
Cloning and mutagenesis of the Rhizobium meliloti isocitrate ...
8
Vol. 174, No. 14 JOURNAL OF BACTERIOLOGY, July 1992, p. 47904797 0021-9193/92/144790-08$02.00/0 Copyright X 1992, American Society for Microbiology Cloning and Mutagenesis of the Rhizobium meliloti Isocitrate Dehydrogenase Gene TIMOTHY R. McDERMOTT' AND MICHAEL L. KAHN1,2* Institute of Biological Chemistry' and Department of Microbiology,2 Washington State University, Pullman, Washington 99164-6340 Received 18 February 1992/Accepted 21 April 1992 The gene encoding Rhizobium meliloti isocitrate dehydrogenase (ICD) was cloned by complementation of an Escherichia coli icd mutant with an R. meliloti genomic library constructed in pUC18. The complementing DNA was located on a 4.4-kb BamHI fragment. It encoded an ICD that had the same mobility as R. meliloti ICD in nondenaturing polyacrylamide gels. In Western immunoblot analysis, antibodies raised against this protein reacted with R. meliloti ICD but not with E. coli ICD. The complementing DNA fragment was mutated with transposon TnS and then exchanged for the wild-type allele by recombination by a novel method that employed the Bacillus subtilis levansucrase gene. No ICD activity was found in the two R. meliloti icd::Tn5 mutants isolated, and the mutants were also found to be glutamate auxotrophs. The mutants formed nodules, but they were completely ineffective. Faster-growing pseudorevertants were isolated from cultures of both R. melloti icd::Tn5 mutants. In addition to lacking all ICD activity, the pseudorevertants also lacked citrate synthase activity. Nodule formation by these mutants was severely affected, and inoculated plants had only callus structures or small spherical structures. In the symbiosis between legumes and rhizobia, the host plant provides the bacteria with reduced carbon as an energy source. The bacteria use this energy to reduce atmospheric nitrogen to ammonia, which they release to the plant. The qualitative nature of this energy source has been the focus of much research. Sucrose is the major photosynthate trans- ported from the shoot to the nodule (41), but biochemical evidence suggests that neither sucrose nor hexoses obtained from sucrose degradation are important sources of energy for the bacteroids (reviewed in references 7 and 34). In support of this conclusion, mutants of various species of rhizobia with defects in sugar metabolism have been found to be effective in symbiosis (7, 34). By contrast, dicarboxylic acids appear to be important carbon sources in the estab- lishment of an effective symbiosis. Succinate and malate are found at high concentrations in nodules (14, 45, 55), are actively transported across the peribacteroid membrane (20, 56), are taken up by bacteroids (11, 20, 42, 50), and are quickly oxidized to CO2 after uptake (47). Dicarboxylic acid transport (dct) mutants of Rhizobium meliloti (5) and R. leguminosarum biovars viciae (3, 12) and trifolii (44) are all Fix-. Succinate and malate are intermediates in the tricarboxy- lic acid (TCA) cycle, and thus the TCA cycle is implicated as a major catabolic sequence. Enzymes of the TCA cycle, such as citrate synthase (CS) (31), isocitrate dehydrogenase (ICD) (22, 28, 31), fumarase (28), malate dehydrogenase (22, 28), and a-ketoglutarate dehydrogenase (48), have been found in bacteroids of various species of Rhizobium and Bradyrhizobium. Radioactive metabolite conversion studies by Stovall and Cole (54) implied a fully functional TCA cycle, but evidence for the decarboxylating leg of the TCA cycle was not obtained until Salminen and Streeter (47) reported that significant amounts of label from [2,3-14C]suc- cinate accumulated in glutamate, indicating that in soybean bacteroids the TCA cycle is complete, at least to oa-ketoglu- * Corresponding author. tarate. Although no data were reported, an R meliloti ax-ketoglutarate dehydrogenase mutant was said to be Fix- (10). We are interested in how the bacteroid TCA cycle is regulated and have selected ICD for our initial studies because it is a regulated enzyme at a branch point in the TCA cycle (15), because it is regulated by aerobiosis in other gram-negative bacteria (23, 24), and because it is a relatively simple enzyme with a single type of subunit that, in other bacteria, is encoded by a single gene (2). In this study, we report the isolation of the gene that encodes ICD in R meliloti and the symbiotic properties of mutants with defects in this gene. MATERIALS AND METHODS Bacterial strains and plasmids. The strains of R. meliloti and Escherichia coli, phage, and plasmids used in this study are shown in Table 1. R meliloti was grown on yeast extract-mannitol medium (YMB [53]) or on minimal manni- tol medium (53) supplemented with arabinose (5 g/liter), glutamate (1.1 g/liter), and filter-sterilized antibiotics as indicated. Strains of E. coli were grown on either LB or M9 mineral salts (49) medium. M9 medium contained glucose as the carbon source and ammonium chloride as the primary nitrogen source plus histidine (15 ,ug/ml), tryptophan (40 p,g/ml), and thiamine (2 ,ug/ml). Filter-sterilized antibiotics and glutamate were added as needed. For long-term storage, strains of R. meliloti and E. coli were grown to mid-log phase in appropriate selective medium, diluted with an equal volume of sterile 50% glycerol, and stored at -70°C. Cloning of R. meliloti icd and isolation of mutants. Con- struction of the R meliloti gene bank and subsequent DNA manipulations followed the protocols of Sambrook et al. (49). Aliquots of chromosomal DNA were partially digested with restriction enzyme Sau3A, and the pooled digests were fractionated by sucrose gradient centrifugation. The 4- to 8-kb fragments were then ligated to pUC18 (60) that had been digested with BamHI and calf intestinal alkaline phos- 4790
Transcript of Cloning and mutagenesis of the Rhizobium meliloti isocitrate ...

Vol. 174, No. 14JOURNAL OF BACTERIOLOGY, July 1992, p. 479047970021-9193/92/144790-08$02.00/0Copyright X 1992, American Society for Microbiology
Cloning and Mutagenesis of the Rhizobium meliloti IsocitrateDehydrogenase Gene
TIMOTHY R. McDERMOTT' AND MICHAEL L. KAHN1,2*Institute ofBiological Chemistry' and Department ofMicrobiology,2 Washington State University,
Pullman, Washington 99164-6340
Received 18 February 1992/Accepted 21 April 1992
The gene encoding Rhizobium meliloti isocitrate dehydrogenase (ICD) was cloned by complementation of anEscherichia coli icd mutant with an R. meliloti genomic library constructed in pUC18. The complementing DNAwas located on a 4.4-kb BamHI fragment. It encoded an ICD that had the same mobility as R. meliloti ICD innondenaturing polyacrylamide gels. In Western immunoblot analysis, antibodies raised against this proteinreacted with R. meliloti ICD but not with E. coli ICD. The complementing DNA fragment was mutated withtransposon TnS and then exchanged for the wild-type allele by recombination by a novel method that employedthe Bacillus subtilis levansucrase gene. No ICD activity was found in the two R. meliloti icd::Tn5 mutantsisolated, and the mutants were also found to be glutamate auxotrophs. The mutants formed nodules, but theywere completely ineffective. Faster-growing pseudorevertants were isolated from cultures of both R. mellotiicd::Tn5 mutants. In addition to lacking all ICD activity, the pseudorevertants also lacked citrate synthaseactivity. Nodule formation by these mutants was severely affected, and inoculated plants had only callusstructures or small spherical structures.
