
Computational Prediction of Pharmaceutical Crystal ... · Computational Prediction of...
36
Control and Prediction of the Organic Solid State A Basic Technology project of the Research Councils UK Computational Prediction of Pharmaceutical Crystal Structures - a severe test of modelling supramolecular assembly Sarah (Sally) L Price Department of Chemistry, UCL www.cposs.org.uk
Transcript of Computational Prediction of Pharmaceutical Crystal ... · Computational Prediction of...

Control and Prediction of the Organic Solid State
A Basic Technology project of the Research Councils UK
Computational Prediction of
Pharmaceutical Crystal Structures -
a severe test of modelling
supramolecular assembly
Sarah (Sally) L Price
Department of Chemistry, UCL www.cposs.org.uk

Contrast Crystals - geological
Durable &
hard as
strong
forces
between
atoms
Grown on
geological
timescales-
10 to 1000
years 2cm
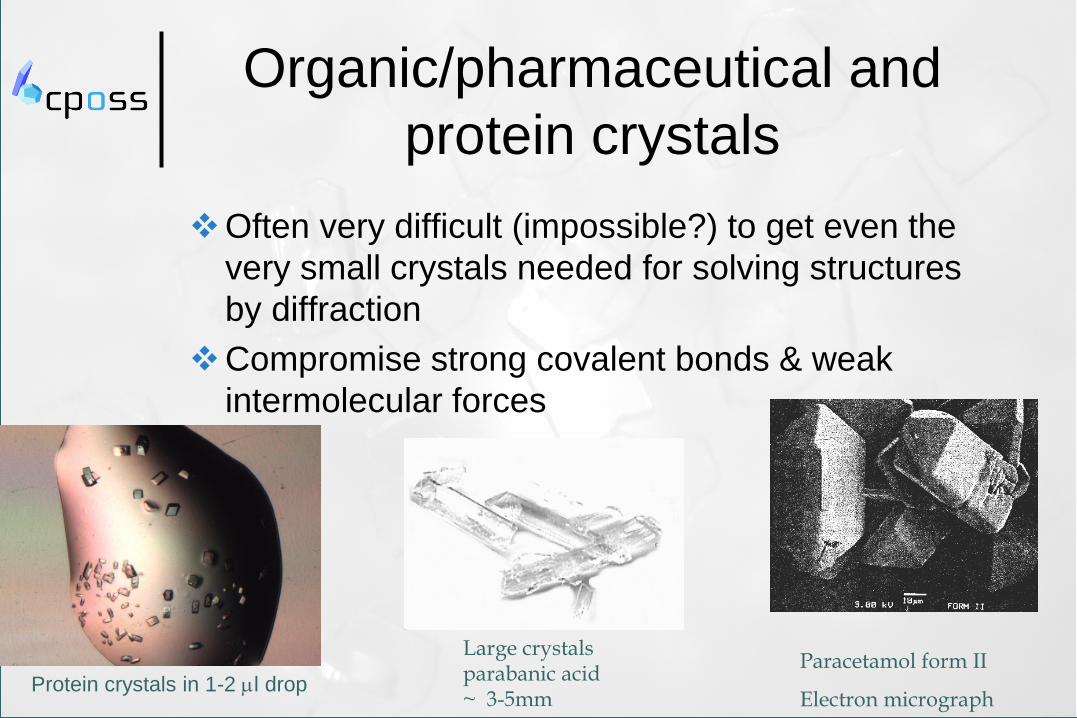
Organic/pharmaceutical and
protein crystals
Often very difficult (impossible?) to get even the
very small crystals needed for solving structures
by diffraction
Compromise strong covalent bonds & weak
intermolecular forces
many
Large crystals parabanic acid ~ 3-5mm
Paracetamol form II
Electron micrograph Protein crystals in 1-2 ml drop
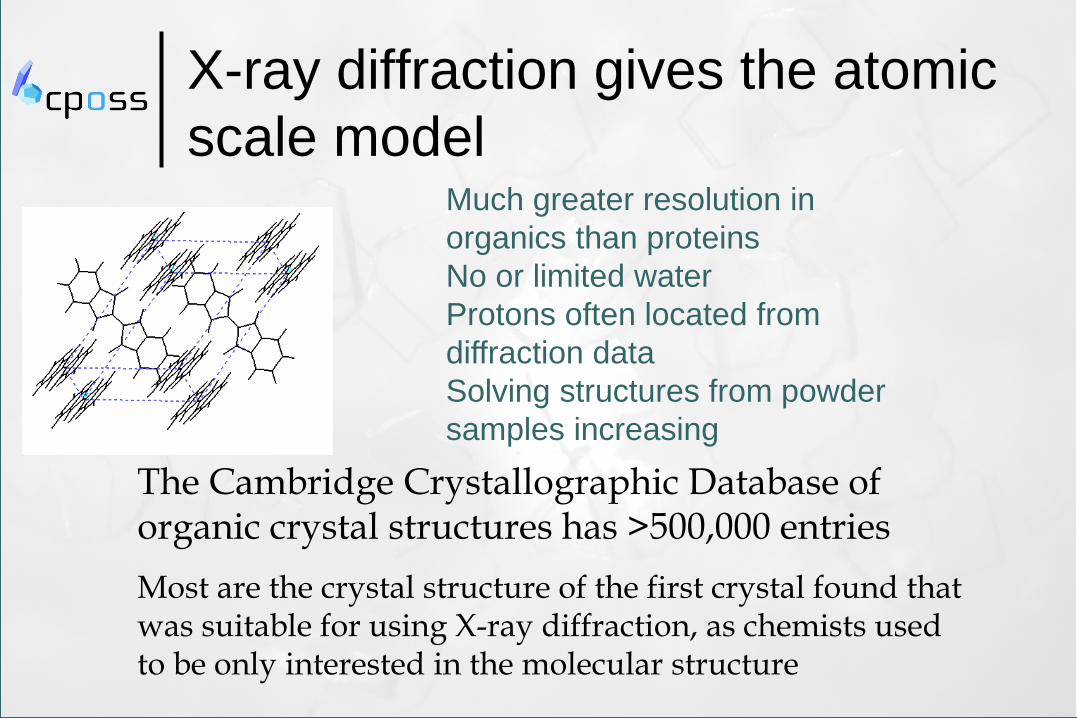
X-ray diffraction gives the atomic
scale model
The Cambridge Crystallographic Database of organic crystal structures has >500,000 entries
Most are the crystal structure of the first crystal found that was suitable for using X-ray diffraction, as chemists used to be only interested in the molecular structure
Much greater resolution in
organics than proteins
No or limited water
Protons often located from
diffraction data
Solving structures from powder
samples increasing

