Multiresolution Monogenic Signal Analysis Using the Riesz–Laplace Wavelet Transform

DIABETES AND PREGNANCY (M-F HIVERT AND CE POWE, SECTION EDITORS)
Clinical Management of Women with MonogenicDiabetes During Pregnancy
Laura T. Dickens1 & Rochelle N. Naylor1,2
# Springer Science+Business Media, LLC, part of Springer Nature 2018
AbstractPurpose of Review Monogenic diabetes accounts for 1–2% of all diabetes cases, but is frequently misdiagnosed as type 1,type 2, or gestational diabetes. Accurate genetic diagnosis directs management, such as no pharmacologic treatment forGCK-MODY, low-dose sulfonylureas for HNF1A-MODY and HNF4A-MODY, and high-dose sulfonylureas for KATP
channel-related diabetes. While diabetes treatment is defined for the most common causes of monogenic diabetes,pregnancy poses a challenge to management. Here, we discuss the key issues in pregnancy affected by monogenicdiabetes.Recent Findings General recommendations for pregnancy affected by GCK-MODY determine need for maternal insulintreatment based on fetal mutation status. However, a recent study suggests macrosomia and miscarriage rates may beincreased with this strategy. Recent demonstration of transplacental transfer of sulfonylureas also raises questions as towhen insulin should be initiated in sulfonylurea-responsive forms of monogenic diabetes.Summary Pregnancy represents a challenge in management of monogenic diabetes, where factors of maternal glycemiccontrol, fetal mutation status, and transplacental transfer of medication must all be taken into consideration. Guidelinesfor pregnancy affected by monogenic diabetes will benefit from large, prospective studies to better define the need forand timing of initiation of insulin treatment.
Keywords Monogenic diabetes . MODY . Glucokinase genemutation . Hepatocyte nuclear factor-1A . Pregnancy
Introduction
Monogenic diabetes refers to a heterogeneous group ofinherited forms of diabetes caused by mutations in genes in-volved in beta cell development, function, and regulation.These mutations result in beta cell dysfunction with abnormal-ities in glucose sensing or insulin secretion and lead to
diabetes [1]. Monogenic diabetes includes maturity-onset di-abetes of the young (MODY), neonatal diabetes, andsyndromic forms of diabetes. The most common form ofmonogenic diabetes is MODY, which is estimated to accountfor 1–2% of all diabetes worldwide [2]. However, MODY isfrequently misdiagnosed as types 1 or 2 diabetes, and studiesestimate that as many as 95% of MODY cases in the USA goundiagnosed [3]. It is important to distinguish MODY fromtypes 1 or 2 diabetes because targeted treatments for MODYcan improve glycemic control and reduce the burden of ther-apy. Furthermore, appropriate genetic diagnosis can identifyat-risk family members for genetic testing. Therapies for themost common subtypes of monogenic diabetes are wellestablished; however, there is less data about managementduring pregnancy. Pregnancy introduces several importantconsiderations for monogenic diabetes and a complex inter-play between maternal glycemic control and mutation statusof the fetus. Here, we will review the pathophysiology of thecommon forms of monogenic diabetes and recent evidenceand recommendations for management during pregnancy.
This article is part of the Topical Collection on Diabetes and Pregnancy
* Laura T. [email protected]
1 Department of Medicine, Section of Adult and PediatricEndocrinology, Diabetes, and Metabolism, University of Chicago,5841 S. Maryland Ave., MC 1027, Chicago, IL 60637, USA
2 Department of Pediatrics, Section of Adult and PediatricEndocrinology, Diabetes, and Metabolism, University of Chicago,Chicago, IL, USA
Current Diabetes Reports (2018) 18:12 https://doi.org/10.1007/s11892-018-0982-8

Pathophysiology and Clinical Presentationof Monogenic Diabetes
Four subtypes account for a majority of MODY cases with agenetic diagnosis: HNF1A, GCK, HNF4A, and HNF1B. Thefrequencies of these subtypes vary in different populations inpart due to differences in recruitment for genetic testing. Alarge study in the United Kingdom of 564 probands observedHNF1A mutations to be the most common (52%), followedby GCK (32%), HNF4A (10%), and HNF1B (6%) [4]. Anadditional class of monogenic diabetes which warrants discus-sion is persistent neonatal diabetes due to KATP channel mu-tations. We will begin by discussing the pathophysiology,clinical presentation, and recommended treatment outside ofpregnancy for the most common subtypes of monogenicdiabetes.
GCK-MODY
GCK-MODY is caused by mutations in the glucokinase gene,which catalyzes the conversion of glucose to glucose-6-phosphate and functions as the beta cell’s glucose sensor.This results in an increase set-point for glucose stimulatedinsulin release which manifests clinically with mild, stablefasting hyperglycemia that is present from birth (fasting glu-cose 98–150 mg/dl, HbA1c 5.6–7.6%) [5•]. Patients withGCK-MODY have low rates of clinically significant micro-vascular and macrovascular complications which are not dif-ferent from control populations. One study observed higherrates of retinopathy in patients with GCK-MODY (30% com-pared to 14% in controls and 63% with type 2 diabetes).However, this difference was exclusively due to backgroundretinopathy, and no patients with GCK-MODY required lasertherapy [6]. Treatment with oral hypoglycemic agents or in-sulin does not significantly change glycemic control [7].Therefore, treatment for GCK-MODY outside of pregnancyis not recommended.
HNF1A-MODY
HNF1A-MODY is caused by mutations in hepatocyte nuclearfactor 1-alpha, a transcription factor that regulates the tissue-specific expression of many genes in pancreatic islet cells andthe liver. Clinically, HNF1A-MODY presents in adolescenceor early adulthood with hyperglycemia, a large rise in 2-hglucose level on oral glucose tolerance test (OGTT, >90 mg/dL), and a lowered renal threshold for glucosuria dueto the role of HNF1A in SGLT2 gene expression [8, 9].Development of diabetic complications in HNF1A-MODYis strongly related to glycemic control. Older studies haveshown complications develop at similar frequency as patientswith types 1 and 2 diabetes [10]. However, the rate of micro-vascular complications and cardiovascular disease was shown
to be lower in a recent study of HNF1A-MODYin a dedicatedMODY clinic [11]. A distinguishing feature of HFN1A issensitivity to treatment with sulfonylureas, which are recom-mended as first line therapy. A majority of patients who havebeen previously treated with insulin can be transitioned offinsulin to sulfonylureas with equal or improved glycemic con-trol. Patients who are not able to successfully transition offinsulin tend to have longer duration of diabetes and have ex-perienced progressive loss of beta cell function [12]. Lowdoses of sulfonylureas are typically sufficient for treatment,and the recommended starting dose is one-fourth of typicaldoses for type 2 diabetes. Though sulfonylureas can remaineffective for many years, obesity and loss of beta cell functionover time can cause worsening glycemic control [11]. Secondline therapies such as meglitinides (nateglinide) and GLP1receptor agonists (liraglutide) have been shown to effectivelylower glucose in patients with HNF1A-MODY with lowerrates of hypoglycemia compared to sulfonylureas [13, 14].
