
Congenital skeletal abnormalities: an introduction...
16
European Journal of Radiology 40 (2001) 168 – 183 Congenital skeletal abnormalities: an introduction to the radiological semiology Filip M. Vanhoenacker a, *, Wim Van Hul b , Jan Gielen a , Arthur M. De Schepper a a Department of Radiology, Uniersity Hospital Antwerp, Wilrijkstraat 10, B-2650, Edegem, Belgium b Department of Medical Genetics, Uniersity of Antwerp, Uniersiteitsplein 1, B-2610, Antwerp, Belgium Received 16 July 2001; received in revised form 17 July 2001; accepted 18 July 2001 Abstract Despite the recent advances in the molecular diagnosis of congenital abnormalities, the initial identification and the decision to refer a patient for further molecular analysis and expensive genetic tests still relies frequently on clinical and radiological criteria. The radiological identification of syndromes, dwarfs and dysplasias is a difficult task, because there are so many findings to consider and so many syndromes to remember that the problem is overwhelming. There is a definite need for an easy and systematic analysis system, in order to try to categorize a skeletal dysplasia in a certain group. In this brief review, we suggest an approach to the evaluation of skeletal syndromes, based on the analysis of cardinal criteria, from which the most useful information is derived, and additional criteria, making further differentiation possible. Generally, cardinal information is derived from analysis of the long bones, hands, pelvis and the spine, whereas the analysis of other skeletal elements, like the skull, feet, and other flat bones is of additional value. © 2001 Elsevier Science Ireland Ltd. All rights reserved. Keywords: Bones; Osteochondrodysplasia www.elsevier.com/locate/ejrad 1. Introduction The radiological identification of syndromes, dwarfs and dysplasias is still an important part in the practice of the pediatric radiologist. Although the progress of molecular genetics in the diagnosis of these group of disorders is considerable, the information provided by clinical and radiological examination is still of utmost importance in the selec- tion of candidate-patients to undergo frequently expen- sive genetic tests. Early recognition of osteochondrodysplasias will not only obviate the need for unnecessary and costly genetic and endocrine tests, but will also provide accurate information about man- agement, prognosis and genetic counseling to the pa- tient and his/her family [1]. The radiological identification of syndromes, dwarfs and dysplasias is a difficult task, even in experienced hands. For this reason, there is a definite need for an easy, systematic and reproducible analysis system, in order to try to categorize skeletal dysplasias in well defined (sub)groups. In this brief review, we suggest an approach to the evaluation of skeletal syndromes, based on the analysis of cardinal criteria, from which the most useful infor- mation is derived, and additional criteria, making fur- ther differentiation possible. Generally, cardinal information is derived from anal- ysis of the long bones, hands, pelvis and the spine, whereas the analysis of other skeletal elements, like the skull, feet, other flat bones, and miscellaneous features is of additional value. 2. Analysis of cardinal criteria 2.1. Long bones The long bones have to be analyzed in a double way. First, the length of the long bones has to be considered, followed by a detailed analysis of their individual seg- * Corresponding author. Tel.: +32-3-821-3532; fax: +32-3-825- 2026. 0720-048X/01/$ - see front matter © 2001 Elsevier Science Ireland Ltd. All rights reserved. PII:S0720-048X(01)00398-9
Transcript of Congenital skeletal abnormalities: an introduction...

