
Connective Tissue Growth Factor CCN2 Interacts with and Activates ...
12
Connective Tissue Growth Factor CCN2 Interacts with and Activates the Tyrosine Kinase Receptor TrkA Nadia Abdel Wahab, Benjamin S. Weston, and Roger M. Mason Cell and Molecular Biology Section, Division of Biomedical Sciences, Faculty of Medicine, Imperial College London, South Kensington, London, United Kingdom Connective tissue growth factor (CTGF) is implicated as a factor promoting tissue fibrosis in several disorders, including diabetic nephropathy. However, the molecular mechanism(s) by which it functions is not known. CTGF rapidly activates several intracellular signaling molecules in human mesangial cells (HMC), including extracellular signal–related kinase 1/2, Jun NH 2 -terminal kinase, protein kinase B, CaMK II, protein kinase C, and protein kinase C, suggesting that it functions via a signaling receptor. Treating HMC with CTGF stimulated tyrosine phosphorylation of proteins 75 to 80 and 140 to 180 kD within 10 min, and Western blot analysis of anti-phosphotyrosine immunoprecipitates identified the neurotrophin receptor TrkA (molecular weight approximately 140 kD). Cross-linking rCTGF to cell surface proteins with 3,3-dithiobis(sulfosuccin- imidylpropionate) revealed that complexes formed with TrkA and with the general neurotrophin co-receptor p75 NTR . rCTGF stimulated phosphorylation of TrkA (tyr 490, 674/675). K252a, a known selective inhibitor of Trk, blocked this phosphoryla- tion, CTGF-induced activation of signaling proteins, and CTGF-dependent induction of the transcription factor TGF-– inducible early gene in HMC. It is concluded that TrkA serves as a tyrosine kinase receptor for CTGF. J Am Soc Nephrol 16: ???-???, 2005. doi: 10.1681/ASN.2003100905 C onnective tissue growth factor (CTGF; CCN2) is a 38-kD secreted protein with multiple domains, en- coded by an immediate-early gene, and is a member of the CCN protein family (1). An increasing number of studies in the past few years indicate that CTGF is strongly implicated in the pathogenesis of a variety of fibrotic disor- ders, including diabetic nephropathy (2). Functionally, the growth factor has been shown to regulate many aspects of cell behavior, including cell adhesion, migration, growth, chemotaxis, and the synthesis and accumulation of matrix proteins (3). However, the molecular mechanism(s) by which it functions is not clear. The presence of multiple domains in CTGF suggests a role for the growth factor as an integrator of several other signaling molecules such as growth factors, integrins, and extracellular matrix proteins. CTGF has been reported to bind directly bone morphogenic protein and TGF- through its von Willebrand type C domain, leading to inhibition of bone morphogenic protein and enhancement of TGF- signaling (4). CTGF also has been shown to bind to integrins (5–10), and this interaction may mediate some of the cellular phenomena mentioned above. Recently, we proposed that CTGF directly enhances the TGF- /Smad signaling pathway. The mechanism underlying this seems to be via the rapid induction of the transcription factor TGF-–inducible early gene (TIEG) (11). TIEG has been shown to bind to the promoter of the Smad7 gene and represses its tran- scription (12). Because Smad7 is a potent inhibitory Smad, it seems that CTGF simply blocks the negative feedback loop of the TGF- signaling pathway, allowing its continued activation. The rapid activation of several intracellular signaling pathways by CTGF, together with its ability to induce the expression of several genes in mesangial cells, including the early gene TIEG, suggested that a specific signaling receptor must exist. Previous cross-linking studies revealed CTGF-receptor complexes with an apparent mo- lecular weight of 280 kD, present in chondrocytes, osteoblasts, and endothelial cells (13). CTGF was also found to bind to LDL recep- tor–related protein. However, this may facilitate clearance rather than signal transduction (14). In the present report, we provide for the first time evidence that CTGF interacts with TrkA and p75 NTR , a dual-receptor system that is known to transduce neurotrophin signals. There are three Trk receptor tyrosine kinase genes (TrkA, TrkB, and TrkC) and a single gene encoding the neurotrophin receptor, p75 NTR . On ligand binding, Trk dimerize and autophosphory- late, leading to the activation of several small G proteins, in- cluding Ras, Rap-1, and the Cdc 42-Rac-Rho family, as well as of pathways regulated by mitogen activated protein kinase (MAPK), phosphatidylinositol 3-kinase, and phospholipase C- (15). On activation, Trk also promote a rapid increase in cyto- plasmic calcium level. This seems to arise from both release of intracellular stores and uptake of extracellular calcium (16,17). Moreover, activated Trk interact, directly or indirectly, with Received October 31, 2003. Accepted October 25, 2004. Published online ahead of print. Publication date available at www.jasn.org. Address correspondence to: Dr. Nadia Wahab, Renal Section, Division of Med- icine, Imperial College London, Hammersmith Hospital, Du Cane Road, London, W12 ONN, UK. Phone: 44-20-838-32718; Fax: 44-20-838-32062; E-Mail: [email protected] The current affiliation for N.A.W. and R.M.M. is Renal Section, Division of Medicine, Imperial College London, Hammersmith Hospital, London, United Kingdom. Copyright © 2005 by the American Society of Nephrology ISSN: 1046-6673/1602-0001 . Published on December 15, 2004 as doi: 10.1681/ASN.2003100905 JASN Express
Transcript of Connective Tissue Growth Factor CCN2 Interacts with and Activates ...

Connective Tissue Growth Factor CCN2 Interacts with andActivates the Tyrosine Kinase Receptor TrkA
Nadia Abdel Wahab, Benjamin S. Weston, and Roger M. MasonCell and Molecular Biology Section, Division of Biomedical Sciences, Faculty of Medicine, Imperial College London,South Kensington, London, United Kingdom
Connective tissue growth factor (CTGF) is implicated as a factor promoting tissue fibrosis in several disorders, includingdiabetic nephropathy. However, the molecular mechanism(s) by which it functions is not known. CTGF rapidly activatesseveral intracellular signaling molecules in human mesangial cells (HMC), including extracellular signal–related kinase 1/2,Jun NH2-terminal kinase, protein kinase B, CaMK II, protein kinase C�, and protein kinase C�, suggesting that it functionsvia a signaling receptor. Treating HMC with CTGF stimulated tyrosine phosphorylation of proteins 75 to 80 and 140 to 180 kDwithin 10 min, and Western blot analysis of anti-phosphotyrosine immunoprecipitates identified the neurotrophin receptorTrkA (molecular weight approximately 140 kD). Cross-linking rCTGF to cell surface proteins with 3,3�-dithiobis(sulfosuccin-imidylpropionate) revealed that complexes formed with TrkA and with the general neurotrophin co-receptor p75NTR. rCTGFstimulated phosphorylation of TrkA (tyr 490, 674/675). K252a, a known selective inhibitor of Trk, blocked this phosphoryla-tion, CTGF-induced activation of signaling proteins, and CTGF-dependent induction of the transcription factor TGF-�–inducible early gene in HMC. It is concluded that TrkA serves as a tyrosine kinase receptor for CTGF.
J Am Soc Nephrol 16: ???-???, 2005. doi: 10.1681/ASN.2003100905
C onnective tissue growth factor (CTGF; CCN2) is a38-kD secreted protein with multiple domains, en-coded by an immediate-early gene, and is a member
of the CCN protein family (1). An increasing number ofstudies in the past few years indicate that CTGF is stronglyimplicated in the pathogenesis of a variety of fibrotic disor-ders, including diabetic nephropathy (2). Functionally, thegrowth factor has been shown to regulate many aspects ofcell behavior, including cell adhesion, migration, growth,chemotaxis, and the synthesis and accumulation of matrixproteins (3). However, the molecular mechanism(s) by whichit functions is not clear. The presence of multiple domains inCTGF suggests a role for the growth factor as an integrator ofseveral other signaling molecules such as growth factors,integrins, and extracellular matrix proteins. CTGF has beenreported to bind directly bone morphogenic protein andTGF-� through its von Willebrand type C domain, leading toinhibition of bone morphogenic protein and enhancement ofTGF-� signaling (4). CTGF also has been shown to bind tointegrins (5–10), and this interaction may mediate some ofthe cellular phenomena mentioned above.