HNF4A-MODY
HNF4A-MODY is caused by mutations in hepatocyte nuclearfactor 4-alpha, an upstream regulator of HNF1A transcriptionfactor. Clinical characteristics of HNF4A-MODY are similarto HNF1A-MODYand include progressive defects in insulinsecretion with presentation in adolescence or early adulthood.Patients with HNF4A-MODY may have large birth weightwith macrosomia in ~ 50% of affected babies and transientneonatal hypoglycemia due to fetal hyperinsulinism [8, 15,16]. In contrast, occurrence of fetal hyperinsulinism inHNF1A-MODY has only been described in rare cases [17].Diabetic complications occur at rates similar to types 1 and 2diabetes and are linked to glycemic control. Treatment withlow dose sulfonylureas is first line and is similarly effective asfor HNF1A-MODY.
HNF1B-MODY
HNF1B-MODY is caused by mutations in hepatocyte nu-clear factor 1-beta, a transcription factor expressed in em-bryonic development of the kidney, pancreas, liver, and GUtract. In addition to diabetes, HNF1B-MODY is associatedwith developmental renal disease (typically cystic and notdiabetes-related), genital tract malformations, abnormal liv-er function, hyperuricemia, and gout. Renal disease is par-ticularly common with a 66% incidence of renal cysts and86% incidence of renal impairment [18]. Patients withHNF1B-MODY have decreased insulin sensitivity com-pared to HNF1A-MODYpatients. Only rarely are sulfonyl-ureas successful; insulin treatment is required in the major-ity of cases for glycemic control [19, 20].
12 Page 2 of 9 Curr Diab Rep (2018) 18:12

KATP Channel Mutation Diabetes
Permanent neonatal diabetes is most commonly caused bymutations in the ATP-sensitive potassium channel includingactivating mutations in the genes KCNJ11 and ABCC8. Thesemutations result in failure of KATP channel closure and insuf-ficient beta cell insulin secretion [21, 22]. Sulfonylureas causeKATP channel closure through an ATP-independent mecha-nism and are effective treatments for a majority of patientswith KCNJ11 and ABCC8-related diabetes. In contrast toHNF1A- and HNF4A-MODY, high doses of sulfonylureasare typically required to treat patients with KATP channel dia-betes with average doses of 0.45 mg/kg/day [23].
Management of Monogenic Diabetesin Pregnancy
Table 1 summarizes the management of each subtype ofmonogenic diabetes during pregnancy including the effect offetal genotype, treatment and monitoring during pregnancy,and postpartum considerations.
GCK-MODY
Background
Prevalence of GCK-MODY in Pregnancy Several research stud-ies have focused on identification of GCK-MODY in womenwith gestational diabetes (GDM). Since all pregnant womenundergo glycemic screening, this is an opportunity to identifywomen with GCK-MODY who will require different treat-ment during and after pregnancy. The Atlantic Diabetes inPregnancy cohort estimated the prevalence of GCK-MODYto be 1% in gestational diabetes and identified body massindex (BMI) and fasting glucose to be useful clinical charac-teristics to distinguish GCK-MODY from GDM. In this pop-ulation, the combination of BMI less than 25 and fasting glu-cose > = 100 was 68% sensitive and 99% specific fordistinguishing GCK-MODY from GDMwith a number need-ed to undergo genetic testing of 2.7 to diagnose one case ofGCK-MODY [25•]. These screening cutoffs have been vali-dated in an Australian cohort with a similar sensitivity of 75%and specificity of 96.1%; however, concern was raised in thisstudy about the performance of these cutoffs in a multiethnicpopulation [26]. Additional research is ongoing to determinethe applicability of these parameters in the US multiethnicpopulation.
Influence of Fetal Genotype In GCK-MODY, pregnancy out-comes depend on whether the fetus has inherited the GCKmutation or not. The unaffected fetus of a mother withGCK-MODY will increase insulin secretion in response to
mild maternal hyperglycemia, resulting in increasedbirthweight and risk for macrosomia. In contrast, an affectedfetus of a mother with GCK-MODY would sense mild mater-nal hyperglycemia to be normal, and growth and birth weightwill be normal. Fetuses with paternally-inherited GCK muta-tions born to unaffected mothers have lower birth weight of ~400 g, and rates of weight below the 10th percentile are in-creased threefold [27]. In the case of GCK-unaffected off-spring, it has been suggested that insulin treatment to lowerglucose could reduce the risk of macrosomia and associatedcomplications. However, aggressive insulin therapy in womenwith GCK-MODY carrying an affected fetus may negativelyimpact fetal growth by decreasing fetal insulin secretion andthereby reducing insulin-mediated growth [27, 28].
The largest study to assess the effect of mutation status onbirth weight was carried out by Spyer et al. In this study of 98live births (genotype data was obtained in 82 offspring) in 42women with GCK-MODY, fetal birth weight was significant-ly greater in unaffected offspring compared to affected off-spring (3.9 versus 3.2 kg), and a higher rate of macrosomiawas seen in unaffected offspring compared to affected off-spring (39 versus 7%) [27]. In the aforementioned study,38% of mothers with GCK-MODY were treated with insulinwhich was initiated at variable times and in variable doses.There was no difference in birth weight of offspring betweeninsulin- and non-insulin-treated mothers or between fetal ge-notypes within treatment groups [27]. The authors suggest thatthe lack of effect may have been due to late timing of insulininitiation and relatively low doses of insulin, since it has beenshown that high doses of insulin up to 1 unit/kg/day are re-quired to normalize glucose in GCK-MODY women duringpregnancy.
