
Coexistence of Takayasu’s arteritis with familial Mediterranean fever
4
Rheumatol Int (2012) 32:1675–1678 DOI 10.1007/s00296-011-1853-7 123 ORIGINAL ARTICLE Coexistence of Takayasu’s arteritis with familial Mediterranean fever Figen Yargucu Zihni · Melike Kalfa · PÂnar Talu Ocakç · Figen Tarhan · Mustafa Parildar · Gokhan Keser · Kenan Aksu Received: 3 November 2010 / Accepted: 18 February 2011 / Published online: 18 March 2011 © Springer-Verlag 2011 Abstract Familial Mediterranean fever (FMF) is the most common autoinXammatory disease characterized by recur- rent self-limited attacks of fever, accompanied with perito- nitis, pleuritis or arthritis. It is well known that FMF may coexist with vasculitic pathologies, especially with those involving small and medium vessels. Among the vasculitic pathologies reported to be associated with FMF, Henoch– Schönlein purpura and polyarteritis nodosa come the Wrst, possibly followed up by protracted febrile myalgia. How- ever, coexistence of FMF with any large vessel vasculitis has not been reported to date. Here, we present a case with FMF who later developed Takayasu arteritis, with a severe disease course, being resistant to corticosteroids and con- ventional immunosuppressive agents, and requiring inXix- imab treatment. Keywords Familial Mediterranean fever · Takayasu’s arteritis Introduction Familial Mediterranean fever (FMF) is the most common autoinXammatory disorder having an autosomal recessive inheritance pattern and mostly seen in people of Middle East and Mediterranean region, including Non-Ashkenazi Jews, Middle Eastern Arabs, Turks and Armenians. It is characterized by recurrent attacks of fever with peritonitis, pleuritis, arthritis, or erysipela-like skin lesions. During these attacks, an intense biological inXammatory syndrome, including elevated acute-phase markers and leucocytosis is observed. MEFV gene mutations, playing an important role in pathogenesis are strongly associated with FMF, and most of these mutations are located in exon 10 at the carboxy-ter- minal portion of the pyrin protein [1]. Coexistence of FMF with small and/or medium size vas- culitic pathologies, including Henoch–Schönlein purpura (HSP), polyarteritis nodosa (PAN) and protracted febrile myalgia is well known, and there is largely accepted belief that vasculitis may indeed be a characteristic feature of FMF [1–6]. However, to our knowledge, coexistence of any large vessel vasculitis with FMF has not been reported in literature so far. Hereby, we present an FMF patient who later developed TA, with a severe disease course, being resistant to corticosteroids and conventional immunosup- pressive agents, and requiring anti-TNF treatment. Case report A 22-year-old male patient was referred to our clinic in June 2003 with the complaints of malaise, vertigo and diz- ziness for the last few months and additional syncope occurring at the end of this period. His past history went back to 1990, when he had been diagnosed as FMF at the age of 9 years, because of recurrent abdominal pain and fever attacks. Colchicine treatment 1.5 mg daily had been started, with an excellent response and with no compliance problem. Years later, when FMF gene mutation analysis became available, this patient had also been shown to have F. Y. Zihni · M. Kalfa · P. T. Ocakç · F. Tarhan · G. Keser · K. Aksu (&) Division of Rheumatology, Department of Internal Medicine, Ege University School of Medicine, 80. Sk. No: 27/3, 35040 Bornova, Izmir, Turkey e-mail: [email protected]; [email protected] M. Parildar Department of Radiology, Ege University School of Medicine, Bornova, Izmir, Turkey
Transcript of Coexistence of Takayasu’s arteritis with familial Mediterranean fever

Rheumatol Int (2012) 32:1675–1678
DOI 10.1007/s00296-011-1853-7ORIGINAL ARTICLE
Coexistence of Takayasu’s arteritis with familial Mediterranean fever
Figen Yargucu Zihni · Melike Kalfa · PÂnar Talu Ocakç · Figen Tarhan · Mustafa Parildar · Gokhan Keser · Kenan Aksu
Received: 3 November 2010 / Accepted: 18 February 2011 / Published online: 18 March 2011© Springer-Verlag 2011
Abstract Familial Mediterranean fever (FMF) is the mostcommon autoinXammatory disease characterized by recur-rent self-limited attacks of fever, accompanied with perito-nitis, pleuritis or arthritis. It is well known that FMF maycoexist with vasculitic pathologies, especially with thoseinvolving small and medium vessels. Among the vasculiticpathologies reported to be associated with FMF, Henoch–Schönlein purpura and polyarteritis nodosa come the Wrst,possibly followed up by protracted febrile myalgia. How-ever, coexistence of FMF with any large vessel vasculitishas not been reported to date. Here, we present a case withFMF who later developed Takayasu arteritis, with a severedisease course, being resistant to corticosteroids and con-ventional immunosuppressive agents, and requiring inXix-imab treatment.
