
Coats-like retinopathy in an infant with preclinical facioscapulohumeral dystrophy
3
Coats-like retinopathy in an infant with preclinical facioscapulohumeral dystrophy Anuradha Ganesh, MD, a Swathi Kaliki, MD, b and Carol L. Shields, MD b Facioscapulohumeral dystrophy (FSHD) is an autosomal-dominant disorder characterized by weakness of the face, upper arm, shoul- der, and lower limb musculature, with an onset between the first and third decades. Coats disease is a congenital disorder of retinal vascular development characterized by unilateral peripheral retinal telangiectasia and progressive subretinal and intraretinal exudation. This condition has a predilection for children and is usually isolated. Retinal vascular changes similar to those seen in Coats disease have been demonstrated by fluorescein angiogra- phy in 40% to 75% of patients with FSHD. Most patients have asymptomatic retinal telangiectasia found at ocular screening in adulthood after diagnosis of FSHD. We report a 7-month-old infant with bilateral Coats-like retinopathy in which the eye disease was discovered before findings of FSHD were clinically evident. To our knowledge, this patient represents the youngest reported case of preclinical FSHD with ocular disease. Case Report A 7-month-old infant girl diagnosed with congenital bilateral moderate sensorineural hearing loss, detected on newborn screening, was referred for ophthalmic evaluation. She was the first born child of nonconsanguineous parents and was the product of a normal pregnancy, labor, and delivery. Medical history, developmental milestones, and family history were unre- markable. Ophthalmic examination revealed central, steady, and maintained fixation in both eyes. No strabismus or nystag- mus was noted, and pupillary responses were normal. Anterior segment examination was unremarkable in both eyes. Dilated fundus examination revealed macular exuda- tive retinopathy with shallow retinal detachment in the right eye and distinctly dilated and tortuous retinal vessels in both eyes (Figure 1). Peripheral retinal examination disclosed the presence of retinal telangiectasia in both eyes with “light bulb” microaneurysms in the right eye. Intravenous fluorescein angiography confirmed the retinal vascular abnormalities as well as 360 degrees of peripheral retinal nonperfusion in both eyes. There was no retinal mass in either eye. The child was diagnosed with Coats- like retinopathy in both eyes, stage 3 in the right eye and stage 1 in left eye. 1 The constellation of bilateral congenital hearing loss and bilateral Coats-like retinopathy suggested the possibility of underlying facioscapulohumeral dystrophy (FSHD; MIM 158900). 2 The findings of the neurological examination and electromyography were normal. Direct testing for FSHD deletion mutation on chromosome 4q35 with the use of southern blot analysis of the EcoRI-digested genomic DNA revealed a heterozygous deletion with an abnormal FSHD allele of 13 kb size (normal .42 kb), consistent with FSHD. Both parents tested normal, implying de novo mutation in the infant. Laser photocoagulation and cryotherapy were delivered to the areas of leakage and nonperfusion in both eyes at the age of 8 months and 12 months, respectively. At age 16 months, 4 intravitreal bevacizumab injections of 1.25 mg/0.05 mL each were administered in the right eye at 6-month intervals to treat macular edema. By 2 years of age, there was complete resolution of exudative retinop- athy (Figure 2), and at 5 years of age, visual acuity was recorded at 20/30 in the right eye and 20/20 in the left eye with stable fundus findings. Discussion Coats disease is typically unilateral in 95% of patients, and 76% of patients are males. 1 The earliest changes involve the equatorial and peripheral retina, and the condition progresses through 5 stages from retinal telangiectasia alone (stage 1) to end-stage phthisis bulbi (stage 5). 1 Bilateral presentation of Coats-like retinopathy suggests the presence of an underlying systemic or hereditary process such as FSHD, incontinentia pigmenti, CRB1- associated retinal dystrophy, or familial exudative vitreore- tinopathy. 