De novo assembly and analysis of RNA-seq data - Nature · De novo assembly and analysis of RNA-seq...
35
Nature Methods De novo assembly and analysis of RNA-seq data Gordon Robertson, Jacqueline Schein, Readman Chiu, Richard Corbett, Matthew Field, Shaun D Jackman, Karen Mungall, Sam Lee, Hisanaga Mark Okada, Jenny Q Qian, Malachi Griffith, Anthony Raymond, Nina Thiessen, Timothee Cezard, Yaron Butterfield, Richard Newsome, Simon K Chan, Rong She, Richard Varhol, Baljit Kamoh, Anna-Liisa Prabhu, Angela Tam, YongJun Zhao, Richard Moore, Martin Hirst, Marco A Marra, Steven J M Jones, Pamela A Hoodless & Inanc Birol Supplementary Figure 1 Schematic of ABySS assembly steps Supplementary Figure 2 Assembly properties for k values of 26 to 50 Supplementary figure 3 Partitioning of MAQ-aligned reads relative to Ensembl transcript models. Supplementary figure 4 Fraction of Ensembl transcripts with at least 80% of exon length covered by unmerged contig alignments, as a function of normalized WTSS coverage threshold Supplementary figure 5 Assembled contigs across multiple k-values are merged to obtain a nonredundant set of contigs for analysis Supplementary figure 6 Coverage of Ensembl v54 transcripts by contig alignments, as a function of mean read transcript cover Supplementary figure 7 Splice site support for 149,877 Trans-ABySS parent (v1.1.1) contig alignments, considering GT-AG, GC-AG and AT-AC donor-acceptor types Supplementary figure 8 Schematic of comparing a transcript model (top) with contig alignments to identify annotated and novel transcripts and transcript structures Supplementary figure 9 RT-PCR validation of Insr’s 36-bp novel exon Supplementary figure 10 Novel UTR candidate for Nlrp6 Supplementary figure 11 Novel transcript candidate Supplementary figure 12 Shank2’s contig alignment supports both an RT-PCR-validated 21-bp skipped exon, and a novel, H3K4me3-supported TSS that is upstream of the 5’-most Ensembl TSS Supplementary figure 13 Sfrs3: assembly can extend contigs through exons that have low to- genome aligned-read densities Supplementary figure 14 Empirical distribution functions for mean normalized read coverage, C, for ENSMUSTs and transcripts with novel retained introns Supplementary figure 15 Coverage metrics for known and novel retained introns Supplementary figure 16 Schematic of identifying novel short and long 3’UTRs with EJ- and PAM- reads Supplementary figure 17 a) PAM-reads identify a novel polyadenylation site in the 3’ UTR of Dmgdh b) PAM-reads and contig alignments identify a novel long 3’UTR for Sult3a1 Supplementary figure 18 Schematic of detecting a fusion gene with a contig alignment Nature Methods: doi:10.1038/nmeth.1517
Transcript of De novo assembly and analysis of RNA-seq data - Nature · De novo assembly and analysis of RNA-seq...

Nature Methods
De novo assembly and analysis of RNA-seq data
Gordon Robertson, Jacqueline Schein, Readman Chiu, Richard Corbett, Matthew Field, Shaun D Jackman,
Karen Mungall, Sam Lee, Hisanaga Mark Okada, Jenny Q Qian, Malachi Griffith, Anthony Raymond,
Nina Thiessen, Timothee Cezard, Yaron Butterfield, Richard Newsome, Simon K Chan, Rong She, Richard
Varhol, Baljit Kamoh, Anna-Liisa Prabhu, Angela Tam, YongJun Zhao, Richard Moore, Martin Hirst,
Marco A Marra, Steven J M Jones, Pamela A Hoodless & Inanc Birol
Supplementary Figure 1 Schematic of ABySS assembly steps
Supplementary Figure 2 Assembly properties for k values of 26 to 50
Supplementary figure 3 Partitioning of MAQ-aligned reads relative to Ensembl transcript models.
Supplementary figure 4 Fraction of Ensembl transcripts with at least 80% of exon length covered by unmerged contig alignments, as a function of normalized WTSS coverage threshold
Supplementary figure 5 Assembled contigs across multiple k-values are merged to obtain a nonredundant set of contigs for analysis
Supplementary figure 6 Coverage of Ensembl v54 transcripts by contig alignments, as a function of mean read transcript cover
Supplementary figure 7 Splice site support for 149,877 Trans-ABySS parent (v1.1.1) contig alignments, considering GT-AG, GC-AG and AT-AC donor-acceptor types
Supplementary figure 8 Schematic of comparing a transcript model (top) with contig alignments to identify annotated and novel transcripts and transcript structures
Supplementary figure 9 RT-PCR validation of Insr’s 36-bp novel exon
Supplementary figure 10 Novel UTR candidate for Nlrp6
Supplementary figure 11 Novel transcript candidate
Supplementary figure 12 Shank2’s contig alignment supports both an RT-PCR-validated 21-bp skipped exon, and a novel, H3K4me3-supported TSS that is upstream of the 5’-most Ensembl TSS
Supplementary figure 13 Sfrs3: assembly can extend contigs through exons that have low to-genome aligned-read densities
Supplementary figure 14 Empirical distribution functions for mean normalized read coverage, C, for ENSMUSTs and transcripts with novel retained introns
Supplementary figure 15 Coverage metrics for known and novel retained introns
Supplementary figure 16 Schematic of identifying novel short and long 3’UTRs with EJ- and PAM-reads
Supplementary figure 17 a) PAM-reads identify a novel polyadenylation site in the 3’ UTR of Dmgdh b) PAM-reads and contig alignments identify a novel long 3’UTR for Sult3a1
Supplementary figure 18 Schematic of detecting a fusion gene with a contig alignment
Nature Methods: doi:10.1038/nmeth.1517