In the symbiosis between legumes and rhizobia, the hostplant provides the bacteria with reduced carbon as an energysource. The bacteria use this energy to reduce atmosphericnitrogen to ammonia, which they release to the plant. Thequalitative nature of this energy source has been the focus ofmuch research. Sucrose is the major photosynthate trans-ported from the shoot to the nodule (41), but biochemicalevidence suggests that neither sucrose nor hexoses obtainedfrom sucrose degradation are important sources of energyfor the bacteroids (reviewed in references 7 and 34). Insupport of this conclusion, mutants of various species ofrhizobia with defects in sugar metabolism have been foundto be effective in symbiosis (7, 34). By contrast, dicarboxylicacids appear to be important carbon sources in the estab-lishment of an effective symbiosis. Succinate and malate arefound at high concentrations in nodules (14, 45, 55), areactively transported across the peribacteroid membrane (20,56), are taken up by bacteroids (11, 20, 42, 50), and arequickly oxidized to CO2 after uptake (47). Dicarboxylic acidtransport (dct) mutants of Rhizobium meliloti (5) and R.leguminosarum biovars viciae (3, 12) and trifolii (44) are allFix-.
Succinate and malate are intermediates in the tricarboxy-lic acid (TCA) cycle, and thus the TCA cycle is implicated asa major catabolic sequence. Enzymes of the TCA cycle,such as citrate synthase (CS) (31), isocitrate dehydrogenase(ICD) (22, 28, 31), fumarase (28), malate dehydrogenase (22,28), and a-ketoglutarate dehydrogenase (48), have beenfound in bacteroids of various species of Rhizobium andBradyrhizobium. Radioactive metabolite conversion studiesby Stovall and Cole (54) implied a fully functional TCAcycle, but evidence for the decarboxylating leg of the TCAcycle was not obtained until Salminen and Streeter (47)reported that significant amounts of label from [2,3-14C]suc-cinate accumulated in glutamate, indicating that in soybeanbacteroids the TCA cycle is complete, at least to oa-ketoglu-
* Corresponding author.
tarate. Although no data were reported, an R melilotiax-ketoglutarate dehydrogenase mutant was said to be Fix-(10).We are interested in how the bacteroid TCA cycle is
regulated and have selected ICD for our initial studiesbecause it is a regulated enzyme at a branch point in the TCAcycle (15), because it is regulated by aerobiosis in othergram-negative bacteria (23, 24), and because it is a relativelysimple enzyme with a single type of subunit that, in otherbacteria, is encoded by a single gene (2). In this study, wereport the isolation of the gene that encodes ICD in Rmeliloti and the symbiotic properties of mutants with defectsin this gene.
MATERIALS AND METHODS
Bacterial strains and plasmids. The strains of R. melilotiand Escherichia coli, phage, and plasmids used in this studyare shown in Table 1. R meliloti was grown on yeastextract-mannitol medium (YMB [53]) or on minimal manni-tol medium (53) supplemented with arabinose (5 g/liter),glutamate (1.1 g/liter), and filter-sterilized antibiotics asindicated. Strains of E. coli were grown on either LB or M9mineral salts (49) medium. M9 medium contained glucose asthe carbon source and ammonium chloride as the primarynitrogen source plus histidine (15 ,ug/ml), tryptophan (40p,g/ml), and thiamine (2 ,ug/ml). Filter-sterilized antibioticsand glutamate were added as needed. For long-term storage,strains ofR. meliloti and E. coli were grown to mid-log phasein appropriate selective medium, diluted with an equalvolume of sterile 50% glycerol, and stored at -70°C.
Cloning of R. meliloti icd and isolation of mutants. Con-struction of theR meliloti gene bank and subsequent DNAmanipulations followed the protocols of Sambrook et al.(49). Aliquots of chromosomal DNA were partially digestedwith restriction enzyme Sau3A, and the pooled digests werefractionated by sucrose gradient centrifugation. The 4- to8-kb fragments were then ligated to pUC18 (60) that hadbeen digested with BamHI and calf intestinal alkaline phos-
4790

R MELILOTI ISOCITRATE DEHYDROGENASE GENE 4791
TABLE 1. Strains of E. coli and R melioti, phage, and plasmids used
Strain, phage, Relevant genotype or characteristics Relevant phenotype" Reference or sourceor plasmid
RI meliloti104A14 Wild type 53A39S 104A14::TnSicd ICD- Glut- Kanr This studyB49S 104A14::TnSicd ICD- Glut- Kanr This studyA39L 104A14::TnSicd gItA (?) ICD- Glut- Kanr CS- This studyB49L 104A14::TnSicdgkA (?) ICD- Glut- Kan' CS This study
E. coliS17-1 Pro- Mob' 52DL39 ilvE12 tyrB507 aspC13 Sup' 4DEK2038 F- A- his-4 thi-1 rspL31 lacBKI rfiB recA ICD- Glut- Peter Thorsness
aceKi icd-11RR1 supE44 hsdS20 ara-14 proA2 lacYI galK2 High-efficiency transformation 49
rpsL20 xyl-5 mtl-l
Phage X467 TnS Kanr 8
PlasmidspUC18 Penr 60pTK509 pBR322 with E. coli icd Penr Peter ThorsnesspTM1 pUC18 withR meliloti icd Pen' This studypRK311 tetA Tetr 9pUM24 nptI sacB sacR Kanr Suc, 43pMK413 tetA sacB sacR Tetr Suc, This studypTM2 pMK413 with R. meliloti icd Tetr Suc, This studypTM3 pRK311 with R melioti icd Tetr This studypTM2::TnSA39 icd::TnS mutant of pTM2 Suc5 Tetr Kanr This studypTM2::Tn5B49 icd::TnS mutant of pTM2 Suc5 Tetr Kanr This studyI Glut, glutamate; Pro, proline; Suc, sucrose.
phatase (Boehringer). Transformation of E. coli RR1 yieldedapproxmately 10,000 independent transformants that con-tained inserts. Plasmid DNA was purified from these colo-nies and used as the gene bank in the initial selection. Fortransposon TnS mutagenesis, we used X467::TnS as outlinedby de Bruijn and Lupski (8).A mutated allele is often recombined into R meliloti by
first using a broad-host-range plasmid to bring the allele intothe strain and then using a second plasmid of the sameincompatibility group to destabilize the first plasmid (46). Bycontinuing to select for the mutated allele, the desiredrecombinant can be recovered, since it will stably maintainthe selected marker. Although the technique works in Rmeliloti 104A14, plasmid incompatibility is not very strong,and this procedure can be tedious, especially if the mutantgrows more slowly than the wild type (26). An alternativemethod was developed in which a vector that allows directselection is used to obtain cells that have lost the plasmidcarrying the mutated allele. pMK413 (Fig. 1) contains theBacillus subtilis levansucrase genes, which inhibit thegrowth of gram-negative bacteria when they are incubated inmedia that contain sucrose (17, 21). To construct pMK413,the 1.7-kb cos fragment of pRK311 (9) was replaced by the3.8-kb BamHI fragment from pUM24 (43), which containsthe B. subtilis sacR and sacB genes, which encode levansu-crase, and the nptl gene, which confers kanamycin resis-tance (KanT). The nptI gene was subsequently deleted byusing PstI. E. coli DEK2038(pMK413) and R meliloti104A14(pMK413) do not grow on media that contain tetra-cycline (to select for plasmid retention) and 5% sucrose.Enzyme assays. (i) ICD. ICD activity in cell extracts was
measured spectrophotometrically. The rate of NADP+ re-duction was measured at 340 nm. Assay mixes contained 0.5
mM DL-isocitrate, 0.4 mM NADP+, 3.3 mM MgCl2, 10 mMpotassium phosphate buffer (pH 7.4), and enzyme extract ina final reaction volume of 1.0 ml.