European Journal of Radiology 40 (2001) 168–183
Congenital skeletal abnormalities: an introduction to theradiological semiology
Filip M. Vanhoenacker a,*, Wim Van Hul b, Jan Gielen a, Arthur M. De Schepper a
a Department of Radiology, Uni�ersity Hospital Antwerp, Wilrijkstraat 10, B-2650, Edegem, Belgiumb Department of Medical Genetics, Uni�ersity of Antwerp, Uni�ersiteitsplein 1, B-2610, Antwerp, Belgium
Received 16 July 2001; received in revised form 17 July 2001; accepted 18 July 2001
Abstract
Despite the recent advances in the molecular diagnosis of congenital abnormalities, the initial identification and the decision torefer a patient for further molecular analysis and expensive genetic tests still relies frequently on clinical and radiological criteria.The radiological identification of syndromes, dwarfs and dysplasias is a difficult task, because there are so many findings toconsider and so many syndromes to remember that the problem is overwhelming. There is a definite need for an easy andsystematic analysis system, in order to try to categorize a skeletal dysplasia in a certain group. In this brief review, we suggest anapproach to the evaluation of skeletal syndromes, based on the analysis of cardinal criteria, from which the most usefulinformation is derived, and additional criteria, making further differentiation possible. Generally, cardinal information is derivedfrom analysis of the long bones, hands, pelvis and the spine, whereas the analysis of other skeletal elements, like the skull, feet,and other flat bones is of additional value. © 2001 Elsevier Science Ireland Ltd. All rights reserved.
Keywords: Bones; Osteochondrodysplasia
www.elsevier.com/locate/ejrad
1. Introduction
The radiological identification of syndromes, dwarfsand dysplasias is still an important part in the practiceof the pediatric radiologist.
Although the progress of molecular genetics in thediagnosis of these group of disorders is considerable,the information provided by clinical and radiologicalexamination is still of utmost importance in the selec-tion of candidate-patients to undergo frequently expen-sive genetic tests. Early recognition ofosteochondrodysplasias will not only obviate the needfor unnecessary and costly genetic and endocrine tests,but will also provide accurate information about man-agement, prognosis and genetic counseling to the pa-tient and his/her family [1].
The radiological identification of syndromes, dwarfsand dysplasias is a difficult task, even in experiencedhands. For this reason, there is a definite need for an
easy, systematic and reproducible analysis system, inorder to try to categorize skeletal dysplasias in welldefined (sub)groups.
In this brief review, we suggest an approach to theevaluation of skeletal syndromes, based on the analysisof cardinal criteria, from which the most useful infor-mation is derived, and additional criteria, making fur-ther differentiation possible.
Generally, cardinal information is derived from anal-ysis of the long bones, hands, pelvis and the spine,whereas the analysis of other skeletal elements, like theskull, feet, other flat bones, and miscellaneous featuresis of additional value.
2. Analysis of cardinal criteria
2.1. Long bones
The long bones have to be analyzed in a double way.First, the length of the long bones has to be considered,followed by a detailed analysis of their individual seg-
* Corresponding author. Tel.: +32-3-821-3532; fax: +32-3-825-2026.
0720-048X/01/$ - see front matter © 2001 Elsevier Science Ireland Ltd. All rights reserved.PII: S 0 7 2 0 -048X(01 )00398 -9

F.M. Vanhoenacker et al. / European Journal of Radiology 40 (2001) 168–183 169
Fig. 1. Dwarfism. Depending on the predominant involved segment, dwarfism can be divided into three main categories. (a) Rhizomelic Dwarfismin a patient with Thanatophoric Dysplasia: there is preferential shortening of the proximal bones (humerus and femur). (b) Mesomelic Dwarfismin a patient with dyschondrosteosis or Leri–Weill’s disease: the middle segments of the appendicular skeleton are shortened (ulna and radius).There is also a characteristic Madelung deformity of the wrist. (c–d) Acro(-meso)melic Dwarfism. Shortening of the middle (ulna and radius) anddistal segments (long bones of the hands) of the appendicular skeleton (c), whereas the pelvis is normal (d).

F.M. Vanhoenacker et al. / European Journal of Radiology 40 (2001) 168–183170
ments, to determine which segment (epiphysis, metaph-ysis or diaphysis) bears the brunt of abnormality.
The bone length is usually too short in skeletaldysplasia, although occasionally the bones are too long,as in Marfan syndrome and homocystinuria.
The approach for shortening of long bones indwarfism is a very complex problem, but for the sake ofsimplicity, dwarfism can be divided into three differentgroups, based on their preferential involvement of thedifferent segments of the extremities.
Rhizomelic dwarfism consists of predominant in-volvement of the proximal segments (humerus, femur)(Fig. 1a). The middle segments (tibia, fibula, radius-ulna) are affected in mesomelic dwarfism (Fig. 1b),whereas the distal segments (acromelic) are mostly in-volved in acromelic dwarfism. Isolated acromelicdwarfism is rare and is also referred to as peripheraldysostosis. Usually acromelic dwarfism occurs in com-bination with mesomelic shortening (Fig. 1c and d) [2].
After the length of the long bones has been assessed,one should try to decide whether the problem in theindividual bone is diaphyseal, metaphyseal, or epiphy-seal [2].
Diaphyseal abnormalities can be subclassified in:1. Thin and overtubulated bones (Table 1): the most
common cause is an underlying neuromuscular dis-order (Fig. 2a).
Fig. 2. Diaphyseal abnormalities in long bones. (a) Overtubulation:thin and long ulna and radius in a patient with a neuromusculardisease. (b) Undertubulation: short and squat tibia and fibula inachondroplasia.
Table 1Abnormal length of long bones (modified from 2, with permission)
Overtubulation: thin, gracile Undertubulation: short, squat bonesbones
Neurologic–neuromuscular Achondroplasiadisease
Storage diseasesOsteogenesis imperfectaArthrogryposis multiplex Metaphyseal dysostosisMarfan syndrome PseudoachondroplasiaHomocystinuria Vitamin D-resistant rickets-type BCockayne syndrome Neonatal dwarfsWinchester syndrome Chondrodystrophy with immune
deficiencyProgeria Patterson–Lowry rhizomelic
dysplasiaRhizomelic punctate epiphysealKenny–Caffey syndromedysplasiaDiastrophic dwarfismStickler syndrome
Hallermann–Streiff Hypophosphatasiasyndrome
Metatropic dwarfismSeckel bird-headed dwarfKniest syndromeDyggve–Melchior–ClausensyndromeCamptomelic dwarfismLarsen syndromeWeissenbacher–Zweymullersyndrome
Diseases in italic: most common.