Keywords Familial Mediterranean fever · Takayasu’s arteritis
Introduction
Familial Mediterranean fever (FMF) is the most commonautoinXammatory disorder having an autosomal recessiveinheritance pattern and mostly seen in people of Middle
East and Mediterranean region, including Non-AshkenaziJews, Middle Eastern Arabs, Turks and Armenians. It ischaracterized by recurrent attacks of fever with peritonitis,pleuritis, arthritis, or erysipela-like skin lesions. Duringthese attacks, an intense biological inXammatory syndrome,including elevated acute-phase markers and leucocytosis isobserved. MEFV gene mutations, playing an important rolein pathogenesis are strongly associated with FMF, and mostof these mutations are located in exon 10 at the carboxy-ter-minal portion of the pyrin protein [1].
Coexistence of FMF with small and/or medium size vas-culitic pathologies, including Henoch–Schönlein purpura(HSP), polyarteritis nodosa (PAN) and protracted febrilemyalgia is well known, and there is largely accepted beliefthat vasculitis may indeed be a characteristic feature ofFMF [1–6]. However, to our knowledge, coexistence of anylarge vessel vasculitis with FMF has not been reported inliterature so far. Hereby, we present an FMF patient wholater developed TA, with a severe disease course, beingresistant to corticosteroids and conventional immunosup-pressive agents, and requiring anti-TNF treatment.
Case report
A 22-year-old male patient was referred to our clinic inJune 2003 with the complaints of malaise, vertigo and diz-ziness for the last few months and additional syncopeoccurring at the end of this period. His past history wentback to 1990, when he had been diagnosed as FMF at theage of 9 years, because of recurrent abdominal pain andfever attacks. Colchicine treatment 1.5 mg daily had beenstarted, with an excellent response and with no complianceproblem. Years later, when FMF gene mutation analysisbecame available, this patient had also been shown to have
F. Y. Zihni · M. Kalfa · P. T. Ocakç · F. Tarhan · G. Keser · K. Aksu (&)Division of Rheumatology, Department of Internal Medicine, Ege University School of Medicine, 80. Sk. No: 27/3, 35040 Bornova, Izmir, Turkeye-mail: [email protected]; [email protected]
M. ParildarDepartment of Radiology, Ege University School of Medicine, Bornova, Izmir, Turkey
123
1676 Rheumatol Int (2012) 32:1675–1678
compound heterozygous MEFV mutation (M694V/V726A). His sister also had the diagnosis of FMF. Hepointed out that since commence of colchicine treatment,the frequency and severity of FMF attacks reduced consid-erably, and he had been well until a few months ago, whenthe above-mentioned complaints, mainly vertigo, dizzinessand syncope occured.
