
Combined Nuclear Magnetic Resonance and Molecular Dynamics...
10
Combined Nuclear Magnetic Resonance and Molecular Dynamics Study of Methane Adsorption in M 2 (dobdc) Metal−Organic Frameworks Velencia J. Witherspoon, †,# Rocío Mercado, ‡,# Efrem Braun, † Amber Mace, § Jonathan Bachman, † Jeffrey R. Long, ‡,† Bernhard Blü mich, ∥ Berend Smit, §,† and Jeffrey A. Reimer* ,†,⊥ † Department of Chemical and Biomolecular Engineering and ‡ Department of Chemistry, University of California, Berkeley, Berkeley, California 94720, United States § Institut des Sciences et Ingé nierie Chimiques, Valais, E ́ cole Polytechnique Fé dé rale de Lausanne (EPFL), Rue de l’Industrie 17, CH-1951 Sion, Switzerland ∥ Institut fü r Technische und Makromolekulare Chemie (ITMC), RWTH Aachen University, 52062 Aachen, Germany ⊥ Materials Science Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States * S Supporting Information ABSTRACT: We examine the diffusion of methane in the metal−organic frameworks M 2 (dobdc) (M = Mg, Ni, Zn; dobdc 4− = 2,5-dioxido-1,4-benzenedicarboxylate) as a function of methane loading through a combination of nuclear magnetic resonance and molecular dynamics simulations. At low gas densities, our results suggest that favorable CH 4 −CH 4 interactions lower the free energy barrier for methane hopping between coordinatively unsaturated metal sites and thus enhance the translational motion of methane down the c- axis. At higher gas densities, CH 4 −CH 4 interactions become more significant, CH 4 −CH 4 collisions become more frequent, and the gas self-diffusion begins to decrease. Finally, we observe that the self-diffusion coefficient of methane is inversely related to the binding energy at the coordinatively unsaturated metal sites, such that diffusion is most rapid in the Zn 2 (dobdc) framework. ■ INTRODUCTION Metal−organic frameworks (MOFs) are porous, three-dimen- sional solids consisting of metal nodes connected by organic linkers, and their immense synthetic and topological diversity has rendered them exceptional candidates for the selective capture and separation of small molecules, including carbon dioxide, methane, and xenon. 1−6 For a number of such materials, known as isoreticular frameworks, it is possible to vary the metal ion and/or linker while maintaining the parent topology, therefore enabling systematic variation of material composition and properties such as pore size. One such family of isoreticular structures is the M 2 (dobdc) frameworks (also known as M-MOF-74; M = Mg, Mn, Fe, Co, Ni, Cu, or Zn; dobdc 4− = 2,5-dioxido-1,4-benzenedicarbox- ylate; Figure 1), which has been widely studied for applications in gas storage and separation because of the presence of coordinatively unsaturated or “open” metal sites that are able to strongly bind and polarize various gases. 7−13 Further tailoring of this class of materials to exhibit selective CH 4 interactions is of interest for establishing highly efficient adsorptive separation processes for applications in natural gas enrichment and the separation of CH 4 from power plant flue gas mixtures, 10,14,15 as well as toward the development of alternatives for natural gas storage in transportation vehicles. 10, 16 Motivated by these applications, previous research has investigated the potential of M 2 (dobdc) frame- works for the storage and separation of methane and demonstrated that the choice of transition metal can significantly influence the material adsorptive capacity. 10,16 In particular, the Ni 2 (dobdc) analogue displays the highest volumetric methane uptake of 230 v STP/v at 35 bar, followed by Mg 2 (dobdc) and Zn 2 (dobdc) (200 v STP/v and 188 v STP/v at 35 bar, respectively). 10,16 Neutron scattering characterization of CH 4 adsorption in M 2 (dobdc) has shed light on this result, revealing that the primary adsorption site is located directly above the coordinatively unsaturated metal Received: February 22, 2019 Revised: April 21, 2019 Published: April 23, 2019 Article pubs.acs.org/JPCC Cite This: J. Phys. Chem. C 2019, 123, 12286-12295 © 2019 American Chemical Society 12286 DOI: 10.1021/acs.jpcc.9b01733 J. Phys. Chem. C 2019, 123, 12286−12295 Downloaded via LAWRENCE BERKELEY NATL LABORATORY on July 29, 2019 at 00:55:27 (UTC). See https://pubs.acs.org/sharingguidelines for options on how to legitimately share published articles.
Transcript of Combined Nuclear Magnetic Resonance and Molecular Dynamics...

Combined Nuclear Magnetic Resonance and Molecular DynamicsStudy of Methane Adsorption in M2(dobdc) Metal−OrganicFrameworksVelencia J. Witherspoon,†,# Rocío Mercado,‡,# Efrem Braun,† Amber Mace,§
Jonathan Bachman,† Jeffrey R. Long,‡,† Bernhard Blumich,∥ Berend Smit,§,†
and Jeffrey A. Reimer*,†,⊥
†Department of Chemical and Biomolecular Engineering and ‡Department of Chemistry, University of California, Berkeley,Berkeley, California 94720, United States§Institut des Sciences et Ingenierie Chimiques, Valais, Ecole Polytechnique Federale de Lausanne (EPFL), Rue de l’Industrie 17,CH-1951 Sion, Switzerland∥Institut fur Technische und Makromolekulare Chemie (ITMC), RWTH Aachen University, 52062 Aachen, Germany⊥Materials Science Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States
*S Supporting Information
ABSTRACT: We examine the diffusion of methane in themetal−organic frameworks M2(dobdc) (M = Mg, Ni, Zn;dobdc4− = 2,5-dioxido-1,4-benzenedicarboxylate) as a functionof methane loading through a combination of nuclearmagnetic resonance and molecular dynamics simulations. Atlow gas densities, our results suggest that favorable CH4−CH4interactions lower the free energy barrier for methane hoppingbetween coordinatively unsaturated metal sites and thusenhance the translational motion of methane down the c-axis. At higher gas densities, CH4−CH4 interactions becomemore significant, CH4−CH4 collisions become more frequent,and the gas self-diffusion begins to decrease. Finally, weobserve that the self-diffusion coefficient of methane isinversely related to the binding energy at the coordinativelyunsaturated metal sites, such that diffusion is most rapid in the Zn2(dobdc) framework.
■ INTRODUCTION
Metal−organic frameworks (MOFs) are porous, three-dimen-sional solids consisting of metal nodes connected by organiclinkers, and their immense synthetic and topological diversityhas rendered them exceptional candidates for the selectivecapture and separation of small molecules, including carbondioxide, methane, and xenon.1−6 For a number of suchmaterials, known as isoreticular frameworks, it is possible tovary the metal ion and/or linker while maintaining the parenttopology, therefore enabling systematic variation of materialcomposition and properties such as pore size.One such family of isoreticular structures is the M2(dobdc)
frameworks (also known as M-MOF-74; M = Mg, Mn, Fe, Co,Ni, Cu, or Zn; dobdc4− = 2,5-dioxido-1,4-benzenedicarbox-ylate; Figure 1), which has been widely studied for applicationsin gas storage and separation because of the presence ofcoordinatively unsaturated or “open” metal sites that are ableto strongly bind and polarize various gases.7−13 Furthertailoring of this class of materials to exhibit selective CH4interactions is of interest for establishing highly efficient
adsorptive separation processes for applications in natural gasenrichment and the separation of CH4 from power plant fluegas mixtures,10,14,15 as well as toward the development ofalternatives for natural gas storage in transportationvehicles.10,16 Motivated by these applications, previousresearch has investigated the potential of M2(dobdc) frame-works for the storage and separation of methane anddemonstrated that the choice of transition metal cansignificantly influence the material adsorptive capacity.10,16 Inparticular, the Ni2(dobdc) analogue displays the highestvolumetric methane uptake of 230 v STP/v at 35 bar, followedby Mg2(dobdc) and Zn2(dobdc) (200 v STP/v and 188 vSTP/v at 35 bar, respectively).10,16 Neutron scatteringcharacterization of CH4 adsorption in M2(dobdc) has shedlight on this result, revealing that the primary adsorption site islocated directly above the coordinatively unsaturated metal
Received: February 22, 2019Revised: April 21, 2019Published: April 23, 2019
Article
pubs.acs.org/JPCCCite This: J. Phys. Chem. C 2019, 123, 12286−12295
© 2019 American Chemical Society 12286 DOI: 10.1021/acs.jpcc.9b01733J. Phys. Chem. C 2019, 123, 12286−12295
Dow
nloa
ded
via
LA
WR
EN
CE
BE
RK
EL
EY
NA
TL
LA
BO
RA
TO
RY
on
July
29,
201
9 at
00:
55:2
7 (U
TC
).Se
e ht
tps:
//pub
s.ac
s.or
g/sh
arin
ggui
delin
es f
or o
ptio
ns o
n ho
w to
legi
timat
ely
shar
e pu
blis
hed
artic
les.