Principles of crystal packing
small molecules to proteins
Close packed
solvent may fill small voids
dynamic water surrounds most protein molecules
Forms hydrogen bonds, p-p stacking, X...X
Intermolecular, more diversity
Intramolecular, amino acids
Conformation vital
~ isolated molecule (Y), some torsions vary
dominant issue & major constraint
Emphasis on inter - vs intra- molecular forces differ

Exercises in prognostication:
Crystal structures and protein folding JD Duntiz & HA Scheraga, 2004 PNAS 101, 14309
global optimisation problems to identify the
structure(s) of lowest potential (or free)
energy
Search challenge
Accuracy of energy evaluation
Kinetic factors ~ preferred pathways to
assembly, may be involved
Objective blind tests “need to be maintained
so they can continue to document progress
and monitor excessive claims”

Polymorphism - a common
phenomenon??
Polymorphism - the ability of a substance to adopt
more than one crystal structure
since different physical properties, now a major
cause for concern when products transform from one
polymorph to another.
•Pigments - change colour.
•Chocolate - need polymorph of
cocoa butter that melts at 37 oC.
• Explosives - change of
polymorphic form leads to
different detonation properties &
industrial accidents.
L. Yu et al. 2000, J. Am. Chem. Soc. 122, 585.

Pharmaceuticals must be
marketed in one controlled
polymorphic form
Change of polymorph changes effective
dose
Want to choose the crystalline form for
optimum properties & control production
Regulatory requirement for
pharmaceuticals that all reasonable
experiments are performed in order to
identify the maximum number of crystalline
forms
Disaster if new polymorph appears during
production or storage, or in rival’s labs

Difficulty in establishing that all
polymorphs are known
McCrone (1963) “the number of
polymorphs of a material depends
on the amount of time and money
spent in research on that
compound”
- Some appear after decades of
crystallisation work on compound
- Some “disappear” after a more stable
polymorph is discovered.

Which drugs may have
undiscovered polymorphs?
1998 Abbott Laboratories anti-HIV drug
Ritonavir produced new polymorph during
manufacture after 2 years
Problem affected plants in different countries
Required reformulation “ Unfortunately, there is nothing we can do today to prevent
a hurricane from striking any community or polymorphism
from striking any drug” Sun, Abbott Laboratories, press conference.
Can we computationally predict whether the drug is
in the polymorphism equivalent of Louisiana or
Hertfordshire, Herefordshire or Hampshire ?

Why calculate crystal energy landscapes?
~ the thermodynamically feasible crystal structures
to confirm that most stable polymorph is known
to design new molecular materials prior to synthesis
to see what structures are plausible undiscovered
polymorphs
Thermodynamics vs. crystallization conditions
(T, P, solvent, supersaturation, impurities, …..)
to help solve structures from powder XRD or other
experimental evidence
as a complement to polymorph screening and “Quality
by Design” crystallization processes in pharmaceutical
development.

