
Comparison of liposomal drug formulations for transdermal iontophoretic drug delivery · 1 Title:...
27
Tampere University of Technology Comparison of liposomal drug formulations for transdermal iontophoretic drug delivery Citation Malinovskaja-Gomez, K., Espuelas, S., Garrido, M. J., Hirvonen, J., & Laaksonen, T. (2017). Comparison of liposomal drug formulations for transdermal iontophoretic drug delivery. European Journal of Pharmaceutical Sciences, 106, 294-301. https://doi.org/10.1016/j.ejps.2017.06.025 Year 2017 Version Early version (pre-print) Link to publication TUTCRIS Portal (http://www.tut.fi/tutcris) Published in European Journal of Pharmaceutical Sciences DOI 10.1016/j.ejps.2017.06.025 Copyright © 2017. This manuscript version is made available under the CC-BY-NC-ND 4.0 license http://creativecommons.org/licenses/by-nc-nd/4.0/ License CC BY-NC-ND Take down policy If you believe that this document breaches copyright, please contact [email protected], and we will remove access to the work immediately and investigate your claim. Download date:03.07.2020
Transcript of Comparison of liposomal drug formulations for transdermal iontophoretic drug delivery · 1 Title:...

Tampere University of Technology
Comparison of liposomal drug formulations for transdermal iontophoretic drugdelivery
CitationMalinovskaja-Gomez, K., Espuelas, S., Garrido, M. J., Hirvonen, J., & Laaksonen, T. (2017). Comparison ofliposomal drug formulations for transdermal iontophoretic drug delivery. European Journal of PharmaceuticalSciences, 106, 294-301. https://doi.org/10.1016/j.ejps.2017.06.025Year2017
VersionEarly version (pre-print)
Link to publicationTUTCRIS Portal (http://www.tut.fi/tutcris)
Published inEuropean Journal of Pharmaceutical Sciences
DOI10.1016/j.ejps.2017.06.025
Copyright© 2017. This manuscript version is made available under the CC-BY-NC-ND 4.0 licensehttp://creativecommons.org/licenses/by-nc-nd/4.0/
LicenseCC BY-NC-ND
Take down policyIf you believe that this document breaches copyright, please contact [email protected], and we will remove accessto the work immediately and investigate your claim.
Download date:03.07.2020

1
Title: Transdermal iontophoretic delivery of diclofenac sodium from various liposome-encapsulated 1
formulations 2
3
Author names and affiliations: 4
K. Malinovskaja-Gomez a, S. Espuelas b, M.J. Garrido b, J. Hirvonen a, T. Laaksonen c,d 5
6
a Division of Pharmaceutical Chemistry and Technology, Faculty of Pharmacy, University of Helsinki, 7
P.O. Box 56, FIN-00014 Helsinki, Finland 8
b Department of Pharmacy and Pharmaceutical Technology, School of Pharmacy, University of Navarra 9
31080, Pamplona, Spain 10
c Division of Pharmaceutical Biosciences, Centre for Drug Research, Faculty of Pharmacy, University of 11
Helsinki, P.O. Box 56, FIN-00014 Helsinki, Finland 12
d Department of Chemistry and Bioengineering, Tampere University of Technology, P. O. Box 541 13
FI-33101 Tampere, Finland 14
15
E-mail addresses: 16
[email protected] (Kristina Malinovskaja-Gomez), [email protected] (Socorro 17
Espuelas), [email protected] (Maria J. Garrido), [email protected] (Jouni Hirvonen), 18
[email protected] (Timo Laaksonen). 19
20
Corresponding author: 21
Kristina Malinovskaja-Gomez 22
Division of Pharmaceutical Chemistry and Technology, Faculty of Pharmacy, University of Helsinki, 23
P.O. Box 56, FIN-00014 Helsinki, Finland 24
Tel.: +358504480724 25
Abstract 26
This study was aimed to evaluate the in vitro transdermal direct/pulsed current iontophoretic delivery of 27
hydrophilic model compound from various lipid vesicle-encapsulated formulations compared to free-28
drug formulation. Conventional, pegylated, ultradeformable liposomes (transfersomes) and ethosomes 29
loaded with negatively charged drug diclofenac sodium (DS) were prepared and characterized. All the 30
liposomes possessed an average size of ≈100-150 nm and negative zeta potential. No changes in colloidal 31

2
stability were detected after 8 h incubation of any vesicle formulation under constant or pulsed 1
iontophoretic current. DS was released from all the liposome formulations with a similar, limited rate 2
(≈50% in 24 h), leading therefore to significantly lower transdermal fluxes across full-thickness porcine 3
skin compared to the respective free drug formulation. From the tested lipid vesicle formulations, 4
transfersomes resulted in the highest passive flux and ethosomes in the highest iontophoretic flux under 5
direct constant current treatment. Higher negative surface charge of the vesicle led to better transport 6
efficiency due to the higher mobility of the drug carrier under electric field. Pulsed current iontophoresis 7
had no advantage over constant current treatment in combination with any type of lipid vesicular 8
nanocarriers, in contrast to what has been described earlier with drug-loaded polymeric nanocarriers. 9
10
11
Keywords: Liposome; NSAID delivery; Iontophoresis; Transdermal drug delivery; Diclofenac sodium; 12
Skin permeation 13
14
15
16
17
18
19
20
21
22
23
24

3
1
1. Introduction 2
Transdermal delivery of drugs across the skin to the systemic circulation provides a convenient 3
administration route for a variety of clinical indications [1]. In addition to avoiding the hepatic first-pass 4
effect and chemical degradation of drug in the gastrointestinal tract, patient compliance can be improved 5
by reducing the frequency of dosing due to the continuous input of drug. Despite of being an attractive 6
alternative to oral and parenteral administration, however, transdermal and topical delivery of 7
therapeutics has been clinically realized only for a handful of drugs, owing to the formidable barrier 8
properties of stratum corneum, the outermost layer of skin. Therefore, only a limited number of 9
molecules with appropriate balance of hydro-/lipophilicity, small size, no charge, and relatively high 10
potency are able to pass this layer passively in therapeutic amounts [2]. In order the expand the range of 11
molecules being able to overcome such resistance, strategies have been developed to improve transport 12
across or into the skin by enhancing the permeability properties of the stratum corneum or providing a 13
driving force acting directly on the drug [3, 4]. 14
15
One such technique, iontophoresis, involves an application of mild electric current to deliver ionized or 16
polar molecules across biological membranes [5]. The drug dose delivered by iontophoresis is directly 17
proportional to the amount of charge passed through the skin and can be therefore controlled by the 18
electric input of the iontophoretic system: current density, type of the current and the application time of 19
iontophoretic treatment. The efficiency of drug transport by iontophoresis can therefore be described by 20
a transport number (td) that reflects the proportion of the current carried by the drug, as compared to other 21
migrating species in formulation, and is determined by its mobility (µd), charge (zd) and concentration 22
(cd; )[6]: 23
i
iii
dddd
cz
czt
24
Iontophoresis is usually carried out by a continuous direct current (DC) that is typically considered to be 25
the most efficient current type in transdermal delivery. However, it has been suggested that the long-term 26
use of DC may have the problem of creating a polarizing current that will decrease the efficiency of DC 27
applied [7]. This could be overcome by a current delivered in a periodic manner (pulsed current; PC) that 28
(1)

