a major pathway. Despite having less activity
than the lead CCG-205432, this information allowed us to move CCG-209023 into
mouse studies, on the basis that enhanced metabolic stability would translate into
improved bioavailability. Gratifyingly, a ten mouse preliminary study demonstrated a
40% survival rate when infected mice were treated with the compound, marking CCG-
209023 as our most efficacious compound to-date.
Several fluorinated analogs (fluoropyrroles CCG-209020 and CCG-209021, and
Janice Sindac’s fluoroindoles CCG-208918 and CCG-208919) were synthesized to
probe potential metabolic soft spots around the pyrrole and indole rings (Table 19).
Incorporation of fluorine into compounds is a well-known and powerful tactic for
improving stability towards Phase I metabolism as fluorine is similar in size to hydrogen
(classical isostere) but is less reactive due to the strength of the C-F bond.177-179
Interestingly, these analogs, while active in the WEEV replicon assay, were also
surprisingly toxic and were therefore not promoted to MLM stability studies.

97
Table 19. WEEV Replicon Data for Fluorinated Analogs.
CCG No. R IC50 (µM)a CC50 (µM)
b
205432†
0.53 ± 0.1 65
206381
0.68 ± 0.04 >100
209021
0.93 ± 0.02 8.0
208918†
0.73 ± 0.05 24
208919†
0.92 ± 0.05 52
aInhibition of luciferase expression in WEEV replicon assay.
bCell viability
determined by the MTT reduction assay. Values are mean of at least n=3 independent
experiments. †Synthesized by Janice Sindac.

98
Synthesis
Scheme 35. Preparation of 4-Fluoropyrrole Analogs.a
aReagents and conditions: (a) TEMPO, TCCA, DCM, 0 °CRT, 20 min, 99%; (b) DAST,
DCM, -78 °CRT, 12, h, 86%; (c) 4 M HCl/1,4-dioxane, THF, RT, 4 h, 100%; (d) MnO2,
THF, reflux, 3 h, 20%; (e) 4-chlorobenzyl chloride, K2CO3, DMF, 60 °C, 30 h, 83%; (f)
10% aq. NaOH, EtOH, RT, 18 h, 78%; (g) ) NH2R1, EDCHCl, HOBT, TEA, DCM, rt,
15 h, 52%.
Fluoropyrroles CCG-209020 and CCG-209021 were prepared in a manner
similar to that previously described (Scheme 35).180
The 4-hydroxyproline 133 was
oxidized to 4-ketoproline 134 under TEMPO catalysis, and this was subsequently
fluorinated with DAST to provide the germinal fluoroproline 135. After acid-mediated
deprotection, the resulting 1H-proline was aromatized to the 4-fluoropyrrole 127 with

99
MnO2.181
Alkylation with 4-chlorobenzyl choride gave the N-benzyl intermediate, which
was saponified and coupled with the appropriate amine.
Scheme 36. Preparation of 6-Azaindole Analog CCG-211751.a
aReagents and conditions: (a) TFAA, 0 °C, 2 h; then Fum. HNO3, RT, 12 h; then
Na2S2O5, H2O, RT, 24 h, 9%; (b) Na0, diethyl oxalate, EtOH, tol, RT, overnight, 40%; (c)
Pt/C, 1 ATM H2, EtOH, AcOH, RT, 24 h; (d) EtOH, K2CO3, RT, 18 h, 51% (2 steps); (e)
2a, t-BuOK, DMF, RT, 14 h, 65%; (f) 10% aq. NaOH, EtOH, RT, 24 h, 84%; (g) 36,
EDCHCl, HOBT, TEA, DCM, RT, 20 h, 55%.
Azaindole analog CCG-211751 was prepared using largely known procedures
(Scheme 36).182
First, 4-picoline was nitrated under inefficient but mild conditions as
reported by Katritzky.183
The resulting 3-nitro-4-picoline 141 was then treated with
sodium in EtOH to form the anion, which was subsequently treated with diethyloxalate to

100
give the β-ketoester 142. The nitro was then fully reduced to the amine under
heterogeneous catalysis, and, upon introduction of basic conditions, this cyclized via
addition/elimination to yield the desired 6-azaindole core 143. Alkylation with 2a was
accomplished by fully deprotonating the azole nitrogren with tBuOK before addition of
the electrophile. Weaker bases, such as alkali metal carbonates (e.g. Cs2CO3) returned
mostly pyridinium salts. Saponification of the ester 144 and amidation with the amine
dihydrochloride 36 provided the desired azaindole analog CCG-211751.
Conclusion
The primary goal of this chapter was the development of a mouse-liver
microsome assay and LC/MS/MS protocol suitable for prioritizing compounds based
upon their stability towards Phase I metabolism. To this end, we designed a relatively
high-throughput MLM method that was successful in assessing and identifying
compound stabilities, and that correlated well with in vivo pharmacokinetic data.
Moreover, it facilitated identification of important modifications (e.g. pyrrole-to-aniline
substitution and chlorine-to-hydrogen substitution) that significantly improved stability,
leading to the discovery of our most stable compound to-date (CCG-209023).

101
Future Directions Chapter VI.
Overall, our design and development of inhibitors of neurotropic alphavirus
replication has been successful in identifying and optimizing potent compounds both in
vitro and in vivo against a broad spectrum of alphaviruses. Furthermore, we have
successfully improved physicochemical properties both predictive and demonstrable of
enhanced bioavailability and BBB-penetration. This is a significant step towards the
development of a successful antiviral for neurotropic alphaviruses, as there are no known
antivirals that possess CNS-permeability that are also therapeutically useful in the
treatment of these viruses.15, 40
Furthermore, the general mechanism of action is unique as there are few known
antivirals that target host machinery. Classically, such targets were avoided due to
assumed toxicity issues, but the low toxicity of our compounds places them firmly within
a small promising class of new host-targeting antivirals, and the only ones specific for
alphaviruses.184-187
The advantages of such a compound include enhanced broad-
spectrum activity and – more importantly – significantly reduced incidence of resistance.
Due to relatively rapid viral replication and poor fidelity, resistance-conferring mutations
in viral proteins may arise after treatment with viral-targeting drugs, which places a large
emphasis on the development of new drugs. On the other hand, due to the relatively low
turnover of host cells, resistance-conferring mutations are expected to occur much more
slowly, if at all, with host-targeting antivirals.

102
However, an important aspect of medicinal chemistry is the establishment of a
pharmacophore model – the collection of structural and electronic features requisite for
engaging with binding site contacts and maximizing activity. Importantly, such a model
facilitates the design and development of future analogs, including those based on
fundamentally different scaffolds. Over the course of the WEEV inhibitor project, we
identified multiple attributes critical to activity, such as the potency-length dependence
(Chapter II) and the disposition of the right-hand secondary amide (Chapter IV). With
the aid of our collaborator Paul Kirchhoff, we have attempted to computationally develop
a three-dimensional pharmacophore model of our compounds and apply such a model to
an in silico high-throughput screen to discover new chemical templates. Unfortunately,
we have been unable to generate a predictive pharmacophore model; models built from
diverse WEEV inhibitor training sets could not discriminate between known active and
inactive compounds. Moving forward, we would like to direct studies toward
identification of the pharmacophore through the design, synthesis, and evaluation of new
analogs.
Related to pharmacophore identification, we have also noted conformation-
dependent recognition by MDR1 in this project. Despite the magnitude of on-going work
in the field of medicinal chemistry, this observation has been noted only rarely in the
literature. Historically, tremendous efforts have been expended by medicinal chemists in
identifying structure-activity relationships for small molecule interactions with MDR1,
but the SAR is still largely undefined due to the promiscuity of the transporter. Therefore,
research regarding conformation-activity relationships could significantly enhance the
design of transporter-evasive compounds.

103
Another important aspect of drug development is the identification of the biological
target, and by extension, the mechanism of action through which the compounds exert
their pharmacological effect. Our collaborators have made tremendous strides towards
this end,104
and Janice Sindac has developed several photo-affinity probes to assist in
target identification. In contrast to the phenotype-based approach to analog design
employed throughout the project, such information could facilitate a target-based avenue
to future drug development. In this way, knowledge of the target could lead to models of
the binding site (e.g. homology) that could allow us to predict and incorporate structural
features tailored specifically to engage with potential binding site contacts. Also, because
toxicity is often a product of off-target binding to structurally similar biological
macromolecules, this could also allow us to improve the toxicity profile of our analogs,
especially in light of their host-based mechanism of action. Therefore, the continued
design and development of probes – no trivial endeavor – is an important aspect of the
project’s future.

104
Experimental Data Chapter VII.
All reagents were used as received from commercial sources unless otherwise noted. 1H
and 13
C spectra were obtained in DMSO-d6 or CDCl3 at room temperature, unless
otherwise noted, on a Varian Inova 400 MHz, Varian Inova 500 MHz, or Bruker Avance
DRX 500 instruments. Chemical shifts for the 1H NMR and
13C NMR spectra were
recorded in parts per million (ppm) on the δ scale from an internal standard of residual
tetramethylsilane (0 ppm). Rotamers are described as a ratio of rotamer A and B if
possible. Otherwise, if the rotamers cannot be distinguished, the NMR peaks are
described as multiplets. Mass spectroscopy data were obtained on a Waters Corporation
LCT. HPLC retention times were recorded in minutes (min) using an Agilent 1100 series
with an Agilent Zorbax Eclipse Plus–C18 column with the gradient 10% ACN/water (1
min), 10–90% ACN/water (6 min), and 90% ACN/water (2 min).
Reagent abbreviations and acronyms: ACAC (acetylacetone), AcOH (acetic acid), Ac2O
(acetic anhydride), AlCl3 (aluminum chloride), Ar (argon), BnBr (benzyl bromide),
Boc2O (di-tert-butyl dicarbonate), Cs2CO3 (cesium carbonate), Cu0 (elemental copper),
Cu2O (copper (I) oxide), DAST (diethylaminosulfur trifluoride), DCM
(dichloromethane), DIPA (diisopropylamine), DIPEA (N,N-diisopropylethylamine,
Hünig’s base), DMF (N,N-dimethylformamide), DPPP (1,3-

105
bis(diphenylphosphino)propane, EDCHCl (1-Ethyl-3-(3-
dimethylaminopropyl)carbodiimide hydrochloride), EGDME (ethylene glycol dimethyl
ether), Et2O (diethyl ether), EtOAc (ethyl acetate), EtOH (ethanol), H2 (hydrogen),
HATU (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid
hexafluorophosphate), H2O (water), HBr (hydrobromic acid), HCl (hydrogen chloride or
hydrochloric acid), Hex (hexanes), HMDS (hexamethyldisilazane), HOBT (1-
hydroxybenzotriazole), ind (indane), IPA (isopropanol), K2CO3 (potassium carbonate),
KOH (potassium hydroxide), LiAlH4 (lithium aluminum hydride), LiBr (lithium
bromide), m-CPBA (meta-chloroperoxybenzoic acid), MeCN (acetonitrile), MeI (methyl
iodide), MeOH (methanol), MgCl2 (magnesium chloride), MgSO4 (magnesium sulfate),
MnO2 (manganese (IV) oxide), N2 (nitrogen), Na0 (elemental sodium), NaBH4 (sodium
borohydride), NaBH3CN (sodium cyanoborohydride), NaBH(OAc)3 (triacetoxy sodium
borohydride), Na2CO3 (sodium carbonate), Na2SO4 (sodium sulfate), NAPDH
(nicotinamide adenine dinucleotide phosphate), NaH (sodium hydride), NaHCO3 (sodium
bicarbonate), NaNO2 (sodium nitrite), NaOH (sodium hydroxide), Na2S2O5 (sodium
metabisulfite), NH3 (ammonia), n-BuLi (n-butyl lithium), Pd/C (palladium on carbon),
PhCl (chlorobenzene), PhLi (phenyllithium), PhNH2 (aniline), PhN(Tf)2 (N,N-
bis(trifluoromethylsulfonyl)anilie, PhSeCl (phenylselenyl chloride), PPh3
(triphenylphosphine), Pt/C (platinum on carbon), PtO2 (platinum (IV) oxide), pyr
(pyridine), s-BuLi (sec-butyl lithium), SmI2 (samarium (II) iodide), SnCl2 (tin (II)
chloride), t-BuOH (tert-butanol), t-BuOK (potassium tert-butoxide), TCCA
(trichloroisocyanuric acid), TEA (triethylamine), TEMPO ((2,2,6,6-tetramethylpiperidin-
1-yl)oxy), TFA (trifluoracetic acid), TFE (trifluoroethanol), TFFA (trifluoroacetic

106
anhydride), TFE (2,2,2-trifluoroethanol); THF (tetrahydrofuran), tol (toluene), TsOH
(toluenesulfonic acid), Zn0 (elemental zinc), ZnCl2 (zinc chloride).
Condition abbreviations and acronyms: ATM (atmosphere), aq. (aqueous), conc.
(concentrated), d (days), fum. (fuming), g (grams), h (hours), M (molar), mg
(milligrams), mL (milliliters), µL (microliters), mM (millimolar), min (minutes), psi
(pressure per square inch), RT (room temperature), wt. (weight).
Biology
Mouse Microsomal Metabolic Stability Procedure. Balb-C mouse liver
microsomes were purchased from Invitrogen. MgCl2 and NADPH were obtained from
Sigma. Solvents were of HPLC grade or better. The HPLC-MS/MS system consisted of a
ThermoElectron Finnigan TSQ Quantum Ultra AM.
Microsomal incubations were done in triplicate. Incubation mixtures contained 2
µL of 20mg/mL microsomes (approximately 0.04 mg microsomal protein), 4 µL of 100
µM DMSO-dissolved substrate (1.0 µM final), in 475 µL potassium phosphate buffer
(0.1 M, pH 7.5, containing 3.3 mM MgCl2.) The incubation mixture was allowed to shake
at 37 °C for 5 min, and a T=0 aliquot was removed. Reaction was initiated by the
addition of 20 µL of NaDPH (22 mM under the same buffer conditions) and the mixture
was allowed to shake at 37 °C until the final aliquot was removed. 30 µL aliquots were
taken after 0 minutes, 5 minutes, and 15 minutes. All aliquots were quenched by dilution
in 90 µL MeCN containing an internal standard and the precipitate was pelleted via
centrifugation. 20 µL of supernatant was injected onto the HPLC-MS/MS. Mobile phase

107
A was 95:5 H2O:MeCN with 0.1% formic acid, and mobile phase B was MeCN with
0.1% formic acid. Column used was a Luna C18(2) 4.6 X 30 mm column with 3µM
particle size. The flow rate was 2 mL/min and a gradient mobile phase composition was
used: isocratic hold for 1 min at 90% A (10% B), 1 min gradient to 10% A (90% B), 1
min gradient back to 90% A (10% B), and 1 min isocratic hold at 90% A (10% B.)
Ionization method consisted of positive electrospray. Source parameters were optimized
for each individual substrate. Substrates and internal standards were followed by selected
reaction monitoring. Substrate/internal standard area ratios were determined and
converted into percent substrate remaining. The natural log of the percent remaining was
plotted against time, the slope of the linear regression was determined, and the equation
T1/2 = – ln(2)/k was used to calculate half-life.
Chemistry
Ethyl 1-(4-chlorobenzyl)-1H-indole-2-carboxylate (3a). Ethyl 1H-indole-2-
carboxylate 1 (4.50 g, 23.78 mmol) was dissolved in anhydrous DMF (60 mL) under N2
at RT, then cooled to -10 °C. Granular t-BuOK (2.94 g, 26.20 mmol) was added to the
solution, eliciting a slight yellow color. 4-Chlorobenzyl chloride 2a (4.02 g, 24.97 mmol)
– pre-dissolved in anhydrous DMF (5 mL) – was added via syringe pump at 0.1 mL/min
at -10 °C. After this time, was allowed to stir at RT for 12 h. The reaction was then
diluted with 1:1 EtOAc:Hex and quenched by the addition of sat. aq. NaHCO3. The
organic phase was then washed with H2O (10 X) and brine (1X), dried (MgSO4), and
concentrated to give the pure compound as a white solid that was subsequently washed
thoroughly with hexanes. Yield: 6.80 g, 21.67 mmol, 91%. 1H NMR (500 MHz,

108
Chloroform-d) δ 7.78 (d, J = 8.0 Hz, 1H), 7.48 (d, J = 1.6 Hz, 1H), 7.38 – 7.34 (m, 2H),
7.25 (ddd, J = 15.1, 8.4, 2.8 Hz, 3H), 7.07 – 7.01 (m, 2H), 5.83 (s, 2H), 4.39 (qd, J = 7.1,
1.6 Hz, 2H), 1.42 (td, J = 7.1, 1.6 Hz, 3H).
Ethyl 1-(4-fluorobenzyl)-1H-indole-2-carboxylate (3b). Ethyl 1H-indole-2-
carboxylate 1 (500 mg, 2.64 mmol) was dissolved in anhydrous DMF (8 ml), followed by
1-(chloromethyl)-4-fluorobenzene 2b (0.380 ml, 3.17 mmol) and the addition of solid
K2CO3 (730 mg, 5.29 mmol). This was stirred at 60 °C for 20 h, at which time the
reaction was allowed to cool to room temperature. Precipitate formed upon the addition
of water (40 mL), was isolated over a filter, and dried under high vacuum overnight to
give 550 mg of the title compound as a white solid which was recrystallized from Et2O.
Yield: 550 mg, 1.850 mmol, 70%. 1H NMR (500 MHz, Chloroform-d) δ 7.72 (d, J = 7.9
Hz, 1H), 7.40 (s, 1H), 7.37 – 7.31 (m, 2H), 7.18 (ddd, J = 7.7, 6.5, 1.3 Hz, 1H), 7.05 (dd,
J = 8.4, 5.3 Hz, 2H), 6.94 (t, J = 8.5 Hz, 2H), 5.81 (s, 2H), 4.34 (q, J = 7.1 Hz, 2H), 1.38
(t, J = 7.1 Hz, 3H).
1-(4-Chlorobenzyl)-1H-indole-2-carboxylic acid (4a). Ethyl 1-(4-chlorobenzyl)-
1H-indole-2-carboxylate 3a (5.95 g, 18.96 mmol) was added to a mixture of 10% aq.
NaOH (13.7 mL, 37.9 mmol), EtOH (15 mL), and THF (15 mL), but did not completely
dissolve. This suspension stirred at RT for 14 h, over which time all material dissolved
and the reaction proceeded to completion. Solvent was stripped off in vacuo, the residue
was taken up in H2O and acidified to pH<1 with conc. HCl at 0 °C. The resulting
precipitate was collected over a filter and washed with cold 1 M HCl, air-dried, then

109
dried thoroughly under high vacuum to give a white powder. No further purification
necessary. Yield: 5.20 g, 18.20 mmol, 96%. 1H NMR (500 MHz, Chloroform-d) δ 7.76
(d, J = 8.1 Hz, 1H), 7.56 (s, 1H), 7.40 – 7.31 (m, 2H), 7.27 – 7.16 (m, 3H), 7.00 (d, J =
8.2 Hz, 2H), 5.81 (s, 2H).
1-(4-Fluorobenzyl)-1H-indole-2-carboxylic acid (4b). Ethyl 1-(4-fluorobenzyl)-
1H-indole-2-carboxylate 3b (409 mg, 1.376 mmol) was suspended in ethanol (5 mL) and
7M NaOH (5 mL) and stirred at 50 °C for 3 h. Ethanol content was reduced in vacuo.
The solution was then acidified to pH 2 and the precipitate was collected over a filter,
washed with a small amount of ice cold-water, air dried on the filter, and then finally
vacuum dried to give the title compound as a fine, white powder. Yield: 360 mg, 1.337
mmol, 97%. 1H NMR (500 MHz, Chloroform-d) δ 8.41 (d, J = 5.8 Hz, 1H), 7.81 – 7.71
(m, 4H), 7.70 – 7.60 (m, 3H), 7.18 (d, J = 5.7 Hz, 1H), 5.90 (d, J = 5.5 Hz, 2H).
Tert-Butyl 4-(hydroxymethyl)piperidine-1-carboxylate (7). Piperidine-4-methanol
6 (3.74 g, 32.5 mmol) and Boc2O (8.5 g, 38.9 mmol) were added to THF (50 mL) at RT,
followed by Na2CO3 (6.88 g, 64.9 mmol) and H2O (30 mL). Addition was mildly
exothermic. The reaction was refluxed at 100 °C for 2 h, after which it was allowed to
cool to RT. The reaction was then extracted with EtOAc, and the extract was washed
with brine (1X), dried (MgSO4), and concentrated. The residue was purified by silica
flash chromatography (150 g silica, 50% EtOAc:Hex). Concentration provided a white
solid. Yield: 6.09 g, 28.3 mmol, 87%. 1H NMR (500 MHz, Chloroform-d) δ 4.10 (s, 2H),

110
3.47 (t, J = 5.2 Hz, 1H), 2.69 (s, 2H), 1.70 (d, J = 13.2 Hz, 2H), 1.62 (ddp, J = 15.2, 6.7,
4.0 Hz, 1H), 1.44 (s, 9H), 1.12 (qd, J = 12.5, 4.4 Hz, 2H).
Tert-Butyl 4-formylpiperidine-1-carboxylate (8). Tert-butyl 4-
(hydroxymethyl)piperidine-1-carboxylate 7 (1.50 g, 6.97 mmol) and anhydrous DMSO
(1.24 mL, 17.42 mmol) were dissolved in DCM (100 mL) at RT, then cooled to -78 °C.
(COCl)2 (1.22 mL, 13.93 mmol) was then added dropwise under N2 and the reaction was
allowed to stir for 30 min at -78 °C. Still at this temperature, TEA (4.86 mL, 34.8 mmol)
was added dropwise and a slight yellow color change was observed. The reaction was
allowed to stir for 30 min at -78 °C, then 3 h at 0 °C. After this time, the reaction was
quenched by the addition of 0.1 M HCl and material was extracted with EtOAc. The
extract was washed with 0.1 M HCl (2X), H2O (3X), and brine (1X), then dried (MgSO4)
and concentrated in vacuo. This was immediately purified via silica flash
chromatography (100 g silica, 40% EtOAc:Hex) to provide the desired product as a white
crystal after removal of solvent. Yield: 1.32 g, 6.19 mmol, 89%. 1H NMR (400 MHz,
Chloroform-d) δ 9.58 (s, 1H), 3.99 – 3.82 (m, 2H), 2.85 (t, J = 11.9 Hz, 2H), 2.35 (dt, J =
10.5, 6.0 Hz, 1H), 1.82 (d, J = 12.4 Hz, 2H), 1.52 – 1.44 (m, 2H), 1.37 (s, 9H).
Tert-Butyl 4-(piperidin-1-ylmethyl)piperidine-1-carboxylate (9). Piperidine (1.2
mL, 11.73 mmol) was dissolved in TFE (10 mL) over 3 Å MS, then a drop of glacial
AcOH (~ 50 µL) was added. Tert-butyl 4-formylpiperidine-1-carboxylate 8 (500 mg,
2.35 mmol) was added slowly with stirring at RT. NaCNBH3 (192 mg, 4.69 mmol) was
added and the reaction was stirred at RT for 19 h. After this time, the reaction was diluted

111
with EtOAc and washed with 10% aq. Na2CO3 (3X), dried (MgSO4), concentrated under
high vacuum. The residue was then purified by silica flash chromatography (60 g silica,
70% to 100% EtOAc:Hex). Yield: 477 mg, 1.69 mmol, 72%. 1H NMR (500 MHz,
Chloroform-d) δ 4.03 (bs, 2H), 2.64 (bs, 2H), 2.28 (bs, 3H), 2.08 (d, J = 6.4 Hz, 2H),
1.71 – 1.45 (m, 10H), 1.42 (s, 9H), 1.03 (m, 2H).
Tert-Butyl 4-(cyclohexyl(hydroxy)methyl)piperidine-1-carboxylate (10). Tert-
butyl 4-formylpiperidine-1-carboxylate 8 (250 mg, 1.172 mmol) was dissolved in
anhydrous Et2O (20 mL) and a 2 M solution of cyclohexylmagnesium chloride (0.62 mL,
1.23 mmol) in Et2O was added dropwise to this under N2. The reaction was allowed to
stir at RT for 2 h, after which time the reaction was quenched by the addition of EtOH
then sat. aq. NaHCO3. Material was extracted with EtOAc, and the extract was dried
(MgSO4) and concentrated to give the desired alcohol as a solid, which was used directly
in the subsequent reaction without purification. Yield: 296 mg, 0.20 mmol, 85%.
Tert-Butyl 4-(dibenzylamino)piperidine-1-carboxylate (13). Tert-butyl 4-
aminopiperidine-1-carboxylate 12 (200 mg, 1.00 mmol) was dissolved in DCM (3 mL),
followed by the addition of H2O (1.5 mL) and granular Na2CO3 (318 mg, 3.00 mmol),
and finally benzyl bromide (0.30 mL, 2.50 mmol). The reaction was stirred at reflux (55
°C) for 3 h, after which H2O (10 mL) was added and the material was extracted with
EtOAc. The extract was dried (MgSO4) and concentrated, and the resulting residue was
purified by silica flash chromatography (20 g silica, 5% EtOAc:Hex) to give the desired
amine as a white solid. Yield: 330 mg, 0.87 mmol, 87%. 1H NMR (500 MHz,

112
Chloroform-d) δ 7.40 (d, J = 7.2 Hz, 4H), 7.33 (t, J = 8.2, Hz, 4H), 7.29 – 7.23 (m, 2H),
4.18 (s, 2H), 3.67 (s, 4H), 2.69 (tt, J = 11.8, 3.5 Hz, 1H), 2.59 (bs, 2H), 1.84 (d, J = 12.4
Hz, 2H), 1.63 – 1.52 (m, 2H), 1.52 – 1.46 (m, 9H).
4-(3-Bromopropyl)pyridine (16). 3-(Pyridin-4-yl)propan-1-ol (500 mg, 3.64
mmol) was dissolved in conc. HBr (1 mL) in a pressure vessel stirred at 130 °C for 2h,
after which the reaction was allowed to cool. The reaction was then basified to pH>14
with 10% aq. NaOH at 0 °C and extracted with EtOAc (3X). The combined extracts were
dried (MgSO4) and concentrated. The residue was then purified on a silica plug (20 g
silica, 100% EtOAc) to give a crude oil that was taken into the next reaction without
further purification. Yield: 292 mg, 1.46 mmol, 40%. ). TOF ES+ MS: (M + H, Br79
)
200.0, (M + H, Br81
) 202.0.
Tert-Butyl 4-(3-(pyridin-4-yl)propyl)piperazine-1-carboxylate (17). t-Butyl
piperazine-1-carboxylate (186 mg, 1.00 mmol) was dissolved in anhydrous DMF (10
mL) under N2, and a 60% oil suspension of NaH (120 mg, 3.00 mmol) was added. After
stirring at RT under N2 for 1 h, 4-(3-bromopropyl)pyridine (200 mg, 1.00 mmol) was
added and the reaction was allowed to stir for 19 h. After this time, the reaction was
quenched with sat. aq. NaHCO3 then diluted with EtOAc and washed with H2O (3X),
10% aq. Na2CO3 (3X), and brine (1X), then dried (MgSO4) and concentrated. The residue
was purified by silica flash chromatography (25 g silica, 1% to 10% methanolic
ammonia:EtOAc). Yield: 201 mg, 0.660 mmol, 66%. 1H NMR (500 MHz, Chloroform-d)

113
δ 8.49 (d, J = 5.9 Hz, 2H), 7.11 (d, J = 5.8 Hz, 2H), 3.45 – 3.39 (m, 4H), 2.65 (t, J = 7.7
Hz, 2H), 2.42 – 2.31 (m, 6H), 1.83 (p, J = 7.6 Hz, 2H), 1.46 (s, 9H).
(1-(4-Fluorobenzyl)-1H-indol-2-yl)(4-(hydroxymethyl)piperidin-1-yl)methanone
(CCG-206462). The following was added sequentially to DMF (10 mL): 1-(4-
fluorobenzyl)-1H-indole-2-carboxylic acid (1.175 g, 4.36 mmol), DIPEA (2.29 mL,
13.09 mmol), EDCHCl (1.00 g, 5.24 mmol), and HOBT (802 mg, 5.24 mmol). This
solution was allowed to stir at RT for 1.5 h, at which time 4-piperidinemethanol (754 mg,
6.55 mmol) was added. Stirring continued for 22 h at RT, at which time the solution was
partitioned between H2O and 1:1 solution of EtOAc:Et2O. The organic extract was then
washed with saturated aq. Na2CO3, dried with MgSO4, and concentrated in vacuo.
Purification was accomplished via silica flash chromatography (100 g silica, 80%
EtOAc/Hexanes.) Resulting residue was crystallized via Et2O:Hex trituration to give the
title compound as a white solid. Yield: 1.51 g, 4.11 mmol, 94%. 1H NMR (400 MHz,
Chloroform-d) δ 7.63 (d, J = 7.9 Hz, 1H), 7.37 (d, J = 8.3 Hz, 1H), 7.29 – 7.22 (m, 1H),
7.14 (t, J = 7.5 Hz, 1H), 7.10 – 7.04 (m, 2H), 6.91 (t, J = 8.2 Hz, 2H), 6.60 (s, 1H), 5.46
(s, 2H), 4.64 (bs, 1H), 4.14 (bs, 1H), 3.42 (t, J = 5.1 Hz, 2H), 2.76 (s, 2H), 1.44 (t, J = 5.0
Hz, 1H), 1.05 (bs, 1H), 0.71 (bs, 1H).
1-(1-(4-Fluorobenzyl)-1H-indole-2-carbonyl)piperidine-4-carbaldehyde (20). (1-
(4-fluorobenzyl)-1H-indol-2-yl)(4-(hydroxymethyl)piperidin-1-yl)methanone (762 mg,
2.08 mmol) was dissolved in DCM (80 mL) in dry glassware under N2. The solution was
chilled to -78 °C and anhydrous DMSO (0.37 mL, 5.20 mmol) and (COCl)2 (0.36 mL,

114
4.16 mmol) were added. The reaction was allowed to stir for 30 min, after which TEA
(1.45 mL, 10.40 mmol) was added dropwise and stirring continued for 1 h, after which
the temperature was elevated to 0 °C and stirring continued for 30 min. At this time, the
solution was taken up in EtOAc while still cold and immediately washed with 0.1 M HCl
(2X), dried (MgSO4), and concentrated to give a white solid. Yield: 527 mg, 1.46 mmol,
70%. 1H NMR (500 MHz, Chloroform-d) δ 9.61 (s, 1H), 7.66 (d, J = 7.9 Hz, 1H), 7.40
(d, J = 8.2 Hz, 1H), 7.32 – 7.26 (m, 1H), 7.19 – 7.14 (m, 1H), 7.12 – 7.06 (m, 2H), 6.93
(t, J = 8.6 Hz, 2H), 6.63 (s, 1H), 5.48 (s, 2H), 4.55 (bs, 1H), 4.07 (s, 1H), 2.50 – 2.41 (m,
1H), 2.16 – 1.93 (m, 2H), 1.84 – 1.47 (m, 4H).
Methyl 1-(4-chlorobenzyl)-1H-pyrrole-2-carboxylate (23a). Methyl 1H-pyrrole-2-
carboxylate (2.77 g, 22.14 mmol) was dissolved in anhydrous DMF (15 mL). Granular
K2CO3 (1.94 g, 26.6 mmol) was added, followed by the addition of 4-chlorobenzyl
chloride (1.80 mL, 26.6 mmol). The reaction was permitted to stir at 60 °C for 24 h, then
again 90 °C for 48 h. Cs2CO3 (21.6 g, 66.4 mmol) was then added, and the reaction was
allowed to stir at 90 °C for 24 h, then NaI (332 mg, 2.214 mmol) was added and stirring
continued at 90 °C for 24 h. At this time, the reaction was allowed to cool to RT, diluted
with EtOAc, and washed with brine (3×). The organic phase was then dried (MgSO4),
concentrated in vacuo, and purified by silica flash chromatography (150 g silica, 5%
EtOAc:Hex) to provide methyl 1-(4-chlorobenzyl)-1H-pyrrole-2-carboxylate as colorless
crystals. Yield: 4.91 g (89%). 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J = 4.7 Hz, 1H),
7.26 (d, J = 3.5 Hz, 1H), 7.05–6.98 (m, 3H), 6.92–6.86 (m, 1H), 6.20 (dd, J = 3.9, 2.6 Hz,
1H), 5.52 (s, 2H), 3.76 (s, 3H).

115
Ethyl 1-(4-chlorobenzyl)-1H-imidazole-2-carboxylate (23b). Ethyl 1H-imidazole-
2-carboxylate (4 g, 28.5 mmol), 4-chlorobenzyl chloride (4.38 mL, 34.3 mmol), and
Na2CO3 (3.63 g, 34.3 mmol), was dissolved in anhydrous DMF (8 mL). The solution was
stirred at RT for 24 h, at which time H2O was added and material was extracted with
EtOAc. The organic phase was collected, dried over MgSO4, and decanted. Purification
accomplished via silica flash chromatography (150 g silica, 10% EtOAc/Hexanes to 80%
EtOAc/Hex.) The title compound was obtained as a clear, yellow-tinted oil. Yield: 7.28 g,
27.5 mmol, 96%. 1H NMR (400 MHz, CDCl3) δ 7.29 (d, J = 8.4 Hz, 2H), 7.18 (d, J = 0.9
Hz, 1H), 7.09 (d, J = 8.4 Hz, 2H), 7.06 (d, J = 0.9 Hz, 1H), 5.59 (s, 2H), 4.37 (q, J = 7.2
Hz, 2H), 1.39 (t, J = 7.1 Hz, 3H).
Ethyl 1-(1-(4-chlorobenzyl)-1H-pyrrole-2-carbonyl)piperidine-4-carboxylate
(24a). To methyl 1-(4-chlorobenzyl)-1H-pyrrole-2-carboxylate (4.5 g, 18.02 mmol) was
added to EtOH (50 mL) and 10% aq. NaOH (20 mL) and stirred overnight at RT, then
again overnight at 50 °C to give completion. The EtOH was stripped by rotary
evaporation until material began to precipitate, at which point the aqueous mixture was
cooled in an ice bath and acidified with conc. HCl, which elicited further precipitation.
The precipitate was collected over a filter and washed with a small amount of cold 1 M
HCl and Hex. The material was then dried under high vacuum to provide the carboxylic
acid as a white powder. Yield: 4.20 g, (99%). 1H NMR (400 MHz, DMSO-d6) δ 7.34
(d, J = 8.2 Hz, 2H), 7.19 (s, 1H), 7.06 (d, J = 8.2 Hz, 2H), 6.82 (s, 1H), 6.16–6.08 (m,
1H), 5.53 (s, 2H).

116
The following were added sequentially to anhydrous DMF (20 mL), 1-(4-
chlorobenzyl)-1H-pyrrole-2-carboxylic acid (2.0 g, 8.5 mmol), DIEA (4.45 mL, 25.5
mmol), EDCI (1.79 g, 9.34 mmol), and HOBt (1.43 g, 9.34 mmol), and this solution was
allowed to stir at RT for 30 min. Finally, ethyl isonipecotate (1.44 mL, 9.34 mmol) was
added and the reaction was permitted to stir at RT for 24 h. At this time, DCM was
removed in vacuo and the residue was dissolved in ethyl acetate, which was subsequently
washed with 1 M HCl (3X), H2O (2X), 10% aq. Na2CO3 (2X), and at last brine (1X). The
organic layer was then dried (MgSO4) and concentrated in vacuo. The residue was
crystallized from EtOAc:Hex via the slow evaporation of solvent at RT to give ethyl 1-
(1-(4-chlorobenzyl)-1H-pyrrole-2-carbonyl)piperidine-4-carboxylate as large translucent
crystals. Yield: 2.16 g (68%). 1HNMR (400 MHz, CDCl3) δ 7.24 (d, J = 8.4 Hz, 2H),
7.03 (d, J = 8.4 Hz, 2H), 6.84−6.74 (m, 1H), 6.31 (dd, J = 3.7, 1.5 Hz, 1H), 6.18−6.06 (m,
1H), 5.27 (s, 2H), 4.21 (d, J = 11.5 Hz, 2H), 4.14 (q, J = 7.1 Hz, 2H), 2.94 (t, J = 11.5 Hz,
2H), 2.46 (tt, J = 10.7, 4.0 Hz, 1H), 1.79 (d, J = 12.7 Hz, 2H), 1.56−1.30 (m, 2H), 1.25 (t,
J = 7.1 Hz, 3H).
1-(1-(4-Fluorobenzyl)-1H-pyrrole-2-carbonyl)piperidine-4-carboxylic acid (25a).
Ethyl 1-(1-(4-chlorobenzyl)-1H-pyrrole-2-carbonyl)piperidine-4-carboxylate (700 mg,
1.867 mmol) was dissolved in EtOH (60 mL) and H2O (20 mL). Granular LiOH (134 mg,
5.60 mmol) was added to this, and the reaction was allowed to stir at RT for 24 h. After
this time, most of the solvent was removed in vacuo, and the remaining aqueous solution
was chilled to 0 °C and acidified by concentrated HCl, which elicited a thick, sticky
precipitate that adhered to the flask. The aqueous solution was decanted, and a small

117
amount of 1 N HCl was added, and the material sonicated. The aqueous solution was
again decanted, and a small amount of ice-cold water was added, mixed, and decanted.
Remaining water and HCl was then removed by rotary evaporation, followed by high-
vacuum drying to afford the title compound as a white powder. Yield: 620 mg (96%).
1HNMR (400 MHz, DMSO-d6) δ 7.33 (dd, J = 9.0, 2.7 Hz, 2H), 7.14−6.97 (m, 2H), 6.27
(dd, J = 3.6, 1.5 Hz, 1H), 6.10−5.94 (m, 1H), 5.25 (s, 2H), 3.98 (d, J = 11.0 Hz, 2H),
3.03−2.80 (m, 2H), 2.35−2.19 (m, 1H), 1.64 (d, J = 11.1 Hz, 2H), 1.24 (d, J = 11.5 Hz,
2H).
Methyl 1-(1-(4-chlorobenzyl)-1H-pyrrole-2-carbonyl)azetidine-3-carboxylate
(29). 1-(4-chlorobenzyl)-1H-pyrrole-2-carboxylic acid (500 mg, 2.12 mmol) was added
to DCM (20 mL), followed sequentially by DIPEA (1.1 mL, 6.36 mmol), EDCHCl (447
mg, 2.33 mmol), HOBT (357 mg, 2.33 mmol), and methyl azetidine-3-carboxylate
hydrochloride (387 mg, 2.55 mmol). The reaction was allowed to stir at rt for 18 h, at
which time the DCM was removed in vacuo and the residue was taken up in 2:1
EtOAc:Et2O and washed with 1 M aq. HCl (5X), H2O (5X), 10% aq. Na2CO3 (5X), and
brine (1X), then dried (MgSO4) and concentrated. The resulting residue was purified by
silica flash chromatography (25 g silica, 5% to 30% EtOAc/Hex) to give the desired
compound as a clear, slightly yellow oil. Yield: 508 mg, 1.53 mmol, 72%. 1H NMR (500
MHz, Chloroform-d) δ 7.25 (d, J = 8.3 Hz, 2H), 7.04 (d, J = 8.2 Hz, 2H), 6.81 (s, 1H),
6.58 – 6.44 (m, 1H), 6.20 – 6.13 (m, 1H), 5.52 (s, 2H), 4.36 (bs, 4H), 3.75 (s, 3H), 3.43
(p, J = 7.5 Hz, 1H).

118
Ethyl 1-((4-chlorobenzyl)carbamoyl)piperidine-4-carboxylate (31a). (4-
chlorobenzyl)isocyanate (0.30 mL, 2.26 mmol) was dissolved in DCM (10 mL) at RT,
then ethyl isonipecotate 26 (0.38 mL, 2.49 mmol) was added dropwise with stirring,
resulting in precipitation and discoloration. This slurry was allowed to stir at RT for 1 h,
at which time the precipitate was collected over a filter and dried via aspirator to afford
an off-white granular solid. No further purification was necessary. Yield: 690 mg, 2.13
mmol, 94%. 1H NMR (500 MHz, Chloroform-d) δ 7.30 – 7.25 (m, 2H), 7.25 – 7.20 (m,
2H), 4.84 (s, 1H), 4.37 (d, J = 5.6 Hz, 2H), 4.14 (q, J = 7.1 Hz, 2H), 3.87 (dt, J = 13.3,
3.7 Hz, 2H), 2.96 – 2.87 (m, 2H), 2.46 (ddd, J = 14.6, 10.7, 3.8 Hz, 1H), 1.91 (dd, J =
13.4, 3.1 Hz, 2H), 1.71 – 1.62 (m, 2H), 1.25 (t, J = 7.2 Hz, 3H).
Ethyl 1-((4-chlorophenethyl)carbamoyl)piperidine-4-carboxylate (31b). 1-
Chloro-4-(2-isocyanatoethyl)benzene 19 (0.45 mL, 2.94 mmol) was dissolved in DCM
(10 mL) at RT. Ethyl isonipecotate 26 was then added dropwise, which elicited
precipitation. The slurry was allowed to stir at RT for 1 h, at which time the precipitate
was collected and dried over a filter to afford ethyl 1-((4-chlorophenethyl)-
carbamoyl)piperidine-4-carboxylate as an off-white granular solid. This material was
taken into the next reaction in this crude form. Yield: 916 mg (92%). 1H NMR (500
MHz, Chloroform-d) δ 7.26 (d, J = 8.2 Hz, 2H), 7.11 (d, J = 8.3 Hz, 2H), 4.52 (t, J = 5.3
Hz, 1H), 4.13 (q, J = 7.1 Hz, 2H), 3.79 (dt, J = 13.4, 3.8 Hz, 2H), 3.44 (q, J = 6.7 Hz,
2H), 2.90 – 2.80 (m, 2H), 2.78 (t, J = 6.9 Hz, 2H), 2.44 (tt, J = 10.8, 3.9 Hz, 1H), 1.92 –
1.83 (m, 2H), 1.62 (qd, J = 11.2, 4.1 Hz, 2H), 1.25 (t, J = 7.1 Hz, 3H).