(ii) CS. CS was assayed by measuring the formation ofcoenzyme A-sulfhydryl by the increase in A412 as describedby Sere (51). Assay mixes contained 200 mM Tris (pH 8.1),
EOR p_iEcoRI/ ~~~~onV ~% Hindlll
sac Ps
pMK413 SetALSmal
23kb
\_oriT/ESolla a2SaiHlPstIHindlilFIG. 1. Restriction map of pMK413. The cos fragment was
deleted from pRK311 (9) and replaced with the sac cassette frompUM24 (49) as described in Materials and Methods. Arrow indicatesorientation of lacZ relative to the polylinker site.
VOL. 174, 1992

4792 McDERMOTT AND KAHN
0.2 mM 5,5'-dithiobis(2-nitrobenzoic acid) (DTNB), 0.1 mMacetyl-S-CoA, 0.5 mM potassium oxaloacetate, and enzymeextract in a final volume of 1.0 ml. DTNB and acetyl-S-CoAwere prepared fresh daily in 0.5 M potassium phosphatebuffer.
Activity stains in nondenaturing polyacrylamide gels. Theprotocol of Kittell et al. (29) was modified to include stackingand resolving phases in the separation system. Nondenatur-ing (native) gels contained 10% acrylamide (9.7% acryl-amide, 0.3% bisacrylamide) in the resolving phase and 5%acrylamide (4.85% acrylamide, 0.15% bisacrylamide) in thestacking phase. Gels were made up in 0.25 M Tris (pH 8.3)and polymerized by using ammonium persulfate andTEMED (N,N,N',N'-tetramethylethylenediamine). Gelswere prerun in 0.375 M Tris (pH 8.3) for 2 h and then, justprior to sample loading, the prerun buffer was replaced by arunning buffer containing 25 mM Tris base and 190 mMglycine, pH 8.5. Cell extracts were mixed 3:1 (vol/vol) withsample buffer (2 mg of xylene cyanol diluted in 5 ml of 40%glycerol), loaded into the gel, and electrophoresed for ap-proximately 1.5 h at 60 mA constant current. Gels wereremoved from the glass plates and submerged in a stainingsolution containing 180 mM Tris (pH 7.9), 0.1 mM NADP+,15 mM MgCl2, 8 mM sodium isocitrate, 0.4 mM 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide,and 0.5 mM phenazine methosulfate (1). Depending on thespecific activity of ICD in the samples, activity bands wereseen within 1 to 20 min.
Protein purification and antibody production. For purifica-tion ofR meliloti ICD, we modified the purification methodof Nambiar and Shethna (36). DEK2038(pTM1) was inocu-lated into 18 liters of LB-penicillin (200 mg/liter) broth,grown to late log phase, and then harvested and washed byfiltration and centrifugation. Cells resuspended in breakingbuffer (25 mM Tris, 3 mM dithiothreitol [pH 7.5]) were lysedby sonication and cleared by centrifugation at 100,000 x gfor 1 h. Protamine sulfate was added to the supernatant(1:10, protamine sulfate to total protein), and after 1 h theprecipitate was removed by centrifugation. The supernatantwas brought to 45% (wt/vol) ammonium sulfate and centri-fuged. The supematant was then brought to 70% (wt/vol)ammonium sulfate and recentrifuged, and the pellet wasresuspended in 0.05 M Tris-HCl (pH 7.5) and desaltedthrough a column (2.5 by 40 cm) of Bio-Gel P-6DG (Bio-Rad,Richmond, Calif.). Further purification was accomplished bychromatography on DEAE-Sepharose (eluted with a 0.0 to0.5 M KCl gradient) and Affi-Gel Blue (Bio-Rad) (eluted witha single step of 2 mM NADP). The protein was thenfractionated by nondenaturing polyacrylamide gel electro-phoresis (PAGE) (see below), and the gels were stained forICD activity. The band of ICD activity was cut out andelectrophoresed in a sodium dodecyl sulfate (SDS)-poly-acrylamide gel. The gel was then stained with Coomassieblue, which revealed two protein bands with molecularmasses of approximately 25 and 53 kDa. The 53-kDa bandwas judged to be ICD from the similarity of the molecularmass to that of ICD from E. coli (16) and because the 53-kDaprotein was clearly the predominant protein in all purifica-tion steps (determined by SDS-PAGE), which would beexpected if the protein was encoded by a highly expressedgene. The 53-kDa band was cut out and used to immunize afemale New Zealand White rabbit. Blood was harvested,allowed to clot, and then cleared by centrifugation. Anti-ICDpolyclonal antibodies were then partially purified by precip-itation in 40% ammonium sulfate, dialyzed against a buffer
containing 0.02 M Tris base and 0.1 M NaCl (pH 8.0), andstored at -20°C.Western immunoblots. Wild-type and ICD- mutant strains
of R meliloti were grown in minimal mannitol-glutamate-arabinose medium, with appropriate antibiotic selection.Cells were collected by centrifugation, washed once inbreaking buffer, and then lysed by sonication. Centrifugedextracts were subjected to SDS-PAGE (12% acrylamide[49]), transferred to nitrocellulose by electrophoresis,probed with anti-ICD antibodies, and visualized by a chemi-luminescent detection system (Amersham). Purified E. coliICD protein was a generous gift from Peter Thorsness.Prestained SDS-PAGE standards (Bio-Rad) were used toestimate molecular weight. The protein concentration ofextracts used in this experiment, as well as those listedabove, was determined with the Bio-Rad protein assay kit.
Plant growth and inoculation. Alfalfa (Medicago sativa)cultivar Ladak was used for all symbiotic performancestudies. Seeds were surface sterilized by soaking in 5%sodium hypochlorite for 5 min, rinsed several times in steriledistilled water, and then germinated on YMB agar. Seedlingsshowing no signs of contamination were transferred to sterilegrowth box units (four seedlings per box) and inoculatedimmediately. Each growth box unit consisted of three Ma-genta (Sigma GA-7 vessel) plant tissue culture boxes ar-ranged as a Leonard jar (58), with the top box inverted toprovide an aseptic barrier.
Plants were cultured in a walk-in growth room at atemperature of 28°C with a 16-h photoperiod. Light wassupplied by Philips 40W Agro-Lites at an intensity of 100,umol of photons m-1 s-. The nutrient solution was modi-fied from that described by Wych and Rains (59) andcontained 1.0 mM K2SO4, 0.5 mM KH2PO4, 0.25 mMK2HPO4, 0.5 mM MgSO4- 7H20, 2.0mM CaSO4. 2H20, 25,uM KCl, 13 FM H3BO3, 1.0 ,uM MnSO4. H20, 1.0 ,uMZnS04 7H20, 0.25 I'M CUSO4- 5H20, 2.5 ,uMCoCl2 6H20, 20 ,uM FeCl2 6H20, and 0.25 ,uMNa2MoO4. 2H20. The solution pH was adjusted to 6.5 withKOH and HCl.
Nodulation and nitrogen fixation. Plants were harvested 5to 6 weeks after inoculation. Nitrogen fixation was estimatedby acetylene reduction, using methods similar to thoseoutlined by Somerville and Kahn (53). Whole plants weretransferred from the growth boxes to 40-ml glass tubes. Eachtube was plugged with a rubber stopper outfitted with arubber septum, and then 10% of the air was replaced withacetylene. After 1 h, gas samples were removed and storedin Vacutainer tubes (Becton Dickinson) until analyzed bygas chromatography. Nodules were removed, counted, andweighed, and plant shoots were detached, dried at 65°C, andweighed.
