
Computer System Validation - annualrdforum.org.uk · 03.03.2015 · Computer System Validation...
28
1 Computer System Validation Balall Naeem, GCP Inspector May 2018 2 © Crown copyright 2018 About copyright All material created by the MHRA, including materials featured within these MHRA presentation notes and delegate pack, is subject to Crown copyright protection. We control the copyright to our work (which includes all information, database rights, logos and visual images), under a delegation of authority from the Controller of Her Majesty’s Stationery Office (HMSO). The MHRA authorises you to make one free copy, by downloading to printer or to electronic, magnetic or optical storage media, of these presentations for the purposes of private research, study and reference. Any other copy or use of Crown copyright materials featured on this site, in any form or medium is subject to the prior approval of the MHRA. Further information, including an application form for requests to reproduce our material can be found at www.mhra.gov.uk/crowncopyright Material from other organisations The permission to reproduce Crown copyright protected material does not extend to any material in this pack which is subject to a separate licence or is the copyright of a third party. Authorisation to reproduce such material must be obtained from the copyright holders concerned.
Transcript of Computer System Validation - annualrdforum.org.uk · 03.03.2015 · Computer System Validation...

1
Computer System Validation
Balall Naeem, GCP InspectorMay 2018
2
© Crown copyright 2018About copyrightAll material created by the MHRA, including materials featured within these MHRA presentation notes and delegate pack, is subject to Crown copyright protection. We control the copyright to ourwork (which includes all information, database rights, logos and visual images), under a delegationof authority from the Controller of Her Majesty’s Stationery Office (HMSO).
The MHRA authorises you to make one free copy, by downloading to printer or to electronic, magnetic or optical storage media, of these presentations for the purposes of private research, study and reference. Any other copy or use of Crown copyright materials featured on this site, in any form or medium is subject to the prior approval of the MHRA.
Further information, including an application form for requests to reproduce our material can be found at www.mhra.gov.uk/crowncopyright
Material from other organisationsThe permission to reproduce Crown copyright protected material does not extend to any material in this pack which is subject to a separate licence or is the copyright of a third party. Authorisation to reproduce such material must be obtained from the copyright holders concerned.

2
3
Agenda1. Regulations and Guidance2. Inspection Team3. Inspection Overview4. Inspection Finding Areas5. Data Integrity6. Good Documentation Process7. Data Transformation8. Computer Systems Validation9. Trial Master Files10. Audit Trails11. Example Data Management/Statistics/Reporting Findings12. Risk Proportionate Approaches
4
UK SI 2004 No.1031 [as amended], Regulation 28:
(1)No person shall –
(a)Conduct a clinical trial; or
(a)Perform the functions of the sponsor of a clinical trial (whether the person is the sponsor or is acting under arrangements made with that sponsor), otherwise than in accordance with the conditions and principles of good clinical practice.
(3) The sponsor of a clinical trial shall put and keep in place arrangements for the purpose of ensuring that with regard to that trial the conditions and principles of GCP are satisfied or adhered to.
CSV: UK LEGISLATION

3
5
UK SI 2004 No.1031 [as amended], Schedule 1, Part 2:
CONDITIONS AND PRINCIPLES WHICH APPLY TO ALL CLINICAL TRIALS
Principles based on Articles 2 to 5 of the GCP Directive
(3)Clinical trials shall be scientifically sound and guided by ethical principles in all
their aspects
(4)The necessary procedures to secure the quality of every aspect of the trial shall
be complied with
(8)The investigator and sponsor shall consider all relevant guidance with respect to
commencing and conducting a clinical trial.
(9)All clinical information shall be recorded, handled and stored in such a way that
it can be accurately reported, interpreted and verified, while the confidentiality of
records of the trial subjects remain respected.
CSV: UK LEGISLATION
6
UK SI 2004 No.1031 [as amended], Regulation 29:
Subject to regulation 30, no person shall conduct a clinical trial otherwise than in accordance with -
(a)the protocol relating to that trial, as may be amended from time to time in accordance with regulations 22 to 25;
(b)(b) the terms of -
(i) the request for authorisation to conduct that trial,
(ii) the application for an ethics committee opinion in relation to that trial, and
(iii) any particulars or documents, other than the protocol, accompanying that request or that application,
as may be amended from time to time in accordance with regulations 22 to 25; and (c) any conditions imposed by the licensing authority under regulation 18(2) or (6), 19(8), 20(5), 24( 4 5) or Schedule 5.
CSV: UK LEGISLATION

