
Determination Transcription Initiation Site and
7
Vol. 163, No. 3 JOURNAL OF BACTERIOLOGY, Sept. 1985, p. 863-869 0021-9193/85/090863-07$02.00/0 Copyright © 1985, American Society for Microbiology Determination of the Transcription Initiation Site and Identification of the Protein Product of the Regulatory Gene xylR for xyl Operons on the TOL Plasmid SACHIYE INOUYE,' ATSUSHI NAKAZAWA,' AND TERUKO NAKAZAWA2* Department of Biochemistry, School of Medicine,l and School of Allied Health Sciences,2 Yamaguchi University, Ube 755, Japan Received 4 March 1985/Accepted 29 May 1985 The xylR gene is a regulatory gene on the TOL plasmid, which acts in a positive manner on xyl operons for degradation of toluene and xylenes in Pseudomonas putida. A DNA fragment containing the xylR promoter region was cloned on promoter-probing vectors, and its nucleotide sequence was determined. The transcription initiation site of the xylR gene was determined in cells of P. putida and Escherichia coli by Si nuclease and reverse transcriptase mapping. Two initiation sites were detected which were identical in both P. putida and E. coli. The amounts of mRNA synthesized in both bacterial cells were alh'ost the same and independent of the inducers for xyl operons. The consensus sequences for E. coli promoters were found in the region preceding the respective transcription initiation sites. The product of the xylR gene was identified by the maxicell system as a protein with an approximate molecular weight of 67,000. The TOL plasmid of Pseudomonas putida mt-2 contains the genes encoding inducible enzymes which are required for the degradation of toluene, m-xylene, and p-xylene (26). The regulation of the gene expression has been revealed by a molecular cloning technique with Escherichia coli (11, 12) and P. putida (5, 6, 9) as host bacteria. The genes for the degradative enzymes are organized into two operons. The first operon (xylABC) encodes upper-pathway enzymes for the oxidation of the aromatic compounds to corresponding carboxylic acids, and the second operon (xylDEGF) encodes lower-pathway enzymes for the oxidation of the carboxylic acids to pyruvate and acetaldehyde. Two regulatory genes, xylR and xylS, positively control these operons. The xylR gene plays a role in the induction of both operons by m-xylene or m-methylbenzyl alcohol, whereas the xylS gene is responsible for the induction of only the second operon by m-toluate. Both xylR and xylS are necessary for the induc- tion of the second operon by m-xylene or m-methylbenzyl alcohol. The levels of expression of the xyl operons in E. coli were lower than those in P. putida, although the induction patterns in these bacteria were quite similar (20). We have recently reported the nucleotide sequences of the operator-promoter regions of the xylABC (13) and the xylDEGF (14) operons. The transcription start sites in vivo of these operons were determined by Si nuclease and reverse transcriptase mapping. The induced synthesis of mRNAs started at the same sites in both P. putida and E. coli, although the amounts of mRNAs in E. coli cells were less than those in P. putida cells. The consensus nucleotide sequences for E. coli promoters were not found in the region preceding the transcription start sites of both operons. The low expression of the xyl operons in E. coli, therefore, was thought to be a reflection of an inefficient transcription of the operons by E. coli RNA polymerase. However, a possibility remained that the levels of the activators encoded by xylR and xylS are lower in E. coli than in P. putida. * Corresponding author. In the present work, we analyzed the in vivo transcription of the xylR gene by S1 nuclease and reverse transcriptase mapping. The levels of xylR transcripts were almost the same in both E. coli and P. putida and independent of the TABLE 1. Bacterial strains and plasmids Strain/plasmid Characteristics Source or reference E. coli 20S0 thi lac mat mtl ara xyl rpsL 1 C600 F- leu tonA thi thr lacY supE Laboratory stock CSR603 F- phr-J recAl uvrA6 24 GM31 thr leu dcm his ara thi lac gal 15 xyl mtl rpsL tonA tsx supE P. putida TN2100 Prototroph 21 TN1126 met trp, TN2100 derivative This study Plasmids pACYC184 Tcr Cmr 3 pTS145 pACYC184 derivative, XyIR+ 12 Cmr pTS146 pTS145 derivative, XylR- Cmr 12 pTS162 pTS145 derivative, XylR+ Cmr 12 Kmr pTS163 pTS145 derivative, XylR- Cmr 12 Kmr pTS171 pTS145 derivative, XylR+ Cmr 12 pTS174 pTS145 derivative, XylR+ Cmr 12 pMCR600 Apr Kmr 18 pTS206 pMCR600 derivative carrying This study xylR promoter, Apr Kmr pACYC177 Apr Kmr 3 pTS1133 Promoter-proving vector, Apr This study Smr pTS1145 pTS1133 derivative carrying This study xylR promoter Apr Smr pTN2 RP4::TOL, Apr Kmr Tcr 19 863 Downloaded from https://journals.asm.org/journal/jb on 02 February 2022 by 190.124.30.105.
Transcript of Determination Transcription Initiation Site and

Vol. 163, No. 3JOURNAL OF BACTERIOLOGY, Sept. 1985, p. 863-8690021-9193/85/090863-07$02.00/0Copyright © 1985, American Society for Microbiology
Determination of the Transcription Initiation Site and Identificationof the Protein Product of the Regulatory Gene xylR for xyl Operons
on the TOL PlasmidSACHIYE INOUYE,' ATSUSHI NAKAZAWA,' AND TERUKO NAKAZAWA2*
Department of Biochemistry, School of Medicine,l and School of Allied Health Sciences,2 Yamaguchi University,Ube 755, Japan
Received 4 March 1985/Accepted 29 May 1985
The xylR gene is a regulatory gene on the TOL plasmid, which acts in a positive manner on xyl operons fordegradation of toluene and xylenes in Pseudomonas putida. A DNA fragment containing the xylR promoterregion was cloned on promoter-probing vectors, and its nucleotide sequence was determined. The transcriptioninitiation site of the xylR gene was determined in cells of P. putida and Escherichia coli by Si nuclease andreverse transcriptase mapping. Two initiation sites were detected which were identical in both P. putida and E.coli. The amounts of mRNA synthesized in both bacterial cells were alh'ost the same and independent of theinducers for xyl operons. The consensus sequences for E. coli promoters were found in the region preceding therespective transcription initiation sites. The product of the xylR gene was identified by the maxicell system as
a protein with an approximate molecular weight of 67,000.