2010 5th CCDC Blind Test results –
can we predict a crystal structure?
x/y x = # correct within 3 submitted
y = # groups submitting *own success
Main issue is accuracy of calculating relative energies of
different crystal structures
O
O
NH
N
S
CH3
SO2
CH3
N+ N
-O
O2N
NO2
Cl
Cl
S
Cl
O
N+
N-
CH3
O O
OH
OH
OHHOOC
OH2
Polymorphs 3 and 4
N NH
+
COOH
COO-
A salt
2/15 2/13* 1/13 2/11
2/10* 0 or 2 excl H* /10

Successful approaches to calculating
lattice energy (biological force-fields rarely adequate)
Plane wave density functional theory (i.e. crystal Y) supplemented by empirically damped
-C6/R6 dispersion
Elatt=Eelectronic+Edisp developed by fitting to
crystal structures
Model for intermolecular forces with electrostatic model derived from isolated molecule Y
DEintra from Y
Elatt = Uinter+ DEintra Non-spherical atoms
Use theory of intermolecular forces, moving toward non- empirical models Success in 4th for C6Br2ClFH2
with no experimental input
Neumann, M. A.; Perrin, M. A. J.Phys.Chem.B 2005, 109, 15531
Misquitta AJ, Welch GWA, Stone AJ, Price SL 2008.Chem Phys Lett 456
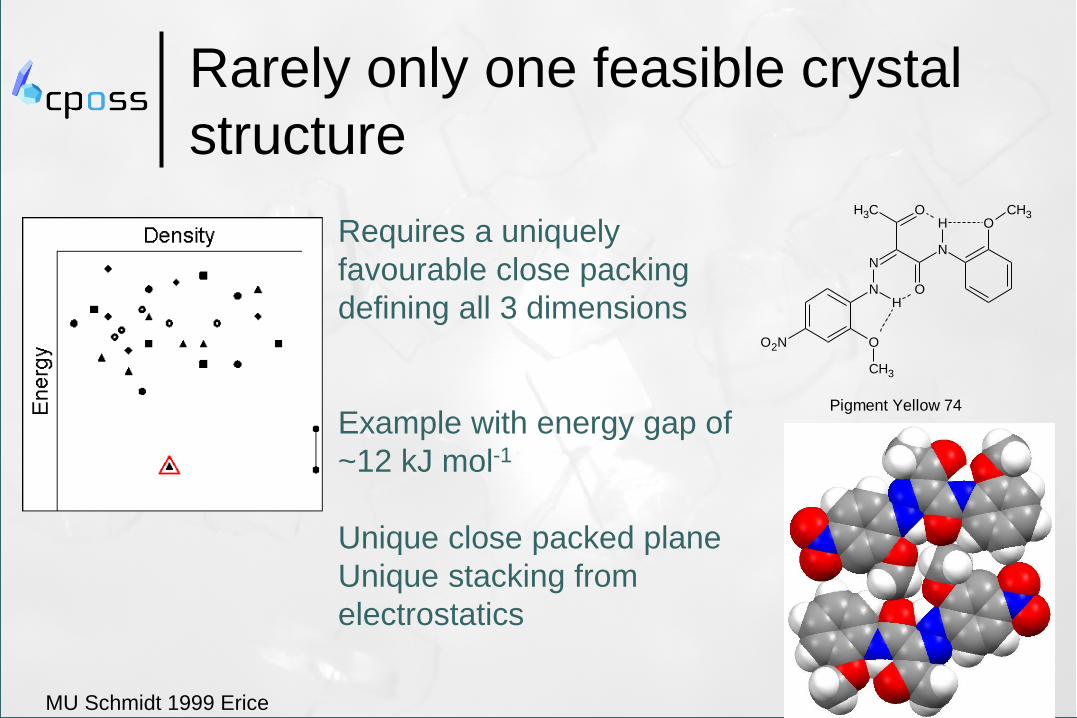
Rarely only one feasible crystal
structure
O
CH3
N
O2N
N
OH
CH3 O
N
H OCH3
Pigment Yellow 74
Requires a uniquely
favourable close packing
defining all 3 dimensions
Example with energy gap of
~12 kJ mol-1
Unique close packed plane
Unique stacking from
electrostatics
MU Schmidt 1999 Erice

More typical
Cl
BrBr
F
HH
from 2007 blind test
Landscapes will show the
expected hydrogen bond
motifs defining
ribbons/layers
BUT different
•packings of ribbons
•stackings of layers
More predicted
structures than known
polymorphs
Relative energies
sensitive to method

Basic method for crystal energy landscapes ~ thermodynamically feasible crystal structures
Use quantum mechanics to predict molecular structure and represent the charge distribution within the molecule (repeat with multiple conformers for flexible molecules, using intramolecular energy penalty DEintra)
Use search method to generate plausible crystal structures (~3000 MOLPAK or ~105 CrystalPredictor for each rigid conformation, or >106 for flexible CrystalPredictor) for Z’=1,...
Use advanced models of the intermolecular forces (distributed multipoles to represent lone pair & p electron density) to minimize the intermolecular lattice energy Uinter of each crystal structure.
Refine conformation within crystal to minimize Elatt= Uinter + DEintra
> Basic Crystal (Lattice) Energy Landscape
Estimate lattice modes, elastic tensor & harmonic free energies for rigid molecules and confidence in relative stabilities.
Calculate other properties: PXRD, morphologies
Karamertzanis PG, Kazantsev AV, Issa N, Welch GWA, Adjiman CS, Pantelides CC, Price SL 2009. J Chem
Theory Comput 5, 1432 Price SL, Leslie M, Welch GWA, Habgood M, Price LS, Karamertzanis PG, Day GM
2010. Phys Chem Chem Phys 12:8478-8490.