4
7
UK SI 2004 No.1031 [as amended], Regulation 29A
(1) The sponsor of a clinical trial shall notify the licensing authority in writing of any serious breach of—
(a) the conditions and principles of good clinical practice in connection with that trial; or
(b) the protocol relating to that trial, as amended from time to time in accordance with regulations 22 to 25,
within 7 days of becoming aware of that breach.
(2) For the purposes of this regulation, a “serious breach” is a breach which is likely to effect to a significant degree —
(a) the safety or physical or mental integrity of the subjects of the trial; or
(b) the scientific value of the trial.
CSV: UK LEGISLATION
8
UK SI 2004 No.1031 [as amended], Regulation 50:
(1) Any person who in the course of -making an application for an ethics committee opinion;(a) making a request for authorisation to conduct a clinical trial; or(b) making an application for the grant or variation of a manufacturing authorisation,provides to the licensing authority or an ethics committee any relevant information which is false or misleading in a material particular shall be guilty of an offence.
(2) Any person who -(a) is conducting a clinical trial authorised in accordance with these Regulations;(b) is a sponsor of such a clinical trial;(c) while acting under arrangements made with a sponsor of such a clinical trial, performs the functions of that sponsor; or(d) holds a manufacturing authorisation,and who, for the purposes of these Regulations, provides to the licensing authority or an ethics committee any relevant information which is false or misleading in a material particular shall be guilty of an offence.
CSV: UK LEGISLATION

5
9
(3) Any person who, for the purpose of being engaged as a qualified person in accordance with regulation 43, provides to the licensing authority or to the holder of a manufacturing authorisation any information which is false or misleading in a material particular shall be guilty of an offence.
(4) In this regulation, "relevant information" means any information which is relevant to an evaluation of -
(a) the safety, quality or efficacy of an investigational medicinal product;(b) the safety or scientific validity of a clinical trial; or(c) whether, with regard to a clinical trial, the conditions and principles of good clinical practice are being satisfied or adhered to.
CSV: UK LEGISLATION
10
UK SI 2004 No.1031 [as amended], Regulation 31A:
(1)The sponsor shall keep a trial master file for a clinical trial.
(2)The sponsor shall ensure that the trial master file is readily available at all reasonable times for inspection by the licensing authority or any person appointed by the sponsor to audit the arrangements for the trial.
(3)The master file shall at all times contain the essential documents relating to that clinical trial.
(4)The essential documents relating to a clinical trial are those which—
(a)enable both the conduct of the clinical trial and the quality of the data produced to be evaluated; and(b)show whether the trial is, or has been, conducted in accordance with the applicable requirements of Directive 2001/83/EC, the Directive, the GCP Directive and Commission Directive 2003/94/EC.
CSV: UK LEGISLATION

6
11
EU Guidance ICH E6 Addendum
IWG Questions and Answers on Computer System Contracts
Structure and Content of Clinical Study Reports(CPMP/ICH/137/95) ICH E3
General Considerations for Clinical Trials(CPMP/ICH/291/95) ICH E8
Statistical Principles for Clinical Trials(CPMP/ICH/363/96) ICH E9
Trial Management, Data Handling, and Record Keeping5.5.3 When using electronic trial data handling and/or remote electronic trial data systems, the sponsor should:(a) Ensure and document that the electronic data processing system(s) conforms to the sponsor’s established requirements for completeness, accuracy, reliability, and consistent intended performance (i.e. validation).
12
Unit Manager Inspectorate Operations
Head of GLPMA
Andrew Gray
Operations Manager GLPMA / Labs Group
Stephen Vinter*
Operations Manager GCP
Paula Walker
Operations Manager GPvP
Mandeep Rai
Leading Senior Inspectors
Andy Fisher
Jennifer Martin
Senior Inspector
Kathleen Meely
Inspectors
Mandy Budwal-Jagait
Hayley Dixey
Balall Naeem
Leading Senior Inspectors
Jason Wakelin-Smith**
Senior Inspector
Emma Whale**
Inspectors
Michael McGuiness*
Inspectors
Catherine Raitt***
Unit Manager
Inspectorate Strategy & Innovation
Ian Rees
Expert Inspectors
Gail Francis
Head of Inspectorate
Deputy Director Inspection, Enforcement & Standards
Mark Birse
Director
Gerald Heddell
* GCP/Labs Inspector
** GCP/GLP Inspector
***GCP/PV Inspector
Inspection, Enforcement & Standards Division (GCP)