Recently, we proposed that CTGF directly enhances the TGF-�/Smad signaling pathway. The mechanism underlying thisseems to be via the rapid induction of the transcription factorTGF-�–inducible early gene (TIEG) (11). TIEG has been shown tobind to the promoter of the Smad7 gene and represses its tran-scription (12). Because Smad7 is a potent inhibitory Smad, it seemsthat CTGF simply blocks the negative feedback loop of the TGF-�signaling pathway, allowing its continued activation. The rapidactivation of several intracellular signaling pathways by CTGF,together with its ability to induce the expression of several genesin mesangial cells, including the early gene TIEG, suggested thata specific signaling receptor must exist. Previous cross-linkingstudies revealed CTGF-receptor complexes with an apparent mo-lecular weight of 280 kD, present in chondrocytes, osteoblasts, andendothelial cells (13). CTGF was also found to bind to LDL recep-tor–related protein. However, this may facilitate clearance ratherthan signal transduction (14).
In the present report, we provide for the first time evidencethat CTGF interacts with TrkA and p75NTR, a dual-receptorsystem that is known to transduce neurotrophin signals. Thereare three Trk receptor tyrosine kinase genes (TrkA, TrkB, andTrkC) and a single gene encoding the neurotrophin receptor,p75NTR. On ligand binding, Trk dimerize and autophosphory-late, leading to the activation of several small G proteins, in-cluding Ras, Rap-1, and the Cdc 42-Rac-Rho family, as well asof pathways regulated by mitogen activated protein kinase(MAPK), phosphatidylinositol 3-kinase, and phospholipase C-�(15). On activation, Trk also promote a rapid increase in cyto-plasmic calcium level. This seems to arise from both release ofintracellular stores and uptake of extracellular calcium (16,17).Moreover, activated Trk interact, directly or indirectly, with
Received October 31, 2003. Accepted October 25, 2004.
Published online ahead of print. Publication date available at www.jasn.org.
Address correspondence to: Dr. Nadia Wahab, Renal Section, Division of Med-icine, Imperial College London, Hammersmith Hospital, Du Cane Road, London,W12 ONN, UK. Phone: �44-20-838-32718; Fax: �44-20-838-32062; E-Mail:[email protected]
The current affiliation for N.A.W. and R.M.M. is Renal Section, Division ofMedicine, Imperial College London, Hammersmith Hospital, London, UnitedKingdom.
Copyright © 2005 by the American Society of Nephrology ISSN: 1046-6673/1602-0001
. Published on December 15, 2004 as doi: 10.1681/ASN.2003100905JASN Express

many cytoplasmic adaptor proteins, leading to a variety ofbiologic responses, including, cell proliferation and survival;axonal and dendritic growth and remodeling; assembly andremodeling of cytoskeleton; membrane trafficking and fusion;and synapse formation, function, and plasticity (18).
The pan-neurotrophin receptor p75NTR is a member of asuperfamily that includes the TNF, B cell antigen CD40, and Fasreceptors (19). p75NTR signals via pathways involved with ac-tivation of sphingomyelinase and ceramide production (20),NF-�B (21), and Jun NH2-terminal kinase (JNK) (22). Althoughboth receptors mediate the biologic effects of neurotrophins, itseems that Trk play the central role in signaling. However, thecontribution of p75NTR is not fully understood. It has beenproposed that p75NTR can act as a co-receptor with Trk to createhigh-affinity receptors for different neurotrophins (23) either bypresenting the ligand to the Trk receptor, or by altering theconformation of the Trk receptors through allosteric interaction(24). It can also function on its own (25). Yet another interestingfinding is that p75NTR can function as a receptor for the imma-ture unprocessed forms of neurotrophins, whereas Trk func-tions as a receptor for the mature forms (26). Cross-talk be-tween the Trk and other membrane receptors, such as Gprotein–coupled receptor, vannilloid receptor, and c-Ret, havealso been reported (18).
Ligand engagement stimulates the endocytosis of the ligand–Trk complex into vesicles via a clathrin-dependent mechanism(27). The receptors remain catalytically active within the sortingvesicles, which are called “signaling endosomes” (28). Inhibit-ing endocytosis inhibits extracellular signal–related kinase(ERK) 1/2 activation in response to neurotrophin stimulation(29).
As Trk receptors have been found to bind a large number ofadaptor proteins and multiple intracellular signaling pathwaysare activated by them, as well as modulated by p75NTR, thismay explain the multifunctional properties of CTGF. The re-sults presented in this study show that Trk tyrosine kinaseactivity is required for the CTGF-dependent induction of thetranscription factor TIEG.
Materials and MethodsCell Cultures, Antibodies, and Reagents
Primary normal adult human mesangial cells (HMC; CC-2259, lot3F1510) were purchased from BioWhittaker (Wokingham, Berkshire,UK), maintained in culture as described previously (2), and used atpassages 9 to 10. Phospho-Akt (P-Ser 472/473/474) antibody was fromPharmingen (San Diego, CA). Phospho-Akt (P-Thr 308) and ERK5antibodies were from Sigma (Gillingham, Dorset, UK). Phospho-ERK1/2 pathway sampler, phospho-JNK pathway sampler, phosphoP38 MAPK pathway sampler, phospho-PKC�, phospho-PKC�, phos-pho-TrkA (Tyr674/675), and phospho-TrkA (Tyr490) antibodies werefrom New England BioLabs (Hitchen, Herts, UK). Phospho-CaMKII(P-Thr286) antibody was from Promega (Southampton, Hants, UK),and anti-phosphotyrosine antibody was from Santa Cruz (AutogenBioclear, Calne, Wilts, UK). Anti-TrkA antibody was obtained fromUpstate Biotechnology (Milton Keynes, UK). Anti–TIEG-1 antibodywas a gift from Dr. Steven Johnson (Mayo Foundation, Rochester, MN).K-252a was purchased from Calbiochem (Nottingham, UK). Recombi-
nant CTGF (CTGF/V5 fusion protein) was expressed in transformedHMC and purified from the medium using Talon metal affinity resin,as reported previously (2). Alternatively, r-CTGF (nonfusion protein)was expressed in the baculovirus system and was a gift from FibroGenInc. (South San Francisco, CA). Rabbit anti-CTGF (pAb2) and chickenanti-CTGF (pIgY3) were also supplied by FibroGen Inc. Phosphothioateantisense and control oligonucleotides directed to TrkA (GTGAAGAT-GAAGCTGGT; ACTACTACACTAGACTAC) and p75NTR (TTCTGCT-TGTCCTG; GCTCTATGACTCCCAG) were designed and manufac-tured by Biognostik GmbH (Gottingen, Germany), who own theintellectual property rights to the sequences.
Cross-Linking and Membrane PreparationCell layers were washed twice with cold binding buffer (PBS and
0.5% glucose) and incubated with CTGF in binding buffer for 2 h at 4°C.After incubation, the cell layers were washed five times with coldbinding buffer and incubated with 1 mM 3,3�-dithiobis(sulfosuccinimi-dylpropionate) (DTSSP) or disuccinimidyl suberate (Pierce Biotechnol-ogy, Tattenhall, Cheshire, UK) in PBS for 30 min at room temperature.The reaction was quenched for 15 min at room temperature by theaddition of 50 mM Tris buffer (pH 7.5). Cell layers were washed withwash buffer (10 mM Tris buffer [pH 7.5], 5 mM MgCl, and 150 mMNaCl), scraped in homogenizing buffer (10 mM Tris buffer [pH 7.5], 250mM sucrose, 1 mM EDTA, 5 mM MgCl, 150 mM NaCl, and 1� proteaseinhibitor cocktail; Roche Applied Science, Mannheim, Germany),passed through a 25-G needle, and homogenized on ice with 30 to 40cycles in a Dounce homogenizer. The homogenate was centrifuged for10 min at 2500 � g at 4°C. The resulting supernatant was centrifugedfor 90 min at 45,000 � g at 4°C. The membrane-enriched pellet wassolubilized for 1 h in solubilizing buffer (10 mM Tris buffer [pH 7.5], 5mM MgCl, 150 mM NaCl, 1% Triton-X100, and 1� protease inhibitorcocktail). Soluble membrane proteins were collected after further cen-trifugation for 1 h at 45,000 � g at 4°C. When rCTGF/V5 fusion proteinwas used, CTGF cross-linked proteins were either immunoprecipitatedwith rabbit anti-CTGF antibody or captured on a Pull-Down PolyHiscolumn (Pierce). When rCTGF was used, CTGF cross-linked proteinswere captured on a goat anti–CTGF-C terminal domain–sepharoseimmunoaffinity column, using an IgG-sepharose column as a control(FibroGen Inc.). After extensive washing of the columns with solubi-lizing buffer, bound proteins were solubilized in reducing SDS-PAGEloading buffer, boiled for 5 min, and resolved on 4 to 12% gradient gelsby SDS-PAGE. Gels were either stained with Coomassie blue or usedfor Western blotting.
