
Control # 848 Title: “Polkadots and Moonbeams” Neurocutaneous Syndromes Made Easy eEdE#...
40
Control # 848 Title: “Polkadots and Moonbeams” Neurocutaneous Syndromes Made Easy eEdE# eEdE-197
-
Upload
junior-montgomery -
Category
Documents
-
view
217 -
download
0
Transcript of Control # 848 Title: “Polkadots and Moonbeams” Neurocutaneous Syndromes Made Easy eEdE#...

Control # 848
Title: “Polkadots and Moonbeams” Neurocutaneous Syndromes Made Easy
eEdE# eEdE-197

Nothing To Disclose

“Polkadots and Moonbeams” Neurocutaneous Syndrome Made Easy
Nucharin Supakul, MD1
Chang Y Ho, MD2
1 Ramathibodi Hospital, Mahidol UniversityBangkok, Thailand
2 Riley Hospital for Children, Indiana University School of MedicineIndianapolis, Indiana, USA

Purpose
• To recognize characteristic imaging findings for the most common neurocutaneous syndromes
• To develop a search pattern for each disease
• To understand and be able to suggest a proper imaging technique for further evaluation and follow-up.

Introduction
• Neurocutaneous syndromeaka phakomatosesGroup of neuroectodermal disorders
• Central nervous system disorder associated with lesions in the skin, eye and other visceral organs

Case list
• Neurofibromatosis type 1
• Neurofibromatosis type 2
• Tuberous sclerosis• Ataxia telangiectasia
• Sturge -Weber syndrome
• Von Hipple-Lindau syndrome
• Neurocutaneous melanosis
• Basal nevous syndrome

NEUROFIBROMATOSIS TYPE 1

Neurofibromatosis type 1 • von Recklinghausen disease
• Most common neurocutaneous disorder• 1: 1,500 - 3,000
• 50% autosomal dominant, 50% spontaneous mutation
• Chromosome 17 (17q11.2)
• NF1 gene Neurofibromin protein
• Multisystemic manifestations (skin, CNS and orthopedic) Neurofibromas Café-au-lait spots Axillary or inguinal freckles Lisch nodule Skeletal dysplasia Malignant nervous system tumor
• Highly related to learning disability/autismwww.medicalobserver.com.auhttp://emedicine.medscape.com/article/1177266-overview#showallhttp://emedicine.medscape.com/article/1219222-overview

Neurofibromatosis type 1 • Diagnostic Criteria (≥ 2
findings for diagnosis)
≥ 6 café-au-lait spots
> 2 axillary or inguinal freckles
≥ 2 typical neurofibromatosis or 1 plexiform neurofibroma
Optic nerve glioma
≥ 2 Lisch nodules (Iris harmartomas)
Sphenoid dysplasia or typical long bone abnormalities
1st degree relative with NF1
• Classic Radiographic Findings Plexiform neurofibromas (target
appearance)
Sphenoid wing dysplasia
Optic nerve gliomas
Deep gray and white matter changes (myelin vacuolization / unidentified bright objects)
• Moya Moya pattern of vasculopathy
• Imaging Recommendation MRI with contrast

Neurofibromatosis type 1 10-year-old boy with café-au-late spots
A-B: Axial and coronal T2 images demonstrate extensive, ropy T2 hyperintense lesions with target appearance consistent with plexiform neurofibromas (orange arrows) along the right side of the face extending to the intra- and extraconal space of the right orbit and along the right Meckel cave.
Noted is right sphenoid wing dysplasia (blue star).
C: Coronal T1 post contrast image demonstrates diffuse heterogeneous enhancement of the plexiform neurofibromas (orange arrows).
D: Axial T2 image of a different patient (2 year-old girl with NF1) shows multiple bilateral foci of abnormal signal intensity in the deep cerebellar white matter and dentate nuclei consistent with NF related dysplasia/myelin vacuolization (pink arrows).
A B
C DBACK to case list