119
Ethyl 1-(2-(4-chlorophenyl)acetyl)piperidine-4-carboxylate (33a). 4-
chlorophenylacetic acid (900 mg, 5.28 mmol) was dissolved in DCM (20 mL), followed
by the sequential addition of TEA (2.21 mL, 15.83 mmol), EDCHCl (1110 mg, 5.80
mmol), HOBT (889 mg, 5.80 mmol), and ethyl isonipecotate (0.894 mL, 5.80 mmol).
The reaction was stirred at RT for 28 h, after which time the DCM was evaporated off.
The residue was taken up in EtOAc and washed with 1M HCl (3X), H2O (5X), 10% aq.
Na2CO3 (3X), and brine (1X), then dried (MgSO4) and concentrated. The residue was
then purified through a plug of silica (30 g silica, 5% EtOAc:Hex). Yield: 1.31 g, 4.22
mmol, 80%. 1H NMR (500 MHz, Chloroform-d) δ 7.29 (d, J = 8.3 Hz, 2H), 7.17 (d, J =
8.2 Hz, 2H), 4.40 (dt, J = 12.5, 4.0 Hz, 1H), 4.13 (q, J = 7.1 Hz, 2H), 3.79 (dt, J = 13.5,
3.7 Hz, 1H), 3.69 (s, 2H), 3.12 – 3.05 (m, 1H), 2.89 – 2.82 (m, 1H), 2.52 – 2.45 (m, 1H),
1.96 – 1.87 (m, 2H), 1.83 – 1.77 (m, 1H), 1.61 – 1.57 (m, 1H), 1.49 (ddd, J = 24.1, 10.9,
3.8 Hz, 1H), 1.25 (t, J = 7.1 Hz, 3H).
Ethyl 1-(3-(4-chlorophenyl)propanoyl)piperidine-4-carboxylate (33b). 3-(4-
chlorophenyl)propionic acid (0.90 g, 4.87 mmol) was dissolved in DCM (20 mL),
followed by TEA (2.04 mL, 14.62 mmol), EDCHCl (1.03 g, 5.36 mmol), HOBT (0.82 g,
5.36 mmol), and ethyl isonipecotate (0.83 mL, 5.36 mmol). The solution was stirred at
RT for 28 h, after which time the DCM was removed in vacuo and the residue was
dissolved in 2:1 EtOAc:Et2O and washed with 1 M aq. HCl (3X), H2O (2X), 10% aq.
Na2SO3 (3X), and brine (1X), then dried (MgSO4) and concentrated. The residue was
then purified by silica flash chromatography (100 g silica, 5% to 50% EtOAc:Hex). 1H
NMR (500 MHz, Chloroform-d) δ 7.20 (d, J = 8.2 Hz, 2H), 7.11 (d, J = 8.2 Hz, 2H), 4.41

120
– 4.34 (m, 1H), 4.10 (q, J = 7.1 Hz, 2H), 3.71 (d, J = 13.7 Hz, 1H), 3.04 – 2.96 (m, 1H),
2.89 (t, J = 7.8 Hz, 2H), 2.80 – 2.72 (m, 1H), 2.58 – 2.53 (m, 2H), 2.46 (tt, J = 10.7, 4.0
Hz, 1H), 1.90 – 1.80 (m, 2H), 1.54 (ddd, J = 24.1, 13.3, 3.5 Hz, 2H), 1.22 (t, J = 7.1 Hz,
3H).
1-Tert-Butyl 4-ethyl piperidine-1,4-dicarboxylate (34). Ethyl isonipecotate 26
(3.53 mL, 22.91 mmol), Boc2O (6.0 g, 27.50 mmol), and Na2CO3 (4.86 g, 45.80 mmol)
were added to a mixture of H2O (8 mL) and THF (20 mL) at RT (mild exotherms). This
was subsequently refluxed at 80 °C for 2 h, over which time the reaction proceeded from
a white suspension to a clear biphasic solution. The reaction was allowed to cool and was
extracted with EtOAc (2X) with no dilution of the aqueous phase. The organic extracts
were combined, dried (MgSO4), and concentrated in vacuo. The clear residue was then
purified by silica flash chromatography (150 g, 10% EtOAc:Hex) to give a thin, clear,
colorless liquid. Yield: 5.07 g, 19.70 mmol, 86%. 1H NMR (500 MHz, Chloroform-d) δ
4.12 (q, J = 7.1 Hz, 2H), 3.99 (s, 2H), 2.81 (t, J = 11.4 Hz, 2H), 2.41 (tt, J = 11.0, 3.6 Hz,
1H), 1.85 (d, J = 12.3 Hz, 2H), 1.60 (qd, J = 12.6, 12.1, 4.4 Hz, 2H), 1.43 (s, 9H), 1.23 (t,
J = 6.8 Hz, 3H).
Tert-Butyl 4-((2-(pyridin-4-yl)ethyl)carbamoyl)piperidine-1-carboxylate (35). The
boc-protected ethyl isonipecotate 34 (3.00 g, 11.66 mmol) was dissolved in EtOH (20
mL) and 10% aq. NaOH (10 mL) and stirred at RT for 4 h, after which time the EtOH
was stripped off in vacuo and the solution neutralized with conc. HCl at 0 ºC as quickly
as possible, and the resulting precipitate was collected over a filter and washed with cold

121
H2O, then dried via aspirator, and finally high vacuum to provide the desired carboxylic
acid as a white powder. Yield: 2.43 g, 10.60 mmol, 91%.
The following was added to DCM: 1-(tert-butoxycarbonyl)piperidine-4-
carboxylic acid (600 mg, 2.62 mmol), DIPEA (1.37 mL, 7.85 mmol), EDC (552 mg, 2.88
mL), HOBT (441 mg, 2.88 mmol), and pyridylethylamine (0.34 mL, 2.88 mL). The
solution was stirred at RT for 17 h, at which time the DCM was stripped off and 10% aq.
sodium carbonate was added. Material was extracted out with EtOAc (3X). The organic
extractions were pooled, dried over magnesium sulfate, and concentrated in vacuo. The
residue was taken up in a small amount of EtOAc and diethyl ether was added. The
precipitate was collected over a filter and washed with diethyl ether to give the title
compound as an off-white solid. Yield: 634 mg, 1.9 mmol, 73%. 1H NMR (400 MHz,
Chloroform-d) δ 8.49 (d, J = 5.9 Hz, 2H), 7.09 (d, J = 5.9 Hz, 2H), 5.59 (t, J = 5.3 Hz,
1H), 4.22 – 3.95 (m, 2H), 3.52 (q, J = 6.8 Hz, 2H), 2.81 (t, J = 6.9 Hz, 2H), 2.68 (t, J =
12.5 Hz, 2H), 2.14 (tt, J = 11.6, 3.8 Hz, 1H), 1.77 – 1.65 (m, 2H), 1.56 (qd, J = 12.1, 4.3
Hz, 2H), 1.43 (s, 9H).
N-(2-(Pyridin-4-yl)ethyl)piperidine-4-carboxamide dihydrochloride (36). The
Boc-protected 35 (575 mg, 1.72 mmol) was suspended in Et2O at RT, and 4M HCl in
dioxane (6 mL, 24 mmol) was added. The mixture was stirred at RT for 30 min, at which
time the organic solution was decanted off and the solid material collected and dried in
vacuo. The title compound was thus obtained as a tan powder. Yield: 448 mg, 1.6 mmol,
97%. 1H NMR (400 MHz, DMSO-d6) δ 9.22 (bs, 1H), 8.91 (bs, 1H), 8.79 (d, J = 6.4 Hz,
2H), 8.18 (t, J = 5.6 Hz, 1H), 7.89 (d, J = 6.3 Hz, 2H), 3.40 (q, J = 6.2 Hz, 2H), 3.15 (d, J

122
= 12.6 Hz, 2H), 3.00 (t, J = 6.4 Hz, 2H), 2.83 – 2.69 (m, 2H), 2.39 – 2.26 (m, 1H), 1.80 –
1.59 (m, 4H). TOF ES+ MS: (M + H) 234.1, (M + Na) 256.1.
(R)-1-(4-Chlorobenzyl)pyrrolidine-2-carboxylic acid (R-38). L-proline (500 mg,
4.34 mmol) was dissolved in IPA (20 mL) in a dry flask, followed by the addition of
KOH pellets (730 mg, 13.03 mmol). Reaction was stirred at 40 °C while an IPA (5 mL)
solution of 4-chlorobenzyl chloride (770 mg, 4.78 mmol) was added via syringe pump
over 3 h. Reaction was allowed to stir an additional 6 h, after which time the reaction was
allowed to cool to RT. The solution was then acidified to pH 5 and DCM was added and
the reaction stirred for 16 hours at RT. After this time, the precipitate was filtered off and
washed with DCM (2X), and the combined organic filtrate extracts were concentrated to
provide a light yellow solid. Dried under high vacuum. Yield: 956 mg, 3.77 mmol, 87%.
1H NMR (400 MHz, Chloroform-d) δ 10.24 (s, 1H), 7.89 (d, J = 9.0 Hz, 1H), 7.35 (s,
4H), 6.62 (s, 2H), 4.75 (s, 4H), 1.58 (s, 2H). TOF ES– MS: (M – H) 238.1.
(S)-1-(4-Chlorobenzyl)pyrrolidine-2-carboxylic acid (S-38). D-proline (500 mg,
4.34 mmol) was dissolved in IPA (20 mL) in a dry flask, followed by the addition of
KOH pellets (730 mg, 13.03 mmol). Reaction was stirred at 40 °C while an IPA (5 mL)
solution of 4-chlorobenzyl chloride (770 mg, 4.78 mmol) was added via syringe pump
over 3 h. Reaction was allowed to stir an additional 6 h, after which time the reaction was
allowed to cool to RT. The solution was then acidified to pH 5 and DCM was added and
the reaction stirred for 16 hours at RT. After this time, the precipitate was filtered off and
washed with DCM (2X), and the combined organic filtrate extracts were concentrated to

123
provide a light yellow solid. Taken directly into the subsequent step without further
characterization. Yield: 923 mg, 3.64 mmol, 84%. TOF ES– MS: (M – H) 238.1.
(R)-1-(4-Chlorobenzoyl)pyrrolidine-2-carboxylic acid (R-39). L-proline (800 mg,
6.95 mmol) was dissolved in IPA (30 mL) in a dry flask, followed by the addition of
KOH pellets (1.17 g, 20.85 mmol). Reaction was stirred at RT while an IPA solution of
4-chlorobenzoyl chloride (1.34 g, 7.64 mmol) was added via syringe pump over 1 h.
Reaction was allowed to stir an additional 1 h, after which time the reaction was cooled
to 0°C. The solution was then acidified to pH 5, DCM was added, and the reaction stirred
for 12 hours at RT. After this time, the precipitate was filtered off and washed with DCM
(2X), and the combined organic filtrate extracts were concentrated to provide an off-
white solid without further purification. Taken directly into the subsequent step without
further characterization. Yield: 1.58 g, 6.23 mmol, 90%. TOF ES– MS: (M – H) 252.0.
(S)-1-(4-Chlorobenzoyl)pyrrolidine-2-carboxylic acid (S-39). D-proline (800 mg,
6.95 mmol) was dissolved in IPA (30 mL) in a dry flask, followed by the addition of
KOH pellets (1.17 g, 20.85 mmol). Reaction was stirred at RT while an IPA solution of
4-chlorobenzoyl chloride (1.34 g, 7.64 mmol) was added via syringe pump over 1 h.
Reaction was allowed to stir an additional 1 h, after which time the reaction was cooled
to 0°C. The solution was then acidified to pH 5, DCM was added, and the reaction stirred
for 13 hours at RT. After this time, the precipitate was filtered off and washed with DCM
(2X), and the combined organic filtrate extracts were concentrated to provide an off-

124
white solid without further purification. Taken directly into the subsequent step without
further characterization. Yield: 1.62 g, 6.39 mmol, 92%. TOF ES– MS: (M – H) 252.0.
(R)-Ethyl 1-(1-(4-chlorobenzoyl)pyrrolidine-2-carbonyl)piperidine-4-carboxylate
(R-40). (R)-1-(4-chlorobenzoyl)pyrrolidine-2-carboxylic acid (430 mg, 1.79 mmol) and
TEA (750 µL, 5.38 mmol) were dissolved in DCM (10 mL) followed by HATU (720 mg,
1.88 mmol), ethyl isonipecotate (290 µL, 1.88 mmol), and 3Å MS. The reaction was
allowed to stir at RT for 17 h, at which time the DCM was removed and the residue taken
up in a 1:1 solution of EtOAc:Et2O and washed with 0.5 M aq. HCl (2X), H2O (1X), 10%
aq. Na2CO3 (3X), and brine (1X). The solution was then dried (MgSO4) and
concentrated, and the residue was purified via silica flash chromatography (100 g silica,
50% to 100% EtOAc:Hex) to give the desired product as a clear oil. Yield: 420 mg, 1.11
mmol, 62%. 1H NMR (500 MHz, Chloroform-d) δ 7.52 (d, J = 8.1 Hz, 2H), 7.35 – 7.30
(m, 2H), 5.04 (dt, J = 14.6, 7.4 Hz, 1H), 4.50 – 4.44 (m, 1H), 4.40 (m, 0.5H), 4.27 (dd, J
= 9.6, 3.9 Hz, 0.5H), 4.17 – 4.09 (m, 2H), 4.04 (d, J = 13.6 Hz, 0.5H), 3.94 (d, J = 13.7
Hz, 0.5H), 3.80 (tt, J = 11.5, 5.9 Hz, 0.5H), 3.67 (q, J = 7.8 Hz, 1H), 3.50 (ddd, J = 10.5,
7.2, 4.1 Hz, 1H), 3.41 – 3.30 (m, 0.5H), 3.17 (dt, J = 13.8, 6.9 Hz, 0.5H), 3.05 – 2.95 (m,
0.5H), 2.90 – 2.76 (m, 0.5H), 2.62 – 2.48 (m, 1H), 2.22 (ddt, J = 22.4, 15.3, 7.9 Hz, 1H),
2.06 – 1.81 (m, 7H), 1.64 (ddd, J = 27.7, 15.2, 8.6 Hz, 2H), 1.27 – 1.21 (m, 3H).
(S)-Ethyl 1-(1-(4-chlorobenzoyl)pyrrolidine-2-carbonyl)piperidine-4-carboxylate
(S-40). (S)-1-(4-chlorobenzoyl)pyrrolidine-2-carboxylic acid (490 mg, 2.05 mmol) and
TEA (860 µL, 6.15 mmol) were dissolved in DCM (10 mL) followed by HATU (820 mg,

125
2.15 mmol), ethyl isonipecotate (330 µL, 2.15 mmol), and 3Å MS. The reaction was
allowed to stir at RT for 16 h, at which time the DCM was removed and the residue taken
up in a 1:1 solution of EtOAc:Et2O and washed with 0.5 M aq. HCl (2X), H2O (1X), 10%
aq. Na2CO3 (3X), and brine (1X). The solution was then dried (MgSO4) and
concentrated, and the residue was purified via silica flash chromatography (100 g silica,
50% to 100% EtOAc:Hex) to give the desired product as a clear oil. Yield: 340 mg, 0.91
mmol, 44%. 1H NMR (500 MHz, Chloroform-d) δ 7.52 (d, J = 8.1 Hz, 2H), 7.33 (m, 2H),
5.04 (dt, J = 14.6, 7.4 Hz, 1H), 4.49 – 4.40 (m, 1H), 4.27 (d, J = 9.6 Hz, 0.5H), 4.13 (dp,
J = 13.9, 7.4 Hz, 2H), 4.04 (d, J = 13.6 Hz, 0.5H), 3.94 (d, J = 13.7 Hz, 0.5H), 3.80 (tt, J
= 11.5, 5.9 Hz, 0.5H), 3.67 (q, J = 7.8 Hz, 1H), 3.50 (ddd, J = 10.5, 7.2, 4.1 Hz, 1H), 3.39
– 3.29 (m, 0.5H), 3.17 (dt, J = 13.8, 6.9 Hz, 0.5H), 3.05 – 2.94 (m, 0.5H), 2.88 – 2.80 (m,
0.5H), 2.61 – 2.48 (m, 1H), 2.22 (ddt, J = 22.4, 15.3, 7.9 Hz, 1H), 2.10 – 1.80 (m, 4H),
1.64 (m, 2H), 1.30 – 1.19 (m, 3H).
Methyl 2-(4-chlorophenoxy)benzoate (42). Methyl salicylate (0.40 mL, 3.86
mmol), pyridine (0.75 mL, 9.26 mmol), (4-chlorophenyl)boronic acid (531 mg, 3.40
mmol), and Cu(OAc)2 (617 mg, 3.40 mmol) were added to MeOH (10 mL) with 4Å MS
and allowed to stir at RT for 5 d. After this time, the reaction mixture was filtered over
celite and the filtrate was concentrated. The resulting residue was taken up in EtOAc and
washed with 5% aq. NaOH (3X) to remove residual copper, then H2O (3X), 1 M aq. HCl
(3X), and brine (1X), then the organic phase as dried (MgSO4) and concentrated, and the
resulting residue was purified via silica flash chromatography (1% to 10% EtOAc:Hex)
to provide the desired ether as colorless clear oil. Yield: 233 mg, 0.89 mmol, 23%. 1H

126
NMR (500 MHz, Chloroform-d) δ 7.90 (dd, J = 7.8, 1.6 Hz, 1H), 7.46 (ddd, J = 9.0, 7.9,
1.7 Hz, 1H), 7.29 – 7.24 (m, 2H), 7.23 – 7.17 (m, 1H), 6.96 (d, J = 8.3 Hz, 1H), 6.92 –
6.84 (m, 2H), 3.94 (s, 3H).
Methyl 2-((4-chlorophenyl)amino)benzoate (43a). Methyl salicylate (1.50 mL,
11.57 mmol), N,N-Bis(triflyl)aniline (4.76 g, 13.31 mmol), and DIPEA (6.06 mL, 34.70
mmol) were dissolved into anhydrous DMF (20 mL) in dry glassware under N2 at RT.
The solution was stirred for 18 h at RT, at which time the reaction was diluted with 2:1
EtOAc:Et2O and washed with H2O (5X) and brine (1X), then dried (MgSO4) and
concentrated. The residue was purified via silica flash chromatography (80 g silica, 5%
EtOAc:Hex) to give methyl 2-(((trifluoromethyl)sulfonyl)oxy)benzoate as a clear,
colorless oil. Yield: 2.76 g, 9.72 mmol, 84%. 1H NMR (500 MHz, Chloroform-d) δ 8.09
(d, J = 7.8 Hz, 1H), 7.63 (t, J = 7.8 Hz, 1H), 7.48 (t, J = 7.6 Hz, 1H), 7.31 (d, J = 8.3 Hz,
1H), 3.97 (s, 3H).
A Schlenck flask was filled with toluene (60 mL, not anhydrous) and then
charged sequentially with the following reagents: methyl 2-
(((trifluoromethyl)sulfonyl)oxy)benzoate (900 mg, 3.17 mmol), 4-chloroaniline (485 mg,
3.80 mmol), Cs2CO3 (1444 mg, 4.43 mmol), Pd(OAc)2 (36 mg, 0.16 mmol), and DPPP
(104 mg, 0.25 mmol). The mixture was then thoroughly sparged with argon, and the flask
was fitted with a condenser and then purged and filled with an argon atmosphere. The
reaction was stirred at reflux under argon for 6 h, then allowed to cool to RT and stir for
12 h. At this time, the reaction was diluted with EtOAc and washed with 1M aq. HCl
(3X) and brine (1X), dried (MgSO4), and concentrated. The residue was purified via

127
silica flash chromatography (40 g silica, 1% to 5% EtOAc:Hex gradient) to yield the
desired compound as a clear, vaguely yellow oil. Yield: 710 mg, 2.71 mmol, 86%. 1H
NMR (500 MHz, Chloroform-d) δ 9.46 (s, 1H), 7.98 (d, J = 9.0 Hz, 1H), 7.37 – 7.26 (m,
3H), 7.24 – 7.16 (m, 3H), 6.77 (t, J = 7.6 Hz, 1H), 3.92 (d, J = 1.1 Hz, 3H).
Methyl 2-(phenylamino)benzoate (43b). Methyl salicylate (1.50 mL, 11.57
mmol), N,N-Bis(triflyl)aniline (4.76 g, 13.31 mmol), and DIPEA (6.06 mL, 34.70 mmol)
were dissolved into anhydrous DMF (20 mL) in dry glassware under N2 at RT. The
solution was stirred for 18 h at RT, at which time the reaction was diluted with 2:1
EtOAc:Et2O and washed with H2O (5X) and brine (1X), then dried (MgSO4) and
concentrated. The residue was purified via silica flash chromatography (80 g silica, 5%
EtOAc:Hex) to give methyl 2-(((trifluoromethyl)sulfonyl)oxy)benzoate as a clear,
colorless oil. Yield: 2.76 g, 9.72 mmol, 84%. 1H NMR (500 MHz, Chloroform-d) δ 8.09
(d, J = 7.8 Hz, 1H), 7.63 (t, J = 7.8 Hz, 1H), 7.48 (t, J = 7.6 Hz, 1H), 7.31 (d, J = 8.3 Hz,
1H), 3.97 (s, 3H).
A Schlenck flask was filled with toluene (30 mL, not anhydrous) and then
charged sequentially with the following reagents: methyl 2-
(((trifluoromethyl)sulfonyl)oxy)benzoate (440 mg, 1.63 mmol), aniline (178 µL, 1.94
mmol), Cs2CO3 (743 mg, 2.28 mmol), Pd(OAc)2 (18 mg, 0.08 mmol), and DPPP (67 mg,
0.16 mmol). The mixture was then thoroughly sparged with argon, and the flask was
fitted with a condenser and then purged and filled with an argon atmosphere. The
reaction was stirred at reflux under argon for 6 h, then allowed to cool to RT and stir for
12 h. At this time, the reaction was diluted with EtOAc and washed with 1M aq. HCl

128
(3X) and brine (1X), dried (MgSO4), and concentrated. The residue was purified via
silica flash chromatography (40 g silica, 1% to 5% EtOAc:Hex gradient) to yield the
desired compound as a clear, somewhat yellow oil. Yield: 377 mg, 1.66 mmol, 57%. 1H
NMR (500 MHz, Chloroform-d) δ 9.47 (s, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.33 (d, J =
16.2, 7.3 Hz, 3H), 7.26 (d, J = 7.8 Hz, 3H), 7.10 (t, J = 7.3 Hz, 1H), 6.74 (t, J = 7.5 Hz,
1H), 3.91 (s, 3H).
Ethyl 1-(2-((4-chlorophenyl)amino)benzoyl)piperidine-4-carboxylate (44a).
Methyl 2-((4-chlorophenyl)amino)benzoate (650 mg, 2.48 mmol) was dissolved in 200
proof EtOH (15 mL) at RT and 10% aq. NaOH (6 mL) was added. The reaction was
allowed to stir at RT for 5 h, at which time the solvent was stripped off in vacuo and 1M
aq. HCl was added to the residue. The solution was then chilled in an ice bath and
acidified to pH 1, and the resulting white precipitate was collected over a filter, washed
with 1M aq. HCl, and dried via aspirator and high vacuum to yield a white powder. No
further purification necessary. Yield: 581 mg, 2.35 mmol, 94%. 1H NMR (500 MHz,
Chloroform-d) δ 9.27 (s, 1H), 8.06 (dd, J = 8.1, 1.5 Hz, 1H), 7.38 (ddd, J = 8.6, 7.1, 1.6
Hz, 1H), 7.35 – 7.31 (m, 2H), 7.23 – 7.19 (m, 2H), 7.17 (d, J = 8.5 Hz, 1H), 6.79 (t, J =
7.5 Hz, 1H).
2-((4-chlorophenyl)amino)benzoic acid (370 mg, 1.49 mmol) was then dissolved
in DCM (15 mL), followed by ethyl isonipecotate (253 µL, 1.64 mmol), TEA (625 µL,
4.48 mmol), EDCHCl (315 mg, 1.64 mmol), and HOBT (252 mg, 1.64 mmol). The
reaction was stirred at RT for 16 h, at which time the reaction was diluted with 2:1
EtOAc:Et2O and washed with 1 M aq. HCl (2X), 10% aq. Na2CO3 (3X), and brine (1X),

129
dried (MgSO4) and concentrated. The residue was purified via silica flash
chromatography (12 g silica, 5% to 20% EtOAc:Hex) to provide the product as a yellow
oil. Yield: 129 mg, 0.333 mmol, 22%. 1H NMR (500 MHz, Chloroform-d) δ 7.32 (d, J =
8.2 Hz, 1H), 7.25 – 7.15 (m, 4H), 7.02 (d, J = 8.7 Hz, 2H), 6.89 (t, J = 7.4 Hz, 1H), 4.30
(bs, 2H), 4.15 (q, J = 7.1 Hz, 2H), 3.09 (t, J = 11.1 Hz, 2H), 2.56 (tt, J = 10.6, 4.0 Hz,
1H), 1.95 (bs, 2H), 1.75 – 1.67 (m, 2H), 1.25 (t, J = 7.1 Hz, 3H).
Methyl 2-((4-chlorophenyl)(methyl)amino)benzoate (45). Methyl 2-((4-
chlorophenyl)amino)benzoate (150 mg, 0.57 mmol) was dissolved in anhydrous DMF (8
mL) under N2. The solution was then charged with granular K2CO3 (158 mg, 1.15 mmol).
MeI (40 µL, 0.63 mmol), dissolved in 1 mL anhydrous DMF, was added dropwise to the
solution at RT under N2, and the reaction was allowed to stir for 12 h at RT. After this
time, the reaction was diluted in 2:1 EtOAc:Et2O and washed with H2O (3X), 10% aq.
Na2S2O3 (1X), and brine (1X), then dried (MgSO4) and concentrated. The residue was
purified via silica flash chromatography (25 g silica, 1% to 10% EtOAc:Hex) to afford
the desired compound as a colorless oil taken crude into the next reaction. Yield: 139 mg,
0.50 mmol, 88%. TOF ES+ MS: (M + H) 276.1
Methyl 2-((4-chlorobenzyl)amino)benzoate (47a). Methyl 2-aminobenzoate (500
mg, 3.31 mmol) was dissolved in anhydrous DMF (15 mL) at RT under N2. Tert-BuOK
(408 mg, 3.64 mmol) was then added and the solution became yellow, and this was
allowed to stir at RT for 30 min, at which point 4-chlorobenzyl chloride (0.45 mL, 3.47
mmol) was added, and the solution lost color. The reaction was allowed to stir for 18 h

130
under N2, at which time the reaction was diluted in EtOAc and washed with 10% aq.
NaHCO3 (1X), H2O (5X), and brine (1X), then dried (MgSO4) and concentrated. The
residue was purified by silica flash chromatography (40 g silica, 1% to 30% EtOAc:Hex)
to provide the desired compound as a colorless oil. Yield: 1.37 g, 2.88 mmol, 87%. 1H
NMR (500 MHz, Chloroform-d) δ 8.20 (t, J = 5.7 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H), 7.31
(q, J = 7.8, 7.1 Hz, 5H), 6.63 (t, J = 7.6 Hz, 1H), 6.58 (d, J = 8.5 Hz, 1H), 4.43 (d, J = 5.7
Hz, 2H), 3.88 (s, 3H). TOF ES+ MS: (M + H) 276.1.
Methyl 2-(benzylamino)benzoate (47b). To anhydrous DMF in dry glassware was
added methyl 2-aminobenzoate (280 mg, 1.85 mmol), benzyl bromide (330 mg, 1.94
mmol), and Cs2CO3 (1.50 g, 4.62 mmol). The reaction was stirred at 85 °C for 5 days,
after which time the reaction was cooled to RT and diluted with 1:1 EtOAc:Et2O and
washed with H2O (3X) and brine (1X), then dried (MgSO4) and concentrated. The
residue was purified via silica flash chromatography (5% EtOAc:Hex isochratic) to
separate product from unreacted starting material. Yield: 138 mg, 0.57 mmol, 31%. 1H
NMR (500 MHz, Chloroform-d) δ 8.18 (s, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.40 – 7.25 (m,
6H), 6.65 (d, J = 8.5 Hz, 1H), 6.61 (t, J = 7.6 Hz, 1H), 4.46 (d, J = 5.6 Hz, 2H), 3.87 (s,
3H).
2-((4-Chlorophenethyl)amino)benzoic acid (49). 2-Bromobenzoate (200 mg, 1.00
mmol), 2-(4-chlorophenyl)ethanamine (290 mg, 1.86 mmol), K2CO3 (260 mg, 1.86
mmol), copper powder (12 mg, 0.19 mmol), and copper (I) oxide (13 mg, 0.09 mmol)
were added to ethylene glycol dimethyl ether (5 mL) in a dry pressure tube. The mixture

131
was sparged and the headspace purged with argon, and the reaction was stirred at 130 °C
for 24 h. After cooling to RT, the reaction was diluted with EtOAc and H2O and filtered
through celite. The filtrate was acidified with conc. HCl and material was extracted with
EtOAc (3X). The combined extracts were dried (MgSO4) and concentrated. Purification
of the resulting residue was accomplished via silica flash chromatography (25 g silica,
10% EtOAc:Hex with 1% formic acid) to give a white powder. Yield: 120 mg, 0.44
mmol, 44%. 1
H NMR (500 MHz, Chloroform-d) δ 8.06 (d, J = 8.4 Hz, 1H), 8.00 (d, J =
8.0 Hz, 1H), 7.41 (t, J = 7.8 Hz, 1H), 7.32 (d, J = 8.2 Hz, 2H), 7.21 (d, J = 8.2 Hz, 2H),
6.71 (d, J = 8.5 Hz, 1H), 6.64 (t, J = 7.5 Hz, 1H), 3.47 (t, J = 7.1 Hz, 2H), 2.97 (t, J = 7.0
Hz, 2H).
2-(4-Chlorobenzoyl)benzoic acid (51). Aluminum chloride (9.0 g, 67.5 mmol)
was carefully added at RT to chlorobenzene (50 mL) in dry glassware with a water
separator for dense solvents, followed by phthalic anhydride (5.0 g, 33.8 mmol). The
reaction was then refluxed under N2 for 1.5 h, over which time it took on a deep red
color. Note: longer reaction times result in rapid accumulation of a second, internal
acylation. The reaction was then allowed to cool to RT over 1 h and was quenched by the
slow and careful addition of 1 M aq. HCl and allowed to stir another 1 h, over which time
color was lost and a white precipitate formed. The reaction was then taken up in EtOAc
and washed with 1M aq. HCl (1X), then the solvent was removed in vacuo. The residue
was then dissolved in 10% aq. NaOH, the resulting precipitate was filtered off, and the
filtrate was washed with EtOAc (2X) before being chilled in an ice bath and acidified to
pH 1 with conc. HCl. The resulting precipitate was collected over a filter to give the

132
desired product as a white powder. No further purification necessary. Yield: 6.34 g, 24.31
mmol, 72%. 1H NMR (500 MHz, Chloroform-d) δ 8.08 (d, J = 7.8 Hz, 1H), 7.70 – 7.61
(m, 3H), 7.58 (tt, J = 7.7, 1.2 Hz, 1H), 7.36 (dd, J = 12.2, 7.9 Hz, 3H). TOF ES– MS: (M
– H) 259.0.
2-(4-Chlorobenzyl)benzoic acid (52). 2-(4-chlorobenzoyl)benzoic acid 51 (200
mg, 0.77 mmol) was added to 28% aq. ammonia (10 mL), followed by zinc (600 mg,
9.21 mmol) and CuSO45H2O (80 mg, 0.31 mmol). This mixture was stirred at reflux for
48 h, with 2 mL recharges of 28% aq. ammonia every 6 h, barring 12 h at night. After
this time, the reaction was allowed to cool to RT and the solution was decanted off the
solid precipitate and concentrated in vacuo. The residue was taken up in H2O and
acidified to pH < 1 by conc. HCl, then extracted 1X with EtOAc. The extract was dried
(MgSO4) and concentrated, and the residue was crystallized from EtOAc to give tan
crystals that were used immediately in the subsequent reaction. Yield: 62 mg, 0.25 mmol,
33%. TOF ES– MS: (M – H) 245.0.
Methyl 6-chloro-9H-carbazole-1-carboxylate (55). 9H-carbazole-1-carboxylic
acid 54 (200 mg, 0.95 mmol) – prepared in a manner similar to that reported120
– was
stirred in refluxing sat. methanolic HCl for 1 h. The reaction was then allowed to cool
and was concentrated. The residue was dissolved in EtOAc and washed with sat. aq.
Na2CO3 (1X), then dried (MgSO4), and concentrated. The residue was then purified by
silica flash chromatography (10 g silica, 10% EtOAc/Hex) to give a white powder. Yield:
196 mg, 0.87 mmol, 92%. 1H NMR (500 MHz, Chloroform-d) δ 9.94 (s, 1H), 8.26 (d, J =

133
7.6 Hz, 1H), 8.14 – 8.04 (m, 2H), 7.57 – 7.46 (m, 2H), 7.34 – 7.28 (m, 1H), 7.26 (t, J =
7.7 Hz, 1H), 4.03 (s, 3H).
Methyl 9H-carbazole-1-carboxylate (90 mg, 0.40 mmol) was stirred in DCM (15
mL) at 0 °C under N2 while SO2Cl2 was added dropwise. The reaction was allowed to stir
at 0 °C for 4 h. At this time, the reaction was diluted with DCM and washed with 10%
aq. Na2CO3 (2X) and brine (1X), then dried (Na2SO4) and concentrated. The residue was
purified via silica flash chromatography (10 g silica, 10 to 30% EtOAc/Hex) to give a
white powder. Yield: 75 mg, 0.29 mmol, 72%. 1H NMR (500 MHz, Chloroform-d) δ 9.93
(s, 1H), 8.21 (d, J = 7.7 Hz, 1H), 8.10 (d, J = 7.6 Hz, 1H), 8.04 (s, 1H), 7.44 – 7.41 (m,
2H), 7.27 (t, J = 7.7 Hz, 1H), 4.03 (s, 3H). 1H NMR (500 MHz, Benzene-d6) δ 9.70 (s,
1H), 8.03 (d, J = 7.6 Hz, 1H), 7.84 (d, J = 1.9 Hz, 1H), 7.70 (d, J = 7.6 Hz, 1H), 7.17 (dd,
J = 8.6, 2.0 Hz, 1H), 6.92 (t, J = 7.7 Hz, 1H), 6.46 (d, J = 8.6 Hz, 1H), 3.48 (s, 3H).
Ethyl 1-(2-oxo-1,2-dihydroquinolin-4-yl)piperidine-4-carboxylate (57). 4-
chloroquinolin-2(1H)-one was prepared according to literature precedent.124, 125
4-
chloroquinolin-2(1H)-one (500 mg, 2.78 mmol), ethyl isonipecotate 26 (0.52 mL), NaI
(420 mg, 2.78 mmol), and Na2CO3 (885 mg, 8.35 mmol) were added to DMF (10 mL) at
RT under N2 and stirred at 80 °C for 48 h. The reaction was then cooled to RT, at which
time it was diluted in 1:1 EtOAc:Et2O and washed with 1 M HCl (3X), H2O (3X), 10%
aq. Na2CO3 (3X), and brine (1X). The organic phase was dried (MgSO4) and
concentrated. The residue was purified by silica flash chromatography (45 g silica, 20 to
80% EtOAc/Hex) to give a white microcrystalline solid. Yield: 567 mg, 1.89 mmol, 68%.
1H NMR (500 MHz, Chloroform-d) δ 12.43 (s, 1H), 7.69 (d, J = 8.2 Hz, 1H), 7.48 – 7.40

134
(m, 2H), 7.17 (t, J = 7.2 Hz, 1H), 6.13 (s, 1H), 4.18 (q, J = 7.1 Hz, 2H), 3.54 (d, J = 12.4
Hz, 2H), 2.82 (t, J = 10.8 Hz, 2H), 2.54 (ddd, J = 15.0, 10.8, 4.1 Hz, 1H), 2.13 – 2.07 (m,
2H), 2.06 – 1.98 (m, 2H), 1.28 (t, J = 7.1 Hz, 3H).
Ethyl 1-(1-(4-chlorophenyl)-2-oxo-1,2-dihydroquinolin-4-yl)piperidine-4-
carboxylate (58). Ethyl 1-(2-oxo-1,2-dihydroquinolin-4-yl)piperidine-4-carboxylate 57
(490 mg, 1.64 mmol) was dissolved in DCM (10 mL) with 4 Å MS, followed by the
addition of 4-chlorophenylboronic acid (510 mg, 3.27 mmol), pyridine (0.26 mL, 3.27
mmol), TEA (0.46 mL, 3.27 mmol), and Cu(OAc)2 (590 mg, 3.27 mmol). The reaction
was stirred at RT for 4 d, over which time the reaction became increasingly green with
precipitate. After this time, the reaction was diluted with more DCM and washed with
10% aq. NaOH (3X), 10% aq. NaHCO3 (1X), and brine (1X), then dried (MgSO4) and
concentrated. The resulting residue was purified by silica flash chromatography (25 g, 5
to 50% EtOAc/Hex) to give a white solid. Yield: 480 mg, 1.18 mmol, 72%. 1H NMR
(500 MHz, Chloroform-d) δ 7.79 (d, J = 8.0 Hz, 1H), 7.56 (d, J = 8.3 Hz, 2H), 7.32 (t, J =
7.8 Hz, 1H), 7.23 (d, J = 8.3 Hz, 2H), 7.19 (t, J = 7.6 Hz, 1H), 6.66 (d, J = 8.5 Hz, 1H),
6.21 (s, 1H), 4.21 (q, J = 7.1 Hz, 2H), 3.55 (d, J = 12.2 Hz, 2H), 2.84 (t, J = 11.5 Hz, 2H),
2.57 (ddd, J = 15.1, 10.9, 4.1 Hz, 1H), 2.16 – 2.09 (m, 2H), 2.09 – 1.96 (m, 2H), 1.31 (t, J
= 7.1 Hz, 3H).
Ethyl 1-(1-(4-chlorobenzyl)-2-oxo-1,2-dihydroquinolin-4-yl)piperidine-4-
carboxylate (59). Ethyl 1-(2-oxo-1,2-dihydroquinolin-4-yl)piperidine-4-carboxylate 58
(95 mg, 0.32 mmol) was dissolved in anhydrous DMF (5 mL) under nitrogen and cooled

135
to 0 °C. KOtBu (43 mg, 0.38 mmol) was added, which elicited a slight color change. 4-
chlorobenzyl chloride 4a (49 µL, 0.38 mmol) was then added and the reaction was
allowed to stir at RT for 15 h. At this time, the reaction was diluted with 2:1 EtOAc:Et2O
and washed with 1M aq. HCl (3X), H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X),
then dried (MgSO4) and concentrated. Residue was purified via silica flash
chromatography (10 g silica, 1% to 30% EtOAc:Hex). Yield: 117 mg, 0.28 mmol, 87%.
1H NMR (500 MHz, Chloroform-d) δ 7.79 (d, J = 7.9 Hz, 1H), 7.40 (t, J = 7.7 Hz, 1H),
7.28 – 7.23 (m, 2H), 7.17 (dd, J = 13.3, 8.2 Hz, 4H), 6.24 (s, 1H), 5.48 (s, 2H), 4.20 (q, J
= 7.3 Hz, 2H), 3.52 (d, J = 12.4 Hz, 2H), 2.82 (t, J = 11.8 Hz, 2H), 2.55 (tt, J = 11.0, 8.8,
4.0 Hz, 1H), 2.06 (ddd, J = 31.9, 19.5, 6.8 Hz, 4H), 1.30 (t, J = 7.4 Hz, 3H).
Ethyl 1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)piperidine-4-carboxylate (70).
The indole carboxylic acid 4a (982 mg, 3.44 mmol) was added to DCM (15 mL),
followed by ethyl isonipecotate 26 (0.80 mL, 5.16 mmol), DIPEA (1.80 mL, 10.31
mmol), EDCHCl (800 mg, 4.12 mmol), and HOBT (630 mg, 4.12 mmol). The reaction
was allowed to stir at RT for 24 h, after which time it was diluted with 1:1 EtOAc:Et2O
and washed with 1M HCl (5X), H2O (5X), 10% aq. Na2CO3 (5X) and brine (1X), then
dried (MgSO4) and concentrated. The residue can then be crystallized from a two-phase
organic solution of EtOAc and Hex to give crystals. Yield: 1.22 g, 2.88 mmol, 83%. 1H
NMR (500 MHz, Chloroform-d) δ 7.66 (d, J = 9.1 Hz, 1H), 7.36 (d, J = 8.3 Hz, 1H), 7.30
– 7.27 (m, 1H), 7.25 – 7.20 (m, 2H), 7.19 – 7.14 (m, 1H), 7.08 – 6.97 (m, 2H), 6.68 –
6.61 (m, 1H), 5.49 (d, J = 2.5 Hz, 2H), 4.49 (bs, 2H), 4.17 (qd, J = 7.1, 2.5 Hz, 2H), 3.08

136
– 2.95 (m, 2H), 2.51 (tq, J = 9.4, 3.6 Hz, 1H), 1.97 – 1.55 (m, 4H), 1.28 (td, J = 7.2, 2.5
Hz, 3H).
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)piperidine-4-carboxylic acid (71).
The ethyl ester 70 (1.22 g, 2.88 mmol) was dissolved in EtOH (20 mL) and 5M aq.
NaOH (20 mL) and stirred at 50 °C for 48 h. Most of the solvent was then removed in
vacuo and the remaining aqueous solution was washed with Et2O (1X), then acidified to
pH<1 at 0 °C with conc. HCl. The material was extracted with 1:1 EtOAc:Et2O (3X), and
the combined extracts were dried (MgSO4) and concentrated. The resulting residue was
dissolved in EtOAc and triturated with Hex, collected over a filter, and dried to give the
desired carboxylic acid as white, fluffy microcrystals. Yield: 1.03 g, 2.59 mmol, 90%. 1H
NMR (500 MHz, Chloroform-d) δ 7.59 (d, J = 7.9 Hz, 1H), 7.32 (d, J = 8.3 Hz, 1H), 7.28
– 7.23 (m, 1H), 7.19 – 7.11 (m, 3H), 6.99 (d, J = 8.0 Hz, 2H), 6.59 (s, 1H), 5.42 (s, 2H),
4.38 (bs, 1H), 4.08 (bs, 1H), 2.87 (bs, 2H), 2.38 (bs, J = 11.0 Hz, 1H), 1.90 – 1.37 (m,
4H).
2,3-Dihydro-1H-indene-2-carboxamide (74). 2-indancarboxylic acid 73 (1.4 g,
8.63 mmol) was dissolved in anhydrous DMF (20 mL), and the following was added
sequentially: DIPEA (3.02 mL, 17.26 mmol), HATU (3.94 g, 10.36 mmol), and HMDS
(2.17 mL, 10.36 mmol) after 30 min of stirring at RT. After stirring for 20 h at RT, the
solution was taken up in EtOAc and washed with 0.5 M HCl (3X), H2O (3X), and 10%
aq. Na2CO3 (3X). The organic phase was collected and dried over MgSO4s, and
concentrated in vacuo. The solid residue was recrystallized from EtOH to afford the title

137
compound as sharp, colorless crystals. Yield: 861 mg, 5.06 mmol, 59%. 1H NMR (400
MHz, DMSO-d6) δ 7.40 (bs, 1H), 7.22 – 7.15 (m, 2H), 7.14 – 7.09 (m, 2H), 6.86 (bs,
1H), 3.19 – 3.10 (m, 1H), 3.06 – 3.02 (m, 4H).
(2,3-Dihydro-1H-inden-2-yl)methanamine (75). 2,3-dihydro-1H-indene-2-
carboxamide 74 (55 mg, 0.341 mmol) was added to anhydrous THF (8 mL), and cooled
in an ice bath under nitrogen. LAH (0.34 mL, 0.341 mmol, 1M THF soln) was added
under nitrogen, and the reaction was allowed to warm to rt and stir for 7 h. At this time,
the reaction was quenched by the Fieser method and the precipitate removed over a filter.
The filtrate was collected and concentrated in vacuo, and then taken up in a small amount
of THF. 4M HCl/dioxane was added, the solvent removed in vacuo, and the residue
sonicated in Et2O to afford the easily handled hydrochloride salt. Yield: 1H NMR (400
MHz, Chloroform-d) δ 7.22 – 7.16 (m, 2H), 7.16 – 7.10 (m, 2H), 3.08 (dd, J = 15.6, 8.0
Hz, 2H), 2.80 (d, J = 7.1 Hz, 2H), 2.66 (dd, J = 15.6, 7.0 Hz, 2H), 2.54 (p, J = 7.4 Hz,
1H). TOF ES+ MS: (M + H) 148.1, (M + Na) 170.1.
1-(2,3-Dihydro-1H-inden-2-yl)ethanone (77). Acetylacetate (97 µL, 0.95 mmol)
was dissolved in anhydrous DMF under N2, followed by the addition of 60% oil-
suspended NaH (95 mg, 2.37 mmol). This was allowed to stir for 30 min, at which time
1,2-bis(bromoethyl)benzene 76 (250 mg, 0.95 mmol) was added. The reaction was
allowed to stir for 1 h at RT, then 8 h at 100 °C, then allowed to cool to RT. The reaction
was quenched with sat. aq. NH4Cl, diluted with EtOAc, and washed with 10% aq.
NaHCO3 (3X) and brine (1X), then dried (MgSO4) and concentrated. The residue was

138
then purified via silica flash chromatography (50 g silica, 1% to 5% EtOAc:Hex) to give
a very greasy solid invisible to electrospray MS. Yield: 1H NMR (400 MHz, Chloroform-
d) δ 7.21 – 7.17 (m, 2H), 7.17 – 7.12 (m, 2H), 3.47 – 3.37 (m, 1H), 3.20 – 3.11 (m, 4H),
2.22 (d, J = 2.6 Hz, 3H).
1-(2,3-Dihydro-1H-inden-2-yl)ethanamine (78). 1-(2,3-dihydro-1H-inden-2-
yl)ethanone 77 (44 mg, 0.275 mmol) was dissolved in methanol (2 mL), and 7 M
methanolic ammonia was added (1 mL, 7 mmol). This was allowed to stir at RT for 4 h,
at which time NaBH4 (31 mg, 0.824 mmol) and a catalytic drop of glacial AcOH was
added. Stirring continued for 14 h at RT, after which time the solvent was removed in
vacuo. The residue was taken up in EtOAc and washed with a small amount of 10% aq.
Na2CO3, dried with MgSO4, and concentrated. The residue was then taken on in
subsequent reactions without further purification or characterization. TOF ES+ MS: (M +
H) 162.2.
Ethyl 7-methyl-1H-indole-2-carboxylate (81). To a 0 °C flask charged with
crushed ice (~10 g) was added o-toluidine 79 (6.00 mL, 55.4 mmol), followed by conc.
HCl (10 mL) which elicited the precipitation of the hydrochloride salt. Dissolved in H2O
(30 mL), NaNO2 (3.82 g, 55.4 mmol) was added dropwise slowly to the flask, and more
ice was added as needed. The reaction was allowed to stir at 0 °C for 30 min, after which
~15 g of crushed ice and a 0 °C slurry of SnCl2 (32 g, 166.0 mmol) in conc. HCl (10 mL)
was added. The reaction was then allowed to sit in a fridge (4 °C) for 11 h, after which
time the precipitate was filtered off and collected, and washed with cold brine and

139
hexanes. The resulting precipitate was collected over a filter and washed with cold brine
and 1:1 EtOAc:Hex. Unlike other reports, no attempt was made to basify, wash, and re-
convert the hydrazine to the hydrazine hydrochloride salt (due to observed stability
issues). The white solid was dried thoroughly under high vacuum. Yield: 4.92 g, 31.02
mmol, 56%.
Ethyl pyruvate 80 (1.17 mL, 10.51 mmol), o-tolylhydrazine hydrochloride (1.5 g,
9.46 mmol), ZnCl2 (7.16 g, 52.55 mmol) and TsOH (9.05 g, 52.55 mmol) were dissolved
in anhydrous EtOH (15 mL) at RT in dry glassware. The reaction was refluxed for 8 h,
after which the reaction was allowed to cool to RT and the resulting precipitate was
filtered off and washed with EtOH. The filtrate was concentrated to give a residue that
was dissolved in 1:1 EtOAc:Et2O and washed with 10% aq. Na2CO3 (2X), dried
(MgSO4), and concentrated. The residue was then purified by silica flash chromatography
(100 g silica, 20% EtOAc:Hex) to give the desired indole as a foul-smelling oil. Yield:
1.37 g, 6.72 mmol, 71%. 1H NMR (400 MHz, Chloroform-d) δ 8.88 (bs, 1H), 7.53 (d, J =
8.0 Hz, 1H), 7.27 – 7.21 (m, 1H), 7.14 – 7.02 (m, 2H), 4.42 (qd, J = 7.1, 1.3 Hz, 2H),
2.52 (s, 3H), 1.42 (td, J = 7.1, 1.3 Hz, 3H).
Ethyl 1-(4-chlorobenzyl)-7-methyl-1H-indole-2-carboxylate (82). Ethyl 7-methyl-
1H-indole-2-carboxylate 81 (500 mg, 2.46 mmol) was dissolved in anhydrous DMF (20
mL) and granular K2CO3 (425 mg, 3.08 mmol) and 4-chlorobenzyl chloride (0.39 mL,
3.08 mmol) were added. The mixture was stirred for 3 d at 60 °C, after which time the
incomplete reaction was terminated by dilution with EtOAc and subsequent washing with
brine (3X). The organic phase was dried over MgSO4, concentrated, and purified vial