Treatment/Management Recommendations
Based on these findings, current recommendations advise thatmanagement of GCK-MODY in pregnancy should be basedon fetal mutation status. Invasive sampling for the sole pur-pose of determining fetal genotype is not recommended [29].The emergence of non-invasive prenatal genetic testingthrough cell-free circulating DNA (cfDNA) extracted frommaternal plasma presents an intriguing option for determiningfetal genotype. However, to date, there have been no recom-mendations about the use of cfDNA in this setting. Thus,second trimester fetal growth scans are used to infer fetalgenotype. The recommendations state that insulin should notbe used pre-conception or in early pregnancy. Starting at 26-week gestation, fetal ultrasound should be performed every2 weeks to identify accelerated fetal growth as indicated byabdominal circumference > 75th percentile. If accelerated fe-tal growth is detected, this suggests that the fetus does notcarry a GCK mutation. Insulin therapy is recommended toreduce the risk ofmacrosomia, and delivery should be induced
Curr Diab Rep (2018) 18:12 Page 3 of 9 12
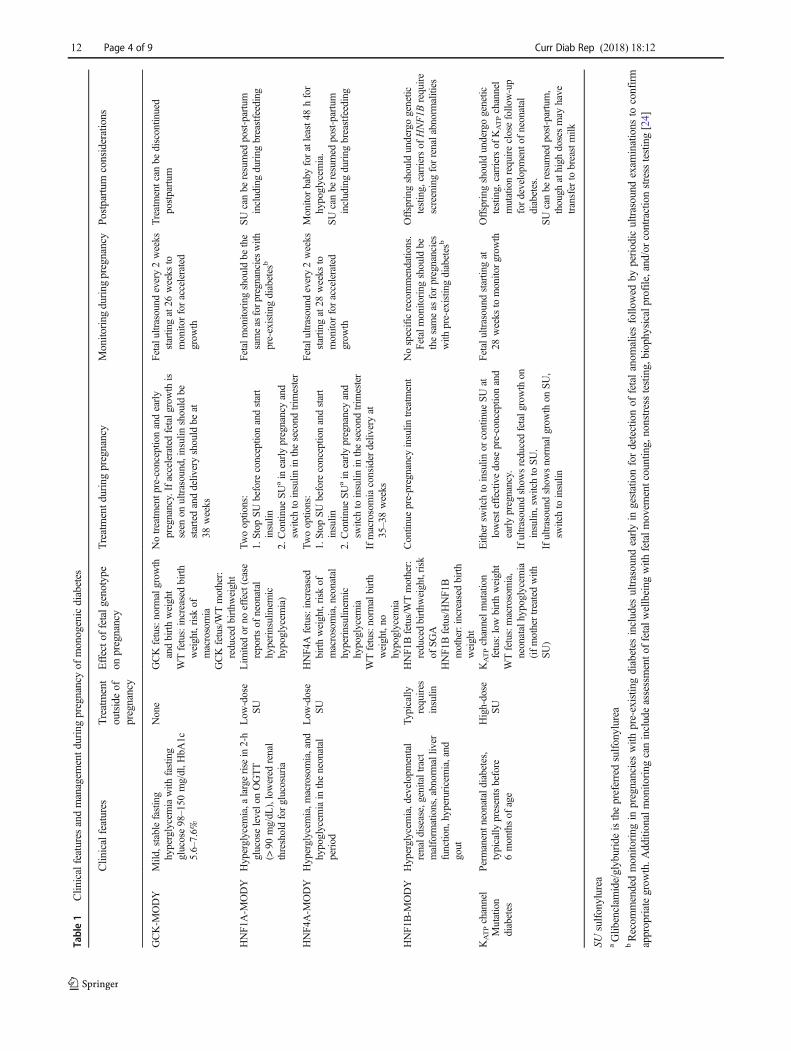
Table1
Clin
icalfeatures
andmanagem
entd
uringpregnancyof
monogenicdiabetes
Clin
icalfeatures
Treatment
outsideof
pregnancy
Effecto
ffetalg
enotype
onpregnancy
Treatmentd
uringpregnancy
Monito
ring
during
pregnancy
Postpartum
considerations
GCK-M
ODY
Mild
,stablefasting
hyperglycemiawith
fasting
glucose98–150
mg/dl,H
bA1c
5.6–7.6%
None
GCKfetus:norm
algrow
thandbirthweight
Notreatm
entp
re-conceptionandearly
pregnancy.Ifacceleratedfetalg
rowth
isseen
onultrasound,insulin
should
bestartedanddeliv
eryshould
beat
38weeks
Fetalultrasound
every2weeks
startin
gat26
weeks
tomonito
rforaccelerated
grow
th
Treatmentcan
bediscontinued
postpartum
WTfetus:increasedbirth
weight,risk
ofmacrosomia
GCKfetus/WTmother:
reducedbirthw
eight
HNF1
A-M
ODY
Hyperglycem
ia,a
largerise
in2-h
glucoselevelo
nOGTT
(>90
mg/dL
),lowered
renal
thresholdforglucosuria
Low
-dose
SULim
itedor
noeffect(case
reportsof
neonatal
hyperinsulinem
ichypoglycem
ia)
Twooptio
ns:
1.Stop
SUbefore
conceptio
nandstart
insulin
2.Continue
SUain
earlypregnancyand
switchto
insulin
inthesecond
trim
ester
Fetalm
onitoring
should
bethe
sameas
forpregnancies
with
pre-existin
gdiabetes
b
SUcanbe
resumed
post-partum
includingduring
breastfeeding
HNF4
A-M
ODY
Hyperglycem
ia,m
acrosomia,and
hypoglycem
iain
theneonatal
period
Low
-dose
SUHNF4
Afetus:increased
birthweight,risk
ofmacrosomia,neonatal
hyperinsulinem
ichypoglycem
ia
Twooptio
ns:
1.Stop
SUbefore
conceptio
nandstart
insulin
2.Continue
SUain
earlypregnancyand
switchto
insulin
inthesecond
trim
ester
Ifmacrosomiaconsider
deliveryat
35–38weeks
Fetalultrasound
every2weeks
startin
gat28
weeks
tomonito
rforaccelerated
grow
th
Monito
rbaby
foratleast4
8hfor
hypoglycem
ia.