NEUROFIBROMATOSIS TYPE 2

Neurofibromatosis type 2
• 1: 25,000 – 30, 000
• 50% autosomal dominant, 50% spontaneous mutation
• Chromosome 22 (22q12)
• NF2 gene Merlin protein
• Manifestations (skin and CNS) Café-au-lait spots (few or
absent)
MISME (Multiple Inherited Schwannomas Meningiomas and Ependymomas)
• Late presentation and diagnosis less cutaneous manifestations compared to NF 1
• Clinical presentations: Hearing loss/ vertigo
http://flipper.diff.org/app/items/3683

Neurofibromatosis type 2 • Diagnostic Criteria:
Bilateral vestibular schwannomas or
1st degree relative with NF2 and 1 vestibular schwannoma or
1st degree relative with NF2 and 2 of the following findings
Neurofibroma Meningioma Glioma Schwannoma Posterior subcapsular
lenticular opacity
• Classic Radiographic Findings: MISME Multiple Inherited
Schwannomas Meningiomas Ependymomas
• Carefully check other cranial nerves in any new diagnosis of schwannoma or meningioma in a child or young adult
• Imaging Recommendation MRI with contrast, high
resolution T1+C to evaluate cranial nerves

Neurofibromatosis type 2 30-year-old female with history of headache and dizziness.A-B: Axial T1 postconstrast images show bilateral enhancing masses in the internal auditory canals, consistent with vestibular schwannomas (orange arrows). A large extra-axial mass with homogeneous enhancement along the posterior falx cerebri, consistent with a meningioma (blue star).
C-D: Axial T1 (C) and sagittal T1 (D) postcontrast images show a mixed enhancing mass in the medulla with a dorsal cyst, consistent with a ependymoma (pink arrows).
A homogeneous intensely enhancing mass is noted at the cranial cervical junction, consistent with a cervical meningioma (blue arrow).
A B
DCBACK to case list

TUBEROUS SCLEROSIS

Tuberous Sclerosis• 50% autosomal
dominant• 50% sporadic mutation• 1 : 10,000 • Clinical triad
Facial angiofibromas (90%)
Mental retardation (50-80%)
Seizure (80-90%)
http://www.e-ijd.org/viewimage.asp?img=IndianJDermatol_2013_58_5_346_117297_f7.jpg
A: Ash leaf maculeB: Adenoma sebaceum C: Periungual fibromaD: Shagreen patch

Tuberous SclerosisDiagnostic criteria: 2 major or 1 major+2 minor• Major:
Facial angiofibroma/forehead plaque sub-/periungual fibroma ≥ 3 hypomelanotic macules shagreen patch multiple retinal nodular hamartomas cortical tuber, Subependymal nodule (SEN) Subependymal giant cell astrocystoma (SEGA) Cardiac rhabdomyoma Lymphangioleiomyomatosis Renal angiomyolipoma
• Minor: Dental enamel pits Hamartomatous rectal polyps Bone cysts Cerebral WM radial migration lines (> 3 = major
sign) Gingival fibromas Nonrenal hamartoma Retinal achromic patch Confetti skin lesions Multiple renal cysts
• Classic Radiographic Findings: Cortical tuber Subependymal nodule
(<1.3 cm) Subepedymal giant cell
astrocytoma (> 1.3 cm)
• Imaging Recommendations: MRI brain with contrast FLAIR is the most
sensitive for white matter abnormalities

Tuberous Sclerosis5-year-old girl with seizure
A-B: Axial FLAIR images shows multiple areas of FLAIR hyper intensity involving bifrontoparietal and occipital cortical and juxtacortical white matter, consistent with cortical tuber (orange arrows).
C: Axial post contrast T1 image shows an enhancing lesion within the right foramen of Monro, measuring approximately 1.5 cm in size, consistent with subependymal giant cell astrocytoma (blue arrow).
D: Axial T2 image shows few T2 hypointense foci along the ependymal lining of the left lateral ventricle, consistent with subependymal nodules (pink arrows).
A B
C D
BACK to case list