RNA Extraction and Reverse Transcriptase–PCR AnalysisTotal RNA was extracted from 6 � 106 mesangial cells using the
RNAzol B method (AMS Biotechnology [UK] Ltd., Oxfordshire, UK).Equal amounts of total RNA (2 �g) from each sample were reverse-transcribed into cDNA using SuperScript II RNase H� reverse tran-scriptase (Life Technologies BRL, Paisley, Scotland, UK) and randomprimers. Equal amounts (0.5 �l) of the reverse transcription reaction (20�l) were subjected to PCR amplification in a 100-�l volume that con-tained 10 �l of 10� PCR buffer, 16 �l of dNTP (1.25 mM each), 2 mMMgCl2, 5 M betaine (Sigma), 0.5 �M of each specific primer, and 1.25 Uof Amplitaq DNA polymerase (Life Technologies BRL). Amplificationwas started with 5 min of denaturation at 94°C followed by 30 PCRcycles for all genes. Each cycle consisted of 60 s at 94°C, 60 s at 55°C,and 60 s at 72°C. The final extension was for 10 min at 72°C. A controlomitting the reverse transcriptase step was also performed. The se-quences of primers were as described by Anderson et al. (30).
2 Journal of the American Society of Nephrology J Am Soc Nephrol 16: ???-???, 2005

Western BlottingCells were lysed in reducing SDS-PAGE loading buffer and imme-
diately scraped off the plate. Cell lysates were sonicated for 10 s toshear DNA. Samples were boiled for 5 min and resolved on 4 to 12%gradient gels by SDS-PAGE. Proteins were transferred onto a polyvi-nylidene difluoride membrane filter (Immobilin-P; Millipore, Bedford,UK) using a BioRad transfer apparatus. Blots were incubated in block-ing buffer that contained 1� TBS and 0.1% Tween-20 with 5% (wt/vol)nonfat dry milk for 1 h. Immunodetection was performed by incubatingthe blots in primary antibody at the appropriate dilution in antibodydilution buffer (1� TBS and 0.1% Tween-20 with 5% BSA) overnight at4°C. Blots then were washed three times with washing buffer (1� TBSand 0.1% Tween-20) and incubated with secondary horseradish peroxi-dase–conjugated antibodies for 1 h at room temperature. Bound anti-bodies were visualized using the enhanced chemiluminescence reagentLuminol (Autogen Bioclear UK Ltd, Wiltshire, UK). Prestained molec-ular weight standards (Amersham International PLC, Amersham, UK)were used to monitor protein migration.
Immunofluorescence StainingCells were fixed with 3.7% paraformaldehyde and permeabilized
with 0.5% Triton X-100 in PBS for 10 min at room temperature. Cov-erslips then were incubated overnight at 4°C with serum (5% in PBS)from the same species as that in which the secondary antibody wasraised. After this, they were incubated with primary antibodies (atoptimum dilution in PBS that contained 3% BSA) for 1 h at 37°C.Coverslips then were washed and incubated in the dark for 1 h withfluorescein-conjugated secondary antibody (Sigma Aldrich, Dorset,UK). After staining, the coverslips were mounted on glass slides withantifade mounting media (Vector Labs, Peterborough, UK) and exam-ined using a fluorescence microscope.
ResultsCTGF Activates Several Intracellular Signaling Pathways
To understand the molecular mechanisms by which CTGFfunctions, we used purified rCTGF-V5 fusion protein to iden-tify the intracellular signal pathways that are activated in re-sponse to the growth factor in HMC. We found that CTGFrapidly triggers the activation of the classical MAPK (ERK1/2)and JNK pathways (Figure 1, A and B) but not the p38 MAPK(data not shown). The figure shows maximal activation of thesekinases after 15 min of CTGF stimulation. CTGF stimulationalso led to the activation of Akt, also known as protein kinaseB (PKB), at both the known phosphorylation sites: thr-308 andser-473 (Figure 1C). It is interesting that the activation of thr-308(by a phosphoinositide-dependent kinase 1 whose activity isstrictly dependent on 3-phosphorylated inositol lipids [31])seems to be rapid and sustained in comparison with the acti-vation of ser-473. Phosphorylation of the latter (by integrin-linked kinase [32]) seems to be transient with a maximal level at15 min and return to a level close to the basal one within 30 minof CTGF exposure. CTGF stimulation also led to the transientactivation of CaMK II (Figure 1C). Other kinases that seem to beactivated in response to CTGF are PKC� and PKC� (Figure 1D).These results indicate that CTGF signals through a receptor,most probably a receptor tyrosine kinase (RTK), because RTKare commonly known to activate these kinases. To test thishypothesis, we exposed HMC to CTGF for various periods oftime, prepared cell lysates, and performed Western Blot anal-
ysis using an anti-phosphotyrosine antibody. Short-term expo-sure of the blot (�5 s) showed that CTGF fusion protein (40ng/ml) stimulates the tyrosine phosphorylation within 10 minof at least two major proteins with apparent molecular weightof approximately 75 to 80 and 140 to 150 kD in HMC (Figure 2).Another phosphotyrosine protein (molecular weight 45 kD)was detected in control cell lysate and seems to be reduced inresponse to the CTGF treatment.
CTGF Interacts with HMC Surface ProteinsTo investigate whether CTGF interacts with HMC surface
proteins, we allowed CTGF to bind to the cell surface, per-formed a subsequent cross-linking procedure, and isolated amembrane-enriched fraction from the cells. After solubilization,this was immunoprecipitated with a rabbit anti-CTGF anti-body. Covalently linked CTGF complexes then were analyzedby PAGE and Western blotting with a chicken anti-CTGF an-tibody. As shown in Figure 3, lane 2, CTGF seems to be cross-linked with membrane proteins to form complexes of apparentmolecular weight 180 kD and �220 kD, the latter being a largediffuse band. These complexes were not immunoprecipitatedfrom the membrane-enriched fraction when the cross-linkingstep was eliminated (lane 1).