4
allows skin to depolarize and return to its initial state before the onset of next pulse [8]. PC is also 1
considered to be less damaging to skin and cause less patient discomfort [9]. Furthermore, compared to 2
conventional DC, PC has been more effective in promoting transdermal transport of large drug like 3
peptides or proteins [10-14], or when the compound of interest has been loaded into polymeric 4
nanoparticles prior administration [15]. 5
6
Alternatively, in the search of improved dermal or transdermal delivery, attempts have been made to 7
design new nanosized carrier systems, such as liposomes, micelles, nanoparticles, nanoemulsions and 8
dendrimers, to ensure adequate penetration into or across the skin [16]. Among those, liposomal carriers 9
have shown to be promising drug-delivery systems to transport therapeutics mainly to the different layers 10
of skin, and by special vesicular systems (e.g. niosomes, ethosomes, transfersomes etc.) also for systemic 11
drug delivery purposes [17]. Potential advantages of liposome use include enhanced drug delivery, 12
solubilization of poorly soluble drugs, drug protection against proteolytic degradation, local skin depot 13
for sustained release, reduction of side-effects and incompatibilities, or formation of rate-limiting barrier 14
for systemic absorption [18]. There are many reports on the separate use of liposomes and iontophoresis 15
for skin penetration enhancement, however, the combined use of both approaches has gained little 16
attention [19-26]. Combining transdermal iontophoresis with liposome-encapsulated formulations could 17
offer some additional benefits, including improvement of drug delivery by liposome membrane/surface 18
charge modifications and more predictable and controlled drug transport, resulting in drug fluxes less 19
dependent on skin variables. 20
21
The model drug used in this study was diclofenac sodium (DS), the most widely prescribed non-steroidal 22
anti-inflammatory drug (NSAID) worldwide for the management of acute conditions of inflammation 23
and pain, musculoskeletal disorders, arthritis and dysmenorrhea [27]. Although widely used by oral 24
administration, alternative delivery approaches would desirable due to the gastrointestinal side effects, 25
such as gastric ulcers and bleeding, extensive first pass metabolism, and short biological half-life. 26
Therefore, topical DS preparations have been developed with the aim of treating local pain and 27
inflammation while limiting systemic exposure and potentially minimizing the risk of side effects 28
associated with the treatment of oral NSAIDs. The physicochemical parameters (small molecular weight, 29
lipophilic nature of the DS form while its salts are water soluble at neutral pH) makes it an excellent 30
candidate drug to be used for transdermal delivery. Although effective passive delivery of DS across the 31

5
skin has been demonstrated, the iontophoretic administration of this drug should be investigated as a 1
means to improve both the rate and extent of drug delivery. 2
3
The objective of this study was to test drug delivery systems that combine drug-loaded lipid vesicles and 4
iontophoresis for the controlled transdermal delivery of hydrophilic model compound. In more detail, we 5
aimed: (1) to develop a range of different DS-loaded liposome formulations suitable for transdermal 6
iontophoretic administration, with regards to drug loading, colloidal properties, electrochemical stability 7
and drug release kinetics, (2) to study the effect of the surface charge of liposomal carrier, or (3) liposome 8
type (conventional vs. special vesicular systems) on the permeation of loaded model drug under 9
iontophoretic delivery, and (4) to determine the suitable iontophoretic current (constant vs. pulsed) type 10
to be combined with the DS loaded vesicles. 11
12
2. Materials and methods 13
14
2.1. Chemicals 15
Soya phosphatidylcholine (Emulmetik 930) was obtained from Lucas Meyer Cosmetics (Champlan, 16
France), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] 17
ammonium salt from Avanti Polar Lipids (Alabaster, AL, US), and cholesterol, Tween-80 and diclofenac 18
sodium from Sigma Chemical Co. (St. Louis, MO, USA). All other chemicals were at least of analytical 19
grade. Deionized water (≥ 18.2 MΩ/cm of resistance; Millipore, Molsheim, France) was used to prepare 20
all the solutions. 21
22
2.2. Preparation of liposomes 23
Conventional liposomes, pegylated liposomes and transfersomes were prepared by the classic cast film 24
method. Briefly, phosphatidylcholine (PC), cholesterol (Chol), 1,2-distearoyl-sn-glycero-3-25
phosphoethanolamine-N-methoxy-polyethylene glycol-2000 ammonium salt (DSPE-PEG2000) and 26
Tween-80 were dissolved in absolute ethanol in a clean, dry, round-bottom flask. The organic solvent 27
was removed by evaporation for 20 min at 45 °C to obtain homogenous thin lipid film on the inner 28
surface of the flask. The deposited film was then hydrated with 2.5 mg/ml diclofenac sodium (DS) 29

6
solution in 10 mM Hepes buffered saline pH=7.4 for 1 h at 45 °C. The resulting vesicles were extruded 1
11 times through 100 nm polycarbonate membrane at 45 °C. 2
In order to prepare ethosomes, the lipids and DS were dissolved in absolute ethanol. This mixture was 3
heated to 30 °C ± 1°C in a water bath. Buffer (10 mM Hepes buffered saline pH=7.4), also heated to 30 4
°C ± 1 °C, was added slowly as a fine stream to lipid mixture with constant stirring at 700 rpm in a closed 5
vessel. Mixing was continued for an additional 5 minutes, while maintaining the system at 30 °C ± 1 °C. 6
The size of the vesicles was reduced by sonication for 15 s at 15 W, followed by extrusion for 11 times 7
through 100 nm polycarbonate membrane at ambient temperature. 8
9
2.3. Characterization of the liposomes 10
The colloidal characteristics of DS loaded liposomes were determined by Malvern Zetasizer Nano 11
(Malvern Instruments, Malvern, UK). The liposome-encapsulated DS was separated from unentrapped 12
drug by ultracentrifugation at 30000 rpm for 6 h at 4 °C. Liposomes were lysed with absolute ethanol 13
and the released DS was quantified by HPLC. The percent of encapsulation efficiency (EE%) was then 14
calculated according to the following equation: 15
𝐸𝐸% =𝑎𝑚𝑜𝑢𝑛𝑡 𝑜𝑓 𝑑𝑟𝑢𝑔 𝑖𝑛 𝑙𝑖𝑝𝑜𝑠𝑜𝑚𝑒𝑠
𝑡𝑜𝑡𝑎𝑙 𝑎𝑚𝑜𝑢𝑛𝑡 𝑜𝑓 𝑑𝑟𝑢𝑔× 100 (2) 16
17
2.4. Skin preparation 18
Porcine ears were obtained from a local abattoir within a few hours post-mortem and were cleaned under 19
cold running water. The whole skin was removed carefully from the outer region of the ear and separated 20
from the underlying cartilage with a scalpel. 21
22
2.5. Stability of liposomes under iontophoretic current 23
The stability of liposomes under different current profiles for 8 h was evaluated at 37 °C in small vials 24
as a change in hydrodynamic diameter and polydispersity index (PDI). Following current profiles were 25
used: 100% constant direct current (DC) and 75% on/25% off pulsed current (PC) (cathodal, current 26
density 0.5 mA/cm2, frequency of pulsing 500 Hz). 27