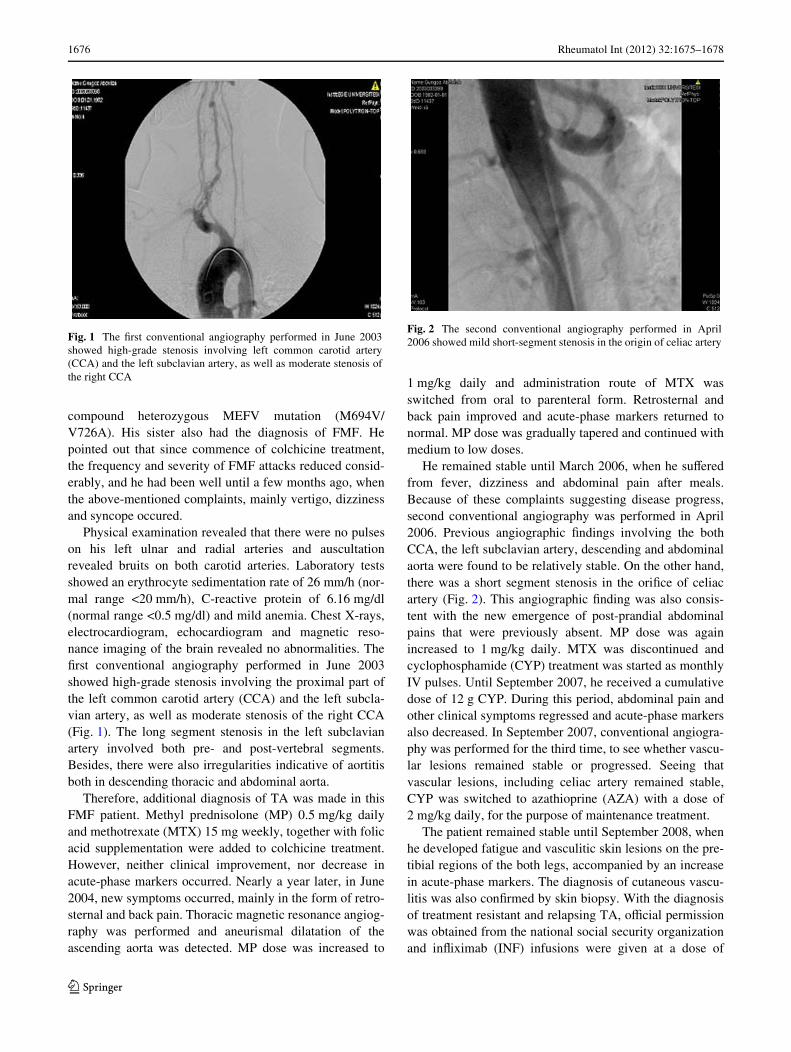
Physical examination revealed that there were no pulseson his left ulnar and radial arteries and auscultationrevealed bruits on both carotid arteries. Laboratory testsshowed an erythrocyte sedimentation rate of 26 mm/h (nor-mal range <20 mm/h), C-reactive protein of 6.16 mg/dl(normal range <0.5 mg/dl) and mild anemia. Chest X-rays,electrocardiogram, echocardiogram and magnetic reso-nance imaging of the brain revealed no abnormalities. TheWrst conventional angiography performed in June 2003showed high-grade stenosis involving the proximal part ofthe left common carotid artery (CCA) and the left subcla-vian artery, as well as moderate stenosis of the right CCA(Fig. 1). The long segment stenosis in the left subclavianartery involved both pre- and post-vertebral segments.Besides, there were also irregularities indicative of aortitisboth in descending thoracic and abdominal aorta.
Therefore, additional diagnosis of TA was made in thisFMF patient. Methyl prednisolone (MP) 0.5 mg/kg dailyand methotrexate (MTX) 15 mg weekly, together with folicacid supplementation were added to colchicine treatment.However, neither clinical improvement, nor decrease inacute-phase markers occurred. Nearly a year later, in June2004, new symptoms occurred, mainly in the form of retro-sternal and back pain. Thoracic magnetic resonance angiog-raphy was performed and aneurismal dilatation of theascending aorta was detected. MP dose was increased to
1 mg/kg daily and administration route of MTX wasswitched from oral to parenteral form. Retrosternal andback pain improved and acute-phase markers returned tonormal. MP dose was gradually tapered and continued withmedium to low doses.
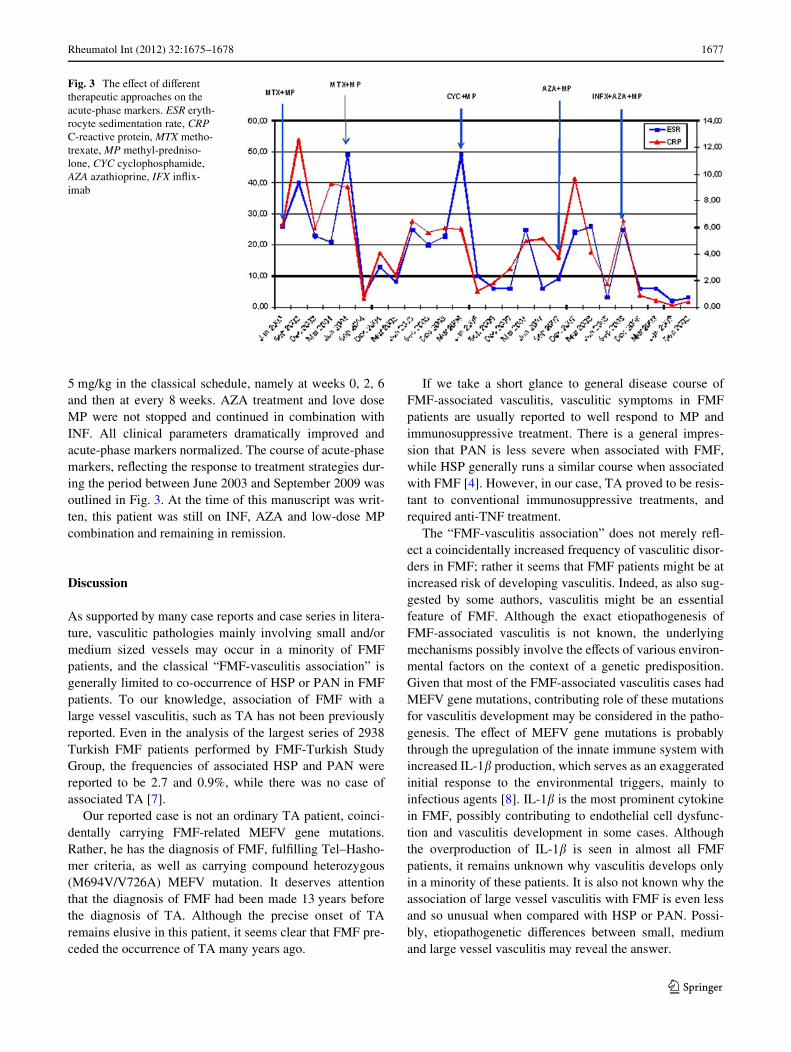
He remained stable until March 2006, when he suVeredfrom fever, dizziness and abdominal pain after meals.Because of these complaints suggesting disease progress,second conventional angiography was performed in April2006. Previous angiographic Wndings involving the bothCCA, the left subclavian artery, descending and abdominalaorta were found to be relatively stable. On the other hand,there was a short segment stenosis in the oriWce of celiacartery (Fig. 2). This angiographic Wnding was also consis-tent with the new emergence of post-prandial abdominalpains that were previously absent. MP dose was againincreased to 1 mg/kg daily. MTX was discontinued andcyclophosphamide (CYP) treatment was started as monthlyIV pulses. Until September 2007, he