Plasmid and transposon stability during symbiosis wasassessed by testing nodule occupants for antibiotic resis-tance and glutamate auxotrophy. Fresh nodules were sur-face sterilized in 1% sodium hypochlorite and washed fivetimes with sterile distilled water. They were then crushed inYMB broth, serially diluted, and plated onto minimal man-nitol medium containing glutamate and arabinose. Colonieswere then screened for resistance to appropriate antibioticsand glutamate auxotrophy.
RESULTS
Cloning and mutagenesis. E. coli DEK2038, an icd mutantand glutamate auxotroph (Table 1), was transformed withthe R. meliloti genomic library and incubated on M9 agar
J. BACTERIOL.
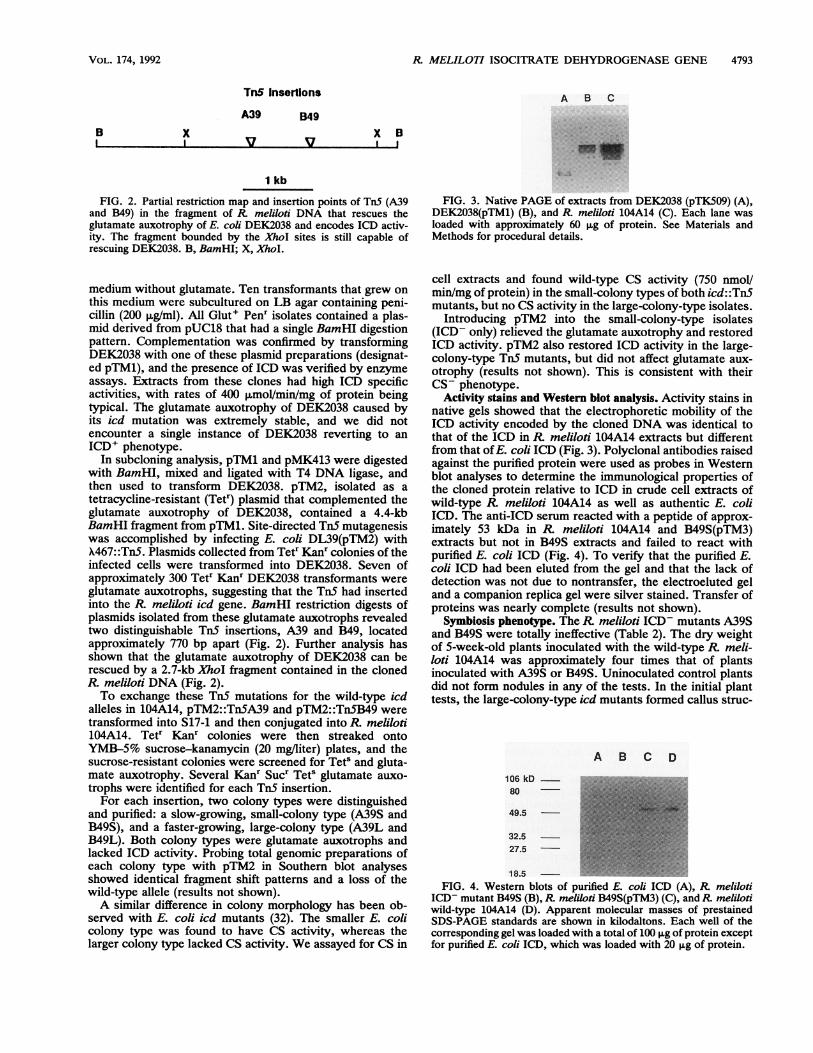
R MELILOTI ISOCITRATE DEHYDROGENASE GENE 4793
Tn5 Insertions
A39B x
V V
A B C
B49x BI I
1 kb
FIG. 2. Partial restriction map and insertion points of TnS (A39and B49) in the fragment of R meliloti DNA that rescues theglutamate auxotrophy of E. coli DEK2038 and encodes ICD activ-ity. The fragment bounded by the XhoI sites is still capable ofrescuing DEK2038. B, BamHI; X, XhoI.
medium without glutamate. Ten transformants that grew onthis medium were subcultured on LB agar containing peni-cillin (200 p,g/ml). All Glut' Penr isolates contained a plas-mid derived from pUC18 that had a single BamHI digestionpattern. Complementation was confirmed by transformingDEK2038 with one of these plasmid preparations (designat-ed pTMl), and the presence of ICD was verified by enzymeassays. Extracts from these clones had high ICD specificactivities, with rates of 400 ,umol/min/mg of protein beingtypical. The glutamate auxotrophy of DEK2038 caused byits icd mutation was extremely stable, and we did notencounter a single instance of DEK2038 reverting to anICD+ phenotype.
In subcloning analysis, pTM1 and pMK413 were digestedwith BamHI, mixed and ligated with T4 DNA ligase, andthen used to transform DEK2038. pTM2, isolated as atetracycline-resistant (Tetr) plasmid that complemented theglutamate auxotrophy of DEK2038, contained a 4.4-kbBamHI fragment from pTMl. Site-directed TnS mutagenesiswas accomplished by infecting E. coli DL39(pTM2) withX467::TnS. Plasmids collected from Tetr Kanr colonies of theinfected cells were transformed into DEK2038. Seven ofapproximately 300 Tetr Kanr DEK2038 transformants wereglutamate auxotrophs, suggesting that the TnS had insertedinto the R meliloti icd gene. BamHI restriction digests ofplasmids isolated from these glutamate auxotrophs revealedtwo distinguishable TnS insertions, A39 and B49, locatedapproximately 770 bp apart (Fig. 2). Further analysis hasshown that the glutamate auxotrophy of DEK2038 can berescued by a 2.7-kb XhoI fragment contained in the clonedR meliloti DNA (Fig. 2).To exchange these TnS mutations for the wild-type icd
alleles in 104A14, pTM2::TnSA39 and pTM2::TnSB49 weretransformed into S17-1 and then conjugated into R. meliloti104A14. Tetr Kanr colonies were then streaked ontoYMB-5% sucrose-kanamycin (20 mg/liter) plates, and thesucrose-resistant colonies were screened for Tets and gluta-mate auxotrophy. Several Kanr Sucr Tet' glutamate auxo-trophs were identified for each TnS insertion.For each insertion, two colony types were distinguished
and purified: a slow-growing, small-colony type (A39S andB49S), and a faster-growing, large-colony type (A39L andB49L). Both colony types were glutamate auxotrophs andlacked ICD activity. Probing total genomic preparations ofeach colony type with pTM2 in Southern blot analysesshowed identical fragment shift patterns and a loss of thewild-type allele (results not shown).A similar difference in colony morphology has been ob-
served with E. coli icd mutants (32). The smaller E. colicolony type was found to have CS activity, whereas thelarger colony type lacked CS activity. We assayed for CS in
FIG. 3. Native PAGE of extracts from DEK2038 (pTK509) (A),DEK2038(pTM1) (B), and R. meliloti 104A14 (C). Each lane wasloaded with approximately 60 pg of protein. See Materials andMethods for procedural details.
cell extracts and found wild-type CS activity (750 nmollmin/mg of protein) in the small-colony types of both icd::TnSmutants, but no CS activity in the large-colony-type isolates.
Introducing pTM2 into the small-colony-type isolates(ICD- only) relieved the glutamate auxotrophy and restoredICD activity. pTM2 also restored ICD activity in the large-colony-type TnS mutants, but did not affect glutamate aux-otrophy (results not shown). This is consistent with theirCS- phenotype.