Why do we overpredict polymorphism ?
1 Neglect of thermal motion
Free energy landscape for benzene has ~ a minimum for each known form in a metadynamics study
Both have many lattice energy minima, and ~ only the observed structures when thermal motion modelled.
Solid state transitions unusually facile for these hydrocarbons
Cyclopentane
C5H10
Torrisi A, Leech CK, Shankland K, David WIF, Ibberson RM, Benet-Buchholz J, Boese R, Leslie M, Catlow CRA,
Price SL 2008. J Phys Chem B 112:3746
MD 30K ~ form III
MD 160K ~ form I
Plastic phases
Raiteri, P. et al. Angew.Chem.,Int.Ed. 2005, 44, 3769
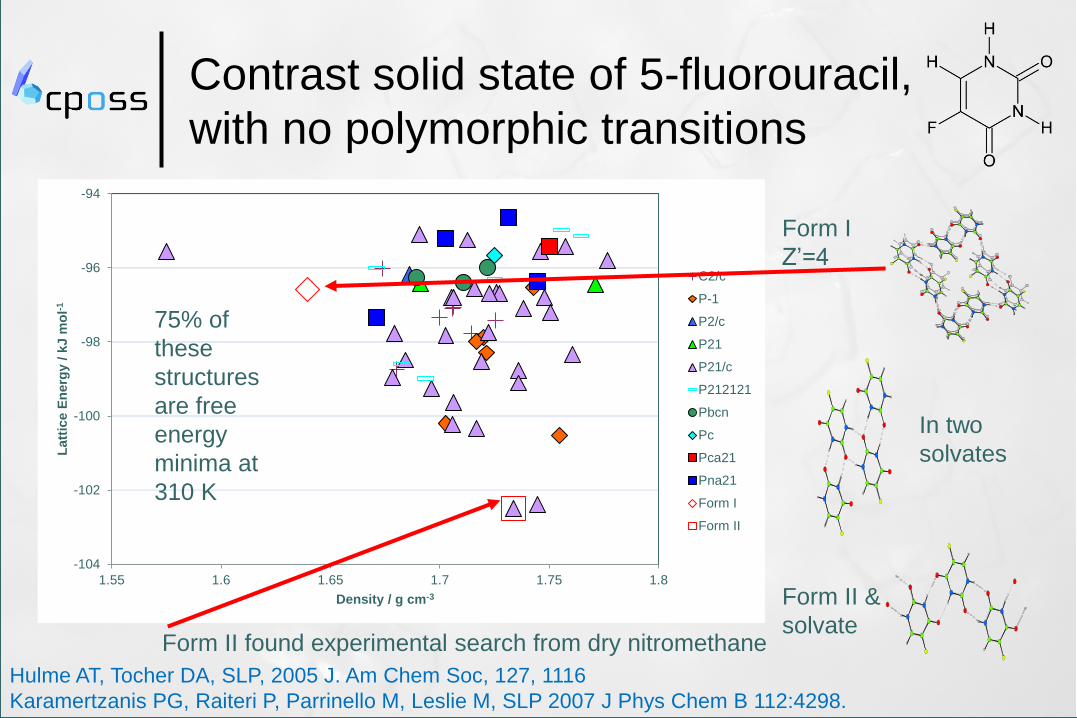
-104
-102
-100
-98
-96
-94
1.55 1.6 1.65 1.7 1.75 1.8
Latt
ice E
nerg
y /
kJ m
ol-
1
Density / g cm-3
C2/c
P-1
P2/c
P21
P21/c
P212121
Pbcn
Pc
Pca21
Pna21
Form I
Form II
Contrast solid state of 5-fluorouracil,
with no polymorphic transitions
Form II found experimental search from dry nitromethane
Form I
Z’=4
Form II &
solvate
In two
solvates
75% of
these
structures
are free
energy
minima at
310 K
Hulme AT, Tocher DA, SLP, 2005 J. Am Chem Soc, 127, 1116
Karamertzanis PG, Raiteri P, Parrinello M, Leslie M, SLP 2007 J Phys Chem B 112:4298.

Do we need to do Molecular
Dynamics to model thermal motion?
Only if expect facile phase transitions.
Dynamics of nucleation & growth will
determine which structures are observed
Hamad, S, Moon, C, Catlow, CRA, Hulme, AT, SLP, 2006 J. Phys. Chem. B, 110 3323
hydration of
uracil in
water gives
close F···F of
form I
in
nitromethane
get R22 (8) of
form II

Solid-State Forms of b-Resorcylic Acid: How Exhaustive Should a Polymorph Screen Be? Braun DE, Karamertzanis PG, Arlin J-B, Florence AJ, Kahlenberg V, Tocher DA,
Griesser UJ, Price SL 2011 Cryst Growth Des 11: 210-220.
New polymorph I predicted,
Added confidence to PXRD
solution and evidence for proton
disorder
Similar
structures,
unlikely to be
distinguishable
polymorphs
How?
Relative stability?
Catemer polymorph?