centers, whereas a secondary site exists along the c-axisbetween the primary sites.13 A tertiary binding site has alsobeen located in the center of the pores for methane and certainother adsorbates.17 At low pressures, the various M2(dobdc)analogues exhibit differences in CH4 uptake that primarily arisebecause of differences in gas binding energies at the open metalsites.Computational investigations have also found that the
relative binding energy of CH4 at the open metal sites inM2(dobdc) (M = Mg, Ni, and Zn) is highest in the Nianalogue, followed by the Mg analogue and finally the Znanalogue, in agreement with experimental results.16,18 Thesestudies also report differences in the binding energiesassociated with the secondary adsorption sites. Further,analysis of spatial probability distributions produced fromgrand canonical Monte Carlo (GCMC) simulation data havepredicted that CH4 molecules spend a larger fraction of theirtime near the open metal sites in frameworks with strongerheats of adsorptionin contrast to frameworks that lack suchstrong adsorption sites where the adsorbate instead spends agreater amount of time in the center of the pores, away fromthe framework walls.16
Whereas many studies have indirectly evaluated the effect ofenergetics on the suitability of a system for gas separations(e.g., by fitting transient breakthrough data), few studies havesystematically studied the effect of host−guest energetics onmass transport in MOFs.19 The transport of CH4 inNi2(dobdc) was investigated previously via permeation experi-ments for mixed-membrane applications, where it was foundthat these macroscopic methods yielded averaged mass transfercoefficients with contributions from both the intercrystallineand intracrystalline regimes.20 However, it was not possible toseparate these contributions using that experimental method.Understanding the transport contributions of small moleculesin MOFs has in general proven challenging, with fewexperimental techniques available to assess mass transfercoefficients associated with the intracrystalline regime.21−23
In this regime, systematic variations in pore features such assize and metal binding site can greatly influence mass transfer.Nuclear magnetic resonance (NMR)24 is a robust technique
that has proven valuable in understanding the motion ofadsorbed gases, for instance by providing access to individualself-diffusion coefficients (Ds) and relaxation rate (R{1,2} = 1/
T{1,2}) constants for a given adsorbate.25−29 Furthermore,molecular dynamics (MD) simulations29,30 performed as afunction of adsorbate loading in a number of MOFs haveindicated that the observed magnitude of intracrystalline Dsdepends strongly on the interaction strength of the adsorbatewith the framework.31 Here, we seek to use these methods intandem to develop an enhanced understanding of methaneadsorption dynamics in M2(dobdc), with MD simulationscomplementing the NMR data at conditions that are notexperimentally accessible. These complementary methodsreveal that at low methane loadings increasing the density ofmethane molecules in the pores leads to a decrease in the freeenergy barrier between metal sites and an increase in moleculediffusivity, whereas at high methane loadings an increase in gasdensity disrupts the paths of other methane molecules anddecreases their diffusivity. Similarly, the open metal sitesdecrease the diffusivity of methane in the pores by interruptingCH4 trajectories in proportion to the metal−CH4 bindingstrength.
■ METHODSNMR Sample Preparation. M2(dobdc) analogues were
prepared via the previously reported routes32 yielding crystalsizes between 10 and 20 μm. For each analogue, the as-synthesized material was activated and transferred to an argonglovebox before being loaded into a valved NMR tube. Thesamples were then activated again for 12 h at 180 °C and 0.01mbar. Following activation,1 the samples were cooled to 40 °Cand held at the desired equilibrium pressure for 30 min to anhour before closing the valve on the NMR tube. The samplewas then allowed to cool to room temperature and transferredto the NMR probe where it was re-heated to 40 °C for 30 minbefore data collection.
NMR Measurements. A 13 Interval Bi-Polar Pulse FieldGradient Stimulated Echo experiment with z-spoiler wasperformed with a Bruker AVANCE III 700 MHz spectrometerusing a Diff30 insert in the Mic5 Bruker imaging probe, asdescribed previously.33 Diffusion times ranged from 1 to 2.5ms, whereas the gradient strength ranged from 0 to 17 T/m in48 steps. The observed attenuation was processed using a one-dimensional inverse Laplace transformation with Tikhonovregularization as implemented in the Kea Prospa software. Thereported experimental Ds values are the average of thelogarithm mean of the resulting bimodal distributions of theinverse Laplace transforms.34 Each loading condition wassampled 3−5 times, with error bars representing the standarddeviation of these multiple measurements. The longitudinal, orspin−lattice, relaxation rate constant (R1) was obtained usingan inversion recovery pulse sequence (180-recover−90-acquire) and the transverse relaxation rate constant (R2) wasmeasured by implementing a Carr−Purcell−Meiboom−Gillpulse sequence with spectroscopic acquisitions.35 For allsample loadings, R1 was smaller than R2 and the stimulatedecho diffusion rate was significantly greater than both R1 andR2. The diffusion time was limited to 2.5 ms because ofevidence of potential diffusive diffraction36 at longer times.
MD Simulations. The self-diffusion coefficients werecomputed for CH4 diffusion in M2(dobdc) (M = Mg, Ni,Zn) from MD simulations performed using the large-scaleatomic/molecular massively parallel simulator (LAMMPS)MD program.37,38 The systems were simulated in the canonicalensemble at loadings corresponding to the equilibrium uptakevalues as determined from GCMC simulations at pressures
Figure 1. Illustration of the M2(dobdc) structure with one-dimensional pores extending along the c-axis (perpendicular to theplane of the page). Gray, red, and green spheres represent C, O, andM atoms, respectively. Hydrogen atoms are omitted for clarity.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.9b01733J. Phys. Chem. C 2019, 123, 12286−12295
12287

ranging from 0.1 to 100 bar and 313 K (Tables S2−S4). Afterequilibration for 1 ns, each system was simulated for 10 ns witha timestep of 1 fs using a Nose−Hoover thermostat. Forframework−CH4 interactions, a previously parameterizeddensity functional theory (DFT)-derived force field16 wasused. Methane−methane interactions were taken fromthe transferrable potentials for phase equilibria (TraPPE)force field.39 Nonbonding interactions were cut off and shiftedat 12.5 Å, and all unit cells were sufficiently replicated so as toavoid interaction between periodic replica.The values of Ds for adsorbed CH4 were computed from
these equilibrium simulations using the Einstein equation,which relates the self-diffusivity of a molecule to its mean-squared displacement (MSD). In particular, Ds values werecalculated from CH4 MSDs in each framework in the diffusiveregime (2−100 ps). The MSDs were computed from MDtrajectories using the order-n algorithm described previ-ously.40,41
At the lowest CH4 loadings considered in this work (∼0.027CH4/M
2+), simulations would consist of boxes containing onlyone or two CH4 molecules if carried out in the same manner asthose simulations at the higher loadings. Such a simulation isproblematic because both the temperature and distribution ofmolecules within the poressix pores per simulation super-cellare not well-defined for only a single (or few) particle(s)per supercell. To address these concerns and to establish thatthe Ds values at these low densities are better defined,simulations at the same low CH4 densities (0.027 CH4/M
2+)were carried out with increasing c-axis lengths, l, of thesimulation supercell (where l ∈ {4, 8, 12, 16, 20, 24, 28, 32}),and with increasing numbers of CH4 atoms, N, in the pores (Nor 2N per pore, uniformly distributed, where N ∈ {1, 2, 3, 4, 5,6, 7, 8}). For each metal framework, the low-density Ds wasthen extrapolated from the converged value corresponding toan infinitely long simulation box.For the lowest density simulations in each M2(dobdc)
framework, which were more expensive because of their size,the error bars for the Ds values were estimated by dividing each10 ns trajectory into ten 1 ns trajectories and calculating thestandard deviation of the ten Ds values computed for each 1 nstrajectory. For all higher density trajectories, the error bars forthe Ds values were estimated by running three differentsimulations at the same set of conditions and calculating thestandard deviation of the three Ds values computed for each 10ns trajectory.Methane Hopping Rates. In order to estimate the CH4
hopping rate between the primary and secondary binding sitesin each M2(dobdc) framework, the average exchange rate wascalculated by counting the number of times a CH4 moleculetransitioned from a primary binding site (2.8−3.0 Å from themetal) to a secondary binding site (3.5−3.7 Å from the metal),or vice versa, per nanosecond of time. The CH4 distances fromeach binding site were determined from the spatial distributionof CH4 around the metals; peaks in the CH4 densitycorresponding to the two densest binding sites occurred near2.9 and 3.6 Å for all three frameworks (see Figures S10−S15 inthe Supporting Information section Methane Binding SiteAnalysis for details). A width of 0.2 Å around the peak densityof each binding site was chosen such that the CH4 binding sitewould be unambiguous. If a CH molecule crossed into theintermediate region (between 3.0 and 3.5 Å), then back to thesame site, this was not counted as an exchange event.