The TOL plasmid of Pseudomonas putida mt-2 containsthe genes encoding inducible enzymes which are required forthe degradation of toluene, m-xylene, and p-xylene (26). Theregulation of the gene expression has been revealed by amolecular cloning technique with Escherichia coli (11, 12)and P. putida (5, 6, 9) as host bacteria. The genes for thedegradative enzymes are organized into two operons. Thefirst operon (xylABC) encodes upper-pathway enzymes forthe oxidation of the aromatic compounds to correspondingcarboxylic acids, and the second operon (xylDEGF) encodeslower-pathway enzymes for the oxidation of the carboxylicacids to pyruvate and acetaldehyde. Two regulatory genes,xylR and xylS, positively control these operons. The xylRgene plays a role in the induction of both operons bym-xylene or m-methylbenzyl alcohol, whereas the xylS geneis responsible for the induction of only the second operon bym-toluate. Both xylR and xylS are necessary for the induc-tion of the second operon by m-xylene or m-methylbenzylalcohol. The levels of expression of the xyl operons in E. coliwere lower than those in P. putida, although the inductionpatterns in these bacteria were quite similar (20).We have recently reported the nucleotide sequences of the
operator-promoter regions of the xylABC (13) and thexylDEGF (14) operons. The transcription start sites in vivoof these operons were determined by Si nuclease andreverse transcriptase mapping. The induced synthesis ofmRNAs started at the same sites in both P. putida and E.coli, although the amounts of mRNAs in E. coli cells wereless than those in P. putida cells. The consensus nucleotidesequences for E. coli promoters were not found in the regionpreceding the transcription start sites of both operons. Thelow expression of the xyl operons in E. coli, therefore, wasthought to be a reflection of an inefficient transcription of theoperons by E. coli RNA polymerase. However, a possibilityremained that the levels of the activators encoded by xylRand xylS are lower in E. coli than in P. putida.
* Corresponding author.
In the present work, we analyzed the in vivo transcriptionof the xylR gene by S1 nuclease and reverse transcriptasemapping. The levels of xylR transcripts were almost thesame in both E. coli and P. putida and independent of the
TABLE 1. Bacterial strains and plasmidsStrain/plasmid Characteristics Source or reference
E. coli20S0 thi lac mat mtl ara xyl rpsL 1C600 F- leu tonA thi thr lacY supE Laboratory stockCSR603 F- phr-J recAl uvrA6 24GM31 thr leu dcm his ara thi lac gal 15
xyl mtl rpsL tonA tsx supE
P. putidaTN2100 Prototroph 21TN1126 met trp, TN2100 derivative This study
PlasmidspACYC184 Tcr Cmr 3pTS145 pACYC184 derivative, XyIR+ 12
CmrpTS146 pTS145 derivative, XylR- Cmr 12pTS162 pTS145 derivative, XylR+ Cmr 12
KmrpTS163 pTS145 derivative, XylR- Cmr 12
KmrpTS171 pTS145 derivative, XylR+ Cmr 12pTS174 pTS145 derivative, XylR+ Cmr 12pMCR600 Apr Kmr 18pTS206 pMCR600 derivative carrying This study
xylR promoter, Apr KmrpACYC177 Apr Kmr 3pTS1133 Promoter-proving vector, Apr This study
SmrpTS1145 pTS1133 derivative carrying This study
xylR promoter Apr SmrpTN2 RP4::TOL, Apr Kmr Tcr 19
863
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
02
Febr
uary
202
2 by
190
.124
.30.
105.

864 INOUYE, NAKAZAWA, AND NAKAZAWA
BamHI HpaI StuIHpaI HindIII BamHIpTS145 j IR k :kACYi.18(9.Skb) A AAb
3.1 0.6 1.6 0.4kb
EcoRl
BglII
BamHI /BglIII
IPy StuI IsAp/ pTS 1 l5 i
I(15.7kb) v
EcoRI
Sm
FIG. 1. Construction of plasmids containing the xylR promoter region. The construction of pTS145 and the location of the xylR gene havebeen described previously (12). Open and hatched segments represent the TOL and pACYC184 regions, respectively. The thin-lined circlerepresents pTS1133 or pMCR600. pT$145 DNA was cleaved with BglII and BamfII, and the resulting fragments were separated on a 5%polyacrylamide gel. The 0.6-kb DNA fragment was recovered and ligated with BamHI-cleaved pTS1133 or BgII-cleaved pMCR600, whichhad been treated with bacterial alkaline phosphatase. After transformation of E. coli C600, pTS1145 was obtained from an ampicillin- andstreptomycin-resistant colony which also showed yellow color after spraying with a catechol solution. pTS206 was obtained from ampicillin-and kanamnycin-resistant colonies. Closed triangles indicate the restriction site of BglII. Small open circles and thick arrows on therecombinant plasmids indicate the promoters and the direction of transcription, respectively.
inducers for xyl operons. We also identified the xylR geneproduct by using the maxicell system.