F.M. Vanhoenacker et al. / European Journal of Radiology 40 (2001) 168–183 171
Table 2Metaphyseal changes in congenital diseases (modified from 2, withpermission)
Widening withoutFlaring, widening, Metaphysealcupping beakingcupping
Achondroplasia Menkes kinkyPyle diseasehair syndrome
Metaphyseal dysostosis Oto-palato-digital Hypophosphatasiasyndrome
Hypochondroplasia Craniometaphyseal Desbuquoissyndromedysostoses
Pseudoachondroplasia Gaucher diseaseThanatophoric dwarfism Weaver syndromeHypophosphatasiaMetatropic dwarfismKniest syndromeEllis–van Creveld
syndromeMesomelic dwarfismShort rib-polydactyly
syndromesDiastrophic dwarfismStippled epiphyses
congenitaHypophophatasiaSpondylometaphyseal
dysplasiaTaybi–Linder syndromeOsteodysplasia
(Melnick–Needles)PhenylketonuriaWeissenbacher–
Zweymuller syndromeAchondrogenesisTrichorhinophalangeal
syndrome
3. Diaphyseal sclerosis: there is a whole range of scle-rosing bone dysplasias, affecting the intramembra-nous bone formation and remodeling, which areresponsible for cortical thickening [3]. The corticalthickening may be caused either by a disturbance inthe periosteal bone formation and apposition or dueto a defective endosteal resorption. According to theinvolved mechanism, the diaphysis may be broad-ened (increased periosteal apposition), or themedullary cavity may be narrowed (defective en-dosteal resorption). The prototype of defective en-dosteal resorption is Van Buchem disease, whereasCamurati–Engelmann disease is a classical exampleof predominant disturbance of the periostealapposition.
Metaphyseal changes consist of splaying, cupping,widening, and irregularity (Table 2 and Fig. 3a).
Epiphyseal abnormalities include smallness, irregu-larity, fragmentation or calcification (Fig. 3b and Table3).
One has to be aware that primary epiphyseal abnor-malities may induce secondary metaphyseal changes,which makes the analysis whether the abnormality ispredominantly epiphyseally or metaphyseally locatedextremely difficult (Fig. 3c). This phenomenon is easyto understand in pseudoachondroplasia syndromes(Fig. 3b) such as multiple epiphyseal dysplasia andspondyloepiphyseal dysplasia.
These entities are primarily characterized by smalland irregularly delineated epiphyses, which make theadjacent growth plate vulnerable to injuries, inducingsecondary metaphyseal deformities (Fig. 3c). In thosecases, where combined epiphyseal and metaphysealchanges are found, it is important to realize that theepiphyseal changes are usually the cause of secondaryacquired metaphyseal changes, in order to avoid anerroneous diagnosis [2].
2. Short, squat and undertubulated bones (Table 1):the prototype here is represented by achondroplasia(Fig. 2b).
Table 3Epiphyseal changes in skeletal dysplasia (modified from 2, with permission)
Stippled epiphysesSmall epiphysesLarge epiphyses Irregular epiphyses
Chondrodystrophies Hypothyroidism Punctate epiphyseal DysplasiaWinchester syndrome Dysplasia Dysplasia Multiple epiphyseal-
Multiple epiphyseal- Spondyloepiphyseal-Trevor’s disease Warfarin embryopathyHypothyroidismMegaepihyseal Spondyloepiphyseal- Fetal alcohol syndrome
dwarfismMorquio diseaseCerebrocostomandibular syndromeMorquio diseaseTrevor’s disease
Smith–Lemli–Opitz syndrome Tricho-rhino-phalangeal syndrome: femoral headDyggve–Melchior–Clausen syndromeMeyer dysplasia (hips)Winchester syndromeZellweger syndrome
Diseases in italic: most common.

F.M. Vanhoenacker et al. / European Journal of Radiology 40 (2001) 168–183172
Fig. 3. Metaphyseal and epiphyseal abnormalities. (a) Metaphyseal changes, consisting of splaying, cupping, widening and irregularity of themetaphyses of the distal femur and proximal tibia in a patient with Janssen metaphyseal chondrodysplasia. (b) Epiphyseal abnormalities, inpseudoachondroplasia syndrome: the epiphyses of the elbow joint are small, irregularly delineated and fragmented. (c) Secondary metaphysealchanges, owing to primary epiphyseal abnormalities in Multiple Epiphyseal Dysplasia (MED). Recurrent metaphyseal trauma, secondary toimpaction of the small epiphysis into the adjacent growth plate, may cause asymetrical premature closure of the growth plate, resulting intibiotalar slanting in an adult patient with MED.