7
13
Types of GCP Inspection
EU Marketing application-related – Triggered/Routine (Centralised co-ordinated by European Medicines Agency (EMA) – conducted by inspectors from 2 Member States). Primarily Trial Specific.
Member State National Inspections (examples provided are for MHRA):• National requested by MS Assessors and occasional ad hoc
collaboration between Member States• National Routine Systems/Routine Trial-Specific (Risk Based
Programme)• National Triggered Systems/Triggered Trial-Specific (requested
by other CA, Ethics Committees, Referrals, Serious Breaches)• Phase 1 Voluntary Accreditation• Inspection in a Third Country (e.g. voluntary inspections in India)
14
Types of Organisations for GCP Inspection
Organisations Inspected (EMA and National)
• Commercial Sponsors (Pharmaceutical Companies etc.)
• Non-Commercial Sponsors (Hospitals, Universities, Charities, Clinical Trials Units)
• Investigator Sites • Subcontractors/Vendors: Niche Providers,
Commercial Laboratories, Phase 1 Units, Contract Research Organisations (CRO), Clinical Trials Units, Computer Systems Providers

8
15
GCP Inspection Activities
Interviews with appropriate staff (e.g. data managers, database programmers, system owners, principal investigators, research nurses, monitors, statisticians, statistical programmers etc.)
Facilities Visits (e.g. storage areas for CRFS, TMFs, Randomisation Codes, Server room)
Paper Document Review (e.g. trial master file, training records, Source Data Verification [hospital notes, test results] etc.)
Electronic Systems/Data Review (e.g. trial master file, database for trial/subject records, audit trails, view programming/stats output/datasets, query process documentation, Paper CRF receipt/tracking/entry, Source Data Verification [hospital notes, test results], Report Contents/CRFs etc.)
16
Source Data Verification
SYSTEMS
STUDY SPECIFIC
•CLINICAL
•STUDY
•REPORT
Monitoring
Data
Management
Statistics
Medical
Writing
CLINICAL STUDY
REPORT

9
17
Inspection Finding Areas Impacted by CSV • Archiving• CRF Data / Source Data• Computer System Validation• Data Integrity• Data Management• Data Monitoring Committee/Trial Steering Committee • IT Systems• Medical Writing• Monitoring• Organisation's Oversight of Clinical Trials of IMP• Record keeping/Essential Documents• Statistics• IMP Management / Pharmacy
18
Good Documentation Practice
Source data and records should meet the ALCOA + CCEA principles:
• Accurate, Legible, Contemporaneous, Original, Attributable
• Complete, Consistent, Enduring, Available when needed

10
19
Good Documentation Practice
Requirements for paper systems are well established, but what about electronic systems?
20
Good Documentation Practice
Meta DataMetadata is data that describe the attributes of other data, and provide context and meaning. Typically, these are data that describe the structure, data elements, inter-relationships and other characteristics of data. It also permits data to be attributable to an individual.

11
21
Good Documentation Practice
42
22
Good Documentation PracticeMeta Data:
42 kg (entered G Tom, 02 March 2015)
41 kg (entered S Bob, 03 March 2015)(changed by S Bob, original record was 42kg (G Tom, 02 March 2015) – reason for change , results for J Smith entered in error)

12
23
Data Transformation
24
Data Transformation
Paper to Paper – Patients Notes to CRF
Paper to Electronic – CRF to Database
Electronic to Electronic – Database to Stats Tables
Electronic to Paper – Stats Tables to Study Report