ATAXIA TELANGIECTASIA

Ataxia telangiectasia• Rare
• Autosomal recessive
• 1: 40,000 - 100,000
• Chromosome 11 (11q33-23)
• ATM gene mutation
• Complex multi systemic disorder Cerebellar ataxia (incoordination and lack of
balance) Mucocutaneous telangiectasia Immune deficiency recurrent
bronchopulmonary and sinonasal infection Sensitive to ionizing radiation
• Increased risk of heart disease and cancer (leukemia/lymphoma in children and solid tumors in adult)
• Most common cause of death: recurrent sinopulmonary infection
http://en.wikipedia.org/wiki/Ataxia_telangiectasiahttp://dermatologyoasis.net/?s=ataxia+telangiectasia

Ataxia telangiectasia• Classic Radiographic Findings:
Progressive cerebellar volume loss Compensatory enlargement of the 4th ventricle Cerebral white matter dysmyelination/demyelination Microhemorrhages/ telangiectasis Decreased/ absent lymphoid tissue: adenoid gland,
thymus, mediastinal lymphoid tissue
• Imaging Recommendation: MRI due to no ionizing radiation and high tissue
contrast
7-year-old boy with history of autism and developmental delay
A and B: Sagittal T1 (A) and coronal T2 (B) images demonstrate severe atrophy of the cerebellar hemisphere with widening of the cerebellar folia and CSF space (orange arrows). Note the hypoplastic adenoid tissue for this age (blue arrow).
A
B
BACK to case list

STURGE-WEBER SYNDROME

Sturge-Weber Syndrome• Synonym: encephalotrigeminal
angiomatosis
• Rare
• Sporadic
• 1: 20,000 - 50,000
• Clinical presentation• Port wine stain (CN V1 98%)• Seizure (75-90%)• Hemiparesis (30-66%)
• Pathophysiology: failure to develop of fetal cortical vein plus persistent primordial vessels venous stasis hypoperfused cortex

Sturge-Weber Syndrome• Classic Radiographic
Findings: Ipsilateral to port wine stain
Gyral/subcortical white matter calcifications (tram-track calcification)
hemispheric brain atrophy
Serpentine leptomeningeal enhancement
Engorged/enlarged enhancing choroid plexi
• Location: Occipital > parietal >
fronto/temporal > midbrain > cerebellum
• 90% of infants with facial port wine stains do not have intracranial lesions and are expected to develop normally.
• Imaging Recommendation: MRI with contrast
leptomeningeal angiomatosis
GRE/SWI sequence gyral/subcortical calcification

Sturge-Weber Syndrome3-year-old girl with history of facial hemangioma and seizure
A: Non contrast CT image of the brain shows severe atrophy of the right cerebral hemisphere with extensive cortical/subcortical calcifications (orange arrows) in the right cerebral hemisphere and left parieto-occipital lobe.
B: GRE image shows hypointensity without blooming involving the right cerebral hemisphere and left occipital cortical/subcortical white matter, consistent with calcifications (orange arrows) seen on the prior CT.
C: Axial FLAIR image shows severe atrophy of the right cerebral hemisphere. Bilateral choroidal cysts are noted (blue arrows).
D: Post contrast T1 image demonstrates extensive leptomeningeal enhancement involving right cerebral cortical sulci and left parieto-occipital sulci (pink arrows) as well as avid enhancement of the choroid plexi bilaterally (yellow arrows).
Relative lack of cortical veins of the atrophied right cerebral hemisphere compared to the contralateral normal brain parenchyma and increased pial enhancement of the left parietal occipital lobe.
A B
C D
BACK to case list

VON HIPPLE-LINDAU SYNDROME

Von Hipple-Lindau Syndrome
• 80% Autosomal dominant (Variable expression and high penetrance; 97% by age of 65)
• 20% sporadic mutation
• Chromosome 3 (3p25-26)
• VHL gene VHL protein
• 1 : 36,000
• Multi Systemic manifestations: Less than 5% with skin manifestration
(Capillary malformation) Hemangioblastomas (Cerebellar 40-70%,
retinal 40-60% and spinal cord 10-60%) Pancreatic lesion (cyst 50-90%,
neuroendocrine tumor/ serous cystadenoma 5-15%)
Renal lesion (cyst 60%, renal cell carcinoma 20-45%)
Pheochromocytoma (0-60%) 15% of VHL patients develop endolymphatic
sac tumor; 30% bilateral • Clinical presentation:
Hemangioblastoma symptoms headache, dizziness, double vision, weakness
Pheochromocytoma symptoms high blood pressure, sweating, rapid/irregular heart rate, tremor, weight loss
http://emedicine.medscape.com/article/1084479-treatment#showallhttp://www.umd.be/VHL/W_VHL/clinic.shtml1