SUcanbe
resumed
post-partum
includingduring
breastfeeding
WTfetus:norm
albirth
weight,no
hypoglycem
iaHNF1
B-M
ODY
Hyperglycem
ia,developmental
renald
isease,genitaltract
malform
ations,abnormalliv
erfunction,hyperuricemia,and
gout
Typically
requires
insulin
HNF1
Bfetus/WTmother:
reducedbirthw
eight,risk
ofSG
A
Continue
pre-pregnancyinsulin
treatm
ent
Nospecificrecommendatio
ns.
Fetalm
onitoring
should
bethesameas
forpregnancies
with
pre-existingdiabetes
b
Offspring
should
undergogenetic
testing,carriersof
HNF1B
require
screeningforrenalabnormalities
HNF1
Bfetus/HNF1
Bmother:increasedbirth
weight
KATPchannel
Mutation
diabetes
Perm
anentn
eonatald
iabetes,
typically
presentsbefore
6monthsof
age
High-dose
SUKATPchannelm
utation
fetus:lowbirthweight
Either
switchto
insulin
orcontinue
SUat
lowesteffectivedose
pre-conceptio
nand
earlypregnancy.
Ifultrasound
show
sreducedfetalgrowthon
insulin
,switchto
SU.
Ifultrasound
show
snorm
algrow
thon
SU,
switchto
insulin
Fetalu
ltrasound
startingat
28weeks
tomonitorgrow
thOffspring
should
undergogenetic
testing,carriersof
KATPchannel
mutationrequireclosefollo
w-up
fordevelopm
ento
fneonatal
diabetes.
SUcanbe
resumed
post-partum,
though
athigh
dosesmay
have
transfer
tobreastmilk
WTfetus:macrosomia,
neonatalhypoglycem
ia(ifmothertreatedwith
SU)
SUsulfonylurea
aGlib
enclam
ide/glyburideisthepreferredsulfonylurea
bRecom
mendedmonito
ring
inpregnancieswith
pre-existin
gdiabetes
includes
ultrasound
earlyin
gestationfordetectionof
fetalanom
aliesfollo
wed
byperiodic
ultrasound
exam
inations
toconfirm
appropriategrow
th.A
dditionalmonito
ring
canincludeassessmento
ffetalw
ellbeing
with
fetalm
ovem
entcounting,nonstresstesting,biophysicalp
rofile,and/orcontractionstress
testing[24]
12 Page 4 of 9 Curr Diab Rep (2018) 18:12

at 38 weeks. If no accelerated fetal growth is seen, the fetuscan be inferred to have inherited theGCKmutation and wouldnot be at risk for macrosomia, so no treatment is indicated [5•].In two cases where invasive sampling was indicated for otherreasons, inheritance of a GCK mutation was confirmed in thefetuses and neither mother received treatment for their hyper-glycemia. Birth weight was normal, and there were no peri-partum complications in the offspring [30]. Unfortunately, da-ta to support these recommendations are limited, and at leastone study raises questions regarding this approach.
A retrospective study by Bacon et al. in 2015 examined 56pregnancies in 12 women with GCK-MODY. In this cohort,insulin was used in 26.6% of pregnancies and initiated at anaverage of 14-week gestation. The GCK-unaffected offspringwhose mothers were treated with insulin had a lower rate ofmacrosomia of 33.3% compared with 62.5% in the non-insulin-treated GCK-unaffected group, though this differencewas not statistically significant. There was no SGA observedin insulin-treated, GCK-affected offspring (n = 3), and no sig-nificant adverse effects of insulin therapy were observed in-cluding no severe hypoglycemia [31••]. The authors conclud-ed that “the lack of available clinical studies…necessitates theuse of guidelines designed for the management of GDM”.They suggest that all women with GCK-MODY should betreated with insulin early during pregnancy since it is not clearif fetal genotype can be accurately predicted or if insulin ini-tiated late in gestation can prevent macrosomia. No prospec-tive studies about the use of ultrasound to predict fetal geno-type have been done, nor have studies specifically addressedthe efficacy of insulin in reducing macrosomia when initiatedin the late second/early third trimester according to currentrecommendations. The major limitations of this study are theretrospective nature and small sample size. Anecdotal experi-ence and unpublished data from the University of ChicagoMonogenic Diabetes Registry based on 130 pregnancies in55 women with GCK-MODY suggest that hypoglycemia, in-cluding severe hypoglycemia, may occur commonly duringpregnancy in women with GCK-MODY treated with insulinand requires consideration.
Currently, more data exist to support the use of fetal GCKmutation status (confirmed from genetic testing or inferredfrom fetal ultrasound) to direct management of maternal hy-perglycemia. Studies from GDM also offer support of thisapproach [32]. However, findings by Bacon et al. show theneed for additional studies.
Other Considerations
Miscarriage Rates in GCK-MODY Concerns have also beenraised about the effects of maternal hyperglycemia on miscar-riage rate and long-term glycemic control in offspring. Thesame retrospective study by Bacon et al. discussed above ob-served a higher rate of miscarriages in women with GCK-
MODY of 33%, compared to 14% in women with HNF1A-MODY and a background population rate of 15%.Miscarriages in women with GCK-MODY tended to occurearly in gestation at an average of 7.5 weeks [31••]. This studyis the first to raise the concern about increased miscarriagewith GCK-MODY, and confirmation of these findings isneeded. Unpublished data from the University of ChicagoMonogenic Diabetes Registry observed a miscarriage rate of18% in a total of 130 pregnancies in women with GCK-MODY, which is not significantly different from backgroundpopulation miscarriage rates.
Long-Term Outcomes in Offspring Data show that despite in-crease in birth weight of children born to mothers with GCK-MODY, there appear to be no long-term effects of exposure tomild maternal hyperglycemia in utero. A study in 2007showed no evidence of impaired glucose tolerance onOGTT or reduced beta cell function in unaffected offspringof GCK-MODY mothers compared to the control group ofoffspring of GCK-MODY fathers [33].