Supplementary figure 19 Comparisons of gene-level expression metrics for Trans-ABySS and ALEXA-Seq
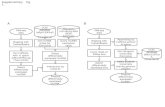
Supplementary figure 20 Overview of the transcriptome assembly and analysis pipeline workflow
Supplementary figure 21 Length-normalized profiles of Burrows-Wheeler Aligner read alignment densities
Supplementary Table 1 Summary of read-to-genome alignments
Supplementary Table 2 Run times for Trans-ABySS, Tophat, Cufflinks and Scripture
Supplementary Table 3 Summary of candidate transcript events that were identified as novel relative to all UCSC, RefSeq, Ensembl and AceView transcript models
Supplementary Table 4 Summary results for identifying annotated and novel polyadenylation sites
Supplementary Note 1 De novo transcriptome assembly
Issues for de novo and reference-based transcriptome assembly
Comparing de novo and reference-based assembly
Detecting novel polyadenylation sites
Identifying fusion genes
Quantifying gene-level expression
Validating novel transcripts and transcript events
WTSS aligned-read pipeline
Generating splice graph visualizations
Nature Methods: doi:10.1038/nmeth.1517

1
Supplementary Figures
Supplementary Figure 1. Schematic of ABySS assembly steps illustrating the origin of main, junction, and bubble contigs, and the manner in which the contig alignments are used for analysis. a) Bubble contig branch pairs (green) typically capture heterozygous SNVs. For each bubble, ABySS writes the higher coverage branch (mid green) into the single end (SE) contig set, and writes the branch pair into the global set of bubble contigs. b) SE contigs are constructed from unambiguous (k-1)-bp overlaps between k-mers. c) Mate pairs identify overlapping contig neighbors, and alternate contig-joining paths may be identified. The shorter, pale blue contig represents a candidate junction contig. Because such a contig typically corresponds of two (k-1) overlaps, it is expected
Nature Methods: doi:10.1038/nmeth.1517

2
to be (2k-2) bp long, in an assembly generated for a k-mer length of k bp. For a given assembly (and so k value), contigs that are at least (2k-2) bp long are expected to be the most informative of transcript structure. Dependent on assembly parameters and the strength of supporting mate pair information, one of the two alternate contigs may be joined to the flanking contigs to construct a longer PE contig; however, it is also possible that neither or both alternative paths will be constructed. d) The path containing the longer alternate contig is constructed, with the shorter contig retained as a junction contig. e) Example of possible outcomes for alignments of main (dark blue), junction (light blue), and bubble pair (light and mid-green) contigs to the reference genome. Comparison of their alignments to that of two transcript isoforms (gray) is shown. The alignment blocks of the main contig support the lower isoform, while the junction contig alignment supports the presence of the upper alternative isoform. The alignment of the bubble contig pair identifies a heterozygous SNV.
Supplementary Figure 2. Assembly properties for k values of 26 to 50. a) Curves show N50 length (the contig length for which the contigs larger than N50 have 50% of the bases of the assembly), the total number of contigs, and the number of contigs longer than 100 bp.
Nature Methods: doi:10.1038/nmeth.1517

3
Supplementary Figure 3. Partitioning of BWA-aligned reads relative to Ensembl v54 transcript models.
Supplementary Figure 4. Fraction of Ensembl transcripts with at least 80% of exon length covered by unmerged contig alignments, as a function of normalized WTSS (Supplementary Note) coverage threshold. Results are shown for the 34,400 Ensembl v54 transcripts (corresponding to 19,508 unique gene IDs) that had a nonzero length-normalized WTSS mean coverage. Curves show results for the single longest contig (blue) and for all contigs (green). For single contigs, 64% and 72% of nonzero-coverage transcripts were covered to at least 80% of the exon length for WTSS coverage thresholds of 10 and 20; considering all contig alignments, the percentages were 88% and 92%.
Nature Methods: doi:10.1038/nmeth.1517

4
Supplementary Figure 5. Assembled contigs across multiple k-values are merged to obtain a non-redundant set of contigs for analysis. a) The contig merging process is shown schematically for eight hypothetical assemblies (k1, k2, …, k8). Contig sets from pairs of assemblies with adjacent k values are reciprocally compared. Those contigs having an exact match to a longer contig from the paired assembly are “buried”. Where contigs are equivalent, the contig from the assembly with the lower k is retained. From the output of this stage, adjacent pairs of contig sets are again merged (e.g. k12 and k34). Merging continues until only one contig set remains. Retained contigs are identified as “parent” contigs. Contigs that are neither buried nor parent are “untouched”. The merging process is applied to both the main and extended junction contigs. See Fig. 1b.
Nature Methods: doi:10.1038/nmeth.1517

5
Supplemental Figure 6. Coverage of Ensembl v54 transcripts by contig alignments, as a function of mean read transcript coverage. Mean transcript read coverage, C, was calculated for each transcript by aligning reads to the NCBI37 reference genome which had been extended by exon-exon junctions, and normalizing the number of aligned reads for a transcript by the sum of exon lengths in the transcript. Distributions are shown for all transcripts with nonzero read-alignment coverage (gray), and for transcripts with de novo contig alignments (Trans-ABySS, for even-k assemblies) or reference-based contigs (Cufflinks, Scripture) representing at least 80% of the total exon length, either considering all contigs for that transcript (red) or the single longest contig (blue).
Nature Methods: doi:10.1038/nmeth.1517

6
Supplementary Figure 7. Splice site support for 149,877 Trans-ABySS parent (v1.1.1) contig alignments, considering GT-AG, GC-AG and AT-AC donor-acceptor types1. An „ss2‟ contig alignment (97.9%) has at least one alignment „intron‟ with both acceptor and donor sites, an „ss1‟ contig alignment (1.8%) have at least one intron with only an acceptor or donor, and an „ss0‟ contig alignment (0.2%) lacks such support.
Nature Methods: doi:10.1038/nmeth.1517

7
Supplementary Figure 8. Schematic of comparing a transcript model (top) with contig alignments to identify annotated and novel transcripts and transcript structures. For each main and extended junction contig we compared coordinates of contig alignment blocks to coordinates of exons in each best-fitting transcript model, considering all mm9 UCSC gene, RefSeq, Ensembl and AceView transcripts. For a „full match‟, edges of all internal blocks and transcript exons match, as do inside edges of the outer or terminal blocks and exons. Because contig ends do not necessarily correlate with transcript ends, outer edges of terminal alignment blocks may not match outer edges of corresponding exons, and so are not considered to represent novel events. A multi-block alignment that matches no known transcript models represents a potential novel transcript (not shown). For schematics for identifying candidate novel short and long 3‟UTRs and candidate fusion genes see Supplementary Figs. 16 and 18.
Nature Methods: doi:10.1038/nmeth.1517