F.M. Vanhoenacker et al. / European Journal of Radiology 40 (2001) 168–183 173
Fig. 4. Hand abnormalities: overall morphology. (a) Spade hand inHurler syndrome. (b) Trident hand in achondroplasia: note diver-gence of the middle three fingers at the level of the proximal interpha-langeal joints.
positive metacarpal sign is seen in several congenitalabnormalities (Table 4). This sign is positive, when aline drawn along the heads of the fourth and fifthmetacarpals intersects the head of the third metacarpal(Fig. 5a).
Other syndromes associated with shortening of oneor more metacarpals or metatarsals are listed in Table4.
Abnormalities in the number of hand bones arereferred to as oligodactyly (Fig. 5b), in case of lack offingers, and polydactyly, if there too many fingers(Table 5). A surnumerary finger can be either preaxial(on the radial side) or postaxial (on the ulnar side) (Fig.5c). Polydactyly can occur in association with shortribs, in a number of polydactyly-short ribs syndromes(Fig. 5d and e)
Absence of digits on one or the other side of thehand is termed either the ‘radial’ or ‘ulnar ray’ syn-drome (Fig. 5f) [2] (Table 6).
Acro-osteolysis is the term used for distal phalangealresorption (Fig. 5). This is usually due to acquiredcauses, some of the most frequent congenital cause aresummarized in Table 7.
Fusion (Table 8) occurring between two (adjacent)fingers is called syndactyly (Fig. 6a), whereas sympha-langism (Fig. 6b) refers to fusion between two pha-langes within the same ray. Syndactyly can bemembranous, bony or combined. Symphalangism canbe proximal (at the proximal interphalangeal joint) ordistal (at the distal interphalangeal joint) or variable(distally and proximally located within the samepatient).
Fusion between carpal (or tarsal) bones is a frequentfinding, which is usually isolated, but can be associatedwith several syndromes (Table 8).
Table 4Congenital diseases characterized by a shortened metacarpals (andmetatarsals) (modified from 2, with permission)
Positive metacarpal sign Shortening of (other) metacarpals
Basal cell nevus syndromeAchondroplasiaBeckwith–Wiedemann syndromeTurner syndromeBiedmon syndromeVarious DwarfismLarsen syndromeGigantismMultiple exostoses syndromeHyperparathyroidism
Pseudohypoparathyroidism Epiphyseal dysplasiaTricho-rhino-phalangeal syndromePseudo–pseudo-
hyperparathyroidismCri-du-chat syndromeRussell–Silver dwarfismPatterson–Lowry rhizomelicdysplasia
Diseases in italic: most common.
2.2. Hand abnormalities
The first observation to be made concerns the overallmorphology of the hand. A spade hand is seen instorage diseases (Fig. 4a), while a characteristic tridentmorphology may be seen in achondroplasia (Fig. 4b).
Secondly, the length, number, and morphology of theindividual bones has to determined.
Shortening of various phalanges, thumb, andmetacarpals are seen in a number of congenital dis-eases. Shortening of metacarpal 4 and 5, resulting in a

F.M. Vanhoenacker et al. / European Journal of Radiology 40 (2001) 168–183174
Fig. 5. Hand abnormalities: shortening and absence of individual bones. (a) Shortening of metacarpal bone 4 and 5, causing a positive metacarpalsign in a patient with pseudohypoparathyroidism (see text). (b) Oligodactyly. (c) (Postaxial) polydactyly: a surnumerary finger is seen on the ulnarside. (d–e) Polydactyly-short rib syndrome: association of polydactyly (d) and short ribs (e). (f) Ulnar ray syndrome: absence of the fifth fingerand ulna. (g) Acro-osteolysis in progeria: resorption of the tufts of the distal phalanges, with associated calcifications.

F.M. Vanhoenacker et al. / European Journal of Radiology 40 (2001) 168–183 175
Fig. 5. (Continued)
The most frequent abnormalities regarding the mor-phology of the individual bones consist of cone shapedepiphyses of the phalanges, smallness, scalloping, andirregularity of the carpal bones.
Macrodactyly refers to enlargement of one of moredigits and is associated with macrodystrophia lipo-matosa, Proteus syndrome, neurofibromatosis, heman-giomatous or lymphangiomatous tumors of the handsor feet.
Positional abnormalities of the fingers (and toes) canbe due to an abnormal form of a phalanx or due tomechanical intrauterine stress. Overlapping fingersare seen in some chromosomal abnormalities (trisomy-18).
2.3. Pel�is
The following characteristics, regarding the pelvis,may be useful in the identification of chondrodystro-phies, storage disease and certain chromosomalabnormalities:
The pelvic configuration in skeletal dysplasias can bedivided in two main types, mainly according to theshape of the iliac wings (Fig. 7a and b). In type A, theiliac wings are short and squared off, whereas in type Bthe iliac wings have a narrow waist (Table 9).
Measurements of the acetabular angles, iliac anglesand indices are useful in the assessment of chromoso-mal abnormalities (Fig. 7c).