2-6 The presence of bilateral Coats-like retinopathy in conjunction with congenital deafness in our patient led to the suspicion for and subsequent genetic confirmation of FSHD, despite lack of lack of abnormalities on neurological examination and electro- myography. Typically, individuals with FSHD become symptomatic with facial, upper arm, and shoulder muscle weakness in the second to fourth decades of life; this leads to genetic and subsequent hearing and ophthalmic testing. Rarely, Coats-like retinopathy is the presenting feature of Author affiliations: a Pediatric Ophthalmology and Ocular Genetics Services and b Ocular Oncology Service, Wills Eye Institute, Thomas Jefferson University, Philadelphia, Pennsylvania Support provided by the Eye Tumor Research Foundation, Philadelphia, PA (CLS). The funders had no role in the design and conduct of the study, in the collection, analysis, and interpretation of the data, and in the preparation, review or approval of the manuscript. Submitted August 18, 2011. Revision accepted November 14, 2011. Correspondence: Carol L. Shields, MD, Ocular Oncology Service, Suite 1440, Wills Eye Institute, 840 Walnut Street, Philadelphia, PA 19107 (email: carol.shields@shieldsoncology. com). J AAPOS 2012;16:204-206. Copyright Ó 2012 by the American Association for Pediatric Ophthalmology and Strabismus. 1091-8531/$36.00 doi:10.1016/j.jaapos.2011.11.005 204 Journal of AAPOS
-
Upload
anuradha-ganesh -
Category
Documents
-
view
213 -
download
0
Transcript of Coats-like retinopathy in an infant with preclinical facioscapulohumeral dystrophy

Coats-like retinopathy in an infant with preclinicalfacioscapulohumeral dystrophyAnuradha Ganesh, MD,a Swathi Kaliki, MD,b and Carol L. Shields, MDb
Facioscapulohumeral dystrophy (FSHD) is an autosomal-dominantdisorder characterized by weakness of the face, upper arm, shoul-der, and lower limb musculature, with an onset between the firstand third decades. Coats disease is a congenital disorder of retinalvascular development characterized by unilateral peripheral retinaltelangiectasia and progressive subretinal and intraretinalexudation. This condition has a predilection for children and isusually isolated. Retinal vascular changes similar to those seenin Coats disease have been demonstrated by fluorescein angiogra-phy in 40% to 75% of patients with FSHD. Most patients haveasymptomatic retinal telangiectasia found at ocular screening inadulthood after diagnosis of FSHD. We report a 7-month-old infantwith bilateral Coats-like retinopathy in which the eye disease wasdiscovered before findings of FSHD were clinically evident. To ourknowledge, this patient represents the youngest reported case ofpreclinical FSHD with ocular disease.
Case Report
A7-month-old infant girl diagnosed with congenitalbilateral moderate sensorineural hearing loss,detected on newborn screening, was referred for
ophthalmic evaluation. She was the first born child ofnonconsanguineous parents and was the product of anormal pregnancy, labor, and delivery. Medical history,developmental milestones, and family history were unre-markable.
Ophthalmic examination revealed central, steady, andmaintained fixation in both eyes. No strabismus or nystag-mus was noted, and pupillary responses were normal.Anterior segment examination was unremarkable in botheyes. Dilated fundus examination revealed macular exuda-tive retinopathy with shallow retinal detachment in theright eye and distinctly dilated and tortuous retinal vesselsin both eyes (Figure 1). Peripheral retinal examinationdisclosed the presence of retinal telangiectasia in botheyes with “light bulb” microaneurysms in the right eye.
Author affiliations: aPediatric Ophthalmology and Ocular Genetics Services and bOcularOncology Service, Wills Eye Institute, Thomas Jefferson University, Philadelphia,PennsylvaniaSupport provided by the Eye Tumor Research Foundation, Philadelphia, PA (CLS). The
funders had no role in the design and conduct of the study, in the collection, analysis, andinterpretation of the data, and in the preparation, review or approval of the manuscript.Submitted August 18, 2011.Revision accepted November 14, 2011.Correspondence: Carol L. Shields, MD, Ocular Oncology Service, Suite 1440, Wills Eye
Institute, 840Walnut Street, Philadelphia, PA 19107 (email: [email protected]).J AAPOS 2012;16:204-206.Copyright � 2012 by the American Association for Pediatric Ophthalmology and
Strabismus.1091-8531/$36.00doi:10.1016/j.jaapos.2011.11.005
204
Intravenous fluorescein angiography confirmed the retinalvascular abnormalities as well as 360 degrees of peripheralretinal nonperfusion in both eyes. There was no retinalmass in either eye. The child was diagnosed with Coats-like retinopathy in both eyes, stage 3 in the right eye andstage 1 in left eye.1
The constellation of bilateral congenital hearing loss andbilateral Coats-like retinopathy suggested the possibility ofunderlying facioscapulohumeral dystrophy (FSHD; MIM158900).2 The findings of the neurological examinationand electromyography were normal. Direct testing forFSHD deletion mutation on chromosome 4q35 with theuse of southern blot analysis of the EcoRI-digested genomicDNA revealed a heterozygous deletion with an abnormalFSHD allele of 13 kb size (normal .42 kb), consistentwith FSHD. Both parents tested normal, implying denovo mutation in the infant.