13
25
2Investigator completes
CRF
3NCR
Copies the CRF
4Investigator
keeps copy of CRF
5Sponsor receives
Original CRF
6Sponsor adds
data to trial database
1Observations made
and recorded in clinical notes
Data verification can occur by:Comparing 1 with 4 (Source Data Verification)
Comparing 1 with 5 and 6 (Trial re-construction)
The Old Paper Way
Contemporaneous Copy!
26
2Investigator completes
e-CRF
3e-CRF data
Storage
1Observations made
and recorded in clinical notes/work
books
4Sponsor copies
data
5Investigator given
a copy of their data
Data verification can occur by:Comparing 1 with 3 and/or 5
Electronic Capture of Transcribed Data
Non- Contemporaneous Copy!
14
27
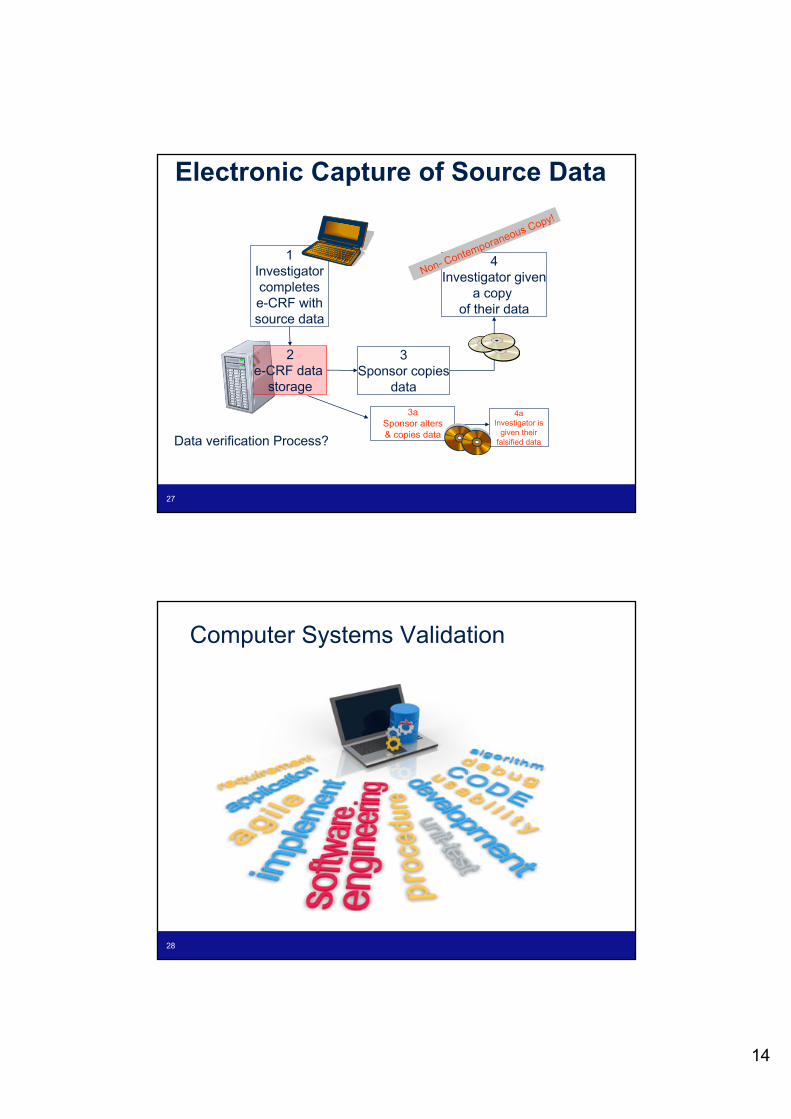
1Investigator completes e-CRF with source data
2e-CRF data
storage
3Sponsor copies
data
4Investigator given
a copy of their data
Data verification Process?
3a Sponsor alters & copies data
4aInvestigator is
given their falsified data
Electronic Capture of Source Data
Non- Contemporaneous Copy!
28
Computer Systems Validation

15
29
Computer System?
What is a computer system?• Computer hardware• Computer software• Peripherals• Documentation (manuals, SOPs)• Personnel
Collectively defines and controls the performance of one or more automated business processes
30
System Development Lifecycle
Similar idea to data lifecycle
Concept
• Business need / Benefits• Requirements• Potential solutions
Project
• Design• Build• Verification
Operation
• Routine use• Maintenance of validated state / control• Change management
Retirement
• Data Retention / migration / destruction• Management of retirement processes

16
31
System Development LifecycleDo you know what you want it to do?Do you know what are the must-haves vs nice-to-haves?Have you defined what is acceptable to you?How will you ‘do’ each of the steps?Once in place how will it be maintained and controlled?Refinements? Exit strategy?
32
Computer System Validation
CSV forms part of the system development lifecycle
‘Establishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting its pre-determined specifications and quality attributes’ (FDA Process Validation: General Principles and Practices - 2011)
‘Achieving and maintaining compliance with applicable GXP regulations and fitness for intended use by: • The adoption of principles, approaches, and life cycle
activities within the framework of validation plans and reports• The application of appropriate operational controls
throughout the life of the system’ (GAMP 5 – 2008)

17
33
“Fit for Purpose” Validation (ICH 5.5.3(a))
It is expected that systems are validated to accepted standards, additionally, principles of computer system validation should be applied to study specific builds/programs/applications, particularly:-
• Specifications• Testing against specifications (traceability)• Approval & release• Change control
34
Why does it matter?
Can I trust the data generated?• Accuracy• Reliability• Integrity• Availability• Authenticity
Does the system do what it is meant to do?