F.M. Vanhoenacker et al. / European Journal of Radiology 40 (2001) 168–183176
Delayed or defective ossification of the pubic bones,resulting in widening of the interpubic distance is seenin most of the neonatal and infantile chondrodystro-phies, but is most characteristic for cleidocranial dysos-tosis [4,5]. Protusio acetabuli can be seen with Turnersyndrome, osteogenesis imperfecta, and Marfan syn-drome [2].
2.4. Spine
Spinal abnormalities, that may be helpful in the(differential) diagnosis of syndromes are:1. Abnormalities of the C1–C2 area, especially with
anomalies of the dens. The atlantodental distance isincreased in Morquio’s disease, as well as in otherstorage diseases.
2. Changes in the overall morphology: kyphoscoliosisis a nonspecific, but very frequent finding in severalcongenital skeletal abnormalities, especially neuro-muscular and neurologic diseases.
Table 6Radial and ulnar ray syndrome
Radial ray syndrome Ulnar ray syndrome
Acromesomelic dysplasiaKlippel–Feil deformityEctodermal dysplasia Boomerang dysplasiaFanconi anemia Cardiomelic syndrome with
ulnar agenesisHolt–Oram syndrome Chondrodysplasia punctata,
tibial-metacarpal typeCraniosynostosis-ulnar aplasiaTAR syndrome
(thrombocytopenia-absentradius syndrome)
Thalidomide embryopathy De la Chapelle syndromeVATER syndrome Distal osteosclerosisTrisomy-18 Hereditary multiple exostosesBaller–Gerold syndrome Familial ulnar aplasia and
lobster claw syndromeDe la Chapelle syndromeAbsent radius and anogenital Femur-fibula-ulna syndrome
anomaliesFacioauriculoradial dysplasia Fibuloulnar aplasia :hypoplasia
and renal dysplasiaIsolated radial deficiency Grebe chondrodysplasiaIVIC syndrome Ives–Houston syndromeLaurin–Sandrow syndrome Klippel–Feil syndrome-absent
ulnaNager acrofacial dysostosis Mesomelic dysplasia, different
typesMetaphyseal-sella turcicaOkihiro syndromedysplasia
Radiodigitofacial dysplasia Osebold–Remondini dysplasiaPostaxial acrofacial dysostosisVater associationsyndrome
WT limb-blood syndrome Roberts syndromeThalidomide embryopathy Spondyloperipheral dysplasia
Ulnar-mammary syndromeTreacher Collins syndromeRoberts syndrome Cornelia de Lange syndrome
Nievergelt dysplasiaPfeiffer ulnofibular dysplasiasWegner syndromeWeyers oligodactyly
Table 5Congenital diseases characterized by an abnormal number of fingers/toes (modified from 2, with permission)
Polydactyly Oligodactyly
Preaxial Aglossia-adactylyAcrocephalosyndactyly Mobius syndrome
ThalidomideBlackfan–Diamond anemiaembryopathy
Dubowitz anemia Amniotic bandsyndromeRadial rayHolt–Oram syndromesyndrome (seeTable 6)
VATER syndrome Ulnar ray syndrome(see Table 6)
Acro-pectoro-vertebral dysplasiaMobius syndromeMohr syndromeFibrodysplasia ossificans progressivaNager syndromeOro-facio-digital syndromePoland syndromeShort-rib polydactyly syndromeTrisomy-13Werner syndrome
PostaxialRubinstein–Taybi syndromeAsphyxiating thoracic dystrophyBiemond syndromeGoltz syndromeGrieg syndromeHeriditary hydrometrocolpos
(McKusick–Kaufman syndrome)Mohr syndromeShort-rib polydactyly syndromeSmith–Lemli–Opitz syndromeWeyer syndrome
Diseases in italic: most common.
3. Clefting abnormalities (Fig. 8a).4. Shape of the vertebral bodies: frequently encoun-
tered abnormal shapes consist of flat vertebrae(platyspondyly; Table 10), cuboid vertebrae, round
Table 7Congenital causes of acro-osteolysis
Acro-osteolysis
Idiopathic acro-osteolysisProgeriaPycnodysostosisOsteopetrosisEhlers–Danlos syndromePseudoxanthoma elasticumRothmund syndromeCongenital insensitivity to painEpidermolysis bullosa