8
Supplementary Figure 9. RT-PCR validation of a 36-bp novel exon prediction in the Insr gene, which was subsequently reported in a shorter full-length RIKEN cDNA clone for adult male testis, in a more recent set of known gene transcript models. a) UCSC genome browser mm9 screenshot showing (top to bottom) Tag-seq data (unpublished), H3K4me3 ChIP-seq data2, exonerate alignments for main contigs, read-alignment pileup, RT-PCR primers (blue arrow) and a range of transcript and other annotations. b,c) Detailed view of the RT-PCR primers on the exons flanking the novel exon. While the pileup coverage is greater than 100 on the flanking exons, the 36-bp novel exon is so much shorter than the 50-bp reads that only two BWA-aligned reads support the novel exon (not shown). d) RT-PCR gel image showing the expected 185-bp product, but not the annotated 149-bp product. e) The approximate alignment coverage for the gene (vertical red line) shown relative to cumulative distributions of transcript coverage for all Ensembl mouse transcripts (gray line) and all contigs whose alignments covered at least 80% of the total exon length of a transcript (see Fig. 1a). The novel exon
Nature Methods: doi:10.1038/nmeth.1517

9
corresponded to 12 amino acids, and overlapped exons in human and rat RefSeq transcript alignments (not shown). All three contigs in the region contain this exon, suggesting that only one isoform is expressed. Despite the gene being relatively highly expressed (read coverage for flanking exons is ~130-fold), the novel exon is shorter than the 50-bp reads, and so has only two reported read alignments. In contrast, read alignments to the assembled contigs indicate a ~90-fold coverage over this detected novel exon (data not shown).
Nature Methods: doi:10.1038/nmeth.1517

10
Supplementary Figure 10. Novel UTR prediction for the Nlrp6 gene. a) UCSC genome browser mm9 screenshot showing (top to bottom) Tag-seq data for the positive and negative strands, an H3K4me3 enrichment profile, exonerate alignments for main contigs, read-alignment pileup, RT-PCR primer positions (blue arrow), and a range of transcript annotations. b,c) Details of the RT-PCR primer locations. d) RT-PCR gel image showing the expected 856-bp product. e) The approximate alignment coverage for the two annotated genes (vertical red lines, ~400 and ~2100) relative to distributions shown in Fig. 1a. The evidence for the detected novel UTR on Nlrp6 includes the following. The main H3K4me3 enrichment signal2 extends across a short UCSC or AceView transcript, while
Nature Methods: doi:10.1038/nmeth.1517

11
weaker H3K4me3 enrichment is consistent with short UCSC and AceView Nlrp6 transcripts. Numerous shorter and particularly longer contigs suggest that the gene model for Nlrp6 is incomplete, and that transcripts extend between this locus and the main enriched H3K4me3 region. Read coverage is approximately 560 for the Nlrp6 transcripts, and higher (approximately 1175) for the upstream transcripts; consistent with this high expression, there is widespread low-level intergenic or (novel) intronic transcription that is reflected in many unspliced contigs. The longest contig exactly reconstructs the ORF part of the RefSeq transcript. The set of contig alignments at the upper left extend ~148kb upstream to a very highly expressed (~6500 pileup) cytochrome P450 Cyp2e1.
Supplementary Figure 11. A novel transcript prediction. a) UCSC genome browser mm9 screenshot showing (top to bottom) exonerate alignments for main contigs, PE reads, a read-alignment pileup, RT-PCR primer positions (blue arrow), a range of transcript annotations and mammalian conservation. b,c) Details of the RT-PCR primer locations. d) RT-PCR gel image showing the expected 264-bp product. e) The approximate alignment coverage for the novel transcript (vertical red line, ~31) relative to distributions shown in Fig. 1a.
Nature Methods: doi:10.1038/nmeth.1517

12
Supplementary Figure 12. Alignments of contigs representing the Shank2 gene support both an RT-PCR-validated 21-bp skipped exon (red arrow), and a novel, H3K4me3-supported2 TSS that is upstream of the 5‟-most Ensembl TSS. a) mm9 UCSC genome browser view of Shank2 showing (top to bottom) Tag-seq data for the positive strand, an H3K4me3 enrichment profile, exonerate alignments for
Nature Methods: doi:10.1038/nmeth.1517

13
main contigs, BWA read-alignment pileup, RT-PCR primer positions (blue arrow), and a range of transcript annotations. b) Detail of RT-PCR primers, with a red arrow indicating the skipped exon. c) Detail of the skipped exon. d) RT-PCR gel, showing the 200-bp annotated and 179-bp novel products. e) The vertical red line shows the approximate read alignment coverage for the gene relative to distributions shown in Fig. 1a.
Supplementary Figure 13. Assembly can generate contigs for exons with low read alignment densities. Sfrs3 is a member of the SR splicing factor family, which has 11 and 10 members in human and mouse, respectively3. In human, SFRS3 shares a splicing pattern with six other family members: a cassette exon that introduces a premature stop codon is skipped in the reference isoform but included in an alternative isoform3. a) For the mouse Sfrs3 shown, exons overlap chained self-alignment blocks. Consistent with this, aligned-read coverage is low on exons flanking the retained intron; however, de novo assembly generates informative contigs. Contig k values and normalized k-mer coverages are consistent with transcripts having a wide range of expression levels (viz. k45:11.2 vs. k31:3.0). A relatively highly expressed 1629-bp k45:11.2 contig is consistent with the RefSeq reference isoform, while k37:14.3 and k33:17.9 contigs show the retained intron. This gene‟s retained intron is one of the three known cases shown as red circles in Supplementary Fig. 15. b) A Sircah4 splice graph representation of the main contig alignments.
Nature Methods: doi:10.1038/nmeth.1517

14
Coverage metrics for known and novel retained introns
Supplementary Figure 14. Empirical distribution functions for mean normalized read coverage, C, for ENSMUSTs and transcripts with novel retained introns. The graph shows 34,400 ENSMUSTs with nonzero coverage (gray), and 181 of the 250 transcripts with novel retained introns (red) that had UCSC gene IDs or ENSMUST IDs. Approximately 75% of transcripts with novel retained introns had mean normalized read coverage that was at or above the 90th percentile coverage for the Ensembl transcripts.
Supplementary Figure 15. Coverage metrics for known and novel retained introns. The axes are the mean read coverage for a retained intron‟s flanking exons, and the ratio of the mean coverage of the retained intron to the mean
Nature Methods: doi:10.1038/nmeth.1517