To ascertain whether CTGF activates an RTK, we incubatedserum-starved HMC in the presence or absence of CTGF for 15min, lysed the cells, and immunoprecipitated phosphotyrosineproteins. The immunoprecipitated proteins were analyzed byWestern blotting using antibodies against TrkA (Figure 4A, B,C, and E) and EGF receptor (EGFR; data not shown), knowntyrosine kinase receptors. Only the anti-TrkA antibody cross-reacted strongly with a band of approximately 140 kD (Figure4A). The intensity of this band was stronger when cells wereincubated with CTGF (lane 2), indicating its activation by thegrowth factor. The interaction of CTGF with the TrkA receptorwas confirmed by different experiments in which either His-tagged CTGF/V5 fusion protein or rCTGF expressed in thebaculovirus system was allowed to bind to the cell surface andthen cross-linked to its ligand(s) using the reversible cross-linker DTSSP. The latter is cleaved by reducing agents. Subse-quently, a membrane fraction was prepared and cross-linkedCTGF complexes were captured on affinity metal beads or onanti–C-terminus CTGF antibody affinity beads. The capturedcomplexes were subjected to SDS-PAGE under reducing con-ditions and analyzed by Western blots. The results in Figure 4,B through D, clearly indicate that CTGF interacts with the TrkAreceptor. As Trk receptors have been shown to interact with thepan neurotrophin receptor p75NTR, blots were stripped andreprobed using an anti-p75NTR antibody. As shown in Figure4E, the antibody cross-reacted with a protein of the correctmolecular weight for P75NTR. Thus, our results indicate thatCTGF interacts with TrkA and p75NTR, two receptors that areknown to be activated by the neurotrophin nerve growth factor(NGF). To confirm this interaction, MC were depleted of TrkAor p75NTR receptors using antisense oligonucleotides (2 �M for3 d), as described previously by Wahab et al. (2). Equal amountof r-CTGF were allowed to bind to the cell surface and thencross-linked with DTSSP as above. Membrane preparations
J Am Soc Nephrol 16: ???-???, 2005 CTGF Activates the TrkA Receptor 3
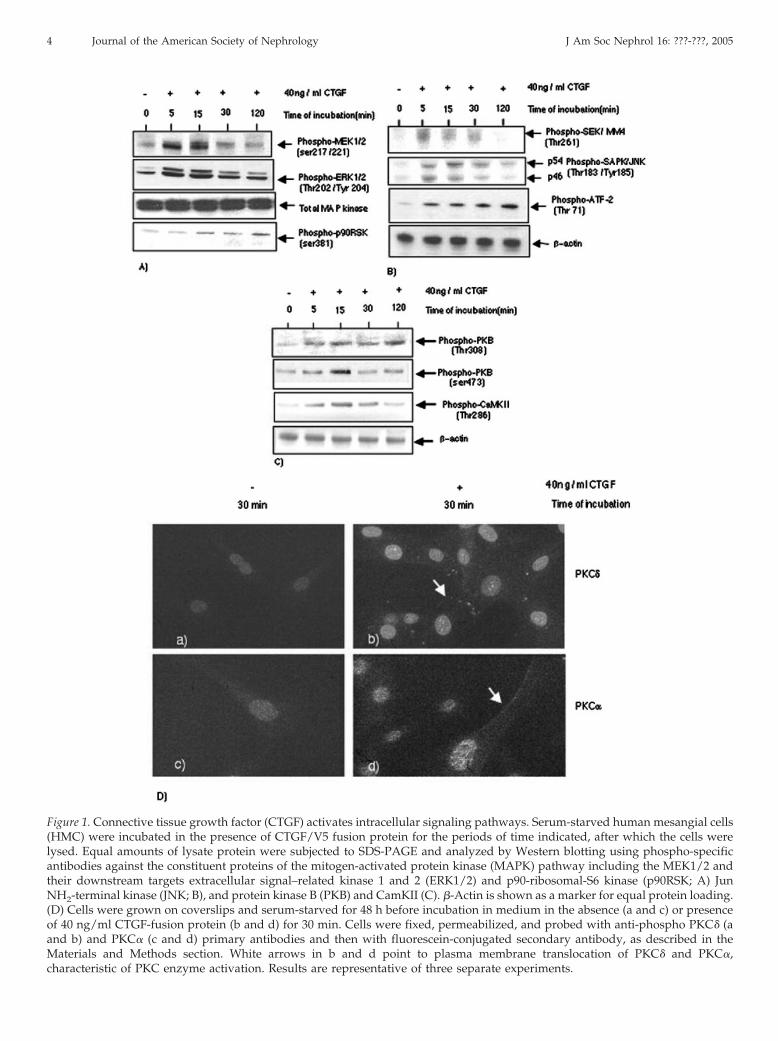
Figure 1. Connective tissue growth factor (CTGF) activates intracellular signaling pathways. Serum-starved human mesangial cells(HMC) were incubated in the presence of CTGF/V5 fusion protein for the periods of time indicated, after which the cells werelysed. Equal amounts of lysate protein were subjected to SDS-PAGE and analyzed by Western blotting using phospho-specificantibodies against the constituent proteins of the mitogen-activated protein kinase (MAPK) pathway including the MEK1/2 andtheir downstream targets extracellular signal–related kinase 1 and 2 (ERK1/2) and p90-ribosomal-S6 kinase (p90RSK; A) JunNH2-terminal kinase (JNK; B), and protein kinase B (PKB) and CamKII (C). �-Actin is shown as a marker for equal protein loading.(D) Cells were grown on coverslips and serum-starved for 48 h before incubation in medium in the absence (a and c) or presenceof 40 ng/ml CTGF-fusion protein (b and d) for 30 min. Cells were fixed, permeabilized, and probed with anti-phospho PKC� (aand b) and PKC� (c and d) primary antibodies and then with fluorescein-conjugated secondary antibody, as described in theMaterials and Methods section. White arrows in b and d point to plasma membrane translocation of PKC� and PKC�,characteristic of PKC enzyme activation. Results are representative of three separate experiments.
4 Journal of the American Society of Nephrology J Am Soc Nephrol 16: ???-???, 2005

were made, and equal amounts of solubilized protein wereincubated with anti–C-terminus CTGF antibody affinity beads.Bound complexes were subjected to SDS-PAGE under reducingconditions and analyzed by Western blots. Figure 5 shows thatreducing the expression level of the TrkA receptor by treatmentwith antisense oligonucleotide (lane 2) resulted in a reductionin the amount of TrkA receptors that bound to CTGF (Figure2B), but it had no significant effect on the amount of p75NTR
bound to CTGF (Figure 2C). In contrast, reducing the expres-sion level of p75NTR (lane 4) reduced the amount of both
receptors interacting with CTGF (Figure 4, B and C). Both TrkAand p75NTR control antisense oligonucleotides did not effect theamount of the interacting receptors (compare lanes 3 and 5).Attempts to confirm chemically the identity of these receptorsusing MALDI-TOF-MS analysis were unsuccessful because oftheir relatively low abundance and, probably, high level ofglycosylation of these receptors.
HMC Express Trk ReceptorsTo investigate whether HMC express any members of the
Trk, we extracted total RNA from HMC and performed reversetranscriptase–PCR analysis. The results (Figure 6) show thatHMC express the three members of the neurotrophin receptorfamily—TrkA, TrkB, and TrkC—as well as the pan receptorp75NTR. It is interesting that HMC also express the neurotro-phin factors NGF and brain-derived growth factor.
CTGF Activates TrkA in HMCOn binding its ligand, TrkA autophosphorylates on several
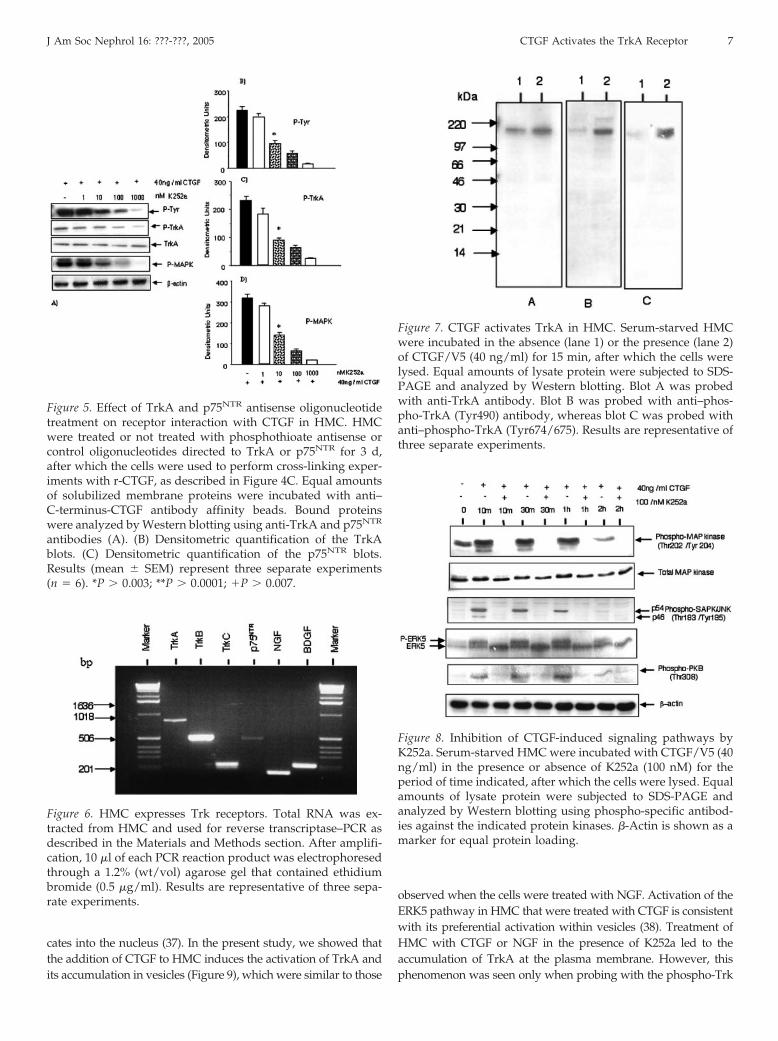
tyrosine residues, leading to the association and activation ofmultiple effectors. Phosphorylation at Tyr490 is required forShc association and activation of the Ras-MAPK cascade. Phos-phorylations at Tyr674/675 lie within the catalytic domain andreflect Trk kinase activity. Therefore, we tested whether stim-ulating cells with CTGF leads to the phosphorylation of TrkA atthese residues. The results in Figure 7 show that CTGF inducesan increase in the phosphorylation of the receptor at the Tyr490and Tyr674/675 residues by 1.8-fold and 2.5-fold, respectively.
Inhibition of CTGF-Induced Signaling Pathways by K252aK252a is an alkaloid-like kinase inhibitor that is known to
selectively inhibit Trk (33). K252a blocked the rapid phosphor-ylation of ERK1/2/JNK/ERK5 and PKB in HMC stimulatedwith CTGF (Figure 8), indicating that the phosphorylation ofthese kinases is indeed induced by ligand activation of the Trkreceptor. Furthermore, treatment with K252a led to the accu-mulation of TrkA at the plasma membrane of HMC that wereexposed to CTGF (Figure 9). This indicates that internalizationand sorting of receptor–CTGF complexes was blocked as aresult of inhibition of the receptor tyrosine kinase activity byK252a. This is consistent with the hypothesis that TrkA activa-tion is an obligatory step in the recruitment of activated Trkreceptors to clathrin-coated pits and entry into the endocyticpathway (34). The results in Figure 9 also indicate that K252acompletely blocks TrkA phosphorylation at Tyr490 but is lesseffective in blocking phosphorylation at Tyr674/675. Tyr674/675 are within the catalytic domain of TrkA but are not asso-ciated with signaling. Phosphorylation of these tyrosines mightturn over at a slower pace than those such as tyr 490, which areassociated with effector binding (35). Thus, tyr 674/675 may bemore readily detected in the presence of K252a than tyr 490.