MATERIALS AND METHODS
Bacterial strains and plasmids. The bacterial strains andplasmids used in this study are listed in Table 1.Enzymes and reagents. Restriction endonucleases were
obtained from Bethesda Research Laboratories, New Eng-land BioLabs, Takara Shuzo, and Nippon Gene and used
TABLE 2. Enzyme activities in crude extracts of cells carryingrecombinant plasmidsa
Sp act (mU/mg) in:Host Plasmid Catechol 2,
3-dioxygenase
E. coli C600 pTS1133 1 530pTS1145 470 550
P. putida TN1126 pTS1133 44 106pTS1145 2,200 57
a Cells were grown in L broth for 6 h with shaking at 37°C for E. coli and27°C for P. putida.
according to the directions of the suppliers. T4 DNA poly-merase, T4 polynucleotide kinase, and T4 ligase were theproducts of Takara Shuzo. Si nuclease was purchased fromBoehringer Mannheim Biochemicals, and reverse transcrip-tase was purchased from Life Sciences. [.y-32P]ATP (specificactivity, >5,000 Ci/mmol), [35S]methionine (specific activity,>800 Ci/mmol) were obtained from Amersham Corp.Media and culture conditions. The medium used through-
out the experiments was L broth (1% tryptone, 0.5% yeastextract, 0.5% NaCl). Agar was added to 1.5% in the L brothmedium for L agar plates. When needed, ampicillin (100,ug/ml), kanamycin (25 ,ug/ml), streptomycin (10 ,ug/ml),tetracycline (10 ,ug/ml), or chloramphenicol (10 ,g/ml) wasadded to the medium. Incubations were carried out at 270Cfor P. putida and at 37°C for E. coli. For enzyme inductionexperiments, the E. coli cells were grown at 30°C.Enzyme assay. Catechol 2,3-dioxygenase was assayed as
described previously (20). One milliunit corresponds to theformation of 1 nmol of 2-hydroxymuconic semialdehyde permin at 27°C. The enzyme synthesized in the cells wasvisualized by yellow color on the colonies developed after asolution of 0.1 M catechol was sprayed over the colonies; theyellow color is due to the reaction product formed fromcatechol by catechol 2,3-dioxygenase.DNA manipulations. The procedures for restriction endo-
J. BACTERIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
02
Febr
uary
202
2 by
190
.124
.30.
105.

PROTEIN PRODUCT OF xylR FOR xyl OPERONS 865
0 100 200 300 400 500 base ping, and reverse transcriptase mapping were petformed asdescribed previously (13, 14). Quantitative analysis of Sinuclease-protected fragments was described previously (14).DNA sequencing was done by the methods of Maxam and
F-'' ' Gilbert (16)., . Labeling of plasmid-encoded proteins in maxicell system.. , . ,, Plasmid-encoded proteins were labeled in maxicells as de-<,,< , scribed by Sancar et al. (24). The procedures for sodium
dodecyl sulfate-polyacrylamide gel electrophoresis andfluorography were described previously (27).
primer_RA-. -
mRNA- 1mRNA-2 _ -
FIG. 2. Restriction map and sequencing strategy for the xylRpromoter region. Restriction enzyme cleavage sites are representedby short vertical lines. Thin arrows show the extent and direction ofsequence analysis. Distance is given in bp from the left BgIlI site.Open boxes in the lower part show the DNA fragments used asprobes for Si nuclease mapping and as a primer for reversetranscriptase mapping. Thick arrows with closed circles indicate thedirection of transcription and the promoter for xyIR.
nuclease cleavage, ligation, transformation, DNA isolation,and gel electrophoresis have been described previously (12).
S1 nuclease and reverse transcriptase mapping and DNAsequencing. Preparation of crude RNA, Si nuclease map-
(a) (b)1 2 3 4 5 6
RESULTSCloning of the xylR promoter. The xylR gene was previ-
ously located in the 2.5-kilobase pairs (kb) HpaI fragment onpTS145 (Fig. 1) (12). To search for the xylR promoter, twopromoter-probing vectors, pTS1133 and pMCR600, wereused. The pTS1133 plasmid is a broad-host-range vectorwhich was constructed in our laboratory from pACYC177(3), pTS87 (13), and a derivative of the Inc P-4 plasmidRlb679 (2) (Fig. 1) (details will be published elsewhere). Aunique BamHI site exists in the upstream region of xylEwhich has no promoter. If a DNA fragment having a pro-moter is inserted into the BamHI site in the proper orienta-tion, the xylE gene is expressed. The pMCR600 plasmid hasthe pBR322 replicon and gives the host cells the resistance toboth ampicillin and kanamycin (18). The insertion of a DNAfragment into a unique BglII site on the plasmid inactivatesthe promoter for the kanamycin phosphotransferase gene(kan). If a DNA fragment having a promoter activity isinserted into the BgIII site, the transformant shows theresistance to kanamycin due to the inserted promoter.
1 2 3 4 5 6 7 8 9
475- * * 9 lb* * *
* -280
220 - * ,00 .~ ~ ~ 4
,.0-.0 * *
FIG. 3. S1 nuclease protection analysis. The DNA probe was the single-stranded 475-base TaqI-BglII fragment (Fig. 2, S1 probe A) labeledat the BglII site. (a) The RNA preparation (20 ,ug) was subjected to hybridization with the DNA probe (0.02 pmol). The mixture was treatedwith 1,000 U of S1 nuclease. The products were electrophoresed on an 8% polyacrylamide-8 M urea gel. Lanes 1 and 2 are marker DNAfragments. RNA was extracted from cells of TN1126(pTS1133) (lane 3), TN1126(pTS1145) (lane 4), C600(pTS1133) (lane 5), andC600(pTS1145) (lane 6). Specific activities of catechol 2,3-dioxygenase, the xylE gene product, of the cells are shown in Table 2. (b) The RNApreparation (40 ,ug) was subjected to hybridization with the DNA probe (0.05 pmol). Other conditions were the same as in (a). Lanes 1, 8,and 9 are marker DNA fragments. RNA was extracted from cells of TN2100(pTN2) (lanes 2 through 4) and 20S0(pTN2) (lanes 5 through 7).Cells were grown in the absence (lanes 2 and 5) and presence of m-toluate (lanes 3 and 6) or m-methylbenzyl alcohol (lanes 4 and 7). A bandcorresponding to 475 nucleotides probably represents the probe DNA protected by the contaminated anti-sense strand.
