cystic fibrosis
51
CYSTIC FIBROSIS Dr. Gunjan Malviya
-
Upload
gunjan-malviya -
Category
Health & Medicine
-
view
205 -
download
1
Transcript of cystic fibrosis

CYSTIC FIBROSIS
Dr. Gunjan Malviya

INTRODUCTION
cystic fibrosis is a disorder of ion transport in epithelial cells that affects fluid secretion in exocrine gland and epithelial lining of respiratory, gastrointestinal and reproduction tract.

PATHOPHYSIOLOGY

The Cystic Fibrosis Associated Gene
In normal duct epithelia, chloride is transported by plasma membrane channels(chloride channel).
The primary defect in cystic fibrosis, result from abnormal function of an epithelial chloride channel protein encoded by the cystic fibrosis transmembrane conductance regulator(CFTR) gene on chromosome 7.
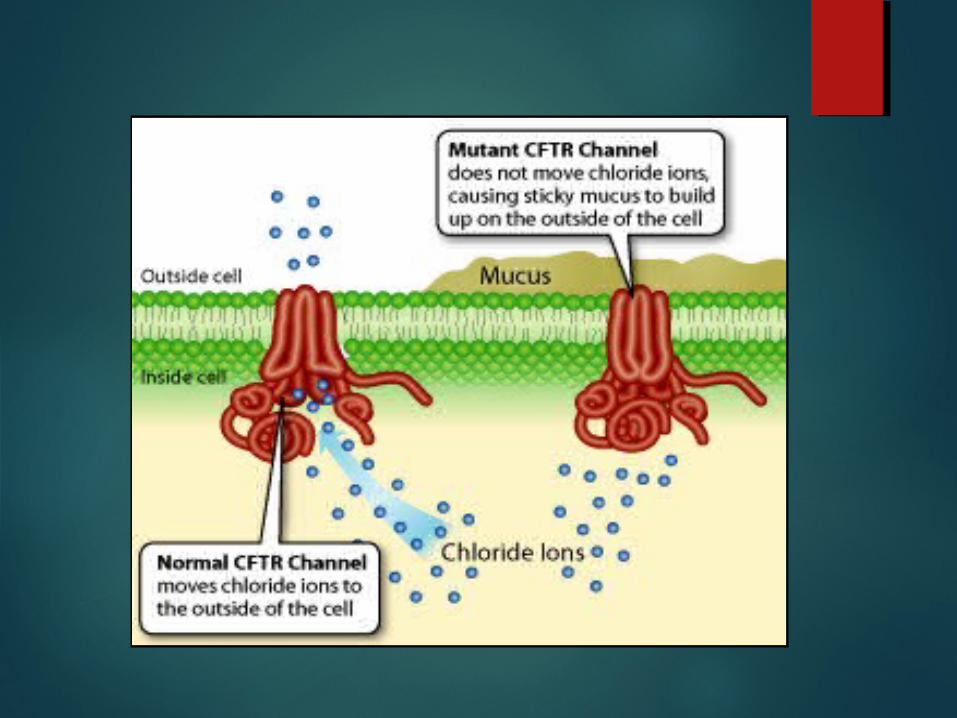

CFTR also regulates epithelial sodium channel
The ENaC(epithelial sodium channel) is situated on the apical surface of exocrine epithelial cells and is responsible for sodium uptake from luminal fluid, rendering it (luminal fluid) hypotonic.
The ENac is inhibited by normally functioning CFTR
In cystic fibrosis ENaC activity increases, markedly augmenting sodium uptake across the apical membrane.

PATHOGENESIS
CF gene present on 7 th chromosome
CF gene codes for CFTR protein
CFTR is expressed largely in epithelial cells of airway, GIT tract, sweat glands and genitourinary system
it functions as chloride channel
dysfunction of CFTR leads to inability to secrete chloride ions,excessive amount of sodium absorption and water retention.
dysfunction of CFTR, limits ability to secrete chloride and absorb sodium in excessive amount, limiting the water available to hydrate secretions.

