
Conversion of Amino Acids 21 to Specialized...
14
Conversion of Amino Acids to Specialized Products I. OVERVIEW In addition to serving as building blocks for proteins, amino acids are precursors of many nitrogen-containing compounds that have important physiologic functions (Figure 21.1). These molecules include porphyrins, neurotransmitters, hormones, purines, and pyrimidines. II. PORPHYRIN METABOLISM Porphyrins are cyclic compounds that readily bind metal ions—usually Fe 2+ or Fe 3+ . The most prevalent metalloporphyrin in humans is heme, which consists of one ferrous (Fe 2+ ) iron ion coordinated in the center of the tetrapyrrole ring of protoporphyrin IX (see p. 280). Heme is the pros- thetic group for hemoglobin, myoglobin, the cytochromes, catalase, nitric oxide synthase , and peroxidase . These hemeproteins are rapidly synthesized and degraded. For example, 6–7 g of hemoglobin are synthesized each day to replace heme lost through the normal turnover of erythrocytes. Coordinated with the turnover of heme- proteins is the simultaneous synthesis and degradation of the associ- ated porphyrins, and recycling of the bound iron ions. A. Structure of porphyrins Porphyrins are cyclic molecules formed by the linkage of four pyrrole rings through methenyl bridges (Figure 21.2). Three structural fea- tures of these molecules are relevant to understanding their medical significance. 1. Side chains: Different porphyrins vary in the nature of the side chains that are attached to each of the four pyrrole rings. Uroporphyrin contains acetate (–CH 2 –COO – ) and propionate (–CH 2 –CH 2 –COO – ) side chains, coproporphyrin contains methyl (–CH 3 ) and propionate groups, and protoporphyrin IX (and heme) contains vinyl (–CH=CH 2 ), methyl, and propionate groups. [Note: The methyl and vinyl groups are produced by decarboxylation of acetate and propionate side chains, respectively.] Figure 21.1 Amino acids as precursors of nitrogen-containing compounds. Synthesis of nonessential amino acids varies Varies Body protein Amino acid pool (100 g) ~400 g/day Synthesis of: • Porphyrins • Creatine • Neuro- transmitters • Purines • Pyrimidines • Other nitrogen- containing compounds Body protein ~400 g/day Ketone bodies, fatty acids, steroids Glucose, glycogen CO 2 + H 2 O ~30 g/day Amino acid pool Dietary protein 100 g/day typical of U.S. diet 21 277
Transcript of Conversion of Amino Acids 21 to Specialized...

Conversion of Amino Acidsto Specialized Products
I. OVERVIEW
In addition to serving as building blocks for proteins, amino acids areprecursors of many nitrogen-containing compounds that have importantphysiologic functions (Figure 21.1). These molecules include porphyrins, neurotransmitters, hormones, purines, and pyrimidines.
II. PORPHYRIN METABOLISM
Porphyrins are cyclic compounds that readily bind metal ions—usuallyFe2+ or Fe3+. The most prevalent metalloporphyrin in humans is heme,which consists of one ferrous (Fe2+) iron ion coordinated in the center ofthe tetrapyrrole ring of proto porphyrin IX (see p. 280). Heme is the pros-thetic group for hemoglobin, myoglobin, the cytochromes, catalase,nitr ic oxide synthase, and peroxidase. These hemeproteinsare rapidly synthesized and degraded. For example, 6–7 g ofhemoglobin are synthesized each day to replace heme lost through thenormal turnover of erythrocytes. Coordinated with the turnover of heme-proteins is the simultaneous synthesis and degradation of the associ-ated porphyrins, and recycling of the bound iron ions.
A. Structure of porphyrins
Porphyrins are cyclic molecules formed by the linkage of four pyrrolerings through methenyl bridges (Figure 21.2). Three structural fea-tures of these molecules are relevant to understanding their medicalsignificance.
1. Side chains: Different porphyrins vary in the nature of the sidechains that are attached to each of the four pyrrole rings.Uroporphyrin contains acetate (–CH2–COO–) and prop ionate(–CH2–CH2–COO–) side chains, coproporphyrin contains methyl(–CH3) and propionate groups, and protoporphyrin IX (and heme)contains vinyl (–CH=CH2), methyl, and propionate groups. [Note:The methyl and vinyl groups are produced by decarboxylation ofacetate and propionate side chains, respectively.]
Figure 21.1Amino acids as precursors ofnitrogen-containing compounds.
Synthesis ofnonessentialamino acids
varies
Varies
Bodyprotein
Amino acidpool (100 g)
~400 g/day
Synthesis of:• Porphyrins• Creatine• Neuro- transmitters• Purines• Pyrimidines• Other nitrogen- containing compounds
Bodyprotein
~400 g/day
Ketone bodies,fatty acids,
steroids
Glucose,glycogen
CO2 + H2O
~30 g/day
Amino acid pool
Dietaryprotein
100 g/day typical of U.S. diet
21
277
168397_P277-290.qxd7.0:21 Specialized products5-27-04 2010.4.4 6:17 PM Page 277

Figure 21.3Pathway of porphyrin synthesis:Formation of porphobilinogen. ALAS = δ-aminolevulinic acid synthase.(Continued in Figures 21.4 and 21.5.)
Glycine
CH2 COO–
NH3+
Succinyl CoA
CH2
COO–
O C CoA
CH2
CoA
ALAS1
ALAS2
CO2
2
COO–
CH2
CH2
OC
CH2
NH3+
N
C C
C CH
H
COO–
CH2
CH2
NH2
COO–
CH2
CH2
Porphobilinogen
Heme
Iron
Lead
δ-Aminolevulinicacid dehydratase
(cytosolic enzyme)
(Two moleculescondensed)
δ-Aminolevulinic acid (ALA)
2 H2O
2. Distribution of side chains: The side chains of porphyrins can beordered around the tetrapyrrole nucleus in four different ways,designated by Roman numerals I to IV. Only Type III porphyrins,which contain an asymmetric substitution on ring D (see Figure21.2), are physiologically impor tant in humans. [Note:Protoporphyrin IX is a member of the Type III series.]
3. Porphyrinogens: These porphyrin precursors (for example, uro-porphyrinogen) exist in a chemically reduced, colorless form, andserve as intermediates between porphobilinogen and the oxidized, colored protoporphyrins in heme biosynthesis.
B. Biosynthesis of heme
The major sites of heme biosynthesis are the liver, which synthe-sizes a number of heme proteins (particularly cytochrome P450 pro-teins), and the erythrocyte-producing cells of the bone marrow,which are active in hemoglobin synthesis. [Note: Over 85% of allheme synthesis occurs in erythroid tissue.] In the liver, the rate ofheme synthesis is highly variable, responding to alterations in thecellular heme pool caused by fluctuating demands for heme pro-teins. In contrast, heme synthesis in erythroid cells is relatively con-stant, and is matched to the rate of globin synthesis. The initialreaction and the last three steps in the formation of porphyrins occurin mitochondria, whereas the intermediate steps of the biosyntheticpathway occur in the cytosol (see Figure 21.8). [Note: Mature redblood cells lack mitochondria and are unable to synthesize heme.]