BgZIITaqlBstEIIFnu4HIDdeIAvaIlHpaIINciIStuIHaeII
SI probe A
SI probe B
340 -*
. - t80
VOL. 163, 1985
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
02
Febr
uary
202
2 by
190
.124
.30.
105.

866 INOUYE, NAKAZAWA, AND NAKAZAWA
1 2 3 4 A >C G A>G C T+C 5 6 7*~~~~~~~~~~~.FIG. 4. Determination of transcription initiation site ofxy_R by
Si nuclease and reverse transcriptase mappi'ng. The RNA prepara-
tion (50 g) was hybridized with the Stul-Ncil fragment (Fig. 1,
primer) labeled at the Ncil site (1 pmol). After incubation with 24 U
of reverse transcriptase, the extended products were subjected to
coelectrophoresis (lan'es 5 and 6) together with sequencing ladder of
the BstEII-Ncil fragment label'ed at the NciI site (lanes A>C to
T +QC. The single-stranded HaeII-Ncil fragment (Fig. 1, Si probe
B) labeled 'at the Ncil site (0.05 pmol) was used for Si nuclease
mapping (lanes through 4 and 7). RNA preparations (lanes 1
through 6) were obtained from cells of m-methylbenzyl alcohol-
ind'uced TN1 126(pTN2), 20S0(pTN2), TN1126(pTS11'45),C600(pTS1145), TN1126(pTS1145), a'nd C600(pTS114S), respec-
tively. The same sample was used in lanes 1 and 7. The amount of
the applied samnple in lanes 1 through 4 and 7 was one-half the
amount in lanes 5 and 6.
pTS145 DNA was digested with BamHI and BglIl, and
fo'ur PNA fragmoents (3.1, 0.6, 1.6, 0.4 kb) derived from the
TOL plasmid were purified. When each of the fragments wa'sligated with pTS1133 and introduced into E. ccli C600, onlythe 0.6-kb BglII fragmnent produced transformants showing a
high xylE expression. From one of such clones, PTS1145 was
obtained. Simtilarly, kanamycin-resistant transformants of E.
ccli were obtained on ligation of the 0.6-kb BglII fragm'entwith pMCR600, and pTS206 was isolated (Fig. 1). The
orientation of the BgllI fragment to the vectors was deter-
mined by the restriction site of Stul in the fragment.To see whether the promoter on 'the Bglll frag'ment is also
active in P. putida, pTSii45 was introduced into P. putida
TN1126, and the levels of catechol 2,3-dioxygenase in crude
extracts were determined. As shown in Table 2, the xylE
gene expression conferred by pTSi145 DNA is approxi-
mately five times higher in P. putida than in E. ccli, whereas
the levels of .-lactamase were lower in P. putida. The
conitent of pTSii45 DNA in both bacteria did not change
significantly as judged by the amount of iplasmid DNA
purified by rapid alkaline methods and stained by ethidium
bromide (12). Based on these results, we conclude that the
xylR gene promoter is located in the 0.6-kb Bglll fragment.Si nuclease protection analysis. The in vivo transcription of
the xylR gene in P. putida and E. ccli was analyzed by Si
nuclease mapping. RNAs were prepared from cells of P.
putida or E. ccli' carrying pTSii33 or pTS1145. The 475-
base-pair (bp) TaqI-Bglii fragment (Fig. 2, Si probe A) was
purified and labeled at the 5' ends. A single-stranded frag-ment was isolated and hybridized with RNAs prepared fromP. putida and E. coli cells. The hybridized materials were
digested with Si nuclease, and the protected DNA waselectrophoresed. As shown in Fig. 3a, two bands corre-sponding to 230 and 200 nucleotides were detected whenRNAs from cells of E. coli C600 or P. putida TN1126carrying pTS1145 were used. In either case, the intensity ofthe large fragment band was more than three times that ofthe small one. On the other hand, no band was detected withRNA from cells carrying vector plasmid pTS1133. Protec-tion by RNA was not observed when another strand wasused as a DNA probe (data not shown). These resultsindicate that the transcription of the xylR gene started at theregion 230 (mRNA-1) and 200 (mRNA-2) nucleotides up-stream from the right BglII site in Fig. 2 in both P. putida andE. coli. Although the present experiment cannot afford thequantitative estimation, amounts of the xylR mRNA synthe-sized in E. coli and P. putida are not much different.The mRNA levels of xylR conferred by pTN2 in E. coli
and P, putida were also determined by Si nuclease mapping.RNA preparations from induced or noninduced cells of P.putida and E. coli carrying pTN2 gave two protected bandswith all the preparations (Fig. 3b). The patterns were essen-tially the same as with RNAs from the cells carryingpTS1145. Thus the transcription of the xylR gene occurredconstitutively in both bacteria and was not affected in thepresence of inducers.