Activity stains and Western blot analysis. Activity stains innative gels showed that the electrophoretic mobility of theICD activity encoded by the cloned DNA was identical tothat of the ICD in R. meliloti 104A14 extracts but differentfrom that of E. coli ICD (Fig. 3). Polyclonal antibodies raisedagainst the purified protein were used as probes in Westernblot analyses to determine the immunological properties ofthe cloned protein relative to ICD in crude cell extracts ofwild-type R meliloti 104A14 as well as authentic E. coliICD. The anti-ICD serum reacted with a peptide of approx-imately 53 kDa in R. meliloti 104A14 and B49S(pTM3)extracts but not in B49S extracts and failed to react withpurified E. coli ICD (Fig. 4). To verify that the purified E.coli ICD had been eluted from the gel and that the lack ofdetection was not due to nontransfer, the electroeluted geland a companion replica gel were silver stained. Transfer ofproteins was nearly complete (results not shown).
Symbiosis phenotype. The R meliloti ICD- mutants A39Sand B49S were totally ineffective (Table 2). The dry weightof 5-week-old plants inoculated with the wild-type R meli-loti 104A14 was approximately four times that of plantsinoculated with A39S or B49S. Uninoculated control plantsdid not form nodules in any of the tests. In the initial planttests, the large-colony-type icd mutants formed callus struc-
A B C D
106 kD80
49.5
32.5 -27.5
18.5 -FIG. 4. Western blots of purified E. coli ICD (A), R meliloti
ICD- mutant B49S (B),R meliloti B49S(pTM3) (C), and R melilotiwild-type 104A14 (D). Apparent molecular masses of prestainedSDS-PAGE standards are shown in kilodaltons. Each well of thecorresponding gel was loaded with a total of 100 Lg of protein exceptfor purified E. coli ICD, which was loaded with 20 F±g of protein.
VOL. 174, 1992
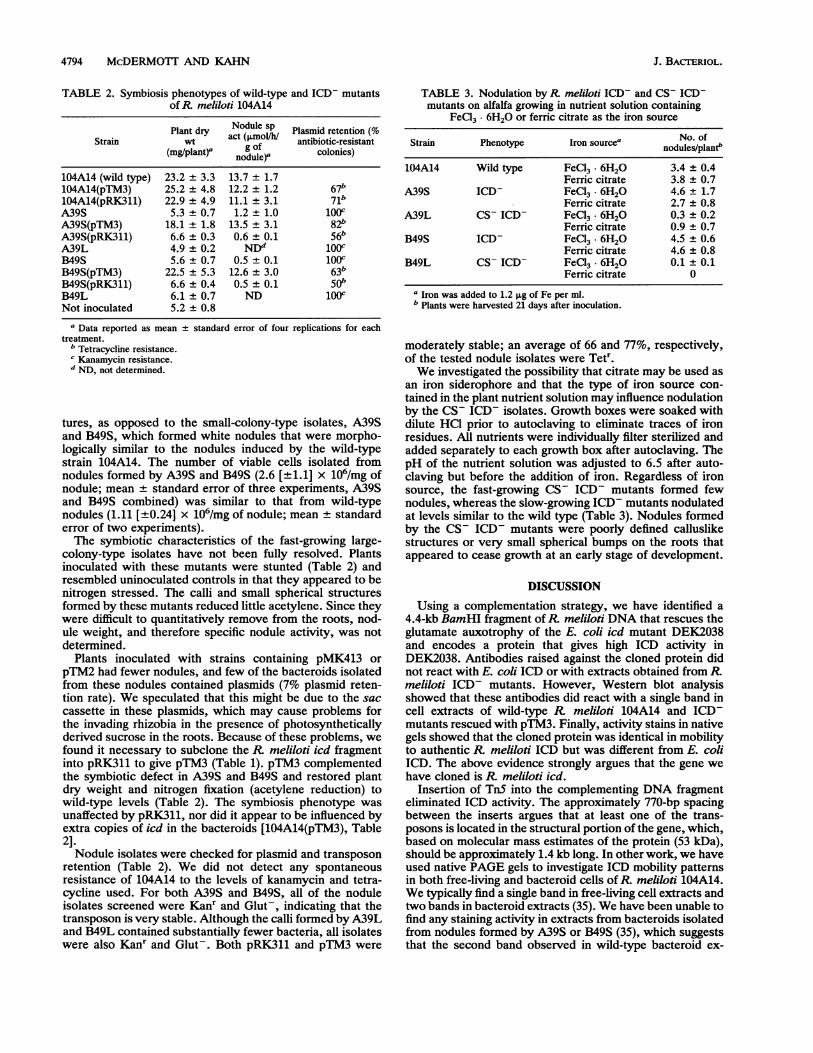
4794 McDERMOTI AND KAHN
TABLE 2. Symbiosis phenotypes of wild-type and ICD- mutantsof R. meliloti 104A14
Plant dry Nodule sp Plasmid retention (%Strain wt act (jLmol/h/ antibiotic-resistant
(mg/plant)' g of colonies)nodule)'104A14 (wild type) 23.2 ± 3.3 13.7 ± 1.7104A14(pTM3) 25.2 ± 4.8 12.2 ± 1.2 67b104A14(pRK311) 22.9 ± 4.9 11.1 ± 3.1 71bA39S 5.3 ± 0.7 1.2 + 1.0 100cA39S(pTM3) 18.1 ± 1.8 13.5 ± 3.1 82"A39S(pRK311) 6.6 + 0.3 0.6 ± 0.1 56bA39L 4.9 + 0.2 NDd 100CB49S 5.6 ± 0.7 0.5 ± 0.1 100cB49S(pTM3) 22.5 ± 5.3 12.6 ± 3.0 63bB49S(pRK311) 6.6 ± 0.4 0.5 ± 0.1 50bB49L 6.1 ± 0.7 ND 100cNot inoculated 5.2 ± 0.8
a Data reported as mean + standard error of four replications for eachtreatment.
b Tetracycline resistance.c Kanamycin resistance.d ND, not determined.
tures, as opposed to the small-colony-type isolates, A39Sand B49S, which formed white nodules that were morpho-logically similar to the nodules induced by the wild-typestrain 104A14. The number of viable cells isolated fromnodules formed by A39S and B49S (2.6 [±1.1] x 106/mg ofnodule; mean + standard error of three experiments, A39Sand B49S combined) was similar to that from wild-typenodules (1.11 [+0.24] x 106/mg of nodule; mean + standarderror of two experiments).The symbiotic characteristics of the fast-growing large-
colony-type isolates have not been fully resolved. Plantsinoculated with these mutants were stunted (Table 2) andresembled uninoculated controls in that they appeared to benitrogen stressed. The calli and small spherical structuresformed by these mutants reduced little acetylene. Since theywere difficult to quantitatively remove from the roots, nod-ule weight, and therefore specific nodule activity, was notdetermined.