HNF1A-MODY
Background
In pregnancies affected by HNF1A-MODY, maternal glyce-mic control is the major determinant of fetal outcomes. In themajority of cases, fetal inheritance of HNF1A mutations doesnot result in increased birth weight or incidence of hypogly-cemia [15]. However, there are case reports of congenital hy-perinsulinism due to HNF1A mutations associated withMODY [17]. Though optimal treatment of HNF1A-MODYoutside of pregnancy is with sulfonylureas, there is limiteddata about best management during pregnancy. The sulfonyl-urea glyburide (glibenclamide) has been commonly used totreat gestational diabetes, and previous publications suggestedthis would be reasonable treatment for HNF1A-MODY dur-ing pregnancy [29]. However, recent studies have shown thatglyburide crosses the placenta and can be measured in fetalumbilical venous samples. In one study, a majority (79%) ofumbilical vein glyburide levels were below the typical limit ofdetection of 10 ng/mL, though in 37% of cases, the fetalglyburide levels were higher than maternal levels [34•].Another study using an LC/MS assay found umbilical cordplasma glyburide concentrations to average 70% of maternalblood concentrations [35]. In addition to these findings thattransplacental glyburide transfer does occur, evidence has alsoshown that glyburide may negatively impact fetal and neona-tal outcomes. A meta-analysis in 2014 found significant in-crease in the risk of macrosomia (RR 3.07) and neonatal hy-poglycemia (RR 2.30) in pregnant women with gestationaldiabetes treated with glyburide compared with insulin treat-ment [36]. These findings have led some to suggest that
Curr Diab Rep (2018) 18:12 Page 5 of 9 12

sulfonylureas should be avoided in pregnancies with mono-genic diabetes, particularly in the third trimester when risk forfetal hyperinsulinism and macrosomia is greatest. In light ofthese recent data, insulin therapy appears to be the most con-servative approach.
Treatment and Monitoring Recommendations
Recent recommendations for management of HNF1A-MODY pregnancies attempt to balance the risk of uncon-trolled hyperglycemia during the first trimester at the time oforganogenesis with the risk for macrosomia and neonatal hy-poglycemia with sulfonylurea therapy in the third trimester.These recommendations present two potential managementoptions: stop sulfonylurea before pregnancy and switch toinsulin or continue sulfonylurea treatment pre-conceptionand in early pregnancy and switch to insulin in the secondtrimester. The second option is suggested only for patientswith excellent pre-pregnancy glycemic control on sulfonyl-ureas. If a woman on a sulfonylurea presents during earlypregnancy, the risk of deteriorating glycemic control duringorganogenesis should be considered when determining treat-ment strategy. If glycemic control is excellent on sulfonylureatherapy, it may be reasonable to continue sulfonylureas untilthe end of the first trimester and then switch to insulin [37••].
Glyburide is specifically recommended as the sulfonylureaof choice if this class of medication is used because it has beenmost extensively studied in pregnancy. Patients on alternativesulfonylureas who are continuing on sulfonylureas in preg-nancy should be switched to equivalent doses of glyburide.To have transition from glyburide to insulin, first basal insulinshould be initiated, and then bolus insulin added, andglyburide discontinued. This transition should be completedprior to 26-week gestation. In exceptional circumstanceswhere glyburide is continued in the third trimester, the lowestdose that provides adequate glycemic control should be used.Fetal monitoring in pregnancies with HNF1A-MODY shouldbe the same as pregnancies with pre-existing diabetes [37••].After delivery, glyburide can be resumed and continued dur-ing breastfeeding. Studies have shown that low-doseglyburide is not excreted in breast milk nor is it associatedwith neonatal hypoglycemia [38].
HNF4A-MODY
Background
In contrast to HNF1A-MODY, fetal outcomes in HNF4A-MODY pregnancies are highly dependent upon fetal geno-type. Offspring who inherit HNF4A mutations demonstratesmedian increases in birthweight of 790 g compared to wildtype family members. Incidence of macrosomia is significant-ly increased in HNF4A mutation carriers at 56% compared to
13% in wild type family members, and neonatalhyperinsulinemic hypoglycemia has been seen in 15% ofHFN4A mutation carriers [15]. There is an additive effect ofmaternal hyperglycemia on rates of macrosomia wherebyHNF4A mutation carriers born to HNF4A-affected mothershave increased birth weight compared to HNF4A mutationcarriers who inherited the mutation from their father.
Treatment and Monitoring Recommendations
In light of these significant risks of fetal and neonatal morbid-ity related to macrosomia and hypoglycemia, tight maternalglycemic control is critical. No treatments have been shown toimprove fetal outcomes or macrosomia in HNF4A-MODYpregnancies. The therapeutic strategies are the same as previ-ously described for HNF1A-MODY pregnancies: stop sulfo-nylurea before pregnancy and switch to insulin or continuesulfonylurea treatment before and in early pregnancy andswitch to insulin in the second trimester [37••]. Fetal monitor-ing during pregnancy with HNF4A-affected mothers shouldinclude ultrasounds for growth assessment at least every2 weeks starting at 28 weeks. If macrosomia is detected, in-duction of labor or elective cesarean section should be consid-ered at 35 to 38 weeks. After delivery, the baby should bemonitored for at least 48 h for hypoglycemia. Postpartumand during breastfeeding, glyburide can be resumed.Recommendations are similar for pregnancies with HNF4A-affected fathers and include ultrasound monitoring beginningat 28 weeks with consideration of early delivery at 37 weeksbased on fetal size and close monitoring for neonatal hypo-glycemia [37••].
HNF1B-MODY
There is very limited data published about pregnancies affect-ed by HNF1B-MODY. Patients with HNF1B-MODY willtypically require insulin for glycemic control and should con-tinue this during pregnancy. The effects of maternal treatmenton fetal outcomes have not been studied, though one smallcohort study did demonstrate a significant impact of maternal/fetal genotype on birth weight. Babies with HNF1B-MODYborn to unaffected mothers had significantly reducedbirthweight, and 69% incidence of SGA while birth weightwas increased in babies with HNF1B-MODY born to motherswith HNF1B-MODYand diabetes [39]. Offspring of affectedparents should undergo genetic testing to identify carriers ofthe HNF1B mutation for close follow-up and screening forrenal abnormalities.
KATP Channel Mutation Diabetes
Background In pregnancies affected by KATP channel mutationdiabetes, fetal birth weight also depends on fetal genotype.
