
Clinical Toxicology Chapter 35
80
T oxicology is a broad, multidisciplinary science whose goal is to determine the effects of chemical agents on living systems. Innumerable potential toxins can inflict harm, including pharmaceuticals, herbals, household products, environmental agents, occupational chemicals, drugs of abuse, and chemical terrorism threats. Each year millions of human exposure cases are reported to the American Association of Poison Control Centers. 96 e Centers for Disease Control has reported that poisoning (both intentional and unintentional) is one of the top 10 causes of injury-related death in the United States in all adult age groups. From the beginnings of written history, poisons and their effects have been well described. Paracelsus (1493- 1541) correctly noted that “Alle Ding sind Giſt, und nichts ohn Giſt; allein die Dosis macht, daß ein Ding kein Giſt ist,” which means, “Everything is a poison; there is nothing which is not. e dose differentiates a poison.” As life in the modern era has become more complex, so has the study of poisons and their treatments. is chapter provides a general overview of Clinical Toxi- cology and the laboratory services necessary to support the care of poisoned patients. Because a comprehensive discus- sion of all aspects of toxicology is beyond the scope of this chapter, the clinical significance and toxicity of only a select number of common drugs, drugs of abuse, and other chemi- cals are discussed. BASIC INFORMATION In practice, it is neither possible nor necessary to test for all of the hundreds or thousands of clinical toxins that may be encountered. In reality up to 24 drugs or agents account for 80% or more of cases of intoxication treated in most emer- gency departments. 588 Moreover, some drugs are encountered very infrequently in some locations but with relatively high frequency in others. For example, phencyclidine (PCP) use is almost nonexistent in some areas but is responsible for a rela- tively high number of intoxications in a few large metropoli- tan cities. us the scope of clinical toxicology testing provided by the laboratory will depend on the pattern of local drug use and on the available resources of the institution and should be developed in consultation with the appropriate clinical staff. e value of drug and/or substance testing (screening) is well established (1) in the workplace, (2) for some athletic competitions, (3) to monitor drug use during pregnancy, (4) to evaluate drug exposure and/or withdrawal in new- borns, (5) to monitor patients in pain management and drug abuse treatment programs, and (6) to aid in the prompt diag- nosis of toxicity for a select number of drugs or agents for which a specific antidote or treatment modality is required (Table 35-1). In many other instances of drug toxicity, the value of drug screening, especially on an emergency basis, is more controversial. 94,411,552,695 Approaches to drug testing vary from the provision of just a few specific tests (e.g., acetaminophen, salicylate, ethanol, digoxin, iron) to testing for additional targeted groups of drugs (e.g., stimulant panel and coma panel) or to a more comprehensive general drug screen that might include hun- dreds of drugs and/or substances. For all of these situations, it is imperative that the laboratory communicate with the physician concerning the scope (and limitation) of the service and the proper timing and selection of specimens; when possible, the laboratory should assist with interpretation of results. At a minimum, the laboratory request slip should clearly state the drugs that it has the capability of detecting. Otherwise, the report of a “negative” result for a drug screen could be misleading. Clinical Considerations To operate effectively, the laboratory should be closely associ- ated with the healthcare team directly managing the patient. rough close and collaborative work, clinical information provided will help to guide appropriate ordering of tests and to ensure that interpretation of results is complete and accurate. For example, the team caring for the patient should provide the following information with the laboratory request: 1. e time and date of the suspected exposure along with the time and date of sample collection. 2. History from the patient or witnesses that might aid in identification of the toxin. 3. Assessment of the physical state of the patient at the time of presentation. Such information is useful to guide test selection and interpretation of results. CHAPTER 35 1109 Clinical Toxicology Loralie Langman, Ph.D., F.C.A.C.B., D.A.B.C.C.(C.C., M.B., T.C.), D.A.B.F.T., Laura Bechtel, Ph.D., D.A.B.C.C., and Christopher P. Holstege, M.D.
-
Upload
osama-bakheet1971 -
Category
Documents
-
view
122 -
download
21
description
Clinical Toxicology chapter 35
Transcript of Clinical Toxicology Chapter 35

Toxicology is a broad, multidisciplinary science whose goal is to determine the effects of chemical agents on living systems. Innumerable potential toxins can
inflict harm, including pharmaceuticals, herbals, household products, environmental agents, occupational chemicals, drugs of abuse, and chemical terrorism threats. Each year millions of human exposure cases are reported to the American Association of Poison Control Centers.96 The Centers for Disease Control has reported that poisoning (both intentional and unintentional) is one of the top 10 causes of injury-related death in the United States in all adult age groups. From the beginnings of written history, poisons and their effects have been well described. Paracelsus (1493-1541) correctly noted that “Alle Ding sind Gift, und nichts ohn Gift; allein die Dosis macht, daß ein Ding kein Gift ist,” which means, “Everything is a poison; there is nothing which is not. The dose differentiates a poison.” As life in the modern era has become more complex, so has the study of poisons and their treatments.
This chapter provides a general overview of Clinical Toxi-cology and the laboratory services necessary to support the care of poisoned patients. Because a comprehensive discus-sion of all aspects of toxicology is beyond the scope of this chapter, the clinical significance and toxicity of only a select number of common drugs, drugs of abuse, and other chemi-cals are discussed.
BASIC INFORMATIONIn practice, it is neither possible nor necessary to test for all of the hundreds or thousands of clinical toxins that may be encountered. In reality up to 24 drugs or agents account for 80% or more of cases of intoxication treated in most emer-gency departments.588 Moreover, some drugs are encountered very infrequently in some locations but with relatively high frequency in others. For example, phencyclidine (PCP) use is almost nonexistent in some areas but is responsible for a rela-tively high number of intoxications in a few large metropoli-tan cities. Thus the scope of clinical toxicology testing provided by the laboratory will depend on the pattern of local drug use and on the available resources of the institution and should be developed in consultation with the appropriate clinical staff.
The value of drug and/or substance testing (screening) is well established (1) in the workplace, (2) for some athletic competitions, (3) to monitor drug use during pregnancy, (4) to evaluate drug exposure and/or withdrawal in new-borns, (5) to monitor patients in pain management and drug abuse treatment programs, and (6) to aid in the prompt diag-nosis of toxicity for a select number of drugs or agents for which a specific antidote or treatment modality is required (Table 35-1). In many other instances of drug toxicity, the value of drug screening, especially on an emergency basis, is more controversial.94,411,552,695
Approaches to drug testing vary from the provision of just a few specific tests (e.g., acetaminophen, salicylate, ethanol, digoxin, iron) to testing for additional targeted groups of drugs (e.g., stimulant panel and coma panel) or to a more comprehensive general drug screen that might include hun-dreds of drugs and/or substances. For all of these situations, it is imperative that the laboratory communicate with the physician concerning the scope (and limitation) of the service and the proper timing and selection of specimens; when possible, the laboratory should assist with interpretation of results. At a minimum, the laboratory request slip should clearly state the drugs that it has the capability of detecting. Otherwise, the report of a “negative” result for a drug screen could be misleading.
Clinical ConsiderationsTo operate effectively, the laboratory should be closely associ-ated with the healthcare team directly managing the patient. Through close and collaborative work, clinical information provided will help to guide appropriate ordering of tests and to ensure that interpretation of results is complete and accurate. For example, the team caring for the patient should provide the following information with the laboratory request:1. The time and date of the suspected exposure along with the
time and date of sample collection.2. History from the patient or witnesses that might aid in
identification of the toxin.3. Assessment of the physical state of the patient at the time
of presentation.Such information is useful to guide test selection and
interpretation of results.
CHAPTER 35
1109
Clinical ToxicologyLoralie Langman, Ph.D., F.C.A.C.B., D.A.B.C.C.(C.C.,
M.B., T.C.), D.A.B.F.T., Laura Bechtel, Ph.D., D.A.B.C.C., and Christopher P. Holstege, M.D.

1110 SectionIII ■ Analytes
and chromatographic and/or mass spectrometric techniques (see Chapters 13 and 14), including thin-layer chromatogra-phy (TLC), high-performance liquid chromatography (HPLC), gas chromatography (GC), gas chromatography-mass spectrometry (GC-MS or GC-MS/MS), and liquid chromatography-mass spectrometry (LC-MS or LC-MS/MS).772 Currently, GC-MS is the most widely used definitive confirmatory procedure, although LC-MS/MS is becoming more popular. Confirmatory testing is mandatory for forensic drug testing (e.g., workplace drug testing).
Speed of analysis, or turnaround time (TOT), and avail-ability are critical issues in clinical toxicology. A drug analysis that requires several hours to complete or that is not available at all hours of the day is of little value in a clinical emergency. Alternatively, a rapid test that provides false information could result in erroneous diagnostic and therapeutic deci-sions. For numerous agents, quantitative determinations guide management during a clinical emergency. These agents include (1) acetaminophen, (2) carbamazepine, (3) digoxin, (4) ethanol, (5) ethylene glycol, (6) iron, (7) isopropanol, (8) lithium, (9) methanol, (10) phenobarbital, (11) phenytoin, (12) salicylate, (13) valproic acid, and (14) theophylline, and in whole blood, (15) carboxyhemoglobin and (16) methemo-globin. Results for these determinations should be available within 1 hour of specimen receipt.860
Proper selection of analytical methods and interpretation of results require knowledge of the pharmacology and phar-macokinetics of the toxins of interest. For example, the potential hepatotoxicity of acetaminophen is related to the concentration of unmetabolized drug. Conversely, delta-9-tetrahydrocannabinol (THC)—a metabolite of marijuana—is measured in urine as an indication of marijuana use.
Clinical EvaluationPrimary SurveyWhen a healthcare team initially evaluates a patient who pre-sents with a potential toxicologically induced health problem, the final diagnosis is often determined by (1) reviewing the history, (2) performing a directed physical examination, (3) using ancillary tests (e.g., electrocardiogram, radiology), and (4) applying a rational approach to laboratory testing. Often no specific antidote or treatment is available for a poi-soned patient and careful supportive care is the most impor-tant intervention.234
All patients who present with potential toxicity should be thoroughly assessed, and it is imperative that the clinician follow a standard “ABC” approach with attention to “airway, breathing, and circulation” respectively. The patient’s airway should be open and unblocked and adequate ventilation ensured. If the patient’s airway is not secure and endotracheal tube intubation is considered, the first diagnostic test that should be performed is a rapid bedside glucose concentra-tion. Hypoglycemia can result in coma or new-onset seizures, thereby mimicking a toxic etiology. In addition, numerous toxins are clinically associated with hypoglycemia (e.g., sul-fonylureas, iron, Mentha pulegium). Clinical effects induced by hypoglycemia can be rapidly reversed with intravenous
Analytical ConsiderationsBecause of the wide range of drugs of interest, no single ana-lytical technique is adequate for broad-spectrum drug detec-tion. Therefore several analytical approaches in combination are generally required. These may include simple, inexpen-sive, and rapid spot tests; immunoassays (see Chapter 16);
TABLE 35-1AntidoteorSpecificTreatmentforIntoxication
ToxinAntidote/Treatment14,68,185,251,722
Acetaminophen N-AcetylcysteineAluminum, iron DeferoxamineAnticholinergic agents PhysostigmineArsenic Dimercaprol;
2,3-dimercaptosuccinic acid; d-penicillamine
Barbiturates Multiple-dose oral activated charcoal; alkaline diuresis (phenobarbital only)
Benzodiazepines FlumazenilBeta-blockers GlucagonCalcium channel
blockersCalcium; glucagon; high-dose
insulin infusionCarbamazepine Multiple-dose oral activated
charcoal; charcoal hemoperfusion
Carbon monoxide OxygenCyanide Amyl nitrite, sodium nitrite,
sodium thiosulfate; hydroxocobalamin
Digoxin Anti-digoxin Fab fragmentsEthylene glycol,
methanolFomepizole (4-methylpyrazol)
or ethanol; hemodialysisIsoniazid PyridoxineLead Calcium disodium edetate;
dimercaprol; 2,3-dimercaptosuccinic acid
Lithium HemodialysisMercury Dimercaprol;
2,3-dimercaptosuccinic acid; d-penicillamine
Methanol Fomepizole (4-methylpyrazol) or ethanol; hemodialysis; folate
Nitrites, nitrates Methylene blueOpioids NaloxoneOrganophosphate or
carbamateAtropine; pralidoxime
(controversial for carbamates)
Salicylates Bicarbonate; hemodialysisTheophylline Multiple-dose oral activated
charcoal; hemodialysisTricyclic
antidepressantsBicarbonate; benzodiazepines

Chapter35 ■ Clinical Toxicology 1111
glucose, thus preventing unnecessary and costly procedures and testing.
Too often healthcare providers are lulled into a false sense of security when a patient presents with altered mental status and oxygen saturations on pulse oximetry that are adequate on high-flow oxygen. If the patient has inadequate ventilation or a poor gag reflex, then the patient may be at risk for pro-gressive CO2 narcosis or aspiration, respectively, and yet may be maintaining adequate oxygen saturation on supplemental oxygen. It is imperative that the healthcare team avoid common complications of poisonings such as aspiration pneumonitis.138,359 An arterial blood gas (ABG) can rapidly aid the healthcare team in determining the need for intuba-tion and mechanical ventilation. The ABG can also provide valuable information regarding the patient’s acid-base status and can help the clinician begin to generate a differential diagnosis. For example, in the scenario of a febrile toxic patient who presents with an altered mental status, a normal ABG (lack of acidosis) immediately rules-out uncoupling of oxidative phosphorylation as a cause of that patient’s fever. Finally, an ABG with co-oximetry can rapidly assist in deter-mining other toxic etiologies, such as carbon monoxide poi-soning (depending on the timing of the blood draw in relation to the exposure) and methemoglobinemia.
The initial treatment of hypotension in all toxic patients consists of the administration of intravenous fluids.340 The patient’s pulmonary status should be closely monitored to ensure that pulmonary edema does not develop as fluids are infused. Symptomatic toxic patients should be placed on con-tinuous cardiac monitoring with pulse oximetry, and the healthcare team must perform frequent neurologic checks to ensure continued protection of the airway. Acutely poisoned patients should receive a large-bore peripheral intravenous line, and all symptomatic patients should have a second line placed in the peripheral or central venous system, depending on the severity of their clinical status. At this time in patient care, blood can be drawn and sent for appropriate laboratory diagnostic testing. Placement of a urinary catheter should be considered early in the care of hemodynamically unstable poisoned patients to monitor urinary output as an indicator of adequate perfusion. A rapid bedside urine dipstick (pho-tometric chemical assay) provides helpful information rapidly as healthcare team members await further laboratory testing. For example, a urine specific gravity will give insight into the patient’s initial hydration status, and the appearance of tea-colored urine positive for blood may indicate the presence of myoglobinuria in a comatose patient with rhabdomyolysis.
Secondary SurveyUpon completion of the primary survey, the healthcare team should be assured that the patient’s airway is open and unblocked (also described as a patent airway), the ventilatory effort is adequate, and blood pressure is sustained in an appropriate range. At this point in management, the second-ary survey can be performed. The secondary survey involves a thorough examination of the entire patient. For adequate access to a toxic patient, the patient must be completely
undressed. Exposure of the patient ensures that a complete physical examination is performed.234 If the patient is not completely undressed, an important diagnostic clue may be missed. For example, skin lesions consistent with pressure necrosis on the back of a comatose patient may indicate the need to obtain a blood creatine phosphokinase activity or a urine myoglobin concentration. A comatose drug abuser may have attached transdermal drug patches (e.g., fentanyl, cloni-dine) in atypical locations (e.g., gluteal sulcus) that when found can rapidly lead to a diagnosis, avoiding the need for further laboratory testing. Besides completing a thorough physical review of all organ systems, the secondary survey involves reviewing items brought with the patient (e.g., medication bottles, drug paraphernalia). Searching carefully through the patient’s clothing may assist in providing clues that change the plan for specific laboratory tests or explain specific laboratory findings. For example, the discovery of a cough and cold product in the patient’s pocket that contains dextromethorphan could explain the clinical presentation of an agitated patient with hyperreflexia whose initial urine toxi-cology screen was positive for phencyclidine but later was found negative on confirmation.
Toxic SyndromesToxic syndromes (“toxidromes”) are clinical syndromes that are essential for the successful recognition of poisoning pat-terns. A toxidrome is the constellation of clinical signs and symptoms that suggests a specific class of poisoning. An important component of the secondary survey is to deter-mine whether a specific toxic syndrome is present.234 The most commonly encountered toxidromes include (1) anti-cholinergic, (2) cholinergic, (3) opioid, (4) sedative-hypnotic, and (5) sympathomimetic (Table 35-2). Many toxidromes have several overlapping features. For example, anticholiner-gic findings are highly similar to sympathomimetic findings, with one exception being the effects on sweat glands: anticho-linergic agents produce warm, flushed dry skin, but sympa-thomimetic agents produce diaphoresis. Toxidrome findings may also be affected by individual variability, comorbid con-ditions, and coingestants. For example, tachycardia associated with sympathomimetic or anticholinergic toxidromes may be absent in a patient who is concurrently taking beta-antagonist medications. Additionally, although toxidromes may be applied to classes of drugs, one or more toxidrome findings may be absent for some individual agents within these classes. For instance, meperidine is an opioid analgesic, but it does not induce miosis, which helps to define the “classic” opioid toxidrome. When accurately identified, the toxidrome may provide invaluable information for diagnosis and subsequent treatment, although the many limitations impeding acute toxidrome diagnosis must be carefully considered.
AnticholinergicCharacteristics of the anticholinergic syndrome have long been taught using the old medical adage, “dry as a bone, blind as a bat, red as a beet, hot as a hare, and mad as a hatter,” which corresponds with a symptomatic person’s anhidrosis,

1112 SectionIII ■ Analytes
mydriasis, flushing, fever, and delirium, respectively. Depend-ing on the dose and time post exposure, various central nervous system effects may manifest from an anticholinergic agent. Restlessness, apprehension, abnormal speech, confu-sion, agitation, tremor, picking movements, ataxia, stupor, and coma all have been described following exposure to various anticholinergics. When manifesting delirium, the individual will often stare into space and mutter, fluctuating between occasional lucid intervals with appropriate responses and then descriptions of vivid hallucinations. Phantom behaviors, such as plucking or picking in the air or at gar-ments, are characteristic. Hallucinations are prominent, and they may be benign, entertaining, or terrifying to the patient experiencing them. Exposed patients may have conversations with hallucinated figures and/or they may misidentify persons they typically know well. Simple tasks typically performed well by the exposed person may become difficult. Motor coor-dination, perception, cognition, and new memory formation are altered.
Mydriasis causes photophobia. Impairment of near vision occurs because of loss of accommodation and reduced depth of field secondary to ciliary muscle paralysis and pupillary enlargement. Tachycardia and exacerbated heart rate responses to exertion are expected. Systolic and diastolic blood pressure may show moderate elevation. A decrease in capillary tone may cause skin flushing. Intestinal motility slows, resulting in nausea, vomiting, and decreased bowel sounds. All glandular cells become inhibited, resulting in dry mucous membranes of the mouth and inhibition of sweating with resultant dry skin. Urination may be difficult, and urinary retention may occur. The exposed patient’s tempera-ture may become elevated from an inability to sweat and dissipate heat. In warm climates, this may result in marked hyperthermia.
Numerous substances can cause the anticholinergic syndrome. More common agents include antihistamines, atropine, cyclic antidepressant drugs, phenothiazines, anti-Parkinson’s drugs, cyclobenzaprine, scopolamine, and several plants such as Datura stramonium (Jimson weed).
CholinergicAcetylcholine is a neurotransmitter found throughout the central nervous system, including (1) the sympathetic and parasympathetic autonomic ganglia, (2) the postganglionic parasympathetic nervous system, and (3) the skeletal muscle motor end plate. Acetylcholine binds to and activates musca-rinic and nicotinic receptors. Activating muscarinic receptors stimulates or inhibits cellular function at visceral smooth muscle, cardiac muscle, and secretory glands. Alternatively, nicotinic receptors are present at postsynaptic membranes in autonomic ganglia and at skeletal muscle motor end plates. The enzyme acetylcholinesterase (AChE) regulates the activ-ity of acetylcholine within the synaptic cleft. Acetylcholine binds to the active site of AChE, where the enzyme rapidly hydrolyzes acetylcholine to choline and acetic acid. These hydrolyzed products rapidly dissociate from AChE, so that the enzyme is free to act on another molecule.
TABLE 35-2 SymptomsoftheImportantToxidromes
Toxidrome Symptom
Anticholinergic AgitationBlurred visionDecreased bowel soundsDry skinFeverFlushingHallucinationsIleusLethargy/comaMydriasisMyoclonusPsychosisSeizuresTachycardiaUrinary retention
Cholinergic DiarrheaUrinationMiosisBradycardiaBronchorrheaEmesisLacrimationSalivation
Opioid BradycardiaDecreased bowel soundsHypotensionHypothermiaLethargy/comaMiosisShallow respirationsSlow respiratory rate
Sedative-hypnotic AtaxiaBlurred visionConfusionDiplopiaDysesthesiasHypotensionLethargy/comaNystagmusRespiratory depressionSedationSlurred speech
Sympathomimetic AgitationDiaphoresisExcessive motor activityExcessive speechHallucinationsHypertensionHyperthermiaInsomniaRestlessnessTachycardiaTremor

Chapter35 ■ Clinical Toxicology 1113
responses to activation of the adrenergic system are complex and depend on the type of receptor (α1, α2, β1, β2) activated; some are excitatory and others have opposing inhibitory responses. Stimulation of the sympathetic nervous system produces central nervous system excitation (agitation, anxiety, tremors, delusions, and paranoia), tachycardia, seizures, hypertension, mydriasis, hyperpyrexia, and diapho-resis. In severe cases, cardiac arrhythmias and coma may occur. Examples of drugs that produce a sympathomimetic response include amphetamines, cocaine, phencyclidine, ephedrine, cathine, methcathinone, and pseudoephedrine.
Hyperthermic SyndromesToxin-induced hyperthermic syndromes are potentially dev-astating and require rapid management. Even though the patient’s temperature is one of the vital signs, the temperature often is not obtained in clinical practice. Fever in a poisoned patient can be associated with several hyperthermic syn-dromes: sympathomimetic toxicity, uncoupling of oxidative phosphorylation, serotonin syndrome, neuroleptic malignant syndrome, malignant hyperthermia, anticholinergic poison-ing, and withdrawal syndromes.692 Sympathomimetics, such as amphetamines and cocaine, may produce hyperthermia as the result of excess serotonin and dopamine, leading to thermal deregulation.372 Uncoupling of oxidative phosphory-lation, as seen in severe salicylate poisoning, occurs when the process of oxidative phosphorylation is disrupted, leading to heat generation and a reduced ability to aerobically generate adenosine-5′-triphosphate (ATP).409 Serotonin syndrome occurs when a relative excess of serotonin is present at both peripheral and central serotonergic receptors.360 Patients may present with hyperthermia, alterations in mental status, and neuromuscular abnormalities (rigidity, hyperreflexia, clonus), although individual variability is noted in these findings. Serotonin syndrome is associated with drug interactions such as those associated with the combination of monoamine oxidase inhibitors and meperidine, but it may also occur with single-agent therapeutic dosing or overdosing of serotonergic agents. Neuroleptic malignant syndrome is a condition caused by relative deficiency of dopamine within the central nervous system.717 It has been associated with dopamine receptor antagonists and the withdrawal of dopamine ago-nists such as levodopa/carbidopa products. Clinically, it may be difficult to distinguish from serotonin syndrome and other hyperthermic emergencies. Malignant hyperthermia occurs when genetically susceptible individuals are exposed to depo-larizing neuromuscular blocking agents or volatile general anesthetics. Anticholinergic poisoning may result in hyper-thermia through impairment of normal cooling mechanisms such as sweating. Withdrawal syndromes can produce exces-sive adrenergic responses (e.g., sedative-hypnotic withdrawal, ethanol withdrawal) and subsequent heat generation. It should be noted that opioid withdrawal is not associated with fever or altered mental status. Overall, differentiating between the toxic hyperthermic syndromes may be challenging, and additional causes of hyperthermia such as heat stroke and infection should be explored.
The respiratory effects of cholinergic poisoning tend to be dramatic and are considered to be the major factor leading to the death of its victims. Respiratory failure typically occurs as a triad of increased airway resistance, neuromuscular failure, and depression of central respiratory centers. Profuse watery nasal discharge, marked salivation, bronchorrhea, and bron-choconstriction result in a prolonged expiratory phase, cough, and wheezing. Because of the widespread presence of cholinergic receptors in the brain, cholinergic poisoning can produce great variation in neurologic signs and symptoms, including centrally mediated respiratory failure, coma, and seizures. Cholinergic cardiotoxicity can result in two clinical scenarios: a period of intense sympathetic activity that results in sinus tachydysrhythmias, or a period of increased para-sympathetic tone that leads to bradydysrhythmias, prolonga-tion of the PR interval, and atrioventricular block. Muscular symptoms may be vague and may consist of muscular weak-ness and difficulty with ambulation that can progress to mus-cular fasciculations and subsequent paralysis. Cholinergic agents cause constriction of both the sphincter muscle of the iris and the ciliary muscle of the lens, as well as stimulation of the lacrimal gland, resulting in lacrimation and miosis. The dermal sweat glands are innervated by sympathetic musca-rinic receptors. When these receptors are stimulated, profuse sweating occurs. Cholinergic gastrointestinal and genitouri-nary symptoms may result in nausea, vomiting, abdominal cramps, tenesmus, and involuntary defecation and urination. Two mnemonics have been developed to help recall cholin-ergic clinical effects: DUMB BELS (diarrhea, urination, miosis, bradycardia, and bronchorrhea-bronchoconstriction, emesis, lacrimation, sweating-salivation)415 and SLUDGE (salivation, lacrimation, urination, defecation, GI distress, emesis-eye findings, miosis).145
Agents that cause these cholinergic clinical effects include organophosphate and carbamate insecticides (cholinesterase inhibitors), certain species of mushrooms that contain mus-carine, and the nictonic receptor agonists nicotine, lobeline, and conine.
OpioidOpioids can induce coma, respiratory depression, bradycar-dia, hypotension, hypothermia, miosis, pulmonary edema, decreased bowel sounds, and decreased reflexes. Common causes of this syndrome, in which central nervous system depression and miosis are the cardinal two signs, include morphine, codeine, diacetylmorphine, oxycodone, hydroco-done, hydromorphone, and methadone. Meperidine and pro-poxyphene toxicity has been associated with mydriasis and not with miosis. Numerous drugs can mimic the opioid syn-drome by inducing coma, respiratory depression, and miosis, including clonidine, oxymetazoline, and antipsychotics.
SympathomimeticNorepinephrine is the neurotransmitter for postganglionic sympathetic fibers (adrenergic) that enervate skin, eyes, heart, lungs, gastrointestinal tract, exocrine glands, and some neuronal tracts in the central nervous system. Physiologic

1114 SectionIII ■ Analytes
Weiner and colleagues examined the application of these two bedside tests in patients presenting to the emergency department with suspected drug overdose with or without unexplained metabolic acidosis.827 Both reagents were added to urine samples from all 180 patients enrolled, and confirma-tory serum salicylate concentrations were drawn. Different from the previous studies, however, any darkening of color was regarded as a positive test. Twenty of these patients (11%) had salicylate concentrations ≥5 mg/dL. Both tests were 100% sensitive for recognizing these patients. The specificities for both tests were relatively low (Trinder with 73% and ferric chloride with 71%).
Overall both of these tests can be used for rapid bedside testing. Each is relatively inexpensive and, when color change is interpreted as suggested by Weiner and coworkers, has a low limit of detection. Positive tests indicate the possible presence of salicylate and not toxicity. Both tests should be followed with determination of serum salicylate concen-trations to confirm toxicity and quantitate the salicylate concentration.
Urine FluorescenceAutomotive antifreeze is a major source of ethylene glycol exposure. Sodium fluorescein is added to the antifreeze to aid in identifying cooling system leaks. Some have suggested that fluorescein excreted in the urine of a patient who has ingested antifreeze would fluoresce with the aid of a Wood’s lamp.818 Unfortunately, it seems that little advantage is derived by checking for urinary fluorescence by Wood’s lamp in those suspected of ethylene glycol poisoning.120,818
Anion GapObtaining a basic metabolic panel in all poisoned patients is recommended and is an important initial screening test. When low serum bicarbonate is discovered on a metabolic panel, the clinician should determine whether an elevated anion gap exists. The formula most commonly used for the anion gap (AG) calculation is as follows124:
AG Na Cl HCO= − +[ ]+ − −[ ] 3
This equation allows one to determine whether serum electroneutrality is being maintained. The primary cation (sodium) and anions (chloride and bicarbonate) are repre-sented in the equation.364 Other contributors to this equation are “unmeasured.”267 Other serum cations are not commonly included in this calculation because either their concentra-tions are relatively low (e.g., potassium) or assigning a number to represent their respective contribution is difficult (e.g., magnesium, calcium).267 Similarly, a multitude of other serum anions (e.g., sulfate, phosphate, organic anions) are also dif-ficult to measure and quantify in an equation.267,364 These “unmeasured” ions represent the anion gap calculated using the previous equation. The reference interval for this anion gap is accepted to be 8 to 16 mmol/L,267 but it has been sug-gested that because of changes in the technique used to measure chloride, the interval should be lowered to 6 to 14 mmol/L.364 Practically speaking, an increase in the anion
SCREENING PROCEDURES FOR DETECTION OF DRUGSScreening procedures are designed for the relatively rapid and generally qualitative detection of drugs or other toxic sub-stances. In general, screening tests have adequate clinical sen-sitivity but may not be highly specific. Thus, a negative result yielded by a screening procedure may rule out with reason-able certainty the presence of clinically significant concentra-tions of a particular analyte. Because of possible interferences, a positive result should be considered “presumptive positive” and should be confirmed by an alternate procedure of greater specificity. Screening procedures may be designed to detect a particular drug or drug class. Tests for such purposes include simple visual color tests (spot tests) and immunoassays.
Spot TestsSpot tests are rapid, easily performed, noninstrumental quali-tative procedures that provide presumptive evidence for the presence of tested drugs. Any positive response must be followed by testing with a more specific method. They are potentially valuable to rule out the presence of drugs or to suggest (but not prove) the presence of a drug of a particular group. Spot tests are less frequently employed now because many have been largely replaced by rapid immunoassays that may be performed at the point of care or in the central laboratory.
Ferric Chloride and Trinder TestsTwo bedside tests have been proposed for rapid identification of the patient with salicylate toxicity. Both involve simply applying a few drops of a prepared reagent to a small sample of a patient’s urine and watching for a characteristic color change. The first such reagent is ferric chloride (FeCl3). Applying a few drops of a 10% solution of ferric chloride to 1 mL of urine containing even very small amounts of salicy-late will produce a characteristic purple color caused by the formation of an iron-salicylate complex.337 This color change also will occur if the urine in question contains acetoacetic acid and phenylpyruvic acid.250 The urine Trinder spot test uses a reagent composed of mercuric chloride, ferric nitrate, concentrated hydrogen chloride, and deionized water.421 Applying 1 mL of this solution to 1 mL of urine with salicy-lates present will also lead to a purple color change.250
One study exploring the salicylate question254 examined the use of the ferric chloride test in 187 patients presenting to the emergency department. These ferric chloride tests were subsequently followed with serum salicylate concentrations. The sensitivity of the ferric chloride was 93.2% for salicylate concentrations ≥3.0 mg/dL and 93.8% for concentrations ≥30.0 mg/dL. Specificity was 88.8% and 75.4%, respectively. Three false negatives were reported, one of which had a toxic salicylate concentration of 34 mg/dL. King and associates evaluated the clinical utility of the Trinder reagent.421 Inves-tigators enlisted 12 volunteers who ingested 975 mg of aspirin. The sensitivity of the test in this study was 100% with two false positives from controls.

Chapter35 ■ Clinical Toxicology 1115
been advocated to detect the presence of potentially radi-opaque poisons. For example, O’Brien and associates studied the detectability of 459 different tablets and capsules using plain radiography.596 Investigators used a ferrous sulfate tablet as a control in grading the radiopacity of other tablets. Overall, of the wide variety of pills tested, only 6.3% were graded as having radiopacity the same as or greater than ferrous sulfate; 29.6% were regarded as having at least moder-ate opacity; and the largest remaining portion of pills (64%) was regarded as no more than minimally detectable. Based on this and other studies, the indiscriminate use of plain abdominal x-rays is not justified, and a negative film should not be relied upon to rule out potential toxic pill ingestion, especially if enough time is given to allow the pills to dissolve.
Osmol GapThe main osmotically active constituents of serum are Na+, Cl−, HCO3
−, glucose, and urea. Several empirical formulas based on measurement of these substances have been used to estimate the serum osmolality.276,539,606,617,662 In practice, one has not shown itself to be superior to the others, yet each equation demonstrates significant differences in the osmol gap reference interval.443 Therefore, reference intervals must be validated on appropriate patient populations. Two com-monly used formulas (in conventional and SI units) are pre-sented here:
OSMc mOsm/kg Na mmol/L glucose mg/dL /
urea mg/dL /( ) ( ) ( )
( ) .= +
+2 18
2 88
OSMc mOsm/kg Na mmol/L glucose mmol/L
urea mmol/L( ) ( ) ( )
( )= +
+2
orOSMc mOsm/kg Na mmol/L glucose mg/dL /
urea mg/dL( ) . ( ) ( )
( )= +
+1 86 18
//2 8 9. +
OSMc mOsm/kg Na mmol/L glucose mmol/L
urea mmol/L( ) . ( ) ( )
( )= +
+ +1 86
99The difference between the actual osmolality (OSMm),
measured by freezing-point depression, and the calculated osmolality (OSMc) is referred to as delta-osmolality, or the osmol gap (OSMg).
OSMg OSMm OSMc= −
Elevated OSMg implies the presence of unmeasured osmotically active substances.427 Volatile alcohols (ethanol, methanol, isopropanol, acetone, and ethylene glycol) when present at significant concentrations, increase serum osmo-lality, thus resulting in an increased OSMg. The calculation of OSMg is commonly used as a screen.276 However, it is important to remember that volatile alcohols are not detected when osmolality is measured with a vapor pressure osmom-eter. Therefore, for the purpose of determining the OSMg, only osmolality measurements based on freezing-point depression are acceptable.
What constitutes a normal osmol gap is widely debated. Traditionally, a normal gap has been defined as 10 mOsm/kg
gap beyond an accepted reference interval, accompanied by a metabolic acidosis, represents an increase in unmeasured endogenous (e.g., lactate) or exogenous (e.g., salicylates) anions.124 A list of the more common causes of this phenom-enon is organized in the classic MUDILES pneumonic (Box 35-1). Note: The “P” has been removed from the older acronym of MUDPILES, because paraldehyde is no longer available.
It is imperative that clinicians who admit poisoned patients initially presenting with an increased anion gap metabolic acidosis investigate the cause of that acidosis. Many symp-tomatic poisoned patients may have an initial mild metabolic acidosis upon presentation caused by processes resulting in elevated serum lactate. However, with adequate supportive care including hydration and oxygenation, the anion gap aci-dosis should improve. If, despite adequate supportive care, an anion gap metabolic acidosis worsens in a poisoned patient, the clinician should consider continued absorption of exog-enous acids (e.g., salicylate), formation of acidic metabolites (e.g., ethylene glycol, methanol, toluene metabolites), and cel-lular ischemia with worsening lactic acidosis (e.g., cyanide) as potential causes.
ElectrocardiogramInterpretation of the electrocardiogram (ECG) in the poi-soned patient can significantly facilitate appropriate labora-tory testing, diagnosis, and management of the poisoned patient, because numerous drugs can cause ECG changes.832 Despite the fact that drugs have widely varying indications for therapeutic use, many unrelated drugs share common cardiac electrocardiographic effects if taken in overdose. Potential toxins can be placed into broad classes on the basis of their cardiac effects. For example, agents that block cardiac potassium efflux channels and agents that block cardiac fast sodium channels can lead to characteristic changes in cardiac indices consisting of QRS prolongation and QT prolongation, respectively. The recognition of specific ECG changes associ-ated with other clinical data (toxidromes) can be potentially lifesaving.341
Radiographic StudiesRadiologic testing is sometimes used to diagnose complica-tions associated with poisonings, such as aspiration pneumo-nitis and anoxic brain injury. The use of radiology has also
MethanolUremiaDiabetic ketoacidosisIron, inhalants (i.e., carbon monoxide, cyanide, toluene),
isoniazid, ibuprofenLactic acidosisEthylene glycol, ethanol ketoacidosisSalicylates, starvation ketoacidosis, sympathomimetics
BOX 35-1 Common Causes of Increased Anion Gap (MUDILE)

1116 SectionIII ■ Analytes
concentrations of ethylene glycol (<50 mg/dL), and methanol (<30 mg/dL).616
ImmunoassayDifferent types of immunoassays are useful in screening specimens for drugs (see Chapters 16 and 34). In some cases, these assays are relatively specific for a single drug (LSD), but in others, several drugs of a similar class are detected (e.g., opiates). The detection limit for various members of a class of drugs or the degree of cross-reactivity for similar drugs varies, and each manufacturer of immuno-assay reagents should be consulted for specific information. These assays are easy to perform; many are available for use on automated instrumentation and may be able to provide “semiquantitative” results. Several portable, noninstrumental, immunoassay-based drug detection devices are available for use in point-of-care testing (POCT). For the vast majority of drugs of abuse, immunoassays are the methods of choice for initial screening. However, for a more comprehensive drug screening, chromatographic procedures complement immunoassays.
Planar ChromatographyPlanar chromatography, commonly known as thin-layer chromatography (TLC), is a versatile procedure that requires no instrumentation and thus is operationally relatively simple and inexpensive (see Chapter 13). With this technique, a large number of drugs may be detected; it may be applied to the analysis of serum, gastric contents, or urine. Urine, however, is the specimen of choice because most drugs and drug metabolites are present in urine in relatively high concentra-tions. However, application of TLC to drug screening requires considerable experience and skill to recognize drug and metabolite patterns and various color hues for detection; it has largely been replaced by other chromatographic tech-niques. For a more comprehensive description, refer to previ-ous editions of this textbook.78
Gas ChromatographyAlso known as gas liquid chromatography, GC is relatively rapid, is capable of resolving a broad spectrum of drugs, and is widely used for qualitative and quantitative drug analysis.392 Capillary columns, because of their high efficiency, are ana-lytical columns that are commonly used for drug detection by GC (see Chapter 13). In many instances, nonderivatized
or less. The original source of this value is an article by Smithline and Gardner,742 which declared that this number was pure convention. Further clinical study has not shown this assumption to be correct. However, large variability is seen in the normal population.456,558 Researchers have found the OSMg to vary from −9 to +5 mOsm/kg288; from −13.5 to +8.9536; and from −10 to +20 mOsm/kg,1 depending on the population studied. An important point to consider is that the day-to-day coefficient of variance of sodium was 1%. This analytical variance alone may account for the variation found in patients’ osmol gaps.1
One would expect that each 100 mg/dL (21.7 mmol/L) of ethanol (molecular weight = 46.068 g/mol) in serum results in an approximate increase of 21.7 mOsm/kg.640 However, this is not found to be the case. Applying a correction factor of 0.83 to the ethanol value will more closely approximate the contribution of ethanol to the OSMg.276 By considering this effect of ethanol on the serum osmolality, it is possible to determine what portion of an increased osmol gap is due to ethanol. The contribution of ethanol to the measured osmo-lality can be calculated (ethanol, mg/dL/4.6 × 0.83) and included in the preceding formula for delta osmolality calcu-lation. However, it has been observed that ethanol and metha-nol do not follow a completely predictable relationship with OSMg. In severe ethanol and methanol intoxication, OSMg increases with increasing concentration, making it appear that something is present besides the alcohol.276,469,539,606,662
A significant residual osmol gap (>10 mOsm/kg) after the correction for ethanol would suggest the possible presence of isopropanol, methanol, acetone, or ethylene glycol. This information, in conjunction with the presence or absence of metabolic acidosis or serum acetone, is helpful to the clini-cian when specific measurements of alcohols other than ethanol and of ethylene glycol are not available on an emer-gency basis (Table 35-3). It must be realized that ketones (diabetics) and substances administered to patients such as polyethylene glycol (burn cream),98 mannitol (osmotic diuretic), and propylene glycol (solvent for diazepam and phenytoin) may increase serum osmolality.
For the diagnosis of ethanol intoxication, OSMg poisoning has lost its usefulness because ethanol is measured quickly on most chemistry analyzers. However, because other toxic alcohols can be measured only by chromatographic tech-niques, it is still useful. Unfortunately, OSMg as a screening method is insensitive to low, yet clinically significant,
TABLE 35-3 LaboratoryFindingsCharacteristicofIngestionofAlcohols
AlcoholSerum
Osmol GapMetabolic Acidosis With Anion Gap
Serum Acetone
Urine Oxalate
Ethanol + − − −Methanol + + − −Isopropanol + − + −Ethylene glycol + + − +

Chapter35 ■ Clinical Toxicology 1117
A more detailed description of these methods can be found in Chapter 16 and in the package insert for each spe-cific test kit.
PHARMACOLOGY AND ANALYSIS OF SPECIFIC DRUGS AND TOXIC AGENTSThe toxic, pharmacologic, biochemical, and analytical char-acteristics of several individual drugs and toxins are discussed in this section.
Agents That Cause Cellular HypoxiaCarbon monoxide and methemoglobin-forming agents inter-fere with oxygen transport, resulting in cellular hypoxia. Cyanide interferes with oxygen use and therefore causes an apparent cellular hypoxia.
Carbon MonoxideCarbon monoxide (CO) is a colorless, odorless, tasteless gas that is a product of incomplete combustion of carbonaceous material. Common exogenous sources of carbon monoxide include cigarette smoke, gasoline engines, and improperly ventilated home heating units. Small amounts of carbon monoxide are produced endogenously in the metabolic con-version of heme to biliverdin.458 This endogenous production of carbon monoxide is accelerated in hemolytic anemias.496
Toxic EffectsWhen inhaled, carbon monoxide combines tightly with the heme Fe2+ of hemoglobin to form carboxyhemoglobin. The binding affinity of hemoglobin for carbon monoxide is about 250 times greater than that for oxygen. Therefore high con-centrations of carboxyhemoglobin limit the oxygen content of blood. Moreover, the binding of carbon monoxide to a hemoglobin subunit increases the oxygen affinity for the remaining subunits in the hemoglobin tetramer. Thus at a given tissue PO2 value, less oxygen dissociates from hemoglo-bin when carbon monoxide is also bound, shifting the hemoglobin-oxygen dissociation curve to the left. Conse-quently, carbon monoxide not only decreases the oxygen content of blood, it also decreases oxygen availability to tissue, thereby producing a greater degree of tissue hypoxia than would result from an equivalent reduction in oxyhemo-globin due to hypoxia alone.368,778 Carbon monoxide may also bind to other heme proteins, such as myoglobin and mito-chondrial cytochrome oxidase a3; this may limit oxygen use when tissue PO2 is very low.368,778
The toxic effects of carbon monoxide are a result of hypoxia. Organs with high oxygen demand, such as heart and brain, are most sensitive to hypoxia and thus account for the major clinical sequelae of carbon monoxide poisoning. It must be emphasized that the carboxyhemoglobin concentra-tion, although helpful in diagnosis, does not always correlate with the clinical findings or prognosis.569,702 Factors other than carboxyhemoglobin concentration that contribute to toxicity include length of exposure, metabolic activity, and under-lying disease, especially cardiac or cerebrovascular disease.
drugs have good GC properties when capillary columns are used; in some instances, derivatization to a less polar or more volatile compound is necessary. Common detectors for drug detection by GC are flame ionization and alkali flame ionization (nitrogen phosphorus) detectors and mass spec-trometers, which provide the greatest accuracy of identifica-tion. Numerous methods for general drug screening by GC-MS spectrometry have been published, but one compre-hensive method can be adapted to multiple body fluids and tissues.392
High-Performance Liquid ChromatographyThe resolving power of HPLC (see Chapter 13) for separating widely divergent chemical constituents has been applied to the complex challenge of comprehensive drug screening in biological fluids. Advantages of HPLC over GC include the ability to analyze polar and thermally labile drugs without derivatization. The advent of diode array detectors that provide a spectral scan of compounds as they elute from the column greatly increased the discriminatory power of this technique.82,500,661,785 LC-MS or LC-MS/MS currently plays a limited role in comprehensive screening but is rapidly gaining in popularity.*
Point of Care (POC) Drug TestingNumerous POC drug test devices for urine (and oral fluid) are designed for easy, rugged, and portable use by nontechni-cal personnel. Although these devices are relatively simple to use, proper training of nonlaboratory users is important for optimal performance (see Chapter 20).279,445 These noninstru-mental immunoassay test devices are designed for use at the site of collection; results are available within minutes and are variously configured to detect only one drug or many drugs simultaneously. The spectrum of drugs tested includes tricy-clic antidepressants, barbiturates, benzodiazepines, metha-done, MDMA (methylenedioxymethamphetamine), MDA (methylenedioxyamphetamine), MDEA (methylenedioxy-ethylamphetamine), oxycodone, and the traditional SAMHSA (Substance Abuse and Mental Health Services Administra-tion) or NIDA (National Institute on Drug Abuse) 5 (amphet-amine, cocaine, marijuana, opiates, and phencyclidine). As previously cited, such devices are also available for measure-ment of acetaminophen and salicylate in serum or whole blood. Evaluations for some of these test devices for urine and oral fluid558,639 have been published. A comprehensive review of on-site drug testing is also available.629 The assay principles of these POC test devices include the following:• Sequential competitive binding microparticle capture
immunoassay• Homogeneous microparticle capture immunochromato-
graphy• Solid-phase competitive sequential enzyme immunoassay• Latex-agglutination-inhibition immunoassay
*References 281, 282, 446, 511, 523, and 794.

1118 SectionIII ■ Analytes
Among several such methods, the most popular are based on automated, multiwavelength measurements of several hemoglobin species. These methods are rapid and convenient for the determination of carboxyhemoglobin and other hemoglobin species. Spectrophotometric methods generally compare favorably with gas chromatographic procedures at carboxyhemoglobin concentrations greater than 2 to 3%, but their precision is poor below these concentrations.809 Therefore, they are sufficiently accurate and precise for measurement of carbon monoxide after exogenous exposure but are too insensitive to detect the increased endogenous production of carbon monoxide that occurs in hemolytic anemia.
Fetal hemoglobin has slightly different spectral properties than adult hemoglobin. Consequently, falsely high carboxy-hemoglobin values of 4 to 7% may occur when blood from neonates is measured by some spectrophotometric methods utilizing fewer wavelengths.810 Moreover, erroneous results may occur with lipemic specimens, with bilirubin, and in the presence of methylene blue (see section on “Methemoglobin-Forming Agents”).
CyanideCyanide is a chemical group that consists of one atom of carbon bound to one atom of nitrogen by three molecular bonds (C≡N). Inorganic cyanides (also known as cyanide salts) contain cyanide in the anion form (CN−) and are used in numerous industries, such as metallurgy, photographic developing, plastic manufacturing, fumigation, and mining. Organic compounds that have a cyano group bonded to an alkyl residue are called nitriles. For example, methyl cyanide is also known as acetonitrile (CH3CN). Hydrogen cyanide (HCN) is a colorless gas at standard temperature and pressure with a reported bitter odor. Cyanogen gas, a dimer of cyanide, reacts with water and breaks down into the cyanide anion. Many plants, such as Manihot spp. (cassava), Linum spp., Lotus spp., Prunus spp., Sorghum spp., and Phaseolus spp., contain cyanogenic glycosides. Iatrogenic cyanide poisoning may occur during use of nitroprusside as a vasodilator given to reduce blood pressure and afterload. Each nitroprusside molecule contains five cyanide molecules, which are slowly released in vivo. If endogenous sulfate stores are depleted, as in the malnourished or postoperative patient, cyanide may accumulate even with therapeutic nitroprusside infusion rates (2 to 10 mcg/kg/min).
Toxic EffectsHydrocyanic acid binds to hemoglobin. The hydrocyanic acid bound in the erythrocyte is in equilibrium with free hydrocyanic acid in the serum at a ratio of 10 : 1. Cyanide in serum readily crosses all biological membranes and avidly binds to heme iron (Fe3+) in the cytochrome a-a3 complex within mitochondria.715,803 When bound to cytochrome a-a3, cyanide is a competitive inhibitor that causes decoupling of oxidative phosphorylation. Patients exposed to toxic concen-trations of cyanide exhibit rapid onset of symptoms typical of cellular hypoxia—flushing, headache, tachypnea, dizziness,
Moreover, low carboxyhemoglobin concentrations relative to the severity of poisoning may be observed if the patient was removed from the carbon monoxide–contaminated environ-ment several hours before blood sampling.318
An insidious effect of carbon monoxide poisoning is the delayed development of neuropsychiatric sequelae, which may include personality changes, motor disturbances, and memory impairment. These manifestations do not correlate with the length of exposure or with the maximum blood carboxyhemoglobin concentration.45
Treatment for carbon monoxide poisoning involves removal of the individual from the contaminated area and administration of oxygen. The half-life (t1/2) of carboxyhemo-globin in the body is variable, and attempts to determine the exact elimination t1/2 for CO based on the inhaled oxygen concentration have not been validated. Hyperbaric oxygen therapy for CO is highly debated, and current position papers have found no evidence to support its use.101
Analytical MethodsCarbon monoxide may be released from hemoglobin and then measured by GC, or it may be determined indirectly as carboxyhemoglobin by spectrophotometry. Gas chromato-graphic methods are accurate and precise even for very low concentrations of carbon monoxide. Spectrophotometric methods are rapid, convenient, accurate, and precise, except at very low concentrations of carboxyhemoglobin (<2 to 3%).
Gas chromatographic methods measure the carbon mon-oxide content of blood. When blood is treated with potassium ferricyanide, carboxyhemoglobin is converted to methemo-globin, and carbon monoxide is released into the gas phase. Measurement of the released carbon monoxide may be per-formed by GC using a molecular sieve column and a thermal conductivity detector.213 A lower detection limit is achieved by incorporating a reducing catalyst (e.g., nickel) between the GC column and the detector to convert carbon monoxide to methane. The methane may then be detected with a flame ionization detector.307 A very low detection limit may be achieved with the use of a heated mercuric oxide reaction chamber between the GC column and an ultraviolet light detector. As carbon monoxide elutes from the column, it reacts with mercuric oxide to form mercury gas, which has a high molar absorptivity at 254 nm.809 In practice, the carbon monoxide binding capacity is also determined after an aliquot of the blood specimen is treated with carbon monoxide to saturate the hemoglobin. The results are then expressed as percent of carboxyhemoglobin:
%HbCO COCO
content
capacity= ×100
GC methods are accurate and precise and are considered to be reference procedures. Normal values for carboxyhemo-globin in rural nonsmokers are about 0.5%; for urban non-smokers, 1 to 2%; and for smokers, 5 to 6%.49 Values may be increased by about 3% in cases of hemolytic anemia.496
Spectrophotometric methods rely on the characteristic spectral absorption properties of carboxyhemoglobin.197,879

Chapter35 ■ Clinical Toxicology 1119
of CN−. A good quality spectrophotometer is required to measure the absorbance. Quick and easy methods for plasma thiocyanate analysis have been described.802
Methemoglobin-Forming AgentsThe heme iron in hemoglobin is normally present in the ferrous state (Fe2+). When oxidized to the ferric state (Fe3+), methemoglobin is formed, and this form of hemo-globin cannot bind oxygen (Figure 35-1). The principal physiologic system that maintains hemoglobin iron in the reduced state is nicotinamide adenine dinucleotide (NADH)-methemoglobin reductase. The NADH for this enzyme is supplied by normal glycolysis (Embden-Meyerhof pathway). A minor pathway for methemoglobin reduction involves nicotinamide adenine dinucleotide phosphate (NADPH)-methemoglobin reductase, and the NADP for this enzyme reaction is derived from the hexose-monophosphate shunt. Congenital methemoglobinemia may result from a deficiency of NADH-methemoglobin reductase or, more rarely, from hemoglobin variants (hemoglobin M) in which heme iron is both more susceptible to oxidation and more resistant to reduction by the methemoglobin reductase system.
Toxic EffectsAn acquired (toxic) methemoglobinemia may be caused by various drugs and chemicals (Table 35-4). The normal per-centage of methemoglobin is <1.5% of total hemoglobin. In otherwise healthy individuals, methemoglobin percentages up to 20% may cause slate-gray cutaneous discoloration, cya-nosis, and chocolate-brown blood. Percentages between 20% and 50% may cause dyspnea, exercise intolerance, fatigue, weakness, and syncope. More severe symptoms of dysrhyth-mias, seizures, metabolic acidosis, and coma are associated with methemoglobin percentages of 50 to 70%, and >70% may be lethal.659,858 All of these symptoms are a consequence
and respiratory depression—which progress rapidly to coma, seizures, complete heart block, and death if the dose is suf-ficiently large.
Hydroxycobalamin or the cyanide antidote kit should be administered as soon as cyanide poisoning is suspected. Hydroxocobalamin, a vitamin B12 precursor, is a metallopro-tein with a central cobalt atom that complexes cyanide, forming cyanocobalamin (vitamin B12). Cyanocobalamin is eliminated in the urine or releases the cyanide moiety at a rate sufficient to allow detoxification by rhodanese. The cyanide antidote kit contains amyl nitrite, sodium nitrite, and sodium thiosulfate. Thiosulfate donates the sulfur atoms nec-essary for rhodanese-mediated cyanide biotransformation to thiocyanate. The mechanism of nitrite is less clear. The tradi-tional rationale relies on the ability of nitrite to generate methemoglobin. Because cyanide has a higher affinity for methemoglobin than for cytochrome a3, cytochrome oxidase function is restored.
Analytical MethodsFollowing microdiffusion, whole blood CN− is measured by photometric analysis179 or by headspace gas chromato-graphy.111
With the spectrophotometric method, a sealed, two-well microdiffusion cell is used to separate hydrocyanic acid from blood by mixing a sample of whole blood with strong acid in a sealed chamber and allowing the hydrocyanic acid gas gen-erated to be absorbed into a strong base located in another part of the sealed chamber. One well of the cell contains the blood specimen and strong acid (unmixed until the cell is sealed), and the other well contains a strong base to absorb the hydrocyanic acid gas. After the hydrocyanic acid is col-lected in the aqueous base medium, pyridine, barbituric acid, and chloramine-T are added to generate a red complex, with the intensity of the color proportional to the concentration
Figure 35-1 Enzymatic pathways for methemoglobin reduction.
NADP Leukomethyleneblue
Methemoglobin (Fe�3) 1,3 DPG
Glyceraldehyde-3-P
Glucose
Glycolysis
Hemoglobin (Fe�2)
Methyleneblue
NADPH
H
G6PD GDHNADPH MetHb
ReductaseNADPH MetHb
Reductase
Hexose shunt
Glucose
Glucose-6-P
6-Phosphogluconate
�
NAD��
NADH
H�
�
�

1120 SectionIII ■ Analytes
EthanolEthanol is the most widely used and often abused chemical substance. Consequently, measurement of ethanol is one of the more frequently performed tests in the toxicology labora-tory. Although less frequently encountered, it is important to include methanol, isopropanol, acetone (a metabolite of iso-propanol), and ethylene glycol in a test battery for alcohols for proper evaluation of the acutely intoxicated patient.
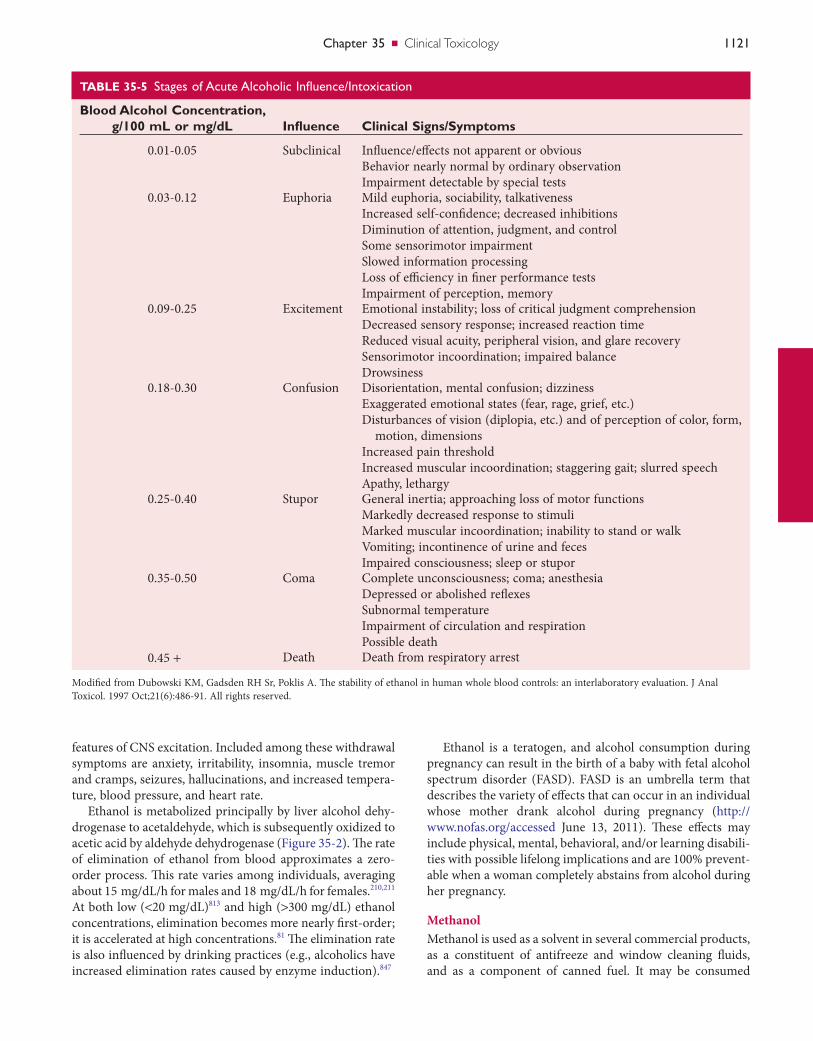
The principal pharmacologic action of ethanol is central nervous system (CNS) depression. CNS effects vary depend-ing on the blood ethanol concentration (Table 35-5) but are also heavily influenced by an individual’s tolerance. Symp-toms vary from euphoria and decreased inhibitions, to increased disorientation and incoordination, and then to coma and death. A blood alcohol concentration of 80 mg/dL (0.08%) has been established as the per se limit for operation of a motor vehicle in most countries.
Because of many factors, not all individuals experience the same degree of CNS dysfunction at similar blood alcohol concentrations. Moreover, the CNS actions of ethanol are more pronounced when the blood ethanol concentration is increasing (absorptive phase) than when it is declining (elim-ination phase), in part because of the phenomenon of acute tolerance.291 In addition, heavy alcohol use leads to a more chronic form of tolerance. When consumed with other CNS depressant drugs, ethanol exerts a potentiation or synergistic depressant effect. This can occur at relatively low alcohol concentrations, and numerous deaths have resulted from combined ethanol and drug ingestion.274
The pharmacologic mechanisms for the CNS depressant actions of ethanol are complex and incompletely understood, but probably involve both enhancement of major inhibitory neurons and impairment of excitatory neurons. The principal CNS inhibitory neuronal system is mediated by the neu-rotransmitter γ-aminobutyric acid (GABA). When GABA binds to its postsynaptic receptor subtype GABAA, this oligo-meric ion-gated complex “opens” to allow inward flux of Cl, leading to membrane hyperpolarization and subsequent decreased electrical response. This GABA-mediated inhibi-tory response is enhanced by ethanol and sedative, hypnotic, and anesthetic agents, including barbiturates, benzodiaze-pines, and volatile anesthetics.248 Neuronal nicotinic acetyl-choline receptors also may be prominent molecular targets of alcohol.576 Both enhancement and inhibition of nicotinic ace-tylcholine receptor function have been reported depending on receptor subunit concentration and the concentrations of ethanol tested. Ethanol also inhibits the function of the N-methyl-d-aspartate (NMDA)- and kainate-receptor sub-types; AMPA receptors are largely resistant to alcohol.119
The aforementioned chronic tolerance to ethanol is con-sidered to be mediated by ethanol-induced increased respon-siveness and upregulation in the synthesis of NMDA receptors, attained by concomitant downregulation and desensitization through phorphorylation of GABAA and glu-tamate receptors.248,452,819 Largely because of these adaptive changes, abrupt withdrawal from chronic, heavy ethanol use leads to a physical abstinence syndrome that has prominent
of hypoxia associated with the diminished O2 content of the blood, and with a decreased O2 dissociation from hemoglobin species in which some, but not all, subunits contain heme iron in the ferric state (i.e., shift of dissociation curve to the left). The PO2 is normal in these patients, and therefore so is the calculated hemoglobin oxygen saturation. Thus, a normal PO2 in a cyanotic patient is a significant indication for the possible presence of methemoglobinemia. Direct measure-ment of methemoglobin is important in these cases and may be performed by the manual spectrophotometric method of Evelyn and Malloy235 or by automated multiwavelength measurements with a co-oximeter (see section on “Carbon Monoxide”).
Specific therapy for toxic methemoglobinemia involves the administration of methylene blue, which acts as an elec-tron transfer agent in the NADPH-methemoglobin reductase reaction, thereby increasing the activity of this system several-fold.649,658 Methylene blue and sulfhemoglobin cause spectral interference in the measurement of methemoglobin with some co-oximeters412,875 but not with the Evelyn-Malloy method.412
Analytical MethodsMethemoglobin is measured in blood manually,235,412 or by automated multiwavelength measurements with a co- oximeter.325 Methemoglobin interferes with the noninvasive pulse oximetry method, measuring the absorbance of light at 660 nm (oxyhemoglobin) and 940 nm (deoxyhemoglobin). Because methemoglobin is not stable at room temperature, specimens should be kept on ice or refrigerated but not frozen.412 The stability of methemoglobin at 4 °C has not been well studied. Some sources indicate significant decreases in methemoglobin concentration after 4 to 8 hours,470 whereas others report little or no change after 24 hours.412 Freezing results in an increase in methemoglobin concentration.412
Alcohols of Toxicologic InterestSeveral alcohols are toxic and medically important.197A They include ethanol, methanol, isopropanol, acetone, and ethyl-ene glycol.
TABLE 35-4 ExamplesofAcquiredCausesofMethemoglobinemia
Drugs Chemical Agents
Amyl nitrite AnilineBenzocaine Amyl nitriteChloroquine Butyl nitriteDapsone ChlorobenzeneNitroglycerin NaphthalenePhenacetin NitratesPhenazopyridine NitritesPrimaquine NitrophenolSulfonamides Nitrous oxide
Chapter35 ■ Clinical Toxicology 1121
TABLE 35-5 StagesofAcuteAlcoholicInfluence/Intoxication
Blood Alcohol Concentration, g/100 mL or mg/dL Influence Clinical Signs/Symptoms
0.01-0.05 Subclinical Influence/effects not apparent or obviousBehavior nearly normal by ordinary observationImpairment detectable by special tests
0.03-0.12 Euphoria Mild euphoria, sociability, talkativenessIncreased self-confidence; decreased inhibitionsDiminution of attention, judgment, and controlSome sensorimotor impairmentSlowed information processingLoss of efficiency in finer performance testsImpairment of perception, memory
0.09-0.25 Excitement Emotional instability; loss of critical judgment comprehensionDecreased sensory response; increased reaction timeReduced visual acuity, peripheral vision, and glare recoverySensorimotor incoordination; impaired balanceDrowsiness
0.18-0.30 Confusion Disorientation, mental confusion; dizzinessExaggerated emotional states (fear, rage, grief, etc.)Disturbances of vision (diplopia, etc.) and of perception of color, form,
motion, dimensionsIncreased pain thresholdIncreased muscular incoordination; staggering gait; slurred speechApathy, lethargy
0.25-0.40 Stupor General inertia; approaching loss of motor functionsMarkedly decreased response to stimuliMarked muscular incoordination; inability to stand or walkVomiting; incontinence of urine and fecesImpaired consciousness; sleep or stupor
0.35-0.50 Coma Complete unconsciousness; coma; anesthesiaDepressed or abolished reflexesSubnormal temperatureImpairment of circulation and respirationPossible death
0.45 + Death Death from respiratory arrest
Modified from Dubowski KM, Gadsden RH Sr, Poklis A. The stability of ethanol in human whole blood controls: an interlaboratory evaluation. J Anal Toxicol. 1997 Oct;21(6):486-91. All rights reserved.
features of CNS excitation. Included among these withdrawal symptoms are anxiety, irritability, insomnia, muscle tremor and cramps, seizures, hallucinations, and increased tempera-ture, blood pressure, and heart rate.
Ethanol is metabolized principally by liver alcohol dehy-drogenase to acetaldehyde, which is subsequently oxidized to acetic acid by aldehyde dehydrogenase (Figure 35-2). The rate of elimination of ethanol from blood approximates a zero-order process. This rate varies among individuals, averaging about 15 mg/dL/h for males and 18 mg/dL/h for females.210,211 At both low (<20 mg/dL)813 and high (>300 mg/dL) ethanol concentrations, elimination becomes more nearly first-order; it is accelerated at high concentrations.81 The elimination rate is also influenced by drinking practices (e.g., alcoholics have increased elimination rates caused by enzyme induction).847
Ethanol is a teratogen, and alcohol consumption during pregnancy can result in the birth of a baby with fetal alcohol spectrum disorder (FASD). FASD is an umbrella term that describes the variety of effects that can occur in an individual whose mother drank alcohol during pregnancy (http://www.nofas.org/accessed June 13, 2011). These effects may include physical, mental, behavioral, and/or learning disabili-ties with possible lifelong implications and are 100% prevent-able when a woman completely abstains from alcohol during her pregnancy.
MethanolMethanol is used as a solvent in several commercial products, as a constituent of antifreeze and window cleaning fluids, and as a component of canned fuel. It may be consumed

1122 SectionIII ■ Analytes
it. Veterinarians are often familiar with ethylene glycol toxic-ity because of cases involving dogs or cats that drank radiator fluid.
Ethylene glycol itself is relatively nontoxic, and its initial CNS effects resemble those of ethanol.386 However, metabo-lism of ethylene glycol by alcohol dehydrogenase (ADH) results in the formation of numerous acid metabolites, including lactate, oxalic acid and glycolic acid.53,386 These acid metabolites are responsible for much of the toxicity of ethylene glycol.334,371 Serum concentrations associated with death from ethylene glycol ingestion have been observed to vary from 0.06 to 4.3 g/L,53,651 highlighting the lack of correla-tion between ethylene glycol concentration and severity of toxicity. It is thus impossible to define a serum ethylene glycol concentration associated with a high probability of death. The serum concentration of glycolic acid correlates more closely with clinical symptoms and mortality than does the concentration of ethylene glycol.334,651 Because of the rapid elimination of ethylene glycol (t1/2, 2 to 5 hours),53 its serum concentration may be low or undetectable at a time when glycolic acid remains elevated.257,334,651 Thus the determination of ethylene glycol and glycolic acid provides useful clinical and confirmatory analytical information in cases of ethylene glycol ingestion. The mainstay of therapy for ethylene glycol toxicity includes administration of ethanol or fomepizole as a competitive alcohol dehydrogenase inhibitor and dialysis.
Analysis of EthanolSerum, plasma, and whole blood are suitable blood-related specimens for the determination of ethanol. The venipunc-ture site should be cleansed with an alcohol-free disinfectant, such as aqueous benzalkonium chloride.
Serum/Plasma and Blood EthanolAlcohol distributes into the aqueous compartments of blood; because the water content of serum is greater than that of whole blood, higher alcohol concentrations are obtained with serum as compared with whole blood. Experimentally, the serum-to-whole blood ethanol ratio is 1.18 (1.10 to 1.35)628 and varies slightly with hematocrit.846 Therefore, laboratories that perform alcohol determinations should make clear the choice of specimen.
Because of the volatile nature of alcohols, specimens should be kept capped to avoid evaporative loss. Blood may be stored, when properly sealed, for 14 days at room tempera-ture or at 4 °C, with or without preservative.848 For longer storage or for nonsterile postmortem specimens, sodium fluoride should be used as a preservative to prevent a decrease or occasionally an increase (via fermentation) in ethanol concentration.
To measure ethanol in serum/plasma, enzymatic analysis is the method of choice for many laboratories. In this method, ethanol is measured by oxidation to acetaldehyde with NAD, a reaction catalyzed by ADH. With this reaction, the forma-tion of NADH, measured at 340 nm, is proportional to the amount of ethanol in the specimen268:
intentionally by alcoholics as an ethanol substitute or acci-dentally when present as a contaminant in illegal whiskey. Accidental ingestions have occurred in children.
The CNS effects of methanol are substantially less severe than those of ethanol. Methanol is oxidized by liver alcohol dehydrogenase (at about one tenth the rate of ethanol) to formaldehyde. Formaldehyde in turn is rapidly oxidized by aldehyde dehydrogenase to formic acid, which may cause serious acidosis and optic neuropathy, resulting in blindness or death.513,534 Serum formate concentrations correlate better with the degree of acidosis and the severity of CNS and ocular toxicity than do serum methanol concentrations.718 There-fore, some investigators recommend the measurement of serum formate to assess the severity of toxicity and to guide appropriate therapy in cases of methanol ingestion. The mainstay of therapy for methanol toxicity includes the admin-istration of ethanol or fomepizole as a competitive alcohol dehydrogenase inhibitor, either folate or folinic acid, and dialysis.
Isopropanol and AcetoneIsopropanol is readily available to the general population as a 70% aqueous solution for use as rubbing alcohol. It has about twice the CNS depressant action as ethanol, but it is not as toxic as methanol.58 Isopropanol has a short t1/2 of 2.5 to 3.0 hours,58 as it is rapidly metabolized by alcohol dehy-drogenase to acetone, which is eliminated much more slowly (t1/2, 3 to 6 hours).47 Therefore concentrations of acetone in serum often exceed those of isopropanol during the elimina-tion phase following isopropanol ingestion. Acetone has CNS depressant activity similar to that of ethanol, and because of its longer t1/2, it may prolong the apparent CNS effects of isopropanol.47,58 Supportive care is the mainstay of treatment, with rare reports of dialysis in severe intoxication.
Ethylene GlycolEthylene glycol, present in antifreeze products, may be ingested accidentally or for the purpose of inebriation or suicide. Because it tastes sweet, some animals are attracted to
Figure 35-2 Metabolism of ethanol.
Alcoholdehydrogenas
90-95%
CH3CH2OH
Ethyl alcohol
NAD
NADH
CH3CHO
Acetaldehyde
Aldehydedehydrogenas
NAD
NADH
CH3COOH
Acetic acid

Chapter35 ■ Clinical Toxicology 1123
a blood alcohol concentration measured at a later time. This process may be applied when certain assumptions are made concerning absorption rates, elimination rates, and patterns of alcohol consumption, including drinking duration and volume consumed. Unfortunately, to be forensically useful and scientifically valid, such extrapolations may require facts about the person and that person’s alcohol consumption, as well as related information that often is not available. Conse-quently, significant legal debate surrounds the validity and accuracy of retrograde extrapolation.
Breath EthanolStatutory laws for driving under the influence of alcohol were originally based on the concentration of ethanol in venous whole blood. Because the collection of blood is invasive and requires intervention by medical personnel, the determina-tion of alcohol in expired air (breath) has long been the main-stay of evidential alcohol measurements.321,389,516 Clinical interest in determination of breath alcohol at the point of care is growing. The fundamental principle for use of breath analy-sis is that alcohol in capillary alveolar blood rapidly equili-brates with alveolar air in a ratio of approximately 2100 : 1 (blood : breath). This blood-to-breath ratio may actually be closer to 2300 : 1 but in any case is variable. Nevertheless, in the United States, evidential breath alcohol measurements are based on the ratio of 2100 : 1. The lower blood-to-breath ratio will predict a slightly lower than actual blood alcohol concen-tration; its use therefore is not prejudicial. To alleviate confu-sion and uncertainty surrounding the conversion from breath to blood alcohol concentration, the traffic laws in many coun-tries specify per se limits for blood and/or breath.
Before breath alcohol analysis, a deprivation period of 15 minutes is required to allow for clearance of any residual alcohol that may have been present in the mouth (e.g., very recent drinking, use of alcohol-containing mouthwash, vomiting of alcohol-rich gastric fluid). Duplicate tests, per-formed 5 to 10 minutes apart and within 20 mg/dL (0.02%) are used as an additional safeguard against mouth alcohol contamination.
During the period of active alcohol absorption, generally 30 to 60 minutes depending on a variety of factors,592,728 and before peak blood alcohol concentration is obtained, the alcohol concentration in arterial blood will be initially higher than that in peripheral venous blood, and the converse is true in the postabsorptive phase.391 Because end-expiratory air equilibrates with pulmonary alveolar/capillary blood, the breath alcohol concentration more closely reflects that of arterial alcohol483; however, the difference between arterial and venous blood is within the analytical error of most assays.
Determination of ethanol in expired air requires special-ized breath alcohol analyzers. Several commercial evidential breath alcohol measurement devices are available. Principles of measurement used in such analyzers include (1) infrared absorption spectrometry (most common), (2) dichromate-sulfuric acid oxidation-reduction (photometric), (3) GC (flame ionization or thermal conductivity detection), (4) elec-trochemical oxidation (fuel cell), and (5) metal oxide
Ethanol NAD Acetaldehyde NADHADH+ → +
Under most assay conditions, ADH is reasonably specific for ethanol, with interferences by isopropanol, acetone, meth-anol, and ethylene glycol of typically <1%. However, spuri-ously increased results for ethanol have been described in the presence of high concentrations of lactate dehydrogenase (LDH) and lactate.34,590 This phenomenon is a result of the production of NADH by LDH:
Lactate NAD Pyruvate NADHLDH+ → +
Serum (or plasma) is the most common specimen for ethanol analysis by ADH methods; this method also performs well with urine or oral fluid, although in some methods, whole blood may be used directly117 or a precipitation step may be required before analysis to avoid interference from hemoglobin.269 Results from these methods generally compare closely with those from gas chromatographic methods.117,291 For more information about these methods, see “Analysis of Volatile Alcohols” section, later.
Estimation of Blood AlcoholDuring the early part of the twentieth century, Dr. Erik M.P. Widmark, a Swedish physician, did much of the foundational research regarding alcohol pharmacokinetics in the human body. In addition, he developed an algebraic equation that allows one to estimate the amount of alcohol consumed by an individual or the associated blood alcohol concentration when the values of the other variables are given489,840:
N W C t / d Zt= • • + • •ρ β[ ] ( )
N = number of drinksW = body weight (kg)ρ (rho) = volume of distribution (L/kg) (0.68 for males,
0.55 for females)Ct = blood alcohol concentration (kg/L)β = rate of ethanol elimination (0.15 g/L/h)t = time since first drink (h)d = specific gravity of alcohol (0.8)Z = amount of ethanol alcohol per drink (L) (15 mL of
ethanol in a standard drink)Typically one wants to calculate the amount of ethanol
consumed or the associated ethanol concentration. Note that it may be necessary to convert the units from those more commonly reported. It is important to remember that this formula is applicable only after completion of alcohol absorp-tion, and when equilibrium has been reached between blood and body tissue.
Frequently the time since the first drink is unknown; the formula can be modified to estimate the number of drinks in an individual’s system at the time of the test.
N W C / d Zt= • • •ρ [ ] ( )
The rate of elimination in the average person is commonly estimated at 0.015 g/100 mL/h (range, 0.010 to 0.030 g/ 100 mL/h).553 Retrograde extrapolation is an estimation of a subject’s alcohol concentration at a prior time, derived from

1124 SectionIII ■ Analytes
Specimens are prepared by a variety of methods; the two most common are direct injection and headspace analysis. Direct injection involves injection of a sample prepared by diluting it with an aqueous solution of internal standard (thus reduc-ing the amount of matrix introduced into the GC). Repeated injection of biological aqueous matrix into the GC will cause buildup on the injector and front of the analytical column, requiring frequent maintenance and column replacement. This can be alleviated by the use of headspace injection. The volatility of the alcohols is used to separate them from the matrix. Specifically, the “Gas Law” states that at a given tem-perature, the amount of volatile substance in the air space above the liquid—headspace—is proportional to the concen-tration of the volatile alcohol in the solution. Therefore, the sample in the headspace allows calculation of the concentra-tion in the specimen.
Headspace gas chromatographic analysis is another excel-lent method for the measurement of methanol, isopropanol, acetone, and ethanol. In addition, an adaptation of this tech-nique may be used to measure formate, the toxic metabolite of methanol, after esterification to methyl formate. Con-versely, direct injection GC is the method of choice for eth-ylene glycol, because it has a higher boiling point and is not as amenable to headspace analysis. A modification of the GC procedure described in 2005 has the potential of combining both toxic alcohols in a single GLC analysis.405 Methods that simultaneously measure ethylene glycol and glycolic acid have the advantage of being free from interference by propyl-ene glycol (a diluent for parenteral drugs) or 2,3-butanediol (may be present in serum from some alcoholics).652,867 Similar techniques are used to measure volatile alcohols in blood, serum, oral fluid, urine, other clinical specimens, and post-mortem specimens (e.g., vitreous fluid, skeletal muscle).
Analgesics (Nonprescription)Analgesics are substances that relieve pain without causing loss of consciousness. When used in excess, analgesics such as acetaminophen and salicylate can result in a toxic response.
AcetaminophenAcetaminophen has analgesic and antipyretic actions. In common with the group of drugs referred to as nonsteroidal anti-inflammatory drugs (NSAIDs; e.g., aspirin, ibuprofen, indomethacin), the pharmacologic actions of acetaminophen are related to its competitive inhibition of cyclooxygenase enzymes. This results in decreased production of prostaglan-dins, which are important mediators of inflammation, pain (low to moderate), and fever.106 Contrary to other NSAIDs, acetaminophen has very weak anti-inflammatory activity—a consequence of its weak inhibition of peripheral tissue cyclo-oxygenase compared with that in the brain. In normal doses, acetaminophen is safe and effective, but it may cause severe hepatic toxicity or death when consumed in overdose quanti-ties. Less frequently, nephrotoxicity may also occur. The initial clinical findings in acetaminophen toxicity are relatively mild and nonspecific (nausea, vomiting, and
semiconductor sensors.321,389 Breath alcohol devices also may be used for the medical evaluation of patients at the point of care (e.g., emergency department).
Oral Fluid EthanolBecause oral fluid (saliva) may be easily and noninvasively collected, interest is growing in its use for ethanol measure-ments and for the detection of drugs of abuse, but it is not a frequently used sample for ethanol determinations (see section below on “Detection of Drugs of Abuse Using Other Types of Specimens”).
Urine EthanolUrine has been used as an alternative, less invasive specimen for the determination of alcohol use. During the postabsorp-tive phase following alcohol ingestion, the concentration of alcohol in urine is roughly 1.3 times that in blood.52,115 However, the use of urine alcohol measurements to estimate blood concentrations is discouraged because the ratio of 1.3 is highly variable, and, perhaps more important, the urine alcohol concentration may better reflect an average of the blood alcohol concentration during the period in which urine is collected in the bladder. The detection of alcohol in urine represents ingestion of alcohol within the previous 8 to 12 hours.
EthylglucuronideEthylglucuronide (EtG) is a phase II metabolite of ethanol formed through the UDP-glucuronosyl transferase–catalyzed conjugation of ethanol with glucuronic acid.182 Because of its long urinary elimination time, its specificity for ethanol expo-sure, and the low detection limits of assays, the use of EtG has been proposed as a marker of recent ethanol intake in a variety of clinical and legal settings, including medical moni-toring for relapse, emergency department patient evaluation, postmortem assessment, and transportation accident investi-gation.613,864 However, challenges associated with factors such as establishing appropriate cutoff concentrations capable of distinguishing between drinking and nonbeverage sources of ethanol exposure, nonuniform laboratory reporting limits, sample stability, and microbial activity substantially compli-cate accurate interpretation of results.613
Analysis of Volatile Alcohols (Methanol, Isopropanol, and Acetone)Methanol poisoning can be lethal if not recognized early. Unfortunately, in some instances, a latent period can be as long as 12 to 24 hours728 before toxicity is recognized, making laboratory identification of this poisoning critical. Develop-ment of gas chromatographic methods for volatiles in 1964217,333 was a significant step in the recognition and treat-ment of this very toxic alcohol.
Flame ionization GC remains the most common method for the detection and quantitation of volatile alcohols in bio-logical samples.52 Not only does it distinguish between ethanol, methanol, isopropanol, and acetone, it has the capa-bility to measure concentrations as low as 10 mg/dL (0.01%).
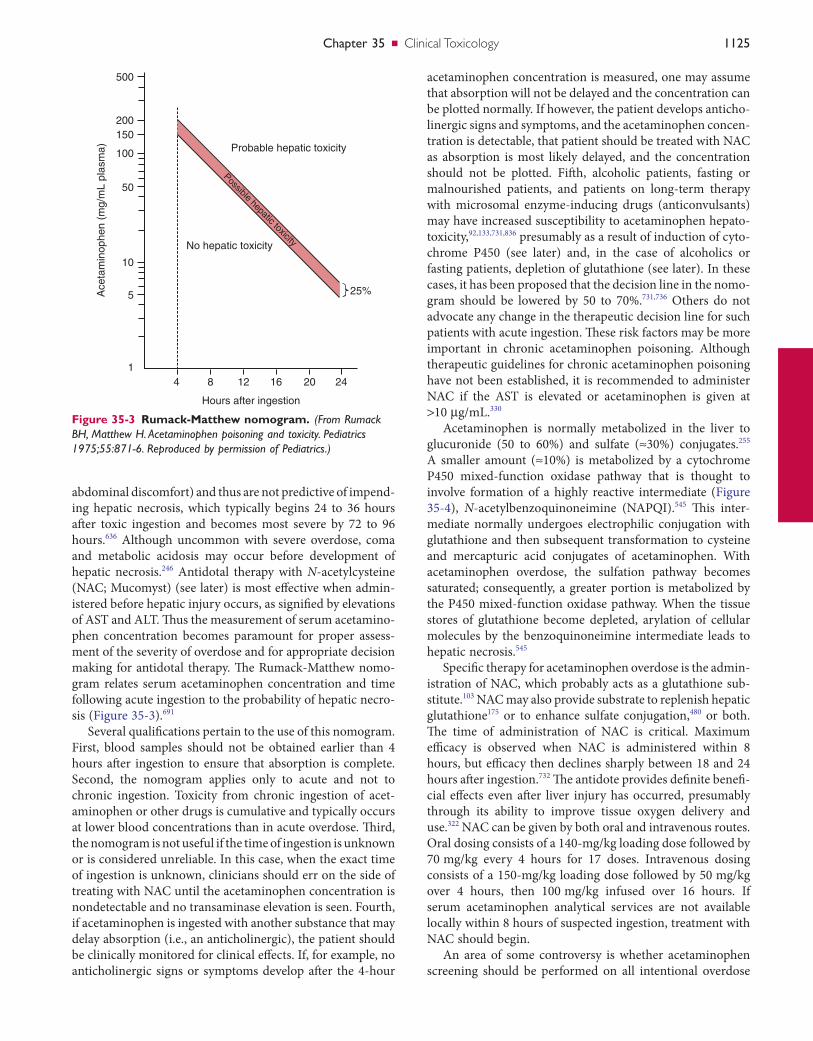
Chapter35 ■ Clinical Toxicology 1125
acetaminophen concentration is measured, one may assume that absorption will not be delayed and the concentration can be plotted normally. If however, the patient develops anticho-linergic signs and symptoms, and the acetaminophen concen-tration is detectable, that patient should be treated with NAC as absorption is most likely delayed, and the concentration should not be plotted. Fifth, alcoholic patients, fasting or malnourished patients, and patients on long-term therapy with microsomal enzyme-inducing drugs (anticonvulsants) may have increased susceptibility to acetaminophen hepato-toxicity,92,133,731,836 presumably as a result of induction of cyto-chrome P450 (see later) and, in the case of alcoholics or fasting patients, depletion of glutathione (see later). In these cases, it has been proposed that the decision line in the nomo-gram should be lowered by 50 to 70%.731,736 Others do not advocate any change in the therapeutic decision line for such patients with acute ingestion. These risk factors may be more important in chronic acetaminophen poisoning. Although therapeutic guidelines for chronic acetaminophen poisoning have not been established, it is recommended to administer NAC if the AST is elevated or acetaminophen is given at >10 µg/mL.330
Acetaminophen is normally metabolized in the liver to glucuronide (50 to 60%) and sulfate (≈30%) conjugates.255 A smaller amount (≈10%) is metabolized by a cytochrome P450 mixed-function oxidase pathway that is thought to involve formation of a highly reactive intermediate (Figure 35-4), N-acetylbenzoquinoneimine (NAPQI).545 This inter-mediate normally undergoes electrophilic conjugation with glutathione and then subsequent transformation to cysteine and mercapturic acid conjugates of acetaminophen. With acetaminophen overdose, the sulfation pathway becomes saturated; consequently, a greater portion is metabolized by the P450 mixed-function oxidase pathway. When the tissue stores of glutathione become depleted, arylation of cellular molecules by the benzoquinoneimine intermediate leads to hepatic necrosis.545
Specific therapy for acetaminophen overdose is the admin-istration of NAC, which probably acts as a glutathione sub-stitute.103 NAC may also provide substrate to replenish hepatic glutathione175 or to enhance sulfate conjugation,480 or both. The time of administration of NAC is critical. Maximum efficacy is observed when NAC is administered within 8 hours, but efficacy then declines sharply between 18 and 24 hours after ingestion.732 The antidote provides definite benefi-cial effects even after liver injury has occurred, presumably through its ability to improve tissue oxygen delivery and use.322 NAC can be given by both oral and intravenous routes. Oral dosing consists of a 140-mg/kg loading dose followed by 70 mg/kg every 4 hours for 17 doses. Intravenous dosing consists of a 150-mg/kg loading dose followed by 50 mg/kg over 4 hours, then 100 mg/kg infused over 16 hours. If serum acetaminophen analytical services are not available locally within 8 hours of suspected ingestion, treatment with NAC should begin.
An area of some controversy is whether acetaminophen screening should be performed on all intentional overdose
abdominal discomfort) and thus are not predictive of impend-ing hepatic necrosis, which typically begins 24 to 36 hours after toxic ingestion and becomes most severe by 72 to 96 hours.636 Although uncommon with severe overdose, coma and metabolic acidosis may occur before development of hepatic necrosis.246 Antidotal therapy with N-acetylcysteine (NAC; Mucomyst) (see later) is most effective when admin-istered before hepatic injury occurs, as signified by elevations of AST and ALT. Thus the measurement of serum acetamino-phen concentration becomes paramount for proper assess-ment of the severity of overdose and for appropriate decision making for antidotal therapy. The Rumack-Matthew nomo-gram relates serum acetaminophen concentration and time following acute ingestion to the probability of hepatic necro-sis (Figure 35-3).691
Several qualifications pertain to the use of this nomogram. First, blood samples should not be obtained earlier than 4 hours after ingestion to ensure that absorption is complete. Second, the nomogram applies only to acute and not to chronic ingestion. Toxicity from chronic ingestion of acet-aminophen or other drugs is cumulative and typically occurs at lower blood concentrations than in acute overdose. Third, the nomogram is not useful if the time of ingestion is unknown or is considered unreliable. In this case, when the exact time of ingestion is unknown, clinicians should err on the side of treating with NAC until the acetaminophen concentration is nondetectable and no transaminase elevation is seen. Fourth, if acetaminophen is ingested with another substance that may delay absorption (i.e., an anticholinergic), the patient should be clinically monitored for clinical effects. If, for example, no anticholinergic signs or symptoms develop after the 4-hour
Figure 35-3 Rumack-Matthew nomogram. (From Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975;55:871-6. Reproduced by permission of Pediatrics.)
Possible hepatic toxicity
Probable hepatic toxicity
No hepatic toxicity
25%Ace
tam
inop
hen
(mg/
mL
plas
ma)
200
500
150
100
50
10
5
14 8 12 16 20 24
Hours after ingestion

1126 SectionIII ■ Analytes
patients.75 One of the most worrisome aspects of acetamino-phen poisoning is that initial clinical symptoms (e.g., nausea, vomiting, abdominal pain) may be vague or even absent in the first 24 hours.76 This possible delay in diagnosis is particu-larly problematic because the antidote, NAC, has been shown to be most effective when initiated within the first 8 hours.732 Studies looking at the issue of universal acetaminophen screening recommend screening all patients with suicidal ingestion and those with altered mental status in whom inges-tion is suspected.31,750
Many spectrophotometric methods are available for the determination of acetaminophen.95,842 In general, these methods are relatively easy to perform but are subject to various interferences such as bilirubin or bilirubin bypro-ducts absorbing at similar wavelengths.95,648 Some methods measure the nontoxic metabolites and the potentially toxic parent acetaminophen, and thus may produce especially mis-leading results. Therefore, only methods specific for parent acetaminophen should be used.650 Immunoassays are widely used for this purpose, as they are rapid, easily performed, and accurate. A different spectrophotometric approach uses ary-lacylamide amidohydrolase to hydrolyze acetaminophen (but not conjugates) to p-aminophenol and acetate. Subsequent formation of the absorbing species depends on the reaction of generated p-aminophenol with 8-hydroxyquinoline560 or o-cresol.657 Arylacylamide amidohydrolase methods are
Figure 35-4 Pathways of acetaminophen metabolism. APAP (N-acetyl-p-aminophenol/acetaminophen), NAPQI (N-acetyl-p-benzo-quinone imine), and NAC (N-acetyl-l-cysteine).
NHH3C
O
OHAPAP
CYP2E1CYP1A2CYP3A4CYP2A6
NHH3C
O
ONAPQI
Cellular Toxicity
5-15%
NAC
APAP
APAP-glucuronide
APAP-sulfate
Non-toxicMetabolic products
NHH3C
O
OH
S Glutathione
Glutathionecysteine
mercapturic acid
NAC
Glutathione
Excreted
UDP-Glucuronosyl transferase40-67%
Renal, unchanged5%
UDP-Sulfate transferase20-46%
susceptible to interference by NAC,526 bilirubin, and immu-noglobulin (Ig)M monoclonal immunoglobulins.351 Most chromatographic methods are very accurate and are consid-ered reference procedures.748 A qualitative, one-step lateral flow immunoassay (cutoff of 25 µg/mL) may be suitable for point-of-care application, yet it has a low positive predictive value.184
SalicylateAcetylsalicylic acid (aspirin) has analgesic, antipyretic, and anti-inflammatory properties. These therapeutic benefits derive from its ability to inhibit biosynthesis of prostaglan-dins by acetylation of active site serine and subsequent irre-versible inhibition of cyclooxygenase enzymes (COX-1; COX-2 isoenzymes).106 Salicylate, the metabolite of aspirin, also reduces prostaglandin synthesis by uncertain mecha-nisms. Because of these therapeutic benefits and the general lack of serious side effects at normal doses, aspirin is widely available and frequently consumed. Therapeutic serum salic-ylate concentrations are generally lower than 60 mg/L for analgesic-antipyretic effects, and 150 to 300 mg/L for anti-inflammatory actions.106
Aspirin also interferes with platelet aggregation and thus prolongs bleeding time. The platelet inhibitory effect is a consequence of the ability of aspirin to acetylate and irrevers-ibly inhibit platelet cyclooxygenase, thereby reducing the

Chapter35 ■ Clinical Toxicology 1127
consequence of increased brain uptake of nonionized sali-cylic acid. Respiratory acidosis, a result of severe CNS depres-sion or pulmonary edema, may sometimes occur and is indicative of a poor prognosis.
Salicylates remain readily available in numerous over-the-counter products. In any patient with a history of salicylate ingestion or possessing characteristic signs or symptoms of salicylate poisoning, a serum salicylate concentration should be obtained. Early identification of salicylate toxicity can be lifesaving.
Following acute salicylate overdose, patients initially may be asymptomatic, especially if that product is enteric coated. Salicylate toxic patients may develop nausea, vomiting, abdominal pain, tinnitus, tachypnea, oliguria, and altered mental status ranging from lethargy to coma.767 Chronic intoxication can present in a similar fashion as acute expo-sures, yet such exposures typically are more insidious and therefore are often misdiagnosed.250
Interpretation of salicylate concentrations as a guide for clinical management decisions can be difficult. Perhaps the most well-known attempt at utilizing salicylate concentra-tions to predict the severity of salicylate toxicity was the nomogram developed by Done.200 After examining both the clinical symptoms and the salicylate concentrations in patients who had a single acute overdose, Done created a nomogram that predicted severity of poisoning based on the salicylate concentration drawn at a given time from ingestion. This tool has significant limitations. Because this nomogram as originally developed was based on only 38 pediatric patients, its utility for acute adult overdose is not known. One of the assumptions allowing the creation of this nomogram was that salicylates are eliminated by first-order kinetics. It has since been well established that some of the pathways for elimination of salicylates become saturated in overdose and follow zero-order kinetics.579 One study demonstrated signifi-cant disagreement between the clinical severity predicted by the nomogram and the severity judged by physicians.214 Therefore, we no longer recommend its use in the manage-ment of the salicylate-poisoned patient.
Use of salicylate concentrations to guide management must be done cautiously and only in conjunction with careful evaluation of a patient’s clinical status. One group of investi-gators examined 97 patients who experienced significant exposure to this agent. Patients who did not survive the inges-tion and patients with reasonably high serum concentrations (≥700 mg/L) were included in this study.129 Although toxic concentrations alone were of poor prognostic value, the investigators did identify certain clinical findings that pre-dicted a poor prognosis, including pulmonary edema, fever, coma, and acidosis. The absorptive phase of salicylates can be unpredictable (delayed or erratic) as a result of bezoar forma-tion, enteric-coated product, gastric outlet obstruction, or pylorospasm.250 Therefore, a concentration drawn soon after the original ingestion may not be reflective of the potential peak concentration. Initial serial concentrations should be performed every 2 hours while the patient is monitored clini-cally. When the concentrations begin to decline and the
formation of thromboxane A2, a potent mediator of platelet aggregation. Platelets have little or no capacity for protein synthesis; therefore, the duration of this enzyme inhibition is the normal life span of the platelets (8 to 11 days).686 Because of this platelet inhibitory activity, low-dose aspirin has been recommended as prophylactic therapy for some individuals at risk for thromboembolic disease.621,753 An epidemiologic association has been noted between aspirin ingestion and Reye syndrome in children and adolescents with viral infec-tion (e.g., varicella, influenza).353 Therefore, aspirin use is contraindicated in these patients.
Absorption of normal doses of regular aspirin from the GI tract is generally rapid, with peak serum concentration achieved within 2 hours.106 This peak value may be delayed for 12 hours or longer for enteric-coated or slow-release for-mulations.447 Moreover, toxic doses of aspirin may form con-cretions or bezoars and produce pylorospasm, thereby delaying absorption. Serum salicylate in such instances may not reach maximum concentration for 6 hours or longer200—an important consideration when assessment of the severity of toxicity is based on such measurements.
Once absorbed, aspirin has a very short half-life (t1/2 = 15 minutes) because of its rapid hydrolysis to salicylate. Salicylate is eliminated mainly by conjugation with glycine to form salicyluric acid, and to a lesser extent with gluc-uronic acid to form phenol and acyl glucuronides.649 A very small amount is hydroxylated to gentisic acid. These meta-bolic pathways may become saturated even at high thera-peutic doses. Consequently, serum salicylate concentration may increase disproportionately with dosage. At high thera-peutic or toxic doses, the salicylate elimination half-life is prolonged (15 to 30 hours vs. 2 to 3 hours at low dose) and a much larger portion of the dose is excreted in urine as salicylate.106
Salicylates directly stimulate the central respiratory center and thereby cause hyperventilation and respiratory alkalosis. Moreover, salicylates cause uncoupling of oxidative phos-phorylation. As a result, heat production (hyperthermia), oxygen consumption, and metabolic rate may be increased. In addition, salicylates enhance anaerobic glycolysis but inhibit the Krebs cycle and transaminase enzymes, all of which lead to accumulation of organic acids and thus to metabolic acidosis.798
The primary acid-base disturbance observed with salicy-late overdosage depends on age and severity of intoxication. Respiratory alkalosis predominates in children over age 4 and in adults, except in very severe cases that may progress through a mixed respiratory alkalosis–metabolic acidosis to metabolic acidosis. Among 97 adult patients who had plasma salicylate concentrations greater than 700 mg/L, 19% were found to have respiratory alkalosis, 61% had combined respi-ratory alkalosis and metabolic acidosis, and 15% had meta-bolic acidosis. Mortality was associated with acidemia.129 In children younger than age 4, the initial period of respiratory alkalosis is very brief and therefore may not be observed; in such cases, metabolic acidosis predominates.798 CNS depres-sion is more pronounced when acidemia is severe, which is a

1128 SectionIII ■ Analytes
Classic methods for the measurement of salicylate in serum are based on the method of Trinder (see “Spot Tests”).782 These procedures rely on the reaction between salicylate and Fe3+ to form a colored complex that is measured at 540 nm. To lessen endogenous background interference, a protein precipitation step or a serum blank is necessary. Nevertheless, blank readings equivalent to about 20 to 25 mg/L are gener-ally observed. Moreover, interference by salicylate metabo-lites, endogenous compounds, and some drugs, especially structurally related drugs such as diflunisal (difluorophenyl salicylate),704 may occur. Azide, present as a preservative in some commercial control sera, also causes interference. Despite these limitations, photometric methods continue to be successfully used to assess salicylate overdose. The Trinder method results agreed very closely with those of a reference HPLC procedure.375 However, significant interference with the Trinder method was observed for one patient, who con-sumed an overdose of dichloralphenazone. Thus for best interpretation of test results, as much information as possible should be obtained regarding drug ingestion history.
Other methods for salicylate quantitation include fluores-cent polarization immunoassay406 and a salicylate hydroxylase–mediated photometric procedure.561,649 These procedures are subject to some of the same interferences as the Trinder method, but the salicylate hydroxylase method is considered more specific561 and has been adapted to automated analyzers. Gas and liquid chromatographic methods are the most spe-cific methods for salicylate,125,665 but their general availability, especially for emergency use, is limited and probably is not necessary. A qualitative, one-step lateral flow immunoassay (cutoff of 100 µg/mL) is commercially available for point-of-care application but has a low positive predictive value (0.47).184
Agents Related to the Anticholinergic Toxidrome125
The tricyclic antidepressants, the phenothiazines, and the antihistamines have divergent therapeutic applications; however in overdose, they often share similar anticholinergic and antihistaminic toxidromes as principal components of their overall toxic effects.
Tricyclic AntidepressantsTricyclic antidepressants (TCAs), so named because of their three-ring structure (Figure 35-5), represent a class of drugs frequently prescribed for the treatment of depression (see Chapter 34). The TCAs have been largely supplanted by the newer, less toxic selective serotonin reuptake inhibitors (SSRIs) and other atypical agents, which now are accepted broadly as drugs of first choice, particularly for medically ill or potentially suicidal patients and for the elderly and the young.97,249,605,730,838 Fatalities are much less common since modern antidepressants have widely replaced these drugs. However, because of their continued use and narrow thera-peutic range, and because of the nature of the illness for which they are typically prescribed, TCAs are frequently associated with severe or fatal toxicity.187,479,632
patient’s clinical status is improved, concentrations can be measured less frequently.
The units reported with each concentration should be documented before management decisions are made. Labo-ratories may alternatively report concentrations in terms of mg/dL and mg/L. This important distinction, which involves a tenfold difference in concentration, is infamous for causing confusion. In extreme cases of these miscommunications, hemodialysis has been ordered for patients thought to have astronomically high salicylate concentrations that were later proven to be nontoxic.312
The need to screen all intentional overdose patients for salicylates is highly debated.127,128,750 Diagnosis of salicylate poisoning based solely on clinical examination is not without pitfalls. Although large, acute ingestions are usually detected through history and clinical symptoms, chronic salicylate toxicity often is more difficult to diagnose. Numerous cases have been reported pertaining to a delayed or mistaken diag-nosis in the face of significant salicylate toxicity. In these cases, patients presented with nonspecific symptoms such as fever, abdominal pain, and encephalopathy and subsequently were misdiagnosed with surgical abdomen, myocardial infarction, sepsis, encephalitis, and alcoholic ketoacido-sis.142,250,465,626 One study revealed that delayed diagnosis (at times up to 72 hours) of chronic salicylate poisoning is associ-ated with higher morbidity and mortality rates compared with diagnosis on admission.18 Another study involving salicylate-related fatalities in Ontario revealed that symptoms and signs of salicylate poisoning were apparently missed even in patients who were alert on presentation.533 The “classic” finding of ototoxicity was described by one group of investi-gators to be neither sensitive nor specific of serum salicylate concentration.316 Characteristic laboratory findings as well may not be reliable. Although a wide anion gap metabolic acidosis with respiratory alkalosis is often encountered in association with salicylate poisoning, one study involving 20 elderly patients with chronic salicylate poisoning revealed that 35% cases presented with a normal anion gap and PCO2.36
Because products containing salicylates are readily avail-able, the clinical effects of salicylate toxicity are nonspecific, and lack of metabolic acidosis does not rule out the potential for salicylate toxicity, clinicians should have a low threshold for obtaining serum salicylate concentrations.
Treatment for salicylate intoxication is directed toward (1) decreasing further absorption, (2) increasing elimination, and (3) correcting acid-base and electrolyte disturbances. Activated charcoal binds aspirin and prevents its absorption. Elimination of salicylate may be enhanced by alkaline diure-sis and in severe cases by hemodialysis.798 Sodium bicarbon-ate may be given to alleviate metabolic acidosis. Indications for hemodialysis include serum salicylate >1000 mg/L, severe CNS depression, intractable metabolic acidosis, hepatic failure with coagulopathy, and renal failure.252
A urine drug screen may be helpful in detecting the pres-ence of drugs included as part of combination medications with aspirin (e.g., antihistamines, sympathomimetic amines, propoxyphene) or that otherwise are coingested.

Chapter35 ■ Clinical Toxicology 1129
monoamine transport, and its clinical actions remain unexplained.38
The TCAs have many other pharmacologic actions that apparently do not contribute to the therapeutic effects but do contribute to the side effects. For example, most TCAs have at least moderate affinity for α1-adrenergic receptors, much less for α2, and virtually none for β-receptors,38 leading to hypotension, dizziness, and sedation.19 TCAs also have seda-tive effects that may be related to antihistamine activity.19 Tertiary amines produce greater sedation than secondary amines. TCAs also exert central and peripheral anticholiner-gic effects (dry skin and mouth, flushing, hyperpyrexia, dilated pupils, constipation, urinary retention, and decreased GI motility) through their interaction with M1 muscarinic receptors.19
Cardiovascular toxicity, the most serious manifestation of TCA overdose, accounts for the majority of fatalities. Cardio-vascular effects include orthostatic hypotension, sinus tachy-cardia, and variable prolongation of cardiac conduction times with the potential for arrhythmias, particularly with over-dose.38 The anticholinergic effect, mediated through M1 blockade, and sympathomimetic effects contribute to cardiac dysrhythmias.19 In mild overdose, these effects result in tachycardia and a slight increase in blood pressure. With more severe overdose, serious arrhythmias and conduction delays may develop, of which the most distinct feature is prolongation of the QRS interval in the electrocardiogram. Cardiac output decreases coupled with peripheral vasodilata-tion (α1-adrenergic blockade) lead to life-threatening hypo-tension. Death often results from arrhythmias or hypotension. Cardiotoxic manifestations may occur within a few hours of overdose, or they may be delayed. It is important to recognize that a patient’s symptoms (perhaps initially only mild anti-cholinergic effects) are due to tricyclic antidepressants, so that a proper period of monitoring for delayed and possibly catastrophic cardiotoxicity is followed.
In general antidepressants are associated with several clin-ically important drug interactions,471 and they potentiate the effects of alcohol and probably other sedatives.38 Virtually any agent with serotonin-potentiating activity, including TCAs, can interact dangerously or even fatally with monoamine oxidase (MAO) inhibitors (particularly long-acting MAO inhibitors).233 The resulting reactions are referred to as sero-tonin syndrome.754
Some tolerance to the sedative and autonomic effects of TCAs tends to develop with continued drug use.38 Occasion-ally, patients show physical dependence, with malaise, chills, muscle aches, and sleep disturbance following abrupt discon-tinuation, particularly of high doses.38 Some withdrawal effects may reflect increased cholinergic activity following its inhibition.38 Some of these reactions have been confused with clinical worsening of depressive symptoms. Emergence of agitated or manic reactions has been observed after abrupt discontinuation of TCAs.38
TCAs are oxidized by hepatic cytochrome P450 (CYP) microsomal enzymes, followed by conjugation with gluc-uronic acid.38 The N-demethylated metabolites of several
Tricyclic antidepressants block neuronal uptake of sero-tonin and/or norepinephrine.19 In general, TCAs with secondary-amine side chains or the N-demethylated (nor) metabolites of agents with tertiary-amine moieties (e.g., desipramine, norclomipramine, nordoxepin, nortriptyline) are relatively selective inhibitors of norepinephrine trans-port.19,38 However, tertiary TCAs (amitriptyline, doxepin, and imipramine) are less selective and inhibit the reuptake of serotonin.19,38 Clomipramine, a notable exception, is relatively selective against serotonin.38 Among the TCAs, trimipramine is exceptional in that it lacks prominent inhibitory effects at
Figure 35-5 Structure of tricyclic antidepressants and related drugs.
CH
CH2
Amitryptyline
CH2
NCH3H3C
CH
CH2
Cyclobenzaprine
CH2
NCH3H3C
CH
CH2
Nortriptyline
CH2
NCH3H
CH
O
CH2
Doxepin
CH2
NCH3H3C
CH2
N
CH2
Imipramine
CH2
NCH3H3C
CH2
N
CH2
Desipramine
CH2
NCH3H
CH2Cl
N
CH2
Clomipramine
CH2
NCH3H3C
CH2
N
HC CH3
Trimipramine
CH2
NCH3H3C

1130 SectionIII ■ Analytes
ion channels.100,109,751,758 The atypical antipsychotics on the other hand have a different mechanism that my involve other dopamine receptors, serotonin receptors, or both.456
The principal manifestations of phenothiazine toxicity involve the CNS and the cardiovascular system.38,65,494 The presentation for most of these drugs is qualitatively similar to that following TCA overdose, but in general they are less toxic. The most common effects in significant phenothiazine overdose include (1) sedation, (2) hypotension, (3) small pupils, (4) anticholinergic effects, and (5) ECG changes.100,102,109 Phenothiazines are relatively safe and rarely cause death when ingested alone. More severe toxicity occurs when phenothi-azines are coingested with tricyclic antidepressant drugs or other CNS depressant drugs, such as ethanol, opioids, barbi-turates, or benzodiazepines.
All of the neuroleptic drugs are metabolized in the liver. Many have active metabolites and complex metabolic path-ways. The main enzymes involved in metabolism are cyto-chrome P450 (CYP) enzymes, specifically CYP1A2, CYP2D6, and CYP3A4.109,183,780 Many sources of variation are found in CYP-mediated metabolism; however, where multiple enzymes are involved, such variability has only a relatively small effect on clearance and drug concentrations.
Toxicity is strongly correlated with peak serum concentra-tions and thus usually occurs within the first 4 to 6 hours after
tricyclic antidepressants are pharmacologically active and may accumulate in concentrations approaching or exceeding those of the parent drug, to contribute variably to overall pharmacodynamic activity (see Chapter 34).
Analytical MethodsTricyclic antidepressants are measured by chromatographic or immunoassay methods (see Chapters 13 and 16). Immu-noassays are rapid and relatively easy to perform but may be subject to interference by other drugs, such as chlorproma-zine,615 thioridazine,694 cyclobenzaprine,615 and diphenhydr-amine,746 and are not able to necessarily identify which TCA is being quantitated. In cases of overdose, qualitative identi-fication (serum or urine) is sufficient, because the severity of intoxication is more reliably indicated by an increase in the QRS interval than by the serum concentration.79
Cyclobenzaprine, a tricyclic amine structurally very similar to amitriptyline (see Figure 35-5), is used as a cen-trally acting skeletal muscle relaxant. Similar to amitriptyline, cyclobenzaprine causes sedation, produces central and peripheral muscarinic blockade, and potentiates adrenergic actions. In overdose, cyclobenzaprine may cause a typical anticholinergic toxidrome and cardiac arrhythmias, hypoten-sion, and coma. The analytical distinction between amitrip-tyline and cyclobenzaprine is often difficult. Cyclobenzaprine cross-reacts with immunoassays for tricyclic antidepressants and can coelute or comigrate with amitriptyline in HPLC and TLC. However, cyclobenzaprine and amitriptyline have dif-ferent ultraviolet spectra; therefore, they may be distinguished by HPLC using a diode array detector by multiwavelength scanning or dual-wavelength discrimination.196,644 However, amitriptyline and cyclobenzaprine are well resolved using capillary column GC and may be distinguished by careful examination of their respective mass spectra.853
Antipsychotic DrugsThe antipsychotic drugs are generally used for primary psychiatric disorders such as (1) schizophrenia, (2) bipolar disorder, (3) schizoaffective disorder, and (4) psychotic depression. In addition to their psychotherapeutic effects, these drugs have a number of other actions, so that certain members of this group are used as antiemetics (prochlorpera-zine), as antihistaminics (promethazine),685 and for sedation or potentiation of analgesia and general anesthesia.38 Anti-psychotic compounds are traditionally divided and sub-divided according to their chemical structure (Table 35-6 and Figure 35-6).456,685
The exact mechanism of action of antipsychotic drugs is not known; however, the primary pharmacologic effect of all antipsychotic drugs is thought to be blockade of D2 receptors in the central nervous system.38,456,673 The idea comes from studies showing that the ability of classical antipsychotics to reduce psychotic symptoms correlates with their affinity for the D2 receptor.456 However, the drugs are pharmacologically “dirty” and bind to many other receptors, including histamine (H1, H2), GABAA, muscarinic receptors, α1- and α2-adrenoreceptors, and sodium and potassium voltage-gated
TABLE 35-6 ExamplesofClassicalandAtypicalAntipsychotics
Antispychotics Examples39,100,456,685
Classical AntipsychoticsPhenothiazines Chlorpromazine
PromethazineTrichlorperazinePerphenazineFluphenazineThioridazineMesoridazineTrifluoperazine
Thioxanthines FlupenthizolZuclopenthixol
Dibenzoxazepine LoxapineDihydroindoles MolindoneButyrophenones Droperidol
HaloperidolDiphenylbutylpiperidines PimozideBenzamides Sulpride
Atypical AntipsychoticsDibenzdiazepine derivatives Clozapine
OlanzapineBenzothiapine derivatives Quetiapine
ZotepineBezisoxazole derivatives RisperidoneBenzoisthioazoyl piperazine ZiprasidoneImidazolindone derivatives Setindole
Chapter35 ■ Clinical Toxicology 1131
Figure 35-6 Classification and structure of select antipsychotic drugs.
A B
N
S
NH3C CH3
CH3
Phenothiazines
Flupenthixol
Thioxanthines
S
N
N
OH
F
F
F
N
O
Cl
N
N
CH3
Dibenzoxazepines
Loxapine
Dihydroindoles
Molindone
O
N
HN
O CH3
CH3
Butyrophenones
HaloperidolF
O
N
Cl
OH
Diphenylbutylpiperidines
PimozideF
N
F
N
HN
O
Typical Antipsychotics
Benzamides
Sulipride
OCH3
O
NHHN
CH3
SO
O
H2N
N
NH
N
N
CH3
Cl
Dibenzodiazepinederivatives
Clozapine
Bezisoxazolederivatives
Risperidone
Atypical Antipsychotics
N
S
N
N
O
OH
Benzodiazepinederivatives
Quetiapine
N
N
O
CH3
N
N O
F
Benzisothioazoylpiperazine
Ziprasidone
Cl
N
N
N S
HN
O
Imidazolindonederivatives
Sertindole
Cl
N
F
N N NH
O
Promethazine

1132 SectionIII ■ Analytes
markedly increased with coingestion of alcohol or other sedative-hypnotic drugs. Antihistamines are available in combination with analgesics such as acetaminophen and salicylate; therefore detection of these analgesics by urine screen and subsequent serum quantification should be per-formed in a symptomatic patient to assess their potential toxicity (see sections on salicylate and acetaminophen).
Common clinically available antihistamines can be detected qualitatively and quantitatively in blood and urine specimens; this is done primarily by using gas chromatogra-phy and mass spectrometry or liquid chromatography tandem mass spectrometry. Yet, the clinical necessity of quantitative serum antihistamine concentrations is questionable. Poor correlation has been noted between patient age, dose, blood concentration, clinical effects, and death.37,260,654 Antihista-mines are detected by forensic laboratories as agents poten-tially used in facilitating sexual assault (see section, “Drugs Used in Sexual Assault”).
Although detection of antihistamines is not typically clini-cally relevant in the acutely toxic patient, it is important to note that very high concentrations of first-generation antihis-tamines, such as promethazine and diphenhydramine, have been documented to cross-react with urine drug immuno-screen analyses. Therefore, physicians and medical review officers (MROs) should be aware of potential false-positive results from on-site drug testing devices, as well as immuno-screens specifically documented in amphetamine,199 pro-poxyphene,646,711 and tricyclic antidepressants.247,795,839
Antimuscarinic AgentsSeveral plant and mushroom species contain antimuscarinic compounds that mimic anticholinergic symptoms when ingested. These compounds are competitive antagonists at central and peripheral muscarinic receptors. Muscarinic receptors are expressed predominantly within the parasym-pathetic nervous system. Common centrally acting agents are atropine and scopolamine. These tropane alkaloid agents contain a tertiary amine structure permitting penetration of the CNS. These compounds have been isolated from numer-ous plants such as Atropa belladonna (Deadly nightshade) and Datura stramonium (Jimson weed) and are abused for their hallucinogenic potential. Hallucinogenic mushrooms such as Amanita muscaria contain several bioactive compo-nents such as muscarine (mucarine agonist), ibotenic acid (NMDA agonist), and muscimol (GABAA agonist). Unlike acetylcholine, muscarine is a quaternary ammonium com-pound that does not cross the blood-brain barrier. Scopol-amine is used clinically for motion sickness. Atropine is an antidote for cholinesterase inhibitors such as insecticides and chemical warfare nerve agents.
Plants are commonly ingested or brewed as tea. Clinical effects include (1) tachycardia, (2) hypertension, (3) hyper-thermia, (4) dry skin and mucous membranes, (5) skin flushing, (6) diminished bowel sounds, (7) urinary retention, (8) agitation, (9) disorientation, and (10) hallucination.612,671 The effects of muscarine often last longer than those of acetylcholine, because it lacks an ester bond required
ingestion of these rapidly absorbed drugs.100 Neuroleptics may be detected by chromatographic methods or by immu-noassay (see Chapters 13 and 16). GC is the primary chro-matographic method used to measure antipsychotic drugs, and nitrogen phosphorus (NP), electron capture (EC), and MS detectors are the detection systems of choice. However, HPLC, LC-MS, and LC-MS/MS methods are being used more commonly.
AntihistaminesAntihistamines are popular medications used by the general public for treatment of allergic reactions and as common sleep aids. Antihistamines are widely available and many do not require a prescription. Although antihistamines are rela-tively safe, these agents are responsible for 19% of human exposures and 6% of fatalities reported in 2008 to Poison Control Centers in the United States.96
Histamine is released from mast cells and plays an impor-tant physiologic role in immediate hypersensitivity and aller-gic responses. Histamine functions as a neurotransmitter in the CNS and stimulates gastric acid secretion. Antihistamine drugs currently available clinically antagonize H1 and H2 his-tamine receptors. First-generation liphophilic antihistamines, such as diphenhydramine (Benadryl), bind H1 receptors and exhibit peripheral and CNS system effects; they can also bind to muscarinic and adrenergic receptors, resulting in their anticholinergic activity. H1 and H2 receptors are coupled via G-proteins to phospholipase C and adenylyl cyclase, respec-tively.777 The principal H2 receptor response is stimulation of gastric acid secretion, whereas other actions of histamine (e.g., smooth muscle contraction, vasodilation, increased capillary permeability, pain, itching) are primarily mediated by H1 receptors.
The broad binding affinities of H1 receptor antagonists are attributed to a common substituted ethylene amine (—CH2CH2NR2—) moiety found in H1 receptor antagonists and in acetylcholine.649 “Second-generation” antihistamines, such as fexofenadine (Allegra), are highly specific for periph-eral H1 receptors and do not penetrate the CNS. Therefore, second-generation H1 receptor antagonists display minimal sedative and anticholinergic effects.
The therapeutic actions of H1 antagonists include (1) smooth muscle relaxation, (2) decreased bronchial secre-tions, (3) decreased allergic response, and (4) sedation. They are therefore used to treat immediate hypersensitivity reac-tions, as cold remedies, to suppress motion sickness, and for sedation. The H2 antagonists are widely used to treat peptic ulcer disease. Overdose with first-generation H1 antihista-mines presents clinically with CNS depression or stimulation and peripheral anticholinergic effects. Signs and symptoms may include somnolence, coma, confusion, agitation, hallu-cinations, convulsions, visual disturbance, dry flushed skin, dry mouth, urinary retention, decreased bowel sounds, hypertension, and supraventricular arrhythmias.777,866
Acute toxic effects are rarely observed when extremely high doses of second-generation H1 and H2 antihistamine are ingested. However, clinical effects such as sedation can be