Plants inoculated with strains containing pMK413 orpTM2 had fewer nodules, and few of the bacteroids isolatedfrom these nodules contained plasmids (7% plasmid reten-tion rate). We speculated that this might be due to the saccassette in these plasmids, which may cause problems forthe invading rhizobia in the presence of photosyntheticallyderived sucrose in the roots. Because of these problems, wefound it necessary to subclone the R meliloti icd fragmentinto pRK311 to give pTM3 (Table 1). pTM3 complementedthe symbiotic defect in A39S and B49S and restored plantdry weight and nitrogen fixation (acetylene reduction) towild-type levels (Table 2). The symbiosis phenotype wasunaffected by pRK311, nor did it appear to be influenced byextra copies of icd in the bacteroids [104A14(pTM3), Table2].Nodule isolates were checked for plasmid and transposon
retention (Table 2). We did not detect any spontaneousresistance of 104A14 to the levels of kanamycin and tetra-cycline used. For both A39S and B49S, all of the noduleisolates screened were Kanr and Glut-, indicating that thetransposon is very stable. Although the calli formed by A39Land B49L contained substantially fewer bacteria, all isolateswere also Kanr and Glut-. Both pRK311 and pTM3 were
TABLE 3. Nodulation by R meliloti ICD- and CS- ICD-mutants on alfalfa growing in nutrient solution containing
FeC13- 6H20 or ferric citrate as the iron source
Strain Phenotype Iron sourcea No. of
nodules/plant"104A14 Wild type FeC13 * 6H20 3.4 ± 0.4
Ferric citrate 3.8 + 0.7A39S ICD- FeC13- 6H20 4.6 + 1.7
Ferric citrate 2.7 ± 0.8A39L CS- ICD- FeC13 6H20 0.3 ± 0.2
Ferric citrate 0.9 + 0.7B49S ICD- FeCl3 6H20 4.5 ± 0.6
Ferric citrate 4.6 ± 0.8B49L CS- ICD- FeCl3 * 6H20 0.1 ± 0.1
Ferric citrate 0a Iron was added to 1.2 ,g of Fe per ml."Plants were harvested 21 days after inoculation.
moderately stable; an average of 66 and 77%, respectively,of the tested nodule isolates were Tetr.We investigated the possibility that citrate may be used as
an iron siderophore and that the type of iron source con-tained in the plant nutrient solution may influence nodulationby the CS- ICD- isolates. Growth boxes were soaked withdilute HCl prior to autoclaving to eliminate traces of ironresidues. All nutrients were individually filter sterilized andadded separately to each growth box after autoclaving. ThepH of the nutrient solution was adjusted to 6.5 after auto-claving but before the addition of iron. Regardless of ironsource, the fast-growing CS- ICD- mutants formed fewnodules, whereas the slow-growing ICD- mutants nodulatedat levels similar to the wild type (Table 3). Nodules formedby the CS- ICD- mutants were poorly defined calluslikestructures or very small spherical bumps on the roots thatappeared to cease growth at an early stage of development.
DISCUSSION
Using a complementation strategy, we have identified a4.4-kb BamHI fragment ofK meliloti DNA that rescues theglutamate auxotrophy of the E. coli icd mutant DEK2038and encodes a protein that gives high ICD activity inDEK2038. Antibodies raised against the cloned protein didnot react with E. coli ICD or with extracts obtained fromRmeliloti ICD- mutants. However, Western blot analysisshowed that these antibodies did react with a single band incell extracts of wild-type R meliloti 104A14 and ICD-mutants rescued with pTM3. Finally, activity stains in nativegels showed that the cloned protein was identical in mobilityto authentic R meliloti ICD but was different from E. coliICD. The above evidence strongly argues that the gene wehave cloned is R meliloti icd.
Insertion of TnS into the complementing DNA fragmenteliminated ICD activity. The approximately 770-bp spacingbetween the inserts argues that at least one of the trans-posons is located in the structural portion of the gene, which,based on molecular mass estimates of the protein (53 kDa),should be approximately 1.4 kb long. In other work, we haveused native PAGE gels to investigate ICD mobility patternsin both free-living and bacteroid cells of R. meliloti 104A14.We typically find a single band in free-living cell extracts andtwo bands in bacteroid extracts (35). We have been unable tofind any staining activity in extracts from bacteroids isolatedfrom nodules formed by A39S or B49S (35), which suggeststhat the second band observed in wild-type bacteroid ex-
J. BACTERIOL.

R. MELILOTI ISOCITRATE DEHYDROGENASE GENE 4795
tracts is not due to a second icd that is expressed undersymbiotic conditions. Consistent with this interpretation,Southern blots with pTM2 as the probe show only a singleband of hybridization (35). We speculate that the secondICD band found in bacteroid extracts might be due toposttranslational modification.The R meliloti ICD- mutants isolated in this study
completely lacked ICD activity, were glutamate auxotrophs,and formed ineffective nodules. While the Fix- phenotype ofthese mutants was not totally unexpected, we would nothave been surprised by a Fix' or partially Fix' phenotype.Johnson et al. (25) found that R. meliloti bacteroids showlittle isocitrate lyase activity, but we speculated that bydisrupting the TCA cycle at ICD, such a lesion might"force" carbon through the glyoxylate shunt. When E. coliis grown in acetate, carbon flow is directed through theglyoxalate shunt as a consequence of phosphorylation ofICD by ICD kinase-phosphatase (15). Under these condi-tions, ICD activity is reduced, increasing the intracellularisocitrate concentration to a level at which isocitrate lyase,which has a higher Km for isocitrate, can compete againstICD for the substrate (37). While we do not know if R.meliloti 104A14 uses the same mechanism, this strain willgrow on acetate as the sole source of carbon (39), suggestingthat it is capable of operating this anaplerotic sequence.Another inference that can be drawn from the symbiosis
phenotype of these mutants concerns the role of glutamate inthe symbiosis. It has been proposed that amino acids play akey role in the exchange of nutrients between the host andbacteroids (27). In this hypothesis, glutamate catabolism andthe malate-aspartate shuttle were suggested to be importantin transferring reducing equivalents from host to bacteroid.The R. meliloti icd mutants isolated in this study are strictglutamate auxotrophs. Since they are Nod', the host mustbe directly or indirectly providing enough glutamate for cellgrowth and maintenance. However, the Fix- phenotypesuggests that either a-ketoglutarate or glutamate is notsupplied in large quantities or that glutamate catabolism is ofrelatively little importance. This is consistent with studiesshowing that the rates of glutamate transport across thesoybean (57) and common bean (20) peribacteroid membraneare much slower than those of dicarboxylic acids. Anotherpossibility might be that ICD is required to supply substratefor the bacteroid aspartate aminotransferase reaction, whichhas recently been shown to be essential for an effectivealfalfa symbiosis (40). Glutamate obtained via aspartateaminotransferase might be significant. This explanationcould account for both the Fix- phenotype of R. meliloti-y-aminobutyric acid shunt mutants (13) and the Fix' pheno-type of R. meliloti glutamate dehydrogenase and glutamatesynthase mutants (30, 38). This pathway for glutamateproduction requires a-ketoglutarate, and icd mutants cannotsynthesize a-ketoglutarate.R. meliloti icd mutants spontaneously gave rise to CS-
mutants and thus were very similar to E. coli icd mutants(32). Lakshmi and Helling (32) hypothesized that the lack ofICD activity might lead to an accumulation of toxic levels ofcitrate or isocitrate but that accumulation of these metabo-lites could be eliminated by a mutation in the gene for CS. Atthis point, we do not know whether theR meliloti mutationis in gltA or perhaps in some regulatory gene, nor do weknow whether the Nod- phenotype of the CS- ICD-mutants is due solely to the lack of CS activity.The reason for the different nodulation phenotypes is not
obvious since the TCA cycle is not functioning in eithermutant and the double mutant grows better than the single
mutant. Citrate can be used in iron uptake, but a majority ofthe Bradyrhizobium japonicum (19) and R. leguminosarumbiovar trifolii (52) strains tested do not use citrate as asiderophore. R. meliloti mutants defective in the productionand uptake of rhizobactin can still form nodules (18), whichsuggests that R. meliloti may use other pathways to acquireiron. High concentrations of citrate have been found inalfalfa nodule cytosol (14), and this would argue that abacteroid CS- phenotype would be of little consequence iniron acquisition. However, the alfalfa peribacteroid mem-brane may be impermeable to citrate, and iron transportacross the peribacteroid membrane may require extracellu-lar reduction and complex dissociation, as is required foriron uptake by soybean roots (6). If this were the case, thenbacteroid synthesis of citrate may be very important inacquiring iron from the peribacteroid space.An alternative explanation for the Nod- phenotype of the
CS- icd mutants might be that the intracellular pool ofacetate accumulates to concentrations at which the acetylmodifications in the extracellular polysaccharide are quanti-tatively affected. This could be important given that extra-cellular polysaccharide composition is critical for successfulinvasion (33). We are currently trying to characterize thenodulation phenotype of these putative g1tA mutants, cloneR. meliloti git4, and study the regulation of this gene.