Why do we overpredict polymorphism ?
2 The right crystallization
experiment has yet to be performed
Huge range of crystallization methods which have generated new polymorphs
– deliberate to failed cocrystallization
Experimental conditions vary kinetics of nucleation & growth
Can we use crystal energy landscapes to find the right crystallization conditions?
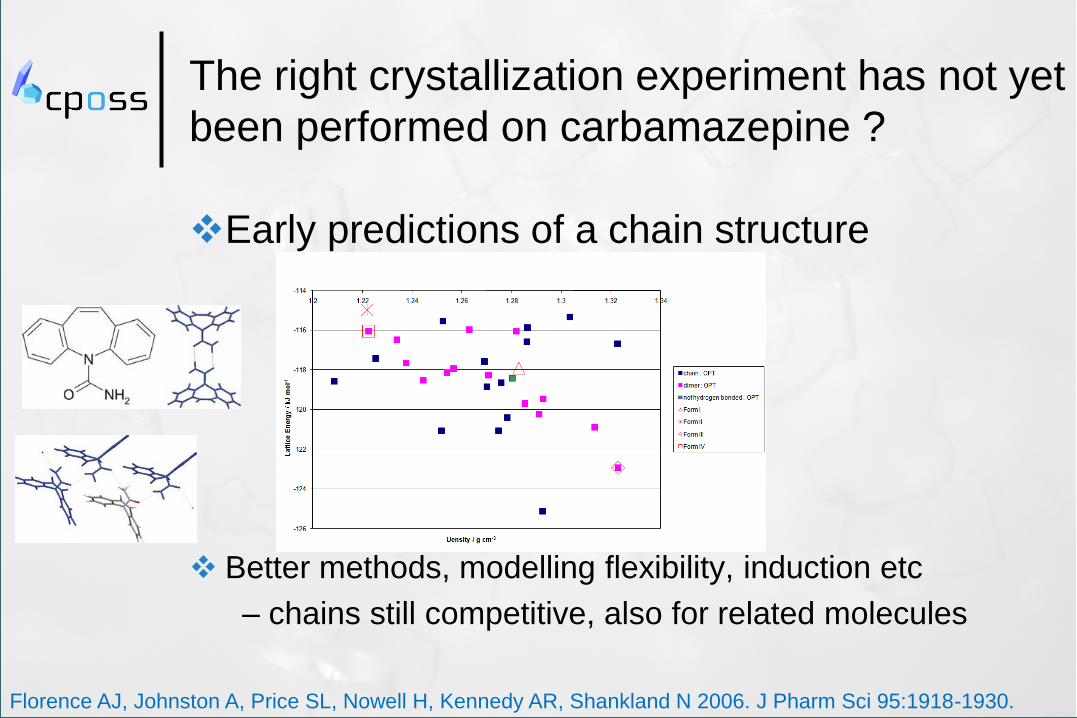
The right crystallization experiment has not yet
been performed on carbamazepine ?
Early predictions of a chain structure
Better methods, modelling flexibility, induction etc
– chains still competitive, also for related molecules
Florence AJ, Johnston A, Price SL, Nowell H, Kennedy AR, Shankland N 2006. J Pharm Sci 95:1918-1930.
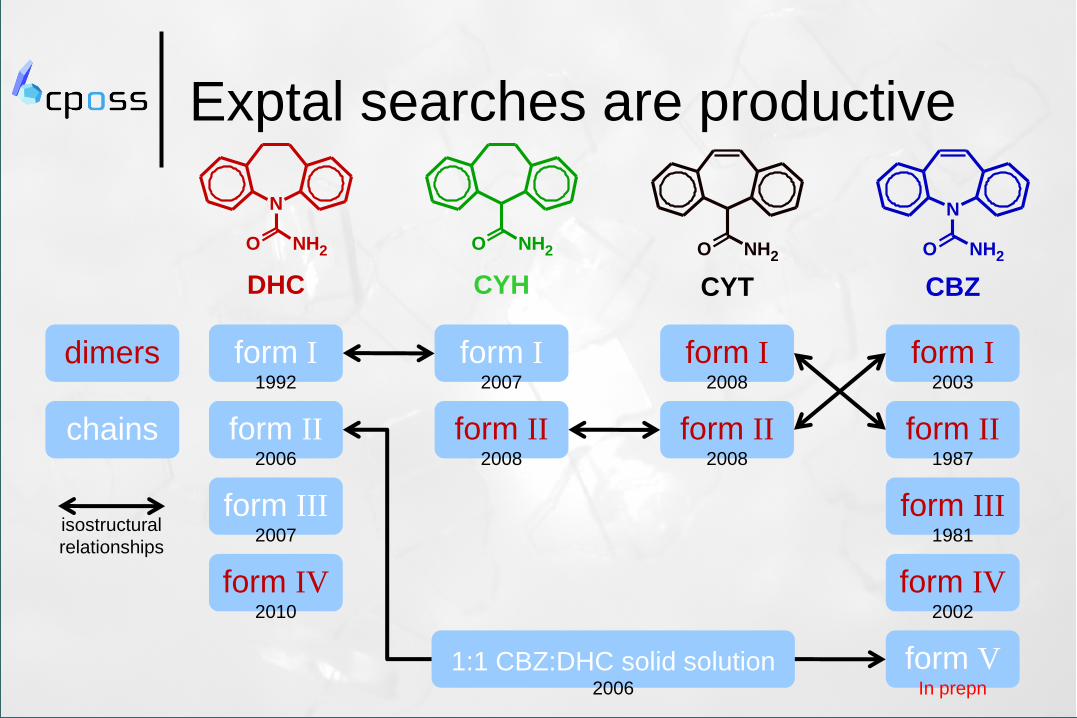
Exptal searches are productive
dimers
form II 2008
form I 2008
chains
form IV 2002
form III 1981
form II 1987
form I 2003
form V In prepn
form IV 2010
form III 2007
form II 2006
form I 1992
form II 2008
form I 2007
1:1 CBZ:DHC solid solution 2006
N
O NH2
CBZ
N
O NH2
DHC
O NH2
CYH
O NH2
CYT
isostructural
relationships