15
coverage of the flanking exons. Contours summarize 5314 retained introns from the mouse ASTD v1.1 database5. Blue squares show 250 non-redundant novel retained introns from the current work. Lower coverage for the flanking exons and higher intron-to-flanking exon coverage ratios were consistent for three examples of retained introns for SR slicing factor genes, which undergo unproductive splicing as part of a regulatory mechanism6 (red circles, see also Supplementary Fig. 13). Detailed work may prioritize focus on the retained introns that are associated with less highly expressed genes and have larger coverage ratios (upper left quadrant), while those in the lower right quadrant may be less biologically relevant.
Nature Methods: doi:10.1038/nmeth.1517

16
Supplementary Figure 16. Schematic of method for identifying novel short and long 3‟UTRs. a) A cDNA with a poly(A) tail. End-junction (EJ) reads and poly(A)-mate (PAM) reads that were generated from the cDNA are identified from the read sequence file. b) 50-bp sequences were added to 3‟ ends of reference transcript sequences (gray). Contig sequences (blue) are expected to terminate in a poly(A) sequence whose length is less than the assembly k; contig sequences were padded with 50-bp poly(A) sequences on their 3‟ ends and 50-bp poly(T) sequences on their 5‟ ends. c) The fragment length distribution, i.e. the measured insert length for paired end reads, was determined from distances between mate pairs mapped to contigs (shown here for k=38). d) The distribution of the number of T‟s in 147.1 M 50-bp reads. Sequence reads with very high proportions T are likely to belong to cDNA poly(A) tails (right edge of the graph). e) Aligning the transcript-read (short blue rectangles) from EJ and PAM mate-pairs to reference transcript sequences (gray) to confirm annotated 3‟UTR ends (e1) and identify novel short 3‟UTR ends (e2). (e3) Refining estimates of ends of novel long 3‟UTRs by aligning, to contigs (blue), reads that do not map to transcripts.
Nature Methods: doi:10.1038/nmeth.1517

17
Supplementary Figure 17. a) PAM-reads identify approximate known and novel polyadenylation sites in the 3‟ UTR of Dmgdh (Supplementary Fig. 16e1,2). The origin of the insert length distribution (Supplementary Fig. 16c) is located at the left-most edges of signal peaks in the stringent evidence pileup track (second from top), and the shaded rectangles correspond to the width of the peak in the insert length distribution. The predicted novel polyadenylation site (left) is consistent with EST evidence. b) PAM-reads identify three candidate polyadenylation sites in the 3‟UTR of Sult3a1 (Supplementary Fig. 16e3). 3‟UTRs that are longer than annotated 3‟UTRs are supported by contig alignments (horizontal blue bars) and read alignments.
Nature Methods: doi:10.1038/nmeth.1517

18
Supplementary Figure 18. Schematic of detecting a fusion gene. a,b) The contig aligns to two genomic regions. The regions may be on different chromosomes, or on one chromosome but separated by a distance that is much longer than the ~200-bp PE insert length (Supplementary Fig. 16a). The contig breakpoint (a, red line) must be supported by reads that align with no mismatches to the contig and cross the breakpoint. The contig alignments may also have mate-pair support from reads aligned to the EEJ-extended genome (b). Annotated transcripts are shown in gray.
Nature Methods: doi:10.1038/nmeth.1517

19
Supplementary Figure 19. Comparisons of gene-level expression metrics for Trans-ABySS, ALEXA-Seq7 and a whole transcriptome shotgun sequencing (WTSS) pipeline (Supplementary Note). Results are shown for the 8190 Ensembl mouse genes that had fractional gene-level contig-to-exon coverage of at least 0.8. The Pearson‟s correlation coefficient was 0.921.
Nature Methods: doi:10.1038/nmeth.1517

20
Supplementary Figure 20. Overview of the transcriptome assembly and analysis pipeline workflow, outlining the steps from initial transcriptome assembly, contig processing and analysis outcomes. Boxes with rounded corners indicate operations, boxes with square corners represent results and blue boxes represent outcome results. a) When a genome sequence is not available, assembly make contigs available for functional or phylogenetic analyses by methods that are not part of the Trans-ABySS pipeline. b) When a genome sequence is available but gene models have not been annotated, contig alignments to the genome can identify a range of transcript structures, as well as chimeric transcripts and variants like indels and SNVs. c) When transcript models are available for comparison to contig alignments, models can be refined and updated to include transcript variants.
Nature Methods: doi:10.1038/nmeth.1517

21
Supplementary Figure 21. Length-normalized profiles of BWA read alignment densities, showing 20th, 50th and 80th quantiles.
Nature Methods: doi:10.1038/nmeth.1517

22
Supplementary tables Supplementary Table 1. Summary of read alignments for 147.1M 50-bp paired end (PE) Illumina reads (7.36Gb). We retained only aligned reads that had a MAQ mapping quality ≥10; these had unique genomic alignment positions and few mismatches to the mm9 reference genome sequence or constructed exon-exon junction sequences. Junctions were constructed for consecutive exons from UCSC, RefSeq, Ensembl and AceView transcripts. Read counts relative to genes were calculated using Ensembl v54. Percentages in “MAPQ filter” columns are relative to “Total mapped” numbers, and those in “Aligned to” columns are relative to the number of retained read sequences.
Total mapped
MAPQ filter Aligned to
Filtered Retained Exons/EEJ Introns Intergenic
# reads 136,685,932 17,999,164 (13.17%)
118,686,768 (86.83%)
91,935,338 (77.46%)
2,901,894 (2.45%)
7,678,810 (6.47%)
Gb 6.83 0.90 5.93 4.60 0.15 0.38
Supplementary Table 2. Run times.
Trans-ABySS
Assembly Using ABySS 1.2.1, assemblies for k=26 to 50 completed in 4.7 hours of wallclock time and 370 CPU-hours using 25 machines, each of which had 8 hyperthreaded cores in two Intel E5540 2.53 GHz CPUs, and 16 GB of RAM.
Analysis Merging a total of 22 million contigs across 25 assemblies completed in about 5-6 hours. Blat alignments completed in about 10-15 minutes of wallclock time per 1000 contigs. Exonerate alignments completed in about 100 minutes of wallclock time per 1000 contigs. Novelty detection completed in about 5-6 hours wallclock time for 1.2 million alignments.
Tophat/Cufflinks/Scripture
Tophat This was run as 8 parallel jobs (one per lane of data), each of which took an average of 6.75 hours. Time to sort, sam2bam, merge, was about 4 hours total CPU time. Total Tophat run time: ~60 CPU hours, which was required for both Cufflinks and Scripture.
Cufflinks 1 job, 12 CPU hours
Scripture 24 jobs, ~30 minutes each on average: 12 CPU hours.
Nature Methods: doi:10.1038/nmeth.1517