ACKNOWLEDGMENTS
We thank Larry Wilhelm for construction of the R. melilotigenomic library, and Derek Wood and Karen Liljebjelke for experttechnical assistance. We also thank Peter Thorsness for generouslyproviding us with purified E. coli ICD protein and Peter Thorsnessand Tony Dean for sending us E. coli icd mutants, includingDEK2038.
This work was supported by grants from the USDA CompetitiveResearch Grant Office and by the Agricultural Research Center atWashington State University.
REFERENCES1. Aebersold, P. B., G. A. Winans, D. J. Teel, G. B. Milner, and
F. M. Utter. 1987. Manual for starch gel electrophoresis: amethod for the detection of genetic variation, p. 1-19. NationalOceanic and Atmospheric Administration, U.S. Department ofCommerce, National Technical Information Service, Spring-field, Va.
2. Apostolakos, D., P. A. Menter, B. J. Rampsch, H. C. Reeves, andE. A. Birge. 1982. Genetic map position of the cistron coding forisocitrate dehydrogenase in Escherichia coli K-12. Curr. Micro-biol. 7:45-47.
3. Arwas, R., I. A. McKay, F. R. P. Rowney, M. J. Dilworth, andA. R. Glenn. 1985. Properties of organic acid utilization mutantsof Rhizobium leguminosarum strain 300. J. Gen. Microbiol.131:2059-2066.
4. Berg, C. M., M. Wang, N. B. Vartak, and L. Lui. 1988.Acquisition of new metabolic capabilities: multicopy suppres-sion by cloned transaminase genes in Escherichia coli K-12.Gene 65:195-202.
5. Bolton, E., B. Higgisson, A. Harrington, and F. O'Gara. 1986.Dicarboxylic acid transport in Rhizobium meliloti: isolation ofmutants and cloning of dicarboxylic acid transport genes. Arch.Microbiol. 144:142-146.
6. Chaney, R. L., J. C. Brown, and L. 0. Tiffin. 1972. Obligatoryreduction of ferric chelates in iron uptake by soybean. PlantPhysiol. 50:208-213.
7. Day, D. A., and L. Copeland. 1991. Carbon metabolism andcompartmentation in nitrogen-fixing legume nodules. PlantPhysiol. Biochem. 29:185-201.
8. de Bruin, F. J., and J. R. Lupski. 1984. The use of transposonTn5 mutagenesis in the rapid generation of correlated physicaland genetic maps of DNA segments cloned into multicopy
VOL. 174, 1992

4796 McDERMOTT AND KAHN
plasmids-a review. Gene 27:131-149.9. Ditta, G., T. Schmidhauser, E. Yakobson, P. Lu, X. Liang, D. R.
Finlay, D. Guiney, and D. R. Helinski. 1985. Plasmids related tothe broad host range vector, pRK290, useful for gene cloningand for monitoring gene expression. Plasmid 13:149-153.
10. Duncan, M. J., and D. G. Fraenkel. 1979. a-Ketoglutaratedehydrogenase mutant of Rhizobium meliloti. J. Bacteriol.137:415-419.
11. Engelke, T. H., M. N. Jagadish, and A. Puhler. 1987. Biochem-ical and genetical analysis of Rhizobium meliloti mutants defec-tive in C4-dicarboxylate transport. J. Gen. Microbiol. 133:3019-3029.
12. Finan, T. M., J. M. Wood, and D. C. Jordan. 1983. Symbioticproperties of C4-dicarboxylic acid transport mutants of Rhizo-bium leguminosawmn. J. Bacteriol. 154:1403-1413.
13. Fitzmaurice, A. M., and F. O'Gara. 1988. Involvement ofglutamate as a carbon source in supporting nitrogen fixationactivity in Rhizobium meliloti, p. 558. In H. Bothe, F. J. deBruijn, and W. E. Newton (ed.), Nitrogen fixation: hundredyears after. Gustav Fischer, New York.
14. Fougere, F., D. Le Rudulier, and J. G. Streeter. 1991. Effects ofsalt stress on amino acid, organic acid, and carbohydratecomposition of roots, bacteroids, and cytosol of alfalfa (Medi-cago sativa L.). Plant Physiol. 96:1228-1236.
15. Garnak, M., and H. C. Reeves. 1979. Phosphorylation of isoci-trate dehydrogenase of Eschenichia coli. Science 203:1111-1112.
16. Garnak, M., and H. C. Reeves. 1979. Purification and propertiesof phosphorylated isocitrate dehydrogenase of Eschenichia coli.J. Biol. Chem. 254:7915-7920.
17. Gay, P., D. Le Coq, M. Steinmetz, T. Berkelman, and C. I.Kado. 1985. Positive selection procedure for entrapment ofinsertion sequence elements in gram-negative bacteria. J. Bac-teriol. 164:918-921.
18. Gill, P. R., Jr., L. L. Barton, M. D. Scoble, and J. B. Neilands.1991. A high affinity iron transport system ofRhazobium melilotimay be required for efficient nitrogen fixation in planta. PlantSoil 130:211-217.
19. Guerinot, M. L., E. J. Meidi, and 0. Plessner. 1990. Citrate as asiderophore in Bradyrhizobium japonicum. J. Bacteriol. 172:3298-3303.
20. Herrada, G., A. Puppo, and J. Rigaud. 1989. Uptake of metab-olites by bacteroid-containing vesicles and by free bacteroidsfrom French bean nodules. J. Gen. Microbiol. 135:3165-3171.
21. Hynes, M. F., J. Quandt, M. P. O'Connell, and A. Puhler. 1989.Direct selection for curing and deletion of Rhizobium plasmidsusing transposons carrying the Bacillus subtilis sacB gene. Gene78:111-120.
22. Irigoyen, J. J., M. Sanchez-Diaz, and D. W. Emerich. 1990.Carbon metabolism enzymes ofRhizobium meliloti cultures andbacteroids and their distribution within the alfalfa nodule. Appl.Environ. Microbiol. 56:2587-2589.
23. Iuchi, S., and E. C. C. Un. 1988. arcA (dye), a global regulatorygene in Escherchia coli mediating repression of enzymes inaerobic pathways. Proc. Natl. Acad. Sci. USA 85:1888-1892.
24. Jackson, F. A., and E. A. Dawes. 1976. Regulation of thetricarboxylic acid cycle and poly-p-hydroxybutyrate metabo-lism inAzotobacter beijerinckii grown under nitrogen or oxygenlimitation. J. Gen. Microbiol. 97:303-312.