12 Page 6 of 9 Curr Diab Rep (2018) 18:12

Offspring who inherit thesemutations from either their mother orfather will have reduced fetal insulin secretion resulting in lowbirth weight [40]. Outside of pregnancy, treatment with high-dose sulfonylureas is standard of care; however, during pregnan-cy the benefit of sulfonylurea treatment depends on fetal geno-type. If the fetus has inherited the KATP channel mutation, casereports indicate that sulfonylurea treatment will restore fetal in-sulin secretion and result in normal birth weight [41]. If the fetushas not inherited the mutation, sulfonylurea treatment can resultin excess fetal insulin secretion, macrosomia, and neonatal hy-poglycemia [42]. Fetal genotyping can be performed using cho-rionic villous sampling or amniocentesis, though these invasiveprocedures carry a risk of miscarriage. Non-invasive cfDNA is apromising option for prenatal genetic testing. One case reportdescribed effective use of cfDNA for prenatal genetic testingfor a fetus at risk of inheriting a KCNJ11 mutation causing per-manent neonatal diabetes [43]. Further study is needed beforethis new technology can be routinely used in this setting. Currentrecommendations advise that fetal genotype be inferred fromserial ultrasound growth monitoring starting at 28 weeks.Reduced growth indicates the fetus has inherited the KATP chan-nel mutation while normal growth suggests the fetus is wild type.
Treatment and Monitoring Recommendations Maternal treat-ment should again take into consideration the risk of adverseeffects from deteriorating glycemic control during critical timesin fetal development and the risk of macrosomia and neonatalhypoglycemia in an unaffected offspring exposed to high-dosesulfonylurea treatment. Recommendations advise that it is rea-sonable to switch to insulin prior to conception or to continuesulfonylurea treatment through the first trimester at the lowestdose possible to maintain good glycemic control with A1c <6.5. If ultrasound scans show reduced fetal growth, suggestingthe fetus has inherited the KATP channel mutation, then glyburideis the treatment of choice and should be continued orreintroduced if the patient was switched to insulin previously. Ifultrasound scans show normal fetal growth, transfer fromglyburide to insulin is advised to avoid excessive fetal growth,macrosomia, and neonatal hypoglycemia [37••]. After delivery,fetal genetic testing should be completed if it was not done duringpregnancy. If the offspring has inherited the KATP channel muta-tion, neonatal diabetes will typically present before 6 months ofage and close follow-up with a pediatrician is recommended.Maternal glyburide treatment can be resumed after delivery andis generally considered to be safe during breastfeeding, though athighmaternal doses theremay be excretion of glyburide to breastmilk [42].
Conclusions
This review summarizes available evidence and recommenda-tions for management of monogenic diabetes in pregnancy.
These recommendations are based on limited data due to mul-tiple challenges for controlled studies in this field. These di-agnoses are relatively rare, and many women do not have agenetic diagnosis at the time of pregnancy and are treated astype 1, type 2, or gestational diabetes. Thus, data about preg-nancy monitoring, effects of therapy, and outcomes are pri-marily retrospective. The available evidence does highlightthe importance of understanding the impact of fetal genotypeon in utero growth and the effect of maternal glycemic control.Prospective studies in pregnancies affected by GCK-MODYcan help to determine if ultrasound monitoring is indeed aneffective tool for inferring fetal genotype and making treat-ment decisions. Furthermore, the effects of maternal hypergly-cemia on miscarriage rates require further study to confirmand better understand this observation. The recent data ontransplacental transfer of sulfonylurea represents a paradigmshift from prior thinking about management of HNF1A/HNF4A and KATP channel diabetes during pregnancy, sug-gesting insulin therapy may be the preferred therapy.Theoretically, the use of non-invasive cell free DNA for pre-natal genotyping could help to clarify management strategiesmoving forward for many forms of monogenic diabetes.
Funding information This work was supported by the National Instituteof Diabetes and Digestive and Kidney Diseases of the National Institutesof Health (grant numbers R01 DK104942, P30 DK020595) and theCTSA (grant number UL1 TR002389).
Compliance with Ethical Standards
Conflict of Interest Laura T. Dickens and Rochelle N. Naylor declarethat they have no conflict of interest.
Human and Animal Rights and Informed Consent This article containsunpublished data from retrospective studies with human subjects per-formed by Laura T. Dickens and Rochelle N. Naylor. Informed consentwas obtained from all subjects.
ReferencesPapers of particular interest, published recently, have beenhighlighted as:• Of importance•• Of major importance
1. Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clin-ical pathophysiology of maturity-onset diabetes of the young. NEngl J Med. 2001;345(13):971–80. https://doi.org/10.1056/NEJMra002168.
2. Ledermann HM. Is maturity onset diabetes at young age (MODY)more common in Europe than previously assumed? Lancet.1995;345(8950):648. https://doi.org/10.1016/S0140-6736(95)90548-0.
3. Kleinberger JW, Pollin TI. Undiagnosed MODY: time for action.Curr Diab Rep. 2015;15(12):110. https://doi.org/10.1007/s11892-015-0681-7.
Curr Diab Rep (2018) 18:12 Page 7 of 9 12

4. Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT,Ellard S.Maturity-onset diabetes of the young (MODY): howmanycases are we missing? Diabetologia. 2010;53(12):2504–8. https://doi.org/10.1007/s00125-010-1799-4.
5.• Chakera AJ, Steele AM, Gloyn AL, Shepherd MH, Shields B,Ellard S, et al. Recognition and management of individuals withhyperglycemia because of a heterozygous glucokinase mutation.Diabetes Care. 2015;38(7):1383–92. https://doi.org/10.2337/dc14-2769. An important article summarizing recommendations formanagement of GCK-MODY in pregnancy based on suspectedfetal genotype.
6. Steele AM, Shields BM, Wensley KJ, Colclough K, Ellard S,Hattersley AT. Prevalence of vascular complications among pa-tients with glucokinase mutations and prolonged, mild hyperglyce-mia. JAMA. 2014;311(3):279–86. https://doi.org/10.1001/jama.2013.283980.