Transcription start site of the xylR gene. The precisetranscription start site was determined by coelectrophoresisof Si nuclease-protected fragments on a sequence gel withchemically cleaved reaction products of the BstEII-NciIfragment labeled at the Ncl site (Fig. 4, lanes 1, 2, 3, 4, and7). A single-stranded DNA of the HaeII-NciI fragmentlabeled at the NciI site (Fig. 2, Si probe B) was hybridizedwith RNA preparations from P. putida and E. coli, carryingeither pTN2 or pTS1145. Essentially the same results wereobtained in Si mapping with P. putida and E. coli RNApreparations. The patterns of the protected fragments didnot change with two different quantities of Si nuclease, 500and 1,000 U (data not shown). A certain degree ofmicroheterogeneity, which was reported to be inherent tothe Si mapping assay, was seen in the protected DNAbands.Another approach, reverse transcriptase mapping, was
also used to assign the start site. The 530-bp NciI fragmentwas purified, labeled at the 5' ends, and cleaved with StuI.The 50-bp StuI-NciI fragment labeled at the NciI site (Fig. 2,primer) was isolated, hybridized with the RNA preparations,and extended with reverse transcriptase. The extended DNAwas electrophoresed on a sequence gel (Fig. 4, lanes 5 and6). Corresponding to the transcription start site of mRNA-1determined by Si-nuclease mapping, a single major bandwas observed in the reverse transcriptase mapping witheither P. putida or E. coli RNA preparations. The length ofthe extended DNA fragment was one to three nucleotidesshorter than that of Si nuclease-protected DNA fragments.After a correction of 1.5 bases in determnining the length ofthe extended fragment (8), the band matches up with adenineof the coding strand at position 342 (Fig. 5a). For themRNA-2 start site, two bands were observed which corre-spond to adenine and thymine of the coding strand atpositions 370 and 371, respectively (Fig. Sa). The length ofmRNA-2 determined by reverse transcriptase mapping wasalso shorter than that obtained by Si nuclease mapping.Incubation with twice the amount of reverse transcriptasedid not extend the length of mRNA-1 and mRNA-2 (data notshown).
Nucleotide sequence. The nucleotide sequence of the
J. BACTERIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
02
Febr
uary
202
2 by
190
.124
.30.
105.

PROTEIN PRODUCT OF xylR FOR xyl OPERONS 867
(a)
AGATCTGACTTTCTCGTTCAATAAGCAAAAATCCATAGTTCACGGTTCTCTTATTTTAATGTGGGCTGCTTGGTGTGATTCTAGACTGAAAAGAGCAAGTTATTCGTTTTTAGGTATCAAGTGCCAAGAGAATAAAATTACACCCGACGAACCACACTA
81GTAGAAAGGCGCCAAGTCGATGAAAATGCATCTCGACGTGATGCGTATACGGGTTACCCCCATTGCCACGTTGCGCCATCCATCTTTCCGCGGTTCAGCTACTTTTACGTAGAGCTGCACTACGCATATGCCCAATGGGGGTAACGGTGCAACGCGGTAG
161CTTTTTGCAATCAGTGACCACTTTTCCAAGCAAAAATAACGCCAAGCAGAACGAAGACG TCTT1mAAGAAGCGAGAACGAAAAACGTTAGTCACTGGTGAAAAGGTTCGTTTTTATTGCGGTTCGTCTTGCTTCTGCAAGAAAAATTCITCGCTCTTG
241 -35ACCAGAAGTTCGTGCTGTCGGGGCATGCGGCGACGAATTGCCGGATAAAGGGGATCTGCGTTGAG di?ATTCAGTTAATGGTCTTCAAGCACGACAGCCCCGTACGCCGCTGCTTAACCGCCTATTTCCCCTAGACGCAACTCCACCTAAAGTCAATT
321 -10 35J mRNA-1 -10 ;RNA- 2TCAATTGG CAa3f1FiS9:ACCACCTAAGCAAATGc1i4iGCAGATGGAATGCTGAGCCGGCAAGCACAGGCCTTAGTTAACCAATTAGAAAGTCCTGGTGGATTCGTTTACGATTTCACCGTCTACCTTACGACTCGGCCGTTCGTGTCCGGAA
401GACGTTGCAAGGTAGTCATGACCGCAGTGAGCCTCTGATGTTCCGCCGGGTGGATCATCCCGATAAAAACAAGAGQGAAAACTGCAACGTTCCATCAGTACTGGCGTCACTCGGAGACTACAAGGCGGCCCACCTAGTAGGGCTATTTTTrGTTCTCCTTTT
481CAAE3GCTTACATACAiACCCAAGATGCAGCATGAGGA AAGACCTTAGCAGCCAGATCCGTTTCGTTGCTCGGTTTACAGCGAATGTATGTTTGGGTTCTACGTCGTACTCCTATACGTTCTGGAATCGTCGGTCTAGGCAAAGCAACGAGC
561CCGAAGGCAAGATCTGGCITCCGTTCTAGA
(b)-70 -60 -50 -40 -30 -20 -10 +1
xylABC AAAATCAATAATTTAGATGAAATAAGGGGATCGGTATAAGCAATGGCATGGCGGTTGCTAGCTATACGAGA
xylR+1
GACGAATTGGCGGATAAAGGGGATCTGCGTTGAGGTGGATTTCAGTTAATCAATTGGTTAATCTTTCAGGAmRNA-1
+1xyIDEGF TTGCAAGAAGCGGATACAGGAGTGCAAAAAATGGCTATCTCTAGAAAGGCCTACCCCTTAGGCTTTATGCA
FIG. 5. (a) Nucleotide sequence of the xylR promoter region. Nucleotides are numbered from the BglII site. Arrows indicate thetranscription start sites of xyIR. Sequences which are homologous to the consensus sequences of E. coli promoters are indicated by bracketsabove the sequence. The underlined bases are complementary to the 3' end of the 16S rRNA of P. aeruginosa. The initiation codon is shownin boxes. (b) Sequence homology of the promoter region ofxylR and the operator-promoter regions of the xylABC and xylDEGF operons. Thetranscription start site is designated as + 1. Homologous bases are underlined.
