
Crystal Structure of Alkaline Phosphatase from the ... · Crystal Structure of Alkaline Phosphatase...
14
Crystal Structure of Alkaline Phosphatase from the Antarctic Bacterium TAB5 Ellen Wang 1 , Dimitris Koutsioulis 2 , Hanna-Kirsti S. Leiros 3 Ole Andreas Andersen 3 , Vassilis Bouriotis 2 Edward Hough 1,3 and Pirkko Heikinheimo 1,4 ⁎ 1 Institutt for Kjemi, University of Tromsø, N-9037 Tromsø, Norway 2 Department of Biology , University of Crete, Vasilika Vouton 71409, Heraklion, Crete, Greece 3 Norstruct, University of Tromsø, N-9037 Tromsø, Norway 4 Institute of Biotechnology, PO Box 65, FIN-00014 University of Helsinki, Finland Alkaline phosphatases (APs) are non-specific phosphohydrolases that are widely used in molecular biology and diagnostics. We describe the structure of the cold active alkaline phosphatase from the Antarctic bacterium TAB5 (TAP). The fold and the active site geometry are conserved with the other AP structures, where the monomer has a large central β-sheet enclosed by α-helices. The dimer interface of TAP is relatively small, and only a single loop from each monomer replaces the typical crown domain. The structure also has typical cold-adapted features; lack of disulfide bridges, low number of salt-bridges, and a loose dimer interface that completely lacks charged interactions. The dimer interface is more hydrophobic than that of the Escherichia coli AP and the interactions have tendency to pair with backbone atoms, which we propose to result from the cold adaptation of TAP. The structure contains two additional magnesium ions outside of the active site, which we believe to be involved in substrate binding as well as contributing to the local stability. The M4 site stabilises an interaction that anchors the substrate-coordinating R148. The M5 metal-binding site is in a region that stabilises metal coordination in the active site. In other APs the M5 binding area is supported by extensive salt-bridge stabilisation, as well as positively charged patches around the active site. We propose that these charges, and the TAP M5 binding, influence the release of the product phosphate and thus might influence the rate-determining step of the enzyme. © 2006 Elsevier Ltd. All rights reserved. *Corresponding author Keywords: alkaline phosphatase; X-ray structure; cold adaptation; psychrophiles Introduction Alkaline phosphatases (AP; EC 3.1.3.1) are found in organisms ranging from bacteria to mammals. They are fairly non-specific, reacting with a variety of substrates and are widely used in molecular biology and clinical diagnostics. Alkaline phosphatases are generally very stable enzymes, the mesophilic Escher- ichia coli enzyme (ECAP) remaining active at 80 °C, 1 and that from Pyrococcus abyssi even at 105 °C. 2 The psychrophilic alkaline phosphatases are invaluable in DNA cloning and amplification, since they can be inactivated easily by heat denaturation. 3 The AP from Antarctic strain TAB5 (TAP) is inactivated completely at 50 °C (15 min incubation), while the traditional calf intestinal AP (CIP) retains 90% of its activity when treated in the same way. 4 In addition to TAP, cold- adapted APs from Atlantic cod (Gadus morhua), 5 a marine Vibrio sp, 6 the bacterial strain HK47, 7 a Shewanella sp, 8 and shrimp (Pandalus borealis, SAP), 9,10 have been isolated. Both SAP and TAP are commercially available and are used for in vitro reactions. Alkaline phosphatase catalyses the hydrolysis to inorganic phosphate and an alcohol or, in the presence of an alcohol, the transphosphorylation of Present addresses: E. Wang, Department of Molecular Biophysics, PO. Box 124, University of Lund, SE-22100 Lund, Sweden; O. A. Andersen, Division of Biological Chemistry and Molecular Biology, School of Life Sciences, University of Dundee, Dundee DD1 5EH, Scotland. Abbreviations used: AP, alkaline phosphatase; TAP, alkaline phosphatase from TAB5; ECAP, E. coli alkaline phosphatase; PLAP, human placental alkaline phosphatase; SAP, shrimp (Pandalus borealis) alkaline phosphatase. E-mail address of the corresponding author: [email protected] doi:10.1016/j.jmb.2006.11.079 J. Mol. Biol. (2007) 366, 1318–1331 0022-2836/$ - see front matter © 2006 Elsevier Ltd. All rights reserved.
Transcript of Crystal Structure of Alkaline Phosphatase from the ... · Crystal Structure of Alkaline Phosphatase...

doi:10.1016/j.jmb.2006.11.079 J. Mol. Biol. (2007) 366, 1318–1331
Crystal Structure of Alkaline Phosphatase from theAntarctic Bacterium TAB5
Ellen Wang1, Dimitris Koutsioulis2, Hanna-Kirsti S. Leiros3
Ole Andreas Andersen3, Vassilis Bouriotis2
Edward Hough1,3 and Pirkko Heikinheimo1,4⁎
1Institutt for Kjemi,University of Tromsø,N-9037 Tromsø, Norway2Department of Biology,University of Crete,Vasilika Vouton 71409,Heraklion, Crete, Greece3Norstruct,University of Tromsø,N-9037 Tromsø, Norway4Institute of Biotechnology,PO Box 65, FIN-00014University of Helsinki, FinlandPresent addresses: E. Wang, DepaBiophysics, PO. Box 124, UniversityLund, Sweden; O. A. Andersen, DivChemistry and Molecular Biology, SUniversity of Dundee, Dundee DD1Abbreviations used: AP, alkaline
alkaline phosphatase from TAB5; ECphosphatase; PLAP, human placentphosphatase; SAP, shrimp (Pandalusphosphatase.E-mail address of the correspondi
0022-2836/$ - see front matter © 2006 E
Alkaline phosphatases (APs) are non-specific phosphohydrolases that arewidely used in molecular biology and diagnostics. We describe the structureof the cold active alkaline phosphatase from the Antarctic bacterium TAB5(TAP). The fold and the active site geometry are conserved with the otherAP structures, where the monomer has a large central β-sheet enclosed byα-helices. The dimer interface of TAP is relatively small, and only a singleloop from each monomer replaces the typical crown domain. The structurealso has typical cold-adapted features; lack of disulfide bridges, low numberof salt-bridges, and a loose dimer interface that completely lacks chargedinteractions. The dimer interface is more hydrophobic than that of theEscherichia coliAP and the interactions have tendency to pair with backboneatoms, which we propose to result from the cold adaptation of TAP. Thestructure contains two additional magnesium ions outside of the active site,which we believe to be involved in substrate binding as well as contributingto the local stability. The M4 site stabilises an interaction that anchors thesubstrate-coordinating R148. The M5 metal-binding site is in a region thatstabilises metal coordination in the active site. In other APs the M5 bindingarea is supported by extensive salt-bridge stabilisation, as well as positivelycharged patches around the active site. We propose that these charges, andthe TAP M5 binding, influence the release of the product phosphate andthus might influence the rate-determining step of the enzyme.
© 2006 Elsevier Ltd. All rights reserved.
Keywords: alkaline phosphatase; X-ray structure; cold adaptation;psychrophiles
*Corresponding authorIntroduction
Alkaline phosphatases (AP; EC 3.1.3.1) are found inorganisms ranging from bacteria to mammals. Theyare fairly non-specific, reacting with a variety ofsubstrates and are widely used in molecular biology
rtment of Molecularof Lund, SE-22100ision of Biologicalchool of Life Sciences,5EH, Scotland.
phosphatase; TAP,AP, E. coli alkaline
al alkalineborealis) alkaline
ng author:
lsevier Ltd. All rights reserve
and clinical diagnostics. Alkaline phosphatases aregenerally very stable enzymes, the mesophilic Escher-ichia coli enzyme (ECAP) remaining active at 80 °C,1
and that from Pyrococcus abyssi even at 105 °C.2 Thepsychrophilic alkaline phosphatases are invaluable inDNA cloning and amplification, since they can beinactivated easily by heat denaturation.3 TheAP fromAntarctic strain TAB5 (TAP) is inactivated completelyat 50 °C (15 min incubation), while the traditional calfintestinal AP (CIP) retains 90% of its activity whentreated in the same way.4 In addition to TAP, cold-adapted APs from Atlantic cod (Gadus morhua),5 amarine Vibrio sp,6 the bacterial strain HK47,7 aShewanella sp,8 and shrimp (Pandalus borealis,SAP),9,10 have been isolated. Both SAP and TAP arecommercially available and are used for in vitroreactions.Alkaline phosphatase catalyses the hydrolysis to
inorganic phosphate and an alcohol or, in thepresence of an alcohol, the transphosphorylation of
d.