7
1
2
2.6. Drug release and permeation across full-thickness porcine skin in vitro 3
The release and in vitro permeation studies were carried out in occlusive (closed) conditions in static 4
Franz diffusion cells. Pieces of full-thickness porcine skin were clamped between the donor and receiver 5
compartments of the diffusion cells. The area of exposed epidermis was 0.785 cm2. To mimic 6
physiological conditions, the cells were thermostated at 37 °C. 0.5 ml of DS solution (2 mg/ml) or drug 7
loaded liposomes with the equivalent amount of drug in 10 mM pH=7.4 HEPES-buffer containing 154 8
mM NaCl was placed in the donor compartment of the diffusion cells. 5 ml of the same buffer was used 9
in the receiver compartment. As DS is negatively charged at pH=7.4, it was iontophoresed from the 10
cathodic compartment. 300 µl-sized samples were collected from the receiver compartment at 0.5, 1, 2, 11
3, 4, 6, 8 (current off) and 24 h for analysis. Release experiments were conducted similarly, but the 12
regenerated cellulose membrane (MWCO=6-8 kD; Spectra/Por, Spectrum Laboratories, US) was used 13
to separate donor and receiver compartment of the diffusion cells. 14
15
2.7. Iontophoretic apparatus 16
Platinum electrodes were used in all the iontophoretic experiments. During the experiments the 17
electrodes were separated from the donor and receiver compartments by salt-bridges consisting of 1 M 18
KCl gelled with 2% agarose inside silicone tubing (inner diameter 2 mm, length ca. 15 cm). Salt bridges 19
prevented direct contact and possible reactions and/or pH-effects of the drug with the electrodes. The 20
electrolyte that surrounded the electrodes was 2 M KCl. Current was on for 8 h (Ministat potentiostat, 21
Sycopel Scientific Ltd., Boldon, UK), whereafter passive flux was followed up to 24 h. Two different 22
current profiles – 100% constant direct current (DC) or 75% on/25% off pulsed current (PC) – were used 23
in iontophoretic experiments (current density 0.5 mA/cm2; pulsing frequency 500 Hz). The current 24
profiles were adjusted by CFG253 function generator and TDS210 oscilloscope (Tektronix Inc., 25
Beaverton, OR, USA) and monitored by Fluke 8808A multimeter (Hewlett Packard, WA, USA). 26
27
2.8. Drug analysis 28

8
The DS concentrations were analyzed by HPLC using the following conditions: column Supelco 1
Discovery® C18, 5 µm, 150*4.6 mm; mobile phase: phosphate buffer pH=3: methanol (30:70); flow rate: 2
1 ml/min; wavelength: 284 nm; injection volume: 50 µl; column temperature: 25 °C; retention time: 3.3 3
± 0.2 min. The linear calibration curve (r2=0.9999) was established in the range of 0.1-100 µg/ml of drug 4
concentration. 5
6
2.9. Data analysis 7
The cumulative amount of drug permeated across the skin was calculated and plotted against time. The 8
linear part of the curve was used for calculating the steady-state flux values (nmol/h*cm2). All the results 9
are expressed as means ± SD. 10
11
3. Results and discussion 12
13
3.1. Properties of liposomes 14
In this study, our goal was to study the drug delivery from lipid vesicle formulations combined with 15
transdermal iontophoresis. Therefore, we aimed at producing liposomes possessing a surface charge of 16
the same sign as the drug loaded inside the vesicles as then the transport of the drug across the skin barrier 17
would benefit from the iontophoretic current treatment in double. It is generally agreed that liposomes 18
do not penetrate the skin but rather remain in the upper layers of stratum corneum. According to our 19
hypothesis, firstly, the electric current would push the liposomes into hair follicles, that has been 20
recognized as one the possible routes in transdermal absorption of drugs [28]. Thereafter, iontophoresis 21
would pump the drug released from the depot in the skin or on the surface of the skin further into the 22
bloodstream. 23
24
In order to optimize this delivery technique, the effect of various liposome formulations had to be studied 25
in a systematic way. The compositions of our final formulations used in the transdermal permeation 26
experiments were based on the findings from literature with minor modifications. In order to study the 27
effect of surface charge of vesicles on DS transport under iontophoresis, pegylating agent (2.5% of 28
molarity of total phospholipids) was used to decrease the highly negative surface charge of conventional 29

9
liposomes. DSPE-PEG2000 forms a shielding layer on the outside of the membrane of the liposome, 1
which would cover the negative charge of the other constituents − PC and Chol [29]. Tween-80 (in 15% 2
w/w concentration) was chosen as edge activator for the production of transfersomes as it has been 3
demonstrated to provide the maximum deformability to the DS vesicle membrane and transdermal flux 4
compared to bile salts and Spans [30]. For the preparation of ethosomes, 20% of ethanol v/v was 5
incorporated into the vesicles as this ratio has proven earlier to provide the highest transdermal fluxes of 6
DS from ethosomes, phenomenon claimed to be related to viscous state of that dispersion that remained 7
best adhered to skin surface [31]. 8
9
The average hydrodynamic diameters of the prepared liposomes ranged from 116.5 to 147.0 nm, with 10
the conventional and pegylated liposomes possessing slightly higher sizes than the transfersomes and 11
ethosomes (Table 1). These results were expected, considering the differences in composition and manual 12
extrusion, and were not believed to lead to significant changes in transdermal drug fluxes. Still, there is 13
lot of controversy among reports on the influence of liposome particle size on the drug transport across 14
the skin. While some studies have not found higher disposition of drug either in lower skin strata or 15
receiver compartments whether the drug was applied in small or large vesicles [24, 32], others claim the 16
opposite [33, 34]. The particle size distribution of all our liposomes was monomodal with a polydispersity 17
index of ≈0.1, indicating narrow size distribution and excellent dispersion homogeneity. 18
19
Zeta potentials were measured with purified liposomes, after being dispersed in 1M KCl solution. All 20
the liposomes were negatively charged with the conventional liposomes possessing the highest negative 21
zeta potential value (-34.5 mV). The addition of pegylated phospholipid (2.5 % of phospholipid molarity) 22
to the formulation decreased the negative zeta potential value (to -27.7 mV). Also the transfersomes and 23
ethosomes had slightly lower negative surface charges compared to the conventional liposomes. 24
25
The encapsulation efficiency shows the percentage of the initial drug incorporated into the liposomes. 26
Results showed that all the vesicles were able to encapsulate DS with good yields (EE% values ranged 27
from 46.6% to 79.9%), with the highest EE% reached in transfersomes. 28
29
3.2. Electrochemical stability of liposomes 30
31