140
silica flash chromatography (80 g silica, 40% EtOAc:Hex) to provide the title compound
as white crystals. Yield: 390 mg, 1.19 mmol, 46%. 1H NMR (400 MHz, Chloroform-d) δ
7.59 – 7.53 (m, 1H), 7.43 – 7.40 (m, 1H), 7.20 (d, J = 8.4 Hz, 2H), 7.06 – 6.97 (m, 2H),
6.78 (d, J = 8.2 Hz, 2H), 6.08 (s, 2H), 4.28 (q, J = 7.1 Hz, 2H), 2.54 (s, 3H), 1.33 (t, J =
7.1 Hz, 3H).
1-(4-Chlorobenzyl)-7-methyl-1H-indole-2-carboxylic acid (83). Ethyl 1-(4-
chlorobenzyl)-7-methyl-1H-indole-2-carboxylate 82 (212 mg, 0.647 mmol) was
dissolved in EtOH (10 mL) and 10% aq. KOH (10 mL) was added. The reaction stirred
for 4 h at 70 °C, after which time as much solvent was stripped off as possible in vacuo.
The mostly aqueous mixture was cooled in an ice bath and acidified with conc. HCl to
obtain a precipitate that was collected over a filter and washed with 1 M HCl and cold
H2O. No further purification necessary. Yield: 182 mg, 0.607 mmol, 94%. 1H NMR (400
MHz, DMSO-d6) δ 12.94 (s, 1H), 7.36 – 7.24 (m, 4H), 7.01 – 6.95 (m, 2H), 6.76 (d, J =
8.1 Hz, 2H), 6.08 (s, 2H), 2.44 (s, 3H).
Ethyl 3-methyl-1H-indole-2-carboxylate (86). Phenylhydrazine was prepared in a
manner similar to that reported.138
Ethyl 2-oxobutanoate 85 (500 mg, 4.90 mmol),
phenylhydrazine (0.44 mL, 4.41 mmol), and TsOH (2.3 g, 12.24 mmol) were dissolved in
anhydrous EtOH (15 mL) at RT in dry glassware. The reaction was refluxed for 12 h,
over which time it took on a dark hue. After the reaction time, the reaction was allowed
to cool to RT and the resulting precipitate was filtered off and washed with EtOH. The
combined filtrate was concentrated to give a residue that was dissolved in 1:1

141
EtOAc:Et2O and washed with 10% aq. Na2CO3 (2X), dried (MgSO4), and concentrated.
The residue was then purified by silica flash chromatography (45 g silica, 20%
EtOAc:Hex) to give the desired indole as a foul-smelling oil. Yield: 646 mg, 3.18 mmol,
65%. 1H NMR (500 MHz, Chloroform-d) δ 8.83 (s, 1H), 7.69 (d, J = 8.1 Hz, 1H), 7.39
(d, J = 8.3 Hz, 1H), 7.35 (d, J = 7.8 Hz, 1H), 7.16 (t, J = 7.4 Hz, 1H), 4.45 (qd, J = 7.1,
1.1 Hz, 2H), 2.64 (d, J = 1.1 Hz, 3H), 1.46 (td, J = 7.2, 1.1 Hz, 3H).
Ethyl 1-(4-chlorobenzyl)-3-methyl-1H-indole-2-carboxylate (87). Anhydrous
DMF (12 mL) in dry glassware was charged with ethyl 3-methyl-1H-indole-2-
carboxylate 86 (646 mg, 3.18 mmol), 4-chlorobenzyl chloride (0.50 mL, 3.97 mmol), and
granular K2CO3, and the mixture was stirred at 60 °C for 14 h, allowed to cool for 1 h,
then diluted in 1:1 EtOAc:Et2O and washed with 10% aq. Na2CO3 (3X), dried (MgSO4),
and concentrated. The residue was purified by silica flash chromatography ( 40 g
silica, 20% EtOAc:Hex) to give the desired compound as an oil. Yield: 750 mg, 2.29
mmol, 72%. 1H NMR (400 MHz, Chloroform-d) δ 7.71 (dd, J = 8.1, 1.1 Hz, 1H), 7.33 –
7.23 (m, 2H), 7.22 – 7.12 (m, 3H), 6.99 – 6.90 (m, 2H), 5.73 (s, 2H), 4.34 (qd, J = 7.2,
1.1 Hz, 2H), 2.62 (d, J = 1.1 Hz, 3H), 1.35 (td, J = 7.1, 1.1 Hz, 3H).
5-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)octahydro-1H-pyrrolo[3,4-
c]pyridin-1-one (91). The following were added sequentially to DMF (10 mL): 1-(4-
chlorobenzyl)-1H-indole-2-carboxylic acid 4a (250 mg, 0.875 mmol), DIPEA (0.46 mL,
2.64 mmol), EDCHCl (202 mg, 1.050 mmol), and HOBT (162 mg, 1.050 mmol). This
solution was allowed to stir at RT for 1 h, at which time crude octahydro-1H-pyrrolo[3,4-

142
c]pyridin-1-one 90 (123 mg, 0.875 mmol) was added, having been prepared through the
known route.140
Stirring continued for 12 h at RT, at which time the solution was
partitioned between water and 1:1 solution of EtOAc:Et2O. The organic extract was then
washed with water (1X) and saturated aq. Na2CO3 (1X), dried with MgSO4, and
concentrated in vacuo. Purification was accomplished via silica flash chromatography
(100 g silica, 100% EtOAc.) Resulting residue was crystallized from the in vacuo
removal of DCM to give the title compound as a slightly pink solid. Yield: 200 mg, 0.49
mmol, 56%. 1H NMR (400 MHz, Chloroform-d) δ 7.72 (d, J = 7.9 Hz, 1H), 7.37 – 7.24
(m, 4H), 7.19 (t, J = 7.4 Hz, 1H), 7.14 (d, J = 8.1 Hz, 2H), 6.93 (s, 1H), 6.31 (bs, 1H),
5.51 (s, 2H), 4.57 (d, J = 13.2 Hz, 1H), 4.18 (bs, 1H), 3.56 – 3.46 (m, 1H), 3.39 (dd, J =
13.7, 5.1 Hz, 1H), 3.05 (d, J = 8.6 Hz, 1H), 2.63 (s, 1H), 2.50 (s, 1H), 1.96 – 1.73 (m,
2H), 1.54 (bs, 1H).
Tert-Butyl 4-(3-(pyridin-4-yl)propanamido)piperidine-1-carboxylate (94). t-Butyl
4-aminopiperidine-1-carboxylate (500 mg, 2.50 mmol), 3-(pyridin-4-yl)propanoic acid
(377 mg, 2.50 mmol), TEA (1.04 mL, 7.49 mmol), EDCHCl (574 mg, 3.00 mmol), and
HOBT (459 mg, 3.00 mmol) were all dissolved in DCM (30 mL) and stirred at RT for 30
h, after which time the DCM was removed in vacuo and the residue was taken up in
EtOAc and washed with H2O (1X), 10% Na2CO3 (3X), and brine (1X), then dried
(MgSO4) and concentrated. The resulting residue was purified via silica flash
chromatography (40 g silica, 1% to 10% methanolic ammonia:EtOAc) to provide a white
solid. Yield: 591 mg, 1.773 mmol, 71%.

143
Ethyl 2-(1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)piperidin-4-yl)acetate (96).
Indole carboxylic acid 4a (520 mg, 1.82 mmol), ethyl 2-(piperidin-4-yl)acetate 95 (345
mg, 1.65 mmol), TEA (1.15 mL, 8.27 mmol), EDCHCl (380 mg, 1.98 mmol), and
HOBT (304 mg, 1.98 mmol) were all dissolved in DCM (50 mL) and stirred at RT for 24
h, after which time the DCM was removed in vacuo and the residue was taken up in
EtOAc and washed with 10% aq. citric acid (3X), H2O (2X), 10% Na2CO3 (3X), and
brine (1X), then dried (MgSO4) and concentrated. The resulting residue was purified via
silica flash chromatography (40 g silica, 10% to 50% EtOAc:Hex) to provide a white
powder. Yield: 460 mg, 1.05 mmol, 63%. 1H NMR (500 MHz, Chloroform-d) δ 7.67 –
7.62 (m, 1H), 7.36 (d, J = 8.3 Hz, 1H), 7.29 – 7.25 (m, 1H), 7.24 – 7.20 (m, 2H), 7.18 –
7.13 (m, 1H), 7.06 – 7.01 (m, 2H), 6.62 (s, 1H), 5.48 (s, 2H), 4.63 (bs, 1H), 4.13 (q, J =
7.2 Hz, 3H), 2.88 (bs, 1H), 2.74 (bs, 1H), 2.18 (d, J = 7.0 Hz, 2H), 1.98 (ttt, J = 11.0, 7.1,
3.7 Hz, 1H), 1.75 (bs, 1H), 1.57 (bs, 1H), 1.26 (t, J = 7.1 Hz, 3H), 1.11 (bs, 1H), 0.65 (bs,
1H).
4-(3-(Triphenylphosphoranyl)propyl)pyridine bromide (98). 4-(3-
hydroxypropyl)pyridine 97 (1.42 g, 10.35 mmol) and PPh3 (4.07 g, 15.53 mmol) were
dissolved in conc. HBr (8 mL) in a pressure vessel. The reaction was sealed and the
suspension as stirred at 135 °C for 2 h, over which time a homogenous solution was
achieved. After this time, the reaction was allowed to cool to RT, eliciting precipitation.
The reaction was taken up in toluene and transferred to a flask equipped with a Dean-
Stark apparatus and water condenser, and H2O was removed with heating of 150 °C for 3
h, at which time more conc. HBr (8 mL) was added. H2O was again removed at 150 °C

144
for 6 h. Once sufficient water had been removed, a precipitate began to appear and the
reaction was allowed to cool to RT, at which point it was diluted with H2O (50 mL) and
washed with Et2O (5X). The pH was raised to pH 7 by the addition of 10% aq. NaOH,
and the resulting solution was washed with Et2O to remove PPh3 which caused
significant precipitation of product. The aqueous slurry was then washed with Et2O (3X)
and cooled in a fridge for 2 h, after which time the precipitate was collected over a filter
and washed with cold H2O and Et2O. The highly hygroscopic crystalline material was
dried under high vacuum for 1 d and stored in a desiccator. No further purification
necessary. Off-white crystalline solid. Yield: 1.601 g, 3.46 mmol, 34%. 1H NMR (500
MHz, Chloroform-d) δ 8.41 (d, J = 5.9 Hz, 2H), 7.82 – 7.74 (m, 9H), 7.70 – 7.64 (m,
6H), 7.17 (d, J = 5.9 Hz, 2H), 3.98 – 3.90 (m, 2H), 3.11 (t, J = 7.4 Hz, 2H), 1.93 (dq, J =
15.7, 7.8 Hz, 2H). TOF ES+ MS: (M+) 382.2.
(Z)-tert-Butyl 4-(4-(pyridin-4-yl)but-1-en-1-yl)piperidine-1-carboxylate (99a).
Due to the poor ethereal solubility of the phosphonium bromide, the reaction was
conducted at RT. 4-(3-(bromotriphenylphosphoranyl)propyl)pyridine 98 (330 mg, 0.714
mmol) was added to anhydrous THF in a flame-dried flask under N2 at RT. To the
suspension, n-BuLi (2.5 M in hexane) was added dropwise at RT until a light yellow
color persisted, at which point a full equivalent was added (285 µL, 0.714 mmol) to give
a red solution. The reaction then stirred for 10 min at RT under N2 to allow for ylide
dissolution. Predissolved in anhydrous THF (10 mL), t-butyl 4-formylpiperidine-1-
carboxylate 8 (138 mg, 0.649 mmol) was added dropwise at RT to the reaction, and the
reaction was allowed to stir at RT under N2 for 14 h, at which time the reaction was

145
quenched with sat. NH4Cl and extracted with a 1:1 solution of EtOAc:Et2O . This extract
was then washed with 10% aq. Na2CO3 (1X) and brine (1X), dried (MgSO4) and
concentrated. The residue was purified via silica flash chromatography (12 g silica,
isocratic 60% EtOAc:Hex) to give a clear, colorless oil, >50:1 Z:E. Yield: 117 mg, 0.370
mmol, 57%. 1H NMR (500 MHz, Chloroform-d) δ 8.47 (d, J = 4.9 Hz, 2H), 7.08 (d, J =
5.1 Hz, 2H), 5.38 (t, J = 6.1 Hz, 1H), 5.36 (d, J = 5.9 Hz, 1H), 4.04 (s, 2H), 2.67 (q, J =
10.3, 7.7 Hz, 4H), 2.31 (q, J = 7.1 Hz, 2H), 2.08 – 1.97 (m, 1H), 1.64 – 1.55 (m, 2H),
1.45 (s, 9H), 1.19 (tt, J = 13.2, 5.9 Hz, 2H). TOF ES+ MS: (M + H, no Boc) 217.2.
(E)-tert-Butyl 4-(4-(pyridin-4-yl)but-1-en-1-yl)piperidine-1-carboxylate (99b).
LiBr (340 mg, 3.92 mmol) was dried by flame-heating a 2-neck flask under high vacuum
until the molten salt ceased bubbling. The flask was then purged with N2 and a stir bar
was added (so the bar would not char during the flame-dry) and the flask was allowed to
cool to RT. The flask was then charged with anhydrous THF (40 mL) and 4-(3-
(triphenylphosphoranyl)propyl)pyridine bromide 98 (823 mg, 1.78 mmol) under N2.
Without cooling – due to the poor ethereal solubility of the phosphonium bromide – 2 M
PhLi in (Bu)2O was added at RT until a slight yellow color persisted, at which point a full
1.1 equivalent of PhLi (0.98 mL, 1.96 mmol) was added, and the suspension gave way to
mostly homogenous red solution after 20 min of RT stirring. This solution was then
chilled to -78 °C, and t-butyl 4-formylpiperidine-1-carboxylate 8 (380 mg, 1.78 mmol) –
pre-dissolved in anhydrous THF (10 mL) – was added dropwise to the solution, causing a
loss of color. The light yellow solution was allowed to stir at -78 °C for 30 min, at which
time the second equivalent of PhLi (0.98 mL, 1.96 mmol) was added dropwise, resulting

146
in the solution becoming a very dark cherry-red color. The cooling bath was then
removed, and the reaction was allowed to stir for 30 min as it approached RT. The
reaction was then re-chilled to -78 °C and t-BuOH (0.204 mL, 2.14 mmol) – predissolved
in anhydrous THF (5 mL) – was added dropwise, eliciting precipitation and
decolorization. The reaction was allowed to warm to RT and stir for 36 h, after which
time the reaction was quenched with a small amount of sat. aq. NH4Cl and diluted with a
2:1 EtOAc:Et2O solution and washed with 10% aq. Na2CO3 (1X) and brine (1X), dried
(MgSO4), and concentrated in vacuo. Purification accomplished by silica flash
chromatography (40 g silica, 50% to 80% EtOAc:Hex) to provide the desired olefin as a
clear colorless oil, ~5:1 E:Z. Yield: 231 mg, 0.730 mmol, 41%. 1H NMR (400 MHz,
Chloroform-d) δ 8.43 (d, J = 4.4 Hz, 2H), 7.06 (d, J = 4.5 Hz, 2H), 5.25 (t, J = 8.9 Hz,
1H), 5.16 (t, J = 10.0 Hz, 1H), 3.98 (s, 2H), 2.67 – 2.55 (m, 4H), 2.34 (q, J = 7.3 Hz, 2H),
2.27 – 2.12 (m, 1H), 1.40 (s, 9H), 1.30 (s, 2H), 1.13 (d, J = 11.8 Hz, 2H). TOF ES+ MS:
(M + H, no Boc) 217.2, (M + Na, no Boc) 239.2.
Tert-Butyl 4-((2-(pyridin-4-yl)ethyl)carbamoyl)piperazine-1-carboxylate (101). t-
Butyl piperazine-1-carboxylate (400 mg, 2.15 mmol) was dissolved in DCM (15 mL) at
RT under N2 and cooled to 0 °C. 15% wt. phosgene (360 µL, 0.50 mmol) in toluene was
added to the solution dropwise, followed by the dropwise addition of TEA (0.90 mL, 6.44
mmol), and then the dropwise addition of 4-(2-aminoethyl)pyridine (565 µL, 4.72 mmol).
The reaction was allowed to stir for 1 h at 0 °C, at which point it was diluted with EtOAc
and washed with 10% aq. NaOH (1X), H2O (3X), and brine (1X), dried (MgSO4), and
concentrated. The residue was then purified via silica flash chromatography (40 g silica,

147
1% to 20% methanolic ammonia:EtOAc) to give a white solid after concentration. Yield:
582 mg, 1.74 mmol, 81%. 1H NMR (500 MHz, Chloroform-d) δ 8.44 (d, J = 5.6 Hz, 2H),
7.09 (d, J = 5.4 Hz, 2H), 4.92 (t, J = 5.8 Hz, 1H), 3.47 (q, J = 6.7 Hz, 2H), 3.39 – 3.34
(m, 5H), 3.30 – 3.27 (m, 3H), 2.81 (t, J = 7.0 Hz, 2H), 1.43 (d, J = 1.6 Hz, 12H).
1-Tert-Butyl 4-ethyl 4-(phenylselanyl)piperidine-1,4-dicarboxylate (103). DIPA
(2.49 mL, 17.49 mmol) was dissolved into anhydrous THF (10 mL) in dry glassware
under N2, then chilled to -78 °C. Then, 2.5 M n-BuLi (5.60 mL, 13.99 mmol) was added
dropwise, and the reaction was allowed to stir for 10 min. 1-Tert-butyl 4-ethyl piperidine-
1,4-dicarboxylate 102 (3.00 g, 11.66 mmol) – pre-dissolved in THF (10 mL) – was added
dropwise to the reaction and the reaction was allowed to stir at -78 °C for 15 min. PhSeCl
(2.68 g, 13.99 mmol) – pre-dissolved in anhydrous THF (10 mL) – was added dropwise
to the reaction under the same conditions. The cooling bath was removed to allow for
warming to RT, and the reaction was stirred for 3 h. After this time, the reaction was
quenched by the addition of sat. aq. NH4Cl and extracted with 1:1 EtOAc:Et2O. The
extract was subsequently washed with H2O (2X) and brine (1X), dried (MgSO4) and
concentrated. The residue was purified by silica flash chromatography (80 g, 5%
EtOAc:Hex) to give a clear, slightly yellow oil. Yield: 2.80 g, 6.78 mmol, 58%. 1H NMR
(500 MHz, Chloroform-d) δ 7.56 (d, J = 7.5 Hz, 2H), 7.39 (t, J = 7.4 Hz, 1H), 7.30 (t, J =
7.7 Hz, 2H), 4.10 (q, J = 7.1 Hz, 2H), 3.74 (s, 2H), 3.11 – 3.03 (m, 2H), 2.13 – 2.06 (m,
2H), 1.84 (s, 2H), 1.44 (s, 9H), 1.18 (t, J = 7.1 Hz, 3H).

148
Ethyl 4-(phenylselanyl)piperidine-4-carboxylate hydrochloride (104). 1-tert-butyl
4-ethyl 4-(phenylselanyl)piperidine-1,4-dicarboxylate (1.70 g, 4.12 mmol) was dissolved
in Et2O (100 mL) at RT and 4 M HCl in 1,4-dioxane (15.46 mL, 61.80 mmol) was added
dropwise. The reaction was stirred at RT for 4 d until complete by TLC. The solvent was
removed in vacuo and Et2O was added and stripped off five times (5X) to remove excess
HCl and solidify the salt. Further removal of solvent and HCl via rotovap and, lastly, high
vacuum furnished the hydrochloride salt. Yield, 1.44, g, 4.12 mmol, 100%. TOF ES+
MS: (M + H) 314.1, (M + Na) 336.0.
Ethyl 1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)-4-(phenylselanyl)piperidine-
4-carboxylate (105). Ethyl 4-(phenylselanyl)piperidine-4-carboxylate hydrochloride (200
mg, 0.574 mmol) was dissolved in DCM (10 mL) and TEA (0.48 mL, 3.44 mmol),
followed by 1-(4-chlorobenzyl)-1H-indole-2-carboxylic acid (164 mg, 0.574 mmol),
EDCHCl (121 mg, 0.631 mmol), and HOBT (97 mg, 0.631 mmol), and the reaction was
allowed to stir at RT for 15 h. After this time, the reaction was diluted with a 1:1 solution
of EtOAc:Et2O and washed with 1M HCl (3X), H2O (3X), 10% Na2CO3 (3X), and brine
(1X), then dried (MgSO4) and concentrated in vacuo. The resulting residue was purified
via silica flash chromatography (12 g silica, 5% to 20% EtOAc:Hex) to yield an oil that
may triturated to provide a fine white powder. Yield: 208 mg, 0.359 mmol, 63%. 1H
NMR (500 MHz, Chloroform-d) δ 7.64 (d, J = 7.9 Hz, 1H), 7.54 (d, J = 7.9 Hz, 2H), 7.43
– 7.36 (m, 2H), 7.30 (dt, J = 15.2, 7.8 Hz, 3H), 7.23 (d, J = 8.3 Hz, 2H), 7.16 (t, J = 7.5
Hz, 1H), 7.04 (d, J = 8.2 Hz, 2H), 6.61 (s, 1H), 5.48 (s, 2H), 4.12 (q, J = 7.1 Hz, 3H),

149
3.84 (s, 1H), 3.33 – 3.20 (m, 2H), 2.12 (s, 1H), 1.81 (s, 2H), 1.45 (s, 1H), 1.19 (t, J = 7.1
Hz, 3H).
Ethyl 1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)-1,2,3,6-tetrahydropyridine-4-
carboxylate (106). Ethyl 1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)-4-
(phenylselanyl)piperidine-4-carboxylate (160 mg, 0.276 mmol) was dissolved in DCM
(10 mL) at RT then cooled to -78 °C. Suspended in DCM (10 mL), ~60% mCPBA (80
mg, 0.276 mmol) was added to the solution dropwise and the reaction was allowed to stir
for 30 min. Still at -78 °C, TEA (115 µL, 0.828 mmol) was added dropwise and the
reaction was allowed to stir for another 30 min. At this time, the reaction was warmed to
RT, then the reaction was then diluted with 2:1 EtOAc:Et2O and washed with 10% aq.
Na2CO3 (3X), H2O (1X), 1 M HCl (2X), H2O (1X) again, and sat. aq. Na2CO3 (1X). The
organic phase was dried with MgSO4 and concentrated. The resulting solid residue was
taken up in Hex and the yellow supernatant was decanted off to remove selenoxides.
Further purification was accomplished by silica flash chromatography (12 g silica, 5% to
20% EtOAc:Hex) to provide the desired unsaturated ester as a white solid. Yield: 96 mg,
0.227 mmol, 82%. 1H NMR (500 MHz, Chloroform-d) δ 7.66 (d, J = 7.9 Hz, 1H), 7.37
(d, J = 8.3 Hz, 1H), 7.29 (t, J = 7.7 Hz, 1H), 7.22 – 7.11 (m, 3H), 7.00 (d, J = 7.5 Hz,
2H), 6.68 (s, 1H), 5.48 (s, 2H), 4.23 (q, J = 7.1 Hz, 4H), 3.68 (s, 2H), 2.32 (s, 2H), 1.31
(t, J = 7.1 Hz, 3H).
1-Tert-Butyl 4-ethyl 4-methylpiperidine-1,4-dicarboxylate (107). DIPA (0.62 mL,
4.37 mmol) was dissolved into anhydrous THF (10 mL) in dry glassware under N2, then

150
chilled to -78 °C. Then, 2.5 M n-BuLi (1.40 mL, 3.50 mmol) was added dropwise, and
the reaction was allowed to stir for 15 min. Boc-ethyl isonipecotate 102 (750 mg, 2.91
mmol) – pre-dissolved in THF (5 mL) – was added dropwise to the reaction and the
reaction was allowed to stir at -78 °C for 15 min. MeI (220 µL, 3.50 mmol) – pre-
dissolved in anhydrous THF (3 mL) – was added dropwise to the reaction under the same
conditions. The cooling bath was removed to allow for warming to RT, and the reaction
was stirred for 8 h. After this time, the reaction was quenched by the addition of sat. aq.
NH4Cl and diluted with 1:1 EtOAc:Et2O, then washed with H2O (3X) and brine (1X),
dried (MgSO4) and concentrated. The residue was purified by silica flash
chromatography (40 g, 5 to 20% EtOAc:Hex) to give a clear colorless oil. 1H NMR (500
MHz, Chloroform-d) δ 4.14 (q, J = 7.1 Hz, 2H), 3.74 (s, 2H), 2.96 (s, 2H), 2.04 (d, J =
13.3 Hz, 2H), 1.42 (s, 9H), 1.32 (t, J = 12.5 Hz, 2H), 1.24 (t, J = 7.5 Hz, 3H), 1.17 (s,
3H).
Ethyl 4-methylpiperidine-1,4-dicarboxylate hydrochloride (108). The Boc-
protected amino ester 107 (500 mg, 1.84 mmol) was dissolved into 4M HCl dioxane (4
mL) and stirred for 15 min. After this time, the dioxane was removed in vacuo and the
material was sonicated in Et2O to give a tan solid. The Et2O was decanted off and the
precipitate was dried via aspirator, then high vacuum, to afford the desired amine
hydrochloride as a tan, hygroscopic solid. No further purification necessary. Yield: 350
mg, 1.68 mmol, 100%. TOF ES+ MS: (M + H) 172.1, (M + Na) 194.1.

151
Ethyl 1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)-4-methylpiperidine-4-
carboxylate (109). The amine hydrochloride 108 (240 mg, 1.16 mmol), indole carboxylic
acid 4a (300 mg, 1.05 mmol), TEA (585 µL, 4.20 mmol), EDCHCl (242 mg, 1.26
mmol), and HOBT (193 mg, 1.26 mmol) were dissolved in DCM (30 mL) and stirred at
RT for 22 h. After this time, the reaction as diluted with EtOAc and washed with 1M HCl
(3X), H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X). The organic extract was dried
(MgSO4) and concentrated, and the residue purified via silica flash chromatography (40 g
silica, 30% to 90% EtOAc:Hex) to give a white solid. Yield: 281 mg, 0.64 mmol, 61%.
1H NMR (500 MHz, Chloroform-d) δ 7.65 (d, J = 7.9 Hz, 1H), 7.38 (d, J = 8.3 Hz, 1H),
7.31 – 7.26 (m, 1H), 7.21 (d, J = 7.3 Hz, 2H), 7.16 (t, J = 7.5 Hz, 1H), 7.03 (d, J = 7.8
Hz, 2H), 6.61 (s, 1H), 5.48 (s, 2H), 4.29 (bs, 1H), 4.18 (q, J = 7.4, 7.0 Hz, 2H), 3.86 (bs,
1H), 3.10 (bs, 1H), 2.98 (bs, 1H), 2.12 (bs, 1H), 1.85 (bs, 1H), 1.31 – 1.20 (m, 4H), 1.14
(s, 3H), 0.77 (s, 2H).
(1R,5S,6r)-Ethyl 3-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)-3-
azabicyclo[3.1.0]hexane -6-carboxylate (113). The amine exo-112 (170 mg, 0.89 mmol)
– prepared in a manner similar to that reported150, 151
– indole carboxylic acid 4a (304 mg,
1.06 mmol), TEA (495 µL, 3.55 mmol), EDCHCl (204 mg, 1.06 mmol), and HOBT (163
mg, 1.06 mmol) were dissolved in DCM (10 mL) and stirred at RT for 18 h. After this
time, the reaction as diluted with EtOAc:Et2O (1:1) and washed with 1M HCl (1X), H2O
(2X), 10% aq. Na2CO3 (3X), and brine (1X). The organic extract was dried (MgSO4) and
concentrated, and the residue purified via silica flash chromatography (25 g silica, 5% to
20% EtOAc:Hex) to give a white solid. Yield: 244 mg, 0.58 mmol, 78%. 1H NMR (500

152
MHz, Chloroform-d) δ 7.66 (d, J = 7.9 Hz, 1H), 7.37 (d, J = 8.4 Hz, 1H), 7.32 – 7.28 (m,
1H), 7.26 – 7.22 (m, 2H), 7.17 (dd, J = 8.2, 6.7 Hz, 1H), 6.99 (d, J = 8.1 Hz, 2H), 6.74 (s,
1H), 5.56 (d, J = 49.7 Hz, 2H), 4.22 (bd, J = 12.4 Hz, 1H), 4.15 (q, J = 7.2 Hz, 2H), 3.77
(bd, J = 13.0 Hz, 2H), 3.52 (s, 1H), 2.18 – 2.04 (m, 2H), 1.29 (td, J = 7.1, 1.2 Hz, 3H),
1.06 (t, J = 3.1 Hz, 1H).
(1R,3s,5S)-Tert-Butyl 3-((2-(pyridin-4-yl)ethyl)carbamoyl)-8-
azabicyclo[3.2.1]octane-8-carboxylate (115). The Boc-protected amino ester 114 (175
mg, 0.65 mmol) was dissolved in anhydrous THF (10 mL) with KOTMS (83 mg, 0.65
mmol) and stirred under dry conditions under N2 for 18 h, at which time a precipitate was
observed and the solvent was removed in vacuo to give the potassium salt as a white
powder. No further purification necessary. Yield: 190 mg, 0.65 mmol, 100%.
The potassium salt (191 mg, 0.65 mmol), amine 27 (93 µL, 0.78 mmol),
EDCHCl (150 mg, 0.78 mmol), HOBT (119 mg, 0.78 mmol), and TEA (362 µL, 2.60
mmol) were dissolved in DCM (10 mL) and stirred at RT for 24 h, at which time the
DCM was removed in vacuo and the residue was taken up in EtOAc and washed with
H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X). The organic extract was dried (MgSO4)
and concentrated, and the residue purified via silica flash chromatography (25 g silica,
1% to 20% methanolic ammonia:EtOAc) to give a tan powder. Yield: 41 mg, 0.083
mmol, 57%. Yield: 146 mg, 0.41 mmol, 63%. 1H NMR (500 MHz, Chloroform-d) δ 8.49
(d, J = 5.8 Hz, 2H), 7.10 (d, J = 5.7 Hz, 2H), 5.66 (t, J = 5.9 Hz, 1H), 4.23 (d, J = 40.3
Hz, 2H), 3.54 – 3.48 (m, 2H), 2.81 (t, J = 7.0 Hz, 2H), 2.58 (tt, J = 11.8, 5.3 Hz, 1H),
1.97 – 1.84 (m, 5H), 1.61 – 1.56 (m, 3H), 1.46 (d, J = 1.1 Hz, 9H).

153
N-(2-(Pyridin-4-yl)ethyl)-2-tosyl-2-azaspiro[3.3]heptane-6-carboxamide (123).
The tosyl amide carboxylic acid 122 (2.0 g, 6.77 mmol) – prepared in a manner similar to
that reported152, 153
– amine 27 (0.97 mL, 8.13 mmol), HATU (309 g, 8.13 mmol), and
TEA (2.83 mL, 20.31 mmol) were dissolved in DMF (10 mL) and stirred at RT for 18 h,
at which time the reaction was diluted with EtOAc and washed with H2O (5X), 10% aq.
Na2CO3 (3X), and brine (1X). The organic extract was dried (MgSO4) and concentrated,
and the residue purified via silica flash chromatography (80 g silica, 1% to 20%
methanolic ammonia:EtOAc) to give an extremely thick, clear, yellow-tinged oil. Yield:
1.95 g, 4.88 mmol, 72%. 1H NMR (500 MHz, Chloroform-d) δ 8.31 (d, J = 5.4 Hz, 2H),
7.59 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 6.99 (d, J = 5.4 Hz, 2H), 6.30 (t, J =
5.8 Hz, 1H), 3.61 (d, J = 5.5 Hz, 4H), 3.37 (q, J = 6.8 Hz, 2H), 2.69 (t, J = 7.1 Hz, 2H),
2.62 (p, J = 7.8 Hz, 1H), 2.37 (s, 3H), 2.12 (dd, J = 11.1, 8.7 Hz, 2H), 2.05 – 1.98 (m,
2H). TOF ES+ MS: (M + H) 400.1, (M + Na) 422.1.
Methyl 1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)azetidine-3-carboxylate
(125). The following reagents were dissolved in DCM (10 mL) at RT: indole carboxylic
acid 4a (150 mg, 0.53 mmol), methyl 4-azetidinecarboxylate hydrochloride (88 mg, 0.58
mmol), TEA (366 µL, 2.26 mmol), EDCHCl (121 mg, 0.63 mmol), and HOBT (96 mg,
0.63 mmol). The reaction was allowed to stir for 26 h at RT, after which time the solvent
was remove in vacuo and the residue was dissolved in EtOAc and washed with 1M HCl
(3X), H2O (2X), 10% aq. Na2CO3 (3X), and brine (1X), then dried (MgSO4) and
concentrated. The residue was purified by silica flash chromatography (25 g silica, 5 to

154
40% EtOAc:Hex). Yield: 117 mg, 0.30 mmol, 58%. 1H NMR (500 MHz, Chloroform-d)
δ 7.67 (d, J = 8.0 Hz, 1H), 7.34 – 7.27 (m, 2H), 7.22 – 7.14 (m, 3H), 7.04 – 6.99 (m, 2H),
6.83 (d, J = 1.2 Hz, 1H), 5.73 (s, 2H), 4.53 (bd, J = 36.9 Hz, 2H), 4.32 (bd, J = 26.0 Hz,
2H), 3.78 (d, J = 1.4 Hz, 3H), 3.47 (p, J = 7.1 Hz, 1H).
Ethyl 1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)azepane-4-carboxylate (132).
The carboxylic acid 4a (757 mg, 2.65 mmol), azapane ester hydrochloride 131 (550 mg,
2.65 mmol) – prepared in a manner similar to that reported160
– EDCHCl (558 mg, 2.91
mmol), HOBT (446 mg, 2.91 mmol), and TEA (1.48 mL, 10.59 mmol) were dissolved in
DCM (20 mL) and stirred at RT for 18 h, at which time the DCM was removed in vacuo
and the residue was taken up in EtOAc and washed with 1M HCl (2X), H2O (3X), 10%
aq. Na2CO3 (3X), and brine (1X). The organic extract was dried (MgSO4) and
concentrated, and the residue purified via silica flash chromatography (45 g silica, 1% to
20% methanolic ammonia:EtOAc) to give a colorless oil. Yield: 756 mg, 1.72 mmol,
65%. 1H NMR (500 MHz, Chloroform-d) δ 8.09 (d, J = 8.4 Hz, 1H), 7.88 (d, J = 8.2 Hz,
1H), 7.74 (d, J = 8.1 Hz, 0.5H), 7.66 (d, J = 8.0 Hz, 0.5H), 7.59 (ddd, J = 8.2, 7.0, 1.1 Hz,
1H), 7.47 (ddd, J = 8.2, 7.0, 1.1 Hz, 1H), 7.36 (t, J = 7.6 Hz, 1H), 7.30 – 7.25 (m, 0.5H),
7.23 – 7.16 (m, 1.5H), 7.04 (d, J = 8.0 Hz, 1H), 6.99 (d, J = 8.1 Hz, 0.5H), 6.68 (d, J =
11.9 Hz, 0.5H), 6.37 (d, J = 1.2 Hz, 2H), 5.59 – 5.42 (m, 1H), 4.16 (ddt, J = 19.2, 14.0,
7.2 Hz, 2H), 3.98 – 3.60 (m, 2.5H), 3.51 (d, J = 12.2 Hz, 1H), 3.42 – 3.25 (m, 1H), 2.79
(s, 0.5H), 2.53 (d, J = 47.4 Hz, 0.5H), 2.41 (s, 0.5H), 2.20 – 2.04 (m, 1H), 1.94 (bs,
1.5H), 1.77 (bd, J = 12.1 Hz, 1H), 1.63 (m, 0.5H), 1.51 (bs, 0.5H), 1.38 (m, J = 10.0 Hz,
0.5H), 1.33 – 1.01 (m, 3H).

155
Methyl 1-(4-chlorobenzyl)-4-fluoro-1H-pyrrole-2-carboxylate (138). Methyl 4-
fluoro-1H-pyrrole-2-carboxylate 137 (130 mg, 0.91 mmol) was dissolved in anhydrous
DMF at room temperature, followed by the addition of potassium carbonate (151 mg,
1.09 mmol) and 2a (0.14 mL, 1.09 mmol). The reaction was then stirred for 30 h at 60
°C, after which time it was allowed to cool to room temperature. The reaction was diluted
with a 1:1 solution of ethyl acetate/diethyl ether, washed with water (3×) and brine (1×),
dried with magnesium sulfate, and concentrated in vacuo. The resulting brown residue
was purified via flash chromatography (60 g silica, 10% ethyl acetate/hexanes) to afford
methyl 1-(4-chlorobenzyl)-4-fluoro-1H-pyrrole-2-carboxylate as a light-yellow oil.
Yield: 203 mg (83%). 1H NMR (400 MHz, CDCl3) δ 7.27 (d, J = 8.3 Hz, 2H), 7.03 (d, J
= 8.3 Hz, 2H), 6.69−6.59 (m, 2H), 5.44 (s, 2H), 3.75 (s, 3H).
1-(4-Chlorobenzyl)-4-fluoro-1H-pyrrole-2-carboxylic acid (139). Methyl 1-(4-
chlorobenzyl)-4-fluoro-1H-pyrrole-2-carboxylate 138 (150 mg, 0.56 mmol) was
dissolved in ethanol (10 mL) at room temperature. Then 10% aqueous sodium hydroxide
(2 mL) was added, and the reaction was stirred for 18 h at room temperature. At this time,
solvent was stripped off in vacuo until material began to precipitate. Additional water
was added (2 mL), and the solution was cooled in an ice bath, then acidified with
concentrated HCl. The resulting precipitate was collected over a filter and washed with
cold 1 M HCl, then dried under high vacuum to afford the desired carboxylic acid as a
white powder. Yield: 111 mg (78%). 1H NMR (400 MHz, CDCl3) δ 7.29 (d, J = 8.3 Hz,
2H), 7.04 (d, J = 8.3 Hz, 2H), 6.79 (d, J = 2.0 Hz, 1H), 6.71−6.65 (m, 1H), 5.43 (s, 2H).

156
Ethyl 1-(4-chlorobenzyl)-1H-pyrrolo[2,3-c]pyridine-2-carboxylate (144). Ethyl 1-
(4-chlorobenzyl)-1H-pyrrolo[2,3-c]pyridine-2-carboxylate 143 (300 mg, 0.953 mmol)
was dissolved into ethanol (8 mL) at RT, followed by the addition of 10% aqueous NaOH
(8 mL), which initially caused precipitation, but homogeneity was achieved over time.
The reaction was allowed to stir at RT for 12 h, at which time the solvent was removed in
vacuo. The residue was taken up in a small amount of water (5 mL), the pH was adjusted
to ∼4, and material was extracted with ethyl acetate (8×). The extracts were combined
and the solvent removed again in vacuo to provide the title compound as a fine white
powder. Yield: 226 mg, 83%. 1H NMR (400 MHz, DMSO-d6) δ 9.64 (s, 1H), 8.40 (d, J =
6.3 Hz, 1H), 8.26 (d, J = 6.3 Hz, 1H), 7.61 (s, 1H), 7.35 (d, J = 8.5 Hz, 2H), 7.10 (d, J =
8.5 Hz, 2H), 6.06 (s, 2H).
(4-(Cyclohexylmethyl)piperazin-1-yl)(1-(4-fluorobenzyl)-1H-indol-2-
yl)methanone (CCG-203941). The following reagents were dissolved in anhydrous DMF
(1.5 mL) at RT: 1-(4-fluorobenzyl)-1H-indole-2-carboxylic acid 4b (50 mg, 0.186
mmol), 1-(cyclohexylmethyl)piperazine (34 mg, 0.186 mmol), DIPEA (97 µL, 0.557
mmol), EDCHCl (46 mg, 0.241 mmol), and HOBT (37 mg, 0.241 mmol). The reaction
was allowed to stir for 24 h at RT, after which time it was diluted with 1:1 EtOAc:Et2O
and washed with 10% aq. Na2CO3 (2X), then dried (MgSO4) and concentrated. The
residue was purified by silica flash chromatography (20 g silica, 60% EtOAc:Hex).
Yield: 45 mg, 0.10 mmol, 53%. 1H NMR (500 MHz, Chloroform-d) δ 7.67 (d, J = 7.9
Hz, 1H), 7.41 (d, J = 8.1 Hz, 1H), 7.32 – 7.29 (m, 1H), 7.20 – 7.16 (m, 1H), 7.11 (dd, J =

157
8.6, 5.4 Hz, 2H), 6.95 (t, J = 8.7 Hz, 2H), 6.64 (s, 1H), 5.51 (s, 2H), 3.65 (bd, J = 74.0
Hz, 4H), 2.36 (s, 2H), 2.11 – 1.95 (m, 4H), 1.79 – 1.67 (m, 5H), 1.31 – 1.16 (m, 4H), 0.89
– 0.80 (m, 2H). HPLC tR = 6.65 min, >95% purity.
(4-Benzylpiperazin-1-yl)(1-(4-fluorobenzyl)-1H-indol-2-yl)methanone (CCG-
203942). The following was added sequentially to anhydrous DMF: 1-(4-fluorobenzyl)-
1H-indole-2-carboxylic acid (50 mg, 0.186 mmol), DIPEA (65 µL, 0.371 mmol),
EDCHCl (46 mg, 0.241 mmol), HOBT (37 mg, 0.241 mmol), and 1-benzylpiperazine
(33 mg, 0.186 mmol). The solution was allowed to stir at room temperature for 22 h. At
this time, a 1:1 solution of EtOAc:Et2O (5 mL) was added, and this was washed with
aqueous 10% Na2CO3 (3X). The extract was then dried with anhydrous MgSO4, filtered,
and the filtrate was concentrated in vacuo. The crude residue was purified by column
chromatography (20 g silica, 80% EtOAC/Hexanes) to provide the title compound. Yield:
57 mg, 0.13 mmol, 72 %. 1H NMR (500 MHz, Chloroform-d) δ 7.68 (d, J = 7.9 Hz, 1H),
7.43 (d, J = 8.3 Hz, 1H), 7.39 – 7.31 (m, 6H), 7.20 (t, J = 7.5 Hz, 1H), 7.12 (dd, J = 7.8,
5.6 Hz, 2H), 6.98 (t, J = 8.5 Hz, 2H), 6.66 (s, 1H), 5.53 (s, 2H), 3.68 (bd, J = 67.9 Hz,
4H), 3.49 (s, 2H), 2.21 (bd, 4H). HPLC tR = 5.91 min, > 95%.
(4-Benzoylpiperidin-1-yl)(1-(4-fluorobenzyl)-1H-indol-2-yl)methanone (CCG-
203943). The following reagents were dissolved in anhydrous DMF (2 mL) at RT: 1-(4-
fluorobenzyl)-1H-indole-2-carboxylic acid (50 mg, 0.186 mmol), phenyl(piperidine-4-
yl)methanone hydrochloride (42 mg, 0.186 mmol), DIPEA (72 µL, 0.557 mmol),
EDCHCl (46 mg, 0.241 mmol), and HOBT (37 mg, 0.241 mmol). The reaction was

158
allowed to stir for 24 h at RT, after which time it was diluted with 1:1 EtOAc:Et2O and
washed with 10% aq. Na2CO3 (3X), then dried (MgSO4) and concentrated. The residue
was purified by silica flash chromatography (20 g silica, 80% EtOAc:Hex). Yield: 68 mg,
0.154 mmol, 83%. 1H NMR (500 MHz, Chloroform-d) δ 7.96 (dt, J = 8.4, 1.2 Hz, 2H),
7.72 – 7.65 (m, 1H), 7.64 – 7.58 (m, 1H), 7.54 – 7.49 (m, 2H), 7.41 (d, J = 8.3 Hz, 1H),
7.30 (ddd, J = 8.5, 7.1, 1.1 Hz, 1H), 7.19 (td, J = 7.4, 1.0 Hz, 1H), 7.16 – 7.11 (m, 2H),
7.02 – 6.93 (m, 2H), 6.69 (d, J = 0.9 Hz, 1H), 5.52 (s, 2H), 4.63 (bs, 1H), 4.20 (bs, 1H),
3.51 (tt, J = 10.7, 3.7 Hz, 1H), 3.06 (s, 2H), 2.05 – 1.48 (m, 4H). HPLC tR = 8.32 min,
>95%.
(4-(Benzyloxy)piperidin-1-yl)(1-(4-fluorobenzyl)-1H-indol-2-yl)methanone
(CCG-203944). The following reagents were dissolved in anhydrous DMF (3 mL) at RT:
1-(4-fluorobenzyl)-1H-indole-2-carboxylic acid (50 mg, 0.186 mmol), 4-
(benzyloxy)piperidine hydrochloride (42 mg, 0.186 mmol), DIPEA (72 µL, 0.557 mmol),
EDCHCl (46 mg, 0.241 mmol), and HOBT (37 mg, 0.241 mmol). The reaction was
allowed to stir for 24 h at RT, after which time it was diluted with 1:1 EtOAc:Et2O and
washed with 10% aq. Na2CO3 (3X), then dried (MgSO4) and concentrated. The residue
was purified by silica flash chromatography (20 g silica, 80% EtOAc:Hex). Yield: 29 mg,
0.066 mmol, 35%. 1H NMR (500 MHz, Chloroform-d) δ 7.70 (d, J = 7.9 Hz, 1H), 7.45 –
7.37 (m, 5H), 7.36 – 7.28 (m, 2H), 7.21 (ddd, J = 7.9, 7.0, 0.9 Hz, 1H), 7.16 – 7.11 (m,
2H), 7.00 – 6.94 (m, 2H), 6.68 (d, J = 0.8 Hz, 1H), 5.53 (s, 2H), 4.58 (s, 2H), 3.92 (bd, J
= 66.0 Hz, 2H), 3.66 (tt, J = 7.2, 3.5 Hz, 1H), 3.47 (bd, J = 48.5 Hz, 2H), 1.97 – 1.42 (m,
4H). HPLC tR = 8.86 min, >95% purity.