1319Structure of TAB5 Alkaline Phosphatase
phosphomonoesters to new phosphoesters. ECAPhas been widely used as a model system and is wellknown both structurally and functionally.11–15 Struc-tures of ECAP,12,15 human placental alkaline phos-phatase (PLAP),16 and shrimp alkaline phosphatase(SAP)10 are known. They are dimeric with extendedβ-sheets stretching through the two monomers andscreened by α-helices of different lengths to form alarge α-β-α-sandwich. A crown domain at the dimerinterface built of strands from both monomers variesin size from species to species.16 In vivo, the alkalinephosphatase reaction mechanism involves threemetal ions that bind to sites M1–M3 in the activesite. The most common combination is a mixture ofZn2+ andMg2+, occasionallyCo2+ andMg2+.17,18 Thereaction proceeds in two steps, with the firstgenerating a covalent phosphoserine intermediateand releasing the alcohol. In the second step, thisintermediate is attacked by a nucleophilic watermolecule or alcohol to release inorganic phosphate orgenerate a new phosphoester, respectively. Thoughthe natural pH environment for alkaline phospha-tases is around neutral, their catalytic optima areabove pH 8.4 At optimal pH, the rate-limiting step isthe release of non-covalent product phosphate,whereas in acidic conditions (pH<6), dephosphor-ylation of the covalent enzyme intermediate is rate-limiting.11
The M1 andM2metal ions are involved in bindingof the substrate/product and in generation of thenucleophiles required in the two steps of the reaction.The hydroxyl group of the active site serine is firstactivated by Zn2+ at theM2 site with formation of thecovalent enzyme intermediate. In the second step, awater molecule activated by Zn2+ at M1 hydrolysesthe covalent intermediate to yield a free serine residueand a phosphate ion. The M3 metal ion activates theactive site serine by deprotonation, as well as affectsthe product release from the enzyme.15,19
Twonon-conserved residues havebeenproposed tobe responsible for defining the APmetal specificity atthe M3 site. Mutagenesis of the ECAP residuecorresponding to TAP H135 (ECAP D153H) resultsin a decrease of metal affinity, but in high concentra-tions ofMg2+ and combinedwith amutation at D330,the enzyme is 17-fold more active than the wild-type.19,20 The reverse, H135D mutation in TAPresulted in a more stable but less active enzyme,21
but the H135E mutation increased the kcat although italso destabilised the enzyme (D. K., unpublishedresults). The TAP residue 260 is histidine in mosteukaryotic alkaline phosphatases, but it is lysine inECAP (K328), and it has been suggested to affect themetal ion specificity of APs. The wild-type ECAP hasvery little activity in the presence of Co2+, while theK328W and D153H/K328W variants can use Co2+
efficiently for catalysis.22 The D153H/K328W varianthas approximately equal activity in the presence ofCo2+ or Zn2+, but is most active in combinations ofthese metals with Mg2+.22
Mammalian APs share a number of physiochem-ical features with the bacterial enzymes, including25–30% amino acid sequence identity, and the active
site is particularly well conserved. The mammalianAPs are still, in general, 20–30-fold more active thanECAP.23 These differences have been explained intwo ways. Firstly, changes in a few key active siteresidues influence metal ion usage and the activa-tion of water for hydrolysis of the covalentintermediate.20,22,24 Secondly, since almost all thewell-known APs function optimally as dimers, it ispossible that sequence variations in the dimerinterface affect both the catalytic efficiency andallostery of the enzyme.16 The specific activity ofTAP is lower than that of mammalian APs butconsiderably higher than that for native ECAP.4
Enzymes showing cold-adapted properties arecatalytically more efficient at low temperatures thantheir mesophilic and thermophilic homologues.25,26
Their temperature optima is often above the ambienttemperature for the organism, so that they functionwell also at temperatures similar to their mesophiliccounterparts, but they retain much of their catalyticactivity at temperatures approaching 0 °C.27 Thisarises from a reduction in activation energy,27,28andis often achieved at the cost of reduced thermalstability and increased flexibility.29–31
The proportion of hydrophobic residues or theextent of intramolecular networks of polar and/orcharged residues in the protein core are ways toadjust the stability of the protein structure.27,32–34 Ahighly hydrophobic core gives tight packing of theprotein, minimizes the surface area and decreasesinteraction with the solvent.34 However, the stabil-ity contribution of hydrophobic forces weakens atlow temperature,32 and the contribution from polarand ionic interactions become more apparent. Anoverall increase in the flexibility of subunits is oftendue to fewer salt-bridges, networks or clusters ofsuch,27,34,35 and in particular those located at thesurface.36,37 The increase in flexibility may alsoinvolve local effects, e.g. in or adjacent to the activesite, while other areas of the protein are wellstabilised.38 For example, introduction of a stabilis-ing Ca2+-binding site into the psychrophilic subtil-isin from the Antarctic Bacillus TA39 increases thespecific activity twofold and at the same timestabilises the protease dramatically.39
TAP shows many characteristics typical ofenzymes from psychrophiles; it is thermolabile, butalso has cold-adapted enzymatic properties, anincreased kcat and lower Ea compared to, forexample, ECAP.40 TAP retains 38% of the activityat 25 °C also at 0 °C,4 where ECAPhas nomeasurableactivity.8,40 Moreover, TAP kcat is 1212 s−1 at 10 °Cand 3500 s−1 at 25 °C,40 where the ECAP kcat is closeto zero and 80 s−1, respectively.41 Furthermore, TAPhas a strikingly low temperature optimum, 25 °C,4 ascompared to approximately 40 °C for SAP9 andCIP,4
and close to 80 °C for ECAP.8 The TAP sequencecontains fewer proline residues and a reduced Arg/(Arg+Lys) ratio in comparison to ECAP.4 Thecatalysis and cold adaptation of TAP have beenstudied by mutagenesis in the active site (D. K.,unpublished results).21,40 The results underline theeffect of local changes on catalysis, and uncouple the
1320 Structure of TAB5 Alkaline Phosphatase
effects of thermal stabilisation and activation energy.Single additional mutations can restore the lowactivation energies, which were increased initiallyby mutagenesis (D. K., unpublished results).21
The crystallised TAP dimer contains almost 200residues less than ECAP, and is the smallest of theknown AP structures, and the smallest of allcharacterized enzymes of this family. It is the firststructure of a bacterial alkaline phosphatase withcold-active properties. The fold shows strongsimilarity to its mesophilic counterpart (ECAP)and other known AP structures, but the structurecontains two additional magnesium ions permonomer in addition to the Mg2+ in M3 in theactive site. Although these magnesium ions arelocalized outside the active site, they are possiblyinvolved in substrate binding and contribute tolocal stability.
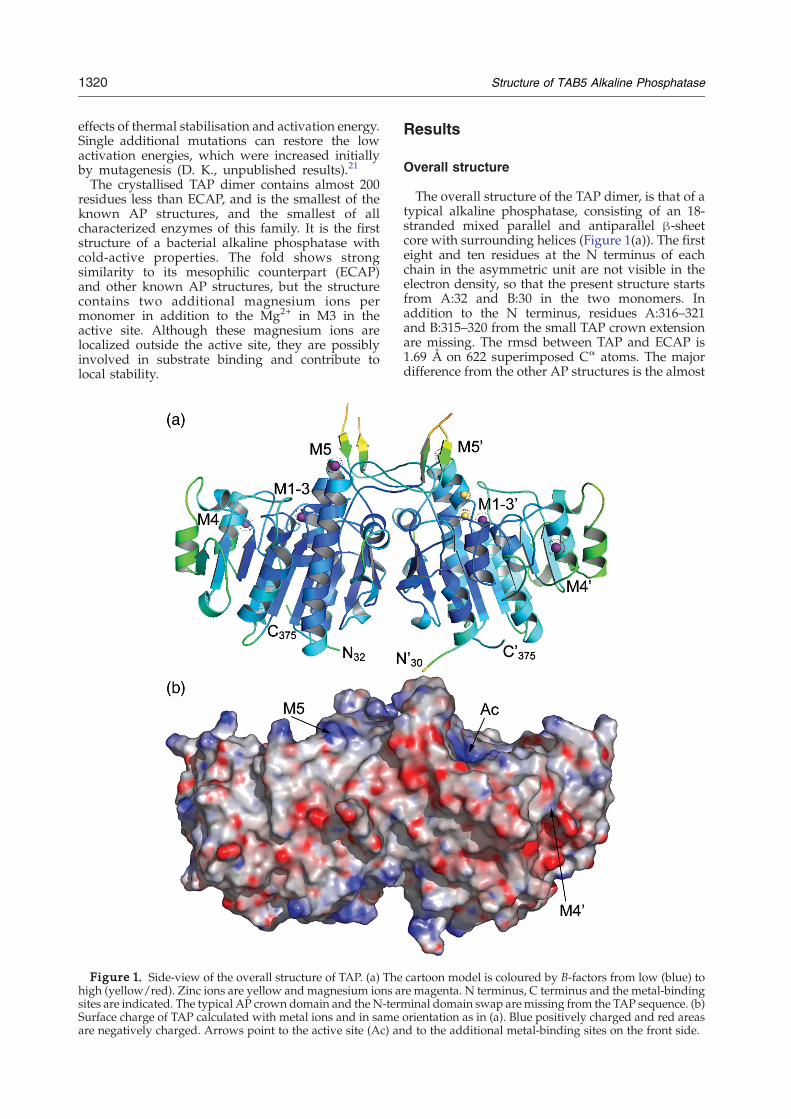
Figure 1. Side-view of the overall structure of TAP. (a) Thehigh (yellow/red). Zinc ions are yellow and magnesium ions asites are indicated. The typical AP crown domain and theN-terSurface charge of TAP calculated with metal ions and in sameare negatively charged. Arrows point to the active site (Ac) an
Results
Overall structure
The overall structure of the TAP dimer, is that of atypical alkaline phosphatase, consisting of an 18-stranded mixed parallel and antiparallel β-sheetcore with surrounding helices (Figure 1(a)). The firsteight and ten residues at the N terminus of eachchain in the asymmetric unit are not visible in theelectron density, so that the present structure startsfrom A:32 and B:30 in the two monomers. Inaddition to the N terminus, residues A:316–321and B:315–320 from the small TAP crown extensionare missing. The rmsd between TAP and ECAP is1.69 Å on 622 superimposed Cα atoms. The majordifference from the other AP structures is the almost
cartoon model is coloured by B-factors from low (blue) tore magenta. N terminus, C terminus and the metal-bindingminal domain swap aremissing from the TAP sequence. (b)orientation as in (a). Blue positively charged and red areasd to the additional metal-binding sites on the front side.