Laser photocoagulation and cryotherapy were deliveredto the areas of leakage and nonperfusion in both eyes at theage of 8 months and 12 months, respectively. At age16 months, 4 intravitreal bevacizumab injections of1.25 mg/0.05 mL each were administered in the right eyeat 6-month intervals to treat macular edema. By 2 yearsof age, there was complete resolution of exudative retinop-athy (Figure 2), and at 5 years of age, visual acuity wasrecorded at 20/30 in the right eye and 20/20 in the lefteye with stable fundus findings.
Discussion
Coats disease is typically unilateral in 95% of patients, and76% of patients are males.1 The earliest changes involvethe equatorial and peripheral retina, and the conditionprogresses through 5 stages from retinal telangiectasiaalone (stage 1) to end-stage phthisis bulbi (stage 5).1
Bilateral presentation of Coats-like retinopathy suggeststhe presence of an underlying systemic or hereditaryprocess such as FSHD, incontinentia pigmenti, CRB1-associated retinal dystrophy, or familial exudative vitreore-tinopathy.2-6 The presence of bilateral Coats-likeretinopathy in conjunction with congenital deafnessin our patient led to the suspicion for and subsequentgenetic confirmation of FSHD, despite lack of lack ofabnormalities on neurological examination and electro-myography.
Typically, individuals with FSHD become symptomaticwith facial, upper arm, and shoulder muscle weakness inthe second to fourth decades of life; this leads to geneticand subsequent hearing and ophthalmic testing. Rarely,Coats-like retinopathy is the presenting feature of
Journal of AAPOS
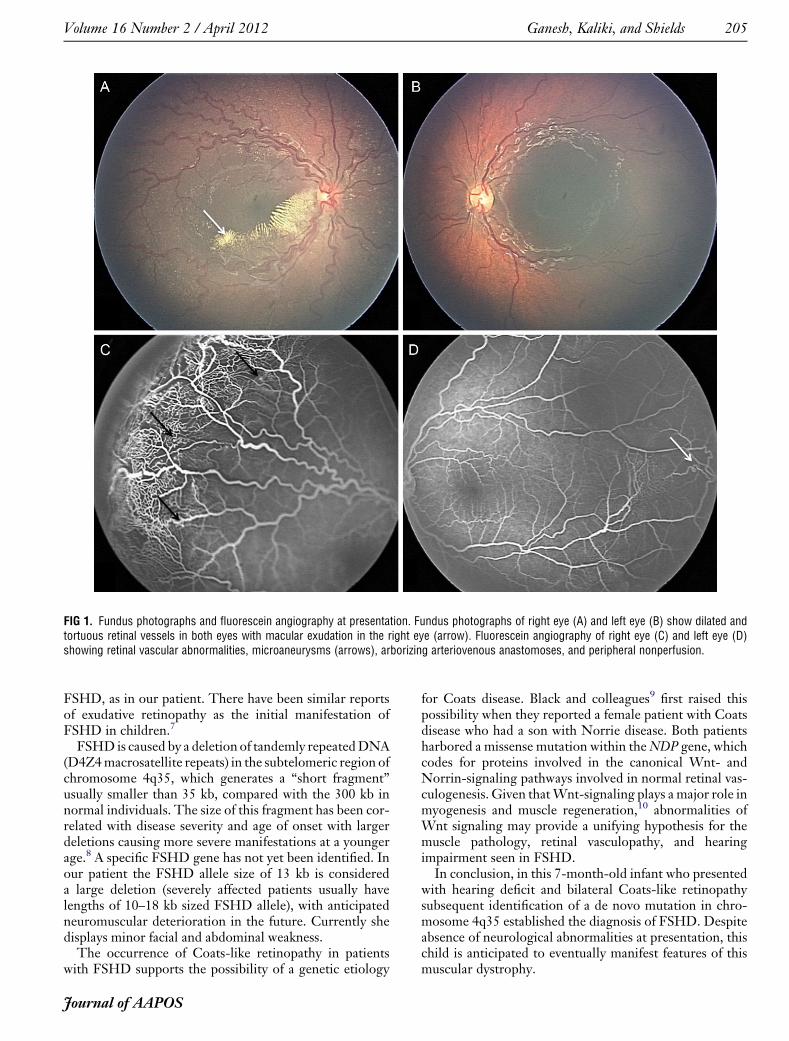
FIG 1. Fundus photographs and fluorescein angiography at presentation. Fundus photographs of right eye (A) and left eye (B) show dilated andtortuous retinal vessels in both eyes with macular exudation in the right eye (arrow). Fluorescein angiography of right eye (C) and left eye (D)showing retinal vascular abnormalities, microaneurysms (arrows), arborizing arteriovenous anastomoses, and peripheral nonperfusion.