1. Formation of δ-aminolevulinic acid (ALA): All the carbon andnitrogen atoms of the porphyrin molecule are provided by glycine(a nonessential amino acid) and succinyl coenzyme A (an inter-mediate in the citric acid cycle) that condense to form ALA in areaction catalyzed by ALA synthase (ALAS) (Figure 21.3) Thisreaction requires pyridoxal phosphate (PLP) as a coenzyme, andis the committed and rate-limiting step in porphyrin biosynthesis.[Note: There are two isoforms of ALAS, 1 and 2, each controlledby different mechanisms. Erythroid tissue produces only ALAS2,the gene for which is located on the X-chromosome. Loss of func-tion mutations in ALAS2 result in X-linked sideroblastic anemia.]
a. End-product inhibition of ALAS1 by hemin: When porphyrinproduction exceeds the availability of the apoproteins that
278 21. Conversion of Amino Acids to Specialized Products
Figure 21.2Structures of uroporphyrin I and uroporphyrin III. A = acetate and P = propionate.
P
N N
N N
A
P
Uroporphyrin I
P
A
A
PA
A B
D C
P
A
Porphyrins contain side chains attached to each of the four pyrrole rings. In Type I porphyrins, the side chains are arranged symmetrically, that is, for uroporphyrin I, A (acetate) alternates with propionate (P) around the tetrapyrrole ring.
Porphyrins contain four pyrrole rings (A, B, C, and D) joined through methenyl bridges.
N N
N N
Uroporphyrin III
A B
D C
P
A
P
P
A
A
P
A
A
A
Acetate (A) andpropionate (P) arereversed in ring Dof uroporphyrin IIIcompared with uroporphyrin I. Only Type III porphyrins are physiologically important in humans.
168397_P277-290.qxd7.0:21 Specialized products5-27-04 2010.4.4 6:17 PM Page 278

require it, heme accumulates and is converted to hemin bythe oxidation of Fe2+ to Fe3+. Hemin decreases the activity ofhepatic ALAS1 by causing decreased synthesis of theenzyme, through inhibition of mRNA synthesis and use (hemedecreases stability of the mRNA), and by inhibiting mitochon-drial import of the enzyme. [Note: In erythroid cells, ALAS2 iscontrolled by the availability of intracellular iron.]
b. Effect of drugs on ALA synthase activity: Administration ofany of a large number of drugs results in a significant increasein hepatic ALAS1 activity. These drugs are metabolized by themicro somal cyto chrome P450 monooxygenase system—aheme protein oxidase system found in the liver (see p. 149). Inresponse to these drugs, the synthesis of cytochrome P450proteins increases, leading to an enhanced consumption ofheme—a component of cytochrome P450 proteins. This, inturn, causes a decrease in the concentration of heme in livercells. The lower intracellular heme concentration leads to anincrease in the synthesis of ALAS1 (derepression), andprompts a corresponding increase in ALA synthesis.
2. Formation of porphobilinogen: The condensation of twomolecules of ALA to form porphobilinogen by Zn-containing ALAdehydratase (porphobilinogen synthase) is extremely sensitive toinhibition by heavy metal ions, for example, lead that replace thezinc (see Figure 21.3). This inhibition is, in part, responsible for theelevation in ALA and the anemia seen in lead poisoning.
3. Formation of uroporphyrinogen: The condensation of fourporpho bilinogens produces the linear tetrapyrrole, hydroxymethyl -bilane, which is isomerized and cyclized by uroporphyrinogen IIIsynthase to produce the asymmetric uroporphyrinogen III. Thiscyclic tetrapyrrole undergoes decarboxylation of its acetategroups, generating coproporphyrinogen III (Figure 21.4). Thesereactions occur in the cytosol.
4. Formation of heme: Coproporphyrinogen III enters the mitochon-drion, and two propionate side chains are decarboxylated to vinylgroups generating protoporphyrinogen IX, which is oxidized toprotoporphyrin IX. The introduction of iron (as Fe2+) into protopor-phyrin IX occurs spontaneously, but the rate is enhanced by ferro -chelatase, an enzyme that, like ALA dehydratase, is inhibited bylead (Figure 21.5).
C. Porphyrias
Porphyrias are rare, inherited (or occasionally acquired) defects inheme synthesis, resulting in the accumulation and increased excre-tion of porphyrins or porphyrin precursors (see Figure 21.8). [Note:With few exceptions, porphyrias are inherited as autosomal dominantdisorders.] The mutations that cause the porphyrias are hetero -genous (not all are at the same DNA locus), and nearly everyaffected family has its own mutation. Each porphyria results in theaccumulation of a unique pattern of intermediates caused by thedeficiency of an enzyme in the heme synthetic pathway. [Note:“Porphyria” refers to the purple color caused by pigment-like por-phyrins in the urine of some patients with defects in heme synthesis.]
II. Porphyrin Metabolism 279
Figure 21.4Pathway of porphyrin synthesis:Formation of protoporphyrin IX. (Continued from Figure 21.3.)
CH2CH2–OOC
CH2–OOC
Uroporphyrinogen III
Coproporphyrinogen III
CH2CH2 COO–
4 CO2
CH2 COO–
CH2–OOC
CH2CH2–OOC
CH2 COO–
CH2CH2 COO–
N N
N NH
HHH
CH2CH2–OOC
CH2CH2 COO–
CH2CH2–OOC CH2CH2 COO–
N N
N NH
HHH
CH2CH2–OOC
CH3
CH3
CH2
H2C CH
CH
CH3 CH3
CH2CH2 COO–
N N
N N
HH
Protoporphyrin IX
COO–
CH2
COO–
CH2
CH2
4 NH3
4
(Four moleculescondensed)
Hydroxymethyl-bilane synthase
Porphobilinogen
Hydroxymethylbilane
Uroporphyrinogen IIIsynthase
(Ring closure andisomerization)
(Decarboxylation)
N
C C
C CH
HCH2
NH2
CH3
CH3
CH3CH3
Uroporphyrinogen decarboxylase
168397_P277-290.qxd7.0:21 Specialized products5-27-04 2010.4.4 6:17 PM Page 279

1. Clinical manifestations: The porphyrias are classified as erythro-poietic or hepatic, depending on whether the enzyme deficiencyoccurs in the erythropoietic cells of the bone marrow or in theliver. Hepatic porphyrias can be further classified as chronic oracute. In general, individuals with an enzyme defect prior to thesynthesis of the tetrapyrroles manifest abdominal and neuro -psychiatric signs, whereas those with enzyme defects leading tothe accumulation of tetrapyrrole intermediates show photosensi-tivity—that is, their skin itches and burns (pruritus) when exposedto visible light. [Note: Photosenstivity is a result of the oxidation ofcolorless porphyrinogens to colored porphyrins, which are photo-sensitizing molecules that are thought to participate in the forma-tion of superoxide radicals from oxygen. These reactive oxygenspecies can oxidatively damage membranes, and cause therelease of destructive enzymes from lysosomes.]