Figure 2. Stereo-view of the TAP active site with potential hydrogen bonding for the reaction intermediate. Zinc ionsare yellow and magnesium ions are magenta. Hydrogen bonds and metal coordination are shown as dotted andcontinuous lines, respectively. The structure can accommodate a phosphorylated intermediate on S84. This is modelledinto the figure in white.
Table 1. Conservation of the active site residues betweenTAP and ECAP
TAP ECAP
D83 D101W260 K328R148 R166Y325 –
M1(Zn) D259 D327H263 H331H337 H412
M2(Zn) D43 D51S84 S102D301 D369H302 H370
M3(Mg) D 43 D51(H135) (D153)T137 T155E254 E322
1321Structure of TAB5 Alkaline Phosphatase
complete absence in TAP of the ”crown domain”. InTAP, this consists of only a single long loop (residues305–335) from each monomer. Residues 312–323 inthis loop are highly flexible and have the highest B-factors in the whole structure (Figure 1(a)).The core is well ordered with low B-factors,
whereas surface loops protruding into the solvent(residue 57–60, 176–182, 197–207 and 244–247), andone short helix at the tip of the “wing” of eachmonomer (residues 183–191) have higher tempera-ture factors (Figure 1(a)). However, the range of B-factors is not larger than that for other AP-structurescollected at low temperature and at a similarresolution. It is thus not possible to draw anygeneral conclusions about the flexibility of theoverall TAP structure on the basis of B-factors alone.The overall charge surface of TAP is neutral, but
due to the metal-binding sites and R148, the activesite forms a positively charged area on the surface ofthe enzyme (Figure 1(b)).
Active site
Zn2+ in the M1 site is coordinated by H263(Nε2),H337(Nε2) and a bidentate coordination by D259,and Zn2+ in the M2 site by D43(Oδ1), S84(Oγ), D301(Oδ2) and H302(Nε2). The M3 site is occupied by amagnesium ion coordinated to oxygen atoms of D43(Oδ2), E254(Oε2) and T137(Oγ1) and three watermolecules (Figure 2). Most of the key active siteresidues are conserved between TAP and ECAP(Table 1).An active site serine, S84 in TAP, is phosphorylat-
ed during the catalysis. In our structure, S84 carriesan unidentified covalently linkedmolecule, showingelongated positive Fo–Fc density pointing towardsH337 (Figure 3). The density is too small to fit theunhydrolysed 4-nitrophenyl phosphate substratefrom the crystallisation solution. Arsenic-containing
compounds are known to react with cysteineresidues,42 and we demonstrated 50% inhibition ofTAP by 100 mM cacodylate (data not shown). Thus,we wanted to test the possibility that covalentmodification of S84 occurred in the crystallisationmixture by placing an As atom into the density. Thislowered the Rfree by 0.7%, but the As B-factorsrefined to ∼40 Å2 indicating that there was too littledensity for full occupancy of this atom and thesurrounding cacodylate molecule. Modelling of Trisand glycerol were tried, but these clash with H337(Cε1). H337(Nε2) is involved in coordination of theM1 metal ion. Hence, H337(Nε2) is not available as ahydrogen bonding partner by rotation ofχ2 by 180°,at least when Zn2+ is bound (Figure 2).Since the density protrudes from the residue that
is phosphorylated during catalysis, we modelled theunidentified density as a covalently linked phos-phorus atom placed at the centre of the density. Thedistance from S84(Oγ) is 2.1 Å and the B-factors are

Figure 3. Electron density in the active site. The 3σFo–Fc difference density is shown in cyan, illustrating apotential chemical modification of S84. The map wascalculated with phases from a structure with S84 unmo-dified. The final 2 Fo–Fc map at 1.5σ in is shown in grey atthe active site. Metal ions, S84 and H337 are labeled.
1322 Structure of TAB5 Alkaline Phosphatase
25 Å2 and 29 Å2 for the two monomers. However, aphosphorylated serine does not sufficiently explainthe shape of the density, which indicates a biggermolecule attached covalently. Unexplained densityat the active site is typical for alkaline phosphatasestructures, as PLAP has a tendency to have densityattached to the active site serine (1zef, 1zed, 1zeb)24and elsewhere in the active site for SAP (1k7h).10
In the known AP structures, the side-chain of S84has several possible conformations. The mostcommon (χ=−60°) places the serine Oγ atom inthe coordination sphere of the M2 metal ion.Roughly the same conformation occurs in allECAP and PLAP structures with transition statemimics or covalently modified serine (e.g. the ECAPvanadate complex, 1b8j),14 but not exclusively inthese structures. When phosphate is bound into theactive site in the AP structures, a conserved arginine(R148 in TAP) coordinates two of the PO4 oxygenatoms with its Nη1 and Nη2. When a SO4 ion iscoordinated or the site is empty, as for example inECAP 1kh7,19 and SAP 1shq and 1k7h,43 thisarginine turns away. In TAP, R148 also pointstowards the built phosphorus atom, and the R148position is stabilised by D83(Oδ2) (Figure 2). Thedistance between R148 on the P atom (3.6 Å) and theconformation of S84 and R148 imply that S84 iscovalently modified, and that the attached moietyhas a shape and hydrogen bonding capacity similarto that of a phosphate ion.
In the final stages of refinement, another cacody-late was added to the structure, away from theactive site (see Materials and Methods). Since thebinding site for this molecule is polar but completelyneutral, we feel that this site is purely a crystallo-graphic artefact and does not reflect a true phos-phate-binding site.Similar to the sequence of PLAP and SAP, TAP has
histidine at the first active site variable position,H135, which has two possible conformations whenMg2+ is bound in M3 that are indistinguishable atthis resolution. In the first, Nε2 points towards R148and this conformation was built, for example, toPLAP (ex. 1ew2).16 However, the hydrogen bondingangles make better sense if the head group is turned180° and Nε2 takes part in M3 water shellcoordination (Figure 2), analogous to the functionof the ECAP aspartate in this position.A second non-conserved position in the AP active
sites is W260. In TAP, W260 and D259 locate a watermolecule that is 4.3 Å from the S84(Oγ) (Figure 2). Thiswater molecule corresponds to one of the mechanis-tically conserved water molecules described forECAP, which bridges the phosphate and W260(K328 and Wat2 in ECAP).19 Two other watermolecules which are conserved are Wat3 and Wat4(ECAP numbering). Wat3 is one of the M3 coordina-tion sphere waters and hydrogen bonded to H135 inour structure, Wat4 is hydrogen bonded to R148(Figure 2). The role for Wat3 is to act as a general acidand base and generate/regenerate S84 during thecatalytic cycle.19 In addition to these conserved watermolecules, the TAP structure has a unique watercoordination chain continuing from Y325, which inthe free enzyme complexmost likely continues with athird water molecule, filling the M1 coordinationsphere (Figure 2) as suggested for ECAP.15
Additional metal-binding sites
The TAP monomer contains two additional metal-binding sites, termed M4 and M5, outside the activesite (Figure 1(a)). Neither of these overlaps with theexternal metal-binding site in PLAP.16,24 Viewingfrom the top, the external metal-binding sites arealmost equidistant from the active site (19 Å and21 Å from S84(Oγ)), on loop areas surrounding thecore β sheet (Figure 1(a)). The M4 binding site isformed by E153(Oε2), D157(Oδ2 and Oδ1), H144(O)and three water molecules. The M4 site stabilises aninteraction between the α-helix (151–160) and thebackbone of H144 and, at the same time, anchors thesubstrate coordinating R148 (Figure 4). H144 isfurther hydrogen bonded to E116(Oε2) and G103(O)(Figure 4).The M5 Mg2+ site is formed by S268(Oγ), and
N266(Oδ1) and four water molecules. The metal-binding site starts the positive N-terminal end of along α-helix and is followed by residue Y269, whichwas earlier mutated to 269A to restore the activity ofa stability mutation at G261A (see below).40 Thislong surface helix at the front side is stabilised alsoby two surface salt-bridges to the adjacent helices

Figure 4. The TAP M4 site (red) superimposed on theECAP structure (light blue). TAP residues are labelled inblack and ECAP residues are labelled in blue. Metalcoordination is shown with continuous lines and hydro-gen bonding is shown with dotted lines. The binding sitestabilises an area supporting the active site residue R148.Metal coordination is replaced by a hydrogen bond in theother APs. In addition, these structures have a longersequence forming a loop towards the active site carrying apositive charge and stabilised by a disulfide bond inECAP. This area may have a role in substrate coordination.