received a cumulativedose of 12 g CYP. During this period, abdominal pain andother clinical symptoms regressed and acute-phase markersalso decreased. In September 2007, conventional angiogra-phy was performed for the third time, to see whether vascu-lar lesions remained stable or progressed. Seeing thatvascular lesions, including celiac artery remained stable,CYP was switched to azathioprine (AZA) with a dose of2 mg/kg daily, for the purpose of maintenance treatment.
The patient remained stable until September 2008, whenhe developed fatigue and vasculitic skin lesions on the pre-tibial regions of the both legs, accompanied by an increasein acute-phase markers. The diagnosis of cutaneous vascu-litis was also conWrmed by skin biopsy. With the diagnosisof treatment resistant and relapsing TA, oYcial permissionwas obtained from the national social security organizationand inXiximab (INF) infusions were given at a dose of
Fig. 1 The Wrst conventional angiography performed in June 2003showed high-grade stenosis involving left common carotid artery(CCA) and the left subclavian artery, as well as moderate stenosis ofthe right CCA
Fig. 2 The second conventional angiography performed in April2006 showed mild short-segment stenosis in the origin of celiac artery
123
Rheumatol Int (2012) 32:1675–1678 1677
5 mg/kg in the classical schedule, namely at weeks 0, 2, 6and then at every 8 weeks. AZA treatment and love doseMP were not stopped and continued in combination withINF. All clinical parameters dramatically improved andacute-phase markers normalized. The course of acute-phasemarkers, reXecting the response to treatment strategies dur-ing the period between June 2003 and September 2009 wasoutlined in Fig. 3. At the time of this manuscript was writ-ten, this patient was still on INF, AZA and low-dose MPcombination and remaining in remission.
Discussion
As supported by many case reports and case series in litera-ture, vasculitic pathologies mainly involving small and/ormedium sized vessels may occur in a minority of FMFpatients, and the classical “FMF-vasculitis association” isgenerally limited to co-occurrence of HSP or PAN in FMFpatients. To our knowledge, association of FMF with alarge vessel vasculitis, such as TA has not been previouslyreported. Even in the analysis of the largest series of 2938Turkish FMF patients performed by FMF-Turkish StudyGroup, the frequencies of associated HSP and PAN werereported to be 2.7 and 0.9%, while there was no case ofassociated TA [7].