The one exception to this rule happens to be the human sweat duct, where ENaC activity decreases as a result of CFTR mutation therefore, a hypertonic luminal fluid containing both high sweat chloride and high sodium.
This is the basis for salty sweat the mothers can often detect in their affected infants.

CFTR mediates transport of bicarbonate ions
CFTR dysfunction result in an altered microenvironment with low bicarbonate ions.
alkaline fluid are secreted by normal tissue, while acidic fluids (due to absence of bicarbonate ions) are secreted by epithelia harboring these mutant CFTR alleles.
The decrease luminal PH can lead to a variety of adverse effects such as increase mucin precipitation and plugging of ducts and increased binding of bacteria to plugging mucins.
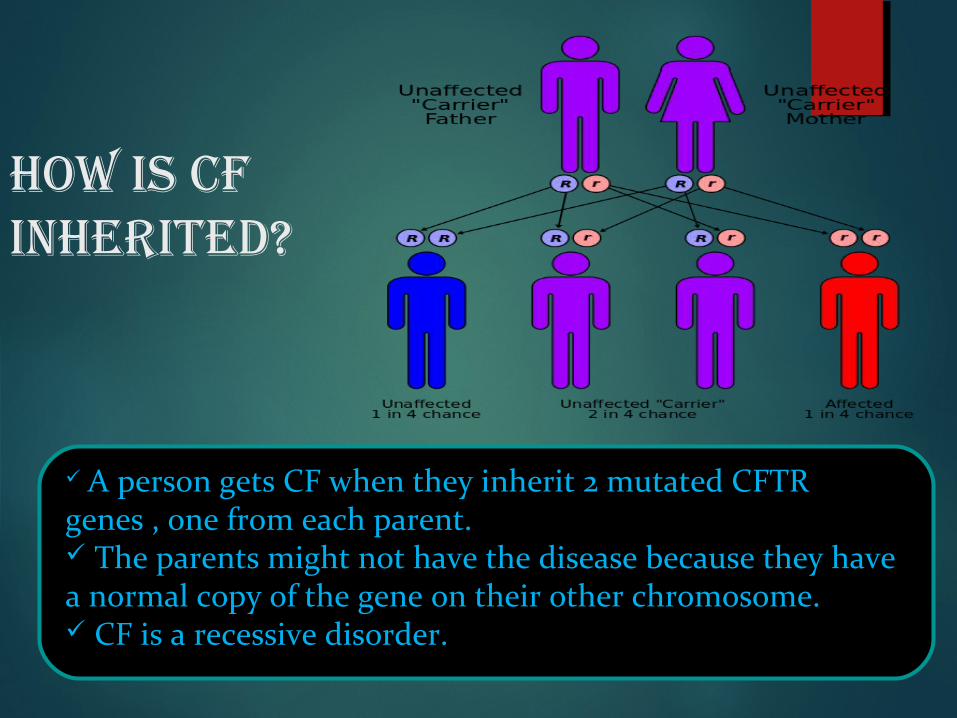
How is CF inHerited?
A person gets CF when they inherit 2 mutated CFTR genes , one from each parent. The parents might not have the disease because they have a normal copy of the gene on their other chromosome. CF is a recessive disorder.
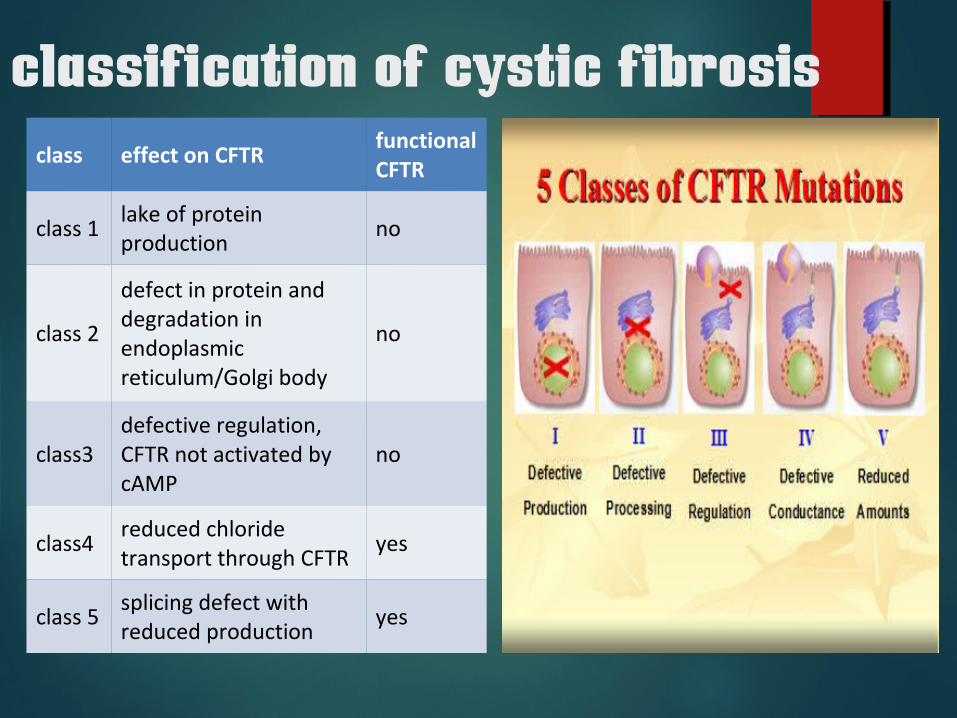
classification of cystic fibrosisclass effect on CFTR
functional CFTR
class 1lake of protein production
no
class 2
defect in protein and degradation in endoplasmic reticulum/Golgi body
no
class3 defective regulation, CFTR not activated by cAMP
no
class4reduced chloride transport through CFTR
yes
class 5splicing defect with reduced production
yes