1323Structure of TAB5 Alkaline Phosphatase
(Figure 5), which are the only salt-bridges in thisregion. The other AP structures have multiplecharge-mediated surface interactions in the corres-ponding area (Figure 5). The conserved stabilisationof this area in addition to the inhibitor binding site inPLAP (Figure 5) suggests that this area is importantfor the AP function. The preceding sequencecontains several active site metal-coordinating resi-dues including H263, W260, D259 and E254 (Figure6) and the glycine cluster (G261 and G262), whichwere earlier mutated to test the effect on the coldactivation of TAP.40 Possibly the function of the M5binding is mediated through optimisation of theactive site M1 and M3 metal coordination. Inaddition, we propose that this area speeds therelease of the product phosphate by routing thenegatively charged phosphate ion away from theactive site.
Interface analysis
The buried surface area at the TAP interface is1830 Å2, which is only 14.2% of the total accessiblesurface area (ASA), whereas dimerisation buries 21–22% of the ASA of the other APs (Table 2). This ispartially because the TAP structure lacks thedimerising crown domain and the domain swap at
the N termini (Figure 1(a)). In PLAP, SAP and ECAPthe N terminus makes a strong dimer interaction,including formation of one (PLAP and SAP) or more(four in ECAP) salt-bridges. In TAP, the N-terminalsequence contains a signal sequence for secretion,4
and is thus cleaved from the mature protein. In thepresent construct, the 22 N-terminal residues weredeleted and a further ten are disordered completelyin the crystal structure Therefore, we consider thatthe N termini of TAP are quite unlikely to beinvolved in significant dimer interactions.One way of describing the tightness and comple-
mentarity of a protein–protein interface is tocompare the cavity volume divided by the wholeinterface area (gap volume index, GV) in differentstructures. In an average dimer interface this indexis 2.1 Å,44 but all alkaline phosphatases seem to packbetter to give scores around 1 Å (Table 2). The worstpacking score is for TAP, which gives a GV index of1.39 Å. A further indication of the looser nature ofthe TAP interface is that, unlike the other APs, TAPcompletely lacks charged interactions on the dimerinterface (Table 2). The TAP dimer interface isstabilised by 26 hydrogen bonds, which is a similarproportion per Å2 to in ECAP, but 20 (77%) of theseinvolve backbone atoms as the other partner andonly six are side-chain to side-chain interactions. Thedimer interface of ECAP is stabilized by 58hydrogen bonds, of which 24 are side-chain toside-chain interactions, thus 59% involve backboneatoms as the other partner. This feature mightfurther support TAP cold adaptation. The backboneatoms have less freedom in the solvated conforma-tion than the side-chains, and so the entropy cost ofdimerisation has already been paid upon folding.Thus, reducing the number of side-chain interac-tions, the free energy difference upon dimerisation isless affected by the TΔS term and temperature andΔG=ΔH–TΔS approaches ΔH. The stability of thedimer is thus less temperature-sensitive and couldcontribute to the cold adaptation of TAP withotherwise looser and softer interface.
Discussion
Structure in general
Although the TAP dimer contains more than 200fewer residues than PLAP it retains the topologyfound in PLAP, SAP and ECAP with a large centralβ-sheet enclosed by α-helices (Figure 1(a)). Theactive site geometry is conserved, as are the basicenzymological properties of the family.The APs have low substrate specificity, which is
consistent with the exposed active site found in allcases where the 3D structure is available. With itsthree metal ions, the active site carries an excesspositive charge, thus attracting phosphate groupsfor hydrolysis at rates almost independent of therest of the substrate molecule. Cleavage occurs viaa phosphorylated enzyme intermediate and, final-ly, the release of the reaction products from the

Figure 5. The M5 binding site. (a) The M5 binding on the start of helix A267–D291 and the salt-bridges stabilising thehelix to the rest of the structure. In other APs this area is stabilised by a network of salt-bridges, displayed here for (b)ECAP (c) SAP and (d) PLAP. Residues participating in a salt-bridge are in yellow. The location of the active site is shownby the metal ions in the background.
1324 Structure of TAB5 Alkaline Phosphatase
enzyme; where release of the phosphate is the rate-determining step for ECAP at alkaline pH.11,15 Thehighly negative surface of the shrimp enzyme wasproposed to direct the substrate towards the activesite, and thus to be part of its cold adaptation.10
However, even if similarly cold-adapted, TAP hasan overall neutral surface charge (Figure 1(b)),which is similar to the mesophilic PLAP andECAP.
The active site
Among the divalent metal ions examined for theirinfluence on the TAP activity at pH 8.5, Mg2+ is themost effective.4 The current structure with Mg2+ atthe M3 site thus presents TAP in the most activecatalytic form.The conformation of H135 is linked to the metal
contents of the active site. The histidine side-chain

Figure 6. The M5 binding site in TAP (red, blacklabels) superimposed on the PLAP structure (green). Themetal binding site is in an area carrying several activesite residues and on the opposite side from R148,possibly to aid in product release from the active site.In PLAP, the same area is covered with positive chargefrom R residues. Table 2. Structural analysis on the related AP proteins
and the dimer interface
TAP ECAP PLAP SAP
A. Dimer analysisResiduesa 689 898 962 952Strong ionic interactionsb
All 16 38 42 38His 0 8 4 2Intersubunit 0 8 4 8
Dimer ASAc (Å2) 22 336 27 912 30 672 31 024ASA/number of atoms (Å2) 3.0 2.8Number of disulfide bridges 0 2 2 2Number of buried
charged residuesd10 14 24 18
Negative buried residues 10 14 20 12Positive buried residues 0 0 4 6
B. Interface analysisInterface residues/monomer 51 103 121 119Ion pairs (PISA)d 0(0) 8 (10) 6 (16) 8 (16)Hydrogen bonds
(side-chain to side-chain)26 (6) 58 (24) 30 (10) 56 (17)
Interface BSAc (Å2)(%total)
1830(14.2)
3801(21.6)
4150(21.3)
4288(21.8)
Gap volume indexe 1.39 1.00 1.14 1.06Polar atoms (%) 35.5 41.3 30.3 36.8Non-polar atoms (%) 64.4 58.6 69.7 63.2
Statistics are from a representative structure for each AP: ECAP(1ed9),15 PLAP (1zef)25and SAP (1shn).10
a Number of built residues in the PDB structure, calculated forthe dimer in PLAP.
b Strong ionic interactions defined as an interaction of chargedresidues <4 Å. The numbers are reported for: all interactions;those involving a histidine residue as the second partner; and thenumber located between the two subunits.
c The accessible surface area (ASA) is defined with a 1.4 Åprobe. The interface buried surface area (BSA) is reported for asingle subunit and the dimer ASA calculated as 2×(monomerASA–interface BSA).
d Manually checked number of ion pairs. The number inparentheses is the distant-based analysis reported by PISA.
e Gap volume index=gap volume/interface ASA.
1325Structure of TAB5 Alkaline Phosphatase
atom Nε2 takes part in organising the first watercoordination sphere of Mg2+ at position M3 (forexample in PLAP structures,16,24 D153H/D330Nmutated ECAP 1khk,19 or SAP 1shq43). Analogously,the corresponding residue in native ECAP (D153)binds to one of the metal-coordinated water mole-cules and thus stabilises the octahedral geometry tofavour Mg2+ binding at M3.41 When a zinc ion bindsto M3, H135 is reoriented to bring its Nε2 intoposition to coordinate the Zn2+ directly (for exampleSAP 1k7h,43 or D153H/D330N mutated ECAP(1khn).19
The two variable residues corresponding to TAPH135 and W260, in ECAP, are proposed to have arole in determining the M3 metal specificity.22 WhenK328 is mutated to tryptophan, (corresponding toW260 in TAP), ECAP prefers cobalt for catalysis.22
The effect is even more pronounced in the doublevariant D153H/K328W.22 However, the mutatedtryptophan has a different orientation in the ECAPstructure, whereas the orientation of W260 in TAP ismore similar to that of the wild-type K328 in ECAP(Figure 7). Steric hindrance from the asparaginereplacing TAPA219 does not allow tryptophan to fitinto the active site of ECAP. On the basis of the TAPstructure and the fact that W260 is not even linkedindirectly toM3 coordination, the role ofW260 inM3specificity seems questionable to us. Together withD259, W260 coordinates one of the conserved watermolecules (Figure 2) in the active site. It represents amore native view of the W260 position, than amutated ECAP structure, and suggests a more directrole in the catalysis or reaction coordination.