a. Chronic hepatic porphyria: Porphyria cutanea tarda, the mostcommon porphyria, is a chronic disease of the liver. The dis-ease is associated with a deficiency in uro porphyrinogendecarboxylase, but clinical expression of the enzyme defi-ciency is influenced by various factors, such as hepatic ironoverload, exposure to sunlight, alcohol ingestion, and the pres-ence of hepatitis B or C, or HIV infections. Clinical onset is typ-ically during the fourth or fifth decade of life. Porphyrinaccumulation leads to cutaneous symptoms (Figure 21.6), andurine that is red to brown in natural light (Figure 21.7), andpink to red in fluorescent light.
b. Acute hepatic porphyrias: Acute hepatic porphyrias (ALAdehydratase deficiency, acute intermittent porphyria, heredi-tary coproporphyria, and variegate porphyria) are character-ized by acute attacks of gastro intestinal, neuro psychiatric, andmotor symptoms that may be accompanied by photosensitivity.Porphyrias leading to accumulation of ALA and porphobilino-gen, such as acute intermittent por phyria, cause abdominalpain and neuro psychiatric disturbances, ranging from anxietyto delirium. Symptoms of the acute hepatic porphyrias areoften precipitated by administration of drugs such as barbitu-rates and ethanol, which induce the synthesis of the heme-containing cytochrome P450 microsomal drug oxidationsystem. This further decreases the amount of available heme,which, in turn, promotes the increased synthesis of ALAS1.
c. Erythropoietic porphyrias: The erythropoietic porphyrias (congenital erythropoietic porphyria and erythropoietic proto -porphyria) are characterized by skin rashes and blisters thatappear in early childhood. The diseases are complicated bycholestatic liver cirrhosis and progressive hepatic failure.
2. Increased ALA synthase activity: One common feature of the por-phyrias is a decreased synthesis of heme. In the liver, heme nor-mally functions as a repressor of the gene for ALAS1. Therefore,the absence of this end product results in an increase in the syn-thesis of ALA synthase1 (derepression). This causes anincreased synthesis of intermediates that occur prior to thegenetic block. The accumulation of these toxic intermediates isthe major pathophysiology of the porphyrias.
280 21. Conversion of Amino Acids to Specialized Products
Figure 21.7Urine from a patient with porphyria cutanea tarda (right) and from a patient with normal porphyrin excretion (left).
Figure 21.6Skin eruptions in a patient with porphyria cutanea tarda.
Figure 21.5Pathway of porphyrin synthesis: Formation of heme. (Continued from Figures 21.3 and 21.4)
CH2CH2–OOC
CH3
CH3
CH2
H2C CH
CH
CH3 CH3
CH2CH2 COO–
N N
N N
HH
Protoporphyrin IX
2 H+
Fe2+
Heme (Fe2+ protoporphyrin IX)
Fe
Ferrochelatase(mitochondrial enzyme)
Lead
CH2CH2–OOC
CH3
CH3
CH2
H2C CH
CH
CH3 CH3
CH2CH2 COO–
N N
N N
168397_P277-290.qxd7.0:21 Specialized products5-27-04 2010.4.4 6:17 PM Page 280

Figure 21.8Summary of heme synthesis. 1Also referred to as porphobilinogen deaminase.
Coproporphyrinogen I
Protoporphyrinogen IX
Protoporphyrin IX
Heme
Fe2+
Uroporphyrin III
Coproporphyrin III
Coproporphyrinogen III
Coproporphyrin I
MITOCHONDRIA
CYTOSOL
δ-Aminolevulinic acid
Succinyl CoA + Glycine
Spontaneous
XXXXIIn nnnnirirryryyyyphy
eFeFFFFeeee2+VARIEGATE PORPHYRIA
• An acute disease caused by a deficiency in protoporphyrinogen oxidase.
• Protoporphyrinogen IX and other intermediates prior to the block accumulate in the urine.
• Patients are photosensitive.
ERYTHROPOIETIC PROTOPORPHYRIA
• This disease is caused by to a deficiency in ferrochelatase.
• Protoporphyrin accumulates in erythrocytes, bone marrow, and plasma.
• Patients are photosensitive.
n IX eneeeeggggogoooonnnnniihyr
III nnnneneeeeggggogoooononyrin
• An acute disease caused by a deficiency in coproporphyrinogen oxidase.
• Coproporphyrinogen III and other intermediates prior to the block accumulate in the urine.
• Patients are photosensitive.
HEREDITARYCOPROPORPHYRIA
Uroporphyrinogen IIIHydroxymethylbilane(enzyme bound)
Uroporphyrinogen I
Coproporphyrinogen III
Uroporphyrin I
Porphobilinogen
δ-Aminolevulinic acid Spontaneous
Spontaneous
Spontaneous
p p p ygggggggggggy
Spontaneous Uroporphyrin III
Coproporphyrin Intaneous
en IIIegegegegeggggggogogogogooooonononononnnnnninininriririririryryryryryyyyhyhyhyhyhyUroporph
Uroporphyrin Is
• This disease is caused by a deficiency in uroporphyrinogen decarboxylase.
• Uroporphyrin accumulates in the urine.
• It is the most common porphyria.
• Patients are photosensitive.
PORPHYRIA CUTANEA TARDA
• This disease is caused by a deficiency in uroporphyrinogen III synthase.
• Uroporphyrinogen I and coproporphyrinogen I accumulate in the urine.
• Patients are photosensitive.
CONGENITAL ERYTHROPOIETICPORPHYRIA
Hepaticporphyria
KEY:
Erythro-poietic
porphyria
opppppppppprrrrrroooooooooooppppppppppooooooooooo
PPPrrroooootttoooooppppp
PPPPPrrroooootttooo
opppprrrooooopppppooooo
• Ferrochelatase and ALA dehydratase are particularly sensitive to inhibition by lead.
• Protoporphyrin and ALA accumulate in urine.
LEAD POISONING
nnnnnnnnnneeeeeeeeeeegPPPPPPPPooooooooooorrrrrrrpppppppppphhhhhhhhhhobilinogobilinogobilinogobilinogobilinogobilinogobilinogobilinog
Hepatic
KEY:
Co
Co
δ-AA ac dm ac dm ac dm ac dm ac dm acim acii aci acn acn acn an ano ao ao o o c l cl ce cevulinic
Succin CoA + cineuccin CoA + cuccin CoA + cuccin CoA + ycuccin CoA + yccin CoA + yccin CoA + yccin CoA + yccin CoA + lcin CoA + lcin CoA + Gcin CoA + Gcin CoA + Gin CoA + Gin CoA + Gn CoA + Gn l CoA + nyl CoA + nyl CoA + yl CoA +yl CoA +y
δ-AAAminolevulinic ac dAminolevulinic ac dminolevulinic ac dminolevulinic acidminolevulinic acidminolevulinic acidminolevulinic acidminolevulinic acidminolevulinic acidminolevulinic acidminolevulinic acidminolevulinic aciminolevulinic aciminolevulinic aciminolevulinic aciminolevulinic aciminolevulinic acinolevulinic acinolevulinic acinolevulinic acinolevulinic acinolevulinic acnolevulinic acnolevulinic acnolevulinic acnolevulinic acnolevulinic acnolevulinic anolevulinic anolevulinic anolevulinic anolevulinic anolevulinic aolevulinic aolevulinic aolevulinic aolevulinic aolevulinic aolevulinic olevulinic olevulinic olevulinic olevulinic olevuli c olevuli clevuli clevuliniclevuliniclevulinicevulinicevulinicevulinicevulinic
• An acute disease caused by a deficiency in hydroxymethylbilane synthase1.