159
(1-(4-Fluorobenzyl)-1H-indol-2-yl)(4-(piperidine-1-carbonyl)piperidin-1-
yl)methanone (CCG-203945). The following was added sequentially to anhydrous DMF
(2 mL): 1-(4-fluorobenzyl)-1H-indole-2-carboxylic acid (50 mg, 0.186 mmol), DIPEA
(0.065 ml, 0.371 mmol), EDCHCl (46.3 mg, 0.241 mmol), HOBT (37.0 mg, 0.241
mmol), and piperidin-1-yl(piperidin-4-yl)methanone (36.4 mg, 0.186 mmol). The
solution was allowed to stir at RT for 24 h. At this time, a 1:1 solution of EtOAc:Et2O (5
mL) was added, and this was washed with aqueous 10% Na2CO3 (3 x 2 mL). The extract
was then dried with anhydrous MgSO4, filtered, and the filtrate was concentrated in
vacuo. The crude residue was purified by column chromatography (20 g silica, 80%
EtOAc/Hex) to provide the title compound. Yield: 35 mg, 0.065 mmol, 35%. 1H NMR
(500 MHz, Chloroform-d) δ 7.70 – 7.64 (m, 1H), 7.37 (d, J = 8.4 Hz, 1H), 7.28 (dt, J =
5.9, 1.3 Hz, 1H), 7.17 (td, J = 7.5, 7.0, 1.0 Hz, 1H), 7.12 (dd, J = 8.5, 5.4 Hz, 2H), 6.99 –
6.93 (m, 2H), 6.67 (s, 1H), 5.49 (s, 2H), 4.61 (bs, 1H), 4.24 (bs, J = 8.8, 5.8 Hz, 1H), 3.58
(t, J = 5.5 Hz, 2H), 3.49 – 3.37 (m, 2H), 2.93 (d, J = 12.7 Hz, 2H), 2.75 (dq, J = 14.3, 7.7,
7.1 Hz, 1H), 1.77 – 1.58 (m, 8H), 1.31 – 1.25 (m, 1H), 0.94 – 0.81 (m, 1H). HPLC tR =
7.55 min, >95% purity.
(4-(Dibenzylamino)piperidin-1-yl)(1-(4-fluorobenzyl)-1H-indol-2-yl)methanone
(CCG-204020). Tert-butyl 4-(dibenzylamino)piperidine-1-carboxylate (330 mg, 0.867
mmol) was dissolved in DCM (5 mL), followed by the addition of trifluoroacetic acid
(0.200 mL, 2.60 mmol). The reaction was allowed to stir for 18 h at RT, after which time

160
the solvent was removed in vacuo and the crude residue of N,N-dibenzylpiperidin-4-
amine bis(trifluoroacetate) was used directly in the subsequent reaction
The following reagents were dissolved in anhydrous DMF (5 mL) at RT: 1-(4-
fluorobenzyl)-1H-indole-2-carboxylic acid (233 mg, 0.867 mmol), N,N-
dibenzylpiperidin-4-amine bis(trifluoroacetate) (243 mg, 0.867 mmol), DIPEA (757 µL,
4.34 mmol), EDCHCl (183 mg, 0.954 mmol), and HOBT (146 mg, 0.954 mmol). The
reaction was allowed to stir for 36 h at RT, after which time it was diluted with 1:1
EtOAc:Et2O and washed with 10% aq. Na2CO3 (2X), then dried (MgSO4) and
concentrated. The residue was triturated with Et2O to yield a light brown solid. Yield:
380 mg, 0.715 mmol, 82%. 1H NMR (500 MHz, Chloroform-d) δ 7.69 (dd, J = 8.0, 3.9
Hz, 1H), 7.43 – 7.38 (m, 5H), 7.37 – 7.31 (m, 5H), 7.29 – 7.25 (m, 2H), 7.21 (t, J = 7.3,
3.4 Hz, 1H), 7.11 – 7.07 (m, 2H), 6.94 (t, J = 11.1, 8.7, 2.0 Hz, 2H), 6.65 (s, 1H), 5.53 (s,
2H), 4.78 (bs, 1H), 4.17 (bs, 1H), 3.59 (s, 4H), 2.85 – 2.67 (m, 2H), 2.58 (bs, 1H), 1.92
(bs, 1H), 1.67 (bs, 1H), 1.49 (bs, 1H), 1.04 (bs, J = 30.5 Hz, 1H). HPLC tR = 6.48 min,
>95%.
N-Benzyl-1-(1-(4-chlorobenzyl)-1H-pyrrole-2-carbonyl)piperidine-4-
carboxamide (CCG-204054). The following was dissolved in anhydrous DMF (3 mL): 1-
(1-(4-chlorobenzyl)-1H-pyrrole-2-carbonyl)piperidine-4-carboxylic acid (115 mg, 0.332
mmol), Hunig's Base (0.174 ml, 0.995 mmol), EDCHCl (76 mg, 0.398 mmol), HOBt
(60.9 mg, 0.398 mmol), and benzylamine (0.040 ml, 0.365 mmol). This was stirred at
room temperature for 1 day with 3Å molecular sieves. At this time, a 1:1 solution of
EtOAc:Et2O (20 mL) was added and the organic layer was washed with 10% aq.

161
Na2CO3 (2 x 10 mL) and brine (1 x 10 mL). The organic solution was then dried
(anhydrous MgSO4) and concentrated in vacuo. Trituration in Et2O provided 101 mg of
the title compound as a fine, ruddy brown solid. 1H NMR (500 MHz, Chloroform-d) δ
7.39 – 7.34 (m, 2H), 7.33 – 7.26 (m, 5H), 7.06 (dd, J = 8.9, 2.5 Hz, 2H), 6.82 (dd, J = 2.7,
1.7 Hz, 1H), 6.35 (dd, J = 3.7, 1.6 Hz, 1H), 6.15 (dd, J = 3.7, 2.7 Hz, 1H), 5.76 (s, 1H),
5.30 (s, 2H), 4.47 (d, J = 5.6 Hz, 2H), 4.39 (bd, J = 13.3 Hz, 2H), 2.86 (t, J = 12.7 Hz,
2H), 2.33 (tt, J = 11.3, 3.8 Hz, 1H), 1.81 (d, J = 13.0 Hz, 2H), 1.51 (d, J = 12.9 Hz, 2H).
HPLC tR = 7.06 min, > 95%.
(1-(4-Fluorobenzyl)-1H-indol-2-yl)(4-(piperidin-1-ylmethyl)piperidin-1-
yl)methanone (CCG-204055). Tert-butyl 4-(piperidin-1-ylmethyl)piperidine-1-
carboxylate (100 mg, 0.469 mmol) was dissolved in DCM (5 mL), and TFA (1 mL) was
added. The reaction was allowed to stir for 12 h, after which time the solvent was
stripped off in vacuo, then thoroughly removed under high vacuum. The crude residue
was then taken directly into the next step.
The following was added to anhydrous DMF (2 mL) sequentially: 1-(4-
fluorobenzyl)-1H-indole-2-carboxylic acid (66.8 mg, 0.248 mmol), DIPEA (0.217 ml,
1.240 mmol), EDCHCl (52.3 mg, 0.273 mmol), HOBT (41.8 mg, 0.273 mmol), and 1-
(piperidin-4-ylmethyl)piperidine trifluoroacetate (45.2 mg, 0.248 mmol). This was stirred
at room temperature for 24 h with 3Å MS. At this time, a 1:1 solution of EtOAc:Et2O
was added and the solution was washed with 10% aq. Na2CO3. The organic phase was
dried with MgSO4 and concentrated in vacuo. The residue was then purified by silica gel

162
chromatography (20g silica, 80% EtOAc/Hexanes) to give an oil. Yield: 58 mg , 0.134
mmol, 54%. HPLC tR = 5.36 min, > 95%. 1H NMR (500 MHz, Chloroform-d) δ 7.66 (d,
J = 7.9 Hz, 1H), 7.40 (d, J = 8.3 Hz, 1H), 7.29 (ddd, J = 8.3, 6.9, 1.2 Hz, 1H), 7.18 (ddd,
J = 8.0, 7.0, 0.9 Hz, 1H), 7.14 – 7.09 (m, 2H), 6.99 – 6.92 (m, 2H), 6.63 (d, J = 0.7 Hz,
1H), 5.50 (s, 2H), 4.66 (bs, 1H), 4.10 (bs, 1H), 2.77 (d, J = 64.4 Hz, 2H), 2.41 – 2.27 (m,
4H), 2.11 – 2.02 (m, 2H), 1.83 – 1.53 (m, 5H), 1.48 – 1.39 (m, 2H), 1.05 (bs, 1H), 0.66
(bs, 1H). HPLC tR = 5.36 min, >95% purity.
(1-(4-Fluorobenzyl)-1H-indol-2-yl)(4-(hydroxy(phenyl)methyl)piperidin-1-
yl)methanone (CCG-204056). The following was added sequentially to anhydrous DMF
(5 mL): 1-(4-fluorobenzyl)-1H-indole-2-carboxylic acid (0.119 g, 0.443 mmol),
Hunig'sBase (0.232 ml, 1.329 mmol), EDCHCl (0.093 g, 0.487 mmol), HOBT (0.075 g,
0.487 mmol), and phenyl(piperidin-4-yl)methanol (0.085 g, 0.443 mmol). The solution
was stirred at room temperature for 30 h, at which time a 1:1 solution of EtOAc:Et2O
was added and washed with 10% aq. Na2CO3. The organic solution was dried with
MgSO4 and concentrated in vacuo to give a residue that was purified by silica gel
chromatography (45 g silica, 80% EtOAc/Hex). Yield: 67 mg, . 1H NMR (500 MHz,
Chloroform-d) δ 7.66 (d, J = 7.9 Hz, 1H), 7.38 (dd, J = 14.5, 7.8 Hz, 3H), 7.32 – 7.26 (m,
4H), 7.17 (t, J = 7.5 Hz, 1H), 7.09 (dd, J = 8.5, 5.3 Hz, 2H), 6.98 – 6.92 (m, 2H), 6.61 (s,
1H), 5.48 (d, J = 6.5 Hz, 2H), 4.67 (d, J = 54.5 Hz, 1H), 4.30 – 4.23 (m, 1H), 4.22 – 3.93
(m, 1H), 2.86 – 2.52 (m, 2H), 2.43 – 1.83 (m, 2H), 1.79 (dtd, J = 11.5, 7.7, 3.7 Hz, 1H),
1.28 – 0.94 (m, 2H). HPLC tR = 7.85 min, > 95%.

163
(4-(Benzylamino)piperidin-1-yl)(1-(4-fluorobenzyl)-1H-indol-2-yl)methanone
(CCG-205420). (4-(Dibenzylamino)piperidin-1-yl)(1-(4-fluorobenzyl)-1H-indol-2-
yl)methanone CCG-204020 was dissolved in EtOH (10 mL) and THF (10 mL), along
with a few drops of conc. HCl. This solution was sparged with N2 and Pd(OH)2 was
added. The reaction was then shaken on a hydrogenator at 45 psi H2 at RT for 1 h (longer
reaction times cause rapid accumulation of complete debenzylation.) The reaction was
filtered over celite and the filtrate was diluted with EtOAc and washed with sat. aq.
Na2CO3 (1X), dried (MgSO4) and concentrated to give the desired compound as an oil.
Yield: 77%. 1H NMR (500 MHz, Chloroform-d) δ 7.70 – 7.65 (m, 1H), 7.41 – 7.32 (m,
5H), 7.32 – 7.27 (m, 2H), 7.22 – 7.16 (m, 1H), 7.12 (dd, J = 8.3, 5.3 Hz, 2H), 6.96 (ddd, J
= 9.7, 7.5, 1.1 Hz, 2H), 6.65 (s, 1H), 5.51 (s, 2H), 4.49 (bs, 1H), 4.07 (bs, 1H), 3.83 (s,
2H), 3.00 (bs, 2H), 2.77 (tt, J = 9.9, 4.0 Hz, 1H), 2.04 – 1.64 (m, 2H), 1.32 (m, 2H). TOF
ES+ MS: 442.1 (M + H), 464.1 (M + Na). HPLC tR = 5.83 min, > 90%.
(4-(Cyclohexyl(hydroxy)methyl)piperidin-1-yl)(1-(4-fluorobenzyl)-1H-indol-2-
yl)methanone (CCG-205421). Tert-butyl 4-(cyclohexyl(hydroxy)methyl)piperidine-1-
carboxylate (100 mg, 0.336 mmol) was dissolved in anhydrous THF (5 mL) and TFA (52
µL, 0.672 mmol) and stirred at RT for 15 h. After this time, the solvent was removed in
vacuo via rotovap, then thoroughly removed under high vacuum. The resulting residue
was taken directly into the next step.
4-(Cyclohexyl(hydroxy)methyl)piperidine trifluoroacetate (~105 mg, 0.336
mmol) was dissolved in anhydrous DMF (3 mL), followed by 1-(4-fluorobenzyl)-1H-
indole-2-carboxylic acid (100 mg, 0.370 mmol), DIPEA (0.352 mL, 2.016 mmol),

164
EDCHCl (77 mg, 0.403 mmol), and HOBT (62 mg, 0.403 mmol). The reaction was
allowed to stir at RT for 24 h, after which time the reaction was diluted with 1:1
EtOAc:Et2O and washed with 10% aq. Na2CO3 (3X), dried (MgSO4), and concentrated.
The resulting residue was purified by silica flash chromatography (20 g silica, 80%
EtOAc:Hex). Yield: 90 mg, 0.202 mmol, 60%. 1H NMR (500 MHz, Chloroform-d) δ
7.66 (d, J = 7.9 Hz, 1H), 7.40 (d, J = 8.3 Hz, 1H), 7.31 – 7.27 (m, 1H), 7.18 (ddd, J = 7.9,
7.0, 1.0 Hz, 1H), 7.14 – 7.08 (m, 2H), 6.98 – 6.92 (m, 2H), 6.64 (d, J = 0.8 Hz, 1H), 5.51
(s, 2H), 4.75 (bs, 1H), 4.15 (bs, 1H), 3.05 (d, J = 5.6 Hz, 1H), 2.83 (bs, 1H), 2.68 (bs,
1H), 1.82 – 1.58 (m, 7H), 1.41 – 1.04 (m, 9H). ES+ TOF MS: 449.2 (M + H), 471.2 (M +
Na). HPLC tR = 8.72 min, > 90%.
N-Benzyl-1-(1-(4-chlorobenzyl)-3-methyl-1H-indole-2-carbonyl)piperidine-4-
carboxamide (CCG-205431). Ethyl 1-(4-chlorobenzyl)-3-methyl-1H-indole-2-
carboxylate (310 mg, 0.946 mmol) was dissolved in THF (20 mL) and 5 M aq. NaOH (20
mL) and stirred at 55 °C for 18 h. The reaction was then cooled to 0 °C and acidified to
pH<1 with conc. HCl and material was extracted from the aqueous phase with 1:1
EtOAc:Et2O. The extract was dried (MgSO4) and concentrated, giving 1-(4-
chlorobenzyl)-3-methyl-1H-indole-2-carboxylic acid as a yellow solid. Yield: 273 mg,
0.911 mmol, 96%.
The following reagents were dissolved in anhydrous DMF (3 mL) at RT: 1-(4-
chlorobenzyl)-3-methyl-1H-indole-2-carboxylic acid (90 mg, 0.315 mmol), N-
benzylpiperidine-4-carboxamide hydrochloride (70 mg, 0.315 mmol), DIPEA (170 µL,
0.945 mmol), and HATU (144 mg, 0.378 mmol). The reaction was allowed to stir for 20

165
h at RT, after which time it was diluted with 1:1 EtOAc:Et2O and washed with 10% aq.
Na2CO3 (3X), then dried (MgSO4) and concentrated. The residue was purified by silica
flash chromatography (20 g silica, 50% EtOAc:Hex). Yield: 77 mg, 0.153 mmol, 51%.
1H NMR (400 MHz, Chloroform-d) δ 7.59 (d, J = 8.0 Hz, 1H), 7.33 (d, J = 8.3 Hz, 7H),
7.18 (t, J = 7.8 Hz, 3H), 7.03 (t, J = 8.2 Hz, 2H), 5.73 (s, 1H), 5.57 (s, 1H), 5.38 (d, J =
11.8 Hz, 2H), 4.77 (d, J = 10.9 Hz, 1H), 4.44 (d, J = 5.9 Hz, 2H), 3.76 (d, J = 14.8 Hz,
1H), 3.08 (d, J = 17.3 Hz, 1H), 2.80 (s, 3H), 2.29 (d, J = 21.5 Hz, 3H), 1.96 (d, J = 11.2
Hz, 1H), 1.76 (d, J = 17.2 Hz, 1H), 1.52 (s, 1H). ES+ TOF MS: 500.3 (M + H), 522.2 (M
+ Na). HPLC tR = 7.95 min, > 95%.
(1-(4-Chlorobenzyl)-1H-indol-2-yl)(4-(isoindoline-2-carbonyl)piperidin-1-
yl)methanone. (CCG-205470) The following was added sequentially to DCM (2 mL): 1-
(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)piperidine-4-carboxylic acid (50 mg, 0.126
mmol), DIPEA (0.07 mL, 0.378 mmol), EDCHCl (30 mg, 0.151 mmol), HOBT (24 mg,
0.151 mmol), and isoindoline (0.018 mL, 0.151 mmol). The mixture was stirred for 18 h
at RT, at which time the solution diluted with a 1:1 solution of EtOAc:Et2O and washed
with 1M HCl (1X), 10% aq. Na2CO3 (1X) and brine (1X). The organic phase was dried
with magnesium sulfate and concentrated in vacuo. The resulting residue was dissolved
in EtOAc and precipitated out with Hex, and then collected over a filter and washed with
a 10:1 solution of Hex:EtOAc. Yield: 22 mg, 0.044 mmol, 35%. 1H NMR (400 MHz,
Chloroform-d) δ 7.65 (d, J = 7.9 Hz, 1H), 7.40 – 7.21 (m, 8H), 7.15 (t, J = 7.4 Hz, 1H),
7.05 (d, J = 8.3 Hz, 2H), 6.68 (d, J = 1.6 Hz, 1H), 5.47 (s, 2H), 4.88 (s, 2H), 4.81 (s, 2H),

166
4.42 (bs, 4H), 2.97 (bs, 2H), 2.72 (td, J = 14.7, 12.7, 4.7 Hz, 1H), 1.78 (bs, 2H). ES+
TOF MS: 498.2 (M + H), 520.2 (M + Na). HPLC tR = 5.85 min, > 95%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(2,3-dihydro-1H-inden-2-yl)piperidine-
4-carboxamide. (CCG-205471) The following was added sequentially to DCM (2 mL):
1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)piperidine-4-carboxylic acid (50 mg, 0.126
mmol), DIPEA (0.07 mL, 0.378 mmol), EDCHCl (30 mg, 0.151 mmol), HOBT (24 mg,
0.151 mmol), and 2-aminoindane (0.020 mL, 0.151 mmol). The mixture was stirred for
36 h at rt, at which time the solution diluted with a 1:1 solution of EtOAc:Et2O and
washed with 1M HCl (1X), 10% aq. Na2CO3 (1X) and brine (1X), which resulted in a
precipitate that failed to go into either phase. The precipitate was collected and washed
with water and Et2O to afford the title compound as a white solid. Yield: 52 mg, 0.102
mmol, 81%. 1H NMR (400 MHz, Chloroform-d) δ 7.64 (d, J = 7.9 Hz, 1H), 7.35 – 7.29
(m, 1H), 7.25 – 7.11 (m, 8H), 7.03 (d, J = 8.1 Hz, 2H), 6.64 (s, 1H), 5.62 (d, J = 7.6 Hz,
1H), 5.46 (s, 2H), 4.83 – 4.69 (m, 1H), 4.32 (bs, 4H), 3.33 (dd, J = 16.3, 7.0 Hz, 2H),
2.89 – 2.72 (m, 4H), 2.27 – 2.14 (m, 1H), 1.74 (bs, 2H). ES+ TOF MS: 512.2 (M + H),
534.2 (M + Na). HPLC tR = 5.96 min, > 95%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(2,3-dihydro-1H-inden-1-
yl)piperidine-4-carboxamide (CCG-205472). The following was added sequentially to
DCM (2 mL): 1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)piperidine-4-carboxylic acid
71 (50 mg, 0.13 mmol), DIPEA (0.07 mL, 0.38 mmol), EDCHCl (30 mg, 0.15 mmol),
HOBT (24 mg, 0.15 mmol), and 1-aminoindane (20 µL, 0.15 mmol). The mixture was

167
stirred for 18 h at rt, at which time the solution diluted with a 2:1 solution of EtOAc:Et2O
and washed with 1M HCl (2X), 10% aq. Na2CO3 (1X) and brine (1X). The organic phase
was dried with MgSO4 and concentrated in vacuo. The resulting residue was dissolved in
EtOAc and precipitated out Hex, and then collected over a filter and washed with Et2O to
give the title compound as a white solid. Yield: 33 mg, 0.06 mmol, 51%. 1H NMR (500
MHz, Chloroform-d) δ 7.66 (d, J = 7.9 Hz, 1H), 7.34 (d, J = 8.3 Hz, 1H), 7.29 – 7.20 (m,
7H), 7.17 (t, J = 7.4 Hz, 1H), 7.04 (d, J = 8.0 Hz, 2H), 6.66 (s, 1H), 5.66 (d, J = 8.4 Hz,
1H), 5.47 (s, 2H), 4.49 (bs, 1H), 4.33 (bs, 1H), 3.04 – 2.95 (m, 1H), 2.89 (dt, J = 15.9, 8.0
Hz, 3H), 2.62 (dtd, J = 12.2, 7.8, 3.8 Hz, 1H), 2.33 (tt, J = 11.1, 3.9 Hz, 1H), 1.94 – 1.54
(m, 6H). ES+ TOF MS: 512.2 (M + H), 534.2 (M + Na). HPLC tR = 5.93 min, > 95%.
(1-(4-Chlorobenzyl)-1H-indol-2-yl)(4-(2-phenylpyrrolidine-1-carbonyl)piperidin-
1-yl)methanone. (CCG-205473) The following was added sequentially to DCM (2 mL):
1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)piperidine-4-carboxylic acid 71 (50 mg,
0.126 mmol), DIPEA (0.07 mL, 0.378 mmol), EDCHCl (30 mg, 0.151 mmol), HOBT
(24 mg, 0.151 mmol), and 2-phenylpyrrolidine (0.023 mL, 0.151 mmol). The mixture
was stirred for 18 h at rt, at which time the solution diluted with a 1:1 solution of
EtOAc:Et2O and washed with 1M HCl (1X), 10% aq. sodium carbonate (1X) and brine
(1X). The organic phase was dried with magnesium sulfate and concentrated in vacuo.
The resulting residue was dissolved in EtOAc and precipitated out hexanes, and then
collected over a filter and washed with diethyl ether to give the title compound as a white
solid. Yield: 20 mg, 0.038 mmol, 30%. 1H NMR (400 MHz, Chloroform-d) δ 7.66 – 7.57
(m, 1H), 7.33 – 7.14 (m, 10H), 7.00 (d, J = 8.1 Hz, 2H), 6.58 (s, 1H), 5.41 (s, 2H), 4.34

168
(s, 4H), 3.73 (dd, J = 19.2, 11.3 Hz, 2H), 3.07 – 2.88 (m, 1H), 2.73 (s, 1H), 2.51 – 2.26
(m, 3H), 2.04 – 1.44 (m, 5H). ES+ TOF MS: 526.2 (M + H), 548.2 (M + Na). HPLC tR =
6.62 min, > 95%.
(1-(4-Chlorobenzyl)-1H-indol-2-yl)(4-(3-phenylpyrrolidine-1-carbonyl)piperidin-
1-yl)methanone. (CCG-205474) The following was added sequentially to DCM (2 mL):
1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)piperidine-4-carboxylic acid (50 mg, 0.126
mmol), DIPEA (0.07 mL, 0.378 mmol), EDCHCl (30 mg, 0.151 mmol), HOBT (24 mg,
0.151 mmol), and 3-phenylpyrrolidine (0.023 mL, 0.151 mmol). The mixture was stirred
for 36 h at rt, at which time the solution diluted with a 1:1 solution of EtOAc:Et2O and
washed with 1M HCl (1X), 10% aq. sodium carbonate (1X) and brine (1X). The organic
phase was dried with magnesium sulfate and concentrated in vacuo. The resulting residue
was dissolved in EtOAc and precipitated out hexanes, and then collected over a filter and
washed with diethyl ether to give the title compound as a slightly yellow solid. Yield: 21
mg, 0.040 mmol, 32%. 1H NMR (500 MHz, Chloroform-d) δ 7.66 (dd, J = 7.8, 2.8 Hz,
1H), 7.34 (dq, J = 14.5, 7.4 Hz, 3H), 7.29 – 7.20 (m, 6H), 7.16 (t, J = 7.8 Hz, 1H), 7.06
(dd, J = 7.9, 5.4 Hz, 2H), 6.68 (d, J = 6.3 Hz, 1H), 5.48 (d, J = 5.0 Hz, 2H), 4.57 (s, 1H),
4.33 (s, 1H), 4.03 (dd, J = 11.9, 7.5 Hz, 1H), 3.98 – 3.91 (m, 1H), 3.85 (ddd, J = 11.6,
8.3, 2.4 Hz, 1H), 3.73 (td, J = 9.1, 8.3, 2.9 Hz, 1H), 3.60 (td, J = 9.6, 6.8 Hz, 1H), 3.49
(ddd, J = 21.3, 9.6, 4.0 Hz, 2H), 3.38 (p, J = 7.9 Hz, 1H), 2.92 (s, 2H), 2.62 (dp, J = 21.4,
7.5 Hz, 1H), 2.46 – 2.36 (m, 1H), 2.31 (dq, J = 13.1, 4.1, 2.6 Hz, 1H), 2.16 – 2.00 (m,
2H). ES+ TOF MS: 526.3 (M + H), 548.2 (M + Na). HPLC tR = 6.37 min, > 95%.

169
(4-(Cyclohexanecarbonyl)piperidin-1-yl)(1-(4-fluorobenzyl)-1H-indol-2-yl)methanone
(CCG-205475). 4-(Cyclohexyl(hydroxy)methyl)piperidin-1-yl)(1-(4-fluorobenzyl)-1H-
indol-2-yl)methanone (85 mg, 0.189 mmol) was dissolved in DCM (4 mL) and cooled to
-78 °C. Anhydrous DMSO (34 µL, 0.474 mmol) and (COCl)2 (34 µL, 0.379 mmol) were
added, and the solution was stirred for 30 min, at which time TEA (130 µL, 0.947 mmol)
was added dropwise and the reaction was allowed to stir for an additional 30 min, then
the bath was removed and it was allowed to stir until it reached RT. The reaction was
diluted with 1:1 EtOAc:Et2O and washed with H2O (2X) and brine (1X), dried (MgSO4),
and concentrated in vacuo. The residue was purified via silica flash chromatography (50
g silica, 10% EtOAc:Hex). Yield: 49 mg, 0.106 mmol, 58%. 1H NMR (500 MHz,
Chloroform-d) δ 7.64 (d, J = 7.8 Hz, 1H), 7.37 (d, J = 8.4 Hz, 1H), 7.29 – 7.24 (m, 1H),
7.15 (t, J = 7.5 Hz, 1H), 7.08 (dd, J = 8.2, 5.5 Hz, 2H), 6.99 – 6.89 (m, 2H), 6.62 (s, 1H),
5.47 (s, 2H), 4.54 (bs, 1H), 4.15 (bs, 1H), 2.94 – 2.81 (m, 2H), 2.68 (tt, J = 11.2, 3.6 Hz,
1H), 2.46 (tt, J = 11.1, 2.9 Hz, 1H), 1.82 – 1.64 (m, 7H), 1.38 – 1.17 (m, 7H). TOF ES+
MS: (M + Na) 469.2. HPLC tR = 8.93 min, > 90%.
(1-(4-Chlorobenzyl)-1H-indol-2-yl)(4-(piperidin-1-ylmethyl)piperidin-1-
yl)methanone (CCG-205476). Tert-Butyl 4-(piperidin-1-ylmethyl)piperidine-1-
carboxylate (238 mg, 0.843 mmol) was dissolved in DCM (10 mL), and TFA (2 mL) was
added. The reaction was allowed to stir for 12 h, after which time the solvent was
stripped off in vacuo, then thoroughly removed under high vacuum. The crude residue
was then taken directly into the next step.

170
The following was added to DCM (6 mL): 1-(4-chlorobenzyl)-1H-indole-2-
carboxylic acid (110 mg, 0.380 mmol), DIPEA (0.50 mL, 2.95 mmol), EDCHCl (100
mg, 0.506 mmol), HOBT (80 mg, 0.506 mmol), and 1-(piperidin-4-ylmethyl)piperidine
(150 mg, 0.843 mmol). The solution was stirred at RT for 15 h, at which time the solution
was diluted with a 1:1 solution of EtOAc:Et2O and washed with H2O (1X), 10% aq.
Na2CO3 (1X), and brine (1X). The organic phase was then dried over MgSO4 and
concentrated in vacuo. The resulting residue was purified via silica flash chromatography
(40 g silica, 10% EtOAc:Hex) and triturated with EtOAc and Hex to afford the title
compound as a white powder. Yield: 109 mg, 0.242 mmol, 63%. 1H NMR (400 MHz,
Chloroform-d) δ 7.63 (d, J = 7.9 Hz, 1H), 7.35 (d, J = 8.3 Hz, 1H), 7.28 – 7.11 (m, 4H),
7.03 (d, J = 8.4 Hz, 2H), 6.59 (s, 1H), 5.46 (s, 2H), 4.59 (bs, 2H), 4.09 (bs, 2H), 2.73 (bs,
2H), 2.34 (bs, 2H), 2.08 (d, J = 6.8 Hz, 2H), 1.67 (s, 6H), 1.44 – 1.32 (m, 2H), 0.95 (bs,
1H), 0.56 (bs, 1H). ES+ TOF MS: 450.2 (M + H). HPLC tR = 6.18 min, > 95%.
(4-Benzylpiperazin-1-yl)(1-(4-chlorobenzyl)-1H-indol-2-yl)methanone (CCG-
206327). The following was added to DCM (5 mL): 1-(4-chlorobenzyl)-1H-indole-2-
carboxylic acid (70 mg, 0.245 mmol), DIPEA (0.130 mL, 0.735 mmol), EDCHCl (57
mg, 0.294 mmol), HOBT (45 mg, 0.294 mmol), and 1-benzylpiperazine (0.085 mL,
0.490 mmol). The solution was stirred at RT for 16 h, at which time the solution was
diluted with a 1:1 solution of EtOAc:Et2O and washed with H2O (1X), 10% aq. Na2CO3
(1 x), and brine (1 x). The organic phase was then dried over MgSO4 and concentrated in
vacuo. The resulting residue was purified via silica flash chromatography (30 g silica,
10% EtOAc:Hex) and triturated with EtOAc and Hex to afford the title compound as a

171
white powder. Yield: 52 mg, 0.117 mmol, 48%. 1H NMR (400 MHz, Chloroform-d) δ
7.62 (d, J = 7.9 Hz, 1H), 7.36 (d, J = 8.3 Hz, 1H), 7.32 – 7.19 (m, 8H), 7.14 (t, J = 7.5
Hz, 1H), 7.02 (d, J = 8.2 Hz, 2H), 6.60 (s, 1H), 5.48 (s, 2H), 3.63 (bs, 4H), 3.42 (s, 2H),
2.35 (bs, 2H), 2.04 (bs, J = 11.7 Hz, 2H). TOF ES+ MS: (M + H) 444.2, (M + Na) 466.2.
HPLC tR = 6.12 min, > 95%.
(1-(4-Chlorobenzyl)-1H-indol-2-yl)(4-(indoline-1-carbonyl)piperidin-1-
yl)methanone. (CCG-206328) The following was added sequentially to DCM (5 mL): 1-
(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)piperidine-4-carboxylic acid (100 mg, 0.252
mmol), DIPEA (1.4 mL, 0.756 mmol), EDCHCl (60 mg, 0.302 mmol), HOBT (48 mg,
0.302 mmol), and indoline (0.040 mL, 0.302 mmol). The mixture was stirred for 24 h at
rt, at which time the solution diluted with EtOAc and washed with water (2X) and brine
(1X). The organic phase was dried with magnesium sulfate and concentrated in vacuo.
The resulting residue was dissolved in EtOAc and precipitated out hexanes, and then
collected over a filter and washed with diethyl ether to give the title compound as a white
solid. Yield: 1H NMR (400 MHz, Chloroform-d) δ 7.66 (d, J = 7.8 Hz, 1H), 7.33 (t, J =
8.8 Hz, 1H), 7.30 – 7.13 (m, 8H), 7.04 (d, J = 8.2 Hz, 2H), 6.65 (s, 1H), 5.48 (s, 2H),
4.36 (s, 2H), 3.39 (t, J = 5.8 Hz, 2H), 3.08 (q, J = 9.6, 9.1 Hz, 2H), 2.99 – 2.82 (m, 2H),
2.71 – 2.64 (m, 2H), 2.26 (tt, J = 11.3, 3.9 Hz, 1H), 1.76 (s, 2H), 1.50 (s, 2H). TOF ES+
MS: (M + H) 498.2. HPLC tR = 8.58 min, >95% purity.
1-(1-(4-Chlorobenzyl)-1H-pyrrole-2-carbonyl)-N-(2-(pyridin-4-
yl)ethyl)piperidine-4-carboxamide (CCG-206381). The following was added

172
sequentially to DCM: 1-(1-(4-chlorobenzyl)-1H-pyrrole-2-carbonyl)piperidine-4-
carboxylic acid (600 mg, 1.73 mmol), TEA (0.725 mL, 5.19 mmol), EDCHCl (365 mg,
1.903 mmol), and HOBt (291 mqg, 1.903 mmol). This was allowed to stir at rt for 30
min, at which time pyridylethylamine ( 0.227 mL, 1.903 mmol) was added. Stirring
continued for 18 h. At this time, the DCM and TEA were stripped off, and the residue
was taken up in EtOAc and washed with 10% aqueous sodium carbonate (3X). The
organic phase was collected, dried over magnesium sulfate, and concentrated in vacuo.
The resulting solid/oil mixture was recrystallized from EtOAc to afford the title
compound as small, white crystals. Yield: 586 mg, 1.29 mmol, 75%. 1HNMR (400 MHz,
CDCl3) δ 8.50 (d, J = 5.7 Hz, 2H), 7.21 (d, J = 8.3 Hz, 2H), 7.09 (d, J = 5.6 Hz, 2H), 7.02
(d, J = 8.2 Hz, 2H), 6.78 (s, 1H), 6.30 (dd, J = 3.6, 1.2 Hz, 1H), 6.13−6.09 (m, 1H), 5.48
(t, J = 5.4 Hz, 1H), 5.26 (s, 2H), 4.32 (d, J = 12.7 Hz, 2H), 3.53 (q, J = 6.8 Hz, 2H), 2.82
(t, J = 7.0 Hz, 4H), 2.19 (tt, J = 11.2, 3.6 Hz, 1H), 1.68 (d, J = 14.4 Hz, 2H), 1.35 (q, J =
11.0, 9.8 Hz, 2H). Anal. Calcd for C25H27ClN4O2: C, 66.58%; H, 6.04%; N, 12.42%.
Found: C, 66.68%; H, 6.32%; N, 12.38%. TOF ES+ MS: (M + H) 451.2, (M + Na) 473.2.
HPLC tR = 5.03 min, >95% purity.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-((2,3-dihydro-1H-inden-2-
yl)methyl)piperidine-4-carboxamide (CCG-206382). The following was added
sequentially to DCM (8 mL): 1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)piperidine-4-
carboxylic acid (180 mg, 0.454 mmol), DIPEA (0.317 mL, 1.814 mmol), EDCHCl (96
mg, 0.499 mmol), HOBT (76 mg, 0.499 mmol), and crude (2,3-dihydro-1H-inden-2-
yl)methanamine (105 mg, 0.713 mmol). The mixture was stirred for 18 h at RT, at which

173
time the solution diluted with a 1:1 solution of EtOAc:Et2O and washed with 1 M HCl
(1X), 10% aq. Na2CO3 (1X). The organic phase was dried with MgSO4 and concentrated
in vacuo. Purification was accomplished via silica flash chromatography (60 g silica,
50% EtOAc:Hex), followed by EtOAc/Hex tritutation to give the title compound as a
white powder. Yield: 137 mg, 0.260 mmol, 57%. 1H NMR (400 MHz, Chloroform-d) δ
7.66 (d, J = 7.8 Hz, 1H), 7.33 (t, J = 8.8 Hz, 1H), 7.30 – 7.13 (m, 8H), 7.04 (d, J = 8.2
Hz, 2H), 6.65 (s, 1H), 5.48 (s, 2H), 4.36 (s, 2H), 3.39 (t, J = 5.8 Hz, 2H), 3.08 (q, J = 9.6,
9.1 Hz, 2H), 2.99 – 2.82 (m, 2H), 2.71 – 2.64 (m, 2H), 2.26 (tt, J = 11.3, 3.9 Hz, 1H),
1.76 (s, 2H), 1.50 (s, 2H). TOF ES+ MS: (M + H) 526.2, (M + Na) 548.2. HPLC tR =
8.25 min, > 95%.
N-Benzyl-1-(1-(4-chlorobenzyl)-7-methyl-1H-indole-2-carbonyl)piperidine-4-
carboxamide (CCG-206447). The following was added to sequentially to anhydrous
DMF (2 mL): 1-(4-chlorobenzyl)-7-methyl-1H-indole-2-carboxylic acid (50 mg, 0.167
mmol), DIPEA (0.10 mL, 0.584 mmol), EDCHCl (38 mg, 0.200 mmol), and HOBT (31
mg, 0.200 mmol). This was allowed to stir for 30 min at RT, after which time the crude
N-benzylpiperidine-4-carboxamide (36 mg, 0.20 mmol) was added. The reaction was
stirred at RT for 18 h, at which time the reaction was diluted with 1:1 EtOAc:Et2O and
washed with 10% aq. Na2CO3 (2x), dried over MgSO4, and concentrated. The residue
was purified via silica flash chromatography (20 g silica, 50% EtOAc:Hex) to afford the
title compound as a white powder. Yield: 39 mg, 0.078 mmol, 47%. 1H NMR (500 MHz,
Chloroform-d) δ 7.51 (d, J = 7.7 Hz, 1H), 7.37 – 7.33 (m, 2H), 7.31 – 7.26 (m, 3H), 7.19
(dd, J = 10.0, 3.6 Hz, 2H), 7.08 – 6.99 (m, 2H), 6.82 (d, J = 8.2 Hz, 2H), 6.64 (s, 1H),

174
5.73 (s, 2H), 4.58 (bs, 1H), 4.45 (t, J = 5.9 Hz, 2H), 4.11 (bs, 1H), 2.83 (bs, 2H), 2.58 (s,
3H), 2.34 – 2.27 (m, 1H), 1.80 (bs, 2H), 1.37 (bs, 2H). TOF ES+ MS: (M + H) 500.2, ( M
+ Na) 522.2. HPLC tR = 8.01 min, > 95%.
(1-(4-Fluorobenzyl)-1H-indol-2-yl)(4-(hydroxymethyl)piperidin-1-yl)methanone
(CCG-206462). The following was added sequentially to DMF (10 mL): 1-(4-
fluorobenzyl)-1H-indole-2-carboxylic acid (1.175 g, 4.36 mmol), DIPEA (2.29 mL,
13.09 mmol), EDCHCl (1.00 g, 5.24 mmol), and HOBT (802 mg, 5.24 mmol). This
solution was allowed to stir at RT for 1.5 h, at which time 4-piperidinemethanol (754 mg,
6.55 mmol) was added. Stirring continued for 22 h at RT, at which time the solution was
partitioned between H2O and 1:1 solution of EtOAc:Et2O. The organic extract was then
washed with saturated aq. Na2CO3, dried with MgSO4, and concentrated in vacuo.
Purification was accomplished via silica flash chromatography (100 g silica, 80%
EtOAc/Hexanes.) Resulting residue was crystallized via Et2O:Hex trituration to give the
title compound as a white solid. Yield: 1.51 g, 4.11 mmol, 94%. 1H NMR (400 MHz,
Chloroform-d) δ 7.63 (d, J = 7.9 Hz, 1H), 7.37 (d, J = 8.3 Hz, 1H), 7.29 – 7.22 (m, 1H),
7.14 (t, J = 7.5 Hz, 1H), 7.10 – 7.04 (m, 2H), 6.91 (t, J = 8.2 Hz, 2H), 6.60 (s, 1H), 5.46
(s, 2H), 4.64 (bs, 1H), 4.14 (bs, 1H), 3.42 (t, J = 5.1 Hz, 2H), 2.76 (s, 2H), 1.44 (t, J = 5.0
Hz, 1H), 1.05 (bs, 1H), 0.71 (bs, 1H). HPLC tR = 6.62 min, >95% purity.
2-Benzyl-5-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)octahydro-1H-pyrrolo[3,4-
c]pyridin-1-one. (CCG-206485) 5-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)octahydro-
1H-pyrrolo[3,4-c]pyridin-1-one (64 mg, 0.245 mmol) was dissolved in anhydrous DMF

175
(5 mL). A 60% oil suspension of NaH (29 mg, 0.735 mmol) was added at RT under N2
and stirred for 1 h. BnBr (0.035 mL, 0.294 mmol) was added and stirring continued for
18 h under the same conditions. After this time, the solution was diluted with EtOAc and
washed with 10% aq. Na2CO3 (2X), dried with MgSO4, and concentrated in vacuo.
Purification was done via silica flash chromatography (25 g silica, 80% EtOAc:Hex.) The
residue was dissolved in DCM, and rapid solvent removal afforded the title compound as
a white powder. Yield: 56 mg, 0.112 mmol, 72%. 1H NMR (500 MHz, Chloroform-d) δ
7.71 (d, J = 7.9 Hz, 1H), 7.37 – 7.26 (m, 5H), 7.26 – 7.20 (m, 5H), 7.15 (t, J = 7.4 Hz,
1H), 7.10 (d, J = 8.2 Hz, 2H), 5.49 (d, J = 3.7 Hz, 2H), 4.55 (d, J = 14.6 Hz, 2H), 4.39 (d,
J = 14.5 Hz, 1H), 4.12 (bs, 1H), 3.43 – 3.30 (m, 2H), 2.98 – 2.89 (m, 1H), 2.84 (d, J = 1.6
Hz, 1H), 2.63 – 2.44 (m, 2H), 1.75 (bs, 1H), 1.41 (bs, J = 13.5 Hz, 1H). TOF ES+ MS:
(M + H) 498.2, (M + Na) 520.2. HPLC tR = 8.23 min, >95% purity.
(1-(4-Fluorobenzyl)-1H-indol-2-yl)(4-(((4-methylphenethyl)amino)methyl)-
piperidin-1-yl)methanone (CCG-206486). 1-(1-(4-fluorobenzyl)-1H-indole-2-
carbonyl)piperidine-4-carbaldehyde (46 mg, 0.126 mmol) was dissolved in THF (2 mL)
and 2-(p-tolyl)ethanamine (0.030 mL, 0.189 mmol) was added. The solution was stirred
at rt for 10 h, at which time the THF was removed in vacuo and the residue dissolved in
EtOH (5 mL), and sodium cyanoborohydride (24 mg, 0.349 mmol) and a catalytic drop
of glacial acetic acid were added. Stirring was permitted for 14 h, at which time the
solvent was removed in vacuo, the residue was taken up in EtOAc. The organic phase
was washed with 10% aq. sodium carbonate, dried over magnesium sulfate, and
concentrated. Purification was accomplished via silica gel flash chromatography (30 g

176
silica gel, 5:95 7M methanolic ammonia:ethyl acetate.) The title compound was obtained
as a yellow oil. Yield: 26 mg, 0.054 mmol, 43%. 1H NMR (500 MHz, Chloroform-d) δ
7.64 (d, J = 8.4 Hz, 1H), 7.38 (d, J = 8.3 Hz, 1H), 7.27 (t, J = 7.7 Hz, 2H), 7.18 – 7.13
(m, 1H), 7.12 – 7.06 (m, 5H), 6.91 (t, J = 8.5 Hz, 2H), 6.60 (s, 1H), 5.47 (s, 2H), 4.62 (bs,
1H), 4.09 (bs, 1H), 2.84 (t, J = 7.0 Hz, 2H), 2.76 (t, J = 7.0 Hz, 2H), 2.70 (bs, 2H), 2.44
(d, J = 5.9 Hz, 2H), 2.32 (s, 3H), 1.79 – 1.62 (m, 5H). TOF ES+ MS: (M + H) 484.1, (M
+ Na) 506.1. HPLC tR = 6.43 min, >95% purity.
(R)-(1-(4-Fluorobenzyl)-1H-indol-2-yl)(4-(((1-phenylethyl)amino)methyl)-
piperidin-1-yl)methanone (CCG-206499). 1-(1-(4-fluorobenzyl)-1H-indole-2-
carbonyl)piperidine-4-carbaldehyde (91 mg, 0.250 mmol) was dissolved in THF (4 mL)
and (R)-α-methylbenzylamine (0.048 mL, 0.375 mmol) was added. The solution was
stirred at rt for 10 h, at which time the THF was removed in vacuo and the residue
dissolved in EtOH (5 mL), and sodium cyanoborohydride (47 mg, 0.749 mmol) and a
catalytic drop of glacial acetic acid were added. Stirring was permitted for 15 h, at which
time the solvent was removed in vacuo, the residue was taken up in EtOAc. The organic
phase was washed with 10% aq. sodium carbonate, dried over magnesium sulfate, and
concentrated. Purification was accomplished via silica gel flash chromatography (30 g
silica gel, 5:95 7M methanolic ammonia:ethyl acetate.) The title compound was obtained
as a yellow oil. Yield: 52 mg, 0.111 mmol, 44%. 1H NMR (500 MHz, Chloroform-d) δ
7.64 (d, J = 7.9 Hz, 1H), 7.45 – 7.25 (m, 7H), 7.16 (t, J = 7.5 Hz, 1H), 7.10 – 7.01 (m,
2H), 6.91 – 6.80 (m, 2H), 6.60 (s, 1H), 5.46 (s, 2H), 4.60 (s, 1H), 4.05 (s, 1H), 3.80 (q, J