The reaction intermediate in AP catalysis wassimulated earlier by the AlF3-complexed ECAPvariants, where AlF3 mimics the transition state inthe hydrolysis of the phosphorylated intermediate(1kh5).19 A water molecule occupies the axialposition in line with the phosphoseryl bond toform five-coordinated geometry.14 The TAP activesite has an unexplained positive difference electrondensity extending from S84 towards the phosphate-binding site and H337 (Figure 3). On the basis of theanalogous structures of ECAP (1b8j)45 and PLAP(1zef),24 we can also model a covalent enzyme-phosphate complex with one oxygen atom bridgingthe zinc ions at positions M1 and M2 as proposed(Figure 2) but we have no density for a watermolecule at the axial position. To be conservative inour structural interpretation, we chose to leave thecrystal structure with the central P atom only, tolocate the centre of a covalently attached phosphatemolecule. The shape of our unassigned density alsoindicates that the moiety is bigger than just a PO3ion, as the density continues further towards H337(Figure 3). Thus, though we believe that thechemistry of the covalently attached molecule at

Figure 7. The active site of TAP highlighting thestructural role of the engineered residues H135, A219,W260–G262 and Y269. Corresponding residues fromECAP (PDB, 1ed9)15 are shown in light blue. Mutatedglycine residues are shown in white and other colours areas in Figure 4.
1326 Structure of TAB5 Alkaline Phosphatase
S84 is mimicking a substrate for AP, it is not a truenative covalently phosphorylated intermediate.
TAP mutations
The cold-adapted properties of TAP have beenengineered by mutagenesis.21,40 The double muta-tion W260K/A219N removed the cold-adaptedcharacter by increasing the activation energy (Eadetermined from an Arrhenius plot in the temper-ature range 5–25 °C) of the catalysis.21 The singlemutation at W260K resulted in a TAP enzyme withlower activity over a wide temperature range, andthe additional mutation A219N decreased theactivity even further at low temperatures, butincreased the activity at 20–25 °C.21
ECAP has a lysine residue at the positioncorresponding to W260 in TAP, and the mutationwas targeted to model the differences betweenECAP and TAP. In ECAP, this lysine forms ahydrogen bond to a nearby aspartate that replacesH135 in the TAP active site. This interaction isexpected to appear also in the TAP double mutantW260K/H135D. A similar interaction was proposedto form in the TAP double mutant W260K/A219N,where it is possible for 260K to hydrogen bond to219N. This would restrict the conformational free-dom of 260K and thus possibly affect the efficiencyof the reaction coordination. Reduced flexibility inthe active site may explain both the decrease inactivity at low temperatures (5–15 °C) and increasein activity at high temperatures (20–25 °C) where the
hydrogen bond weakens and possibly breaks. Wepropose that the W260 position might also have arole in product release. It coordinates one of theconserved active site water molecules close to thephosphate-binding site (Figure 2). The subsequenttwo glycine residues suggest that it must be flexible.In addition, it might take part in product releaseaided by the near by positive charge from M5 (seebelow).The W260K/A219N/H135D triple TAP mutation
restored the energy of activation to the wild-typelevel and, in addition, increased the stability of theenzyme.21 The corresponding residue in ECAP(D153) to the engineered aspartate (135D) in TAPcoordinates the M3 water sphere, thus contributingto the chemistry and shape of the metal-binding site.Stronger binding of Mg2+ by O atom coordinationinstead of N atoms at this site, could explain theincreased stability for the enzyme.Two glycine residues occupy the sequence be-
tween the active site W260 and the Zn2+ coordinat-ing H263 (Figure 7). The mutation G262A fullyinactivates the enzyme.40 The TAP structure has nospace to accommodate the alanine side-chain andthe mutation probably induces a displacement ofH263 and the M1 binding site. The side-chainmoreover clashes with the hydrogen atoms at T304(Cγ2), which is (in the opposite direction) tightlypacked against the metal coordinating H302 of M2metal binding site (Figure 7). The G261A mutationdestabilises the enzyme, but stability can be partiallyrestored by a second mutation, Y269A.40 Similar toresidue 262, there is no space to fit a side-chain inposition 261, but when the side-chain of Y269 is alsoremoved, efficient packing of 261A is again possible(Figure 7). The double mutation Y269A/G261A alsohas restored the low activation energy (calculatedfrom the slope of an Arrhenius plot in thetemperature range 5–25 °C)40 of the wild-typeenzyme, suggesting that the local flexibility of theG261–262 connection is not alone important forcatalysis, but also requires tight packing of the turnto the rest of the structure.
Dimerisation and interface effects
Most alkaline phosphatases are active as dimers,but a few have been reported as a mixture of activemonomeric and homodimeric forms,2 or as uniquelymonomeric.6,7 Mutagenesis, which prevents dimer-isation of ECAP, suggests that a conformationalchange, which enhances thermal stability, metalbinding and catalysis, occurs upon dimerisation ofECAP.46 Compared to the other bacterial enzyme,ECAP, the TAP dimer interface is much looser andlacks all strong inter-subunit interactions (Table 2).Unlike ECAP, human PLAP is an allostericenzyme.47 The allostery has been attributed to thepresence of an active site residue that does not existin either ECAP or TAP, and to the PLAP hydropho-bic dimer interface.16 A total of 70% of the PLAPinterface is hydrophobic compared to the 59% ofECAP (Table 2). Replacement of specific hydrogen

1327Structure of TAB5 Alkaline Phosphatase
bonds by less specific van der Waals contactspossibly enables easier rearrangement of the mono-mers,16 whereas in ECAP the dimer is stabilised bymatching polar interfaces that do not allow smoothrearrangement of the interactions.16 The TAP inter-face is less hydrophobic than that of PLAP, but moreso than ECAP (Table 2). However, since TAP is notallosteric, we propose that this rather contributes tothe cold adaptation, which requires interface flexi-bility. SAP has a very similar proportion of non-polar atoms at the interface (Table 2). In addition, theTAP interface packs with larger cavities, more waterand without charged interactions, which furthersuggests that the interface flexibility is an importantproperty for cold-activation of the psychrophilicAPs.The TAP interface also has a higher proportion of
interactions to the backbone residues than themesophilic ECAP. This could be a sign of coldadaptation. A similar tendency is visible in the triosephosphate isomerase (TIM) family, where TIM fromthe mesophilic E. coli48 involves backbone atoms in59% of the interface hydrogen bonding but TIMfrom the psychrophilic bacterium Vibrio marinus49
has 63% of interface hydrogen bonding to backboneatoms, thus showing a similar preference forbackbone interactions by the cold-adapted enzymeas in the AP family.
Local structural flexibility and thermal stability:adaptation to cold
Cold adaptation of enzymes is generally attribut-ed to greater structural flexibility and higherturnover at low temperature. The optimal temper-ature for TAP catalysis (25 °C) is considerably lowerthan for the mesophilic homologues and as much as37% of TAP activity is retained at 0 °C.4 In contrast,the more thermostabile ECAP retains only 6% of itsactivity even at 20 °C.8 However, there is a price forTAP to pay. The greater flexibility allows catalysis inthe cold but renders the enzyme less thermostable,so that TAP is among the most thermolabile of theAPs.4
Many of the general expectations regardingprotein stability are met when we compare TAPwith the other bacterial AP, ECAP. The accessiblesurface area of a TAP dimer is smaller than that forECAP due to the difference in size (Table 2).However, the ratio of surface area per surfaceatom is higher in TAP (3.0 compared to 2.8), sothat the water-exposed area of individual surfaceatoms is, on average, a little larger. Also, the extentof intramolecular salt-bridge networking in APsfollows the general assumptions of cold adaptation.The TAP monomer has eight strong intramolecularsalt-bridges (16/dimer), whereas the more stableECAP monomer has 15 such bridges. Only two ofthese interactions are conserved between TAP andECAP (TAP R148(Nη1)-D83(Oδ2) and K96(Nζ)-E359(Oε1)). The first is involved in positioning of R148.The latter stabilises the back side of the active siteloop carrying S84.