25. Johnson, G. V., H. J. Evans, and T. Ching. 1966. Enzymes ofthe glyoxylate cycle in rhizobia and nodules of legumes. PlantPhysiol. 41:1330-1336.
26. Kahn, M. L. Unpublished data.27. Kahn, M. L., J. Kraus, and J. E. Sommerville. 1985. A model of
nutrient exchange in the Rhizobium-legume symbiosis, p. 193-199. In H. J. Evans, P. J. Bottomley, and W. E. Newton (ed.),Nitrogen fixation research progress. Martinus Nijhoff,Dordrecht, The Netherlands.
28. Karr, D. B., K. K. Waters, F. Suzuki, and D. W. Emerich. 1984.Enzymes of the poly-13-hydroxybutyrate and citric acid cyclesof Rhizobium japonicum bacteroids. Plant Physiol. 75:1158-1162.
29. Kittell, B. L., D. R. Helinski, and G. S. Ditta. 1989. Aromaticaminotransferase activity and indoleacetic acid production inRhizobium meliloti. J. Bacteriol. 171:5458-5466.
30. Kondorosi, A., Z. Svab, G. B. Kiss, and R. A. Dixon. 1977.Ammonia assimilation and nitrogen fixation in Rhizobium meli-loti. Mol. Gen. Genet. 151:221-226.
31. Kurz, W. G. W., and T. A. LaRue. 1977. Citric acid cycleenzymes and nitrogenase in nodules of Pisum sativa. Can. J.Microbiol. 23:1197-1200.
32. Lakshmi, T. M., and R. B. Helling. 1976. Selection for citratesynthase deficiency in icd mutants of Escherichia coli. J.Bacteriol. 127:76-83.
33. Leigh, J. A., J. W. Reed, J. F. Hanks, A. M. Hirsch, and G. C.Walker. 1987. Rhizobium meliloti mutants that fail to succiny-late their calcofluor-binding exopolysaccharide are defective innodule invasion. Cell 51:579-587.
34. McDermott, T. R., S. M. Griffith, C. P. Vance, and P. H.Graham. 1989. Carbon metabolism in Bradyrhizobium japoni-cum bacteroids. FEMS Microbiol. Rev. 63:327-340.
35. McDermott, T. R., and M. L. Kahn. Unpublished data.36. Nambiar, P. T. C., and Y. I. Shethna. 1976. Purification and
properties of an NADP+-specific isocitrate dehydrogenase fromRhizobium meliloti. Antonie van Leeuwenhoek J. Microbiol.Serol. 42:471-482.
37. Nimmo, H. G. 1984. Control of Escherichia coli isocitratedehydrogenase: an example of protein phosphorylation in aprokaryote. Trends Biochem. Sci. 9:475-478.
38. Osburne, M. S. 1982. Rhizobium meliloti mutants altered inammonium utilization. J. Bacteriol. 151:1633-1636.
39. Perez-Galdona, R., and M. L. Kahn. Unpublished data.40. Rastogi, V. K., and R. J. Watson. 1991. Aspartate aminotrans-
ferase activity is required for aspartate catabolism and symbi-otic nitrogen fixation in Rhizobium meliloti. J. Bacteriol. 173:2879-2887.
41. Reibach, P. H., and J. G. Streeter. 1983. Metabolism of 4C-labeled photosynthate and distribution of enzymes of glucosemetabolism in soybean nodules. Plant Physiol. 72:634-640.
42. Reibach, P. H., and J. G. Streeter. 1984. Evaluation of activeversus passive uptake of metabolites by Rhizobium japonicumbacteroids. J. Bacteriol. 159:47-52.
43. Ried, J. L., and A. Collmer. 1987. An nptI-sacB-sacR cartridgefor constructing directed, unmarked mutations in gram-negativebacteria by marker exchange-eviction mutagenesis. Gene 57:239-246.
44. Ronson, C. W., P. Lyttleton, and J. G. Robertson. 1981. C4-dicarboxylate transport mutants of Rhizobium tnfolii form inef-fective nodules on Trifolium repens. Proc. Natl. Acad. Sci.USA 78:4284-4288.
45. Rosendahl, L., C. P. Vance, and W. B. Pedersen. 1990. Productsof dark CO2 fixation in pea root nodules support bacteroidmetabolism. Plant Physiol. 93:12-19.
46. Ruvkun, G. B., and F. M. Ausubel. 1980. A general method forsite-directed mutagenesis in prokaryotes. Nature (London) 289:85-89.
47. Salminen, S. O., and J. G. Streeter. 1987. Involvement ofglutamate in the respiratory metabolism of Bradyrhizobiumjaponicum bacteroids. J. Bacteriol. 169:495499.
48. Salminen, S. O., and J. G. Streeter. 1990. Factors contributingto the accumulation of glutamate in Bradyrhizobium japonicumbacteroids under microaerobic conditions. J. Gen. Microbiol.136:2119-2126.
49. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecularcloning: a laboratory manual, 2nd ed. Cold Spring HarborLaboratory, Cold Spring Harbor, N.Y.
50. San Fransisco, M. J. D., and G. R. Jacobson. 1985. Uptake ofsuccinate and malate in cultured cells and bacteroids of twoslow-growing species of Rhizobium. J. Gen. Microbiol. 131:1441-1448.
51. Sere, P. A. 1969. Citrate synthase. Methods Enzymol. 13:3-11.52. Skorupska, A., M. Derylo, and Z. Lorkiewicz. 1989. Siderophore
production and utilization by Rhizobium tnifolii. Biol. Metals2:4549.
53. Somerville, J. E., and M. L. Kahn. 1983. Cloning of the
J. BACTERIOL.

VOL. 174, 1992 R. MELILOTI ISOCITRATE DEHYDROGENASE GENE 4797
glutamine synthetase I gene from Rhizobium meliloti. J. Bacte-riol. 156:168-176.
54. Stovall, I., and M. Cole. 1978. Organic acid metabolism byisolated Rhizobium japonicum bacteroids. Plant Physiol. 61:787-790.
55. Streeter, J. G. 1987. Carbohydrate, organic acid, and amino acidcomposition of bacteroids and cytosol from soybean nodules.Plant Physiol. 85:768-773.
56. Udvardi, M. K., G. D. Price, P. M. Gresshoff, and D. A. Day.1988. A dicarboxylate transporter on the peribacteroid mem-brane of soybean nodules. FEBS Lett. 231:36-40.
57. Udvardi, M. K., C. L. Salom, and D. A. Day. 1988. Transport of
L-glutamate across the bacteroid membrane but not the peri-bacteroid membrane from soybean root nodules. Mol. Plant-Microbe Interact. 1:250-254.
58. Vincent, J. M. 1970. A manual for the practical study ofroot-nodule bacteria (IBP handbook no. 15), p. 86. BlackwellScientific Publications, Oxford.
59. Wych, R. D., and D. W. Rains. 1978. Simultaneous measure-ment of nitrogen fixation estimated by acetylene-ethylene assayand nitrate absorption by soybean. Plant Physiol. 62:443-448.
60. Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. ImprovedM13 phage cloning vectors and host strains: nucleotide se-quences of the M13mpl8 and pUC19 vectors. Gene 33:103-119.

![Comparison between the reference [i]Rhizobium … · Common bean plants were inoculated with rhizobia strains: Rhizobium tropici CIAT899 or Rhizobium etli 12a3, ... +216 94120093.](https://static.fdocuments.in/doc/165x107/5b915a7d09d3f26a278b6ea5/comparison-between-the-reference-irhizobium-common-bean-plants-were-inoculated.jpg)
