Probability Densities. The probability densities (FigureS16) of adsorbed CH4 in all three M2(dobdc) frameworkswere computed by binning CH4 positions from the 10 nstrajectories onto a 0.2 × 0.2 × 0.2 Å3 grid using PEGrid42 andplotted in VisIt.43
Free Energy Calculations. To estimate the free energybarrier for hopping between adjacent metal sites, both downthe c-axis (supercell axis in line with the z-axis for a single unitcell) and along a given ab-plane (orthogonal to the c-axis), MDsimulations were carried out at loadings of 2−14 CH4molecules per pore in each M2(dobdc) framework. The MDsimulations were run for 10 ns, saving the positions of theadsorbates every MD cycle.The positions of the adsorbates in a single pore were
mapped to one unit cell along the c-axis and binned to a 0.4 ×0.4 × 0.4 Å3 grid. The topology of the free energy landscapewas analyzed using the TuTraSt algorithm.44 This algorithmpartitions the free energy grid into potential energy wells andtransition states separating the wells by (1) identifying gridpoint coordinates of energy minima, (2) growing thesesubsequently with increasing energy values using a connectedcomponent search, and (3) detecting merging points. Thisprovides the necessary information to compute the free energybarriers and estimate Ds analytically by applying transition statetheory (Bennett−Chandler method45,46) for the transitionsalong the c-axis and within the ring. Here, Ds along the c-axisindicates a “true” diffusion of the CH4 traveling through thematerial, whereas the Ds for the “ring-like” diffusion does not.To test the hypothesis that the presence of a CH4 lowers the
energy barriers for CH4 diffusion in surrounding diffusionchannels parallel with the c-axis, we compare the potentialenergy surface (PES) of CH4 within the field of the frameworkalone and with that with a CH4 particle placed at a primaryadsorption site. The PES is derived from Monte Carlosimulations where 100 million single particle insertions arecarried out in the canonical ensemble to sample the energeticspace. The coordinates are saved at every step and treated thesame way as the MD coordinates to create a 3D gridquantifying the PES. This procedure is first carried out on anempty framework and from the resulting PES grid, thecoordinates corresponding to the minimum of a potentialenergy well of a primary adsorption site are identified, and aCH4 particle is positioned at one of these sites within theframework; the PES is then computed for this derivedframework. The energy barriers for the c-axis transitions arecomputed from the two PES grids using the partitioningalgorithm to compare how these are altered in the field ofanother adsorbate. The procedure is described in detail inMace et al.44
■ RESULTS AND DISCUSSIONNMR relaxation rates of free and adsorbed molecules within anMOF are determined by fluctuating magnetic fields associatedwith molecular motion. Knowledge of the correlation times formolecular motion, and the origin and magnitude of localmagnetic fields, are thus required to quantitatively model theserelaxation rates. Ignoring chemical shift anisotropy (forprotons this effect is vanishingly small), the fluctuatingmagnetic fields driving NMR relaxation derive from time-varying hetero- and homonuclear dipole interactions, as well asthe coupling of the nuclear spin to the rotational angularmomentum of the molecule. The magnitude of fluctuatingintramolecular dipolar interactions in gas-phase methane has
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.9b01733J. Phys. Chem. C 2019, 123, 12286−12295
12288
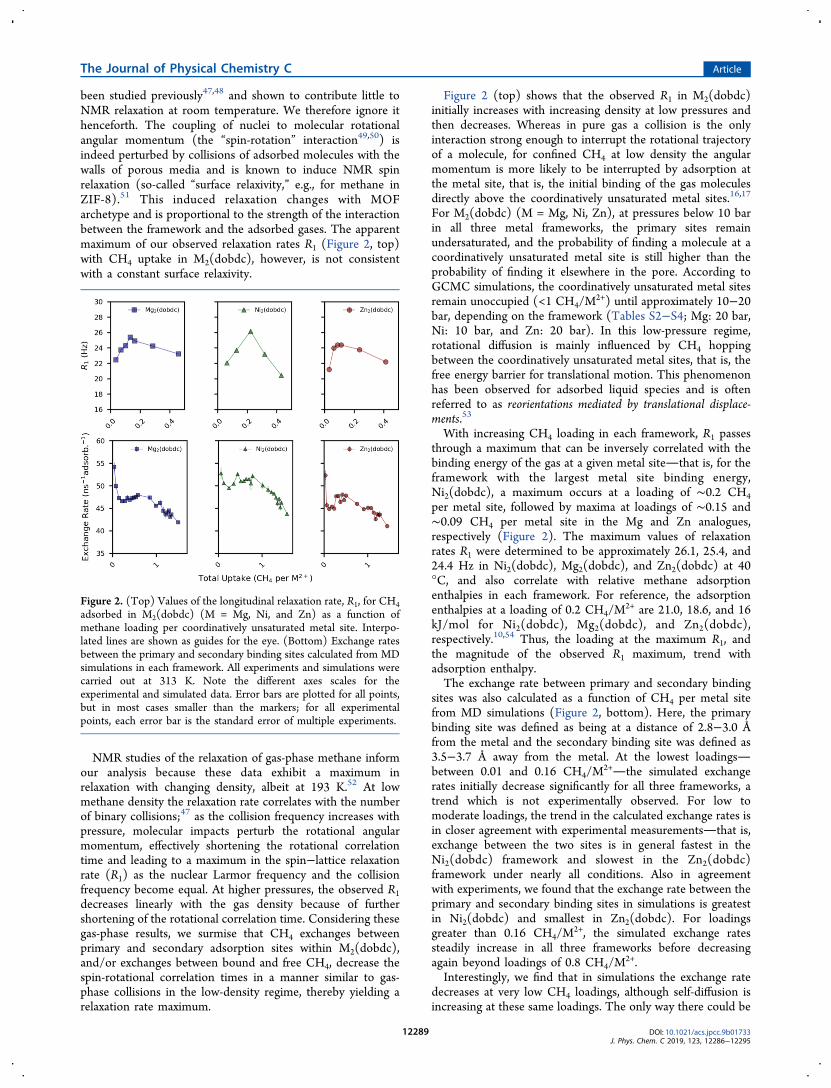
been studied previously47,48 and shown to contribute little toNMR relaxation at room temperature. We therefore ignore ithenceforth. The coupling of nuclei to molecular rotationalangular momentum (the “spin-rotation” interaction49,50) isindeed perturbed by collisions of adsorbed molecules with thewalls of porous media and is known to induce NMR spinrelaxation (so-called “surface relaxivity,” e.g., for methane inZIF-8).51 This induced relaxation changes with MOFarchetype and is proportional to the strength of the interactionbetween the framework and the adsorbed gases. The apparentmaximum of our observed relaxation rates R1 (Figure 2, top)with CH4 uptake in M2(dobdc), however, is not consistentwith a constant surface relaxivity.