• Porphobilinogen and δ-amino- levulinic acid accumulate in the urine.
• Urine darkens on exposure to light and air.
• Patients are NOT photosensitive.
ACUTE INTERMITTENTPORPHYRIA
II. Porphyrin Metabolism 281
168397_P277-290.qxd7.0:21 Specialized products5-27-04 2010.4.4 6:17 PM Page 281
3. Treatment: During acute porphyria attacks, patients require medi-cal support, particularly treatment for pain and vomiting. Theseverity of symptoms of the porphyrias can be diminished byintravenous injection of hemin and glucose, which decreases thesynthesis of ALAS1. Avoidance of sunlight and ingestion ofβ-carotene (a free-radical scavenger) are helpful in porphyriaswith photosensitivity.
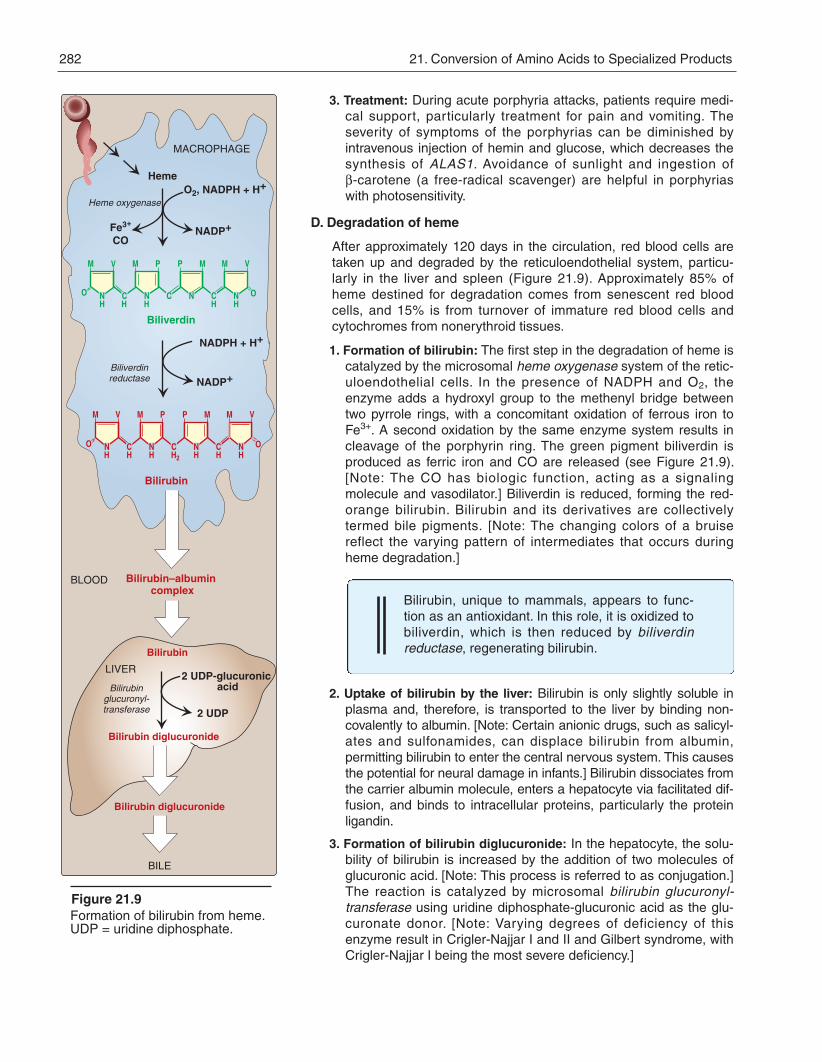
D. Degradation of heme
After approximately 120 days in the circulation, red blood cells aretaken up and degraded by the reticuloendothelial system, particu-larly in the liver and spleen (Figure 21.9). Approximately 85% ofheme destined for degradation comes from senescent red bloodcells, and 15% is from turnover of immature red blood cells andcytochromes from nonerythroid tissues.
1. Formation of bilirubin: The first step in the degradation of heme iscatalyzed by the microsomal heme oxygenase system of the retic-uloendothelial cells. In the presence of NADPH and O2, theenzyme adds a hydroxyl group to the methenyl bridge betweentwo pyrrole rings, with a concomitant oxidation of ferrous iron toFe3+. A second oxidation by the same enzyme system results incleavage of the porphyrin ring. The green pigment biliverdin isproduced as ferric iron and CO are released (see Figure 21.9).[Note: The CO has biologic function, acting as a signalingmolecule and vasodilator.] Biliverdin is reduced, forming the red-orange bilirubin. Bilirubin and its derivatives are collectivelytermed bile pigments. [Note: The changing colors of a bruisereflect the varying pattern of intermediates that occurs duringheme degradation.]
Bilirubin, unique to mammals, appears to func-tion as an antioxidant. In this role, it is oxidized tobiliverdin, which is then reduced by biliverdinreductase, regenerating bilirubin.
2. Uptake of bilirubin by the liver: Bilirubin is only slightly soluble inplasma and, therefore, is transported to the liver by binding non-covalently to albumin. [Note: Certain anionic drugs, such as salicyl-ates and sulfonamides, can displace bilirubin from albumin,permitting bilirubin to enter the central nervous system. This causesthe potential for neural damage in infants.] Bilirubin dissociates fromthe carrier albumin molecule, enters a hepatocyte via facilitated dif-fusion, and binds to intracellular proteins, particularly the proteinligandin.
3. Formation of bilirubin diglucuronide: In the hepatocyte, the solu-bility of bilirubin is increased by the addition of two molecules ofglucuronic acid. [Note: This process is referred to as conjugation.]The reaction is catalyzed by microsomal bilirubin glucur onyl -transferase using uridine diphosphate-glucuronic acid as the glu-curonate donor. [Note: Varying degrees of deficiency of thisenzyme result in Crigler-Najjar I and II and Gilbert syndrome, withCrigler-Najjar I being the most severe deficiency.]
282 21. Conversion of Amino Acids to Specialized Products
Figure 21.9Formation of bilirubin from heme.UDP = uridine diphosphate.