Von Hipple-Lindau Syndrome• Diagnostic Criteria
≥ 1 hemangioblastomas in the CNS (brain and spinal cord) or retina or
A hemangioblastoma in the CNS or retina plus a visceral manifestation or
Positive family history plus any of above manifestations
• Classic Radiographic Findings:
Mixed cystic lesion with peripheral nodular enhancement
Near pial surface
• Imaging Recommendation: MRI brain and spine with contrast
• NIH recommendation for screening:
MRI brain and spine with contrast from age of 11 then every 2 years
US abdomen from age of 11, then every year
CT abdomen from age of 20, then every year
MRI temporal bone if hearing loss/ tinnitus/ vertigo

Von Hipple-Lindau Syndrome20-year-old male with history of headache and back pain
A: Non contrast CT image shows a cystic lesion (orange asterisk) in the left cerebellar hemisphere with surrounding vasogenic edema. Mass effect on the 4th ventricle (red arrow), causes obstructive hydrocephalus (blue arrows).
B: Axial T1 post contrast image shows cystic lesions with mural nodular enhancement (pink arrows) in the left cerebellum and right-sided pons. Also noted are few nodular enhancing foci in the right cerebellar hemisphere (pink arrows).
C: Sagittal T1 post contrast image of the thoracic spine demonstrates several enhancing lesions in the spinal cord (yellow arrows).
D: Coronal contrasted CT image of the abdomen shows multiple heterogeneous enhancing lesions scattered through both kidneys (green arrows), pathologically proven renal cell carcinoma.
A B
C D
BACK to case list

NEUROCUTANEOUS MELANOSIS

Neurocutaneous melanosis
• Rare genetic disorder
• 100+ case reports
• Mutation of NRAS gene
• Clinical presentation: melanocytic deposition within Skin Giant or multiple
melanocytic nevi CNS
• Diagnostic Criteria: Melanocytic deposition within the CNS and
skin lesions Important to establish that cutaneous
lesions are benign or malignant to rule out metastasis of cutaneous melanoma
• 5-15% lifetime risk of malignant transformation
• Can have poor prognosis with median survival of 6.5 months after symptom onset
• Classic Radiographic Findings: T1 shortening with susceptibility artifact on
GRE or SWI No contrast enhancement Locations: amygdala, cerebellum,
brainstem, inferior frontal lobes and thalami
• Imaging Recommendation: MRI with contrast of the brain and spine GRE/ SWI sequences
http://pediatricneuro.com/alfonso/pg303.htm

Neurocutaneous melanosis13-day-old term infant with multiple congenital nevi
A-B: Axial T1 noncontrast images demonstrate a wedge-shaped T1 hyperintensity in the right caudal thalamic groove and foci of T1 hyperintensity in the right medial temporal lobe/ amygdala (orange arrows).
C-D: Post contrast T1 image demonstrates no contrast enhancement within these lesions.
A B
C D
BACK to case list

BASAL CELL NEVUS SYNDROME

Basal cell nevus syndrome• Synonym: Gorlin syndrome• 2/3 Autosomal dominant with
complete penetrance and variable expressivity
• 1/3 sporadic mutation• 1 : 31,000• Chromosome 9 (9q22.1-q31)• PICH1 gene patched-1 protein
http://www.nlm.nih.gov/medlineplus/ency/imagepages/3190.htm