Chapter35 ■ Clinical Toxicology 1133
the cholinesterase-active site serine hydroxyl group. Acetyl-cholinesterase enzyme activity can be reactivated by admin-istering an “oxime” drug, or through de novo cellular synthesis of enzyme.
Organophosphate and Carbamate CompoundsOrganophosphate insecticides are toxic because they inacti-vate acetylcholinesterases that are required for hydrolyzing acetylcholine at nerve junctions. Nerve agents are also organophosphorus inhibitors of acetylcholinesterases, yet these agents are more potent than their pesticide counter-parts.566 Nerve agents share a similar structural backbone observed in organophosphate insecticides (see Figure 35-7).
Excess synaptic acetylcholine stimulates muscarinic recep-tors (peripheral and CNS) and stimulates but then depresses or paralyzes nicotinic receptors. Activation of peripheral muscarinic receptors causes signs and symptoms described by the mnemonics SLUDGE or DUMB BELS, defined earlier. CNS neurotoxic effects include (1) restlessness, (2) agitation, (3) lethargy, (4) confusion, (5) slurred speech, (6) seizures, (7) coma, (8) cardiorespiratory depression, and (9) death. Stimulation or paralysis of nicotinic receptors at the neuro-muscular junction causes muscle fasciculations, cramping, weakness, and respiratory muscle paralysis; stimulation of nicotinic receptors at sympathetic ganglia results in hyperten-sion, tachycardia, pallor, and mydriasis.
The actual signs and symptoms observed with these toxins depend on the balance of muscarinic and nicotinic receptor activation. Although miosis (muscarinic action) is most common, it may not always be present, and indeed mydriasis (nicotinic action) may occur. Likewise, tachycardia (nicotinic effect) may be present rather than bradycardia (muscarinic action). Death most commonly results from respiratory failure, a consequence of nicotinic receptor–mediated muscle paralysis, combined with muscarinic-facilitated bronchor-rhea, bronchoconstriction, and CNS depression.
Organophosphate inhibition is a consequence of phos-phorylation of the serine hydroxyl group at the active site of the cholinesterase enzyme catalytic triad (Ser-Glu-His). The partially electropositive phosphorus is attracted to the par-tially electronegative serine (see Figure 35-7). The negatively charged glutamate present in the catalytic triad attracts the organophosphate leaving group and forms an alkyl-phosphoryl serine bond, creating a “transition state.” Subse-quent hydrolysis results in irreversible dealkylation (“aging”) of the AChE.9 Aging proceeds at variable rates depending on the size and branching of the alkyl groups (24 to 48 hours) to form a phosphoryl oxyanion serine bond that is completely resistant to even pharmacologically mediated hydrolysis.
Although carbamates are structurally different from organophosphates (see Figure 35-7), carbamates exert their toxicity at the active site of AChE but inhibit enzyme activity through carbamylation rather than phosphorylation of the serine hydroxyl group. In contrast to the phosphoryl-serine bond, the carbamyl-serine bond undergoes spontaneous hydrolysis, and regeneration of enzyme activity occurs in hours rather than days.265 Carbamates exhibit poor CNS
for acetylcholinesterase hydrolysis. Patients are clinically managed on the basis of clinical presentation, rather than by identification of ingested plants or drug-specific testing.482 Testing for atropine and scopolamine can be performed by liquid chromatography–tandem mass spectrometry and is available in some clinical laboratories. Although HPLC tech-niques may be used to identify muscarine, no clinical tech-niques are yet available.
Agents Related to Cholinergic SyndromeAgents inducing cholinergic syndrome are diverse and act by producing uncontrolled acetylcholine transmission through inactivation of cholinesterase enzymes or direct stimulation of acetylcholine receptors. Acetylcholine is an essential neu-rotransmitter that affects parasympathetic synapses (auto-nomic and CNS), sympathetic preganglionic synapses, and the neuromuscular junction (see also prior section, “Toxic Syndromes”). Clinical manifestations in cholinergic syn-drome include muscarinic, nicotinic, and central effects (see Table 35-2). The duration of acetylcholine action is controlled by acetylcholinesterase and butyrylcholinesterase (pseudo-cholinesterase). Acetylcholinesterase is found in red blood cells, nervous tissue, and skeletal muscle. Butyrylcholinester-ase is found in plasma, liver, heart, pancreas, and brain.
Organophosphate (e.g., Malathion, Parathion, Diazinon, Dursban) and carbamate (e.g., Sevin, Furadan) insecticides (Figure 35-7), as well as military nerve agents [e.g., Sarin (GB), Soman (GD), Tabun (GA) (VX)], exert their toxicity by inhibiting the action of acetylcholinesterase and thereby causing a pronounced cholinergic response.43,118,145 Acetyl-cholinesterase inhibition is the consequence of phosphoryla-tion (organophosphates) or carbamylation (carbamates) of
Figure 35-7 General chemical structure for organophosphate, nerve gas, and carbamate insecticides.
O
O
orX X
R1
R2
R1
Organophosphate insecticide
Carbamate insecticide
R2
S
PP
R1, R2 � for example,
X � for example,
X � for example,
F, CN, O NO2
CH3 NH C
O
O X
CH3, CH2 CH3O

1134 SectionIII ■ Analytes
enzyme.844,845 2-PAM is not effective on aged AChE, so it must be administered as soon as possible after intoxication. Reac-tivation of aged AChE requires de novo cellular synthesis of enzyme (days).
Diagnosis of organophosphate and carbamate toxicity depends mainly on exposure history, physical presentation, clinical suspicion, and laboratory support. Treatment requires immediate attention and should not rely on laboratory con-firmation. Yet cholinesterase activity is often monitored by clinicians in occupational exposure, acute intentional and accidental exposure, and response to therapy.
Three neurologic sequelae of organophosphate poisoning may occur after the initial cholinergic crisis has responded to atropine and oxime therapy in what is referred to as the inter-mediate syndrome.719 Paralysis of proximal limb muscles, neck flexors, cranial nerves, and respiratory muscles may occur 24 to 96 hours following cholinergic resolution. Respiratory muscle paralysis may be severe enough to result in death. This phenomenon, caused by excessive nicotinic receptor stimula-tion, may result from redistribution of lipophilic organophos-phates from adipose tissue and/or from inadequate oxime therapy. In another syndrome, organophosphate-induced delayed neuropathy (OPIDN),3 weakness of extremities, ataxia, and eventually paralysis may occur 1 to 3 weeks fol-lowing severe intoxication. Respiratory muscles are not affected. It is believed that this peripheral neuropathy is the consequence of phosphorylation and inhibition of an axonal membrane enzyme that is designated neuropathy target ester-ase. Alternatively, phosphorylation and activation of a Ca2+/calmodulin kinase may in turn enhance proteolysis of neuronal cytoskeletal proteins and cause structural changes
penetration and have a shorter duration of action; therefore neurotoxicity is usually less severe. Oximes are not recom-mended for carbamate toxicity and may exacerbate symp-toms by stabilizing the carbamylation of acetylcholinesterase enzymes.220
Specific therapy for organophosphate and carbamate insecticide poisoning includes administration of atropine to block the muscarinic (but not nicotinic) actions of acetyl-choline. In addition, pralidoxime is given to reactivate cho-linesterase. Pralidoxime binds to the cholinesterase catalytic site and, via nucleophilic attack by its oxime group, dephos-phorylates or decarbamylates the serine group (see Figures 35-7 and 35-8). Pralidoxime is ineffective in reactivating the “aged” form of the phosphorylated enzyme. Administration of pralidoxime may not be necessary in cases of carbamate insecticide poisoning because carbamylated cholinesterase spontaneously reactivates within a few hours. In fact, prali-doxime is considered contraindicated in these cases by some authors, because cholinesterase inhibition by carbaryl (Sevin), but not by other carbamates, may be enhanced by prali-doxime. Others administer pralidoxime in either case because the particular insecticide ingested may not be known.118,145 A more potent bisquaternary oxime, obidoxime, is available outside the United States.
Administration of a site-directed nucleophile [pyridine-2-aldoxime chloride (2-PAM)] targets AChE reactivation during the transition state. Oxime antidotes such as 2-PAM contain a quaternary nitrogen that binds to the choline-binding site of AChE, positioning the oxime for nucleophilic attack, and by transferring the phosphoryl group from the serine hydroxyl group to itself, it releases free, active
Figure 35-8 Reactivation of inactivated acetylcholinesterase by pralidoxime. The partially electropositive phosphorus of the organophosphate/nerve agent is attracted to the partially electronegative serine present in the catalytic triad of the acetylcholinesterase enzyme. The negatively charged glutamate in the catalytic triad attracts the leaving group of the organophosphate, creating a “transition state” compound. Pralidoxime can mediate regeneration of acetylcholinesterase in the transition state. Formation of the dealkylated “aged” acetylcholinesterase does not reactivate.
Inactivated cholinesterase
Pralidoxime
Pralidoxime
Ser
O�
GluHis
Transition state
Ser
O��H�
R2
��
R1O or S
Leavinggroup
P
R2
R1O or S
P
GluHis
Active cholinesterase
Ser
HO��
�
�
Glu
His

Chapter35 ■ Clinical Toxicology 1135
Testing for drugs of abuse usually involves testing a single urine specimen for various drugs. It should be noted, however, that a single urine drug test detects only fairly recent drug use and it does not differentiate casual use from chronic drug abuse. The latter requires sequential drug testing and clinical evaluation. Moreover, urine drug testing alone cannot deter-mine the degree of impairment, the dose of drug taken, or the exact time of use. Many of these issues were described in detail at the 1987 Arnold O. Beckman Conference212 and in a report by the Committee on Substance Abuse Testing.453 Because of these and other limitations of testing for drugs in urine, integrating the use of alternate biological specimens for drug testing is a matter of growing interest (see section on alternate specimens).
Drug testing results for nonmedical purposes may provide the sole evidence for punitive action or denial of individual rights. Therefore this testing should be considered a forensic toxicology activity, requiring the highest standards of analyti-cal methods, specimen security, and documentation.239 Moreover, laboratories engaged in this testing should be appropriately certified by the Substance Abuse and Mental Health Service Administration (SAMHSA) of the U.S. Department of Health and Human Services (DHHS) or the Forensic Urine Drug Testing program sponsored jointly by the American Association for Clinical Chemistry and the College of American Pathologists.
Several techniques are used by persons attempting to mask or adulterate drugs to avoid detection. These tactics may include the exchange of urine from a drug-free individual or dilution of the urine specimen by excessive consumption of water, use of a diuretic, or simple addition of water to the specimen to reduce drug concentrations to below cutoff limits. Also, readily available adulterants, such as detergent, bleach, salt, alkali, ammonia, tetrahydrozoline, or acid, may be added to the specimen after collection in an attempt to interfere with immunoassay screening procedures. Other more sophisticated adulterants specifically marketed to avoid drug detection include glutaraldehyde (Urine Aid; Clear Choice), nitrite (Klear; Whizzies), chromate (Urine Luck; Sweet Pee’s Spoiler), and a combination of peroxide and per-oxidase (Stealth). These adulterants also interfere with immu-noassays to variable degrees, and the oxidizing agents (nitrite, chromate, and peroxide/peroxidase) may result in destruc-tion of morphine, codeine, and the principal metabolite resulting from marijuana use, thus interfering with their GC-MS confirmation and with immunoassays.154
Direct observation of urine collection is the most stringent means to guard against specimen exchange or adulteration. However, an individual’s right to privacy and dignity must be weighed against the need for the highest degree of certainty of specimen integrity. Alternative measures to prevent speci-men adulteration include (1) limitations on clothing or other personal belongings allowed in the specimen collection area, (2) addition of coloring agent to toilet water, and (3) inactiva-tion of the hot water tap. In addition, several validity checks for specimen integrity may be made at the collection site and at the testing site. Validity testing criteria have been
in neurofilaments, resulting in impaired axonal transport.3 Finally, extrapyramidal symptoms similar to those of Parkin-son’s disease have very rarely occurred several days after cho-linergic crisis resolution. A favorable response was observed with an antiparkinsonian agent.27
Cholinesterase activity is measured to assess exposure and to monitor reactivation during treatment. Acetylcholinester-ase and butyrylcholinesterase enzyme activity are typically monitored using spectrophotometric analyses (see Chapter 22). Acetylcholinesterase activity present at nerve junctions is similar to that present in red blood cells and is an appropri-ate index of neurotoxicity.773 This assay is more sensitive than serum cholinesterase activity and often is used to confirm exposure and to predict enzyme reactivation during treat-ment. A different cholinesterase, butyrylcholinesterase (pseu-docholinesterase), is present in serum and is also inhibited by these insecticides. The activity of butyrylcholinesterase declines then returns to normal more rapidly than is observed for the red cell enzyme. Serum butyrylcholinesterase can be readily measured on hemolyzed samples and in clinical labo-ratories without isolation of red blood cells. However, inter-individual variability is high; therefore pre-exposure activities are optimal when butyrylcholinesterase activities are inter-preted.515 Because butyrylcholinesterases are synthesized in the liver, this assay is particularly sensitive to conditions such as pregnancy and liver disease (acute and chronic hepatitis, cirrhosis, malignancy).535 Thus red cell cholinesterase activity theoretically should correlate more closely with the degree of neurotoxicity. In acute poisoning, symptoms generally begin when cholinesterase activity is inhibited by about 50% of the lower limits of normal, and this degree of inhibition is of diagnostic value. However, the degree of cholinesterase inhi-bition generally does not correlate well with the clinical sever-ity of poisoning. Interpretation of test results is made more difficult by considerable individual variability of normal activities. The presence of urinary organophosphate and car-bamate metabolites is generally measured by GC- MS670 and GC-MS/MS.91 These methods are labor intensive and are typically reserved for monitoring chronic occupa-tional exposure to specific agents rather than for emergency management of acute toxicity.
Drugs of AbuseDrug use and abuse are widespread in society, and public awareness has been heightened as to their impact on public safety and on lost productivity in industry.197A To resolve these issues, governmental, industrial, educational, and sports agencies are increasingly requiring drug testing of prospec-tive and existing employees, students, and participants in professional and amateur athletics. Moreover, drug abuse during pregnancy is a matter of concern, both medically and socially.15 Testing for drugs of abuse may be a medical require-ment for (1) organ transplantation candidates, (2) pain management clinics, (3) drug abuse treatment programs, and (4) psychiatric programs.635 Drug testing for these purposes represents a significant activity for toxicology laboratories.

1136 SectionIII ■ Analytes
and processing of specimens for drug testing has been pre-sented in the federal rules for employee drug testing239 and in the federal regulations promulgated by the Depart-ment of Transportation10 and the Nuclear Regulatory Com-mission (http://www.nrc.gov/reading-rm/doc-collections/cfr/part026/full-text.html; accessed on July 20, 2010).
Workplace drug testing generally is restricted to alcohol (see section on alcohols) and a few drugs that have high abuse potential, some of which are illicit (Tables 35-7 and 35-8). Depending on the nature of the testing program, testing may be provided for a select number of the following drug classes: amphetamines, barbiturates, benzodiazepines, canna-binoids, cocaine, LSD, opiates, synthetic opioids, and PCP (drugs in italics are required for testing by the National Insti-tute of Drug Abuse). Testing programs for participants86 engaged in athletic competition typically are much more extensive and include assays for a larger group of drugs, including stimulants, β-blockers, diuretics, and anabolic steroids.5
Initial screening tests for the previously listed drugs are typically immunoassays (see Chapter 16). These assays are calibrated at established cutoff concentrations. Specimens yielding responses greater than the cutoff (threshold) value are considered positive, whereas values below the cutoff are considered negative. Cutoff values are not synonymous with assay detection limits. Instead, the cutoff is established higher than the detection limit (to ensure reliable measurement) but
established by the DHHS for the drug testing program man-dated for U.S. federal employees.239 According to these crite-ria, the specimen must be examined for unusual color, odor, foaming, or precipitate, and its temperature should be 90 to 100 °F (32 to 38 °C) when determined within 4 minutes of collection. A specimen is reported as dilute when the specific gravity is >1.0010 but <1.0030 and the creatinine is >2 mg/dL but ≤20 mg/dL. A substituted specimen is defined by a specific gravity ≤1.0010 or ≥1.0200 and a creatinine <2 mg/dL. Adulterated urine has pH <3 or ≥11 or nitrite >500 µg/mL (much lower concentrations occur with some urinary tract infections), or may be confirmed if a specific adulterant is detected and confirmed. A specimen is invalid if the pH is ≥3 and <4.5 or ≥9 and <11, if the creatinine and specific gravity are inconsistent, if nitrite is >200 and <500 µg/mL, or if the presence of other adulterants is suspected. In such cases, the urine specimen is rejected and generally is not tested for drugs. The finding of a substituted or an adulterated specimen is deemed equivalent to a refusal to test and would result in removal of the individual from safety-sensitive duties. Numer-ous commercial reagents for validity testing are available in both test strip and liquid forms.
Urine should be collected in tamper-proof specimen cups, and a chain of custody maintained to identify all indi-viduals involved in specimen collection, transfer, and testing. Specimens that test positive should be stored frozen for a minimum of 1 year. Detailed information on the collection
TABLE 35-7 U.S.GovernmentDrugDetectionCutoffConcentrations105
SCREENING, ng/mL CONFIRMATION, ng/mLDrug or Drug Class HHS/DOT239 DOD597 HHS/DOT239 DOD597
Amphetamines 1000 500Designer amphetamines 500Amphetamine 500 100†
Methamphetamine 500* 100*†
MDA 500MDMA 500MDEA 500Cannabinoids 50 50THC-COOH 15 15Cocaine metabolites 300 150Benzoylecgonine 150 100Opiates 2000 2000Morphine 2000 4000Codeine 2000 20006-Acetylmorphine 10 10 10Oxycodone/Oxymorphone 100Oxycodone 100Oxymorphone 100PCP 25 25 25 25
*Also requires the presence of amphetamine (≥200 ng/mL).†Requires chiral analysis; S(+)-amphetamine/methamphetamine.DOD, Department of Defense; DOT, Department of Transportation; GC-MS, gas chromatography–mass spectrometry; HHS, U.S. Department of Health and Human Services.
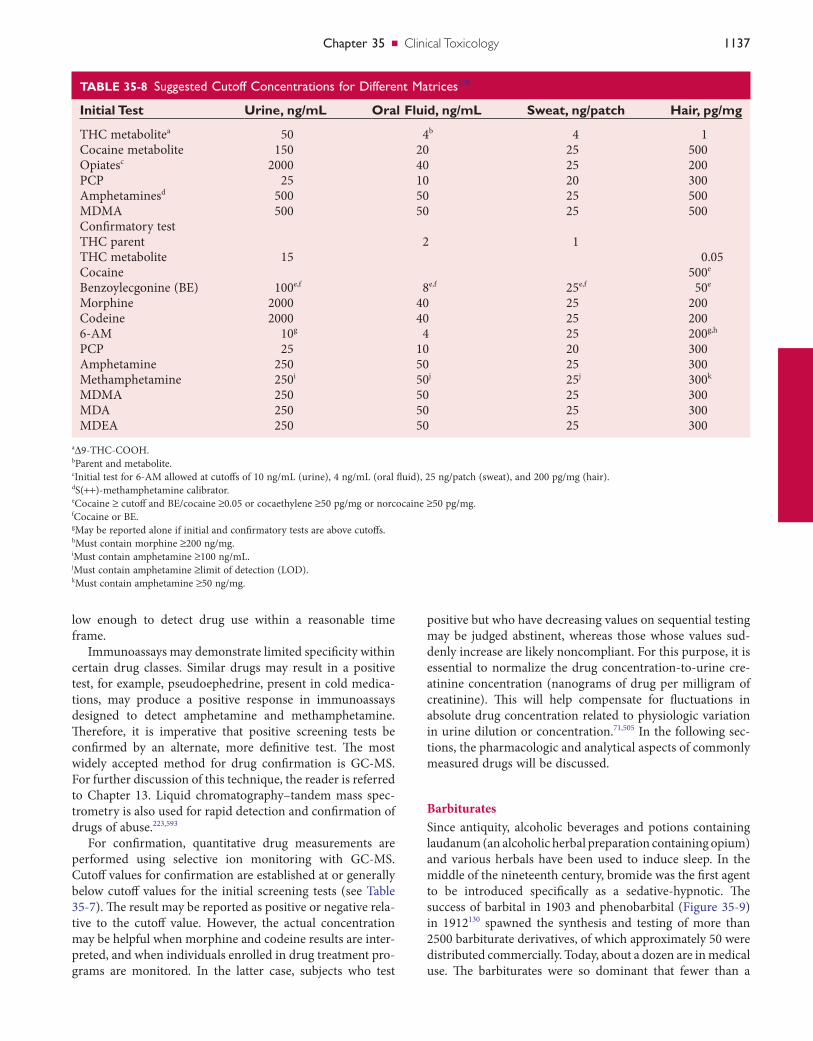
Chapter35 ■ Clinical Toxicology 1137
positive but who have decreasing values on sequential testing may be judged abstinent, whereas those whose values sud-denly increase are likely noncompliant. For this purpose, it is essential to normalize the drug concentration-to-urine cre-atinine concentration (nanograms of drug per milligram of creatinine). This will help compensate for fluctuations in absolute drug concentration related to physiologic variation in urine dilution or concentration.71,505 In the following sec-tions, the pharmacologic and analytical aspects of commonly measured drugs will be discussed.
BarbituratesSince antiquity, alcoholic beverages and potions containing laudanum (an alcoholic herbal preparation containing opium) and various herbals have been used to induce sleep. In the middle of the nineteenth century, bromide was the first agent to be introduced specifically as a sedative-hypnotic. The success of barbital in 1903 and phenobarbital (Figure 35-9) in 1912130 spawned the synthesis and testing of more than 2500 barbiturate derivatives, of which approximately 50 were distributed commercially. Today, about a dozen are in medical use. The barbiturates were so dominant that fewer than a
low enough to detect drug use within a reasonable time frame.
Immunoassays may demonstrate limited specificity within certain drug classes. Similar drugs may result in a positive test, for example, pseudoephedrine, present in cold medica-tions, may produce a positive response in immunoassays designed to detect amphetamine and methamphetamine. Therefore, it is imperative that positive screening tests be confirmed by an alternate, more definitive test. The most widely accepted method for drug confirmation is GC-MS. For further discussion of this technique, the reader is referred to Chapter 13. Liquid chromatography–tandem mass spec-trometry is also used for rapid detection and confirmation of drugs of abuse.223,593
For confirmation, quantitative drug measurements are performed using selective ion monitoring with GC-MS. Cutoff values for confirmation are established at or generally below cutoff values for the initial screening tests (see Table 35-7). The result may be reported as positive or negative rela-tive to the cutoff value. However, the actual concentration may be helpful when morphine and codeine results are inter-preted, and when individuals enrolled in drug treatment pro-grams are monitored. In the latter case, subjects who test
TABLE 35-8 SuggestedCutoffConcentrationsforDifferentMatrices110
Initial Test Urine, ng/mL Oral Fluid, ng/mL Sweat, ng/patch Hair, pg/mg
THC metabolitea 50 4b 4 1Cocaine metabolite 150 20 25 500Opiatesc 2000 40 25 200PCP 25 10 20 300Amphetaminesd 500 50 25 500MDMA 500 50 25 500Confirmatory testTHC parent 2 1THC metabolite 15 0.05Cocaine 500e
Benzoylecgonine (BE) 100e,f 8e,f 25e,f 50e
Morphine 2000 40 25 200Codeine 2000 40 25 2006-AM 10g 4 25 200g,h
PCP 25 10 20 300Amphetamine 250 50 25 300Methamphetamine 250i 50j 25j 300k
MDMA 250 50 25 300MDA 250 50 25 300MDEA 250 50 25 300
aΔ9-THC-COOH.bParent and metabolite.cInitial test for 6-AM allowed at cutoffs of 10 ng/mL (urine), 4 ng/mL (oral fluid), 25 ng/patch (sweat), and 200 pg/mg (hair).dS(++)-methamphetamine calibrator.eCocaine ≥ cutoff and BE/cocaine ≥0.05 or cocaethylene ≥50 pg/mg or norcocaine ≥50 pg/mg.fCocaine or BE.gMay be reported alone if initial and confirmatory tests are above cutoffs.hMust contain morphine ≥200 ng/mg.iMust contain amphetamine ≥100 ng/mL.jMust contain amphetamine ≥limit of detection (LOD).kMust contain amphetamine ≥50 ng/mg.

1138 SectionIII ■ Analytes
necessary to monitor serum pentobarbital concentrations in these circumstances.
Barbiturates continue, although much less frequently than in the past, to be subject to abuse. Because of their rapid onset and short duration of action, the short- to intermediate-acting barbiturates that are used as sedative-hypnotics (amobarbital, butabarbital, butalbital, pentobarbital, and secobarbital) are most commonly abused. The longer-acting barbiturates (mephobarbital and phenobarbital), used pri-marily for their anticonvulsant properties, are rarely abused. The detection period in urine following ingestion of barbitu-rates varies with different assays and depends on the pharma-cologic properties of the drugs. Short- to intermediate-acting barbiturates generally may be detected for 1 to 4 days follow-ing use; long-acting barbiturates, such as phenobarbital, may be detected for several weeks after long-term use.299
Barbiturates act throughout the CNS; nonanesthetic doses preferentially suppress polysynaptic responses, suppress CNS neuronal activity, and thus have sedative and hypnotic prop-erties.310 The site of inhibition occurs primarily at synapses where neurotransmission is mediated by GABA acting at GABAA receptors. This CNS suppression is a result of barbiturate-enhanced activation of the inhibitory GABA-ergic neuronal system.130 Postsynatic GABAA receptors are multisubunit transmembrane Cl conductance channels that when activated by GABA open to allow flow of Cl into the neuron, with subsequent hyperpolarization and inhibition of electrical transmission. High doses of barbiturates increase neural chloride conductance independent of GABA.130 Mechanisms underlying the actions of barbiturates on GABAA receptors appear to be distinct from those of GABA or the benzodiazepines, and they promote the binding of benzodiazepines.16,130 In addition, barbiturates suppress excit-atory glutamate-responsive AMPA (alpha-amino-3-OH-4-isoxozole propionic acid) ion-gated receptor subtypes. Taken together, the findings that barbiturates activate inhibitory GABAA receptors and inhibit excitatory AMPA receptors explain their CNS-depressant effects.706
The barbiturates produce all degrees of depression of the CNS, ranging from mild sedation to general anesthesia. Bar-biturates reversibly depress the activity of all excitable tissues. The CNS is exquisitely sensitive, and even when barbiturates are given in anesthetic concentrations, direct effects on peripheral excitable tissues are weak. However, serious defi-cits in cardiovascular and other peripheral functions occur in acute barbiturate intoxication.130 Severe intoxication results in coma, hypothermia, hypotension, and cardiorespiratory arrest.130 Pharmacodynamic (functional) and pharmacoki-netic tolerance to barbiturates can occur. With long-term administration of gradually increasing doses, pharmacody-namic tolerance continues to develop over a period of weeks to months, depending on the dosage schedule, whereas phar-macokinetic tolerance reaches its peak in a few days to a week. Tolerance to effects on mood, sedation, and hypnosis occurs more readily and is greater than tolerance to anticon-vulsant and lethal effects; thus, as tolerance increases, the therapeutic index decreases.130
dozen other sedative-hypnotics were marketed successfully before 1960.
The antianxiety properties of the barbiturates are less than those exerted by the benzodiazepines.130 Because of their low therapeutic index and high potential for abuse, they have been largely replaced by the much safer benzodiazepines. Nevertheless, barbiturates continue to be available as sedative-hypnotics or for use in combination with analgesic, anti-hypertensive, antiasthmatic, antispasmodic, or antidiuretic drugs. The combination of barbiturates, such as butalbital, with analgesic preparations is ironic. Not only do barbiturates lack analgesic properties, but at low doses they antagonize the effects of analgesics.130 Phenobarbital is effective as an anticonvulsant drug (see Chapter 34), and short- and ultra-short-acting barbiturates are used for IV anesthesia. The classification of barbiturates as “ultra-short-acting,” “short-acting,” “intermediate-acting,” and “long-acting” refers to the duration of effect and not to the elimination half-life (Table 35-9). The duration of action is determined by the rate of distribution into brain and subsequent redistribution to other tissues.130 Anesthetic doses of barbiturates, such as pentobar-bital, are used to reduce intracranial pressure from cerebral edema associated with head trauma, surgery, or cerebral ischemia.512 Therefore appropriate analytical methods are
Figure 35-9 Structure of phenobarbital.
NH
HN
O
O OH3C
TABLE 35-9 Half-lifeandSignificantActiveMetabolitesofSelectBarbiturates
Drug725 Half-life46Active Metabolite
Ultra-Short-Actingthiopental 6-46 h pentobarbitalmethohexital 1.2-2.1 hthiamylal 0.6-0.8 h initial
12-34 h terminal
Short-Acting and Intermediate-Actingpentobarbital 15-48 hsecobarbital 22-29 hbutalbital 35-88 haprobarbital 14-34 hamobarbital 15-40 h (dose
dependent)butabarbital 34-42 h
Long-Actingphenobarbital 2-6 dmephobarbital 48-52 h phenobarbital

Chapter35 ■ Clinical Toxicology 1139
phenobarbital cannot be distinguished from mephobarbital after methylation.
BenzodiazepinesBenzodiazepines are any of a group of compounds having a common molecular structure and acting similarly as depres-sants of the CNS. The term benzodiazepine refers to the portion of the structure composed of a benzene ring fused to a seven-membered diazepine ring and a phenyl ring attached to the 5-position of the diazepine ring.130,475 The prototype benzodiazepines are diazepam and nordiazepam (N-desmethyl diazepam) (Figure 35-10). Fifteen members of this group are presently marketed in the United States, and about 20 additional benzodiazepines are marketed in other coun-tries725; the most common of these are listed in Table 35-10.
Pharmacologic ResponseAs a class of drugs, benzodiazepines are among the most commonly prescribed drugs in the Western hemisphere because of their (1) efficacy, (2) safety, (3) low addiction potential, (4) minimal side effects, and (5) high public demand for sedative and anxiolytic agents. They have largely replaced barbiturates for sedative-hypnotic use because they have fewer side effects and liver enzyme inductions and are safer in overdose.130,238,475,499 New-generation sedative-hypnotics such as zolpidem (Ambien), eszopiclone (Lunesta), and zaleplon (Sonata) modulate the GABAA receptor, as do ben-zodiazepines, yet they are structurally different, permitting unique physiologic properties that will be discussed in a sub-sequent section (see “Drugs Used in Sexual Assault”).
Long-term benzodiazepine use poses a risk for the devel-opment of dependence and abuse,852,855,856 particularly for those agents with the shortest half-life, the highest potency (alprazolam, triazolam), and the greatest lipophilicity (diaz-epam).727,789 Regular use will produce tolerance to most of the adverse effects of benzodiazepines.207 Consequently, some of the sedative and other adverse effects of benzodiazepines dis-cussed earlier may wane with repeated drug use.207 Tolerance may take weeks or months to develop, although this will depend on the dose of drugs used, the frequency of admin-istration, and the pharmacokinetic half-life of the drug. Drugs with short half-lives are more likely to produce a quicker onset of tolerance.207
The benzodiazepines given by themselves or in com-bination with other drugs, particularly narcotic analgesics
The barbiturates undergo extensive hepatic metabolism. The metabolic elimination of barbiturates is more rapid in young people than in the elderly and in infants, and half-lives are increased during pregnancy in part because of the expanded volume of distribution. Chronic liver disease, espe-cially cirrhosis, often increases the half-life of the biotrans-formable barbiturates. Repeated administration, especially of phenobarbital, shortens the half-life of barbiturates that are metabolized as a result of induction of microsomal enzymes.130 Oxidation of radicals at C5 is the most important biotransformation that terminates biological activity.130 Oxi-dation results in the formation of alcohols, ketones, phenols, or carboxylic acids, which may appear in the urine as such or as glucuronic acid conjugates.130 For phenobarbital and amobarbital, N-glycosylation is an important metabolic pathway. Other biotransformations include N-hydroxylation, desulfuration of thiobarbiturates to oxybarbiturates, opening of the barbituric acid ring, and N-dealkylation of N-alkylbarbiturates to active metabolites (e.g., mephobarbital to phenobarbital). Except for the less lipid-soluble aprobarbital and phenobarbital, nearly complete metabolism and/or con-jugation of barbiturates in the liver precedes their renal excre-tion.130 As a result, only a relatively small amount of an administered barbiturate dose is excreted in urine as a parent drug; notable exceptions are phenobarbital and aprobarbital. About 25% of phenobarbital and nearly all of aprobarbital are excreted unchanged in the urine. Their renal excretion can be increased greatly by osmotic diuresis and/or alkalinization of urine.130 Nevertheless, the parent drugs, rather than hydroxy or carboxylic acid metabolites, are targeted for detection in urine screening and confirmation procedures. This analytical approach is generally successful for barbiturates because these drugs are ingested in sufficiently high doses to allow detection of unmetabolized drug in urine.
Analytical MethodsScreening. Numerous commercial immunoassays for
barbiturates are available. Most use antibodies directed toward secobarbital, and although the degree of cross-reactivity of other barbiturates varies with each assay, most have sufficient cross-reactivity to detect the major therapeuti-cally used barbiturates.475,485
Confirmation Testing. Numerous confirmation methods for barbiturates have been described. These include GC with flame ionization detection,358,833,843 nitrogen phos-phorous detection769 and MS,* capillary electrophoresis-ultraviolet (UV),195,383,825 liquid chromatography using ultraviolet (LC-UV) detection,413,506 and LC-MS328,394 and mass spectrometry.546 GC-MS has merits attributable to high resolution and precise retention times with sharp peaks; however, the detection limit of GC-MS for barbiturates is compromised by adsorption at its NH group.698 To overcome this problem, derivatization prior to injection is widely used, but this procedure is time-consuming.42,537,872 On-column methylation is a rapid and sensitive method,769 but
Figure 35-10 Structure of (A) diazepam and (B) nordiazepam.
N
N
Cl
H3CO
Diazepam
N
HN
Cl
O
NordiazepamA B
*References 42, 366, 537, 574, 575, 641, and 872.

1140 SectionIII ■ Analytes
benzodiazepine are commonly used to administer drugs. The most common benzodiazepines used in drug-facilitated sexual assaults are flunitrazepam, midazolam, temazepam, and clonazepam, although almost all members of the class are suitable.207
Although the benzodiazepines exert qualitatively similar clinical effects, important quantitative differences in their pharmacodynamic spectra and pharmacokinetic properties have led to varying patterns of therapeutic application. Several distinct mechanisms of action are thought to contribute to the sedative-hypnotic, muscle relaxant, anxiolytic, and anti-convulsant effects of the benzodiazepines, and specific sub-units of the GABAA receptor are responsible for specific pharmacologic properties of benzodiazepines.130
All the benzodiazepines are well absorbed, with the excep-tion of clorazepate; this drug is decarboxylated rapidly in gastric juice to N-desmethyldiazepam (nordazepam), which subsequently is absorbed.130 Benzodiazepines are rapidly distributed to the CNS. Subsequently, benzodiazepines are more slowly redistributed from the CNS to more poorly perfused tissue, such as adipose tissue and muscle. The rate of this redistribution is an important determinant of the duration of action of benzodiazepines and, similar to that for GI absorption, is largely determined by drug lipophilicity, with the more lipophilic drugs, such as midazolam and triazolam, having the shortest duration of action. These drugs cross the placental barrier and are secreted into breast milk.130
Benzodiazepines may be divided into four categories based on their elimination half-lives: (1) ultra-short-acting; (2) short-acting agents, with half-lives less than 6 hours; (3) intermediate-acting agents, with half-lives of 6 to 24 hours; and (4) long-acting agents, with half-lives greater than 24 hours.130 These pharmacokinetic properties in part deter-mine the primary clinical applications for some benzodiaz-epines. For instance, midazolam (t1/2, 1 to 4 hours) is used for preanesthetic sedation or for sedation for endoscopic proce-dures because of its rapid onset and short duration of action. Benzodiazepines useful in treating anxiety generally have intermediate to long elimination half-lives (alprazolam and diazepam), and those primarily used as anticonvulsants (clonazepam) have the longest. Elimination half-life clearly is not the sole determinant of duration of action of benzodiaz-epines, and in some cases, the rate of drug redistribution from the CNS may be a more important factor.26
Benzodiazepines undergo hepatic oxidation (phase I) and conjugation (phase II), often forming metabolites with phar-macologic activity (see Table 35-10). Cytochrome P450 enzymes, particularly CYP3A4 and CYP2C19, are frequently involved.130 Following these reactions, conjugation with glucuronic acid occurs; these glucuronidated metabolites constitute the major urinary products of benzodiaze-pines.130,475,688,789,855 Drugs and other agents that are inhibitors of CYP3A4 (erythromycin, clarithromycin, ritonavir, itracon-azole, ketoconazole, nefazodone, and grapefruit juice) affect the metabolism of benzodiazepines.203 However, benzodiaz-epines apparently do not significantly induce the synthesis of
(opioids), are among the most widely abused drugs. Their ability to suppress or dampen withdrawal symptoms and to boost the effects of heroin has made them a favored drug type among the drug-using population.207 They are also widely used by the cocaine-using population, especially clonazepam to increase the seizure threshold.207 They are commonly seen in drug-facilitated sexual assault cases. Drinks spiked with a
TABLE 35-10 Half-lifeofSelectBenzodiazepines
Drug725 Half-life, h46
Significant Phase I Metabolites
Short-ActingMidazolam 1-4 α-hydroxy-
midazolamEstazolam 10-24 3-hydroxy-
estazolamFlurazepam 1-3
47-100 (N-desalkyl-flurazepam)
hydroxy-ethyl-flurazepam
N-desalkyl-flurazepam*
Temazepam 3-13Triazolam 1.8-3.9 α-hydroxy-
triazolam
Intermediate-ActingFlunitrazepam† 9-25 7-amino-
flunitrazepam
Long-Acting AgentsDiazepam 21-37 Nordiazepam*
Oxazepam*Temazepam*
Quazepam 39-53 3-hydroxy-quazepam
N-desalkyl-2-oxo-quazepam
2-oxo-3-hydroxy-quazepam
Alprazolam 6-27 α-hydroxy-alprazolam
Chlordiazepoxide 6-27 nordiazepam*oxazepam*
Clonazepam 19-60 7-amino-clonazepam
Clorazepate‡ 231-97
(nordiazepam)
nordiazepam*oxazepam*
Lorazepam 9-16Oxazepam 4-11
*Active metabolite.†Not available in the United States.‡Converted to nordiazepam by gastric HCl.

Chapter35 ■ Clinical Toxicology 1141
directly to a specific site that is distinct from that of GABA binding. Multiple GABAA receptors are known, and benzodi-azepines seem to interact with many of these subtypes, which could account for the varied pharmacologic use of these drugs.475 Unlike barbiturates, benzodiazepines do not activate GABAA receptors directly but rather require GABA to express their effects as they only modulate the effects of GABA. Ben-zodiazepines modulate GABA binding, and GABA alters benzodiazepine binding in an allosteric fashion.130 The remarkable safety of benzodiazepines compared with barbi-turates is probably related to this effect.
Benzodiazepines and related compounds also act as ago-nists, partial agonists, inverse agonists, or antagonists. Ago-nists and partial agonists increase the amount of chloride current generated by GABAA receptor activation, and inverse agonists decrease it. The vast majority of effects of agonists and inverse agonists can be reversed or prevented by the benzodiazepine antagonist flumazenil, which competes for binding to the GABAA receptor.
Benzodiazepines occasionally have paradoxical effects and sometimes cause garrulousness, anxiety, irritability, tachycar-dia, and sweating. Amnesia, euphoria, restlessness, hallucina-tions, and hypomanic behavior have been reported to occur during use of various benzodiazepines. The release of bizarre uninhibited behavior has been noted in some users, whereas hostility and rage may occur in others; collectively, these are sometimes referred to as disinhibition or dyscontrol reac-tions. Paranoia, depression, and suicidal ideation occasion-ally may accompany the use of these agents. Such paradoxical or disinhibition reactions are rare and appear to be dose related.130 Valproate and benzodiazepines given in combina-tion may cause psychotic episodes.130
One benzodiazepine, flunitrazepam (Rohypnol), is appro-ved for use in many countries but not the United States. However, it has illegally entered the United States and has been illicitly sold to the drug-abusing community. In addi-tion, because of its potent sedative-hypnotic action, especially in combination with alcohol, and its ability to induce short-term (anterograde) amnesia, it has gained notoriety for drug-facilitated crimes.
Long-acting benzodiazepines (diazepam, chlordiazepox-ide, and clorazepate) are given in relatively large doses and may be detected for several days to weeks or even months following long-term use. Short-acting benzodiazepines (alprazolam and triazolam) are used in lower doses and might be detected only for a few days.
The treatment of benzodiazepine toxicity is primarily sup-portive. Flumazenil may be used in select cases and is a com-petitive inhibitor of the benzodiazepine site on the GABA complex. It finds its greatest utility in the reversal of benzodiazepine-induced sedation from minor surgical pro-cedures. However, flumazenil should not be administered as a nonspecific coma-reversal drug and should be used with extreme caution after intentional benzodiazepine overdose because it has the potential to precipitate withdrawal in benzodiazepine-dependent individuals and/or to induce sei-zures in those at risk.
hepatic cytochrome P450 enzymes; their long-term adminis-tration usually does not result in accelerated metabolism of other drugs.
Nordazepam is a major metabolite common to the bio-transformation of diazepam, clorazepate, and prazepam; it is formed from chlordiazepoxide via an intermediate metabo-lite demoxepam.130 Some benzodiazepines, such as oxazepam and lorazepam, are conjugated directly and do not undergo phase I metabolism. In some cases, metabolic transforma-tions occur before the drug reaches significant concentrations in the systemic circulation. For example, clorazepate is decar-boxylated to nordiazepam by stomach acid, and flurazepam and prazepam are converted to active metabolites by hepatic first-pass metabolism.130,238
Because active metabolites of some benzodiazepines are biotransformed more slowly than are the parent compounds, the duration of action of many benzodiazepines bears little relationship to the half-life of elimination of the drug that has been administered (see Table 35-10). For example, the half-life of flurazepam in plasma is 1 to 3 hours, but that of a major active metabolite (N-desalkylflurazepam) is 50 hours or longer.54 Conversely, with benzodiazepines that lack active metabolites (oxazepam, lorazepam, temazepam, triazolam, and midazolam), the half-life is an important determinant of their duration of action. Additional factors that influence the duration of benzodiazepine action are hepatic metabolism and acute tolerance, resulting in decreased response to ben-zodiazepines with continued drug exposure.
Virtually all results of the pharmacologic effects of benzo-diazepines are caused by their actions on the CNS. The most prominent of these effects are (1) sedation, (2) hypnosis, (3) decreased anxiety, (4) muscle relaxation, (5) anterograde amnesia, and (6) anticonvulsant activity. Only two effects of these drugs result from peripheral actions: (1) coronary vaso-dilation, seen after intravenous administration of therapeutic doses of certain benzodiazepines; and (2) neuromuscular blockade, seen only with very high doses. Ethanol increases both the rate of absorption of benzodiazepines and the asso-ciated CNS depression. Except for additive effects with other sedative or hypnotic drugs, reports of clinically important pharmacodynamic interactions between benzodiazepines and other drugs have been infrequent.
Benzodiazepines are believed to exert most of their effects by interacting with inhibitory neurotransmitter receptors directly activated by GABA. GABA receptors are membrane-bound proteins that are divided into two major subtypes: GABAA and GABAB receptors. The ionotropic GABAA recep-tors are responsible for most inhibitory neurotransmission in the CNS. Binding enhances GABA-mediated chloride trans-membrane conductance, which results in hyperpolarization and diminished neural electrical discharge. Ultimately, this reduces the arousal of the cortical and limbic systems in the CNS.207 Benzodiazepines also depress the electrical after-discharge in the amygdala, hippocampus, and septum com-ponents of the limbic system that affect emotions.207 In contrast are the metabotropic GABAB receptors. Benzodiaz-epines act at GABAA but not GABAB receptors by binding

1142 SectionIII ■ Analytes
and alprazolam. Drugs that are more polar, such as those with hydroxyl groups (oxazepam, temazepam, and lorazepam) or a nitro group (clonazepam, nitrazepam), display poor chro-matographic characteristics and require derivatization.475 Chlordiazepoxide is thermally unstable and may degrade at high temperatures in the GC.475 Some consider GC-MS as the definitive confirmation method207; however, LC with UV detection (240 nm) has been used to detect benzodiazepines and metabolites without derivatization. LC-MS and LC-MS/MS are becoming increasingly useful and popular methods for benzodiazepines.433,466,530,546,800,801,828,869
CannabinoidsCannabinoids are a group of C21 compounds found in the marijuana plant Cannabis sativa. Cannabis is the most exten-sively abused drug in the world176 and it has been used as a medicinal and an illicit psychotropic agent for centuries. The main psychotropic effects are (1) euphoria, (2) distorted per-ceptions, (3) relaxation, and (4) a feeling of well-being.298,306 Since 1996, 13 states have legalized cannabis for medical con-ditions such as glaucoma, chemotherapy-related nausea and vomiting, migraine, and anorexia. In 2005, the Gonzales vs. Raich ruling permitted the federal government to ban the nonmedical and medical use of cannabis.
Delta-9-tetrahydrocannabinol (THC), the primary psy-choactive component of the C. sativa plant (Figure 35-11), binds to endogenous cannabinoid receptors, CB1 (neuronal) and CB2 (immune cells).305,614,637 These transmembrane recep-tors are G-protein–coupled receptors that mediate signal transduction through inhibition of adenylate cyclase and calcium ions, and activation of potassium ion channels.259,345 The distribution pattern of CB1 receptors in the CNS accounts for most of the clinical effects of THC such as mood, memory, cognition, pain, and appetite.365,467,759 CB2 may regulate immune and inflammatory processes.
THC is typically consumed by smoking the plant leaves, flower buds, and sometimes stems. THC also has been extracted from the glandular hairs of cannabis flowers and produced as a resin (hashish). Hashish is often a more potent form and has been mixed into foods, brewed as tea then ingested, or smoked. Hemp oil also has been extracted from cannabis seeds for use in soaps, body care products, and dietary supplements and is used because of its high essential fatty acid content, but negligible THC content.
Pharmacologic ResponseWhen marijuana is smoked, THC rapidly diffuses into the plasma in seconds and is distributed multiphasically. First, it distributes to highly vascularized tissues in minutes because of its lipophilic nature.12 THC then is redistributed back into the bloodstream, undergoes hepatic metabolism, and slowly accumulates into less vascularized and fatty tissues.304,448 After cessation of marijuana smoking, THC and its metabolites are slowly released from fat stores.570
The main psychotropic effects after inhalation of mari-juana occur within minutes and persist for several hours. The peak plasma concentration of THC is dependent on the dose
Analytical MethodsBenzodiazepines are measured using a variety of techniques. However, their structural diversity and wide variations in potency provide a challenge for laboratories to detect all rel-evant members in one analytical scheme. Reviews of analysis of benzodiazepines have been published.205,206,522 These cover the techniques used to screen for the presence of the class of drugs and to confirm the presence of one or more members.
Screening. Screening techniques using immunoassay kits will rarely be able to detect all members of the class because of differing immunoreactivities among active drug and metabolites. This seems to apply to the more potent members (i.e., lorazepam, triazolam, clonazepam).205 Several commer-cial immunoassay systems are available for the detection of a wide variety of benzodiazepines and metabolites, but they differ somewhat in their ability to detect the various benzo-diazepines, their metabolites, and glucuronide conjugates. Cross-reactivity in screening immunoassays of the various benzodiazepines and their metabolites varies considerably from manufacturer to manufacturer, and screening assays are not able to distinguish between the individual benzodiaze-pines. Most assays are calibrated to the common metabolite oxazepam, temazepam, or nordiazepam.300 However, the large number of different functional groups that may be present on the benzodiazepine nucleus makes it difficult to detect all drugs in this class, and some compounds such as midazolam, chlordiazepoxide, and flunitrazepam may not be detected by many assays.* Other factors, such as low doses and short half-lives, make the detection of some benzodiaz-epines especially challenging. In the absence of sufficiently sensitive or specific immunoassays, direct analysis by a con-firmatory method is warranted in suspected cases.
It should be noted that benzodiazepines may be identified and quantified in serum, but such quantitative information is not warranted in cases of benzodiazepine overdose because serum concentrations are not predictive of severity of intoxi-cation.238 However, a urine or serum immunoassay screening test for benzodiazepines is valuable in the evaluation of patients with an unknown cause of CNS depression.
Confirmation Testing. Analysts need to be aware that the specimen type will dictate the target substance. Blood analy-ses invariably will target the parent benzodiazepine and perhaps the major active metabolite (e.g., nordiazepam for diazepam and other analogs metabolized to nordiazepam). This applies similarly to analyses targeted for saliva.638A In urine, a metabolite is often the required target species.207
Benzodiazepines and their metabolites have been extracted from biological specimens by liquid-liquid extraction (LLE) or solid-phase extraction (SPE). When urine specimens are analyzed, a hydrolysis step is necessary to cleave the glucuro-nide conjugates.475 Enzymatic hydrolysis is preferred over acid hydrolysis because some benzodiazepines are unstable and rearrange to form benzophenones.475
Many benzodiazepines are analyzed without deriva tization by GC; these include diazepam, nordiazepam, flurazepam,
*References 156, 273, 699, 743, 821, and 824.

Chapter35 ■ Clinical Toxicology 1143
Figure 35-11 Principal metabolic route for delta-9-tetrahydrocannabinol (THC) in humans.
OH
8-Hydroxy-THC
C5H11
CH3
HO
O
H3C
H3C
OH
�9-THC
C5H11
CH3
O
H3C
H3C
OH
8,11-Dihydroxy-THC
Conjugation
C5H11
CH2OH
HO
O
H3C
H3C
OH
11-Hydroxy-THC
C5H11
CH2OH
O
H3C
H3C
OH
Carboxy-THC
C5H11
COOH
O
H3C
H3C
Dronabinol (Marinol) contains synthetic THC and is used to treat anorexia and nausea in patients with acquired immuno-deficiency syndrome (AIDS) and those with nausea and vomiting associated with chemotherapy, or asthma and glaucoma.532 Measurement in urine of the principle THC metabolite THC-COOH, present in cannabis but not in dronabinol, has been proposed as a means to distinguish ingestion of marijuana from ingestion of Marinol.228
Analytical MethodsAn immunoassay method is typically used to screen for potential cannabinoid use in workplace drug testing, athlete drug testing, and clinical specimens. A presumptive positive sample should be confirmed by quantitative GC-MS. Confir-mation of quantitative concentrations of the parent com-pound, THC, is typically reserved for forensic samples.
Screening. Legitimate concern has been raised concern-ing the potential for false-positive results from dietary sources and “passive inhalation” of sufficient sidestream marijuana smoke from nearby users, resulting in a positive urine can-nabinoid test. Hemp seeds and oil are produced from the same Cannabis sativa plant that is harvested for drug use.
and occurs during the early acute phase (6 to 10 minutes). Numerous factors contribute to the variability in dose, such as (1) method of consumption, (2) depth of inhalation, (3) exposure frequency, and (4) cannabis potency.231,567,647 Onset of clinical symptoms and peak plasma concentrations after oral ingestion of THC is slower (2 to 6 hours) than after inhalation, primarily as the result of first-pass hepatic clear-ance.6,816 The intensity of clinical effects described for smoked cannabis occurs during multiple phases: acute (0 to 60 minutes), postacute (60 to 150 minutes), and residual (>150 minutes). THC blood concentrations accurately reflect clini-cal psychotropic effects observed during the early postacute phase after smoking cannabis.304,598,811 Therefore, plasma concentrations of THC can be monitored to discriminate between intoxication and prior use of cannabis. The ratio of THC to 11-nor-delta-9-tetrahydrocannabinol-9-carboxylic acid (THC-COOH) metabolite has been used to estimate the time of exposure to marijuana.548 This approach may be useful in naive users but is unreliable in chronic abusers of marijuana.
Although marijuana is the most frequently used illicit drug, it does have some limited legitimate medicinal use.