F.M. Vanhoenacker et al. / European Journal of Radiology 40 (2001) 168–183 177
or bullet-shape (Fig. 8b), beaked shape and tall(tower-) vertebrae and hemivertebrae. Tower verte-brae are seen in trisomy-21. Beaked vertebrae areseen in neuromuscular diseases, storage diseases,achondroplasia and neurofibromatosis.
5. Scalopping of the posterior wall of the vertebrae(Table 10 and Fig. 8c and d).
6. Narrowing of the spinal canal, with decrease of theinterpedicular distance towards the sacrum (Fig.8e–g).
Fig. 6. Hand abnormalities: fusion of individual bones. (a) Syn-dactyly: complex bony fusion between adjacent fingers. (b) Sympha-langism: fusion between two phalanges within the same ray.
Table 8Abnormal fusion (modified from 2, with permission)
Carpal (tarsal)SymphalangismSyndactylyfusion
Isolated IsolatedAcrocephalosyndactylyEllis–�an Cre�eldCornelia de Lange Acrocephalosynda
ctyly syndromessyndrome syndromeHolt–OramFanconi anemia DiastrophicsyndromedwarfismAcrocephalosyndactHolt–Oram syndrome Isolated
brachydactyly ylyArthrogryposisTAR syndrome Poplitealcongenitapterygium
syndromeTrisomy-13 Turner syndromeTrisomy-18 DyschondrosteosisSyndromes with Diastrophic
polydactyly dwarfismIsolated Hand–foot–uterus
syndromeAarskog syndrome Kniest syndromeAglossia–adactyly Nievergelt
mesomelicsyndromedwarfism
Bloom syndrome Otopalato-digitalsyndromeFrontometaphysealCarpenter syndromedysplasia
Punctate epiphyseal Stickler syndromedysplasia (Conradisyndrome)
Goltz syndromeLaurence–Moon–Biedl
syndromeMobius syndromeNager syndromeMesomelic dwarfismOtopalatodigital syndromePopliteal pterygium
syndromeRobinov–Silvermann
syndromeRothmund–Thomson
syndromeRubinstein–Taybi
syndromeSmith–Lemi–Opitz
syndromeTricho–rhino–phalangeal
syndrome
Diseases in italic: most common.
3. Analysis of additional criteria
3.1. Skull abnormalities
The following characteristics regarding the skullbones may narrow the differential diagnosis of varioussyndromes:1. The presence of macro- or microcephaly.2. An abnormal shape of the calvaria, premature clo-
sure of sutures, the presence of impressiones digiti-formes can be indicative for craniosynostosis (Fig.9a).

F.M. Vanhoenacker et al. / European Journal of Radiology 40 (2001) 168–183178
Fig. 7. Abnormalities of the pelvis. (a) Type A pelvis in achondroplasia: the iliac wings are short and squared off. (b) Type B pelvis in Morquiosyndrome: there is a narrow waist of the iliac wings. (c) Acetabular and iliac angles and indices are useful in the assessment of chromosomalabnormalities. The acetabular angle is the angle between the line intersecting the Y-cartilage and the line tangential to the acetabular roof. Theiliac angle is the angle between the horizontal line intersecting the Y-cartilage and the line tangential to the lateral border of the ilium. The iliacindex is the sum of the acetabular angle and the iliac angle. An index below 60° is very suggestive of an underlying chromosomal abnormality.
Fig. 8.

F.M. Vanhoenacker et al. / European Journal of Radiology 40 (2001) 168–183 179
Fig. 8. Spinal abnormalities. (a) Abnormal clefts. Coronal clefts in the bodies of L2 and L4 are seen on this lateral radiograph of the spine. (b)Bullet shape of L1 and beaked shape of L2 and L3 in a patient with Hurler syndrome. (c–d) Scalopping of the posterior wall of the vertebraein Marfan syndrome, as seen on a lateral radiograph (c). The scalopping is caused by liquor pulsations in arachnoidocoeles, causing chronicpressure erosions on the posterior wall of the vertebral bodies. The enlargement of the dural sac is well appreciated on the sagittal T1-weightedMR image, in another Marfan patient (d). (e–g) Narrowing of the spinal canal in achondroplasia. On a lateral radiograph of the spine, theshortness of the pedicles is well appreciated (e). On an AP view, there is gradual decrease of the interpedicular distance towards the sacrum (f).The narrowing of the spinal canal, especially in transverse direction, is best evaluated on a CT-scan (g). Note also the scalopping of the posteriorwall on the lateral radiograph, and the characteristic pelvis abnormalities (type A pelvis).
F.M. Vanhoenacker et al. / European Journal of Radiology 40 (2001) 168–183180
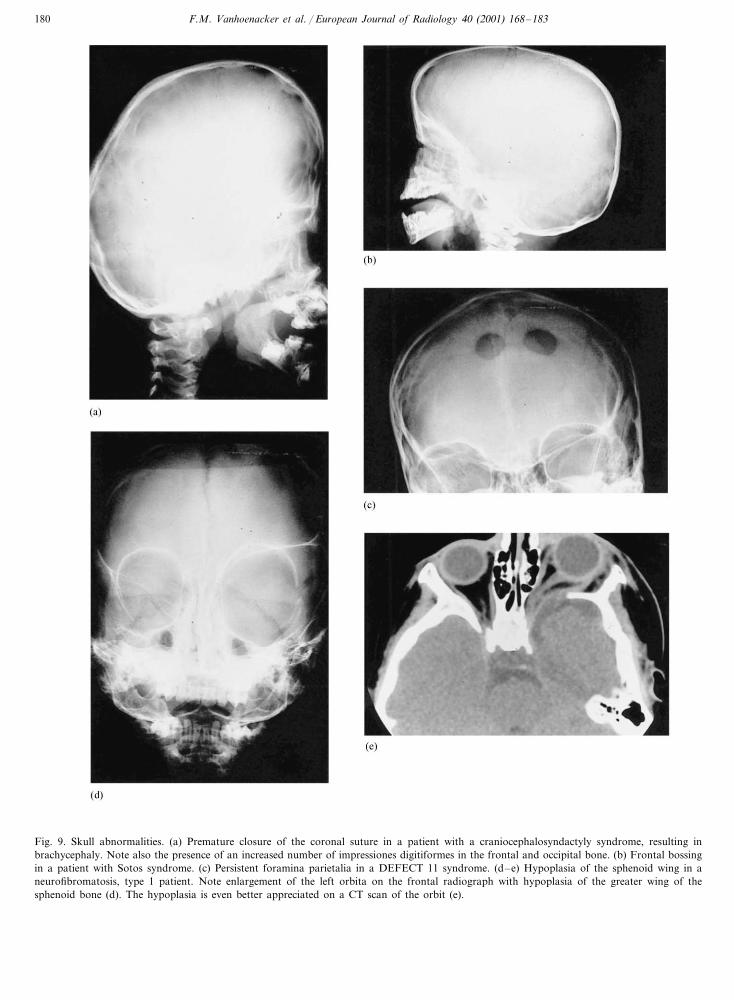
Fig. 9. Skull abnormalities. (a) Premature closure of the coronal suture in a patient with a craniocephalosyndactyly syndrome, resulting inbrachycephaly. Note also the presence of an increased number of impressiones digitiformes in the frontal and occipital bone. (b) Frontal bossingin a patient with Sotos syndrome. (c) Persistent foramina parietalia in a DEFECT 11 syndrome. (d–e) Hypoplasia of the sphenoid wing in aneurofibromatosis, type 1 patient. Note enlargement of the left orbita on the frontal radiograph with hypoplasia of the greater wing of thesphenoid bone (d). The hypoplasia is even better appreciated on a CT scan of the orbit (e).