classes 1 to 3 mutations are considered severe
class 4 and 5 mutations- mild.

clinical manifestation
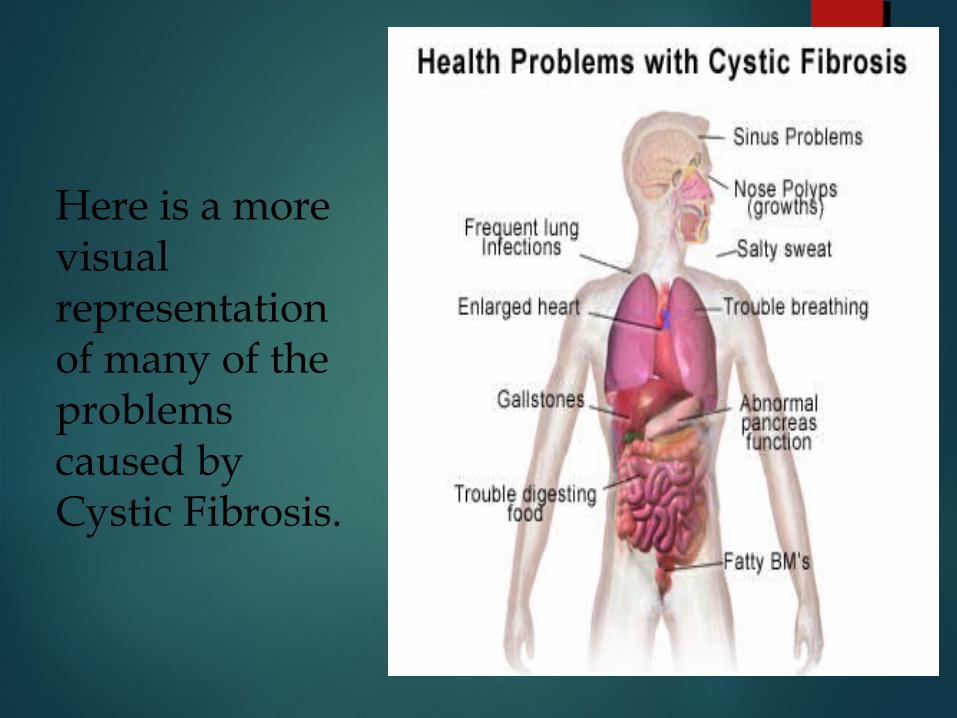
Here is a more visual representation of many of the problems caused by Cystic Fibrosis.