1144 SectionIII ■ Analytes
interpreted as THC-COOH equivalents. The National Insti-tute on Drug Abuse guidelines specify that a 50-ng/mL cutoff should be used for immunoscreens.
A positive result from a urine cannabinoid screen or con-firmation does not indicate intoxication or degree of expo-sure. The window of detection for the urine concentration of THC-COOH varies among casual (2 to 7 days)567 and chronic abusers (up to 73 days)226,457 of marijuana and is dose depen-dent. Variables affecting the duration of detection include (1) dose, (2) frequency of exposure, (3) route of exposure, (4) body composition, (5) fluid excretion, and (6) method of detection. Therefore, monitoring of abstinence is particularly challenging. Dilution of urine due to normal biological fluc-tuations (hydration) or ingested adulterants has caused a negative result one day and a positive on the next. To correct for hydration fluctuations, urine concentrations of THC-COOH per milligram creatinine are normalized for monitor-ing individuals who are resuming cannabis use. Using these normalized THC-COOH:creatinine concentrations, a ratio is calculated by comparing any normalized urine specimen (U2) with a previously collected normalized urine specimen (U1). “New use” is defined as a U2/U1 ratio of ≥0.5 to 1.5 collected from urine specimens taken more than 24 hours apart and containing THC-COOH concentrations >15 ng/mL.166,258,738 Using the 1.5 cutoff rate results in decreased false-positives, but increased false-negative decisions.738
Confirmation. A positive screening result for THC obtained by immunoassay is confirmed by GC-MS analysis of the urine specimen. In the United States, the Division of Workplace Programs (DWP) in SAMHSA set the cutoff for confirming the presence of TCH-COOH metabolite at 15 ng/mL (GC-MS).110
Opiates (Opioids)The term opioid describes a wide range of compounds encom-passing the natural and semisynthetic opiates—essentially variations on the structure of morphine—and fully synthetic opioids with minimal structural homology to the natural alkaloids (Figure 35-12).630 The defining characteristic of this class of drugs is their morphine-like antinociceptive activity stemming from interaction with opioid receptors, which play a major role in pain perception.309,745 Other compounds that are somewhat loosely referred to as “opioids” include receptor antagonists and mixed agonist/antagonists, as well as other opium-derived alkaloids such as papaverine that are not known to bind opioid receptors.874
Pharmacologic ResponseFor pain management, opioid therapy is a mainstay in treat-ing acute needs such as postsurgical analgesia, and in reliev-ing moderate to severe chronic pain.320 In the latter case, opioids are well accepted in the setting of cancer-related pain, but the propriety and effectiveness of their use in nonmalig-nant chronic pain are controversial.320 Most opioids have both substantial addictive capacity and potentially life-threatening side effects; thus the benefits of their use in non–end-stage patients must be carefully weighed against the chance of
Hemp has been used to make soaps, lotions, rope, and cloth-ing, and as an ingredient in a wide variety of food products. In the mid-1990s, several studies reported that ingestion of single or multiple doses of hemp products caused positive results in cannabinoid screens and confirmatory analyses. Since 1998, the U.S. Federal Government has prohibited the importation of Cannabis sativa seeds and oil containing greater than 0.3% THC to reduce human exposure to THC. The concentration of THC consumed in drug use is 2 to 20%.231,473 Subsequent studies have suggested that these mea-sures were successful in reducing potential positive cannabi-noid drug screen results from dietary sources.473 Yet, immunoscreens and GC-MS analyses of urine specimens from volunteers exposed to very low doses of THC (0.39 mg/d) have tested positive for cannabinoids.293,308
Numerous studies have been conducted to investigate exposure to THC from second-hand smoke, concluding that the SAMHSA cutoff is sufficient to separate moderate passive exposure from first-hand inhalation exposure to THC. Several of these studies demonstrated that significant concentrations of TCH-COOH (<10 ng/mL) could be detected in passive inhalers housed in unventilated confined facilities, but most were below the assay cutoff.463,565 Individuals that tested posi-tive (>20 ng/mL cutoff, immunoassay) were exposed to mul-tiple marijuana cigarettes in an unventilated car containing ≤3500 L of air.559,633 Therefore, it is improbable that a passive inhaler would be able to sustain exposure to significant THC concentrations long enough to produce a positive drug screen. Nevertheless, as a precaution against passive inhalations resulting in a positive test, some laboratories screen for urine cannabinoids at a cutoff concentration of 100 ng/mL THC-COOH equivalents. However, at this cutoff value, test sensitivity in one study was only 47% when com-pared with that for GC-MS (cutoff value, 15 ng/mL THC-COOH). Test sensitivity increased to 93% at a cutoff value of 20 ng/mL THC-COOH equivalents.735 The U.S. federally mandated screening cutoff was reduced from 100 ng/mL to 50 ng/mL THC-COOH equivalents.761 One study demon-strated that such a reduction in screening cutoff resulted in a 23 to 54% increase in test sensitivity, depending on the immunoassay, with only a slight decrease (1.0 to 2.6%) in test specificity.349 A 1997 study suggests that consideration should be given to lowering the values listed for THC-COOH (see Table 35-7).849
TCH is metabolized by CYP2D6 liver enzymes to greater than 100 metabolites. The main active metabolite, 11-hydroxy-delta-9-THC, is further oxidized to the most abundant inac-tive THC-COOH (see Figure 35-11).348,567 Immunoassay screens have been designed to detect cannabis use in urine samples using antibody reagents developed against the inac-tive THC-COOH metabolite; these reagents cross-react with numerous other THC metabolites. Therefore the presence of multiple cannabinoid metabolites in a patient specimen will have an additive effect in immunoscreen analyses. Quantita-tive results based on these metabolites are 1.5 to 8 times greater than the actual concentration of THC-COOH as determined by GC-MS.387 Therefore immunoassay results are
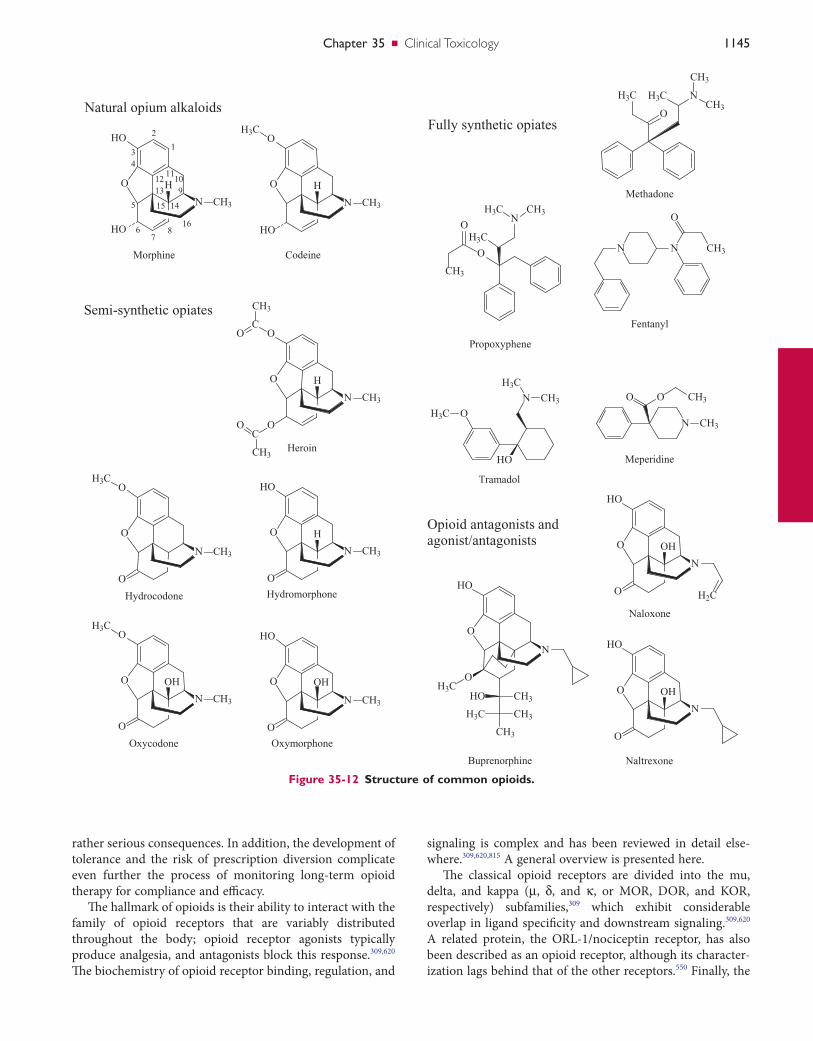
Chapter35 ■ Clinical Toxicology 1145
Figure 35-12 Structure of common opioids.
O
HO
O
N CH3
H
Hydromorphone
Morphine
O
HO
O
N
OH
H2C
Naloxone
O
O
HO
N CH3
H
H3C
Codeine
O
O
O
N CH3
OH
H3C
Oxycodone
Meperidine
H3C
O
N
CH3
CH3
H3C
Methadone
Propoxyphene
NH3C CH3
O
CH3
O
H3C
Hydrocodone
O
O
O
N CH3
H3C
Heroin
O
O
O
N CH3
H
C
CH3
O
C
CH3
O
O
HO
HO
N CH3
H
1
2
34
5
67
8
910
111213
1415
16
Natural opium alkaloids
Semi-synthetic opiates
O
HO
O
N CH3
OH
Oxymorphone
Fully synthetic opiates
HO
OH3C
N CH3
H3C
Tramadol
Fentanyl
N N CH3
O
N CH3
O O CH3
Opioid antagonists andagonist/antagonists
O
HO
O
N
H3C
CH3
CH3H3C
CH3HO
Buprenorphine
O
HO
O
N
OH
Naltrexone
signaling is complex and has been reviewed in detail else-where.309,620,815 A general overview is presented here.
The classical opioid receptors are divided into the mu, delta, and kappa (µ, δ, and κ, or MOR, DOR, and KOR, respectively) subfamilies,309 which exhibit considerable overlap in ligand specificity and downstream signaling.309,620 A related protein, the ORL-1/nociceptin receptor, has also been described as an opioid receptor, although its character-ization lags behind that of the other receptors.550 Finally, the
rather serious consequences. In addition, the development of tolerance and the risk of prescription diversion complicate even further the process of monitoring long-term opioid therapy for compliance and efficacy.
The hallmark of opioids is their ability to interact with the family of opioid receptors that are variably distributed throughout the body; opioid receptor agonists typically produce analgesia, and antagonists block this response.309,620 The biochemistry of opioid receptor binding, regulation, and

1146 SectionIII ■ Analytes
by other drugs, herbal supplements, or endogenous com-pounds that are substrates of the same enzyme. For example, methadone concentrations may be lower than expected in a patient taking St. John Wort—a noted CYP3A4 inducer—but higher in a patient ingesting a CYP3A4 inhibitor such as grapefruit juice.243
TypesTypes of opiates include natural opium alkaloids, semisyn-thetic opiates, fully synthetic opioids, and opioid antagonists and mixed agonist/antagonists.
Natural Opium AlkaloidsMorphine and codeine are examples of natural opiates. The juice and seeds of the poppy plant are their primary source.
Source. Opium is obtained from the unripe seed capsules of the poppy plant, Papaver somniferum. The milky juice is dried and powdered to make powdered opium, which con-tains several alkaloids. Only a few—morphine, codeine, and papaverine—have clinical usefulness. These alkaloids are divided into two distinct chemical classes: phenanthrenes and benzylisoquinolines. The principal phenanthrenes are mor-phine (10% of opium), codeine (0.5%), and thebaine (0.2%). The principal benzylisoquinolines are papaverine (1%), which is a smooth muscle relaxant and noscapine (6%).309
Poppy seeds contain morphine and to a lesser extent codeine.631 Ingestion of bakery products containing poppy seeds leads to excretion of morphine (and codeine) in urine.324,757 Because of first-pass metabolism, no pharmaco-logic effect is experienced from poppy seed ingestion. Con-sumption of large amounts has been known to result in urine morphine concentrations up to 2000 ng/mL for a period of 6 to 12 hours after ingestion. In practice, it is obvious that caution is required when the results of a positive urine test for morphine and codeine are interpreted.
Morphine. The archetypical opiate, morphine, is used as the basis of comparison for relative characterizations of the opioid class. Morphine interacts primarily with MOR to mediate its effects, but it also shows some affinity for KOR.309 Its major metabolites are glucuronide conjugates, including inactive morphine-3-glucuronide (M3G; ≈60%), active morphine-6-glucuronide (M6G; ≈10%), and a small amount of morphine-3,6-diglucuronide.155,850 Free hydroxyl groups, such as the 3- and 6-hydroxy moieties of morphine, are frequently glucuronidated by enzymes of the uridine diphosphate glucuronyl transferase (UGT) family.309,492 UGT2B7 is the isoform primarily responsible for morphine glucuronidation in humans155; other UGT enzymes such as UGT1A1 and UGT1A8 metabolize morphine in vitro, but their relevance in vivo remains uncertain.599 Most morphine glucuronides are excreted in the feces, where substantial enterohepatic circulation of conjugated and intestinally deconjugated morphine occurs. The detection time for mor-phine is usually 48 hours, but this varies with individual differences in metabolism excretion and route and frequency of use.414
sigma receptor family will interact with some opioids but produces very different physiologic responses, including cardiac excitation and tachypnea; sigma receptors are now considered to be completely distinct from the classical opioid receptors.620
Opioids also have preferential or selective binding to one or more of the different receptor classes. It is possible for a compound to stimulate one opioid receptor subtype while inhibiting another, as with mixed agonist/antagonist com-pounds.309,620 The effect of ligand binding varies between receptor classes. Morphine-like analgesia is thought to be mediated primarily through stimulation of MOR, although compounds with preferential binding to DOR or KOR also produce analgesia.309,620 Other classical sequelae of opioid treatment are also attributable to MOR, including sedation and inhibition of respiratory function and gastrointestinal transit.270,309,591 In contrast, neither DOR nor KOR is thought to affect respiration; DOR agonists do not produce sedation or reduce gastrointestinal motility.309 KOR and its endoge-nous ligand dynorphin are implicated in response to addic-tion to numerous drugs such as opioids; KOR gene polymorphisms have been linked to susceptibility to alcohol dependence, supporting a role for this receptor in addictive behavior.501,502,790,791
In addition to undesirable side effects, a major concern in long-term opioid therapy is the development of toler-ance.215,309,620 Tolerant individuals may require many-fold increases in dose to achieve the same concentration of anal-gesia, which can greatly complicate interpretation of serum results and establishment of a therapeutic window. Tolerance to a particular opioid is thought to be a consequence of altered regulation of the opioid receptor(s) to which that compound binds; for this reason, cross-tolerance can occur when multiple drugs interact with the same receptor.215,309,620,689 In addition, several of the enzymes involved in opioid metab-olism (see later) display substrate-dependent alterations in activity. Although substrate inhibition and induction repre-sent different phenomena than tolerance, the clinical effect can be similar and may necessitate modification of the thera-peutic regimen.
The metabolism of opioids is varied, but numerous bio-transformations are common to these drugs. Several of the most commonly used opiates are formed in vivo by metabo-lism of other compounds, as is seen with codeine demethyl-ation resulting in conversion to morphine.309,492 This interconversion is a frequent source of confusion and must be considered when the results of opiate screens are inter-preted; specific details will be outlined later for key opioids with active metabolites.
One of the more important CYP enzymes, CYP2D6, is particularly notable for its role in variable clinical response to opioids; it will be discussed in greater detail in a later section. Many additional CYP enzymes are involved in opioid metabolism, including CYP3A and CYP2C isoforms, among others.492 It is important to note that several of these enzymes are subject to substrate inhibition and/or induction.243 Substrate-dependent changes in metabolic activity are affected

Chapter35 ■ Clinical Toxicology 1147
of pharmacogenetic effects is therefore important to consider when appropriate dosing, patient compliance, and potential diversion or illicit use are assessed.
Semisynthetic OpiatesHeroin, hydrocodone, hydromorphone, oxycodone, and oxy-morphone are examples of semisynthetic opiates.
Heroin. Heroin is a synthetic opiate that is made from morphine and is also called diacetylmorphine or diamorphine; it has an analgesic potency two to three times that of mor-phine414 because of its better penetration across the blood-brain barrier. Heroin is no longer legally produced in the United States, but it is still used elsewhere for fast-acting analgesia.284 The two acetyl groups enhance CNS distribu-tion,684 providing a rapid effect when first-pass metabolism is bypassed (e.g., intravenous administration). Heroin itself is rarely found in body fluids because of its extremely short half- life (2 to 6 minutes).55,414 The metabolite, 6-acetylmorphine, is hydrolyzed to morphine,55,357 and although it has a longer half-life (6 to 25 minutes),55 it is detectable in urine only for about 8 hours after administration.414 Both 6-acetylmorphine and morphine are pharmacologically active, with 6-MAM being four to six times more potent than morphine.414 Other than the presence of its unique metabolite 6- monoacetylmorphine (6-MAM), which is definitive for heroin use, the metabolic profile of heroin resembles that of morphine.683 Given that acetylcodeine is a common contami-nant of heroin, both morphine and low concentrations of codeine are frequently detected in urine following heroin use.
Hydrocodone. Hydromorphone has about six times the potency and greater oral bioavailability than codeine,57 but it is thought to be more toxic than codeine.756 Hydrocodone is O-demethylated to hydromorphone, N-demethylated to form norhydrocodone, and C6-keto-reduced to form approxi-mately equal amounts of 6-alpha- and 6-beta-hydrocol.37,38 Similar to codeine, hydrocodone is metabolized by CYP2D6 to an active metabolite (hydromorphone) and therefore may be subject to pharmacogenetic variability in patients with abnormal CYP2D6 activity.744
It has been suggested that most of the pharmacologic effects of hydrocodone actually result from the hydromor-phone formed during metabolism.756 However, studies are somewhat contradictory. Hydrocodone may provide effective pain relief even in the absence of CYP2D6- mediated conver-sion to hydromorphone.29 It remains unclear whether this is due primarily to the activity of hydrocodone itself or to that of other active metabolites.56
Hydromorphone. Oral hydromorphone is five to seven times more potent than morphine.57 Although it is used as an analgesic in its own right with potency somewhat higher than hydrocodone,29 hydromorphone is also an active metab-olite of hydrocodone.163 Similar to morphine, hydromor-phone is metabolized in large part to a 3-glucuronide by UGT2B7, but also to a lesser extent by UGT1A3.28,401 Hydro-morphone lacks a free hydroxyl group at the 6-position, thus there is no metabolite analogous to M6G.28,401 Two minor metabolites of hydromorphone—dihydromorphine
With long-term administration and when morphine concentrations are high, a minor fraction is converted to hydromorphone (up to 2.5% of the urine morphine con-centration).163 M6G has greater MOR agonist activity than morphine and appears to contribute less to unwanted side effects.309,492,604 However, the relative importance of morphine and M6G in analgesia and adverse responses remains con-troversial.850 The elimination half-life for glucuronides is longer than for morphine.287 Therefore, glucuronides accu-mulate in serum to greater concentrations than morphine, and in patients with renal insufficiency, morphine gluc-uronides are thought to significantly contribute to opioid toxicity, as patients are unable to excrete the water-soluble metabolites.309,638
Codeine. Because of its antitussive and analgesic proper-ties, codeine is one of the most frequently prescribed opiates in the world; it is frequently combined with nonopiate anal-gesic agents such as aspirin and acetaminophen. Therefore, detection of salicylate or acetaminophen along with codeine in the urine of patients who display an opiate toxidrome should lead to the measurement of salicylate or acetamino-phen in serum to assess its toxicity. Alternatively, empirical quantitative serum acetaminophen and salicylate determina-tions are appropriate for patients with the opioid toxidrome. Codeine has only about one tenth the analgesic potency of morphine and shows poor affinity for MOR, with only a frac-tion of the pain-relieving capacity of morphine; therefore, it is generally considered a prodrug.50 Analgesia is attributed to the small fraction (<10%) of codeine converted to morphine by CYP2D6 via O-demethylation, although some studies suggest that the predominant (≈80%) metabolite, codeine-6-glucuronide, may be capable of mediating CNS effects inde-pendently of morphine.492 Both codeine and morphine may be detected in urine following codeine ingestion; however, after 30 hours only morphine may be detectable.170 Codeine is also converted to an inactive metabolite, norcodeine (10%), and long-term high-dose administration leads to metabolism to the active compound hydrocodone (up to 11% of the urine codeine concentration).492,610 During the early phase of excre-tion, codeine and conjugates predominate, but after this time, morphine conjugates are the major product. Approximately 3 days after codeine use, morphine and its conjugates are the only metabolites detected.50,414
Genetic variation may play a significant role in the metab-olism of codeine and several other opioids. More than 60 alleles have been described for CYP2D6, with resultant enzy-matic activity varying from essentially zero, in the case of null alleles, to many times higher than normal, in the case of amplified alleles (http://www.cypalleles.ki.se/cyp2d6.htm; accessed on July 23, 2010).73,355 Thus, at the same codeine dose, patients with minimal CYP2D6 activity (poor metabo-lizers) would likely receive inadequate analgesia because of lack of conversion to morphine; however, patients with very high CYP2D6 activity (ultra-rapid metabolizers) would be at risk for adverse responses to excessive mor-phine.221,309 Without knowledge of the CYP2D6 genotype, these clinical presentations can be confusing; the possibility

1148 SectionIII ■ Analytes
meperidine, particularly in patients receiving concomitant monoamine oxidase inhibitors.309,462 Meperidine use has declined in recent years in favor of alternatives such as fentanyl.
Methadone. A relatively long-acting opiate, methadone is used both for analgesia and in the treatment of opioid addiction.309 It is thought to provide (1) milder withdrawal, (2) somewhat lower potential for abuse, and (3) reduced exposure to the risks of illicit intravenous drug use.216 Metha-done has affinity for both MOR and DOR,504 the latter of which may explain its apparent utility in patients whose pain no longer responds to other opioids.177 Substantial interindi-vidual and intraindividual variability in metabolism and elimination has been noted; both urine pH and seemingly self-inducible metabolism substantially influence the phar-macokinetics of this compound, as do commonly coadmin-istered drugs such as benzodiazepines and antiretrovirals.309 Although a large fraction of methadone is excreted unchanged, measurement of a metabolite such as EDDP (2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine) in the setting of addic-tion treatment provides evidence for patient compliance rather than an exogenously spiked sample.271,278,280 EDDP excretion is less pH dependent than is clearance of the parent drug.59,309,414 Use of the methadone/EDDP ratio to assess com-pliance has been suggested but is complicated by the phar-macokinetic variability already described.271,278,280
Propoxyphene. A relatively weak analgesic, propoxy-phene is less potent than codeine but carries the significant risk of atypical adverse effects such as cardiac arrhythmia and seizure. The incidence of such negative responses is particu-larly high in the elderly.44 In July 2009, the U.S. Food and Drug Administration (FDA) required manufacturers to strengthen the black box warning to address the increased risk of overdose (http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm170769.htm, accessed on June 14, 2011), and in November of 2010 recommends against the continued use of the drug (http://www.fda.gov/Drugs/DrugSafety/ucm234338.htm; accessed on June 14, 2011) and announced that prescription containing medications were being withdrawn form the US market (http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2010/ucm234350.htm, accessed on June 14, 2011) However, its nonmedical abuse remains common.264
Tramadol. Unlike the majority of opioid agonists, trama-dol has low abuse potential and therefore is unscheduled.663 It has low affinity for opioid receptors and mediates analgesia through opioid-independent regulation of neurotransmitter uptake; however, its main active metabolite (O-desmethyl- tramadol, or M1) is a potent opioid receptor agonist.492 These mechanisms are thought to work synergistically to provide greater total pain relief than the sum of each individual component. Metabolism to M1 occurs via CYP2D6; thus opioid-like effects are subject to genetic vari-ability, as with codeine.663 However, because of its effects on neurotransmission, tramadol has the potential to cause serotonergic toxicity even in patients lacking CYP2D6.492 In fact, several synthetic phenylpiperidine opioids (tramadol,
and dihydroisomorphine—have demonstrated pharmaco-logic activity, but their contribution may be minimal because of the small amount formed.57,756
Oxycodone. Oxycodone is a potent analgesic with high oral bioavailability62,309 that is frequently formulated in com-bination with aspirin or acetaminophen. Therefore, the detec-tion of salicylate or acetaminophen along with oxycodone in the urine of patients who display an opiate toxidrome should lead to measurement of serum salicylate or acetaminophen concentration to assess toxicity. Noncombination oxycodone is also available in immediate- and extended-release dosage forms. The latter (OxyContin) is a very effective oral analgesic for patients with chronic pain (e.g., cancer patients). The pills may be chewed, crushed, snorted, or solubilized for IV injec-tion to permit immediate availability of the entire dose, which is intended for extended release over a 12-hour period. This misuse has led to widespread misuse, more frequent emer-gency department visits, and increased mortality in the United States.756
Although its own strong analgesic activity precludes oxy-codone from being considered a prodrug, it is converted to a highly active metabolite, oxymorphone, through the activity of CYP2D6.674 This conversion appears to be less of a concern for CYP2D6 poor metabolizers, in whom oxycodone itself still provides analgesia, than for ultra-rapid metabolizers, who could be at increased risk for adverse effects.191
Oxymorphone. Oxymorphone provides potent analgesia with minimal interaction with CYP enzymes, although it is also a substrate for CYP2C9 and CYP3A4.126 The majority of oxymorphone is metabolized by UGT2B7 to the 3- glucuronide; a minor metabolite, 6-hydroxyoxymorphone, is an active analgesic with a steady-state area under the curve (AUC) similar to the parent compound. Oxymorphone is a metabolite of oxycodone that is formed via CYP2D6.28,401
Fully Synthetic OpioidsFentanyl, meperidine, methadone, propoxyphene, and tra-madol are examples of fully synthetic opioids.
Fentanyl. Fentanyl is an alipophilic drug with numerous routes of administration that is used in applications ranging from anesthesia to rapid management of breakthrough pain.619 Fentanyl provides the structural backbone for a number of related, ultra-short-acting opioids, including remifentanil and sufentanil. Norfentanyl, the primary metab-olite, is generated by CYP3A and is inactive765; the high potency of fentanyl and the clinical insignificance of its metabolites make it a preferred analgesic for patients with major organ failure.619 Transdermal fentanyl patches are used for longer-term administration and are gaining popularity among drug abusers, although nonstandard application of the patch (e.g., chewing, extraction) carries substantial risk for overdose.774
Meperidine. Originally synthesized as an anticholinergic, meperidine has analgesic potency comparable with or some-what lower than that of morphine.462 One major metabolite, normeperidine, also has analgesic activity; normeperidine is thought to be responsible for the serotonergic toxicity of

Chapter35 ■ Clinical Toxicology 1149
at risk for recurrence of narcotic effect. This is particularly true for patients exposed to opioids with long elimination half-lives, such as methadone and sustained-release opioid products. Patients should be observed for resedation for at least 4 hours after reversal with naloxone. Because naloxone is via the kidney eliminated, patients with renal dysfunction may have delayed resedation past the 4 hours and should therefore be observed for a longer period of time.
Naltrexone. Commonly used for the treatment of alco-holism, naltrexone is a potent antagonist of all three opioid receptors.781 Its combined formulation with opioid agonists is less common than are naloxone/opioid combinations; however, the greater oral bioavailability of naltrexone sug-gests that it may be useful in applications where poor oral delivery limits the utility of naloxone.264
Analytical Methods. Many different immunoassay methods are used to screen for opiates. Gas chromatography (GC) with mass spectroscopic detection (GC-MS) is the tech-nique of choice for confirmation of a positive screening test.
Screening Assays. Given their relatively rapid turnaround time and ability to identify several opiates, immunoassays are the methods of choice to screen urine samples for their opiate content. For clinical application, a cutoff of 300 ng/mL mor-phine (or morphine equivalents) is commonly used to distin-guish negative from positive urine specimens, whereas a cutoff of 2000 ng/mL is mandated by SAMHSA for workplace drug screening. Antibodies in opiate abuse screens com-monly target morphine, because commercial immunoassay development has largely been driven by detection of illicit heroin use. Wide variability in cross-reactivity to other con-geners has been noted; thus some opiates or opioids (see Figure 35-12) with high abuse potential such as oxycodone are often poorly detected.485,669,739 To address this problem, several immunoassays are commercially available for indi-vidual synthetic opioids, such as fentanyl. Finally, analytical interferences are also a problem with opiate immunoassays.* Other general opiate screening methods are available, includ-ing thin-layer chromatography, but these techniques are more labor intensive and may not provide adequate turnaround time for stat or emergency testing. In this setting, point-of-care devices are being used more frequently.178,547,861
In pain management programs, urine drug testing is often used to monitor compliance, diversion, or substitution for prescribed drugs. Based on the results of such tests, an indi-vidual may be dismissed from the program. It is important for drug-testing laboratories to communicate relevant aspects of the metabolic interconversion of opiates to physicians responsible for these programs. Monitoring compliance for oxycodone in pain management programs is problematic because of the low cross-reactivity of oxycodone in most opiate immunoassays. In this instance, a false-negative opiate immunoassay test may lead to an accusation of oxycodone diversion. Direct determination of oxycodone by a confirma-tory method (GC-MS, LC-MS, LC-MS/MS) is more appro-priate to monitor compliance for this drug.
methadone, dextromethorphan, and propoxyphene) have been associated with increased risk of serotonin toxicity caused by weak reuptake inhibition of monoamines when used in combination with serotonin reuptake inhibitors, monoamine oxidase inhibitors, and amphetamine-type stimulants.40,89,149,283
Opioid Antagonists and Mixed Agonist/AntagonistsThese clinically useful compounds produce very different physiologic responses, depending on the situation. For example, in opioid-naive patients, mixed agonist/antagonists (MAAs) provide MOR-mediated analgesia with less risk of an adverse reaction, but the same dose in an opioid- tolerant patient may precipitate immediate withdrawal. In medical usage, coadministration of low-dose antagonists or MAAs alleviates minor opioid-induced side effects and appears useful in preventing opioid tolerance. In opioid addiction treatment, the addition of a low-dose antagonist to maintenance therapy seems to minimize subjective “feel-good” effects without substantially worsening withdrawal symptoms.
Buprenorphine, naloxone, and naltrexone are examples of opioid antagonists and mixed agonist/antagonists.
Buprenorphine. A semisynthetic derivative of thebaine, buprenorphine is a MOR partial agonist and a KOR antago-nist. Low doses provide analgesia through MOR activation, but unlike full agonists, pain relief has a maximal threshold or “ceiling effect.”820 Buprenorphine is available as sublingual tablets (with or without naloxone) for the treatment of opioid dependence.264 Buprenorphine is metabolized via N-dealkylation by CYP3A4 to the active compound, norbu-prenorphine, both of which can be further conjugated to inactive glucuronides by UGT1A1.28,29 CYP3A4 and UGT1A1 are subject to environmental and genetic variability, although the effects of these factors on buprenorphine are not well characterized.309 The drug is eliminated primarily in feces, with only a small amount in urine, and is usually detectable for 1 to 3 days.414
Naloxone. The prototypical opioid antagonist naloxone binds nonspecifically to all three receptor types, with the greatest effect at MOR and the least effect at DOR.309,781 Its efficacy is much greater by intravenous administration as compared with oral and sublingual routes.263,264 This charac-teristic is advantageous in deterring misuse of prescribed opioids: oral or sublingual opioid/naloxone formulations provide the desired benefit when taken properly, but when diverted for intravenous use cause opioid antagonism and may precipitate withdrawal.263,264
Naloxone is commonly used in comatose patients as a therapeutic and diagnostic agent. The standard dosage regimen is 0.4 mg/mL administered slowly, preferably intra-venously, with the dose increased until the desired end point is achieved, namely, restoration of respiratory function, ability to protect the airway, and improved level of consciousness. Naloxone has been known to precipitate profound with-drawal symptoms in opioid-dependent patients. Its clinical efficacy lasts for as little as 45 minutes. Therefore, patients are *References 35, 181, 538, 669, 740, and 793.

1150 SectionIII ■ Analytes
available and have been used for GC applications. Various GC-MS methods have been described for the identification and determination of opiates. Some investigators use chemi-cal ionization,144,161,204,627 but electron impact mode is more common. The GC is typically equipped with a 12- or 15-m fused-silica capillary column with a polar stationary phase of cross-linked dimethylsilicone, phenyl methyl silicone, or 95% dimethyl-5% polysiloxane.72,90,131,227,240 Because of structural similarities between many opiates, particularly natural and semisynthetic opiates, assays must be evaluated for interfer-ence from metabolites and congeners. The degree of overlap is such that the fragmentation patterns of various opioids can resemble one another greatly, as is seen with the mass spectra of the trimethylsilane (TMS) derivatives of hydromorphone, morphine, and norcodeine.290 Chromatographic resolution of these compounds must be carefully optimized to provide reli-able characterization, particularly because many structurally related opiates are commercially available and are part of the same metabolic pathways.
Although acetyl derivatives have the advantage of being stable for up to 72 hours when stored at room temperature in ethyl acetate, incomplete derivatization may occur when acetyl-donating agents are used.90,240 Both morphine and 6-MAM are converted to diacetylmorphine (heroin); thus acetyl derivatization does not permit distinction between morphine, 6-MAM, and heroin. In addition to diacetylmor-phine, a small amount of 3-monoacetylmorphine (3-MAM) is formed by acetylating agents; although clinically insignifi-cant, 3-MAM shares the m/z 285 ion with deuterated (d3) d3-acetylcodeine and interferes with analysis of these compounds.303
In contrast to acetylating agents, TMS creates single deriv-atives for most opiates, although TMS derivatives are sensi-tive to moisture.131 Several analytical interferences are associated with TMS: codeine and norcodeine derivatives coelute on gas chromatography, while 6-MAM produces an additional peak that coelutes with morphine and increases with room temperature storage.140 Like TMS, pentafluoropro-pionic anhydride (PFP) derivatives are moisture-sensitive; however, no breakdown products are detected after storage for 24 hours.303 The addition of pentafluoropropanol (PFPOH) improves the yield of PFP derivatives and allows morphine and 6-MAM to be clearly distinguished.240,714
Liquid Chromatography. Despite the long-standing role of GC in opiate analysis, LC methods are common and are often analytically advantageous. One notable example is that LC provides the ability to analyze glucuronide-conjugated metabolites as well as parent compounds. In addition, LC methods are able to measure polar metabolites without prior derivatization,86 and on-column extraction is possible with some LC systems. As with GC, a variety of detectors are avail-able for LC. For example, HPLC methods for opioid analysis have been described using fluorescence (FD), ultraviolet-visible (UV), electrochemical (EC), and diode array detection (DAD), alone and in various combinations. In addition, several analytical methods for morphine and its glucuronide
Confirmation Testing. For compound-specific confirma-tion assays, GC with mass spectroscopic detection (GC-MS) has historically been considered the method of choice. Analy-sis of specific opioids is typically performed using GC or LC. GC generally results in longer run times and is often incom-patible with larger metabolites such as glucuronide conju-gates. LC systems require large quantities of organic solvents and are not considered acceptable for federal testing. A wide variety of detectors are available for both GC and LC; MS or tandem MS is often preferred for the structural and mass-specific information provided. Analytical and technical con-siderations are discussed in detail later.
Sample Preparation and Extraction. The matrix and rationale for opioid testing influence the choice of method. Analysis of urine requires hydrolysis to recover glucuronide- or sulfate-conjugated metabolites of various opioids. Hydrolysis is performed by acidification (e.g., concentrated hydrochloric acid at 115 to 120 °C for 15 minutes)162,623,757 or by enzymatic treatment with β-glucuronidase alone90,157,491 or in com-bination with arylsulfatase.72 Acid hydrolysis is simpler and more rapid, and typically provides greater recovery than enzymatic methods, although a few studies have shown better recovery of some analytes with glucuronidase.194 Acidification, however, destroys the metabolite 6-MAM, preventing conclusive determination of heroin use; it also partially degrades morphine.244 For this reason, drugs-of-abuse testing for opiates typically employs enzymatic hydrolysis, regardless of its generally poorer analytical performance.
Serum analysis is performed with or without a hydrolysis step; if a hydrolysis step is included, results reflect the sum of parent drug and metabolites, that is, “total” drug concentra-tion. For detection of illicit drug use, total concentrations are typically sufficient. However, omitting hydrolysis to preserve conjugated metabolites can be useful, for example, when both the parent and the metabolite are active compounds, as with morphine and M6G.
Methods of analysis from serum or urine were initially developed using liquid-liquid extraction (LLE),* although solid-phase extraction (SPE)72,286,347,714,826 is now often pre-ferred. Some methods do not derivatize prior to GC analy-sis,113,519 but this typically results in poor chromatographic properties. Although the number of derivatizing agents described in the literature is relatively limited, great variabil-ity in experimental conditions has been noted.†
Gas Chromatography. Several GC-MS methods have been developed to quantitate various combinations of morphine, other opiates, and their metabolites from extracts of human urine.162,266,551 GC-MS is considered the reference method for determination of most natural and semisynthetic opiates, particularly in forensic settings, although other detectors are
†References 72, 90, 131, 157, 193, 240, 303, 324, 564, 623, 625, and 757.
*References 90, 157, 218, 390, 468, 623, and 625.

Chapter35 ■ Clinical Toxicology 1151
norepinephrine transporters. Once in the cell, they interfere with the vesicular monoamine transporter (VMAT) and MAO,296,835 depleting synaptic vesicles of their neurotransmit-ter content. As a consequence, concentrations of dopamine (or other transmitter amines) in the cytoplasm increase and quickly become sufficient to cause release into the synapse by reversal of the plasma membrane dopamine transporter (DAT/ SLC6A3). Normal vesicular release of dopamine con-sequently is decreased (because synaptic vesicles contain less transmitter), while nonvesicular release is increased. Similar mechanisms apply to other biogenic amines (serotonin and norepinephrine).497,762
Amphetamine cardiovascular activation is thought to be due to the release of norepinephrine from sympathetic nerve endings.292,835 Stereotyped repetitive behavior and some aspects of locomotor activity induced by amphetamine prob-ably are a consequence of the release of dopamine from dopaminergic nerve terminals, particularly in the neostria-tum.835 The anorectic effect and at least a component of its locomotor-stimulating action are mediated by release of norepinephrine.835 With higher doses, dopamine release in the mesolimbic system and enhanced release of 5- hydroxytryptamine (5-HT; serotonin) in tryptaminergic neurons may be responsible for disturbances of perception and frank psychotic behavior.224,835 High doses also lead to decreases in brain concentrations of the neurotransmitters dopamine and 5-HT, as well as a reduction in the activity of enzymes responsible for their synthesis (tyrosine dehydroxy-lase and tryptophan hydroxylase, respectively).487
Amphetamine and methamphetamine (1) increase blood pressure, heart rate, body temperature, and motor activity, (2) relax bronchial muscle, and (3) depress the appetite. Abuse of these drugs may lead to strong psychologic depen-dence, marked tolerance, and mild physical dependence asso-ciated with tachycardia, increased blood pressure, restlessness, irritability, insomnia, personality changes, and a severe form of chronic intoxication psychosis similar to schizophrenia. These unpleasant responses reinforce repetitive use of the
metabolites exist for LC-MS or LC-MS/MS with different MS interfaces.*
Analytical methods also include common opioids such as methadone682 or buprenorphine,8 or other nonopioid drugs of abuse such as cocaine, amphetamines, and lysergic acid diethylamide (LSD).86,158,830
For TDM testing, several reports have focused on quanti-tation of multiple opioids used therapeutically (e.g., in pallia-tive care). For example, in one study, an LC-MS/MS method was developed that was capable of measuring 11 opioids and 5 metabolites, namely, buprenorphine, codeine, fentanyl, hydromorphone, methadone, morphine, oxycodone, oxy-morphone, piritramide, tilidine, and tramadol, with the metabolites bisnortilidine, morphine glucuronides, norfen-tanyl, and nortilidine.568 In another study, a combination screening and confirmation method was developed that could be used to identify fentanyl, alfentanil, remifentanil, and sufentanil and their respective N-dealkylated or de-esterified metabolites by LC-MS/MS.771 Metabolite profil-ing is another growing area in TDM testing, especially for compounds with known active metabolites such as tramadol.313
Drugs of Abuse Related to the Sympathomimetic SyndromeSeveral stimulants and hallucinogens chemically related to phenylethylamine are referred to collectively as amphetamine-type stimulants (ATSs). They are considered to be sympatho-mimetic drugs, meaning that they mimic endogenous transmitters in the sympathetic nervous system.487 Other drugs related to the sympathomimetric syndrome include cocaine and LSD.
AmphetaminesAmphetamine and methamphetamine (Figure 35-13) are CNS stimulant drugs that have limited legitimate pharmaco-logic use,343 including narcolepsy, obesity, and attention-deficit hyperactivity disorders. They produce an initial euphoria and have a high abuse potential. Other sympatho-mimetic amines that have high potential for abuse include the “designer” amphetamines—ephedrine, pseudoephedrine, phenylpropanolamine, and methylphenidate (Ritalin).
Amphetamine and MethamphetamineThese drugs are sympathomimetic amines that have a stimu-lating effect on both the central and peripheral nervous systems. In the brain, a primary action is to elevate the con-centrations of extracellular monoamine neurotransmitters (dopamine, serotonin, norepinephrine) by promoting pre-synaptic release from the nerve endings497,687,835 rather than blockade of reuptake.595 Amphetamine and methamphet-amine are substrates for the dopamine, serotonin, and
Figure 35-13 Select amphetamine-type stimulants.
CH3
NH2Amphetamine
CH3
HNCH3
CH3
HNCH3
O
O
MDMA
CH3
O
O
NH2MDA
Methamphetamine
*References 77, 83-85, 407, 562, 571, 611, 707, 724, 786, 873, and 876.

1152 SectionIII ■ Analytes
methamphe tamine.33,442,464,481 However, the effects of meth-amphetamine are not reliably predicted from serum concentrations.676
Designer AmphetaminesThe terms “designer drugs” and “club drugs” originated in the 1980s.488 These drugs include derivatives of amphetamines and the new benzylpiperazine, phenylpiperazine; pyrolidino-phenone types have gained popularity and notoriety among people who participate in all-night dance parties (raves) and who visit nightclubs.22,525,779 Most designer drugs produce feelings of euphoria and energy and a desire to socialize402; they also promote social and physical interactions. They are used at these events to enhance energy for prolonged partying and/or dancing, and to distort or enhance visual and auditory sensations. The moniker “club drug” does not imply that rec-reational use is restricted to this social environment. In this context, designer drugs mistakenly have the reputation of being safe; several experimental studies in rats and humans and epidemiologic studies have revealed risks to humans such as life-threatening serotonin syndrome, hepatotoxicity, neu-rotoxicity, psychopathology, and the abuse potential of such drugs.402,525,529
Some of the more common designer amphetamines are listed in Box 35-2; however, only a few will be discussed here.
MDMA (3,4-Methylenedioxymethamphetamine) and MDA (3,4-Methylenedioxyamphetamine). MDMA (also known as “Ecstasy”) is categorized as a stimulant as a result of its sympathomimetic effects, including (1) peripheral vaso-constriction, (2) bronchodilation, (3) cardiorespiratory stim-ulation, (4) pupillary dilation, and (5) appetite suppression. The drug is a sympathomimetic; however, it has significantly fewer CNS stimulant properties than methamphetamine.488 It also is categorized as an empathogen-entactogen.488,563
Similar to amphetamine and methamphetamine, MDMA causes release of biogenic amines by reversing the action of their respective transporters. It has a preferential affinity for the serotonin transporter and therefore most strongly increases the extracellular concentration of serotonin.497 This release is so profound that marked presynaptic intra-cellular depletion occurs for 24 hours after a single dose. With repetitive administration, concentrations of 5-HT, 5-hydroxyindoleacetic acid (5-HIAA), and tryptophan hydroxylase, and serotonin transporter density are reduced.618 Some suggest that serotonin depletion may become perma-nent; this has triggered a debate on its neurotoxicity. Although direct proof from animal models for neurotoxicity remains weak, several studies have reported long-term cognitive impairment in heavy users of MDMA.497
MDMA is a chiral compound in which the S(+)-enantio-mer possesses greater pharmacologic activity. MDMA under-goes demethylation to MDA,542 with the rate of conversion of S(+)-MDMA to S(+)-MDA exceeding that of R(−)-MDMA to R(−)-MDA. Consequently, the concentrations in urine of R(−)-MDMA and S(+)-MDA are greater than those for S(+)-MDMA and R(−)-MDA subsequent to ingestion of racemic MDMA.236,331
drugs to maintain the “high.” Tolerance and psychologic dependence develop with repeated use of amphetamines.343 Long-term effects may include depression and impaired memory and motor skills, probably caused by a decrease in dopamine transporters and by damage to dopaminergic and serotonergic neurons. Methamphetamine has greater CNS efficacy, most likely because of its greater ability to penetrate the CNS.556
The optical isomers of amphetamine and methamphet-amine exhibit stereoselective pharmacologic properties. The CNS activity of S(+) amphetamine (d-amphetamine) is three to four times greater than that of R(−) amphetamine (l-amphetamine), but the latter drug has more potent cardio-vascular effects than the former.48,343 The CNS effects of S(+) methamphetamine (d-methamphetamine) are about 10 times greater than those of R(−) methamphetamine (l-methamphetamine), but the latter drug has greater vaso-constrictive properties than the former.245,343 Because of minimal CNS activity and thus low abuse potential, R(−) methamphetamine is included in some nonprescription nasal inhalants (e.g., Vick’s) for its vasoconstrictive properties.
The main metabolic pathways of amphetamine and meth-amphetamine include (1) aromatic hydroxylation, (2) ali-phatic hydroxylation, (3) N-demethylation, (4) oxidative deamination, (5) N-oxidation, and (6) conjugation of nitro-gen.442 Amphetamine itself is extensively metabolized to a variety of metabolites, including norephedrine and p-hydroxyamphetamine, both of which are pharmacologically active, and may be glucuronidated prior to excretion.487 Amphetamine is metabolized in a stereoselective manner such that the elimination half-life for R(−) amphetamine may be as much as 40% longer than that for S(+) amphet-amine.112,823 Methamphetamine is metabolized in liver pri-marily by hydroxylation and, to a lesser extent, by N-demethylation to amphetamine. Overall metabolism, including formation of amphetamine, is enantioselective.70,112 Thus when racemic methamphetamine is ingested, urine specimens contain relatively more R(−) methamphetamine than S(+) methamphetamine, but a greater amount of S(+) amphetamine than R(−) amphetamine.151,350
In addition to hepatic metabolism, amphetamine is elimi-nated as unchanged drug in urine. Elimination is dependent on urine pH, and although typically about 30% of a dose is excreted unchanged, this may vary from as much as 74% in acid urine to as little as 1% in alkaline urine.48 Therefore, elimination half-life (renal excretion and hepatic metabo-lism) also varies with urine pH from 7 to 14 hours at acid pH to 18 to 34 hours at alkaline pH.253,556 These effects of urine pH on the elimination of unchanged amphetamines are a consequence of tubular reabsorption of nonionized amphet-amine (pKa, 9.9). Similarly methamphetamine is eliminated in urine in a pH-dependent manner similar to that used for amphetamine.
Pharmacogenetics may play a role in the differences seen in the metabolism/elimination of these drugs. CYP2D6 is responsible for the 4-hydroxylation of amphetamine and methamphetamine and the N-demethylation of

Chapter35 ■ Clinical Toxicology 1153
“Love.” MDEA undergoes oxidative cleavage of the methoxy rings but also N-de-ethylation.60,442 MDEA also undergoes de-ethylation to MDA, with the rate of conversion of S(+)-MDEA to S(+)-MDA exceeding that of R(−)-MDEA to R(—)-MDA.541
The MDEA enantiomers have different pharmacokinetic properties. They include S-MDEA, which produces elevated mood and impairment in conceptually driven cognition, and R-MDEA, which produces increased depression and enhanced visual feature processing541; a generally higher affinity toward S-MDEA than R-MDEA is seen.541
PMA (Paramethoxyamphetamine) and PMMA (Para-methoxymethamphetamine). PMA and PMMA are meth-oxylated phenylethylamine derivatives with effects similar to but more potent than those of MDMA; they are frequently sold on this basis.385 PMA is a metabolite of PMMA,680 but it is also an especially toxic designer amphetamine that has resulted in several deaths from its unsuspected ingestion as an ecstasy substitute.444
PMA is 10 times more active than MDMA in elevating brain serotonin concentrations and inhibiting serotonin uptake, but it has only a few effects on the dopamine system.186,327,783,784 Inhibition of MAO A is a further pharma-cologic property of PMA.297 These pharmacologic properties are thought by some to be responsible for the higher rate of death seen with PMA compared with other substituted amphetamines.404 Multiple deaths have been associated with its use; symptoms usually mimic serotonin syndrome and include hyperthermia, tachycardia, seizures, cardiac dys-rhythmias, and coma.143,241,374
Ephedrine and PseudoephedrineThese amines are diastereoisomers that possess two asym-metrical carbon atoms and exist as four isomers designated as 1R,2S- and 1S,2R-ephedrine and 1R,2R- and 1S,2S-pseudoephedrine.302 The 1R,2S-ephedrine (ephedrine) and 1S,2S-pseudoephedrine (pseudoephedrine) isomers occur naturally in various plants of the Ephedra genus.
Ephedrine and pseudoephedrine have been used as nasal decongestants, bronchodilators, and CNS stimulants, and for the treatment of obesity.25,403,486,835 Ephedrine is both an α- and a β-adrenergic receptor agonist; in addition, it enhances the release of norepinephrine from sympathetic neurons and is considered a mixed-acting sympathomimetic drug.835 Many dietary supplements contain ephedra, the herbal form of ephedrine. These products are widely marketed for energy enhancement or weight loss51,395 and are used by some ath-letes to enhance performance. Adverse effects such as hyper-tension, tremors, myocardial infarction, seizures, and stroke have resulted in fatalities.141,317,837 Because of this, the FDA banned the sale of dietary supplements containing ephedra in 2004.835 However, herbal products containing ephedra remain in use in other countries.
Pseudoephedrine is used primarily as a decongestant because of its vasoconstrictive properties (α-adrenergic action).64 It also is used as a nasal decongestant and precursor for the illicit synthesis of methamphetamine. Because of this,
MDMA is N-demethylated in humans to MDA, via CYP1A2, and to a significantly lesser extent by CYP2D6.488 Although extensive and poor MDMA metabolizers have been identified, the contribution of these polymorphisms to MDMA toxicity is unclear, because the metabolism may be saturable even at normal doses, resulting in greater dose-proportional excretion of the parent drug.189 The saturable kinetics does, however, suggest that beyond a certain thresh-old, small increases in dose may result in larger increases in plasma concentration, and consequently greater risk of toxicity.488
MDEA (3,4-Methylenedioxyethylamphetamine). MDEA is an empathogen-entactogen drug of the phenethylamine family that produces distinctive emotional and social effects similar to those of MDMA. On the street, it is known as
Phenylethylamines• 3,4-Methylenedioxymethamphetamine (MDMA; Ecstasy)• 3,4-Methylenedioxyethylamphetamine (MDEA; “Eve”)• 3,4-Methylenedioxyamphetamine (MDA), which is also a
metabolite of MDMA• Paramethoxyamphetamine (PMA)• Paramethoxymethamphetamine (PMMA)• 2,5-Dimethoxy-4-methylamphetamine (DOM)• 2,5-Dimethoxy-4-methylthioamphetamine (DOT)• 4-Iodo-2,5-dimethoxyamphetamine (DOI)• 2,5-Dimethoxy-4-bromo-amphetamine (DOB)• 2,5-Dimethoxy-4-bromo-methamphetamine (MDOB)• 3,4-(Methylenedioxyphenyl)-2-butanamine (BDB)• N-Methyl-1-(3,4-methylenedioxy-phenyl)-2-butanamine
(MBDB)• 6-Chloro-3,4-methylenedioxymethamphetamine
(Cl-MDMA)• 3,4-Methylenedioxymethcathinone• 4-Bromo-2,5-diemthoxy-phenylethylamine (2C-B)• 2,5-Dimethoxy-4ethylthio-phenylethylamine (2C-T-2)• 2,5-Dimethoxy-4 propylthio-phenylethylamine (2C-T-7)
Benzylpiperazines• 1-Benzylpiperazine (BZP)• 1-(3,4-Methylenedioxybenzyl)-piperazine (MDBP)
Phenylpiperazines• 1-(3-Trifluoromethylphenyl)piperazine (TFMPP)• 1-(3-Chlorophenyl)piperazine (mCPP)• 1-(4-Methoxyphenyl)piperazine (MeOPP)
Pyrrolidinophenone• α-Pyrrolidinopropiophenone (PPP)• 4-Methoxy-α-pyrrolidinopropiophenone (MOPPP)• 3,4-Methylenedioxy-α-pyrrolidinopropiophenone (MDPPP)• 4-Methyl-α-pyrrolidinopropiophenone (MPPP)• 4-Methyl-α-pyrrolidinohexanophenone (MPHP)
BOX 35-2 Designer Drugs Related to Phenylethylamine, Benzylpiperazine, Phenylpiperazine, and Pyrrolidinophenone87,139,385,509,525,770

1154 SectionIII ■ Analytes
manufacturers can have very different “interference” profiles, which the pathologist and the laboratory scientist must understand and relay to clinicians.
Regarding methylphenidate, it should be noted that its detection by urine drug immunoassay is problematic, as it does not cross-react well with amphetamine immunoassays; detection of the parent drug is made difficult by its generally low concentration; and ritalinic acid, present in much higher concentration, is difficult to extract and analyze by GC; it is unstable upon storage even when frozen.61
Confirmatory Methods. All positive immunoassay results should be confirmed by a second independent method, but what may be more significant is that if the other designer amphetamines (see Box 35-2) are suspected, a negative immunoassay screen cannot rule out the presence of these drugs. Fortunately, numerous GC- and LC-based methods for identification and quantitation of these drugs in biological samples have been put forth.418,640,808
Amphetamine-type stimulants are considered volatile and are lost during a dry-down or evaporation step, if this is part of the procedure. This loss is avoided by the addition of a small amount of hydrochloric acid during the evaporation step, or the addition of a less volatile “keeper” solvent such as dimethylformamide (DMF).487 Also, because of their extreme volatility at the high temperatures encountered in GC-MS, derivatization prior to analysis lowers the limit of detection. Although many derivatives are available for GC-MS use, the most commonly used include heptafluorobuic anhydride (HFBA), pentafluoropropionic anhydride (PFPA), trifluoro-acetic anhydride (TFAA), and 4-carbethoxyhexfluorobutyryl chloride (4-CB). However, the 4-CB derivative in the pres-ence of ephedrine/pseudoephedrine may generate metham-phetamine,775 which leads to the DHHS rule to have amphetamine also detected to report a positive methamphet-amine result.556
Methamphetamine is a prototypical basic drug (pKa 9.9) that is readily extracted from biological material into organic solvents at alkaline pH. It is readily soluble in chloroform, N-butyl chloride, ethyl acetate, and diethyl ether, and is extracted in most common protocols designed to isolate alkaloidal and basic drugs. It also readily extracts back into acid, and back into organic solvents without significant loss.487 Most published methods for analysis of members of the amphetamine class in urine, plasma, and blood use LLE or SPE.488
Methamphetamine is readily analyzed by GC; this is the most popular method in use today for analysis of metham-phetamine in biological material. Its poor UV absorption properties make it an unsuitable candidate for HPLC with UV detection, and it has no native fluorescence and no sig-nificant oxidative electrochemical properties at low volt-ages.487 Liquid chromatography may be used for MDMA analysis and offers an advantage over GC in that MDMA and its polar metabolites can be quantified simultaneously without derivatization.488
The molecular weight of methamphetamine, the low intensity of its mass fragments in electron impact mode, and
the quantity per purchase of products containing these drugs is now restricted in many places.
PPA (Phenylpropanolamine)PPA was widely available in a number of nonprescription cold medications and diet control products. Adverse effects are similar to those described for ephedrine. In response to an FDA warning of increased risk of hemorrhagic stroke, especially in women, PPA has been withdrawn from the market by most manufacturers.63 PPA is also a metabolite of ephedrine and pseudoephedrine.51,64
Methylphenidate (Ritalin)Methylphenidate (MPH) is a phenethylamine derivative with psychostimulant properties similar to S(+) amphetamine. It is commonly used to treat attention-deficit hyperactivity disorder (ADHD) and narcolepsy.61,497 Its pharmacologic properties are essentially the same as those of the amphet-amines.835 Like many of its related amphetamine-type stimu-lants, it exists as an isomer, as (R,R)-methylphenidate (d-MPH), and as (S,S)-methylphenidate (l-MPH).510 The pharmacologic actions of MPH are almost solely performed by the d-isomer.510 Methylphenidate is rapidly metabolized favoring l-MPH over d-MPH,510 such that the more potent d-MPH has a half-life of about 6 hours, and the less potent l-MPH has a half-life of about 4 hours.835 Diversion and abuse of methylphenidate have been increasing among children and adults because of its stimulant and purported aphrodisiac properties. In overdose, the clinical effects of methylphenidate are similar to those of amphetamine and produce signs of generalized CNS stimulation that may lead to convulsions.497
Analytical Methods. The initial screening test for amphet-amines and related drugs is typically immunoassay. For con-firmation of a presumptive positive test, a quantitative drug measurement is performed using GC-MS.
Immunoassay. Most “amphetamine” immunoassays have been designed to detect amphetamine/methamphetamine; others have been designed to detect MDMA and MDA; and others to more broadly capture the ATS group—all with varying cross-reactivities.346,799 Many older immunoassays lacked the ability to distinguish between the isoforms.645 Currently, many use antibodies specific for S(+) amphet-amine (d-amphetamine) and/or S(+) methamphetamine (d-methamphetamine). The degree of cross-reactivity of these antibodies varies, with antibodies raised to immuno-gens protein-linked to the amphetamine molecule through the phenyl ring having better cross-reactivity than those linked through the sidechain.488
Not all amphetamine immunoassays were suitable for detection of the amphetamine-derived designer drugs PMA, PMMA, and MDEA,367,472,766,871 and especially not for the new piperazine-derived substances.188,752 Alternatively, other chemically related compounds such as pseudoephedrine have been shown to produce positive results.860 Additionally, many psychotropic medications have been reported to interfere with immunoassays.556,734 Immunoassays from different