575-bp BglII fragment was determined according to thesequence strategy presented in Fig. 2, and the resultingsequence is shown in Fig. 5a. The transcription start sites areindicated by vertical arrows as determined by reverse tran-scriptase mapping. In the region downstream from the startsites, there are two AUG start codons in the same codingframe, from which a continuous reading frame exists at leastup to the BglII site at the end. Preceding each initiationcodon there is a sequence AGGA which is complementary tothe 3' end of 16S rRNA of either E. coli or Pseudomonasaeruginosa (25). Preceding both the transcription start sites,the sequences homologous to the -10 and -35 consensussequences for E. coli promoters have been found (22, 23).
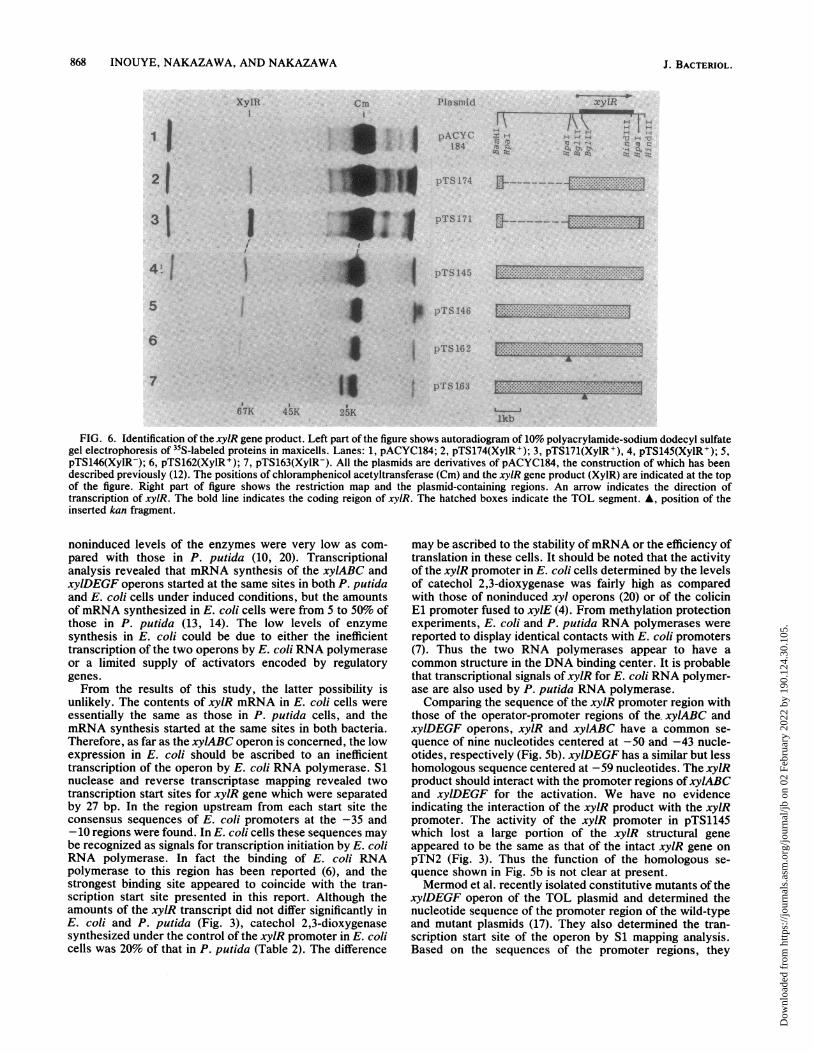
Identification of xylR gene product. Plasmid-encoded pro-teins were analyzed in the maxicell system using[35S]methionine as a tracer (Fig. 6). Cells of E. coli CSR603carrying pACYC184 produced one protein band correspond-ing to chloramphenicol acetyltransferase with a molecular
weight of 24,000. All the XylR+ plasmids, pTS174, pTS171,pTS145, and pTS162 derived from pACYC184 gave a bandof a 67,000-dalton polypeptide in addition to chlorampheni-col acetyltransferase. The XylR- plasmid pTS163 did notdirect the synthesis of the 67,000-dalton protein. Instead, aband corresponding to kanamycin phosphotransferase with amolecular weight of 26,000 was observed that is encoded bythe inserted DNA fragment. Therefore, the 67,000-daltonprotein was considered to be the product of the xylR gene.Plasmid pTS146 was also XylR-, but it encoded a polypep-tide with a molecular weight very close to 67,000. Theplasmid may have lost a small COOH-terminal portion ofxylR which is necessary for the activity.
DISCUSSIONWe previously reported that induction of toluene-
degrading enzymes from pTN2, a derivative of the TOLplasmid, occurred in E. coli, but both induced and
VOL. 163, 1985
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
02
Febr
uary
202
2 by
190
.124
.30.
105.