NMR studies of the relaxation of gas-phase methane informour analysis because these data exhibit a maximum inrelaxation with changing density, albeit at 193 K.52 At lowmethane density the relaxation rate correlates with the numberof binary collisions;47 as the collision frequency increases withpressure, molecular impacts perturb the rotational angularmomentum, effectively shortening the rotational correlationtime and leading to a maximum in the spin−lattice relaxationrate (R1) as the nuclear Larmor frequency and the collisionfrequency become equal. At higher pressures, the observed R1decreases linearly with the gas density because of furthershortening of the rotational correlation time. Considering thesegas-phase results, we surmise that CH4 exchanges betweenprimary and secondary adsorption sites within M2(dobdc),and/or exchanges between bound and free CH4, decrease thespin-rotational correlation times in a manner similar to gas-phase collisions in the low-density regime, thereby yielding arelaxation rate maximum.
Figure 2 (top) shows that the observed R1 in M2(dobdc)initially increases with increasing density at low pressures andthen decreases. Whereas in pure gas a collision is the onlyinteraction strong enough to interrupt the rotational trajectoryof a molecule, for confined CH4 at low density the angularmomentum is more likely to be interrupted by adsorption atthe metal site, that is, the initial binding of the gas moleculesdirectly above the coordinatively unsaturated metal sites.16,17
For M2(dobdc) (M = Mg, Ni, Zn), at pressures below 10 barin all three metal frameworks, the primary sites remainundersaturated, and the probability of finding a molecule at acoordinatively unsaturated metal site is still higher than theprobability of finding it elsewhere in the pore. According toGCMC simulations, the coordinatively unsaturated metal sitesremain unoccupied (<1 CH4/M
2+) until approximately 10−20bar, depending on the framework (Tables S2−S4; Mg: 20 bar,Ni: 10 bar, and Zn: 20 bar). In this low-pressure regime,rotational diffusion is mainly influenced by CH4 hoppingbetween the coordinatively unsaturated metal sites, that is, thefree energy barrier for translational motion. This phenomenonhas been observed for adsorbed liquid species and is oftenreferred to as reorientations mediated by translational displace-ments.53
With increasing CH4 loading in each framework, R1 passesthrough a maximum that can be inversely correlated with thebinding energy of the gas at a given metal sitethat is, for theframework with the largest metal site binding energy,Ni2(dobdc), a maximum occurs at a loading of ∼0.2 CH4per metal site, followed by maxima at loadings of ∼0.15 and∼0.09 CH4 per metal site in the Mg and Zn analogues,respectively (Figure 2). The maximum values of relaxationrates R1 were determined to be approximately 26.1, 25.4, and24.4 Hz in Ni2(dobdc), Mg2(dobdc), and Zn2(dobdc) at 40°C, and also correlate with relative methane adsorptionenthalpies in each framework. For reference, the adsorptionenthalpies at a loading of 0.2 CH4/M
2+ are 21.0, 18.6, and 16kJ/mol for Ni2(dobdc), Mg2(dobdc), and Zn2(dobdc),respectively.10,54 Thus, the loading at the maximum R1, andthe magnitude of the observed R1 maximum, trend withadsorption enthalpy.The exchange rate between primary and secondary binding
sites was also calculated as a function of CH4 per metal sitefrom MD simulations (Figure 2, bottom). Here, the primarybinding site was defined as being at a distance of 2.8−3.0 Åfrom the metal and the secondary binding site was defined as3.5−3.7 Å away from the metal. At the lowest loadingsbetween 0.01 and 0.16 CH4/M
2+the simulated exchangerates initially decrease significantly for all three frameworks, atrend which is not experimentally observed. For low tomoderate loadings, the trend in the calculated exchange rates isin closer agreement with experimental measurementsthat is,exchange between the two sites is in general fastest in theNi2(dobdc) framework and slowest in the Zn2(dobdc)framework under nearly all conditions. Also in agreementwith experiments, we found that the exchange rate between theprimary and secondary binding sites in simulations is greatestin Ni2(dobdc) and smallest in Zn2(dobdc). For loadingsgreater than 0.16 CH4/M
2+, the simulated exchange ratessteadily increase in all three frameworks before decreasingagain beyond loadings of 0.8 CH4/M
2+.Interestingly, we find that in simulations the exchange rate
decreases at very low CH4 loadings, although self-diffusion isincreasing at these same loadings. The only way there could be
Figure 2. (Top) Values of the longitudinal relaxation rate, R1, for CH4adsorbed in M2(dobdc) (M = Mg, Ni, and Zn) as a function ofmethane loading per coordinatively unsaturated metal site. Interpo-lated lines are shown as guides for the eye. (Bottom) Exchange ratesbetween the primary and secondary binding sites calculated from MDsimulations in each framework. All experiments and simulations werecarried out at 313 K. Note the different axes scales for theexperimental and simulated data. Error bars are plotted for all points,but in most cases smaller than the markers; for all experimentalpoints, each error bar is the standard error of multiple experiments.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.9b01733J. Phys. Chem. C 2019, 123, 12286−12295
12289
less exchange events at these loadings would be if the methanemolecules remained (on average) a fixed or nearly-fixeddistance from the metal site for longer periods throughouttheir trajectories. As such, a possible explanation for the initialdecrease in the exchange rate is that as the number of methanemolecules at the primary binding sites increases at very lowloadings, any other methane molecules in the pores then taketrajectories “around” the primary adsorbed methane molecules(along the minimum free energy path proposed herein). Thisexplanation is supported by the histograms in Figures S11, S13,and S15 in the Supporting Information, which correspond touptakes of 0.1−0.25 CH4/M
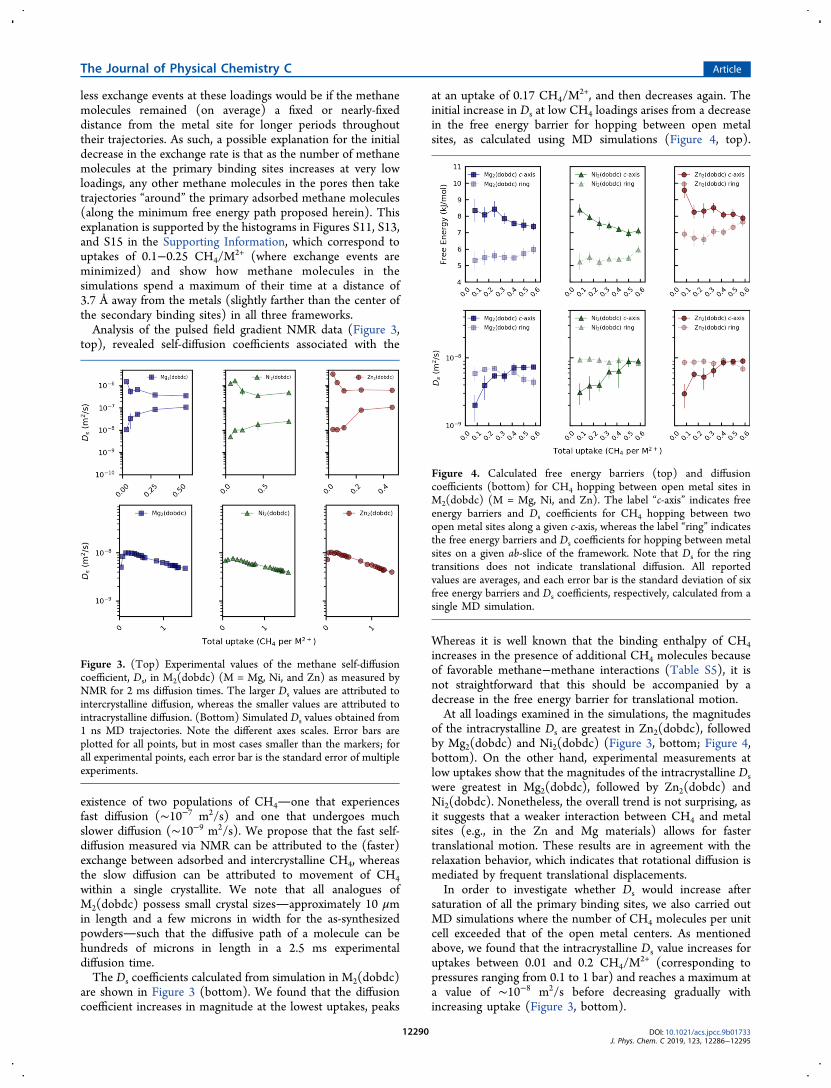
2+ (where exchange events areminimized) and show how methane molecules in thesimulations spend a maximum of their time at a distance of3.7 Å away from the metals (slightly farther than the center ofthe secondary binding sites) in all three frameworks.Analysis of the pulsed field gradient NMR data (Figure 3,
top), revealed self-diffusion coefficients associated with the
existence of two populations of CH4one that experiencesfast diffusion (∼10−7 m2/s) and one that undergoes muchslower diffusion (∼10−9 m2/s). We propose that the fast self-diffusion measured via NMR can be attributed to the (faster)exchange between adsorbed and intercrystalline CH4, whereasthe slow diffusion can be attributed to movement of CH4within a single crystallite. We note that all analogues ofM2(dobdc) possess small crystal sizesapproximately 10 μmin length and a few microns in width for the as-synthesizedpowderssuch that the diffusive path of a molecule can behundreds of microns in length in a 2.5 ms experimentaldiffusion time.The Ds coefficients calculated from simulation in M2(dobdc)
are shown in Figure 3 (bottom). We found that the diffusioncoefficient increases in magnitude at the lowest uptakes, peaks
at an uptake of 0.17 CH4/M2+, and then decreases again. The
initial increase in Ds at low CH4 loadings arises from a decreasein the free energy barrier for hopping between open metalsites, as calculated using MD simulations (Figure 4, top).