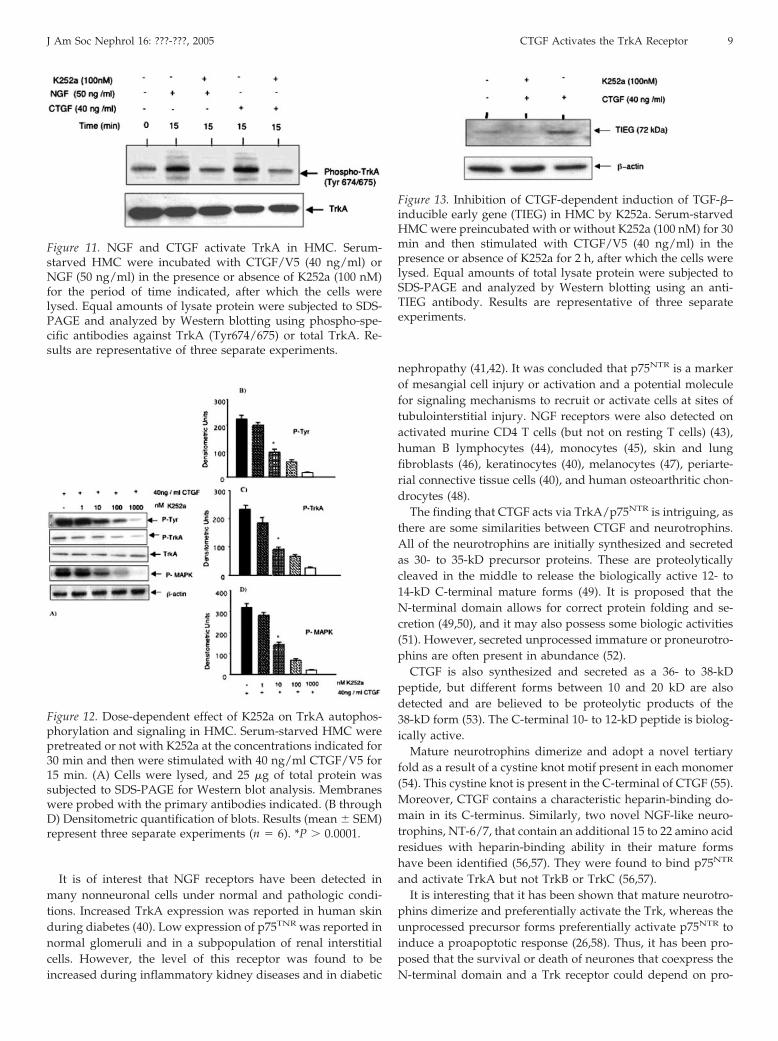
It is interesting that similar results were obtained with HMCthat were treated with 50 ng/ml NGF (Figure 10). The activa-tion of TrkA by both CTGF and NGF were confirmed byWestern blot analysis, as shown in Figure 11.
For further evaluating the effect of K252a on TrkA autophos-phorylation and signaling, HMC were serum starved for 24 h,
Figure 2. CTGF induces tyrosine phosphorylation of differentproteins. Serum-starved HMC were incubated in the presenceof 40 ng/ml CTGF/V5 fusion protein for the periods of timeindicated, after which the cells were lysed. Equal amounts oflysate protein were subjected to SDS-PAGE and analyzed byWestern blotting using anti-phosphotyrosine antibody. Resultsare representative of three separate experiments.
Figure 3. CTGF interacts with HMC surface proteins. CTGF/V5fusion protein was allowed to bind to the cell surface and thenchemically cross-linked to its ligands with BS3, after which amembrane-enriched fraction was prepared from the cells, asdescribed in the Materials and Methods section. Cross-linkedCTGF complexes were immunoprecipitated using rabbit anti-CTGF antibody, resolved by SDS-PAGE, and analyzed byWestern blotting using chicken anti-CTGF antibody (lane 2).The cross-linking step was omitted for some cultures (lane 1).Results are representative of three separate experiments.
J Am Soc Nephrol 16: ???-???, 2005 CTGF Activates the TrkA Receptor 5

pretreated with increasing concentrations of the inhibitor for 30min, and then stimulated with 40 ng/ml CTGF/V5 for 15 min.Cells were lysed and analyzed for phosphorylation of TrkA andthe downstream effector MAPK. Figure 12 shows that CTGF-induced total tyrosine phosphorylation of TrkA, phosphoryla-tion at TrkA tyr-490 residue, and p-MAPK all are significantlyinhibited by K252a at 10-nM concentration. This result is ingood agreement with the published IC50 for Trk (approxi-mately 3 nM) (33,36), whereas inhibitory effects on other ki-nases require much higher concentrations (36).
TrkA Tyrosine Kinase Activity is Required forCTGF-Dependent Induction of TIEG
To determine whether Trk tyrosine kinase activity is requiredfor CTGF-dependent induction of the transcription factorTIEG-1, we treated HMC with K252a 30 min before incubationwith CTGF for 2 h. As shown in Figure 13, CTGF enhanced theexpression of TIEG-1 by 3.4-fold (P � 0.0001). K252a was apotent inhibitor for TIEG induction at a concentration that isknown specifically to inhibit Trk kinase activity in other cells(33,36).
DiscussionIt is becoming clear that CTGF is implicated in the pathogen-
esis of diabetic nephropathy and possibly of other fibroticdisorders. Thus, therapeutic approaches that could selectivelyblock CTGF activity would be beneficial. However, it is impor-
tant first to understand fully the precise role and the moleculareffector mechanisms of this growth factor.
In this report, we provide evidence that CTGF triggers therapid simultaneous activation of the signaling pathways MAPK(ERK 1/2), JNK, ERK5, phosphatidylinositol 3-K, CaM-KII,PKC�, and PKC� in HMC. By subsequent binding, cross-link-ing, and immunoprecipitation studies, we identified the ty-rosine kinase receptor TrkA and p75NTR as two receptors thatinteract with CTGF. TrkA and p75NTR are known to be acti-vated by the neurotrophin factor NGF. Neurotrophins are sur-vival and differentiation factors in the nervous system, andalthough both receptors mediate the biologic effects of neuro-trophins, it seems that TrkA plays the central role in signaling,whereas the contribution of p75NTR is not fully understood.Activation of TrkA in HMC in response to CTGF treatment wasfurther supported by the ability of the Trk selective inhibitorK252a to block the rapid phosphorylation of ERK1/2, JNK,PKB, and ERK5.
On binding and activation of Trk by neurotrophins, the ligand–receptor complex is endocytosed into vesicles via a clathrin-dependent mechanism (27,29), where receptors remain catalyti-cally active within these vesicles (28). Inhibiting endocytosis withspecific inhibitors, or chemical inhibition of Trk kinase, inhibitsERK1/2 activation in response to neurotrophin stimulation (29).Previously, we demonstrated that CTGF is endocytosed from thecell surface in endosomes, from which the growth factor translo-
Figure 4. CTGF interacts with TrkA and p75NTR in HMC. (A) Serum-starved HMC were incubated in the absence (lane 1) orpresence (lane 2) of CTGF/V5 fusion protein (40 ng/ml) for 15 min, after which cell lysates were prepared in RIPA buffer. Equalamounts of lysate protein were immunoprecipitated using anti-phosphotyrosine beads. Bound proteins were resolved bySDS-PAGE and analyzed by Western blotting using an antibody against TrkA. (B) HMC were incubated in the absence (lane 1)or presence (lane 2) of His tagged-CTGF/V5 fusion protein (200 ng/ml) for 2 h at 4°C to allow it to bind to cell surface receptors,after which it was chemically cross-linked with 3,3�-dithiobis(sulfosuccinimidylpropionate). A membrane-enriched fraction wasprepared and solubilized. Equal amounts of solubilized protein were incubated with metal affinity beads. Bound proteins weresubjected to SDS-PAGE under reducing conditions and Western blotting using an antibody against TrkA. (C) HMC wereincubated with 200 ng/ml rCTGF (FibroGen Inc.) for 2 h at 4°C. Bound CTGF was cross-linked as above, and a membrane-enriched fraction was prepared and solubilized. Cross-linked CTGF complexes in the solubilized fraction were captured onanti–C-terminus-CTGF antibody affinity beads (lane 1), or the fraction was incubated with control IgG affinity beads (lane 2).Bound proteins were analyzed by Western blotting using anti-TrkA antibody. (D) The sample shown in C, lane 1, was boiled fora longer time and then Western blotted using anti-TrkA antibody. The same blot (D) was stripped and reprobed using anti-p75NTR
antibody (E). Results are representative of four separate experiments.