18
35
Where does risk fit in?Expectation is that appropriate controls are in place to support and safeguard the data generated
Expect systems to be designed and operated in a manner which provides an acceptable state of control based upon the data integrity risk
Expect that effort and resource to ensure appropriate control and governance is commensurate with the criticality of the data being generated
36
Where does risk fit in?
The data integrity risk assessment should be fully documented
• System requirements level• Computerised system level• Business process level
Part of a wider control and assurance strategy

19
37
CSV Requirements
Off The Shelf
CustomisedBespoke
Infrastructure as a service (Iaas)Platform as a service (Paas)Software as a service (Saas)
InterfacesSynchronisationProgramsSpreadsheetsCloud
38
Vendor Supplied Software
If you purchase software from a vendor and also purchase their validation package from them do you need to conduct any further validation activities?
1 = Yes2 = No

20
39
Vendor Supplied Software
If you purchase software from a vendor and also purchase their validation package from them do you need to conduct any further validation activities?
1 = Yes
40
Vendor Supplied Software
Vendor likely to have only performed functional verification (IQ and OQ) activitiesCan you demonstrate fitness for intended use?• Performance qualification (fitness for use within business
processes and operational environment)• Configuration (which bits turned on and off?)• SOPs (use, data review, control, training)• Staff training

21
41
Validation Models
Expectation is that validation process is formalised to include appropriate specifications and requirements, robust testing, accurate reporting of outcomes with suitable quality ‘gates’ and ‘gate keepers’Many models available (or as a mixture):• V-Model• Agile development • Waterfall• Fountain model
• Build and Fix • Synchronise & Stabilise
42
Validation ModelsExpectation is that validation process is formalised to include appropriatespecifications and requirements, robust testing, accurate reporting ofoutcomes with suitable quality ‘gates’ and ‘gate keepers’Many models available (or as a mixture):
• V-Model• Agile development • Waterfall• Fountain model• Build and Fix • Synchronise & Stabilise

22
43
V-Model
44
Agile

23
45
Agile
46
Agile

24
47
Whose Responsibility is Validation?
• CSV Representative• Quality Representative• Technical Representative• Key User
48
Inspecting GCP eSystems
What does the documentation say?
• Specifications and risk assessments• Plan for validation• Test planning and execution• Traceability• User Manuals/Training/SOPs• Validation Report• System Release into Production• Change control

25
49
Inspecting GCP eSystems
What does the system say?
• Release• Access & Security• Users• Audit trails• Reporting• Error Logs
Evidence
50
Inspecting GCP e-Systems
What do the people say?• System understanding• Awareness• Risk awareness• Electronic infrastructure• Supporting frameworks and quality structures

26
51
Audit Trails
52
Point Audit Trail vs Audit Trail Dataset
• Ability to access the audit trail on a field in the live system• Electronic Data Query Form
The change to electronic systems provided opportunity to review the audit trail AS A WHOLE DATASET. The eSystems appear to have been developed without this benefit being exploited
Basis of opinion? – poor quality presentation of audit trial datasets

27
53
Quality of Data• Very variable how data are/could be presented (pdf, WORD ®,
Excel ®, SAS, screen dumps of CRF pages etc.)
• Issues in type, formatting, variable naming etc., can make use difficult
• Data can be a dataset, log, or a document
• Accessibility of the data – often locked for editing, printing, saving etc. or provided as an image rather so hard to interrogate further
• Poor quality indicative of fact sponsors do not generally (from asking at inspections) hold or review this metadata.
54
CSV Issues
• No processes for CSV and control of the system• Records not approved and not issued in a timely manner
(e.g. validation report issued after system release)• Lack of detailed risk assessment• Failure to document testing of functional unit coding
development prior to release to user acceptance testing• Lack of traceability of the testing of requirements and
verification of passes• Testing documentation incomplete or not retained• Deferring “critical” test failures for future release with no
documented rationale• Poor documentation of change control

28
55
CSV Issues
• Installation into a production environment prior to confirmation system/study build has been validated (e.g. eCRF released on draft edit checks specification)
• Poor documentation of the go live decision (e.g. engineer installation reports, emails etc.)
• User manuals / training not produced in timely manner • Change control backlogs• Incomplete documentation of programming validation (e.g.
for data validation, data extraction or analysis)• Lack of Sponsor control of the system• Access controls
56
Questions?


