23
Supplementary Table 3. Summary of candidate transcript events that were identified as novel relative to all UCSC, RefSeq, Ensembl and AceView transcript models.
Event type
Contigs with
eventsa
Unique contig
eventsb Genes
affected
Novel exons 123 94 88
Novel skipped exons 111 117 87
Novel introns 62 56 56
Alternative exon splicing 220 184 160
Novel UTRs 133 122 78
Retained introns 335 293 209
Novel transcripts 65 65 65
Novel polyadenylation sites 84 84 84
aTotal number of contigs containing novel events relative to annotated transcript models. In some cases multiple contigs identify the same event.
bThe number of unique genomic locations represented by the contig events. These identify unique transcript events.
Nature Methods: doi:10.1038/nmeth.1517

24
Supplementary Table 4. Summary results for identifying annotated and novel polyadenylation sites. EJ-reads and PAM-reads were mapped to NCBI37 (mm9) UCSC8, RefSeq9, Ensembl10 and AceView11 transcript models, and to GenBank12 mRNAs. a) EJ-read mappings
EJ-reads that mapped to transcript models
EJ-reads that did not map to transcript models
Reads All transcripts Reads Contigs >50 bp (novel short) 6,505 >50 bp 13,016 <= 50 bp (known) 11,060 <= 50 bp 5,221 Unmapped 200,676 Unmapped 182,439 Total 218,242 Total 200,676
b) PAM-read mappings
PAM-reads that mapped to transcript models
PAM-reads that did not map to transcript models
Reads All transcripts Reads Contigs >300 bp (novel short) 4,424 >300 bp 327 <= 300 bp (known) 34,699 <= 300 bp 2,243 Unmapped 10,240 Unmapped 7,670 Total 49,363 Total 10,240
c) Transcripts identified by EJ-reads
Filter Known Novel short
Novel long Total
All transcripts mapped by EJ-reads Na 4,667 8,885 13,552
Novel short (>50 bp), novel long (<=50 bp) 2,774 2,664 2,807 5,471 Mate read maps within range on same
transcript 2,225 1,952 956 2,908
Stretch of T prefix > 10 bp of read 338 255 133 388
Transcripts with at least 2 EJ-reads of support 71 36 22 58
d) Transcripts identified by PAM-reads
Filter Known Novel short
Novel long Total
All transcripts with mapped PAM-reads na 7,496 1,069 8,565
Novel short (>300 bp), novel long (<=300 bp) 6,672 1,478 972 2,450
Has at least 1 PAM-read with a 49/50 T mate 2800 191 92 283 Filtered for high AT content (80%) and
antisense 2,110 92 92 184 Transcripts with at least 2 PAM-reads of
support 1377 30 22 52
Filtered for reads with genomic mapping 1273 28 19 47
Manually reviewed 20 10 30
Nature Methods: doi:10.1038/nmeth.1517

25
Supplementary Note
De novo transcriptome assembly Non-normalized transcriptome shotgun libraries differ from whole genome shotgun libraries in presenting a very wide range of sequence representations to an assembler. We address expression level differences by using a wide range of k values to assemble contigs that represent cDNAs, then merging the contig lists from independent assemblies into a smaller set of meta-assembly contigs for analysis. Transcriptome shotgun libraries also differ from whole genome shotgun libraries in that many genes express multiple transcript isoforms, and so present multiple correct, overlapping paths to an assembler. In contrast, in genome assembly, a single „correct‟ assembly path is expected through any genomic region, with the exception of repetitive and duplicated sequences and those representing haplotypic variation or mutational alterations. ABySS captures single nucleotide variation within a sample as pairs of short sequences, which are referred to as „bubble‟ contigs (Supplementary Fig. 1). The variant with the highest coverage is represented in the assembled contigs, but both variants are written out to a separate file as a bubble contig pair that can be analyzed independently to identify allelic variation within the sample and SNVs relative to known variants. ABySS typically handles heterozygous indel variants by creating a pair of short contigs for each variant in the initial assembly stages (Supplementary Fig. 1b,c). The contig representing a deletion variant is usually comprised of sequences of length k-1 flanking the insertion point, and thus is characteristically (2k-2) bp in length. The contig representing an insertion variant is comprised of the same (2k-2) bp sequence, plus the additional sequence representing the insertion, and is therefore somewhat longer than the (2k-2) bp deletion variant. We refer to these contigs as „junction‟ contigs. Depending on assembly parameters, individual junction contigs may or may not be incorporated into longer contigs in later stages of the assembly (Supplementary Fig. 1d) (see Methods). As we reported previously13, in transcriptome assembly these junction contigs also capture exon content differences between transcript isoforms. While results for SNVs and indels are not reported here, our pipeline therefore includes methods for bubble and junction contigs. Given the above considerations, the Trans-ABySS workflow consists of the following stages: 1) assembling reads into contigs using ABySS, 2) aligning contigs to the reference genome, and 3) analyzing the contig alignments to correlate with known transcript annotations and to identify SNVs, indels, novel transcripts and transcript structures, and gene rearrangements and fusions.
From each assembly, we considered all contigs of length L (2k-2) bp, and all bubble contigs; summed across all assemblies, there were 9.5 M of the former
and 346,787 of the latter. To reduce the number of L (2k-2) bp contigs analyzed, while maintaining the transcript representation provided by all
Nature Methods: doi:10.1038/nmeth.1517