M V
NO C
M P
N CH
P M
N CHH
M V
N O
Heme
Fe3+
O2, NADPH + H+
NADP+
NADPH + H+
NADP+
M V
N CH
M P
N CH2
P M
N CH
M
HH
Bilirubin
Biliverdin
CO
Bilirubin–albumin complex
Bilirubin
BILE
Bilirubin diglucuronide
2 UDP-glucuronic acid
2 UDP
MACROPHAGE
BLOOD
Bilirubin diglucuronide
LIVER
Biliverdinreductase
Heme oxygenase
Bilirubinglucuronyl-transferase
H H
OO
V
NHH
168397_P277-290.qxd7.0:21 Specialized products5-27-04 2010.4.4 6:17 PM Page 282

4. Secretion of bilirubin into bile: Bilirubin diglucuronide (conjugatedbilirubin) is actively transported against a concentration gradientinto the bile canaliculi and then into the bile. This energy-depen-dent, rate-limiting step is susceptible to impairment in liver dis-ease. [Note: A deficiency in the protein required for transport ofconjugated bilirubin out of the liver results in Dubin-Johnson syn-drome.] Unconjugated bilirubin is normally not secreted.
5. Formation of urobilins in the intestine: Bilirubin diglucuronide ishydrolyzed and reduced by bacteria in the gut to yield uro -bilinogen, a colorless compound. Most of the urobilinogen is oxi-dized by intestinal bacteria to ster co bilin, which gives feces thecharacteristic brown color. However, some of the urobilinogen isreabsorbed from the gut and enters the portal blood. A portion of
Figure 21.10
BLOOD VESSEL
GALLBLADDERKIDNEY
INTESTINE
Senescent red cells are a major source of hemeproteins.
Unconjugated bilirubin is transported throughthe blood (complexed toalbumin) to the liver.
Urobilinogen is oxidized by intestinal bacteria tothe brown stercobilin.
3
Breakdown of heme to bilirubinoccurs in macrophages of the reticulo-endothelial system (tissue macro-phages, spleen, and liver).
2
1
To feces
To urine
U
B
B
B
B
B
B
S
B
BU
U
U
U
Ub
U
10
LIVER
9Bilirubin is taken up via facilitated diffusion by the liver and conjugated with glucuronic acid.
4
MACROPHAGE
UbCatabolism of heme = bilirubin; = bilirubin diglucuronide; = urobilinogen; = urobilin; = stercobilin.BG U SB
BG
BG
BG
In the intestine, glucuronic acid is removed by bacteria.The resulting bilirubin is converted tourobilinogen.
6
Conjugated bilirubin is actively secreted into bile and then the intestine.
5
8 A portion of this urobilinogen participates in the enterohepatic urobilinogen cycle.
Some of the urobilinogen is reabsorbed from the gut and enters the portal blood.
7
The remainder of the urobilinogen is transported by the blood to the kidney, where it is converted to yellow urobilin and excreted, giving urine its characteristic color.
II. Porphyrin Metabolism 283
168397_P277-290.qxd7.0:21 Specialized products5-27-04 2010.4.4 6:17 PM Page 283

this urobilinogen participates in the enterohepatic urobilinogencycle in which it is taken up by the liver, and then resecreted intothe bile. The remainder of the urobilinogen is transported by theblood to the kidney, where it is converted to yellow uro bilin andexcreted, giving urine its characteristic color. The metabolism ofbilirubin is summarized in Figure 21.10.
E. Jaundice
Jaundice (also called icterus) refers to the yellow color of skin, nailbeds, and sclerae (whites of the eyes) caused by deposition of bilirubin, secondary to increased bilirubin levels in the blood (hyper-bili rubinemia, Figure 21.11). Although not a disease, jaundice is usu-ally a symptom of an underlying disorder.
1. Types of jaundice: Jaundice can be classified into three majorforms described below. However, in clinical practice, jaundice isoften more complex than indicated in this simple classification.For example, the accumulation of bilirubin may be a result ofdefects at more than one step in its metabolism.
a. Hemolytic jaundice: The liver has the capacity to conjugate andexcrete over 3,000 mg of bilirubin per day, whereas the normalproduction of bilirubin is only 300 mg/day. This excess capacityallows the liver to respond to increased heme degradation witha corresponding increase in conjugation and secretion of bili -rubin diglucuronide. However, massive lysis of red blood cells(for example, in patients with sickle cell anemia, pyruvatekinase or glucose 6-phosphate dehydrogenase deficiency) mayproduce bilirubin faster than it can be conjugated. Unconju-gated bilirubin levels in the blood become elevated, causingjaundice (Figure 21.12A). [Note: More conjugated bilirubin isexcreted into the bile, the amount of urobilinogen entering theenterohepatic circulation is increased, and urinary urobilinogenis increased.]
b. Hepatocellular jaundice: Damage to liver cells (for example,in patients with cirrhosis or hepatitis) can cause unconjugatedbilirubin levels in the blood to increase as a result ofdecreased conjugation. Urobilinogen is increased in the urinebecause hepatic damage decreases the enterohepatic circu-lation of this compound, allowing more to enter the blood,from which it is filtered into the urine. The urine thus darkens,whereas stools may be a pale, clay color. Plasma levels ofAST and ALT (see p. 251) are elevated. [Note: If conjugatedbilirubin is not efficiently secreted from the liver into bile (intra-hepatic cholestasis), it can diffuse (“leak”) into the blood,causing a conjugated hyperbilirubinemia.]
c. Obstructive jaundice: In this instance, jaundice is not causedby overproduction of bilirubin or decreased conjugation, butinstead results from obstruction of the bile duct (extrahepaticcholestasis). For example, the presence of a tumor or bilestones may block the bile ducts, preventing passage of biliru-bin into the intestine. Patients with obstructive jaundice experi-ence gastrointestinal pain and nausea, and produce stools thatare a pale, clay color, and urine that dark ens upon standing.The liver “regurgitates” conjugated bilirubin into the blood
284 21. Conversion of Amino Acids to Specialized Products
Figure 21.11Jaundiced patient, with the scleraeof his eyes appearing yellow.
Figure 21.12Alterations in the metabolism of heme. A. Hemolytic jaundice. B. Neonatal jaundice.BG = bilirubin glucuronide; B = bilirubin; U = urobilinogen; S = stercobilin.
BBB
BB B
BG
BG
BG
B
S
LIVER
INTESTINE
Hemolyticjaundice
U
BG
BG
BB S
S
LIVER
INTESTINE
Hemolysis
Bilirubin glucuronyl- transferase
Unconjugatedbilirubin
Unconjugatedbilirubin
A
B
UU
B
B B
Neonataljaundice
B
B
168397_P277-290.qxd7.0:21 Specialized products5-27-04 2010.4.4 6:17 PM Page 284

Figure 21.14Phototherapy in neonatal jaudice
(hyperbilirubinemia). The compound is eventually excreted inthe urine. Urinary urobiloinogen is absent. [Note: Pro longedobstruction of the bile duct can lead to liver damage and asubsequent rise in unconjugated bilirubin.]
2. Jaundice in newborns: Newborn infants, particularly if premature,often accumulate bilirubin, because the activity of hepatic bilirubinglucuronyltransferase is low at birth—it reaches adult levels inabout 4 weeks (Figures 21.12B and 21.13). Elevated bili rubin, inexcess of the binding capacity of albumin, can diffuse into thebasal ganglia and cause toxic encephalopathy (kernicterus).Thus, newborns with significantly elevated bilirubin levels aretreated with blue fluorescent light (Figure 21.14), which convertsbilirubin to more polar and, hence, water-soluble isomers. Thesephotoisomers can be excreted into the bile without conjugation toglucuronic acid.