6 Journal of the American Society of Nephrology J Am Soc Nephrol 16: ???-???, 2005
cates into the nucleus (37). In the present study, we showed thatthe addition of CTGF to HMC induces the activation of TrkA andits accumulation in vesicles (Figure 9), which were similar to those
observed when the cells were treated with NGF. Activation of theERK5 pathway in HMC that were treated with CTGF is consistentwith its preferential activation within vesicles (38). Treatment ofHMC with CTGF or NGF in the presence of K252a led to theaccumulation of TrkA at the plasma membrane. However, thisphenomenon was seen only when probing with the phospho-Trk
Figure 5. Effect of TrkA and p75NTR antisense oligonucleotidetreatment on receptor interaction with CTGF in HMC. HMCwere treated or not treated with phosphothioate antisense orcontrol oligonucleotides directed to TrkA or p75NTR for 3 d,after which the cells were used to perform cross-linking exper-iments with r-CTGF, as described in Figure 4C. Equal amountsof solubilized membrane proteins were incubated with anti–C-terminus-CTGF antibody affinity beads. Bound proteinswere analyzed by Western blotting using anti-TrkA and p75NTR
antibodies (A). (B) Densitometric quantification of the TrkAblots. (C) Densitometric quantification of the p75NTR blots.Results (mean � SEM) represent three separate experiments(n � 6). *P � 0.003; **P � 0.0001; �P � 0.007.
Figure 6. HMC expresses Trk receptors. Total RNA was ex-tracted from HMC and used for reverse transcriptase–PCR asdescribed in the Materials and Methods section. After amplifi-cation, 10 �l of each PCR reaction product was electrophoresedthrough a 1.2% (wt/vol) agarose gel that contained ethidiumbromide (0.5 �g/ml). Results are representative of three sepa-rate experiments.
Figure 7. CTGF activates TrkA in HMC. Serum-starved HMCwere incubated in the absence (lane 1) or the presence (lane 2)of CTGF/V5 (40 ng/ml) for 15 min, after which the cells werelysed. Equal amounts of lysate protein were subjected to SDS-PAGE and analyzed by Western blotting. Blot A was probedwith anti-TrkA antibody. Blot B was probed with anti–phos-pho-TrkA (Tyr490) antibody, whereas blot C was probed withanti–phospho-TrkA (Tyr674/675). Results are representative ofthree separate experiments.
Figure 8. Inhibition of CTGF-induced signaling pathways byK252a. Serum-starved HMC were incubated with CTGF/V5 (40ng/ml) in the presence or absence of K252a (100 nM) for theperiod of time indicated, after which the cells were lysed. Equalamounts of lysate protein were subjected to SDS-PAGE andanalyzed by Western blotting using phospho-specific antibod-ies against the indicated protein kinases. �-Actin is shown as amarker for equal protein loading.
J Am Soc Nephrol 16: ???-???, 2005 CTGF Activates the TrkA Receptor 7

(Tyr674/675) antibody, which is specific for phosphorylated ty-rosine residues that lie within the catalytic domain. In contrast, thephospho-Trk (Tyr490) antibody, which is specific for the tyrosineresidue required for Shc association and activation of the Ras-MAPK cascade, did not show accumulation of phospho-TrkA atthe plasma membrane. This suggests that K252a is more potenttoward selective residues and indicates that internalization andsorting of the receptor–ligand complex is blocked as a result of theoverall inhibition of the receptor tyrosine kinase activity by K252a.This is consistent with the hypothesis that TrkA activation is anobligatory step in the recruitment to clathrin-coated pits and entry
into the endocytic pathway (34). K252a also blocks the CTGF-dependent induction of the transcription factor TIEG (Figure 11),indicating that Trk tyrosine kinase activity is required for CTGF toinduce expression of this gene.
The principal domains in Trk that determine affinity andspecificity of binding of neurotrophins are the two IgC2 do-mains (39), whereas p75NTR binding is facilitated through fournegatively charged cysteine-rich repeats (19). It is likely thatlocalization of the CTGF binding sites on these receptors willultimately lead to the generation of selective small moleculeantagonists.
Figure 9. Inhibition of CTGF-dependent activation of TrkA by K252a in HMC. HMC were grown on coverslips and serum-starvedfor 24 h, after which they were incubated with CTGF/V5 (40 ng/ml) in the presence or absence of K252a (100 nM) for 30 min. Cellswere fixed, permeabilized, and immunostained using anti–phospho-TrkA (Tyr674/675) or (Tyr490) antibodies. Results arerepresentative of three separate experiments.
Figure 10. Inhibition of nerve growth factor (NGF)-dependent activation of TrkA by K252a in HMC. HMC were grown oncoverslips and serum-starved for 24 h, after which they were incubated with NGF (50 ng/ml) in the presence or absence of K252a(100 nM) for 30 min. Cells were fixed, permeabilized, and immunostained using anti–phospho-TrkA (Tyr674/675) or (Tyr490)antibodies.
8 Journal of the American Society of Nephrology J Am Soc Nephrol 16: ???-???, 2005
It is of interest that NGF receptors have been detected inmany nonneuronal cells under normal and pathologic condi-tions. Increased TrkA expression was reported in human skinduring diabetes (40). Low expression of p75TNR was reported innormal glomeruli and in a subpopulation of renal interstitialcells. However, the level of this receptor was found to beincreased during inflammatory kidney diseases and in diabetic
nephropathy (41,42). It was concluded that p75NTR is a markerof mesangial cell injury or activation and a potential moleculefor signaling mechanisms to recruit or activate cells at sites oftubulointerstitial injury. NGF receptors were also detected onactivated murine CD4 T cells (but not on resting T cells) (43),human B lymphocytes (44), monocytes (45), skin and lungfibroblasts (46), keratinocytes (40), melanocytes (47), periarte-rial connective tissue cells (40), and human osteoarthritic chon-drocytes (48).
The finding that CTGF acts via TrkA/p75NTR is intriguing, asthere are some similarities between CTGF and neurotrophins.All of the neurotrophins are initially synthesized and secretedas 30- to 35-kD precursor proteins. These are proteolyticallycleaved in the middle to release the biologically active 12- to14-kD C-terminal mature forms (49). It is proposed that theN-terminal domain allows for correct protein folding and se-cretion (49,50), and it may also possess some biologic activities(51). However, secreted unprocessed immature or proneurotro-phins are often present in abundance (52).
CTGF is also synthesized and secreted as a 36- to 38-kDpeptide, but different forms between 10 and 20 kD are alsodetected and are believed to be proteolytic products of the38-kD form (53). The C-terminal 10- to 12-kD peptide is biolog-ically active.
Mature neurotrophins dimerize and adopt a novel tertiaryfold as a result of a cystine knot motif present in each monomer(54). This cystine knot is present in the C-terminal of CTGF (55).Moreover, CTGF contains a characteristic heparin-binding do-main in its C-terminus. Similarly, two novel NGF-like neuro-trophins, NT-6/7, that contain an additional 15 to 22 amino acidresidues with heparin-binding ability in their mature formshave been identified (56,57). They were found to bind p75NTR
and activate TrkA but not TrkB or TrkC (56,57).It is interesting that it has been shown that mature neurotro-
phins dimerize and preferentially activate the Trk, whereas theunprocessed precursor forms preferentially activate p75NTR toinduce a proapoptotic response (26,58). Thus, it has been pro-posed that the survival or death of neurones that coexpress theN-terminal domain and a Trk receptor could depend on pro-
Figure 11. NGF and CTGF activate TrkA in HMC. Serum-starved HMC were incubated with CTGF/V5 (40 ng/ml) orNGF (50 ng/ml) in the presence or absence of K252a (100 nM)for the period of time indicated, after which the cells werelysed. Equal amounts of lysate protein were subjected to SDS-PAGE and analyzed by Western blotting using phospho-spe-cific antibodies against TrkA (Tyr674/675) or total TrkA. Re-sults are representative of three separate experiments.
Figure 12. Dose-dependent effect of K252a on TrkA autophos-phorylation and signaling in HMC. Serum-starved HMC werepretreated or not with K252a at the concentrations indicated for30 min and then were stimulated with 40 ng/ml CTGF/V5 for15 min. (A) Cells were lysed, and 25 �g of total protein wassubjected to SDS-PAGE for Western blot analysis. Membraneswere probed with the primary antibodies indicated. (B throughD) Densitometric quantification of blots. Results (mean � SEM)represent three separate experiments (n � 6). *P � 0.0001.