Volume 16 Number 2 / April 2012 Ganesh, Kaliki, and Shields 205
FSHD, as in our patient. There have been similar reportsof exudative retinopathy as the initial manifestation ofFSHD in children.7
FSHD is caused by a deletion of tandemly repeatedDNA(D4Z4macrosatellite repeats) in the subtelomeric region ofchromosome 4q35, which generates a “short fragment”usually smaller than 35 kb, compared with the 300 kb innormal individuals. The size of this fragment has been cor-related with disease severity and age of onset with largerdeletions causing more severe manifestations at a youngerage.8 A specific FSHD gene has not yet been identified. Inour patient the FSHD allele size of 13 kb is considereda large deletion (severely affected patients usually havelengths of 10–18 kb sized FSHD allele), with anticipatedneuromuscular deterioration in the future. Currently shedisplays minor facial and abdominal weakness.The occurrence of Coats-like retinopathy in patients
with FSHD supports the possibility of a genetic etiology
Journal of AAPOS
for Coats disease. Black and colleagues9 first raised thispossibility when they reported a female patient with Coatsdisease who had a son with Norrie disease. Both patientsharbored a missense mutation within theNDP gene, whichcodes for proteins involved in the canonical Wnt- andNorrin-signaling pathways involved in normal retinal vas-culogenesis. Given thatWnt-signaling plays a major role inmyogenesis and muscle regeneration,10 abnormalities ofWnt signaling may provide a unifying hypothesis for themuscle pathology, retinal vasculopathy, and hearingimpairment seen in FSHD.
In conclusion, in this 7-month-old infant who presentedwith hearing deficit and bilateral Coats-like retinopathysubsequent identification of a de novo mutation in chro-mosome 4q35 established the diagnosis of FSHD. Despiteabsence of neurological abnormalities at presentation, thischild is anticipated to eventually manifest features of thismuscular dystrophy.
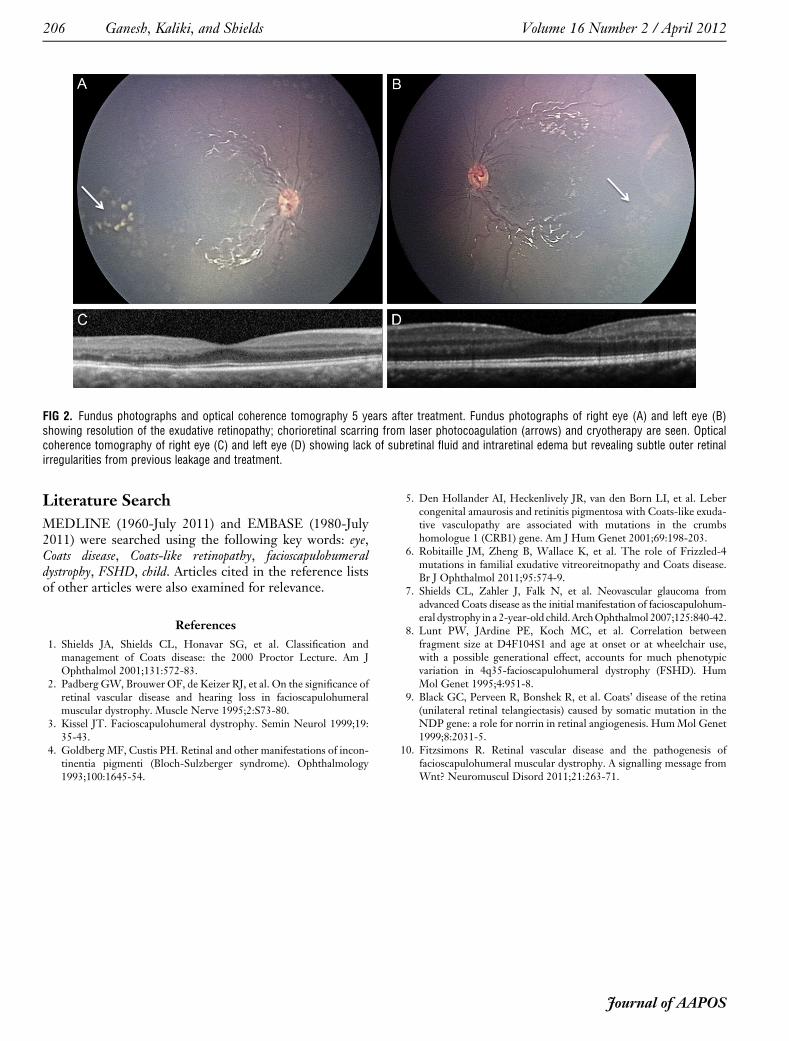
FIG 2. Fundus photographs and optical coherence tomography 5 years after treatment. Fundus photographs of right eye (A) and left eye (B)showing resolution of the exudative retinopathy; chorioretinal scarring from laser photocoagulation (arrows) and cryotherapy are seen. Opticalcoherence tomography of right eye (C) and left eye (D) showing lack of subretinal fluid and intraretinal edema but revealing subtle outer retinalirregularities from previous leakage and treatment.