26
assemblies, we merged the assemblies by removing („burying‟) contigs that were redundant because they were exactly represented within longer („parent‟) contigs in another assembly. To accomplish this, we iteratively and reciprocally aligned contigs between pairs of assemblies, removing redundant contigs at each round (Supplementary Fig. 5). The iterative burying process returned a set of 1,200,130 non-redundant contigs (Fig. 1b), which we refer to as the „main‟ contig set (Supplementary Fig. 20). Preliminary analysis showed that a junction contig shorter than (2k-2) bp can be assembled when there are short homologous sequences on either side of the junction. To ensure that such contigs were included in our dataset for analysis, we identified contigs with length L < (2k-2) bp for which mate pair information indicated overlap with a single candidate contig „neighbor‟ at each end. To support robust genome alignments for these small contigs, we extended them by adding their two neighboring contig sequences. We refer to these as „extended junction‟ contigs (Supplementary Figs. 1, 20). Subsequent merging reduced the 96,019 extended junction contigs across all assemblies to 16,287 contigs for analysis. Alignments of main and extended junction contigs were compared to structures of known transcript models in order to identify novel transcripts and alternative transcript structures. Alignments for all contigs were used to identify SNVs and indels relative to the genome (data not shown), and candidate fusion genes were identified from the main contig and extended junction contig alignments (Supplementary Fig. 20).
Issues for de novo and reference-based transcriptome assembly A number of issues pose challenges to both reference-based and de novo assembly approaches. First, the library protocol that we used generated double-stranded cDNA, and so did not retain the strand of the original transcript. While for spliced contig alignments we inferred the strand of the source transcript from the splice sites in the contig alignments, for some cases confirmation would require orthogonal evidence. It is likely that directional library protocols currently under development will reduce the complexity of such analysis14. Second, while a de novo approach can be robust to sequence similarity between exons, shared sequences that are highly similar will halt contig extension, with repetitive regions assembling into separate contigs, each of which aligns to multiple locations. Third, aligned-read densities are non-uniform along exons due to multi-mapping and other technical biases15-17. Fourth, isoform reconstruction remains problematic for genes that have multiple expressed isoforms. Although suggested transcript models have been reported for both de novo and reference-based assembly algorithms, complex alternative isoforms cannot be reconstructed reliably, due to short read lengths and short fragment lengths for paired end reads. Also, attempts to use expression levels in inference fail due to both theoretical (under-, over- or ill-defined linear mathematical models) and
Nature Methods: doi:10.1038/nmeth.1517

27
practical (3‟/5‟ sequence bias, Supplementary Fig. 21) obstacles. Unless one is supplied with reads that associate longer lengths across transcripts, assembly methods can at best report splice diagrams for genes with alternative isoforms.
Comparing de novo and reference-based assembly We ran TopHat18 1.0.13 Beta on each of the eight lanes of data separately, then sorted and joined the output .bam-format19 files into a single merged file, which we used as input into Cufflinks20 0.8.3 Beta (02 July 2010), and Scripture21 Beta (22 June 2010). For our TopHat analysis we generated the intron result set by merging the resulting BED-format files from each lane, and accumulating scores for identical introns. Unique introns for the other three tools were generated from exonerate alignments for Trans-ABySS contigs, BED files for Scripture contigs, and GTF files for Cufflinks contigs. We then compared the predicted splice sites to the unique coordinates of all the donor-acceptor pairs in the reference annotations, which corresponded to all nonredundant introns for the union of UCSC, RefSeq, Ensembl and AceView transcript models. A splice site was only considered to match between datasets if the coordinates of the donor-acceptor pair matched exactly. Supplementary Table 2 outlines run times.
TopHat identified alignments for 145,798,588 (78.8%) of 184,915,546 reads. Of the aligned reads, 592,864 (0.4%) were gapped or split alignments; these identified 141,846 unique dinucleotide splice sites, which we compared against the unique coordinates of all the donor-acceptor pairs in UCSC, RefSeq, Ensembl and AceView gene annotations. Methods that use split read alignments may have difficulty in detecting exons that are shorter than the read length, particularly when 50-bp reads are used. For TopHat, every detected splice junction is required to be supported by at least one read that “anchors” by a user defined minimum length on either side of a split. This makes it insensitive to exons shorter than the anchor length, but also less sensitive for relatively short exons, especially when these are in isoforms that are weakly expressed. Consequently, using the TopHat spliced read alignments as input, we observed that Cufflinks was strongly biased against detecting shorter exons. To estimate performance differences between contig alignments and spliced read alignments more directly, we compared dinucleotide splice sites detected by Trans-ABySS and TopHat using the splice sites in UCSC gene transcripts as our reference set. We included TopHat because, although the „assembly‟ of exons is deferred to the Cufflinks software, the splice sites are reported by TopHat. Fig. 2 compares sensitivity (SN) and specificity (SP), relative to the reference junctions, are approximate metrics for this comparison. The SN reported is the fraction of all unique splice sites that are detected in the UCSC, RefSeq, Ensembl and AceView transcript models. SN, as reported, is an underestimate,
Nature Methods: doi:10.1038/nmeth.1517

28
because it includes splice sites from unexpressed transcripts. The SP reported is the ratio of the number of reference introns to the total number of introns detected. It too is an underestimate, because apparently non-specific predictions include not only false positives, but also true positive exon-exon junctions that are novel relative to the reference intron set.
Detecting novel polyadenylation sites Alternative polyadenylation sites can affect mRNA stability, translocation and translation22. For fission yeast, polyadenylation sites have been identified from single-end read RNA-seq data through reads that aligned at junctions of transcripts and poly(A) tails (end-junction or EJ reads)23. In a transcriptome assembly, a contig representing a polyadenylated transcript should terminate in a homopolymer-A sequence whose length approaches k. In our study, the read length was 50 bp, while the merged contig set included contigs from assemblies with 26≤k≤50. Given this, we expect that terminal poly(A) sequences for merged contigs will be shorter than the read length, which could interfere with the EJ-read alignments. We addressed this by adding 50-bp poly(A) and poly(T) sequences to 3‟ and 5‟ ends of each contig, respectively. Similarly, we added 50-bp Poly(A) sequences to the 3‟ end of each reference (e.g. RefSeq) mRNA sequence (Supplementary Fig. 16). Contigs that are downstream of such a transcript contig in the de Bruijn graph represent the poly(A) tail, but are not incorporated into any particular transcript contig due to the difficulty of assembling simple sequence. Here, as an initial step towards a future graph-based analysis, we identified and annotated novel polyadenylation sites using end-junction (EJ-) and mate-pair (PAM-) reads in paired-end sequence data (Supplementary Fig. 16). An EJ-read spanned a poly(A) start site23; a PAM-read had one mate mapped to a poly(A) tail, while its mate mapped either to an annotated transcript or to a contig sequence.
We identified candidate EJ-reads spanning poly(A) start sites as reads whose sequence was prefixed by poly(T) runs that were at least 5 bp long. We identified candidate PAM-reads as those in which the mate‟s sequence contained 80% to 98% (40 to 49 of 50 nt) of T‟s. We used BWA24 v0.5.4 to map candidate EJ-reads and PAM-reads to known transcripts annotations from UCSC, RefSeq, Ensembl, AceView, and to Genbank mRNAs. Files for all of these were downloaded from the UCSC mm9 genome browser25. To identify transcripts with candidate novel short 3‟ UTRs, we used the length distribution for PE reads and the distance from each PAM-read to the end of each transcript (Supplemental Fig. 16b,c and Supplemental Table 4). Specifically, we considered that mapping distances longer than 50 bp for EJ-reads, and 300 bp for PAM-reads from a transcript to mark such cases.
Nature Methods: doi:10.1038/nmeth.1517