Figure 13. Inhibition of CTGF-dependent induction of TGF-�–inducible early gene (TIEG) in HMC by K252a. Serum-starvedHMC were preincubated with or without K252a (100 nM) for 30min and then stimulated with CTGF/V5 (40 ng/ml) in thepresence or absence of K252a for 2 h, after which the cells werelysed. Equal amounts of total lysate protein were subjected toSDS-PAGE and analyzed by Western blotting using an anti-TIEG antibody. Results are representative of three separateexperiments.
J Am Soc Nephrol 16: ???-???, 2005 CTGF Activates the TrkA Receptor 9

cessing of the neurotrophin ligands (59). Activation of p75NTR
by proneurotrophins may also promote other functionsthrough pathways involving increased NF-�B or c-Jun kinaseactivities.
We used recombinant full-length CTGF in our experiments.However, our preparations always contain the 10- to 20-kDC-terminal form. Currently, we do not know whether there ispreferential binding of the different forms of CTGF toward eachreceptor. If this proved to be the case, then it would mean thatpostsecretory proteolytic processing of CTGF is essential forefficient Trk receptor binding and activation. At present, thedifferential ability of precursor and mature neurotrophins tobind selective receptors to mediate distinctive biologic actionsis thought to be unique. However, if CTGF has similar proper-ties, then the phenomenon is more common than first thought.
Our finding that CTGF activates the Trk receptor that isknown to associate with numerous cytoplasmic adaptors andsignaling proteins such as the nonreceptor tyrosine kinasec-abI, an ankyrin repeat-rich membrane spanning protein, ionchannels, GRK-2, B-Arrestin I, Sh2B, rAPS, p62 dok, and atyp-ical PKC enzymes (15,18), may explain the multifunctionalproperties of CTGF. Cells may respond differently to CTGFdepending on their expression level of the Trk and p75 recep-tors and the availability of different forms of CTGF in theirenvironment. Different cells may also express different subsetsof adaptor proteins that may compete with each other forbinding to activated receptors, leading to differential reper-toires of signaling proteins, which exhibit differences in signaltransmission.
It is also tempting to speculate that CTGF may act as afine-tuner for the TGF-� signaling pathway by regulating thelevel of Smad7. Thus, under certain pathologic conditions, itmay activate the Trk receptor and induce TIEG expression toenhance TGF-� signaling, whereas under other conditions, itmay induce the expression of Smad7 through the activation ofp75NTR and NF-�B. This would lead to diminishing the effectsof TGF-�. Further experiments to address these issues shouldprovide insight into the mechanisms by which CTGF regulatescell function under normal and pathologic conditions.
AcknowledgmentsWe thank the Medical Research Council (UK) for the financial sup-
port. We are grateful to FibroGen Inc. for recombinant CTGF, CTGFantibodies, and anti–CTGF-C-terminal domain–sepharose.
References1. Bork P: The modular architecture of a new family of
growth regulators related to connective tissue growth fac-tor. FEBS Lett 327: 125–130, 1993
2. Wahab NA, Yevdokimova N, Weston BS, Roberts T, Li XJ,Brinkman H, Mason RM: Role of connective tissue growthfactor in the pathogenesis of diabetic nephropathy. BiochemJ 359: 77–87, 2001
3. Takigawa M: CTGF/Hcs24 as a multifunctional growthfactor for fibroblasts, chondrocytes and vascular endothe-lial cells. Drug News Perspect 16: 11–21, 2003
4. Abreu JG, Ketpura NI, Reversade B, De Robertis EM: Con-
nective-tissue growth factor (CTGF) modulates cell signal-ing by BMP and TGF-beta. Nat Cell Biol 4: 599–604, 2002
5. Babic AM, Chen CC, Lau LF: Fisp12/mouse connectivetissue growth factor mediates endothelial cell adhesionand migration through integrin �v�3, promotes endothe-lial cell survival, and induces angiogenesis in vivo. Mol CellBiol 19: 2958–2966, 1999
6. Chen N, Chen CC, Lau LF: Adhesion of skin fibroblasts toCyr61 is mediated through integrin �6�1 and cell surfaceheparin sulfate proteoglycans. J Biol Chem 275: 24953–24961,2000
7. Crean JK, Finlay D, Murphy M, Moss C, Godson C, MartinF, Brady HR: The role of p42/44 MAPK and protein kinaseB in connective tissue growth factor induced extracellularmatrix protein production, cell migration, and actin cy-toskeletal rearrangement in human mesangial cells. J BiolChem 277: 44187–44194, 2002
8. Jedsadayanmata A, Chen CC, Kireeva ML, Lau LF, LamSC: Activation-dependent adhesion of human platelets toCyr61 and Fisp12/mouse connective tissue growth factoris mediated through integrin �(IIb)�(3). J Biol Chem 274:24321–24327, 1999
9. Leu SJ, Lam SC, Lau LF: Pro-angiogenic activities of CYR61(CCN1) mediated through integrins �v�3 and �6�1 inhuman umbilical vein endothelial cells. J Biol Chem 277:46248–46255, 2002
10. Schober JM, Chen N, Grzeszkiewicz TM, Jovanovic I, Eme-son EE, Ugarova TP, Ye RD, Lau LF, Lam SC: Identificationof integrin �(M)�(2) as an adhesion receptor on peripheralblood monocytes for Cyr61 (CCN1) and connective tissuegrowth factor (CCN2): Immediate-early gene products ex-pressed in atherosclerotic lesions. Blood 99: 4457–4465, 2002
11. Wahab NA, Mason RM: Connective tissue growth factorand renal disease: Some answers, more questions. CurrOpin Nephrol Hypertens 13: 53–58, 2004
12. Johnsen SA, Subramaniam M, Janknecht R, Spelsberg TC:TGF� inducible early gene enhances TGF�/Smad-depen-dent transcriptional responses. Oncogene 21: 5783–5790,2002
13. Nishida T, Nakanishi T, Shimo T, Asano M, Hattori T,Tamatani T, Tezuka K, Takigawa M: Demonstration ofreceptors specific for connective tissue growth factor on ahuman chondrocytic cell line (HCS-2/8). Biochem BiophysRes Commun 247: 905–909, 1998
14. Segarini PR, Nesbitt JE, Li D, Hays LG, Yates JR 3rd,Carmichael DF: The low density lipoprotein receptor-re-lated protein/�2-macroglobulin receptor is a receptor forconnective tissue growth factor. J Biol Chem 276: 40659–40667, 2001
15. Segal RA: Selectivity in neurotrophin signaling: Theme andvariations. Annu Rev Neurosci 26: 299–330, 2003
16. Lazarovici P, Levi BZ, Lelkes PI, Koizumi S, Fujita K,Matsuda Y, Ozato K, Guroff G: K-252a inhibits the increasein c-fos transcription and the increase in intracellular cal-cium produced by nerve growth factor in PC12 cells. J Neu-rosci Res 23: 1–8, 1989
17. Nikodijevic B, Guroff G: Nerve growth factor-induced in-crease in calcium uptake by PC12 cells. J Neurosci Res 28:192–199, 1991
18. Huang EJ, Reichardt LF: TRK receptors: Roles in neuronalsignal transduction. Annu Rev Biochem 72: 609–642, 2003
19. Smith CA, Farrah T, Goodwin RG: The TNF receptor su-
10 Journal of the American Society of Nephrology J Am Soc Nephrol 16: ???-???, 2005

perfamily of cellular and viral proteins: Activation, co-stimulation, and death. Cell 76: 959–962, 1994
20. Dobrowsky RT, Jenkins GM, Hannun YA: Neurotrophinsinduce sphingomyelin hydrolysis. Modulation by co-ex-pression of p75NTR with Trk receptors. J Biol Chem 270:22135–22142, 1995
21. Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhauser N,Bohm-Matthaei R, Baeuerle PA, Barde YA: Selective acti-vation of NF-kappa B by nerve growth factor through theneurotrophin receptor p75. Science 272: 542–545, 1996
22. Casaccia-Bonnefil P, Kong H, Chao MV: Neurotrophins:The biological paradox of survival factors eliciting apopto-sis. Cell Death Differ 5: 357–364, 1998
23. Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF,Chao MV: High-affinity NGF binding requires coexpres-sion of the trk proto-oncogene and the low-affinity NGFreceptor. Nature 350: 678–683, 1991