Success of catemeric CBZ V
required seeded sublimation
Pbca a/Å b/Å c/Å
Expt 9.1245(5) 10.4518(5) 24.8224(11)
Rigid
prediction
9.3124 10.5979 24.8819
Flex
prediction
9.4816 10.3426 24.7227
DHC form II (seed)
CBZ form V CBZ form V
CBZ form V
Arlin J-B, Price LS, Price SL, Florence AJ, in prepn

Finding the right crystallization
conditions may be even harder
Racemic crystal could not be
formed without racemization
Lancaster, RW; Karamertzanis, PG; Hulme, AT; Tocher, DA; Covey, DF; Price, SL, Chem.Commun., 2006, 47, 4921
- or obliging synthetic chemist
Challenge: what about cases where barrier is high but no so high?
e.g. Changing to an unfavourable conformation – c.f. ritonavir

(Dis)Appearing polymorphs
Only need to nucleate more stable form
once to get seeds
Develop other routes to most stable form
May lead to loss of control of crystallisation
of metastable form
Other forms of seeding/templating May need impurities to producing a polymorph
1 mol% ethamindosulphathiazole stabilizes form I sulphathiazole
Attempts to reproduce form 2 progesterone failed
– could only get moderately unstable samples when
crystallised in presence of pregnenolone
Lancaster RW, Karamertzanis PG, Hulme AT, Tocher DA, Lewis TC, Price SL 2007. J Pharm Sci 96:3419-3431.
N. Blagden, R. J. Davey, R. Rowe and R. Roberts, Int. J. Pharm., 1998, 172, 169-177.
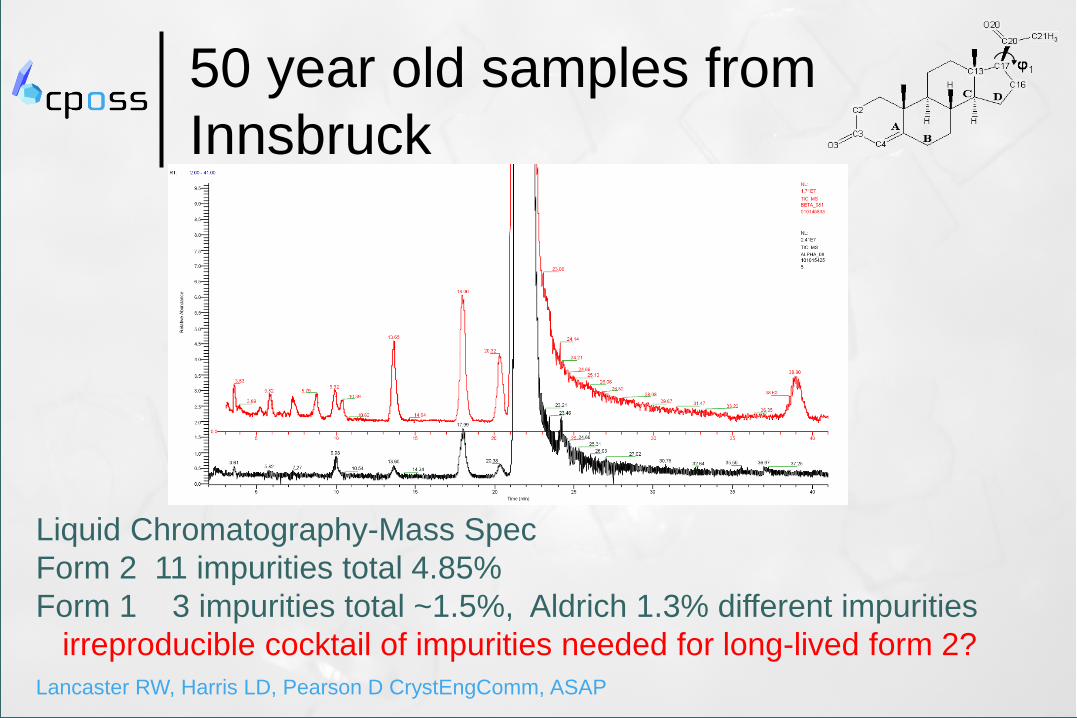
50 year old samples from
Innsbruck
Liquid Chromatography-Mass Spec
Form 2 11 impurities total 4.85%
Form 1 3 impurities total ~1.5%, Aldrich 1.3% different impurities
irreproducible cocktail of impurities needed for long-lived form 2?
Lancaster RW, Harris LD, Pearson D CrystEngComm, ASAP