10
Prior to permeation experiments, the colloidal stability of vesicles was evaluated under direct constant 1
and pulsed current modes. The results presented as a change in hydrodynamic diameter and 2
polydispersity index are shown in Fig. 1. No changes in hydrodynamic diameters were detected after 8 3
h incubation in any vesicle formulation under constant or pulsed current. The polydispersity indices of 4
formulations F1, F2 and F3 had increased slightly, but these changes did not reach statistical significance 5
(p>0.05 when compared with unpaired t-test before and after current treatment). As all the tested 6
liposomes were stable under iontophoretic current, the formulations were deemed suitable for subsequent 7
transdermal permeation studies. 8
9
Only one earlier study, with regards to the combined use of liposomes and transdermal iontophoresis, 10
has been reported on the electrochemical stability of the liposomes under iontophoretic current [20]. The 11
antimicrobial drug enoxacin-loaded vesicles showed no changes in encapsulation under constant current 12
treatment for 6 h and the size of the current-treated liposomes was generally smaller than of the untreated 13
liposomes after incubation in buffer. Although pore widening of lipid bilayers under electric field has 14
been associated with breakdown of liposomes and leakage of loaded drug, the magnitude of current used 15
in typical transdermal iontophoretic treatment regimens is too small (maximum current density is 16
typically 0.5 mA per cm2 of skin) to cause the rupture of vesicles. Furthermore, under electric current 17
field, the fusion of charged vesicles is inhibited, so the current can even contribute to better colloidal 18
stability of the formulations. 19
20
3.3. Release of DS from liposomes 21
The drug formulation to be combined with transdermal iontophoresis should ideally avoid immediate/ 22
burst drug release and deliver the drug to the permeation site in a gradual yet continuous manner. In this 23
way, more predictable and controlled drug transport could be achieved, resulting in fluxes less dependent 24
on skin variables. In this study, the release kinetics of DS was studied in a similar setting as the in vitro 25
transdermal permeation − at 37°C in static Franz diffusion cells, only using a synthetic dialysis membrane 26
to separate the donor compartment from the receiver compartment. Fig. 2 illustrates the release profiles 27
of DS from different liposome formulations. As a control, the same amount of drug dissolved in the 28
buffer was utilized. From all lipid vesicle formulations DS was released with similar rate, resulting in 29
≈50% of encapsulated drug released in 24 h. 30

11
In general, the most important factors affecting drug release from liposomes are the physicochemical 1
properties of the liposome membrane and the therapeutic agent, and the vesicle size [35]. Typically, the 2
incorporation of cholesterol leads to the formation of more ordered and rigid lipid membranes [36], 3
whereas surfactants (edge activators in transfersomes) and ethanol result in leakier vesicle membranes, 4
which makes the drug release easier [30, 37]. All the study formulations possessed similar vesicle sizes 5
and drug-to-lipid ratios (data not shown)´. Thus, the effect of these composition parameters on drug 6
release were not evident in this study. In summary, all the formulations demonstrated suitable release 7
kinetics to be used in transdermal delivery and any changes in transport efficiency across the skin of DS 8
from different liposome formulations could only be attributed to the differences in liposome – skin - 9
iontophoretic current interactions, not in drug release rates. 10
11
3.4. Transdermal delivery of DS across porcine full-thickness skin in vitro 12
3.4.1 Delivery from solution 13
There have been several reports on transdermal iontophoretic delivery of DS either in vitro [20, 38-40] 14
or in vivo [41-45]. Also combining iontophoresis with other chemical or physical enhancement 15
techniques for the delivery of DS have been utilized [46-49]. Our permeation studies were conducted at 16
pH 7.4, where DS carries a negative charge (pKa=4.15) and can therefore be administered by cathodal 17
iontophoresis. According to the Henderson–Hasselbalch equation, the ionization degree of an acid I% = 18
100/[1 + antilog (pKa – pH)] [50]. At pH 7.4 practically all of DS (99.94%) is in ionized state and can 19
therefore maximally benefit from the iontophoretic current treatment. The total iontophoretic flux of a 20
charged compound is typically the sum of fluxes driven by electrorepulsion, electroosmosis and passive 21
diffusion [2]. The flux of DS under iontophoresis is expected to be the sum of electrorepulsion and 22
passive diffusion, as electroosmosis does not contribute to the transport of negatively charged solutes 23
and can be neglected in our case. 24
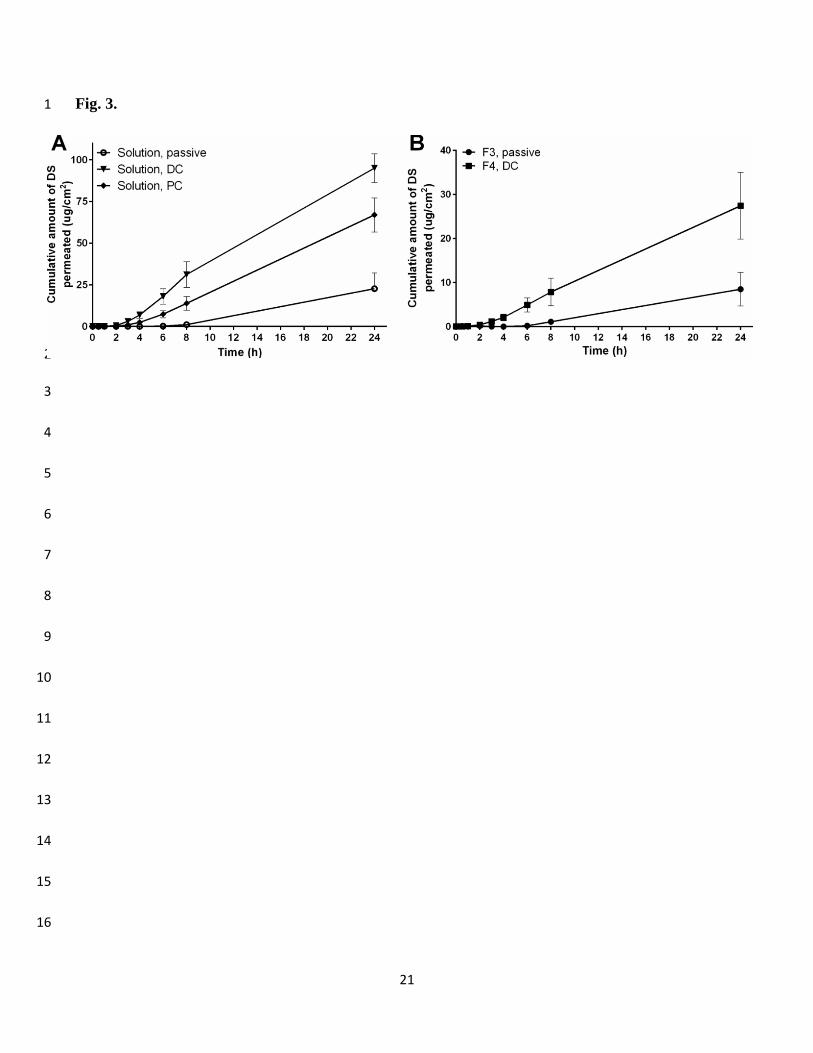
Results from the in vitro transdermal permeation experiments with the DS solution and liposomes are 25
presented on Table 2 and Fig. 3. Experiments with DS solution demonstrated that iontophoresis 26
significantly increased the transport of negatively charged model drug across porcine skin compared to 27
passive transport (e.g. EDC= 5.1). As expected, the direct constant current was more effective in 28
enhancing the delivery of DS to receiver compartment compared to pulsed current that resulted in ≈25% 29