Chapter35 ■ Clinical Toxicology 1155
medicine, it is used mainly for local anesthesia and vasocon-striction in nasal surgery, and to dilate pupils in ophthalmol-ogy. Sigmund Freud famously proposed its use to treat depression and alcohol dependence, but the realities of cocaine addiction quickly brought this idea to an end.497 Cocaine abuse has a long history and is rooted in the drug culture in the United States.21 Cocaine is still one of the most common illicit drugs of abuse.18,361 According to the National Survey on Drug Use and Health, the rate of past year use for cocaine (powder and crack combined) among individuals aged 12 and older has remained stable since 2002; 2.1 million users were reported in 2007.107
Cocaine is sold on the street in two forms: a hydrochloride salt (powder) and a free-base product known as “crack.” The hydrochloride salt form of cocaine is administered by nasal insufflation (“snorting”) or, less frequently, intravenously. “Crack” is a free-base form that has not been neutralized by an acid to make the hydrochloride salt. It comes as a rock crystal that is heated and its vapors smoked. The term refers to the crackling sound heard when it is heated.361
It should be noted that the use of “crack” cocaine is not to be confused with “free-basing,” which is a process in which the user purifies cocaine HCl by mixing an aqueous solution of cocaine with baking soda or ammonia and adding diethyl ether, thereby extracting the free form of the drug into the organic solvent, which is then evaporated to dryness. The drug can then be smoked. However, because of the extremely flammable nature of diethyl ether, and therefore the risk of igniting any remaining ether, “free-basing” is no longer com-monly practiced.361
Chemically, cocaine is methylbenzoylecognine (COC), an ester of benzoic acid and the amino alcohol (methylecog-nine) that contains a tropine moiety.362 Its metabolism is complex (Figure 35-14) and occurs via both nonenzymatic hydrolysis and enzymatic transformation in the plasma and liver, where it is rapidly metabolized to benzoylecgonine (BE) and ecogonine methyl ester, both of which are inactive.361 COC contains two ester moieties; the alkyl ester is hydrolyzed to its major metabolite BE via spontaneous hydrolysis at physiologic and alkaline pH.361 It has been shown that COC is also hydrolyzed to BE by liver carboxylesterases.192 BE is considered to be a pharmacologically inactive metabolite, but because its half-life is longer than that of COC, it is the most commonly monitored analyte in urine for determination of COC use.
BE is further metabolized to minor metabolites such as m-hydroxybenzoylecgonine (m-HOBE) and p- hydroxybenzoylecgonine (p-HOBE).362,441 Of these, m-HOBE has been shown to be an important metabolite in the meco-nium of cocaine-exposed babies.474,555 Positive BE results in urine are sometimes challenged in legal and administrative proceedings on the grounds that the presence of BE is due to the addition of COC to the urine sample with subsequent in vitro hydrolysis to BE. However, m-HOBE is believed to arise exclusively via in vivo metabolism435; therefore, its presence confirms COC use. Additionally, in adults, m-HOBE has a longer half-life and has the potential to be detected for longer
the structural similarity of many endogenous and exogenous compounds result in its mass spectrum not being highly char-acteristic.487 The issue of lack of specificity of the metham-phetamine mass spectrum is resolved by derivatization.792 Many methods have been published for analysis of amphet-amine, methamphetamine, and related compounds.* Unfor-tunately, routine GC-MS also does not distinguish between the two isomers and necessitates the use of chiral chromatog-raphy to differentiate between them. Chiral discrimination of methamphetamine isomers may be necessary to distinguish the use of nonprescription nasal inhalants [R(−) metham-phetamine] from the illicit use of methamphetamine [S(+) and R(−)] or other prescription medications, as indicated in Table 35-11. Some immunoassays have high specificity for S(+)isoforms. However, definitive enantio-discrimination requires the use of a chiral derivatization reagent conven-tional GC-MS,150,245,350,667 or possibly chiral separation by LC-MS or LC-MS/MS. Also, care must be taken in interpret-ing the results of drug screens. Several other prescription drugs available in the United States and Canada that are metabolized to amphetamine or methamphetamine are listed in Table 35-11.
Regarding methylphenidate, its confirmation by GC-MS is complicated by the fact that it does not form a stable N,O-di-trimethylsilyl derivative. However, after sequential reactions with MSTFA [N-methyl-N-(trimethylsilyl)trifluoroacet-amide] and MBTFA [N-methyl-bis (trifluoroacetamide)] to form the N-trifluoroacetyl, O-trimethylsilyl ester,222 it is pos-sible to measure methylphenidate by GC-MS. Ritalinic acid may be isolated from urine using a dehydration procedure, then methylated with dimethylformamide dimethyl acetal, and the resulting methylphenidate analyzed by GC-MS. Last, ritalinic acid may be analyzed directly by LC-tandem mass spectrometry,508 or by GC-MS, after sequential reactions with MSTFA and MBTFA to form the N-trifluoroacetyl, O-trimethylsilyl ester,222 or after methylation to re-form methylphenidate.
CocaineCocaine is an alkaloid found in Erythroxylon coca, which grows principally in the northern South American Andes and to a lesser extent in India, Africa, and Java.361,362 In clinical
TABLE 35-11 PrescriptionDrugsThatAreMetabolizedtoAmphetamineorMethamphetamine
Drug Drugs Detected153,487,681
Adderall amphetamine amphetamineDexedrin d-amphetamine d-amphetamineDeprenyl selegiline l-methamphetamine
l-amphetamineDidrex benzphetamine methamphetamine
amphetamine
*References 2, 198, 275, 331, 487, 488, 524, and 723.
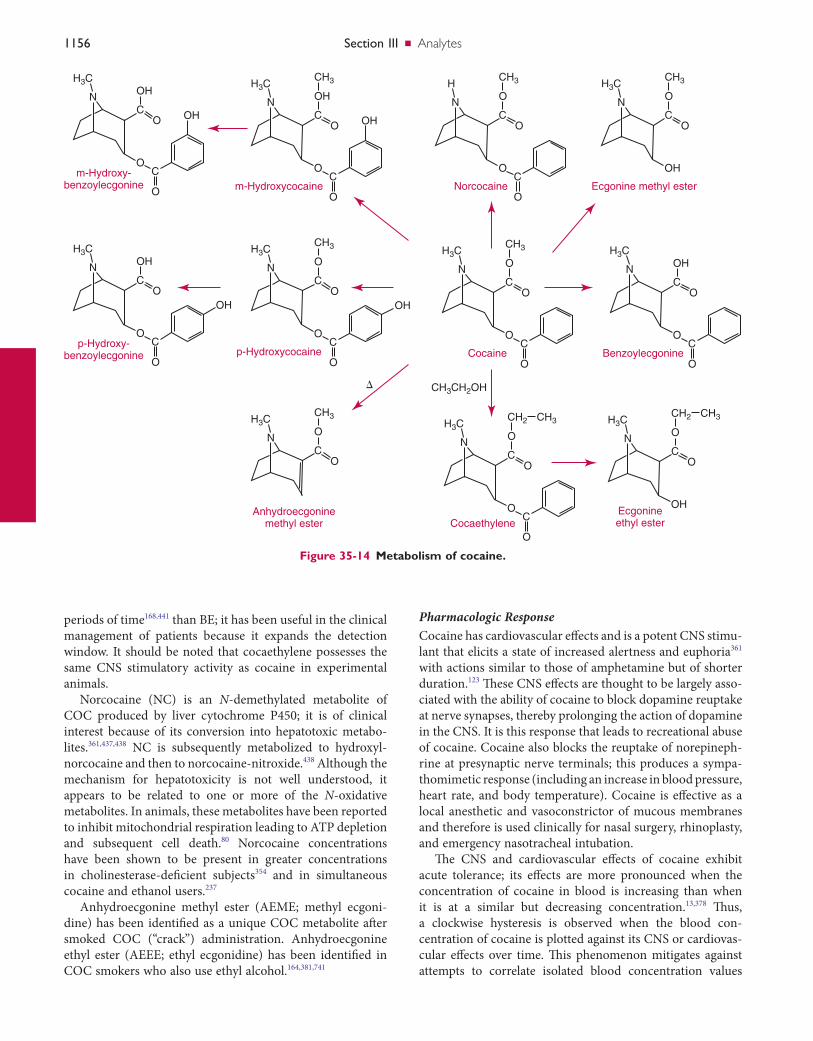
1156 SectionIII ■ Analytes
Pharmacologic ResponseCocaine has cardiovascular effects and is a potent CNS stimu-lant that elicits a state of increased alertness and euphoria361 with actions similar to those of amphetamine but of shorter duration.123 These CNS effects are thought to be largely asso-ciated with the ability of cocaine to block dopamine reuptake at nerve synapses, thereby prolonging the action of dopamine in the CNS. It is this response that leads to recreational abuse of cocaine. Cocaine also blocks the reuptake of norepineph-rine at presynaptic nerve terminals; this produces a sympa-thomimetic response (including an increase in blood pressure, heart rate, and body temperature). Cocaine is effective as a local anesthetic and vasoconstrictor of mucous membranes and therefore is used clinically for nasal surgery, rhinoplasty, and emergency nasotracheal intubation.
The CNS and cardiovascular effects of cocaine exhibit acute tolerance; its effects are more pronounced when the concentration of cocaine in blood is increasing than when it is at a similar but decreasing concentration.13,378 Thus, a clockwise hysteresis is observed when the blood con-centration of cocaine is plotted against its CNS or cardiovas-cular effects over time. This phenomenon mitigates against attempts to correlate isolated blood concentration values
periods of time168,441 than BE; it has been useful in the clinical management of patients because it expands the detection window. It should be noted that cocaethylene possesses the same CNS stimulatory activity as cocaine in experimental animals.
Norcocaine (NC) is an N-demethylated metabolite of COC produced by liver cytochrome P450; it is of clinical interest because of its conversion into hepatotoxic metabo-lites.361,437,438 NC is subsequently metabolized to hydroxyl-norcocaine and then to norcocaine-nitroxide.438 Although the mechanism for hepatotoxicity is not well understood, it appears to be related to one or more of the N-oxidative metabolites. In animals, these metabolites have been reported to inhibit mitochondrial respiration leading to ATP depletion and subsequent cell death.80 Norcocaine concentrations have been shown to be present in greater concentrations in cholinesterase-deficient subjects354 and in simultaneous cocaine and ethanol users.237
Anhydroecgonine methyl ester (AEME; methyl ecgoni-dine) has been identified as a unique COC metabolite after smoked COC (“crack”) administration. Anhydroecgonine ethyl ester (AEEE; ethyl ecgonidine) has been identified in COC smokers who also use ethyl alcohol.164,381,741
Figure 35-14 Metabolism of cocaine.
OH
OH
Om-Hydroxy-
benzoylecgonine
O
O
C
CN
H3C
OH
CH3
OH
O
m-Hydroxycocaine
O
O
C
CN
H3CO
CH3
OH
OC
N
H3CO
CH3
O
Norcocaine Ecgonine methyl ester
O
O
C
CN
H
OH
OH
Op-Hydroxy-
benzoylecgonine
O
O
C
CN
H3CO
CH3
OH
O
p-Hydroxycocaine
O
O
�
C
CN
H3C
O
CH3
Anhydroecgoninemethyl ester
OC
N
H3C
O
CH3
O
Cocaine
O
O
C
CN
H3C
O
CH2
CH3CH2OH
CH3
OCocaethylene
Ecgonineethyl ester
O
O
C
CN
H3CO
CH2 CH3
OH
OC
N
H3C
OH
O
Benzoylecgonine
O
O
C
CN
H3C

Chapter35 ■ Clinical Toxicology 1157
interpreted. A positive urine drug test for benzoylecgonine beyond 3 days after the last dose does not necessarily indicate continued use. For such purposes, it is better to monitor quantitatively the urinary excretion of benzoylecgonine, nor-malized to creatinine, over time.768 Drug abstinence would be indicated by decreasing urinary excretion of cocaine metabo-lites. However, creatinine normalization may not always reli-ably indicate reuse.656
The initial screening test for cocaine (BE) is typically immunoassay. For confirmation of a presumptive positive, BE is quantified by GC-MS.
Screening. The half-life of cocaine is 0.5 to 1.5 hours, of ecgonine methyl ester 3 to 4 hours, and of BE 4 to 7 hours.376 Thus, BE is the analyte of choice in screening for cocaine use.362 The initial screening test for BE is typically immunoas-say, and screening immunoassays frequently apply a 300-ng/mL cutoff.
Confirmation. Most confirmation assays offer quanti-fication of both parent drug and metabolite. Numerous methods have been described for the measurement of COC and various metabolites. GC techniques for analysis of COC and its metabolites require derivatization, especially of polar metabolites. Early detection techniques have included flame ion detection (FID), EC, and nitrogen-phosphorous detector (NPD).* GC-MS is the method of choice for many laborato-ries.† Some methods have included not only COC and BE, but also clinically and forensically relevant secondary metab-olites such as m-HOBE, CE, NC, AEME, and AEEE.164,399,435,622 The use of LC-based separation techniques that detect COC, BE, and CE has been described previously, including LC-UV detection,517,543,760 as well as LC-DAD.147 LC-MS/MS methods have also been described, including COC, BE, and m-HOBE,‡ along with other relevant secondary metabolites such as CE, NC, AEME, and AEEE.460 Reports have suggested that AEME is not a truly unique indicator of smoked cocaine use, because it has been reported to be produced in the injector port of a GC370,495,776 at high temperatures. However, less than 1% generation of AEME occurs if the injector port of the GC is maintained at 250 °C.164 In an LC method, high temperatures are not present in the injector or in any other part of the LC; therefore, AEME is not generated, and its presence identifies a smoked route of COC use.
Lysergic Acid Diethylamide (LSD)LSD shares structural features with serotonin (5- hydroxytryptamine; Figure 35-15), a major CNS neurotrans-mitter and neuromodulator.32,857 LSD is synthesized from d-lysergic acid, a naturally occurring ergot alkaloid found in the fungus Claviceps purpurea, which grows on wheat and other grains. During synthesis, some LSD epimerizes to iso-LSD, which is inactive.649
with psychomotor effects. Because rate of change is probably more significant than absolute concentration, the psychomo-tor stimulant effects of cocaine are dependent both on dose and on route of administration, with IV administration and smoking resulting in the most rapid rates of increase in concentration.
Acute cocaine toxicity produces a sympathomimetic response that may result in (1) mydriasis, (2) diaphoresis, (3) hyperactive bowel sounds, (4) tachycardia, (5) hyper-tension, (6) hyperthermia, (7) hyperactivity, (8) agitation, (9) seizures, or (10) coma. Sudden death due to cardiotoxicity may occur following cocaine use. Death may also occur fol-lowing the sequential development of hyperthermia, agitated delirium, and respiratory arrest. Excited delirium and extreme physical activity may lead to rhabdomyolysis, acute renal failure, and disseminated intravascular coagulopathy.
COC is frequently used with other drugs, most commonly ethanol. In simultaneous COC and ethanol use, liver methyl-esterase catalyzes the conversion of COC to BE and the transesterification of COC to CE in the presence of ethyl alcohol.326,361,377 This reaction occurs about 3.5 times faster than hydrolysis to BE.99 COC administered with ethanol pro-duced greater euphoria and enhanced perception of well-being relative to COC.361,528 CE appears to be equipotent to cocaine with regard to dopamine transporter affinity326 but is less potent than cocaine pharmacologically.323,634 As a consequence, large amounts of COC and ethanol may be ingested, placing users at greater risk for toxicity than if either drug were used alone. The elimination half-life for cocaethylene is longer than that for cocaine.369,378 This longer elimination half-life may contribute to the toxicity of coca-ethylene. Additionally, with simultaneous administration of COC and ethanol, the production of NC may be increased, along with the potential for toxicity.237,362,441 It has been sug-gested that simultaneous COC and ethanol use carries an 18- to 25-fold increase in risk for immediate death over COC alone.20,361,528,796
Analytical MethodsThe elimination half-life for cocaine varies from 0.5 to 1.5 hours, for ecgonine methyl ester from 3 to 4 hours, and for benzoylecgonine from 4 to 7 hours.5-7 The principal urinary metabolites are benzoylecgonine and ecgonine methyl ester. Only small amounts of cocaine are excreted in urine. The elimination half-life for cocaethylene is 2.5 to 6 hours,19,218,221 which is considerably longer than that for cocaine. This longer elimination half-life may contribute to the toxicity of cocaethylene.
BE excretion is detectable for 1 to 3 days following cocaine use. However, for chronic heavy cocaine users, the detection time may extend to 10 to 22 days following the last dose,831 apparently because of tissue storage of cocaine. Ordinarily, cocaine may be detected in urine by chromatographic methods for only about 8 to 12 hours after use, but in heavy chronic users, this detection period may last 4 to 5 days.169 These facts should be considered when the results of urine drug testing for individuals in drug treatment programs are ‡References 242, 380, 384, 436, 578, 677, and 763.
†References 174, 190, 335, 363, 733, and 747.*References 373, 379, 440, 603, 807, and 817.

1158 SectionIII ■ Analytes
man.649 Iso-LSD is not a metabolite but is formed by nonen-zymatic epimerization of LSD during synthesis or storage of urine at alkaline pH and elevated temperature.700
The clinical effects of LSD ingestion are usually benign and require no medical intervention. However, panic attacks may be severe and require treatment with diazepam; LSD-induced psychosis has been treated with haloperidol. Rare cases of massive overdose have resulted in life-threatening hyperther-mia, rhabdomyolysis, acute renal failure, hepatic failure, dis-seminated intravascular coagulation (DIC), respiratory arrest, and coma. Few if any well-documented deaths directly related to LSD ingestion have been reported.
Analytical MethodsBecause of the very high potency of LSD, and therefore a low typical dose (20 to 80 µg) and rapid and extensive metabo-lism, only about 1 to 2% of the drug is excreted unchanged in urine.672 Thus, detection of LSD presents an especially difficult analytical challenge. Even with sensitive assays, the detection window for LSD is generally only 12 to 24 hours.672
Immunoassays are targeted to detect LSD at the usual cutoff concentration of 500 pg/mL. Confirmation is typically performed by GC-MS256,624 at the U.S. Department of Defense established cutoff concentration of 200 pg/mL. Although the metabolites 2-oxo-3-hydroxy-LSD and N-demethyl LSD gen-erally cross-react only when present at about 100 to 200 times the amount in LSD,841 other metabolites may potentially account for some instances of nonconfirmed positive immu-noassay response.152,859 However, true false-positive results due to various therapeutic drugs may occur.152,675,841 The detection window may be extended, perhaps twofold to threefold, by including 2-oxo-3-hydroxy-LSD in the confir-matory test, using sensitive techniques such as GC-MS-MS,672 LC-MS-MS,643 or LC-MS.434,642 Likewise, detection of iso-LSD in addition to LSD may extend the detection interval.146 Urine specimens should be protected from sunlight, bright fluores-cent light, or elevated temperature at alkaline pH to avoid degradation of LSD478 and 2-oxo-3-hydroxy-LSD or epimer-ization of LSD to iso-LSD.478,700
Drugs Used in Sexual AssaultDrug-facilitated sexual assault (DFSA) is defined as voluntary or surreptitious use of alcohol, drugs, and/or chemical agents to incapacitate an individual and facilitate sexual assault.315 In addition to alcohol, the drugs most often impli-cated in drug-facilitated sexual assault include (1) choral hydrate, (2) flunitrazepam, (3) nonbenzodiazepine sedative-hypnotics, (4) gamma-hydroxybutyric acid (GHB), (5) dex-tramethorphan, (6) ketamine, (7) phencyclidine, and (8) benzodiazepines, and nonprescription medications such as (9) antihistamines and (10) anticholinergics (Box 35-3). These drugs share similar characteristics that are desired by an assailant such as fast onset, colorlessness, tastelessness, and easy access. Similar clinical effects permit the victim to be easily incapacitated. They include impaired judgment, confu-sion, reduced inhibitions, sedation, hypnosis, loss of muscle coordination, and sometimes anterograde amnesia. These
Pharmacologic ResponseLSD is an extremely potent psychedelic ergot alkaloid derived from the fungus, Claviceps purpurea.32 The drug LSD binds to serotonin receptors in the CNS and acts as a serotonin agonist. The principal psychological effects of LSD are per-ceptual distortions of color, sound, distance, and shape; de-personalization and loss of body image; and rapidly changing emotions from ecstasy to depression or paranoia. These hal-lucinogenic actions of LSD are stereoselective, elicited only by the d-isomer. A resurgence has occurred in the use of LSD, previously popular as a drug of abuse during the 1960s. The Department of Defense includes LSD among the drugs for which urine testing is required (see Table 35-7).
The physiologic effects of LSD are related to its sympatho-mimetic actions and include mydriasis (most frequent and consistent), tachycardia, increased body temperature, dia-phoresis, and hypertension; at higher doses, parasympatho-mimetic actions may be observed [e.g., salivation, lacrimation, nausea, vomiting (muscarinic actions)]. Neuromuscular effects may include paresthesia, muscle twitches, and incoor-dination (nicotinic actions).32,857
The most common adverse effects of LSD are panic attacks. In addition, unpredictable recurrence of hallucinations (flashbacks) may occur weeks or months after last drug use, and LSD may elicit psychotic reactions (thought disorders, hallucinations, depression, and depersonalization). LSD is used illicitly because of its hallucinogenic effects. No evidence suggests that repeated LSD use results in dependence or with-drawal symptoms.32,857
Popular dosage forms include powder, gelatin capsule, tablet, and LSD-impregnated sugar cubes, filter paper, or postage stamps. The drug is rapidly absorbed from the GI tract; the effects begin within 40 to 60 minutes, peak at about 2 to 4 hours, and subside by 6 to 8 hours. The elimination t1/2 is about 3 hours. The metabolism of LSD in humans is incom-pletely understood, but 2-oxo-3-hydroxy-LSD is present in urine at concentrations 10- to 43-fold greater than LSD.642,643,649,672 N-demethyl-LSD is also present in urine spec-imens, but at concentrations approximately equivalent to those of LSD. The other metabolites are among those identi-fied in animals, but as yet not conclusively identified in
Figure 35-15 Chemical structure of lysergic acid diethylamide (LSD) and serotonin.
HO
O
C
N CH3
N
H
LSD Serotonin
(C2H5)2N
N
NH2
H

Chapter35 ■ Clinical Toxicology 1159
clinical effects, but may also prolong their duration of action.93,587
Analytical MethodsChloral hydrate is not detected on routine, commercially available drug screens. Quantification of chloral hydrate and its metabolites trichloroethanol (TCE), TCE-glucuronide, and trichloroacetic acid is detected in plasma using HPLC-MS and capillary gas chromatography with electron-capture detection (GC-ECD),352,546,710 or GC-flame ion detection (GC-FID).393 Typical therapeutic concentrations are 2 to 12 µg/mL. Chloral hydrate metabolites have been detected as low as 10 ng/mL using GC-ECD.352
FlunitrazepamIt is estimated that 8% of sexual assault cases are positive for benzodiazepines.232,336,582,581 Flunitrazepam (Rohypnol) is the most frequently reported benzodiazepine used in DFSA, par-tially because of the development and implementation of specific toxicologic tests in response to increased public awareness, resulting in a testing bias.219,232,600,705,729 Other ben-zodiazepines that have been reported in sexual assault victims are diazepam, triazolam, temazepam, tetrazepam, and clonazepam.4,134,398,507,581
Flunitrazepam is a fast-acting sedative-hypnotic catego-rized as a Schedule I drug in the United States. Because it is still licensed for use in Europe, Asia, and Latin America for sedation and treatment of insomnia, sexual predators can acquire this drug through illegal trafficking.822 Sexual assault predators use flunitrazepam because it can be easily dissolved into a beverage, it is relatively tasteless and odorless, it will quickly incapacitate their victims, and routine drug screens do not detect its presence.
Pharmacologic EffectsFlunitrazepam is more potent than diazepam because of its slower dissociation from the GABA receptor.137,520,521 It is rapidly absorbed and distributed into tissues upon oral administration. Onset of its sedative, amnesic, hypnotic, and disinhibitory effects can occur within 20 to 30 minutes.520 Flunitrazepam has a long half-life (≈26 hours), permitting an extended window of detection in blood and urine.
Although the effects of flunitrazepam occur rapidly when used alone, it is often coingested with alcohol, which ampli-fies its effects.209,720 Initial symptoms may consist of dizziness, disorientation, lack of coordination, and slurred speech, all of which mimic alcohol intoxication. Another unique effect is anterograde amnesia as early as 15 minutes after oral administration.294 Rapid alternation of hot and cold flashes may be followed precipitously by loss of consciousness. Large doses (>2 g) have produced aspiration, muscular hypotonia, hypotension, bradycardia, coma, and death.134,507,716
Analytical MethodsThe detection of flunitrazepam is especially challenging because of the low therapeutic and illicit doses and the low degree of cross-reactivity of most immunoassays with the
effects are intensified when they are coadministered willingly or involuntarily with other psychotropic medications that produce CNS depression. This is a common occurrence in reported cases of sexual assault or rape.232,336
Choral HydrateAnecdotal reports concerning assailants dosing beverages with incapacitating compounds to assault their victims date back to the early nineteenth century. An infamous example is the saloon proprietor Mickey Finn. He was alleged to have drugged his customers with the addition of chloral hydrate to their ethanol-based beverages and to have subsequently robbed them.
Chloral hydrate is classified as a nonbarbiturate hypnotic. It is an inexpensive transparent crystalline compound that easily dissolves in beverages. It was first synthesized in 1832 and was one of the original “depressants” developed for the specific purpose of inducing sleep. This drug is still used today in pediatric medicine for sedating children before diag-nostic procedures. Abuse and misuse of this drug and subse-quent introduction of newer sedatives (barbiturates and benzodiazepines) led to its decline for medicinal purposes.
Pharmacologic EffectsThe clinical diagnosis of chloral hydrate intoxication is diffi-cult to differentiate from alcohol, benzodiazepine, and barbi-turate intoxication, as all share similar clinical effects. Although the exact mechanism of action of chloral hydrate has not been determined, it is a general CNS depressant that has sedative effects with minimal analgesic effects when administered independently. At low doses (<20 mg/kg), symptoms may include relaxation, dizziness, slurred speech, confusion, disorientation, euphoria, irritability, and hyper-sensitivity rash. At higher doses (>50 mg/kg), chloral hydrate causes hypotension, hypothermia, hypoventilation, tachydys-rhythmias, nausea, vomiting, diarrhea, headache, and amnesia.690 Onset of action is rapid (10 to 20 minutes). The elimination half-life of choral hydrate is 4 to 12 hours.93,690 If coingested with alcohol, the metabolism of chloral hydrate may be seriously impaired. Because both ethanol and chloral hydrate are metabolized by CYP2E1 and alcohol dehydrogenase, coingestion may not only exacerbate their
AnticholinergicsAntihistaminesBarbituratesBenzodiazepinesChloral hydrateDextromethorphanEthanolγ-Hydroxybutyrate (GHB)KetamineOpioidsSedative-hypnotics
BOX 35-3 Examples of Agents Used in Drug-Facilitated Sexual Assault

1160 SectionIII ■ Analytes
following the event and may be delayed in reporting their sexual assault. Commonly utilized drug screens do not test for these substances.
Recognition of these new-generation sleep aids as agents potentially used in facilitating sexual assault has been reported in the United States, the United Kingdom, and France for over a decade.17,232,295,336,716 Yet only two published reports in the United States tested sexual assault victims for the presence of zolpidem.429,805
All may produce additive CNS-depressant effects when coadministered with other psychotropic medications such as anticonvulsants, antihistamines, ethanol, and other drugs that produce CNS depression.
Pharmacologic EffectsExamples of nonbenzodiazepine sedative-hypnotics include (1) zolpidem, (2) zaleplon, (3) eszopiclone, and (4) zopiclone.
Zolpidem. The pharmacologic effects of Zolpidem (Ambien) are believed to result from its interaction with a specific subtype of GABAA receptor complex consisting of α1-subunits.319 It is available as an immediate- or extended-release tablet. After an average oral dose of 10 to 15 mg, onset of effects occurs in 10 and 30 minutes. Clinical effects peak at approximately 1.5 hours for immediate release, duration is about 6 to 8 hours for both immediate- and extended-release preparations, and the t1/2 is approximately 2.5 hours.23 Evidence of minimal respiratory depression is noted when used as a single agent, but it may produce additive CNS-depressive effects and death when coadministered with other sedatives.289
Zaleplon. Zaleplon (Sonata) is available as an immediate-release tablet or capsule. After an average oral dose of 10 to 15 mg onset of effects occurs in approximately 10 to 30 minutes. Although the t1/2 for zaleplon is about 1 hour, the duration of clinical effects may persist for longer than 6 hours. This may be due to the higher affinity of zaleplon for specific α2- and α3-subunits of the GABA receptor, in contrast to zolpidem or zopiclone.277 At higher doses (>40 to 60 mg), its use may cause increased central nervous system effects and impaired motor skills.690
Eszopiclone. The exact mechanism of action of eszopi-clone (Lunesta) is unknown, but its effect is believed to result from interaction with GABAA receptor complexes containing α1- to α5-subunits.319 After an average dose of 2 to 3 mg onset of effects occurs in approximately 30 minutes. Both immedi-ate- and extended-release forms are available. The clinical effects of eszopiclone are longer in duration compared with those of zopiclone or zolpidem, with a t1/2 of 6 hours.703
Zopiclone. Zopiclone is not currently available in the United States. It is the racemic mixture of two stereoisomers; the active stereoisomer is eszopiclone. Therefore, clinical effects are similar to those of eszopiclone.
Analytical MethodsBecause of the amnesic properties of these drugs, victims often may not report their sexual assault for several days. Therefore, sensitive analytical techniques are necessary to
principal urinary metabolite, 7-aminoflunitrazepam.518,699,743 As for other benzodiazepines, prior glucuronidase hydrolysis may improve immunoassay detection. Enzyme-linked immu-nosorbent assay (ELISA) methods with high selectivity for 7-aminoflunitrazepam and low limits of detection have been developed.821,824 Direct analysis or confirmation of 7-aminoflunitrazepam by GC-MS or LC-MS/MS is indicated in suspected cases of flunitrazepam ingestion.230,824,863 Fluni-trazepam metabolites are detectable as early as 7 days in hair samples (HPLC-MS/MS).134 Deposition and stability of a drug in hair samples are variable, depending on the route of exposure and the chemical characteristics.
Nonbenzodiazepine Sedative-HypnoticsZopiclone, eszopiclone (Lunesta), zolpidem (Ambien), and zaleplon (Sonata) belong to a new generation of sedative-hypnotics that are structurally different from benzodiazepines (Figure 35-16). Similar to benzodiazepines, these drugs mod-ulate the GABAA receptor chloride channel by binding to the benzodiazepine (BZ) receptors, otherwise known as the omega1 (ω1) receptors, in the brain812 without binding to peripheral BZ receptors.703,721 Therefore, these drugs have fewer muscle relaxant properties.721
Most of the nonbenzodiazepine sleep aids are available through a prescription as a Schedule IV drug and are readily prescribed, shared, and sold illegally. The rapid-onset and amnesic properties of this class of drugs can result in disin-hibition, passivity, and retrograde amnesia, making it a favored DFSA drug. These drugs require only a low dosage to cause an effect and are rapidly metabolized. Because of the amnesic properties of these drugs, victims are often confused
Figure 35-16 Chemical structures of the nonbenzodiazepine sedative-hypnotics (zolpidem, eszopiclone, zaleplon).
N
N
H3C
CH3
N
O
H3CCH3
Zolpidem (Ambien)
O
N
N
CH3
CH3
F
Eszopiclone (Lunesta)
N
N N
N
N CH3
CH3O
Zaleplon (Sonata)

Chapter35 ■ Clinical Toxicology 1161
properties and the availability of GHB in dietary supplements have led to growing recreational abuse of the drug. GHB has become popular as a euphorigenic club drug, most often used in combination with alcohol, and also with MDMA or cocaine, to “mellow” their adverse stimulant properties. Its rapid onset and hypnotic and short-term amnestic properties have resulted in the use of GHB for drug-facilitated sexual assault (date rape drug).201,295,716,737 Publications estimate that 4% of alleged sexual assault cases in the United States are positive for GHB.225,232,716,729
GHB is a naturally occurring substance produced in the brain. GHB is reversibly metabolized to GABA through mul-tiple endogenous enzymes (Figure 35-17).202,503,797 Illicit con-sumption of GHB, or the synthetic GHB precursor compound 1,4-BD or GBL, will promote GABA activity.202 In addition to increased metabolism to GABA, GHB has direct effects on the CNS by binding GHB-specific receptors and GABAB receptors.74,410,439,862 The latter are G-protein–coupled recep-tors distinct from the GABAA receptors for depressant drugs, such as benzodiazepines and barbiturates. Of note, patients with GHB overdose do not respond to the opioid antagonist naloxone or to the benzodiazepine antagonist flumazenil.
detect these drugs and their metabolites in urine or hair samples after a single dose. Although these drugs do not cross-react with most benzodiazepine immunoassays, spe-cific reagent systems (ELISA) directed against the non-benzodiazepine hypnotics are available.668 Screening and confirmation are performed by GC-MS or LC-MS/MS.
γ-Hydroxybutyrate, 1,4-butanediol, and γ-butyrolactoneγ-Hydroxybutyrate (GHB) and its synthetic precursor com-pounds, 1,4-butanediol (1,4-BD) and γ-butyrolactone (GBL), are Schedule I agents in the United States, and availability is restricted in numerous other countries. GHB is illegally pur-chased as an odorless and colorless liquid form, or as an off-white powder that easily dissolves in liquids. When ingested, GHB stimulates dopamine release, leading to pleasurable effects such as euphoria, muscle relaxation, and heightened sexual desire.74,180 It also has CNS depressant effects, resulting in sedation and hypnosis. Because GHB was reported to enhance growth hormone release, it has been used by body builders and athletes as a steroid alternative. Athletes have used GHB as a sleep aid because they believe it promotes rapid recovery from vigorous repetitive competition. These
Figure 35-17 Metabolism of γ-hydroxybutyrate. γ-Hydroxybutyrate (GHB) and its synthetic precursor compounds, 1,4-butanediol (1,4-BD) and γ-butyrolactone (GBL), are often used illicitly. These drugs are endogenously metabolized to γ-aminobutyric acid (GABA). GHB and GABA mediate GABA receptors.
O OH
H2NOH
O
Glutamate
Glutamic aciddecarboxylase H2N
OH
O
GABA
GABAtransaminase
Succinic semialdehyde
OOH
OSuccinic semialdehydedehydrogenase
Succinic acid
OOH
O
OH
TCAcycle
GHB
HOOH
O
GHBdehydrogenase
Succinicsemialdehyde
reductase
O O
GBL
HOOH
1,4-BD
Lactonase
ADH/ALD
Potentialdrugs ofabuse

1162 SectionIII ■ Analytes
depression. High doses may also cause lethargy, agitation, ataxia, nystagmus, diaphoresis, and hypertension.594,851,870
DXM is present in various over-the-counter (OTC) cough medications, often in combination with antihistamines, nasal decongestants, guaifenesin, aspirin, and acetamino-phen. Potential toxicity from OTC combination medications must be considered when DXM is consumed in large doses to achieve euphoric effects.41,432 Abuse of DXM, especially by adolescents and teenagers, who refer to it as “Dex, Robos, Skittles,” has become widespread. Abusers describe feelings of euphoria, dissociative effects such as a sense of floating, and hallucinations. Discontinuation of the drug is frequently followed by dysphoria and depression.
Pharmacologic EffectsDextromethorphan is metabolized to dextrophan649 by the cytochrome P450 isozyme 2D6 (CYP2D6), which exhibits genetic polymorphisms. Dextrophan may be responsible for the more pleasant psychotropic effects of high-dose dextro-methorphan, whereas the parent drug may cause dysphoria, sedation, and ataxia.870 Thus, poor metabolizers (deficient in CYP2D6 activity) may be less prone and extensive metaboliz-ers more prone to continue the abuse of dextromethorphan. Dextrophan and to a lesser degree DXM bind to the PCP- and ketamine-binding site on the NMDA receptor, causing seda-tion; this may account for their similar dissociative psycho-tropic actions585 (see phencyclidine and ketamine sections).
Analytical MethodsClinically approved doses of DXM are not detected by most clinical opiate immunoassays,755 but larger doses may cross-react.740 However, ELISA assays are now available to detect DXM and its major metabolite, dextrorphan.678 Some toxico-logic laboratories have used the cross-reactivity of DXM with phencyclidine antibodies to screen for the presence of DXM.709 Because most preparations contain dextrome-thorphan as the bromide salt, excessive ingestion of dextro-methorphan may result in bromide poisoning and in a negative serum anion gap consequent to the disproportionate response to bromide with common methods of chloride anal-ysis.586 The presence of DXM or dextrophan in a sample is confirmed by GC-MS or LC-MS/MS.
Dextrophan is the enantiomer of levorphanol, a potent opioid agonist available in the United States (Levo-Dromoran). Unless chiral analytical techniques are used, these enantio-mers are not resolved. Drug testing laboratories that use con-ventional chromatographic techniques should not report a finding of levorphanol only, but should instead report dextrophan/levorphanol, with a comment on their isomeric relationship and on the origin of dextrophan. This is espe-cially important for pain management drug screening, in which a false report of levorphanol may result in dismissal from the program. This report duality is advisable even when parent dextromethorphan is also detected. Savvy abusers of levorphanol conceivably may coingest dextromethorphan to conceal use of levorphanol. If such is suspected, chiral
Fomepizole, an inhibitor of alcohol dehydrogenase, is likely beneficial for patients who ingest 1,4-butanediol.878 GHB is suggested to increase dopamine concentrations in the sub-stantia nigra, to potentiate the endogenous opioid system, and to mediate GABA transmission.202
Pharmacologic EffectsOnset of GHB effects occurs in approximately 15 to 30 minutes, depending on the dose (average, 1 to 5 g) and the chemical purity. The duration of response is short, typically 1 to 3 hours for normal dose and 2 to 4 hours with excessive doses. The clinical effects are dose dependent and typically last 3 to 6 hours. A low dose (<1 g) produces mild symptoms such as CNS depression, amnesia, hypotonia, and reduced inhibitions (similar to alcohol). Larger doses (1 to 2 g) cause increased somnolence, drowsiness, dizziness, bradycardia, and bradypnea. High doses (>2 g) often interfere with motor coordination and balance and may induce significant respira-tory depression and bradypnea, Cheyne-Stokes respiration, nausea, and vomiting, diminished cardiac output, seizures, coma, and death.289,514,804 Periods of agitation may be inter-spersed between times of apnea and unresponsiveness. It is uncertain whether this agitation is a direct GHB effect or a consequence of coingested stimulant drugs. Deaths have been reported but are almost always associated with coingestion of alcohol or other drugs.
Analytical MethodsGHB is metabolized rapidly (t1/2 ≈30 minutes) and currently is not detected on immunoscreens. GHB is identified on urine and serum specimens using GC-FID or GC-MS.225,430,531 Because GBH is metabolized rapidly, timely sample collection is an important facet of GBH assay; plasma samples should be collected within 6 to 8 hours after ingestion, and urine samples within 10 to 12 hours. Urine and plasma concentra-tions may exhibit endogenous concentrations of GHB within 8 to 12 hours after ingestion (<1 mg/dL in urine; <4 mg/L in blood/plasma).868 Samples approaching endogenous concen-trations make it difficult to legally associate GHB doping in sexual assault cases. Exogenous concentrations of GHB have been detected in hair samples at 7 days post intoxication.295 Timely presentation of the patient for medical attention and physician recognition of GHB symptoms presented by sexu-ally assaulted victims are essential for prosecution of sexual offenders.
DextromethorphanDextromethorphan (DXM) is structurally related to the opioids, but it does not bind to opioid receptors at normal dose and thus is devoid of analgesic activity.585 The (−) isomer of dextromethorphan, levorphan (not available in the United States), is a potent opioid analgesic and is an example of the stereoselective nature of opioid receptor binding. DXM lacks analgesic activity but does have antitussive activity compa-rable with that of codeine. At high doses, DXM binds opioid receptors to produce miosis, respiratory depression, and CNS

Chapter35 ■ Clinical Toxicology 1163
(2) ataxia, (3) nystagmus, (4) agitation, (5) anxiety, (6) para-noia, (7) amnesia, (8) seizures, (9) muscle rigidity, (10) hostil-ity, (11) delirium, (12) delusions, and (13) hallucinations is unpredictable. LSD users can experience “flashbacks of the drug experience.” Flashbacks occur suddenly, often without warning, and may occur within a few days or more than a year after LSD use.
The onset of action for PCP is fast for intravenous and inhalation routes (2 to 5 minutes) and slower following oral administration (30 to 60 minutes).172,173 Clinical effects typi-cally last 4 to 6 hours, yet psychotic episodes have been reported to last a month.11,664 The relationship between dosage, clinical effects, and serum concentrations is not a reliable predictor of the degree of PCP intoxication.601 PCP has a pKa between 8.5 and 9.4, is highly lipophilic, and dis-tributes to the brain and fat tissues. An ion-trapping phenom-enon occurs after oral, IV, or inhalation dosing. PCP enters acidic gastric fluid after oral administration, where concen-trations may be 20 to 50 times greater than in serum, then undergoes gastroenterohepatic recirculation.857 Ion trapping also occurs in cerebrospinal fluid (CSF), causing it to cross back into the blood; CSF may accumulate to concentrations six to nine times greater than those observed in serum.544 These properties may contribute to the waxing and waning of clinical effects and prolonged excretion. PCP has a large Vd of 5 to 7 L/kg, a long elimination t1/2 (20 to 50 hours), a long duration of action (24 to 48 hours), and prolonged urinary excretion after the last dose (1 to 2 weeks; longer with long-term use).857
With repeated use of PCP, psychologic dependence may develop, but tolerance or withdrawal syndrome is not pro-found. A sense of superhuman strength coupled with lack of pain perception may lead to excessive physical exertion and accidental or intentionally induced trauma. Thus PCP-related deaths most often are secondary to these adverse behavioral drug effects. Treatment of PCP toxicity is supportive. Severe agitation or seizures may respond to diazepam; severe psy-choses may require a neuroleptic drug, such as haloperidol. For the most serious cases, continuous nasogastric suction to help remove PCP may be beneficial; urine acidification to hasten elimination has been advocated by some but is controversial.601
Ketamine. Ketamine was discovered during subsequent studies characterizing PCP analogs. Liquid ketamine is rapidly injected intramuscularly. The liquid or powder form can be easily disguised in a victim’s beverage; this has resulted in its use in DFSA.716,737 Ketamine powder can even be sprin-kled onto marijuana or tobacco and smoked.
Ketamine produces effects similar to those of phencycli-dine. Onset of clinical effects is rapid and is dependent on dose and route of administration. Anesthesia effects via intra-muscular injection take as little as 20 to 30 seconds, oral ingestion about 30 minutes,301 and nasal insufflation approxi-mately 10 minutes.332,493,690 Its hallucinatory effects may be short-acting (<1 hour) but so intense that the victim may have trouble discerning reality.737
resolution of dextrophan and levorphanol would then be necessary.
Ketamine and PhencyclidineKetamine and phencyclidine (PCP) are potent analgesics and general anesthetics used in veterinary medicine. PCP is listed as a Schedule II drug in the U.S. Federal Controlled Substance Act and is not approved for human use. Ketamine is a Sched-ule III drug, commonly used as an anesthetic in pediatric medicine for short surgical procedures. Both drugs have been used illicitly in human cases of drug abuse, as well as in cases of drug-facilitated sexual assault.
On the street, ketamine and PCP are sold under a variety of names. They are available as a colorless, odorless liquid, or as a white powder. Either form can be easily disguised in a victim’s beverage. More commonly, these powdered agents are sprinkled onto marijuana or tobacco and smoked.
Pharmacologic EffectsKetamine and PCP share similar structural features649 and pharmacologic actions. They are classified as dissociative anesthetics because they produce rapid-acting dissociation of perception, consciousness, movement, and memory.171,342,601 The effects are dose dependent and vary between individuals. Some individuals experience effects similar to the psychosis observed in schizophrenia.11,664 An anesthetic dose produces profound analgesia, but the individual is awake yet incapaci-tated, with limited voluntary limb movement. Ketamine has about one tenth the potency of PCP, a shorter duration of action, and less prominent emergentce reactions, especially in children. Both PCP and ketamine have been associated with psychologic disturbances.
The mechanism of action for these compounds consists of complex integration of neurologic pathways. They bind and antagonize the excitatory glutaminergic system by binding to NMDA receptors. They also decrease GABA transmission, disrupt cortical activity, and increase dopamine and norepi-nephrine synaptic reuptake. These actions can produce clinical effects such as euphoria, elevated blood pressure, tachycardia, and bronchodilation, all of which are conse-quences of inhibition of dopamine and norepinephrine syn-aptic reuptake.461,601 At a higher dose, GABA-ergic and central nicotinic actions may produce sedation, lethargy, coma, and respiratory depression. Additionally, central and peripheral muscarinic and nicotinic responses may cause miosis or mydriasis, diaphoresis, increased salivation, bronchorrhea, blurred vision, and urinary retention.
Phencyclidine. PCP [1-(1-phenylcyclohexyl)-piperidine] was synthesized in 1926 and was clinically utilized as a general anesthetic. Because of adverse side effects experienced by some individuals, such as acute psychosis and dysphoria during emergence from PCP-induced anesthesia, clinical use was discontinued. PCP is used recreationally for its mind-altering “out of body” experience. Recreational use of PCP declined in the 1980s but has re-emerged in recent years. Presentation of adverse effects such as (1) dysphoria,

1164 SectionIII ■ Analytes
that urine will continue to be the only biological fluid approved for testing.239 An additional review conducted by DHHS is expected.
Meconium, oral fluid, hair, and sweat have been investi-gated as alternative types of samples for drug analysis.
MeconiumIllicit drug use during pregnancy is a major social and medical issue. Drug abuse during pregnancy is associated with signifi-cant perinatal complications, including a high incidence of (1) stillbirth, (2) meconium-stained fluid, (3) premature rupture of the membranes, (4) maternal hemorrhage (abrup-tio placentae or placenta previa), and (5) fetal distress.607 In the neonate, the mortality rate and morbidity (e.g., asphyxia, prematurity, low birth weight, hyaline membrane disease, infection, aspiration pneumonia, cerebral infarction, abnor-mal heart rate and breathing patterns, drug withdrawal) are increased.607
Unfortunately, identification of the drug-exposed mother or her neonate is not easy. Maternal admission of the use of drugs is often inaccurate principally because of denial about addiction or fear of the consequences stemming from such admission. Likewise, many infants who have been exposed to drugs in utero may appear normal at birth and show no overt manifestations of drug effects. Thus, identification of the drug-exposed mother or her infant requires a high index of suspicion. Drug testing, on the other hand, is an objective means of determining drug exposure in both mother and infant. In infants, drug testing is necessary to document proof of the infant’s exposure to illicit drugs. Urine testing of the mother or newborn can detect only recent drug use (within a few days before birth), and urine collection from newborns may be problematic.
The first intestinal discharge from newborns is meconium, which is a viscous, dark green substance composed of intes-tinal secretions, desquamated squamous cells, lanugo hair, bile pigments, and blood. Meconium also contains pancreatic enzymes, free fatty acids, porphyrins, interleukin-8, and phospholipase A2 primary bile acids with a small quantity of secondary bile acids. Water is the major liquid constituent, making up 85 to 95% of meconium.7 Meconium is derived from the Greek word “mekonion,” meaning poppy juice or opium. Aristotle is credited with noting the relationship between the presence of meconium in amniotic fluid and a sleepy fetal state in utero.7 Meconium begins to form during the second trimester and continues to accumulate until birth; drugs taken by the mother can be detected in the meconium of the newborn.608
The disposition of drug in meconium is not well under-stood. The proposed mechanism is that the fetus excretes drug into bile and amniotic fluid. Drug accumulates in meco-nium by direct deposition from bile or through swallowing of amniotic fluid.229,608 The first evidence of meconium in the fetal intestine appears at approximately the 10th to 12th week of gestation; meconium slowly moves into the colon by the 16th week of gestation.7 Therefore, the presence of drugs in meconium has been proposed to be indicative of in utero
Ketamine has a t1/2 of 2 to 3 hours.148 Ketamine is metabo-lized to norketamine, which has about one third the activity of ketamine, and to dehydronorketamine, which also may be active.649 Duration of anesthetic effects is dose dependent (usually <1 hour), and effects on the senses, judgment, and coordination can have a longer duration (≈6 to 24 hours). At higher doses, ketamine causes delirium, amnesia, dissociative anesthesia, hallucinations, delirium, hypersalivation, nystag-mus, impaired motor function, hypertension, and potentially fatal respiratory problems. Effects on blood pressure and respiratory depression are significantly enhanced when coin-gested with alcohol.
Analytical MethodsInitial screening for PCP is typically done by immunoassay. Confirmation of a presumptive positive test is performed by GC-MS.253 No immunoassays are available to detect ketamine at this time. Ketamine and its active metabolites are detected in urine samples using GC-MS or LC-MS analyses.557
Immunoassay. Quantification of PCP in serum is not helpful in the diagnosis or management of PCP toxicity because the correlation between drug concentration and drug effects is low.601 However, qualitative identification of PCP in urine is useful to help diagnose PCP toxicity. For this purpose, PCP-specific immunoassays are rapid and generally are more sensitive than thin-layer chromatography. Whether or not PCP is included in a general urine drug screen depends on applicable regulations and on the prevalence of PCP use in the local community. In some locations, the prevalence of PCP use may be too low to warrant routine screening for PCP. Immunoassays for PCP are generally reliable; false positives have been reported because of high concentrations of dextro-methorphan,104,709 diphenhydramine,476 and thioridazine.490,814 Immunoassay-positive specimens should be confirmed using GC-MS.253
Gas Chromatography–Mass Spectrometry. PCP is required to be included in U.S. Government–regulated drug abuse screening programs (see Table 35-7); nongovernmental screening programs may elect to include PCP in drug abuse screens, depending on the local probability of PCP use. Initial screening by immunoassay, if positive, is followed by confir-mation using GC-MS. Ketamine and its active metabolites norketamine and dehydronorketamine are detected in urine samples using GC-MS132,420 or LC-MS analyses.557
Detection of Drugs of Abuse Using Other Types of SpecimensThe collection of biological samples for the purpose of deter-mining exposure to various agents is dominated by blood and urine. Blood is considered invasive, and the collection of urine may require some invasion of privacy and loss of dignity; urine specimens are subject to adulteration or manipulation to evade detection. For these reasons, alternate biological specimens have been investigated.116,197A Cutoff values had been proposed by SAMHSA110 for some of the matrices and are listed in Table 35-8. However, current guide-lines for Federal Workplace Drug Testing have determined

Chapter35 ■ Clinical Toxicology 1165
solvent, such as buffered methanol, may prevent decreases for as long as 72 hours.607 For prolonged storage, meconium should be frozen. Drugs are stable in meconium, frozen at −15 °C, for as long as 9 months.607
Overinterpretation of meconium data is a dangerous prac-tice. It is clear that matrix effects are associated with the analysis of meconium, as they are with each biological fluid or tissue.474 Another important confounding factor is possible contamination of the meconium specimen by urine. Numer-ous reports have described the specificity and sensitivity of different analytes with the use of different testing methods.454,455,527,607,609 A tremendous, and potentially inap-propriate, value has been placed on a meconium result. On occasion, decisions about treatment or custody of the infant have been based solely on meconium drug screen results. It is critical to remember that a positive test could indicate intrauterine drug exposure. However, a negative result does not rule it out. It is clear that additional work is necessary to address these important issues and to improve our under-standing of the disposition of drugs in meconium.474
Oral FluidReports concerning the appearance of organic solutes in saliva have been included in the scientific literature for longer than 70 years.726 Analysis of saliva for drugs was first used almost 30 years ago for the purpose of therapeutic drug monitoring.344 It has since been evaluated for use in forensic
drug exposure up to 5 months before birth—a longer histori-cal measure than is possible by urinalysis.608
Meconium has been used for detection of prenatal drug use, showing an improved drug detection rate compared with urine.114,477,572,608 The collection of meconium is noninvasive, making sample collection easy,693 and is more successful than urine collection.527 The amount collected is usually sufficient for complete analysis, including confirmation. Meconium testing does have some limitations. Meconium is usually passed by full-term newborns within 24 to 48 hours, after which transition from a blackish-green color to a yellow color indicates the beginning of passage of neonatal stool. Infants with low birth weight (<1000 g) have been shown to pass their first meconium at a median age of 3 days. Thus, meco-nium collection is missed because of delayed passage, and meconium may not be available soon after birth for early detection of intrauterine drug exposure.
In the clinical laboratory, meconium is an unfamiliar matrix; it is a sticky material that is more difficult to work with than urine. Meconium drug screening has been adapted to various analytical techniques, including radioimmunoas-say, enzyme immunoassay, and fluorescence polarization immunoassay. Urine drugs-of-abuse screening assays fre-quently use meconium extracts and therefore must be inves-tigated for possible effects of matrix on accuracy, precision, and assay linearity. However, some other immunoassays screening methods have been used.
Radioimmunoassay (RIA), fluorescence polarization immunoassay (FPIA), enzyme multiplied immunoassay tech-nique (EMIT), and enzyme-linked immunosorbent assay (ELISA) have been used for detection of drugs in meconium, but ELISA is rapidly becoming the method of choice for screening.272 The sample preparation for the ELISA screen is usually a simple buffer extraction versus a lengthy and more laborious sample preparation procedure for the other immu-noassay methods.
As with any immunoassay-based drug screen, confirma-tion by MS is critical. Confirmation assays for meconium are more difficult than those for urine. Recovery of drugs from meconium is sometimes low (30 to 50%). A variety of GC-MS, LC-MS, and LC-MS/MS methods and their advantages and disadvantages have been described elsewhere272,477,555 and will not be discussed here.
Many questions remain to be answered about the dis-position of drugs in meconium. Some debate continues as to which are the most appropriate drug analytes to measure in meconium; Table 35-12 attempts to summarize the current knowledge. Meconium drug testing is growing but is far less standardized than urine drug testing. Assay cutoff limits and units (ng/g meconium or ng/mL of extract) may vary; suitable reference or control materials are not yet available.
Meconium should be sent to the laboratory for processing as soon as it is collected to prevent possible loss of drugs. Meconium, allowed to stand at room temperature for 24 hours, showed a decrease in cocaine and cannabinoid con-centrations.607 However, suspending meconium in an organic
TABLE 35-12 DrugsandMetabolitesofSignificanceinMeconium
Drug Class Confirmation Compound69,229,474,555
Cocaine cocainebenzoylecgoninecocaethylenem-hydroxybenzoylecgonine
Opiates morphinecodeine6-monoacetylmorphine (6-MAM)hydromorphonehydrocodoneoxycodone
Cannabinoids 9-carboxy-11-nor-delta-9-THC11-hydroxy-delta-9-
tetrahydrocannabinol8,11-dihydroxy-delta-9-
tetrahydrocannabinolAmphetamines amphetamine
methamphetamineMDMAMDAMDEA
Ethanol fatty acid ethyl estersPCP PCP

1166 SectionIII ■ Analytes
noninvasive procedure, and some of the risks associated with the drawing of blood are avoided. Furthermore, for the patient, fear, anxiety, and discomfort that may accompany the drawing of blood are diminished. Although some training and explanation are necessary to ensure proper gathering of oral fluid samples, the level of training needed for blood sampling is not required. In principle, oral fluid drug concen-tration is related to plasma free drug concentration, except when buccal contamination may have occurred because of oral ingestion, smoking, or snorting of the drug. Therefore oral fluid has the potential to show a relation between behavior/impairment and drug concentration, making it a possible medium for monitoring drug intoxication or for conducting therapeutic drug management.431,459 On a related note, one significant disadvantage of oral fluid is that the window of detection is about equivalent to that of blood or serum and is short compared with that in urine.712,713,749 Another disadvantage is the small volume of sample col-lected. The problem of small sample size can be overcome by using methods that simultaneously extract multiple drug groups.431,865
HairFor more than 30 years, hair has been analyzed for trace metals, including lead, arsenic, and mercury. This was achieved using atomic absorption spectroscopy (see Chapters 10 and 36). At first, the examination of hair for organic substances, specifically drugs, was not possible because the analytical methods were not sensitive enough.424 Baumgartner and associates in 197966 published the first report on the detection of morphine in the hair of heroin abusers using RIA. Since that time, interest in analysis of hair for the purpose of detecting drug use has increased.67,356,416,422
It is generally accepted that drugs enter hair by at least three mechanisms: (1) from blood that supplies the growing hair follicle, (2) through sweat and sebum, and (3) via the external environment.396,424 The exact mechanism by which chemicals are incorporated into hair is not known. It has been suggested that passive diffusion may be augmented by binding of the drug to intracellular components of hair cells such as the hair pigment melanin. Specific binding of basic drugs to hair components is likely to involve both electrostatic attrac-tion and weaker forces, such as van der Waals attraction. Neutral and acidic drugs presumably bind through weaker forces and possibly by other mechanisms.160
Factors that may affect how efficiently drugs are incorpo-rated into hair are not well established but may include rate of hair growth, anatomic location of hair, hair color (melanin content), and hair texture (thick or fine, porous or not); these are determined by genetic factors and by the effects of various hair treatments.424 For example, in vitro substantially higher binding was found with hair from black men than with hair from blond white men, suggesting that melanin pigment plays an important role in drug binding.397 Studies have demonstrated that after the same dosage is given, black hair incorporates much more of the drug than is incorporated
toxicology, with recognition of its advantages over other biological matrices.638A Most studies on saliva in humans use whole saliva. The term “oral fluid” is preferred for the specimen collected from the mouth. Oral fluid is a complex fluid consisting not only of secretions from the three major pairs of salivary glands (parotid, submandibular, and sublin-gual), but also secretions from the minor glands (labial, buccal, and palatal), bacteria, sloughed epithelial cells, gingi-val fluid, food debris, and other particulate matter.338 The concentration of drug from each secretion and the relative contributions of the various glands to the final fluid may vary.726
Biologically, before any drug circulating in plasma can enter the oral fluid, it must pass through the capillary wall, the basement membrane, and the membrane of the salivary gland epithelial cells. However, this fluid is not a simple ultrafiltrate of plasma, as has sometimes been suggested, but rather a complex fluid formed by different mechanisms, including ultrafiltration through pores in the membrane, active transport against a concentration gradient, and passive diffusion.*
Ethanol was the first drug of abuse to be investigated in oral fluid261; since then, many additional studies have expanded on our knowledge of this drug in it. Ethanol appears to reach a higher peak concentration in oral fluid than in peripheral blood. Because distribution of ethanol in the body is considered to occur by passive diffusion, under equilibrium conditions, ethanol content will be dependent upon the water content of the fluid or tissue being measured. The content in saliva therefore will be higher than that found in blood or serum. On a theoretical basis, the saliva-to-blood (S/B) ethanol ratio should be 1.17; however, lower ratios have been found in the postabsorption phase.159,388
In recent years, great interest has been expressed in the use of oral fluid testing for roadside drug screening, for monitor-ing the compliance of individuals on drug maintenance pro-grams, and for workplace drug testing (see Table 35-8). Low concentrations of drugs and metabolites necessitate sensitive screening methods, which typically are immunoassays.431 Again, the low concentrations of drugs have necessitated that confirmatory methods be equally as sensitive. Many confirmatory methods have been developed for oral fluid testing of abused drugs, including GC-MS, LC-MS and LC-MS/MS.† Specimens can be monitored for cannabinoids, cocaine, opiates, amphetamines, phencyclidine, methadone, barbiturates, and benzodiazepines. Interpretation of the presence and concentrations of these drugs in saliva and information on their use have been extensively reviewed elsewhere.159,208,400,417,708
Several advantages are associated with monitoring oral fluid as contrasted with monitoring plasma or serum concen-trations.749 Collection of oral fluid is considered to be a
†References 136, 167, 262, 314, 382, 399, 408, 426, 427, 554, 577, 589, 602, 679, 701, 707, 708, 712, 764, and 854.
*References 24, 105, 311, 338, 419, 498, 666, 806, and 877.