Whereas it is well known that the binding enthalpy of CH4increases in the presence of additional CH4 molecules becauseof favorable methane−methane interactions (Table S5), it isnot straightforward that this should be accompanied by adecrease in the free energy barrier for translational motion.At all loadings examined in the simulations, the magnitudes
of the intracrystalline Ds are greatest in Zn2(dobdc), followedby Mg2(dobdc) and Ni2(dobdc) (Figure 3, bottom; Figure 4,bottom). On the other hand, experimental measurements atlow uptakes show that the magnitudes of the intracrystalline Dswere greatest in Mg2(dobdc), followed by Zn2(dobdc) andNi2(dobdc). Nonetheless, the overall trend is not surprising, asit suggests that a weaker interaction between CH4 and metalsites (e.g., in the Zn and Mg materials) allows for fastertranslational motion. These results are in agreement with therelaxation behavior, which indicates that rotational diffusion ismediated by frequent translational displacements.In order to investigate whether Ds would increase after
saturation of all the primary binding sites, we also carried outMD simulations where the number of CH4 molecules per unitcell exceeded that of the open metal centers. As mentionedabove, we found that the intracrystalline Ds value increases foruptakes between 0.01 and 0.2 CH4/M
2+ (corresponding topressures ranging from 0.1 to 1 bar) and reaches a maximum ata value of ∼10−8 m2/s before decreasing gradually withincreasing uptake (Figure 3, bottom).
Figure 3. (Top) Experimental values of the methane self-diffusioncoefficient, Ds, in M2(dobdc) (M = Mg, Ni, and Zn) as measured byNMR for 2 ms diffusion times. The larger Ds values are attributed tointercrystalline diffusion, whereas the smaller values are attributed tointracrystalline diffusion. (Bottom) Simulated Ds values obtained from1 ns MD trajectories. Note the different axes scales. Error bars areplotted for all points, but in most cases smaller than the markers; forall experimental points, each error bar is the standard error of multipleexperiments.
Figure 4. Calculated free energy barriers (top) and diffusioncoefficients (bottom) for CH4 hopping between open metal sites inM2(dobdc) (M = Mg, Ni, and Zn). The label “c-axis” indicates freeenergy barriers and Ds coefficients for CH4 hopping between twoopen metal sites along a given c-axis, whereas the label “ring” indicatesthe free energy barriers and Ds coefficients for hopping between metalsites on a given ab-slice of the framework. Note that Ds for the ringtransitions does not indicate translational diffusion. All reportedvalues are averages, and each error bar is the standard deviation of sixfree energy barriers and Ds coefficients, respectively, calculated from asingle MD simulation.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.9b01733J. Phys. Chem. C 2019, 123, 12286−12295
12290

Both the NMR data and simulations reveal that themagnitude of Ds is inversely related to the binding energy atthe coordinatively unsaturated metal sites (Ubinding: Ni > Mg >Zn)that is, self-diffusion is fastest in Zn2(dobdc) andMg2(dobdc) and slowest in Ni2(dobdc). We also observethat the CH4 heat of desorption increases with increasingmethane loading in the pores up until a loading of ∼0.8 CH4/M2+ (Table S5; Figure S2). It might readily be interpretedfrom this result that the diffusion coefficient should alsodecrease with increasing CH4 uptake in this regime. However,as can be seen from Figure 3 (bottom), the Ds coefficientsinitially increase in all three metal frameworks at very lowloadings (<0.16 CH4/M
2+).This increase can be explained by considering the free
energy barriers between two metal sites. Because CH4 diffusesdown the pore by hopping along nearby metal sites, diffusionmay occur down the c-axis or in a “ring-like” fashion along agiven ab-slice. However, only a lower free energy barrierbetween two primary adsorption sites located along the c-axiswill lead to a greater Ds coefficient: lower free energy barriersalong the ring lead to negligible translational motion. We havecalculated this free energy barrier from the MD trajectories andindeed found that having more CH4 molecules in the poreslowers the free energy barrier for CH4 hopping between openmetal sites along the c-axis at low loadings (Figure 4, top).To illustrate how, at very low loadings, the presence of a
second CH4 molecule lowers the free energy barrier for CH4hopping from one metal site to another, we have calculated thefree energy profiles from simulations with and without a rigidCH4 already bound at the open metal site (Figure 5). Thevalues of the free energy at the corresponding transition statesfrom these simulations are given in Table 1. Indeed, weobserve that for transitions at angles |ρ| = π/3, the free energybarrier is lower in all three frameworks. Figure 6 zooms in onthe free energy barriers for transitions along the c-axis at angles|ρ| = π/3, illustrating how the two free energy wells meet firstin the Ni framework, followed by the Mg framework, andfinally the Zn framework; it also illustrates further how the freeenergy is overall lower when there is already a CH4 bound atthe open metal site.One might hypothesize that the sharp increase in Ds at low
loadings could be due to more CH4 in the pores effectivelyblocking the metal sites, thus shielding other CH4 moleculesfrom these strong binding sites and allowing them to diffusepast without binding. However, probability density maps showthat the CH4 spends most of its time above the metal sites insimulations at both of these densities (Figure S16), and in fact
the results at both densities look very similar, thus eliminatingthis as a possible mechanism.Although the magnitudes of the diffusivities calculated from
simulations differ by an order of magnitude from the pulsedfield gradient measured values, trends between the two areremarkably similar. For example, Ni2(dobdc) exhibits theslowest diffusion, and Zn2(dobdc) and Mg2(dobdc) haveabout the same diffusivities in both simulation and experiment.In both experimental and simulation data, there is an initial risein diffusivity with increasing CH4 uptake. These initialincreases are rationalized by considering the growing presenceof CH4 molecules in the pores where, at low densities, theyserve to stabilize the transition states for hopping from onemetal site to another. At the high uptakes not accessible withNMR, the simulations show how decreasing diffusivity is aresult of an increasing frequency of methane−methanecollisions within the pores that leads to a decrease in CH4MSD.As discussed in the Methods section, at the lowest CH4
loadings considered in this work (∼0.027 CH4/M2+),
simulations would consist of boxes containing only one ortwo CH4 molecules if carried out in the same way as thesimulations at the higher loadings, which would be problematicfrom the simulation point of view. As such, various fixed-density simulations were carried out that would allow us toextrapolate to an infinitely large simulation box. From theseconstant-density simulations at low uptakes, we find that as thenumber of molecules in the simulation box increases at a fixeddensity, the self-diffusion coefficients slowly increase untilconverging, as illustrated in Figure 7.The magnitude of Ds converges faster for simulations
involving 2N CH4 atoms per pore compared to N CH4 atoms
Figure 5. Free energy distributions in cylindrical coordinates integrated and normalized over radius values derived from MC simulations without(top) and with (bottom) a rigid CH4 located at the minimum energy site at 0 radians. Free energy values are given in kJ/mol.