Respiratory Symptoms
Chronic Cough
Wheezing and Breathlessness
Repeated lung infections
A stuffy nose

In Lung
Lung disease results from clogging of the airways due to mucus build-up, decreased mucociliary clearance, and resulting inflammation.
Inflammation and infection cause injury and structural changes to the lungs, leading to a variety of symptoms. In the early stages, coughing, copious sputum production, and decreased ability to exercise are common.
later stages, changes in the architecture of the lung, such as pathology in the major airways (bronchiectasis), further exacerbate difficulties in breathing. Other symptoms include hemoptysis, pulmonary hypertension, heart failure, difficulties getting enough oxygen to the body (hypoxia), and respiratory failure

Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa are the three most common organisms causing lung infections in Cystic fibrosis patients.
In addition to typical bacterial infections, people with Cystic fibrosis more commonly develop other types of lung disease. Among these is allergic bronchopulmonary aspergillosis, in which the body's response to the common fungus Aspergillus fumigatus causes worsening of breathing problems.
respiratory complications are the most common cause of death (~80%) in patients in cystic fibrosis.

pathology
bronchiolitis -the earliest pathologic lesion in lung is bronchiolitis (mucous plagging and an inflammatory response in walls of small airway)
bronchitis-with time mucus accumulation and inflammation extend to large airway.
bronchiectasis- with long standing disease, evidence of airway destruction such as bronchiectasis.
emphysematous bullae are frequent with advance lung disease. These enlarged air space may rupture and cause pneumothorax
hemoptysis - bronchial arteries are enlarged and tortuous, contributing to propensity for hemoptysis in bronchiectatic airway
RESPIRATORY TRACT

Sinuses
Mucus in the para nasal sinuses is equally thick and may also cause blockage of the sinus passages, leading to infection. This may cause facial pain, fever, nasal drainage, and headaches. Individuals with cystic fibrosis may develop overgrowth of the nasal tissue (nasal polyps) due to inflammation from chronic sinus infections. Recurrent sinonasal polyps can occur in as many as 10% to 25% of Cystic fibrosis patients. These polyps can block the nasal passages and increase breathing difficulties.

Pancreas As a part of normal digestion, HCL is
neutralized by pancreatic bicarbonate, leading to the optimal pH for pancreatic enzyme action. Reduced bicarbonate secretion in response to secretin stimulation has been demonstrated in patients with Cystic fibrosis .
Reduced bicarbonate secretion affects the digestion so that exogenous pancreatic enzymes can not work at their optimal pH.

Other factors, such as reduction of water content of secretions, precipitation of proteins, and plugging of ductules and acini, prevent the pancreatic enzymes from reaching the gut.
Autodigestion of the pancreas occasionally leads to pancreatitis. Most patients with Cystic fibrosis (90-95%) have pancreatic enzyme insufficiency and present with digestive symptoms and/or failure to thrive early in life.

Patients with pancreatic enzymes insufficience typically present with poor weight gain in association with frequent stools that are malodorous, and colicky pain after feeding.
pancreatic enzymes insufficience predisposes patients to poor absorption of fat-soluble vitamins A, D, E, and K.

Intestine Defects in CFTR lead to reduced chloride secretion
with water following into the gut. This may result in meconium ileus at birth and in distal intestinal obstruction syndrome (DIOS) later in life.
The pancreatic insufficiency (PI) decreases the absorption of intestinal contents. Mechanical problems associated with inflammation, scarring, and strictures may predispose the patient to sludging of intestinal contents, leading to intestinal obstruction by fecal impaction or to intussusception. Adhesions may form, leading to complete obstruction.