177
= 6.3 Hz, 1H), 2.80 (s, 2H), 2.38 (dd, J = 11.6, 6.0 Hz, 1H), 2.23 (dd, J = 16.5, 4.8 Hz,
1H), 1.70 (d, J = 54.9 Hz, 4H), 1.42 (d, J = 6.6 Hz, 3H), 0.99 (s, 1H), 0.57 (s, 1H). TOF
ES+ MS: (M + H) 470.3, (M + Na) 492.2. HPLC tR = 5.96 min, >95% purity.
(4-((Benzylamino)methyl)piperidin-1-yl)(1-(4-fluorobenzyl)-1H-indol-2-
yl)methanone. (CCG-206500). 1-(1-(4-fluorobenzyl)-1H-indole-2-carbonyl)piperidine-4-
carbaldehyde (52 mg, 0.143 mmol) was dissolved in THF (2 mL) and benzylamine
(0.025 mL, 0.285 mmol) was added. The solution was stirred at RT for 10 h, at which
time the THF was removed in vacuo and the residue dissolved in EtOH (5 mL), and
NaCNBH3 (18 mg, 0.285 mmol) and a catalytic drop of glacial AcOH were added.
Stirring was permitted for 14 h, at which time the solvent was removed in vacuo, the
residue was taken up in EtOAc. The organic phase was washed with 10% aq. Na2CO3,
dried MgSO4, and concentrated. Purification was accomplished via silica flash
chromatography (30 g silica gel, 5:95 7M methanolic ammonia:EtOAc.) The title
compound was obtained as a yellow oil. Yield: 23 mg, 0.050 mmol, 40%. 1H NMR (500
MHz, Chloroform-d) δ 7.65 (d, J = 7.9 Hz, 1H), 7.39 – 7.27 (m, 7H), 7.16 (t, J = 7.5 Hz,
1H), 7.09 (dd, J = 8.2, 5.5 Hz, 2H), 6.91 (t, J = 8.6 Hz, 2H), 6.61 (s, 1H), 5.48 (s, 2H),
4.65 (bs, 1H), 4.10 (bs, 1H), 3.78 (s, 2H), 2.77 (bd, J = 50.6 Hz, 2H), 2.46 (d, J = 6.4 Hz,
2H), 1.79 – 1.61 (m, 3H), 1.09 (bs, 1H), 0.66 (bs, 1H). TOF ES+ MS: (M + H) 456.2, (M
+ Na) 478.2. HPLC tR = 6.04 min, >90% purity.
(S)-(1-(4-Fluorobenzyl)-1H-indol-2-yl)(4-(((1-phenylethyl)amino)methyl)-
piperidin-1-yl)methanone (CCG-206501). 1-(1-(4-fluorobenzyl)-1H-indole-2-

178
carbonyl)piperidine-4-carbaldehyde (35 mg, 0.096 mmol) was dissolved in THF (2 mL)
and (S)-α-methylbenzylamine (0.013 mL, 0.115 mmol) was added. The solution was
stirred at RT for 10 h, at which time the THF was removed in vacuo and the residue
dissolved in EtOH (5 mL), and sodium cyanoborohydride (13 mg, 0.192 mmol) and a
catalytic drop of glacial AcOH were added. Stirring was permitted for 14 h, at which time
the solvent was removed in vacuo, the residue was taken up in EtOAc. The organic phase
was washed with 10% aq. Na2CO3, dried over MgSO4, and concentrated. Purification was
accomplished via silica flash chromatography (30 g silica gel, 5:95 7M methanolic
ammonia:EtOAc.) The title compound was obtained as a yellow oil. Yield: 21 mg, 0.045
mmol, 47%. 1H NMR (500 MHz, Chloroform-d) δ 7.64 (d, J = 8.0 Hz, 1H), 7.39 – 7.22
(m, 7H), 7.16 (t, J = 7.5 Hz, 1H), 7.08 (dd, J = 8.1, 5.5 Hz, 2H), 6.88 (t, J = 7.9 Hz, 2H),
6.59 (s, 1H), 5.47 (s, 2H), 4.61 (s, 1H), 4.05 (s, 1H), 3.74 – 3.69 (m, 1H), 2.75 (d, J =
49.0 Hz, 2H), 2.41 – 2.33 (m, 1H), 2.22 (dd, J = 11.6, 7.1 Hz, 1H), 1.59 (d, J = 10.4 Hz,
4H), 1.35 (d, J = 6.6 Hz, 3H), 1.01 (s, 1H), 0.63 (s, 1H). TOF ES+ MS: (M + H) 470.3,
(M + Na) 492.2. HPLC tR = 5.98 min, > 95% purity.
(1-(4-Fluorobenzyl)-1H-indol-2-yl)(4-(morpholinomethyl)piperidin-1-
yl)methanone. (CCG-206502) 1-(1-(4-fluorobenzyl)-1H-indole-2-carbonyl)piperidine-4-
carbaldehyde (52 mg, 0.143 mmol) was dissolved in EtOH (5 mL) and morpholine (0.025
mL, 0.285 mmol), sodium cyanoborohydride (18 mg, 0.285 mmol), and a catalytic drop
of glacial acetic acid were added. Stirring was permitted for 13 h, at which time the
solvent was removed in vacuo, the residue was taken up in EtOAc. The organic phase
was washed with 10% aq. sodium carbonate, dried over magnesium sulfate, and

179
concentrated. Purification was accomplished via silica gel flash chromatography (30 g
silica gel, 100% EtOAc.) The title compound was obtained as an oil. Yield: 15 mg, 0.034
mmol, 24%. 1H NMR (500 MHz, Chloroform-d) δ 7.65 (d, J = 8.0 Hz, 1H), 7.40 (d, J =
8.3 Hz, 1H), 7.30 – 7.27 (m, 1H), 7.17 (t, J = 8.3 Hz, 1H), 7.12 – 7.07 (m, 2H), 6.97 –
6.89 (m, 2H), 6.63 – 6.59 (m, 1H), 5.49 (s, 2H), 4.65 (bs, 1H), 4.11 (bs, 1H), 3.69 (s, 4H),
2.76 (bd, J = 43.7 Hz, 2H), 2.39 (s, 4H), 2.11 (s, 2H), 1.83 – 1.57 (m, 3H), 0.99 (bs, 1H),
0.60 (bs, 1H). TOF ES+ MS: (M + H) 436.2, (M + Na) 458.2. HPLC tR = 5.38 min,
>95% purity.
(4-((Benzyl(methyl)amino)methyl)piperidin-1-yl)(1-(4-fluorobenzyl)-1H-indol-2-
yl)methanone (CCG-206503). 1-(1-(4-fluorobenzyl)-1H-indole-2-carbonyl)piperidine-4-
carbaldehyde (86 mg, 0.236 mmol) was dissolved in EtOH (5 mL) and N-
methylbenzylamine (0.027 mL, 0.283 mmol), NaCNBH3 (30 mg, 0.472 mmol), and a
catalytic drop of glacial AcOH were added. Stirring was permitted for 14 h, at which time
the solvent was removed in vacuo, the residue was taken up in EtOAc. The organic phase
was washed with 10% aq. Na2CO3, dried over MgSO4, and concentrated. Purification was
accomplished via silica flash chromatography (30 g silica gel, 100% EtOAc.) The title
compound was obtained as an oil. Yield: 23 mg, 0.049 mmol, 20%. 1H NMR (500 MHz,
Chloroform-d) δ 7.64 (t, J = 7.4 Hz, 1H), 7.39 (t, J = 7.7 Hz, 1H), 7.35 – 7.22 (m, 6H),
7.16 (q, J = 7.3 Hz, 1H), 7.07 (td, J = 8.0, 5.4 Hz, 2H), 6.87 – 6.79 (m, 2H), 6.59 (d, J =
7.0 Hz, 1H), 5.47 (s, 2H), 4.61 (bs, 1H), 4.06 (bs, 1H), 3.46 (s, 2H), 2.70 (bs, 2H), 2.18
(d, J = 7.2 Hz, 3H), 2.10 – 1.60 (m, 5H), 0.95 (bs, 1H), 0.50 (bs, 1H). TOF ES+ MS: (M
+ H) 470.2, (M + Na) 492.2. HPLC tR = 6.04 min, > 95% purity.

180
(1-(4-Fluorobenzyl)-1H-indol-2-yl)(4-(((2-(pyridin-4-yl)ethyl)amino)methyl)-
piperidin-1-yl)methanone (CCG-206504). 1-(1-(4-fluorobenzyl)-1H-indole-2-
carbonyl)piperidine-4-carbaldehyde (100 mg, 0.273 mmol) was dissolved in EtOH (5
mL) and 4-(2-aminoethyl)pyridine (50 µL, 0.410 mmol), NaCNBH3 (51 mg, 0.819
mmol), and a catalytic drop of glacial AcOH were added. Stirring was permitted for 10 h,
at which time the solvent was removed in vacuo and the residue was taken up in EtOAc.
The organic phase was washed with 10% aq. Na2CO3, dried over MgSO4, and
concentrated. Purification was accomplished via silica flash chromatography (30 g silica
gel, 100% EtOAc.) The title compound was obtained as an oil. Yield: 51 mg, 0.109
mmol, 45%. 1H NMR (500 MHz, Chloroform-d) δ 8.51 (d, J = 4.7 Hz, 2H), 7.64 (d, J =
7.9 Hz, 1H), 7.39 (d, J = 8.3 Hz, 1H), 7.29 – 7.26 (m, 1H), 7.17 – 7.11 (m, 3H), 7.10 –
7.06 (m, 2H), 6.97 – 6.89 (m, 3H), 6.60 (s, 1H), 5.47 (s, 2H), 4.65 (bs, 1H), 4.08 (bs, 1H),
2.87 (t, J = 7.1 Hz, 2H), 2.78 (t, J = 7.0 Hz, 2H), 2.70 (bs, 2H), 2.43 (s, 2H), 1.72 – 1.54
(m, 5H). TOF ES+ MS: (M + H) 471.3, (M + Na) 493.2. HPLC tR = 5.51 min, >95%
purity.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(1-(2,3-dihydro-1H-inden-2-
yl)ethyl)piperidine-4-carboxamide (CCG-206549). The following was added
sequentially to DCM (5 mL): 1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)piperidine-4-
carboxylic acid (109 mg, 0.275 mmol), DIPEA (0.144 mL, 0.825 mmol), EDCHCl (63
mg, 0.330 mmol), HOBT (51 mg, 0.330 mmol), and crude 2-(1-aminoethyl)indane (~44

181
mg, 0.275 mmol). The mixture was stirred for 16 h at rt, at which time the solution
diluted with a 1:1 solution of ethyl acetate:diethyl ether and washed with 1M HCl (1X),
10% aq. sodium carbonate (1X) and brine (1X). The organic phase was dried with
magnesium sulfate and concentrated in vacuo. The resulting residue was dissolved in
EtOAc and precipitated out hexanes, and then collected over a filter and washed with
diethyl ether to give the title compound as a white solid. Yield: 59 mg, 0.109 mmol, 40%.
1H NMR (500 MHz, Chloroform-d) δ 7.66 (d, J = 7.9 Hz, 1H), 7.33 (d, J = 8.3 Hz, 1H),
7.29 – 7.13 (m, 8H), 7.04 (d, J = 8.2 Hz, 2H), 6.65 (s, 1H), 5.48 (s, 2H), 5.30 (d, J = 8.9
Hz, 1H), 4.45 (s, 1H), 4.25 – 4.14 (m, 1H), 3.02 (td, J = 16.7, 8.3 Hz, 2H), 2.91 – 2.65
(m, 4H), 2.51 (h, J = 8.1 Hz, 1H), 2.22 (tq, J = 10.8, 4.1 Hz, 1H), 1.63 (d, J = 95.6 Hz,
5H), 1.21 (d, J = 6.7 Hz, 3H). TOF ES+ MS: (M + H) 540.2. HPLC tR = 8.55 min, >95%
purity.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-((2,3-dihydro-1H-inden-1-
yl)methyl)piperidine-4-carboxamide. (CCG-206550) The following was added
sequentially to DCM (6 mL): 1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)piperidine-4-
carboxylic acid (134 mg, 0.340 mmol), DIPEA (0.178 mL, 1.019 mmol), EDCHCl (78
mg, 0.408 mmol), HOBT (62.4 mg, 0.408 mmol), and 2-indanmethanamine
hydrochloride (62 mg, 0.340 mmol). The mixture was stirred for 18 h at rt, at which time
the solution diluted with a 1:1 solution of ethyl acetate:diethyl ether and washed with 1M
HCl (1X), 10% aq. sodium carbonate (1X) and brine (1X). The organic phase was dried
with magnesium sulfate and concentrated in vacuo. The resulting residue was dissolved
in EtOAc and precipitated out hexanes, and then collected over a filter and washed with

182
diethyl ether to give the title compound as a white solid. Yield: 72 mg, 0.137 mmol, 41%.
1H NMR (400 MHz, Chloroform-d) δ 7.63 (d, J = 7.9 Hz, 1H), 7.32 (d, J = 8.2 Hz, 1H),
7.27 – 7.22 (m, 3H), 7.21 – 7.13 (m, 5H), 7.01 (d, J = 8.2 Hz, 2H), 6.62 (s, 1H), 5.45 (s,
2H), 5.38 (t, J = 5.3 Hz, 1H), 4.36 (s, 2H), 3.62 (dt, J = 12.2, 5.8 Hz, 1H), 3.50 – 3.34 (m,
2H), 2.88 (tt, J = 15.8, 8.2 Hz, 4H), 2.23 (ddd, J = 13.3, 9.5, 5.8 Hz, 2H), 1.78 (ddd, J =
13.3, 8.2, 6.4 Hz, 3H), 1.49 (s, 2H). TOF ES+ MS: (M + H) 526.2. HPLC tR = 8.35 min,
>95% purity.
N-Benzyl-1-(1-(4-chlorobenzyl)-1H-imidazole-2-carbonyl)piperidine-4-
carboxamide (CCG-206586). Ethyl 1-(4-chlorobenzyl)-1H-imidazole-2-carboxylate
(7.28 g, 27.5 mmol), was dissolved in EtOH (10 mL) and 10% aq. NaOH (20 mL) and
stirred at rt for 15 h. The solvent was then stripped off, water was added, and the solution
acidified with HCl. The resulting precipitate was collected over a filter and washed with
1M HCl and dried to afford the carboxylic acid as a white powder which was taken
directly into the next step. Yield: 6.506 g, 27.5 mmol, 100%.
The following was added sequentially to DMF: 1-(4-chlorobenzyl)-1H-imidazole-
2-carboxylic acid (71 mg, 0.298 mmol), DIPEA (0.16 mL, 0.894 mmol), EDCHCl (69
mg, 0.358 mmol), and HOBT (55 mg, 0.358 mmol). This was allowed to stir at rt for 30
min, at which time N-benzylpiperidine-4-carboxamide (~ 65 mg, 0.298 mmol) was added
and syirring continued at rt for 14 h. Addition of water caused precipitation, which was
collected over a filter and washed with water and small amount of diethyl ether and
EtOAc to give the title compound as a white solid. Yield: 37 mg, 0.085 mmol, 28%. 1H
NMR (400 MHz, Chloroform-d) δ 7.34 – 7.22 (m, 7H), 7.12 (d, J = 8.3 Hz, 2H), 7.04 (s,

183
1H), 6.93 (s, 1H), 5.72 (t, J = 5.1 Hz, 1H), 5.36 (d, J = 5.1 Hz, 2H), 4.63 (t, J = 15.1 Hz,
2H), 4.43 (d, J = 5.6 Hz, 2H), 3.14 (t, J = 12.6 Hz, 1H), 2.82 (t, J = 12.8 Hz, 1H), 2.38 (tt,
J = 11.3, 3.9 Hz, 1H), 1.95 (d, J = 13.4 Hz, 1H), 1.83 (d, J = 13.3 Hz, 1H), 1.72 – 1.55
(m, 2H). TOF ES+ MS: (M + H) 437.2, (M + Na) 459.2. HPLC tR = 5.59 min, >95%
purity.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(3-phenylpropyl)piperidine-4-
carboxamide (CCG-206587). The following were added sequentially to DCM (10
mL): 1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)piperidine-4-carboxylic acid (100 mg,
0.252 mmol), DIPEA (132 µL, 0.756 mmol), EDCHCl (58 mg, 0.302 mmol), HOBt (46
mg, 0.302 mmol), and then 3-phenylpropylamine (43 µL, 0.302 mmol). The reaction was
allowed to stir at RT for 13 h, at which time it was diluted with EtOAc, washed with 1 M
HCl (3X), H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X), dried over MgSO4, and
concentrated in vacuo. The residue was the purified by silica flash chromatography (20 g
silica, 70% EtOAc:Hex) to provide the desired material as a colorless oil. Yield: 101 mg,
0.197 mmol, 78%. 1H NMR (500 MHz, Chloroform-d) δ 7.65 (d, J = 7.9 Hz, 1H), 7.37 –
7.23 (m, 5H), 7.24 – 7.12 (m, 6H), 6.64 (s, 1H), 5.45 (s, 2H), 5.39 (t, J = 5.4 Hz, 1H),
4.50 (bs, 1H), 4.21 (bs, 1H), 3.29 (q, J = 6.5 Hz, 2H), 2.84 (bs, 2H), 2.65 (t, J = 7.5 Hz,
2H), 2.20 (tt, J = 11.0, 3.5 Hz, 1H), 1.85 (p, J = 7.5 Hz, 2H), 1.72 (bs, 2H), 1.44 (bs, 2H).
TOF ES+ MS: (M + H) 515.1, (M + Na) 537.0. HPLC tR = 8.16 min, > 90%.

184
1-(1-(4-Chlorobenzyl)-1H-pyrrole-2-carbonyl)-N-((2,3-dihydro-1H-inden-2-
yl)methyl)azetidine-3-carboxamide (CCG-208793). Methyl 1-(1-(4-chlorobenzyl)-1H-
pyrrole-2-carbonyl)azetidine-3-carboxylate 29 (300 mg, 0.90 mmol) was dissolved in 200
proof EtOH (10 mL) and 10% aq. NaOH (10 mL) was added. The reaction was allowed
to stir at RT for 24 h, at which time the solvent was stripped off in vacuo and the residue
was taken up in H2O and acidified to pH 1 by addition of conc. HCl at 0°C. The resulting
precipitate was collected over a filter and dried by aspirator then high vacuum to afford
the desired carboxylic acid as a white powder. Yield: 250 mg, 0.78 mmol, 87%.
1-(1-(4-chlorobenzyl)-1H-pyrrole-2-carbonyl)azetidine-3-carboxylic acid (50 mg,
0.16 mmol) was added to DCM (5 mL), followed by indanyl amine 75 (29 mg, 0.17
mmol), TEA (66 µL, 0.47 mmol), EDCHCl (33 mg, 0.17 mmol), and HOBT (26 mg,
0.17 mmol). The reaction was allowed to stir at RT for 21 h, at which time the reaction
was concentrated and the residue taken up in EtOAc. This solution was then washed with
1M HCl (2X), H2O (3X) and 10% aq. Na2CO3 (3X), and brine (1X), dried (MgSO4), and
concentrated. The residue was purified by silica flash chromatography (4 g silica, 10% to
60% EtOAc:Hex) to provide the final compound as a white powder. Yield: 39 mg, 0.09
mmol, 56%. 1H NMR (500 MHz, Chloroform-d) δ 7.28 – 7.24 (m, 3H), 7.21 – 7.18 (m,
2H), 7.16 – 7.13 (m, 2H), 7.09 – 7.04 (m, 2H), 6.81 (dd, J = 2.6, 1.6 Hz, 1H), 6.52 (dd, J
= 4.0, 1.5 Hz, 1H), 6.19 – 6.16 (m, 1H), 5.56 (d, J = 9.0 Hz, 3H), 4.30 (bs, 4H), 3.42 (t, J
= 6.0 Hz, 2H), 3.22 (p, J = 7.4 Hz, 1H), 3.09 (q, J = 10.0, 9.3 Hz, 2H), 2.75 – 2.65 (m,
3H). TOF ES+ MS: (M + H) 448.2, (M + Na) 470.2. HPLC tR = 7.56 min, > 95%.

185
N1-(4-Chlorobenzyl)-N4-(2-(pyridin-4-yl)ethyl)piperidine-1,4-dicarboxamide
(208825). Ethyl 1-((4-chlorobenzyl)carbamoyl)piperidine-4-carboxylate 31a (810 mg,
2.50 mmol) was dissolved in 200 proof EtOH (20 mL) at RT, followed by the addition of
10% aq. NaOH, which elicited precipitation. The mixture was then stirred at 50 °C for 2
h over which time it became a solution. The reaction was then allowed to cool to RT and
the solvent was then removed in vacuo. The residue was taken up in H2O and acidified to
pH 1 with conc. HCl at 0 °C and the resulting precipitate was collected over a filter,
washed with cold 1 M aq. HCl, and dried over the filter then under high vacuum to give
the desired compound as a white powder. No further purification as necessary. Yield: 675
mg, 2.27 mmol, 91%.
1-((4-chlorobenzyl)carbamoyl)piperidine-4-carboxylic acid (60 mg, 0.20 mmol)
and TEA (93 µL, 0.61 mmol) were dissolved in DCM (5 mL), followed by EDCHCl (47
mg, 0.22 mmol), HOBT (37 mg, 0.22 mmol), and amine 27 (30 µL, 0.20 mmol). The
reaction was allowed to stir at RT for 15 h. After this time, the reaction was diluted with
EtOAc and washed with H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X), then dried
with MgSO4 and concentrated. The residue was then crystallized from EtOAc and
washed with Et2O to provide an off-white, slightly tanned solid. Yield: 42 mg, 0.11
mmol, 52%. 1H NMR (500 MHz, Chloroform-d) δ 8.52 (d, J = 4.4 Hz, 2H), 7.32 – 7.20
(m, 4H), 7.11 (d, J = 4.4 Hz, 2H), 5.56 (s, 1H), 4.79 (d, J = 5.9 Hz, 1H), 4.37 (d, J = 5.2
Hz, 2H), 3.96 (d, J = 13.2 Hz, 2H), 3.55 (q, J = 6.4, 5.8 Hz, 2H), 2.85 – 2.78 (m, 4H),
2.19 (t, J = 11.5 Hz, 1H), 1.77 (d, J = 13.0 Hz, 2H), 1.63 (dd, J = 18.8, 5.5 Hz, 2H). TOF
ES+ MS: (M + H) 401.2, (M + Na) 423.2. HPLC tR = 4.61 min, > 95% purity.

186
N1-(4-Chlorobenzyl)-N
4-((2,3-dihydro-1H-inden-2-yl)methyl)piperidine-1,4-
dicarboxamide (CCG-208826). Ethyl 1-((4-chlorobenzyl)carbamoyl)piperidine-4-
carboxylate 31a (810 mg, 2.50 mmol) was dissolved in 200 proof EtOH (20 mL) at RT,
followed by the addition of 10% aq. NaOH, which elicited precipitation. The mixture was
then stirred at 50 °C for 2 h over which time it became a solution. The reaction was then
allowed to cool to RT and the solvent was then removed in vacuo. The residue was taken
up in H2O and acidified to pH 1 with conc. HCl at 0 °C and the resulting precipitate was
collected over a filter, washed with cold 1 M aq. HCl, and dried over the filter then under
high vacuum to give the desired compound as a white powder. No further purification as
necessary. Yield: 675 mg, 2.27 mmol, 91%.
1-((4-chlorobenzyl)carbamoyl)piperidine-4-carboxylic acid (60 mg, 0.20 mmol)
and TEA (93 µL, 0.61 mmol) were dissolved in DCM (5 mL), followed by EDCHCl (47
mg, 0.22 mmol), HOBT (37 mg, 0.22 mmol), and amine 27 (30 µL, 0.20 mmol). The
reaction was allowed to stir at RT for 16 h. After this time, the reaction was diluted with
EtOAc and washed with H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X), then dried
with MgSO4 and concentrated. The residue was then crystallized from EtOAc and
washed with Et2O to provide a tan solid. Yield: 47 mg, 0.11 mmol, 54%. 1H NMR (500
MHz, Chloroform-d) δ 7.32 – 7.28 (m, 2H), 7.25 (d, J = 8.4 Hz, 2H), 7.21 – 7.18 (m,
2H), 7.16 – 7.13 (m, 2H), 5.57 (s, 1H), 4.39 (s, 2H), 3.98 (dt, J = 13.4, 3.7 Hz, 2H), 3.38
(t, J = 6.0 Hz, 2H), 3.08 (q, J = 10.3, 9.4 Hz, 2H), 2.85 (ddd, J = 13.3, 11.8, 2.8 Hz, 2H),
2.70 – 2.64 (m, 3H), 2.23 (ddt, J = 11.4, 7.7, 3.8 Hz, 1H), 1.83 – 1.58 (m, 4H). TOF ES+
MS: (M + H) 426.2. HPLC tR = 6.69 min, > 95%.

187
N1-(4-Chlorophenethyl)-N
4-(2-(pyridin-4-yl)ethyl)piperidine-1,4-dicarboxamide
(CCG-208827). Ethyl 1-((4-chlorophenethyl)carbamoyl)-piperidine-4-carboxylate 31b
(807 mg, 2.382 mmol) was dissolved in EtOH (20 mL), and 10% aq NaOH (15 mL) was
added. The resulting mixture was warmed to 50 °C and stirred for 2 h. The now
homogeneous solution was then concentrated in vacuo, a small amount of water was
added back, and the solution chilled in an ice bath. The solution was then acidified with
concentrated HCl, and the resulting precipitate was collected over a filter, washed with 1
M HCl and ice-cold water, filter-dried, then further dried under high vacuum to afford 1-
((4-chlorophenethyl)carbamoyl)piperidine-4-carboxylic acid as a white powder. Yield:
631 mg (85%). 1H NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 7.30 (d, J = 8.4 Hz, 2H),
7.18 (d, J = 8.4 Hz, 2H), 6.54 (t, J = 5.4 Hz, 1H), 3.80 (d, J = 13.3 Hz, 2H), 3.18 (q, J =
7.0 Hz, 2H), 2.76−2.62 (m, 4H), 2.36 (ddt, J = 11.0, 7.6, 3.9 Hz, 1H), 1.75−1.64 (m, 2H),
1.40−1.25 (m, 2H).
The carboxylic acid (60 mg, 0.193 mmol), TEA (81 μL, 0.579 mmol), EDCHCl
(41 mg, 0.212 mmol), HOBT (33 mg, 0.212 mmol), and 4-(2-aminoethyl)pyridine 27 (25
μL, 0.212 mmol) were all added sequentially to DCM (5 mL) at RT. This solution was
allowed to stir for 16 h at RT, after which time the reaction was diluted with ethyl acetate
and washed with water (3×), 10% aq sodium carbonate (3×), and brine (1×), then dried
(magnesium sulfate) and concentrated in vacuo. The resulting residue was then
crystallized from ethyl acetate and washed with diethyl ether to provide the title
compound as a tan solid. Yield: 46 mg (57%). 1HNMR (400 MHz, CDCl3) δ 8.61−8.26
(m, 2H), 7.30−7.19 (m, 2H), 7.17−7.01 (m, 4H), 5.91−5.66 (m, 1H), 4.59 (dd, J = 13.4,
5.6 Hz, 1H), 3.86 (d, J = 13.2 Hz, 2H), 3.53 (q, J = 6.7 Hz, 2H), 3.42 (q, J = 6.8 Hz, 2H),

188
2.82 (t, J = 6.8 Hz, 2H), 2.79−2.61 (m, 4H), 2.17 (t, J = 9.6 Hz, 1H), 1.73 (d, J = 12.9 Hz,
2H), 1.57 (qd, J = 12.9, 12.5, 3.8 Hz, 2H). TOF ES+ MS: (M + H) 415.2, (M + Na)
437.2. HPLC tR = 4.70 min, > 95%.
N1-(4-Chlorophenethyl)-N4-((2,3-dihydro-1H-inden-2-yl)methyl)piperidine-1,4-
dicarboxamide (CCG-208828). Ethyl 1-((4-chlorophenethyl)carbamoyl)-piperidine-4-
carboxylate 31b (807 mg, 2.382 mmol) was dissolved in EtOH (20 mL), and 10% aq
NaOH (15 mL) was added. The resulting mixture was warmed to 50 °C and stirred for 2
h. The now homogeneous solution was then concentrated in vacuo, a small amount of
water was added back, and the solution chilled in an ice bath. The solution was then
acidified with concentrated HCl, and the resulting precipitate was collected over a filter,
washed with 1 M HCl and ice-cold water, filter-dried, then further dried under high
vacuum to afford 1-((4-chlorophenethyl)carbamoyl)piperidine-4-carboxylic acid as a
white powder. Yield: 631 mg (85%). 1H NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H),
7.30 (d, J = 8.4 Hz, 2H), 7.18 (d, J = 8.4 Hz, 2H), 6.54 (t, J = 5.4 Hz, 1H), 3.80 (d, J =
13.3 Hz, 2H), 3.18 (q, J = 7.0 Hz, 2H), 2.76−2.62 (m, 4H), 2.36 (ddt, J = 11.0, 7.6, 3.9
Hz, 1H), 1.75−1.64 (m, 2H), 1.40−1.25 (m, 2H).
The carboxylic acid (60 mg, 0.19 mmol), TEA (81 μL, 0.58 mmol), EDCHCl (41
mg, 0.21 mmol), HOBT (33 mg, 0.21 mmol), and indanyl amine 75 (39 mg, 0.21 mmol)
were all added sequentially to DCM (5 mL) at RT. This solution was allowed to stir for
15 h at RT, after which time the reaction was diluted with EtOAc and washed with 05M
HCl (3X), H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X), then dried (magnesium
sulfate) and concentrated in vacuo. The resulting residue was then crystallized from

189
EtOAc to give tan crystals. Yield: 55 mg, 0.12 mmol, 64%. 1H NMR (500 MHz,
Chloroform-d) δ 7.28 – 7.27 (m, 2H), 7.21 – 7.18 (m, 2H), 7.16 – 7.12 (m, 4H), 5.54 (s,
1H), 4.45 (s, 1H), 3.88 (d, J = 9.9 Hz, 1H), 3.46 (q, J = 6.7 Hz, 2H), 3.38 (t, J = 6.0 Hz,
2H), 3.08 (q, J = 7.3, 6.5 Hz, 2H), 2.83 – 2.75 (m, 3H), 2.72 – 2.65 (m, 3H), 2.21 (tt, J =
11.5, 3.9 Hz, 1H), 1.79 (dd, J = 13.0, 3.2 Hz, 2H), 1.62 (td, J = 12.7, 12.2, 4.4 Hz, 4H).
TOF ES+ MS: (M + H) 440.2, (M + Na) 462.2. HPLC tR = 7.32 min, > 95%.
N-Benzyl-1-(1-(4-chlorobenzyl)-1H-pyrrole-2-carbonyl)azetidine-3-carboxamide
(CCG-208829). Methyl 1-(1-(4-chlorobenzyl)-1H-pyrrole-2-carbonyl)azetidine-3-
carboxylate 29 (300 mg, 0.90 mmol) was dissolved in 200 proof EtOH (10 mL) and 10%
aq. NaOH (10 mL) was added. The reaction was allowed to stir at RT for 24 h, at which
time the solvent was stripped off in vacuo and the residue was taken up in H2O and
acidified to pH 1 by addition of conc. HCl at 0°C. The resulting precipitate was collected
over a filter and dried by aspirator then high vacuum to afford the desired carboxylic acid
as a white powder. Yield: 250 mg, 0.78 mmol, 87%.
1-(1-(4-chlorobenzyl)-1H-pyrrole-2-carbonyl)azetidine-3-carboxylic acid (50 mg,
0.16 mmol) was added to DCM (5 mL), followed by benzylamine (19 µL, 0.17 mmol),
TEA (66 µL, 0.47 mmol), EDCHCl (33 mg, 0.17 mmol), and HOBT (26 mg, 0.17
mmol). The reaction was allowed to stir at RT for 15 h, at which time the reaction was
concentrated and the residue taken up in EtOAc. This solution was then washed with
0.5M HCl (2X), H2O (3X) and 10% aq. Na2CO3 (3X), and brine (1X), dried (MgSO4),
and concentrated. The residue was purified by silica flash chromatography (4 g silica,
30% to 60% EtOAc:Hex) to provide the final compound as a white powder. Yield: 29

190
mg, 0.07 mmol, 45%. 1H NMR (500 MHz, Chloroform-d) δ 7.38 – 7.23 (m, 7H), 7.06 (d,
J = 8.2 Hz, 2H), 6.80 (s, 1H), 6.53 (d, J = 3.5 Hz, 1H), 6.19 – 6.16 (m, 1H), 5.82 (s, 1H),
5.54 (s, 2H), 4.57 (s, 1H), 4.48 (d, J = 5.7 Hz, 2H), 4.32 (s, 2H), 3.28 (p, J = 7.4 Hz, 1H).
TOF ES+ MS: (M + H) 408.1, (M + Na) 430.1. HPLC tR = 7.06 min, > 95%.
1-(3-(4-Chlorophenyl)propanoyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-
carboxamide (CCG-208846). Ethyl 1-(3-(4-chlorophenyl)propanoyl)piperidine-4-
carboxylate 33b (850 mg, 2.62 mmol) was dissolved in EtOH (10 mL), followed by the
addition of 10% aq. NaOH (10 mL). This was stirred 11 h at RT, after which time a
majority of the solvent was stripped off in vacuo. The remaining concentrated aq.
solution was chilled to 0 °C and acidified to pH<1 with conc. HCl. The aq. solution was
decanted off of the resulting gummy residue. H2O was then added, sonicated, and
decanted. The residue was then dried under high vacuum, and the thick viscous residue
was sonicated in Et2O to afford the carboxylic acid as a white solid that was collected
over a filter. Yield: 730 mg, 2.47 mmol, 94%.
1-(3-(4-chlorophenyl)propanoyl)piperidine-4-carboxylic acid (50 mg, 0.17 mmol)
was added to DCM (2 mL) at RT followed by TEA (71 µL, 0.51 mmol), EDC HCl (36
mg, 0.19 mmol), HOBT (28 mg, 0.19 mmol), and amine 27 (22 µL, 0.19 mmol). The
reaction was allowed to stir at RT for 14 h, after which time the DCM was removed in
vacuo and the residue taken up in EtOAc, which was subsequently washed with 0.5 M aq.
HCl (1X), H2O (2X), 10% aq. Na2CO3 (2X), and brine (1X), then dried (MgSO4) and
decanted. The material was crystallized from the dropwise addition of EtOAc to boiling
hexanes, then cooling of the solution. Yield: 42 mg, 0.11 mmol, 62%. 1H NMR (500

191
MHz, Chloroform-d) δ 8.52 – 8.48 (m, 2H), 7.29 – 7.20 (m, 2H), 7.17 – 7.07 (m, 4H),
5.66 (s, 1H), 4.57 (d, J = 12.9 Hz, 1H), 3.81 (d, J = 13.2 Hz, 1H), 3.54 (q, J = 5.9 Hz,
2H), 2.96 – 2.89 (m, 3H), 2.82 (t, J = 6.1 Hz, 2H), 2.63 – 2.54 (m, 3H), 2.22 (dt, J = 11.2,
5.8 Hz, 1H), 1.79 – 1.69 (m, 2H), 1.60 – 1.46 (m, 2H). TOF ES+ MS: 400.2 (M + H),
422.2 (M + Na). HPLC tR = 4.89 min, > 95% purity.
1-(3-(4-Chlorophenyl)propanoyl)-N-((2,3-dihydro-1H-inden-2-
yl)methyl)piperidine-4-carboxamide (CCG-208847). Ethyl 1-(3-(4-
chlorophenyl)propanoyl)piperidine-4-carboxylate 33b (850 mg, 2.62 mmol) was
dissolved in EtOH (10 mL), followed by the addition of 10% aq. NaOH (10 mL). This
was stirred 11 h at RT, after which time a majority of the solvent was stripped off in
vacuo. The remaining concentrated aq. solution was chilled to 0 °C and acidified to pH<1
with conc. HCl. The aq. solution was decanted off of the resulting gummy residue. H2O
was then added, sonicated, and decanted. The residue was then dried under high vacuum,
and the thick viscous residue was sonicated in Et2O to afford the carboxylic acid as a
white solid that was collected over a filter. Yield: 730 mg, 2.47 mmol, 94%.
The following were added sequentially to DCM (2 mL) at RT: 1-(2-(4-
chlorophenyl)acetyl)piperidine-4-carboxylic acid (50 mg, 0.17 mmol), TEA (71 µL, 0.51
mmol), EDCHCl (36 mg, 0.19 mmol), HOBT (29 mg, 0.19 mmol), and indanyl amine 75
(34 mg, 0.19 mmol). This solution was stirred a RT for 15 h, at which time the DCM was
removed in vacuo and the residue was diluted with EtOAc and washed with 1M HCl
(3X), H2O (2X), 10% aq. Na2CO3 (2X), and brine (1X), then dried (MgSO4) and
concentrated. The crude residue was purified by crystallization from boiling EtOAc to

192
give crystals. Yield: 44 mg, 0.10 mmol, 61%. 1H NMR (500 MHz, Chloroform-d) δ 7.26
(d, J = 8.7 Hz, 2H), 7.21 – 7.18 (m, 2H), 7.17 – 7.12 (m, 4H), 5.54 (t, J = 5.9 Hz, 1H),
4.58 (dd, J = 13.6, 3.6 Hz, 1H), 3.83 (dt, J = 14.4, 3.9 Hz, 1H), 3.38 (t, J = 5.7 Hz, 2H),
3.08 (q, J = 7.5, 6.6 Hz, 2H), 3.00 – 2.89 (m, 3H), 2.74 – 2.65 (m, 3H), 2.60 (dd, J = 8.6,
6.3 Hz, 2H), 2.26 (tt, J = 11.4, 3.9 Hz, 1H), 1.83 – 1.74 (m, 2H), 1.60 – 1.47 (m, 2H).
TOF ES+ MS: (M + H) 425.2, (M + Na) 447.2. HPLC tR = 7.14 min, > 95%.
1-(2-(4-Chlorophenyl)acetyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide
(CCG-208848). Ethyl 1-(2-(4-chlorophenyl)acetyl)piperidine-4-carboxylate 3a (811 mg,
2.62 mmol) was added to EtOH (15 mL) and 10% aq. NaOH (5 mL). This mixture was
heated to 50 °C and stirred for 5 h, then allowed to cool to RT. Solvent was removed in
vacuo until precipitation began to occur, at which point the material was cooled to 0 °C
and acidified to pH<1 by the dropwise addition of conc. HCl. The solution was decanted
off the resulting gum, and the residue was air dried, then dried under high vacuum to
afford a highly gummy material that could be solidified by sonication in Et2O to provide
the carboxylic acid as a white powder without further purification. Yield: 723 mg, 2.57
mmol, 98%.
The following were added sequentially to DCM (5 mL) at RT: 1-(2-(4-
chlorophenyl)acetyl)piperidine-4-carboxylic acid (60 mg, 0.22 mmol), TEA (93 µL, 0.67
mmol), EDCHCl (47 mg, 0.25 mmol), HOBT (37 mg, 0.25 mmol), and amine 27 (29
µL, 0.25 mmol). This solution was stirred a RT for 18 h, at which time the reaction was
diluted with EtOAc and washed with H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X),
then dried (MgSO4) and concentrated. The crude residue was purified by crystallization

193
from the dropwise addition of EtOAc to boiling hexanes, then cooling of the solution, to
give crystals. Yield: 40 mg, 0.105 mmol, 54%. 1H NMR (500 MHz, Chloroform-d) δ
8.52 (d, J = 4.7 Hz, 2H), 7.29 (d, J = 7.8 Hz, 2H), 7.17 (d, J = 8.1 Hz, 2H), 7.10 (d, J =
4.8 Hz, 2H), 5.50 (bs, 1H), 4.57 (d, J = 13.3 Hz, 1H), 3.86 (d, J = 13.5 Hz, 1H), 3.68 (s,
2H), 3.54 (q, J = 6.6 Hz, 2H), 3.03 – 2.96 (m, 1H), 2.82 (t, J = 6.9 Hz, 2H), 2.70 – 2.62
(m, 1H), 2.21 (tt, J = 11.1, 3.6 Hz, 1H), 1.77 (d, J = 12.3 Hz, 2H), 1.57 (qd, J = 12.2, 4.1
Hz, 1H), 1.46 (qd, J = 12.3, 3.9 Hz, 1H). TOF ES+ MS: 386.2 (M + H), 408.2 (M + Na).
HPLC tR = 6.87 min, > 95% purity.
1-(2-(4-Chlorophenyl)acetyl)-N-((2,3-dihydro-1H-inden-2-yl)methyl)piperidine-
4-carboxamide (CCG-208849). Ethyl 1-(2-(4-chlorophenyl)acetyl)piperidine-4-
carboxylate (811 mg, 2.62 mmol) was added to EtOH (15 mL) and 10% aq. NaOH (5
mL). This mixture was heated to 50 °C and stirred for 5 h, then allowed to cool to RT.
Solvent was removed in vacuo until precipitation began to occur, at which point the
material was cooled to 0 °C and acidified to pH<1 by the dropwise addition of conc. HCl.
The solution was decanted off the resulting gum, and the residue was air dried, then dried
under high vacuum to afford a highly gummy material that could be solidified by
sonication in Et2O to provide the carboxylic acid as a white powder without further
purification. Yield: 723 mg, 2.57 mmol, 98%.
The following were added sequentially to DCM (2 mL) at RT: 1-(2-(4-
chlorophenyl)acetyl)piperidine-4-carboxylic acid (50 mg, 0.18 mmol), TEA (74 µL, 0.53
mmol), EDCHCl (37 mg, 0.20 mmol), HOBT (30 mg, 0.20 mmol), and indanyl amine 75
(36 mg, 0.20 mmol). This solution was stirred a RT for 10 h, at which time the DCM was

194
removed in vacuo and the residue was diluted with EtOAc and washed with 0.5M HCl
(2X), H2O (2X), 10% aq. Na2CO3 (2X), and brine (1X), then dried (MgSO4) and
concentrated. The crude residue was purified by crystallization from boiling EtOAc to
give crystals. Yield: 43 mg, 0.11 mmol, 59%. 1H NMR (500 MHz, Chloroform-d) δ 7.32
– 7.24 (m, 2H), 7.23 – 7.02 (m, 6H), 5.52 (t, J = 6.0 Hz, 1H), 4.63 – 4.53 (m, 1H), 3.95 –
3.82 (m, 1H), 3.69 (s, 2H), 3.37 (td, J = 6.0, 1.8 Hz, 2H), 3.13 – 2.97 (m, 3H), 2.70 – 2.57
(m, 4H), 2.24 (tt, J = 11.2, 3.9 Hz, 1H), 1.83 – 1.70 (m, 2H), 1.61 – 1.45 (m, 2H). TOF
ES+ MS: (M + H) 411.2. HPLC tR = 6.87 min, > 95%.
1-(1-(4-Chlorobenzyl)-1H-imidazole-2-carbonyl)-N-((2,3-dihydro-1H-inden-2-
yl)methyl)piperidine-4-carboxamide (CCG-208913). Ethyl 1-(4-chlorobenzyl)-1H-
imidazole-2-carboxylate 23b (7.28 g, 27.5 mmol), was dissolved in EtOH (10 mL) and
10% aq. NaOH (20 mL) and stirred at rt for 15 h. The solvent was then stripped off,
water was added, and the solution acidified with HCl. The resulting precipitate was
collected over a filter and washed with 1M HCl and dried to afford the carboxylic acid as
a white powder which was taken directly into the next step. Yield: 6.506 g, 27.5 mmol,
100%.
1-(4-chlorobenzyl)-1H-imidazole-2-carboxylic acid (50 mg, 0.21 mmol) was
added to DCM (5 mL), followed by N-((2,3-dihydro-1H-inden-2-yl)methyl)piperidine-4-
carboxamide (69 mg, 0.23 mmol), TEA (88 µL, 0.63 mmol), EDCHCl (45 mg, 0.23
mmol), and HOBT (36 mg, 0.23 mmol). The reaction was allowed to stir at RT for 20 h,
at which time the reaction was concentrated and the residue taken up in EtOAc. This
solution was then washed with H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X), dried