3. Determination of bilirubin concentration: Bilirubin is most com-monly determined by the van den Bergh reaction, in which diazo -tized sulfanilic acid reacts with bilirubin to form red azodipyrrolesthat are measured colorimetrically. In aqueous solution, the water-soluble, conjugated bilirubin reacts rapidly with the reagent (withinone minute), and is said to be “direct-reacting.” The unconjugatedbilirubin, which is much less soluble in aqueous solution, reactsmore slowly. However, when the reaction is carried out inmethanol, both conjugated and unconjugated bilirubin are solubleand react with the reagent, providing the total bilirubin value. The“in direct-reacting” bilirubin, which corresponds to the unconju-gated bilirubin, is obtained by subtracting the direct-reacting biliru-bin from the total bilirubin. [Note: In normal plasma, only about 4%of the total bilirubin is conjugated or direct-reacting, because mostis secreted into bile.]
III. OTHER NITROGEN-CONTAINING COMPOUNDS
A. Catecholamines
Dopamine, norepinephrine, and epinephrine are biologically active(biogenic) amines that are collectively termed catecholamines.Dopamine and norepinephrine are synthesized in the brain andfunction as neurotransmitters. Norepinephrine is also synthesized inthe adrenal medulla, as is epinephrine.
1. Function: Outside the nervous system, norepinephrine and itsmethylated derivative, epinephrine, are hormone regulators of car-bohydrate and lipid metabolism. Norepinephrine and epinephrineare released from storage vesicles in the adrenal medulla inresponse to fright, exercise, cold, and low levels of blood glucose.They increase the degradation of glycogen and triacylglycerol, aswell as increase blood pressure and the output of the heart. Theseeffects are part of a coordinated response to prepare the individualfor stress, and are often called the “fight-or-flight” reactions.
2. Synthesis of catecholamines: The catecholamines are synthe-sized from tyrosine, as shown in Figure 21.15. Tyrosine is firsthydroxylated by tyrosine hydroxylase to form 3,4-dihydroxy phenyl -
Figure 21.13Neonatal jaundice. GT = glucuronyl-transferase.
0 6 12 18 24 30Postnatal days
Serum levels of bilirubin rise after birth in full-term infants, although usually not to dangerous concentrations.
Serum levels of bilirubin in premature infants may rise totoxic levels.
1
2
3
Activity of the enzyme that conjugates bilirubin with glucuronic acid, bilirubin glucuronyltransferase, is low in newborns and especially low in premature babies.
III. Other Nitrogen-Containing Compounds 285
168397_P277-290.qxd7.0:21 Specialized products5-27-04 2010.4.4 6:17 PM Page 285

alanine (DOPA) in a reaction analogous to that described for thehydroxylation of phenylalanine (see p. 268). The tetrahydro-biopterin (BH4)-requiring enzyme is abundant in the central ner-vous system, the sympathetic ganglia, and the adrenal medulla,and is the rate-limiting step of the pathway. DOPA is decarboxy-lated in a reaction requiring pyridoxal phosphate (PLP, see p. 378)to form dopamine, which is hydroxylated by dopamine β-hydroxy-lase to yield nor epi nephrine in a reaction that requires ascorbate(vitamin C) and copper. Epinephrine is formed from nore-pinephrine by an N-methylation reaction using S-adenosylmethio-nine (SAM) as the methyl donor (see p. 264).
Parkinson disease, a neurodegenerative move-ment disorder, is due to insufficient dopamineproduction as a result of the idiopathic loss ofdopamine-producing cells in the brain.Administration of L-DOPA (levodopa) is the mostcommon treatment.
3. Degradation of catecholamines: The catecholamines are inacti-vated by oxidative deamination catalyzed by monoamine oxidase(MAO), and by O-methylation carried out by catechol-O-methyl-transferase using SAM as the methyl donor (Figure 21.16). Thetwo reactions can occur in either order. The aldehyde products ofthe MAO reaction are oxidized to the corresponding acids. Themetabolic products of these reactions are excreted in the urine asvanillylmandelic acid (VMA) from epinephrine and norepinephrine,and homovanillic acid from dopamine. [Note: VMA is increasedwith pheochromocytomas, tumors of the adrenal characterized byexcessive production of catecholamines.]
4. MAO inhibitors: MAO is found in neural and other tissues, suchas the intestine and liver. In the neuron, this enzyme oxidativelydeaminates and inactivates any excess neurotransmittermolecules (norepinephrine, dopamine, or serotonin) that may
286 21. Conversion of Amino Acids to Specialized Products
Figure 21.15Synthesis of catecholamines. PLP = pyridoxal phosphate.
HO
CH2CHCOO–
NH3+
Tyrosine
Tyrosinehydroxylase
Tetrahydro-biopterin
+ O2
Dihydro-biopterin+ H2O
HO
CH2CHCOO–
NH3+
3,4-Dihydroxy-phenylalanine
(dopa)
OH CO2
PLP
Cu+2
Ascorbate+ O2
Dehydro-ascorbate
+ H2O
Dopamineβ-hydroxylase
Phenylethanolamine-N-methyl-
transferase
Epinephrine
HO
C N
OH
CH H
OH H
H
CH3
Norepinephrine
HO
C NH2
OH
CH H
OH H
S-Adenosyl-methionine (SAM)
S-Adenosyl-homocysteine
DOPA-decarboxylase
HO
CH2CH2NH2
OH
Dopamine
Figure 21.16Metabolism of the catecholamines by catechol-O-methyltranferase (COMT) and monoamine oxidase (MAO).
Epinephrine Norepinephrine
Metanephrine Normetanephrine
Vanillylmandelic acid (VMA)
Homovanillicacid (HVA)
3-MethoxytyramineDihydroxyphenyl-acetic acid
Dopamine
MAO
COMT COMT
Dihydroxymandelic acid
MAO
COMT
MAO MAO
MAO COMT
COMT MAO
168397_P277-290.qxd7.0:21 Specialized products5-27-04 2010.4.4 6:17 PM Page 286

leak out of synaptic vesicles when the neuron is at rest. MAOinhibitors may irreversibly or reversibly inactivate the enzyme,permitting neurotransmitter molecules to escape degradationand, therefore, to both accumulate within the presynaptic neuronand to leak into the synaptic space. This causes activation ofnorepinephrine and serotonin receptors, and may be responsiblefor the antidepressant action of these drugs.
B. Histamine
Histamine is a chemical messenger that mediates a wide range ofcellular responses, including allergic and inflammatory reactions, gas-tric acid secretion, and possibly neurotransmission in parts of thebrain. A powerful vasodilator, histamine is formed by decarboxylationof histidine in a reaction requiring PLP (Figure 21.17). It is secreted bymast cells as a result of allergic reactions or trauma. Histamine has noclinical applications, but agents that interfere with the action of his-tamine have important therapeutic applications.