Chapter35 ■ Clinical Toxicology 1167
under the influence of a drug, (4) child sedation and abuse, (5) suspicious death, (6) child custody, (7) abuse of drugs in jail, (8) body identification, (9) survey of drug addicts, (10) chemical submission, (11) obtaining a driver’s license, and (12) doping control.423,431,549,834
SweatDrugs may be excreted in sweat, with the parent drug gener-ally present in a greater amount than metabolites.356,417 More-over, sweat excretion may be an important mechanism by which drugs enter hair.165
Sweat patch collection devices that resemble an adhesive bandage may be worn for several days to several weeks; during this time, drug, if present, accumulates in the absor-bent pad in the patch, while water vapor escapes through the semipermeable covering.428 Thus sweat drug testing offers the possibility of monitoring drug use over extended periods of time without the need for frequent collection of urine.108 Sweat drug testing would be particularly advantageous for monitoring drug use in correctional institutions or in drug rehabilitation programs.425
Athletes and Drug Testing“Doping” in athletic competitions has a history of abuse in a variety of sports for centuries. Regulation of performance-enhancing substances was initiated in 1967 in response to the death of Danish cyclist, Knud Jensen, at the 1960 Olympic Games in Rome. After decades of reform between universal governments and sporting agencies, the World Anti-Doping Agency (WDA) was established in 1999. Currently, 36 facili-ties around the world are accredited by the International Organization for Standardization (ISO) for detecting drug use among competitive athletes.122 Prohibited drugs are substances or methods that conform to two of three criteria: (1) performance enhancing, (2) may endanger the athlete’s health, (3) go against the spirit of the sport. “In-competition” testing was established to detect drugs (stimulants, narcotics, cannabinoids, glucocorticosteroids) taken at the time of a competition to temporarily enhance performance. “Out-of-competition” testing is performed to detect substances and methods such as anabolic-androgenic steroids (AAS), hormones, hormone modulators, oxygen transfer enhancement, and gene transfer, whose performance-enhancing effects have a gradual onset to allow for more intense and efficient training; they can be abruptly discontin-ued before competition.5,122
Most testing for prohibited substance abuse is performed by GC-MS. Specialized testing such as isoelectric focusing (“double blotting”) was implemented at the 2002 Salt Lake City Olympics to discriminate between endogenous and recombinant forms of erythropoietin.121 Erythrocyte pheno-typing by flow cytometry is used to detect homologous blood transfusions.30,285,484 Limited windows of detection and indi-vidual biological variability for specific substances (especially hormones) are challenges faced when novel analytical methods are developed and validated. Tracking of specific clinical biomonitors (such as hormone or hormone-responsive
by blond hair.329,339,449 This may lead to bias in hair testing for drugs of abuse and discussions about possible genetic variability of drug deposition in hair and is still under evaluation.
Drugs, when deposited in hair, are generally present in relatively low concentrations (pg/ng/µg/mg); thus sensitive analytical techniques are required for detection. Immunoas-say procedures have been modified for use with hair.424 Although GC-MS is generally the method of choice,696 various GC-MS/MS or LC-MS/MS methods may be used for targeted analysis of low-dose compounds such as fentanyl, buprenor-phine, and flunitrazepam.450,451,697,787,829 These methods are also useful in the detection of some drugs or metabolites typically present in hair at trace concentrations such as THC-COOH135,788 or in retrospective detection of drugs adminis-tered as single doses.430,580,581,583
As mentioned, external exposure to drugs causes them to be detected in hair, and because it is unlikely that anyone would intentionally or accidentally apply drugs of abuse to his or her own hair, the most crucial issue facing hair analysts today is technical and evidentiary false positives.424 False positives may be caused during collection or after collection. Externally deposited substances easily contaminate the hair because of its high surface-to-volume ratio. Substances deposited in hair from the environment are loosely bound to the surface of the hair, and thus are removed by appropriate decontamination procedures. These usually involve a washing step.67,424 It is fundamental to be able to distinguish between passive exposure (environmental contamination) and active consumption; consequently, decontamination procedures for hair are compulsory.88 Needless to say, hair analysis is a complex scientific undertaking, and a comprehensive review on this topic has been published.655
Hair is advantageous as a biological specimen because it is easily obtained, with less embarrassment; it is not easily altered or manipulated to avoid drug detection. Hair also differs from other human materials used for toxicological analysis such as blood or urine in that it has a substantially longer detection window. Once deposited in hair, drugs are very stable, and analysis can be performed even after centuries.653
Hair also differs from other human materials used for toxicological analysis such as blood or urine because of its substantially longer detection window (months to years). Hair grows at a relatively constant rate. The average rate of hair growth is usually stated to be 0.44 mm per day (range, 0.38 to 0.48 mm/d) for men and 0.45 mm per day (range, 0.40 to 0.55/d) for women in the vertex region of the scalp.573 The rate of hair growth depends on anatomic loca-tion, race, gender, and age. Scalp hair grows faster than pubic or axillary hair (≈0.3 mm/d), which in turn grows faster than beard hair (≈0.27 mm/d).88 It is generally accepted that sec-tional hair analysis can be used to prove drug history.88 Numerous forensic applications in which hair analysis was used to document the case have been described in the literature; these include (1) differentiation between a drug dealer and a drug consumer, (2) chronic poisoning, (3) crime

1168 SectionIII ■ Analytes
22. Anonymous. An overview of club drugs, Drug Intelligence Brief, DEA, February 2000.
23. Anonymous. Ambien CR (zolpidem tartrate extended-release) prescribing information. Bridgewater, NJ: Sanofi-Aventis US LLC, January 2008a.
24. Araki Y. Nitrogenous substances in saliva. I. Protein and non-protein nitrogens. Jpn J Physiol 1951;2:69-78.
25. Arch JR, Ainsworth AT, Cawthorne MA, Piercy V, Sennitt MV, Thody VE, et al. Atypical beta-adrenoceptor on brown adipocytes as target for anti-obesity drugs. Nature 1984;309:163-5.
26. Ariano RE, Kassum DA, Aronson KJ. Comparison of sedative recovery time after midazolam versus diazepam administration. Crit Care Med 1994;22:1492-6.
27. Arima H, Sobue K, So M, Morishima T, Ando H, Katsuya H. Transient and reversible parkinsonism after acute organophosphate poisoning. J Toxicol Clin Toxicol 2003;41:67-70.
28. Armstrong SC, Cozza KL. Pharmacokinetic drug interactions of morphine, codeine, and their derivatives: theory and clinical reality, part I. Psychosomatics 2003;44:167-71.
29. Armstrong SC, Cozza KL. Pharmacokinetic drug interactions of morphine, codeine, and their derivatives: theory and clinical reality, part II. Psychosomatics 2003;44:515-20.
30. Arndt PA, Kumpel BM. Blood doping in athletes—detection of allogeneic blood transfusions by flow cytofluorometry. Am J Hematol 2008;83:657-67.
31. Ashbourne JF, Olson KR, Khayam-Bashi H. Value of rapid screening for acetaminophen in all patients with intentional drug overdose. Ann Emerg Med 1989;18:1035-8.
32. Babu KM, Ferm RP. Hallucinogens. In: Goldfrank LR, Flomenbaum NE, Hoffman RS, Howland MA, Lewin NA, Nelson L, eds. Goldfrank’s toxicologic emergencies, 8th edition. New York, NY: McGraw-Hill, 2006:1202-11.
33. Bach MV, Coutts RT, Baker GB. Involvement of CYP2D6 in the in vitro metabolism of amphetamine, two N-alkylamphetamines and their 4-methoxylated derivatives. Xenobiotica 1999;29:719-32.
34. Badcock NR, O’Reilly DA. False-positive EMIT-ST ethanol screen with post-mortem infant plasma. Clin Chem 1992;38:434.
35. Baden LR, Horowitz G, Jacoby H, Eliopoulos GM. Quinolones and false-positive urine screening for opiates by immunoassay technology. JAMA 2001;286:3115-9.
36. Bailey RB, Jones SR. Chronic salicylate intoxication: a common cause of morbidity in the elderly. J Am Geriatr Soc 1989;37:556-61.
37. Baker AM, Johnson DG, Levisky JA, Hearn WL, Moore KA, Levine B, et al. Fatal diphenhydramine intoxication in infants. J Forensic Sci 2003;48:425-8.
38. Baldessarini R. Drug therapy of depression and anxiety disorders (Chapter 17). In: Brunton LL, Lazo JS, Parker KL, eds. Goodman & Gilman’s the pharmacological basis of therapeutics, 11th edition. New York, NY: McGraw-Hill, 2006.
39. Baldessarini RJ, Tarazi FI. Pharmacotherapy of psychosis and mania (Chapter 18). In: Brunton LL, Lazo JS, Parker KL, eds. Goodman & Gilman’s the pharmacological basis of therapeutics, 11th edition. New York, NY: McGraw-Hill, 2006.
40. Bamigbade TA, Davidson C, Langford RM, Stamford JA. Actions of tramadol, its enantiomers and principal metabolite, O-desmethyltramadol, on serotonin (5-HT) efflux and uptake in the rat dorsal raphe nucleus. Br J Anaesth 1997;79:352-6.
41. Banerji S, Anderson IB. Abuse of Coricidin HBP cough and cold tablets: episodes recorded by a poison center. Am J Health Syst Pharm 2001;58:1811-4.
42. Barbour AD. GC/MS analysis of propylated barbiturates. J Anal Toxicol 1991;15:214-5.
43. Bardin PG, van Eeden SF, Moolman JA, Foden AP, Joubert JR. Organophosphate and carbamate poisoning. Arch Intern Med 1994; 154:1433-41.
44. Barkin RL, Barkin SJ, Barkin DS. Propoxyphene (dextropropoxyphene): a critical review of a weak opioid analgesic that should remain in antiquity. Am J Ther 2006;13:534-42.
protein concentrations) over an athlete’s performance career, termed biological passport and longitudinal profiling, is currently under investigation.584,660
REFERENCES1. Aabakken L, Johansen KS, Rydningen EB, Bredesen JE, Ovrebo S,
Jacobsen D. Osmolal and anion gaps in patients admitted to an emergency medical department. Hum Exp Toxicol 1994;13:131-4.
2. Aalberg L, DeRuiter J, Noggle FT, Sippola E, Clark CR. Chromatographic and mass spectral methods of identification for the side-chain and ring regioisomers of methylenedioxymethamphetamine. J Chromatogr Sci 2000;38:329-37.
3. Abou-Donia MB, Lapadula DM. Mechanisms of organophosphorus ester-induced delayed neurotoxicity: type I and type II. Annu Rev Pharmacol Toxicol 1990;30:405-40.
4. Adamowicz P, Kala M. Date-rape drugs scene in Poland. Przegl Lek 2005;62:572-5.
5. Agency WA-D. The 2009 prohibited list. Geneva, Switzerland: International Organization for Standardization, 2009.
6. Agurell S, Halldin M, Lindgren JE, Ohlsson A, Widman M, Gillespie H, et al. Pharmacokinetics and metabolism of delta 1-tetrahydrocannabinol and other cannabinoids with emphasis on man. Pharmacol Rev 1986;38:21-43.
7. Ahanya SN, Lakshmanan J, Morgan BL, Ross MG. Meconium passage in utero: mechanisms, consequences, and management. Obstet Gynecol Surv 2005;60:45-56; quiz 73-4.
8. Al-Asmari AI, Anderson RA. Method for quantification of opioids and their metabolites in autopsy blood by liquid chromatography-tandem mass spectrometry. J Anal Toxicol 2007;31:394-408.
9. Aldridge WN. Some properties of specific cholinesterase with particular reference to the mechanism of inhibition by diethyl p-nitrophenyl thiophosphate (E 605) and analogues. Biochem J 1950;46:451-60.
10. Allen MJ. Procedures for transportation workplace drug testing programs. Fed Reg 1989;54:49854-84.
11. Allen RM, Young SJ. Phencyclidine-induced psychosis. Am J Psychiatry 1978;135:1081-4.
12. Alozie SO, Martin BR, Harris LS, Dewey WL. 3H-Delta-9-tetrahydrocannabinol, 3H-cannabinol and 3H-cannabidiol: penetration and regional distribution in rat brain. Pharmacol Biochem Behav 1980;12:217-21.
13. Ambre JJ, Belknap SM, Nelson J, Ruo TI, Shin SG, Atkinson AJ Jr. Acute tolerance to cocaine in humans. Clin Pharmacol Ther 1988;44: 1-8.
14. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Position statement and practice guidelines on the use of multi-dose activated charcoal in the treatment of acute poisoning. J Toxicol Clin Toxicol 1999;37:731-51.
15. American College of Obstetricians and Gynecologists. Committee opinion no. 422: at-risk drinking and illicit drug use: ethical issues in obstetric and gynecologic practice. Obstet Gynecol 2008;112:1449-60.
16. Amin J, Weiss DS. GABAA receptor needs two homologous domains of the beta-subunit for activation by GABA but not by pentobarbital. Nature 1993;366:565-9.
17. Anderson IB, Kim SY, Dyer JE, Burkhardt CB, Iknoian JC, Walsh MJ, et al. Trends in gamma-hydroxybutyrate (GHB) and related drug intoxication: 1999 to 2003. Ann Emerg Med 2006;47:177-83.
18. Anderson RJ, Potts DE, Gabow PA, Rumack BH, Schrier RW. Unrecognized adult salicylate intoxication. Ann Intern Med 1976;85: 745-8.
19. Anderson WH. Therapeutic drugs II: antidepressants. In: Levine B, ed. Principles of forensic toxicology, 2nd edition. Washington, DC: AACC Press, 2003:297-314.
20. Andrews P. Cocaethylene toxicity. J Addict Dis 1997;16:75-84.21. Andrinolo D, Michea LF, Lagos N. Toxic effects, pharmacokinetics
and clearance of saxitoxin, a component of paralytic shellfish poison (PSP), in cats. Toxicon 1999;37:447-64.

Chapter35 ■ Clinical Toxicology 1169
therapeutic drug monitoring and clinical toxicology, New York: Marcel Dekker Inc, 1992:577-97.
68. Bayer MJ, McKay C. Advances in poison management. Clin Chem 1996;42:1361-6.
69. Bearer CF, Lee S, Salvator AE, Minnes S, Swick A, Yamashita T, Singer LT. Ethyl linoleate in meconium: a biomarker for prenatal ethanol exposure. Alcohol Clin Exp Res 1999;23:487-93.
70. Beckett AH, Rowland M. Urinary excretion of methylamphetamine in man. Nature 1965;206:1260-1.
71. Bell R, Taylor EH, Ackerman B, Pappas AA. Interpretation of urine quantitative 11-nor-delta-9 tetrahydrocannabinol-9-carboxylic acid to determine abstinence from marijuana smoking. J Toxicol Clin Toxicol 1989;27:109-15.
72. Bermejo AM, Ramos I, Fernandez P, Lopez-Rivadulla M, Cruz A, Chiarotti M, et al. Morphine determination by gas chromatography/mass spectroscopy in human vitreous humor and comparison with radioimmunoassay. J Anal Toxicol 1992;16:372-4.
73. Bernard S, Neville KA, Nguyen AT, Flockhart DA. Interethnic differences in genetic polymorphisms of CYP2D6 in the U.S. population: clinical implications. The Oncologist 2006;11: 126-35.
74. Bernasconi R, Mathivet P, Bischoff S, Marescaux C. Gamma-hydroxybutyric acid: an endogenous neuromodulator with abuse potential? Trends Pharmacol Sci 1999;20:135-41.
75. Bertholf RL, Johannsen LM, Bazooband A, Mansouri V. False-positive acetaminophen results in a hyperbilirubinemic patient. Clin Chem 2003;49:695-8.
76. Bizovi K, Smilkstein M. Acetaminophen. In: Goldfrank L, Flomenbaum N, Lewin N, Howland M, Hoffman R, Nelson L, eds. Goldfrank’s toxicologic emergencies, 7th edition. New York, NY: McGraw-Hill, 2002:480-501.
77. Blanchet M, Bru G, Guerret M, Bromet-Petit M, Bromet N. Routine determination of morphine, morphine 3-beta-d-glucuronide and morphine 6-beta-d-glucuronide in human serum by liquid chromatography coupled to electrospray mass spectrometry. J Chromatogr A 1999;854:93-108.
78. Blanke R, Decker W. Analysis of toxic substances. In: Tietz N, ed. Textbook of clinical chemistry, 1st edition. Philadelphia, Pa: WB Saunders, 1986:1670-744.
79. Boehnert MT, Lovejoy FH Jr. Value of the QRS duration versus the serum drug level in predicting seizures and ventricular arrhythmias after an acute overdose of tricyclic antidepressants. N Engl J Med 1985;313:474-9.
80. Boess F, Ndikum-Moffor FM, Boelsterli UA, Roberts SM. Effects of cocaine and its oxidative metabolites on mitochondrial respiration and generation of reactive oxygen species. Biochem Pharmacol 2000; 60:615-23.
81. Bogusz M, Pach J, Stasko W. Comparative studies on the rate of ethanol elimination in acute poisoning and in controlled conditions. J Forensic Sci 1977;22:446-51.
82. Bogusz M, Wu M. Standardized HPLC/DAD system, based on retention indices and spectral library, applicable for systematic toxicological screening. J Anal Toxicol 1991;15:188-97.
83. Bogusz MJ. Liquid chromatography-mass spectrometry as a routine method in forensic sciences: a proof of maturity. J Chromatogr 2000; 748:3-19.
84. Bogusz MJ, Maier RD, Driessen S. Morphine, morphine-3-glucuronide, morphine-6-glucuronide, and 6-monoacetylmorphine determined by means of atmospheric pressure chemical ionization-mass spectrometry-liquid chromatography in body fluids of heroin victims. J Anal Toxicol 1997;21:346-55.
85. Bogusz MJ, Maier RD, Erkens M, Driessen S. Determination of morphine and its 3- and 6-glucuronides, codeine, codeine-glucuronide and 6-monoacetylmorphine in body fluids by liquid chromatography atmospheric pressure chemical ionization mass spectrometry. J Chromatogr 1997;703:115-27.
86. Bogusz MJ, Maier RD, Kruger KD, Kohls U. Determination of common drugs of abuse in body fluids using one isolation procedure
45. Bartlett R. Carbon monoxide poisoning. In: Haddad L, Shannon M, Winchester J, eds. Clinical management of poisoning and drug overdose, 3rd edition. Philadelphia, Pa: WB Saunders, 1998:885-98.
46. Baselt RC. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008.
47. Baselt RC. Acetone. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:15-7.
48. Baselt RC. Amphetamine. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:83-6.
49. Baselt RC. Carbon monoxide. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008.
50. Baselt RC. Codeine. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:355-60.
51. Baselt RC. Ephedrine. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:542-4.
52. Baselt RC. Ethanol. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:561-5.
53. Baselt RC. Ethylene glycol. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:578-82.
54. Baselt RC. Flunitrazepam. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:633-5.
55. Baselt RC. Heroin. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:730-5.
56. Baselt RC. Hydrocodone. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:745-6.
57. Baselt RC. Hydromorphone. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:750-2.
58. Baselt RC. Isopropanol. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:789-91.
59. Baselt RC. Methadone. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:941-5.
60. Baselt RC. Methylenedioxyethylamphetamine. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:995-6.
61. Baselt RC. Methylphenidate. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:1008-11.
62. Baselt RC. Oxycodone. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:1166-8.
63. Baselt RC. Phenylpropanolamine. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:1252-4.
64. Baselt RC. Pseudoephedrine. In: Baselt RC, ed. Disposition of toxic drugs and chemical in man, 8th edition. Foster City, Calif: Biomedical Publications, 2008:1344-6.
65. Bass R. The antipsychotic drugs. In: Haddad L, Shannon M, Winchester J, eds. Clinical management of poisoning and drug overdose, 3rd edition. Philadelphia, Pa: WB Saunders, 1998:780-93.
66. Baumgartner AM, Jones PF, Baumgartner WA, Black CT. Radioimmunoassay of hair for determining opiate-abuse histories. J Nucl Med 1979;20:748-52.
67. Baumgartner WA, Hill V. Hair analysis for drugs of abuse: decontamination issues. In: Sunshine I, ed. Recent developments in

1170 SectionIII ■ Analytes
110. Bush DM. The U.S. mandatory guidelines for federal workplace drug testing programs: current status and future considerations. Forensic Sci Int 2008;174:111-9.
111. Calafat AM, Stanfill SB. Rapid quantitation of cyanide in whole blood by automated headspace gas chromatography. J Chromatogr B Analyt Technol Biomed Life Sci 2002;772:131-7.
112. Caldwell J. The metabolism of amphetamines in mammals. Drug Metab Rev 1976;5:219-80.
113. Caldwell R, Challenger H. A capillary column gas-chromatographic method for the identification of drugs of abuse in urine samples. Ann Clin Biochem 1989;26(Pt 5):430-43.
114. Callahan CM, Grant TM, Phipps P, Clark G, Novack AH, Streissguth AP, et al. Measurement of gestational cocaine exposure: sensitivity of infants’ hair, meconium, and urine. J Pediatr 1992;120:763-8.
115. Caplan YH. Blood, urine, and other fluid and tissue specimens for alcohol analyses. In: Garriott JC, ed. Medicolegal aspects of alcohol. Tucson, Ariz: Lawyers & Judges Publishing, 1996:137-50.
116. Caplan YH, Goldberger BA. Alternative specimens for workplace drug testing. J Anal Toxicol 2001;25:396-9.
117. Caplan YH, Levine B. Evaluation of the Abbott TDx-radiative energy attenuation (REA) ethanol assay in a study of 1105 forensic whole blood specimens. J Forensic Sci 1987;32:55-61.
118. Carlton FB, Simpson WMJ, Haddad LM. The organophosphates and other insecticides. In: Haddad LM, Shannon MW, Winchester JF, eds. Clinical management of poisoning and drug overdose, 3rd edition. Philadelphia, Pa: WB Saunders, 1999:836-45.
119. Carta M, Ariwodola OJ, Weiner JL, Valenzuela CF. Alcohol potently inhibits the kainate receptor-dependent excitatory drive of hippocampal interneurons. Proc Natl Acad Sci U S A 2003;100: 6813-8.
120. Casavant MJ, Shah MN, Battels R. Does fluorescent urine indicate antifreeze ingestion by children? Pediatrics 2001;107:113-4.
121. Catlin DH, Breidbach A, Elliott S, Glaspy J. Comparison of the isoelectric focusing patterns of darbepoetin alfa, recombinant human erythropoietin, and endogenous erythropoietin from human urine. Clin Chem 2002;48:2057-9.
122. Catlin DH, Fitch KD, Ljungqvist A. Medicine and science in the fight against doping in sport. J Intern Med 2008;264:99-114.
123. Catterall WA, Mackie K. Local anesthetics (Chapter 14). In: Brunton LL, Lazo JS, Parker KL, eds. Goodman & Gilman’s the pharmacological basis of therapeutics, 11th edition. New York, NY: McGraw-Hill, 2006.
124. Chabali R. Diagnostic use of anion and osmolal gaps in pediatric emergency medicine. Pediatr Emerg Care 1997;13:204-10.
125. Cham BE, Johns D, Bochner F, Imhoff DM, Rowland M. Simultaneous liquid-chromatographic quantitation of salicylic acid, salicyluric acid, and gentisic acid in plasma. Clin Chem 1979;25:1420-5.
126. Chamberlin KW, Cottle M, Neville R, Tan J. Oral oxymorphone for pain management. Ann Pharmacother 2007;41:1144-52.
127. Chan TY, Chan AY. Use of a plasma salicylate assay service in a medical unit in Hong Kong: a follow-up study. Vet Hum Toxicol 1996;38:278-9.
128. Chan TY, Chan AY, Ho CS, Critchley JA. The clinical value of screening for salicylates in acute poisoning. Vet Hum Toxicol 1995;37:37-8.
129. Chapman BJ, Proudfoot AT. Adult salicylate poisoning: deaths and outcome in patients with high plasma salicylate concentrations. Q J Med 1989;72:699-707.
130. Charney DS, Mihic SJ, Harris RA. Hypnotics and sedatives (Chapter 16). In: Brunton LL, Lazo JS, Parker KL, eds. Goodman & Gilman’s the pharmacological basis of therapeutics, 11th edition. New York, NY: McGraw-Hill, 2006.
131. Chen BH, Taylor EH, Pappas AA. Comparison of derivatives for determination of codeine and morphine by gas chromatography/mass spectrometry. J Anal Toxicol 1990;14:12-7.
132. Cheng PS, Fu CY, Lee CH, Liu C, Chien CS. GC-MS quantification of ketamine, norketamine, and dehydronorketamine in urine specimens and comparative study using ELISA as the preliminary
and liquid chromatography—atmospheric-pressure chemical-ionization mass spectrometry. J Anal Toxicol 1998;22:549-58.
87. Bossong MG, Van Dijk JP, Niesink RJ. Methylone and mCPP, two new drugs of abuse? Addict Biol 2005;10:321-3.
88. Boumba VA, Ziavrou KS, Vougiouklakis T. Hair as a biological indicator of drug use, drug abuse or chronic exposure to environmental toxicants. Int J Toxicol 2006;25:143-63.
89. Bowdle TA. Adverse effects of opioid agonists and agonist-antagonists in anaesthesia. Drug Saf 1998;19:173-89.
90. Bowie LJ, Kirkpatrick PB. Simultaneous determination of monoacetylmorphine, morphine, codeine, and other opiates by GC/MS. J Anal Toxicol 1989;13:326-9.
91. Bravo R, Driskell WJ, Whitehead RD Jr, Needham LL, Barr DB. Quantitation of dialkyl phosphate metabolites of organophosphate pesticides in human urine using GC-MS-MS with isotopic internal standards. J Anal Toxicol 2002;26:245-52.
92. Bray GP, Harrison PM, O’Grady JG, Tredger JM, Williams R. Long-term anticonvulsant therapy worsens outcome in paracetamol-induced fulminant hepatic failure. Hum Exp Toxicol 1992;11:265-70.
93. Breimer DD. Clinical pharmacokinetics of hypnotics. Clin Pharmacokinet 1977;2:93-109.
94. Brett AS. Implications of discordance between clinical impression and toxicology analysis in drug overdose. Arch Intern Med 1988;148: 437-41.
95. Bridges RR, Kinniburgh DW, Keehn BJ, Jennison TA. An evaluation of common methods for acetaminophen quantitation for small hospitals. J Toxicol Clin Toxicol 1983;20:1-17.
96. Bronstein AC, Spyker DA, Cantilena LR Jr, Green JL, Rumack BH, Heard SE. The American Association of Poison Control Centers’ National Poison Data System (NPDS): 25th annual report. Clin Toxicol 2008;46:927-1057.
97. Brown WA, Khan A. Which depressed patients should receive antidepressants? CNS Drugs 1994;1:341-7.
98. Bruns DE, Herold DA, Rodeheaver GT, Edlich RF. Polyethylene glycol intoxication in burn patients. Burns 1982;9:49-52.
99. Brzezinski MR, Abraham TL, Stone CL, Dean RA, Bosron WF. Purification and characterization of a human liver cocaine carboxylesterase that catalyzes the production of benzoylecgonine and the formation of cocaethylene from alcohol and cocaine. Biochem Pharmacol 1994;48:1747-55.
100. Buckley NA. Antipsychotic drugs (neuroleptics). In: Dart RC, ed. Medical toxicology. Philadelphia, Pa: Lippincott Williams & Wilkins, 2004:861-70.
101. Buckley NA, Eddleston M, Szinicz L. Oximes for acute organophosphate pesticide poisoning. Cochrane Database Syst Rev 2005(1):CD005085.
102. Buckley NA, Whyte IM, Dawson AH. Diagnostic data in clinical toxicology—should we use a Bayesian approach? J Toxicol Clin Toxicol 2002;40:213-22.
103. Buckpitt AR, Rollins DE, Mitchell JR. Varying effects of sulfhydryl nucleophiles on acetaminophen oxidation and sulfhydryl adduct formation. Biochem Pharmacol 1979;28:2941-6.
104. Budai B, Iskandar H. Dextromethorphan can produce false positive phencyclidine testing with HPLC. Am J Emerg Med 2002;20:61-2.
105. Burgen AS. The secretion of non-electrolytes in the parotid saliva. J Cell Physiol 1956;48:113-38.
106. Burke A, Smyth E, FitzGerald GA. Analgesic-antipyretic and antiinflammatory agents: pharmacotherapy of gout (Chapter 26). In: Brunton LL, Lazo JS, Parker KL, eds. Goodman & Gilman’s the pharmacological basis of therapeutics, 11th edition. New York, NY: McGraw-Hill, 2006.
107. Burkhart KK. Intravenous propranolol reverses hypertension after sympathomimetic overdose: two case reports. J Toxicol Clin Toxicol 1992;30:109-14.
108. Burns M, Baselt RC. Monitoring drug use with a sweat patch: an experiment with cocaine. J Anal Toxicol 1995;19:41-8.
109. Burns MJ. The pharmacology and toxicology of atypical antipsychotic agents. J Toxicol Clin Toxicol 2001;39:1-14.

Chapter35 ■ Clinical Toxicology 1171
154. Cody JT, Valtier S. Effects of Stealth adulterant on immunoassay testing for drugs of abuse. J Anal Toxicol 2001;25:466-70.
155. Coffman BL, Rios GR, King CD, Tephly TR. Human UGT2B7 catalyzes morphine glucuronidation. Drug Metab Dispos 1997;25: 1-4.
156. Colbert DL. Drug abuse screening with immunoassays: unexpected cross-reactivities and other pitfalls. Br J Biomed Sci 1994;51:136-46.
157. Combie J, Blake JW, Nugent TE, Tobin T. Morphine glucuronide hydrolysis: superiority of beta-glucuronidase from Patella vulgata. Clin Chem 1982;28:83-6.
158. Concheiro M, de Castro A, Quintela O, Lopez-Rivadulla M, Cruz A. Determination of drugs of abuse and their metabolites in human plasma by liquid chromatography-mass spectrometry: an application to 156 road fatalities. J Chromatogr B Analyt Technol Biomed Life Sci 2006;832:81-9.
159. Cone EJ. Saliva testing for drugs of abuse. Ann N Y Acad Sci 1993; 694:91-127.
160. Cone EJ. Mechanisms of drug incorporation into hair. Ther Drug Monit 1996;18:438-43.
161. Cone EJ, Darwin WD. Simultaneous determination of hydromorphone, hydrocodone and their 6alpha- and 6beta-hydroxy metabolites in urine using selected ion recording with methane chemical ionization. Biomed Mass Spectrom 1978;5:291.
162. Cone EJ, Darwin WD, Buchwald WF. Assay for codeine, morphine and ten potential urinary metabolites by gas chromatography–mass fragmentography. J Chromatogr 1983;275:307-18.
163. Cone EJ, Heit HA, Caplan YH, Gourlay D. Evidence of morphine metabolism to hydromorphone in pain patients chronically treated with morphine. J Anal Toxicol 2006;30:1-5.
164. Cone EJ, Hillsgrove M, Darwin WD. Simultaneous measurement of cocaine, cocaethylene, their metabolites, and “crack” pyrolysis products by gas chromatography-mass spectrometry. Clin Chem 1994;40:1299-305.
165. Cone EJ, Hillsgrove MJ, Jenkins AJ, Keenan RM, Darwin WD. Sweat testing for heroin, cocaine, and metabolites. J Anal Toxicol 1994;18: 298-305.
166. Cone EJ, Lange R, Darwin WD. In vivo adulteration: excess fluid ingestion causes false-negative marijuana and cocaine urine test results. J Anal Toxicol 1998;22:460-73.
167. Cone EJ, Oyler J, Darwin WD. Cocaine disposition in saliva following intravenous, intranasal, and smoked administration. J Anal Toxicol 1997;21:465-75.
168. Cone EJ, Sampson-Cone AH, Darwin WD, Huestis MA, Oyler JM. Urine testing for cocaine abuse: metabolic and excretion patterns following different routes of administration and methods for detection of false-negative results. J Anal Toxicol 2003;27:386-401.
169. Cone EJ, Weddington WW Jr. Prolonged occurrence of cocaine in human saliva and urine after chronic use. J Anal Toxicol 1989;13: 65-8.
170. Cone EJ, Welch P, Paul BD, Mitchell JM. Forensic drug testing for opiates, III. Urinary excretion rates of morphine and codeine following codeine administration. J Anal Toxicol 1991;15:161-6.
171. Contreras PC, DiMaggio DA, O’Donohue TL. Evidence for an endogenous peptide ligand and antagonist for PCP receptors. Prog Clin Biol Res 1985;192:495-8.
172. Cook CE, Brine DR, Jeffcoat AR, Hill JM, Wall ME, Perez-Reyes M, et al. Phencyclidine disposition after intravenous and oral doses. Clin Pharmacol Ther 1982;31:625-34.
173. Cook CE, Brine DR, Quin GD, Wall ME, Perez-Reyes M, Di Guiseppi SR. Smoking of phencyclidine: disposition in man and stability to pyrolytic conditions. Life Sci 1981;29:1967-72.
174. Corburt MR, Koves EM. Gas chromatography/mass spectrometry for the determination of cocaine and benzoylecgonine over a wide concentration range (<0.005-5 mg/dL) in postmortem blood. J Forensic Sci 1994;39:136-49.
175. Corcoran GB, Todd EL, Racz WJ, Hughes H, Smith CV, Mitchell JR. Effects of N-acetylcysteine on the disposition and metabolism of acetaminophen in mice. J Pharmacol Exp Ther 1985;232:857-63.
test methodology. J Chromatogr B Analyt Technol Biomed Life Sci 2007;852:443-9.
133. Cheung L, Potts RG, Meyer KC. Acetaminophen treatment nomogram. N Engl J Med 1994;330:1907-8.
134. Cheze M, Duffort G, Deveaux M, Pepin G. Hair analysis by liquid chromatography-tandem mass spectrometry in toxicological investigation of drug-facilitated crimes: report of 128 cases over the period June 2003-May 2004 in metropolitan Paris. Forensic Sci Int 2005;153:3-10.
135. Chiarotti M, Costamagna L. Analysis of 11-nor-9-carboxy-delta(9)-tetrahydrocannabinol in biological samples by gas chromatography tandem mass spectrometry (GC/MS-MS). Forensic Sci Int 2000;114: 1-6.
136. Chikhi-Chorfi N, Pham-Huy C, Galons H, Manuel N, Lowenstein W, Warnet JM, et al. Rapid determination of methadone and its major metabolite in biological fluids by gas-liquid chromatography with thermionic detection for maintenance treatment of opiate addicts. J Chromatogr 1998;718:278-84.
137. Chiu TH, Rosenberg HC. Comparison of the kinetics of [3H]diazepam and [3H]flunitrazepam binding to cortical synaptosomal membranes. J Neurochem 1982;39:1716-25.
138. Christ A, Arranto CA, Schindler C, Klima T, Hunziker PR, Siegemund M, et al. Incidence, risk factors, and outcome of aspiration pneumonitis in ICU overdose patients. Intensive Care Med 2006;32:1423-7.
139. Christophersen AS. Amphetamine designer drugs—an overview and epidemiology. Toxicol Lett 2000;112-113:127-31.
140. Christophersen AS, Biseth A, Skuterud B, Gadeholt G. Identification of opiates in urine by capillary column gas chromatography of two different derivatives. J Chromatogr 1987;422:117-24.
141. Chua SS, Benrimoj SI. Non-prescription sympathomimetic agents and hypertension. Med Toxicol Adverse Drug Exp 1988;3:387-417.
142. Chui PT. Anesthesia in a patient with undiagnosed salicylate poisoning presenting as intraabdominal sepsis. J Clin Anesth 1999;11: 251-3.
143. Cimbura G. PMA deaths in Ontario. Can Med Assoc J 1974;110: 1263-7.
144. Clarke PA, Foltz RL. Quantitative analysis of morphine in urine by gas chromatography-chemical ionization-mass spectrometry, with (N-C2H3)morphine as an internal standard. Clin Chem 1974;20:465-9.
145. Clarke RF. Insecticides: organic phosphorus compounds and carbamates. In: Flomenbaum NE, Goldfrank LR, Hoffman RS, Howland MA, Lewin NA, Nelson LS, eds. Goldfrank’s toxicologic emergencies, 8th edition. New York, NY: McGraw-Hill, 2006:1497-512.
146. Clarkson ED, Lesser D, Paul BD. Effective GC-MS procedure for detecting iso-LSD in urine after base-catalyzed conversion to LSD. Clin Chem 1998;44:287-92.
147. Clauwaert KM, Van Bocxlaer JF, Lambert WE, De Leenheer AP. Liquid chromatographic determination of cocaine, benzoylecgonine, and cocaethylene in whole blood and serum samples with diode-array detection. J Chromatogr Sci 1997;35:321-8.
148. Clements JA, Nimmo WS, Grant IS. Bioavailability, pharmacokinetics, and analgesic activity of ketamine in humans. J Pharm Sci 1982;71: 539-42.
149. Codd EE, Shank RP, Schupsky JJ, Raffa RB. Serotonin and norepinephrine uptake inhibiting activity of centrally acting analgesics: structural determinants and role in antinociception. J Pharmacol Exp Ther 1995;274:1263-70.
150. Cody JT. Determination of methamphetamine enantiomer ratios in urine by gas chromatography-mass spectrometry. J Chromatogr 1992;580:77-95.
151. Cody JT, Schwarzhoff R. Interpretation of methamphetamine and amphetamine enantiomer data. J Anal Toxicol 1993;17:321-6.
152. Cody JT, Valtier S. Immunoassay analysis of lysergic acid diethylamide. J Anal Toxicol 1997;21:459-64.
153. Cody JT, Valtier S. Detection of amphetamine and methamphetamine following administration of benzphetamine. J Anal Toxicol 1998;22: 299-309.

1172 SectionIII ■ Analytes
196. Demorest DM. Distinguishing cyclobenzaprine from amitriptyline and imipramine by liquid chromatography with UV multi-wavelength or full-spectrum detection: blood levels of cyclobenzaprine in emergency screens. J Anal Toxicol 1987;11:133-4.
197. Dennis RC, Valeri CR. Measuring percent oxygen saturation of hemoglobin, percent carboxyhemoglobin and methemoglobin, and concentrations of total hemoglobin and oxygen in blood of man, dog, and baboon. Clin Chem 1980;26:1304-8.
197A. Dasgupa A, ed. Alcohol and Drugs of Abuse Testing. Washington D.C., AACC Press, 2009:1-319.
198. Di Pietra AM, Gotti R, Del Borrello E, Pomponio R, Cavrini V. Analysis of amphetamine and congeners in illicit samples by liquid chromatography and capillary electrophoresis. J Anal Toxicol 2001;25: 99-105.
199. Dietzen DJ, Ecos K, Friedman D, Beason S. Positive predictive values of abused drug immunoassays on the Beckman Synchron in a veteran population. J Anal Toxicol 2001;25:174-8.
200. Done AK. Salicylate intoxication: significance of measurements of salicylate in blood in cases of acute ingestion. Pediatrics 1960;26: 800-7.
201. Dorandeu AH, Pages CA, Sordino MC, Pepin G, Baccino E, Kintz P. A case in south-eastern France: a review of drug facilitated sexual assault in European and English-speaking countries. J Clin Forensic Med 2006;13:253-61.
202. Drasbek KR, Christensen J, Jensen K. Gamma-hydroxybutyrate—a drug of abuse. Acta Neurol Scand 2006;114:145-56.
203. Dresser GK, Spence JD, Bailey DG. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibition. Clin Pharmacokinet 2000;38:41-57.
204. Drost RH, van Ooijen RD, Ionescu T, Maes RA. Determination of morphine in serum and cerebrospinal fluid by gas chromatography and selected ion monitoring after reversed-phase column extraction. J Chromatogr 1984;310:193-8.
205. Drummer OH. Methods for the measurement of benzodiazepines in biological samples. J Chromatogr 1998;713:201-25.
206. Drummer OH. Chromatographic screening techniques in systematic toxicological analysis. J Chromatogr 1999;733:27-45.
207. Drummer OH. Benzodiazepines—effects on human performance and behavior. Forensic Sci Rev 2002;14:1-14.
208. Drummer OH. Review: pharmacokinetics of illicit drugs in oral fluid. Forensic Sci Int 2005;150:133-42.
209. Drummer OH, Syrjanen ML, Cordner SM. Deaths involving the benzodiazepine flunitrazepam. Am J Forensic Med Pathol 1993;14: 238-43.
210. Dubowski KM. Human pharmacokinetics of alcohol. Alcohol Tech Rep 1976:55-63.
211. Dubowski KM. Absorption, distribution and elimination of alcohol: highway safety aspects. J Studies Alcohol 1985;10:98-108.
212. Dubowski KM. Proceedings of the 1987 Arnold O. Beckman Conference. Clin Chem 1987;33:5B-112B.
213. Dubowski KM, Luke JL. Measurement of carboxyhemoglobin and carbon monoxide in blood. Ann Clin Lab Sci 1973;3:53-65.
214. Dugandzic RM, Tierney MG, Dickinson GE, Dolan MC, McKnight DR. Evaluation of the validity of the Done nomogram in the management of acute salicylate intoxication. Ann Emerg Med 1989;18:1186-90.
215. DuPen A, Shen D, Ersek M. Mechanisms of opioid-induced tolerance and hyperalgesia. Pain Manag Nurs 2007;8:113-21.
216. Durakovic Z, Gjurasin M, Plavsic F. [Poisoning by a large dose of digitalis]. Arh Hig Rada Toksikol 1984;35:277-84.
217. Duritz G, Truitt EB Jr. A rapid method for the simultaneous determination of acetaldehyde and ethanol in blood using gas chromatography. Q J Studies Alcohol 1964;25:498-510.
218. Dutt MC, Lo DS, Ng DL, Woo SO. Gas chromatographic study of the urinary codeine-to-morphine ratios in controlled codeine consumption and in mass screening for opiate drugs. J Chromatogr 1983;267:117-24.
219. Dyer JK. Drug facilitated sexual assault: a review of 24 incidents. J Toxicol Clin Toxicol 2004;42:519.
176. Costa A, Chawla S, Me A, le Pichon T. World drug report: United Nations Office on Drugs and Crime. New York, NY: United Nations Publication, 2008.
177. Crews JC, Sweeney NJ, Denson DD. Clinical efficacy of methadone in patients refractory to other mu-opioid receptor agonist analgesics for management of terminal cancer pain: case presentations and discussion of incomplete cross-tolerance among opioid agonist analgesics. Cancer 1993;72:2266-72.
178. Crouch DJ, Frank JF, Farrell LJ, Karsch HM, Klaunig JE. A multiple-site laboratory evaluation of three on-site urinalysis drug-testing devices. J Anal Toxicol 1998;22:493-502.
179. Cruz-Landeira A, Lopez-Rivadulla M, Concheiro-Carro L, Fernandez-Gomez P, Tabernero-Duque MJ. A new spectrophotometric method for the toxicological diagnosis of cyanide poisoning. J Anal Toxicol 2000;24:266-70.
180. Curry SC, Mills KC, Buha A. Neurotransmitters and neuromodulators. In: Flomenbaum G, Hoffman RS, Howland MA, Lewin NA, Nelson LS, eds. Goldfrank’s toxicologic emergencies, 8th edition. New York: McGraw Hill, 2006:214-48.
181. Daher R, Haidar JH, Al-Amin H. Rifampin interference with opiate immunoassays. Clin Chem 2002;48:203-4.
182. Dahl H, Stephanson N, Beck O, Helander A. Comparison of urinary excretion characteristics of ethanol and ethyl glucuronide. J Anal Toxicol 2002;26:201-4.
183. Dahl ML. Cytochrome p450 phenotyping/genotyping in patients receiving antipsychotics: useful aid to prescribing? Clin Pharmacokinet 2002;41:453-70.
184. Dale C, Aulaqi AA, Baker J, Hobbs RC, Tan ME, Tovey C, et al. Assessment of a point-of-care test for paracetamol and salicylate in blood. QJM 2005;98:113-8.
185. Dart RC. Part 1. General approach to the poisoned patient. Section 5. Antidotes and specific therapies. In: Dart RC, ed. Medical toxicology, 3rd edition. Philadelphia: Lippincott Williams & Wilkins, 2004:160-268.
186. Daws LC, Irvine RJ, Callaghan PD, Toop NP, White JM, Bochner F. Differential behavioural and neurochemical effects of para-methoxyamphetamine and 3,4-methylenedioxymethamphetamine in the rat. Prog Neuropsychopharmacol Biol Psychiatry 2000;24:955-77.
187. Dawson AH. Cyclic antidepressants drugs. In: Dart RC, ed. Medical toxicology. Philadelphia, Pa: Lippincott Williams & Wilkins, 2004:834-51.
188. de Boer D, Bosman IJ, Hidvegi E, Manzoni C, Benko AA, dos Reys LJ, et al. Piperazine-like compounds: a new group of designer drugs-of-abuse on the European market. Forensic Sci Int 2001;121: 47-56.
189. de la Torre R, Farre M, Ortuno J, Mas M, Brenneisen R, Roset PN, et al. Non-linear pharmacokinetics of MDMA (‘ecstasy’) in humans. Br J Clin Pharmacol 2000;49:104-9.
190. de la Torre R, Ortuno J, Gonzalez ML, Farre M, Cami J, Segura J. Determination of cocaine and its metabolites in human urine by gas chromatography/mass spectrometry after simultaneous use of cocaine and ethanol. J Pharmaceut Biomed Anal 1995;13:305-12.
191. de Leon J, Dinsmore L, Wedlund P. Adverse drug reactions to oxycodone and hydrocodone in CYP2D6 ultrarapid metabolizers. J Clin Psychopharmacol 2003;23:420-1.
192. Dean RA, Christian CD, Sample RH, Bosron WF. Human liver cocaine esterases: ethanol-mediated formation of ethylcocaine. FASEB J 1991;5:2735-9.
193. Delbeke FT, Debackere M. Urinary concentrations of codeine and morphine after the administration of different codeine preparations in relation to doping analysis. J Pharmaceut Biomed Anal 1991;9: 959-64.
194. Delbeke FT, Debackere M. Influence of hydrolysis procedures on the urinary concentrations of codeine and morphine in relation to doping analysis. J Pharmaceut Biomed Anal 1993;11:339-43.
195. Delinsky DC, Srinivasan K, Solomon HM, Bartlett MG. Simultaneous capillary electrophoresis determination of barbiturates from meconium. J Liquid Chromatogr Relat Technol 2002;25:113-23.

Chapter35 ■ Clinical Toxicology 1173
241. Felgate HE, Felgate PD, James RA, Sims DN, Vozzo DC. Recent paramethoxyamphetamine deaths. J Anal Toxicol 1998;22:169-72.
242. Feng J, Wang L, Dai I, Harmon T, Bernert JT. Simultaneous determination of multiple drugs of abuse and relevant metabolites in urine by LC-MS-MS. J Anal Toxicol 2007;31:359-68.
243. Ferrari A, Coccia CP, Bertolini A, Sternieri E. Methadone—metabolism, pharmacokinetics and interactions. Pharmacol Res 2004;50:551-9.
244. Fish F, Hayes TS. Hydrolysis of morphine glucuronide. J Forensic Sci 1974;19:676-83.
245. Fitzgerald RL, Ramos JM Jr, Bogema SC, Poklis A. Resolution of methamphetamine stereoisomers in urine drug testing: urinary excretion of R(−)-methamphetamine following use of nasal inhalers. J Anal Toxicol 1988;12:255-9.
246. Flanagan RJ, Mant TG. Coma and metabolic acidosis early in severe acute paracetamol poisoning. Hum Toxicol 1986;5:179-82.
247. Fleischman A, Chiang VW. Carbamazepine overdose recognized by a tricyclic antidepressant assay. Pediatrics 2001;107:176-7.
248. Fleming M, Mihic SJ, Harris RA. Ethanol (Chapter 22). In: Brunton LL, Lazo JS, Parker KL, eds. Goodman & Gilman’s the pharmacological basis of therapeutics, 11th edition. New York, NY: McGraw-Hill, 2006.
249. Flint AJ. Choosing appropriate antidepressant therapy in the elderly: a risk-benefit assessment of available agents. Drugs Aging 1998;13:269-80.
250. Flomenbaum NE. Salicylates. In: Goldfrank LR, Flomenbaum NE, Lewin NA, Howland MA, Hoffman RS, Nelson LS, eds. Goldfrank’s toxicologic emergencies, 7th edition. New York, NY: McGraw-Hill, 2002:507-27.
251. Flomenbaum NE. Principles of managing the poisoned or overdosed patient. In: Flomenbaum NE, Goldfrank LR, Hoffman RS, Howland MA, Lewin NA, Nelson LS, eds. Goldfrank’s toxicologic emergencies, 8th edition. New York, NY: McGraw-Hill, 2006:42-50.
252. Flomenbaum NE. Salicylates. In: Flomenbaum NE, Goldfrank LR, Hoffman RS, Howland MA, Lewin NA, Nelson LS, eds. Goldfrank’s toxicologic emergencies, 8th edition. New York, NY: McGraw-Hill, 2006:550-64.
253. Foltz RL, Fentiman AF Jr, Foltz RB. GC/MS assays for abused drugs in body fluids. NIDA Res Monogr 1980;32:1-198.
254. Ford M, Tomaszewski C, Kerns W, Kirk M, Eichhorst A. Bedside ferric chloride urine test to rule out salicylate intoxication. Vet Hum Toxicol 1994;36:364.
255. Forrest JA, Clements JA, Prescott LF. Clinical pharmacokinetics of paracetamol. Clin Pharmacokinet 1982;7:93-107.
256. Francom P, Andrenyak D, Lim HK, Bridges RR, Foltz RL, Jones RT. Determination of LSD in urine by capillary column gas chromatography and electron impact mass spectrometry. J Anal Toxicol 1988;12:1-8.
257. Fraser AD, MacNeil W. Colorimetric and gas chromatographic procedures for glycolic acid in serum: the major toxic metabolite of ethylene glycol. J Toxicol Clin Toxicol 1993;31:397-405.
258. Fraser AD, Worth D. Urinary excretion profiles of 11-nor-9-carboxy-delta9-tetrahydrocannabinol. Study III: a delta9-THC-COOH to creatinine ratio study. Forensic Sci Int 2003;137:196-202.
259. Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 2003;83:1017-66.
260. Frick S, Roos M, Fattinger K. How much is too much? Oligosymptomatic presentation after 11.5 g of diphenhydramine. Hum Exp Toxicol 2007;26:131-3.
261. Friedemann T. The excretion of ingested ethyl alcohol in saliva. J Lab Clin Med 1938;29:1007-14.
262. Fucci N, De Giovanni N, Chiarotti M, Scarlata S. SPME-GC analysis of THC in saliva samples collected with “EPITOPE” device. Forensic Sci Int 2001;119:318-21.
263. Fudala PJ, Bridge TP, Herbert S, Williford WO, Chiang CN, Jones K, et al. Office-based treatment of opiate addiction with a sublingual-tablet formulation of buprenorphine and naloxone. N Engl J Med 2003;349:949-58.
220. Eddleston M, Dawson A, Karalliedde L, Dissanayake W, Hittarage A, Azher S, et al. Early management after self-poisoning with an organophosphorus or carbamate pesticide—a treatment protocol for junior doctors. Crit Care 2004;8:R391-7.
221. Eichelbaum M, Evert B. Influence of pharmacogenetics on drug disposition and response. Clin Exp Pharmacol Physiol 1996;23:983-5.
222. Eichhorst J, Etter M, Lepage J, Lehotay DC. Urinary screening for methylphenidate (Ritalin) abuse: a comparison of liquid chromatography-tandem mass spectrometry, gas chromatography-mass spectrometry, and immunoassay methods. Clin Biochem 2004;37:175-83.
223. Eichhorst JC, Etter ML, Rousseaux N, Lehotay DC. Drugs of abuse testing by tandem mass spectrometry: a rapid, simple method to replace immunoassays. Clin Biochem 2009;42:1531-42.
224. Ellinwood EH Jr, Kilbey MM. Fundamental mechanisms underlying altered behavior following chronic administration of psychomotor stimulants. Biol Psychiatry 1980;15:749-57.
225. Elliott SP, Burgess V. Clinical urinalysis of drugs and alcohol in instances of suspected surreptitious administration (“spiked drinks”). Sci Justice 2005;45:129-34.
226. Ellis GM Jr, Mann MA, Judson BA, Schramm NT, Tashchian A. Excretion patterns of cannabinoid metabolites after last use in a group of chronic users. Clin Pharmacol Ther 1985;38:572-8.
227. ElSohly HN, Stanford DF, Jones AB, ElSohly MA, Snyder H, Pedersen C. Gas chromatographic/mass spectrometric analysis of morphine and codeine in human urine of poppy seed eaters. J Forensic Sci 1988;33: 347-56.
228. ElSohly MA, deWit H, Wachtel SR, Feng S, Murphy TP. Delta9-tetrahydrocannabivarin as a marker for the ingestion of marijuana versus Marinol: results of a clinical study. J Anal Toxicol 2001;25: 565-71.
229. ElSohly MA, Feng S. Delta 9-THC metabolites in meconium: identification of 11-OH-delta 9-THC, 8 beta,11-diOH-delta 9-THC, and 11-nor-delta 9-THC-9-COOH as major metabolites of delta 9-THC. J Anal Toxicol 1998;22:329-35.
230. ElSohly MA, Feng S, Salamone SJ, Wu R. A sensitive GC-MS procedure for the analysis of flunitrazepam and its metabolites in urine. J Anal Toxicol 1997;21:335-40.
231. ElSohly MA, Ross SA, Mehmedic Z, Arafat R, Yi B, Banahan BF 3rd. Potency trends of delta9-THC and other cannabinoids in confiscated marijuana from 1980-1997. J Forensic Sci 2000;45:24-30.
232. ElSohly MA, Salamone SJ. Prevalence of drugs used in cases of alleged sexual assault. J Anal Toxicol 1999;23:141-6.
233. Ener RA, Meglathery SB, Van Decker WA, Gallagher RM. Serotonin syndrome and other serotonergic disorders. Pain Med 2003;4:63-74.
234. Erickson TB, Thompson TM, Lu JJ. The approach to the patient with an unknown overdose. Emerg Med Clin North Am 2007;25:249-81; abstract vii.
235. Evelyn KA, Malloy HT. Microdetermination of oxyhemoblobin, methemoglobin, and sulfhemoglobin in a single sample of blood. J Biol Chem 1938;126:655-62.
236. Fallon JK, Kicman AT, Henry JA, Milligan PJ, Cowan DA, Hutt AJ. Stereospecific analysis and enantiomeric disposition of 3,4-methylenedioxymethamphetamine (Ecstasy) in humans. Clin Chem 1999;45:1058-69.
237. Farre M, de la Torre R, Llorente M, Lamas X, Ugena B, Segura J, et al. Alcohol and cocaine interactions in humans. J Pharmacol Exp Ther 1993;266:1364-73.
238. Farrell SE, Roberts JR. Benzodiazepines. In: Haddad LM, Shannon MW, Winchester JF, eds. Clinical management of poisoning and drug overdose, 3rd edition. Philadelphia, Pa: WB Saunders, 1998: 609-27.
239. Federal Register 49 CFR Part 40. Procedures for transportation workplace drug and alcohol testing programs. Updated as of August 31, 2009.
240. Fehn J, Megges G. Detection of O6-monoacetylmorphine in urine samples by GC/MS as evidence for heroin use. J Anal Toxicol 1985;9: 134-8.

1174 SectionIII ■ Analytes
288. Glasser L, Sternglanz PD, Combie J, Robinson A. Serum osmolality and its applicability to drug overdose. Am J Clin Pathol 1973;60:695-9.
289. Gock SB, Wong SH, Nuwayhid N, Venuti SE, Kelley PD, Teggatz JR, et al. Acute zolpidem overdose—report of two cases. J Anal Toxicol 1999;23:559-62.
290. Goldberger BA, Cone EJ. Confirmatory tests for drugs in the workplace by gas chromatography-mass spectrometry. J Chromatogr A 1994;674:73-86.
291. Goldstein DB. Pharmacology of alcohol. New York, NY: Oxford University Press, 1983.
292. Goldstein DS, Nurnberger J Jr, Simmons S, Gershon ES, Polinsky R, Keiser HR. Effects of injected sympathomimetic amines on plasma catecholamines and circulatory variables in man. Life Sci 1983;32: 1057-63.
293. Goodwin RS, Gustafson RA, Barnes A, Nebro W, Moolchan ET, Huestis MA. Delta(9)-tetrahydrocannabinol, 11-hydroxy-delta(9)-tetrahydrocannabinol and 11-nor-9-carboxy-delta(9)-tetrahydrocannabinol in human plasma after controlled oral administration of cannabinoids. Ther Drug Monit 2006;28:545-51.
294. Goulle JP, Anger JP. Drug-facilitated robbery or sexual assault: problems associated with amnesia. Ther Drug Monit 2004;26:206-10.
295. Goulle JP, Cheze M, Pepin G. Determination of endogenous levels of GHB in human hair: are there possibilities for the identification of GHB administration through hair analysis in cases of drug-facilitated sexual assault? J Anal Toxicol 2003;27:574-80.
296. Green AL, el Hait MA. Inhibition of mouse brain monoamine oxidase by (+)-amphetamine in vivo. J Pharm Pharmacol 1978;30:262-3.
297. Green AL, el Hait MA. p-Methoxyamphetamine, a potent reversible inhibitor of type-A monoamine oxidase in vitro and in vivo. J Pharm Pharmacol 1980;32:262-6.
298. Green B, Kavanagh D, Young R. Being stoned: a review of self-reported cannabis effects. Drug Alcohol Rev 2003;22:453-60.
299. Green KB, Isenschmid DS. Medical review officer interpretation of urine drug test results. In: Liu R, Goldberger B, eds. Handbook of workplace drug testing. Washington, DC: AACC Press, 1995:321-53.
300. Green KB, Isenschmid DS. Medical review officer interpretation of urine drug testing results. Forensic Sci Rev 1995;7:41-60.
301. Green SM, Johnson NE. Ketamine sedation for pediatric procedures. Part 2: review and implications. Ann Emerg Med 1990;19:1033-46.
302. Griffith RK, Johnson EA. Adrenergic drugs. In: Foye WO, Lemke TL, Williams DA, eds. Principles of medicinal chemistry. Baltimore, Md: Williams & Wilkins, 1995:345-65.
303. Grinstead GF. A closer look at acetyl and pentafluoropropionyl derivatives for quantitative analysis of morphine and codeine by gas chromatography/mass spectrometry. J Anal Toxicol 1991;15:293-8.
304. Grotenhermen F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet 2003;42:327-60.
305. Grotenhermen F. Pharmacology of cannabinoids. Neuroendocrinol Lett 2004;25:14-23.
306. Grotenhermen F, Leson G, Berghaus G, Drummer OH, Kruger HP, Longo M, et al. Developing limits for driving under cannabis. Addiction 2007;102:1910-7.
307. Guillot JG, Weber JP, Savoie JY. Quantitative determination of carbon monoxide in blood by head-space gas chromatography. J Anal Toxicol 1981;5:264-6.
308. Gustafson RA, Levine B, Stout PR, Klette KL, George MP, Moolchan ET, et al. Urinary cannabinoid detection times after controlled oral administration of delta9-tetrahydrocannabinol to humans. Clin Chem 2003;49:1114-24.
309. Gutstein HB, Akil H. Opioid analgesics (Chapter 21). In: Brunton LL, Lazo JS, Parker KL, eds. Goodman & Gilman’s the pharmacological basis of therapeutics, 11th edition. New York, NY: McGraw-Hill, 2006.
310. Haddad LM, Winchester JF. Barbiturates. In: Haddad L, Shannon M, Winchester J, eds. Clinical management of poisoning and drug overdose. Philadelphia, Pa: WB Saunders, 1998:521-7.
264. Fudala PJ, Johnson RE. Development of opioid formulations with limited diversion and abuse potential. Drug Alcohol Depend 2006; 83(Suppl 1):S40-7.
265. Fukuto TR. Mechanism of action of organophosphorus and carbamate insecticides. Environ Health Perspect 1990;87:245-54.
266. Fuller DC, Anderson WH. A simplified procedure for the determination of free codeine, free morphine, and 6-acetylmorphine in urine. J Anal Toxicol 1992;16:315-8.
267. Gabow PA. Disorders associated with an altered anion gap. Kidney Int 1985;27:472-83.
268. Gadsden RH Jr, Taylor EH, Steindel SJ. Ethanol in biological fluids by enzymic analysis. In: Frings C, Faulkner W, eds. Selected methods of emergency toxicology. Washington, DC: AACC Press, 1986: 63-5.
269. Gadsden RH Sr. Study of forensic and clinical source hemoglobin interference with the duPont ACA ethanol method. Ann Clin Lab Sci 1986;16:399-406.
270. Gallagher RM, Rosenthal LJ. Chronic pain and opiates: balancing pain control and risks in long-term opioid treatment. Arch Phys Med Rehabil 2008;89:S77-82.
271. Galloway FR, Bellet NF. Methadone conversion to EDDP during GC-MS analysis of urine samples. J Anal Toxicol 1999;23:615-9.
272. Gareri J, Klein J, Koren G. Drugs of abuse testing in meconium. Clin Chim Acta 2006;366:101-11.
273. Garretty DJ, Wolff K, Hay AW, Raistrick D. Benzodiazepine misuse by drug addicts. Ann Clin Biochem 1997;34(Pt 1):68-73.
274. Garriott JC. Pharmacology and toxicology of ethyl alcohol. In: Garriott JC, ed. Medicolegal aspects of alcohol. Tucson, Ariz: Lawyers & Judges Publishing, 1996:35-63.
275. Geiser L, Cherkaoui S, Veuthey JL. Simultaneous analysis of some amphetamine derivatives in urine by nonaqueous capillary electrophoresis coupled to electrospray ionization mass spectrometry. J Chromatogr A 2000;895:111-21.
276. Geller RJ, Spyker DA, Herold DA, Bruns DE. Serum osmolal gap and ethanol concentration: a simple and accurate formula. J Toxicol Clin Toxicol 1986;24:77-84.
277. George CF. Pyrazolopyrimidines. Lancet 2001;358:1623-6.278. George S, Braithwaite RA. A pilot study to determine the usefulness
of the urinary excretion of methadone and its primary metabolite (EDDP) as potential markers of compliance in methadone detoxification programs. J Anal Toxicol 1999;23:81-5.
279. George S, Braithwaite RA. Use of on-site testing for drugs of abuse. Clin Chem 2002;48:1639-46.
280. George S, Parmar S, Meadway C, Braithwaite RA. Application and validation of a urinary methadone metabolite (EDDP) immunoassay to monitor methadone compliance. Ann Clin Biochem 2000;37(Pt 3): 350-4.
281. Gergov M, Ojanpera I, Vuori E. Simultaneous screening for 238 drugs in blood by liquid chromatography-ion spray tandem mass spectrometry with multiple-reaction monitoring. J Chromatogr B Analyt Technol Biomed Life Sci 2003;795:41-53.
282. Gergov M, Robson JN, Ojanpera I, Heinonen OP, Vuori E. Simultaneous screening and quantitation of 18 antihistamine drugs in blood by liquid chromatography ionspray tandem mass spectrometry. Forensic Sci Int 2001;121:108-15.
283. Gillman PK. Monoamine oxidase inhibitors, opioid analgesics and serotonin toxicity. Br J Anaesth 2005;95:434-41.
284. Giovannelli M, Bedforth N, Aitkenhead A. Survey of intrathecal opioid usage in the UK. Eur J Anaesthesiol 2008;25:118-22.
285. Giraud S, Robinson N, Mangin P, Saugy M. Scientific and forensic standards for homologous blood transfusion anti-doping analyses. Forensic Sci Int 2008;179:23-33.
286. Gjerde H, Fongen U, Gundersen H, Christophersen AS. Evaluation of a method for simultaneous quantification of codeine, ethylmorphine and morphine in blood. Forensic Sci Int 1991;51:105-10.
287. Glare PA, Walsh TD. Clinical pharmacokinetics of morphine. Ther Drug Monit 1991;13:1-23.