TAP has an additional metal-binding site (M4)adjacent to the R148 region (Figure 4) and another(M5) on the opposite side of the active site (Figure 6),both occupied by Mg2+. M4 is not conserved in theother APs. However, H144 is conserved and thebackbone forms an interaction with a glutamine(corresponding to D157 in TAP) localized in thefollowing helix and replacing the metal-binding site(Figure 4). The side-chain interaction to anotherglutamine, E116, in TAP, is present only in TAP andECAP (Figure 4). At the M4 site, the TAP sequence isalso shorter than that of the other APs. Between TAPresidues E152 and the M4 coordinating E153, theother AP structures have a 15–20 residue insertion(ECAP, G170–S190 (Figure 4); PLAP, S170–D185; andSAP, N166–D182). The last acidic residue in the loopsof PLAP and SAP makes a salt-bridge in the oppositedirectionwith respect to themetal-binding site in TAP.TheM4metal-coordinating residueD157 correspondsto glutamine in ECAP, PLAP and SAP, and interactsdirectly with the backbone of H144 (Figure 4). Inaddition, loops inECAP, PLAPand SAP are stabilizedbydisulfide bridges (C168–C178 in ECAP,C121–C183in PLAP and C115–C180 in SAP) and by two salt-bridges in ECAP and SAP, and one in PLAP. The roleof R148 in substrate binding may, to some extent,explain why stabilisation in this area is advantageousfor the catalysis, but it needs to be carefully balancedwith flexibility, as demonstrated by the G149Dmutation in TAP. This mutation restricts the confor-mational freedomofG149 andpossiblyR148 and, as aresult, increases the activation energy in TAP (D. K.,unpublished results).A second evidently stabilised area in the AP
monomers is on the opposite side of the active site.On this “front” side, the surface helix layer is coveredby numerous ion pairs that connect the helices. Thisis evident for all structures except TAP (Figure 5),and is a nice example of the proposed strategy toobtain increased stability by additional surface salt-bridges.36,37 In contrast, reduction of the number ofsalt-bridges in the psychrophilic TAP indicates ahigher lability of this area. TheM5metal-binding siteis located in the same region, on top of a long surfacehelix, and the short helix following the M5 sitecontains several active site residues (E254, D259-W260 and H263) (Figures 2 and 6). Analogously toM4, the M5 coordinating residues are not conservedin any other AP, but ECAP has an asparagine residuecorresponding to the metal-coordinating N266 inTAP. In addition, the area in ECAP is followed by aproline and a disulfide bridge connecting thefollowing α-helix to a loop area that is not presentin the TAP structure. In PLAP, the TAP M5 bindingarea is more buried, but the local surface in PLAP islined with four arginine residues (Figure 6). This lineof charged residues into or out of the active site is notunique to PLAP, both SAP and ECAP have similargroups of residues, but in slightly different areas.PLAP covers the area of M5 with charge, on theopposite side of the phosphate-coordinating R148.ECAP has a line of R and K residues on the oppositesite (ECAP R166, K167, K177 Figure 4), which is

Table 3. Quality of the data
Space group P21212
Unit cell dimensionsa (Å) 70.04b (Å) 173.18c (Å) 55.34
Resolution range (Å) 30–1.95 (2.06–1.95)No of unique reflections 48 685Redundancy 4.2 (3.0)Completeness 97.3(84.3)<I>/<σI 12.3 (2.3)Rsym (%) 12.8 (46.2)
The statistics for the highest resolution shell is given inparentheses. Rsym=(ΣhΣi|Ii (h)–<I(h)>|)/(ΣhΣI I(h)), where Ii(h)is the ith measurement of reflection h and <I(h)> is the weightedmean of all measurements of h.
1328 Structure of TAB5 Alkaline Phosphatase
stabilised by M4 in TAP, but is covered by a longerloop in ECAP (Figure 4). Also, SAP has a series ofcharged residues leading away from R148, but herethe grouped arginine residues are spread on bothsides of the active site, thus creating a more diffusecharged area. Since these charge arrangements are allclose to the active site, we propose that the chargehas an effect for the catalytic rate, through phosphatebinding or release. The positive charge close to theactive site could help by directing the releasedinorganic phosphate further away from the activesite; accordingly, it could pull a potential substratecloser to the active site and therefore help it to findthe actual binding site. This hypothesis is supportedalso by the fact that PLAP has an inhibitor bindingsite at the stabilised helix area (Figure 5(d)), althoughthis site has been suggested to be specific only to theallosteric PLAP.24
With half as many salt-bridges and no disulfidebridge, TAP is a very typical cold-adapted enzymewith an overall flexibility higher than that of therelated mesophilic enzymes. On the other hand, tworegions in each monomer are stabilised by addition-al metal-binding sites, M4 and M5, which wepropose enhance the catalytic turnover rate. It hasbeen proposed that in order to increase kcat, the bestcompromise between the two opposing effects ofnegative change in enthalpy (ΔH) or entropy (ΔS) isto allow flexibility in parts of the enzyme that areinvolved in catalysis and increased rigidity in areasthat are not.38 TAP has evolved towards the lowestpossible stability of the overall structure butincreased stability of two regions related to thecatalysis and by stabilising the position of active siteresidues. We do not expect the active site of TAP tobe more stable (or less flexible) than in the other APstructures, but the metal-stabilisation of this regionmight be necessary to guarantee accurate position-ing of the catalytic metal ions in the otherwise moreflexible structure of TAP, as revealed by mutationstudies of both ECAP and TAP (D. K., unpublishedresults).19–21,40,41 The importance of the surfacecharge for the overall catalytic turnover in the APsawaits more mutagenesis to the external metal ion-binding sites in TAP or the corresponding arginineresidues in SAP, PLAP or ECAP.
Materials and Methods
Crystallisation and data collection
TAP was produced and purified as described (D. K.,unpublished results). The current construct contains 353residues and is truncated from its N terminus by 22 aminoacid residues, which contain the signal sequence in theTAB5 bacteria in order to increase the solubility of theenzyme during purification.4 This enzyme eluted with anapparent molecular mass of 72 kDa in the last gel-filtrationstep, suggesting a dimer in solution. Before crystallisation,the protein was concentrated using Centricon tubes(Millipore) with a 10 kDa cutoff and rediluted into20 mM Tris–HCl (pH 8.0), 10 mM MgCl2, 0.01 mMZnCl2. Glycerol from the storage buffer was diluted from
50% (v/v) to about 1% in this process. Crystals wereobtained by the hanging-drop, vapour-diffusion methodat 4 °C, under the following conditions: 17 mg/ml of TAPwas mixed 1:1 (v/v) with 23% (w/v) PEG3350, 0.2 Msodium acetate, 10mM4-nitrophenyl phosphate (the assaysubstrate) and 0.1 M cacodylate at pH 6.5. Crystals shapedas thin, transparent plates of variable size, were obtainedafter two to three weeks. The above solution and 10%glycerol (without substrate), was used as cryo-protectantfor data collection.TAP crystallised in space group P21212 with unit cell
dimensions of a=70.04 Å, b=173.18 Å, and c=55.34 Å (Table3). Data were collected at the European SynchrotronRadiation Facility (ESRF) at ID14-EH2, using an ADSCQ4R CCD-detector. The crystal was cooled to 100K in astream of nitrogen gas and data to 1.95 Å were collected ata wavelength of 0.933 Å. Images corresponding to 120° ofthe reciprocal space were processed with Mosflm,50 andscaled in SCALA and TRUNCATE of the CCP4 programsuite.51
Structure solution and refinement
Molecular replacement was done with Molrep (of theCCP4 suite)51 using one monomer of the wild-type ECAP(1ed9)15 with no ligand as a search model. This gave twosolutions resulting in a dimer in the asymmetric unit. Dueto the larger loops of ECAP, itwas not possible to refine thismodel further. On the basis of the sequence alignment,residues present in ECAP but absent from TAP weredeleted from the search model and the sequence mutatedto polyserine, alanine or glycine. The initial solution withthis ECAPmodel had a correlation coefficient of 46.3% andan R-factor of 47.3%. After the molecular replacement,5.1% of the datawas set aside for the freeR-factor (Table 4).The refinement was continued by checking the protein
manually in Coot.52 The phasing power of the originalmodel was too low for automated structure building usingARP/wARP (CCP4 suite). For the most part, the mainchain was built correctly and the maps permitted manualfitting of the sequence. From very early on it was clearfrom the density that the active site contained three metalions and two of these were more electron-dense than thethird. The latter was also an octahedrally coordinated site.Zinc ions were placed at sites M1 and M2, and Mg2+ wasplaced in the octahedral M3 site (Figure 2). Both metal ionswere included in the crystallisation solution with Mg2+ inexcess, since it is known to bind more weakly than Zn2+.4
Refinement was continued with Refmac5 in theCCP4suite 4.2.2 and 5.0.2. During refinement, medium

†http://www.ebi.ac.uk/msd-srv/prot_int/cgi-bin/piserver‡PP, http://www.biochem.ucl.ac.uk/bsm/PP/server/§http://www.pymol.org
Table 4. Refinement statistics and quality of the structure
Initial model
Correlation coefficient (%) 46.3R-factor (%) 47.3Final modelRefined R-factor (%) 16.2Free R-factor (%) 22.5No. residues 689No. metal ions 4 Zn2+ and 6 Mg2+
No. water molecules 445Average B-factors (Å2)
Main chain (Å2) 17.0Side-chain (Å2) 19.4Water molecules (Å2) 27.5Metal ions (Å2) 21.6
Ramachandran plot,In most favoured area (%) 89.7In additionally allowed area (%) 9.3
rmsd from ideal geometryBond lengths (Å) 0.027Bond angles (deg.) 1.93°
1329Structure of TAB5 Alkaline Phosphatase
non-crystallographic restraints (NCS) were imposed onthe two monomers in the asymmetric unit. Later, therestraints were released on residues 301–329, to allow thesingle loop replacing the AP crown domain to adopt aunique position, and the final refinement round did notinclude NCS. The missing areas were subsequently builtusing the correct sequence, and water was introduced inthe final stages with ARP/wARP. During the waterrefinement, two additional metal sites became apparent.Coordination geometry and electron density from thesubsequent refinements indicated that these were mostlikely to be magnesium ions. A cacodylate molecule wasadded to the surface of the molecule in the last stages ofrefinement. Cacodylate is hydrogen bonded to T334(Oγ1)and G335(N) from the B monomer below the small crownloop formed by monomer A. The shape of the density isclear but the B-factors allowed only a half-occupied siteand refinement to 17–28 Å2. Since this site is polar, notcharged and totally lacking the corresponding interface atthe A to B-crown, we believe it is only a crystallographicartefact and not a true phosphate-binding site.Data collection and refinement statistics are presented
in Tables 3 and 4. The free R-factor53 converged to 22.5%and the Rwork to 16.2% in the final model.