12
lower transdermal flux. This can be considered typical as the iontophoretic flux of charged small 1
molecular compounds is mostly driven by electrorepulsion mechanism that is directly related to the 2
amount of current passed across the permeation membrane (the total amount of current of PC treatment 3
was 75% of DC treatment). 4
5
3.4.2 Delivery from liposomes 6
This is the first report on investigating the transdermal iontophoretic delivery of DS from liposomal 7
carriers. In general, the use of DS loaded vesicles in the donor compartment led to decreased flux values 8
and longer lag-times to reach steady-state fluxes compared to DS delivery from solution. This 9
phenomenon was caused by gradual and limited release of drug from the liposomes during the 24 h 10
permeation experiments. The inability of lipid vesicles to enhance transdermal DS permeation compared 11
to free-drug formulation has been demonstrated also earlier [51, 52]. This could be explained by the 12
hydrophilic nature of the compound that leads to poor drug partitioning from the vesicles into lipid 13
environment of the skin. The application of iontophoretic current enhanced the delivery of the model 14
drug compared to passive delivery in a similar magnitude than from solutions. On Fig. 3B the permeation 15
curves of the liposome formulations that provided the highest passive and iontophoretic flux values are 16
presented. From all the tested lipid vesicles, ultradeformable liposomes (F3) resulted in the highest 17
passive flux and ethosomes (F4) the highest iontophoretic flux under direct constant current treatment. 18
One of the aims of this study was also to study the effect of the surface charge of liposomal nanocarriers 19
on model drug transport under the iontophoretic current. For that purpose we compared the DS transport 20
from pegylated liposomes with reduced negative zeta potential value (F2) to the highly negatively 21
charged conventional liposomes (F1). As expected, the highly negatively charged conventional 22
liposomes led to more efficient iontophoretic delivery of DS, indicating that liposome charge can affect 23
the iontophoretic treatment outcome. Also, the enhancement factors and transport numbers of F2 were 24
lower than of F1. At the same time, passive drug fluxes from both the formulations were in the same 25
range. This indicates that the higher iontophoretic flux of F1 liposomes could be related directly to the 26
more pronounced mobility of the nanocarrier under electric current (electrorepulsion of liposome) 27
leading to improved diffusion of liposomes - not to changes in skin-liposome interactions. In previous 28
reports, other techniques than pegylation have been used to change/ impart charges of/ to drug loaded 29
liposomes. Cationic lipids like stearylamine, DDAB, DODAP, DOTAP, DOSPER, spermine or 30

13
protamine sulfate have been added to formulations to induce positive charge, and negative lipids, such 1
as phospatidylserine, dicetylphosphate or DMPG, have been added to impart negative charge [19, 20, 2
25]. These earlier studies have demonstrated the following: Firstly, the surface charge of the liposome 3
has to be of the same sign as the drug for effective drug transport. Secondly, the charge inducing excipient 4
is more efficient if it is a multi-cation by nature and does not possess too big molecular size as this lowers 5
the mobility under electric current. Thirdly, a possible competition effect between the charge inducing 6
agent and the drug is possible, especially when the excipient is a smaller molecule and therefore more 7
mobile under electric current. Considering the possible competitive ion effect that might have an 8
unwanted effect on drug flux, we chose pegylation in this study for the charge effect comparison. 9
Although pegylation was demonstrated to be a good technique in shielding the negative charge of vesicle, 10
its potential benefits remained somewhat limited. 11
In general, it is agreed, that conventional liposomes have not much value as drug carriers for transdermal 12
delivery [17] as the deep skin penetration is inhibited and the vesicles remain in the upper layer of stratum 13
corneum. With special techniques carrier vesicles can be produced in such a way that enables modulated 14
delivery across the skin layers and provides a systemic action. In this study we investigated the potential 15
of two such vesicle types – ultradeformable liposomes and ethosomes – to be used in combination with 16
iontophoresis and to enhance transdermal DS delivery. Ultradeformable liposomes (transfersomes), 17
having an edge activity in their lipid bilayer, are described as freely moving very flexible vesicles that 18
are able to penetrate the skin due to proposed transdermal hydration force [53]. Ethosomes are known to 19
facilitate drug permeation by a tentative synergistic “ethanol-ethosome effect” in which ethanol disrupts 20
the organization of stratum corneum lipid bilayer, allowing the flexible ethosomes then to penetrate and 21
possibly permeate this layer [54]. From all the tested vesicle formulations, transfersomes led to the 22
highest passive flux of DS across the porcine skin. Due to the longer lag-time (data not shown) to reach 23
the steady-state flux, the total amount of DS delivered during 24 h remained still smaller than from the 24
pegylated liposomes. Interestingly, the iontophoretic current was quite inefficient in further enhancing 25
DS delivery from transfersomes (EDC=2.4). The prepared ethosomes failed to increase DS permeation 26
passively compared to conventional liposomes, but at the same time the iontophoretic enhancement was 27
the highest (EDC=6.4), also leading to the highest steady-state iontophoretic flux and transport number 28
among all the liposome formulations. In contrast to our results, DS formulations based on transfersomes 29
and ethosomes have been demonstrated several times to significantly improve DS transport across the 30
skin in passive delivery, reaching higher fluxes than from free-drug solution or commercial DS gels [30, 31

14
31, 55, 56]. In regards to the composition of the transfersomes, the choice of edge activator and its 1
concentration can have a crucial outcome on drug transport [30]. Also, for transfersomes, open (non-2
occluded) administration has been proposed to be a prerequisite to obtain maximum permeation 3
enhancing effect, as the driving force for penetration − transdermal hydration gradient − is eliminated by 4
occlusive conditions [57]. At the same time, as the application of iontophoretic current requires aqueous 5
media around the electrodes, all our permeation experiments were conducted under occlusion to prevent 6
the evaporation of the buffer from the donor compartment. Interestingly, occlusion did not abolish totally 7
the penetration enhancing effects of the transfersomes compared to other tested lipid vesicles. Similar 8
results were obtained when estradiol transport from transfersomes was studied under occlusion [21]. 9
This is the first study on the effect of the iontophoretic current type on the transdermal delivery of drug 10
loaded liposomes. In all earlier studies, where liposomes were combined with iontophoresis, only 11
constant direct current profile had been utilized [19-26]. Typically, constant current treatment is the most 12
efficient in enhancing transdermal delivery of charged or ionic drugs, as for most compounds the fluxes 13
are proportional to the amount of current passed across the skin. However, the use of constant current 14
has its limitations, especially in long-term use (see Introduction). Also, for some larger drugs (peptides) 15
or even drug-loaded polymeric carrier systems pulsed current has been proven to be more efficient in 16
transport enhancement [13-15]. Interestingly, pulsed current treatment had no clear advantage over 17
constant current treatment in combination with any liposome formulation. The treatment was especially 18
ineffective with liposomes with lower surface charge, such as pegylated or ultradeformable liposomes, 19
as was seen from the % of JPC of JDC value (that shows the percent of steady-state flux of pulsed current 20
treatment from steady-state flux of constant current treatment). This demonstrated that lipid vesicles with 21
less mobility under electric current were benefitting less from the iontophoretic treatment delivered in 22
pulsatile manner. On the contrary, drug delivery from polymeric nanospheres was more enhanced under 23
pulsed current than under constant direct current, indicating that there must be other factors than surface 24
charge underlying the interaction between the drug-loaded nanocarriers and current type [15]. It is also 25
possible, that in combination with lipid vesicles, the pulsed current is more responsible in delivering the 26
drug into deeper layers of skin, but not enhancing the permeation further across the skin to systemic 27
blood circulation. 28
29
Conclusions 30