C. Serotonin
Serotonin, also called 5-hydroxytryptamine (5HT), is synthesizedand stored at several sites in the body (Figure 21.18). By far thelargest amount of serotonin is found in cells of the intestinalmucosa. Smaller amounts occur in the central nervous system,where it functions as a neurotransmitter, and in platelets. Serotoninis synthesized from tryptophan, which is hydroxylated in a BH4-requiring reaction analogous to that catalyzed by phenylalaninehydroxylase. The product, 5-hydroxy tryptophan, is decarboxylated toserotonin, which is also degraded by MAO. Serotonin has multiplephysiologic roles, including pain perception, regulation of sleep,appetite, temperature, blood pressure, cognitive functions, andmood (causes a feeling of well-being). [Note: Serotonin is convertedto melatonin in the pineal gland via acetylation and methylation.]
D. Creatine
Creatine phosphate (also called phosphocreatine), the phosphory-lated derivative of creatine found in muscle, is a high-energy com-pound that provides a small but rapidly mobilized reserve ofhigh-energy phosphates that can be reversibly transferred to ADP(Figure 21.9) to maintain the intracellular level of ATP during the firstfew minutes of intense muscular contraction. [Note: The amount ofcreatine phosphate in the body is proportional to the muscle mass.]
1. Synthesis: Creatine is synthesized from glycine and the guanidinogroup of arginine, plus a methyl group from SAM (see Figure21.19). Creatine is reversibly phosphorylated to creatine phos-phate by creatine kinase, using ATP as the phosphate donor.[Note: The presence of creatine kinase (MB isozyme) in theplasma is indicative of heart damage, and is used in the diagnosisof myocardial infarction (see p. 65).]
2. Degradation: Creatine and creatine phosphate spontaneouslycyclize at a slow but constant rate to form creatinine, which isexcreted in the urine. The amount of creatinine excreted is propor-
III. Other Nitrogen-Containing Compounds 287
CH2
NH2 COOHCH
N N
Histidine
CH2
NH2
N N
CH2
Histamine
Figure 21.17Biosynthesis of histamine. PLP =pyridoxal phosphate.
Decarboxylase
CPLP
O2
Figure 21.18Synthesis of serotonin. PLP =pyridoxal phosphate.
NH
CH2CHCOO–
NH3+
Tryptophan
NH
CH2CHCOO–
NH3+
Decarboxylase
5-Hydroxy-tryptophan
CO2
HO
NH
CH2CH2NH2
Serotonin
HO
O2
Hydroxylase
Tetrahydro-biopterin
PLP
Dihydro-biopterin+ H2O
168397_P277-290.qxd7.0:21 Specialized products5-27-04 2010.4.4 6:17 PM Page 287

tional to the total creatine phosphate content of the body, and thuscan be used to estimate muscle mass. When muscle massdecreases for any reason (for example, from paralysis or musculardystrophy), the creatinine content of the urine falls. In addition,any rise in blood creatinine is a sensitive indicator of kidney mal-function, because creatinine normally is rapidly removed from theblood and excreted. A typical adult male excretes about 15 mmolof creatinine per day.
E. Melanin
Melanin is a pigment that occurs in several tissues, particularly theeye, hair, and skin. It is synthesized from tyrosine in the epidermis bypigment-forming cells called melanocytes. Its function is to protectunderlying cells from the harmful effects of sunlight. [Note: A defect inmelanin production results in albinism, the most common form beingdue to defects in copper-containing tyrosinase (see p. 273).]
IV. CHAPTER SUMMARY
Amino acids are precursors of many nitrogen-containing compoundsincluding porphyrins, which, in combination with ferrous (Fe2+) iron,form heme (Figure 21.20). The major sites of heme biosynthesis arethe liver, which synthesizes a number of heme proteins (particularlycytochrome P450), and the erythrocyte-producing cells of the bonemarrow, which are active in hemoglobin synthesis. In the liver, the rateof heme synthesis is highly variable, responding to alterations in thecellular heme pool caused by fluctuating demands for hemeproteins. Incontrast, heme synthesis in erythroid cells is relatively constant, and ismatched to the rate of globin synthesis. Porphyrin synthesis start withglycine and succinyl CoA. The committed step in heme synthesis isthe formation of δ-aminolevulinic acid (ALA). This reaction is catalyzedby ALA synthase-1 in liver (inhibited by hemin, the oxidized form ofheme that accumulates in the cell when heme is being underutilized)and ALA synthase-2 in erythroid t issues (regulated by iron).Porphyrias are caused by inherited or acquired defects in heme syn-thesis, resulting in the accumulation and increased excretion of por-phyrins or porphyrin precursors. With few exceptions, porphyrias areinherited as autosomal dominant disorders. Degradation of hemepro-teins occurs in the reticuloendothelial system, particularly in the liverand spleen. The first step in the degradation of heme is the productionof the green pigment biliverdin, which is subsequently reduced tobilirubin. Bilirubin is transported to the liver, where its solubility isincreased by the addition of two molecules of glucuronic acid. Bilirubindiglucuronide is transported into the bile canaliculi, where it is firsthydrolyzed and reduced by bacteria in the gut to yield uro bilinogen,then oxidized by intestinal bacteria to ster co bilin. Jaundice refers tothe yellow color of the skin and sclera that is caused by deposition ofbilirubin, secondary to increased bilirubin levels in the blood. Threecommonly encountered type of jaundice are hemolytic jaundice,obstructive jaundice, and hepatocellular jaundice. Other important N-containing compounds derived from amino acids include the catechol -amines (dopamine, norepinephrine, and epinephrine), creatine,histamine, serotonin, and melanin.
288 21. Conversion of Amino Acids to Specialized Products
Figure 21.19Synthesis of creatine.
CH2
ArginineCOO–
HCNH3+
H20
CH2
CH2
NHC
NH2
NH2
H
GlycineCOO–
HCNH3++
Amidino-transferaseOrnithine
GuanidinoacetateCOO–CH2
NHC
NH2
NH2
+
+
Methyltransferase
S-Adenosylmethionine
S-Adenosylhomocysteine
CH2
CreatineCOO–
NCH3
C
NH2
NH2+
ATP
ADP + H+
ATP
ADPO
P–O O–
NH2+
C NH
NCH3
CH2
COO–
C NH
NCH3
CH2
CO
NH
Creatine phosphate
Creatinine
Creatinekinase
Pi
168397_P277-290.qxd7.0:21 Specialized products5-27-04 2010.4.4 6:17 PM Page 288

Glycine +
Succinyl CoA
Figure 21.20Key concept map for heme metabolism. = Block in the pathway.