Structure analysis
The stereochemistry of the structure was analysed byPROCHECK (CCP4 suite).51 In all, 89.7% of the residuesare in the most favoured areas of the Ramachandran plot,and none is in the disallowed region (Table 4). The averageB-factors were analysed in BAVERAGE (CCP4 suite).51The structure was compared to other APs by thesecondary structure matching program (SSM) as imple-mented in Coot.52
Delphi was used for the calculation of surface electro-statics with metal ions.54 Before these calculations, all theside-chains of charged residues were added to thestructure (by mutating in Coot) including those not visiblein the final structure and thus had their side-chainstruncated in the model.Ion pairs were analysed using What if55 to calculate
distances between charged atoms. Salt-bridges are definedas a distance <4 Å between atoms of opposite charge, andinclude histidine. We counted bidentate connectionsbetween arginine residues and acidic residues as a single
salt-bridge, since the shared charge is only equal to 1.Buried charged residues were found by Protein InterfaceSurface and Assemblies server, PISA† and defined bysolvent-accessible area <10 Å2.56
The dimer interface was analysed at the PISA server56
and the Protein-Protein Interface server‡. We manuallychecked all dimer contact salt-bridges reported by PISAand accepted only those that were chemically possible; inother words, had reasonable bond lengths and numbers ofpartners. The results as well as PISA numbers are reportedin Table 2.Accessible surface areas (ASA) were found by the
program SURFACE57,58 in the CCP4 suite, and is thecalculated solvent-accessible surface when using a 1.4 Åprobe. The interface buried surface area (BSA) is reportedfor a single subunit and the dimer ASA calculated as2×(monomer ASA – interface BSA). Gap volume (GV)measures the complementarity of the interacting surfacesand was calculated using the program SURFNET59
included in the PP server. The gap volume index is thegap volume divided by the interface BSA.Figures were prepared with Pymol§ and edited in
Photoshop.
Protein Data Bank accession code
The structure has been deposited in the Protein DataBank with accession code 2IUC.
Acknowledgements
The Norwegian Structural Biology Centre (Nor-Struct) is supported by the national program inFunctional Genomics (FUGE) in the ResearchCouncil of Norway (grant NFR 154197/432). Thisproject was also supported directly by FUGE-NORDand NordForsk. We thank Ronny Helland for help,advice and assistance in data collection. Provision ofbeamtime at the European Synchrotron RadiationFacilities (ESRF) is gratefully acknowledged.
References
1. Janeway, C. M., Xu, X., Murphy, J. E., Chaidaroglou,A. & Kantrowitz, E. R. (1993). Magnesium in the activesite of Escherichia coli alkaline phosphatase is impor-tant for both structural stabilization and catalysis.Biochemistry, 32, 1601–1609.
2. Zappa, S., Rolland, J. L., Flament, D., Gueguen, Y.,Boudrant, J. & Dietrich, J. (2001). Characterization of ahighly thermostable alkaline phosphatase from theeuryarchaeon Pyrococcus abyssi.Appl. Environ. Microbiol.67, 4504–4511.
3. Sauer, S., Lechner, D., Berlin, K., Lehrach, H., Escary,J. L., Fox, N. & Gut, I. G. (2000). A novel procedurefor efficient genotyping of single nucleotide poly-morphisms. Nucl. Acids Res. 28, E13.

1330 Structure of TAB5 Alkaline Phosphatase
4. Rina, M., Pozidis, C., Mavromatis, K., Tzanodaska-laki, M., Kokkinidis, M. & Bouriotis, V. (2000).Alkaline phosphatase from the Antarctic strainTAB5. Properties and psychrophilic adaptations. Eur.J. Biochem. 267, 1230–1238.
5. Asgeirsson, B., Nielsen, B. N. & Hojrup, P. (2003).Amino acid sequence of the cold-active alkaline phos-phatase from Atlantic cod (Gadus morhua). Comp.Biochem. Physiol. B, 136, 45–60.
6. Asgeirsson, B. & Andresson, O. S. (2001). Primarystructure of cold-adapted alkaline phosphatase from aVibrio sp. as deduced from the nucleotide genesequence. Biochim. Biophys Acta, 1549, 99–111.
7. Kobori, H., Sullivan, C. W. & Shizuya, H. (1984). Heat-labile alkaline posphatase from Antarctic bacteria:rapid 5′ end-labeling of nucleic acids. Proc. Natl Acad.Sci. USA, 81, 6691–6695.
8. Suzuki, Y., Mizutani, Y., Tsuji, T., Ohtani, N., Takano,K., Haruki, M., Morikawa, M. & Kanaya, S. (2005).Gene cloning, overproduction, and characterization ofthermolabile alkaline phosphatase from a psychro-trophic bacterium. Biosci. Biotechnol. Biochem, 69,364–373.
9. Olsen, R. L., Øverbø, K. & Myrnes, B. (1991). Alkalinephosphatase from the hepatopancreas of shrimp(Pandalus borealis): a dimeric enzyme with catalyticallyactive subunits. Comp. Biochem. Physiol. B, 99, 755–761.
10. de Backer, M., McSweeney, S., Rasmussen, H. B., Riise,B. W., Lindley, P. & Hough, E. (2002). The 1.9 Å crystalstructure of heat-labile shrimp alkaline phosphatase.J. Mol. Biol. 318, 1265–1274.
11. Hull,W. E., Halford, S. E., Gutfreund, H. & Sykes, B. D.(1976). 31P nuclear magnetic resonance study ofalkaline phosphatase: the role of inorganic phosphatein limiting the enzyme turnover rate at alkaline pH.Biochemistry, 15, 1547–1561.
12. Kim, E. E. & Wyckoff, H. W. (1991). Reactionmechanism of alkaline phosphatase based on crystalstructures. Two-metal ion catalysis. J. Mol. Biol. 218,449–464.
13. Coleman, J. E. (1992). Structure and mechanism ofalkaline phosphatase. Annu. Rev. Biophys. Biomol.Struct. 21, 441–483.
14. Holtz, K. M., Stec, B. & Kantrowitz, E. R. (1999). Amodel of the transition state in the alkaline phospha-tase reaction. J. Biol. Chem. 274, 8351–8354.
15. Stec, B., Holtz, K. M. & Kantrowitz, E. R. (2000). Arevised mechanism for the alkaline phosphatasereaction involving three metal ions. J. Mol. Biol. 299,1303–1311.
16. Le Du, M. H., Stigbrand, T., Taussig, M. J., Menez, A.& Stura, E. A. (2001). Crystal structure of alkalinephosphatase from human placenta at 1.8 Å resolution.Implication for a substrate specificity. J. Biol. Chem.276, 9158–9165.
17. Wojciechowski, C. L., Cardia, J. P. & Kantrowitz, E. R.(2002). Alkaline phosphatase from the hyperthermo-philic bacterium T. maritima requires cobalt foractivity. Protein Sci. 11, 903–911.
18. Tibbitts, T. T., Murphy, J. E. & Kantrowitz, E. R. (1996).Kinetic and structural consequences of replacing theaspartate bridge by asparagine in the catalytic metaltriad of Escherichia coli alkaline phosphatase. J. Mol.Biol. 257, 700–715.
19. Le Du, M. H., Lamoure, C., Muller, B. H., Bulgakov, O.V., Lajeunesse, E., Menez, A. & Boulain, J. C. (2002).Artificial evolution of an enzyme active site: structuralstudies of three highly active mutants of Escherichiacoli alkaline phosphatase. J. Mol. Biol. 316, 941–953.
20. Muller, B. H., Lamoure, C., Le Du, M. H., Cattolico, L.,Lajeunesse, E., Lemaitre, F. et al. (2001). ImprovingEscherichia coli alkaline phosphatase efficacy byadditional mutations inside and outside the catalyticpocket. ChemBiochem, 2, 517–523.