Liver
Absence of functional CFTR in epithelial cells lining the biliary ductules leads to reduced secretion of chloride and reduction in passive transport of water and chloride, resulting in increased viscosity of bile. The biliary ductules may be plugged with secretions. If this process is extensive, obstructive cirrhosis complicated by esophageal varices, splenomegaly, and hypersplenism may occur.

Gallstones are more prevalent in patients with CF than in age-matched control subjects. As many as 15% of young adults with CF have gallstones, irrespective of the status of the pancreatic function.
Abnormal mucin in the gallbladder and malabsorption of bile acids in a patient with PI result in a higher frequency of gallstones.

Genitourinary tract
more than 95 % of male are azoospermic because of failure of development of wolffian duct structures.
the incidence of inguinal hernia, hydrocele and undescended testis is higher than expected.
cervicitis common in female.

Sweat gland
excessive loss of salt in sweat predisposes young children to salt depletion episodes, especially during episode of gastroenteritis and warm weather.
these children present with hypochloremic alkalosis.

Rectal prolapse
caused by increased fecal volume, malnutrition, and increased intra–abdominal pressure due coughing.

Diagnosis and Assessment The diagnosis of cystic fibrosis has been
based on positive sweat test (more than 60mEq/l of chloride in sweat) in conjunction with 1 or more of following features
1. typical chronic obstructive pulmonary disease,
2. documented exocrine pancreatic insufficiency
3. positive family history