Table 1. Energy Barriers for Transitions between PrimaryBinding Sites Along the c-Axisa
Mg Ni Zn
FW 7.6 8.3 8.1FW + 1 CH4, |ρ| = π/3 7.4 8.1 7.7FW + 1 CH4, |ρ| = 2π/3 7.6 8.3 8.0FW + 1 CH4, |ρ| = π 7.7 8.4 8.1
aValues are given in kJ/mol and are averaged over equivalenttransitions. For the empty framework structure (FW) the average isover six transitions and for the CH4 enriched structure (FW + CH4)averages are over 2, 2, and 1 transitions at angles |ρ| = π/3, 2π/3 and πradians, respectively, relative to the added rigid CH4 particle in theframework. Minimum free energy values for each framework are inbold.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.9b01733J. Phys. Chem. C 2019, 123, 12286−12295
12291
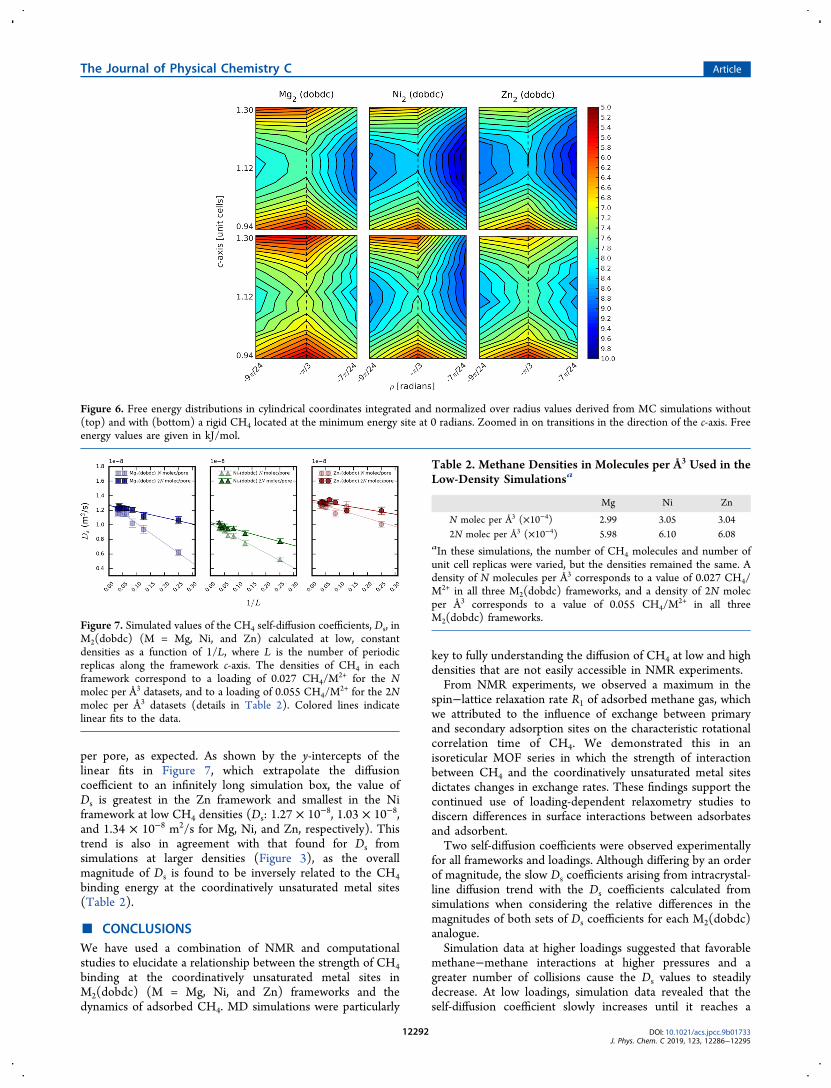
per pore, as expected. As shown by the y-intercepts of thelinear fits in Figure 7, which extrapolate the diffusioncoefficient to an infinitely long simulation box, the value ofDs is greatest in the Zn framework and smallest in the Niframework at low CH4 densities (Ds: 1.27 × 10−8, 1.03 × 10−8,and 1.34 × 10−8 m2/s for Mg, Ni, and Zn, respectively). Thistrend is also in agreement with that found for Ds fromsimulations at larger densities (Figure 3), as the overallmagnitude of Ds is found to be inversely related to the CH4binding energy at the coordinatively unsaturated metal sites(Table 2).
■ CONCLUSIONSWe have used a combination of NMR and computationalstudies to elucidate a relationship between the strength of CH4binding at the coordinatively unsaturated metal sites inM2(dobdc) (M = Mg, Ni, and Zn) frameworks and thedynamics of adsorbed CH4. MD simulations were particularly
key to fully understanding the diffusion of CH4 at low and highdensities that are not easily accessible in NMR experiments.From NMR experiments, we observed a maximum in the
spin−lattice relaxation rate R1 of adsorbed methane gas, whichwe attributed to the influence of exchange between primaryand secondary adsorption sites on the characteristic rotationalcorrelation time of CH4. We demonstrated this in anisoreticular MOF series in which the strength of interactionbetween CH4 and the coordinatively unsaturated metal sitesdictates changes in exchange rates. These findings support thecontinued use of loading-dependent relaxometry studies todiscern differences in surface interactions between adsorbatesand adsorbent.Two self-diffusion coefficients were observed experimentally
for all frameworks and loadings. Although differing by an orderof magnitude, the slow Ds coefficients arising from intracrystal-line diffusion trend with the Ds coefficients calculated fromsimulations when considering the relative differences in themagnitudes of both sets of Ds coefficients for each M2(dobdc)analogue.Simulation data at higher loadings suggested that favorable
methane−methane interactions at higher pressures and agreater number of collisions cause the Ds values to steadilydecrease. At low loadings, simulation data revealed that theself-diffusion coefficient slowly increases until it reaches a
Figure 6. Free energy distributions in cylindrical coordinates integrated and normalized over radius values derived from MC simulations without(top) and with (bottom) a rigid CH4 located at the minimum energy site at 0 radians. Zoomed in on transitions in the direction of the c-axis. Freeenergy values are given in kJ/mol.
Figure 7. Simulated values of the CH4 self-diffusion coefficients, Ds, inM2(dobdc) (M = Mg, Ni, and Zn) calculated at low, constantdensities as a function of 1/L, where L is the number of periodicreplicas along the framework c-axis. The densities of CH4 in eachframework correspond to a loading of 0.027 CH4/M
2+ for the Nmolec per Å3 datasets, and to a loading of 0.055 CH4/M
2+ for the 2Nmolec per Å3 datasets (details in Table 2). Colored lines indicatelinear fits to the data.
Table 2. Methane Densities in Molecules per Å3 Used in theLow-Density Simulationsa
Mg Ni Zn
N molec per Å3 (×10−4) 2.99 3.05 3.042N molec per Å3 (×10−4) 5.98 6.10 6.08
aIn these simulations, the number of CH4 molecules and number ofunit cell replicas were varied, but the densities remained the same. Adensity of N molecules per Å3 corresponds to a value of 0.027 CH4/M2+ in all three M2(dobdc) frameworks, and a density of 2N molecper Å3 corresponds to a value of 0.055 CH4/M
2+ in all threeM2(dobdc) frameworks.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.9b01733J. Phys. Chem. C 2019, 123, 12286−12295
12292

maximum in all three structures; we attribute this increase to astabilization of the free energy barrier for CH4 hoppingbetween two open metal sites because of the presence ofadditional CH4 molecules. This result is interesting because thebinding enthalpy of CH4 in M2(dobdc) also becomes morefavorable as more methane adsorbs in the framework (at lowdensities); however, this does not lead to CH4 becoming morelocalized because of the simultaneous stabilization of the freeenergy barrier for hopping between open metal sites along thec-axis. This increase in Ds is only noticeable until a loadingcorresponding to a single CH4 molecule per six metal sites,where the Ds values are maximized.At very low CH4 loadings (∼0.027 CH4/M
2+), simulationsof CH4 in M2(dobdc) were carried out in increasingly largerand larger simulation boxes (keeping the CH4 density in thepores fixed) to show that as we increase the number ofmolecules in the simulation box while keeping the densitiesfixed, the self-diffusion coefficients slowly increase untilconverging at a constant value. We attribute this behavior toan increase in the number of intermolecular collisions at thesevery dilute densities when there are simply more adsorbents inthe simulation box.Finally, the magnitudes of the intracrystalline CH4 Ds
coefficients are overall smallest in Ni2(dobdc) and largest inZn2(dobdc). This is because the coordinatively unsaturatedmetal sites interact more strongly with adsorbed CH4 in theNi2(dobdc) framework and more weakly in the Zn2(dobdc)framework; the stronger a CH4 molecule adsorbs to theframework, the less distance it can travel over a fixed amount oftime. Simulations and experiments are in agreement with thetrend in the Ds coefficients predicted at the higher CH4loadings studied.We note that none of the computational studies presented
here would have been possible without parameterization of theDFT-derived force field reported previously,16 illustrating thevalue of having inexpensive yet accurate models of adsorptionin promising nanoporous materials for the comprehensivestudy of adsorbate thermodynamics. From an experimentalstandpoint, NMR facilitates a valuable systematic study of theeffect of host−guest energetics on adsorbate diffusion inMOFs. The comprehensive and improved understanding ofadsorbate behavior at the atomic scale using these comple-mentary techniques reinforces the concept that combinedexperimental−computational studies can be very valuable inguiding the improved design of materials for gas separationapplications, especially when seeking to understand adsorptionand diffusion behavior at the atomic scale.