195
(MgSO4), and concentrated. The residue was purified by silica flash chromatography (10
g silica, 60 to 100% EtOAc:Hex) to provide the final compound as a white powder.
Yield: 48 mg, 0.10 mmol, 47%. 1H NMR (500 MHz, Chloroform-d) δ 7.31 (d, J = 8.3
Hz, 2H), 7.21 – 7.12 (m, 6H), 7.07 (s, 1H), 6.96 (s, 1H), 5.67 – 5.61 (m, 1H), 5.38 (d, J =
5.6 Hz, 2H), 4.62 (t, J = 14.0 Hz, 2H), 3.37 (t, J = 5.8 Hz, 2H), 3.15 (t, J = 12.1 Hz, 1H),
3.07 (q, J = 9.6, 8.8 Hz, 2H), 2.87 – 2.79 (m, 1H), 2.71 – 2.65 (m, 3H), 2.34 (tt, J = 11.4,
4.0 Hz, 1H), 1.89 (d, J = 13.4 Hz, 1H), 1.82 – 1.75 (m, 1H), 1.61 (dtd, J = 36.5, 12.2, 3.5
Hz, 2H). TOF ES+ MS: (M + H) 477.2, (M + Na) 499.2. HPLC tR = 6.07 min, > 95%.
1-(1-(4-Chlorobenzyl)-1H-pyrrole-2-carbonyl)-N-((2,3-dihydro-1H-inden-2-
yl)methyl)piperidine-4-carboxamide (CCG-208914). 1-(1-(4-chlorobenzyl)-1H-pyrrole-
2-carbonyl)piperidine-4-carboxylic acid 25a (50 mg, 0.14 mmol) was added to DCM (5
mL), followed by indanyl amine 75 (29 mg, 0.16 mmol), TEA (60 µL, 0.43 mmol),
EDCHCl (30 mg, 0.16 mmol), and HOBT (24 mg, 0.16 mmol). The reaction was
allowed to stir at RT for 20 h, at which time the reaction was concentrated and the residue
taken up in EtOAc. This solution was then washed with 1M HCl (3X), H2O (3X) and
10% aq. Na2CO3 (3X), and brine (1X), dried (MgSO4), and concentrated. The residue
was purified by silica flash chromatography (10 g silica, 60% EtOAc:Hex) to provide the
final compound as a white powder. Yield: 42 mg, 0.09 mmol, 61%. 1H NMR (500 MHz,
Chloroform-d) δ 7.29 – 7.25 (m, 2H), 7.23 – 7.17 (m, 2H), 7.17 – 7.13 (m, 2H), 7.05 (d, J
= 8.4 Hz, 2H), 6.81 – 6.80 (m, 1H), 6.34 (dd, J = 3.7, 1.6 Hz, 1H), 6.14 (d, J = 6.4 Hz,
1H), 5.52 (t, J = 5.0 Hz, 1H), 5.29 (s, 2H), 4.36 (d, J = 11.7 Hz, 2H), 3.38 (t, J = 6.0 Hz,
2H), 3.08 (q, J = 9.4, 8.9 Hz, 2H), 2.84 (t, J = 11.9 Hz, 2H), 2.73 – 2.65 (m, 3H), 2.25 (tq,

196
J = 11.2, 3.8 Hz, 1H), 1.77 – 1.68 (m, 2H), 1.43 (d, J = 11.7 Hz, 2H). TOF ES+ MS: (M
+ H) 476.2, (M + Na) 498.2. HPLC tR = 7.60 min, > 95%.
1-(1-(4-Chlorobenzyl)-1H-pyrrole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)azetidine-
3-carboxamide (CCG-208915). Methyl 1-(1-(4-chlorobenzyl)-1H-pyrrole-2-
carbonyl)azetidine-3-carboxylate 29 (300 mg, 0.90 mmol) was dissolved in 200 proof
EtOH (10 mL) and 10% aq. NaOH (10 mL) was added. The reaction was allowed to stir
at RT for 24 h, at which time the solvent was stripped off in vacuo and the residue was
taken up in H2O and acidified to pH 1 by addition of conc. HCl at 0°C. The resulting
precipitate was collected over a filter and dried by aspirator then high vacuum to afford
the desired carboxylic acid as a white powder. Yield: 250 mg, 0.78 mmol, 87%.
1-(1-(4-chlorobenzyl)-1H-pyrrole-2-carbonyl)azetidine-3-carboxylic acid (50 mg,
0.16 mmol) was added to DCM (5 mL), followed by 4-(2-aminoethyl)pyridine (20 µL,
0.17 mmol), TEA (66 µL, 0.47 mmol), EDCHCl (33 mg, 0.17 mmol), and HOBT (26
mg, 0.17 mmol). The reaction was allowed to stir at RT for 17 h, at which time the
reaction was concentrated and the residue taken up in EtOAc. This solution was then
washed with H2O (3X) and 10% aq. Na2CO3 (3X), and brine (1X), dried (MgSO4), and
concentrated. The residue was purified by silica flash chromatography (4 g silica, 1% to
30% 7 M methanolic ammonia/EtOAc) to provide the final compound as a white powder.
Yield: 32 mg, 0.076 mmol, 48%. 1H NMR (500 MHz, Chloroform-d) δ 8.48 (s, 2H), 7.25
(d, J = 8.3 Hz, 2H), 7.10 (d, J = 4.1 Hz, 2H), 7.04 (d, J = 8.2 Hz, 2H), 6.80 (s, 1H), 6.50
(d, J = 3.9 Hz, 1H), 6.17 (t, J = 3.2 Hz, 1H), 5.97 (t, J = 5.2 Hz, 1H), 5.52 (s, 2H), 4.25

197
(bs, 4H), 3.58 (q, J = 6.6 Hz, 2H), 3.20 (p, J = 7.4 Hz, 1H), 2.84 (t, J = 6.9 Hz, 2H). TOF
ES+ MS: (M + H) 423.1, (M + Na) 445.1. HPLC rt = 5.09 min, >95% purity.
1-(1-(4-Chlorobenzyl)-1H-imidazole-2-carbonyl)-N-(2-(pyridin-4-
yl)ethyl)piperidine-4-carboxamide (CCG-208916). Ethyl 1-(4-chlorobenzyl)-1H-
imidazole-2-carboxylate 23b (7.28 g, 27.5 mmol), was dissolved in EtOH (10 mL) and
10% aq. NaOH (20 mL) and stirred at rt for 15 h. The solvent was then stripped off,
water was added, and the solution acidified with HCl. The resulting precipitate was
collected over a filter and washed with 1M HCl and dried to afford the carboxylic acid as
a white powder which was taken directly into the next step. Yield: 6.506 g, 27.5 mmol,
100%.
The following were added sequentially to DCM (5 mL): 1-(4-chlorobenzyl)-1H-
imidazole-2-carboxylic acid (73 mg, 0.309 mmol), N-(2-(pyridin-4-yl)ethyl)piperidine-4-
carboxamide dihydrochloride (100 mg, 0.371 mmol), DIPEA (162 μL, 0.927 mmol),
EDCHCl (65 mg, 0.340 mmol), and HOBt (52 mg, 0.340 mmol). The solution was
allowed to stir at RT for 24 h, after which time the DCM was stripped off in vacuo and
the resulting residue was taken up on 10% aq. Na2CO3. Material was then extracted with
EtOAc. The extract was dried (MgSO4) and concentrated in vacuo. The residue was then
triturated with EtOAc:Hex and collected over a filter. The impure yellow powder was
then recrystallized from EtOAc to provide the title compound as off-white crystals.
Yield: 89 mg (64%). 1H NMR (400 MHz, CDCl3) δ 8.50 (d, J = 5.2 Hz, 2H), 7.28 (d, J =
8.3 Hz, 2H), 7.15–7.07 (m, 4H), 7.05 (s, 1H), 6.95 (s, 1H), 5.59 (t, J = 5.7 Hz, 1H), 5.35
(d,J = 8.9 Hz, 2H), 4.58 (d, J = 13.4 Hz, 2H), 3.53 (q, J = 6.7 Hz, 2H), 3.10 (t, J = 13.7

198
Hz, 1H), 2.87–2.71 (m, 3H), 2.29 (tt, J = 11.0, 3.4 Hz, 1H), 2.00–1.41 (m, 4H). TOF ES+
MS: (M + H) 452.2, (M + Na) 474.2. HPLC rt = 4.24 min, >90% purity.
1-(1-(4-Chlorobenzyl)-4-fluoro-1H-pyrrole-2-carbonyl)-N-((2,3-dihydro-1H-
inden-2-yl)methyl)piperidine-4-carboxamide (CCG-209020). The following reagents
were dissolved in DCM (3 mL) at RT: 1-(4-chlorobenzyl)-4-fluoro-1H-pyrrole-2-
carboxylic acid 136 (50 mg, 0.197 mmol), N-((2,3-dihydro-1H-inden-2-
yl)methyl)piperidine-4-carboxamide dihydrochloride (70 mg, 0.237 mmol), TEA (110
µL, 0.788 mmol), EDCHCl (45 mg, 0.237 mmol), and HOBT (36 mg, 0.237 mmol). The
reaction was allowed to stir for 16 h, after which time it was diluted with EtOAc and
washed with HCl (3X), H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X), then dried
(MgSO4) and concentrated. The residue was purified by silica flash chromatography (10
g silica, 5% to 50% EtOAc:Hex) to give the desired product as a white solid. Yield: 1H
NMR (400 MHz, Chloroform-d) δ 7.26 (d, J = 8.0 Hz, 2H), 7.21 – 7.09 (m, 4H), 7.05 (d,
J = 8.2 Hz, 2H), 6.56 – 6.50 (m, 1H), 6.05 (s, 1H), 5.54 (t, J = 5.1 Hz, 1H), 5.15 (s, 2H),
4.28 (d, J = 13.2 Hz, 2H), 3.36 (t, J = 5.7 Hz, 2H), 3.05 (q, J = 9.2 Hz, 2H), 2.82 (t, J =
11.7 Hz, 2H), 2.71 – 2.61 (m, 3H), 2.22 (tt, J = 11.1, 3.7 Hz, 1H), 1.71 (d, J = 12.0 Hz,
2H), 1.42 (d, J = 11.4 Hz, 2H). TOF ES+ MS: (M + H) 494.2. HPLC rt = 7.75 min, >95%
purity.
1-(1-(4-Chlorobenzyl)-4-fluoro-1H-pyrrole-2-carbonyl)-N-(2-(pyridin-4-
yl)ethyl)piperidine-4-carboxamide (CCG-209021). The following were added
sequentially to DCM (3 mL): 1-(4-chlorobenzyl)-4-fluoro-1H-pyrrole-2-carboxylic

199
acid 139 (50 mg, 0.20 mmol), TEA (0.11 mL, 0.79 mmol), EDCHCl (45 mg, 0.24
mmol), HOBT (36 mg, 0.24 mmol), and then amine dihydrochloride 36 (64 mg, 0.24
mmol). The reaction was allowed to stir at RT for 14 h, at which time it was diluted with
EtOAc, washed with H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X), dried with
MgSO4, and concentrated in vacuo. The residue was the purified by silica flash
chromatography (10 g silica, 1% 7 M methanolic ammonia/EtOAc) to provide the desired
material as a white powder. Yield: 48 mg (52%). 1H NMR (400 MHz, CDCl3) δ 8.54
(d, J = 4.3 Hz, 2H), 7.25–7.20 (m, 2H), 7.14 (d, J = 4.4 Hz, 2H), 7.06 (d, J = 6.9 Hz, 2H),
6.58–6.54 (m, 1H), 6.05 (s, 1H), 5.46–5.38 (m, 1H), 5.17 (s, 2H), 4.29 (d, J = 6.7 Hz,
2H), 3.55 (q, J = 7.3 Hz, 2H), 2.83 (dt, J = 17.9, 9.0 Hz, 4H), 2.27–2.15 (m, 1H), 1.77–
1.63 (m, 2H), 1.38 (d, J = 13.4 Hz, 2H). TOF ES+ MS: (M + H) 469.2, (M + Na) 491.2.
HPLC rt = 5.18 min, >95% purity.
1-(1-(4-Chlorobenzyl)-1H-pyrrolo[2,3-c]pyridine-2-carbonyl)-N-(2-(pyridin-4-
yl)ethyl)piperidine-4-carboxamide (CCG-211751). Azaindole ethyl ester 144 (300 mg,
0.953 mmol) was dissolved into ethanol (8 mL) at RT, followed by the addition of 10%
aqueous NaOH (8 mL), which initially caused precipitation, but homogeneity was
achieved over time. The reaction was allowed to stir at RT for 12 h, at which time the
solvent was removed in vacuo. The residue was taken up in a small amount of water (5
mL), the pH was adjusted to ∼4, and material was extracted with ethyl acetate (8×). The
extracts were combined and the solvent removed again in vacuo to provide the title
compound as a fine white powder. Yield: 226 mg (83%). 1H NMR (400 MHz, DMSO-

200
d6) δ 9.64 (s, 1H), 8.40 (d, J = 6.3 Hz, 1H), 8.26 (d, J = 6.3 Hz, 1H), 7.61 (s, 1H), 7.35 (d,
J = 8.5 Hz, 2H), 7.10 (d, J = 8.5 Hz, 2H), 6.06 (s, 2H).
The azaindole carboxylic acid (50 mg, 0.174 mmol) was dissolved into DCM (2
mL) at RT, followed by amine dihydrochloride 36 (59 mg, 0.192 mmol) and TEA (122
μL, 0.872 mmol). Once all material was in solution, EDCHCl (37 mg, 0.192 mmol) and
HOBT (29 mg, 0.192 mmol) were added and the reaction was allowed to stir for 18 h at
RT. At this time, the reaction was diluted with a solution of ethyl acetate/diethyl ether
(1:1) and washed with water (3×), 10% aq sodium carbonate (3×), and brine (1×), then
dried (magnesium sulfate) and concentrated in vacuo. The resulting residue was then
purified by flash chromatography (50 g silica, 5% 7.0 M methanolic ammonia/95% ethyl
acetate) to give the title compound as white solid. Yield: 53 mg (61%). 1H NMR (400
MHz, CDCl3) δ 8.78 (s, 1H), 8.49 (d, J = 5.4 Hz, 2H), 8.27 (d, J = 5.5 Hz, 1H), 7.51 (d, J
= 5.5 Hz, 1H), 7.21 (d, J = 8.2 Hz, 2H), 7.09 (d, J = 5.3 Hz, 2H), 7.05 (d, J = 8.2 Hz, 2H),
6.56 (s, 1H), 5.67 (t, J = 5.6 Hz, 1H), 5.48 (s, 2H), 4.57 (s, 1H), 3.91 (s, 1H), 3.53 (q, J =
6.6 Hz, 2H), 2.96−2.69 (m, 4H), 2.21 (tt, J =11.1, 7.5, 3.7 Hz, 1H), 1.88−1.69 (m, 1H),
1.67−1.46 (m, 2H), 1.33−1.22 (m, 1H). TOF ES+ MS: (M + H) 502.2. HPLC tR = 4.01
min, > 95%.
(R)-1-(1-(4-Chlorobenzoyl)pyrrolidine-2-carbonyl)-N-((2,3-dihydro-1H-inden-2-
yl)methyl)piperidine-4-carboxamide (CCG-211753). (R)-Ethyl 1-(1-(4-
chlorobenzoyl)pyrrolidine-2-carbonyl)piperidine-4-carboxylate (R)-40 (300 mg, 0.76
mmol) was dissolved in a 9:1 DCM:MeOH solution (20 mL), and a solution of
methanolic NaOH (92 mg NaOH/2mL MeOH) was added. This was stirred at RT for 2 h,

201
after which time the solvent was stripped off in vacuo to give a white solid that was
dissolved in a small amount of H2O and acidified by the addition of conc. HCl, and the
material was extracted with EtOAc (3X). The combined organic extracts were dried
(MgSO4) and concentrated. The clear oily residue was then dissolved in minimum EtOAc
and precipitated with Hex to afford a white powder. No further purification necessary.
Yield: 260 mg, 0.71 mmol, 93%.
(R)-1-(4-chlorobenzoyl)pyrrolidine-2-carboxylic acid (50 mg, 0.14 mmol) was
added to DCM (5 mL), followed by the sequential addition of TEA (76 µL, 0.55 mmol),
HATU (63 mg, 0.16 mmol), and indanyl amine 75 (24 mg, 0.16 mmol). The reaction
was stirred for 22 h at RT, after which time the reaction was diluted in sat. aq. NH4Cl and
extracted with EtOAc. The organic extract was then washed with 1M HCl (3X), H2O
(3X), 10% aq. Na2CO3 (3X), and sat. aq. Na2CO3 (1X), then dried with MgSO4 and
concentrated. The residue was purified via silica flash chromatography (4 g silica, 5% to
20% 7 M methanolic ammonia:EtOAc) to give the desired compound as a white powder.
Yield: 37 mg, 0.08 mmol, 57%. 1H NMR (400 MHz, Chloroform-d) δ 7.52 (d, J = 8.1
Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.18 – 7.11 (m, 4H), 6.04 (s, 0.5H), 5.75 – 5.67 (m,
0.5H), 5.03 (m, 1H), 4.60 (d, J = 13.3 Hz, 0.5H), 4.47 (d, J = 13.6 Hz, 0.5H), 4.07 (dd, J
= 30.7, 13.8 Hz, 1H), 3.80 (m, 0.5H), 3.68 (m, 1.5H), 3.51 (m, 1H), 3.32 (m, 2H), 3.04
(m, 2H), 2.85 – 2.74 (m, 1H), 2.72 – 2.56 (m, 3H), 2.43 (m, 0.5H), 2.36 – 2.14 (m, 1.5H),
2.14 – 2.00 (m, 2H), 2.02 – 1.76 (m, 5H), 1.65 (dd, J = 29.2, 12.8 Hz, 2H). TOF ES+ MS:
(M + H) 494.2. HPLC tR = 6.37 min, > 95% purity.

202
(S)-1-(1-(4-Chlorobenzoyl)pyrrolidine-2-carbonyl)-N-(2-(pyridin-4-
yl)ethyl)piperidine-4-carboxamide (CCG-211751). (S)-Ethyl 1-(1-(4-
chlorobenzoyl)pyrrolidine-2-carbonyl)piperidine-4-carboxylate (S)-40 (300 mg, 0.76
mmol) was dissolved in a 9:1 DCM:MeOH solution (20 mL), and a solution of
methanolic NaOH (92 mg NaOH/2mL MeOH) was added. This was stirred at RT for 2 h,
after which time the solvent was stripped off in vacuo to give a white solid that was
dissolved in a small amount of H2O and acidified by the addition of conc. HCl, and the
material was extracted with EtOAc (3X). The combined organic extracts were dried
(MgSO4) and concentrated. The clear oily residue was then dissolved in minimum EtOAc
ad precipitated with Hex to afford a white powder. No further purification necessary.
Yield: 265 mg, 0.73 mmol, 95%.
(S)-1-(4-chlorobenzoyl)pyrrolidine-2-carboxylic acid (50 mg, 0.14 mmol) was
added to DCM (5 mL), followed by the sequential addition of TEA (76 µL, 0.55 mmol),
HATU (63 mg, 0.16 mmol), and amine 27 (20 µL, 0.16 mmol). The reaction was stirred
for 17 h at RT, after which time the reaction was diluted in sat. aq. NH4Cl and extracted
with EtOAc. The organic extract was then washed with 10% aq. NH4Cl (2X), 10% aq.
Na2CO3 (3X), and sat. aq. Na2CO3 (1X), then dried with MgSO4 and concentrated. The
residue was purified via silica flash chromatography (4 g silica, 5% to 20% 7 M
methanolic ammonia:EtOAc) to give the desired compound as a white powder. Yield: 42
mg, 0.09 mmol, 65%. 1H NMR (500 MHz, Chloroform-d) δ 8.52 (m, 2H), 7.53 (d, J =
8.0 Hz, 2H), 7.37 (t, J = 7.0 Hz, 2H), 7.16 – 7.09 (m, 2H), 5.91 (d, J = 2.1 Hz, 0.5H),
5.58 – 5.49 (m, 0.5H), 5.07 – 4.97 (m, 1H), 4.63 (d, J = 14.5 Hz, 0.5H), 4.48 (d, J = 13.9
Hz, 1H), 4.09 (dd, J = 41.8, 14.7 Hz, 2H), 3.68 (t, J = 9.9 Hz, 1H), 3.54 (m, 3H), 3.32 –

203
3.21 (m, 0.5H), 3.12 (t, J = 12.8 Hz, 0.5H), 2.83 (dt, J = 14.6, 6.8 Hz, 3H), 2.64 (t, J =
12.6 Hz, 0.5H), 2.47 –1.15 (m, 7H). TOF ES+ MS: (M + H) 469.3, (M + Na) 491.2.
HPLC tR = 4.36 min, > 95% purity.
(S)-1-(1-(4-Chlorobenzoyl)pyrrolidine-2-carbonyl)-N-((2,3-dihydro-1H-inden-2-
yl)methyl)piperidine-4-carboxamide (CCG-211755). (S)-Ethyl 1-(1-(4-
chlorobenzoyl)pyrrolidine-2-carbonyl)piperidine-4-carboxylate (S)-40 (300 mg, 0.76
mmol) was dissolved in a 9:1 DCM:MeOH solution (20 mL), and a solution of
methanolic NaOH (92 mg NaOH/2mL MeOH) was added. This was stirred at RT for 2 h,
after which time the solvent was stripped off in vacuo to give a white solid that was
dissolved in a small amount of H2O and acidified by the addition of conc. HCl, and the
material was extracted with EtOAc (3X). The combined organic extracts were dried
(MgSO4) and concentrated. The clear oily residue was then dissolved in minimum EtOAc
ad precipitated with Hex to afford a white powder. No further purification necessary.
Yield: 265 mg, 0.73 mmol, 95%.
(S)-1-(4-chlorobenzoyl)pyrrolidine-2-carboxylic acid (50 mg, 0.14 mmol) was
added to DCM (5 mL), followed by the sequential addition of TEA (76 µL, 0.55 mmol),
HATU (63 mg, 0.16 mmol), and indanyl amine 75 (24 mg, 0.16 mmol). The reaction
was stirred for 19 h at RT, after which time the reaction was diluted in sat. aq. NH4Cl and
extracted with EtOAc. The organic extract was then washed with 1M HCl (3X), H2O
(3X), 10% aq. Na2CO3 (3X), and sat. aq. Na2CO3 (1X), then dried with MgSO4 and
concentrated. The residue was purified via silica flash chromatography (4 g silica, 5% to
20% 7 M methanolic ammonia:EtOAc) to give the desired compound as a white powder.

204
Yield: 44 mg, 0.09 mmol, 68%. 1H NMR (400 MHz, Chloroform-d) δ 7.61 – 7.54 (m,
1H), 7.54 – 7.49 (m, 1H), 7.44 – 7.37 (m, 1H), 7.37 – 7.28 (m, 2H), 7.25 – 7.06 (m, 4H),
6.14 – 6.02 (m, 0.5H), 5.81 – 5.70 (m, 0.5H), 5.19 – 4.96 (m, 1H), 4.60 (d, J = 13.6 Hz,
0.5H), 4.47 (d, J = 13.7 Hz, 0.5H), 4.06 (dd, J = 30.8, 14.3 Hz, 1H), 3.73 – 3.61 (m, 1H),
3.59 – 3.43 (m, 1H), 3.42 – 3.21 (m, 2H), 3.19 – 2.94 (m, 3H), 2.76 – 2.53 (m, 3H), 2.35
– 2.14 (m, 2H), 2.11 – 1.77 (m, 5H), 1.77 – 1.51 (m, 2H). TOF ES+ MS: (M + H) 494.2.
HPLC tR = 6.37 min, > 95% purity.
(R)-1-(1-(4-Chlorobenzoyl)pyrrolidine-2-carbonyl)-N-(2-(pyridin-4-
yl)ethyl)piperidine-4-carboxamide (CCG-211756). (R)-Ethyl 1-(1-(4-
chlorobenzoyl)pyrrolidine-2-carbonyl)piperidine-4-carboxylate (R)-40 (300 mg, 0.76
mmol) was dissolved in a 9:1 DCM:MeOH solution (20 mL), and a solution of
methanolic NaOH (92 mg NaOH/2mL MeOH) was added. This was stirred at RT for 2 h,
after which time the solvent was stripped off in vacuo to give a white solid that was
dissolved in a small amount of H2O and acidified by the addition of conc. HCl, and the
material was extracted with EtOAc (3X). The combined organic extracts were dried
(MgSO4) and concentrated. The clear oily residue was then dissolved in minimum EtOAc
and precipitated with Hex to afford a white powder. No further purification necessary.
Yield: 260 mg, 0.71 mmol, 93%.
(R)-1-(4-chlorobenzoyl)pyrrolidine-2-carboxylic acid (50 mg, 0.14 mmol) was
added to DCM (5 mL), followed by the sequential addition of TEA (76 µL, 0.55 mmol),
HATU (63 mg, 0.16 mmol), and amine 27 (20 µL, 0.16 mmol). The reaction was stirred
for 18 h at RT, after which time the reaction was diluted in sat. aq. NH4Cl and extracted

205
with EtOAc. The organic extract was then washed with 10% aq. NH4Cl (2X), 10% aq.
Na2CO3 (3X), and sat. aq. Na2CO3 (1X), then dried with MgSO4 and concentrated. The
residue was purified via silica flash chromatography (4 g silica, 5% to 20% 7 M
methanolic ammonia:EtOAc) to give the desired compound as a white powder. Yield: 41
mg, 0.09 mmol, 62%. 1H NMR (500 MHz, Chloroform-d) δ 8.59 – 8.47 (m, 2H), 7.53
(d, J = 8.1 Hz, 2H), 7.38 (t, J = 7.6 Hz, 2H), 7.12 (dd, J = 9.5, 4.8 Hz, 2H), 5.90 (s, 0.5H),
5.50 (s, 0.5H), 5.10 – 4.99 (m, 1H), 4.63 (d, J = 13.3 Hz, 0.5H), 4.48 (d, J = 13.2 Hz,
1H), 4.09 (dd, J = 40.4, 14.2 Hz, 2H), 3.70 (t, J = 8.6 Hz, 1H), 3.54 (m, 3H), 3.29 (t, J =
12.8 Hz, 0.5H), 3.12 (t, J = 12.7 Hz, 0.5H), 2.83 (dt, J = 14.7, 6.6 Hz, 3H), 2.64 (t, J =
12.6 Hz, 0.5H), 2.40-1.20 (m, 7H). TOF ES+ MS: (M + H) 469.3, (M + Na) 491.3. HPLC
tR = 4.26 min, > 95% purity.
(S)-1-(1-(4-Chlorobenzyl)pyrrolidine-2-carbonyl)-N-(2-(pyridin-4-
yl)ethyl)piperidine-4-carboxamide (CCG-211757). (S)-1-(4-chlorobenzyl)pyrrolidine-2-
carboxylic acid (S)-38 (63 mg, 0.27 mmol) was added to DCM (2 mL), followed by the
sequential addition of TEA (150 µL, 1.06 mmol), HATU (111 mg, 0.29 mmol), and
amine dihydrochloride 36 (75 mg, 0.28 mmol). The reaction was stirred for 24 h at RT,
after which time the reaction was diluted in sat. aq. NH4Cl and extracted with EtOAc.
The organic extract was then washed with 10% aq. NH4Cl (2X), 10% aq. Na2CO3 (3X),
and sat. aq. Na2CO3 (1X), then dried with MgSO4 and concentrated. The residue was
purified via silica flash chromatography (4 g silica, 5% to 20% 7 M methanolic
ammonia/EtOAc) to give a slightly yellow oil that was triturated from Et2O/Hex to
provide a white powder. Yield: 64 mg, 0.14 mmol, 53%. 1H NMR (500 MHz,

206
Chloroform-d) δ 8.48 (d, J = 4.6 Hz, 2H), 7.31 – 7.22 (m, 4H), 7.11 (bs, 2H), 5.82 (bs,
1H), 4.52 (bs, 1H), 4.13 (d, J = 11.9 Hz, 1H), 3.89 (dd, J = 18.3, 13.1 Hz, 1H), 3.58 –
3.49 (m, 2H), 3.46 – 3.34 (m, 2H), 3.10 – 3.00 (m, 1H), 2.94 (d, J = 9.3 Hz, 1H), 2.87 –
2.79 (m, 2H), 2.63 – 2.52 (m, 1H), 2.33 – 2.26 (m, 2H), 2.18 – 2.08 (m, 1H), 1.89 – 1.75
(m, 4H), 1.60 – 1.50 (m, 2H). TOF ES+ MS: (M + H) 455.2, (M + Na) 477.2. HPLC tR =
3.39 min, > 90% purity.
(R)-1-(1-(4-Chlorobenzyl)pyrrolidine-2-carbonyl)-N-(2-(pyridin-4-
yl)ethyl)piperidine-4-carboxamide (CCG-211758). (R)-1-(4-chlorobenzyl)pyrrolidine-2-
carboxylic acid (R)-38 (64 mg, 0.27 mmol) was added to DCM (2 mL), followed by the
sequential addition of TEA (150 µL, 1.06 mmol), HATU (111 mg, 0.29 mmol), and
amine dihydrochloride 36 (75 mg, 0.28 mmol). The reaction was stirred for 20 h at RT,
after which time the reaction was diluted in sat. aq. NH4Cl and extracted with EtOAc.
The organic extract was then washed with 10% aq. NH4Cl (2X), 10% aq. Na2CO3 (3X),
and sat. aq. Na2CO3 (1X), then dried with MgSO4 and concentrated. The residue was
purified via silica flash chromatography (4 g silica, 5% to 20% 7 M methanolic
ammonia:EtOAc) to give a slightly yellow oil that was triturated from Et2O/Hex to
provide a white powder. Yield: 75 mg, 0.17 mmol, 62%. 1H NMR (500 MHz,
Chloroform-d) δ 8.52 (d, J = 5.1 Hz, 2H), 7.26 (s, 4H), 7.12 (s, 2H), 5.57 (s, 1H), 4.55 (d,
J = 11.7 Hz, 1H), 4.15 (bs, 1H), 3.90 (dd, J = 18.8, 13.1 Hz, 1H), 3.61 – 3.50 (m, 2H),
3.39 (d, J = 12.0 Hz, 2H), 3.06 (d, J = 7.0 Hz, 1H), 2.98 – 2.91 (m, 1H), 2.84 (q, J = 6.2
Hz, 2H), 2.59 (q, J = 13.9, 13.3 Hz, 1H), 2.33 – 2.21 (m, 2H), 2.14 (s, 1H), 1.80 – 1.77

207
(m, 2H), 1.59 – 1.48 (m, 2H). TOF ES+ MS: (M + H) 455.2, (M + Na) 477.2. HPLC tR =
3.44 min, > 90% purity.
(E)-(1-(4-Chlorobenzyl)-1H-indol-2-yl)(4-(4-(pyridin-4-yl)but-1-en-1-
yl)piperidin-1-yl)methanone (CCG-211822). (E)-tert-butyl 4-(4-(pyridin-4-yl)but-1-en-
1-yl)piperidine-1-carboxylate 99b (100 mg, 0.316 mmol) was dissolved in anhydrous
1,4-dioxane (1 mL) and 4 M HCl in 1,4-dioxane (0.55 mL, 2.21 mmol) was added. The
reaction was allowed to stir for 1 h, at which time the solvent was stripped off and
replaced with Et2O. The oily residue was solidified with sonication, washed (decantation)
with Et2O (3X) and then dried under high vacuum to give the dihydrochloride as a white
powder. Yield: 91 mg, 0.316 mmol, 100%.
(E)-4-(4-(pyridin-4-yl)but-1-en-1-yl)piperidine dihydrochloride (75 mg, 0.259
mmol) was dissolved in DCM (10 mL) and TEA (217 µL, 1.56 mmol), followed by the
indole carboxylic acid 4a (89 mg, 0.311 mmol), EDCHCl (60 mg, 0.311 mmol), and
HOBT (48 mg, 0.311 mmol), and the reaction was allowed to stir at RT for 18 h. After
this time, the reaction was diluted with EtOAc and washed with H2O (3X), 10% Na2CO3
(3X), and brine (1X), then dried (Na2SO4) and concentrated in vacuo. The resulting
residue was purified via silica flash chromatography (10 g silica, 1% to 20% methanolic
ammonia:EtOAc) to provide a clear, colorless oil. Yield: 72 mg, 0.148 mmol, 57%. 1H
NMR (400 MHz, Chloroform-d) δ 8.54 – 8.39 (m, 2H), 7.63 (d, J = 7.8 Hz, 1H), 7.35 (d,
J = 8.2 Hz, 1H), 7.25 (t, J = 7.6 Hz, 1H), 7.21 – 7.10 (m, 3H), 7.08 (s, 2H), 7.01 (d, J =
8.0 Hz, 2H), 6.59 (s, 1H), 5.46 (s, 2H), 5.37 – 5.31 (m, 1H), 5.26 (d, J = 11 Hz, 1H), 4.57
(bs, 1H), 4.04 (bs, 1H), 2.85 (bs, 2H), 2.65 (t, J = 7.3 Hz, 2H), 2.39 – 2.23 (m, 2H), 2.07

208
(s, 1H), 1.64 (bs, 1H), 1.41 (bs, 1H), 1.08 (bs, 1H), 0.64 (bs, 1H). TOF ES+ MS: (M + H)
484.2. HPLC tR = 6.24 min, > 95% purity.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)-1,2,3,6-
tetrahydropyridine-4-carboxamide (CCG-211823). Ethyl 1-(1-(4-chlorobenzyl)-1H-
indole-2-carbonyl)-1,2,3,6-tetrahydropyridine-4-carboxylate (53 mg, 0.125 mmol) was
dissolved in EtOH (5 mL), followed by the addition of 10% aq. NaOH (2 mL). This
reaction was allowed to stir for 4 h, at which time the solvent was removed in vacuo. The
residue was dissolved in H2O and acidified with conc. HCl at 0 °C. The resulting
precipitate was collected over a filter and dried under high vacuum to give a white
powder. Yield: 46 mg, 0.116 mmol, 93%.
1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)-1,2,3,6-
tetrahydropyridine-4-carboxylic acid (33 mg, 0.084 mmol) was dissolved in DCM (5 mL)
and TEA (35 µL, 0.251 mmol), followed by EDCHCl (18 mg, 0.092 mmol), and HOBT
(14 mg, 0.092 mmol), and the reaction was allowed to stir at RT for 21 h. After this time,
the reaction was diluted with EtOAc:Et2O and washed with H2O (2X), 10% Na2CO3
(3X), and brine (1X), then dried (Na2SO4) and concentrated in vacuo. The resulting
residue was purified via silica flash chromatography (4 g silica, 80% to 100%
EtOAc:Hex) to provide a fine white powder. Yield: 38 mg, 0.076 mmol, 91%. 1H NMR
(400 MHz, Chloroform-d) δ 8.51 (d, J = 4.1 Hz, 2H), 7.66 (d, J = 7.9 Hz, 1H), 7.30 –
7.05 (m, 8H), 6.97 (d, J = 8.1 Hz, 2H), 6.78 (s, 1H), 5.77 (s, 1H), 5.51 (s, 2H), 4.83 (bs,
1H), 3.89 (bs, 1H), 3.61 – 3.43 (m, 3H), 2.99 (s, 1H), 2.84 (t, J = 6.9 Hz, 2H), 2.16 (bs,

209
1H), 1.94 (bs, 1H). TOF ES+ MS: (M + H) 499.2, (M + Na) 521.2. HPLC tR = 5.46 min,
> 95%.
1-(2-((4-Chlorophenyl)amino)benzoyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-
carboxamide (CCG-211824). Ethyl 1-(2-((4-chlorophenyl)amino)benzoyl)piperidine-4-
carboxylate (72 mg, 0.19 mmol) was dissolved in EtOH (5 mL), followed by the addition
of 10% aq. NaOH (2 mL). This reaction was allowed to stir at RT for 24 h, after which
time the solvent was stripped off in vacuo and the residue was taken up in H2O and
acidified to pH<1 with conc. HCl at 0 °C. The resulting white precipitate was collected
over a filter, dried via aspirator then high vacuum to afford the carboxylic acid as a white
powder requiring no further purification. Yield: 47 mg, 0.13 mmol, 70%.
1-(2-((4-chlorophenyl)amino)benzoyl)piperidine-4-carboxylic acid (36 mg, 0.10
mmol), 4-(2-aminoethyl)pyridine (13 µL, 0.11 mmol), TEA (42 µL, 0.30 mmol),
EDCHCl (21 mg, 0.11 mmol), and HOBT (17 mg, 0.11 mmol) were all added to DCM
(5 mL) in the listed order and stirred at RT for 20 h, at which time the reaction was
diluted with 1:1 EtOAc:Et2O and washed with 0.5 M aq. HCl (1X), H2O (1X), 10% aq.
Na2CO3 (3X), and brine (1X). The organic phase was then dried (MgSO4) and
concentrated, and the residue was purified via silica flash chromatography (4 g silica,
80% to 100% EtOAc:Hex gradient) to give the title compound as a clear, colorless oil.
Yield: 36 mg, 0.078 mmol, 78%. 1H NMR (500 MHz, Chloroform-d) δ 8.51 (d, J = 4.9
Hz, 2H), 7.32 (d, J = 8.3 Hz, 1H), 7.26 – 7.19 (m, 3H), 7.17 (d, J = 7.6 Hz, 1H), 7.13 –
7.09 (m, 2H), 7.00 (d, J = 8.6 Hz, 2H), 6.89 (t, J = 7.4 Hz, 1H), 5.56 (s, 1H), 4.23 (bs,
2H), 3.53 (q, J = 6.6 Hz, 2H), 2.93 (s, 2H), 2.82 (t, J = 6.9 Hz, 2H), 2.28 (tt, J = 11.2, 3.7

210
Hz, 1H), 1.80 – 1.66 (m, 4H). TOF ES+ MS: (M + H) 463.2, (M + Na) 485.2. HPLC tR =
5.23 min, > 95%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-4-methyl-N-(2-(pyridin-4-
yl)ethyl)piperidine-4-carboxamide (CCG-211825). 1-(1-(4-chlorobenzyl)-1H-indole-2-
carbonyl)-4-methylpiperidine-4-carboxylic acid (100 mg, 0.243 mmol), 4-(2-
aminoethyl)pyridine (32 µL, 0.268 mmol), TEA (100 µL, 0.730 mmol), EDCHCl (51
mg, 0.268 mmol), and HOBT (41 mg, 0.268 mmol) were dissolved in DCM (5 mL) and
stirred at RT for 16 h. After this time, the reaction was diluted with a 2:1 solution of
EtOAc:Et2O and washed with H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X), then
dried (MgSO4) and concentrated. The residue was purified by silica flash
chromatography (4 g silica, 1% to 5% methanolic ammonia:EtOAc) to give a clear
colorless oil. Yield: 113 mg, 0.219 mmol, 90%. 1H NMR (500 MHz, Chloroform-d) δ
8.46 (d, J = 4.7 Hz, 2H), 7.63 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 8.3 Hz, 1H), 7.26 (t, J =
7.7 Hz, 1H), 7.19 – 7.12 (m, 3H), 7.08 (d, J = 4.8 Hz, 2H), 7.00 (d, J = 8.0 Hz, 2H), 6.58
(s, 1H), 5.95 (t, J = 5.6 Hz, 1H), 5.44 (s, 2H), 4.02 (s, 1H), 3.68 (s, 1H), 3.52 (q, J = 6.5
Hz, 2H), 3.22 (s, 2H), 2.81 (t, J = 6.9 Hz, 2H), 2.22 (s, 1H), 1.86 (s, 1H), 1.67 (s, 1H),
1.28 (s, 1H), 1.05 (s, 3H), 0.77 (s, 1H). TOF ES+ MS: (M + H) 515.0. HPLC tR = 5.49
min, > 95%.
(1-(4-Chlorobenzyl)-1H-indol-2-yl)(4-(3-(pyridin-4-yl)propyl)piperazin-1-
yl)methanone (CCG-211826). t-Butyl 4-(3-(pyridin-4-yl)propyl)piperazine-1-carboxylate
(160 mg, 0.524 mmol) was dissolved in 1,4-dioxane (2 mL) at RT, and a 4 M HCl in 1,4-

211
dioxane solution (1.31 mL, 5.24 mmol) was added. The reaction was allowed to stir for
15 min, over which time precipitation occurred. The solvent was removed in vacuo and
dried under high vacuum to give the trishydrochloride salt as a tan powder. Yield: 168
mg, 0.524 mmol, 100%.
1-(4-Chlorobenzyl)-1H-indole-2-carboxylic acid (91 mg, 0.318 mmol), 4-(3-
(pyridin-4-yl)propyl)piperazine trishydrochloride (100 mg, 0.318 mmol), TEA (266 µL,
1.907 mmol), EDCHCl (67 mg, 0.350 mmol), and HOBT (54 mg, 0.350 mmol) were
dissolved in DCM (5 mL) and stirred at RT for 14 h. After this time, the reaction was
diluted with a 2:1 solution of EtOAc:Et2O and washed with H2O (3X), 10% aq. Na2CO3
(3X), and brine (1X), then dried (MgSO4) and concentrated. The residue was purified by
silica flash chromatography (4 g silica, 1% to 5% MeOH:EtOAc) to give a clear colorless
oil. Yield: 94 mg, 0.199 mmol, 63%. 1H NMR (500 MHz, Chloroform-d) δ 8.49 (d, J =
5.2 Hz, 2H), 7.65 (d, J = 7.9 Hz, 1H), 7.38 (d, J = 8.3 Hz, 1H), 7.31 – 7.26 (m, 1H), 7.19
(dd, J = 13.2, 7.9 Hz, 3H), 7.11 (d, J = 5.2 Hz, 2H), 7.03 (d, J = 8.2 Hz, 2H), 6.62 (s, 1H),
5.49 (s, 2H), 3.64 (bs, 4H), 2.64 (t, J = 7.6 Hz, 2H), 2.36 – 2.26 (m, 4H), 2.00 (bs, 2H),
1.79 (p, J = 7.5 Hz, 2H). TOF ES+ MS: (M + H) 473.2, (M + Na) 495.2. HPLC tR = 4.78
min, > 95%.
(1-(4-Chlorobenzyl)-1H-indol-2-yl)(4-phenethylpiperazin-1-yl)methanone (CCG-
211827). 1-(4-Chlorobenzyl)-1H-indole-2-carboxylic acid (100 mg, 0.350 mmol), N-(2-
phenylethyl)piperazine (73 µL, 0.385 mmol), TEA (146 µL, 1.050 mmol), EDCHCl (74
mg, 0.385 mmol), and HOBT (59 mg, 0.385 mmol) were dissolved in DCM (6 mL) and
stirred at RT for 14 h. After this time, the reaction was diluted with a 2:1 solution of

212
EtOAc:Et2O and washed with H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X), then
dried (MgSO4) and concentrated. The residue was purified by silica flash
chromatography (4 g silica, 60% EtOAc:Hex) to give a clear colorless oil. Yield: 109 mg,
0.238 mmol, 68%. 1H NMR (500 MHz, Chloroform-d) δ 7.67 (d, J = 7.9 Hz, 1H), 7.39
(d, J = 8.4 Hz, 1H), 7.33 – 7.26 (m, 3H), 7.20 (dt, J = 19.0, 7.8 Hz, 6H), 7.05 (d, J = 8.3
Hz, 2H), 6.65 (s, 1H), 5.50 (s, 2H), 3.68 (s, 4H), 2.77 (dd, J = 9.6, 6.5 Hz, 2H), 2.57 (dd,
J = 9.7, 6.5 Hz, 2H), 2.28 (d, J = 147.4 Hz, 5H). TOF ES+ MS: (M + H) 458.2. HPLC tR
= 5.76 min, > 95%.
(Z)-(1-(4-Chlorobenzyl)-1H-indol-2-yl)(4-(4-(pyridin-4-yl)but-1-en-1-
yl)piperidin-1-yl)methanone (CCG-211829). (Z)-tert-butyl 4-(4-(pyridin-4-yl)but-1-en-1-
yl)piperidine-1-carboxylate 99a (100 mg, 0.316 mmol) was dissolved in anhydrous 1,4-
dioxane (1 mL) and 4 M HCl in 1,4-dioxane (0.55 mL, 2.21 mmol) was added. The
reaction was allowed to stir for 1 h, at which time the solvent was stripped off and
replaced with Et2O. The oily residue was solidified with sonication, washed (decantation)
with Et2O (3X) and then dried under high vacuum to give the dihydrochloride as a white
powder. Yield: 90 mg, 0.316 mmol, 100%.
(Z)-4-(4-(pyridin-4-yl)but-1-en-1-yl)piperidine dihydrochloride (75 mg, 0.259
mmol) was dissolved in DCM (10 mL) and TEA (217 µL, 1.56 mmol), followed by the
indole carboxylic acid 4a (89 mg, 0.311 mmol), EDCHCl (60 mg, 0.311 mmol), and
HOBT (48 mg, 0.311 mmol), and the reaction was allowed to stir at RT for 16 h. After
this time, the reaction was diluted with EtOAc and washed with H2O (3X), 10% Na2CO3
(3X), and brine (1X), then dried (Na2SO4) and concentrated in vacuo. The resulting

213
residue was purified via silica flash chromatography (10 g silica, 1% to 20% methanolic
ammonia:EtOAc) to provide a clear, colorless oil. Yield: 54 mg, 0.111 mmol, 43%. 1H
NMR (400 MHz, Chloroform-d) δ 8.47 (d, J = 5.5 Hz, 2H), 7.62 (d, J = 7.9 Hz, 1H), 7.35
(d, J = 8.3 Hz, 1H), 7.28 – 7.22 (m, 1H), 7.17 (dd, J = 15.8, 7.9 Hz, 3H), 7.09 (d, J = 5.5
Hz, 2H), 7.01 (d, J = 8.3 Hz, 2H), 6.58 (s, 1H), 5.46 (s, 2H), 5.32 (dt, J = 8.1, 7.4 Hz,
1H), 5.11 (t, J = 8.1 Hz, 1H), 4.54 (bs, 1H), 4.05 (bs, 1H), 2.78 (bs, 2H), 2.64 (t, J = 7.2
Hz, 2H), 2.36 (q, J = 7.4 Hz, 2H), 2.31 – 2.19 (m, 1H), 1.43 (bs, 1H), 1.12 (bs, 2H), 0.64
(bs, 1H). TOF ES+ MS: (M + H) 484.2. HPLC tR = 6.29 min, > 95%.
N-(1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)piperidin-4-yl)-3-(pyridin-4-
yl)propanamide (CCG-212052). t-Butyl 4-(3-(pyridin-4-yl)propanamido)piperidine-1-
carboxylate (250 mg, 0.750 mmol) was dissolved in 1,4-dioxane (1 mL) and 4 M HCl
(1.0 mL, 4.12 mmol) in 1,4-dioxane was added. The reaction was allowed to stir for 1 h,
then the solvent was removed and replaced with Et2O and the oily material was solidified
with sonication. Washing with Et2O and thorough drying under high vacuum afforded the
desired dihydrochloride salt. Yield: 230 mg, 0.750 mmol, 100%.
4-(3-(pyridin-4-yl)propanamido)piperidine dihydrochloride (100 mg, 0.327
mmol), 1-(4-chlorobenzyl)-1H-indole-2-carboxylic acid 4a (112 mg, 0.392 mmol), TEA
(273 µL, 1.959 mmol), EDCHCl (75 mg, 0.392 mmol), and HOBT (60 mg, 0.392 mmol)
were all dissolved in DCM (10 mL) and stirred at RT for 22 h, after which time the DCM
was removed in vacuo and the residue was taken up in EtOAc and washed with H2O
(2X), 10% Na2CO3 (3X), and brine (1X), then dried (MgSO4) and concentrated. The
resulting residue was purified via silica flash chromatography (10 g silica, 1% to 20%

214
methanolic ammonia:EtOAc) to provide a white solid. Yield: 100 mg, 0.20 mmol, 61%.
1H NMR (500 MHz, Chloroform-d) δ 8.50 (d, J = 4.3 Hz, 2H), 7.65 (d, J = 7.9 Hz, 1H),
7.41 (d, J = 8.4 Hz, 1H), 7.30 (t, J = 7.7 Hz, 1H), 7.22 – 7.17 (m, 3H), 7.14 – 7.11 (m,
2H), 7.07 – 7.03 (m, 2H), 6.59 (d, J = 1.8 Hz, 1H), 5.48 (s, 2H), 5.30 (d, J = 8.0 Hz, 1H),
4.50 (s, 1H), 4.09 (s, 1H), 3.89 (d, J = 7.1 Hz, 1H), 2.95 (t, J = 7.3 Hz, 2H), 2.82 (s, 2H),
2.45 (t, J = 7.1 Hz, 2H), 1.83 (bs, 1H), 1.62 (bs, 1H), 1.02 (bs, 1H), 0.36 (bs, 1H). TOF
ES+ MS: (M + H) 501.2, (M + Na) 523.2.
4-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)-
piperazine-1-carboxamide (CCG-212390). t-Butyl 4-((2-(pyridin-4-
yl)ethyl)carbamoyl)piperazine-1-carboxylate (233 mg, 0.70 mmol) was dissolved in
anhydrous 1,4-dioxane (1 mL), and 4 M HCl in 1,4-dioxane (1.0 mL, 4.18 mmol) was
added. The reaction was allowed to stir for 2 h, after which time the solvent was removed
and the residue was solidified via sonication in Et2O. The solid was washed with Et2O
then thoroughly dried under high vacuum to give the dihydrochloride salt as a tan
powder. Yield: 214 mg, 0.70 mmol, 100%.
4-((2-(pyridin-4-yl)ethyl)carbamoyl)piperazine dihydrochloride (214 mg, 0.70
mmol), 1-(4-chlorobenzyl)-1H-indole-2-carboxylic acid (200 mg, 0.70 mmol), TEA
(0.388 mL, 2.79 mmol), EDCHCl (147 mg, 0.766 mmol), and HOBT (117 mg, 0.766
mmol) were dissolved in DCM (10 mL) and stirred for 16 h at RT. After this time, the
reaction was diluted with EtOAc and washed with H2O (3X), 10% Na2CO3 (3X), and
brine (1X), then dried (MgSO4) and concentrated. The resulting residue was purified via
silica flash chromatography (25 g silica, 1% to 20% methanolic ammonia:EtOAc) to

215
provide a white solid. Yield: 224 mg, 0.446 mmol, 64%. 1H NMR (400 MHz,
Chloroform-d) δ 8.47 (d, J = 5.4 Hz, 2H), 7.64 (d, J = 7.9 Hz, 1H), 7.36 (d, J = 8.4 Hz,
1H), 7.31 – 7.26 (m, 1H), 7.17 (dd, J = 12.9, 7.8 Hz, 3H), 7.11 (d, J = 5.4 Hz, 2H), 6.99
(d, J = 8.2 Hz, 2H), 6.63 (s, 1H), 5.47 (s, 2H), 4.72 (t, J = 5.4 Hz, 1H), 3.60 (s, 4H), 3.49
(q, J = 6.6 Hz, 2H), 3.16 (bs, 4H), 2.82 (t, J = 6.9 Hz, 2H). TOF ES+ MS: (M + H) 502.2,
(M + Na) 524.2. HPLC tR = 5.40 min, > 95%.
1-(2-(4-Chlorobenzoyl)benzoyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-
carboxamide (CCG-212391). 2-(4-chlorobenzoyl)benzoic acid (366 mg, 1.404 mmol)
was added to DCM (20 mL), followed by amine dihydrochloride 36 (473 mg, 1.544
mmol), TEA (978 µL, 7.02 mmol), EDCHCl (296 mg, 1.544 mmol), and HOBT (237
mg, 1.544 mmol.) The reaction was allowed to stir at RT for 20 h, at which time the
solvent was removed in vacuo. The residue was taken up in EtOAc and washed with H2O
(1X), 10% aq. Na2CO3 (3X), and brine (1X), then dried (MgSO4) and concentrated. The
residue was then purified by silica flash chromatography (25 g silica, 1% to 20% 7 M
methanolic ammonia:EtOAc.) 1H NMR (400 MHz, Chloroform-d) δ 8.47 (d, J = 5.5 Hz,
2H), 7.72 (d, J = 8.4 Hz, 2H), 7.61 – 7.53 (m, 1H), 7.52 – 7.24 (m, 5H), 7.10 (d, J = 5.4
Hz, 2H), 5.98 (t, J = 5.5 Hz, 1H), 4.47 (d, J = 12.9 Hz, 1H), 3.62 (d, J = 13.5 Hz, 1H),
3.51 (q, J = 6.6 Hz, 2H), 3.02 (t, J = 11.6 Hz, 1H), 2.81 (t, J = 6.9 Hz, 2H), 2.71 (t, J =
11.9 Hz, 1H), 2.28 (ddd, J = 14.9, 11.1, 3.9 Hz, 1H), 1.91 – 1.52 (m, 4H). TOF ES+ MS:
(M + H) 476.2, (M + Na) 498.2. HPLC tR = 4.83 min, > 95%.