CT CHEST
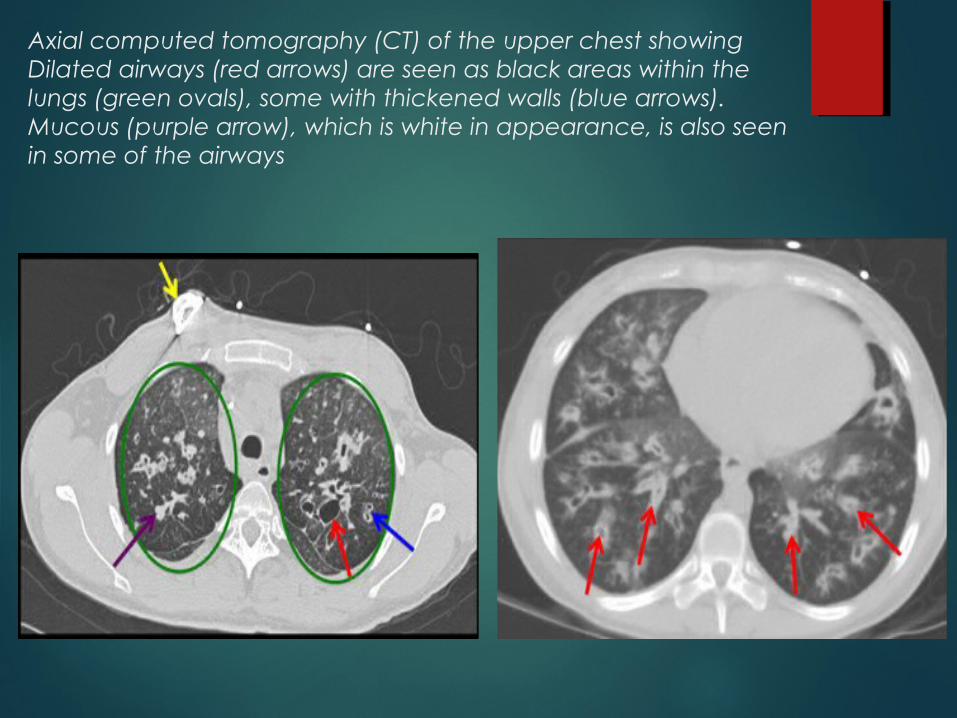
Axial computed tomography (CT) of the upper chest showing Dilated airways (red arrows) are seen as black areas within the lungs (green ovals), some with thickened walls (blue arrows). Mucous (purple arrow), which is white in appearance, is also seen in some of the airways
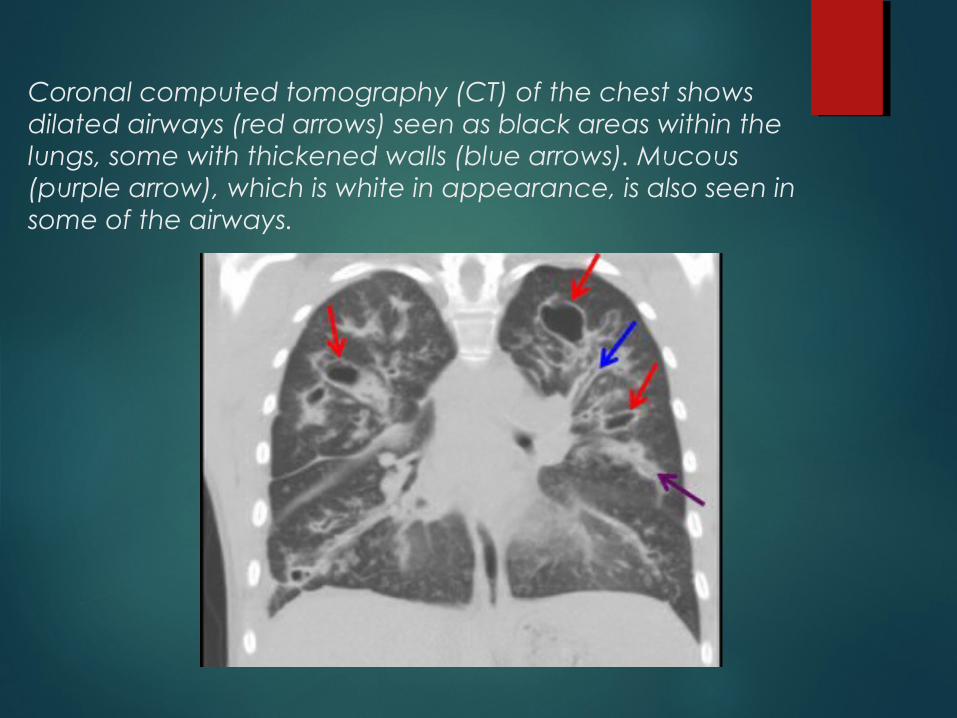
Coronal computed tomography (CT) of the chest shows dilated airways (red arrows) seen as black areas within the lungs, some with thickened walls (blue arrows). Mucous (purple arrow), which is white in appearance, is also seen in some of the airways.

Pulmonary function testing (PFT)
Standard spirometry may not be reliable until patients are aged 5-6 years.
Typically, peripheral airway involvement resulting from CF manifests as an obstructive defect with air trapping and hyperinflation; oxyhemoglobin desaturation may occur because of a ventilation-perfusion mismatch. In the early stages, forced expiratory volume in 1 second (FEV1) may be normal and forced expiratory flow (FEF) after 25-75% of vital capacity has been expelled (FEF 25-75) is reduced, suggesting small airway involvement. As the disease progresses, FEV1 is also reduced out of proportion to FCV(forced vital capacity)

Difference b/w obstructive and Difference b/w obstructive and restrictive lung disease on bases of restrictive lung disease on bases of lung function testlung function test
parameter obstructive lung disease restrictive lung disease
forced vital capacitynormal or slightly decreased
decreased
Forced expiratory volume in 1 sec
decrease out of proportion to FVC
decreased in proportion to FVC
FEV1/FVCdecreased can be as low as 20-30%
near normal(70%) or increased
total lung capacity increased decreased
residual volume increase decreased

The associated air trapping results in an elevated ratio of residual volume to total lung capacity (RV/TLC). With hyperinflation, TLC is also increased. In patients with advanced disease, extensive lung changes with fibrosis are reflected as restrictive changes characterized by declining TLC and vital capacity.