■ ASSOCIATED CONTENT*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/acs.jpcc.9b01733.
Simulation structures; GCMC simulation details; MSDs;self-diffusion coefficients; methane binding site analysis;probability densities; and example experimental data(PDF)
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] J. Witherspoon: 0000-0002-2718-6605
Rocío Mercado: 0000-0002-6170-6088Efrem Braun: 0000-0001-5379-7031Amber Mace: 0000-0002-0323-0210Jonathan Bachman: 0000-0002-3313-2355Jeffrey R. Long: 0000-0002-5324-1321Berend Smit: 0000-0003-4653-8562Jeffrey A. Reimer: 0000-0002-4191-3725Author Contributions#Joint first authors.NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSThis work was supported as part of the Center for GasSeparations Relevant to Clean Energy Technologies, as anEnergy Frontier Research Center funded by the U.S.Department of Energy, Office of Science, Basic EnergySciences under Award DE-SC0001015. V.J.W. and R.M.acknowledge support by the National Science Foundationunder both grant numbers DGE 1752814 and DGE 1106400.This research also used resources of the National EnergyResearch Scientific Computing Center, a DOE Office ofScience User Facility supported by the Office of Science of theU.S. Department of Energy under contract no. DE-AC02-05CH11231. We also thank ACalNet, the Aachen-CaliforniaNetwork of Academic Exchange (DAAD Germany), forsupporting the research visit of V.J.W. to RWTH AachenUniversity, and Dr. Katie R. Meihaus for editorial assistance.
■ REFERENCES(1) Tanabe, K. K.; Wang, Z.; Cohen, S. M. SystematicFunctionalization of a Metal−Organic Framework via a PostsyntheticModification Approach. J. Am. Chem. Soc. 2008, 130, 8508−8517.(2) Wang, Z.; Cohen, S. M. Postsynthetic modification of metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1315−1329.(3) Morris, W.; Doonan, C. J.; Yaghi, O. M. PostsyntheticModification of a Metal-Organic Framework for Stabilization of aHemiaminal and Ammonia Uptake. Inorg. Chem. 2011, 50, 6853−6855.(4) Wang, C.; Liu, D.; Lin, W. Metal−organic frameworks as atunable platform for designing functional molecular materials. J. Am.Chem. Soc. 2013, 135, 13222.(5) Marshall, R. J.; Forgan, R. S. Postsynthetic Modification ofZirconium Metal-Organic Frameworks. Eur. J. Inorg. Chem. 2016,4310−4331.(6) Rungtaweevoranit, B.; Diercks, C. S.; Kalmutzki, M. J.; Yaghi, O.M. Spiers Memorial Lecture: : Progress and prospects of reticularchemistry. Faraday Discuss. 2017, 201, 9−45.(7) Zhou, H.-C.; Long, J. R.; Yaghi, O. M. Introduction to Metal-Organic Frameworks. Chem. Rev. 2012, 112, 673−674.(8) Furukawa, H.; Cordova, K. E.; O’Keeffe, M.; Yaghi, O. M. TheChemistry and Applications of Metal-Organic Frameworks. Science2013, 341, 1230444.(9) Peng, Y.; Krungleviciute, V.; Eryazici, I.; Hupp, J. T.; Farha, O.K.; Yildirim, T. Methane Storage in Metal-Organic Frameworks:Current Records, Surprise Findings, and Challenges. J. Am. Chem. Soc.2013, 135, 11887−11894.(10) Mason, J. A.; Veenstra, M.; Long, J. R. Evaluating metal-organicframeworks for natural gas storage. Chem. Sci. 2014, 5, 32−51.(11) Kuppler, R. J.; Timmons, D. J.; Fang, Q.-R.; Li, J.-R.; Makal, T.a.; Young, M. D.; Yuan, D.; Zhao, D.; Zhuang, W.; Zhou, H.-C.Potential applications of metal-organic frameworks. Coord. Chem. Rev.2009, 253, 3042−3066.(12) Sumida, K.; Rogow, D. L.; Mason, J. A.; McDonald, T. M.;Bloch, E. D.; Herm, Z. R.; Bae, T.-H.; Long, J. R. Carbon Dioxide
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.9b01733J. Phys. Chem. C 2019, 123, 12286−12295
12293

Capture in Metal-Organic Frameworks. Chem. Rev. 2011, 112, 724−781.(13) Queen, W. L.; et al. Comprehensive study of carbon dioxideadsorption in the metal-organic frameworks M2(dobdc) (M = Mg,Mn, Fe, Co, Ni, Cu, Zn). Chem. Sci. 2014, 5, 4569−4581.(14) Simon, C. M.; Kim, J.; Gomez-Gualdron, D. A.; Camp, J. S.;Chung, Y. G.; Martin, R. L.; Mercado, R.; Deem, M. W.; Gunter, D.;Haranczyk, M.; Sholl, D. S.; Snurr, R. Q.; Smit, B. The materialsgenome in action: identifying the performance limits for methanestorage. Energy Environ. Sci. 2015, 8, 1190−1199.(15) Schoedel, A.; Ji, Z.; Yaghi, O. M. The role of metal-organicframeworks in a carbon-neutral energy cycle. Nat. Energy 2016, 1,16034.(16) Mercado, R.; Vlaisavljevich, B.; Lin, L.-C.; Lee, K.; Lee, Y.;Mason, J. A.; Xiao, D. J.; Gonzalez, M. I.; Kapelewski, M. T.; Neaton,J. B.; Smit, B. Force Field Development from Periodic DensityFunctional Theory Calculations for Gas Separation ApplicationsUsing Metal-Organic Frameworks. J. Phys. Chem. C 2016, 120,12590−12604.(17) Wu, H.; Zhou, W.; Yildirim, T. High-Capacity Methane Storagein Metal−Organic Frameworks M2(dhtp): The Important Role ofOpen Metal Sites. J. Am. Chem. Soc. 2009, 131, 4995−5000.(18) Lee, K.; Howe, J. D.; Lin, L.-C.; Smit, B.; Neaton, J. B. Small-Molecule Adsorption in Open-Site Metal-Organic Frameworks: ASystematic Density Functional Theory Study for Rational Design.Chem. Mater. 2015, 27, 668−678.(19) Sutrisno, A.; Huang, Y. Solid-state NMR: A powerful tool forcharacterization of metal-organic frameworks. Solid State Nucl. Magn.Reson. 2013, 49−50, 1−11.(20) Chen, D.-L.; Shang, H.; Zhu, W.; Krishna, R. Reprint of:Transient breakthroughs of CO 2 /CH 4 and C 3 H 6 /C 3 H 8mixtures in fixed beds packed with Ni-MOF-74. Chem. Eng. Sci. 2015,124, 109−117.(21) Karger, J.; Caro, J.; Cool, P.; Coppens, M.-O.; Jones, D.;Kapteijn, F.; Rodríguez-Reinoso, F.; Stoecker, M.; Theodorou, D.;Vansant, E. F.; et al. Benefit of microscopic diffusion measurement forthe characterization of nanoporous materials. Chem. Eng. Technol.2009, 32, 1494−1511.(22) Bernin, D.; Hedin, N. Perspectives on NMR studies of CO2adsorption. Curr. Opin. Colloid Interface Sci. 2018, 33, 53−62.