7. Stride A, Shields B, Gill-Carey O, Chakera AJ, ColcloughK, EllardS, et al. Cross-sectional and longitudinal studies suggest pharmaco-logical treatment used in patients with glucokinase mutations doesnot alter glycaemia. Diabetologia. 2014;57(1):54–6. https://doi.org/10.1007/s00125-013-3075-x.
8. Colclough K, Bellanne-Chantelot C, Saint-Martin C, Flanagan SE,Ellard S. Mutations in the genes encoding the transcription factorshepatocyte nuclear factor 1 alpha and 4 alpha in maturity-onsetdiabetes of the young and hyperinsulinemic hypoglycemia. HumMutat. 2013;34(5):669–85. https://doi.org/10.1002/humu.22279.
9. Pontoglio M, Prié D, Cheret C, Doyen A, Leroy C, Froguel P, et al.HNF1alpha controls renal glucose reabsorption in mouse and man.EMBO Rep. 2000;1(4):359–65. https://doi.org/10.1093/embo-reports/kvd071.
10. Isomaa B,HenricssonM, LehtoM, ForsblomC,Karanko S, SarelinL, et al. Chronic diabetic complications in patients with MODY3diabetes. Diabetologia. 1998;41(4):467–73. https://doi.org/10.1007/s001250050931.
11. Bacon S, Kyithar MP, Rizvi SR, Donnelly E, McCarthy A, BurkeM, et al. Successful maintenance on sulphonylurea therapy and lowdiabetes complication rates in a HNF1A-MODY cohort. DiabetMed. 2016;33(7):976–84. https://doi.org/10.1111/dme.12992.
12. Shepherd M, Shields B, Ellard S, Rubio-Cabezas O, Hattersley AT.A genetic diagnosis of HNF1A diabetes alters treatment and im-proves glycaemic control in the majority of insulin-treated patients.Diabet Med. 2009;26(4):437–41. https://doi.org/10.1111/j.14645491.2009.02690.x.
13. Tuomi T, Honkanen EH, Isomaa B, Sarelin L, Groop LC. Improvedprandial glucose control with lower risk of hypoglycemia withnateglinide than with glibenclamide in patients with maturity-onset diabetes of the young type 3. Diabetes Care. 2006;29(2):189–94. https://doi.org/10.2337/diacare.29.02.06.dc05-1314.
14. Østoft SH, Bagger JI, Hansen T, Pedersen O, Faber J, Holst JJ, et al.Glucose-lowering effects and low risk of hypoglycemia in patientswith maturity-onset diabetes of the young when treated with a GLP-1 receptor agonist: a double-blind, randomized, crossover trial.Diabetes Care. 2014;37(7):1797–805. https://doi.org/10.2337/dc13-3007.
15. Pearson ER, Boj SF, Steele AM, Barrett T, Stals K, Shield JP, et al.Macrosomia and hyperinsulinaemic hypoglycaemia in patients withheterozygous mutations in the HNF4A gene. PLoS Med.2007;4(4):e118. https://doi.org/10.1371/journal.pmed.0040118.
16. Pingul MM, Hughes N, Wu A, Stanley CA, Gruppuso PA.Hepatocyte nuclear factor 4α gene mutation associated with famil-ial neonatal hyperinsulinism and maturity-onset diabetes of theyoung. J Pediatr. 2011;158(5):852–4. https://doi.org/10.1016/j.jpeds.2011.01.003.
17. Stanescu DE, Hughes N, Kaplan B, Stanley CA, De León DD.Novel presentations of congenital hyperinsulinism due tomutationsin the MODY genes: HNF1A and HNF4A. J Clin Endocrinol
Metab. 2012;97(10):E2026–30. https://doi.org/10.1210/jc.2012-1356.
18. Bingham C, Hattersley AT. Renal cysts and diabetes syndromeresulting from mutations in hepatocyte nuclear factor-1beta.Nephrol Dial Transplant. 2004;19(11):2703–8. https://doi.org/10.1093/ndt/gfh348.
19. Pearson ER, Badman MK, Lockwood CR, Clark PM, Ellard S,Bingham C, et al. Contrasting diabetes phenotypes associated withhepatocyte nuclear factor-1alpha and -1beta mutations. DiabetesCare. 2004;27(5):1102–7. https://doi.org/10.2337/diacare.27.5.1102.
20. Dubois-Laforgue D, Cornu E, Saint-Martin C, Coste J, Bellanné-Chantelot C, Timsit J, et al. Diabetes, associated clinical Spectrum,long-term prognosis, and genotype/phenotype correlations in 201adult patients with hepatocyte nuclear factor 1B (HNF1B) molecu-lar defects. Diabetes Care. 2017;40(11):1436–43. https://doi.org/10.2337/dc16-2462.
21. Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ,Slingerland AS, et al. Activating mutations in the gene encodingthe ATP-sensitive potassium-channel subunit Kir6.2 and perma-nent neonatal diabetes. N Engl J Med. 2004;350(18):1838–49.Erratum in: N Engl J Med. 2004;351(14):1470. https://doi.org/10.1056/NEJMoa032922.
22. Babenko AP, Polak M, Cavé H, Busiah K, Czernichow P,Scharfmann R, et al. Activating mutations in the ABCC8 gene inneonatal diabetes mellitus. N Engl J Med. 2006;355(5):456–66.https://doi.org/10.1056/NEJMoa055068.
23. Pearson ER, Flechtner I, Njølstad PR, Malecki MT, Flanagan SE,Larkin B, et al. Switching from insulin to oral sulfonylureas inpatients with diabetes due to Kir6.2 mutations. N Engl J Med.2006;355(5):467–77. https://doi.org/10.1056/NEJMoa061759.
24. ACOG Committee on Practice Bulletins, ACOG Practice Bulletin.Clinical management guidelines for obstetrician-gynecologists.Number 60, March 2005. Pregestational diabetes mellitus. ObstetGynecol. 2005;105(3):675–85.
25.• Chakera AJ, Spyer G, Vincent N, Ellard S, Hattersley AT, DunneFP. The 0.1% of the population with glucokinase monogenic dia-betes can be recognized by clinical characteristics in pregnancy: theAtlantic diabetes in pregnancy cohort. Diabetes Care. 2014;37(5):1230–6. https://doi.org/10.2337/dc13-2248. A study showing thatanthropometric and glycemic data can be used to differentiatepatients with GCK-MODY from gestational diabetes.