15
1
This study tested a system combining iontophoresis and drug-loaded lipid vesicles for the controlled 2
transdermal delivery of a hydrophilic model compound. Therefore, four different types of lipid vesicles 3
− conventional, pegylated, transfersomes and ethosomes − loaded with diclofenac sodium were prepared 4
and the effect of differences in composition was evaluated on drug transport efficiency across the skin. 5
Although all the obtained liposome formulations were deemed as suitable for transdermal iontophoretic 6
administration, regarding the colloidal properties, stability under iontophoretic current and release 7
kinetics, no improvement in drug delivery was seen from nanoencapsulating diclofenac sodium into lipid 8
vesicles compared to free drug formulation. All the liposome formulations led to reduced transdermal 9
fluxes and smaller drug amounts delivered, both in passive and iontophoretic delivery regimens. 10
11
Iontophoretic drug transport from liposomes was significantly affected by the composition and charge of 12
lipid bilayer vesicles, and the iontophoretic current mode utilized. Transfersomes resulted in the highest 13
passive flux and ethosomes in the highest iontophoretic flux under direct constant current treatment. 14
Lipid vesicles with higher negative surface charge led to better transport efficiencies of the loaded model 15
drug due to the higher mobility of the drug carriers under iontophoretic current. This is the first report to 16
study the effect of current type on the transdermal permeation of a drug loaded into lipid vesicles. Based 17
on our findings, pulsed current treatment has no clear advantage over constant current treatment in 18
combination with any type of lipid vesicular nanocarriers, which is in contrast to what has been described 19
earlier with polymeric nanocarriers. 20
21
22
Acknowledgements 23
24
The financial support from Academy of Finland (grants no. 258114 and 264988) is gratefully 25
acknowledged. 26
27
28
29
30
31

16
1
2
3
Abbreviations 4
5
DDAB dimethyldiooctadecylammonium bromide 6
DODAP 1,2- dioleoyl-3-dimethylammonium propane 7
DOSPER (1,3-dioleoyloxy-2-(6-carboxyspemyl) propylamide (tetraammonium tetraacetate) 8
DOTAP 1,2- dioleoyl-3-trimethylammonium propane 9
DC direct current 10
DS diclofenac sodium 11
E iontophoretic enhancement factor 12
F Faraday constant (C/mol) 13
HEPES 4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid 14
HPLC high performance liquid chromatography 15
JSS steady-state flux (µg/h per cm2) 16
MW molecular weight (g/mol) 17
MWCO molecular weight cut off 18
PC pulsed current 19
PDI polydispersity index 20
Tn transport number 21
Z valence 22
23
24
25
26
27
28
29
30
31

17
1
2
3
Tables 4
5
Table 1. Composition, particle size, polydispersity index, zeta potential and mean encapsulation 6
efficiency (EE%) of prepared vesicles. 7
Formulation and code Composition Size (nm) PDI Charge (mV) Mean EE %
Conventional liposomes (F1) 90 mM PC
30 mM Chol 147.0 ± 1.5 0.085 ± 0.013 -34.3 ± 1.7
67.5
Pegylated liposomes (F2)
90 mM PC
30 mM Chol
2.5% (mol/mol%) DSPE-PEG2000
144.2 ± 0.1 0.079 ± 0.029 -27.7 ± 1.0 60.4
Transfersomes (F3)
120 mM PC
15% (w/w%) Tween-80 117.7 ± 0.5 0.073 ± 0.028 -29.8 ± 1.6 79.9
Ethosomes (F4)
90 mM PC
30 mM Chol
20% (v/v%) ETOH
116.5 ± 0.6 0.104 ± 0.005 -26.7 ±1.9 49.6
PC– soya phosphatidylcholine (Emulmetik 930); Chol – cholesterol; DSPE-PEG2000 – 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-8 N-methoxy-polyethylene glycol-2000 ammonium salt; DS – diclofenac sodium. 9 10 11
12
13
14
Table 2. The results from the permeation experiments with DS solution and different liposome 15
formulations (F1-F4). Mean, n=5-6. 16
17
18

18
a Jss – steady-state flux, calculated from the linear slope of the permeation curve 1
b Tn - transport number, calculated using an equation: IMW
zFdQ/dtTn
,
2
where z is the valence of the drug, F is the Faraday`s constant, dQ/dt is the linear slope of the iontophoretic phase (in mass units per 3 time), MW is the molecular weight and I is the total current passed through the skin. 4 c E – enhancement factor; E= Jiontophoresis/Jpassive 5 d Q24h - cumulative amount of drug permeated in 24 h 6 e % of JPC of JDC – percent of steady-state flux of pulsed current treatment from steady-state flux of constant current treatment 7
8
9
10
11
12
13
14
15
Treatment Jss
a
(nmol/h*cm3) Tn
b Ec Q24hd (ug)
% of JPC of
JDCe
Solution
Passive 2.78 ± 1.31 - - 22.63 ± 9.48 -
DC 14.21 ± 7.51 8.13E-04 ± 3.81E-04 5.1 79.68 ± 25.10 -
PC 10.45 ± 4.27 7.47E-04 ± 2.86E-04 3.8 73.92 ± 17.89 73.5
F1
Passive 1.11 ± 0.43 - - 6.39 ± 2.64 -
DC 3.98 ± 1.78 2.13E-04 ± 9.53E-05 3.6 23.76 ± 2.90 -
PC 2.94 ± 0.90 2.10E-04 ± 6.46E-05 2.6 22.96 ± 4.41 73.9
F2
Passive 1.11 ± 0.56 - - 11.62 ± 3.93 -
DC 3.64 ± 1.12 1.95E-04 ± 7.47E-05 3.3 25.25 ± 7.60 -
PC 1.78 ± 0.72 1.27E-04 ± 5.09E-05 1.6 14.89 ± 4.36 48.6
F3
Passive 1.44 ± 0.30 - - 8.48 ± 3.81 -
DC 3.47 ± 1.23 1.86E-04 ± 6.72E-05 2.4 24.37 ± 8.17 -
PC 2.38 ± 0.64 1.70E-04 ± 3.98E-05 1.6 18.50 ± 4.57 68.6
F4
Passive 0.71 ± 0.18 - - 3.68 ± 0.95 -
DC 4.53 ± 1.59 2.43E-04 ± 1.04E-04 6.4 27.42 ± 7.54 -
PC 2.87 ± 1.82 2.05E-04 ± 1.39E-04 4.1
19.07 ±7.24 63.4

19
Captions 1
Fig. 1. Electrochemical stability of liposomes under iontophoretic current for 8 h. Expressed as a change 2
in hydrodynamic diameter and PDI. Mean ± SD, n=3. 3
Fig. 2. Release of diclofenac sodium (DS) from different liposome formulations (F1-F4). The total 4
amount of DS in liposomes was 100 µg. Mean ± SD, n=3. 5
Fig. 3. The in vitro transdermal permeation of DS across porcine full-thickness skin. A. The permeation 6
curves of DS from solution formulation passively and under direct constant (DC) or pulsed current (PC). 7
B. The liposome formulations with best passive (F3, passive) or iontophoretic flux (F4, DC). DC – 100 8
% constant direct current 0.5 mA/cm2; PC – 75% on:25% off pulsed current. The amount of DS in the 9
donor compartment was 1000 µg (mean ± SD, n=5-6). 10
11
12
13
14
15
16
17
18
19
20
21
22
23