Exocrine pancreatic insufficiency
The diagnostic options include
1.indirect measures (ie, 72-hour fecal fat and fecal elastase) or
2. direct measures (secretin–pancreozymin tests)
Fecal elastase testing may be used to demonstrate a lack of endogenous enzyme. It has been found that fecal elastase is 72% sensitive for severe pancreatic insufficiency and 90% specific.
not as useful in diabetics, as fecal elastase decreases with increased duration of diabetes.

In contrast to the above indirect measurements of pancreatic function, direct measurements with the secretin–cerulein or secretin–pancreozymin tests are the gold standard for accurate assessment of the exocrine function of the pancreas
However, the limitations of the direct functional tests are that they are usually performed only at specialized centers, and they are time consuming and expensive.

Bronchoalveolar lavage
recovery of Pseudomonas aeruginosa from bronchoalveolar lavage fluid supports the diagnosis of CF in a clinically atypical case

Sputum microbiology
The most common bacterial pathogens in the sputum of patients with CF are Haemophilus influenzae, Staphylococcus aureus, P aeruginosa. Findings of P aeruginosa, especially the mucoid form, support the diagnosis of CF in children.

Immunoreactive Trypsinogen Test (IRT) is used for newborns who do not produce enough sweat for the sweat test. In the IRT test, blood drawn 2 to 3 days after birth is analyzed for a specific protein called trypsinogen. Positive IRT tests must be confirmed by sweat and other tests.
NEWBORN SCREENING

Treatment

Management-CHEST
Physiotherapy
Antibiotics
Bronchodilators
Mucolytics
Anti inflammatory

Physiotherapy
Chest Physiotherapy Physical activity

Antibiotics
antibiotics are mainstay of therapy to control progression of lung infection.
the goal is to reduce the intensity of endobronchial infection and to delay progressive lung damage.
three way to give antibiotics- 1.intravenous
2. oral
3. aerosolized form

route organism agents
oralstaphylococcus aureus linezolid
amoxicillin-clavulanate
hemophilus infuenzaeamoxicllin
ciplofloxacin
i/v s.aureus vancomycin
p.aeruginosa
pipzo
meropenem
amikacin
aerosol tobramycin

Reducing sputum viscosity inhaled hypertonic Saline- nebulized hypertonic saline
acting as hperosmolar agent, draw water into airway and rehydrate mucus and improve mucociliary clearance.
Recombinant DNAse-degrades DNA in CF sputum and increase air flow.

Anti inflammatory oral glucocorticoids may reduce airway inflammation but their long
term use is limited by adverse side effects.
Inhaled beta adrenergic can be useful to control air ways constriction, but long term benefit has not been shown.
Bronchodilators

GI & Nutrition-management
maintenance of adequate nutrition is critical for health of CF patient.
most (90) of cystic fibrosis patient require pancreatic enzyme replacement. capsule generally contain b/w 4000 -20000 unit of lipase
the dose of enzyme depend on weight, abdominal symptomatology and stool character.
not give more than 2500unit/kg/ meal to avoid risk of fibrosing colonopathy
replacement of fat soluble vitamins(A, D ,E ,K) Hyperglycemia most often becomes manifest in
adult and typically requires insulin treatment.

Lung transplantation The review suggest that transplantation is not
prolong life nor significantly improve its quality .

Psychosocial factors
CF imposes a tremendous burden on patients and depression is common.
health insurance, career option, family planning, and life expectancy become major issue.
thus assisting patient with psychosocial adjustment required by CF is critical.

complications
Atelectasis
Hemoptysis
Pneumothorax
Allergic aspergillosis
Acute respiratory failure
Right side Heart failure
Nasal polys
Diabetes Mellitus
Rectal prolapse
Pancreatitis
Gastroesophageal reflux
Intussusception
Appendicitis
Inguinal hernia
Biliary cirrhosis
Infertility
Delayed puberty