Heme synthesis
Hemolytic jaundice Obstructive jaundice Hepatocellar jaundice
LIVER
Heme degradation
catalyzed by
Hepatic porphyrias
Heme
δ-Amino-levulinate synthase-1
δ-Amino-levulinate synthase-2
Protoporphyrinogen IX
Protoporphyrin IX
Uroporphyrinogen III
Hydroxymethylbilane
Coproporphyrinogen III
Porphobilinogen
δ-Aminolevulinic acid
Rate-limiting step
Lead
Lead
Iron
1
2
3
4
5
6
7
8
catalyzes catalyzes
lead to
Erythropoietic porphyrias
Inherited enzymedeficiencies inbone marrow
lead to 3 Acute intermittent porphyria5 Porphyria cutanea tarda6 Hereditary coproporphyria7 Variegate porphyria
4 Congenital erythropoietic porphyria
8 Erythropoietic protoporphyria
SPLEEN, LIVER
Heme
Bilirubin glucuronide
Bilirubin
Bilirubin-albumin
Biliverdin, CO, Fe2+
Hemoglobin,Cytochromes
Erythrocytes, heptocytes
Amino acids
Bilirubin
Bilirubin glucuronide
Urobilinogen
Bilirubin
Stercobilin(brown pigment in stools)
Urobilin(yellow pigment in urine)
Bilirubin glucuronide
Heme
Biliverdin, CO, Fe2+
Hemoglobin,Cytochromes
Amino acids
Erythrocytes, heptocytes
Bilirubin
Heme
Biliverdin, CO, Fe2+
Hemoglobin,Cytochromes
Amino acids
Erythrocytes, heptocytes
Heme
Biliverdin, CO, Fe2+
Hemoglobin
Amino acids
Bilirubin glucuronide
Lysed erythrocytes
Bilirubin glucuronideBilirubin glucuronide
Bilirubin
Neonatal jaundice
Heme
Biliverdin, CO, Fe2+
Hemoglobin,Cytochromes
Amino acids
Erythrocytes, heptocytes
BilirubinBilirubin
BONE MARROW
Glycine +
Succinyl CoA
Heme
Protoporphyrinogen IX
Protoporphyrin IX
Uroporphyrinogen III
Hydroxymethylbilane
Coproporphyrinogen III
Porphobilinogen
δ-Aminolevulinic acid1
2
3
4
5
6
7
8
Lead
Lead
occurs in occurs in
Inherited enzymedeficiencies in
liver
BLOOD
LIVER
INTESTINE
Urobilinogen
Bilirubin
Gallstone
StercobilinUrobilin
Urobilinogen
StercobilinUrobilin
Urobilinogen
Bilirubin
StercobilinUrobilin
StercobilinUrobilin
Urobilinogen
Bilirubin Bilirubin INTESTINE
catalyzed by
IV. Chapter Summary 289
168397_P277-290.qxd7.0:21 Specialized products5-27-04 2010.4.4 6:17 PM Page 289

Study Questions
Choose the ONE best answer.
21.1 δ-Aminolevulinic acid synthase activity:
A. in liver is frequently decreased in individualstreated with drugs, such as the barbiturate pheno-barbital.
B. catalyzes a rate-limiting reaction in porphyrinbiosynthesis.
C. requires the coenzyme biotin.D. is strongly inhibited by heavy metal ions such as lead.E. occurs in the cytosol.
21.2 The catabolism of hemoglobin:
A. occurs in red blood cells.B. involves the oxidative cleavage of the porphyrin ring.C. results in the liberation of carbon dioxide.D. results in the formation of protoporphyrinogen.E. is the sole source of bilirubin.
21.3 A 50-year-old man presented with painful blisters onthe backs of his hands. He was a golf instructor, andindicated that the blisters had erupted shortly afterthe golfing season began. He did not have recentexposure to poison ivy or sumac, new soaps ordetergents, or new medications. He denied havingprevious episodes of blistering. He had partial com-plex seizure disorder that had begun about threeyears earlier after a head injury. The patient hadbeen taking phenytoin—his only medication—sincethe onset of the seizure disorder. He admitted to anaverage weekly ethanol intake of about eighteen 12-oz cans of beer. The patient’s urine was reddishorange. Cultures obtained from skin lesions failed togrow organisms. A 24-hour urine collection showedelevated uroporphyrin (1,000 mg; normal, <27mg).The most likely presumptive diagnosis is:
A. porphyria cutanea tarda.B. acute intermittent porphyria.C. hereditary coproporphyria.D. congenital erythropoietic porphyria.E. erythropoietic protoporphyria.
21.4 A 10-year-old boy is referred to a specialist becauseof skin that blisters easily, urine that darkens onstanding, and stained teeth. Lab studies are remark-able for high levels of uroporphyrin I and copropor-phyrin I in plasma, with uroporphyrin I being presentin the urine. The most likely biochemical pathology inthis case is:
A. deficiency of ALA synthase.B. deficiency of bilirubin glucuronyltransferase.C. deficiency of uroporphyrinogen III synthase.D. down-regulation of tyrosinase.E. inhibition of ALA dehydratase by lead.
Correct answer = B. The activity of δ-aminolev -ulinic acid synthase controls the rate of por-phyrin synthesis. The hepatic form of theenzyme is increased in patients treated with cer-tain drugs, and requires pyridoxal phosphate asa coenzyme. Another enzyme in the pathway (δ-aminolevulinic acid dehydrase) is extremely sen-sitive to the presence of heavy metals.
Correct answer = A. The disease is associatedwith a deficiency in uroporphyrinogen decar-boxylase, but clinical expression of the enzymedeficiency is influenced by hepatic injury causedby iron overload, chronic ethanol consumption,and the presence of hepatitis B or C and HIVinfections. Exposure to sunlight can also be aprecipitating factor. Clinical onset is typicallyduring the fourth or fifth decade of life. Porphyrinaccumulation leads to cutaneous symptoms andurine that is red to brown. Treatment of thepatient's seizure disorder with phenytoin causedincreased synthesis of ALA synthase, and,therefore, of uroporphyrinogen, the substrate ofthe deficient enzyme. The laboratory and clinicalfindings are inconsistent with other porphyrias.
Correct answer = B. The cyclic heme moleculeis oxidatively cleaved to form biliverdin. Thecatabolism occurs in the cells of the reticulo -endothelial system, particularly the spleen, andresults in the liberation of carbon monoxide.Protoporphyrinogen is an intermediate in thesynthesis, not degradation, of heme.Cytochromes and other non-hemoglobin heme-proteins are also precursors of bilirubin.
290 21. Conversion of Amino Acids to Specialized Products
Correct answer = C. A deficiency of uropor-phyrinogen III synthase results in accumulationof hydroxymethylbilane and the spontaneousconversion of this substrate to porphyrins of theType I series. A deficiency of ALA synthase orinhibition of ALA dehydratase by lead would notallow the synthesis of porphobilinogen, the firstpyrrole product in the heme biosynthetic path-way, and thus would not result in uro- or copro-porphyrin synthesis. Deficiency of theglucuronyltransferase would not present with thesystems described, and lab studies would beremarkable for an elevation of unconjugatedbilirubin. Down-regulation of tyrosinase wouldresult in decreased pigmentation.
168397_P277-290.qxd7.0:21 Specialized products5-27-04 2010.4.4 6:17 PM Page 290


