20
Figures 1
2
Fig.1. 3
4
5
Fig. 2 6
7
21
Fig. 3. 1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16

22
References 1
[1] M.N. Pastore, Y.N. Kalia, M. Horstmann, M.S. Roberts, Transdermal patches: history, development 2
and pharmacology, Br J Pharmacol, 172 (2015) 2179-2209. 3
[2] Y.N. Kalia, A. Naik, J. Garrison, R.H. Guy, Iontophoretic drug delivery, Adv Drug Deliv Rev, 56 4
(2004) 619-658. 5
[3] R. Parhi, P. Suresh, S. Mondal, P.M. Kumar, Novel penetration enhancers for skin applications: a 6
review, Curr Drug Deliv, 9 (2012) 219-230. 7
[4] A. Herwadkar, A.K. Banga, An update on the application of physical technologies to enhance 8
intradermal and transdermal drug delivery, Ther Deliv, 3 (2012) 339-355. 9
[5] J. Hirvonen, Topical iontophoretic delivery, Am J Drug Deliv, 3 (2005) 67-81. 10
[6] B.J. Phipps, J.R. Gyory, Transdermal ion migration, Adv Drug Deliv Rev, 9 (1992) 137-176. 11
[7] J.C. Lawler, M.J. Davis, E.C. Griffith, Electrical characteristics of the skin. The impedance of the 12
surface sheath and deep tissues, J Invest Dermatol, 34 (1960) 301-308. 13
[8] C. Zakzewski, D. Amory, D. Jasaitis, J.-J. Li, Iontophoretically enhanced transdermal delivery of an 14
ACE inhibitor in induced hypertensive rabbits: Preliminary report, Cardiovasc Drug Ther, 6 (1992) 589-15
595. 16
[9] M. Clemessy, G. Couarraze, B. Bevan, F. Puisieux, Preservation of skin permeability during in vitro 17
iontophoretic experiments, Int J Pharm, 101 (1994) 219-226. 18
[10] P. Singh, L. Puchun, S.M. Dinh, in: R.L. Bronaugh, H.I. Maibach (Eds.) Percutaneous Absorption: 19
Drugs--Cosmetics--Mechanisms--Methods, Marcel Dekker Inc., New York, 1998. 20
[11] P. Knoblauch, F. Moll, In vitro pulsatile and continuous transdermal delivery of buserelin by 21
iontophoresis, J Control Release, 26 (1993) 203-212. 22
[12] Y.W. Chien, O. Siddiqui, W.M. Shi, P. Lelawongs, J.C. Liu, Direct current iontophoretic 23
transdermal delivery of peptide and protein drugs, J Pharm Sci, 78 (1989) 376-383. 24
[13] K. Malinovskaja, T. Laaksonen, J. Hirvonen, Controlled transdermal delivery of leuprorelin by 25
pulsed iontophoresis and ion-exchange fiber, Eur J Pharm Biopharm, 88 (2014) 594-601. 26
[14] J. Raiman, M. Koljonen, K. Huikko, R. Kostiainen, J. Hirvonen, Delivery and stability of LHRH 27
and Nafarelin in human skin: the effect of constant/pulsed iontophoresis, Eur J Pharm Sci, 21 (2004) 28
371-377. 29
[15] K. Malinovskaja-Gomez, H. Labouta, M. Schneider, J. Hirvonen, T. Laaksonen, Transdermal 30
iontophoresis of flufenamic acid loaded PLGA nanoparticles, Eur J Pharm Sci, 89 (2016) 154-162. 31

23
[16] G. Cevc, U. Vierl, Nanotechnology and the transdermal route: A state of the art review and critical 1
appraisal, J Control Release, 141 (2010) 277-299. 2
[17] M.B. Pierre, I. Dos Santos Miranda Costa, Liposomal systems as drug delivery vehicles for dermal 3
and transdermal applications, Arch Dermatol Res, 303 (2011) 607-621. 4
[18] N. Weiner, L. Lieb, S. Niemiec, C. Ramachandran, Z. Hu, K. Egbaria, Liposomes: a novel topical 5
delivery system for pharmaceutical and cosmetic applications, J Drug Target, 2 (1994) 405-410. 6
[19] N.B. Vutla, G.V. Betageri, A.K. Banga, Transdermal iontophoretic delivery of enkephalin 7
formulated in liposomes, J Pharm Sci, 85 (1996) 5-8. 8
[20] J.Y. Fang, K.C. Sung, H.H. Lin, C.L. Fang, Transdermal iontophoretic delivery of enoxacin from 9
various liposome-encapsulated formulations, J Control Release, 60 (1999) 1-10. 10
[21] E.A. Essa, M.C. Bonner, B.W. Barry, Electrically assisted skin delivery of liposomal estradiol; 11
phospholipid as damage retardant, J Control Release, 95 (2004) 535-546. 12
[22] E.A. Essa, M.C. Bonner, B.W. Barry, Iontophoretic estradiol skin delivery and tritium exchange in 13
ultradeformable liposomes, Int J Pharm, 240 (2002) 55-66. 14
[23] E.A. Essa, M.C. Bonner, B.W. Barry, Human skin sandwich for assessing shunt route penetration 15
during passive and iontophoretic drug and liposome delivery, J Pharm Pharmacol, 54 (2002) 1481-1490. 16
[24] K. Kajimoto, M. Yamamoto, M. Watanabe, K. Kigasawa, K. Kanamura, H. Harashima, K. Kogure, 17
Noninvasive and persistent transfollicular drug delivery system using a combination of liposomes and 18
iontophoresis, Int J Pharm, 403 (2011) 57-65. 19
[25] I. Han, M. Kim, J. Kim, Enhanced transfollicular delivery of adriamycin with a liposome and 20
iontophoresis, Exp Dermatol, 13 (2004) 86-92. 21
[26] K. Kigasawa, M. Miyashita, K. Kajimoto, K. Kanamura, H. Harashima, K. Kogure, Efficient 22
intradermal delivery of superoxide dismutase using a combination of liposomes and iontophoresis for 23
protection against UV-induced skin damage, Biol Pharm Bull, 35 (2012) 781-785. 24
[27] R. Altman, B. Bosch, K. Brune, P. Patrignani, C. Young, Advances in NSAID development: 25
evolution of diclofenac products using pharmaceutical technology, Drugs, 75 (2015) 859-877. 26
[28] A.C. Lauer, L.M. Lieb, C. Ramachandran, G.L. Flynn, N.D. Weiner, Transfollicular drug delivery, 27
Pharm Res, 12 (1995) 179-186. 28
[29] M.L. Immordino, F. Dosio, L. Cattel, Stealth liposomes: review of the basic science, rationale, and 29
clinical applications, existing and potential, Int J Nanomedicine, 1 (2006) 297-315. 30