26. Rudland VL, Hinchcliffe M, Pinner J, Cole S, Mercorella B,Molyneaux L, et al. Identifying glucokinase monogenic diabetesin a multiethnic gestational diabetes mellitus cohort: new pregnan-cy screening criteria and utility of HbA1c. Diabetes Care.2016;39(1):50–2. https://doi.org/10.2337/dc15-1001.
27. Spyer G, Macleod KM, Shepherd M, Ellard S, Hattersley AT.Pregnancy outcome in patients with raised blood glucose due to aheterozygous glucokinase gene mutation. Diabet Med. 2009;26(1):14–8. https://doi.org/10.1111/j.1464-5491.2008.02622.x.
28. Spyer G, Hattersley AT, Sykes JE, Sturley RH, MacLeod KM.Influence of maternal and fetal glucokinase mutations in gestationaldiabetes. Am Obstet Gynecol. 2001;185(1):240–1. https://doi.org/10.1067/mob.2001.113127.
29. Colom C, Corcoy R. Maturity onset diabetes of the young andpregnancy. Best Pract Res Clin Endocrinol Metab. 2010;24(4):605–15. https://doi.org/10.1016/j.beem.2010.05.008.
30. Chakera AJ, Carleton VL, Ellard S,Wong J, YueDK, Pinner J, et al.Antenatal diagnosis of fetal genotype determines if maternal hyper-glycemia due to a glucokinase mutation requires treatment.Diabetes Care. 2012;35(9):1832–4. https://doi.org/10.2337/dc12-0151.
31.•• Bacon S, Schmid J, McCarthy A, Edwards J, Fleming A, KinsleyB, et al. The clinical management of hyperglycemia in pregnancycomplicated by maturity-onset diabetes of the young. Am J Obstet
12 Page 8 of 9 Curr Diab Rep (2018) 18:12

Gynecol. 2015;213(2):236.e1–7. https://doi.org/10.1016/j.ajog.2015.04.037.A retrospective study of GCK-MODY in pregnan-cy that observed a trend towards lower rates of macrosomia inGCK-unaffected offspring with insulin treatment of the motherand higher miscarriage rates in GCK-MODYpregnancies.
32. Kjos SL, Schaefer-Graf U, Sardesi S, Peters RK, Buley A, XiangAH, et al. A randomized controlled trial using glycemic plus fetalultrasound parameters versus glycemic parameters to determine in-sulin therapy in gestational diabetes with fasting hyperglycemia.Diabetes Care. 2001;24(11):1904–10. https://doi.org/10.2337/diacare.24.11.1904.
33. Singh R, Pearson ER, Clark PM, Hattersley AT. The long-termimpact on offspring of exposure to hyperglycaemia in utero dueto maternal glucokinase gene mutations. Diabetologia.2007;50(3):620–4. https://doi.org/10.1007/s00125-006-0541-8.
34.• Schwartz RA, Rosenn B, Aleksa K, Koren G. Glyburide transportacross the human placenta. Obstet Gynecol. 2015;125(3):583–8.https://doi.org/10.1097/AOG.0000000000000672. A studyshowing transplacental glyburide transfer does occur and ishighly variable between patients.
35. Hebert MF, Ma X, Naraharisetti SB, Krudys KM, Umans JG,Hankins GD, et al. Obstetric-fetal pharmacology research unit net-work. Are we optimizing gestational diabetes treatment withglyburide? The pharmacologic basis for better clinical practice.Clin Pharmacol Ther. 2009;85(6):607–14. https://doi.org/10.1038/clpt.2009.5.
36. Poolsup N, Suksomboon N, Amin M. Efficacy and safety of oralantidiabetic drugs in comparison to insulin in treating gestationaldiabetes mellitus: a meta-analysis. PLoSOne. 2014;9(10):e109985.https://doi.org/10.1371/journal.pone.0109985.
37.•• Shepherd M, Brook AJ, Chakera AJ, Hattersley AT. Managementof sulfonylurea-treated monogenic diabetes in pregnancy: implica-tions of placental glibenclamide transfer. Diabet Med. 2017;34(10):
1332–9. https://doi.org/10.1111/dme.13388. An important articleproviding recommendations for management of sulfonylurea-treated monogenic diabetes in pregnancy.
38. Feig DS, Briggs GG, Kraemer JM, Ambrose PJ, Moskovitz DN,Nageotte M, et al. Transfer of glyburide and glipizide into breastmilk. Diabetes Care. 2005;28(8):1851–5. https://doi.org/10.2337/diacare.28.8.1851.
39. Edghill EL, Bingham C, Slingerland AS, Minton JA, Noordam C,Ellard S, et al. Hepatocyte nuclear factor-1 beta mutations causeneonatal diabetes and intrauterine growth retardation: support fora critical role of HNF-1beta in human pancreatic development.Diabet Med. 2006;23(12):1301–6. https://doi.org/10.1111/j.1464-5491.2006.01999.x.
40. Slingerland AS, Hattersley AT. Activating mutations in the geneencoding Kir6.2 alter fetal and postnatal growth and also causeneonatal diabetes. J Clin Endocrinol Metab. 2006;91(7):2782–8.https://doi.org/10.1210/jc.2006-0201.
41. Gaal Z, Klupa T, Kantor I, Mlynarski W, Albert L, Tolloczko J,et al. Sulfonylurea use during entire pregnancy in diabetes becauseof KCNJ11 mutation: a report of two cases. Diabetes Care.2012;35(6):e40. https://doi.org/10.2337/dc12-0163.
42. Myngheer N, Allegaert K, Hattersley A, McDonald T, Kramer H,Ashcrof t FM, et al . Fetal macrosomia and neonatalhyperinsulinemic hypoglycemia associated with transplacentaltransfer of sulfonylurea in a mother with KCNJ11-related neonataldiabetes. Diabetes Care. 2014;37(12):3333–5. https://doi.org/10.2337/dc14-1247.
43. De Franco E, Caswell R, Houghton JA, Iotova V, Hattersley AT,Ellard S. Analysis of cell-free fetal DNA for non-invasive prenataldiagnosis in a family with neonatal diabetes. Diabet Med.2017;34(4):582–5. https://doi.org/10.1111/dme.13180.
Curr Diab Rep (2018) 18:12 Page 9 of 9 12