24. Esposito D, Patel P, Stephens RM, Perez P, Chao MV,Kaplan DR, Hempstead BL: The cytoplasmic and trans-membrane domains of the p75 and Trk A receptors regu-late high affinity binding to nerve growth factor. J BiolChem 276: 32687–32695, 2001
25. Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV:Death of oligodendrocytes mediated by the interaction ofnerve growth factor with its receptor p75. Nature 383:716–719, 1996
26. Lee R, Kermani P, Teng KK, Hempstead BL: Regulation ofcell survival by secreted proneurotrophins. Science 294:1945–1948, 2001
27. Grimes ML, Beattie E, Mobley WC: A signaling organellecontaining the nerve growth factor-activated receptor ty-rosine kinase, TrkA. Proc Natl Acad Sci U S A 94: 9909–9914, 1997
28. Beattie EC, Zhou J, Grimes ML, Bunnett NW, Howe CL,Mobley WC: A signaling endosome hypothesis to explainNGF actions: Potential implications for neurodegeneration.Cold Spring Harb Symp Quant Biol 61: 389–406, 1996
29. Howe CL, Valletta JS, Rusnak AS, Mobley WC: NGF sig-naling from clathrin-coated vesicles: Evidence that signal-ing endosomes serve as a platform for the Ras-MAPKpathway. Neuron 32: 801–814, 2001
30. Anderson RA, Robinson LL, Brooks J, Spears N: Neurotro-phins and their receptors are expressed in the human fetalovary. J Clin Endocrinol Metab 87: 890–897, 2002
31. Datta SR, Brunet A, Greenberg ME: Cellular survival: Aplay in three Akts. Genes Dev 13: 2905–2927, 1999
32. Delcommenne M, Tan C, Gray V, Rue L, Woodgett J,Dedhar S: Phosphoinositide-3-OH kinase-dependent regu-lation of glycogen synthase kinase 3 and protein kinaseB/AKT by the integrin-linked kinase. Proc Natl Acad SciU S A 95: 11211–11216, 1998
33. Berg MM, Sternberg DW, Parada LF, Chao MV: K-252ainhibits nerve growth factor-induced trk proto-oncogenetyrosine phosphorylation and kinase activity. J Biol Chem267: 13–16, 1992
34. Barker PA, Hussain NK, McPherson PS: Retrograde signal-ing by the neurotrophins follows a well-worn trk. TrendsNeurosci 25: 379–381, 2002
35. Ponzetto C, Bardelli A, Zhen Z, Maina F, dalla Zonca P,Giordano S, Graziani A, Panayotou G, Comoglio PM: Amultifunctional docking site mediates signaling and trans-
formation by the hepatocyte growth factor/scatter factorreceptor family. Cell 77: 261–271, 1994
36. Wang LH, Besirli CG, Johnson EM: Mixed-lineage kinases:A target for the prevention of neurodegeneration. AnnuRev Pharmacol Toxicol 44: 451–474, 2004
37. Wahab NA, Brinkman H, Mason RM: Uptake and intracel-lular transport of the connective tissue growth factor: Apotential mode of action. Biochem J 359: 89–97, 2001
38. Geetha T, Wooten MW: Association of the atypical proteinkinase C-interacting protein p62/ZIP with nerve growthfactor receptor TrkA regulates receptor trafficking andErk5 signaling. J Biol Chem 278: 4730–4739, 2003
39. Perez P, Coll PM, Hempstead BL, Martin-Zanca D, ChaoMV: NGF binding to the trk tyrosine kinase receptor re-quires the extracellular immunoglobulin-like domains. MolCell Neurosci 6: 97–105, 1995
40. Terenghi G, Mann D, Kopelman PG, Anand P: trkA andtrkC expression is increased in human diabetic skin. Neu-rosci Lett 228: 33–36, 1997
41. Alpers CE, Hudkins KL, Ferguson M, Johnson RJ, Schatte-man GC, Bothwell M: Nerve growth factor receptor ex-pression in fetal, mature, and diseased human kidneys. LabInvest 69: 703–713, 1993
42. Alpers CE, Hudkins KL, Floege J, Johnson RJ: Human renalcortical interstitial cells with some features of smooth mus-cle cells participate in tubulointerstitial and crescentic glo-merular injury. J Am Soc Nephrol 5: 201–209, 1994
43. Ehrhard PB, Erb P, Graumann U, Otten U: Expression ofnerve growth factor and nerve growth factor receptor ty-rosine kinase Trk in activated CD4-positive T-cell clones.Proc Natl Acad Sci U S A 90: 10984–10988, 1993
44. Brodie C, Gelfand EW: Functional nerve growth factorreceptors on human B lymphocytes. Interaction with IL-2.J Immunol 148: 3492–3497, 1992
45. Caroleo MC, Costa N, Bracci-Laudiero L, Aloe L: Humanmonocyte/macrophages activate by exposure to LPS over-express NGF and NGF receptors. J Neuroimmunol 113: 193–201, 2001
46. Micera A, Vigneti E, Pickholtz D, Reich R, Pappo O, BoniniS, Maquart FX, Aloe L, Levi-Schaffer F: Nerve growthfactor displays stimulatory effects on human skin and lungfibroblasts, demonstrating a direct role for this factor intissue repair. Proc Natl Acad Sci U S A 98: 6162–6167, 2001
47. Yaar M, Eller MS, DiBenedetto P, Reenstra WR, Zhai S,McQuaid T, Archambault M, Gilchrest BA: The trk familyof receptors mediates nerve growth factor and neurotro-phin-3 effects in melanocytes. J Clin Invest 94: 1550–1562,1994
48. Iannone F, De Bari C, Dell’Accio F, Covelli M, Patella V, LoBianco G, Lapadula G: Increased expression of nervegrowth factor (NGF) and high affinity NGF receptor (p140TrkA) in human osteoarthritic chondrocytes. Rheumatology(Oxford) 41: 1413–1418, 2002
49. Seidah NG, Benjannet S, Pareek S, Savaria D, Hamelin J,Goulet B, Laliberte J, Lazure C, Chretien M, Murphy RA:Cellular processing of the nerve growth factor precursor bythe mammalian pro-protein convertases. Biochem J 314:951–960, 1996
50. Suter U, Heymach JV Jr, Shooter EM: Two conserved do-mains in the NGF propeptide are necessary and sufficientfor the biosynthesis of correctly processed and biologicallyactive NGF. EMBO J 10: 2395–2400, 1991
J Am Soc Nephrol 16: ???-???, 2005 CTGF Activates the TrkA Receptor 11

51. Dicou E, Pflug B, Magazin M, Lehy T, Djakiew D, FerraraP, Nerriere V, Harvie D: Two peptides derived from thenerve growth factor precursor are biologically active. J CellBiol 136: 389–398, 1997
52. Fahnestock M, Michalski B, Xu B, Coughlin MD: The pre-cursor pro-nerve growth factor is the predominant form ofnerve growth factor in brain and is increased in Alzhei-mer’s disease. Mol Cell Neurosci 18: 210–220, 2001
53. Ball DK, Surveyor GA, Diehl JR, Steffen CL, Uzumcu M,Mirando MA, Brigstock DR: Characterization of 16- to20-kilodalton (kDa) connective tissue growth factors(CTGFs) and demonstration of proteolytic activity for 38-kDa CTGF in pig uterine luminal flushings. Biol Reprod 59:828–835, 1998
54. McDonald NQ, Hendrickson WA: A structural superfam-ily of growth factors containing a cystine knot motif. Cell73: 421–424, 1993
55. Bradham DM, Igarashi A, Potter RL, Grotendorst GR: Con-
nective tissue growth factor: A cysteine-rich mitogen se-creted by human vascular endothelial cells is related to theSRC-induced immediate early gene product CEF-10. J CellBiol 114: 1285–1294, 1991
56. Lai KO, Fu WY, Ip FC, Ip NY: Cloning and expression of anovel neurotrophin, NT-7, from carp. Mol Cell Neurosci 11:64–76, 1998
57. Li X, Franz J, Lottspeich F, Gotz R: Recombinant fishneurotrophin-6 is a heparin-binding glycoprotein: Impli-cations for a role in axonal guidance. Biochem J 324: 461–466,1997
58. Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R,Kohn J, Causing CG, Miller FD: The p75 neurotrophinreceptor mediates neuronal apoptosis and is essential fornaturally occurring sympathetic neuron death. J Cell Biol140: 911–923, 1998
59. Chao MV, Bothwell M: Neurotrophins: To cleave or not tocleave. Neuron 33: 9–12, 2002
12 Journal of the American Society of Nephrology J Am Soc Nephrol 16: ???-???, 2005

![EVALUATION OF THE POTENTIAL OF ......SOD-Mn expression is controlled by soxRS genes [6–8], which encode the SoxR receptor. The SoxR receptor interacts with O2.– and activates the](https://static.fdocuments.in/doc/165x107/60dcfc92715ec708510165c8/evaluation-of-the-potential-of-sod-mn-expression-is-controlled-by-soxrs.jpg)
