216
1-(2-(4-chlorophenoxy)benzoyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-
carboxamide (CCG-212392). Methyl 2-(4-chlorophenoxy)benzoate 41 was dissolved in
EtOH (5 mL), followed by the addition of 10% aq. NaOH (2 mL). The reaction was
allowed to stir for 14 h, after which time the solvent was stripped off in vacuo and the
residue re-dissolved in H2O. This aqueous solution was then cooled to 0 °C and acidified
to pH<1 with conc. HCl, and the resulting precipitate was collected over a filter, then
dried via aspirator and high vacuum to provide the desired carboxylic acid as a white
powder. No further purification necessary. Yield: 92 mg, 0.37 mmol, 97%.
All of the previously prepared carboxylic acid (92 mg, 0.37 mmol) was added to
DCM (5 mL), followed by amine dihydrochloride 36 (136 mg, 0.44 mmol), TEA (310
µL, 2.22 mmol), EDCHCl (85 mg, 0.44 mmol), and HOBT (68 mg, 0.44 mmol). The
solution was allowed to stir at RT for 18 h, after which time the reaction was
concentrated in vacuo and the resulting residue was taken up in EtOAc and washed with
H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X), then dried (MgSO4) and concentrated.
The resulting residue was purified by silica flash chromatography (10 g silica, 1% to 20%
methanolic ammonia:EtOAc) to give the desired compound as a thick oil. 1H NMR (500
MHz, Chloroform-d) δ 8.43 (d, J = 4.7 Hz, 2H), 7.28 (dt, J = 29.2, 7.9 Hz, 4H), 7.14 (t, J
= 7.4 Hz, 1H), 7.06 (d, J = 4.8 Hz, 2H), 6.97 – 6.76 (m, 3H), 6.15 (d, J = 47.8 Hz, 1H),
4.57 (d, J = 12.6 Hz, 1H), 3.63 (bs, 1H), 3.48 (q, J = 6.5 Hz, 2H), 3.04 – 2.88 (m, 1H),
2.77 (t, J = 6.9 Hz, 2H), 2.75 – 2.62 (m, 1H), 2.24 (t, J = 11.0 Hz, 1H), 1.81 – 1.73 (m,
1H), 1.72 – 1.61 (m, 2H), 1.61 – 1.51 (m, 1H). TOF ES+ MS: (M + H) 464.1, (M + Na)
486.1. HPLC tR = 4.79 min, > 90%.

217
1-(2-((4-Chlorophenyl)(methyl)amino)benzoyl)-N-(2-(pyridin-4-yl)ethyl)-
piperidine-4-carboxamide (CCG-212393). Methyl 2-((4-
chlorophenyl)(methyl)amino)benzoate 45 (120 mg, 0.44 mmol) was dissolved in EtOH (8
mL), followed by the addition of 10% aq. NaOH (3 mL). The reaction was allowed to stir
at RT for 6 h, after which time the solvent was removed and the residue was taken up in
H2O, cooled to 0 °C, and acidified to pH<1. The resulting white precipitate was collected
over a filter, dried under high vacuum, and used without further purification. Yield: 108
mg, 0.41 mmol, 95%. 1H NMR (400 MHz, Chloroform-d) δ 9.44 (s, 1H), 7.96 (dd, J =
8.0, 1.5 Hz, 1H), 7.35 – 7.24 (m, 3H), 7.17 (t, J = 9.0 Hz, 3H), 6.79 – 6.72 (m, 1H), 3.89
(s, 3H).
2-((4-chlorophenyl)(methyl)amino)benzoic acid (85 mg, 0.33 mmol), amine
dihydrochloride 36 (119 mg, 0.39 mmol), TEA (270 µL, 1.95 mmol), EDCHCl (75 mg,
0.39 mmol), and HOBT (60 mg, 0.39 mmol) were added to DCM (5 mL) sequentially,
and the solution was allowed to stir at RT for 20 h. After this time, the solvent was
removed in vacuo and the residue was dissolved in EtOAc and washed with H2O (3X),
10% aq. Na2CO3 (3X), and brine (1X), then dried (MgSO4) and concentrated. The residue
was then purified via silica flash chromatography (10 g silica, 1% to 20% methanolic
ammonia:EtOAc) to provide the desired compound as a thick colorless oil. Yield: 90 mg,
0.19 mmol, 58%. 1H NMR (400 MHz, Chloroform-d, mixture of rotamers) δ 8.48 (m,
2H), 7.39 (t, J = 8.1 Hz, 1H), 7.34 – 7.19 (m, 3H), 7.09 (d, J = 7.9 Hz, 4H), 6.63 (t, J =
9.6 Hz, 2H), 5.75 (t, J = 5.4 Hz, 0.5H), 5.41 (t, J = 5.4 Hz, 0.5H), 4.64 – 4.44 (m, 1H),
3.61 – 3.39 (m, 3H), 3.22 (s, 1.5H), 3.17 (s, 1.5H), 2.93 – 2.84 (m, 0.5H), 2.83 – 2.74 (m,
2H), 2.74 – 2.63 (m, 1H), 2.53 – 2.44 (m, 0.5H), 2.22 – 2.09 (m, 1H), 1.82 (bd, J = 13.1

218
Hz, 0.5H), 1.78 – 1.51 (m, 3.5H), 1.41 (bd, J = 13.2 Hz, 0.5H), 1.30 (qd, J = 12.9, 4.2 Hz,
0.5H). TOF ES+ MS: (M + H) 477.2, 499.1. HPLC tR = 5.13 min, > 90% purity.
(3aS,7aR)-5-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-2-(2-(pyridin-4-
yl)ethyl)octahydro-1H-pyrrolo[3,4-c]pyridin-1-one (CCG-212394). 5-(1-(4-
chlorobenzyl)-1H-indole-2-carbonyl)octahydro-1H-pyrrolo[3,4-c]pyridin-1-one 91 (35
mg, 0.09 mmol) was dissolved in anhydrous DMF (5 mL). A 60% oil suspension of NaH
(17 mg, 0.43 mmol) was added at RT under N2 and stirred for 1 h. 4-(2-
Bromoethyl)pyridine (19 mg, 0.10 mmol) was added and stirring continued for 22 h
under the same conditions. After this time, the solution was diluted with EtOAc and
washed with H2O (1X), 10% aq. Na2CO3 (2X), and brine (1X), dried with MgSO4, and
concentrated in vacuo. Purification was done via silica flash chromatography (10 g silica,
1 to 20% methanolic ammonia:EtOAc.) The residue was dissolved in DCM, and rapid
solvent removal afforded the title compound as a white powder. Yield: 36 mg, 0.07
mmol, 82%. 1H NMR (400 MHz, Chloroform-d) δ 8.51 (d, J = 4.8 Hz, 2H), 7.68 (d, J =
7.9 Hz, 1H), 7.29 – 7.23 (m, 2H), 7.23 – 7.17 (m, 3H), 7.17 – 7.11 (m, 3H), 7.05 (d, J =
8.0 Hz, 2H), 5.45 (d, J = 7.0 Hz, 2H), 4.43 (d, J = 13.7 Hz, 1H), 4.10 (d, J = 3.8 Hz, 1H),
3.66 – 3.49 (m, 2H), 3.41 – 3.34 (m, 1H), 3.32 – 3.25 (m, 1H), 2.92 – 2.77 (m, 4H), 2.42
(bs, 2H), 2.10 (bs, 1H), 1.75 (bs, 1H), 1.31 (bs, 1H). TOF ES+ MS: (M + H) 513.2, (M +
Na) 535.2. HPLC tR = 5.53 min, > 95% purity.
1-(2-((4-Chlorobenzyl)amino)benzoyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-
carboxamide (CCG-222660). Methyl 2-((4-chlorobenzyl)amino)benzoate 47a (80 mg,

219
0.29 mmol) was dissolved in MeOH (8 mL) and 10% aq. NaOH (4 mL) was added and
the reaction was stirred at RT for 24 h. At this time, the solvent was removed in vacuo
and the residue was dissolved in H2O and acidified with conc. HCl to pH<1 at 0 °C. The
resulting precipitate was collected over a filter and dried via aspirator then high vacuum
to afford the desired carboxylic acid as a white powder. No further purification necessary.
Yield: 75 mg, 0.29 mmol, 99%.
2-((4-chlorobenzyl)amino)benzoic acid (75 mg, 0.29 mmol) was dissolved in
DCM (12 mL) along with TEA (325 µL, 2.32 mmol), EDCHCl (67 mg, 0.35 mmol),
HOBT (53 mg, 0.35 mmol), and N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide
dihydrochloride (133 mg, 0.44 mmol). The solution was stirred at RT for 20 h, after
which time the DCM was stripped off in vacuo and the resulting residue was taken up in
EtOAc and washed with H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X), then dried
(MgSO4) and concentrated. The residue was then purified via silica flash chromatography
(10 g silica, 1% to 30% methanolic ammonia:EtOAc) to give the desired compound.
Yield: 63 mg, 0.132 mmol, 46%. 1H NMR (500 MHz, Chloroform-d) δ 8.60 – 8.51 (m,
2H), 7.30 – 7.25 (m, 4H), 7.22 – 7.15 (m, 1H), 7.14 – 7.10 (m, 2H), 7.08 (dd, J = 7.6, 1.6
Hz, 1H), 6.67 (t, J = 7.4 Hz, 1H), 6.58 (d, J = 8.4 Hz, 1H), 5.58 (t, J = 5.2 Hz, 1H), 5.53
(t, J = 5.7 Hz, 1H), 4.33 (m, 4H), 3.57 (q, J = 6.7 Hz, 2H), 2.95 (t, J = 12.8 Hz, 2H), 2.85
(t, J = 6.9 Hz, 2H), 2.30 (tt, J = 11.1, 3.8 Hz, 1H), 1.82 (m, 2H), 1.74 (dt, J = 15.9, 7.9
Hz, 2H). TOF ES+ MS: (M + H) 477.2, (M + Na) 499.2. HPLC tR = 5.33 min, > 90%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)azepane-4-
carboxamide (CCG-222661). The ethyl ester 132 (160 mg, 0.37 mmol) was dissolved in

220
EtOH (6 mL) and 10% aq. NaOH (4 mL) was added and the reaction was stirred at RT
for 24 h. At this time, the solvent was removed in vacuo and the white residue was
dissolved in H2O and acidified with conc. HCl to pH<1 at 0 °C. The resulting precipitate
was collected over a filter and dried via aspirator then high vacuum to afford the desired
carboxylic acid as a white powder. No further purification necessary. Yield: 144 mg, 0.35
mmol, 96%.
The carboxylic acid (60 mg, 0.15 mmol), amine 27 (21 µL, 0.18 mmol),
EDCHCl (34 mg, 0.18 mmol), HOBT (27 mg, 0.18 mmol), and TEA (61 µL, 0.44
mmol) were dissolved in DCM (10 mL) and stirred at RT for 29 h, at which time the
DCM was removed in vacuo and the residue was taken up in EtOAc and washed with
H2O (1X), 10% aq. Na2CO3 (3X), and brine (1X). The organic extract was dried (MgSO4)
and concentrated, and the residue purified via silica flash chromatography (10 g silica,
1% to 20% methanolic ammonia:EtOAc) to give a colorless oil. Yield: 40 mg, 0.76
mmol, 52%. HPLC tR = 5.53 min, > 95%. 1H NMR (500 MHz, Chloroform-d) δ 8.51 (bs,
2H), 7.65 (d, J = 7.6 Hz, 1H), 7.39 – 7.32 (m, 1H), 7.28 (s, 1H), 7.16 (d, J = 7.4 Hz, 3H),
7.12 (bs, 2H), 7.02 (d, J = 8.0 Hz, 2H), 6.65 (d, J = 6.3 Hz, 1H), 5.48 (d, J = 9.4 Hz, 2H),
3.77 – 3.64 (m, 2H), 3.60 – 3.36 (m, 4H), 3.31 – 3.15 (m, 1H), 2.86 – 2.77 (m, 2H), 2.08
– 1.97 (m, 2H), 1.91 – 1.69 (m, 3H), 1.56 – 1.48 (m, 1H), 1.43 – 1.34 (m, 1H). TOF ES+
MS: (M + H) 515.2, (M + Na) 537.2. HPLC tR = 5.52 min, > 95%.
1-(2-(4-Chlorobenzyl)benzoyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide
(CCG-222821). 2-(4-chlorobenzyl)benzoic acid (40 mg, 0.16 mmol), N-(2-(pyridin-4-
yl)ethyl)piperidine-4-carboxamide dihydrochloride (55 mg, 0.18 mmol), TEA (225 µL,

221
1.62 mmol), EDCHCl (37 mg, 0.20 mmol), and HOBT (30 mg, 0.20 mmol) were
dissolved in DCM (10 mL) and stirred at RT for 24 h. The DCM was then removed in
vacuo and the residue was taken up in EtOAc, then washed with H2O (3X), 10% aq.
Na2CO3 (3X), and brine (1X), then dried (MgSO4) and concentrated. Purification via
silica flash chromatography (4 g silica, 1% to 20% methanolic ammonia:EtOAc)
provided the desired compound as white microcrystals upon concentration. 1H NMR (500
MHz, Chloroform-d, 3:2 mixture of rotamers) δ 8.51 (bs, 2H), 7.33 (t, J = 7.3 Hz, 1H),
7.27 – 7.18 (m, 4H), 7.17 – 7.05 (m, 5H), 5.61 – 5.42 (m, 1H), 4.73 – 4.59 (m, 1H), 4.18
– 4.03 (m, 1H), 3.93 – 3.80 (m, 1H), 3.53 (q, J = 6.5 Hz, 2H), 3.37 – 3.15 (m, 1H), 2.86 –
2.81 (m, 2H), 2.50 (td, J = 12.7, 3.1 Hz, 1H), 2.23 – 1.99 (m, 2H), 1.90 – 1.75 (m, 2H),
1.73 – 1.49 (m, 2H). TOF ES+ MS: (M + H) 462.2, (M + Na) 484.2. HPLC tR = 4.90
min, > 90%.
1-(1-(4-Chlorobenzyl)-2-oxo-1,2-dihydroquinolin-4-yl)-N-(2-(pyridin-4-
yl)ethyl)piperidine-4-carboxamide (CCG-222822). Ethyl 1-(1-(4-chlorobenzyl)-2-oxo-
1,2-dihydroquinolin-4-yl)piperidine-4-carboxylate (75 mg, 0.18 mmol) was dissolved
into a mixture of EtOH (4 mL), THF (4 mL), and 10% aq. NaOH (2 mL) and stirred at
RT for 18 h. After this time, the reaction was concentrated and the residue dissolved in
H2O, chilled to 0 °C, and acidified to pH<1 with conc. HCl. The resulting precipitate was
collected over a filter and washed with cold 1M HCl, air dried via aspirator, then dried
under high vacuum to give 1-(1-(4-chlorobenzyl)-2-oxo-1,2-dihydroquinolin-4-
yl)piperidine-4-carboxylic acid as a white solid. No further purification necessary. Yield:
67 mg, 0.17 mmol, 95%.

222
1-(1-(4-chlorobenzyl)-2-oxo-1,2-dihydroquinolin-4-yl)piperidine-4-carboxylic
acid (48 mg, 0.12 mmol), 4-(2-aminoethyl)pyridine (17 µL, 0.15 mmol), TEA (51 µL,
0.36 mmol), EDCHCl (28 mg, 0.15 mmol), and HOBT (22 mg, 0.15 mmol) were
dissolved in DCM (5 mL) and stirred at RT for 20 h. DCM was then removed in vacuo
and the residue taken up in EtOAc and washed with H2O (3X), 10% aq. Na2CO3 (3X),
and brine (1X), then dried (MgSO4) and concentrated. The residue was purified by silica
flash chromatography (10 g silica, 1% to 20% methanolic ammonia:EtOAc). Yield: 45
mg, 0.05 mmol, 45%. 1H NMR (500 MHz, Chloroform-d) δ 8.54 (s, 2H), 7.78 (d, J = 8.1
Hz, 1H), 7.40 (t, J = 7.8 Hz, 1H), 7.26 (d, J = 8.0 Hz, 2H), 7.21 – 7.00 (m, 6H), 6.22 (s,
1H), 5.81 (t, J = 5.4 Hz, 1H), 5.46 (s, 2H), 3.58 (dt, J = 16.9, 8.7 Hz, 4H), 2.87 (t, J = 6.9
Hz, 2H), 2.74 (t, J = 11.6 Hz, 2H), 2.28 (tt, J = 11.4, 3.7 Hz, 1H), 2.05 (qd, J = 12.5,
12.0, 3.5 Hz, 2H), 1.94 (d, J = 11.2 Hz, 2H). TOF ES+ MS: (M + H) 501.2, (M + Na)
523.2. HPLC tR = 5.25 min, >95% purity.
1-(1-(4-Chlorophenyl)-2-oxo-1,2-dihydroquinolin-4-yl)-N-(2-(pyridin-4-
yl)ethyl)piperidine-4-carboxamide (CCG-222823). Ethyl 1-(1-(4-chlorophenyl)-2-oxo-
1,2-dihydroquinolin-4-yl)piperidine-4-carboxylate 58 (150 mg, 0.37 mmol) was
dissolved in THF (10 mL) followed by the addition of 10% aq. NaOH (5 mL). This
biphasic reaction was stirred at RT for 20 h. The solvent was then stripped off in vacuo
and the resulting residue was dissolved in H2O, chilled to 0°C, then acidified to pH < 1.
The resulting precipitate was collected over a filter, air dried, then high vacuum dried to
provide 1-(1-(4-chlorophenyl)-2-oxo-1,2-dihydroquinolin-4-yl)piperidine-4-carboxylic

223
acid as a white powder. No further purification necessary. Yield: 130 mg, 0.33 mmol,
91%.
1-(1-(4-Chlorophenyl)-2-oxo-1,2-dihydroquinolin-4-yl)piperidine-4-carboxylic
acid (66 mg, 0.17 mmol), 4-(2-aminoethyl)pyridine (25 µL, 0.21 mmol), TEA (72 µL,
0.52 mmol), ED C (40 mg, 0.21 mmol), and HOBT (32 mg, 0.21 mmol) were dissolved
in DCM (5 mL) and stirred at RT for 19 h. The reaction was then diluted with EtOAc and
washed with H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X), then dried (MgSO4) and
concentrated. The residue was purified via silica flash chromatography (10 g silica, 1 to
17% methanolic ammonia/EtOAc) to give the title compound as an off-white powder.
Yield: 47 mg, 0.10 mmol, 56%. 1H NMR (500 MHz, Chloroform-d) δ 8.55 (d, J = 5.1
Hz, 2H), 7.79 (d, J = 7.9 Hz, 1H), 7.55 (d, J = 8.2 Hz, 2H), 7.32 (t, J = 7.5 Hz, 1H), 7.25
– 7.18 (m, 3H), 7.15 (d, J = 4.9 Hz, 2H), 6.66 (d, J = 8.4 Hz, 1H), 6.20 (s, 1H), 5.78 (t, J
= 6.0 Hz, 1H), 5.30 (s, 1H), 3.58 (q, J = 9.6, 6.8 Hz, 4H), 2.85 (t, J = 6.8 Hz, 2H), 2.77 (t,
J = 11.9 Hz, 2H), 2.34 – 2.21 (m, 2H), 2.07 (q, J = 13.3 Hz, 2H), 1.95 (d, J = 13.5 Hz,
2H). TOF ES+ MS: (M + H) 487.2, (M + Na) 509.2. HPLC tR = 5.09 min, >95% purity.
1-(9H-Carbazole-1-carbonyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide
(CCG-222824). 9H-carbazole-1-carboxylic acid was prepared according to Journal of
Organic Chemistry, Vol. 53, No. 4, 1988, 794-799.
9H-Carbazole-1-carboxylic acid (120 mg, 0.57 mmol), N-(2-(pyridin-4-
yl)ethyl)piperidine-4-carboxamide dihydrochloride (190 mg, 0.63 mmol), TEA (475 µL,
3.41 mmol), EDCHCl (120 mg, 0.63 mmol) and HOBT (96 mg, 0.63 mmol) were
dissolved in DCM (10 mL) and stirred at RT for 14 h. At this time, the reaction was

224
diluted with EtOAc and washed with H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X),
then dried (MgSO4) and concentrated. The residue was purified via silica flash
chromatography (10 g silica, 10% to 30% methanolic ammonia/EtOAc) to give a tan
powder. Yield: 170 mg, 0.40 mmol, 70%. 1H NMR (500 MHz, Chloroform-d) δ 9.13 (s,
1H), 8.53 (d, J = 5.6 Hz, 2H), 8.14 (d, J = 7.7 Hz, 1H), 8.08 (d, J = 7.8 Hz, 1H), 7.50 –
7.42 (m, 2H), 7.41 (d, J = 7.5 Hz, 1H), 7.26 – 7.23 (m, 1H), 7.21 (t, J = 7.6 Hz, 1H), 7.11
(d, J = 5.3 Hz, 2H), 5.59 (t, J = 5.5 Hz, 1H), 4.46 (s, 2H), 3.56 (q, J = 6.6 Hz, 2H), 3.04
(t, J = 12.3 Hz, 2H), 2.84 (t, J = 6.9 Hz, 2H), 2.32 (ddd, J = 15.0, 10.9, 4.1 Hz, 1H), 1.91
– 1.72 (m, 4H). TOF ES+ MS: (M + H) 427.2, (M + Na) 449.2. HPLC tR = 4.80 min,
>95% purity.
1-(6-Chloro-9H-carbazole-1-carbonyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-
carboxamide (CCG-222825). Methyl 6-chloro-9H-carbazole-1-carboxylate (54 mg, 0.21
mmol) was dissolved in EtOH (4 mL) and THF (5 mL), then 10% aq. NaOH (2 mL) was
added. The reaction was allowed to stir at RT for 22 h, after which time the solvent was
stripped off in vacuo. The residue was taken up in H2O and acidified to pH<1 with conc.
HCl, and the resulting white precipitate was collected over a filter, air dried, then high
vacuum dried to give 6-chloro-9H-carbazole-1-carboxylic acid as a white powder with no
further purification. Yield: 51 mg, 0.21 mmol, 100%.
6-Chloro-9H-carbazole-1-carboxylic acid (51 mg, 0.21 mmol) was added to DCM
(5 mL), followed by N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide dihydrochloride
(76 mg, 0.25 mmol), TEA (145 µL, 1.04 mmol), EDCHCl (48 mg, 0.25 mmol) and
HOBT (38 mg, 0.25 mmol). This solution was stirred at RT for 16 h, at which time the

225
solvent was removed in vacuo and the residue taken up in EtOAc and washed with H2O
(3X), 10% aq. Na2CO3 (3X), and brined (1X). The extract was dried (MgSO4) and
concentrated, and the residue purified via silica flash chromatography (10 g silica, 1% to
20% methanolic ammonia: EtOAc). Yield: 48 mg, 0.10 mmol, 50%. 1H NMR (500 MHz,
Chloroform-d) δ 8.53 (d, J = 4.7 Hz, 2H), 8.10 – 8.06 (m, 1H), 8.04 – 7.98 (m, 1H), 7.48
– 7.36 (m, 3H), 7.22 (ddd, J = 7.6, 5.9, 1.0 Hz, 1H), 7.13 (d, J = 5.1 Hz, 2H), 5.61 – 5.52
(m, 1H), 4.45 (bs, 2H), 3.57 (q, J = 6.6 Hz, 2H), 3.06 (s, 2H), 2.85 (t, J = 6.9 Hz, 2H),
2.34 (tt, J = 10.9, 4.6 Hz, 1H), 1.91 – 1.69 (m, 4H). TOF ES+ MS: (M + H) 461.2, (M +
Na) 483.2. HPLC tR = 5.23 min, > 90% purity.
Exo-(1R,5S,6r)-3-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)-N-(2-(pyridin-4-
yl)ethyl)-3-azabicyclo[3.1.0]hexane-6-carboxamide (CCG-222980). The ethyl ester 113
(206 mg, 0.49 mmol) was dissolved in anhydrous THF (10 mL), followed by the addition
of KOTMS (66 mg, 0.51 mmol). The reaction stirred at RT for 48 h, solvent was
removed in vacuo, the residue was dried under high vacuum overnight, leaving the
potassium salt residue that was taken directly into the next step. Yield: 213 mg, 0.49
mmol, 100%.
The potassium salt (211 mg, 0.49 mmol), amine 27 (64 µL, 0.54 mmol), HATU
(204 mg, 0.54 mmol), and DIPEA (170 µL, 0.97 mmol) were dissolved in DCM (10 mL)
and stirred at RT for 24 h, at which time the DCM was removed in vacuo and the residue
was diluted in EtOAc and washed with H2O (2X), 10% aq. Na2CO3 (3X), and brine (1X).
The organic extract was dried (MgSO4) and concentrated, and the residue purified via
silica flash chromatography (25 g silica, 1% to 20% methanolic ammonia:EtOAc) to give

226
an off-white powder. Yield: 134 mg, 0.27 mmol, 55%. 1H NMR (500 MHz, Chloroform-
d) δ 8.44 (d, J = 5.0 Hz, 2H), 7.61 (d, J = 7.9 Hz, 1H), 7.33 – 7.24 (m, 2H), 7.17 – 7.10
(m, 3H), 7.06 (d, J = 5.0 Hz, 2H), 6.88 (d, J = 8.1 Hz, 2H), 6.66 (s, 1H), 6.07 (t, J = 5.9
Hz, 1H), 5.55 – 5.34 (m, 2H), 4.02 (d, J = 12.3 Hz, 1H), 3.72 – 3.58 (m, 2H), 3.53 – 3.32
(m, 4H), 2.75 (t, J = 7.0 Hz, 2H), 1.99 (d, J = 9.4 Hz, 2H). TOF ES+ MS: (M + H) 499.2,
(M + Na) 521.2. HPLC tR = 5.36 min, > 95%.
1-(2-(Phenylamino)benzoyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide
(CCG-222981). Methyl 2-(phenylamino)benzoate (74 mg, 0.33 mmol) was dissolved in
10% aq. NaOH (1 mL), THF (5 mL), and EtOH (2 mL), and stirred at RT for 5 h. At this
time, the solvent was stripped off in vacuo and the residue was dissolved in H2O and
acidified with conc. HCl to pH < 1. Material was collected over a filter and air-dried then
dried under high vacuum to give 2-(phenylamino)benzoic acid as a white powder. No
further purification necessary. Yield: 62 mg, 0.29 mmol, 89%.
2-(Phenylamino)benzoic acid (56 mg, 0.26 mmol), N-(2-(pyridin-4-
yl)ethyl)piperidine-4-carboxamide dihydrochloride (97 mg, 0.32 mmol), EDCHCl (60
mg, 0.32 mmol), HOBT (48 mg, 0.32 mmol), and TEA (220 µL, 1.58 mmol) were
dissolved in DCM (10 mL) and stirred at RT for 17 h. After this time, the DCM was
removed in vacuo. The residue was taken up in EtOAc and washed with H2O (3X), 10%
aq. Na2CO3 (3X), and brine (1X), and dried (MgSO4) and concentrated. The residue was
purified via silica flash chromatography (10 g silica, 1% to 20% methanolic NH3/EtOAc)
to give the desired compound as a colorless oil. Yield: 53 mg, 0.12 mmol, 47%. 1H NMR
(500 MHz, Chloroform-d) δ 8.47 (d, J = 5.0 Hz, 2H), 7.37 (d, J = 8.3 Hz, 1H), 7.28 –

227
7.21 (m, 3H), 7.15 (d, J = 7.6 Hz, 1H), 7.10 – 7.02 (m, 4H), 6.93 (t, J = 7.8 Hz, 1H), 6.86
(t, J = 7.4 Hz, 1H), 5.78 (s, 1H), 4.26 (s, 2H), 3.50 (q, J = 6.5 Hz, 2H), 2.92 (bs, 2H), 2.80
(t, J = 6.9 Hz, 2H), 2.28 (ddd, J = 14.7, 11.0, 3.4 Hz, 1H), 1.77 (bs, 2H), 1.70 – 1.57 (m,
2H). TOF ES+ MS: (M + H) 429.2, (M + Na) 451.2. HPLC tR = 5.05 min, > 95%.
1-(2-(Benzylamino)benzoyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide
(CCG-222982). Methyl 2-(benzylamino)benzoate (50 mg, 0.21 mmol) was dissolved in
EtOH (2 mL) and THF (5 mL), then 10% aq. NaOH (2 mL) was added and the reaction
was stirred at RT for 15 h. At this time, the solvent was removed in vacuo and the residue
was dissolved in H2O and acidified with conc. HCl to pH<1. The resulting precipitate
was collected over a filter and dried via aspirator then high vacuum to afford the desired
2-(benzylamino)benzoic acid as a white powder. No further purification necessary. Yield:
41 mg, 0.18 mmol, 88%.
Ethyl 2-(benzylamino)benzoate (30 mg, 0.13 mmol) was dissolved in DCM (5
mL), followed by N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide dihydrochloride (45
mg, 0.15 mmol), TEA (0.11 mL, 0.79 mmol), EDCHCl (30 mg, 0.16 mmol), and HOBT
(24 mg, 0.16 mmol). The reaction was then stirred at RT for 16 h, at which time the
reaction was diluted with EtOAc and washed with H2O (2X), 10% aq. Na2CO3 (2X), and
brine (1X), then dried (MgSO4) and concentrated in vacuo. The resulting residue was
then purified by silica flash chromatography (10 g silica, 1% to 20% methanolic
ammonia:EtOAc) to provide the desired product as a colorless oil. Yield: 26 mg, 0.06
mmol, 48%. 1H NMR (500 MHz, Chloroform-d) δ 8.48 (d, J = 5.7 Hz, 2H), 7.32 (d, J =
6.7 Hz, 4H), 7.26 – 7.21 (m, 1H), 7.17 (t, J = 7.8 Hz, 1H), 7.10 (d, J = 5.7 Hz, 2H), 7.07

228
– 7.02 (m, 1H), 6.63 (dd, J = 7.8, 3.8 Hz, 2H), 5.92 (t, J = 5.6 Hz, 1H), 5.44 (t, J = 5.4
Hz, 1H), 4.33 (d, J = 5.3 Hz, 2H), 4.26 (bs, 2H), 3.52 (q, J = 6.7 Hz, 2H), 2.90 (t, J = 11.7
Hz, 2H), 2.81 (t, J = 6.9 Hz, 2H), 2.28 (tt, J = 11.2, 3.5 Hz, 1H), 1.78 (d, J = 12.1 Hz,
2H), 1.69 (qd, J = 12.9, 12.4, 4.0 Hz, 2H). TOF ES+ MS: (M + H) 443.2, (M + Na)
465.2. HPLC tR = 4.92 min, > 95%.
1-(2-((4-Chlorophenethyl)amino)benzoyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-
carboxamide (CCG-222983). 2-((4-chlorophenethyl)amino)benzoic acid (40 mg, 0.15
mmol), N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide dihydrochloride (53 mg, 0.17
mmol), EDCHCl (33 mg, 0.17 mmol), HOBT (26 mg, 0.17 mmol), and TEA (0.12 mL,
0.87 mmol) were dissolved in DCM (10 mL) and stirred at RT for 24 h, at which time the
DCM was removed in vacuo and the residue was taken up in EtOAc and washed with
H2O (2X), 10% aq. Na2CO3 (3X), and brine (1X). The organic extract was dried (MgSO4)
and concentrated, and the residue purified via silica flash chromatography (10 g silica,
1% to 20% methanolic ammonia:EtOAc) to give a colorless oil. Yield: 41 mg, 0.083
mmol, 57%. HPLC tR = 5.55 min, > 95%. 1H NMR (500 MHz, Chloroform-d) δ 8.52 (d,
J = 5.2 Hz, 2H), 7.30 – 7.21 (m, 3H), 7.16 (d, J = 8.2 Hz, 2H), 7.12 (d, J = 5.2 Hz, 2H),
7.05 (d, J = 7.5 Hz, 1H), 6.69 (d, J = 8.3 Hz, 1H), 6.65 (d, J = 7.4 Hz, 1H), 5.57 (t, J =
5.6 Hz, 1H), 4.97 (bs, 1H), 4.21 (bs, 2H), 3.56 (q, J = 6.5 Hz, 2H), 3.34 (t, J = 7.2 Hz,
2H), 2.89 (t, J = 7.2 Hz, 2H), 2.83 (q, J = 12.6, 9.7 Hz, 4H), 2.24 (tt, J = 11.2, 3.7 Hz,
1H), 1.77 – 1.62 (m, 7H). TOF ES+ MS: (M + H) 491.2, (M + Na) 513.2. HPLC tR =
5.44 min, >95% purity.

229
(1R,3s,5S)-8-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(2-(pyridin-4-
yl)ethyl)-8-azabicyclo[3.2.1]octane-3-carboxamide (CCG-224000). The Boc-protected
right-hand half 115 (100 mg, 0.35 mmol) was dissolved into 4M HCl (5 mL) and stirred
for 15 min at RT. After this time, Et2O (15 mL) was added to precipitate the material and
the reaction was sonicated for 15 min. The resulting residue did not solidify, so the liquid
phase was decanted off and the residue was dried under aspirator vacuum, then high
vacuum, for 48 h. The resulting solid aminde dihydrochloride 116 was then taken directly
into the subsequent reaction. Yield: 115 mg, 0.35 mmol, 99%.
The indole carboxylic acid 4a (52 mg, 0.18 mmol), amine dihydrochloride 116
(60 mg, 0.18 mmol), EDCHCl (45 mg, 0.24 mmol), HOBT (36 mg, 0.24 mmol), and
TEA (252 µL, 0.24 mmol) were dissolved in DCM (3 mL) and stirred at RT for 20 h, at
which time the DCM was removed in vacuo and the residue was diluted in EtOAc and
washed with H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X). The organic extract was
dried (MgSO4) and concentrated, and the residue purified via silica flash chromatography
(10 g silica, 1% to 20% methanolic ammonia:EtOAc) to give an off-white powder. Yield:
59 mg, 0.11 mmol, 61%. 1H NMR (500 MHz, Chloroform-d) δ 8.44 (d, J = 4.8 Hz, 2H),
7.63 (d, J = 7.9 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 7.26 (t, J = 7.7 Hz, 1H), 7.19 – 7.12
(m, 3H), 7.07 (d, J = 5.0 Hz, 2H), 6.98 (d, J = 8.1 Hz, 2H), 6.74 (s, 1H), 6.11 (t, J = 5.7
Hz, 1H), 5.51 (d, J = 9.8 Hz, 2H), 4.82 (bs, 1H), 4.39 (bs, 1H), 3.48 (q, J = 6.6 Hz, 2H),
2.78 (t, J = 7.0 Hz, 2H), 2.60 (tt, J = 11.5, 5.2 Hz, 1H), 2.30 (bs, 2H), 1.76 (dd, J = 143.8,
60.1 Hz, 7H). TOF ES+ MS: (M + H) 527.2, (M + Na) 549.2. HPLC tR = 5.55 min,
>95%.

230
2-(1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)piperidin-4-yl)-N-(pyridin-4-
ylmethyl)acetamide (CCG-224001). The ethyl ester 96 (350 mg, 0.80 mmol) was
dissolved in EtOH (10 mL), THF (10 mL), and 10% aq. KOH (3.22 mL) was added and
the reaction was stirred at RT for 13 h. At this time, the solvent was removed in vacuo
and the thick white residue was diluted in H2O, sonicated, and acidified with conc. HCl to
pH<1 at 0 °C. The resulting precipitate was collected over a filter and dried via aspirator
then high vacuum to afford the desired carboxylic acid as a white powder. No further
purification necessary. Yield: 311 mg, 0.76 mmol, 95%.
The carboxylic acid (62 mg, 0.15 mmol), amine 27 (18 µL, 0.18 mmol),
EDCHCl (35 mg, 0.18 mmol), HOBT (28 mg, 0.18 mmol), and TEA (84 µL, 0.60
mmol) were dissolved in DCM (5 mL) and stirred at RT for 18 h, at which time the DCM
was removed in vacuo and the residue was taken up in EtOAc and washed with H2O
(3X), 10% aq. Na2CO3 (3X), and brine (1X). The organic extract was dried (MgSO4) and
concentrated, and the residue purified via silica flash chromatography (10 g silica, 1% to
20% methanolic ammonia:EtOAc) to give a colorless oil. Yield: 42 mg, 0.08 mmol, 54%.
1H NMR (500 MHz, Chloroform-d) δ 8.51 (d, J = 5.6 Hz, 2H), 7.64 (d, J = 7.9 Hz, 1H),
7.39 (d, J = 8.3 Hz, 1H), 7.29 (t, J = 7.7 Hz, 1H), 7.20 – 7.14 (m, 3H), 7.13 (d, J = 5.3
Hz, 2H), 7.02 (d, J = 8.2 Hz, 2H), 6.59 (s, 1H), 6.34 (t, J = 5.7 Hz, 1H), 5.45 (s, 2H), 4.59
(bs, 1H), 4.39 (s, 2H), 4.06 (bs, 1H), 2.88 (bs, 1H), 2.67 (bs, 1H), 2.13 – 2.02 (m, 3H),
1.71 (bs, 1H), 1.45 (bs, 1H), 0.97 (bs, 1H), 0.32 (bs, 1H). TOF ES+ MS: (M + H) 501.2,
(M + Na) 523.2. HPLC tR = 5.49 min, > 95%.

231
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)azetidine-
3-carboxamide (CCG-224002). The methyl ester 125 (100 mg, 0.26 mmol) was
dissolved in EtOH (5 mL) and 10% aq. NaOH (3 mL) was added and the reaction was
stirred at RT for 24 h. At this time, the solvent was removed in vacuo and the white
residue was dissolved in H2O and acidified with conc. HCl to pH<1 at 0 °C. The resulting
precipitate was collected over a filter and dried via aspirator then high vacuum to afford
the desired carboxylic acid as a white powder. No further purification necessary. Yield:
95 mg, 0.26 mmol, 99%.
The carboxylic acid (50 mg, 0.14 mmol), amine 27 (100 µL, 0.16 mmol),
EDCHCl (31 mg, 0.16 mmol), HOBT (25 mg, 0.16 mmol), and TEA (76 µL, 0.54
mmol) were dissolved in DCM (5 mL) and stirred at RT for 15 h, at which time the DCM
was removed in vacuo and the residue was taken up in EtOAc and washed with H2O
(3X), 10% aq. Na2CO3 (3X), and brine (1X). The organic extract was dried (MgSO4) and
concentrated, and the residue purified via silica flash chromatography (10 g silica, 1% to
20% methanolic ammonia:EtOAc) to give a colorless oil. Yield: 42 mg, 0.09 mmol, 65%.
1H NMR (500 MHz, Chloroform-d) δ 8.53 (d, J = 5.2 Hz, 2H), 7.67 (d, J = 8.0 Hz, 1H),
7.34 – 7.28 (m, 2H), 7.22 (d, J = 8.4 Hz, 2H), 7.19 – 7.15 (m, 1H), 7.13 (s, 2H), 7.03 (d, J
= 8.4 Hz, 2H), 6.83 (s, 1H), 5.73 (s, 2H), 5.67 (s, 1H), 4.62 (s, 1H), 4.31 (d, J = 71.6 Hz,
3H), 3.61 (q, J = 6.6 Hz, 2H), 3.22 (p, J = 7.4 Hz, 2H), 2.87 (t, J = 7.0 Hz, 2H). TOF ES+
MS: (M + H) 473.2, (M + Na) 495.2. HPLC tR = 5.60 min, >95% purity.
2-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)-2-
azaspiro[3.3]heptane-6-carboxamide (CCG-224220). In a dry round bottom flask under

232
argon at RT, naphthalene (220 mg, 1.74 mmol) was dissolved in anhydrous THF (10 mL)
containing hexane-washed sodium (100 mg, 4.35 mmol). The mixture was sonicated until
the reaction became a dark green homogenous solution and no solid material was visible.
This entire solution was then added dropwise to tosyl amide 123 (41 mg, 0.10 mmol),
dissolved in anhydrous THF (1 mL), which caused the reaction to turn turbid brown. This
was allowed to stir for 5 min. After this time, the reaction was diluted with EtOAc and a
few drops of MeOH and washed with H2O (1X), 10% Na2CO3 (3X), and brine (1X), then
dried (MgSO4) and concentrated. The residue of amine 124 was used directly in the
subsequent reaction.
The indole carboxylic acid 4a (29 mg, 0.10 mmol), spiro amine 124 (approx. 30
mg, 0.10 mmol), HATU (49 mg, 0.12 mmol), and TEA (42 µL, 0.30 mmol) were
dissolved in DMF (2 mL) and stirred at RT for 18 h, at which time the reaction was
diluted with EtOAc and washed with H2O (3X), 10% aq. Na2CO3 (3X), and brine (1X).
The organic extract was dried (MgSO4) and concentrated, and the residue purified via
silica flash chromatography (4 g silica, 1% to 20% methanolic ammonia:EtOAc) to give a
colorless oil. Yield: 18 mg, 0.04 mmol, 35%. 1H NMR (500 MHz, Chloroform-d) δ 8.50
(d, J = 4.5 Hz, 2H), 7.65 (d, J = 7.7 Hz, 1H), 7.28 (d, J = 16.1 Hz, 3H), 7.19 – 7.13 (m,
2H), 7.11 (d, J = 5.2 Hz, 2H), 6.99 (d, J = 7.6 Hz, 2H), 6.80 (s, 1H), 5.71 (s, 2H), 5.58
(bs, 1H), 4.28 (bd, J = 32.5 Hz, 2H), 4.06 (s, 2H), 3.52 (q, J = 6.7 Hz, 2H), 2.82 (t, J =
7.0 Hz, 2H), 2.74 (bs, 1H), 2.49 – 2.29 (m, 4H). TOF ES+ MS: (M + H) 513.2, (M + Na)
535.2. HPLC tR = 5.65 min, > 95%.