Basal cell nevus syndromeDiagnostic criteria (2 majors, or 1 major + 2 minor)• Major criteria
Early development of multiple basal cell carcinomas
Greater than 10 basal cell nevi Odontogenic keratocysts, commonly in the
mandible Palmar and plantar pitting Ectopic intracranial calcification, most often
along falx cerebri Family history
• Minor criteria Craniofacial anomalies (macrocephaly,
frontal bossing, hypertelorism) Bifid ribs Early onset desmoplastic medulloblastoma Cardiac or ovarian fibroma, often bilateral Lymphomesenteric cysts Congenital malformation (cleft lip/palate,
polydactyly, eye abnormalties- colobomas, cataract, glaucoma
•
• Classic Radiographic Findings: Multiple odontogenic keratocysts Early dural calcifications Basal cell nevi and carcinomas
• Odontogenic keratocyst (keratocytic odontogenic tumor: KOT) in Gorlin syndrome usually develop earlier than non-syndromic KOT
• Most patients with basal cell nevus syndrome have KOT; however, only 5% of patients with KOT have basal cell nevus syndrome.
• Associated with desmoplastic medulloblastoma
• Imaging Recommendation: MRI to screen for medulloblastoma, cystic
jaw lesion CT of face for oral surgery planning
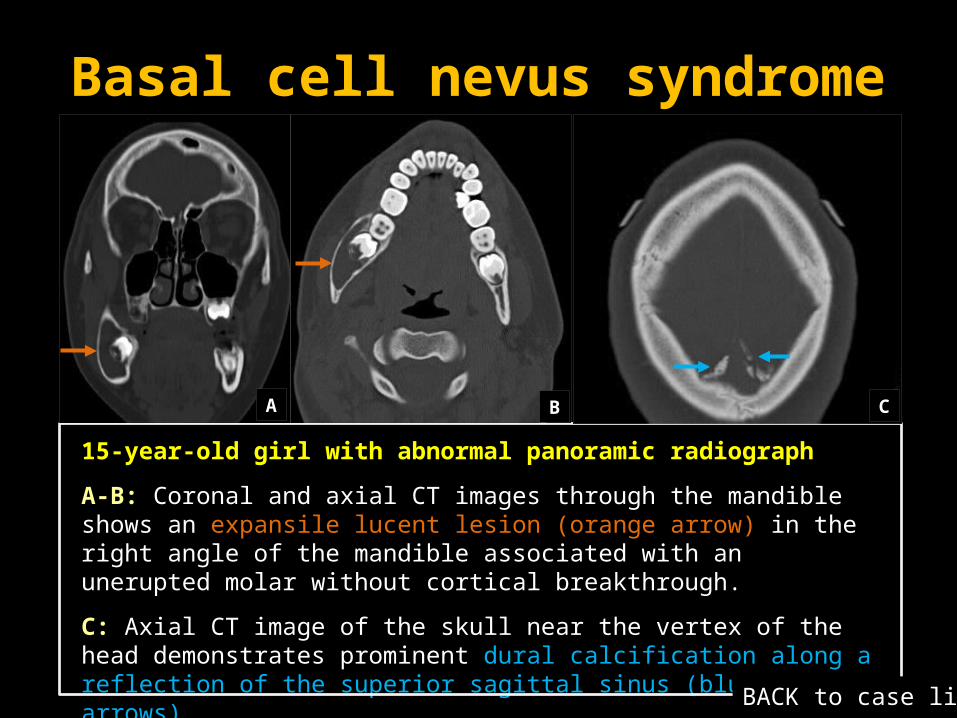
Basal cell nevus syndrome
15-year-old girl with abnormal panoramic radiograph
A-B: Coronal and axial CT images through the mandible shows an expansile lucent lesion (orange arrow) in the right angle of the mandible associated with an unerupted molar without cortical breakthrough.
C: Axial CT image of the skull near the vertex of the head demonstrates prominent dural calcification along a reflection of the superior sagittal sinus (blue arrows).
A B C
BACK to case list