(23) Chen, J. J.; Kong, X.; Sumida, K.; Manumpil, M. A.; Long, J. R.;Reimer, J. A. Ex-situ NMR relaxometry of metal−organic frameworksfor rapid surface-area screening. Angew. Chem. Int. Ed. 2013, 52,12043−12046.(24) Pusch, A.-K.; Splith, T.; Moschkowitz, L.; Karmakar, S.;Biniwale, R.; Sant, M.; Suffritti, G. B.; Demontis, P.; Cravillon, J.;Pantatosaki, E.; Stallmach, F. NMR studies of carbon dioxide andmethane self-diffusion in ZIF-8 at elevated gas pressures. Adsorption2012, 18, 359−366.(25) Stallmach, F.; Groger, S.; Kunzel, V.; Karger, J.; Yaghi, O. M.;Hesse, M.; Muller, U. NMR Studies on the Diffusion of Hydro-carbons on the Metal-Organic Framework Material MOF-5. Angew.Chem., Int. Ed. 2006, 45, 2123−2126.(26) Kong, X.; Scott, E.; Ding, W.; Mason, J. A.; Long, J. R.; Reimer,J. A. CO2 Dynamics in a Metal-Organic Framework with Open MetalSites. J. Am. Chem. Soc. 2012, 134, 14341−14344.(27) Lin, L.-C.; Kim, J.; Kong, X.; Scott, E.; McDonald, T. M.; Long,J. R.; Reimer, J. A.; Smit, B. Understanding CO2Dynamics in Metal-Organic Frameworks with Open Metal Sites. Angew. Chem. 2013, 125,4506−4509.(28) Stallmach, F.; Pusch, A.-K.; Splith, T.; Horch, C.; Merker, S.NMR relaxation and diffusion studies of methane and carbon dioxidein nanoporous ZIF-8 and ZSM-58. Microporous Mesoporous Mater.2015, 205, 36−39.(29) Forse, A. C.; Gonzalez, M. I.; Siegelman, R. L.; Witherspoon, V.J.; Jawahery, S.; Mercado, R.; Milner, P. J.; Martell, J. D.; Smit, B.;Blumich, B.; Long, J. R.; Reimer, J. A. Unexpected DiffusionAnisotropy of Carbon Dioxide in the Metal-Organic FrameworkZn2(dobpdc). J. Am. Chem. Soc. 2018, 140, 1663−1673.
(30) Chmelik, C. Characteristic features of molecular transport inMOF ZIF-8 as revealed by IR microimaging. Microporous MesoporousMater. 2015, 216, 138.(31) Krishna, R.; van Baten, J. M. Influence of adsorptionthermodynamics on guest diffusivities in nanoporous crystallinematerials. Phys. Chem. Chem. Phys. 2013, 15, 7994−8016.(32) Bachman, J. E.; Kapelewski, M. T.; Reed, D. A.; Gonzalez, M.I.; Long, J. R. M2(m-dobdc) (M = Mn, Fe, Co, Ni) Metal-OrganicFrameworks as Highly Selective, High-Capacity Adsorbents forOlefin/Paraffin Separations. J. Am. Chem. Soc. 2017, 139, 15363−15370.(33) Cotts, R. M.; Hoch, M. J. R.; Sun, T.; Markert, J. T. Pulsed fieldgradient stimulated echo methods for improved NMR diffusionmeasurements in heterogeneous systems. J. Magn. Reson. 1989, 83,252−266.(34) Day, I. J. On the inversion of diffusion NMR data: Tikhonovregularization and optimal choice of the regularization parameter. J.Magn. Reson. 2011, 211, 178−185.(35) Casanova, F.; Perlo, J.; Blumich, B. Single-Sided NMR; Springer,2011; pp 1−10.(36) Callaghan, P. T. NMR imaging, NMR diffraction andapplications of pulsed gradient spin echoes in porous media. Magn.Reson. Imaging 1996, 14, 701−709.(37) Plimpton, S. Fast Parallel Algorithms for Short-RangeMolecular Dynamics. J. Comput. Phys. 1995, 117, 1−19.(38) Sandia National Labs. LAMMPS. http://lammps.sandia.gov,using July 30, 2016 release throughout this work.(39) Martin, M. G.; Siepmann, J. I. Transferable Potentials for PhaseEquilibria. 1. United-Atom Description ofn-Alkanes. J. Phys. Chem. B1998, 102, 2569−2577.(40) Dubbeldam, D.; Ford, D. C.; Ellis, D. E.; Snurr, R. Q.;Dubbeldam, D.; Ford, D.; Ellis, D.; Snurr, R. A new perspective on theorder-nalgorithm for computing correlation functions. Mol. Simul.2009, 35, 1084−1097.(41) Frenkel, D.; Smit, B. Understanding Molecular Simulation: FromAlgorithms to Applications; Elsevier, 2001; Vol. 1.(42) Simon, C. PEGrid. 2017, https://github.com/CorySimon/PEGrid (accessed December 1, 2017).(43) Childs, H.; Brugger, E.; Whitlock, B.; Meredith, J.; Ahern, S.;Pugmire, D.; Biagas, K.; Miller, M.; Harrison, C.; Weber, G. H.; et al.VisIt: An End-User Tool for Visualizing and Analyzing Very Large Data;Oak Ridge National Lab, 2012; pp 357−372.(44) Mace, A.; Barthel, S.; Smit, B. Automated multiscale approachto predict self-diffusion from a potential energy field. J. Chem. TheoryComput. 2019, 15, 2127−2141.(45) Bennett, C. H. Molecular dynamics and transition state theory:the simulation of infrequent events. ACS Symposium Series; AmericanChemical Society, 1977; Vol. 46, pp 63−97.(46) Chandler, D. Statistical mechanics of isomerization dynamics inliquids and the transition state approximation. J. Chem. Phys. 1978, 68,2959−2970.(47) Jackowski, K.; Jaszunski, M. Gas Phase NMR; Royal Society ofChemistry: Cambridge, U.K., 2016.(48) Gerritsma, C. J.; Oosting, P. H.; Trappeniers, N. J. Proton-spin-lattice relaxation and self-diffusion in methanes. Physica 1971, 51,381−394.(49) Kowalewski, J. Nuclear Spin Relaxation in Diamagnetic Fluids.Part 1. General Aspects and Inorganic Applications; Elsevier, 1990; Vol.22, pp 307−414.(50) McClung, R. Spin-rotation relaxation theory. Encyclopedia ofMagnetic Resonance; Wiley, 2007.(51) Stallmach, F.; Pusch, A.-K.; Splith, T.; Horch, C.; Merker, S.NMR relaxation and diffusion studies of methane and carbon dioxidein nanoporous ZIF-8 and ZSM-58. Microporous Mesoporous Mater.2015, 205, 36−39.(52) Bloom, M.; Lipsicas, M.; Muller, B. Proton spin-latticerelaxation in polyatomic gases. Can. J. Phys. 1961, 39, 1093.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.9b01733J. Phys. Chem. C 2019, 123, 12286−12295
12294

(53) Stapf, S.; Kimmich, R.; Seitter, R.-O. Proton and DeuteronField-Cycling NMR Relaxometry of Liquids in Porous Glasses:Evidence for Levy-Walk Statistics. Phys. Rev. Lett. 1995, 75, 2855.(54) Xiang, S.; He, Y.; Zhang, Z.; Wu, H.; Zhou, W.; Krishna, R.;Chen, B. Microporous metal−organic framework with potential forcarbon dioxide capture at ambient conditions. Nat. Commun. 2012, 3,954.
The Journal of Physical Chemistry C Article
DOI: 10.1021/acs.jpcc.9b01733J. Phys. Chem. C 2019, 123, 12286−12295
12295


