Chapter35 ■ Clinical Toxicology 1175
332. Hersack RA. Ketamine’s psychological effects do not contraindicate its use based on a patient’s occupation. Aviat Space Environ Med 1994; 65:1041-6.
333. Hessel DW, Modglin FR. The quantitative determination of ethanol and other volatile substances in blood by gas-liquid partition chromatography. J Forensic Sci 1964;9:255-64.
334. Hewlett TP, McMartin KE, Lauro AJ, Ragan FA Jr. Ethylene glycol poisoning: the value of glycolic acid determinations for diagnosis and treatment. J Toxicol Clin Toxicol 1986;24:389-402.
335. Hime GW, Hearn WL, Rose S, Cofino J. Analysis of cocaine and cocaethylene in blood and tissues by GC-NPD and GC-ion trap mass spectrometry. J Anal Toxicol 1991;15:241-5.
336. Hindmarch I, ElSohly M, Gambles J, Salamone S. Forensic urinalysis of drug use in cases of alleged sexual assault. J Clin Forensic Med 2001;8:197-205.
337. Hoffman RJ, Nelson LS, Hoffman RS. Use of ferric chloride to identify salicylate-containing poisons. J Toxicol Clin Toxicol 2002;40:547-9.
338. Hold K, de Boer D, Zuidema J, Maes RA. Saliva as an analytical tool in toxicology. Int J Drug Testing 1995;1:1-36.
339. Hold KM, Borges CR, Wilkins DG, Rollins DE, Joseph RE Jr. Detection of nandrolone, testosterone, and their esters in rat and human hair samples. J Anal Toxicol 1999;23:416-23.
340. Holstege C, Baer A, Brady WJ. The electrocardiographic toxidrome: the ECG presentation of hydrofluoric acid ingestion. Am J Emerg Med 2005;23:171-6.
341. Holstege CP, Dobmeier S. Cardiovascular challenges in toxicology. Emerg Med Clin North Am 2005;23:1195-217.
342. Honey CR, Miljkovic Z, MacDonald JF. Ketamine and phencyclidine cause a voltage-dependent block of responses to l-aspartic acid. Neurosci Lett 1985;61:135-9.
343. Hornbeck CL, Czarny RJ. Retrospective analysis of some l-methamphetamine/l-amphetamine urine data. J Anal Toxicol 1993;17:23-5.
344. Horning MG, Brown L, Nowlin J, Lertratanangkoon K, Kellaway P, Zion TE. Use of saliva in therapeutic drug monitoring. Clin Chem 1977;23:157-64.
345. Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 2002;54:161-202.
346. Hsu J, Liu C, Liu CP, Tsay WI, Li JH, Lin DL, et al. Performance characteristics of selected immunoassays for preliminary test of 3,4-methylenedioxymethamphetamine, methamphetamine, and related drugs in urine specimens. J Anal Toxicol 2003;27:471-8.
347. Huang W, Andollo W, Hearn WL. A solid phase extraction technique for the isolation and identification of opiates in urine. J Anal Toxicol 1992;16:307-10.
348. Huestis MA, Henningfield JE, Cone EJ. Blood cannabinoids. I. Absorption of THC and formation of 11-OH-THC and THCCOOH during and after smoking marijuana. J Anal Toxicol 1992;16:276-82.
349. Huestis MA, Mitchell JM, Cone EJ. Lowering the federally mandated cannabinoid immunoassay cutoff increases true-positive results. Clin Chem 1994;40:729-33.
350. Hughes RO, Bronner WE, Smith ML. Detection of amphetamine and methamphetamine in urine by gas chromatography/mass spectrometry following derivatization with (−)-menthyl chloroformate. J Anal Toxicol 1991;15:256-9.
351. Hullin DA. An IgM paraprotein causing a falsely low result in an enzymatic assay for acetaminophen. Clin Chem 1999;45:155-7.
352. Humbert L, Jacquemont MC, Leroy E, Leclerc F, Houdret N, Lhermitte M. Determination of chloral hydrate and its metabolites (trichloroethanol and trichloracetic acid) in human plasma and urine using electron capture gas chromatography. Biomed Chromatogr 1994;8:273-7.
353. Hurwitz ES, Barrett MJ, Bregman D, Gunn WJ, Schonberger LB, Fairweather WR, et al. Public Health Service study on Reye’s syndrome and medications: report of the pilot phase. N Engl J Med 1985;313:849-57.
311. Haeckel R, Hanecke P. Application of saliva for drug monitoring: an in vivo model for transmembrane transport. Eur J Clin Chem Clin Biochem 1996;34:171-91.
312. Hahn IH, Chu J, Hoffman RS, Nelson LS. Errors in reporting salicylate levels. Acad Emerg Med 2000;7:1336-7.
313. Hakala KS, Kostiainen R, Ketola RA. Feasibility of different mass spectrometric techniques and programs for automated metabolite profiling of tramadol in human urine. Rapid Commun Mass Spectrom 2006;20:2081-90.
314. Hall BJ, Satterfield-Doerr M, Parikh AR, Brodbelt JS. Determination of cannabinoids in water and human saliva by solid-phase microextraction and quadrupole ion trap gas chromatography/mass spectrometry. Anal Chem 1998;70:1788-96.
315. Hall JA, Moore CB. Drug facilitated sexual assault—a review. J Forensic Leg Med 2008;15:291-7.
316. Halla JT, Atchison SL, Hardin JG. Symptomatic salicylate ototoxicity: a useful indicator of serum salicylate concentration? Ann Rheum Dis 1991;50:682-4.
317. Haller CA, Benowitz NL. Adverse cardiovascular and central nervous system events associated with dietary supplements containing ephedra alkaloids. N Engl J Med 2000;343:1833-8.
318. Hampson NB, Hauff NM. Carboxyhemoglobin levels in carbon monoxide poisoning: do they correlate with the clinical picture? Am J Emerg Med 2008;26:665-9.
319. Hanson SM, Morlock EV, Satyshur KA, Czajkowski C. Structural requirements for eszopiclone and zolpidem binding to the gamma-aminobutyric acid type-A (GABAA) receptor are different. J Med Chem 2008;51:7243-52.
320. Harden RN. Chronic pain and opiates: a call for moderation. Arch Phys Med Rehabil 2008;89:S72-6.
321. Harding P. Methods for breath analysis. In: Garriott JC, ed. Medicolegal aspects of alcohol. Tucson, Ariz: Lawyers & Judges Publishing, 1996:181-217.
322. Harrison PM, Wendon JA, Gimson AE, Alexander GJ, Williams R. Improvement by acetylcysteine of hemodynamics and oxygen transport in fulminant hepatic failure. N Engl J Med 1991;324:1852-7.
323. Hart CL, Jatlow P, Sevarino KA, McCance-Katz EF. Comparison of intravenous cocaethylene and cocaine in humans. Psychopharmacology 2000;149:153-62.
324. Hayes LW, Krasselt WG, Mueggler PA. Concentrations of morphine and codeine in serum and urine after ingestion of poppy seeds. Clin Chem 1987;33:806-8.
325. Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005;51:434-44.
326. Hearn WL, Flynn DD, Hime GW, Rose S, Cofino JC, Mantero-Atienza E, et al. Cocaethylene: a unique cocaine metabolite displays high affinity for the dopamine transporter. J Neurochem 1991;56:698-701.
327. Hegadoren KM, Greenshaw AJ, Baker GB, Martin-Iverson MT, Lodge B, Soin S. 4-Ethoxyamphetamine: effects on intracranial self-stimulation and in vitro uptake and release of 3H-dopamine and 3H-serotonin in the brains of rats. J Psychiatry Neurosci 1994;19: 57-62.
328. Heller DN. Liquid chromatography/mass spectrometry for timely response in regulatory analyses: identification of pentobarbital in dog food. Anal Chem 2000;72:2711-6.
329. Henderson GL, Harkey MR, Zhou C, Jones RT, Jacob P 3rd. Incorporation of isotopically labeled cocaine into human hair: race as a factor. J Anal Toxicol 1998;22:156-65.
330. Hendrickson R, Bizovi KE. Acetaminophen. In: Goldfrank LR, Flomenbaum NE, Lewin NA, Howland MA, Hoffman RS, Nelson LS, eds. Goldfrank’s toxicologic emergencies, 8th edition. New York, NY: McGraw-Hill, 2006:523-43.
331. Hensley D, Cody JT. Simultaneous determination of amphetamine, methamphetamine, methylenedioxyamphetamine (MDA), methylenedioxymethamphetamine (MDMA), and methylenedioxyethylamphetamine (MDEA) enantiomers by GC-MS. J Anal Toxicol 1999;23:518-23.

1176 SectionIII ■ Analytes
378. Jatlow PI. Drug of abuse profile: cocaine. Clin Chem 1987;33: 66B-71B.
379. Javaid JI, Dekirmenjian H, Brunngraber EG, Davis JM. Quantitative determination of cocaine and its metabolites benzoylecgonine and ecgonine by gas-liquid chromatography. J Chromatogr 1975;110: 140-9.
380. Jeanville PM, Estape ES, Needham SR, Cole MJ. Rapid confirmation/quantitation of cocaine and benzoylecgonine in urine utilizing high performance liquid chromatography and tandem mass spectrometry. J Am Soc Mass Spectrom 2000;11:257-63.
381. Jenkins AJ, Goldberger BA. Identification of unique cocaine metabolites and smoking by-products in postmortem blood and urine specimens. J Forensic Sci 1997;42:824-7.
382. Jenkins AJ, Oyler JM, Cone EJ. Comparison of heroin and cocaine concentrations in saliva with concentrations in blood and plasma. J Anal Toxicol 1995;19:359-74.
383. Jiang T-F, Wang Y-H, Lu Z-H, Yue M-E. Direct determination of barbiturates in urine by capillary electrophoresis using a capillary coated dynamically with polycationic polymers. Chromatographia 2007;65:611-5.
384. Johansen SS, Bhatia HM. Quantitative analysis of cocaine and its metabolites in whole blood and urine by high-performance liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2007;852:338-44.
385. Johansen SS, Hansen AC, Muller IB, Lundemose JB, Franzmann MB. Three fatal cases of PMA and PMMA poisoning in Denmark. J Anal Toxicol 2003;27:253-6.
386. Jolliff HA, Sivilotti ML. Ethylene glycol. In: Dart RC, ed. Medical toxicology. Philadelphia, Pa: Lippincott Williams & Wilkins, 2004:1223-30.
387. Jones AB, ElSohly HN, Arafat ES, ElSohly MA. Analysis of the major metabolite of delta 9-tetrahydrocannabinol in urine. IV: a comparison of five methods. J Anal Toxicol 1984;8:249-51.
388. Jones AW. Distribution of ethanol between saliva and blood in man. Clin Exp Pharmacol Physiol 1979;6:53-9.
389. Jones AW. Measurement of alcohol in blood and breath for legal purposes. In: Crow KE, Batt RD, eds. Human metabolism of alcohol, vol I. Pharmacokinetics, medicolegal aspects, and general interests. Boca Raton, Fla: CRC Press, 1989:71-99.
390. Jones AW, Blom Y, Bondesson U, Anggard E. Determination of morphine in biological samples by gas chromatography-mass spectrometry: evidence for persistent tissue binding in rats twenty-two days post-withdrawal. J Chromatogr 1984;309:73-80.
391. Jones AW, Lindberg L, Olsson SG. Magnitude and time-course of arterio-venous differences in blood-alcohol concentration in healthy men. Clin Pharmacokinet 2004;43:1157-66.
392. Jones G. Post-mortem toxicology. In: Moffat A, ed. Clarke’s analysis of drugs and poisons, 3rd edition. London, UK: Pharmaceutical Press, 2004:95-108.
393. Jones GR, Singer PP. An unusual trichloroethanol fatality attributed to sniffing trichloroethylene. J Anal Toxicol 2008;32:183-6.
394. Jones JJ, Kidwell H, Games DE. Application of atmospheric pressure chemical ionisation mass spectrometry in the analysis of barbiturates by high-speed analytical countercurrent chromatography. Rapid Commun Mass Spectrom 2003;17:1565-72.
395. Josefson D. Herbal stimulant causes US deaths. BMJ 1996;312:1441.396. Joseph RE Jr, Hold KM, Wilkins DG, Rollins DE, Cone EJ. Drug
testing with alternative matrices II. Mechanisms of cocaine and codeine deposition in hair. J Anal Toxicol 1999;23:396-408.
397. Joseph RE Jr, Su TP, Cone EJ. In vitro binding studies of drugs to hair: influence of melanin and lipids on cocaine binding to Caucasoid and Africoid hair. J Anal Toxicol 1996;20:338-44.
398. Joynt BP. Triazolam blood concentrations in forensic cases in Canada. J Anal Toxicol 1993;17:171-7.
399. Jufer RA, Walsh SL, Cone EJ. Cocaine and metabolite concentrations in plasma during repeated oral administration: development of a human laboratory model of chronic cocaine use. J Anal Toxicol 1998; 22:435-44.
354. Inaba T, Stewart DJ, Kalow W. Metabolism of cocaine in man. Clin Pharmacol Ther 1978;23:547-52.
355. Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenom J 2005;5:6-13.
356. Inoue T, Seta S, Goldberger BA. Analysis of drugs in unconventional samples. In: Liu R, Goldberger B, eds. Handbook of workplace drug testing. Washington, DC: AACC Press, 1995:131-58.
357. Inturrisi CE, Max MB, Foley KM, Schultz M, Shin SU, Houde RW. The pharmacokinetics of heroin in patients with chronic pain. N Engl J Med 1984;310:1213-7.
358. Ioannides C, Chakraborty J, Parke D. Improvement of barbiturate gas chromatography by formic acid: possible mechanism. Chromatographia 1974;7:351-6.
359. Isbister G, Downes F, Sibbritt D, Dawson A, Whyte I. Aspiration pneumonitis in an overdose population: frequency, predictors, and outcomes. Crit Care Med 2004;32:88-93.
360. Isbister GK, Buckley NA, Whyte IM. Serotonin toxicity: a practical approach to diagnosis and treatment. Med J Aust 2007;187:361-5.
361. Isenschmid DS. Cocaine: effects on human performance and behavior. Forensic Sci Rev 2002;14:61-100.
362. Isenschmid DS. Cocaine. In: Levine B, ed. Principles of forensic toxicology, 2nd edition. Washington, DC: AACC Press, 2003:207-28.
363. Isenschmid DS, Levine BS, Caplan YH. A method for the simultaneous determination of cocaine, benzoylecgonine, and ecgonine methyl ester in blood and urine using GC/EIMS with derivatization to produce high mass molecular ions. J Anal Toxicol 1988;12:242-5.
364. Ishihara K, Szerlip HM. Anion gap acidosis. Semin Nephrol 1998;18: 83-97.
365. Iversen L. Cannabis and the brain. Brain 2003;126:1252-70.366. Iwai M, Hattori H, Arinobu T, Ishii A, Kumazawa T, Noguchi H, et al.
Simultaneous determination of barbiturates in human biological fluids by direct immersion solid-phase microextraction and gas chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2004;806:65-73.
367. Iwersen-Bergmann S, Schmoldt A. Direct semiquantitative screening of drugs of abuse in serum and whole blood by means of CEDIA DAU urine immunoassays. J Anal Toxicol 1999;23:247-56.
368. Jackson DL, Menges H. Accidental carbon monoxide poisoning. JAMA 1980;243:772-4.
369. Jacob P 3rd, Jones RT, Benowitz NL. Formation and elimination kinetics of cocaethylene in humans. Clin Pharmacol Ther 1993;53:174.
370. Jacob P 3rd, Lewis ER, Elias-Baker BA, Jones RT. A pyrolysis product, anhydroecgonine methyl ester (methylecgonidine), is in the urine of cocaine smokers. J Anal Toxicol 1990;14:353-7.
371. Jacobsen D, Ovrebo S, Ostborg J, Sejersted OM. Glycolate causes the acidosis in ethylene glycol poisoning and is effectively removed by hemodialysis. Acta Med Scand 1984;216:409-16.
372. Jaehne EJ, Salem A, Irvine RJ. Pharmacological and behavioral determinants of cocaine, methamphetamine, 3,4-methylenedioxymethamphetamine, and para-methoxyamphetamine-induced hyperthermia. Psychopharmacology 2007;194:41-52.
373. Jain NC, Chinn DM, Budd RD, Sneath TS, Leung WJ. Simultaneous determination of cocaine and benzoyl ecgonine in urine by gas chromatography with on-column alkylation. J Forensic Sci 1977;22:7-16.
374. James RA, Dinan A. Hyperpyrexia associated with fatal paramethoxyamphetamine (PMA) abuse. Med Sci Law 1998;38:83-5.
375. Jarvie DR, Heyworth R, Simpson D. Plasma salicylate analysis: a comparison of colorimetric, HPLC and enzymatic techniques. Ann Clin Biochem 1987;24(Pt 4):364-73.
376. Jatlow P. Cocaethylene: pharmacologic activity and clinical significance. Ther Drug Monit 1993;15:533-6.
377. Jatlow P, Elsworth JD, Bradberry CW, Winger G, Taylor JR, Russell R, et al. Cocaethylene: a neuropharmacologically active metabolite associated with concurrent cocaine-ethanol ingestion. Life Sci 1991; 48:1787-94.

Chapter35 ■ Clinical Toxicology 1177
422. Kintz P. Drug testing in hair. Boca Raton, Fla: CRC Press, 1996.423. Kintz P. Hair testing and doping control in sport. Toxicol Lett 1998;
102:109-13.424. Kintz P. Value of hair analysis in postmortem toxicology. Forensic Sci
Int 2004;142:127-34.425. Kintz P, Brenneisen R, Bundeli P, Mangin P. Sweat testing for heroin
and metabolites in a heroin maintenance program. Clin Chem 1997; 43:736-9.
426. Kintz P, Samyn N. Determination of “Ecstasy” components in alternative biological specimens. J Chromatogr 1999;733:137-43.
427. Kintz P, Samyn N. Use of alternative specimens: drugs of abuse in saliva and doping agents in hair. Ther Drug Monit 2002;24:239-46.
428. Kintz P, Tracqui A, Jamey C, Mangin P. Detection of codeine and phenobarbital in sweat collected with a sweat patch. J Anal Toxicol 1996;20:197-201.
429. Kintz P, Villain M, Dumestre-Toulet V, Ludes B. Drug-facilitated sexual assault and analytical toxicology: the role of LC-MS/MS. A case involving zolpidem. J Clin Forensic Med 2005;12:36-41.
430. Kintz P, Villain M, Ludes B. Testing for the undetectable in drug-facilitated sexual assault using hair analyzed by tandem mass spectrometry as evidence. Ther Drug Monit 2004;26:211-4.
431. Kintz P, Villain M, Ludes B. Testing for zolpidem in oral fluid by liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2004;811:59-63.
432. Kirages TJ, Sule HP, Mycyk MB. Severe manifestations of coricidin intoxication. Am J Emerg Med 2003;21:473-5.
433. Kleinschnitz M, Herderich M, Schreier P. Determination of 1,4-benzodiazepines by high-performance liquid chromatography-electrospray tandem mass spectrometry. J Chromatogr B Biomed Appl 1996;676:61-7.
434. Klette KL, Horn CK, Stout PR, Anderson CJ. LC-MS analysis of human urine specimens for 2-oxo-3-hydroxy LSD: method validation for potential interferants and stability study of 2-oxo-3-hydroxy LSD under various storage conditions. J Anal Toxicol 2002;26:193-200.
435. Klette KL, Poch GK, Czarny R, Lau CO. Simultaneous GC-MS analysis of meta- and para-hydroxybenzoylecgonine and norbenzoylecgonine: a secondary method to corroborate cocaine ingestion using nonhydrolytic metabolites. J Anal Toxicol 2000;24:482-8.
436. Klingmann A, Skopp G, Aderjan R. Analysis of cocaine, benzoylecgonine, ecogonine methyl ester, and ecgonine by high-pressure liquid chromatography-API mass spectrometry and application to a short-term degradation study of cocaine in plasma. J Anal Toxicol 2001;25:425-30.
437. Kloss MW, Rosen GM, Rauckman EJ. N-demethylation of cocaine to norcocaine: evidence for participation by cytochrome P-450 and FAD-containing monooxygenase. Mol Pharmacol 1983;23:482-5.
438. Kloss MW, Rosen GM, Rauckman EJ. Cocaine-mediated hepatotoxicity: a critical review. Biochem Pharmacol 1984;33:169-73.
439. Koek W, Carter LP, Lamb RJ, Chen W, Wu H, Coop A, France CP. Discriminative stimulus effects of gamma-hydroxybutyrate (GHB) in rats discriminating GHB from baclofen and diazepam. J Pharmacol Exp Ther 2005;314:170-9.
440. Kogan MJ, Verebey KG, DePace AC, Resnick RB, Mule SJ. Quantitative determination of benzoylecgonine and cocaine in human biofluids by gas-liquid chromatography. Anal Chem 1977;49:1965-9.
441. Kolbrich EA, Barnes AJ, Gorelick DA, Boyd SJ, Cone EJ, Huestis MA. Major and minor metabolites of cocaine in human plasma following controlled subcutaneous cocaine administration. J Anal Toxicol 2006; 30:501-10.
442. Kraemer T, Maurer HH. Toxicokinetics of amphetamines: metabolism and toxicokinetic data of designer drugs, amphetamine, methamphetamine, and their N-alkyl derivatives. Ther Drug Monit 2002;24:277-89.
443. Krahn J, Khajuria A. Osmolality gaps: diagnostic accuracy and long-term variability. Clin Chem 2006;52:737-9.
444. Kraner JC, McCoy DJ, Evans MA, Evans LE, Sweeney BJ. Fatalities caused by the MDMA-related drug paramethoxyamphetamine (PMA). J Anal Toxicol 2001;25:645-8.
400. Kadehjian L. Legal issues in oral fluid testing. Forensic Sci Int 2005; 150:151-60.
401. Kadiev E, Patel V, Rad P, Thankachan L, Tram A, Weinlein M, et al. Role of pharmacogenetics in variable response to drugs: focus on opioids. Exp Opin Drug Metab Toxicol 2008;4:77-91.
402. Kalant H. The pharmacology and toxicology of “ecstasy” (MDMA) and related drugs. CMAJ 2001;165:917-28.
403. Kalix P. The pharmacology of psychoactive alkaloids from ephedra and catha. J Ethnopharmacol 1991;32:201-8.
404. Kaminskas LM, Irvine RJ, Callaghan PD, White JM, Kirkbride P. The contribution of the metabolite p-hydroxyamphetamine to the central actions of p-methoxyamphetamine. Psychopharmacology 2002;160: 155-60.
405. Kapur BM, Vandenbroucke A, Cogionis B, Losner K. Rapid analysis of toxic alcohols. Clin Chem 2005;51:A155.
406. Karnes HT, Beightol LA. Evaluation of fluorescence polarization immunoassay for quantitation of serum salicylates. Ther Drug Monit 1985;7:351-4.
407. Katagi M, Nishikawa M, Tatsuno M, Miki A, Tsuchihashi H. Column-switching high-performance liquid chromatography-electrospray ionization mass spectrometry for identification of heroin metabolites in human urine. J Chromatogr 2001;751:177-85.
408. Kato K, Hillsgrove M, Weinhold L, Gorelick DA, Darwin WD, Cone EJ. Cocaine and metabolite excretion in saliva under stimulated and nonstimulated conditions. J Anal Toxicol 1993;17:338-41.
409. Katz KD, Curry SC, Brooks DE, Gerkin RD. The effect of cyclosporine A on survival time in salicylate-poisoned rats. J Emerg Med 2004;26: 151-5.
410. Kaupmann K, Cryan JF, Wellendorph P, Mombereau C, Sansig G, Klebs K, et al. Specific gamma-hydroxybutyrate-binding sites but loss of pharmacological effects of gamma-hydroxybutyrate in GABA(B)(1)-deficient mice. Eur J Neurosci 2003;18:2722-30.
411. Kellermann AL, Fihn SD, LoGerfo JP, Copass MK. Impact of drug screening in suspected overdose. Ann Emerg Med 1987;16:1206-16.
412. Kelner MJ, Bailey DN. Mismeasurement of methemoglobin (“methemoglobin revisited”). Clin Chem 1985;31:168-9.
413. Kepczynska E, Bojarski J, Haber P, Kaliszan R. Retention of barbituric acid derivatives on immobilized artificial membrane stationary phase and its correlation with biological activity. Biomed Chromatogr 2000;14:256-60.
414. Kerrigan S, Goldberger BA. Opioids. In: Levine B, ed. Principles of forensic toxicology, 2nd edition. Washington, DC: AACC Press, 2003:187-205.
415. Keyes DC, Dart RC. Initial diagnosis and treatment of the poisoned patient. In: Dart RC, ed. Medical toxicology. Philadelphia, Pa: Lippincott Williams & Wilkins, 2004:21-31.
416. Kidwell DA, Blank D. Hair analysis: techniques and potential problems. In: Sunshine I, ed. Recent developments in therapeutic drug monitoring and clinical toxicology. New York, NY: Marcel Dekker, 1992:555-63.
417. Kidwell DA, Holland JC, Athanaselis S. Testing for drugs of abuse in saliva and sweat. J Chromatogr 1998;713:111-35.
418. Kikura-Hanajiri R, Hayashi M, Saisho K, Goda Y. Simultaneous determination of nineteen hallucinogenic tryptamines/beta-calbolines and phenethylamines using gas chromatography-mass spectrometry and liquid chromatography-electrospray ionisation-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2005;825:29-37.
419. Killmann SA, Thaysen JH. The permeability of the human parotid gland to a series of sulfonamide compounds, para-aminohippurate and inulin. Scand J Clin Lab Invest 1955;7:86-91.
420. Kim EM, Lee JS, Choi SK, Lim MA, Chung HS. Analysis of ketamine and norketamine in urine by automatic solid-phase extraction (SPE) and positive ion chemical ionization-gas chromatography-mass spectrometry (PCI-GC-MS). Forensic Sci Int 2008;174:197-202.
421. King JA, Storrow AB, Finkelstein JA. Urine Trinder spot test: a rapid salicylate screen for the emergency department. Ann Emerg Med 1995;26:330-3.

1178 SectionIII ■ Analytes
467. Ledent C, Valverde O, Cossu G, Petitet F, Aubert JF, Beslot F, et al. Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science 1999;283:401-4.
468. Lee HM, Lee CW. Determination of morphine and codeine in blood and bile by gas chromatography with a derivatization procedure. J Anal Toxicol 1991;15:182-7.
469. LeGatt DF, Audette RJ, Blakney G, Vaughan D. Excess serum osmolality after ingestion of methanol: the exception, not the rule. Clin Chem 1991;37:1802-4.
470. Leikin J, Paloucek F. Methemoglobin, blood. In: Leikin J, Paloucek F, eds. Leikin & Paloucek’s poisoning & toxicology handbook, 4th edition. Hudson, Ohio: Lexi-Comp, 2008:1047.
471. Leipzig RM, Mendelowitz A. Adverse psychotropic drug interactions. In: Kane JM, Lieberman JA, eds. Adverse effects of psychotropic drugs. New York, NY: Guilford Press, 1992:13-76.
472. Lekskulchai V, Mokkhavesa C. Evaluation of Roche Abuscreen ONLINE amphetamine immunoassay for screening of new amphetamine analogues. J Anal Toxicol 2001;25:471-5.
473. Leson G, Pless P, Grotenhermen F, Kalant H, ElSohly MA. Evaluating the impact of hemp food consumption on workplace drug tests. J Anal Toxicol 2001;25:691-8.
474. Lester BM, ElSohly M, Wright LL, Smeriglio VL, Verter J, Bauer CR, et al. The maternal lifestyle study: drug use by meconium toxicology and maternal self-report. Pediatrics 2001;107:309-17.
475. Levine B. Central nervous system depressants. In: Levine B, ed. Principles of forensic toxicology, 2nd edition. Washington, DC: AACC Press, 2003:157-72.
476. Levine BS, Smith ML. Effects of diphenhydramine on immunoassays of phencyclidine in urine. Clin Chem 1990;36:1258.
477. Lewis DE, Moore CM, Leikin JB, Koller A. Meconium analysis for cocaine: a validation study and comparison with paired urine analysis. J Anal Toxicol 1995;19:148-50.
478. Li Z, McNally AJ, Wang H, Salamone SJ. Stability study of LSD under various storage conditions. J Anal Toxicol 1998;22:520-5.
479. Liebelt EL, Francis PD. Cyclic antidepressants. In: Goldfrank LR, Flomenbaum NE, Lewin NA, Howland MA, Hoffman RS, Nelson LS, eds. Goldfrank’s toxicologic emergencies, 7th edition. New York, NY: McGraw-Hill, 2002:847-64.
480. Lin JH, Levy G. Sulfate depletion after acetaminophen administration and replenishment by infusion of sodium sulfate or N-acetylcysteine in rats. Biochem Pharmacol 1981;30:2723-5.
481. Lin LY, Di Stefano EW, Schmitz DA, Hsu L, Ellis SW, Lennard MS, et al. Oxidation of methamphetamine and methylenedioxy-methamphetamine by CYP2D6. Drug Metab Dispos 1997;25: 1059-64.
482. Lin TJ, Nelson LS, Tsai JL, Hung DZ, Hu SC, Chan HM, et al. Common toxidromes of plant poisonings in Taiwan. Clin Toxicol 2009;47:161-8.
483. Lindberg L, Brauer S, Wollmer P, Goldberg L, Jones AW, Olsson SG. Breath alcohol concentration determined with a new analyzer using free exhalation predicts almost precisely the arterial blood alcohol concentration. Forensic Sci Int 2007;168:200-7.
484. Lippi G, Banfi G. Blood transfusions in athletes: old dogmas, new tricks. Clin Chem Lab Med 2006;44:1395-402.
485. Liu RH. Evaluation of common immunoassay kits for effective workplace drug testing. In: Liu R, Goldberger B, eds. Handbook of workplace drug testing. Washington, DC: AACC Press, 1995:67-129.
486. Liu YL, Toubro S, Astrup A, Stock MJ. Contribution of beta 3-adrenoceptor activation to ephedrine-induced thermogenesis in humans. Int J Obes Relat Metab Disord 1995;19:678-85.
487. Logan BK. Methamphetamine: effects on human performance and behavior. Forensic Sci Rev 2002;14:133-51.
488. Logan BK, Cooper FA. 3,4-Methylenedioxymethamphetamine: effects on human performance and behavior. Forensic Sci Rev 2003;15:11-28.
489. Logan BK, Gullberg RG. Alcohol, drugs and driving. In: Moffat AC, Osselton MD, Widdop B, eds. Clarke’s analysis of drugs and poisons, 3rd ed. Grayslake, Ill: Pharmaceutical Press, 2004:53-67.
445. Kranzler HR, Stone J, McLaughlin L. Evaluation of a point-of-care testing product for drugs of abuse: testing site is a key variable. Drug Alcohol Depend 1995;40:55-62.
446. Kratzsch C, Peters FT, Kraemer T, Weber AA, Maurer HH. Screening, library-assisted identification and validated quantification of fifteen neuroleptics and three of their metabolites in plasma by liquid chromatography/mass spectrometry with atmospheric pressure chemical ionization. J Mass Spectrom 2003;38:283-95.
447. Krenzelok EP, Kerr F, Proudfoot AT. Salicylate toxicity. In: Haddad LM, Shannon MW, Winchester JD, eds. Clinical management of poisoning and drug overdose, 3rd ed. Philadelphia, Pa: WB Saunders, 1998:675-87.
448. Kreuz DS, Axelrod J. Delta-9-tetrahydrocannabinol: localization in body fat. Science 1973;179:391-3.
449. Kronstrand R, Forstberg-Peterson S, Kagedal B, Ahlner J, Larson G. Codeine concentration in hair after oral administration is dependent on melanin content. Clin Chem 1999;45:1485-94.
450. Kronstrand R, Nystrom I, Josefsson M, Hodgins S. Segmental ion spray LC-MS-MS analysis of benzodiazepines in hair of psychiatric patients. J Anal Toxicol 2002;26:479-84.
451. Kronstrand R, Nystrom I, Strandberg J, Druid H. Screening for drugs of abuse in hair with ion spray LC-MS-MS. Forensic Sci Int 2004;145: 183-90.
452. Kumar S, Fleming RL, Morrow AL. Ethanol regulation of gamma-aminobutyric acid A receptors: genomic and nongenomic mechanisms. Pharmacol Ther 2004;101:211-26.
453. Kwong TC, Chamberlain RT, Frederick DL, Kapur BM, Sunshine I. Critical issues in urinalysis of abused substances: report of the Substance-Abuse Testing Committee. Review. Clin Chem 1988;34: 605-32.
454. Kwong TC, Ryan RM. Detection of intrauterine illicit drug exposure by newborn drug testing. National Academy of Clinical Biochemistry. Clin Chem 1997;43:235-42.
455. Kwong TC, Shearer D. Detection of drug use during pregnancy. Obstet Gynecol Clin North Am 1998;25:43-64.
456. Lacy TL, Nichols JH. Therapeutic drugs III: neuroleptic (antipsychotic) drugs. In: Levine B, ed. Principles of forensic toxicology, 2nd edition. Washington, DC: AACC Press, 2003:315-25.
457. Lafolie P, Beck O, Blennow G, Boreus L, Borg S, Elwin CE, et al. Importance of creatinine analyses of urine when screening for abused drugs. Clin Chem 1991;37:1927-31.
458. Landaw SA, Winchell HS. Endogenous production of carbon-14 labeled carbon monoxide: an in vivo technique for the study of heme catabolism. J Nucl Med 1966;7:696-707.
459. Langman LJ. The use of oral fluid for therapeutic drug management: clinical and forensic toxicology. Ann N Y Acad Sci 2007;1098: 145-66.
460. Langman LJ, Bjergum MW, Williamson CL, Crow FW. Sensitive method for detection of cocaine and associated analytes by liquid chromatography: tandem mass spectrometry in urine. J Anal Toxicol 2009;33:447-455.
461. Large CH. Do NMDA receptor antagonist models of schizophrenia predict the clinical efficacy of antipsychotic drugs? J Psychopharmacol 2007;21:283-301.
462. Latta KS, Ginsberg B, Barkin RL. Meperidine: a critical review. Am J Ther 2002;9:53-68.
463. Law B, Mason PA, Moffat AC, King LJ, Marks V. Passive inhalation of cannabis smoke. J Pharm Pharmacol 1984;36:578-81.
464. Law MY, Slawson MH, Moody DE. Selective involvement of cytochrome P450 2D subfamily in in vivo 4-hydroxylation of amphetamine in rat. Drug Metab Dispos 2000;28:348-53.
465. Leatherman JW, Schmitz PG. Fever, hyperdynamic shock, and multiple-system organ failure: a pseudo-sepsis syndrome associated with chronic salicylate intoxication. Chest 1991;100:1391-6.
466. LeBeau MA, Montgomery MA, Miller ML, Burmeister SG. Analysis of biofluids for gamma-hydroxybutyrate (GHB) and gamma-butyrolactone (GBL) by headspace GC-FID and GC-MS. J Anal Toxicol 2000;24:421-8.

Chapter35 ■ Clinical Toxicology 1179
513. Martin-Amat G, McMartin KE, Hayreh SS, Hayreh MS, Tephly TR. Methanol poisoning: ocular toxicity produced by formate. Toxicol Appl Pharmacol 1978;45:201-8.
514. Marwick C. Coma-inducing drug GHB may be reclassified. JAMA 1997;277:1505-6.
515. Mason HJ, Lewis PJ. Intra-individual variation in plasma and erythrocyte cholinesterase activities and the monitoring of uptake of organo-phosphate pesticides. J Soc Occup Med 1989;39:121-4.
516. Mason MF, Dubowski KM. Breath as a specimen for analysis for ethanol and other low molecular weight alcohols. In: Garriott JC, ed. Medicolegal aspects of alcohol. Tucson, Ariz: Lawyers & Judges Publishing, 1996:171-80.
517. Masoud AN, Krupski DM. High-performance liquid chromatographic analysis of cocaine in human plasma. J Anal Toxicol 1980;4:305-10.
518. Mastrovitch TA, Bithoney WG, DeBari VA, Nina AG. Point-of-care testing for drugs of abuse in an urban emergency department. Ann Clin Lab Sci 2002;32:383-6.
519. Masumoto K, Tashiro Y, Matsumoto K, Yoshida A, Hirayama M, Hayashi S. Simultaneous determination of codeine and chlorpheniramine in human plasma by capillary column gas chromatography. J Chromatogr 1986;381:323-9.
520. Mattila MA, Larni HM. Flunitrazepam: a review of its pharmacological properties and therapeutic use. Drugs 1980;20:353-74.
521. Mattila MA, Saila K, Kokko T, Karkkainen T. Comparison of diazepam and flunitrazepam as adjuncts to general anaesthesia in preventing arousal following surgical stimuli. Br J Anaesth 1979;51: 329-37.
522. Maurer HH. Systematic toxicological analysis of drugs and their metabolites by gas chromatography-mass spectrometry. J Chromatogr 1992;580:3-41.
523. Maurer HH. Current role of liquid chromatography-mass spectrometry in clinical and forensic toxicology. Anal Bioanal Chem 2007;388:1315-25.
524. Maurer HH, Bickeboeller-Friedrich J, Kraemer T, Peters FT. Toxicokinetics and analytical toxicology of amphetamine-derived designer drugs (‘Ecstasy’). Toxicol Lett 2000;112-113:133-42.
525. Maurer HH, Kraemer T, Springer D, Staack RF. Chemistry, pharmacology, toxicology, and hepatic metabolism of designer drugs of the amphetamine (ecstasy), piperazine, and pyrrolidinophenone types: a synopsis. Ther Drug Monit 2004;26:127-31.
526. Mayer M, Salpeter L. More on interference of N-acetylcysteine in measurement of acetaminophen. Clin Chem 1998;44:892-3.
527. Maynard EC, Amoruso LP, Oh W. Meconium for drug testing. Am J Dis Child 1991;145:650-2.
528. McCance-Katz EF, Price LH, McDougle CJ, Kosten TR, Black JE, Jatlow PI. Concurrent cocaine-ethanol ingestion in humans: pharmacology, physiology, behavior, and the role of cocaethylene. Psychopharmacology 1993;111:39-46.
529. McCann UD, Szabo Z, Scheffel U, Dannals RF, Ricaurte GA. Positron emission tomographic evidence of toxic effect of MDMA (“Ecstasy”) on brain serotonin neurons in human beings. Lancet 1998;352:1433-7.
530. McClean S, O’Kane E, Hillis J, Smyth WF. Determination of 1,4-benzodiazepines and their metabolites by capillary electrophoresis and high-performance liquid chromatography using ultraviolet and electrospray ionisation mass spectrometry. J Chromatogr A 1999;838: 273-91.
531. McCusker RR, Paget-Wilkes H, Chronister CW, Goldberger BA. Analysis of gamma-hydroxybutyrate (GHB) in urine by gas chromatography-mass spectrometry. J Anal Toxicol 1999;23:301-5.
532. McGuigan M. Cannabinoids. In: Goldfrank LR, Flomenbaum NE, Hoffman RS, Howland MA, Lewin NA, Nelson LS, eds. Goldfrank’s toxicologic emergencies, 8th edition. New York, NY: McGraw-Hill, 2006:1212-30.
533. McGuigan MA. A two-year review of salicylate deaths in Ontario. Arch Intern Med 1987;147:510-2.
534. McMartin KE, Ambre JJ, Tephly TR. Methanol poisoning in human subjects: role for formic acid accumulation in the metabolic acidosis. Am J Med 1980;68:414-8.
490. Long C, Crifasi J, Maginn D. Interference of thioridazine (Mellaril) in identification of phencyclidine. Clin Chem 1996;42:1885-6.
491. Lora-Tamayo C, Tena T, Tena G. Concentrations of free and conjugated morphine in blood in twenty cases of heroin-related deaths. J Chromatogr 1987;422:267-73.
492. Lotsch J. Opioid metabolites. J Pain Symptom Manage 2005;29:S10-24.493. Louon A, Lithander J, Reddy VG, Gupta A. Sedation with nasal
ketamine and midazolam for cryotherapy in retinopathy of prematurity. Br J Ophthalmol 1993;77:529-30.
494. LoVecchio F, Lewin LA. Antipsychotics. In: Goldfrank LR, Flomenbaum NE, Lewin NA, Howland MA, Hoffman RS, Nelson LS, eds. Goldfrank’s toxicologic emergencies, 7th edition. New York, NY: McGraw-Hill, 2002:875-84.
495. Lukaszewski T, Jeffery W. Impurities and artifacts of illicit cocaine. J Forensic Sci 1980;25:499-508.
496. Lundh B, Cavallin-Stahl E, Mercke C. Heme catabolism, carbon monoxide production and red cell survival in anemia. Acta Med Scand 1975;197:161-71.
497. Lüscher C. Drugs of abuse (Chapter 32). In: Katzung BG, ed. Basic & clinical pharmacology, 11th edition. New York, NY: McGraw-Hill, 2009.
498. Macheras P, Rosen A. Is monitoring of drug in saliva reliable for bioavailability testing of a protein-bound drug? A theoretical approach. Pharm Acta Helv 1984;59:34-6.
499. Mackler SA, Schweizer E. Benzodiazepines as anxiolytic agents: the risks of long-term treatment. Hosp Pract 1992;27:109-12, 15-6.
500. Maier RD, Bogusz M. Identification power of a standardized HPLC-DAD system for systematic toxicological analysis. J Anal Toxicol 1995;19:79-83.
501. Maisonneuve IM, Ho A, Kreek MJ. Chronic administration of a cocaine “binge” alters basal extracellular levels in male rats: an in vivo microdialysis study. J Pharmacol Exp Ther 1995;272:652-7.
502. Maisonneuve IM, Kreek MJ. Acute tolerance to the dopamine response induced by a binge pattern of cocaine administration in male rats: an in vivo microdialysis study. J Pharmacol Exp Ther 1994;268:916-21.
503. Maitre M. The gamma-hydroxybutyrate signalling system in brain: organization and functional implications. Prog Neurobiol 1997;51: 337-61.
504. Mancini I, Lossignol DA, Body JJ. Opioid switch to oral methadone in cancer pain. Curr Opin Oncol 2000;12:308-13.
505. Manno J. Interpretation of urinalysis results. In: Hawks R, Chiang C, eds. Urine testing for drugs of abuse. National Institute on Drug Abuse, Research Monograph 73. Publication (ADM) 87-1481. Washington, DC: U.S. Department of Health and Human Services, 1986:65-71.
506. Mao Y, Carr PW. Separation of barbiturates and phenylthiohydantoin amino acids using the thermally tuned tandem column concept. Anal Chem 2001;73:1821-30.
507. Marc B, Baudry F, Vaquero P, Zerrouki L, Hassnaoui S, Douceron H. Sexual assault under benzodiazepine submission in a Paris suburb. Arch Gynecol Obstet 2000;263:193-7.
508. Marchei E, Farre M, Pellegrini M, Rossi S, Garcia-Algar O, Vall O, et al. Liquid chromatography-electrospray ionization mass spectrometry determination of methylphenidate and ritalinic acid in conventional and non-conventional biological matrices. J Pharmaceut Biomed Anal 2009;49:434-9.
509. Maresova V, Hampl J, Chundela Z, Zrcek F, Polasek M, Chadt J. The identification of a chlorinated MDMA. J Anal Toxicol 2005;29:353-8.
510. Markowitz JS, Patrick KS. Differential pharmacokinetics and pharmacodynamics of methylphenidate enantiomers: does chirality matter? J Clin Psychopharmacol 2008;28:S54-61.
511. Marquet P. Is LC-MS suitable for a comprehensive screening of drugs and poisons in clinical toxicology? Ther Drug Monit 2002;24:125-33.
512. Marshall LF, Smith RW, Shapiro HM. The outcome with aggressive treatment in severe head injuries. Part II: acute and chronic barbiturate administration in the management of head injury. J Neurosurg 1979;50:26-30.

1180 SectionIII ■ Analytes
557. Moore KA, Sklerov J, Levine B, Jacobs AJ. Urine concentrations of ketamine and norketamine following illegal consumption. J Anal Toxicol 2001;25:583-8.
558. Moore L, Wicks J, Spiehler V, Holgate R. Gas chromatography-mass spectrometry confirmation of Cozart RapiScan saliva methadone and opiates tests. J Anal Toxicol 2001;25:520-4.
559. Morland J, Bugge A, Skuterud B, Steen A, Wethe GH, Kjeldsen T. Cannabinoids in blood and urine after passive inhalation of Cannabis smoke. J Forensic Sci 1985;30:997-1002.
560. Morris HC, Overton PD, Ramsay JR, Campbell RS, Hammond PM, Atkinson T, et al. Development and validation of an automated enzyme assay for paracetamol (acetaminophen). Clin Chim Acta 1990;187:95-104.
561. Morris HC, Overton PD, Ramsay JR, Campbell RS, Hammond PM, Atkinson T, et al. Development and validation of an automated, enzyme-mediated colorimetric assay of salicylate in serum. Clin Chem 1990;36:131-5.
562. Mortier KA, Maudens KE, Lambert WE, Clauwaert KM, Van Bocxlaer JF, Deforce DL, et al. Simultaneous, quantitative determination of opiates, amphetamines, cocaine and benzoylecgonine in oral fluid by liquid chromatography quadrupole-time-of-flight mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2002;779:321-30.
563. Mueller PD, Korey WS. Death by “ecstasy”: the serotonin syndrome? Ann Emerg Med 1998;32:377-80.
564. Mule SJ, Casella GA. Rendering the “poppy-seed defense” defenseless: identification of 6-monoacetylmorphine in urine by gas chromatography/mass spectroscopy. Clin Chem 1988;34:1427-30.
565. Mule SJ, Lomax P, Gross SJ. Active and realistic passive marijuana exposure tested by three immunoassays and GC/MS in urine. J Anal Toxicol 1988;12:113-6.
566. Munro N. Toxicity of the organophosphate chemical warfare agents GA, GB, and VX: implications for public protection. Environ Health Perspect 1994;102:18-38.
567. Musshoff F, Madea B. Review of biologic matrices (urine, blood, hair) as indicators of recent or ongoing cannabis use. Ther Drug Monit 2006;28:155-63.
568. Musshoff F, Trafkowski J, Kuepper U, Madea B. An automated and fully validated LC-MS/MS procedure for the simultaneous determination of 11 opioids used in palliative care, with 5 of their metabolites. J Mass Spectrom 2006;41:633-40.
569. Myers RA, Britten JS. Are arterial blood gases of value in treatment decisions for carbon monoxide poisoning? Crit Care Med 1989;17: 139-42.
570. Nahas G, Leger C, Tocque B, Hoellinger H. The kinetics of cannabinoid distribution and storage with special reference to the brain and testis. J Clin Pharmacol 1981;21:208S-14S.
571. Naidong W, Lee JW, Jiang X, Wehling M, Hulse JD, Lin PP. Simultaneous assay of morphine, morphine-3-glucuronide and morphine-6-glucuronide in human plasma using normal-phase liquid chromatography-tandem mass spectrometry with a silica column and an aqueous organic mobile phase. J Chromatogr 1999;735:255-69.
572. Nair P, Rothblum S, Hebel R. Neonatal outcome in infants with evidence of fetal exposure to opiates, cocaine, and cannabinoids. Clin Pediatr 1994;33:280-5.
573. Nakahara Y. Hair analysis for abused and therapeutic drugs. J Chromatogr 1999;733:161-80.
574. Namera A, Yashiki M, Iwasaki Y, Ohtani M, Kojima T. Automated procedure for determination of barbiturates in serum using the combined system of PrepStation and gas chromatography-mass spectrometry. J Chromatogr B Biomed Appl 1998;716:171-6.
575. Namera A, Yashiki M, Okada K, Iwasaki Y, Ohtani M, Kojima T. Automated preparation and analysis of barbiturates in human urine using the combined system of PrepStation and gas chromatography-mass spectrometry. J Chromatogr 1998;706:253-9.
576. Narahashi T, Aistrup GL, Marszalec W, Nagata K. Neuronal nicotinic acetylcholine receptors: a new target site of ethanol. Neurochem Int 1999;35:131-41.
535. McQueen MJ. Clinical and analytical considerations in the utilization of cholinesterase measurements. Clin Chim Acta 1995;237:91-105.
536. McQuillen KK, Anderson AC. Osmol gaps in the pediatric population. Acad Emerg Med 1999;6:27-30.
537. Meatherall R. GC/MS confirmation of barbiturates in blood and urine. J Forensic Sci 1997;42:1160-70.
538. Meatherall R, Dai J. False-positive EMIT II opiates from ofloxacin. Ther Drug Monit 1997;19:98-9.
539. Meatherall R, Krahn J. Excess serum osmolality gap after ingestion of methanol. Clin Chem 1990;36:2004-7.
540. Meng QH, Adeli K, Zello GA, Porter WH, Krahn J. Elevated lactate in ethylene glycol poisoning: true or false? Clin Chim Acta 2010;411: 601-4.
541. Meyer MR, Maurer HH. Enantioselectivity in the methylation of the catecholic phase I metabolites of methylenedioxy designer drugs and their capability to inhibit catechol-O-methyltransferase-catalyzed dopamine 3-methylation. Chem Res Toxicol 2009;22:1205-11.
542. Meyer MR, Peters FT, Maurer HH. The role of human hepatic cytochrome P450 isozymes in the metabolism of racemic 3,4-methylenedioxy-methamphetamine and its enantiomers. Drug Metab Dispos 2008;36:2345-54.
543. Miller RL, DeVane CL. Determination of cocaine, benzoylecgonine and ecgonine methyl ester in plasma by reversed-phase high-performance liquid chromatography. J Chromatogr 1991;570:412-8.
544. Misra AL, Pontani RB, Bartolomeo J. Persistence of phencyclidine (PCP) and metabolites in brain and adipose tissue and implications for long-lasting behavioural effects. Res Commun Chem Pathol Pharmacol 1979;24:431-45.
545. Mitchell JR, Thorgeirsson SS, Potter WZ, Jollow DJ, Keiser H. Acetaminophen-induced hepatic injury: protective role of glutathione in man and rationale for therapy. Clin Pharmacol Ther 1974;16: 676-84.
546. Miyaguchi H, Kuwayama K, Tsujikawa K, Kanamori T, Iwata YT, Inoue H, Kishi T. A method for screening for various sedative-hypnotics in serum by liquid chromatography/single quadrupole mass spectrometry. Forensic Sci Int 2006;157:57-70.
547. Moeller KE, Lee KC, Kissack JC. Urine drug screening: practical guide for clinicians. Mayo Clin Proc 2008;83:66-76.
548. Moeller MR, Doerr G, Warth S. Simultaneous quantitation of delta-9-tetrahydrocannabinol (THC) and 11-nor-9-carboxy-delta-9-tetrahydrocannabinol (THC-COOH) in serum by GC/MS using deuterated internal standards and its application to a smoking study and forensic cases. J Forensic Sci 1992;37:969-83.
549. Moeller MR, Fey P, Sachs H. Hair analysis as evidence in forensic cases. Forensic Sci Int 1993;63:43-53.
550. Mollereau C, Parmentier M, Mailleux P, Butour JL, Moisand C, Chalon P, et al. ORL1, a novel member of the opioid receptor family: cloning, functional expression and localization. FEBS Lett 1994;341: 33-8.
551. Montagna M, Stramesi C, Vignali C, Groppi A, Polettini A. Simultaneous hair testing for opiates, cocaine, and metabolites by GC-MS: a survey of applicants for driving licenses with a history of drug use. Forensic Sci Int 2000;107:157-67.
552. Montague RE, Grace RF, Lewis JH, Shenfield GM. Urine drug screens in overdose patients do not contribute to immediate clinical management. Ther Drug Monit 2001;23:47-50.
553. Montgomery MR, Reasor MJ. Retrograde extrapolation of blood alcohol data: an applied approach. J Toxicol Environ Health 1992;36: 281-92.
554. Moolchan ET, Cone EJ, Wstadik A, Huestis MA, Preston KL. Cocaine and metabolite elimination patterns in chronic cocaine users during cessation: plasma and saliva analysis. J Anal Toxicol 2000;24: 458-66.
555. Moore C, Negrusz A, Lewis D. Determination of drugs of abuse in meconium. J Chromatogr 1998;713:137-46.
556. Moore KA. Amphetamines/sympathomimetic amines. In: Levine B, ed. Principles of forensic toxicology, 2nd edition. Washington, DC: AACC Press, 2003:245-64.