SummaryCutaneous
manifestationsCNS
manifestationsOther organ
manifestations Key images
NF1 • Neurofibroma• Café-au-lait spots• Axillary or inguinal
freckles
• Plexiform neurofibroma
• Optic nerve glioma• myelin vacuolization • Sphenoid wing
dysplasia
• Lisch nodule• Skeletal dysplasia
• MRI with contrast
• T2 target lesion plexiform neurofibroma
NF2 • Few café-au-lait spot • MISME - • MRI brain and spine with contrast
• High resolution cranial nerve
Tuberous sclerosis
• Ash leaf macule• Adenoma sebaceum • Periungual fibroma• Shagreen patch
• Cortical tuber• Subependymal
nodules• SEGA
• Cardiac rhabdomyoma• Lymphangiomyomatosis• Renal AML
• MRI brain with contrast
• FLAIR to detect cortical tuber
Ataxia telangiectasia
• Mucocutaneous telangiectasia
• Progressive cerebellar volume loss
• Atrophy of lymphoid tissue, thymus, adenoid gland
• MRI brain to avoid ionizing radiation
Sturge-Weber Syndrome
• Port wine stain along trigeminal nerve
• Gyral/ subcortical calcification
• Hemispheric atrophy• Sepentine
leptomeningeal enhancement
• Engored/ enlarged choroid plexus
- • MRI brain with contrast leptomeningeal angiomatosis
• SWI/GRE for gyral calcification

SummaryCutaneous
manifestationsCNS
manifestationsOther organ
manifestations Key images
Von Hipple-Lindau Syndrome
• Capillary malformation (less than 5% of patients)
• Hemangioblatoma (retina, cerebellum, spine)
• Pancreas (cyst, neuroendocrine tumor, serous cystadenoma)
• Renal (cyst, RCC)• phreochromocytoma• Endolymphatic sac
tumor
• MRI brain and spine with contrast
• screening - US and CT
abdomen- MRI temporal
bone
Neurocutaneous melanosis
• Giant or multiple melanocytic nevi
• Melanocytic deposition within the CNS with no contrast enhancement
- • MRI brain and spine with contrast
• GRE/SWI sequence
Basal nervous syndrome
• Basal cell nevi • Early dural calficification
• Odontogenic keratosis
• Desmoplastic medulloblastoma
• Bifid ribs• Cardiac or ovarian
firbromas• Lymphomesenteric
cyst• Cleft lip/palate• Polydactyly• Eye abnormalities
(colobomas, cataract, glaucoma)
• MRI /CT to demonstrate odontogenic cyst and dural calcification

Conclusion
• Neurocutaneous syndromes are a heterogeneous group of disorders that primarily affect the central nervous system with cutaneous manifestations.
• Cross-sectional imaging, especially MRI, can be tailored to help the diagnosis and guide further evaluation and treatment.
• Radiologists should be familiar with the characteristic imaging findings of specific neurocutaneous syndromes and develop a search pattern for potential associated lesions once a diagnosis is favored.

References1. www.medicalobserver.com.au
2. http://emedicine.medscape.com/article/1177266-overview#showall
3. http://emedicine.medscape.com/article/1219222-overview
4. http://flipper.diff.org/app/items/3683
5. http://www.e-ijd.org/viewimage.asp?img=IndianJDermatol_2013_58_5_346_117297_f7.jpg
6. http://en.wikipedia.org/wiki/Ataxia_telangiectasia
7. http://dermatologyoasis.net/?s=ataxia+telangiectasia
8. http://emedicine.medscape.com/article/1084479-treatment#showall
9. http://www.umd.be/VHL/W_VHL/clinic.shtml
10. http://pediatricneuro.com/alfonso/pg303.htm
11. http://www.nlm.nih.gov/medlineplus/ency/imagepages/3190.htm
12. Elster AD Radiologic screening in the neurocutaneous syndromes: strategies and controversies. AJNR Am J Neuroradiol. 1992 Jul-Aug;13(4):1078-82.
13. Tortori-Donati, Paolo, et al. "Phakomatoses." Pediatric Neuroradiology. Springer Berlin Heidelberg, 2005. 763-818.
14. Pont MS, and Elster AD. Lesions of skin and brain: modern imaging of the neurocutaneous syndromes. American Journal of Roentgenology 1992 158:6 , 1193-1203
15. Coats, David K., Evelyn A. Paysse, and Moise L. Levy. "PHACE: a neurocutaneous syndrome with important ophthalmologic implications: case report and literature review." Ophthalmology 106.9 (1999): 1739-1741.
16. Roach, E. S. "Neurocutaneous syndromes." Pediatric Clinics of North America 39.4 (1992): 591-620.


