206 Ganesh, Kaliki, and Shields Volume 16 Number 2 / April 2012
Literature Search
MEDLINE (1960-July 2011) and EMBASE (1980-July2011) were searched using the following key words: eye,Coats disease, Coats-like retinopathy, facioscapulohumeraldystrophy, FSHD, child. Articles cited in the reference listsof other articles were also examined for relevance.
References
1. Shields JA, Shields CL, Honavar SG, et al. Classification andmanagement of Coats disease: the 2000 Proctor Lecture. Am JOphthalmol 2001;131:572-83.
2. Padberg GW, Brouwer OF, de Keizer RJ, et al. On the significance ofretinal vascular disease and hearing loss in facioscapulohumeralmuscular dystrophy. Muscle Nerve 1995;2:S73-80.
3. Kissel JT. Facioscapulohumeral dystrophy. Semin Neurol 1999;19:35-43.
4. Goldberg MF, Custis PH. Retinal and other manifestations of incon-tinentia pigmenti (Bloch-Sulzberger syndrome). Ophthalmology1993;100:1645-54.
5. Den Hollander AI, Heckenlively JR, van den Born LI, et al. Lebercongenital amaurosis and retinitis pigmentosa with Coats-like exuda-tive vasculopathy are associated with mutations in the crumbshomologue 1 (CRB1) gene. Am J Hum Genet 2001;69:198-203.
6. Robitaille JM, Zheng B, Wallace K, et al. The role of Frizzled-4mutations in familial exudative vitreoreitnopathy and Coats disease.Br J Ophthalmol 2011;95:574-9.
7. Shields CL, Zahler J, Falk N, et al. Neovascular glaucoma fromadvanced Coats disease as the initial manifestation of facioscapulohum-eral dystrophy in a 2-year-old child.ArchOphthalmol 2007;125:840-42.
8. Lunt PW, JArdine PE, Koch MC, et al. Correlation betweenfragment size at D4F104S1 and age at onset or at wheelchair use,with a possible generational effect, accounts for much phenotypicvariation in 4q35-facioscapulohumeral dystrophy (FSHD). HumMol Genet 1995;4:951-8.
9. Black GC, Perveen R, Bonshek R, et al. Coats’ disease of the retina(unilateral retinal telangiectasis) caused by somatic mutation in theNDP gene: a role for norrin in retinal angiogenesis. HumMol Genet1999;8:2031-5.
10. Fitzsimons R. Retinal vascular disease and the pathogenesis offacioscapulohumeral muscular dystrophy. A signalling message fromWnt? Neuromuscul Disord 2011;21:263-71.
Journal of AAPOS


