29
To identify candidate novel long 3‟UTRs, all EJ-reads and PAM-reads that did not align to annotated transcript sequences were mapped with BWA to ABySS contig sequences. We identified contigs that had EJ-reads mapped to the ends and PAM-reads mapped within 300 bp from a contig end, and mapped the contigs to the mouse mm9 genome to determine the transcript product with the novel elongated 3‟UTR. In such cases the contig alignment already suggested the extended 3‟UTR, and the PAM-reads refined the estimate of the position of the end of the UTR. We then filtered candidate polyadenylation sites, as follows. For shortening and lengthening cases using EJ-reads, we required EJ-reads to satisfy two conditions: that they map to the genome or to transcripts only when their poly(T) prefix or poly(A) suffix had been trimmed; and that their mate pair map 150-300 bp from the opposite strand of the same transcript. We ranked mapping positions of a read, prioritizing positions with the fewest mismatches and then the shortest distance to a transcript end. We then required at least two reads to map to each position. Transcripts from the four annotated sets used were resolved to gene symbols when possible. For both shortening and lengthening cases using PAM-reads, we required that at least one of these had at least 49 T‟s out of 50 bases in the poly(A) tail read. When a PAM-read mapped to more than one genomic location, we ranked mapping positions in the same way as for EJ-reads. To reduce the number of false positives, we rejected transcripts that had one or more 50-bp windows in which 80% of the bases were A or T. We then required at least two reads to map to each position. For the 218,242 potential EJ-reads, requiring at least two reads of support for each transcript event and comparing events to four sets of transcript annotations and to Genbank mRNAs, we confirmed 71 annotated 3‟ UTRs ends, as well as 36 novel short UTRs. Mapping the unmapped reads to ABySS contigs then identified 22 novel long UTRs (Supplementary Table 4a,c). For the 49,363 PAM-reads, 39,123 mapped to the transcript models and Genbank mRNAs. By requiring at least two PAM reads for each event, we confirmed 1277 annotated 3' transcript ends, as well as 20 transcripts with novel short 3‟ UTRs (Supplementary Fig. 17). Mapping the unmapped reads to contigs then identified 10 transcripts with novel long UTRs (Supplementary Table 4b,d). By combining EJ- and PAM-read singletons, we also confirmed 9 annotated UTRs as well as 6 novel short UTRs. Overall, we confirmed polyadenylation start sites in 1299 annotated transcripts, inferred 84 novel polyadenylation sites that corresponded to 56 novel short 3'UTRs and, from contig alignments, 32 novel long 3‟UTRs (Supplementary Table 4, Supplementary Fig. 17). Relatively few novel events were predicted by both methods; in almost all cases a novel event was predicted by only one of the two methods.
Nature Methods: doi:10.1038/nmeth.1517

30
Identifying fusion genes To identify candidate contigs spanning gene fusion breakpoints we apply filters to identify contigs that aligned discretely to distinct genomic regions using BLAT (Supplementary Fig. 18). We parse the top-scoring five alignments and perform the corresponding 10 pairwise comparisons. Initially, we discard any contig that
contained a single alignment that represented 95% of the contig length, as any candidate fusions generated from the relatively short remaining part of the contig were marked as likely to be false positives. Alignments are subsequently filtered for quality by requiring that alignment identity be at least 95%. To ensure that the entire contig was represented in the alignments and to minimize overlap between
alignment pairs, we require that 95% of the entire contig length be covered by the alignments, and that no more than 5% of the contig bases, and none of the reference bases, be shared between alignments. We then filter all candidate fusion alignments. We discard alignments that align to mtDNA or haplotype reference sequences. We reject candidate fusion contigs that are reported as a fusion candidate multiple times. Contig alignments that overlap RepeatMasker RNA repeat elements are also rejected, as are contigs that have fewer than two Bowtie read alignments spanning the candidate breakpoint (Supplementary Fig. 20a). As a final piece of confirmatory evidence we require that the contig alignments be supported by mate-pairs aligned to the EEJ-extended reference genome and that the number of such supporting mate-pairs be within an acceptable range [4, 2000] (Supplementary Fig. 20b).
Quantifying gene-level expression The Trans-ABySS pipeline includes a general method for determining a contig-based expression metric for gene loci, given a reference genome with transcript annotations. The approach considers reads aligned to all contigs whose alignment blocks on a reference genome overlap with exons in transcript model annotations. For Ensembl v54 genes, we compare the expression levels predicted by this approach with those from two methods that align reads to a reference genome that has transcript annotations. The first method was ALEXA-Seq7, whose expression values agree well with those from microarrays and qPCR. The second was a WTSS (whole transcriptome shotgun sequencing, i.e. RNA-Seq) pipeline that extends reference chromosome sequences with exon-exon junction sequences and is used for production-level analysis at the GSC (unpublished). For the 8190 genes with fractional contig-to-exonic coverage of at least 0.8, the expression levels for the two read-alignment methods were highly correlated, with a Pearson‟s coefficient of r2=0.998. Correlation coefficients between Trans-ABySS and ALEXA-seq and the WTSS pipeline were 0.921 and 0.888 respectively.
Nature Methods: doi:10.1038/nmeth.1517