F.M. Vanhoenacker et al. / European Journal of Radiology 40 (2001) 168–183 181
Fig. 10. Miscellaneous abnormalities. (a) Frontal radiograph of the knee joint, demonstrating dislocation of the knee joint in a patient witharthrogryposis multiplex congenita. (b) Frontal radiograph of the pelvis and lower limbs. Demineralization of the skeleton associated with multiplefractures, resulting in an ‘accordeon-like’ shortening of the long bones in a patient with lethal osteogenesis imperfecta. (c) AP radiograph of theshoulder in a patient with Maffucci disease. Multiple enchondromas in the proximal humerus and acromion, associated with soft tissue phlebolits,indicating the presence of haemangiomas. Note also bowing of the proximal humeral diaphysis. (d) Lateral radiographs of the knee joints.Absence of the patellae in a patient with nail-patella syndrome. (e) Fibrodysplasia ossificans progressiva. Multiple band-like calcifications are seenwithin the soft tissues of the chest wall. (f) Frontal radiographs of the shoulders. Absence of the lateral part of the clavicles in cleidocranialdysplasia.

F.M. Vanhoenacker et al. / European Journal of Radiology 40 (2001) 168–183182
Fig. 10. (Continued)
Table 9Congenital diseases characterized by an abnormal shape of the pelvis(modified from 2, with permission)
Type A Type B
Trisomy-21AchondroplasiaAchondrogenesis Mucopolysaccaridoses,
except MorquioMucolipidosesAsphyxiating thoracic dystrophyOther storage diseasesEllis–van Creveld syndrome
Short rib-polydactyly syndromes Cleidocranial dysostosisMetatropic dwarfism Cockayne syndrome
AcrocephalosyndactylyKniest syndromeSpondyloepipyseal dysplasia Aminopterin-induced
syndromecongenitaArthrogryposisCornelia de LangePunctate epiphyseal dysplasia:
rhizomelic form syndromeHypophosphatasia
Thanatophoric dwarfism Popliteal pterygiumsyndrome
Morquio disease Osteo-onychodysplasiaSevere metaphyseal dysostoses Prune–belly syndromeDyggve–Melchior–Clausen syndrome Rubinstein–Taybi
syndromeBladder extrophySacral agenesisTrisomy-13, -18Metaphyseal dysostosesOsteogenesis imperfectaWeissenbacher–Zweymuller syndromeLarsen syndromeMelnick–Needlessyndrome
Table 10Spinal abnormalities (modified from 2, with permission)
Platyspondyly Scalopping posterior wall
NeurofibromatosisOsteogenesis imperfectaAchondroplasiaSpondyloepiphyseal dysplasiaOther chondrodystrophiesThanatophoric dwarfismStorage diseasesMorquio diseaseEhlers–Danlos syndromeAchondrogenesis
Kniest syndrome Marfan syndromeDyggve–Melchior–Clausen syndromePatterson–Lowry rhizomelic dysplasias
3. Contour anomalies and/or associated skull defects,like cystic hygroma, encephalocoele, and frontalbossing (Fig. 9b and c).
4. Abnormal shape of the individual skull bones: e.g.abnormal shape of sella turcica.
5. Absence/hypoplasia of individual bones: e.g. lateossification of nasal bones in trisomy 21; absenceof the sphenoid wing in neurofibromatosis type 1(Fig. 9d and e.).
3.2. Abnormalities of the feet and the other flat bones
The changes seen in the hands are very similar to thefindings seen in the feet, but hand abnormalities areusually more pronounced.