Why do we overpredict polymorphism ?
3 The right crystallization
experiment cannot be performed
Crystal may be unstable relative to other products,
inherent in possible range of crystallization
experiments
Solvates may form
Proton transfer – salt or cocrystal if 0< DpKa <3
Cocrystal may be less stable than components

Why do we overpredict polymorphism ?
4.Plurality of possible structures is
hindering crystallization
Crystallization is difficult
“Commonly found that when good quality large crystals of a substance cannot be grown, the small crystals are poor in quality with substantial mosaic spread” Harding, M. M. J. Synchrotron Radiat. 1996, 3, 250
i.e. structures solved from very small crystals (synchroton) are more likely to be disordered
Can crystal energy landscape can warn of possibilities of disorder = combinations of low energy structures?
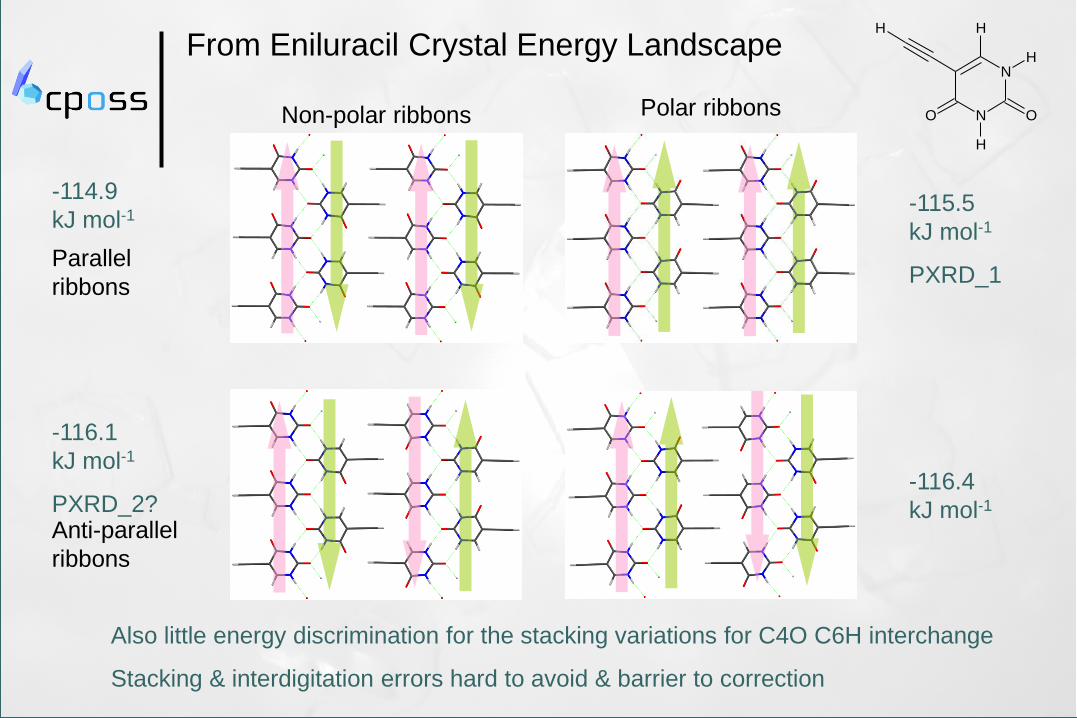
Parallel
ribbons
-114.9
kJ mol-1
Anti-parallel
ribbons
-116.1
kJ mol-1
PXRD_2?
-115.5
kJ mol-1
PXRD_1
-116.4
kJ mol-1
From Eniluracil Crystal Energy Landscape
Non-polar ribbons
Also little energy discrimination for the stacking variations for C4O C6H interchange
Stacking & interdigitation errors hard to avoid & barrier to correction
Polar ribbons
N
N
H
H
OO
HH
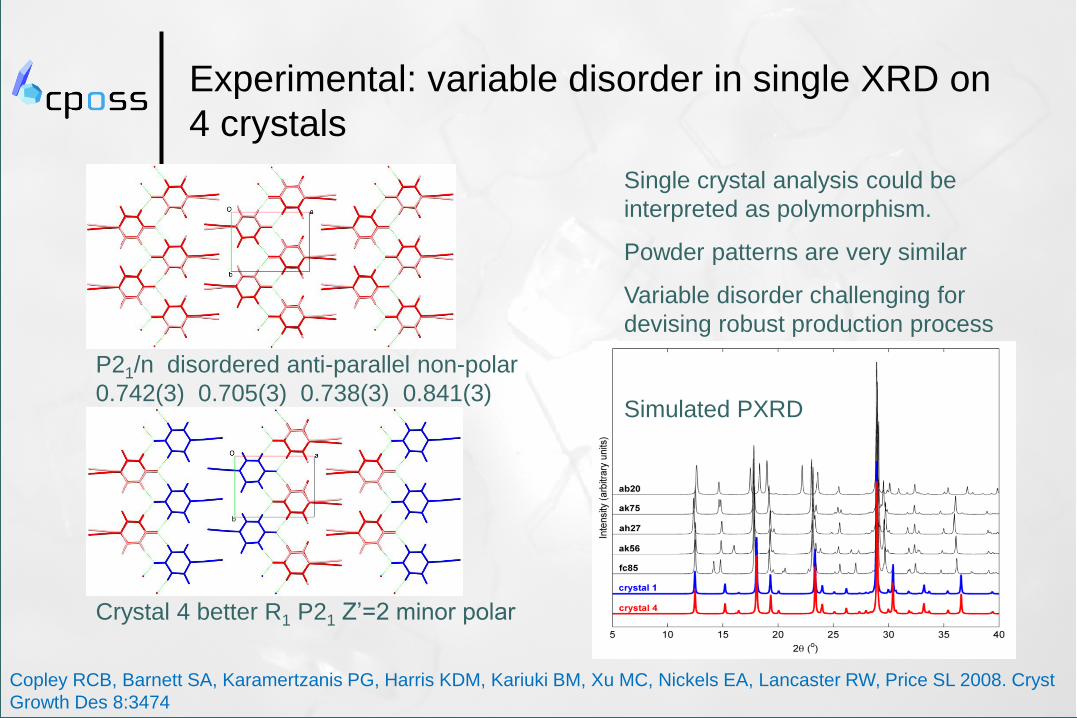
Experimental: variable disorder in single XRD on
4 crystals
P21/n disordered anti-parallel non-polar
0.742(3) 0.705(3) 0.738(3) 0.841(3)
Crystal 4 better R1 P21 Z’=2 minor polar
Single crystal analysis could be
interpreted as polymorphism.
Powder patterns are very similar
Variable disorder challenging for
devising robust production process
Simulated PXRD
Copley RCB, Barnett SA, Karamertzanis PG, Harris KDM, Kariuki BM, Xu MC, Nickels EA, Lancaster RW, Price SL 2008. Cryst
Growth Des 8:3474