233
Bibliography
1. Griffin, D.E. 2001. Alphaviruses. In Fields Virology. D.M. Knipe, Howley, P. M.,
Griffin, D. E., Lamb, R. A., Martin, M. A., Roizman, B., and Straus, S. S., editor.
Philadelphia: Lippincott Williams & Wilkins. 917-962.
2. Morse, S.S. 1997. The public health threat of emerging viral disease. The Journal
of nutrition 127:951S-957S.
3. Satcher, D. 1995. Emerging infections: getting ahead of the curve. Emerging
Infectious Diseases 1:1-6.
4. Enserink, M. 2007. Infectious diseases. Chikungunya: no longer a third world
disease. Science 318:1860-1861.
5. Nash, D., Mostashari, F., Fine, A., Miller, J., O'Leary, D., Murray, K., Huang, A.,
Rosenberg, A., Greenberg, A., Sherman, M., et al. 2001. The outbreak of West
Nile virus infection in the New York City area in 1999. New England Journal of
Medicine 344:1807-1814.
6. Carrera, J.P., Forrester, N., Wang, E.Y., Vittor, A.Y., Haddow, A.D., Lopez-
Verges, S., Abadia, I., Castano, E., Sosa, N., Baez, C., et al. 2013. Eastern Equine
Encephalitis in Latin America. New England Journal of Medicine 369:732-744.
7. 2006. Eastern equine encephalitis--New Hampshire and Massachusetts. MMWR
Morb Mortal Wkly Rep:697-700.
8. 2003. Category A, B, & C Priority Pathogens. NIH/NIAID.
9. Bronze, M.S., Huycke, M.M., Machado, L.J., Voskuhl, G.W., and Greenfield,
R.A. 2002. Viral agents as biological weapons and agents of bioterrorism. The
American journal of the medical sciences 323:316-325.
10. Aguilar, M.J. 1970. Pathological changes in brain and other target organs of infant
and weanling mice after infection with non-neuroadapted Western equine
encephalitis virus. Infection and immunity 2:533-542.
11. Zlotnik, I., Batterha.D, Grant, D.P., and Peacock, S. 1972. Pathogenesis of
Western Equine Encephalitis-Virus (Wee) in Adult Hamsters with Special
Reference to Long and Short-Term Effects on Cns of Tenuated Clone 15 Variant.
British Journal of Experimental Pathology 53:59-&.
12. Liu, C., Voth, D.W., Rodina, P., Shauf, L.R., and Gonzalez, G. 1970. A
Comparative Study of the Pathogenesis of Western Equine and Eastern Equine
Encephalomyelitis Viral Infections in Mice by Intra Cerebral and Sub Cutaneous
Inoculations. Journal of Infectious Diseases 122:53-63.
13. Flint, S.J.E., L.W.; Racaniello, V.R.; Salka, A.M. 2009. Principles of Virology:
American Society for Microbiology Press.
14. Domingo, E. 1997. Rapid evolution of viral RNA genomes. The Journal of
nutrition 127:958S-961S.

234
15. Griffin, D.E. 2008. Neurotropic Alphaviruses. In Neurotropic Viral Infections.
C.S. Reiss, editor. United Kingdom: Cambridge University Press. 94-119.
16. Steele, K.E.R., D.S.; Glass, P.J.; Hart, M.K.; Ludwig, G.V.; Pratt, W.D.; Parker,
M.D.; Smith, J.F. 2007. Alphavirus Encephalitides. In Medical Aspects of
Biological Warfare. Z.F. Dembek, editor. United States of America: Office of the
Surgeon General. 241-270.
17. Mendenhall, I.H., Tello, S.A., Neira, L.A., Castillo, L.F., Ocampo, C.B., and
Wesson, D.M. 2012. Host preference of the arbovirus vector Culex erraticus
(Diptera: Culicidae) at Sonso Lake, Cauca Valley Department, Colombia. Journal
of medical entomology 49:1092-1102.
18. 2005. CDC Fact Sheet: Western Equine Encephalitis. Centers for Disease Control
and Prevention.
19. Zacks, M.A., and Paessler, S. 2010. Encephalitic alphaviruses. Veterinary
Microbiology 140:281-286.
20. Longshore, W.A., Jr., Stevens, I.M., Hollister, A.C., Jr., Gittelsohn, A., and
Lennette, E.H. 1956. Epidemiologic observations on acute infectious encephalitis
in California, with special reference to the 1952 outbreak. American journal of
hygiene 63:69-86.
21. Palmer, R.J., and Finley, K.H. 1956. Sequelae of encephalitis-report of a study
after the California epidemic. Califor Nia Med 84:98-100.
22. Finley, K.H., Longshore, W.A., Palmer, R.J., Cook, R.E., and Riggs, N. 1955.
Western equine and St. Louis encephalitis; preliminary report of a clinical follow-
up study in California. Neurology 5:223-236.
23. Kokernot, R.H., Shinefield, H.R., and Longshore, W.A. 1953. The 1952 outbreak
of encephalitis in California. Differential diagnosis. California Med 79:73-77.
24. Calisher, C.H. 1994. Medically important arboviruses of the United States and
Canada. Clinical microbiology reviews 7:89-116.
25. Medovy, H. 1943. Western equine encephalomyelitis in infants. Journal of
Pediatrics 22:308-318.
26. 2010. Eastern Equine Encephailitis. Centers for Disease Control and Prevention.
27. Feemster, R.F. 1957. Equine Encephalitis in Massachusetts. New England Journal
of Medicine 257:701-704.
28. Deresiewicz, R.L., Thaler, S.J., Hsu, L.G., and Zamani, A.A. 1997. Clinical and
neuroradiographic manifestations of Eastern equine encephalitis. New England
Journal of Medicine 336:1867-1874.
29. Rivas, F., Diaz, L.A., Cardenas, V.M., Daza, E., Bruzon, L., Alcala, A., De La
Hoz, O., Caceres, F.M., Aristizabal, G., Martinez, J.W., et al. 1997. Epidemic
Venezuelan equine encephalitis in La Guajira, Colombia, 1995. Journal of
Infectious Diseases 175:828-832.
30. Ehrenkranz, N.J., and Ventura, A.K. 1974. Venezuelan Equine Encephalitis Virus
Infection in Man.9-14.
31. Ventura, A.K., Buff, E.E., and Ehrenkranz, N.J. 1974. Human Venezuelan Equine
Encephalitis Virus Infection in Florida. American Journal of Tropical Medicine
and Hygiene 23:507-512.

235
32. Kortepeter, M.G., and Parker, G.W. 1999. Potential biological weapons threats.
Emerging Infectious Diseases 5:523-527.
33. Sidwell, R.W., and Smee, D.F. 2003. Viruses of the Bunya- and Togaviridae
families: potential as bioterrorism agents and means of control. Antiviral
Research 57:101-111.
34. Randall, R., Mills, J.W., and Engel, L.L. 1947. The preparation and properties of
a purified equine encephalomyelitis vaccine. Journal of immunology 55:41-52.
35. Edelman, R., Ascher, M.S., Oster, C.N., Ramsburg, H.H., Cole, F.E., and Eddy,
G.A. 1979. Evaluation in humans of a new, inactivated vaccine for Venezuelan
equine encephalitis virus (C-84). The Journal of infectious diseases 140:708-715.
36. Strizki, J.M., and Repik, P.M. 1995. Differential reactivity of immune sera from
human vaccinees with field strains of eastern equine encephalitis virus. The
American journal of tropical medicine and hygiene 53:564-570.
37. Calisher, C.H., Sasso, D.R., and Sather, G.E. 1973. Possible evidence for
interference with Venezuelan equine encephalitis virus vaccination of equines by
pre-existing antibody to Eastern or Western Equine encephalitis virus, or both.
Applied microbiology 26:485-488.
38. McClain, D.J., Pittman, P.R., Ramsburg, H.H., Nelson, G.O., Rossi, C.A.,
Mangiafico, J.A., Schmaljohn, A.L., and Malinoski, F.J. 1998. Immunologic
interference from sequential administration of live attenuated alphavirus vaccines.
The Journal of infectious diseases 177:634-641.
39. Digoutte, J.P., and Girault, G. 1976. [The protective properties in mice of tonate
virus and two strains of cabassou virus against neurovirulent everglades
Venezuelan encephalitis virus (author's transl)]. Annales de microbiologie
127B:429-437.
40. Griffin, D.E. 1991. Therapy of viral infections of the central nervous system.
Antiviral Research 15:1-10.
41. Leung, J.Y.-S., Ng, M.M.-L., and Chu, J.J.H. 2011. Replication of Alphaviruses:
A Review on the Entry Process of Alphaviruses into Cells. Advances in Virology
2011:9.
42. Maramorosch, K., Murphy, F.A., and Shatkin, A.J. 1995. Advances in Virus
Research: Elsevier Science.
43. Griffin, D.E. 1998. A Review of Alphavirus Replication in Neurons.
Neuroscience & Biobehavioral Reviews 22:721-723.
44. Wang, Y.F., Sawicki, S.G., and Sawicki, D.L. 1994. Alphavirus nsP3 functions to
form replication complexes transcribing negative-strand RNA. Journal of
Virology 68:6466-6475.
45. Lundstrom, K. 2005. Biology and application of alphaviruses in gene therapy.
Gene Ther 12:S92-S97.
46. Vogel, P., Kell, W.M., Fritz, D.L., Parker, M.D., and Schoepp, R.J. 2005. Early
Events in the Pathogenesis of Eastern Equine Encephalitis Virus in Mice. The
American journal of pathology 166:159-171.
47. Jahrling, P.B., DePaoli, A., and Powanda, M.C. 1978. Pathogenesis of a
Venezuelan encephalitis virus strain lethal for adult white rats. Journal of Medical
Virology 2:109-116.

236
48. Griffin, D. 1986. Alphavirus Pathogenesis and Immunity. In The Togaviridae and
Flaviviridae. S. Schlesinger, and M. Schlesinger, editors: Springer New York.
209-249.
49. Charles, P.C., Walters, E., Margolis, F., and Johnston, R.E. 1995. Mechanism of
Neuroinvasion of Venezuelan Equine Encephalitis Virus in the Mouse. Virology
208:662-671.
50. Cook, S.H., and Griffin, D.E. 2003. Luciferase Imaging of a Neurotropic Viral
Infection in Intact Animals. Journal of Virology 77:5333-5338.
51. Dropulić, B., and Masters, C.L. 1990. Entry of Neurotropic Arboviruses into the
Central Nervous System: An In Vitro Study Using Mouse Brain Endothelium.
Journal of Infectious Diseases 161:685-691.
52. Gorelkin, L. 1973. Venezuelan equine encephalomyelitis in an adult animal host.
An electron microscopic study. The American journal of pathology 73:425-442.
53. Gleiser, C.A., Gochenour, W.S., Jr., Berge, T.O., and Tigertt, W.D. 1962. The
comparative pathology of experimental Venezuelan equine encephalomyelitis
infection in different animal hosts. The Journal of infectious diseases 110:80-97.
54. Davis, N.L., Grieder, F.B., Smith, J.F., Greenwald, G.F., Valenski, M.L., Sellon,
D.C., Charles, P.C., and Johnston, R.E. 1994. A molecular genetic approach to the
study of Venezuelan equine encephalitis virus pathogenesis. Archives of virology.
Supplementum 9:99-109.
55. Griffin, D.E., Levine, B., Ubol, S., and Hardwick, J.M. 1994. The effects of
alphavirus infection on neurons. Annals of Neurology 35:S23-S27.
56. Lewis, J., Wesselingh, S.L., Griffin, D.E., and Hardwick, J.M. 1996. Alphavirus-
induced apoptosis in mouse brains correlates with neurovirulence. Journal of
Virology 70:1828-1835.
57. Griffin, D.E., and Hardwick, J.M. 1997. Regulators of apoptosis on the road to
persistent alphavirus infection. Annual review of microbiology 51:565-592.
58. Schwartz, O., and Albert, M.L. 2010. Biology and pathogenesis of chikungunya
virus. Nat Rev Micro 8:491-500.
59. Jan, J.-T., and Griffin, D.E. 1999. Induction of Apoptosis by Sindbis Virus Occurs
at Cell Entry and Does Not Require Virus Replication. Journal of Virology
73:10296-10302.
60. Peng, W., Peltier, D.C., Larsen, M.J., Kirchhoff, P.D., Larsen, S.D., Neubig, R.R.,
and Miller, D.J. 2009. Identification of thieno[3,2-b]pyrrole derivatives as novel
small molecule inhibitors of neurotropic alphaviruses. The Journal of infectious
diseases 199:950-957.
61. Schoepp, R.J., Smith, J.F., and Parker, M.D. 2002. Recombinant Chimeric
Western and Eastern Equine Encephalitis Viruses as Potential Vaccine
Candidates. Virology 302:299-309.
62. Castorena, K.M., Peltier, D.C., Peng, W., and Miller, D.J. 2008. Maturation-
dependent responses of human neuronal cells to western equine encephalitis virus
infection and type I interferons. Virology 372:208-220.
63. Petrakova, O., Volkova, E., Gorchakov, R., Paessler, S., Kinney, R.M., and
Frolov, I. 2005. Noncytopathic Replication of Venezuelan Equine Encephalitis

237
Virus and Eastern Equine Encephalitis Virus Replicons in Mammalian Cells.
Journal of Virology 79:7597-7608.
64. Sindac, J.A., Yestrepsky, B.D., Barraza, S.J., Bolduc, K.L., Blakely, P.K., Keep,
R.F., Irani, D.N., Miller, D.J., and Larsen, S.D. 2012. Novel Inhibitors of
Neurotropic Alphavirus Replication That Improve Host Survival in a Mouse
Model of Acute Viral Encephalitis. Journal of Medicinal Chemistry 55:3535-
3545.
65. Venable, J.D., Cai, H., Chai, W., Dvorak, C.A., Grice, C.A., Jablonowski, J.A.,
Shah, C.R., Kwok, A.K., Ly, K.S., Pio, B., et al. 2005. Preparation and Biological
Evaluation of Indole, Benzimidazole, and Thienopyrrole Piperazine
Carboxamides: Potent Human Histamine H4 Antagonists. Journal of Medicinal
Chemistry 48:8289-8298.
66. Blair, J.B., Marona-Lewicka, D., Kanthasamy, A., Lucaites, V.L., Nelson, D.L.,
and Nichols, D.E. 1999. Thieno[3,2-b]- and Thieno[2,3-b]pyrrole Bioisosteric
Analogues of the Hallucinogen and Serotonin Agonist N,N-Dimethyltryptamine.
Journal of Medicinal Chemistry 42:1106-1111.
67. Bonafoux, D., Abibi, A., Bettencourt, B., Burchat, A., Ericsson, A., Harris, C.M.,
Kebede, T., Morytko, M., McPherson, M., Wallace, G., et al. 2011. Thienopyrrole
acetic acids as antagonists of the CRTH2 receptor. Bioorganic & Medicinal
Chemistry Letters 21:1861-1864.
68. Kalgutkar, A.S., and Soglia, J.R. 2005. Minimising the potential for metabolic
activation in drug discovery. Expert Opinion on Drug Metabolism & Toxicology
1:91-142.
69. Kobayashi, T., Sugihara, J., and Harigaya, S. 1987. Mechanism of metabolic
cleavage of a furan ring. Drug Metabolism and Disposition 15:877-881.
70. Steele, K.E., and Twenhafel, N.A. 2010. Pathology of Animal Models of
Alphavirus Encephalitis. Veterinary Pathology 47:790-805.
71. Lustig, S., Halevy, M., Ben-Nathan, D., Rice, C.M., and Kobiler, D. 1999. The
role of host immunocompetence in neuroinvasion of Sindbis virus. Archives of
Virology 144:1159-1171.
72. Goldfield, M.S., O. 1968. The 1959 Outbreak of Eastern Encephailits In New
jersey: I. Introduction and Description of Outbreak. American Journal of
Epidemiology 87:1-10.
73. Calisher, C.H., Elkafrawi, A.O., Mahmud, M.I.A., Darosa, A.P.A.T., Bartz, C.R.,
Brummerkorvenkontio, M., Haksohusodo, S., and Suharyono, W. 1986.
Complex-Specific Immunoglobulin-M Antibody Patterns in Humans Infected
with Alphaviruses. Journal of Clinical Microbiology 23:155-159.
74. Pajouhesh, H., and Lenz, G. 2005. Medicinal chemical properties of successful
central nervous system drugs. NeuroRX 2:541-553.
75. Pardridge, W. 2005. The blood-brain barrier: Bottleneck in brain drug
development. NeuroRX 2:3-14.
76. Pardridge, W.M. 1998. CNS Drug Design Based on Principles of Blood-Brain
Barrier Transport. Journal of Neurochemistry 70:1781-1792.
77. Pardridge, W.M. 2012. Drug transport across the blood-brain barrier. Journal of
Cerebral Blood Flow and Metabolism 32:1959-1972.

238
78. Clark, D.E. 2003. In silico prediction of blood–brain barrier permeation. Drug
Discovery Today 8:927-933.
79. Kerns, E.H.D., L. 2008. Blood-Brain Barrier. In Drug-like Properties: Concepts,
Structure Design and Methods. United States of America: Elsevier. 122-136.
80. Doan, K.M.M., Humphreys, J.E., Webster, L.O., Wring, S.A., Shampine, L.J.,
Serabjit-Singh, C.J., Adkison, K.K., and Polli, J.W. 2002. Passive Permeability
and P-Glycoprotein-Mediated Efflux Differentiate Central Nervous System
(CNS) and Non-CNS Marketed Drugs. Journal of Pharmacology and
Experimental Therapeutics 303:1029-1037.
81. Kerns, E.H., and Di, Li. 2008. Drug-like Properties: Concepts, Structure Design
and Methods. USA: Elsevier.
82. Begley, D.J. 2004. ABC transporters and the blood-brain barrier. Current
pharmaceutical design 10:1295-1312.
83. Sindac, J.A., Barraza, S.J., Dobry, C.J., Xiang, J., Blakely, P.K., Irani, D.N.,
Keep, R.F., Miller, D.J., and Larsen, S.D. 2013. Optimization of Novel Indole-2-
carboxamide Inhibitors of Neurotropic Alphavirus Replication. Journal of
Medicinal Chemistry 56:9222-9241.
84. Larsen, S.D., Wilson, M.W., Abe, A., Shu, L., George, C.H., Kirchhoff, P.,
Showalter, H.D.H., Xiang, J., Keep, R.F., and Shayman, J.A. 2012. Property-
based design of a glucosylceramide synthase inhibitor that reduces
glucosylceramide in the brain. Journal of Lipid Research 53:282-291.
85. Ertl, P., Rohde, B., and Selzer, P. 2000. Fast Calculation of Molecular Polar
Surface Area as a Sum of Fragment-Based Contributions and Its Application to
the Prediction of Drug Transport Properties. Journal of Medicinal Chemistry
43:3714-3717.
86. Hou, T.J., and Xu, X.J. 2003. ADME Evaluation in Drug Discovery. 3. Modeling
Blood-Brain Barrier Partitioning Using Simple Molecular Descriptors. Journal of
Chemical Information and Computer Sciences 43:2137-2152.
87. Palm, K., Stenberg, P., Luthman, K., and Artursson, P. 1997. Polar Molecular
Surface Properties Predict the Intestinal Absorption of Drugs in Humans.
Pharmaceutical Research 14:568-571.
88. Kelder, J., Grootenhuis, P.J., Bayada, D., Delbressine, L.C., and Ploemen, J.-P.
1999. Polar Molecular Surface as a Dominating Determinant for Oral Absorption
and Brain Penetration of Drugs. Pharmaceutical Research 16:1514-1519.
89. Hitchcock, S.A. 2012. Structural Modifications that Alter the P-Glycoprotein
Efflux Properties of Compounds. Journal of Medicinal Chemistry 55:4877-4895.
90. Fernandes, J., and Gattass, C.R. 2009. Topological Polar Surface Area Defines
Substrate Transport by Multidrug Resistance Associated Protein 1
(MRP1/ABCC1). Journal of Medicinal Chemistry 52:1214-1218.
91. Di, L., Kerns, E.H., Fan, K., McConnell, O.J., and Carter, G.T. 2003. High
throughput artificial membrane permeability assay for blood-brain barrier.
European Journal of Medicinal Chemistry 38:223-232.
92. Avdeef, A., Bendels, S., Di, L., Faller, B., Kansy, M., Sugano, K., and Yamauchi,
Y. 2007. PAMPA--critical factors for better predictions of absorption. Journal of
Pharmaceutical Sciences 96:2893-2909.

239
93. Ruell, J.A., Tsinman, O., and Avdeef, A. 2004. Acid-base cosolvent method for
determining aqueous permeability of amiodarone, itraconazole, tamoxifen,
terfenadine and other very insoluble molecules. Chemical & pharmaceutical
bulletin 52:561-565.
94. Bermejo, M., Avdeef, A., Ruiz, A., Nalda, R., Ruell, J.A., Tsinman, O., González,
I., Fernández, C., Sánchez, G., Garrigues, T.M., et al. 2004. PAMPA—a drug
absorption in vitro model: 7. Comparing rat in situ, Caco-2, and PAMPA
permeability of fluoroquinolones. European Journal of Pharmaceutical Sciences
21:429-441.
95. Avdeef, A., Artursson, P., Neuhoff, S., Lazorova, L., Gråsjö, J., and Tavelin, S.
2005. Caco-2 permeability of weakly basic drugs predicted with the Double-Sink
PAMPA method. European Journal of Pharmaceutical Sciences 24:333-349.
96. Mensch, J., Melis, A., Mackie, C., Verreck, G., Brewster, M.E., and Augustijns,
P. 2010. Evaluation of various PAMPA models to identify the most
discriminating method for the prediction of BBB permeability. European Journal
of Pharmaceutics and Biopharmaceutics 74:495-502.
97. Di, L., Kerns, E.H., Bezar, I.F., Petusky, S.L., and Huang, Y. 2009. Comparison
of blood–brain barrier permeability assays: in situ brain perfusion, MDR1-
MDCKII and PAMPA-BBB. Journal of Pharmaceutical Sciences 98:1980-1991.
98. Pardridge, W.M. 2003. Blood-brain barrier drug targeting: the future of brain drug
development. Molecular interventions 3:90-105, 151.
99. Fischer, H., Gottschlich, R., and Seelig, A. 1998. Blood-Brain Barrier
Permeation: Molecular Parameters Governing Passive Diffusion. The Journal of
Membrane Biology 165:201-211.
100. Waring, M.J. 2009. Defining optimum lipophilicity and molecular weight ranges
for drug candidates—Molecular weight dependent lower log D limits based
on permeability. Bioorganic & Medicinal Chemistry Letters 19:2844-2851.
101. Scala, S., Akhmed, N., Rao, U.S., Paull, K., Lan, L.-B., Dickstein, B., Lee, J.-S.,
Elgemeie, G.H., Stein, W.D., and Bates, S.E. 1997. P-Glycoprotein Substrates and
Antagonists Cluster into Two Distinct Groups. Molecular Pharmacology
51:1024-1033.
102. Forster, S., Thumser, A.E., Hood, S.R., and Plant, N. 2012. Characterization of
Rhodamine-123 as a Tracer Dye for Use In In vitro Drug Transport Assays. Plos
One 7.
103. Bevan, C.D., and Lloyd, R.S. 2000. A high-throughput screening method for the
determination of aqueous drug solubility using laser nephelometry in microtiter
plates. Analytical chemistry 72:1781-1787.
104. Delekta, P.C.D., C.J.; Sindac, J.A.; Barraza, S.J.; Blakely, P.K.; Xiang, J.;
Kirchhoff, P.D.; Keep, R.F.; Irani, D.N.; Larsen, S.D.; and Miller, D.J. . 2014.
Novel indole-2-carboxamide compounds are potent broad spectrum antivirals
active against western equine encephalitis virus in vivo. Journal of Virology:In
press.
105. Lin, K., and Gallay, P. 2013. Curing a viral infection by targeting the host: The
example of cyclophilin inhibitors. Antiviral Research 99:68-77.

240
106. Bochkov, Y.A., and Palmenberg, A.C. 2006. Translational efficiency of EMCV
IRES in bicistronic vectors is dependent upon IRES sequence and gene location.
Biotechniques 41:283-+.
107. Traverse, J.F., Zhao, Y., Hoveyda, A.H., and Snapper, M.L. 2005. Proline-Based
N-Oxides as Readily Available and Modular Chiral Catalysts. Enantioselective
Reactions of Allyltrichlorosilane with Aldehydes. Organic Letters 7:3151-3154.
108. Han, Z.-j., Wang, R., Zhou, Y.-f., and Liu, L. 2005. Simple Derivatives of Natural
Amino Acids as Chiral Ligands in the Catalytic Asymmetric Addition of
Phenylacetylene to Aldehydes. European Journal of Organic Chemistry
2005:934-938.
109. Ladziata, U., Carlson, J., and Zhdankin, V.V. 2006. Synthesis and oxidative
reactivity of new chiral hypervalent iodine(V) reagents based on (S)-proline.
Tetrahedron Letters 47:6301-6304.
110. Theodorou, V., Skobridis, K., Tzakos, A.G., and Ragoussis, V. 2007. A simple
method for the alkaline hydrolysis of esters. Tetrahedron Letters 48:8230-8233.
111. Stasi, L.P., Bhimani, K., Borriello, M., Canciani, L., Caselli, G., Colace, F.,
Ferioli, C., Kaswala, M., Mennuni, L., Piepoli, T., et al. 2011. Synthesis,
pharmacophore modeling and in vitro activity of 10,11-
dihydrodibenzo[b,f]oxepine-4-carboxamide derivatives as novel and potent
antagonists of the prostaglandin EP4 receptor. Bioorganic & Medicinal Chemistry
Letters 21:6336-6340.
112. Adeniji, A.O., Twenter, B.M., Byrns, M.C., Jin, Y., Chen, M., Winkler, J.D., and
Penning, T.M. 2012. Development of Potent and Selective Inhibitors of Aldo–
Keto Reductase 1C3 (Type 5 17β-Hydroxysteroid Dehydrogenase) Based on N-
Phenyl-Aminobenzoates and Their Structure–Activity Relationships. Journal of
Medicinal Chemistry 55:2311-2323.
113. Adeniji, A.O., Twenter, B.M., Byrns, M.C., Jin, Y., Winkler, J.D., and Penning,
T.M. 2011. Discovery of substituted 3-(phenylamino)benzoic acids as potent and
selective inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3).
Bioorganic & Medicinal Chemistry Letters 21:1464-1468.
114. Liu, S., Pestano, J.P.C., and Wolf, C. 2007. Regioselective Copper-Catalyzed C-N
and C-S Bond Formation Using Amines, Thiols and Halobenzoic Acids. Synthesis
2007:3519-3527.
115. Li, X., Xiao, D., and Meng, Y. 2010. Synthesis and characterization of multiblock
copolymer ionomers containing phthalazinone ether ketone and fluorene ether
ketone moieties. Journal of Applied Polymer Science 118:1100-1110.
116. Utz, C.G., and Shechter, H. 1985. Kinetics of decomposition of rigid diazo
compounds: substituent effects on the rates of thermolysis of 10-diazoanthrones.
The Journal of Organic Chemistry 50:5705-5709.
117. Witiak, D.T., Abood, N.A., Goswami, S., and Milo, G.E. 1986. The 5-, 6-, and
10-fluoro-1,2,3,4-tetrahydro-7,12-dimethylbenz[a]anthracenes. Comparative acid-
catalyzed isomerization to exomethylene tautomers: a 6-fluoro peri effect. The
Journal of Organic Chemistry 51:4499-4507.
118. Horning, E.C., and Reisner, D.B. 1949. Catalytic Reduction of Aromatic Ketones.
Journal of the American Chemical Society 71:1036-1037.

241
119. Newman, M.S., Dhawan, B., and Khanna, V.K. 1986. Synthesis of 10-
(hydroxymethyl)-7,12-dimethylbenz[a]anthracene. The Journal of Organic
Chemistry 51:1631-1632.
120. Katritzky, A.R., Rewcastle, G.W., and Vazquez de Miguel, L.M. 1988. Improved
syntheses of substituted carbazoles and benzocarbazoles via lithiation of the
(dialkylamino)methyl (aminal) derivatives. The Journal of Organic Chemistry
53:794-799.
121. Gan, Z., Hu, B., Song, Q., and Xu, Y. 2012. Convenient Chlorination of Some
Special Aromatic Compounds Using N-Chlorosuccinimide. Synthesis 44:1074-
1078.
122. Bowyer, P.M., Iles, D.H., and Ledwith, A. 1971. Chlorination of carbazole and its
derivatives with 1-chlorobenzotriazole. Journal of the Chemical Society C:
Organic:2775-2777.
123. Bagriantsev, S.N., Ang, K.-H., Gallardo-Godoy, A., Clark, K.A., Arkin, M.R.,
Renslo, A.R., and Minor, D.L. 2013. A High-Throughput Functional Screen
Identifies Small Molecule Regulators of Temperature- and Mechano-Sensitive
K2P Channels. ACS Chemical Biology 8:1841-1851.
124. Engen, W., O’Brien, T.E., Kelly, B., Do, J., Rillera, L., Stapleton, L.K.,
Youngren, J.F., and Anderson, M.O. 2010. Synthesis of aryl-heteroaryl ureas
(AHUs) based on 4-aminoquinoline and their evaluation against the insulin-like
growth factor receptor (IGF-1R). Bioorganic & Medicinal Chemistry 18:5995-
6005.
125. Rowlett, R.J., and Lutz, R.E. 1946. Antimalarials. Hydrolysis and Methanolysis
of 2,4,7-Trichloroquinoline1. Journal of the American Chemical Society 68:1288-
1291.
126. Mederski, W.W.K.R., Lefort, M., Germann, M., and Kux, D. 1999. N-aryl
heterocycles via coupling reactions with arylboronic acids. Tetrahedron
55:12757-12770.
127. Lemke, T.L.W., D.A., editor. 2008. Foye's Principles of Medicinal Chemistry.
USA: Lippincott Williams and Wilkinds.
128. Vagner, J., Qu, H., and Hruby, V.J. 2008. Peptidomimetics, a synthetic tool of
drug discovery. Current opinion in chemical biology 12:292-296.
129. Lima, L.M.A., and Barreiro, E.J. 2005. Bioisosterism: A useful strategy for
molecular modification and drug design. Current Medicinal Chemistry 12:23-49.
130. Olesen, P.H. 2001. The use of bioisosteric groups in lead optimization. Current
opinion in drug discovery & development 4:471-478.
131. Silverman, R.B. 2004. The Organic Chemistry of Drug Design and Drug Action.
USA: Elsevier Inc.
132. 2004. Organic Medicinal and Pharmaceutical Chemistry. J.H.B. Block, J.M.,
editor. USA: Lippincott Williams and Wilkins.
133. Taylor, R.D., Jewsbury, P.J., and Essex, J.W. 2002. A review of protein-small
molecule docking methods. Journal of Computer-Aided Molecular Design
16:151-166.
134. Wermuth, C.G.M., A. 2003. The Practice of Medicinal Chemistry. C.G.
Wermuth, editor. USA: Elsevier Inc.

242
135. Dwoskin, L.P., and Crooks, P.A. 2001. Competitive Neuronal Nicotinic Receptor
Antagonists: A New Direction for Drug Discovery. Journal of Pharmacology and
Experimental Therapeutics 298:395-402.
136. Kazuta, Y., Hirano, K., Natsume, K., Yamada, S., Kimura, R., Matsumoto, S.-i.,
Furuichi, K., Matsuda, A., and Shuto, S. 2003. Cyclopropane-Based
Conformational Restriction of Histamine. (1S,2S)-2-(2-Aminoethyl)-1-(1H-
imidazol-4-yl)cyclopropane, a Highly Selective Agonist for the Histamine H3
Receptor, Having a cis-Cyclopropane Structure. Journal of Medicinal Chemistry
46:1980-1988.
137. Willhite, C.C., and Dawson, M.I. 1990. Structure-activity relationships of
retinoids in developmental toxicology: IV. Planar cisoid conformational
restriction. Toxicology and applied pharmacology 103:324-344.
138. Hunsberger, I.M., Shaw, E.R., Fugger, J., Ketcham, R., and Lednicer, D. 1956.
The Preparation of Substituted Hydrazines. IV. Arylhydrazines via Conventional
Methods. The Journal of Organic Chemistry 21:394-399.
139. Maddirala, S.J., Gokak, V.S., Rajur, S.B., and Basanagoudar, L.D. 2003. Fischer
indolisation of 2,6-dialkyl and 2,4,6-trialkylphenylhydrazones of diketones and
ketoesters. Tetrahedron Letters 44:5665-5668.
140. Dugar, S.M., D.; Deokar, R.C.; Hollinger, F.P.; Kapoor, K.K. 2012. Preparation
of heteroarylmorpholinotriazine derivatives and analogs for use as
antiproliferative agents. W.I.P. Organization, editor. India: Sphaera Pharma Pvt.
Ltd.
141. Galli, U., Mesenzani, O., Coppo, C., Sorba, G., Canonico, P.L., Tron, G.C., and
Genazzani, A.A. 2012. Identification of a sirtuin 3 inhibitor that displays
selectivity over sirtuin 1 and 2. European Journal of Medicinal Chemistry 55:58-
66.
142. Staab, H.A., Zipplies, M.F., Müller, T., Storch, M., and Krieger, C. 1994.
Pyridinio-isoalloxazinophanes as Model Systems for Active-Site Complexes in
Flavoenzymes: Syntheses, X-Ray Structure Analyses and Spectroscopic
Properties. Chemische Berichte 127:1667-1680.
143. Maryanoff, B.E., and Reitz, A.B. 1989. The Wittig olefination reaction and
modifications involving phosphoryl-stabilized carbanions. Stereochemistry,
mechanism, and selected synthetic aspects. Chemical Reviews 89:863-927.
144. Wang, Q., Deredas, D., Huynh, C., and Schlosser, M. 2003. Sequestered
Alkyllithiums: Why Phenyllithium Alone is Suitable for Betaine-Ylide
Generation. Chemistry – A European Journal 9:570-574.
145. Liberatore, F., Casini, A., Carelli, V., Arnone, A., and Mondelli, R. 1975.
Borohydride reduction of pyridinium salts. V. Thermal dimerization of 1,6-
dihydro-1-methylpyridine-2-carbonitrile. The Journal of Organic Chemistry
40:559-563.
146. Oediger, H., and Joop, N. 1973. Eine neue Synthese von 1-unsubstituierten
1.2.5.6-Tetrahydropyridinen. Justus Liebigs Annalen der Chemie 764:21-27.
147. Reich, H.J., Renga, J.M., and Reich, I.L. 1975. Organoselenium chemistry.
Conversion of ketones to enones by selenoxide syn elimination. Journal of the
American Chemical Society 97:5434-5447.

243
148. Murphy, J.A., Tripoli, R., Khan, T.A., and Mali, U.W. 2005. Novel Phosphorus
Radical-Based Routes to Horsfiline. Organic Letters 7:3287-3289.
149. Callant, P., Ongena, R., and Vandewalle, M. 1981. Iridoids : Novel total synthesis
of (±)- isoiridomyrmecin and of (±)-verbenalol. Tetrahedron 37:2085-2089.
150. Biagetti, M., Leslie, C.P., Mazzali, A., Seri, C., Pizzi, D.A., Bentley, J., Genski,
T., Fabio, R.D., Zonzini, L., and Caberlotto, L. 2010. Synthesis and structure–
activity relationship of N-(3-azabicyclo[3.1.0]hex-6-ylmethyl)-5-(2-pyridinyl)-
1,3-thiazol-2-amines derivatives as NPY Y5 antagonists. Bioorganic & Medicinal
Chemistry Letters 20:4741-4744.
151. Brighty, K.E., and Castaldi, M.J. 1996. Synthesis of (1 alpha,5 alpha,6 alpha)-6-
amino-3-azabicyclo[3.1.0]hexane, a novel achiral diamine. Synlett:1097-&.
152. Radchenko, D., Grygorenko, O., and Komarov, I. 2010. Synthesis of 2-
azaspiro[3.3]heptane-derived amino acids: ornitine and GABA analogues. Amino
Acids 39:515-521.
153. Radchenko, D.S., Pavlenko, S.O., Grygorenko, O.O., Volochnyuk, D.M.,
Shishkina, S.V., Shishkin, O.V., and Komarov, I.V. 2010. Cyclobutane-Derived
Diamines: Synthesis and Molecular Structure. The Journal of Organic Chemistry
75:5941-5952.
154. Allinger, N.L., and Tushaus, L.A. 1965. Conformational Analysis. XLIII.
Stereochemical Studies in the Cyclobutane Ring System1-3. The Journal of
Organic Chemistry 30:1945-1951.
155. Ankner, T., and Hilmersson, G. 2009. Instantaneous Deprotection of Tosylamides
and Esters with SmI2/Amine/Water. Organic Letters 11:503-506.
156. Vedejs, E., and Lin, S. 1994. Deprotection of Arenesulfonamides with Samarium
iodide. The Journal of Organic Chemistry 59:1602-1603.
157. Sabitha, G., Reddy, B.V.S., Abraham, S., and Yadav, J.S. 1999. Deprotection of
sulfonamides using iodotrimethylsilane. Tetrahedron Letters 40:1569-1570.
158. Gethin, D.M., and Simpkins, N.S. 1997. A new synthetic approach to the
polyoxins: Concise stereoselective preparation of protected thymine polyoxin C.
Tetrahedron 53:14417-14436.
159. Ohki, Y., Tanifuji, K., Yamada, N., Imada, M., Tajima, T., and Tatsumi, K. 2011.
Synthetic analogues of [Fe4S4(Cys)3(His)] in hydrogenases and [Fe4S4(Cys)4] in
HiPIP derived from all-ferric [Fe4S4{N(SiMe3)2}4]. Proceedings of the National
Academy of Sciences.
160. Davenport, A.J.H., D.J.; Corsi, M. 2009. Azetidines And Cyclobutanes As
Histamine H3 Receptor Antagonists. W.I.P. Organization, editor: Evotec
Neurosciences Gmbh.
161. Smith, D.A., and Waterbeemd, H.v.d. 1999. Pharmacokinetics and metabolism in
early drug discovery. Current opinion in chemical biology 3:373-378.
162. Meyer, U. 1996. Overview of enzymes of drug metabolism. Journal of
Pharmacokinetics and Biopharmaceutics 24:449-459.
163. Guengerich, F.P. 2006. Cytochrome P450s and other enzymes in drug metabolism
and toxicity. The AAPS Journal 8:E101-E111.
164. Guengerich, F.P. 2003. Cytochromes P450, drugs, and diseases. Molecular
interventions 3:194-204.

244
165. Rydberg, P., Gloriam, D.E., and Olsen, L. 2010. The SMARTCyp cytochrome
P450 metabolism prediction server. Bioinformatics (Oxford) 26:2988-2989.
166. Rydberg, P., Gloriam, D.E., Zaretzki, J., Breneman, C., and Olsen, L. 2010.
SMARTCyp: A 2D Method for Prediction of Cytochrome P450-Mediated Drug
Metabolism. ACS Medicinal Chemistry Letters 1:96-100.
167. Rydberg, P., and Olsen, L. 2012. Predicting Drug Metabolism by Cytochrome
P450 2C9: Comparison with the 2D6 and 3A4 Isoforms. ChemMedChem 7:1202-
1209.
168. Rydberg, P., and Olsen, L. 2012. Ligand-Based Site of Metabolism Prediction for
Cytochrome P450 2D6. ACS Medicinal Chemistry Letters 3:69-73.
169. Rydberg, P., Rostkowski, M., Gloriam, D.E., and Olsen, L. 2013. The
Contribution of Atom Accessibility to Site of Metabolism Models for
Cytochromes P450. Molecular Pharmaceutics 10:1216-1223.
170. Lewis, D.F.V., Jacobs, M.N., and Dickins, M. 2004. Compound lipophilicity for
substrate binding to human P450s in drug metabolism. Drug Discovery Today
9:530-537.
171. Hansch, C. 1972. Quantitative Relationships Between Lipophilic Character and
Drug Metabolism. Drug Metabolism Reviews 1:1-13.
172. Nassar, A.-E.F., Kamel, A.M., and Clarimont, C. 2004. Improving the decision-
making process in the structural modification of drug candidates: enhancing
metabolic stability. Drug Discovery Today 9:1020-1028.
173. Genin, M.J., Poel, T.J., Yagi, Y., Biles, C., Althaus, I., Keiser, B.J., Kopta, L.A.,
Friis, J.M., Reusser, F., Adams, W.J., et al. 1996. Synthesis and Bioactivity of
Novel Bis(heteroaryl)piperazine (BHAP) Reverse Transcriptase Inhibitors:
Structure−Activity Relationships and Increased Metabolic Stability of Novel
Substituted Pyridine Analogs. Journal of Medicinal Chemistry 39:5267-5275.
174. Riley, R.J., Parker, A.J., Trigg, S., and Manners, C.N. 2001. Development of a
Generalized, Quantitative Physicochemical Model of CYP3A4 Inhibition for Use
in Early Drug Discovery. Pharmaceutical Research 18:652-655.
175. Lin, J., and Lu, A.H. 1998. Inhibition and Induction of Cytochrome P450 and the
Clinical Implications. Clinical Pharmacokinetics 35:361-390.
176. Murray, M. 1999. Mechanisms and significance of inhibitory drug interactions
involving cytochrome P450 enzymes (Review). International Journal of
Molecular Medicine 3:227-238.
177. Hagmann, W.K. 2008. The Many Roles for Fluorine in Medicinal Chemistry.
Journal of Medicinal Chemistry 51:4359-4369.
178. Purser, S., Moore, P.R., Swallow, S., and Gouverneur, V. 2008. Fluorine in
medicinal chemistry. Chemical Society Reviews 37:320-330.
179. Park, B.K., Kitteringham, N.R., and O'Neill, P.M. 2001. Metabolism of fluorine-
containing drugs. Annual Review of Pharmacology and Toxicology 41:443-470.
180. Kamal, A., Rajender, Reddy, D.R., Reddy, M.K., Balakishan, G., Shaik, T.B.,
Chourasia, M., and Sastry, G.N. 2009. Remarkable enhancement in the DNA-
binding ability of C2-fluoro substituted pyrrolo[2,1-c][1,4]benzodiazepines and
their anticancer potential. Bioorganic & Medicinal Chemistry 17:1557-1572.

245
181. Leroy, J., Porhiel, E., and Bondon, A. 2002. Synthesis and characterization of
partially β-fluorinated 5,10,15,20-tetraphenylporphyrins and some derivatives.
Tetrahedron 58:6713-6722.
182. Guandalini, L., Martini, E., Gualtieri, F., Romanelli, M.N., and Varani, K. 2004.
Design, synthesis and preliminary pharmacological evaluation of rigid analogues
of the nicotinic agonist 1,1-dimethyl-4-phenylpiperazinium iodide (DMPP).
Arkivoc:286-300.
183. Katritzky, A.R., Scriven, E.F.V., Majumder, S., Akhmedova, R.G., Vakulenko,
A.V., Akhmedov, N.G., Murugan, R., and Abboud, K.A. 2005. Preparation of
nitropyridines by nitration of pyridines with nitric acid. Organic & biomolecular
chemistry 3:538-541.
184. Bonavia, A., Franti, M., Pusateri Keaney, E., Kuhen, K., Seepersaud, M.,
Radetich, B., Shao, J., Honda, A., Dewhurst, J., Balabanis, K., et al. 2011.
Identification of broad-spectrum antiviral compounds and assessment of the
druggability of their target for efficacy against respiratory syncytial virus (RSV).
Proceedings of the National Academy of Sciences 108:6739-6744.
185. Krumm, S.A., Ndungu, J.M., Yoon, J.J., Dochow, M., Sun, A., Natchus, M.,
Snyder, J.P., and Plemper, R.K. 2011. Potent host-directed small-molecule
inhibitors of myxovirus RNA-dependent RNA-polymerases. Plos One 6:e20069.
186. Yoon, J.-J., Chawla, D., Paal, T., Ndungu, M., Du, Y., Kurtkaya, S., Sun, A.,
Snyder, J.P., and Plemper, R.K. 2008. High-Throughput Screening—Based
Identification of Paramyxovirus Inhibitors. Journal of Biomolecular Screening
13:591-608.
187. Gallay, P.A., and Lin, K. 2013. Profile of alisporivir and its potential in the
treatment of hepatitis C. Drug design, development and therapy 7:105-115.