F.M. Vanhoenacker et al. / European Journal of Radiology 40 (2001) 168–183 183
3.3. Miscellaneous findings
The following features may further allow a moreaccurate radiological diagnosis:
1. Abnormal configuration of the joints, with either acongenital dislocation or synostosis of differentjoints (Fig. 10a).
2. Abnormalities in mineralisation, either sclerosis ordemineralization (Fig. 10b).
3. Bowing or twisting of bones.4. The presence of exostoses in Hereditary Multiple
Exostosis syndrome or iliac horns in the nail-patella syndrome (Fig. 10c).
5. The presence of associated bone tumors, like en-chondromes in Ollier disease and soft-tissue tu-mors in Mafucci disease (Fig. 10c).
6. The absence of bones: e.g. the absence of theradius in the so-called radial ray syndrome (Table6); the absence of the patella in the nail-patellasyndrome (Fig. 10d).
7. The presence of a congenital pseudarthrosis.8. The presence of calcifications in the cartilage, epi-
physes, ligaments and aponeurosis (Fig. 10c).9. Abnormalities in the shape of the clavicula and
scapula (Fig. 10f).10. Abnormal number, shape, and length of ribs.11. An abnormal bone age.
4. Conclusion
A systematic analysis of the different bones, willallow to define and localize the abnormalities precisely.This analytic approach will serve as the first step in theidentification of syndromes and their differential diag-nosis. The second step is the integration of the abnor-malities in different skeletal elements with otherassociated abnormalities in other organ systems. In-deed, symptoms in bone dysplasias are only rarelyrestricted to the skeletal system. In the majority of
cases, syndromic associations in organ systems, like theskin, heart, abdominal viscera, or central nervous sys-tem are depicted. Each individual syndrome usuallyconsists of a combination of several abnormalities. Inthis integration process, the use of a number of cur-rently available textbooks [2,6–12], lists of gamuts [10–14] and internet databanks can be of particular help.
References
[1] Mortier GR. Genetic disorders of the skeleton. A clinical, radio-graphic and molecular study of chondrodysplasias. [academicthesis]. Ghent: University of Gent, 1999.
[2] Swischuk LE, John SD. Bones and soft tissues. In: Swischuk LE,John SD, editors. Differential diagnosis in pediatric radiology,2nd ed. Baltimore: Williams & Wilkins, 1995:187–344.
[3] Vanhoenacker FM, De Beuckeleer LH, Van Hul W, BalemansW, Tan GJ, Hill SC, De Schepper AM. Sclerosing bone dys-plasias: genetic and radioclinical features. Eur Radiol2000;10:1423–33.
[4] Bloom RA. The metacarpal sign. Br J Radiol 1970;43:133–5.[5] Cortina H, Vallcanera A, Andres V, Gracia A, Aparici R, Mari
A. The non-ossified pubis. Pediatr Radiol 1979;8:87–92.[6] Muecke EC, Currarino G. Congenital widening of the pubic
symphysis. Am J Radiol 1968;103:179–85.[7] Poznanski AK. The hand in radiologic diagnosis. Philadelphia:
W.B. Saunders, 1984.[8] Smith DW. Recognizable patterns of human malformations:
genetic, embryologic and clinical aspects. Philadelphia: W.B.Saunders, 1970.
[9] Spranger JW, Langer LO Jr., Wiedemann HR. Bone Dysplasias:An atlas of constitutional disorders of skeletal development.Philadelphia: W.B. Saunders, 1974.
[10] Jones KL. Smith’s recognizable patterns of human malforma-tions, 5th ed. Philadelphia: W.B. Saunders Company, 1997.
[11] Baraitser M. Color atlas of congenital malformations. London:Mosby-Wolfe, 1996.
[12] Taybi H, Lachman RS. Radiology of syndromes, 4th ed.Chicago: Year Book Medical Publishers, 1996.
[13] Kozlowski K, Beighton P. Gamut index of skeletal dysplasias.An aid to radiodiagnosis, 3rd ed. London: Spinger-Verlag, 2001.
[14] Reeder MM, Bradley WG. Reeder and Felson’s gamuts inradiology. Comprehensive lists of roentgen differential diagnosis,3rd ed. New York: Springer-Verlag, 1933.


