Where are we now?
Crystal energy landscapes complement experiment, providing the alternatives
likely motifs in solid forms
range of possible target structures
possible types of disorder
Can be calculated with “good enough” accuracy for increasing range of molecules & multi-component systems
from aspirin / paracetamol to modern pharmaceuticals
Database of
computed
crystal
structures
>150 molecules
O
OOHH
N
O
NH2
O
N+
H
H
HO-
O
N
NH
SCH3
N
N
CH3

What are the challenges?
Improving accuracy of relative energies
periodic electronic structure DFT+D
non-empirical anisotropic atom-atom potentials
free energies
Understanding limitations of thermodynamic
predictions ~ kinetic factors that lead to
polymorphism
Move to modern pharmaceuticals
Computational efficiency

Grateful Thanks to
Matthew Habgood, Doris Braun, Nizar Issa, Gareth Welch, Sharmarke Mohamed
Derek Tocher, Louise Price, M Leslie (ex-CCLRC) Bob Lancaster (ex-GSK) (UCL)
Andrei Kazantsev, Panos Karamertzanis, Costas Pantelides, Claire Adijman (IC)
Alastair Florence, Andrea Johnston, Jean-Baptiste Arlin, Phillipe Fernandes (SU)
Other coworkers in CPOSS and many collaborators
Other Programs AJ Stone (Cambridge), H Ammon (Maryland), CCDC
Computing infrastructure: National Grid Service (database), HPC(x), UCL
CCDC & CSP community for blind tests
Funding EPSRC (including E-Science)
Basic Technology Program of RC UK for funding Control and Prediction of the Organic Solid State www.cposs.org.uk, including “Translation” funding for Knowledge Transfer in CPOSS Industrial Alliance from April 2008.

CPOSS Open Day, UCL
Wednesday 30 March 2011
10.00, Coffee and registration in the South Cloisters
Introduction and welcome,
Progress in the fifth International Test of Crystal Structure Prediction,
Industrial problems from polymorphism and how we might avoid them, Dr Colin Groom, CCDC
Mapping Crystallization Processes Using In-Situ SSNMR, Dr Colan Hughes, Cardiff University
First and Second Order Transitions: A Re-appraisal, Dr Terry Threlfall, University of Southampton
12.30, Lunch and poster session
2.00, The role of transformations in pharmaceutical crystallization, Prof. Kieran Hodnett, University of
Limerick
GIPAW: a "Bragg's Law" for solid state NMR, Prof. Chris Pickard, UCL
Experimental screening and characterization of solid forms, Prof. Alastair Florence, University of
Strathclyde
3.45, Coffee and poster session cont.
5.00, Removal of posters
Sponsored by CPOSS Industrial Alliance – visit www.cposs.org.uk to register


