31
Validating novel transcripts and transcript events We generated 50 µL of double stranded cDNA by reverse transcribing 0.2 µg of DNAase-treated RNA from a biological replicate (see Library construction and sequencing, above). We used 1.5 µL of cDNA for each RT-PCR reaction. Primers were designed with PrimerQuest from IDT SciTools26, BatchPrimer327, or Visual OMP (DNA Software, Ann Arbor MI). Each primer pair was checked against the UCSC mouse mm9 assembly to confirm expected RT-PCR products. The following PCR cycle was repeated 40 times: 95oC for 30 min, 53oC for 30 min, and 72oC for 60 min. For Csnk2a2, Fbrs, Foxn2, Kynu, novel transcripts 'Event 17', and 'Event 18', primers were hybridized at 55oC and the reaction was run for 35 cycles. RT-PCR products were resolved on a 1.8% agarose gel. Product sizes for bands were estimated by a custom Matlab (Mathworks, Natick, MA) program that read an image file corresponding to a gel and text file specifying ladder fragment sizes and expected mobilities (Supplemental Note). Product sizes for bands were estimated by a custom Matlab (Mathworks, Natick, MA) program that read an image file corresponding to a gel and text file specifying ladder fragment sizes and expected mobilities (Supplemental Note). The user participated in lane tracking, and, because the shape information for the ladder bands is used for de-noising sample lanes, manually confirmed the automatically identified ladder bands. The user then set a minimum threshold brightness for detecting bands. The program analyzed each sample lane, automatically identifying bands as local profile maxima, calculating a relative profile height at each maximum as an intensity metric, and assigning a product size to each maximum by linearly interpolating a size versus mobility relationship between the ladders. When a peak was saturated by an abundant product, the product size was estimated as the center of the plateau.
WTSS aligned-read pipeline Using a whole shotgun transcriptome sequencing pipeline (WTSS, unpublished), we constructed a sequence resource by extending the NCBI37 reference genome with a pool of non-redundant exon-exon junction sequences. The junction sequences were constructed using Ensembl10, UCSC gene8, RefSeq9, AceView11, and Genscan28 transcript annotations from the UCSC genome browser25, by concatenating (read length-1) nucleotides from each side of each pair of consecutive exons for each transcript, and then eliminating redundant junctions from the pooled set. We aligned the PE reads to the sequence resource using BWA24 v0.5.4, and manipulated the output .bam-format19 file to assign reads that had aligned to exon-exon junctions to their absolute genomic positions. Coverage for Ensembl v54 genes was calculated using the subset of mapped reads that had a mapping quality of at least 10. UCSC wig-format and then bigwig-format files were
Nature Methods: doi:10.1038/nmeth.1517

32
generated using SAMtools, Unix scripts and the UCSC wigToBigWig application, again removing reads with a MAQ mapping quality lower than 10. We determined length-normalized read density profiles along transcripts, from the BWA-aligned .bam file, using custom Java software (Supplementary Fig. 5).
Generating splice graph visualizations Trans-ABySS contigs were aligned to the NCBI37/mm9 assembly using GMAP29, and results were written out in GFF3 EST_match format. Sircah4 was used to associate the contig alignments with genes using annotated gene start and end coordinates, and to draw a splicing diagram for each gene of interest (Supplementary Figure 13).
References 1. Burset, M., Seledtsov, I.A., and Solovyev, V.V., Nucleic Acids Res 28 (21), 4364-4375 (2000). 2. Robertson, A.G. et al., Genome Res 18 (12), 1906-1917 (2008). 3. Lareau, L.F. et al., Nature 446 (7138), 926-929 (2007). 4. Harrington, E.D. and Bork, P., Bioinformatics 24 (17), 1959-1960 (2008). 5. Koscielny, G. et al., Genomics 93 (3), 213-220 (2009). 6. Lareau, L.F. et al., Adv Exp Med Biol 623, 190-211 (2007). 7. Griffith, M. et al., Nature Methods [Epub ahead of print] (2010). 8. Hsu, F. et al., Bioinformatics 22 (9), 1036-1046 (2006). 9. Pruitt, K.D., Tatusova, T., and Maglott, D.R., Nucleic Acids Res 35 (Database issue), D61-65 (2007). 10. Hubbard, T.J. et al., Nucleic Acids Res 37 (Database issue), D690-697 (2009). 11. Thierry-Mieg, D. and Thierry-Mieg, J., Genome Biol 7 Suppl 1, S12 11-14 (2006). 12. Benson, D.A. et al., Nucleic Acids Res 38 (Database issue), D46-51 (2010). 13. Birol, I. et al., Bioinformatics 25 (21), 2872-2877 (2009). 14. Parkhomchuk, D. et al., Nucleic Acids Res 37 (18), e123 (2009). 15. Degner, J.F. et al., Bioinformatics 25 (24), 3207-3212 (2009). 16. Hansen, K.D., Brenner, S.E., and Dudoit, S., Nucleic Acids Res 38 (12), e131 (2010). 17. Li, J., Jiang, H., and Wong, W.H., Genome Biol 11 (5), R50 (2010). 18. Trapnell, C., Pachter, L., and Salzberg, S.L., Bioinformatics 25 (9), 1105-1111 (2009). 19. Li, H. et al., Bioinformatics 25 (16), 2078-2079 (2009). 20. Trapnell, C. et al., Nat Biotechnol 28 (5), 511-515 (2010). 21. Guttman, M. et al., Nat Biotechnol 28 (5), 503-510 (2010).
Nature Methods: doi:10.1038/nmeth.1517

33
22. Millevoi, S. and Vagner, S., Nucleic Acids Res 38 (9), 2757-2774 (2009). 23. Nagalakshmi, U. et al., Science 320 (5881), 1344-1349 (2008). 24. Li, H. and Durbin, R., Bioinformatics 25 (14), 1754-1760 (2009). 25. Rhead, B. et al., Nucleic Acids Res 38 (Database issue), D613-619 (2010). 26. Owczarzy, R. et al., Nucleic Acids Res 36 (Web Server issue), W163-169 (2008). 27. You, F.M. et al., BMC Bioinformatics 9, 253 (2008). 28. Burge, C. and Karlin, S., J Mol Biol 268 (1), 78-94 (1997). 29. Wu, T.D. and Watanabe, C.K., Bioinformatics 21 (9), 1859-1875 (2005).
Nature Methods: doi:10.1038/nmeth.1517