24
[30] G.M. El Zaafarany, G.A. Awad, S.M. Holayel, N.D. Mortada, Role of edge activators and surface 1
charge in developing ultradeformable vesicles with enhanced skin delivery, Int J Pharm, 397 (2010) 164-2
172. 3
[31] S. Jain, N. Patel, P. Madan, S. Lin, Quality by design approach for formulation, evaluation and 4
statistical optimization of diclofenac-loaded ethosomes via transdermal route, Pharm Dev Technol, 20 5
(2015) 473-489. 6
[32] J. Du Plessis, C. Ramacandran, N. Weiner, The influence of particle size of liposomes on the 7
disposition of drugs into the skin, Int J Pharm, 103 (1994) 227-282. 8
[33] J. Schramlova, K. Blazek, M. Bartackova, B. Otova, L. Mardesicova, V. Zizkovsky, D. Hulinska, 9
Electron microscopic demonstration of the penetration of liposomes through skin, Folia biologica, 43 10
(1997) 165-169. 11
[34] D.D. Verma, S. Verma, G. Blume, A. Fahr, Particle size of liposomes influences dermal delivery of 12
substances into skin, Int J Pharm, 258 (2003) 141-151. 13
[35] L.H. Lindner, M. Hossann, Factors affecting drug release from liposomes, Curr Opin Drug Discov 14
Devel, 13 (2010) 111-123. 15
[36] F.M. Cagdas, N. Ertugral, S. Bucak, N.Z. Atay, Effect of preparation method and cholesterol on 16
drug encapsulation studies by phospholipid liposomes, Pharm Dev Technol, 16 (2011) 408-414. 17
[37] R. Rakesh, K.R. Anoop, Formulation and optimization of nano-sized ethosomes for enhanced 18
transdermal delivery of cromolyn sodium, J Pharm Bioallied Sci, 4 (2012) 333-340. 19
[38] P.C. Kasha, C.R. Anderson, R.L. Morris, W.L. Sembrowich, A. Chaturvedula, A.K. Banga, 20
Subcutaneous concentrations following topical iontophoretic delivery of diclofenac, Drug Discov Ther, 21
6 (2012) 256-262. 22
[39] T. Koizumi, M. Kakemi, K. Katayama, H. Inada, K. Sudeji, M. Kawasaki, Transfer of diclofenac 23
sodium across excised guinea pig skin on high-frequency pulse iontophoresis. II. Factors affecting 24
steady-state transport rate, Chem Pharm Bull (Tokyo), 38 (1990) 1022-1023. 25
[40] J. Fang, R. Wang, Y. Huang, P.C. Wu, Y. Tsai, Passive and iontophoretic delivery of three 26
diclofenac salts across various skin types, Biol Pharm Bull, 23 (2000) 1357-1362. 27
[41] I. Garcia, C. Lobo, E. Lopez, J.L. Servan, J.M. Tenias, Comparative effectiveness of 28
ultrasonophoresis and iontophoresis in impingement syndrome: a double-blind, randomized, placebo 29
controlled trial, Clin Rehabil, 30 (2016) 347-358. 30

25
[42] R. Crevenna, A. Burian, Z. Oesterreicher, E. Lackner, W. Jager, G. Rezcicek, M. Keilani, M. 1
Zeitlinger, Iontophoresis driven concentrations of topically administered diclofenac in skeletal muscle 2
and blood of healthy subjects, Eur J Clin Pharmacol, 71 (2015) 1359-1364. 3
[43] R. Clijsen, J.P. Baeyens, A.O. Barel, P. Clarys, In vivo determination of the diclofenac skin 4
reservoir: comparison between passive, occlusive, and iontophoretic application, Drug Des Devel Ther, 5
9 (2015) 835-840. 6
[44] B.F. Riecke, E.M. Bartels, S. Torp-Pedersen, S. Ribel-Madsen, H. Bliddal, B. Danneskiold-Samsoe, 7
L. Arendt-Nielsen, A microdialysis study of topically applied diclofenac to healthy humans: Passive 8
versus iontophoretic delivery, Results Pharma Sci, 1 (2011) 76-79. 9
[45] E. Varghese, R.K. Khar, Enhanced skin permeation of diclofenac by iontophoresis: in vitro and in 10
vivo studies, J Control Release, 38 (1996) 21-27. 11
[46] C. Xin, W. Li-Hong, Y. Yue, G. Ya-Nan, W. Qi-Fang, Y. Yang, L. San-Ming, A novel method to 12
enhance the efficiency of drug transdermal iontophoresis delivery by using complexes of drug and ion-13
exchange fibers, Int J Pharm, 428 (2012) 68-75. 14
[47] K. Kigasawa, K. Kajimoto, M. Watanabe, K. Kanamura, A. Saito, K. Kogure, In vivo transdermal 15
delivery of diclofenac by ion-exchange iontophoresis with geraniol, Biol Pharm Bull, 32 (2009) 684-16
687. 17
[48] K. Sugibayashi, M. Kagino, S. Numajiri, N. Inoue, D. Kobayashi, M. Kimura, M. Yamaguchi, Y. 18
Morimoto, Synergistic effects of iontophoresis and jet injector pretreatment on the in-vitro skin 19
permeation of diclofenac and angiotensin II, J Pharm Pharmacol, 52 (2000) 1179-1186. 20
[49] H. Patel, A. Joshi, A. Joshi, G. Stagni, Effect of microporation on passive and iontophoretic delivery 21
of diclofenac sodium, Drug Dev Ind Pharm, 41 (2015) 1962-1967. 22
[50] A.T. Florence, A.D. Attwood, Physicochemical Principles of Pharmacy, Pharmaceutical Press, 23
London, 2011. 24
[51] A. Fathi-Azarbayjani, K.X. Ng, Y.W. Chan, S.Y. Chan, Lipid Vesicles for the Skin Delivery of 25
Diclofenac: Cerosomes vs. Other Lipid Suspensions, Adv Pharm Bull, 5 (2015) 25-33. 26
[52] C. Caddeo, O.D. Sales, D. Valenti, A.R. Sauri, A.M. Fadda, M. Manconi, Inhibition of skin 27
inflammation in mice by diclofenac in vesicular carriers: liposomes, ethosomes and PEVs, Int J Pharm, 28
443 (2013) 128-136. 29

26
[53] G. Cevc, D. Gebauer, J. Stieber, A. Schatzlein, G. Blume, Ultraflexible vesicles, Transfersomes, 1
have an extremely low pore penetration resistance and transport therapeutic amounts of insulin across 2
the intact mammalian skin, Biochim Biophys Acta, 1368 (1998) 201-215. 3
[54] E. Touitou, N. Dayan, L. Bergelson, B. Godin, M. Eliaz, Ethosomes - novel vesicular carriers for 4
enhanced delivery: characterization and skin penetration properties, J Control Release, 65 (2000) 403-5
418. 6
[55] G. Cevc, G. Blume, New, highly efficient formulation of diclofenac for the topical, transdermal 7
administration in ultradeformable drug carriers, Transfersomes, Biochim Biophys Acta, 1514 (2001) 8
191-205. 9
[56] S. Ghanbarzadeh, S. Arami, Enhanced transdermal delivery of diclofenac sodium via conventional 10
liposomes, ethosomes, and transfersomes, Biomed Res Int, 2013 (2013) 616810. 11
[57] G. Cevc, G. Blume, Lipid vesicles penetrate into intact skin owing to the transdermal osmotic 12
gradients and hydration force, BBA - Biomembranes, 1104 (1992) 226-232. 13
14
15
16
17
18
19
20
21
22


