21. Tsigos, I., Mavromatis, K., Tzanodaskalaki, M.,Pozidis, C., Kokkinidis, M. & Bouriotis, V. (2001).Engineering the properties of a cold active enzymethrough rational redesign of the active site. Eur. J.Biochem. 268, 5074–5080.
22. Wojciechowski, C. L. & Kantrowitz, E. R. (2002).Altering of the metal specificity of Escherichia colialkaline phosphatase. J. Biol. Chem. 277, 50476–50481.
23. Murphy, J. E. & Kantrowitz, E. R. (1994). Why aremammalian alkaline phosphatases much more activethan bacterial alkaline phosphatases? Mol. Microbiol.12, 351–357.
24. Llinas, P., Stura, E. A., Menez, A., Kiss, Z., Stigbrand,T.,Millan, J. L. &LeDu,M.H. (2005). Structural studiesof human placental alkaline phosphatase in complexwith functional ligands. J. Mol. Biol. 350, 441–451.
25. Somero, G. N. & Hochachka, P. W. (1968). The effect oftemperature on catalytic and regulatory functions ofpyruvate kinases of the rainbow trout and theantarctic fish Trematomus bernacchii. Biochem. J. 110,395–400.
26. Hochachka, P.W.& Somero, G.N. (1984). Temperatureadaptation. In Biochemical Adaptations, pp. 355–449Princeton University Press, New Jersey.
27. Smalås, A. O., Leiros, H. K., Os, V. & Willassen, N. P.(2000). Cold adapted enzymes. Biotechnol. Annu. Rev.6, 1–57.
28. Lonhienne, T., Baise, E., Feller, G., Bouriotis, V. &Gerday, C. (2001). Enzyme activity determination onmacromolecular substrates by isothermal titrationcalorimetry: application to mesophilic and psychro-philic chitinases. Biochim. Biophys. Acta, 1545, 349–356.
29. Davail, S., Feller, G., Narinx, E. & Gerday, C. (1994).Cold adaptation of proteins. Purification, character-ization, and sequence of the heat-labile subtilisin fromthe antarctic psychrophile Bacillus TA41. J. Biol. Chem.269, 17448–17453.
30. Zavodszky, P., Kardos, J., Svingor & Petsko, G. A.(1998). Adjustment of conformational flexibility is akey event in the thermal adaptation of proteins. Proc.Natl Acad. Sci. USA, 95, 7406–7411.
31. Feller, G., d'Amico, D. & Gerday, C. (1999). Thermo-dynamic stability of a cold-active alpha-amylasefrom the Antarctic bacterium Alteromonas haloplanctis.Biochemistry, 38, 4613–4619.
32. Kajander, T., Kahn, P. C., Passila, S. H., Cohen, D. C.,Lehtio, L., Adolfsen, W. et al. (2000). Buried chargedsurface in proteins. Struct. Fold. Des. 8, 1203–1214.
33. Vlassi, M., Cesareni, G. & Kokkinidis, M. (1999). Acorrelation between the loss of hydrophobic corepacking interactions and protein stability. J. Mol. Biol.285, 817–827.
34. Russell, R. J., Gerike, U., Danson, M. J., Hough, D. W.& Taylor, G. L. (1998). Structural adaptations of thecold-active citrate synthase from an Antarctic bacte-rium. Structure, 6, 351–361.
35. Kumar, S. & Nussinov, R. (2004). Different roles ofelectrostatics in heat and in cold: adaptation by citratesynthase. Chembiochem, 5, 280–290.
36. Aghajari, N., Feller, G., Gerday, C. & Haser, R. (1998).Structures of the psychrophilicAlteromonas haloplanctisalpha-amylase give insights into cold adaptation at amolecular level. Structure, 6, 1503–1516.
37. Feller, G. & Gerday, C. (2003). Psychrophilic enzymes:

1331Structure of TAB5 Alkaline Phosphatase
hot topics in cold adaptation. Nature Rev. Microbiol. 1,200–208.
38. Lonhienne, T., Gerday, C. & Feller, G. (2000). Psychro-philic enzymes: revisiting the thermodynamic para-meters of activation may explain local flexibility.Biochim. Biophys. Acta, 1543, 1–10.
39. Narinx, E., Baise, E. & Gerday, C. (1997). Subtilisinfrom psychrophilic antarctic bacteria: characterizationand site-directed mutagenesis of residues possiblyinvolved in the adaptation to cold. Protein Eng. 10,1271–1279.
40. Mavromatis, K., Tsigos, I., Tzanodaskalaki, M.,Kokkinidis, M. & Bouriotis, V. (2002). Exploring therole of a glycine cluster in cold adaptation of analkaline phosphatase. Eur. J. Biochem. 269, 2330–2335.
41. Stec, B., Hehir, M. J., Brennan, C., Nolte, M. &Kantrowitz, E. R. (1998). Kinetic and X-ray structuralstudies of three mutant E. coli alkaline phosphatases:insights into the catalytic mechanism without thenucleophile Ser102. J. Mol. Biol. 277, 647–662.
42. Jiang, G., Gong, Z., Li, X. F., Cullen, W. R. & Le, X. C.(2003). Interaction of trivalent arsenicals with metal-lothionein. Chem. Res. Toxicol. 16, 873–880.
43. de Backer, M. M., McSweeney, S., Lindley, P. F. &Hough, E. (2004). Ligand-binding andmetal-exchangecrystallographic studies on shrimp alkaline phospha-tase. Acta Crystallog. sect. D, 60, 1555–1561.
44. Bahadur, R. P., Chakrabarti, P., Rodier, F. & Janin, J.(2004). A dissection of specific and non-specificprotein-protein interfaces. J. Mol. Biol. 336, 943–955.
45. Holtz, K. M. & Kantrowitz, E. R. (1999). Themechanism of the alkaline phosphatase reaction:insights from NMR, crystallography and site-specificmutagenesis. FEBS Letters, 462, 7–11.
46. Boulanger, R. R., Jr. & Kantrowitz, E. R. (2003).Characterization of a monomeric Escherichia colialkaline phosphatase formed upon a single aminoacid substitution. J. Biol. Chem. 278, 23497–23501.
47. Hoylaerts, M. F., Manes, T. &Millan, J. L. (1997). Mam-malian alkaline phosphatases are allosteric enzymes.J. Biol. Chem. 272, 22781–22787.
48. Noble, M. E., Zeelen, J. P., Wierenga, R. K.,Mainfroid, V., Goraj, K., Gohimont, A. C. & Martial,J. A. (1993). Structure of triosephosphate isomerasefrom Escherichia coli determined at 2.6 Å resolution.Acta Crystallog. sect. D, 49, 403–417.
49. Alvarez, M., Zeelen, J. P., Mainfroid, V., Rentier-Delrue, F., Martial, J. A., Wyns, L. et al. (1998). Triose-phosphate isomerase (TIM) of the psychrophilicbacterium Vibrio marinus. Kinetic and structuralproperties. J. Biol. Chem. 273, 2199–2206.
50. Leslie, A. G.W. (1992). Recent changes to theMOSFLMpackage for processing film and image plate data. JointCCP4 ESF-EACMB Newsletter Protein Crystallog. 26.
51. Collaborative Computational Project Number 4(1994). The CCP4 suite: programs for protein crystal-lography. Acta Crystallog. sect. D, 50, 760–763.
52. Emsley, P. & Cowtan, K. (2004). Coot: model-buildingtools for molecular graphics. Acta Crystallog. sect. D,60, 2126–2132.
53. Brünger, A. T. (1992). Free R value: a novel statisticalquantity for assessing the accurancy of crystalstructures. Nature, 355, 472–475.
54. Nicholls, A., Sharp, K. A. & Honig, B. (1991). Proteinfolding and association: insights from the interfacialand thermodynamic properties of hydrocarbons.Proteins: Struct. Funct. Genet. 11, 281–296.
55. Vriend, G. (1990). WHAT IF: a molecular modelingand drug design program. J. Mol. Graph. 8, 52–56.
56. Krissinel, E. & Henrick, K. (2005). Detection of proteinassemblies in crystals. InDetection of Protein Assembliesin Crystals (Berthold, M. R., et al ed.), pp. 163–174,Springer-Verlag, Berlin.
57. Lee, B. & Richards, F. M. (1971). The interpretation ofprotein structures: estimation of static accessibility.J. Mol. Biol. 3, 379–400.
58. Chothia, C. (1975). Structural invariants in proteinfolding. Nature, 254, 304–308.
59. Laskowski, J. S., Liu, Q. & Bolin, N. J. (1991).Polysaccharides in flotation of sulphides. I. Adsorp-tion of polysaccharides onto mineral surfaces. Int. J.Mineral Processing (The Netherlands), 33, 223–234.
Edited by R. Huber
(Received 10 July 2006; received in revised form 21 November 2006; accepted 28 November 2006)
Available online 2 December 2006

