Chapter35 ■ Clinical Toxicology 1181
598. Ohlsson A, Lindgren JE, Wahlen A, Agurell S, Hollister LE, Gillespie HK. Plasma delta-9 tetrahydrocannabinol concentrations and clinical effects after oral and intravenous administration and smoking. Clin Pharmacol Ther 1980;28:409-16.
599. Ohno S, Kawana K, Nakajin S. Contribution of UDP-glucuronosyltransferase 1A1 and 1A8 to morphine-6-glucuronidation and its kinetic properties. Drug Metab Dispos 2008;36:688-94.
600. Ohshima T. A case of drug-facilitated sexual assault by the use of flunitrazepam. J Clin Forensic Med 2006;13:44-5.
601. Olmedo R. Phencyclidine and ketamine. In: Goldfrank L, Flomenbaum N, Howland M, Lewin N, Hoffman R, Nelson L, eds. Goldfrank’s toxicologic emergencies, 8th edition. New York, NY: McGraw Hill, 2006:1231-43.
602. Ortelli D, Rudaz S, Chevalley AF, Mino A, Deglon JJ, Balant L, et al. Enantioselective analysis of methadone in saliva by liquid chromatography-mass spectrometry. J Chromatogr A 2000;871:163-72.
603. Ortuno J, de la Torre R, Segura J, Cami J. Simultaneous detection in urine of cocaine and its main metabolites. J Pharmaceut Biomed Anal 1990;8:911-4.
604. Osborne R, Joel S, Trew D, Slevin M. Morphine and metabolite behavior after different routes of morphine administration: demonstration of the importance of the active metabolite morphine-6-glucuronide. Clin Pharmacol Ther 1990;47:12-9.
605. Oshima A, Higuchi T. Treatment guidelines for geriatric mood disorders. Psychiatry Clin Neurosci 1999;53(Suppl):S55-9.
606. Osterloh JD, Kelly TJ, Khayam-Bashi H, Romeo R. Discrepancies in osmolal gaps and calculated alcohol concentrations. Arch Pathol Lab Med 1996;120:637-41.
607. Ostrea EM Jr. Understanding drug testing in the neonate and the role of meconium analysis. J Perinat Neonat Nurs 2001;14:61-82; quiz 105-6.
608. Ostrea EM Jr, Brady MJ, Parks PM, Asensio DC, Naluz A. Drug screening of meconium in infants of drug-dependent mothers: an alternative to urine testing. J Pediatr 1989;115:474-7.
609. Ostrea EM Jr, Knapp DK, Tannenbaum L, Ostrea AR, Romero A, Salari V, et al. Estimates of illicit drug use during pregnancy by maternal interview, hair analysis, and meconium analysis. J Pediatr 2001;138:344-8.
610. Oyler JM, Cone EJ, Joseph RE Jr, Huestis MA. Identification of hydrocodone in human urine following controlled codeine administration. J Anal Toxicol 2000;24:530-5.
611. Pacifici R, Pichini S, Altieri I, Caronna A, Passa AR, Zuccaro P. High-performance liquid chromatographic-electrospray mass spectrometric determination of morphine and its 3- and 6-glucuronides: application to pharmacokinetic studies. J Chromatogr B Biomed Appl 1995;664:329-34.
612. Palmer M, Betz JM. Plants. In: Goldfrank LR, Flomenbaum NE, Hoffman RS, Howland MA, Lewin NA, Nelson L, eds. Goldfrank’s toxicologic emergencies, 8th edition. New York, NY: McGraw Hill, 2006:1577-602.
613. Palmer RB. A review of the use of ethyl glucuronide as a marker for ethanol consumption in forensic and clinical medicine. Semin Diagn Pathol 2009;26:18-27.
614. Pandey R, Mousawy K, Nagarkatti M, Nagarkatti P. Endocannabinoids and immune regulation. Pharmacol Res 2009;60:85-92.
615. Pankey S, Collins C, Jaklitsch A, Izutsu A, Hu M, Pirio M, et al. Quantitative homogeneous enzyme immunoassays for amitriptyline, nortriptyline, imipramine, and desipramine. Clin Chem 1986;32: 768-72.
616. Pappas AA, Gadsden RH Jr, Porter WH, Mullins RE. Osmolality of serum for evaluating the acutely intoxicated patient. In: Frings C, Faulkner W, eds. Selected methods of emergency toxicology. Washington, DC: AACC Press, 1986:85-8.
617. Pappas AA, Gadsden RH Sr, Taylor EH. Serum osmolality in acute intoxication: a prospective clinical study. Am J Clin Pathol 1985;84: 74-9.
618. Parrott AC. Human psychopharmacology of Ecstasy (MDMA): a review of 15 years of empirical research. Hum Psychopharmacol 2001;16:557-77.
577. Navarro M, Pichini S, Farre M, Ortuno J, Roset PN, Segura J, de la Torre R. Usefulness of saliva for measurement of 3,4-methylenedioxymethamphetamine and its metabolites: correlation with plasma drug concentrations and effect of salivary pH. Clin Chem 2001;47:1788-95.
578. Needham SR, Jeanville PM, Brown PR, Estape ES. Performance of a pentafluorophenylpropyl stationary phase for the electrospray ionization high-performance liquid chromatography-mass spectrometry-mass spectrometry assay of cocaine and its metabolite ecgonine methyl ester in human urine. J Chromatogr 2000;748:77-87.
579. Needs CJ, Brooks PM. Clinical pharmacokinetics of the salicylates. Clin Pharmacokinet 1985;10:164-77.
580. Negrusz A, Bowen AM, Moore CM, Dowd SM, Strong MJ, Janicak PG. Deposition of 7-aminoclonazepam and clonazepam in hair following a single dose of Klonopin. J Anal Toxicol 2002;26:471-8.
581. Negrusz A, Gaensslen RE. Analytical developments in toxicological investigation of drug-facilitated sexual assault. Anal Bioanal Chem 2003;376:1192-7.
582. Negrusz A, Juhascik M, Gaensslen RE. Estimate of the incidence of drug-facilitated sexual assault in the U.S. U.S. Department of Justice, 2005.
583. Negrusz A, Moore CM, Hinkel KB, Stockham TL, Verma M, Strong MJ, et al. Deposition of 7-aminoflunitrazepam and flunitrazepam in hair after a single dose of Rohypnol. J Forensic Sci 2001;46:1143-51.
584. Nelson AE, Ho KK. A robust test for growth hormone doping—present status and future prospects. Asian J Androl 2008;10:416-25.
585. Nelson LS. Opioids. In: Flomenbaum NE, Goldfrank LR, Hoffman RS, Howland MA, Nelson LS, eds. Goldfrank’s toxicologic emergencies, 8th edition. New York, NY: McGraw Hill, 2006:590-613.
586. Ng YY, Lin WL, Chen TW, Lin BC, Tsai SH, Chang CC, et al. Spurious hyperchloremia and decreased anion gap in a patient with dextromethorphan bromide. Am J Nephrol 1992;12:268-70.
587. Ni YC, Wong TY, Lloyd RV, Heinze TM, Shelton S, Casciano D, et al. Mouse liver microsomal metabolism of chloral hydrate, trichloroacetic acid, and trichloroethanol leading to induction of lipid peroxidation via a free radical mechanism. Drug Metab Dispos 1996;24:81-90.
588. Nice A, Leikin JB, Maturen A, Madsen-Konczyk LJ, Zell M, Hryhorczuk DO. Toxidrome recognition to improve efficiency of emergency urine drug screens. Ann Emerg Med 1988;17:676-80.
589. Niedbala RS, Kardos KW, Fritch DF, Kardos S, Fries T, Waga J, et al. Detection of marijuana use by oral fluid and urine analysis following single-dose administration of smoked and oral marijuana. J Anal Toxicol 2001;25:289-303.
590. Nine JS, Moraca M, Virji MA, Rao KN. Serum-ethanol determination: comparison of lactate and lactate dehydrogenase interference in three enzymatic assays. J Anal Toxicol 1995;19:192-6.
591. Noble M, Tregear SJ, Treadwell JR, Schoelles K. Long-term opioid therapy for chronic noncancer pain: a systematic review and meta-analysis of efficacy and safety. J Pain Symptom Manage 2008;35:214-28.
592. Norberg A, Jones AW, Hahn RG, Gabrielsson JL. Role of variability in explaining ethanol pharmacokinetics: research and forensic applications. Clin Pharmacokinet 2003;42:1-31.
593. Nordgren HK, Beck O. Direct screening of urine for MDMA and MDA by liquid chromatography-tandem mass spectrometry. J Anal Toxicol 2003;27:15-9.
594. Nordt SP. “DXM”: a new drug of abuse? Ann Emerg Med 1998;31: 794-5.
595. O’Brien CP. Drug addiction and drug abuse (Chapter 23). In: Brunton LL, Lazo JS, Parker KL, eds. Goodman & Gilman’s the pharmacological basis of therapeutics, 11th edition. New York, NY: McGraw-Hill, 2006.
596. O’Brien RP, McGeehan PA, Helmeczi AW, Dula DJ. Detectability of drug tablets and capsules by plain radiography. Am J Emerg Med 1986;4:302-12.
597. Office of the Assistant Secretary of Defense for Health Affairs: Status of drug use in the Department of Defense personnel: fiscal year 2007. Drug testing statistical report, 2008.

1182 SectionIII ■ Analytes
saliva with Drugwipe and Drugread: a controlled study in recreational users. Clin Chem 2002;48:174-6.
640. Pichini S, Pujadas M, Marchei E, Pellegrini M, Fiz J, Pacifici R, et al. Liquid chromatography-atmospheric pressure ionization electrospray mass spectrometry determination of “hallucinogenic designer drugs” in urine of consumers. J Pharmaceut Biomed Anal 2008;47:335-42.
641. Pocci R, Dixit V, Dixit VM. Solid-phase extraction and GC/MS confirmation of barbiturates from human urine. J Anal Toxicol 1992;16:45-7.
642. Poch GK, Klette KL, Anderson C. The quantitation of 2-oxo-3-hydroxy lysergic acid diethylamide (O-H-LSD) in human urine specimens, a metabolite of LSD: comparative analysis using liquid chromatography-selected ion monitoring mass spectrometry and liquid chromatography-ion trap mass spectrometry. J Anal Toxicol 2000;24:170-9.
643. Poch GK, Klette KL, Hallare DA, Manglicmot MG, Czarny RJ, McWhorter LK, et al. Detection of metabolites of lysergic acid diethylamide (LSD) in human urine specimens: 2-oxo-3-hydroxy-LSD, a prevalent metabolite of LSD. J Chromatogr 1999;724:23-33.
644. Poklis A, Edinboro LE. REMEDi drug profiling system readily distinguishes between cyclobenzaprine and amitriptyline in emergency toxicology urine specimens. Clin Chem 1992;38: 2349-50.
645. Poklis A, Moore KA. Response of EMIT amphetamine immunoassays to urinary desoxyephedrine following Vicks inhaler use. Ther Drug Monit 1995;17:89-94.
646. Poklis A, Poklis JL, Tarnai LD, Backer RC. Evaluation of the Triage PPY on-site testing device for the detection of dextropropoxyphene in urine. J Anal Toxicol 2004;28:485-8.
647. Policy ONDC. Study finds highest levels of THC in U.S. marijuana to date. Washington, DC: Office of National Drug Control Policy, 2007.
648. Polson J, Wians FH Jr, Orsulak P, Fuller D, Murray NG, Koff JM, et al. False positive acetaminophen concentrations in patients with liver injury. Clin Chim Acta 2008;391:24-30.
649. Porter W. Clinical toxicology. In: Burtis CA, Bruns DE, eds. Tietz textbook of clinical chemistry, 4th edition. St Louis, Mo: Elsevier Saunders, 2006:1287-369.
650. Porter WH. In acetaminophen assay, only unconjugated drug should be measured. Clin Chem 1984;30:1884-5.
651. Porter WH, Rutter PW, Bush BA, Pappas AA, Dunnington JE. Ethylene glycol toxicity: the role of serum glycolic acid in hemodialysis. J Toxicol Clin Toxicol 2001;39:607-15.
652. Porter WH, Rutter PW, Yao HH. Simultaneous determination of ethylene glycol and glycolic acid in serum by gas chromatography-mass spectrometry. J Anal Toxicol 1999;23:591-7.
653. Pragst F, Balikova MA. State of the art in hair analysis for detection of drug and alcohol abuse. Clin Chim Acta 2006;370:17-49.
654. Pragst F, Herre S, Bakdash A. Poisonings with diphenhydramine—a survey of 68 clinical and 55 death cases. Forensic Sci Int 2006;161: 189-97.
655. Pragst F, Rothe M, Spiegel K, Sporkert F. Illegal and therapeutic drug concentrations in hair segments: a timetable of drug exposure. Forensic Sci Rev 1998;10:81-111.
656. Preston KL, Epstein DH, Cone EJ, Wtsadik AT, Huestis MA, Moolchan ET. Urinary elimination of cocaine metabolites in chronic cocaine users during cessation. J Anal Toxicol 2002;26:393-400.
657. Price CP, Hammond PM, Scawen MD. Evaluation of an enzymic procedure for the measurement of acetaminophen. Clin Chem 1983;29:358-61.
658. Price D. Methemoglobinemia. In: Goldfrank LR, Flomenbaum NE, Lewin NA, Howland MA, Hoffman RS, Nelson LS, eds. Goldfrank’s toxicologic emergencies, 7th edition. New York, NY: McGraw-Hill, 2002:1438-49.
659. Price D. Methemoglobin inducers. In: Goldfrank LR, Flomenbaum NE, Hoffman RS, Howland MA, Lewin NA, Nelson L, eds. Goldfrank’s toxicologic emergencies, 8th edition. New York, NY: McGraw Hill, 2006:1734-45.
619. Pasero C. Fentanyl for acute pain management. J Perianesth Nurs 2005;20:279-84.
620. Pasternak GW. Multiple opiate receptors: deja vu all over again. Neuropharmacology 2004;47(Suppl 1):312-23.
621. Patrono C. Aspirin as an antiplatelet drug. N Engl J Med 1994;330: 1287-94.
622. Paul BD, Lalani S, Bosy T, Jacobs AJ, Huestis MA. Concentration profiles of cocaine, pyrolytic methyl ecgonidine and thirteen metabolites in human blood and urine: determination by gas chromatography-mass spectrometry. Biomed Chromatogr 2005;19: 677-88.
623. Paul BD, Mell LD Jr, Mitchell JM, Irving J, Novak AJ. Simultaneous identification and quantitation of codeine and morphine in urine by capillary gas chromatography and mass spectroscopy. J Anal Toxicol 1985;9:222-6.
624. Paul BD, Mitchell JM, Burbage R, Moy M, Sroka R. Gas chromatographic-electron-impact mass fragmentometric determination of lysergic acid diethylamide in urine. J Chromatogr 1990;529:103-12.
625. Paul BD, Mitchell JM, Mell LD Jr, Irving J. Gas chromatography/electron impact mass fragmentometric determination of urinary 6-acetylmorphine, a metabolite of heroin. J Anal Toxicol 1989;13:2-7.
626. Paul BN. Salicylate poisoning in the elderly: diagnostic pitfalls. J Am Geriatr Soc 1972;20:387-90.
627. Pawula M, Barrett DA, Shaw PN. An improved extraction method for the HPLC determination of morphine and its metabolites in plasma. J Pharmaceut Biomed Anal 1993;11:401-6.
628. Payne JP, Hill DW, Wood DG. Distribution of ethanol between plasma and erythrocytes in whole blood. Nature 1968;217:963-4.
629. Peace MR, Poklis JL, Tarnai LD, Poklis A. An evaluation of the OnTrak Testcup-er on-site urine drug-testing device for drugs commonly encountered from emergency departments. J Anal Toxicol 2002;26:500-3.
630. Peat M. Workplace drug testing. In: Anthony M, Osselton MD, Brian W, eds. Clarke’s analysis of drugs and poisons, vol 3. London, UK: Pharmaceutical Press, 2004:68-79.
631. Pelders MG, Ros JJ. Poppy seeds: differences in morphine and codeine content and variation in inter- and intra-individual excretion. J Forensic Sci 1996;41:209-12.
632. Pentel PR, Keyler DE, Haddad LM. Tricyclic antidepressants and selective serotonin reuptake inhibitors. In: Haddad LM, Shannon MW, Winchester JF, eds. Clinical management of poisoning and drug overdose. Philadelphia, Pa: WB Saunders, 1998:437-51.
633. Perez-Reyes M, Di Guiseppi S, Mason AP, Davis KH. Passive inhalation of marihuana smoke and urinary excretion of cannabinoids. Clin Pharmacol Ther 1983;34:36-41.
634. Perez-Reyes M, Jeffcoat AR, Myers M, Sihler K, Cook CE. Comparison in humans of the potency and pharmacokinetics of intravenously injected cocaethylene and cocaine. Psychopharmacology 1994;116:428-32.
635. Perrone J, De Roos F, Jayaraman S, Hollander JE. Drug screening versus history in detection of substance use in ED psychiatric patients. Am J Emerg Med 2001;19:49-51.
636. Perry H, Shannon MW. Acetaminophen. In: Haddad LM, Shannon MW, Winchester JF, eds. Clinical management of poisoning and drug overdose, 3rd edition. Philadelphia, Pa: WB Saunders, 1998: 664-74.
637. Pertwee RG. The pharmacology of cannabinoid receptors and their ligands: an overview. Int J Obes 2006;30(Suppl 1):S13-8.
638. Peterson GM, Randall CT, Paterson J. Plasma levels of morphine and morphine glucuronides in the treatment of cancer pain: relationship to renal function and route of administration. Eur J Clin Pharmacol 1990;38:121-4.
638A. Pfaffe T, Cooper-White J, Beyerlein P, Karam Kostner K, Punyadeera C. Diagnostic potential of saliva: current state and future applications. Clin Chem 2011;57:675-87.
639. Pichini S, Navarro M, Farre M, Ortuno J, Roset PN, Pacifici R, et al. On-site testing of 3,4-methylenedioxymethamphetamine (ecstasy) in

Chapter35 ■ Clinical Toxicology 1183
680. Rohanova M, Balikova M. Studies on distribution and metabolism of para-methoxymethamphetamine (PMMA) in rats after subcutaneous administration. Toxicology 2009;259:61-8.
681. Romberg RW, Needleman SB, Snyder JJ, Greedan A. Methamphetamine and amphetamine derived from the metabolism of selegiline. J Forensic Sci 1995;40:1100-2.
682. Rook EJ, Hillebrand MJ, Rosing H, van Ree JM, Beijnen JH. The quantitative analysis of heroin, methadone and their metabolites and the simultaneous detection of cocaine, acetylcodeine and their metabolites in human plasma by high-performance liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2005;824:213-21.
683. Rook EJ, Huitema AD, van den Brink W, van Ree JM, Beijnen JH. Population pharmacokinetics of heroin and its major metabolites. Clin Pharmacokinet 2006;45:401-17.
684. Rook EJ, van Ree JM, van den Brink W, Hillebrand MJ, Huitema AD, Hendriks VM, et al. Pharmacokinetics and pharmacodynamics of high doses of pharmaceutically prepared heroin, by intravenous or by inhalation route in opioid-dependent patients. Basic Clin Pharmacol Toxicol 2006;98:86-96.
685. Ropper A, Samuels M. Disorders of the nervous system caused by drugs, toxins, and other chemical agents (Chapter 43). In: Ropper A, Samuels M, eds. Adams and Victor’s principles of neurology, 9th edition. New York, NY: McGraw-Hill, 2009.
686. Roth GJ, Majerus PW. The mechanism of the effect of aspirin on human platelets. I. Acetylation of a particulate fraction protein. J Clin Invest 1975;56:624-32.
687. Rothman RB, Baumann MH. Monoamine transporters and psychostimulant drugs. Eur J Pharmacol 2003;479:23-40.
688. Rothschild AJ, Shindul R, Viguera A, Murray M, Brewster S. Comparison of the frequency of behavioral disinhibition on alprazolam, clonazepam, or no benzodiazepine in hospitalized psychiatric patients. J Clin Psychopharmacol 2000;20:7-11.
689. Rozenfeld R, Abul-Husn NS, Gomez I, Devi LA. An emerging role for the delta opioid receptor in the regulation of mu opioid receptor function. Sci World J 2007;7:64-73.
690. Rumack B, Toll L, Gelman C. Micromedex healthcare series, vol 129. Englewood, Colo: Micromedex, 2006.
691. Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975;55:871-6.
692. Rusyniak DE, Sprague JE. Hyperthermic syndromes induced by toxins. Clin Lab Med 2006;26:165-84, ix.
693. Ryan RM, Wagner CL, Schultz JM, Varley J, DiPreta J, Sherer DM, et al. Meconium analysis for improved identification of infants exposed to cocaine in utero. J Pediatr 1994;125:435-40.
694. Ryder KW, Glick MR. The effect of thioridazine on the Automatic Clinical Analyzer serum tricyclic anti-depressant screen. Am J Clin Pathol 1986;86:248-9.
695. Rygnestad T, Aarstad K, Gustafsson K, Jenssen U. The clinical value of drug analyses in deliberate self-poisoning. Hum Exp Toxicol 1990;9:221-30.
696. Sachs H, Kintz P. Testing for drugs in hair: critical review of chromatographic procedures since 1992. J Chromatogr 1998;713: 147-61.
697. Sachs H, Uhl M, Hege-Scheuing G, Schneider E. Analysis of fentanyl and sufentanil in hair by GC/MS/MS. Int J Legal Med 1996;109: 213-5.
698. Saka K, Uemura K, Shintani-Ishida K, Yoshida K. Determination of amobarbital and phenobarbital in serum by gas chromatography-mass spectrometry with addition of formic acid to the solvent. J Chromatogr B Analyt Technol Biomed Life Sci 2008;869:9-15.
699. Salamone SJ, Honasoge S, Brenner C, McNally AJ, Passarelli J, Goc-Szkutnicka K, et al. Flunitrazepam excretion patterns using the Abuscreen OnTrak and OnLine immunoassays: comparison with GC-MS. J Anal Toxicol 1997;21:341-5.
700. Salamone SJ, Li Z, McNally AJ, Vitone S, Wu RS. Epimerization studies of LSD using 1H nuclear magnetic resonance (NMR) spectroscopy. J Anal Toxicol 1997;21:492-7.
660. Prommer N, Sottas PE, Schoch C, Schumacher YO, Schmidt W. Total hemoglobin mass—a new parameter to detect blood doping? Med Sci Sports Exerc 2008;40:2112-8.
661. Puopolo PR, Volpicelli SA, Johnson DM, Flood JG. Emergency toxicology testing (detection, confirmation, and quantification) of basic drugs in serum by liquid chromatography with photodiode array detection. Clin Chem 1991;37:2124-30.
662. Purssell RA, Pudek M, Brubacher J, Abu-Laban RB. Derivation and validation of a formula to calculate the contribution of ethanol to the osmolal gap. Ann Emerg Med 2001;38:653-9.
663. Raffa RB. Basic pharmacology relevant to drug abuse assessment: tramadol as example. J Clin Pharm Ther 2008;33:101-8.
664. Rainey JM Jr, Crowder MK. Prolonged psychosis attributed to phencyclidine: report of three cases. Am J Psychiatry 1975;132:1076-8.
665. Rance MJ, Jordan BJ, Nichols JD. A simultaneous determination of acetylsalicylic acid, salicylic acid and salicylamide in plasma by gas liquid chromatography. J Pharm Pharmacol 1975;27:425-9.
666. Rasmussen F. Salivary excretion of sulphonamides and barbiturates by cows and goats. Acta Pharmacol Toxicol (Copenh) 1964;21:11-9.
667. Rasmussen LB, Olsen KH, Johansen SS. Chiral separation and quantification of R/S-amphetamine, R/S-methamphetamine, R/S-MDA, R/S-MDMA, and R/S-MDEA in whole blood by GC-EI-MS. J Chromatogr B Analyt Technol Biomed Life Sci 2006;842:136-41.
668. Reidy L, Gennaro W, Steele BW, Walls HC. The incidence of Zolpidem use in suspected DUI drivers in Miami-Dade Florida: a comparative study using immunalysis Zolpidem ELISA KIT and gas chromatography-mass spectrometry screening. J Anal Toxicol 2008; 32:688-94.
669. Reisfield GM, Salazar E, Bertholf RL. Rational use and interpretation of urine drug testing in chronic opioid therapy. Ann Clin Lab Sci 2007;37:301-14.
670. Remaley AT, Hicks DG, Kane MD, Shaw LM. Laboratory assessment of poisoning with a carbamate insecticide. Clin Chem 1988;34:1933-6.
671. Renner UD, Oertel R, Kirch W. Pharmacokinetics and pharmacodynamics in clinical use of scopolamine. Ther Drug Monit 2005;27:655-65.
672. Reuschel SA, Percey SE, Liu S, Eades DM, Foltz RL. Quantitative determination of LSD and a major metabolite, 2-oxo-3-hydroxy-LSD, in human urine by solid-phase extraction and gas chromatography-tandem mass spectrometry. J Anal Toxicol 1999;23:306-12.
673. Richelson E. Preclinical pharmacology of neuroleptics: focus on new generation compounds. J Clin Psychiatry 1996;57(Suppl 11):4-11.
674. Riley J, Eisenberg E, Muller-Schwefe G, Drewes AM, Arendt-Nielsen L. Oxycodone: a review of its use in the management of pain. Curr Med Res Opin 2008;24:175-92.
675. Ritter D, Cortese CM, Edwards LC, Barr JL, Chung HD, Long C. Interference with testing for lysergic acid diethylamide. Clin Chem 1997;43:635-7.
676. Riviere GJ, Gentry WB, Owens SM. Disposition of methamphetamine and its metabolite amphetamine in brain and other tissues in rats after intravenous administration. J Pharmacol Exp Ther 2000;292:1042-7.
677. Robandt PP, Reda LJ, Klette KL. Complete automation of solid-phase extraction with subsequent liquid chromatography-tandem mass spectrometry for the quantification of benzoylecgonine, m-hydroxybenzoylecgonine, p-hydroxybenzoylecgonine, and norbenzoylecgonine in urine—application to a high-throughput urine analysis laboratory. J Anal Toxicol 2008;32:577-85.
678. Rodrigues WC, Wang G, Moore C, Agrawal A, Vincent MJ, Soares JR. Development and validation of ELISA and GC-MS procedures for the quantification of dextromethorphan and its main metabolite dextrorphan in urine and oral fluid. J Anal Toxicol 2008;32:220-6.
679. Rodriguez-Rosas ME, Medrano JG, Epstein DH, Moolchan ET, Preston KL, Wainer IW. Determination of total and free concentrations of the enantiomers of methadone and its metabolite (2-ethylidene-1,5-dimethyl-3,3-diphenyl-pyrrolidine) in human plasma by enantioselective liquid chromatography with mass spectrometric detection. J Chromatogr A 2005;1073:237-48.

1184 SectionIII ■ Analytes
723. Shima N, Kamata H, Katagi M, Tsuchihashi H, Sakuma T, Nemoto N. Direct determination of glucuronide and sulfate of 4-hydroxy-3-methoxymethamphetamine, the main metabolite of MDMA, in human urine. J Chromatogr B Analyt Technol Biomed Life Sci 2007; 857:123-9.
724. Shou WZ, Pelzer M, Addison T, Jiang X, Naidong W. An automatic 96-well solid phase extraction and liquid chromatography-tandem mass spectrometry method for the analysis of morphine, morphine- 3-glucuronide and morphine-6-glucuronide in human plasma. J Pharmaceut Biomed Anal 2002;27:143-52.
725. Sides GD. QT interval prolongation as a biomarker for torsades de pointes and sudden death in drug development. Dis Markers 2002;18: 57-62.
726. Siegel IA. The role of saliva in drug monitoring. Ann N Y Acad Sci 1993;694:86-90.
727. Simpson RJ, Power KG, Wallace LA, Butcher MH, Swanson V, Simpson EC. Controlled comparison of the characteristics of long-term benzodiazepine users in general practice. Br J Gen Pract 1990;40:22-6.
728. Sivilotti ML. Ethanol, isopropanol, and methanol. In: Dart RC, ed. Medical toxicology. Philadelphia, Pa: Lippincott Williams & Wilkins, 2004:1211-23.
729. Slaughter L. Involvement of drugs in sexual assault. J Reprod Med 2000;45:425-30.
730. Small GW. Treatment of geriatric depression. Depression Anxiety 1998;8(Suppl 1):32-42.
731. Smilkstein MJ, Douglas DR, Daya MR. Acetaminophen poisoning and liver function. N Engl J Med 1994;331:1310-1; author reply 1-2.
732. Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose: analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557-62.
733. Smirnow D, Logan BK. Analysis of ecgonine and other cocaine biotransformation products in postmortem whole blood by protein precipitation-extractive alkylation and GC-MS. J Anal Toxicol 1996; 20:463-7.
734. Smith-Kielland A, Olsen KM, Christophersen AS. False-positive results with Emit II amphetamine/methamphetamine assay in users of common psychotropic drugs. Clin Chem 1995;41:951-2.
735. Smith DE, Gutgesell ME, Schwartz RH, Thorne MM, Bogema S. Federal guidelines for marijuana screening should have lower cutoff levels: a comparison of results from immunoassays and gas chromatography-mass spectrometry. Arch Pathol Lab Med 1989;113: 1299-300.
736. Smith JA, Hine ID, Beck P, Routledge PA. Paracetamol toxicity: is enzyme induction important? Hum Toxicol 1986;5:383-5.
737. Smith KM. Drugs used in acquaintance rape. J Am Pharm Assoc 1999;39:519-25; quiz 81-3.
738. Smith ML, Barnes AJ, Huestis MA. Identifying new cannabis use with urine creatinine-normalized THCCOOH concentrations and time intervals between specimen collections. J Anal Toxicol 2009;33: 185-9.
739. Smith ML, Hughes RO, Levine B, Dickerson S, Darwin WD, Cone EJ. Forensic drug testing for opiates. VI. Urine testing for hydromorphone, hydrocodone, oxymorphone, and oxycodone with commercial opiate immunoassays and gas chromatography-mass spectrometry. J Anal Toxicol 1995;19:18-26.
740. Smith ML, Shimomura ET, Summers J, Paul BD, Nichols D, Shippee R, et al. Detection times and analytical performance of commercial urine opiate immunoassays following heroin administration. J Anal Toxicol 2000;24:522-9.
741. Smith RM. Ethyl esters of arylhydroxy- and arylhydroxymethoxycocaines in the urines of simultaneous cocaine and ethanol users. J Anal Toxicol 1984;8:38-42.
742. Smithline N, Gardner KD Jr. Gaps—anionic and osmolal. JAMA 1976;236:1594-7.
743. Snyder H, Schwenzer KS, Pearlman R, McNally AJ, Tsilimidos M, Salamone SJ, et al. Serum and urine concentrations of flunitrazepam
701. Samyn N, van Haeren C. On-site testing of saliva and sweat with Drugwipe and determination of concentrations of drugs of abuse in saliva, plasma and urine of suspected users. Int J Legal Med 2000;113: 150-4.
702. Sanchez R, Fosarelli P, Felt B, Greene M, Lacovara J, Hackett F. Carbon monoxide poisoning due to automobile exposure: disparity between carboxyhemoglobin levels and symptoms of victims. Pediatrics 1988;82:663-6.
703. Sanna E, Busonero F, Talani G, Carta M, Massa F, Peis M, et al. Comparison of the effects of zaleplon, zolpidem, and triazolam at various GABA(A) receptor subtypes. Eur J Pharmacol 2002;451: 103-10.
704. Sarma L, Wong SH, DellaFera S. Diflunisal significantly interferes with salicylate measurements by FPIA-TDx and UV-VIS aca methods. Clin Chem 1985;31:1922-3.
705. Saum CA, Inciardi JA. Rohypnol misuse in the United States. Subst Use Misuse 1997;32:723-31.
706. Saunders PA, Ho IK. Barbiturates and the GABAA receptor complex: progress in drug research. Fortsch Arzneimittelforsch 1990;34:261-86.
707. Schanzle G, Li S, Mikus G, Hofmann U. Rapid, highly sensitive method for the determination of morphine and its metabolites in body fluids by liquid chromatography-mass spectrometry. J Chromatogr 1999;721:55-65.
708. Schepers RJ, Oyler JM, Joseph RE Jr, Cone EJ, Moolchan ET, Huestis MA. Methamphetamine and amphetamine pharmacokinetics in oral fluid and plasma after controlled oral methamphetamine administration to human volunteers. Clin Chem 2003;49:121-32.
709. Schier J. Avoid unfavorable consequences: dextromethorpan can bring about a false-positive phencyclidine urine drug screen. J Emerg Med 2000;18:379-81.
710. Schmitt TC. Determination of chloral hydrate and its metabolites in blood plasma by capillary gas chromatography with electron capture detection. J Chromatogr B Analyt Technol Biomed Life Sci 2002;780: 217-24.
711. Schneider S, Wennig R. Interference of diphenhydramine with the EMIT II immunoassay for propoxyphene. J Anal Toxicol 1999;23: 637-8.
712. Schramm W, Craig PA, Smith RH, Berger GE. Cocaine and benzoylecgonine in saliva, serum, and urine. Clin Chem 1993;39: 481-7.
713. Schramm W, Smith RH, Craig PA, Kidwell DA. Drugs of abuse in saliva: a review. J Anal Toxicol 1992;16:1-9.
714. Schuberth J, Schuberth J. Gas chromatographic-mass spectrometric determination of morphine, codeine and 6-monoacetylmorphine in blood extracted by solid phase. J Chromatogr 1989;490:444-9.
715. Schulz V. Clinical pharmacokinetics of nitroprusside, cyanide, thiosulphate and thiocyanate. Clin Pharmacokinet 1984;9:239-51.
716. Scott-Ham M, Burton FC. Toxicological findings in cases of alleged drug-facilitated sexual assault in the United Kingdom over a 3-year period. J Clin Forensic Med 2005;12:175-86.
717. Seitz DP, Gill SS. Neuroleptic malignant syndrome complicating antipsychotic treatment of delirium or agitation in medical and surgical patients: case reports and a review of the literature. Psychosomatics 2009;50:8-15.
718. Sejersted OM, Jacobsen D, Ovrebo S, Jansen H. Formate concentrations in plasma from patients poisoned with methanol. Acta Med Scand 1983;213:105-10.
719. Senanayake N, Karalliedde L. Neurotoxic effects of organophosphorus insecticides: an intermediate syndrome. N Engl J Med 1987;316:761-3.
720. Seppala T, Nuotto E, Dreyfus JF. Drug-alcohol interactions on psychomotor skills: zopiclone and flunitrazepam. Pharmacology 1983;27(Suppl 2):127-35.
721. Lunesta (eszopiclone) product information. Marlborough, Mass: Sepracor, 2005.
722. Shannon M, Haddad L. The emergency management of poisoning. In: Haddad L, Shannon M, Winchester J, eds. Clinical management of poisoning and drug overdose, 3rd edition. Philadelphia, Pa: WB Saunders, 1998:2-31.

Chapter35 ■ Clinical Toxicology 1185
766. Taylor EH, Oertli EH, Wolfgang JW, Mueller E. Accuracy of five on-site immunoassay drugs-of-abuse testing devices. J Anal Toxicol 1999;23:119-24.
767. Temple AR. Acute and chronic effects of aspirin toxicity and their treatment. Arch Intern Med 1981;141:364-9.
768. Tennant F, Shannon J. Quantitative urine testing: a new tool for diagnosing and treating cocaine use. Postgrad Med 1989;86:107-14.
769. Terada M, Shinozuka T, Bai H, Islam MN, Tun Z, Honda K, et al. Simultaneous determination of barbiturate drugs in human serum by wide-bore capillary gas chromatography with nitrogen-phosphorus detection. Jpn J Forensic Toxicol 1995;13:223-31.
770. Theobald DS, Fehn S, Maurer HH. New designer drug, 2,5-dimethoxy-4-propylthio-beta-phenethylamine (2C-T-7): studies on its metabolism and toxicological detection in rat urine using gas chromatography/mass spectrometry. J Mass Spectrom 2005;40: 105-16.
771. Thevis M, Geyer H, Bahr D, Schanzer W. Identification of fentanyl, alfentanil, sufentanil, remifentanil and their major metabolites in human urine by liquid chromatography/tandem mass spectrometry for doping control purposes. Eur J Mass Spectrom 2005;11:419-27.
772. Thevis M, Opfermann G, Schanzer W. Liquid chromatography/electrospray ionization tandem mass spectrometric screening and confirmation methods for beta2-agonists in human or equine urine. J Mass Spectrom 2003;38:1197-206.
773. Thiermann H, Szinicz L, Eyer P, Zilker T, Worek F. Correlation between red blood cell acetylcholinesterase activity and neuromuscular transmission in organophosphate poisoning. Chem Biol Interact 2005;157-158:345-7.
774. Thomas S, Winecker R, Pestaner JP. Unusual fentanyl patch administration. Am J Forensic Med Pathol 2008;29:162-3.
775. Thurman EM, Pedersen MJ, Stout RL, Martin T. Distinguishing sympathomimetic amines from amphetamine and methamphetamine in urine by gas chromatography/mass spectrometry. J Anal Toxicol 1992;16:19-27.
776. Toennes SW, Fandino AS, Kauert G. Gas chromatographic-mass spectrometric detection of anhydroecgonine methyl ester (methylecgonidine) in human serum as evidence of recent smoking of crack. J Chromatogr 1999;735:127-32.
777. Tomassoni AW. Antihistamines and decongestants. In: Goldfrank LR, Flomenbaum NE, Hoffman RS, Howland MA, Lewin NA, Nelson L, eds. Goldfrank’s toxicologic emergencies, 8th edition. New York, NY: McGraw-Hill, 2005.
778. Tomaszewski C. Carbon monoxide. In: Goldfrank LR, Flomenbaum NE, Hoffman RS, Howland MA, Lewin NA, Nelson L, eds. Goldfrank’s toxicologic emergencies, 8th edition. New York, NY: McGraw Hill, 2006:1689-704.
779. Tong T, Boyer EW. Club drugs, smart drugs, raves, and circuit parties: an overview of the club scene. Pediatr Emerg Care 2002;18:216-8.
780. Trenton A, Currier G, Zwemer F. Fatalities associated with therapeutic use and overdose of atypical antipsychotics. CNS Drugs 2003;17:307-24.
781. Trescot AM, Datta S, Lee M, Hansen H. Opioid pharmacology. Pain Physician 2008;11:S133-53.
782. Trinder P. Rapid determination of salicylate in biological fluids. Biochem J 1954;57:301-3.
783. Tseng LF. Effects of para-methoxyamphetamine and 2,5-dimethoxyamphetamine on serotonergic mechanisms. Arch Pharmacol 1978;304:101-5.
784. Tseng LF, Menon MK, Loh HH. Comparative actions of monomethoxyamphetamines on the release and uptake of biogenic amines in brain tissue. J Pharmacol Exp Ther 1976;197:263-71.
785. Turcant A, Premel-Cabic A, Cailleux A, Allain P. Toxicological screening of drugs by microbore high-performance liquid chromatography with photodiode-array detection and ultraviolet spectral library searches. Clin Chem 1991;37:1210-15.
786. Tyrefors N, Hyllbrant B, Ekman L, Johansson M, Langstrom B. Determination of morphine, morphine-3-glucuronide and morphine-6-glucuronide in human serum by solid-phase extraction and liquid
and metabolites, after a single oral dose, by immunoassay and GC-MS. J Anal Toxicol 2001;25:699-704.
744. Somogyi AA, Barratt DT, Coller JK. Pharmacogenetics of opioids. Clin Pharmacol Ther 2007;81:429-44.
745. Sora I, Takahashi N, Funada M, Ujike H, Revay RS, Donovan DM, et al. Opiate receptor knockout mice define mu receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc Natl Acad Sci U S A 1997;94:1544-9.
746. Sorisky A, Watson DC. Positive diphenhydramine interference in the EMIT-ST assay for tricyclic antidepressants in serum. Clin Chem 1986;32:715.
747. Spanbauer AC, Moody DE, Foltz RL, Walsh SL. A gas chromatographic-positive ion chemical ionization-mass spectrometric method for determination of cocaine, benzoylecgonine, ecgonine methyl ester, and norcocaine in plasma: detection of norcocaine in plasma after oral administration of cocaine. J Anal Toxicol 2000;24: 453-5.
748. Speed DJ, Dickson SJ, Cairns ER, Kim ND. Analysis of paracetamol using solid-phase extraction, deuterated internal standards, and gas chromatography-mass spectrometry. J Anal Toxicol 2001;25:198-202.
749. Spiehler V. Drugs in saliva. In: Moffat A, Osselton MD, Widdop B, eds. Clarke’s analysis of drugs and poisons, vol 3. London, UK: Pharmaceutical Press, 2004:109-23.
750. Sporer KA, Khayam-Bashi H. Acetaminophen and salicylate serum levels in patients with suicidal ingestion or altered mental status. Am J Emerg Med 1996;14:443-6.
751. Squires RF, Saederup E. Mono N-aryl ethylenediamine and piperazine derivatives are GABAA receptor blockers: implications for psychiatry. Neurochem Res 1993;18:787-93.
752. Staack RF, Fritschi G, Maurer HH. Studies on the metabolism and toxicological detection of the new designer drug N-benzylpiperazine in urine using gas chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2002;773:35-46.
753. Steering Committee of the Physicians’ Health Study Research Group. Final report on the aspirin component of the ongoing Physicians’ Health Study. N Engl J Med 1989;321:129-35.
754. Sternbach H. The serotonin syndrome. Am J Psychiatry 1991;148: 705-13.
755. Storrow AB, Wians FH Jr, Mikkelsen SL, Norton J. Does naloxone cause a positive urine opiate screen? Ann Emerg Med 1994;24:1151-3.
756. Stout PR, Farrell LJ. Opioids: effects on human performance and behavior. Forensic Sci Rev 2003;15:30-59.
757. Struempler RE. Excretion of codeine and morphine following ingestion of poppy seeds. J Anal Toxicol 1987;11:97-9.
758. Studenik C, Lemmens-Gruber R, Heistracher P. Proarrhythmic effects of antidepressants and neuroleptic drugs on isolated, spontaneously beating guinea-pig Purkinje fibers. Eur J Pharm Sci 1999;7:113-8.
759. Sugiura T, Waku K. Cannabinoid receptors and their endogenous ligands. J Biochem 2002;132:7-12.
760. Sukbuntherng J, Walters A, Chow HH, Mayersohn M. Quantitative determination of cocaine, cocaethylene (ethylcocaine), and metabolites in plasma and urine by high-performance liquid chromatography. J Pharm Sci 1995;84:799-804.
761. Sullivan M. Mandatory guidelines for federal workplace drug testing programs, vol 53. Federal Register 1988;119:70-89.
762. Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol 2005;75:406-33.
763. Sun QR, Xiang P, Yan H, Shen M. [Simultaneous analyses of cocaine and its metabolite benzoylecgonine in urine by LC-MS/MS]. Fa Yi Xue Za Zhi 2008;24:268-72.
764. Suzuki S, Inoue T, Hori H, Inayama S. Analysis of methamphetamine in hair, nail, sweat, and saliva by mass fragmentography. J Anal Toxicol 1989;13:176-8.
765. Tateishi T, Krivoruk Y, Ueng YF, Wood AJ, Guengerich FP, Wood M. Identification of human liver cytochrome P-450 3A4 as the enzyme responsible for fentanyl and sufentanil N-dealkylation. Anesth Analg 1996;82:167-72.

1186 SectionIII ■ Analytes
810. Vreman HJ, Ronquillo RB, Ariagno RL, Schwartz HC, Stevenson DK. Interference of fetal hemoglobin with the spectrophotometric measurement of carboxyhemoglobin. Clin Chem 1988;34:975-7.
811. Wachtel SR, ElSohly MA, Ross SA, Ambre J, de Wit H. Comparison of the subjective effects of delta(9)-tetrahydrocannabinol and marijuana in humans. Psychopharmacology 2002;161:331-9.
812. Wagner J, Wagner ML, Hening WA. Beyond benzodiazepines: alternative pharmacologic agents for the treatment of insomnia. Ann Pharmacother 1998;32:680-91.
813. Wagner JG, Wilkinson PK, Sedman AJ, Kay DR, Weidler DJ. Elimination of alcohol from human blood. J Pharm Sci 1976;65: 152-4.
814. Walberg CB, Gupta RC. Quantitation of phencyclidine in urine by enzyme immunoassay. J Anal Toxicol 1982;6:97-9.
815. Waldhoer M, Bartlett SE, Whistler JL. Opioid receptors. Annu Rev Biochem 2004;73:953-90.
816. Wall ME, Sadler BM, Brine D, Taylor H, Perez-Reyes M. Metabolism, disposition, and kinetics of delta-9-tetrahydrocannabinol in men and women. Clin Pharmacol Ther 1983;34:352-63.
817. Wallace JE, Hamilton HE, King DE, Bason DJ, Schwertner HA, Harris SC. Gas-liquid chromatographic determination of cocaine and benzoylecgonine in urine. Anal Chem 1976;48:34-8.
818. Wallace KL, Suchard JR, Curry SC, Reagan C. Diagnostic use of physicians’ detection of urine fluorescence in a simulated ingestion of sodium fluorescein-containing antifreeze. Ann Emerg Med 2001;38:49-54.
819. Wallner M, Hanchar HJ, Olsen RW. Ethanol enhances alpha 4 beta 3 delta and alpha 6 beta 3 delta gamma-aminobutyric acid type A receptors at low concentrations known to affect humans. Proc Natl Acad Sci U S A 2003;100:15218-23.
820. Walsh SL, Preston KL, Stitzer ML, Cone EJ, Bigelow GE. Clinical pharmacology of buprenorphine: ceiling effects at high doses. Clin Pharmacol Ther 1994;55:569-80.
821. Walshe K, Barrett AM, Kavanagh PV, McNamara SM, Moran C, Shattock AG. A sensitive immunoassay for flunitrazepam and metabolites. J Anal Toxicol 2000;24:296-9.
822. Waltzman ML. Flunitrazepam: a review of “roofies.” Pediatr Emerg Care 1999;15:59-60.
823. Wan SH, Matin SB, Azarnoff DL. Kinetics, salivary excretion of amphetamine isomers, and effect of urinary pH. Clin Pharmacol Ther 1978;23:585-90.
824. Wang PH, Liu C, Tsay WI, Li JH, Liu RH, Wu TG, et al. Improved screen and confirmation test of 7-aminoflunitrazepam in urine specimens for monitoring flunitrazepam (Rohypnol) exposure. J Anal Toxicol 2002;26:411-8.
825. Wang QL, Fan LY, Zhang W, Cao CX. Sensitive analysis of two barbiturates in human urine by capillary electrophoresis with sample stacking induced by moving reaction boundary. Anal Chim Acta 2006;580:200-5.
826. Wasels R, Belleville F, Paysant P, Nabet P, Krakowski I. Determination of morphine in plasma by gas chromatography using a macrobore column and thermoionic detection after Extrelut column extraction: application to follow-up morphine treatment in cancer patients. J Chromatogr 1989;489:411-18.
827. Weiner AL, Ko C, McKay CA Jr. A comparison of two bedside tests for the detection of salicylates in urine. Acad Emerg Med 2000;7: 834-6.
828. Weinmann W, Lehmann N, Muller C, Wiedemann A, Svoboda M. Identification of lorazepam and sildenafil as examples for the application of LC/ionspray-MS and MS-MS with mass spectra library searching in forensic toxicology. Forensic Sci Int 2000;113:339-44.
829. Weinmann W, Muller C, Vogt S, Frei A. LC-MS-MS analysis of the neuroleptics clozapine, flupentixol, haloperidol, penfluridol, thioridazine, and zuclopenthixol in hair obtained from psychiatric patients. J Anal Toxicol 2002;26:303-7.
830. Weinmann W, Svoboda M. Fast screening for drugs of abuse by solid-phase extraction combined with flow-injection ionspray-tandem mass spectrometry. J Anal Toxicol 1998;22:319-28.
chromatography-mass spectrometry with electrospray ionisation. J Chromatogr A 1996;729:279-85.
787. Uhl M. Determination of drugs in hair using GC/MS/MS. Forensic Sci Int 1997;84:281-94.
788. Uhl M, Sachs H. Cannabinoids in hair: strategy to prove marijuana/hashish consumption. Forensic Sci Int 2004;145:143-7.
789. Uhlenhuth EH, Balter MB, Ban TA, Yang K. International study of expert judgment on therapeutic use of benzodiazepines and other psychotherapeutic medications. IV. Therapeutic dose dependence and abuse liability of benzodiazepines in the long-term treatment of anxiety disorders. J Clin Psychopharmacol 1999;19:23S-9S.
790. Unterwald EM, Ho A, Rubenfeld JM, Kreek MJ. Time course of the development of behavioral sensitization and dopamine receptor up-regulation during binge cocaine administration. J Pharmacol Exp Ther 1994;270:1387-96.
791. Unterwald EM, Rubenfeld JM, Kreek MJ. Repeated cocaine administration upregulates kappa and mu, but not delta, opioid receptors. Neuroreport 1994;5:1613-6.
792. Valtier S, Cody JT. Evaluation of internal standards for the analysis of amphetamine and methamphetamine. J Anal Toxicol 1995;19:375-80.
793. van As H, Stolk LM. Rifampicin cross-reacts with opiate immunoassay. J Anal Toxicol 1999;23:71.
794. Van Bocxlaer JF, Clauwaert KM, Lambert WE, Deforce DL, Van den Eeckhout EG, De Leenheer AP. Liquid chromatography-mass spectrometry in forensic toxicology. Mass Spectrom Rev 2000;19: 165-214.
795. Van Hoey N. Effect of cyclobenzaprine on tricyclic antidepressant assays. Ann Pharmacother 2005;39:1314-7.
796. Vanek VW, Dickey-White HI, Signs SA, Schechter MD, Buss T, Kulics AT. Concurrent use of cocaine and alcohol by patients treated in the emergency department. Ann Emerg Med 1996;28:508-14.
797. Vayer P, Mandel P, Maitre M. Conversion of gamma-hydroxybutyrate to gamma-aminobutyrate in vitro. J Neurochem 1985;45:810-4.
798. Veltri JC, Thompson MIB. Salicylates. In: Stoutakis V, ed. Clinical toxicology of drugs: principles and practice. Philadelphia, Pa: Lea & Febiger, 1982:227-43.
799. Verstraete AG, Heyden FV. Comparison of the sensitivity and specificity of six immunoassays for the detection of amphetamines in urine. J Anal Toxicol 2005;29:359-64.
800. Verweij AM, Hordijk ML, Lipman PJ. Liquid chromatographic-thermospray tandem mass spectrometric quantitative analysis of some drugs with hypnotic, sedative and tranquillising properties in whole blood. J Chromatogr B Biomed Appl 1996;686:27-34.
801. Verweij AM, Lipman PJ, Zweipfenning PG. Quantitative liquid chromatography, thermospray/tandem mass spectrometry (LC/TSP/MS/MS) analysis of some thermolabile benzodiazepines in whole-blood. Forensic Sci Int 1992;54:67-74.
802. Vesey CJ, Kirk CJ. Two automated methods for measuring plasma thiocyanate compared. Clin Chem 1985;31:270-4.
803. Vesey CJ, Wilson J. Red cell cyanide. J Pharm Pharmacol 1978;30: 20-6.
804. Viera AJ, Yates SW. Toxic ingestion of gamma-hydroxybutyric acid. South Med J 1999;92:404-5.
805. Villain M, Cheze M, Tracqui A, Ludes B, Kintz P. Windows of detection of zolpidem in urine and hair: application to two drug facilitated sexual assaults. Forensic Sci Int 2004;143:157-61.
806. Vining RF, McGinley RA. Hormones in saliva. Crit Rev Clin Lab Sci 1986;23:95-146.
807. von Minden DL, D’Amato NA. Simultaneous determination of cocaine and benzoylecgonine in urine by gas-liquid chromatography. Anal Chem 1977;49:1974-7.
808. Vorce SP, Sklerov JH. A general screening and confirmation approach to the analysis of designer tryptamines and phenethylamines in blood and urine using GC-EI-MS and HPLC-electrospray-MS. J Anal Toxicol 2004;28:407-10.
809. Vreman HJ, Mahoney JJ, Van Kessel AL, Stevenson DK. Carboxyhemoglobin as measured by gas chromatography and with the IL 282 and 482 co-oximeters. Clin Chem 1988;34:2562-6.

Chapter35 ■ Clinical Toxicology 1187
857. Wright R, Woolf A. Phencyclidine. In: Haddad L, Shannon M, Winchester J, eds. Clinical management of poisoning and drug overdose, 3rd edition. Philadelphia, Pa: WB Saunders, 1998:552-9.
858. Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999;34: 646-56.
859. Wu AH, Feng YJ, Pajor A, Gornet TG, Wong SS, Forte E, Brown J. Detection and interpretation of lysergic acid diethylamide results by immunoassay screening of urine in various testing groups. J Anal Toxicol 1997;21:181-4.
860. Wu AH, McKay C, Broussard LA, Hoffman RS, Kwong TC, Moyer TP, et al. National Academy of Clinical Biochemistry Laboratory Medicine practice guidelines: recommendations for the use of laboratory tests to support poisoned patients who present to the emergency department. Clin Chem 2003;49:357-79.
861. Wu AH, Wong SS, Johnson KG, Callies J, Shu DX, Dunn WE, et al. Evaluation of the triage system for emergency drugs-of-abuse testing in urine. J Anal Toxicol 1993;17:241-5.
862. Wu Y, Ali S, Ahmadian G, Liu CC, Wang YT, Gibson KM, et al. Gamma-hydroxybutyric acid (GHB) and gamma-aminobutyric acid B receptor (GABABR) binding sites are distinctive from one another: molecular evidence. Neuropharmacology 2004;47: 1146-56.
863. Wu YH, Tan JY, Xia Y. [Determination of 7-aminoflunitrazepam, the major metabolite of flunitrazepam in urine by high performance thin-layer chromatography]. Se Pu 2002;20:182-4.
864. Wurst FM, Skipper GE, Weinmann W. Ethyl glucuronide—the direct ethanol metabolite on the threshold from science to routine use. Addiction 2003;98(Suppl 2):51-61.
865. Wylie FM, Torrance H, Anderson RA, Oliver JS. Drugs in oral fluid. Part I. Validation of an analytical procedure for licit and illicit drugs in oral fluid. Forensic Sci Int 2005;150:191-8.
866. Wyngaarden JB, Seevers MH. The toxic effects of antihistaminic drugs. JAMA 1951;145:277-82.
867. Yao HH, Porter WH. Simultaneous determination of ethylene glycol and its major toxic metabolite, glycolic acid, in serum by gas chromatography. Clin Chem 1996;42:292-7.
868. Yeatman DT, Reid K. A study of urinary endogenous gamma-hydroxybutyrate (GHB) levels. J Anal Toxicol 2003;27:40-2.
869. Yuan H, Mester Z, Lord H, Pawliszyn J. Automated in-tube solid-phase microextraction coupled with liquid chromatography-electrospray ionization mass spectrometry for the determination of selected benzodiazepines. J Anal Toxicol 2000;24:718-25.
870. Zawertailo LA, Kaplan HL, Busto UE, Tyndale RF, Sellers EM. Psychotropic effects of dextromethorphan are altered by the CYP2D6 polymorphism: a pilot study. J Clin Psychopharmacol 1998;18:332-7.
871. Zhao H, Brenneisen R, Scholer A, McNally AJ, ElSohly MA, Murphy TP, et al. Profiles of urine samples taken from Ecstasy users at Rave parties: analysis by immunoassays, HPLC, and GC-MS. J Anal Toxicol 2001;25:258-69.
872. Zhao H, Wang L, Qiu Y, Zhou Z, Zhong W, Li X. Multiwalled carbon nanotubes as a solid-phase extraction adsorbent for the determination of three barbiturates in pork by ion trap gas chromatography-tandem mass spectrometry (GC/MS/MS) following microwave assisted derivatization. Anal Chim Acta 2007;586:399-406.
873. Zheng M, McErlane KM, Ong MC. High-performance liquid chromatography-mass spectrometry-mass spectrometry analysis of morphine and morphine metabolites and its application to a pharmacokinetic study in male Sprague-Dawley rats. J Pharmaceut Biomed Anal 1998;16:971-80.
874. Zollner C, Stein C. Opioids: handbook of experimental pharmacology. New York, NY: Springer-Verlag, 2007:31-63.
875. Zoppi F, Brenna S, Fumagalli C, Marocchi A. Discrimination among dyshemoglobins: analytical approach to a toxicological query. Clin Chem 1996;42:1300-2.
876. Zuccaro P, Ricciarello R, Pichini S, Pacifici R, Altieri I, Pellegrini M, et al. Simultaneous determination of heroin 6-monoacetylmorphine,
831. Weiss RD, Gawin FH. Protracted elimination of cocaine metabolites in long-term high-dose cocaine abusers. Am J Med 1988;85:879-80.
832. Wells K, Williamson M, Holstege CP, Bear AB, Brady WJ. The association of cardiovascular toxins and electrocardiographic abnormality in poisoned patients. Am J Emerg Med 2008;26:957-9.
833. Welton B. Analysis of nanogram levels of barbiturates. Chromatographia 1970;3:211-5.
834. Wennig R. Potential problems with the interpretation of hair analysis results. Forensic Sci Int 2000;107:5-12.
835. Westfall TC, Westfall DP. Adrenergic agonists and antagonists (Chapter 10). In: Brunton LL, Lazo JS, Parker KL, eds. Goodman & Gilman’s the pharmacological basis of therapeutics, 11th edition. New York, NY: McGraw-Hill, 2006.
836. Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994;272:1845-50.
837. White LM, Gardner SF, Gurley BJ, Marx MA, Wang PL, Estes M. Pharmacokinetics and cardiovascular effects of ma-huang (Ephedra sinica) in normotensive adults. J Clin Pharmacol 1997;37:116-22.
838. Whittington CJ, Kendall T, Fonagy P, Cottrell D, Cotgrove A, Boddington E. Selective serotonin reuptake inhibitors in childhood depression: systematic review of published versus unpublished data. Lancet 2004;363:1341-5.
839. Wians FH Jr, Norton JT, Wirebaugh SR. False-positive serum tricyclic antidepressant screen with cyproheptadine. Clin Chem 1993;39: 1355-6.
840. Widmark EMP. Principles and applications of medicolegal alcohol determination. Davis, Calif: Biomedical Publications, 1981.
841. Wiegand RF, Klette KL, Stout PR, Gehlhausen JM. Comparison of EMIT II, CEDIA, and DPC RIA assays for the detection of lysergic acid diethylamide in forensic urine samples. J Anal Toxicol 2002;26:519-23.
842. Wiener K. A review of methods for plasma paracetamol estimation. Ann Clin Biochem 1978;15:187-96.
843. Williams AJ, Jones TW, Cooper JD. A rapid method for the determination of therapeutic barbiturate levels in serum using gas-liquid chromatography. Clin Chim Acta 1973;43:327-32.
844. Wilson IB, Ginsburg B. A powerful reactivator of alkylphosphate-inhibited acetylcholinesterase. Biochim Biophys Acta 1955;18:168-70.
845. Wilson IB, Ginsburg S. Reactivation of acetylcholinesterase inhibited by alkylphosphates. Arch Biochem 1955;54:569-71.
846. Winek CL, Carfagna M. Comparison of plasma, serum, and whole blood ethanol concentrations. J Anal Toxicol 1987;11:267-8.
847. Winek CL, Murphy KL. The rate and kinetic order of ethanol elimination. Forensic Sci Int 1984;25:159-66.
848. Winek CL, Paul LJ. Effect of short-term storage conditions on alcohol concentrations in blood from living human subjects. Clin Chem 1983;29:1959-60.
849. Wingert WE. Lowering cutoffs for initial and confirmation testing for cocaine and marijuana: large-scale study of effects on the rates of drug-positive results. Clin Chem 1997;43:100-3.
850. Wittwer E, Kern SE. Role of morphine’s metabolites in analgesia: concepts and controversies. AAPS J 2006;8:E348-52.
851. Wolfe TR, Caravati EM. Massive dextromethorphan ingestion and abuse. Am J Emerg Med 1995;13:174-6.
852. Wolff K, Hay A, Raistrick D. Methadone in saliva. Clin Chem 1991; 37:1297-8.
853. Wong EC, Koenig J, Turk J. Potential interference of cyclobenzaprine and norcyclobenzaprine with HPLC measurement of amitriptyline and nortriptyline: resolution by GC-MS analysis. J Anal Toxicol 1995;19:218-24.
854. Wood M, De Boeck G, Samyn N, Morris M, Cooper DP, Maes RA, De Bruijn EA. Development of a rapid and sensitive method for the quantitation of amphetamines in human plasma and oral fluid by LC-MS-MS. J Anal Toxicol 2003;27:78-87.
855. Woods JH, Katz JL, Winger G. Benzodiazepines: use, abuse, and consequences. Pharmacol Rev 1992;44:151-347.
856. Woods JH, Winger G. Abuse liability of flunitrazepam. J Clin Psychopharmacol 1997;17:1S-57S.

1188 SectionIII ■ Analytes
morphine, and its glucuronides by liquid chromatography—atmospheric pressure ionspray-mass spectrometry. J Anal Toxicol 1997;21:268-77.
877. Zuidema J, van Ginneken CA. Clearance concept in salivary drug excretion. Part I. Theory. Pharm Acta Helv 1983;58: 88-93.
878. Zvosec DL, Smith SW, McCutcheon JR, Spillane J, Hall BJ, Peacock EA. Adverse events, including death, associated with the use of 1,4-butanediol. N Engl J Med 2001;344:87-94.
879. Zwart A, van Kampen EJ, Zijlstra WG. Results of routine determination of clinically significant hemoglobin derivatives by multicomponent analysis. Clin Chem 1986;32:972-8.


