868 INOUYE, NAKAZAWA, AND NAKAZAWA
XyIR
I
CmI
*... ...'elk,
1-AI R _
a67K 45K 25K
Plasmid
| pACYC E
184E
ppTS174
ppTS 146 E
Ij)TS 162
I pTS 163
lJ1kjb
FIG. 6. Identification of the xyIR gene product. Left part of the figure shows autoradiogram of 10% polyacrylamide-sodium dodecyl sulfategel electrophoresis of 35S-labeled proteins in maxicells. Lanes: 1, pACYC184; 2, pTS174(XylR+); 3, pTS171(XylR+), 4, pTS145(XylR+); 5,pTS146(XylR-); 6, pTS162(XylR+); 7, pTS163(XylR-). All the plasmids are derivatives of pACYC184, the construction of which has beendescribed previously (12). The positions of chloramphenicol acetyltransferase (Cm) and the xylR gene product (XylR) are indicated at the topof the figure. Right part of figure shows the restriction map and the plasmid-containing regions. An arrow indicates the direction oftranscription of xylR. The bold line indicates the coding reigon of xylR. The hatched boxes indicate the TOL segment. A, position of theinserted kan fragment.
noninduced levels of the enzymes were very low as com-pared with those in P. putida (10, 20). Transcriptionalanalysis revealed that mRNA synthesis of the xylABC andxylDEGF operons started at the same sites in both P. putidaand E. coli cells under induced conditions, but the amountsofmRNA synthesized in E. coli cells were from 5 to 50% ofthose in P. putida (13, 14). The low levels of enzymesynthesis in E. coli could be due to either the inefficienttranscription of the two operons by E. coli RNA polymeraseor a limited supply of activators encoded by regulatorygenes.From the results of this study, the latter possibility is
unlikely. The contents of xylR mRNA in E. coli cells wereessentially the same as those in P. putida cells, and themRNA synthesis started at the same sites in both bacteria.Therefore, as far as the xylABC operon is concerned, the lowexpression in E. coli should be ascribed to an inefficienttranscription of the operon by E. coli RNA polymerase. Sinuclease and reverse transcriptase mapping revealed twotranscription start sites for xylR gene which were separatedby 27 bp. In the region upstream from each start site theconsensus sequences of E. coli promoters at the -35 and-10 regions were found. In E. coli cells these sequences maybe recognized as signals for transcription initiation by E. coliRNA polymerase. In fact the binding of E. coli RNApolymerase to this region has been reported (6), and thestrongest binding site appeared to coincide with the tran-scription start site presented in this report. Although theamounts of the xylR transcript did not differ significantly inE. coli and P. putida (Fig. 3), catechol 2,3-dioxygenasesynthesized under the control of the xylR promoter in E. colicells was 20% of that in P. putida (Table 2). The difference
may be ascribed to the stability ofmRNA or the efficiency oftranslation in these cells. It should be noted that the activityof the xylR promoter in E. coli cells determined by the levelsof catechol 2,3-dioxygenase was fairly high as comparedwith those of noninduced xyl operons (20) or of the colicinEl promoter fused to xylE (4). From methylation protectionexperiments, E. coli and P. putida RNA polymerases werereported to display identical contacts with E. coli promoters(7). Thus the two RNA polymerases appear to have acommon structure in the DNA binding center. It is probablethat transcriptional signals of xylR for E. coli RNA polymer-ase are also used by P. putida RNA polymerase.Comparing the sequence of the xylR promoter region with
those of the operator-promoter regions of the xylABC andxylDEGF operons, xylR and xylABC have a common se-quence of nine nucleotides centered at -50 and -43 nucle-otides, respectively (Fig. Sb). xylDEGF has a similar but lesshomologous sequence centered at -59 nucleotides. The xylRproduct should interact with the promoter regions ofxylABCand xylDEGF for the activation. We have no evidenceindicating the interaction of the xylR product with the xylRpromoter. The activity of the xylR promoter in pTS1145which lost a large portion of the xylR structural geneappeared to be the same as that of the intact xylR gene onpTN2 (Fig. 3). Thus the function of the homologous se-quence shown in Fig. Sb is not clear at present.Mermod et al. recently isolated constitutive mutants of the
xylDEGF operon of the TOL plasmid and determined thenucleotide sequence of the promoter region of the wild-typeand mutant plasmids (17). They also determined the tran-scription start site of the operon by Si mapping analysis.Based on the sequences of the promoter regions, they
1
2
45I
5
6
7
,.7-77-71I.I .I.,.I.-,I.I ............ " ."" ''..j......A.,.,...... ..-...........,......... ...ig
J. BACTERIOL.
.. n_
xylR
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
02
Febr
uary
202
2 by
190
.124
.30.
105.

PROTEIN PRODUCT OF xylR FOR xyl OPERONS 869
proposed consensus sequences for P. putida promoters,which are the sequences A-AGGC-T and GCTATA or
GCAATA centered at -25 and -8 nucleotides, respectively.Such sequences were not found in the upstream region of themRNA-1 start site, but the homologous sequences were
found in the region preceding the mRNA-2 start site.The maxicell system showed that a 67,000-dalton polypep-
tide was encoded by the XylR+ plasmids. The size of theDNA fragment which encodes a 67,000-dalton protein isabout 1.8 kb. Since the translation might start at position 484or 523 (Fig. 5a), the coding region of the xylR gene wasdeduced as shown in Fig. 6.
ACKNOWLEDGMENTS
We thank T. Horiuchi for the bacterial strain and S. lyobe for a
plasmid. This work was supported in part by grants-in-aid forScientific Research from the Ministry of Education, Science, andCulture, Japan. A part of the work was carried out under a contractwith the Science and Technology Agency in Japan.
LITERATURE CITED1. Bachmann, B. J. 1972. Pedigrees of some mutant strains of
Escherichia coli K 12. Bacteriol. Rev. 36:525-557.2. Barth, P. T., and N. J. Grinter. 1974. Comparison of the
deoxyribonucleic acid molecular weights and homologies ofplasmids conferring linked resistance to streptomycin andsulfonamides. J. Bacteriol. 120:618-630.
3. Chang, A. C. Y., and S. N. Cohen. 1978. Construction andcharacterization of amplifiable multicopy DNA cloning vehiclesderived from the P1SA cryptic miniplasmid. J. Bacteriol.134:1141-1156.
4. Ebina, Y., Y. Takahara, K. Shirabe, M. Yamada, T. Nakazawa,and A. Nakazawa. 1983. Plasmid-encoded regulation of colicinEl gene expression. J. Bacteriol. 156:487-492.