Our reported case is not an ordinary TA patient, coinci-dentally carrying FMF-related MEFV gene mutations.Rather, he has the diagnosis of FMF, fulWlling Tel–Hasho-mer criteria, as well as carrying compound heterozygous(M694V/V726A) MEFV mutation. It deserves attentionthat the diagnosis of FMF had been made 13 years beforethe diagnosis of TA. Although the precise onset of TAremains elusive in this patient, it seems clear that FMF pre-ceded the occurrence of TA many years ago.
If we take a short glance to general disease course ofFMF-associated vasculitis, vasculitic symptoms in FMFpatients are usually reported to well respond to MP andimmunosuppressive treatment. There is a general impres-sion that PAN is less severe when associated with FMF,while HSP generally runs a similar course when associatedwith FMF [4]. However, in our case, TA proved to be resis-tant to conventional immunosuppressive treatments, andrequired anti-TNF treatment.
The “FMF-vasculitis association” does not merely reX-ect a coincidentally increased frequency of vasculitic disor-ders in FMF; rather it seems that FMF patients might be atincreased risk of developing vasculitis. Indeed, as also sug-gested by some authors, vasculitis might be an essentialfeature of FMF. Although the exact etiopathogenesis ofFMF-associated vasculitis is not known, the underlyingmechanisms possibly involve the eVects of various environ-mental factors on the context of a genetic predisposition.Given that most of the FMF-associated vasculitis cases hadMEFV gene mutations, contributing role of these mutationsfor vasculitis development may be considered in the patho-genesis. The eVect of MEFV gene mutations is probablythrough the upregulation of the innate immune system withincreased IL-1� production, which serves as an exaggeratedinitial response to the environmental triggers, mainly toinfectious agents [8]. IL-1� is the most prominent cytokinein FMF, possibly contributing to endothelial cell dysfunc-tion and vasculitis development in some cases. Althoughthe overproduction of IL-1� is seen in almost all FMFpatients, it remains unknown why vasculitis develops onlyin a minority of these patients. It is also not known why theassociation of large vessel vasculitis with FMF is even lessand so unusual when compared with HSP or PAN. Possi-bly, etiopathogenetic diVerences between small, mediumand large vessel vasculitis may reveal the answer.
Fig. 3 The eVect of diVerent therapeutic approaches on the acute-phase markers. ESR eryth-rocyte sedimentation rate, CRP C-reactive protein, MTX metho-trexate, MP methyl-predniso-lone, CYC cyclophosphamide, AZA azathioprine, IFX inXix-imab
123

1678 Rheumatol Int (2012) 32:1675–1678
In conclusion, to our knowledge, this is the Wrst casereport of TA associated with FMF. Although this FMFpatient, later developing treatment-resistant TA, may sim-ply be an anecdotal case, in future new similar cases mayalso be reported. In a minority group of FMF patients, notonly small or medium vessel vasculitis may occur, but alsolarge vessel vasculitis, such as TA may complicate FMF.
References
1. Onen F (2006) Familial Mediterranean Fever. Rheumatol Int26:489–496
2. Ozcakar ZB, Yalcinkaya F, Cakar N et al (2008) MEFV mutationsmodify the clinical presentation of Henoch–Schönlein purpura.J Rheumatol 35:2427–2429
3. Ozdogan H, Arisoy N, Kasapçapur O et al (1997) Vasculitis infamilial Mediterranean fever. J Rheumatol 24:323–327
4. Hatemi G, Masatlioglu S, Gogus F, Ozdogan H (2004) Necrotizingvasculitis associated with familial Mediterranean fever. Am J Med117:516–519
5. Ozen S, Ben-Chetrit E, Bakkaloglu A et al (2001) Polyarteritis no-dosa in patients with familial Mediterranean fever (FMF): a con-comitant disease or a feature of FMF? Semin Arthritis Rheum30:281–287
6. Sidi G, Shinar Y, Livneh A et al (2000) Protracted febrile myalgiaof familial Mediterranean fever Mutation analysis and clinical cor-relations. Scand J Rheumatol 29:174–176
7. Tunca M, Akar S, Onen F et al (2005) Turkish FMF Study Group.Familial Mediterranean fever (FMF) in Turkey: results of a nation-wide multicenter study. Medicine (Baltimore) 84:1–11
8. Cattan D (2005) MEFV mutation carriers and diseases other thanfamilial Mediterranean fever: proved and non-proved associations;putative biological advantage. Current Drug Targets InXamm Aller-gy 4:105–112
123


