5. Franklin, F. C. H., M. Bagdasarian, M. M. Bagdasarian, andK. N. Timmis. 1981. Molecular and functional analysis of theTOL plasmid pWWO from Pseudomonas putida and cloning ofgenes for the entire regulated aromatic ring meta cleavagepathway. Proc. Natl. Acad. Sci. U.S.A. 78:7458-7462.
6. Franklin, F. C. H., P. R. Lehrbach, R. Lurz, B. Rueckert, M.Bagdasarian, and K. N. Timmis. 1983. Localization and func-tional analysis of transposon mutations in regulatory genes ofthe TOL catabolic pathway. J. Bacteriol. 154:676-685.
7. Gragerov, A. I., A. A. Chenchik, V. A. Aivasashvilli, R. S.Beabealashvilli, and V. G. Nikiforov. 1984. Escherichia coli andPseudomonas putida RNA polymerases display identical con-tacts with promoters. Mol. Gen. Genet. 195:511-515.
8. Green, M. R., and R. G. Roeder. 1980. Definition of a novelpromoter for the major adenovirus-associated virus mRNA.Cell 22:231-242.
9. Harayama, S., P. R. Lehrbach, and K. N. Timmis. 1984.Transposon mutagenesis analysis of meta-cleavage pathwayoperon genes of the TOL plasmid of Pseudomonas putida mt-2.J. Bacteriol. 160:251-255.
10. Inouye, S., A. Nakazawa, and T. Nakazawa. 1981. Molecular
cloning of TOL genes xylB and xylE in Escherichia coli. J.Bacteriol. 145:1137-1143.
11. Inouye, S., A. Nakazawa, and T. Nakazawa. 1981. Molecularcloning of gene xylS of the TOL plasmid: evidence for positiveregulation of xylDEGF operon by xylS. J. Bacteriol. 148:413-418.
12. Inouye, S., A. Nakazawa, and T. Nakazawa. 1983. Molecularcloning of regulatory gene xylR and operator-promoter regionsof the xylABC and xylDEGF operons of the TOL plasmid. J.Bacteriol. 155:1192-1199.
13. Inouye, S., A. Nakazawa, and T. Nakazawa. 1984. Nucleotidesequence surrounding the transcription initiation site ofxylABCoperon on TOL plasmid of Pseudomonas putida. Proc. Natl.Acad. Sci. U.S.A. 81:1688-1691.
14. Inouye, S., A. Nakazawa, and T. Nakazawa. 1984. Nucleotidesequence of the promoter region of the xylDEGF operon onTOL plasmid of Pseudomonas putida. Gene 29:323-330.
15. Marinus, M. G. 1973. Location of DNA methylation genes onthe Escherichia coli K-12 genetic map. Mol. Gen. Genet.127:47-55.
16. Maxam, A. M., and W. Gilbert. 1980. Sequencing end-labeledDNA with base-specific chemical cleavages. Methods Enzymol.65:499-560.
17. Mermod, N., P. R. Lehrbach, W. Reineke, and K. N. Timmis.1984. Transcription of the TOL plasmid toluate catabolic path-way operon of Pseudomonas putida is determined by a pair ofco-ordinately and positively regulated overlapping promoters.EMBO J. 3:2461-2466.
18. Miki, T., H. Kumahara, and A. Nakazawa. 1981. Constructionof a fused operon consisting of the recA and kan (kanamycinresistance) genes and regulation of its expression by the lexAgene. Mol. Gen. Genet. 183:25-31.
19. Nakazawa, T., E. Hayashi, T. Yokota, Y. Ebina, and A.Nakazawa. 1978. Isolation of TOL and RP4 recombinants byintegrative suppression. J. Bacteriol. 134:270-277.
20. Nakazawa, T., S. Inouye, and A. Nakazawa. 1980. Physical andfunctional mapping of RP4-TOL plasmid recombinants: analysisof insertion and deletion mutants. J. Bacteriol. 144:222-231.
21. Nakazawa, T., and T. Yokota. 1977. Isolation of a mutant TOLplasmid with increased activity and transmissibility from Pseu-domonas putida (arvilla) mt-2. J. Bacteriol. 129:39-46.
22. Pribnow, D. 1975. Nucleotide sequence of an RNA polymerasebinding site at an early T7 promoter. Proc. Natl. Acad. Sci.U.S.A. 72:784-788.
23. Rosenberg, M., and D. Court. 1979. Regulatory sequencesinvolved in the promotion and termination of RNA transcrip-tion. Annu. Rev. Genet. 13:319-353.
24. Sanacar, A., A. M. Hack, and W. D. Rupp. 1979. Simple methodfor identification of plasmid-coded proteins. J. Bacteriol.137:692-693.
25. Shine, J., and L. Dalgarno. 1975. Determination of cistronspecificity in bacterial ribosome. Nature (London) 254:34-38.
26. Worsey, M. J., and P. A. Williams. 1975. Metabolism of tolueneand xylenes by Pseudomonas putida (arvilla) mt-2: evidence fora new function of the TOL plasmid. J. Bacteriol. 124:7-13.
27. Yamada, M., and A. Nakazawa. 1984. Factors necessary for theexport process of colicin El across cytoplasmic membrane ofEscherichia coli. Eur. J. Biochem. 140:249-255.
VOL. 163, 1985
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
02
Febr
uary
202
2 by
190
.124
.30.
105.








![[V]. Process of Transcription and Transcriptional Control of Gene Expression 1 RNA polymerases and Initiation of transcription Transcriptional elongation.](https://static.fdocuments.in/doc/165x107/56649e595503460f94b52b31/v-process-of-transcription-and-transcriptional-control-of-gene-expression.jpg)









